 Shogo Sunaga
Shogo Sunaga Junya Tsunoda
Junya Tsunoda Toshiaki Teratani1
Toshiaki Teratani1 Yohei Mikami
Yohei Mikami Takanori Kanai
Takanori Kanai- 1Division of Gastroenterology and Hepatology, Department of Internal Medicine, Keio University School of Medicine, Shinjuku-ku, Tokyo, Japan
- 2Department of Surgery, Keio University School of Medicine, Tokyo, Japan
- 3AMED-CREST, Japan Agency for Medical Research and Development, Tokyo, Japan
Group 2 innate lymphoid cells (ILC2s) were identified in 2010 as a novel lymphocyte subset lacking antigen receptors, such as T-cell or B-cell receptors. ILC2s induce local immune responses characterized by producing type 2 cytokines and play essential roles for maintaining tissue homeostasis. ILC2s are distributed across various organs, including the intestine where immune cells are continuously exposed to external antigens. Followed by luminal antigen stimulation, intestinal epithelial cells produce alarmins, such as IL-25, IL-33, and thymic stromal lymphopoietin, and activate ILC2s to expand and produce cytokines. In the context of parasite infection, the tuft cell lining in the epithelium has been revealed as a dominant source of intestinal IL-25 and possesses the capability to regulate ILC2 homeostasis. Neuronal systems also regulate ILC2s through neuropeptides and neurotransmitters, and interact with ILC2s bidirectionally, a process termed “neuro-immune crosstalk”. Activated ILC2s produce type 2 cytokines, which contribute to epithelial barrier function, clearance of luminal antigens and tissue repair, while ILC2s are also involved in chronic inflammation and tissue fibrosis. Recent studies have shed light on the contribution of ILC2s to inflammatory bowel diseases, mainly comprising ulcerative colitis and Crohn’s disease, as defined by chronic immune activation and inflammation. Modern single-cell analysis techniques provide a tissue-specific picture of ILC2s and their roles in regulating homeostasis in each organ. Particularly, single-cell analysis helps our understanding of the uniqueness and commonness of ILC2s across tissues and opens the novel research area of ILC2 heterogeneity. ILC2s are classified into different phenotypes depending on tissue and phase of inflammation, mainly inflammatory and natural ILC2 cells. ILC2s can also switch phenotype to ILC1- or ILC3-like subsets. Hence, recent studies have revealed the heterogeneity and plasticity of ILC2, which indicate dynamicity of inflammation and the immune system. In this review, we describe the regulatory mechanisms, function, and pathological roles of ILC2s in the intestine.
Introduction
The intestine is one of the largest organs continually exposed to the external environment and it harbors an immune system to protect the host from pathobionts (1). Innate lymphoid cells (ILCs) are newly classified lymphocyte subsets that serve as a frontline defense, particularly in the mucosal tissues (2, 3). Unlike T- and B-cells, ILCs do not express adaptive antigen recognition receptors, and as such their expansion and activation are not driven in an antigen-specific manner, but rather by cytokine signals in the local microenvironment in each tissue. Although ILCs cannot induce antigen-specific reactions, they quickly respond to external antigen from the local microenvironment and rapidly produce various cytokines including interleukins (IL) and interferon (IFN) to maintain tissue homeostasis. ILCs are classified into three groups based on lineage-determining transcription factors and cytokine production, mirroring T helper cell subsets (2, 3). Group 2 ILCs (ILC2s) require transcription factors GATA3 (4, 5) and RORα (6, 7) for differentiation and produce signature “Type 2” cytokines, such as IL-4, IL-5, IL-9, and IL-13, as well as IL-6, IL-10, IL-17, and amphiregulin (AREG) (8–12). ILC2s were first reported as natural helper cells, nuocytes, and innate type 2 helper cells, and were detected in mesenteric adipose tissue, mesenteric lymph nodes, spleen, liver, lung, and small intestine (13–15). More recently, ILC2s have been found in various organs that are confronted with external antigen, such as the intestine, respiratory system, and skin, and also in those that are not continually challenged, such as liver, heart, muscle, and brain (16). Recent advancement of single cell omics and mass cytometry technologies have revealed that ILC2s possess tissue-specific phenotypes and contribute to the tissue-specific regulation of inflammation, allergic immunity, parasite infection, metabolism, and tissue repair (16–19).
In the intestine, epithelial cells respond to bacteria, parasites, and allergen within the intestinal lumen, and produce alarmins, such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), which subsequently activate ILC2s to proliferate and produce cytokines (16). Recent studies have reported abundant regulation of ILC2s beyond alarmins, including neuro-immune crosstalk, which is mediated by neurotransmitters and cytokines. Activation of tissue-resident ILC2s causes not only local inflammation but also subsequent tissue remodeling and organ fibrosis associated with intestinal chronic inflammatory conditions, such as inflammatory bowel disease (IBD), including ulcerative colitis (UC) or Crohn’s disease (CD) (17). However, negative clinical trial results regarding targeting type 2 immune responses have encouraged us to explore the complexity of ILC2 and other type 2 immune cells, and their cytokine production. New technologies, including single-cell analysis, have been used to better decipher the functions and heterogeneity of ILC2s. Initially, ILC2s were thought to have roles in defending against parasitic infection and promoting allergic pathology (20), whereas studies of ILC2s in the context of IBD are developing. In this review, we focus on the roles of ILC2s in the intestine and discuss their regulation, neuroimmunology, fibrosis, and contribution to IBD.
Regulation of ILC2
ILC2s are localized in the lamina propria below the epithelial layer and are activated following epithelial damage by parasites and allergens in the mucosal tissues. This process is mediated by alarmins, such as IL-25, IL-33, and TSLP, which initially activate expansion and cytokine production in ILC2s when triggered by mucosal barrier damage (Figure 1). ILC2s responding to IL-33 produce the growth factor AREG, which binds to epithelium-expressed epidermal growth factor receptor (11). AREG has a critical role for epithelial cell proliferation and differentiation through the epidermal growth factor receptor pathway (21, 22). A recent study demonstrated that secretion of IL-33 was significantly accelerated in the colons of mice treated with dextran sulfate sodium (DSS) and injecting recombinant murine IL-33 improved epithelial damage, pro-inflammatory cytokine secretion, and loss of barrier function in DSS-induced colitic mice (23). Anti-colitic effect of IL-33 was observed in RAG2-/- or diphtheria toxin-treated DEREG mice where whole T cells or Tregs are depleted respectively (23). This suggests that ILC2 has significant roles in anti-colitic effect upon stimulation of IL-33 which is also known to enhance suppressive function of Foxp3+ regulatory T cells (Tregs) through its receptor ST2 (24) or stimulate CD103+ dendritic cells (DCs) to produce IL-2 and expand Tregs (25). In the small intestine, tuft cells, which exist in the epithelial layer of the intestinal tract and project microvilli into the lumen, produce IL-25 to sustain ILC2 homeostasis in the resting lamina propria (26). Together with high expression of IL-17RB (27), tuft cell-derived IL-25 activates ILC2s to produce IL-13, which affects epithelial crypt progenitors to promote differentiation of tuft and goblet cells, resulting in further activation of ILC2s in a positive feedback circuit of type 2 inflammation (26). This feed-forward pathway is constrained by CISH, a suppressor of cytokine signaling family member (28) and CISH-deficient ILC2s show excessive proliferation and cytokine production, resulting in increased tuft cells in the small intestinal (29). TSLP, which belongs to the IL-2 family with structural similarities to IL-7, is released from epithelial cells (30). TSLP incorporates the TSLP receptor (R) and the α-subunit of the IL-7R, and this ternary molecular complex activates multiple signaling pathways, such as the JAK1 and 2, STAT3 and 5, MAPK, PI3K, and NF-κB pathways (31–33). TSLP enhances the type 2 immune response, in particular the activation of ILC2s, resulting in increased type 2 cytokines IL-4, IL-5, and IL-13 (34–39). These effector cytokines are also regulated at the post-transcriptional level. Tristetraprolin, encoded by Zfp36, is an RNA-binding protein that destabilize mRNA. In the Zfp36-/- mice, ILC2 produces excessive Il5 and Il13 in the small intestine and other organs (40). Taken together, ILC2 in the intestine is regulated at the transcriptional and post-transcriptional level upon cytokine stimulation.
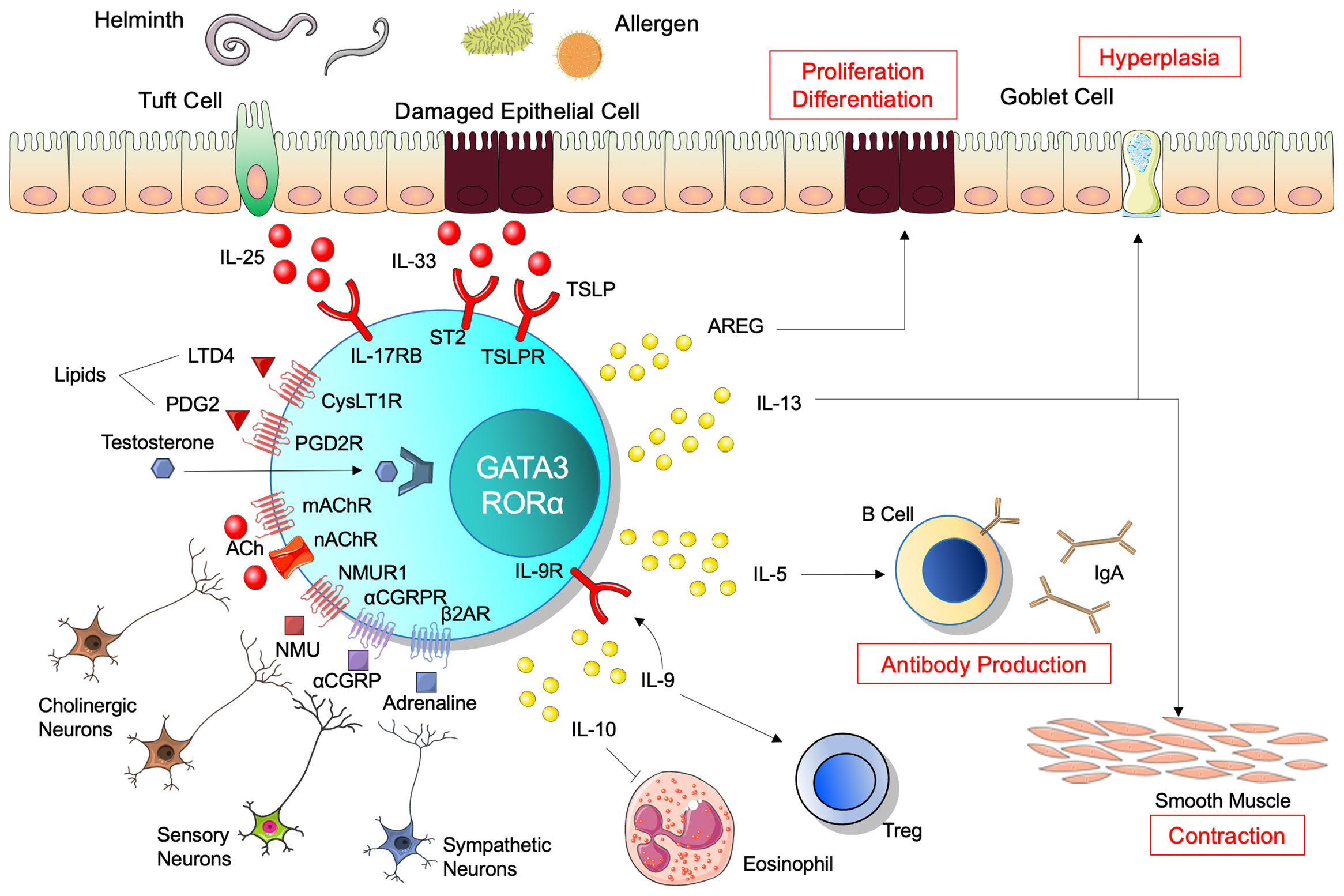
Figure 1 Regulation and function of ILC2. Alarmins, IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), activate innate lymphoid cells 2 (ILC2) to expand and produce cytokines. Neural systems regulate ILC2s bidirectionally through neuropeptides and neurotransmitters. ILC2s require transcription factors GATA3 and RORα, and subsequently produce various cytokines. IL-13 induces hyperplasia of goblet cells and contraction of smooth muscle in the intestine, involved in clearance of antigens in the lumen. IL-5 regulates B-cell antibody production and enhances IgA production. Amphiregulin (AREG) promotes epithelial cell proliferation and differentiation. IL-9 and IL-10 contribute to resolution of inflammation, while IL-9 also promotes regulatory T-cell activation and IL-10 decreases eosinophil recruitment. ILC2 expresses IL-9 receptor and thus receives IL-9 autocrine feedback. Regarding ligands and receptors, red indicates activation, blue indicates inhibition, and purple indicates both functions.
Neuroimmunology: ILC2 Neuro-Immune Crosstalk
The gastrointestinal tract is one of the most innervated organs, particularly by enteric neurons and extrinsic sympathetic and parasympathetic nerves, such as the vagus nerve (41). This neuronal regulation shapes the levels of inflammation and homeostasis in the gut via controlling the epithelia, stroma, and immune cell compartments. The immune and neuronal systems interact bidirectionally, namely through neuropeptides and neurotransmitters that regulate immune cell functions, while inflammatory mediators from immune cells enhance neuronal activation. This “neuro-immune crosstalk” plays critical roles in tissue homeostasis (42–44). In addition to the intestinal macrophage (45), T-cell (46, 47), and ILC3 (48), ILC2s have been investigated for neuro-immune crosstalk from the early stages following identification (49, 50). ILC2s express receptors for neuropeptides and neurotransmitters, and are regulated through these receptors. Neuromedin U (NMU), a neuropeptide secreted from sensory cholinergic neurons, is detected in the intestine with high levels of expression and exerts biological activities through two G protein-coupled receptors: NMU receptor 1 (NMUR1) and 2 (NMUR2). NMUR1 is distributed in the peripheral tissues while NMUR2 is mainly observed in the central nervous system (51). Among the immune cells reported to express NMUR1 at a significant level, ILC2s predominantly express NMUR1 compared to other immune cell subsets, such as T cell, mast cell, and other groups of ILCs (52–56). NMU induces activation, proliferation, and type 2 cytokine secretion in ILC2s through NMUR1 (54–56). Although NMU regulation of ILC2s has been elucidated mainly in the field of allergic respiratory diseases, this relationship has also been revealed in the mouse gastrointestinal tract, indicating that NMU induces ILC2 activation, proliferation, and secretion of the type 2 cytokines IL-5, IL-9, and IL-13 (55). Calcitonin gene-related peptide (CGRP) is a later-identified neuropeptide that regulates ILC2s and is expressed and released by sensory neurons and ILC2s themselves (57–59). ILC2s express the receptor for α-CGRP in homeostatic and inflammatory conditions, and α-CGRP suppresses ILC2 proliferation by activating a cAMP response module, while promoting IL-5 expression (59). Single-cell RNA sequencing has revealed that expression of Calca, which encodes α-CGRP, is induced in intestinal killer-cell lectin like receptor G1 (KLRG1)-positive ILC2s in a food allergy model, but it is expressed in choline O-acetyltransferase (ChAT)+ sensory neurons in the steady state (59). These paradoxical functions of CGRP in terms of pro- and anti-inflammatory influence on immune responses may represent key roles for maintenance of epithelial cell homeostasis by adjusting immune responses to neuronal signals. In particular, IL-5 enhanced by α-CGRP promotes repair of epithelial cell damage, while α-CGRP prevents excessive type 2 inflammation by suppression of ILC2 proliferation (59). Of note, ChAT+ ILC2s are strongly induced by type 2 inflammatory conditions, such as helminth infection, Alternaria sensitization, and IL-25 and IL-33 treatment (38). In addition, ILC2s purified from the small intestine or cultured under IL-2, IL-7, and IL-33, express both muscarinic (Chrm4 and Chrm5) and nicotinic (Chrna2, Chrna5, Chrna9, and Chrna10, Chrnb1 and Chrnb2) acetylcholine (ACh) receptors. Therefore, ILC2s can respond to ACh to produce IL-5 and IL-13, and induce goblet cell hyperplasia, eosinophil accumulation, and helminth expulsion in the small intestine, which are partially abrogated in ILC-specific deletion of ChAT. Tuft cells also have the capacity to produce ACh and contribute to the regulation of ILC2s (26, 60). Studies suggest that the expression of CGRP and ChAT in ILC2s is similarly induced by type 2 inflammation and positive autocrine loops of ILC2-ACh or ILC2-CGRP potentially modify intestinal inflammation and homeostasis, and also raise an interesting question to identify the distinct roles of neuropeptides like CGRP and ACh released by ILC2s and sensory neurons. Although ILC2s respond to ACh both in the intestine and lung, ILC2s in the lung express the α7-nicotinic ACh receptor in contrast to intestinal ILC2s, suggesting tissue specificity of ACh receptor usage in ILC2s (61). Similar to the CGRP, another vasodilative neuropeptide, vasoactive intestinal polypeptide (VIP) is also involved in the regulation of ILC2s. Intestinal ILC2s express the VIP receptor and produce IL-5 when incubated with IL-7 and VIP (62). Reciprocally, IL-5 from ILC2s directly activates nociceptors, such as TRPV1 and TRPA1, on afferent NaV1.8+ neurons and upregulates the release of VIP, which induces ILC2s and T-cells to release more IL-5 and form a positive feedback loop of type 2 inflammation (63).
ILC2s are regulated by not only cholinergic neurons but also adrenergic neurons. The β2-adrenergic receptor, a catecholamine receptor expressed on ILC2s, recognizes noradrenaline released from sympathetic neurons and downregulates ILC2 function and type 2 inflammatory responses (64). Hence, both parasympathetic neurons releasing ACh and sympathetic neurons releasing noradrenaline affect suppression of ILC2-derived type 2 inflammation.
ILC2 in Inflammatory Bowel Diseases
IBD, mainly comprising UC and CD, is chronic inflammatory disease of the gastrointestinal tract, although the etiology of IBD remains unclear. To date, more than 200 IBD-associated genes have been identified and impinge on the pathways associated with cytokine signaling, bacterial recognition, and barrier function (65–67). Accumulation of many studies reveals that abnormal immune responses against microorganisms of the gut flora initiates chronic intestinal inflammation in genetically susceptible individuals (68). Furthermore, dysregulation of both innate and adaptive immune pathways contributes to the pathogenesis of IBD (69) (Figure 2). Detailed elucidation of the innate immune system, including ILCs, is required to gain new insights into the immunologic mechanisms of intestinal inflammation.
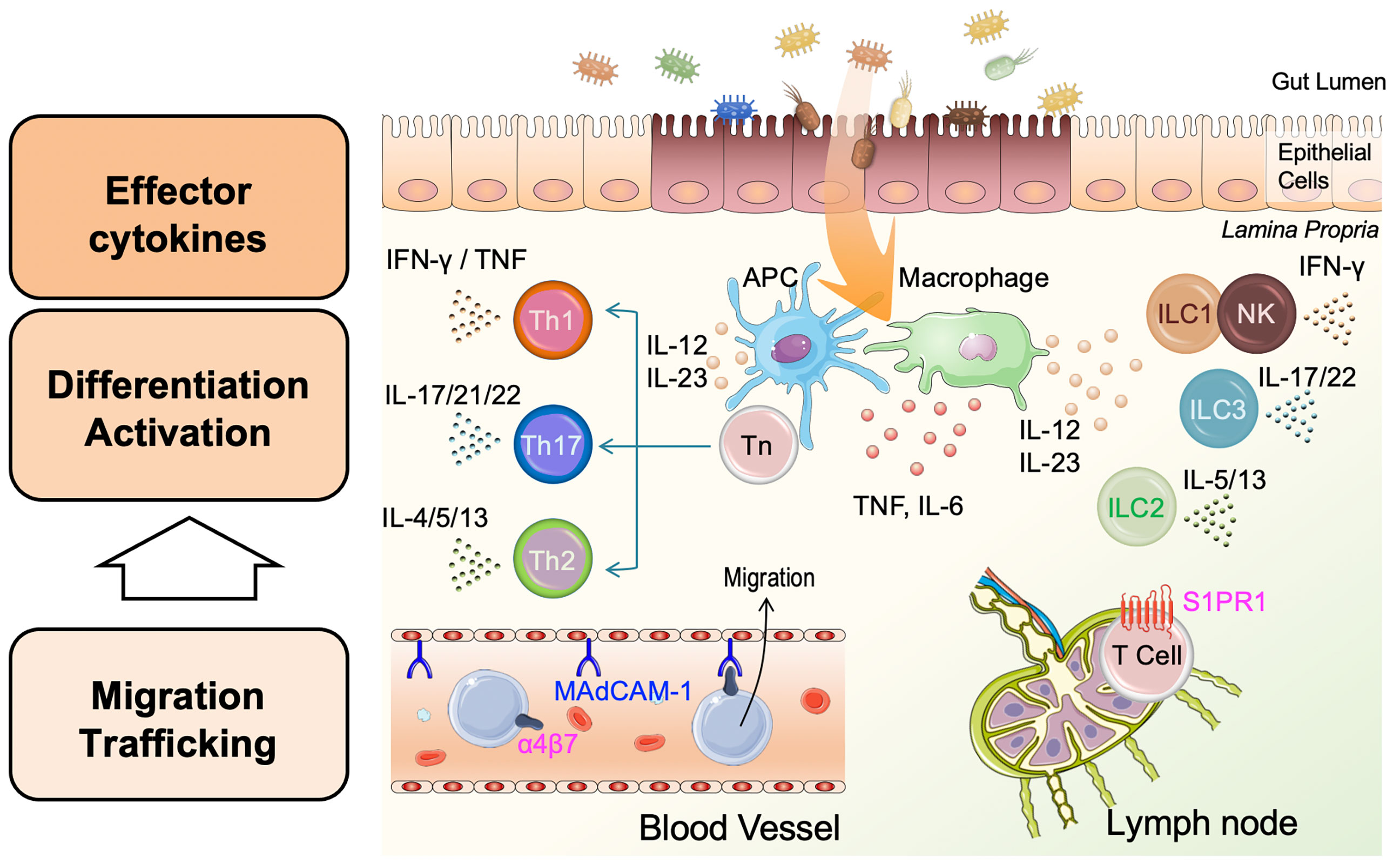
Figure 2 Pathophysiology of inflammatory bowel disease (IBD). Damaged epithelial cells enhance antigen presenting cell (APC) and macrophage uptake of antigens such as bacteria in the gut lumen, leading to APC/macrophage activation. APCs and macrophages produce pro-inflammatory cytokines, including tumor necrosis factor (TNF), IL-6, IL-12, and IL-23. Activated APCs present processed antigens to naïve helper T-cells (Tn) and promote the differentiation of Tn to effector T-cells, helper T-1 (Th1), Th2, and Th17 cells. Th1 and Th2 release type 1 cytokines (interferon [IFN]-γ and TNF), and type 2 cytokines (IL-4, IL-5, and IL-13), respectively. Independent of these, Th17 cells release IL-17, IL-21, and IL-22. Innate lymphoid cells 1 (ILC1s) and natural killer (NK) cells, ILC2s, and ILC3s are activated in a non-antigen-specific manner in tissues and produce cytokines corresponding to adaptive Th cell phenotypes, Th1, Th2, and Th17, respectively. Inflammation in the lamina propria is involved in the migration and trafficking of lymphoid cells from blood vessels and lymph nodes. Circulating lymphoid cells bearing integrin-α4β7 bind to the mucosal vascular epithelium through mucosal addressin-cell adhesion molecule 1 (MAdCAM-1) and migrate to the inflamed intestine. T-cells expressing shingosine-1-phosphase receptor 1 (S1PR1) in the lymph node are recruited to the site of inflammation by stimulation with S1P. Each cytokine or molecule has been targeted for IBD treatment.
Compared with ILC1s and ILC3s, the role of ILC2s in IBD patients is less well understood (19). This might be attributed to the very low frequency of ILC2s in the entire human gastrointestinal tract compared with the relatively high abundance of ILC1s in the upper gastrointestinal tract and ILC3s in the ileum and colon (70). At the time of IBD diagnosis, the frequency of ILC1s is increased in patients with CD, and the frequency of NKp44+ ILC3s in inflamed tissue is decreased in both CD and UC patients (71), consistent with the previous literature showing that NKp44+ ILC3s produce IL-22 and IL-22-producing ILC3s are decreased in IBD (72–76). In contrast, the frequency of ILC2s is increased in patients with UC at diagnosis (71), while both ILC1s and ILC2s are increased in patients with IBD established for at least 1 year (71). Although reports of increased ILC2 frequency are traditionally present in CD but not UC (77), a recent study suggests the involvement of ILC2s with mucosal inflammation in both CD and UC. Impressively, ILC2s show plasticity towards an ILC1 cytokine profile with IL-12 stimulation, and some ILC2s in the mucosa of CD patients acquire capacity to produce IFN-γ in addition to IL-13, which could potentially contribute to intestinal inflammation (78). IL-12 is expressed and actively released in CD intestinal tissues (79), and is the therapeutic target of ustekinumab, which is used to treat CD and UC (80, 81). Notably, in IBD patients receiving vedolizumab, a monoclonal antibody that targets integrin α4-β7 and blocks gut-homing of activated immune cells, the frequencies of ILCs in peripheral blood remained unchanged, suggesting that distribution of ILCs is due to local proliferation or plasticity rather than recruitment of ILCs to the intestine (71). Therefore, the finding of increased frequency of ILC2s secreting IFN-γ may indicate that the plasticity of ILCs depends on the local mucosal microenvironment.
IL-33 expression is enhanced in the inflamed mucosa of IBD patients (24, 82) and experimental models of colitis (83), and has been previously shown to play both protective and detrimental roles in colitis, based on different models of colitis and analyses of cell types. Genetic ablation of ST2, a receptor of IL-33, resulted in amelioration of colitis induced by DSS or trinitrobenzene sulfonic acid (84). In addition, ILC2s expand and produce more Th2 cytokines during DSS-induced colitis, which is repressed in the steady state by E-cadherin on colonic epithelial cells and KLRG1 on ILC2s (85). Conversely, treatment with IL-33 or transfer of ILC2s improve intestinal mucosal damage through the AREG pathway in the DSS-induced colitis model (11). A recent study has reported that the intracellular pattern recognition receptor NOD2 drives early IL-33-dependent expansion of ILC2s during CD ileitis, based on CD patient samples and an established murine model of CD-like ileitis, the SAMP1/YitFc mouse strain (86, 87). In addition to alarmins, CC chemokine ligand 1 (CCL1) exerts unique roles on ILC2s in the intestine. ILC2s express high levels of the Th2-type chemokine receptor, C-C motif chemokine receptor 8 (CCR8), both in mouse intestine and human peripheral blood. In addition, the expression of CCR8 and its ligand CCL1 is upregulated in patients with UC and in the DSS-induced colitis model (88). In a helminth infection model, mice lacking CCR8 exhibit reduced type 2 cytokines IL-5, IL-13, and IL-9, and greater worm burden in the small intestine (89). This is not attributed to aberrant migration but to impaired proliferation and cytokine production in ILC2s in the lung and intestine, although CCL1/CCR8 signaling contributes to mediating monocyte and lymphocyte chemoattraction and is implicated in vascular regulatory T-cell recruitment and function (90). Of note, ILC2s are the major producers of CCL1, which forms a paracrine CCL1/CCR8 feed-forward loop during helminth clearance (89). Unlike parasite infection, the major source of CCL1 during DSS-induced colitis is macrophages rather than ILC2s, but CCL1/CCR8 signaling similarly protects hosts from both parasite infection and acute intestinal damage in a DSS colitis model (88). In addition, mice lacking CCR8 exhibit comparable numbers of ILC2 and tissue-repairing cytokines, IL-10 and AREG, but reduced numbers of intestinal IFN-γ-producing ILCs (88). However, these IFN-γ-producing ILCs may also have dual roles in colitis as discussed above. Further studies are needed to disentangle the complex results of previous reports regarding the roles of ILC2s in colitis and clinical trials targeting Th2 cytokines, and to further enhance our comprehension of the contribution of ILC2s to immune mechanisms in IBD.
ILC2 Contribution to Intestinal Fibrosis
Inflammation and impaired tissue repair induce accumulation of myofibroblasts, which produce extracellular matrix components, resulting in organ fibrosis (91, 92). In the intestine, fibrosis can lead to stenosis or perforation. Th2 cells produce type 2 cytokines, IL-4, IL-5, and IL-13, generating various pathological changes, such as infiltration of eosinophils, increased mucus production, and fibrosis (93). Recent studies have revealed that not only Th2 cells but also ILC2s producing type 2 cytokines in an antigen non-specific manner play an important role in immune-mediated fibrosis and modulation of tissue remodeling, causing dysfunction in various organs. Regarding the lung, expression of IL-25 and the ILC2 population increase in the lungs of idiopathic pulmonary fibrosis patients (94). Other alarmin cytokines, IL-33 and TSLP, are elevated in idiopathic pulmonary fibrosis, cystic fibrosis, and steroid-resistant asthma sufferers (95–98). These studies suggest that alarmin cytokines have critical roles in lung fibrosis and remodeling. In patients suffering from liver fibrosis of various etiologies, such as virus infection, alcoholic liver disease, non-alcoholic steatohepatitis, autoimmune hepatitis, and primary cholangitis (99), numbers of liver-resident ILC2s are activated and expanded followed by expression of IL-33 (100). A recent study has revealed the contribution of ILC2s in skin fibrosis within systemic sclerosis (101). Following activation by IL-33, ILC2s express the growth factor AREG and participate in epithelial barrier function and tissue repair in the intestine (11).
IL-13 produced by ILC2s is involved in expression of the tumor necrosis factor family cytokine TL1A, overexpression of which brings about intestinal fibrosis (102). Constitutive expression of TL1A in lymphoid and myeloid cells leads to spontaneous inflammation and fibrosis in the small intestine and colon (103, 104). TL1A is a ligand for death receptor 3 and enhances secretion of pro-inflammatory cytokines through multiple cell lineages (105). ILC2s highly express death receptor 3 and overexpression of TL1A activates ICL2 expansion, independent of IL-25 or IL-33 (106). Notably inhibition of TL1A function by either anti-TL1A neutralizing antibody or deletion of death receptor 3 reduces numbers of intestinal fibroblasts and myofibroblasts in murine DSS colitis, the model of human IBD (107). Deficiency of another tumor necrosis factor family cytokine, LIGHT, in mice exacerbates DSS colitis compared with controls and accumulates ILCs, suggesting LIGHT plays roles in regulating inflammation in the colon (108). Signaling through LIGHT receptor, lymphotoxin β receptor, in epithelial cells and dendritic cells protects against mucosal damage by inducing IL-22 from ILC3s (109). Although Tnfsf14, the gene encoding LIGHT, is highly expressed in not only ILC3s but also ILC2s (108, 110), the role of the LIGHT-lymphotoxin β receptor interaction in ILC2s has not yet been revealed and further research is needed.
Blocking IL-13 production from ILC2s by IL-25 neutralization enhances IL-22 production from ILC3s, which repair epithelial damage (111, 112). In murine models, IL-13 is associated with chronic gut inflammation caused by trinitrobenzene sulfonic acid (113) and triggers transforming growth factor β 1-dependent fibrosis (114). Notably, IL-13 was identified as the key effector cytokine in UC by affecting epithelial apoptosis, tight junctions, and restitution velocity (115) and a promotor of collagen accumulation in CD by inhibiting fibroblast matrix metalloproteinase synthesis, resulting in fibrosis of intestine tissue (116). These studies indicate that blockade of IL-13 improves inflammation and subsequent fibrosis in IBD patients. However, clinical trials evaluating tralokinumab, an anti-IL-13 neutralizing antibody for moderate-to-severe UC (117), and anrukinzumab, an anti-IL-13 monoclonal antibody for mild-to-moderate UC (118), could not demonstrate statistically significant therapeutic effects compared with placebo controls. Although the effect of inhibiting IL-13 for IBD patients remains controversial, a recent study that found a high frequency of autoantibodies against integrin αvβ6 in UC patients suggests the possible contribution of type 2 immune responses in the pathogenesis in IBD (119).
The Roles of ILC2 for infection and Allergy
As mentioned above, the exposure to pathogens such as parasites and allergens triggers ILC2 activation in mucosal tissue. Parasites and allergens contain catalytic enzymes that digest the mucosal barrier and provoke massive epithelial cell death, leading to release of IL-33, which rapidly activates ILC2s in the lung (120) and colon (121, 122). Since IL-33 rescues RAG2-/-, but not RAG2-/- γc-/-, mice from Clostridioides difficile (121) and amebic (122) infection, IL-33-ILC2 exerts host protection from these intestinal infections. In the nucleus of epithelial cells, endogenous IL-33 is highly expressed upon tissue inflammation (123). Additionally, lipid chemical mediators play critical roles in ILC2 activation (124, 125). ILC2s in the lung from wild-type, RAG2-/-, and STAT6-/- mice express cysteinyl leukotriene receptor 1 (CYSLTR1), and are induced to produce IL-4, IL-5, and IL-13 followed by stimulation of leukotriene D4 (124). Similar to the lung ILC2, small intestine ILC2 expresses CYSLTR1 and CYSLTR2, and produces IL-13 upon stimulation of leukotriene C4 and D4 (126). In small intestine, tuft cells become the essential source of cysteinyl leukotriene and activate ILC2s in cooperation with IL-25 following helminth infection (126). An in vitro study of ILC2s isolated from human skin showed that prostaglandin D2 induces ILC2 migration, production of type 2 cytokines and other pro-inflammatory cytokines, and upregulation of the expression of IL-33R and IL-25R (125). A subsequent study demonstrated that testosterone attenuates ILC2 function, and this result may explain the sex difference in prevalence of allergic disease (127). However, these ILC2 regulatory mechanisms have not yet been demonstrated in the intestinal tract.
Activated ILC2s exert inflammatory responses mainly via type 2 cytokines. IL-5, IL-6, and IL-13 are ILC2-derived cytokines that were identified when ILC2s were first discovered (13). IL-5 regulates B-cell antibody production and enhances IgA production from B-cells, while IL-5 and IL-13 are implicated in allergic inflammation and protection against helminth infection (13, 128). The recent study demonstrates that ILC2s predominate in the stomach, are induced by commensal bacteria, and protect against H. pylori infection through B cell activation and IgA production (129). IL-13 promotes intestinal smooth muscle contractility for exclusion of enteric nematode parasites and is required for expression of STAT6 (130). IL-13 derived from ILC2s induces hyperplasia of goblet cells, the columnar epithelial cell that lines gastrointestinal mucous membrane and contains abundant mucin, and participates in clearance of luminal antigens (131, 132). While activated ILC2s produce large amounts of IL-5 and IL-13, the level of IL-4 is generally low except in specific inflammatory conditions or disease models (14, 133, 134). IL-4 released from ILC2s promotes food allergy by blocking allergen-specific regulatory T-cells (135), and is required for type 2 helper T-cell (Th2) differentiation following helminth infection (136). Alternatively, ILC2s can also respond to IL-4 derived from eosinophils or basophils and accelerate proliferation and activation of ILC2s themselves. This feed-forward loop contributes to amplification of type 2 inflammation (137, 138). ILC2s also produce IL-9 following activation by IL-33, but not IL-25 (139). IL-9 derived from ILC2s promotes regulatory T-cell activation and effectively induces resolution of inflammation (140). Moreover, ILC2 simultaneously expresses IL-9R during helminth infection, suggesting an autocrine feedback of ILC2-derived IL-9 (139). Conversely, a molecularly distinct subset of ILC2s produce IL-10 following IL-2 activation and subsequently decrease eosinophil recruitment, suggesting downregulation of inflammation (12). Consequently, ILC2s interact with other immune cells through various cytokine crosstalk pathways and form amplification loops of type 2 immune responses with Th2 cells, eosinophils, and basophils.
Heterogeneity and Plasticity of ILC2
Recent studies have shown the heterogeneity of ILC2 subsets between tissues and implicated environmental factors in this variability. In the lung, the existence of two different ILC2 subsets—inflammatory ILC2 (iILC2) and natural ILC2 (nILC2)—have been identified, and these have different phenotypes, such as ST2 (a heterodimer of IL-33R), Thy1, KLRG1, and IL-17RB (10). iILC2 cells express more IL-25R and develop into nILC2-like cells, producing IL-5 and IL-13 after stimulation with IL-33 during worm infection. Moreover, iILC2 migrate from the intestinal lamina propria to other organs, including lung and liver, dependent on chemotaxis mediated by sphingosine 1-phosphate after injection of IL-25 or helminth infection (141). Although ILC2s are largely tissue-resident (142, 143), the ability of ILC2s to migrate suggests that ILC2s complement adaptive immunity by protecting both local and distant tissue against infection.
In the small intestine, IL-33 promotes the generation of iILC2s by induction of tryptophan hydroxylase 1, deletion of which results in increased susceptibility to helminth infection (144). However, in the colon, ILC2s express ST2. Following administration of IL-33, these cells proliferate and demonstrate high expression of IL-5 and IL-13, with lower expression of IL-17 (145). Compared with other organs such as lung or skin, ILC2s in the small intestine express higher levels of IL-17RB, which forms the IL-25R together with IL-17RA (146, 147). Enriched IL-17RB in intestinal ILC2s suggests that IL-25 derived from tuft cells promotes efficient activation of ILC2s and defense against infection with helminths or other pathogens (26, 27, 148). Although TSLP is primarily expressed in skin keratinocytes, lung, and gut epithelial cells (31), the function of TSLP in the gut has not yet been clearly identified in contrast to its role in allergy and infection in the lung and skin (149).
The first cell population-specific RNA sequence study to characterize murine ILC subsets in the lamina propria of the small intestine identified the expression of genes associated with lipid metabolism, such as Dgat2, Pparg, and Lpcat2, and a gene associated with enteric neuron communication, Bmp2 (147). In another single-cell sequencing study, graded expression of GATA3 characterized four different groups of intestinal ILC2s (150). ILC2s, which express high levels of marker genes, such as Klrg1, Klf4, Ly6a, and Il2ra, uniquely expressed high levels of Il5, Csf2, and Areg (150). As described above, single-cell RNA sequencing assists to determine the heterogeneity of ILC2s, particularly in the field of neuroimmunology. In studies of the lung, ILC2s in Nippostrongylus brasiliensis-infected mice are clustered into four subsets: resting nILC2s, Il5-high nILC2s, Il13-high nILC2s, and iILC2s (57). The expression of α-CGRP receptor is enriched within an Il5-high subpopulation of ILC2s and α-CGRP promotes IL-5 production only at early time point stimulations (57). Intestinal ILC2s express the components of the α-CGRP receptor at steady state, while α-CGRP suppresses the proliferation of ILC2s, but increases IL-5 levels during the early inflammatory phase (59). Furthermore, ILC2s of the small intestine express abundant NMUR1 gene, while adaptive immune cells, ILC1s, and ILC3s do not (55).
In vitro studies demonstrate the plasticity of human ILC2s, which switch phenotype between subsets such as ILC1s and ILC3s. IL-12 promotes the conversion of ILC2s into ILC1-like cells, characterized by expression of T-bet and production of IFN-γ (78, 138, 151). Conversely, IL-4 derived from eosinophils promotes ILC2 maintenance and proliferation by preventing IL-12-mediated ILC2 differentiation into the ILC1 phenotype (138). Furthermore, the ILC2 subpopulation that expresses c-Kit can convert into ILC3-like cells, producing IL-17 in response to IL-1β and IL-23 (152). Removal of the aryl hydrogen receptor, a transcription factor for ILC3, activates intestinal ILC2s, whereas increased aryl hydrogen receptor expression suppresses ILC2 function and enhances ILC3 function (153). As ILC3-to-ILC1 conversion has been reported (19, 154), ILC2s also demonstrate plasticity, resulting in ILC2 heterogeneity in the inflammatory gut.
Conclusion
ILC2s play important roles not only for protection against infection but also for promotion of chronic inflammation and tissue fibrosis. A variety of cytokines and cellular interactions with other immune cells and neuronal systems are involved in the homeostasis of ILC2s, suggesting complexity of ILC2 regulation. Recent studies revealed the potential of intestinal ILC2s, such as migration to other organs and plasticity of conversion to ILC1s or ILC3s. In particular, single-cell analysis can help our understanding of heterogeneity of ILC2s potentially attributed to pathological mechanisms and aid in discovery of therapeutic targets for chronic inflammation, including IBD. Understanding uniqueness and commonness of ILC2 between mice and humans, between the gut and other organs, and between health and disease may help answer important questions in gut biology: What role does ILC2 play in the contexts such as IBD, infectious disease, colorectal cancer, food allergy and intestinal fibrosis? What is the unique role of each ILC2 subset in the clinical settings? How are these ILC2 subsets dynamically regulated during the course of intestinal disease? How does each ILC2 subset interact with the other ILC2 subsets and other types of immune cells? What factors contribute to diversification of ILC2 subsets? Can these ILC2 subsets be targeted to develop effective therapeutic strategies for human intestinal diseases? Further research on ILC2s in different environments at different phases of intestinal inflammation will provide a clearer view on the roles of ILC2 during colitis, tissue regeneration, fibrosis, and cancer.
Author Contributions
SS wrote the first draft of the manuscript and figures. JT wrote sections of the manuscript and drafted the figures. YM conceived, supervised, revised the text and figures. TK and TT supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Japan Society for the Promotion of Science (JSPS) KAKENHI (B) 20H03666 to YM, and (A) 20H00536 to TK; JSPS Grant-in-Aid for Transformative Research Areas(B): 21H05123 to YM; Advanced Research and Development Programs for Medical Innovation (AMED-CREST: 16gm1010003h0001 and 21gm1510002h0001to TK, and 20gm1210001h0002 to YM; the Practical Research Project for Rare/Intractable Disease: 21ek0109556h0001 to YM); the Japan Foundation for Applied Enzymology; and Keio University Medical Fund. The authors declared no conflicts of interest.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all Kanai lab members for suggestions and discussions. We thank Gillian Campbell, PhD, from Edanz (https://www.jp.edanz.com/ac) for editing a draft of this manuscript.
References
1. Zheng D, Liwinski T, Elinav E. Interaction Between Microbiota and Immunity in Health and Disease. Cell Res (2020) 30:492–506. doi: 10.1038/s41422-020-0332-7
2. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells–a Proposal for Uniform Nomenclature. Nat Rev Immunol (2013) 13:145–9. doi: 10.1038/nri3365
3. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
4. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The Transcription Factor GATA-3 Controls Cell Fate and Maintenance of Type 2 Innate Lymphoid Cells. Immunity (2012) 37:634–48. doi: 10.1016/j.immuni.2012.06.020
5. Furusawa J, Moro K, Motomura Y, Okamoto K, Zhu J, Takayanagi H, et al. Critical Role of P38 and GATA3 in Natural Helper Cell Function. J Immunol (2013) 191:1818–26. doi: 10.4049/jimmunol.1300379
6. Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-Acid-Receptor-Related Orphan Nuclear Receptor Alpha Is Required for Natural Helper Cell Development and Allergic Inflammation. Immunity (2012) 37:463–74. doi: 10.1016/j.immuni.2012.06.012
7. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription Factor Rorα Is Critical for Nuocyte Development. Nat Immunol (2012) 13:229–36. doi: 10.1038/ni.2208
8. Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The Transcription Factor GATA3 Is Essential for the Function of Human Type 2 Innate Lymphoid Cells. Immunity (2012) 37:649–59. doi: 10.1016/j.immuni.2012.08.015
9. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A Role for IL-25 and IL-33-Driven Type-2 Innate Lymphoid Cells in Atopic Dermatitis. J Exp Med (2013) 210:2939–50. doi: 10.1084/jem.20130351
10. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-Responsive, Lineage-Negative KLRG1(hi) Cells Are Multipotential 'Inflammatory' Type 2 Innate Lymphoid Cells. Nat Immunol (2015) 16:161–9. doi: 10.1038/ni.3078
11. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 Promotes an Innate Immune Pathway of Intestinal Tissue Protection Dependent on Amphiregulin-EGFR Interactions. Proc Natl Acad Sci USA (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
12. Seehus CR, Kadavallore A, Torre B, Yeckes AR, Wang Y, Tang J, et al. Alternative Activation Generates IL-10 Producing Type 2 Innate Lymphoid Cells. Nat Commun (2017) 8:1900. doi: 10.1038/s41467-017-02023-z
13. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate Production of T(H)2 Cytokines by Adipose Tissue-Associated C-Kit(+)Sca-1(+) Lymphoid Cells. Nature (2010) 463:540–4. doi: 10.1038/nature08636
14. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature (2010) 464:1367–70. doi: 10.1038/nature08900
15. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically Dispersed Innate IL-13-Expressing Cells in Type 2 Immunity. Proc Natl Acad Sci USA (2010) 107:11489–94. doi: 10.1073/pnas.1003988107
16. Kiniwa T, Moro K. Localization and Site-Specific Cell-Cell Interactions of Group 2 Innate Lymphoid Cells. Int Immunol (2021) 33:251–9. doi: 10.1093/intimm/dxab001
17. Mikami Y, Takada Y, Hagihara Y, Kanai T. Innate Lymphoid Cells in Organ Fibrosis. Cytokine Growth Factor Rev (2018) 42:27–36. doi: 10.1016/j.cytogfr.2018.07.002
18. Meininger I, Carrasco A, Rao A, Soini T, Kokkinou E, Mjösberg J. Tissue-Specific Features of Innate Lymphoid Cells. Trends Immunol (2020) 41:902–17. doi: 10.1016/j.it.2020.08.009
19. Saez A, Gomez-Bris R, Herrero-Fernandez B, Mingorance C, Rius C, Gonzalez-Granado JM. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int J Mol Sci (2021) 22:7618. doi: 10.3390/ijms22147618
20. Kobayashi T, Motomura Y, Moro K. Discovery of Group 2 Innate Lymphoid Cells has Changed the Concept of Type 2 Immune Diseases. Int Immunol (2021) 33:705–9. doi: 10.1093/intimm/dxab063
21. Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, Mosmann TR. Amphiregulin, a TH2 Cytokine Enhancing Resistance to Nematodes. Science (2006) 314:1746. doi: 10.1126/science.1133715
22. Shao J, Sheng H. Amphiregulin Promotes Intestinal Epithelial Regeneration: Roles of Intestinal Subepithelial Myofibroblasts. Endocrinology (2010) 151:3728–37. doi: 10.1210/en.2010-0319
23. Ngo Thi Phuong N, Palmieri V, Adamczyk A, Klopfleisch R, Langhorst J, Hansen W, et al. IL-33 Drives Expansion of Type 2 Innate Lymphoid Cells and Regulatory T Cells and Protects Mice From Severe, Acute Colitis. Front Immunol (2021) 12:669787. doi: 10.3389/fimmu.2021.669787
24. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The Alarmin IL-33 Promotes Regulatory T-Cell Function in the Intestine. Nature (2014) 513:564–8. doi: 10.1038/nature13577
25. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an Unconventional Alarmin That Stimulates IL-2 Secretion by Dendritic Cells to Selectively Expand IL-33r/ST2+ Regulatory T Cells. J Immunol (2014) 193:4010–20. doi: 10.4049/jimmunol.1400481
26. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-Cell-Derived IL-25 Regulates an Intestinal ILC2-Epithelial Response Circuit. Nature (2016) 529:221–5. doi: 10.1038/nature16161
27. Schneider C, O'Leary CE, von Moltke J, Liang HE, Ang QY, Turnbaugh PJ, et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell (2018) 174:271–284 e14. doi: 10.1016/j.cell.2018.05.014
28. Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, et al. A Novel Cytokine-Inducible Gene CIS Encodes an SH2-Containing Protein That Binds to Tyrosine-Phosphorylated Interleukin 3 and Erythropoietin Receptors. EMBO J (1995) 14:2816–26. doi: 10.1002/j.1460-2075.1995.tb07281.x
29. Kotas ME, Mroz NM, Koga S, Liang HE, Schroeder AW, Ricardo-Gonzalez RR, et al. CISH Constrains the Tuft-ILC2 Circuit to Set Epithelial and Immune Tone. Mucosal Immunol (2021) 14:1295–305. doi: 10.1038/s41385-021-00430-6
30. Corren J, Ziegler SF. TSLP: From Allergy to Cancer. Nat Immunol (2019) 20:1603–9. doi: 10.1038/s41590-019-0524-9
31. Varricchi G, Pecoraro A, Marone G, Criscuolo G, Spadaro G, Genovese A, et al. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front Immunol (2018) 9:1595. doi: 10.3389/fimmu.2018.01595
32. Marković I, Savvides SN. Modulation of Signaling Mediated by TSLP and IL-7 in Inflammation, Autoimmune Diseases, and Cancer. Front Immunol (2020) 11:1557. doi: 10.3389/fimmu.2020.01557
33. Matera MG, Rogliani P, Calzetta L, Cazzola M. TSLP Inhibitors for Asthma: Current Status and Future Prospects. Drugs (2020) 80:449–58. doi: 10.1007/s40265-020-01273-4
34. Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic Stromal Lymphopoietin Induces Corticosteroid Resistance in Natural Helper Cells During Airway Inflammation. Nat Commun (2013) 4:2675. doi: 10.1038/ncomms3675
35. Klose CS, Artis D. Innate Lymphoid Cells as Regulators of Immunity, Inflammation and Tissue Homeostasis. Nat Immunol (2016) 17:765–74. doi: 10.1038/ni.3489
36. Gauvreau GM, Sehmi R, Ambrose CS, Griffiths JM. Thymic Stromal Lymphopoietin: Its Role and Potential as a Therapeutic Target in Asthma. Expert Opin Ther Targets (2020) 24:777–92. doi: 10.1080/14728222.2020.1783242
37. Kabata H, Flamar AL, Mahlakõiv T, Moriyama S, Rodewald HR, Ziegler SF, et al. Targeted Deletion of the TSLP Receptor Reveals Cellular Mechanisms That Promote Type 2 Airway Inflammation. Mucosal Immunol (2020) 13:626–36. doi: 10.1038/s41385-020-0266-x
38. Chu C, Parkhurst CN, Zhang W, Zhou L, Yano H, Arifuzzaman M, et al. The ChAT-Acetylcholine Pathway Promotes Group 2 Innate Lymphoid Cell Responses and Anti-Helminth Immunity. Sci Immunol (2021) 6:eabe3218. doi: 10.1126/sciimmunol.abe3218
39. Maggi L, Mazzoni A, Capone M, Liotta F, Annunziato F, Cosmi L. The Dual Function of ILC2: From Host Protection to Pathogenic Players in Type 2 Asthma. Mol Aspects Med (2021) 80:100981. doi: 10.1016/j.mam.2021.100981
40. Hikichi Y, Motomura Y, Takeuchi O, Moro K. Posttranscriptional Regulation of ILC2 Homeostatic Function via Tristetraprolin. J Exp Med (2021) 218:e20210181. doi: 10.1084/jem.20210181
41. Furness JB, Rivera LR, Cho HJ, Bravo DM, Callaghan B. The Gut as a Sensory Organ. Nat Rev Gastroenterol Hepatol (2013) 10:729–40. doi: 10.1038/nrgastro.2013.180
42. Godinho-Silva C, Cardoso F, Veiga-Fernandes H. Neuro-Immune Cell Units: A New Paradigm in Physiology. Annu Rev Immunol (2019) 37:19–46. doi: 10.1146/annurev-immunol-042718-041812
43. Chu C, Artis D, Chiu IM. Neuro-Immune Interactions in the Tissues. Immunity (2020) 52:464–74. doi: 10.1016/j.immuni.2020.02.017
44. Huh JR, Veiga-Fernandes H. Neuroimmune Circuits in Inter-Organ Communication. Nat Rev Immunol (2020) 20:217–28. doi: 10.1038/s41577-019-0247-z
45. Matteoli G, Gomez-Pinilla PJ, Nemethova A, Di Giovangiulio M, Cailotto C, van Bree SH, et al. A Distinct Vagal Anti-Inflammatory Pathway Modulates Intestinal Muscularis Resident Macrophages Independent of the Spleen. Gut (2014) 63:938–48. doi: 10.1136/gutjnl-2013-304676
46. Teratani T, Mikami Y, Nakamoto N, Suzuki T, Harada Y, Okabayashi K, et al. The Liver-Brain-Gut Neural Arc Maintains the T(reg) Cell Niche in the Gut. Nature (2020) 585:591–6. doi: 10.1038/s41586-020-2425-3
47. Yan Y, Ramanan D, Rozenberg M, McGovern K, Rastelli D, Vijaykumar B, et al. Interleukin-6 Produced by Enteric Neurons Regulates the Number and Phenotype of Microbe-Responsive Regulatory T Cells in the Gut. Immunity (2021) 54:499–513.e5. doi: 10.1016/j.immuni.2021.02.002
48. Fonseca-Pereira D, Arroz-Madeira S, Rodrigues-Campos M, Barbosa IA, Domingues RG, Bento T, et al. The Neurotrophic Factor Receptor RET Drives Haematopoietic Stem Cell Survival and Function. Nature (2014) 514:98–101. doi: 10.1038/nature13498
49. Kabata H, Artis D. Neuro-Immune Crosstalk and Allergic Inflammation. J Clin Invest (2019) 129:1475–82. doi: 10.1172/JCI124609
50. Klose CSN, Veiga-Fernandes H. Neuroimmune Interactions in Peripheral Tissues. Eur J Immunol (2021) 51:1602–14. doi: 10.1002/eji.202048812
51. Ye Y, Liang Z, Xue L, Neuromedin U. Potential Roles in Immunity and Inflammation. Immunology (2021) 162:17–29. doi: 10.1111/imm.13257
52. Johnson EN, Appelbaum ER, Carpenter DC, Cox RF, Disa J, Foley JJ, et al. Neuromedin U Elicits Cytokine Release in Murine Th2-Type T Cell Clone D10.G4.1. J Immunol (2004) 173:7230–8. doi: 10.4049/jimmunol.173.12.7230
53. Moriyama M, Sato T, Inoue H, Fukuyama S, Teranishi H, Kangawa K, et al. The Neuropeptide Neuromedin U Promotes Inflammation by Direct Activation of Mast Cells. J Exp Med (2005) 202:217–24. doi: 10.1084/jem.20050248
54. Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, et al. Neuronal Regulation of Type 2 Innate Lymphoid Cells via Neuromedin U. Nature (2017) 549:277–81. doi: 10.1038/nature23469
55. Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The Neuropeptide Neuromedin U Stimulates Innate Lymphoid Cells and Type 2 Inflammation. Nature (2017) 549:282–6. doi: 10.1038/nature23676
56. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The Neuropeptide NMU Amplifies ILC2-Driven Allergic Lung Inflammation. Nature (2017) 549:351–6. doi: 10.1038/nature24029
57. Nagashima H, Mahlakoiv T, Shih HY, Davis FP, Meylan F, Huang Y, et al. Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity (2019) 51:682–695 e6. doi: 10.1016/j.immuni.2019.06.009
58. Wallrapp A, Burkett PR, Riesenfeld SJ, Kim SJ, Christian E, Abdulnour RE, et al. Calcitonin Gene-Related Peptide Negatively Regulates Alarmin-Driven Type 2 Innate Lymphoid Cell Responses. Immunity (2019) 51:709–723 e6. doi: 10.1016/j.immuni.2019.09.005
59. Xu H, Ding J, Porter CBM, Wallrapp A, Tabaka M, Ma S, et al. Transcriptional Atlas of Intestinal Immune Cells Reveals That Neuropeptide Alpha-CGRP Modulates Group 2 Innate Lymphoid Cell Responses. Immunity (2019) 51:696–708 e9. doi: 10.1016/j.immuni.2019.09.004
60. Schneider C, O'Leary CE, Locksley RM. Regulation of Immune Responses by Tuft Cells. Nat Rev Immunol (2019) 19:584–93. doi: 10.1038/s41577-019-0176-x
61. Galle-Treger L, Suzuki Y, Patel N, Sankaranarayanan I, Aron JL, Maazi H, et al. Nicotinic Acetylcholine Receptor Agonist Attenuates ILC2-Dependent Airway Hyperreactivity. Nat Commun (2016) 7:13202. doi: 10.1038/ncomms13202
62. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 Innate Lymphoid Cells Control Eosinophil Homeostasis. Nature (2013) 502:245–8. doi: 10.1038/nature12526
63. Talbot S, Abdulnour RE, Burkett PR, Lee S, Cronin SJ, Pascal MA, et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron (2015) 87:341–54. doi: 10.1016/j.neuron.2015.06.007
64. Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. β(2)-Adrenergic Receptor-Mediated Negative Regulation of Group 2 Innate Lymphoid Cell Responses. Science (2018) 359:1056–61. doi: 10.1126/science.aan4829
65. Khor B, Gardet A, Xavier RJ. Genetics and Pathogenesis of Inflammatory Bowel Disease. Nature (2011) 474:307–17. doi: 10.1038/nature10209
66. Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD Genetics: Common Pathways With Other Diseases. Gut (2011) 60:1739–53. doi: 10.1136/gut.2009.199679
67. Graham DB, Xavier RJ. Pathway Paradigms Revealed From the Genetics of Inflammatory Bowel Disease. Nature (2020) 578:527–39. doi: 10.1038/s41586-020-2025-2
68. Abraham C, Cho JH. Inflammatory Bowel Disease. N Engl J Med (2009) 361:2066–78. doi: 10.1056/NEJMra0804647
69. Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and Adaptive Immunity in Inflammatory Bowel Disease. Autoimmun Rev (2014) 13:3–10. doi: 10.1016/j.autrev.2013.06.004
70. Krämer B, Goeser F, Lutz P, Glässner A, Boesecke C, Schwarze-Zander C, et al. Compartment-Specific Distribution of Human Intestinal Innate Lymphoid Cells is Altered in HIV Patients Under Effective Therapy. PloS Pathog (2017) 13:e1006373. doi: 10.1371/journal.ppat.1006373
71. Forkel M, van Tol S, Höög C, Michaëlsson J, Almer S, Mjösberg J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn's Disease and Ulcerative Colitis. J Crohns Colitis (2019) 13:67–78. doi: 10.1093/ecco-jcc/jjy119
72. Chinen H, Matsuoka K, Sato T, Kamada N, Okamoto S, Hisamatsu T, et al. Lamina Propria C-Kit+ Immune Precursors Reside in Human Adult Intestine and Differentiate Into Natural Killer Cells. Gastroenterology (2007) 133:559–73. doi: 10.1053/j.gastro.2007.05.017
73. Takayama T, Kamada N, Chinen H, Okamoto S, Kitazume MT, Chang J, et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) Natural Killer Cells in the Intestinal Mucosa of Patients With Crohn's Disease. Gastroenterology (2010) 139:882–92, 892.e1-3. doi: 10.1053/j.gastro.2010.05.040
74. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX3CR1+ Mononuclear Phagocytes Support Colitis-Associated Innate Lymphoid Cell Production of IL-22. J Exp Med (2014) 211:1571–83. doi: 10.1084/jem.20140678
75. Mizuno S, Mikami Y, Kamada N, Handa T, Hayashi A, Sato T, et al. Cross-Talk Between Rorγt+ Innate Lymphoid Cells and Intestinal Macrophages Induces Mucosal IL-22 Production in Crohn's Disease. Inflamm Bowel Dis (2014) 20:1426–34. doi: 10.1097/MIB.0000000000000105
76. Powell N, Lo JW, Biancheri P, Vossenkämper A, Pantazi E, Walker AW, et al. Interleukin 6 Increases Production of Cytokines by Colonic Innate Lymphoid Cells in Mice and Patients With Chronic Intestinal Inflammation. Gastroenterology (2015) 149:456–67.e15. doi: 10.1053/j.gastro.2015.04.017
77. Geremia A, Arancibia-Carcamo CV. Innate Lymphoid Cells in Intestinal Inflammation. Front Immunol (2017) 8:1296. doi: 10.3389/fimmu.2017.01296
78. Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, et al. IL-12 Drives Functional Plasticity of Human Group 2 Innate Lymphoid Cells. J Exp Med (2016) 213:569–83. doi: 10.1084/jem.20151750
79. Monteleone G, Biancone L, Marasco R, Morrone G, Marasco O, Luzza F, et al. Interleukin 12 Is Expressed and Actively Released by Crohn's Disease Intestinal Lamina Propria Mononuclear Cells. Gastroenterology (1997) 112:1169–78. doi: 10.1016/S0016-5085(97)70128-8
80. Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as Induction and Maintenance Therapy for Crohn's Disease. N Engl J Med (2016) 375:1946–60. doi: 10.1056/NEJMoa1602773
81. Sands BE, Sandborn WJ, Panaccione R, O'Brien CD, Zhang H, Johanns J, et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med (2019) 381:1201–14. doi: 10.1056/NEJMoa1900750
82. Kobori A, Yagi Y, Imaeda H, Ban H, Bamba S, Tsujikawa T, et al. Interleukin-33 Expression Is Specifically Enhanced in Inflamed Mucosa of Ulcerative Colitis. J Gastroenterol (2010) 45:999–1007. doi: 10.1007/s00535-010-0245-1
83. Waddell A, Vallance JE, Moore PD, Hummel AT, Wu D, Shanmukhappa SK, et al. IL-33 Signaling Protects From Murine Oxazolone Colitis by Supporting Intestinal Epithelial Function. Inflamm Bowel Dis (2015) 21:2737–46. doi: 10.1097/MIB.0000000000000532
84. Sedhom MA, Pichery M, Murdoch JR, Foligné B, Ortega N, Normand S, et al. Neutralisation of the Interleukin-33/ST2 Pathway Ameliorates Experimental Colitis Through Enhancement of Mucosal Healing in Mice. Gut (2013) 62:1714–23. doi: 10.1136/gutjnl-2011-301785
85. Lu J, Liu D, Tan Y, Deng F, Li R. M1 Macrophage Exosomes MiR-21a-5p Aggravates Inflammatory Bowel Disease Through Decreasing E-Cadherin and Subsequent ILC2 Activation. J Cell Mol Med (2021) 25:3041–50. doi: 10.1111/jcmm.16348
86. De Salvo C, Buela KA, Creyns B, Corridoni D, Rana N, Wargo HL, et al. NOD2 Drives Early IL-33-Dependent Expansion of Group 2 Innate Lymphoid Cells During Crohn's Disease-Like Ileitis. J Clin Invest (2021) 131:e140624. doi: 10.1172/JCI140624
87. Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, et al. SAMP1/YitFc Mouse Strain: A Spontaneous Model of Crohn's Disease-Like Ileitis. Inflamm Bowel Dis (2011) 17:2566–84. doi: 10.1002/ibd.21638
88. Kang L, Schmalzl A, Leupold T, Gonzalez-Acera M, Atreya R, Neurath MF, et al. CCR8 Signaling via CCL1 Regulates Responses of Intestinal IFN-γ Producing Innate Lymphoid CelIs and Protects From Experimental Colitis. Front Immunol (2020) 11:609400. doi: 10.3389/fimmu.2020.609400
89. Knipfer L, Schulz-Kuhnt A, Kindermann M, Greif V, Symowski C, Voehringer D, et al. A CCL1/CCR8-Dependent Feed-Forward Mechanism Drives ILC2 Functions in Type 2-Mediated Inflammation. J Exp Med (2019) 216:2763–77. doi: 10.1084/jem.20182111
90. Vila-Caballer M, González-Granado JM, Zorita V, Abu Nabah YN, Silvestre-Roig C, Del Monte-Monge A, et al. Disruption of the CCL1-CCR8 Axis Inhibits Vascular Treg Recruitment and Function and Promotes Atherosclerosis in Mice. J Mol Cell Cardiol (2019) 132:154–63. doi: 10.1016/j.yjmcc.2019.05.009
91. Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host Responses in Tissue Repair and Fibrosis. Annu Rev Pathol (2013) 8:241–76. doi: 10.1146/annurev-pathol-020712-163930
92. Wick G, Grundtman C, Mayerl C, Wimpissinger TF, Feichtinger J, Zelger B, et al. The Immunology of Fibrosis. Annu Rev Immunol (2013) 31:107–35. doi: 10.1146/annurev-immunol-032712-095937
93. Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, et al. Th2 Cells in Health and Disease. Annu Rev Immunol (2017) 35:53–84. doi: 10.1146/annurev-immunol-051116-052350
94. Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. IL-25 and Type 2 Innate Lymphoid Cells Induce Pulmonary Fibrosis. Proc Natl Acad Sci U S A (2014) 111:367–72. doi: 10.1073/pnas.1315854111
95. Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, Croft M. Tumor Necrosis Factor Superfamily 14 (LIGHT) Controls Thymic Stromal Lymphopoietin to Drive Pulmonary Fibrosis. J Allergy Clin Immunol (2015) 136:757–68. doi: 10.1016/j.jaci.2014.12.1936
96. Lee JU, Chang HS, Lee HJ, Jung CA, Bae DJ, Song HJ, et al. Upregulation of Interleukin-33 and Thymic Stromal Lymphopoietin Levels in the Lungs of Idiopathic Pulmonary Fibrosis. BMC Pulm Med (2017) 17:39. doi: 10.1186/s12890-017-0380-z
97. Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, et al. Steroid Resistance of Airway Type 2 Innate Lymphoid Cells From Patients With Severe Asthma: The Role of Thymic Stromal Lymphopoietin. J Allergy Clin Immunol (2018) 141:257–268.e6. doi: 10.1016/j.jaci.2017.03.032
98. Roussel L, Farias R, Rousseau S. IL-33 is Expressed in Epithelia From Patients With Cystic Fibrosis and Potentiates Neutrophil Recruitment. J Allergy Clin Immunol (2013) 131:913–6. doi: 10.1016/j.jaci.2012.10.019
99. Schuppan D, Afdhal NH. Liver Cirrhosis. Lancet (2008) 371:838–51. doi: 10.1016/S0140-6736(08)60383-9
100. McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-Dependent Innate Lymphoid Cells Mediate Hepatic Fibrosis. Immunity (2013) 39:357–71. doi: 10.1016/j.immuni.2013.07.018
101. Laurent P, Allard B, Manicki P, Jolivel V, Levionnois E, Jeljeli M, et al. Tgfβ Promotes Low IL10-Producing ILC2 With Profibrotic Ability Involved in Skin Fibrosis in Systemic Sclerosis. Ann Rheum Dis (2021) 80:1594–603. doi: 10.1136/annrheumdis-2020-219748
102. Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, et al. The TNF-Family Cytokine TL1A Promotes Allergic Immunopathology Through Group 2 Innate Lymphoid Cells. Mucosal Immunol (2014) 7:958–68. doi: 10.1038/mi.2013.114
103. Shih DQ, Barrett R, Zhang X, Yeager N, Koon HW, Phaosawasdi P, et al. Constitutive TL1A (TNFSF15) Expression on Lymphoid or Myeloid Cells Leads to Mild Intestinal Inflammation and Fibrosis. PloS One (2011) 6:e16090. doi: 10.1371/journal.pone.0016090
104. Zheng L, Zhang X, Chen J, Ichikawa R, Wallace K, Pothoulakis C, et al. Sustained Tl1a (Tnfsf15) Expression on Both Lymphoid and Myeloid Cells Leads to Mild Spontaneous Intestinal Inflammation and Fibrosis. Eur J Microbiol Immunol (Bp) (2013) 3:11–20. doi: 10.1556/EuJMI.3.2013.1.2
105. Migone TS, Zhang J, Luo X, Zhuang L, Chen C, Hu B, et al. TL1A is a TNF-Like Ligand for DR3 and TR6/DcR3 and Functions as a T Cell Costimulator. Immunity (2002) 16:479–92. doi: 10.1016/S1074-7613(02)00283-2
106. Yu X, Pappu R, Ramirez-Carrozzi V, Ota N, Caplazi P, Zhang J, et al. TNF Superfamily Member TL1A Elicits Type 2 Innate Lymphoid Cells at Mucosal Barriers. Mucosal Immunol (2014) 7:730–40. doi: 10.1038/mi.2013.92
107. Shih DQ, Zheng L, Zhang X, Zhang H, Kanazawa Y, Ichikawa R, et al. Inhibition of a Novel Fibrogenic Factor Tl1a Reverses Established Colonic Fibrosis. Mucosal Immunol (2014) 7:1492–503. doi: 10.1038/mi.2014.37
108. Krause P, Zahner SP, Kim G, Shaikh RB, Steinberg MW, Kronenberg M. The Tumor Necrosis Factor Family Member TNFSF14 (LIGHT) is Required for Resolution of Intestinal Inflammation in Mice. Gastroenterology (2014) 146:1752–62.e4. doi: 10.1053/j.gastro.2014.02.010
109. Macho-Fernandez E, Koroleva EP, Spencer CM, Tighe M, Torrado E, Cooper AM, et al. Lymphotoxin Beta Receptor Signaling Limits Mucosal Damage Through Driving IL-23 Production by Epithelial Cells. Mucosal Immunol (2015) 8:403–13. doi: 10.1038/mi.2014.78
110. Heng TS, Painter MW. The Immunological Genome Project: Networks of Gene Expression in Immune Cells. Nat Immunol (2008) 9:1091–4. doi: 10.1038/ni1008-1091
111. Camelo A, Barlow JL, Drynan LF, Neill DR, Ballantyne SJ, Wong SH, et al. Blocking IL-25 Signalling Protects Against Gut Inflammation in a Type-2 Model of Colitis by Suppressing Nuocyte and NKT Derived IL-13. J Gastroenterol (2012) 47:1198–211. doi: 10.1007/s00535-012-0591-2
112. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Bérard M, Kleinschek M, et al. Rorγt+ Innate Lymphoid Cells Regulate Intestinal Homeostasis by Integrating Negative Signals From the Symbiotic Microbiota. Nat Immunol (2011) 12:320–6. doi: 10.1038/ni.2002
113. Lawrance IC, Wu F, Leite AZ, Willis J, West GA, Fiocchi C, et al. A Murine Model of Chronic Inflammation-Induced Intestinal Fibrosis Down-Regulated by Antisense NF-Kappa B. Gastroenterology (2003) 125:1750–61. doi: 10.1053/j.gastro.2003.08.027
114. Fichtner-Feigl S, Fuss IJ, Young CA, Watanabe T, Geissler EK, Schlitt HJ, et al. Induction of IL-13 Triggers TGF-Beta1-Dependent Tissue Fibrosis in Chronic 2,4,6-Trinitrobenzene Sulfonic Acid Colitis. J Immunol (2007) 178:5859–70. doi: 10.4049/jimmunol.178.9.5859
115. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 Is the Key Effector Th2 Cytokine in Ulcerative Colitis That Affects Epithelial Tight Junctions, Apoptosis, and Cell Restitution. Gastroenterology (2005) 129:550–64. doi: 10.1016/j.gastro.2005.05.002
116. Bailey JR, Bland PW, Tarlton JF, Peters I, Moorghen M, Sylvester PA, et al. IL-13 Promotes Collagen Accumulation in Crohn's Disease Fibrosis by Down-Regulation of Fibroblast MMP Synthesis: A Role for Innate Lymphoid Cells? PloS One (2012) 7:e52332. doi: 10.1371/journal.pone.0052332
117. Danese S, Rudziński J, Brandt W, Dupas JL, Peyrin-Biroulet L, Bouhnik Y, et al. Tralokinumab for Moderate-to-Severe UC: A Randomised, Double-Blind, Placebo-Controlled, Phase IIa Study. Gut (2015) 64:243–9. doi: 10.1136/gutjnl-2014-308004
118. Reinisch W, Panés J, Khurana S, Toth G, Hua F, Comer GM, et al. Anrukinzumab, an Anti-Interleukin 13 Monoclonal Antibody, in Active UC: Efficacy and Safety From a Phase IIa Randomised Multicentre Study. Gut (2015) 64:894–900. doi: 10.1136/gutjnl-2014-308337
119. Kuwada T, Shiokawa M, Kodama Y, Ota S, Kakiuchi N, Nannya Y, et al. Identification of an Anti-Integrin αvβ6 Autoantibody in Patients With Ulcerative Colitis. Gastroenterology (2021) 160:2383–2394.e21. doi: 10.1053/j.gastro.2021.02.019
120. Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental Allergens Induce Allergic Inflammation Through Proteolytic Maturation of IL-33. Nat Immunol (2018) 19:375–85. doi: 10.1038/s41590-018-0067-5
121. Frisbee AL, Saleh MM, Young MK, Leslie JL, Simpson ME, Abhyankar MM, et al. IL-33 Drives Group 2 Innate Lymphoid Cell-Mediated Protection During Clostridium Difficile Infection. Nat Commun (2019) 10:2712. doi: 10.1038/s41467-019-10733-9
122. Uddin MJ, Leslie JL, Burgess SL, Oakland N, Thompson B, Abhyankar M, et al. The IL-33-ILC2 Pathway Protects From Amebic Colitis. Mucosal Immunol (2022) 15:165–75. doi: 10.1038/s41385-021-00442-2
123. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is Highly Expressed in Mouse Epithelial Barrier Tissues, Lymphoid Organs, Brain, Embryos, and Inflamed Tissues: In Situ Analysis Using a Novel Il-33-LacZ Gene Trap Reporter Strain. J Immunol (2012) 188:3488–95. doi: 10.4049/jimmunol.1101977
124. Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung Type 2 Innate Lymphoid Cells Express Cysteinyl Leukotriene Receptor 1, Which Regulates TH2 Cytokine Production. J Allergy Clin Immunol (2013) 132:205–13. doi: 10.1016/j.jaci.2013.03.048
125. Xue L, Salimi M, Panse I, Mjösberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 Activates Group 2 Innate Lymphoid Cells Through Chemoattractant Receptor-Homologous Molecule Expressed on TH2 Cells. J Allergy Clin Immunol (2014) 133:1184–94. doi: 10.1016/j.jaci.2013.10.056
126. McGinty JW, Ting HA, Billipp TE, Nadjsombati MS, Khan DM, Barrett NA, et al. Tuft-Cell-Derived Leukotrienes Drive Rapid Anti-Helminth Immunity in the Small Intestine But Are Dispensable for Anti-Protist Immunity. Immunity (2020) 52:528–541.e7. doi: 10.1016/j.immuni.2020.02.005
127. Cephus JY, Stier MT, Fuseini H, Yung JA, Toki S, Bloodworth MH, et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep (2017) 21:2487–99. doi: 10.1016/j.celrep.2017.10.110
128. Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, et al. Generation of Gut-Homing IgA-Secreting B Cells by Intestinal Dendritic Cells. Science (2006) 314:1157–60. doi: 10.1126/science.1132742
129. Satoh-Takayama N, Kato T, Motomura Y, Kageyama T, Taguchi-Atarashi N, Kinoshita-Daitoku R, et al. Bacteria-Induced Group 2 Innate Lymphoid Cells in the Stomach Provide Immune Protection Through Induction of IgA. Immunity (2020) 52:635–649.e4. doi: 10.1016/j.immuni.2020.03.002
130. Zhao A, McDermott J, Urban JF Jr., Gause W, Madden KB, Yeung KA, et al. Dependence of IL-4, IL-13, and Nematode-Induced Alterations in Murine Small Intestinal Smooth Muscle Contractility on Stat6 and Enteric Nerves. J Immunol (2003) 171:948–54. doi: 10.4049/jimmunol.171.2.948
131. Madden KB, Yeung KA, Zhao A, Gause WC, Finkelman FD, Katona IM, et al. Enteric Nematodes Induce Stereotypic STAT6-Dependent Alterations in Intestinal Epithelial Cell Function. J Immunol (2004) 172:5616–21. doi: 10.4049/jimmunol.172.9.5616
132. Kondo M, Tamaoki J, Takeyama K, Nakata J, Nagai A. Interleukin-13 Induces Goblet Cell Differentiation in Primary Cell Culture From Guinea Pig Tracheal Epithelium. Am J Respir Cell Mol Biol (2002) 27:536–41. doi: 10.1165/rcmb.4682
133. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-Responsive Lineage- CD25+ CD44(hi) Lymphoid Cells Mediate Innate Type 2 Immunity and Allergic Inflammation in the Lungs. J Immunol (2012) 188:1503–13. doi: 10.4049/jimmunol.1102832
134. Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, et al. Pulmonary Innate Lymphoid Cells are Major Producers of IL-5 and IL-13 in Murine Models of Allergic Asthma. Eur J Immunol (2012) 42:1106–16. doi: 10.1002/eji.201142018
135. Noval Rivas M, Burton OT, Oettgen HC, Chatila T. IL-4 Production by Group 2 Innate Lymphoid Cells Promotes Food Allergy by Blocking Regulatory T-Cell Function. J Allergy Clin Immunol (2016) 138:801–811.e9. doi: 10.1016/j.jaci.2016.02.030
136. Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Rückerl D, Seddon B, et al. IL-4-Producing ILC2s are Required for the Differentiation of T(H)2 Cells Following Heligmosomoides Polygyrus Infection. Mucosal Immunol (2016) 9:1407–17. doi: 10.1038/mi.2016.4
137. Motomura Y, Morita H, Moro K, Nakae S, Artis D, Endo TA, et al. Basophil-Derived Interleukin-4 Controls the Function of Natural Helper Cells, a Member of ILC2s, in Lung Inflammation. Immunity (2014) 40:758–71. doi: 10.1016/j.immuni.2014.04.013
138. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 Control the Fate of Group 2 Innate Lymphoid Cells in Human Airway Inflammation in the Lungs. Nat Immunol (2016) 17:636–45. doi: 10.1038/ni.3444
139. Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-Mediated Survival of Type 2 Innate Lymphoid Cells Promotes Damage Control in Helminth-Induced Lung Inflammation. J Exp Med (2013) 210:2951–65. doi: 10.1084/jem.20130071
140. Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of Inflammation by Interleukin-9-Producing Type 2 Innate Lymphoid Cells. Nat Med (2017) 23:938–44. doi: 10.1038/nm.4373
141. Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-Dependent Interorgan Trafficking of Group 2 Innate Lymphoid Cells Supports Host Defense. Science (2018) 359:114–9. doi: 10.1126/science.aam5809
142. Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 Antagonize the Function of Group 2 Innate Lymphoid Cells and Type 2 Innate Immune Responses. Nat Immunol (2016) 17:76–86. doi: 10.1038/ni.3309
143. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue Residency of Innate Lymphoid Cells in Lymphoid and Nonlymphoid Organs. Science (2015) 350:981–5. doi: 10.1126/science.aac9593
144. Flamar AL, Klose CSN, Moeller JB, Mahlakõiv T, Bessman NJ, Zhang W, et al. Interleukin-33 Induces the Enzyme Tryptophan Hydroxylase 1 to Promote Inflammatory Group 2 Innate Lymphoid Cell-Mediated Immunity. Immunity (2020) 52:606–619.e6. doi: 10.1016/j.immuni.2020.02.009
145. You Y, Zhang X, Wang X, Yue D, Meng F, Zhu J, et al. ILC2 Proliferated by IL-33 Stimulation Alleviates Acute Colitis in Rag1(-/-) Mouse Through Promoting M2 Macrophage Polarization. J Immunol Res (2020) 2020:5018975. doi: 10.1155/2020/5018975
146. Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue Signals Imprint ILC2 Identity With Anticipatory Function. Nat Immunol (2018) 19:1093–9. doi: 10.1038/s41590-018-0201-4
147. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional Programs Define Molecular Characteristics of Innate Lymphoid Cell Classes and Subsets. Nat Immunol (2015) 16:306–17. doi: 10.1038/ni.3094
148. Angkasekwinai P, Sodthawon W, Jeerawattanawart S, Hansakon A, Pattanapanyasat K, Wang YH. ILC2s Activated by IL-25 Promote Antigen-Specific Th2 and Th9 Functions That Contribute to the Control of Trichinella Spiralis Infection. PloS One (2017) 12:e0184684. doi: 10.1371/journal.pone.0184684
149. Olguín-Martínez E, Ruiz-Medina BE, Licona-Limón P. Tissue-Specific Molecular Markers and Heterogeneity in Type 2 Innate Lymphoid Cells. Front Immunol (2021) 12:757967. doi: 10.3389/fimmu.2021.757967
150. Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell (2016) 166:1231–1246.e13. doi: 10.1016/j.cell.2016.07.043
151. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory Triggers Associated With Exacerbations of COPD Orchestrate Plasticity of Group 2 Innate Lymphoid Cells in the Lungs. Nat Immunol (2016) 17:626–35. doi: 10.1038/ni.3443
152. Bernink JH, Ohne Y, Teunissen MBM, Wang J, Wu J, Krabbendam L, et al. C-Kit-Positive ILC2s Exhibit an ILC3-Like Signature That may Contribute to IL-17-Mediated Pathologies. Nat Immunol (2019) 20:992–1003. doi: 10.1038/s41590-019-0423-0
153. Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF Jr., et al. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity (2018) 49:915–928.e5. doi: 10.1016/j.immuni.2018.09.015
Keywords: ILC2 - group 2 innate lymphoid cell, IBD - inflammatory bowel disease, Crohn’s disease, ulcerative colitis, mucosal immunology
Citation: Sunaga S, Tsunoda J, Teratani T, Mikami Y and Kanai T (2022) Heterogeneity of ILC2s in the Intestine; Homeostasis and Pathology. Front. Immunol. 13:867351. doi: 10.3389/fimmu.2022.867351
Received: 01 February 2022; Accepted: 05 May 2022;
Published: 30 May 2022.
Edited by:
Cyril Seillet, The University of Melbourne, AustraliaReviewed by:
Naoko Satoh-Takayama, RIKEN Center for Integrative Medical Sciences, JapanElia Tait Wojno, University of Washington, United States
Copyright © 2022 Sunaga, Tsunoda, Teratani, Mikami and Kanai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yohei Mikami, eW9oZWltaWthbWlAa2Vpby5qcA==; Takanori Kanai, dGFrYWdhc3RAa2Vpby5qcA==
†These authors contributed equally to this work