
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 16 March 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.866747
This article is part of the Research TopicInsights in Molecular Innate Immunity: 2021View all 13 articles
 János G. Filep1,2*
János G. Filep1,2*Acute inflammation is a localized and self-limited innate host-defense mechanism against invading pathogens and tissue injury. Neutrophils, the most abundant immune cells in humans, play pivotal roles in host defense by eradicating invading pathogens and debris. Ideally, elimination of the offending insult prompts repair and return to homeostasis. However, the neutrophils` powerful weaponry to combat microbes can also cause tissue damage and neutrophil-driven inflammation is a unifying mechanism for many diseases. For timely resolution of inflammation, in addition to stopping neutrophil recruitment, emigrated neutrophils need to be disarmed and removed from the affected site. Accumulating evidence documents the phenotypic and functional versatility of neutrophils far beyond their antimicrobial functions. Hence, understanding the receptors that integrate opposing cues and checkpoints that determine the fate of neutrophils in inflamed tissues provides insight into the mechanisms that distinguish protective and dysregulated, excessive inflammation and govern resolution. This review aims to provide a brief overview and update with key points from recent advances on neutrophil heterogeneity, functional versatility and signaling, and discusses challenges and emerging therapeutic approaches that target neutrophils to enhance the resolution of inflammation.
Acute inflammation is a localized, self-limited, multicellular innate host-defense mechanism against invading pathogens and tissue injury. Polymorphonuclear neutrophil granulocytes play pivotal roles in host defense and are rapidly deployed to the affected sites, where they engage in immediate and intense antimicrobial responses (1–3). Elimination of the offending insult ideally prompts repair of the collateral tissue damage, restoration of tissue function and return to homeostasis (4). However, the neutrophils` powerful weaponry to combat pathogens can cause collateral damage to the host (5). This will then amplifies the initial response through feed-forward inflammatory mechanisms, leading to loss of functional tissue and ultimately to organ dysfunction (6). Neutrophil-driven inflammation is a common mechanism for many diseases, including reperfusion injury, atherosclerosis, cancer, autoimmune diseases, neurodegeneration, and obesity (1, 5, 7). The capacity of neutrophils to augment tissue damage beyond that evoked by the initial infection or tissue injury itself, suggest that early checkpoints control neutrophil kinetics and fate within the inflamed tissue to prevent secondary tissue damage by these effector cells. To assure timely resolution of inflammation, neutrophil influx needs to be stopped, and emigrated neutrophils need to be disarmed and removed from the affected sites.
Neutrophils sense and integrate signals from the inflammatory microenvironment, which modulate their survival and function, and generate cues that can orchestrate innate or adaptive immune effector responses (1, 8). These include secretion of granular proteins (9, 10) cytokines (11), extracellular vesicles (12), neutrophil extracellular traps (13) and formation of membrane tethers (named cytonemes) (14). The role of neutrophils in initiation and progression of a wide range of pathologies makes neutrophils attractive therapeutic targets. However, the critical requirement of neutrophils for antibacterial host defense limits the usefulness of therapies that globally reduce neutrophil numbers or functional responses. Current treatments that target single mediators of inflammation may have limited efficacy because of the redundancy within the innate immune system and many eventually become immunosuppressive (15). Arguably, an ideal therapeutic strategy would be to prevent or reverse neutrophil-mediated tissue injury without impairing their ability to control microbial invasion. One way to develop innovative approaches for the treatment of inflammatory pathologies is to exploit neutrophil biology to enhance the resolution of inflammation. We provide here a brief overview and update with key points from recent advances on neutrophil heterogeneity, functional versatility and signaling, which can be exploited to enhance resolution of inflammation.
The acute inflammatory response is protective and resolves on their own. Neutrophils are the most abundant leukocytes in blood and form the first line of cellular defense against invading pathogens (1, 2). Neutrophils deploys a potent enzymatic and chemical arsenal to neutralize and clear invaders and necrotic tissues (2, 3) and to facilitate repair (16). Neutrophil trafficking into tissues is a multistep, tightly controlled process (17–19). Aberrant neutrophil recruitment and activation causes tissue damage that amplifies the initial inflammatory response and may continue to chronicity (3, 5, 9).
Preclinical data indicate that impaired neutrophil removal from inflamed tissues results in aggravation and prolongation of the inflammatory responses (5). Accumulating evidence indicates that ongoing inflammation is a prominent component of many diseases, including cardiovascular, acute respiratory, neurodegenerative, metabolic, and autoimmune diseases, arthritis, inflammatory bowel disease, periodontitis and sepsis (7, 18).
The resolution of inflammation is an active process, integrating mechanisms that lead to the restoration of normal tissue function. This process is governed by specialized pro-resolving lipid mediators (lipoxins, resolvins, protectins and maresins), proteins (e.g., annexin A1and galectins) and gaseous mediators (e.g. hydrogen sulfite and carbon monoxide) produced during resolution of self-limited inflammation (15, 20, 21).These mediators act predominantly on phagocytes and other immune cells to instruct repair. Their biosynthesis, receptors, cellular targets, signaling pathways and networks have been described in several excellent reviews (15, 20), and mapped into the searchable Atlas of Inflammation Resolution (21), hence will not be reviewed here. Low grade ongoing inflammation is thought to impair activation of the resolution process (5, 22). Defect in resolution mechanism is increasingly being recognized as an important trigger for acute exacerbation of chronic inflammatory conditions as reported for atherosclerotic plaque rupture (23, 24) or propagation of bacterial infection in mice (25).
While pro-resolving mediators signal through several distinct receptors, two receptors, the β2 integrin Mac-1 (CD11b/CD18) and formyl peptide receptor 2/lipoxin A4 receptor (ALX/FPR2) have emerged as master regulators of neutrophil responses and fates.
Mac-1 functions as a bidirectional allosteric “signaling machine” (26). Mac-1 is best known for mediating neutrophil adherence to the activated endothelium and the extracellular matrix (17, 18) and phagocytosis of complement C3b-opsonized bacteria (27). Mac-1 binding to platelets, immune complexes or myeloperoxidase generates survival signals (28), leading to preservation of Mcl-1, the central regulator of lifespan of human neutrophils (29). Phagocytosis of opsonized bacteria induces ROS-dependent activation of caspase-8, which overrides Mac-1 ligation-activated survival signals, resulting in apoptosis (30, 31). Caspase-8 forms a complex with FLIP (FLICE-inhibitory protein), which inhibits RIPK3-dependent necrosis and prevents degranulation (32, 33). Mac-1 also binds neutrophil elastase that directs reverse transendothelial migration (34).
ALX/FPR2 is a member of the formyl peptide receptor family, consisting of three class A G-protein-coupled receptors that share significant sequence homology (35). Formyl peptide receptors recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) to initiate innate immunity. ALX/FPR2 binds an unusually large number of structurally diverse ligands, including proteins, peptides and lipids, and conveys contrasting biological effects (35, 36). For example, ligation of ALX/FPR2 with the acute-phase protein serum amyloid A or the antimicrobial peptide LL-37 activates proinflammatory circuits (37, 38). Annexin A1, annexin A1-derived peptide Ac2-26, lipoxin A4, aspirin-triggered 15-epi-LXA4 and 17-epi-RvD1 also signal through ALX/FPR2 to limit neutrophil trafficking and lifespan and to promote efferocytosis (25, 39, 40), critical events in the resolution of inflammation. Interestingly, opposing ALX/FPR2 ligands, such as serum amyloid A and lipoxin A4, allosterically inhibit each other to bias ALX/FPR2 signaling to promote either inflammation or resolution (37, 41). ALX/FPR2 can form homodimers and heterodimers with FPR1 receptor in ligand-dependent manner, resulting in alternate patterns of downstream signal coupling that dictate neutrophil functional responses (42–45). Recent data suggest that lipoxin A4 may act as a biased allosteric modulator, exerting a dual regulatory mechanism on intracellular cAMP accumulation and Ca2+ mobilization (45). Binding of serum amyloid A to ALX/FPR2 decreases formation of homodimers and induces phosphorylation of ERK and Akt, whereas lipoxin A4 engagement increases heterodimerization with FPR1 with activation of the JNK-caspase-3 pathway, leading to apoptosis in neutrophils (42, 43). While the structural basis of diverse downstream signaling remains largely unexplored, ALX/FPR2 contains a C-terminal motif that mediates receptor recycling following endocytosis and provides protection against apoptosis (46).
Evolving evidence suggest that neutrophils also contribute to wound healing, revascularization and tissue repair (16, 47, 48). Infiltrating neutrophils provide fibronectin as “emergency extracellular matrix” to promote early bone fracture healing (49) and neutrophil-derived matrix metalloprotease 9 facilitates tissue repair in acute lung injury (50). Neutrophil gelatinase-associated lipocalin (NGAL) was reported to orchestrate post-myocardial infarction by increasing the capacity of cardiac macrophages to clear apoptotic cells in mice (51). Conversely, defect in phagocytosis or neutrophil-induced genomic instability in epithelial cells impedes resolution of inflammation and wound healing (12).
Neutrophils are traditionally viewed as a relatively homogeneous cell population with highly conserved function. This perception is, however, rapidly evolving as accumulating data indicate heterogeneity in morphology, phenotype or function under homeostatic and a variety of pathological conditions (52–57). Neutrophil classification has traditionally relied on morphology, gradient separation or surface markers. However, the exact function of some neutrophil subpopulations remain elusive. Single cell RNA sequencing revealed transcriptomically distinct neutrophil populations, even amongst mature peripheral neutrophils (58, 59) and neutrophils associated with chronic inflammatory states (58, 60–62). Different neutrophil states in healthy mice and humans can be projected onto a signal development continuum (termed neutrotime) characterized by clearly defined poles separated by a smooth transcriptome shift (63). Linking the neutrophil transcriptome to the neutrophil phenotype or functional properties will, however, require further investigations. For instance, expression of CD177+ (together with membrane-bound proteinase 3) on a subset of human neutrophils facilitates their transmigration (64), hence antimicrobial defense, whereas the elevated frequency of CD117+ neutrophils is associated with increased risk of relapse in patients with ANCA-dependent vasculitis (65, 66). VEGF-A recruits a distinct subset of neutrophils with proangiogenic properties into transplanted hypoxic tissues to facilitate restoration of blood supply (67). Reduced CD62L expression on circulating neutrophils defines “senescent” or “aged” neutrophils, which are destined for clearance (68). Neutrophil senescence is controlled by circadian oscillations in the hematopoietic niche (69), the microbiome (70) and the clock-related genes, such as Bmal1 and the CXCR2 signaling pathway (68). Functional heterogeneity, such as competitive phagocytosis (71), in the human circulating neutrophil pool has also been reported, though linking functional responses to phenotype remains challenging.
The widely used term “low density neutrophils” (“low density granulocytes” or “granulocytic myeloid-derived suppressor cells”) also refers to a heterogeneous population of CD66b+mature and immature neutrophils with both proinflammatory and immunosuppressive properties (53). Both immature (banded neutrophils) and hypersegmented neutrophils have been identified in this neutrophil subset (53). Hence, the buoyant density of neutrophils is partially coupled to maturation and may rather reflect a spectrum of different densities found in healthy individuals (72). Since mature neutrophils decrease their density when activated in vitro, circulating low density neutrophils have been suggested to acquire the activated phenotype within the tissue, perhaps indicating neutrophils that underwent reverse transmigration (72, 73). Consistently, homing neutrophils to the lung was found to switch to an activated phenotype irrespective of the inflammatory disease (74). Low density neutrophils have been implicated in the pathogenesis of systemic lupus erythematosus, albeit it is uncertain whether they are premature neutrophils released from the bone marrow (75) or represent a distinct lineage of neutrophils caused by genomic damage (76). CD10 was suggested as a marker to distinguish proinflammatory and immunosuppressive neutrophils within heterogeneous neutrophil populations in patients with acute or chronic inflammatory diseases (77). A recent study has identified two neutrophil subsets, CD123+ immature neutrophils and programmed death-ligand 1 (PD-L1)+/CD10- neutrophils as potential biomarkers for patients with sepsis (78). Future studies are needed to elucidate the contribution of these subsets to the pathogenesis of sepsis.
Distinct subsets of tumor infiltrating neutrophils have also been identified. The N1 subset, characterized by hypersegmented nuclei possesses potent tumor killing capacity, whereas the N2 subset that displays an immature phenotype favors tumor growth in mice (79). Although the origin of the N1 and N2 populations is uncertain (80), TGF-β and IFN-β have been implied in polarizing neutrophils toward the N1 phenotype (81, 82). Another subset of tumor-associated neutrophils with antigen-presenting cell features has been found to trigger an anti-tumor T cell response in early-stage of human lung cancer (83). Tumor growth is associated with loss of this neutrophil subset and functional divergence of tumor-associated neutrophils (83, 84).
Mature neutrophils exhibit transcriptional and translational plasticity in response to signals from the inflammatory environment (11) and display a cell-specific pattern of non-coding regulatory regions (85). Thus, de novo synthesis of cytokines and membrane receptors, e.g. program death ligand 1 (PD-L1), may alter neutrophil function and contribute to heterogeneity. The importance of gene expression regulation of neutrophils is illustrated by the association of altered methylation profiles with susceptibility to lupus erythematosus (86). Metabolic reprogramming also occurs during the neutrophil life cycle. Neutrophils may utilize glycogen for fuel during phagocytosis or under hypoglycemic conditions (87, 88), and glycogen levels may directly control their lifespan (89). Furthermore, neutrophils exposed to PGE2 or PGD2 induces a phenotype switch from LTB4 production to lipoxin production, which marks the resolution phase (15, 90), further highlighting the functional diversity of these cells.
Neutrophils recruited to the site of infection or tissue injury engage in different activities to respond to the initial insult, which will also determine that fate and govern their ultimate removal from the inflamed area, critical for protective inflammation and return to homeostasis. By contrast, suppression of certain neutrophil functions or excessive neutrophil responses contribute to uncontrolled inflammation and may lead to chronicity. These responses are discussed in the following sections and summarized on Figure 1.
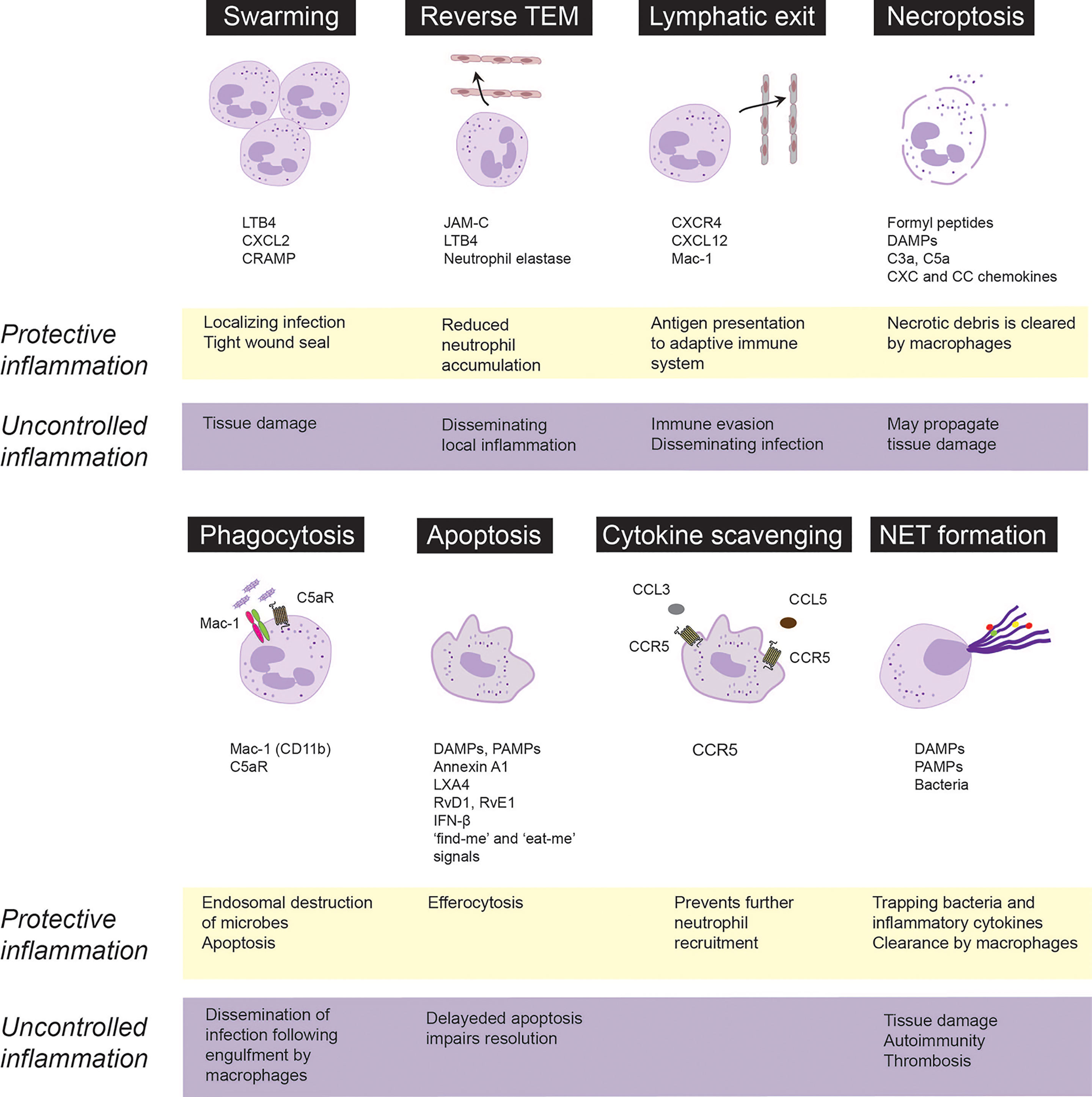
Figure 1 Fate and roles of emigrated neutrophils in protective vs. uncontrolled inflammation. Neutrophils are rapidly recruited from the circulation to the infected or injured tissues. Following extravasation, neutrophils swarm toward the infected sites to localize infection and from a tight wound seal. Neutrophils may trap, neutralize and kill invading pathogens through necroptosis, phagocytosis or release of extracellular traps (NETs). Phagocytosis of opsonized bacteria usually induces apoptosis followed by phagocytosis of apoptotic cells by macrophages via efferocytosis. Neutrophils integrate pro-survival and apoptosis-promoting cues from the inflammatory environment, which governs their lifespan. Apoptotic neutrophils express CCR5, which by binding chemokines prevents further neutrophil recruitment. Excessive swarming and necroptosis may aggravate and perpetuate tissue damage. Neutrophils may egress from the inflammatory locus through reverse transendothelial migration (TEM) or lymphatic vessels, which dampen neutrophil accumulation, but may also lead to immune evasion and dissemination of local inflammation. Neutrophils carrying bacteria that they cannot destroy, may serve as “Trojan horses” to disseminate the infection on phagocytosis of apoptotic neutrophils by macrophages. CRAMP, cathelin-related antimicrobial peptide; DAMPs, damage-associated molecular patterns, JAM-C, junctional adhesion molecules C; LTB4, leukotriene B4; LXA4, lipoxin A4; PAMPs, pathogen-associated molecular patterns; RvD1, resolvin D1; RvE1, resolvin E1.
Following transendothelial migration, neutrophils congregate or swarm in tissues, forming clusters around the infected or damaged core to seal off the affected site (90–93). Neutrophil contact with necrotic cells is critical to initiate swarming (94), followed by coordinated LTB4 release form neutrophils (91, 95), leading to formation of a stable LTB4 gradient that drives concerted waves of neutrophil migration (91, 95). Microscale protein arrays have identified numerous protein mediators, including galectin-3, CXCL8, lipocalin-2 and pentraxin-3that can further enhance LTB4-driven neutrophil swarming (95). Mac-1 (CD11b) and LFA-1 (CD11a) mediate neutrophil accumulation in the collagen-free injury center (91). Chemokine receptor trafficking and the LTB4 receptor BLT1 coordinate dense neutrophils clusters to form a tight wound seal (91, 96). Development of human neutrophil swarms is associated with lipid mediator class-switching, leading to generation of lipoxin A4 and resolvin E3, which, in turn, can limit swarm size (95). By cloaking the injured area (sensing and removing debris as well as damage-associated alarmins), tissue-resident macrophages also contribute to sealing off the damage by preventing initiation of the LTB4-driven feedforward signaling cascade that results in neutrophil swarms (94).
Neutrophil swarming has been found to limit tissue damage and contain pathogens in a variety of preclinical models (91, 94, 97, 98), indicating a protective role. Swarming behavior of neutrophils from patients following major trauma or patients receiving immunosuppressive therapy is deficient and is associated with increased susceptibility and reduced ability to clear bacterial (99) or fungal infections (100). Excessive neutrophil swarming leads to collateral tissue damage through release of neutrophil granule content via frustrated phagocytosis or necrosis (3, 101) as exemplified by pulmonary ischemia reperfusion injury in mice (97) and inflammation flares around uric acid crystals in gout (102).
Advances in intravital imaging technologies have revealed that transmigrated neutrophils can also exhibit motility away from inflamed sites, return across the endothelium and re-enter circulation (34, 103–106). Neutrophil reverse transmigration is most prevalent in tissues subjected to ischemia-reperfusion injury (106, 107). Luminal to abluminal transendothelial migration is regulated by various junctional proteins, including VE-cadherin, platelet endothelial cell adhesion molecule-1 and CD99, whereas reverse transmigration predominantly depends on junctional adhesion molecule-C (JAM-C) (106). Under ischemic conditions, excessive production of LTB4 induces neutrophil degranulation, expression of neutrophil elastase on Mac-1, which leads to JAM-C degradation and reverse transmigration (34). Reverse transmigrated neutrophils display a different phenotype, characterized by high ICAM-1 and low CXCR1 expression, increased capacity to produce superoxide and prolonged lifespan (103, 106), thereby contributing to the heterogeneity of circulating neutrophils. The biological consequences of reverse transmigration are unclear and may depend on the circumstances. As reverse transendothelial migration leads to removal of neutrophils from the inflamed site, it may function as a protective mechanism that limits the inflammatory response (104, 108). On the contrary, reverse transmigration could lead to systemic propagation of inflammation or distant organ injury in mice subjected to cremaster muscle or lower-limb ischemia-reperfusion injury (34, 106).
An alternative way of neutrophil egress from the inflamed site may involve the exit through the lymphatic vessels. During infection, neutrophils were detected carrying living bacteria from the infected tissue to draining lymph nodes in mice (109–112). Skin egress of neutrophils via lymphatic vessels depends on CXCR4 and its ligand CXCL12 expressed by lymphatic endothelial cells as well as on Mac-1 (111, 113), though the counter-ligand for Mac-1 remains to be identified. Staphylococcus aureus-pulsed neutrophils recruited into the lymph node acquire the phenotype (expression of major histocompatibility complex (MHC) II and the costimulatory molecules CD80 and CD86) and functionality of antigen-presenting cells to initiate adaptive immunity (111, 114). By contrast, other studies have proposed that neutrophils carrying bacteria or viruses may lead to immune evasion and permit the dissemination of the infection upon engulfment by macrophages (115, 116).
Mature neutrophils have a short half-life in the circulation (117, 118) and die rapidly via apoptosis (119). Following recruitment to inflamed tissues, neutrophil lifespan is increased through delaying apoptosis in response to PAMPs, DAMPs and environmental signals, though the extent of increased lifespan remains unknown (120, 121). Neutrophils contribute to interstitial acidosis, which serves as a danger signal (121) that extends neutrophil lifespan by preserving the expression of the anti-apoptotic protein Mcl-1 (122). Hypoxia or bacterial infections even under normoxia were shown to induce release of HIF-1α and HIF-2α, which generates survival cues for neutrophils and enhances their bactericidal activity to restrict systemic spread of infection (123, 124). Activated neutrophils release myeloperoxidase that activates a Mac-1-centered feed-forward loop to induce degranulation and generate survival signals, thereby perpetuating the inflammatory response (28). Conversely, genetic deletion of myeloperoxidase or disruption of the myeloperoxidase-triggered feedforward loop with 15-epi-LXA4 limits neutrophil-evoked tissue damage and facilitates resolution (40, 125, 126).
Extended neutrophil lifespan through delayed apoptosis is a common feature of many inflammatory diseases, including sepsis (127, 128), acute respiratory distress syndrome (129), severe asthma (130) and acute coronary syndrome (131), and is associated with disease severity. In experimental models, suppressing neutrophil apoptosis prolongs and aggravates the inflammatory response (28, 132), whereas promoting neutrophil apoptosis with cyclin-dependent kinase inhibitors (133), 15-epi-LXA4 (40), or IFN-β (134) accelerates the resolution of inflammation. Consistently, genetic deletion of the pro-apoptotic ARTS protein hinders the execution of the intrinsic apoptosis program in neutrophils and delays activation of resolution programs (135).
Phagocytosis of complement-opsonized bacteria or necrotic cells overrides survival signals generated by Mac-1 ligation and accelerates neutrophil apoptosis (also known as phagocytosis-induced cell death or PICD) (30, 31). Complement-mediated phagocytosis is governed by a delicate balance between Mac-1 and the complement C5a receptor (C5aR or CD88) (136, 137). Thus, reduced Mac-1 expression or genetic deletion of C5aR disables phagocytosis and reduces bacterial killing (136, 138). Bacterial or mitochondrial DNA signaling through TLR-9 upregulates Mac-1 expression and induces neutrophil elastase and proteinase 3-mediated shedding of C5aR, leading to reduced phagocytosis of E. coli, suppressed PICD and efferocytosis, thereby prolonging acute lung injury in mice (25). Conversely, by preventing TLR9 activation-mediated Mac-1 upregulation and C5aR shedding, aspirin-triggered 15-epi-LXA4 and 17-epi-RvD1 restore the balance between Mac-1 and C5aR and consequently enhance phagocytosis, bacterial killing, PICD and the resolution of lung injury (25). Of note, the pro-resolving lipid mediators resolvin E1 and resolvin D5, which signals through the LTB4 receptor BLT1 (31) and GPR32 (139), respectively, can also enhance phagocytosis of bacteria by naïve neutrophils.
Removal of apoptotic neutrophils (and other cell types) by macrophages is critical for restoring tissue homeostasis. The detection and elimination of apoptotic cells are orchestrated by “find-me” and “keep-out” signals that regulates recruitment of phagocytes to the vicinity of apoptotic cells and “eat-me” signals that allow recognition and engulfment (139–143). Apoptotic cells release nucleotides, such as ATP and UTP through caspase-mediated activation of pannexin 1 channels, which act as key “find-me” signals (144, 145). By contrast, lactoferrin released by apoptotic cells inhibits neutrophil chemotaxis without hindering monocyte recruitment (146), thereby assuring the recruitment of appropriate phagocytes for clearance of apoptotic cells and limit inflammation. Efferocytosis induces a metabolic switch in engulfing macrophages, leading to glycolysis and lactate release through SLC16A1 and reprograms macrophages from the inflammatory phenotype to an anti-inflammatory phenotype (22, 147, 148) and subsequently to a CD11blow subset with minimal phagocytic activity, increased oxidative phosphorylation and expression of IFN-β-related gene signature (134, 149). By promoting neutrophil apoptosis and efferocytosis as well as reprogramming macrophages to the CD11blow phenotype, IFN-β orchestrates bidirectional cross-talk between neutrophils and macrophages to accelerate resolution (134). Genetic deletion or pharmacological inhibition of cyclin-dependent kinases 5 and 9 drives neutrophil apoptosis and reprograms macrophages, thereby facilitating neutrophil clearance and resolution (133, 150).
Neutrophils carrying Toxoplasma gondii or Leishmania donovani, which they cannot destroy, may serve as “Trojan horses” to disseminate the infection following macrophage engulfment (115, 116). The Gram-negative intracellular coccobacillus Francisella tularensis can evade phagosomal elimination and replicates in the cytosol (151) parallel with sustaining mitochondrial integrity and delaying neutrophil apoptosis (152, 153). Continued accumulation of dysfunctional neutrophils at the infection site is thought to contribute to disease exacerbation.
Among the neutrophil defense armory is the release of extracellular traps (NETs), consisting of a nucleic acid scaffold decorated with histones and granular proteins to entrap and kill bacteria, viruses and fungi (13, 154, 155). Suicidal NET release (commonly referred to as NETosis) occurs in response to various stimuli and classically involves activation of protein kinase C and the Raf-MEK-ERK pathway, NADPH-dependent translocation of neutrophil elastase and myeloperoxidase from cytosolic granules into the nucleus, leading to the breakdown of chromatin and the nuclear envelop. NETs are extruded following the rupture of the neutrophil cell membrane (13). Hence, NETosis may be considered as a distinct form of necrotic cell death (156). Differences in NET composition have also been reported (157, 158), though the implications of these differences remain to be investigated. NET release may also occur in the absence of cellular suicide (also known as vital NETosis) in response to recognition of certain bacteria or PAMPs (154). For example, HMGB1 released from activated platelets or necrotic cells evokes NET release through interactions with TLR4, independent of NADPH oxidase (159), while suppressing phagocytosis (160). Vital NETosis requires vesicular trafficking of DNA for delivering the NET out of the cell without requiring membrane perforation (161). NET caused by extrusion of mitochondrial rather than nuclear DNA does not cause lytic cell death (162). Reports also exist that neutrophils that had already underwent vital NETosis were still capable of chasing and imprisoning live Staphylococcus aureus or Candida albicans, whereas NETs recruited additional neutrophils in a swarming-like behavior (161, 163). Although limited information is available on the molecular switches that trigger phagocytosis, NETosis or degranulation, it is plausible that selective activation of these processes assures the most effective neutrophil response to an insult. One possible control mechanism is ALX/FPR2, as genetic deletion of Fpr2 (the equivalent of human ALX/FPR2) in mice is associated with excess NET production and more severe lung injury following bacterial infection (164).
NETs are eventually degraded by macrophages and dendritic cells through DNase 1 that cleaves chromatin within NETs (165) or the cytosolic exonuclease TREX1 (DNase III) following endocytosis (166, 167). The antibacterial protein LL-37 facilitates NET uptake by macrophages, while protecting NETs against degradation by bacterial nucleases (167). A recent study reported that the thirteen-series (or T-series) resolvins, present in resolution exudates, enhance NET uptake by macrophages through the cAMP-PKA-MAPK pathway (168). The receptor for T-series resolvins remains to be identified.
NETs effectively capture a large range of microbes, exert direct antimicrobial activities and demarcate the infected locus (169, 170). However, since the effects of NET components are not restricted to invading pathogens, excessive or uncontrolled NET formation can inflict damage to the surrounding tissue, maintaining a pro-inflammatory and pro-thrombotic environment that underlies various pathologies. For example, extracellular histone components through TLR-mediated generation of thrombin can evoke microaggregation, and endothelial and tissue injury (169, 171, 172), whereas neutrophil granule constituents expressed on NETs, such as proteinase 3 or myeloperoxidase can trigger autoimmunity when NET degradation is impaired (165, 173). Clinical studies have also reported an association between NET generation and disease severity in sepsis-induced (164, 174) or COVID-19-associated acute respiratory distress syndrome (175–177). Albeit wide-ranging differences in intrapulmonary neutrophils were reported in COVID-19 autopsies (178), intense neutrophilic inflammation and NET release contribute to progression of the disease and higher mortality (175, 179–181). NETs infiltrate the airways, pulmonary interstitial space and vasculature in severe COVID-19 (182), leading to tissue damage and formation of microthrombi in pulmonary capillaries (175, 177, 179–181). Activated neutrophils secrete ROS and proteases, which in turn, enhance NETosis and inactivate plasma antiproteases that protect against neutrophil proteases (183). These create a vicious cycle to propagate tissue destruction. Enhancing NET degradation by DNase I or partial genetic deletion of peptidyl arginine deiminase 4 (PAD4+/-) reduced the severity of bacterial lung injury in mice (164). Complete PAD4 deficiency markedly suppressed NET formation and lung injury, but increased bacterial burden, indicating a shift in the balance between the protective and deleterious actions of NETs during bacterial infections.
Accumulating evidence indicates dual role for NETs in cancer (184). NETs were found to inhibit proliferation of colon carcinoma cells (185) and exert cytotoxic activity on malignant melanoma cells (186). Accumulation of NET producing CD16high CD62Ldim neutrophils in tumor sites was reported to predict improved survival in patients with head and neck squamous cell carcinoma (187). In contrast, tumor cells can prime neutrophils to release NETs (188), forming an amplifying loop that links NET formation to tumor progression. As an example, NET-associated neutrophil elastase and matrix metallopeptidase 9 (MMP9) could awaken dormant cancer cells, thereby promoting invasion and metastases (189, 190). NETs may shield tumor cells against NK cells and cytotoxic T cells (191), exert pro-angiogenic activities that support tumor growth (192), and contribute to tumor-associated thrombosis (193, 194) and hypercoagulability (195). Furthermore, NETs can also capture tumor cells and carry them in the circulation, thus favoring tumor dissemination (196). NET-DNA was also shown to act as a chemotactic factor to attract tumor cells through binding to the transmembrane protein CCDC25 expressed on primary cancer cells, thereby promoting metastasis (197).
At sites of inflammation, neutrophils can undergo necrotic cell death, which occurs in a disorderly manner following cell injury, or necroptosis, a programmed form of necrosis (198). Necrotic cell death is associated with the release of DAMPs and cell debris, which are potent inducers of inflammation (198). TNFα or ligation of the adhesion receptors Mac-1, CD18, CD15 or CD44 in GM-CSF-primed neutrophils activates the receptor-interacting protein kinase 1 (RIPK1)-RIPK3- mixed lineage kinase domain-like protein (MLKL) signaling pathway (199, 200), leading to translocation of MLKL1 to the inner leaflet of plasma membrane and membrane permeabilization (201). X-linked IAP (XIAP) ubiquitinylates RIPK1 (202) and thus functions as a switch to direct neutrophils to either necroptosis or apoptosis (201). NADPH oxidase-mediated generation of ROS is essential for necroptosis (199, 200). Consistently, neutrophils from patients with chronic granulomatous disease (caused by a genetic defect in NADPH oxidase) do not undergo necroptosis (200). Some studies reported association of necroptosis with NET formation in a mouse model of gouty arthritis (203), though NET release occurred independently of RIPK3 and MLKL signaling (204). Phagocytosis of methicillin-resistant Staphylococcus aureus redirects neutrophils from phagocytosis-induced apoptosis to necroptosis, which may allow the escape of viable bacteria from dead neutrophils, thereby persisting infection (205, 206). This requires RIPK3, but not RIPK1 and MLKL, and is associated with RIPK3- and protease-mediated production of IL-1β (205, 207). Neutrophil necroptosis, evidenced by the activation of RIPK3 and MLKL, was detected in tissue samples from patients with neutrophilic diseases (200). The pathological significance of these observations remains elusive.
Dying cells release DAMPs, including mitochondrial formyl peptides, purines, LTB4, cytokines and chemokines, and triggers generation of C3a and C5a, which collectively function as “find-me” signals for phagocytes (208–210). Similar to apoptotic cells, necrotic cells also express “eat-me” signals, such as externalization of phosphatidylserine and LTB4, which facilitate their clearance by macrophages (209). “Eat-me” cues unique to necrotic cells include deposition of complement C1q on the cell membrane (211) and cell surface externalization of annexin A1 (212). The importance of removing necrotic neutrophils is illustrated by the role of the neutrophil granule constituent proteinase 3 in autoimmune vasculitis (65, 66) and chronic obstructive pulmonary disease (213).
Given the central role of neutrophils in inflammation, it is paramount to seek novel therapeutic approaches controlling neutrophil-mediated collateral tissue damage and/or facilitating clearance of neutrophils from the inflamed site upon fulfillment of their immediate mission. Indeed, a variety of strategies have been developed to prevent the detrimental effects of neutrophils, with some approaches entering clinical trials (Figure 2).
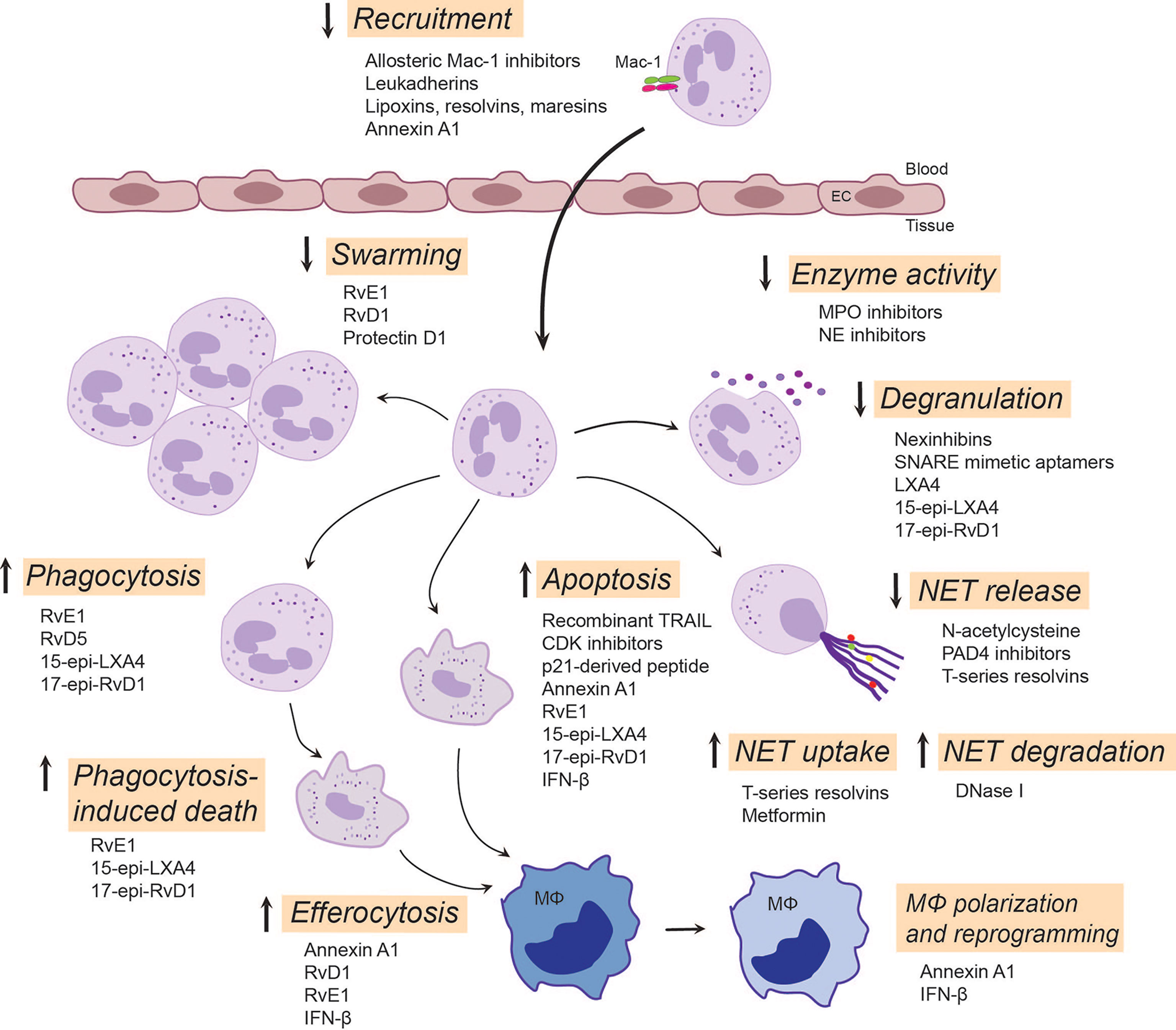
Figure 2 Emerging neutrophil-targeted therapeutic approaches to promote the resolution of inflammation. The strategies include blocking, restoring or activating neutrophil functions. Thus, blocking function of Mac-1 or upregulation of Mac-1expression dampens neutrophil accumulation, a critical component of terminating the inflammatory response. LXA4, RvE3 and protectin D1 serve as stop signals for swarming. Inhibition of degranulation or the activity of secreted enzymes, such as MPO and NE, could reduce tissue injury and alter composition of NETs. Enhancing NET degradation by DNase I or promoting NET uptake by T-series resolvins or metformin may prevent the deleterious actions of excessive NET formation. RvD5 and RvE1 facilitates phagocytosis, whereas 15-epi-LXA4 and 17-epi-RvD1 restore impaired phagocytosis, facilitate clearance of bacteria and phagocytosis-induced apoptotic cell death. By countering survival cues, many molecules, including CDK inhibitors, annexin A1, IFN-β and lipid SPMs, can redirect neutrophils to apoptosis and promote their uptake by macrophages through efferocytosis. This leads to reprogramming and polarization of macrophages toward a pro-resolution, regenerative phenotype that promotes further removal of neutrophils. Annexin A1, RvE1, RvD1 and IFN-β play pivotal roles in mediating feedforward resolution programs. Of note, although most of these data are from experimental models, some strategies (e.g. LXA4 mimetics, NE inhibitors or DNase I) are currently being investigated in clinical trials. C5aR, complement C5a receptor; CDK, cyclin-dependent kinase; EC, endothelial cell; IFN-β, interferon-β; LXA4, lipoxin A4; 15-epi-LXA4, 15-epi-lipoxin A4; MPO, myeloperoxidase; NE, neutrophil elastase; NET, neutrophil extracellular traps; PAD4, peptidyl arginine deiminase 4; 17-epi-RvD1, 17-epi-resolvin D1; RvE1, resolvin E1; RvE3, resolvin E3; RvD5, resolvin D5; SNARE, soluble N-ethylmaleimide-sensitive-factor attachment protein receptor; SPMs, specialized pro-resolving mediators; TRAIL, TNF-related apoptosis-inducing ligand.
Mac-1 conformations and broad ligand recognition specificity shape neutrophil responses and contribute to neutrophil functional heterogeneity (30, 214). Hence, β2 integrins have attracted considerable interest as potential therapeutic targets. Indeed, currently available monoclonal antibodies and small molecule inhibitors that block the ligand-binding site and a broad repertoire of β2 integrin functionality efficiently reduced neutrophil-driven inflammation in numerous experimental models (7, 18, 214). However, global β2 integrin blockade lacks functional selectivity, and can impair phagocytosis and antibacterial defense (215). Conventional β2 integrin blockade may also increase the risk of development of LAD-like symptoms. Alternative approaches include targeting Mac-1 conformation or ligand-specific signaling mechanisms without compromising host defense. Selective inhibition of Mac-1 binding of its ligand CD40L with the M7 monoclonal antibody reduced inflammation without affecting protective immunity (19, 216). Allosteric inhibitors that stabilize β2 integrins in the high affinity bent conformation efficiently blocked neutrophil adherence (217) and restricted neutrophil accumulation in murine models (218, 219). Selective targeting of discrete glycan motifs present on Mac-1 with plant lectins was reported to reduce neutrophil adhesion and trans-epithelial migration, while enhancing phagocytosis and neutrophil apoptosis (220). Other studies have reported that activation of Mac-1 with the small molecule agonists leukadherins reduced neutrophil trafficking into the kidney, while augmented leukocyte adherence to the endothelium in murine models (221). These resulted in attenuation of arterial narrowing and improved renal function. Leukadherin-1 was reported to activate microRNA Let7a and induce polarization of M0 macrophages toward the pro-inflammatory M1 phenotype that drives anti-tumor immunity (222). Thus, leukadherins likely exert context-dependent actions. Hence, additional studies are required to explore their effects on macrophage polarization and the resolution of inflammation.
Among the mechanisms by which specialized pro-resolving mediators (SPMs) facilitate resolution of inflammation is inhibition of neutrophil trafficking into the inflamed site by decreasing their adhesion and transmigration. SPMs signal through stereospecific binding to cellular receptors (223) to prevent upregulation of Mac-1 expression on neutrophils and to reduce Mac-1-mediated neutrophil adhesion and transendothelial migration, consequently limiting their tissue accumulation (15, 21). Thus, annexin A1, lipoxin A4 and resolvin D1 interact with ALX/FPR2, resolvin E1 binds ERV1 and resolvin D2 binds to DRV2 to repress Mac-1 expression (20, 223). In a feedforward mechanism for resolution, SPM receptor signaling by one mediator can trigger mobilization or synthesis of other SPMs for other receptors, exemplified by LXA4 mobilization of annexin A1 to limit neutrophil trafficking into the inflamed microvasculature (224), resolvin D1-triggered LXA4 generation in periodontal wound healing (225), and resolvin E1-ERV1-induced biosynthesis of LXA4 for ALX/FPR2-mediated resolution of allergic lung inflammation (226). Of note, the gaseous mediator hydrogen sulfide and mast cell-stabilizing drug nedocronil also mobilize annexin A1 to control leukocyte trafficking in the mouse mesenteric circulation (224, 227).
As the pleiotropic receptor ALX/FPR2 conveys ligand-specific pro- or anti-inflammatory actions, it has been proposed to function as a master switch to initiate the resolution of inflammation. A unique feature of ALX/FPR2 is that ligation of this receptor can to activate several, if not all of the processes that are critical for inflammation resolution, including blocking neutrophil trafficking into tissues, promoting neutrophil apoptosis and macrophage efferocytosis (15, 20, 25, 31, 40). As an example, annexin A1 and its mimetic peptide Ac2-26 induce the detachment of adherent neutrophils from the endothelium and inhibit neutrophil chemotaxis, thereby controlling neutrophil accumulation within the inflammatory locus (228), regulate phagocytosis of bacteria and fungi (229) and accelerate neutrophil apoptosis (39). Annexin A1 released from apoptotic neutrophils recruits monocytes to clear apoptotic cells (230), promotes their polarization towards the M2 phenotype (231), thereby protecting the surrounding healthy tissue and accelerating muscle regeneration through AMPK activation (232). Activated neutrophils releases annexin A1-containing microparticles and exosomes, which mediate its anti-inflammatory activity (233) and orchestrate epithelial wound repair through ALX/FPR2 and FPR1 (234). Neutrophil-derived microvesicles can enter cartilage and protect the joint in inflammatory arthritis (235). These findings raise the possibility of harnessing annexin A1-loaded microvesicles as a therapeutic strategy for reducing neutrophil infiltration and protection against tissue damage.
Ligation of ALX/FPR2 with LXA4, resolvin D1 and aspirin-triggered 15-epi-LXA4 and 17-epi-RvD1 activates mechanisms that partially overlap those stimulated by annexin A1, but also distinct patterns of activation for intracellular pathways, including ERK and NF-κB phosphorylation (15, 31). In addition to ALX/FPR2, to date, three other surface receptors, ERV1, DRV1 and DRV2 have been identified mediate cell-specific actions of SPMs (15, 223). A common feature of ligation of these receptors is attenuation of neutrophil activation and trafficking into inflamed tissues (15, 20, 223). SPM binding to ALX/FPR2 decreases NF-κB activity and cytokine production (31, 236), disrupts the myeloperoxidase-centered feedforward loop and redirects neutrophils to apoptosis (40), and restores TLR9-impaired phagocytosis of bacteria and promotes phagocytosis-induced neutrophil death (25). Consistently, 15-lipoxin A4 was found to accelerate the resolution of inflammation in a variety of experimental models, including asthma (237), peritonitis (238, 239), cystic fibrosis (240), ischemia-reperfusion (139), and myeloperoxidase and E. coli-induced acute lung injury in mice (25, 40). By reducing bacterial burden, ALX/FPR2 agonists may also be used to lower reduce antibiotic requirements or used in conjunction with antibiotics to strengthen host defense against infections (241, 242). Most SPMs are metabolically inactivated within the inflammatory site, some SPMs reach circulation (242, 243). To circumvent rapid inactivation, several metabolically stable analogs, such as benzo-RvD1, were synthesized (15) and nanomedicines were designed to deliver SPMs and their analogs to promote wound healing (244). Furthermore, the lipoxin A4 analog BLXA4-ME is currently in trial for periodontal inflammation (245), whereas other molecules are in clinical development program (15). Nanomedicine delivery of SPMs to correct resolution deficits represents a fascinating novel avenue for preventing the progression of chronic diseases, as exemplified by the decreases in tissue SPMs immediately before plaque rupture (23, 24).
The intriguing biology of the ALX/FPR2 receptor has initiated numerous medicinal chemistry programs to develop small-molecule agonists to activate resolution programs (15, 246). Relevant examples here are the beneficial actions of synthetic lipoxin mimetics and the prototype peptide agonist WKYMVM in various preclinical models (247, 248). Phase I clinical trials reported promising tissue protective actions with other small-molecule ALX/FPR2 agonists, such as compound ACT-389949 (Actelion) (249), and compound BMS986235 (Bristol –Myers Squibb) (250) in heart failure. However, since ALX/FPR2 expression is not restricted to myeloid cells, further studies are required to identify the cellular targets (e.g. endothelium, smooth muscle cells or fibroblasts) mediating the beneficial actions of these compounds.
Several preclinical studies indicate the therapeutic potential of targeting neutrophil apoptosis for facilitating the resolution of inflammation. Thus, pharmacologic blockade of cyclin-dependent kinases (CDKs), which principally inhibit CDK9-mediated transcription of Mcl-1 (150, 251) has been shown to exert potent anti-inflammatory effects in experimental models of neutrophil-dominated inflammation and enhance resolution of severe lung injury models (133, 251, 252). Interestingly, the CDK inhibitor drug R-roscovitine also increased bacterial clearance (251) through a yet unidentified mechanism. Ex vivo studies showed that the CDK inhibitor AT7519 efficiently overrides the delayed neutrophil apoptosis in patients with sepsis-associated ARDS concurrent with reduced expression of Mcl-1 (253). Studies in preclinical models indicate that several SPMs, including 15-epi-lipoxin A4 and resolvin E1, signaling through ALX/FPR2 and the LTB4 receptor BLT1, respectively, can also override pro-survival cues and redirect neutrophils to apoptosis in part by reducing Mcl-1 expression (31, 40). In line with these observations, the annexin A1 mimetic peptide Ac2-26 induces neutrophil apoptosis (254, 255). Furthermore, IFN-β, produced by resolution phase macrophages, drives neutrophil apoptosis through the IFNαR1-STAT3 signaling pathway and acceleration of Mcl-1 degradation (134). These findings identify Mcl-1as a promising target for resolution therapy.
Another potential mechanism to accelerate neutrophil apoptosis is restoring impaired phagocytosis. Indeed, bacterial and mitochondrial DNA were shown to reduce phagocytosis and consequently bacterial clearance as well as phagocytosis-induced death by inducing the cleavage of complement C5a receptor, which acts in concert with Mac-1 to mediate phagocytosis (10, 256). By preventing cleavage of C5a receptor, aspirin-triggered 15-epi-lipoxin A4 and 17-epi-resolvin D1 restore impaired phagocytosis, enhances bacterial clearance, drive phagocytosis-induced death and consequently attenuate E. coli-evoked lung injury in mice (25).
Other strategies to modulate neutrophil apoptosis include activating the extrinsic pathway of apoptosis by TNF-related apoptosis-inducing ligand (TRAIL) and the use of peptides derived from the cyclin-dependent kinase inhibitor p21. While TRAIL appears to have no role in constitutive neutrophil apoptosis, treatment with recombinant TRAIL was shown to enhance neutrophil apoptosis and limit the inflammatory response to LPS in mice (257). The p21 peptide binds and sequesters proliferating cell nuclear antigen (PCNA), which acts as a cytoplasmic platform to control the lifespan of human neutrophils (258). Consistently, a p21-derived peptide was shown to induce apoptosis in neutrophils isolated from patients with Pseudomonas aeruginosa infection (259), highlighting PCNA as a novel target to modulate pathological inflammation. Apoptotic neutrophils and T cells sequester chemokines, such as CCL3 and CCL5, through modulation of CCR5 expression, thereby reducing the availability of proinflammatory cytokines for other neutrophils and preventing further neutrophil recruitment (260). An interesting approach emerged from these observations is that administration of apoptotic cells markedly reduced the cytokine/chemokine storm and protected against acute lung and kidney injury in a mouse model of severe sepsis (261).
Macrophages play a crucial role in the clearance of apoptotic and necrotic cells, including neutrophils (140, 209, 262). Various pathologies, including ARDS is associated with impaired macrophage phagocytic function (263, 264). Thus, restoring macrophage function represents another avenue of potential therapy. As an example, IFN-β, produced by resolution phase macrophages, mediates a feedforward loop to promote neutrophil apoptosis and efferocytosis, which contributes to macrophage reprogramming and production of additional IFN-β (134). Several clinical trials reported favorable response to early IFN-β use to mitigate SARS-CoV2 infection-associated severe ARDS and other studies are underway testing the clinical efficacies of type I interferons (265).
As excessive or aberrant NET formation has been implicated in the pathogenesis of many pathologies, inhibiting NET release or enhancing NET clearance open promising avenues for therapy. Preclinical studies showed that ROS scavengers, such as N-acetyl cysteine (266), myeloperoxidase inhibitors (267) and PAD4 inhibitors (268–270) could inhibit NET release and dampen tissue injury in experimental models of arthritis, arteriosclerosis and autoimmune diseases. Likewise, the reversible PAD4 inhibitor GSK484 was shown to inhibit suicidal NETosis (271) and to prevent cancer-associated neutrophil-mediated renal injury in mice (272). Studies in PAD4-knockout mice suggest that bacterial infections may shift the balance of the protective and deleterious effects of NETs in host defense (273, 274). Select lipid SPMs, such as resolvin D4 (275) and T-series resolvins (168) also limit NET formation, though the underlying molecular mechanisms are incompletely understood.
Another potential therapeutic approach is blocking neutrophil degranulation or the effects of granule enzymes. Neutrophil-specific exocytosis inhibitors, termed Nexinhibs, and SNARE domain-derived peptide aptamers have been developed. Nexinhibs selectively inhibit release of azurophil granule contents by interrupting the Rab27a-JFC1 interaction without affecting phagocytosis (276, 277), and reduce neutrophil accumulation in the kidney and liver in a mouse model of systemic inflammation (277). Intrapulmonary delivery of Nexinhib20-loaded nanoparticles, which release Nexinhib20 upon cleavage by neutrophil elastase, was shown to dampen neutrophil recruitment and degranulation within the lower airways (278). SNARE mimicking peptide aptamers exhibit varying selectivity towards neutrophil granule subsets (276). As an example, TAT-SNAP-23 (a fusion protein containing the N-terminal SNAP-23 SNARE domain fused with the cell penetrating HIV peptide TAT) was reported to attenuate lung injury evoked by pulmonary immune complex deposition (279) or sepsis (280). However, since SNARE expression is not restricted to neutrophils, further studies are needed to distinguish their actions of neutrophils and other cell types in vivo. Several SPMs, including lipoxin A4, resolvin D1 and aspirin-triggered 15-epi-LXA4 and 17-epi-RvD1, acting through ALX/FPR2, also block myeloperoxidase- or TLR9 activation evoked release of myeloperoxidase, neutrophil elastase and proteinase 3 and consequently accelerate resolution of sterile (40) and E. coli-induced lung injury in mice (25). Several synthetic inhibitors and natural compounds became available over the past years, in particular compounds that target neutrophil elastase (281). Neutrophil elastase inhibition was reported to prevent progression of lung injury in various experimental models (282). However, the synthetic selective neutrophil elastase inhibitor, sivelastat failed to improve 28-day mortality in patients with ARDS (283). It remains to be investigated whether this was due to its toxic or off-target effects.
Promoting NET degradation by treatment with DNase I attenuated tissue injury and increased survival in mouse models of severe bacterial pneumonia/acute lung injury (164, 174), transplantation-associated lung injury (284), tumor (285) and lupus (286). Furthermore, an ongoing phase III clinical trial is investigating the effectiveness of inhaled dornase-α (recombinant human DNase I) in reducing the incidence of ARDS in severe trauma patients (287). Other bioengineered DNases, such as actin-resistant DNase (alidornase-α) (288, 289) or DNase 1-like 3 (290) have been developed and some are currently being tested in phase I and II trials (288, 289). Macrophages from ARDS patients exhibit reduced capacity to clear NETs as well as apoptotic cells (263). Treatment of bronchoalveolar lavage fluid macrophages from ARDS patients ex vivo with the AMPK activator metformin (264) enhanced NET clearance and efferocytosis (263). In addition to blocking suicidal NET formation, T-series resolvins, and RvT2 in particular, were shown to facilitate NET uptake by monocyte-derived M0 macrophages in vitro as well as by peritoneal macrophages in mice (168), indicating the therapeutic potential of RvT2 in the in vivo setting.
Timely removal of neutrophils from the inflamed area is of outmost importance to efficient resolution of inflammation and return to homeostasis. Failure to clear neutrophils may lead to perpetuation of the inflammatory response and persisting tissue damage. Thus, exploring the fate of emigrated neutrophils and their contribution to mechanisms that distinguish self-limiting or protective inflammation from aggravated and chronic inflammation is critical to improve current therapies. The phenotypic heterogeneity and functional versatility of neutrophils and their diverse roles in innate and adaptive immune responses provide important cues for development of neutrophil-targeting therapies. However, whether the neutrophil`s actions are mediated by different polarization states of mature neutrophils or distinct neutrophil subsets remains unclear. A simple “one size fit all” anti-neutrophil approach is perhaps naïve and outdated (282). Indeed, over the past years numerous strategies have been developed, which show promising results in preclinical models to prevent the detrimental effects of neutrophils. These include molecules that can inhibit, restore or enhance specific neutrophil functions. Use of pro-resolving agonists, such as lipoxins, resolvins and annexin A1, which activate endogenous resolution programs and would serve as immunoresolvants rather than immunosuppressant (15), represent a conceptual change for the treatment of inflammatory pathologies as well as the emergence of “resolution pharmacology” (291). Although large-scale clinical studies with these compounds seem distant, some strategies, e.g. topical application of a LXA4 mimetic and degrading NETs within the lung with inhaled dornase-α, are currently being investigated in clinical trials. This ongoing research highlights the importance of targeting neutrophils, and distinct neutrophil subsets in particular, and will likely spur further advances in neutrophil-targeted therapies to dampen inflammation to favor reparative processes without comprising antimicrobial host defense.
The author confirms being the sole contributor of this work and has approved it for publication.
This study was supported by grants from the Canadian Institutes of Health Research (MOP-97742 and MOP-102619).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat Rev Immunol (2011) 11:519–31. doi: 10.1038/nri3024
2. Nauseef WM, Borregaard N. Neutrophils at Work. Nat Immunol (2014) 15:602–11. doi: 10.1038/ni.2921
3. Gordon S. Phagocytosis: An Immunobiologic Process. Immunity (2016) 44:463–75. doi: 10.1016/j.immuni.2016.02.026
4. Medzhitov R. Origin and Physiological Roles of Inflammation. Nature (2008) 454:428–35. doi: 10.1038/nature07201
5. Nathan C, Ding A. Nonresolving Inflammation. Cell (2010) 140:871–82. doi: 10.1016/j.cell.2010.02.029
6. Brandes M, Klauschen F, Kuchen S, Germain RN. A Systems Analysis Identifies a Feedforward Inflammatory Circuit Leading to Lethal Influenza Infection. Cell (2013) 154:197–212. doi: 10.1016/j.cell.2013.06.013
7. Liew PX, Kubes P. The Neutrophil’s Role During Health and Disease. Physiol Rev (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
8. Nicolás-Ávila JÁ, Adrover JM, Hidalgo A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity (2017) 46:15–28. doi: 10.1016/j.immuni.2016.12.012
9. Klebanoff SJ. Myeloperoxidase: Friend and Foe. J Leukoc Biol (2005) 77:598–625. doi: 10.1189/jlb.1204697
10. Othman A, Sekheri M, Filep JG. Roles of Neutrophil Granule Protein in Orchestrating Inflammation and Immunity. FEBS J (2021) 2021. doi: 10.1111/febs.15803
11. Cassatella MA, Östberg NK, Tamassia N, Soehnlein O. Biological Roles of Neutrophil-Derived Granule Proteins and Cytokines. Trends Immunol (2019) 40:648–64. doi: 10.1016/j.it.2019.05.003
12. Butin-Israeli V, Bui TM, Wiesolek HL, Mascarenhas L, Lee JJ, Mehl LC, et al. Neutrophil-Induced Genomic Instability Impedes Resolution of Inflammation and Wound Healing. J Clin Invest (2019) 129:712–26. doi: 10.1172/JCI122085
13. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
14. Galkina SI, Fedorova NV, Serebryakova MV, Romanova JM, Golyshev SA, Stadnichuk VI, et al. Proteome Analysis Identified Human Neutrophil Membrane Tubulovesicular Extensions (Cytonemes, Membrane Tethers) as Bactericide Trafficking. Biochim Biophys Acta (2012) 1820:1705–14. doi: 10.1016/j.bbagen.2012.06.016
15. Serhan CN, Levy BD. Resolvins in Inflammation: Emergence of the Pro-Resolving Superfamily of Mediators. J Clin Invest (2018) 128:2657–69. doi: 10.1172/JCI97943
16. Peiseler M, Kubes P. More Friend Than Foe: The Emerging Role of Neutrophils in Tissue Repair. J Clin Invest (2019) 129:2629–39. doi: 10.1171/JCI124616
17. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
18. Kolaczkowska E, Kubes P. Neutrophil Recruitment and Function in Health and Inflammation. Nat Rev Immunol (2013) 13:159–75. doi: 10.1038/nri3399
19. Nourshargh S, Alon R. Leukocyte Migration Into Inflamed Tissues. Immunity (2014) 41:694–707. doi: 10.1016/j.immuni.2014.10.008
20. Perretti M, Cooper D, Dalli J, Norling LV. Immune Resolution Mechanisms in Inflammatory Arthritis. Nat Rev Rheumatol (2017) 13:87–99. doi: 10.1038/nrrheum.2016.193
21. Serhan CN, Gupta SK, Perretti M, Godson C, Brennan E, Li Y, et al. The Atlas of Inflammation Resolution (AIR). Mol Aspects Med (2020) 74:100894. doi: 10.1016/j.mam.2020.100894
22. Serhan CN, Savill J. Resolution of Inflammation: The Beginning Programs the End. Nat Immunol (2005) 6:1191–7. doi: 10.1038/ni1276
23. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An Imbalance Between Specialized Pro-Resolving Lipid Mediators and Pro-Inflammatory Leukotrienes Promotes Instability of Atherosclerotic Plaques. Nat Commun (2016) 7:12859. doi: 10.1038/ncomms12859
24. Fredman G, Tabas I. Boosting Inflammation Resolution in Atherosclerosis: The Next Frontiers for Therapy. Am J Pathol (2017) 187:1211–21. doi: 10.1016/j.ajpath.2017.01.018
25. Sekheri M, El Kebir D, Edner N, Filep JG. 15-Epi-LXA 4 and 17-Epi-RvD1 Restore TLR9-Mediated Impaired Neutrophil Phagocytosis and Accelerate Resolution of Lung Inflammation. Proc Natl Acad Sci USA (2020) 117:7971–80. doi: 10.1073/pnas.1920193117
26. Hynes RO. Integrins: Bidirectional, Allosteric Signaling Machines. Cell (2002) 110:673–87. doi: 10.1016/S0092-8674(02)00971-6
27. Dupuy AG, Caron E. Integrin-Dependent Phagocytosis: Spreading From Microadhesion to New Concepts. J Cell Sci (2008) 121:1773–83. doi: 10.1242/jcs.018036
28. El Kebir D, József L, Pan W, Filep JG. Myeloperoxidase Delays Neutrophil Apoptosis Through CD11b/CD18 Integrins and Prolongs Inflammation. Circ Res (2008) 103:352–9. doi: 10.1161/01.RES.0000326772.76822.7a
29. Dzhagalov I, St. John A, He YW. The Antiapoptotic Protein Mcl-1 Is Essential for the Survival of Neutrophils But Not Macrophages. Blood (2007) 109:1620–6. doi: 10.1182/blood-2006-03-013771
30. Mayadas TN, Cullere X. Neutrophil β2 Integrins: Moderators of Life or Death Decisions. Trends Immunol (2005) 26:388–95. doi: 10.1016/j.it.2005.05.002
31. El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 Promotes Phagocytosis-Induced Neutrophil Apoptosis and Accelerates Resolution of Pulmonary Inflammation. Proc Natl Acad Sci USA (2012) 109:14983–8. doi: 10.1073/pnas.1206641109
32. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic Activity of the Caspase-8-FLIP(L) Complex Inhibits RIPK3-Dependent Necrosis. Nature (2011) 471:363–7. doi: 10.1038/nature09852
33. Oberst A, Green DR. It Cuts Both Ways: Reconciling the Dual Roles of Caspase 8 in Cell Death and Survival. Nat Rev Mol Cell Biol (2011) 12:757–63. doi: 10.1038/nrm3214
34. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity (2015) 42:1075–86. doi: 10.1016/j.immuni.2015.05.010
35. Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the Formyl Peptide Receptor (FPR) Family. Pharmacol Rev (2009) 61:119–61. doi: 10.1124/pr.109.001578
36. Chiang N, Serhan CN, Dahlén SE, Drazen JM, Hay DWP, Rovati GE, et al. The Lipoxin Receptor ALX: Potent Ligand-Specific and Stereoselective Actions in Vivo. Pharmacol Rev (2006) 58:463–87. doi: 10.1124/pr.58.3.4
37. El Kebir D, József L, Khreiss T, Pan W, Petasis NA, Serhan CN, et al. Aspirin-Triggered Lipoxins Override the Apoptosis-Delaying Action of Serum Amyloid A in Human Neutrophils: A Novel Mechanism for Resolution of Inflammation. J Immunol (2007) 179:616–22. doi: 10.4049/jimmunol.179.1.616
38. Wan M, Godson C, Guiry PJ, Agerberth B, Haeggstrom JZ. Leukotriene B4/antimicrobial Peptide LL-37 Pro-Inflammatory Circuits are Mediated by BLT1 and FPR2/ALX and are Counterregulated by Lipoxin A4 and Resolvin E1. FASEB J (2011) 25:1697–705. doi: 10.1096/fj.10-175687
39. Leoni G, Nusrat A. Annexin A1: Shifting the Balance Towards Resolution and Repair. Biol Chem (2016) 397:971–9. doi: 10.1515/hsz-2016-0180
40. El Kebir D, József L, Pan W, Wang L, Petasis NA, Serhan CN, et al. 15-Epi-Lipoxin A4 Inhibits Myeloperoxidase Signaling and Enhances Resolution of Acute Lung Injury. Am J Respir Crit Care Med (2009) 180:311–9. doi: 10.1164/rccm.200810-1601OC
41. Bozinovski S, Uddin M, Vlahos R, Thompson M, McQualter JL, Merritt AS, et al. Serum Amyloid A Opposes Lipoxin A4 to Mediate Glucocorticoid Refractory Lung Inflammation in Chronic Obstructive Pulmonary Disease. Proc Natl Acad Sci USA (2012) 109:935–40. doi: 10.1073/pnas.1109382109
42. Cooray SN, Gobbetti T, Montero-Melendez T, McArthur S, Thompson D, Clark AJL, et al. Ligand-Specific Conformational Change of the G-Protein-Coupled Receptor ALX/FPR2 Determines Proresolving Functional Responses. Proc Natl Acad Sci USA (2013) 110:18232–7. doi: 10.1073/pnas.1308253110
43. Filep JG. Biasing the Lipoxin A4/formyl Peptide Receptor 2 Pushes Inflammatory Resolution. Proc Natl Acad Sci USA (2013) 110:18033–4. doi: 10.1073/pnas.1317798110
44. Chen T, Xiong M, Zong X, Ge Y, Zhang H, Wang M, et al. Structural Basis of Ligand Binding Modes at the Human Formyl Peptide Receptor 2. Nat Commun (2020) 11:1208. doi: 10.1038/s41467-020-15009-1
45. Ge Y, Zhang S, Wang J, Xia F, Wan J, Lu J, et al. Dual Modulation of Formyl Peptide Receptor 2 by Aspirin-Triggered Lipoxin Contributes to Its Anti-Inflammatory Activity. FASEB J (2020) 34:6920–33. doi: 10.1096./fj.201903206R
46. Thompson D, McArthur S, Hislop JN, Flower RJ, Perretti M. Identification of a Novel Recycling Sequence in the C-Tail of FPR2/ALX: Association With Cell Protection From Apoptosis. J Biol Chem (2014) 289:36166–78. doi: 10.1074/jbc.M114.612630
47. Soehnlein O, Wantha S, Simsekyilmaz S, Döring Y, Megens RT, Mause SF, et al. Neutrophil-Derived Cathelicidin Protects From Neointimal Hyperplasia. Sci Transl Med (2011) 3:103a98. doi: 10.1126/scitranslmed.3002531
48. Jones HR, Robb CT, Perretti M, Rossi AG. The Role of Neutrophils in Inflammation Resolution. Semin Immunol (2016) 28:137–45. doi: 10.1016/j.smim.2016.03.007
49. Bastian OW, Koenderman L, Alblas J, Leenen LP, Blokhuis TJ. Neutrophils Contribute to Fracture Healing by Synthesizing Fibronectin+ Extracellular Matrix Rapidly After Injury. Clin Immunol (2016) 164:78–84. doi: 10.1016/j.clim.2016.02.001
50. Blázquez-Prieto J, López-Alonso I, Amado-Rodríguez L, Huidobro C, González-López A, Kuebler WM, et al. Impaired Lung Repair During Neutropenia Can be Reverted by Matrix Metalloproteinase-9. Thorax (2018) 73:321–30. doi: 10.1136/thoraxjnl-2017-210105
51. Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, et al. Neutrophils Orchestrate Post-Myocardial Infarction Healing by Polarizing Macrophages Towards a Reparative Phenotype. Eur Heart J (2017) 38:187–97. doi: 10.1093/eurheartj/ehw002
52. Kruger P, Saffarzadeh M, Weber AN, Rieber W, Radsak M, von Bernuth H, et al. Neutrophils: Between Host Defence, Immune Modulation, and Tissue Injury. PloS Pathog (2015) 11:e1004651. doi: 10.1371/journal.ppat.1004651
53. Scapini P, Marini O, Tecchio C, Cassatella MA. Human Neutrophils in the Saga of Cellular Heterogeneity: Insights and Open Questions. Immunol Rev (2016) 273:48–60. doi: 10.1111/imr.12448
54. Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil Heterogeneity: Implications for Homeostasis and Pathogenesis. Blood (2016) 127:2173–81. doi: 10.1182/blood-2016-01-688887
55. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of Neutrophils. Nat Rev Immunol (2019) 19:255–65. doi: 10.1038/s41577-019-0141-8
56. Hidalgo A, Casanova-Ascebes M. Dimensions of Neutrophil Life and Fate. Semin Immunol (2021) 25:101506. doi: 10.1016/j.smim.2021.101506
57. Silvestre-Roig C, Friedlander ZG, Glogauer M, Scapini P. Neutrophil Diversity in Health and Disease. Trends Immunol (2019) 40:565–83. doi: 10.1016/j.it.2019.04.012
58. Xie X, Shi Q, Wu P, Zhang X, Kambara H, Su J, et al. Single-Cell Transcriptome Profiling Reveals Neutrophil Heterogeneity in Homeostasis and Infection. Nat Immunol (2020) 21:1119–33. doi: 10.1038/s41590-020-0736-z
59. Garratt LW. Current Understanding of the Neutrophil Transcriptome in Health and Disease. Cells (2021) 10:2406. doi: 10.3390/cells10092406
60. Jiang K, Sun X, Chen Y, Shen Y, Jarvis JN. RNA Sequencing From Human Neutrophils Reveals Distinct Transcriptional Differences Associated With Chronic Inflammatory States. BMC Med Genomics (2015) 8:55. doi: 10.1186/s12920-015-0128-7
61. Gomez GC, Dang H, Kanke M, Hagan RS, Mock JR, Kelada SNP, et al. Predicted Effects of Observed Changes in the mRNA and microRNA Transcriptome of Lung Neutrophils During S. Pneumoniae Pneumonia in Mice. Sci Rep (2017) 7:11258. doi: 10.1038/s41598-017-11638-7
62. Ai Z, Udalova IA. Transcriptional Regulation of Neutrophil Differentiation and Function During Inflammation. J Leukoc Biol (2020) 107:419–30. doi: 10.1002/JLB.1RU1219-504RR
63. Grieshaber-Bouyer R, Radtke FA, Cunin P, Stifano G, Levescot A, Vijaykumar B, et al. The Neutrotime Transcriptional Signature Defines a Single Continuum of Neutrophils Across Biological Compartments. Nat Commun (2021) 12:2856. doi: 10.1038/s41467-021-22973-9
64. Kuckleburg CJ, Tilkens SB, Santoso S, Newman PJ. Proteinase 3 Contributes to Transendothelial Migration of NB1-Positive Neutrophils. J Immunol (2012) 188:2419–26. doi: 10.4049/jimmunol.1102540
65. Hu N, Westra J, Huitema MG, Bijl M, Brouwer E, Stegeman CA, et al. Coexpression of CD177 and Membrane Proteinase 3 on Neutrophils in Antineutrophil Cytoplasmic Autoantibody-Associated Systemic Vasculitis: Anti-Proteinase 3-Mediated Neutrophil Activation Is Independent of the Role of CD177-Expressing Neutrophils. Arthritis Rheum (2009) 60:1548–57. doi: 10.1002/art.24442
66. Rarok AA, Stegeman CA, Limburg PC, Kallenberg CG. Neutrophil Membrane Expression of Proteinase 3 (PR3) is Related to Relapse in PR3-ANCA-Associated Vasculitis. J Am Soc Nephrol (2002) 13:2232–8. doi: 10.1097/01.ASN.0000028642.26222.00
67. Christofferson G, Vågesjö E, Vandooren J, Lidén M, Massena S, Reinert RB, et al. VEGF-A Recruits a Proangiogenic MMP-9-Delivering Neutrophil Subset That Induces Angiogenesis in Transplanted Hypoxic Tissue. Blood (2012) 120:4653–62. doi: 10.1182/blood-2012-04-421040
68. Adrover JM, del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity (2019) 50:390–402. doi: 10.1016/j.immuni.2019.01.002
69. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, et al. Rhythmic Modulation of the Hematopoietic Niche Through Neutrophil Clearance. Cell (2013) 153:1025–35. doi: 10.1016/j.cell.2013.04.040
70. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil Ageing is Regulated by the Microbiome. Nature (2015) 525:528–32. doi: 10.1038/nature15367
71. Hellebrekers P, Hietbrink F, Vrisekoop, Leenen LPH, Koenderman L. Neutrophil Functional Heterogeneity: Identification of Competitive Phagocytosis. Front Immunol (2017) 8:1498. doi: 10.3389/fimmu.2017.01498
72. Hassani M, Hellebrekers P, Chen N, van Aalst C, Bongers S, Hietbrink F, et al. On the Origin of Low Density Neutrophils. J Leukoc Biol (2020) 107:809–18. doi: 10.1002/JLB.5HR0120-459R
73. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a Phenotypically and Functionally Distinct Population of Long-Lived Neutrophils in a Model of Reverse Endothelial Transmigration. J Leukoc Biol (2006) 79:303–11. doi: 10.1189/jlb.0905496
74. Fortunati E, Kazemier KM, Grutters JC, Koenderman L, Van den Bosch van JMM. Human Neutrophils Switch to an Activated Phenotype After Homing to the Lung Irrespective of Inflammatory Disease. Clin Exp Immunol (2009) 155:559–66. doi: 10.1111/j.1365-2249.2008.03791.x
75. Carmona-Rivera C, Kaplan MJ. Low Density Granulocytes a Distinct Class of Neutrophils in Systemic Autoimmunity. Semin Immunopathol (2013) 35:455–63. doi: 10.1007/s00281-013-0375-7
76. Singh N, Traisak P, Martin KA, Kaplan MJ, Cohen PL, Denny MF. Genomic Alterations in Abnormal Neutrophils Isolated From Adult Patients With Systemic Lupus Erythematosus. Arthritis Res Ther (2014) 16:R165. doi: 10.1186/ar4681
77. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10+ and Immature CD1- Neutrophils Present in G-CSF-Treated Donors Display Opposite Effects on T Cells. Blood (2017) 129:1343–56. doi: 10.1182/blood-2016-04-713206
78. Meghraoui-Kheddar A, Chousterman BG, Guillou N, Barone SM, Granjeaud S, Vallet H, et al. Two New Neutrophil Subsets Define a Discriminating Sepsis Signature. Am J Respir Crit Care Med (2022) 205:46–59. doi: 10.1164/rccm.202104-1027OC
79. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” Versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
80. Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, et al. Transcriptomic Analysis of Comparing Tumor-Associated Neutrophils With Granulocytic Myeloid-Derived Suppressor Cells and Normal Neutrophils. PloS One (2012) 7:e31524. doi: 10.1371/journal.pone.0031524
81. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils Responsive to Endogenous IFN-Beta Regulate Tumor Angiogenesis and Growth in a Mouse Tumor Model. J Clin Invest (2010) 120:1151–64. doi: 10.1172/JCI37223
82. Andzinski L, Kasnitz N, Stahnke S, Wu CF, Gereke M, von Köckritz-Blickwede M, et al. Type I IFNs Induce Anti-Tumor Polarization of Tumor-Associated Neutrophils in Mice and Human. Int J Cancer (2016) 138:1982–93. doi: 10.1002/ijc.29945
83. Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, et al. Origin and Role of a Subset of Tumor-Associated Neutrophils With Antigen-Presenting Cell Features in Early Stage Human Lung Cancer. Cancer Cell (2016) 30:120–35. doi: 10.1016/j.ccell.2016.06.001
84. Saha S, Biswas SK. Tumor-Associated Neutrophils Show Phenotypic and Functional Divergence in Human Lung Cancer. Cancer Cell (2016) 30:11–3. doi: 10.1016/j.ccell.2016.06.016
85. Naranbhai V, Fairfax BP, Makino S, Humburg P, Wong D, Ng E, et al. Genomic Modulators of Gene Expression in Human Neutrophils. Nat Commun (2015) 6:7545. doi: 10.1038/ncomms8545
86. Coit P, Yalavarthi S, Ognenovski M, Zhao W, Hasni S, Wren JD, et al. Epigenome Profiling Reveals Significant DNA Demethylation of Interferon Signature Genes in Lupus Neutrophils. J Autoimmun (2015) 58:59–66. doi: 10.1016/j.jaut.2015.01.004
87. Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil Metabolic Shift During Their Lifecycle: Impact on Their Survival and Activation. Int J Mol Sci (2019) 21:287. doi: 10.3390/ijms21010287
88. Curi R, Levada-Pires AC, Silva EBD, Poma SO, Zambonatto RF, Domenech P, et al. The Critical Role of Cell Metabolism for Essential Neutrophil Functions. Cell Physiol Biochem (2020) 54:629–47. doi: 10.33594/000000245
89. Sadiku P, Willson JA, Ryan EM, Sammut D, Coelho P, Watts ER, et al. Neutrophils Fuel Effective Immune Responses Through Gluconeogenesis and Glycogenesis. Cell Metab (2021) 33:1062–4. doi: 10.1016/j.cmet.2020.11.016
90. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid Mediator Class Switching During Acute Inflammation: Signals in Resolution. Nat Immunol (2001) 2:612–9. doi: 10.1038/89759
91. Lämmermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, Parent CA, et al. Neutrophil Swarms Require LTB4 and Integrins at Sites of Cell Death In Vivo. Nature (2013) 498:371–5. doi: 10.1038/nature12175
92. Kienle K, Lämmermann T. Neutrophil Swarming: An Essential Process of the Neutrophil Tissue Response. Immunol Rev (2016) 273:76–93. doi: 10.1111/imr.12458
93. Lämmermann T. In the Eye of the Neutrophil Swarm-Navigation Signals That Bring Neutrophils Together in Inflamed and Infected Tissues. J Leukoc Biol (2016) 100:55–63. doi: 10.1189/jlb.1MR0915-403
94. Uderhardt S, Martins AJ, Tsang JS, Lämmermann T, Germain RN. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil Driven Inflammatory Damage. Cell (2019) 177:541–55.e17. doi: 10.1016/j.cell.2019.02.028
95. Reátegui E, Jalali F, Khankhel AH, Wong E, Cho H, Lee J, et al. Microscale Arrays for the Profiling of Start and Stop Signals Coordinating Human-Neutrophil Swarming. Nat BioMed Eng (2017) 1:0094. doi: 10.1038/s41551-017-0094
96. Coombs C, Georgantzoglou A, Walker HA, Patt J, Merten N, Poplimont H, et al. Chemokine Receptor Trafficking Coordinates Neutrophil Clustering and Dispersal at Wounds in Zebrafish. Nat Commun (2019) 10:5166. doi: 10.1038/s41467-019-13107-3
97. Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, et al. In Vivo Two-Photon Imaging Reveals Monocyte-Dependent Neutrophil Extravasation During Pulmonary Inflammation. Proc Natl Acad Sci USA (2010) 107:18073–8. doi: 10.1073/pnas.1008737107
98. Kamenyeva O, Boularan C, Kabat J, Cheung GY, Cicala C, Yeh AJ, et al. Neutrophil Recruitment to Lymph Nodes Limits Local Humoral Response to Staphylococcus Aureus. PloS Pathog (2015) 11:e1004827. doi: 10.1371/journal.ppat.1004827
99. Leliefeld PHC, Wessels CM, Leenen LPH, Koenderman L, Pillay J. The Role of Neutrophils in Immune Dysfunction During Severe Inflammation. Crit Care (2016) 20:73. doi: 10.1186/s13054-016-1250-4
100. Jones CN, Dimisko L, Forrest K, Judice K, Poznansky MC, Markmann JF, et al. Human Neutrophils are Primed by Chemoattractant Gradients for Blocking the Growth of Aspergillus Fumigatus. J Infect Dis (2016) 213:465–75. doi: 10.1093/infdis/jiv419
101. Malawista SE, de Boisfleury Chevance A, van Damme J, Serhan CN. Tonic Inhibition of Chemotaxis in Human Plasma. Proc Natl Acad Sci USA (2008) 105:17949–54. doi: 10.1073/pnas.0802572105
102. Malawista SE, de Boisfleury AC, Naccache PH. Inflammatory Gout: Observations Over a Half-Century. FASEB J (2011) 25:4073–8. doi: 10.1096/fj.11-1201ufm
103. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a Phenotypically and Functionally Distinct Population of Long-Lived Neutrophils in a Model of Reverse Endothelial Migration. J Leukoc Biol (2006) 79:303–11. doi: 10.1189/jlb.0905496
104. Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of Inflammation by Retrograde Chemotaxis of Neutrophils in Transgenic Zebrafish. J Leukoc Biol (2006) 80:1281–8. doi: 10.1189/jlb.0506346
105. Lambris JD, Ricklin D, Geisbrecht BV. Complement Evasion by Human Pathogens. Nat Rev Microbiol (2008) 6:132–42. doi: 10.1038/nrmicro1824
106. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The Junctional Adhesion Molecule JAM-C Regulates Polarized Transendothelial Migration of Neutrophils In Vivo. Nat Immunol (2011) 12:761–9. doi: 10.1038/ni.2062
107. Scheiermann C, Colom B, Meda P, Patel NS, Voisin MB, Marrelli A, et al. Junctional Adhesion Molecule-C Mediates Leukocyte Infiltration in Response to Ischemia Reperfusion Injury. Arterioscler Thromb Vasc Biol (2009) 29:1509–15. doi: 10.1161/ATVBAHA.109.187559
108. Nourshargh S, Renshaw SA, Imhof BA. Reverse Migration of Neutrophils: Where, When, How and Why? Trends Immunol (2016) 37:273–86. doi: 10.1016/j.it.2016.03.006
109. Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, et al. Neutrophils Rapidly Migrate via Lymphatics After Mycobacterium Bovis BCG Intradermal Vaccination and Shuttle Live Bacilli to the Draining Lymph Nodes. Blood (2005) 106:1843–50. doi: 10.1182/blood-2005-03-1281
110. Chtanova T, Schaeffer M, Han SJ, van Dooren GG, Nollmann M, Herzmark P, et al. Dynamics of Neutrophil Migration in Lymph Nodes During Infection. Immunity (2008) 29:487–96. doi: 10.1016/j.immuni.2008.07.012
111. Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. Microbe Dependent Lymphatic Migration of Neutrophils Modulates Lymphocyte Proliferation in Lymph Nodes. Nat Commun (2015) 6:7139. doi: 10.1038/ncomms8139
112. Hampton HR, Chtanova T. The Lymph Node Neutrophil. Semin Immunol (2016) 28:129–36. doi: 10.1016/j.smim.2016.03.008
113. Rigby DA, Ferguson DJ, Johnson LA, Jackson DG. Neutrophils Rapidly Transit Inflamed Lymphatic Vessel Endothelium via Integrin-Dependent Proteolysis and Lipoxin-Induced Junctional Retraction. J Leukoc Biol (2015) 98:897–912. doi: 10.1189/jlb.1HI0415-149R
114. Takashima A, Yao Y. Neutrophil Plasticity: Acquisition of Phenotype and Functionality of Antigen-Presenting Cell. J Leukoc Biol (2015) 98:489–96. doi: 10.1189/jlb.1MR1014-502R
115. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, et al. In Vivo Imaging Reveals an Essential Role for Neutrophils in Leishmaniosis Transmitted by Sand Flies. Science (2008) 321:970–4. doi: 10.1126/science.1159194
116. Coombes JL, Charsar BA, Han SJ, Halkias J, Chan SW, Koshy AA, et al. Motile Invaded Neutrophils in the Small Intestine of Toxoplasma Gondii-Infected Mice Reveal a Potential Mechanism for Parasite Spread. Proc Natl Acad Sci USA (2013) 110:E1913–22. doi: 10.1073/pnas.1220272110
117. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human Neutrophil Kinetics: Modeling of Stable Isotope Labeling Data Supports Short Blood Neutrophil Half-Lives. Blood (2016) 127:3431–8. doi: 10.1182/blood-2016-03-700336
118. Tak T, Tesselaar K, Pillay J, Borghans JA, Koenderman L. What’s Your Age Again? Determination of Human Neutrophil Half-Lives Revisited. J Leukoc Biol (2013) 94:595–601. doi: 10.1189/jlb.1112571
119. Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage Phagocytosis of Aging Neutrophils in Inflammation. Programmed Cell Death in the Neutrophil Leads to its Recognition by Macrophages. J Clin Invest (1989) 83:865–75. doi: 10.1172/JCI113970
120. József L, Khreiss T, Filep JG. CpG Motifs in Bacterial DNA Delay Apoptosis of Neutrophil Granulocytes. FASEB J (2004) 18:1776–8. doi: 10.1096/fj.04-2048fje
121. Rajamäki K, Nordström T, Nurmi K, Åkerman KE, Kovanen PT, Öörni K, et al. Extracellular Acidosis is a Novel Danger Signal Alerting Innate Immunity via the NLRP3 Inflammasome. J Biol Chem (2013) 288:13410–9. doi: 10.1074/jbc.M112.426254
122. El Kebir D, de Oliveira Lima Dos Santos E, Mansouri S, Sekheri M, Filep JG. Mild Acidosis Delays Neutrophil Apoptosis via Multiple Signaling Pathways and Acts in Concert With Inflammatory Mediators. J Leukoc Biol (2017) 102:1389–400. doi: 10.1189/jlb.3A0117-041R
123. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1α Expression Regulates the Bactericidal Capacity of Phagocytes. J Clin Invest (2005) 115:1806–15. doi: 10.1172/JCI23865
124. Thompson AA, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, et al. Hypoxia-Inducible Factor 2α Regulates Key Neutrophil Functions in Humans, Mice, and Zebrafish. Blood (2014) 123:366–76. doi: 10.1182/blood-2013-05-500207
125. Matthijsen RA, Huugen D, Hoebers NT, de Vries B, Peutz-Kootstra CJ, Aratani Y, et al. Myeloperoxidase is Critically Involved in the Induction of Organ Damage After Renal Ischemia Reperfusion. Am J Pathol (2007) 171:1743–52. doi: 10.2353/ajpath.2007.070184
126. Brovkovych V, Gao XP, Ong E, Brovkovych S, Brennan ML, Su X, et al. Augmented Inducible Nitric Oxide Synthase Expression and Increased NO Production Reduce Sepsis-Induced Lung Injury and Mortality in Myeloperoxidase-Null Mice. Am J Physiol Lung Cell Mol Physiol (2008) 295:L96–L103. doi: 10.1152/ajplung.00450.2007
127. Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, et al. Pre-B Cell Colony-Enhancing Factor Inhibits Neutrophil Apoptosis in Experimental Inflammation and Clinical Sepsis. J Clin Invest (2004) 113:1318–27. doi: 10.1172/JCI19930
128. Keel M, Ungethüm U, Steckholzer U, Niederer E, Hartung T, Trentz O, et al. Interleukin-10 Counterregulates Proinflammatory Cytokine Induced Inhibition of Neutrophil Apoptosis During Severe Sepsis. Blood (1997) 90:3356–63. doi: 10.1182/blood.V90.9.3356
129. Matute-Bello G, Liles WC, Radella F II, Steinberg KP, Ruzinski JT, Jonas M, et al. Neutrophil Apoptosis in the Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (1997) 156:1969–77. doi: 10.1164/ajrccm.156.6.96-12081
130. Uddin M, Nong G, Ward J, Seumois G, Prince LR, Wilson SJ, et al. Prosurvival Activity for Airway Neutrophils in Severe Asthma. Thorax (2010) 65:684–9. doi: 10.1136/thx.2009.120741
131. Garlichs CD, Eskafi S, Cicha I, Schmeisser A, Walzog B, Raaz D, et al. Delay of Neutrophil Apoptosis in Acute Coronary Syndromes. J Leukoc Biol (2004) 75:828–35. doi: 10.1189/jlb.0703358
132. Jonsson H, Allen P, Peng SL. Inflammatory Arthritis Requires Foxo3a to Prevent Fas Ligand-Induced Neutrophil Apoptosis. Nat Med (2005) 11:666–71. doi: 10.1038/nm1248
133. Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, et al. Cyclin-Dependent Kinase Inhibitors Enhance the Resolution of Inflammation by Promoting Inflammatory Cell Apoptosis. Nat Med (2006) 12:1056–64. doi: 10.1038/nm1468
134. Kumaran Satyanarayanan S, El Kebir D, Soboh S, Butenko S, Sekheri M, Saadi J, et al. IFN-β Is a Macrophage-Derived Effector Cytokine Facilitating the Resolution of Bacterial Inflammation. Nat Commun (2019) 10:3471. doi: 10.1038/s41467-019-10903-9
135. Maimon N, Zamir ZZ, Kalkar P, Zeytuni-Timor O, Schif-Zuck S, Larisch S, et al. The Pro-Apoptotic ARTS Protein Promotes Neutrophil Apoptosis, Efferocytosis and Macrophage Reprogramming in Resolving Inflammation. Apoptosis (2020) 25:558–73. doi: 10.1007/s10495-020-01615-3
136. Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, et al. Essential Role of the C5a Receptor in E. Coli-Induced Oxidative Burst and Phagocytosis Revealed by a Novel Lepirudin-Based Human Whole Blood Model of Inflammation. Blood (2002) 100:1869–77.
137. Freeman SA, Goyette J, Furuya W, Woods EC, Bertozzi CR, Bergmeier W, et al. Integrins Form an Expanding Diffusion Barrier That Coordinates Phagocytosis. Cell (2016) 164:128–40. doi: 10.1016/j.cell.2015.11.048
138. Berger M, Sorensen RU, Tosi MF, Dearborn DG, Döring G. Complement Receptor Expression on Neutrophils at an Inflammatory Site, the Pseudomonas-Infected Lung in Cystic Fibrosis. J Clin Invest (1989) 84:1302–13. doi: 10.1172/JCI114298
139. Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA, et al. Infection Regulates Pro-Resolving Mediators That Lower Antibiotic Requirements. Nature (2012) 484:524–8. doi: 10.1038/nature11042
140. Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in Health and Disease. Nat Rev Immunol (2020) 20:254–67. doi: 10.1038/s41577-019-0240-6
141. Kourtzelis I, Hajishengallis G, Chavakis T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front Immunol (2020) 11:553. doi: 10.3389/fimmu.2020.00553
142. Morioka S, Maueröder C, Ravichandran KS. Living on the Edge: Efferocytosis at the Interface of Homeostasis and Pathology. Immunity (2019) 50:1149–62. doi: 10.1016/j.immuni.2019.04.018
143. Sugimoto MA, Vago JP, Perretti M, Teixeira MM. Mediators of the Resolution of the Inflammatory Response. Trends Immunol (2019) 40:212–27. doi: 10.1016/j.it.2019.01.007
144. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides Released by Apoptotic Cells Acts as a Find-Me Signal to Promote Phagocytic Clearance. Nature (2009) 461:282–6. doi: 10.1038/nature08296
145. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 Channels Mediate ‘Find-Me’ Signal Release and Membrane Permeability During Apoptosis. Nature (2010) 467:863–7. doi: 10.1038/nature09413
146. Bournazou I, Pound JD, Duffin R, Bournazos S, Melville LA, Brown SB, et al. Apoptotic Human Cells Inhibit Migration of Granulocytes via Release of Lactoferrin. J Clin Invest (2009) 119:20–32. doi: 10.1172/JCI36226
147. Ariel A, Serhan CN. New Lives Given by Cell Death: Macrophage Differentiation Following Their Encounter With Apoptotic Leukocytes During the Resolution of Inflammation. Front Immunol (2012) 3:4. doi: 10.3389/fimmu.2012.00004
148. Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, et al. Efferocytosis Induces a Novel SLC Program to Promote Glucose Uptake and Lactate Release. Nature (2018) 563:714–8. doi: 10.1038/s41586-018-0735-5
149. Schif-Zuck S, Gross N, Assi S, Rostoker R, Serhan CN, Ariel A. Saturated-Efferocytosis Generates Pro-Resolving CD11blow Macrophages: Modulation by Resolvins and Glucocorticoids. Eur J Immunol (2011) 41:366–79. doi: 10.1002/eji.201040801
150. Hoodless LJ, Lucas CD, Duffin R, Denvir MA, Haslett C, Tucker CS, et al. Genetic and Pharmacological Inhibition of CDK9 Drives Neutrophil Apoptosis to Resolve Inflammation in Zebrafish In Vivo. Sci Rep (2016) 5:36980. doi: 10.1038/srep36980
151. Kinkead LC, Allen LA. Multifaceted Effects of Francisella Tularensis on Human Neutrophil Function and Lifespan. Immunol Rev (2016) 273:266–81. doi: 10.1111/imr.12445
152. Schwartz JT, Bandyopadhyay S, Kobayashi SD, McCracken J, Whitney AR, De Leo FR, et al. Francisella Tularensis Alters Human Neutrophil Gene Expression: Insights Into the Molecular Basis of Delayed Neutrophil Apoptosis. J Innate Immun (2013) 5:124–36. doi: 10.1159/000342430
153. McCracken JM, Kinkead LC, McCaffrey RL, Allen L-AH. Francisella Tularensis Modulates a Distinct Subset of Regulatory Factors and Sustains Mitochondrial Integrity to Impair Human Neutrophil Apoptosis. J Innate Immun (2016) 8:299–313. doi: 10.1159/000443882
154. Yipp BG, Kubes P. NETosis: How Vital Is It? Blood (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
155. Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The Balancing Act of Neutrophils. Cell Host Microbe (2014) 15:526–36. doi: 10.1016/j.chom.2014.04.011
156. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A. Papayannopoulos V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics During NETosis. Cell Rep (2014) 8:883–96. doi: 10.1016/j.celrep.2014.06.044
157. Sørensen OE, Borregaard N. Neutrophil Extracellular Traps - the Dark Side of Neutrophils. J Clin Invest (2016) 126:1612–20. doi: 10.1172/JCI84538
158. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18:134–47. doi: 10.1038/nri.2017.105
159. Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1promotes Neutrophil Extracellular Trap Formation Through Interactions With Toll-Like Receptor 4. Am J Physiol Lung Cell Mol Physiol (2013) 304:L342–9. doi: 10.1152/ajplung.00151.2012
160. Banerjee S, de Freitas A, Friggeri A, Zmijewski JW, Liu G, Abraham E. Intracellular HMGB1 Negatively Regulates Efferocytosis. J Immunol (2011) 187:4686–94. doi: 10.4049/jimmunol.1101500
161. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J Immunol (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
162. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
163. Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An Extracellular Matrix-Based Mechanism of Rapid Neutrophil Extracellular Trap Formation in Response to Candida Albicans. J Immunol (2013) 190:4136–48. doi: 10.4049/jimmunol.1202671
164. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive Role of Neutrophil Extracellular Traps in Pathogen-Induced Lung Injury. JCI Insight (2018) 3:e98178. doi: 10.1172/jci.insight.98178
165. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of Neutrophil Extracellular Trap Degradation Is Associated With Lupus Nephritis. Proc Natl Acad Sci USA (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
166. Farrera C, Fadeel B. Macrophage Clearance of Neutrophil Extracellular Traps is a Silent Process. J Immunol (2013) 191:2647–56. doi: 10.4049/jimmunol.1300436
167. Lazzaretto B, Fadeel B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J Immunol (2019) 203:2276–90. doi: 10.4049/jimmunol.1800159
168. Chiang N, Sakuma M, Rodriguez AR, Spur BW, Irimia D, Serhan CN. Resolvin T-Series Reduce Neutrophil Extracellular Traps. Blood (2022) 139:1222–33. doi: 10.1182/blood.2021013422
169. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat Med (2007) 13:463–9. doi: 10.1038/nm1565
170. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA Traps Promote Thrombosis. Proc Natl Acad Sci USA (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
171. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular Histones Promote Thrombin Generation Through Platelet-Dependent Mechanisms: Involvement of Platelet TLR2 Andtlr4. Blood (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
172. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular Histones are Mediators of Death Through TLR2 and TLR4 in Mouse Fatal Liver Injury. J Immunol (2011) 187:2626–31. doi: 10.4049/jimmunol.1003930
173. Gupta S, Kaplan MJ. The Role of Neutrophils and NETosis in Autoimmune and Renal Diseases. Nat Rev Nephrol (2016) 12:402–13. doi: 10.1038/nrneph.2016.71
174. Li H, Zhou X, Tan H, Hu Y, Zhang L, Liu S, et al. Neutrophil Extracellular Traps Contribute to the Pathogenesis of Acid-Aspiration-Induced ALI/ARDS. Oncotarget (2018) 9:1772–84. doi: 10.18632/oncotarget.22744
175. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217:e20200652. doi: 10.1084/jem.20200652
176. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5:e138999. doi: 10.1172/jci.insight.138999
177. Middleton EA, He X-Y, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136:1169–79. doi: 10.1182/blood.2020007008
178. Nathan C. Neutrophils and COVID-19: Nots, NETs, and Knots. J Exp Med (2020) 217:e20201439. doi: 10.1084./jem.20201439
179. Tomar B, Anders H-J, Desai J, Mulay SR. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells (2020) 9:1383. doi: 10.3390/cells9061383
180. Schönrich G, Raftery MJ, Samstag Y. Devilishly Radical NETwork in COVID-19: Oxidative Stress, Neutrophil Extracellular Traps (NETs), and T Cell Suppression. Rev Adv Biol Regul (2020) 77:100741. doi: 10.1016/j.jbior.2020.100741
181. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe (2020) 27:883–90.e2. doi: 10.1016/j.chom.2020.04.017
182. Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d'Emal C, et al. Neutrophil Extracellular Traps Infiltrate the Lung Airway, Interstitial, and Vascular Compartments in Severe COVID-19. J Exp Med (2020) 217:e20201012. doi: 10.1084/jem.20201012
183. Nathan C. Neutrophils and Immunity: Challenges and Opportunities. Nat Rev Immunol (2006) 6:173–82. doi: 10.1038/nri1785
184. Cristinziano L, Modestino L, Antonelli A, Marone G, Simon H-U, Varricchi G, et al. Neutrophil Extracellular Traps in Cancer. Semin Cancer Biol (2022) 79:91–204. doi: 10.1016/j.semcancer.2021.07.011
185. Arelaki S, Arampatzioglou A, Kambas K, Papagoras C, Miltiades P, Angelidou I, et al. Gradient Infiltration of Neutrophil Extracellular Traps in Colon Cancer and Evidence for Their Involvement in Tumour Growth. PloS One (2016) 11:e0154484. doi: 10.1371/journal.pone.0154484
186. Schedel F, Mayer-Hain S, Pappelbaum KI, Metze D, Stock M, Goerge T, et al. Evidence and Impact of Neutrophil Extracellular Traps in Malignant Melanoma. Pigment Cell Melanoma Res (2020) 33:63–73. doi: 10.1111/pcmr.12818
187. Millrud CR, Kågedal Å, Kumlien Georén S, Winqvist O, Uddman R, Razavi R, et al. NET-Producing CD16high CD62Ldim Neutrophils Migrate to Tumor Sites and Predict Improved Survival in Patients With HNSCC. Int J Cancer (2017) 140:2557–67. doi: 10.1002/ijc.30671
188. Erpenbeck L, Schon MP. Neutrophil Extracellular Traps: Protagonists of Cancer Progression? Oncogene (2017) 36:2483–90. doi: 10.1038/onc.2016.406
189. Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, et al. Metastatic Growth From Dormant Cells Induced by a Col-I Enriched Fibrotic Environment. Cancer Res (2010) 70:5706–16. doi: 10.1158/0008-5472.CAN-09-2356
190. Pierce BL, Ballard-Barbash R, Bernstein L, Baumgartner RN, Neuhouser ML, Wener MH, et al. Elevated Biomarkers of Inflammation Are Associated With Reduced Survival Among Breast Cancer Patients. J Clin Oncol (2009) 27:3437–44. doi: 10.1200/JCO.2008.18.9068
191. Teijeira Á, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps That Interfere With Immune Cytotoxicity. Immunity (2020) 52:856–71.e8. doi: 10.1016/j.immuni.2020.03.001
192. Cristinziano L, Modestino L, Loffredo S, Varricchi G, Braile M, Ferrara AL, et al. Anaplastic Thyroid Cancer Cells Induce the Release of Mitochondrial Extracellular DNA Traps by Viable Neutrophils. J Immunol (2020) 204:1362–72. doi: 10.4049/jimmunol.1900543
193. Seo JD, Gu J-Y, Jung HS, Kim YJ, Kim HK. Contact System Activation and Neutrophil Extracellular Trap Markers: Risk Factors for Portal Vein Thrombosis in Patients With Hepatocellular Carcinoma. Clin Appl Thromb Hemost (2019) 25:1076029618825310. doi: 10.1177/1076029618825310
194. Bang OY, Chung J-W, Cho YH, Oh MJ, Seo W-K, Kim G-M, et al. Circulating DNAs, a Marker of Neutrophil Extracellular Traposis and Cancer-Related Stroke: The OASIS-Cancer Study. Stroke (2019) 50:2944–7. doi: 10.1161/STROKEAHA.119.026373
195. Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, et al. Chloroquine Reduces Hypercoagulability in Pancreatic Cancer Through Inhibition of Neutrophil Extracellular Traps. BMC Cancer (2018) 18:678. doi: 10.1186/s12885-018-4584-2
196. McDonald B, Spicer J, Giannais B, Fallavollita L, Brodt P, Ferri LE. Systemic Inflammation Increases Cancer Cell Adhesion to Hepatic Sinusoids by Neutrophil Mediated Mechanisms. Int J Cancer (2009) 125:1298–305. doi: 10.1002/ijc.24409
197. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of Neutrophil Extracellular Traps Promotes Cancer Metastasis via CCDC25. Nature (2020) 583:133–8. doi: 10.1038/s41586-020-2394-6
198. Pasparakis M, Vandenabeele P. Necroptosis and its Role in Inflammation. Nature (2015) 517:311–20. doi: 10.1038/nature14191
199. Benarafa C, Simon HU. Role of Granule Proteases in the Life and Death of Neutrophils. Biochem Biophys Res Commun (2017) 482:473–81. doi: 10.1016/j.bbrc.2016.11.086
200. Wang X, He Z, Liu H, Yousefi S, Simon HU. Neutrophil Necroptosis is Triggered by Ligation of Adhesion Molecules Following GM-CSF Priming. J Immunol (2016) 197:4090–100. doi: 10.4049/jimmunol.1600051
201. Wicki S, Gurzeler U, Wei-Lynn Wong W, Jost PJ, Bachmann D, Kaufmann T. Loss of XIAP Facilitates Switch to Tnfα-Induced Necroptosis in Mouse Neutrophils. Cell Death Dis (2016) 7:e2422. doi: 10.1038/cddis.2016.311
202. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in Development, Inflammation, and Disease. Nat Rev Mol Cell Biol (2017) 18:127–36. doi: 10.1038/nrm.2016.149
203. Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and Crystal-Induced Neutrophil Extracellular Trap Formation Involves RIPK1-RIPK3-MLKL Signaling. Eur J Immunol (2016) 46:223–9. doi: 10.1002/eji.201545605
204. Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW, et al. NET Formation can Occur Independently of RIPK3 and MLKL Signaling. Eur J Immunol (2016) 46:178–84. doi: 10.1002/eji.201545615
205. Greenlee-Wacker MC, Kremserová S, Nauseef WM. Lysis of Human Neutrophils by Community-Associated Methicillin-Resistant Staphylococcus Aureus. Blood (2017) 129:3237–44. doi: 10.1182/blood-2017-02-766253
206. Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, De Leo FR, Nauseef WM. Phagocytosis of Staphylococcus Aureus by Human Neutrophils Prevents Macrophage Efferocytosis and Induces Programmed Necrosis. J Immunol (2014) 192:4709–17. doi: 10.4049/jimmunol.1302692
207. Kremserova S, Nauseef WM. Frontline Science: Staphylococcus Aureus Promotes Receptor-Interacting Protein Kinase 3- and Protease-Dependent Production of IL-1β in Human Neutrophils. J Leukoc Biol (2019) 105:437–47. doi: 10.1002/JLB.4HI0918-346R
208. Chen GY, Nuñez G. Sterile Inflammation: Sensing and Reacting to Damage. Nat Rev Immunol (2010) 10:826–37. doi: 10.1038/nri2873
209. Westman J, Grinstein S, Marques PE. Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front Immunol (2020) 10:3030. doi: 10.3389/fimmu.2019.03030
210. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular Danger Signals Guide Neutrophils to Sites of Sterile Inflammation. Science (2010) 330:362–6. doi: 10.1126/science.1195491
211. Gaipl US, Kuenkele S, Voll RE, Beyer TD, Kolowos W, Heyder P, et al. Complement Binding is an Early Feature of Necrotic and a Rather Late Event During Apoptotic Cell Death. Cell Death Differ (2001) 8:327–34. doi: 10.1038/sj.cdd.4400826
212. Blume KE, Soeroes S, Waibel M, Keppeler H, Wesselborg S, Herrmann M, et al. Cell Surface Externalization of Annexin A1 as a Failsafe Mechanism Preventing Inflammatory Responses During Secondary Necrosis. J Immunol (2009) 183:8138–47. doi: 10.4049/jimmunol.0902250
213. Crisford H, Sapey E, Stockley RA. Proteinase 3; a Potential Target in Chronic Obstructive Pulmonary Disease and Other Chronic Inflammatory Diseases. Respir Res (2018) 19:180. doi: 10.1186/s12931-018-0883-z
214. Ley K, Rivera-Nieves J, Sandborn WJ, Shattil S. Integrin-Based Therapeutics: Biological Basis, Clinical Use, and New Drugs. Nat Rev Drug Discov (2016) 15:173–83. doi: 10.1038/nrd.2015.10
215. Wolf D, Anto-Michel N, Blankenbach H, Wiedermann A, Buscher K, Hohmann JD, et al. A Ligand-Specific Blockade of the Integrin Mac-1selectively Targets Pathologic Inflammation While Maintaining Protective Host-Defense. Nat Commun (2018) 9:525. doi: 10.1038/s41467-018-02896-8
216. Wolf D, Hohmann J-D, Wiedermann A, Bledzka K, Blankenbach H, Marchini T, et al. Binding of CD40L to Mac-1’s I-Domain Involves the EQLKKSKTL Motif and Mediates Leukocyte Recruitment and Atherosclerosis–But Does Not Affect Immunity and Thrombosis in Mice. Circ Res (2011) 109:1269–79. doi: 10.1161/CIRCRESAHA.111.247684
217. Fan Z, McArdle S, Marki A, Mikulski Z, Gutierrez E, Engelhardt B, et al. Neutrophil Recruitment Limited by High Affinity Bent β2 Integrin Binding Ligand in Cis. Nat Commun (2016) 7:12658. doi: 10.1038/ncomms12658
218. Wilson ZS, Ahn LB, Serratelli WS, Belley MD, Lomas-Nera J, Sen M, et al. Activated β2 Integrins Restrict Neutrophil Recruitment During Murine Acute Pseudomonal Pneumonia. Am J Respir Cell Mol Biol (2017) 56:620–7. doi: 10.1165/rcmb.2016-0215OC
219. Saggu G, Okubo K, Chen Y, Vattepu R, Tsuboi N, Rosetti F, et al. Cis Interaction Between Sialylated FcgRIIA and the aI-Domain of Mac-1 Limits Antibody-Mediated Neutrophil Recruitment. Nat Commun (2018) 9:5058. doi: 10.1038/s41467-018-07506-1
220. Kelm M, Lehoux S, Azcutia V, Cummings RD, Nusrat A, Parkos CA, et al. Regulation of Neutrophil Function by Selective Targeting of Glycan Epitopes Expressed on the Integrin CD11b/Cd18. FASEB J (2020) 34:2326–43. doi: 10.1096/fj.201902542R
221. Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D, et al. Small Molecule–Mediated Activation of the Integrin CD11b/CD18 Reduces Inflammatory Disease. Sci Signal (2011) 4:ra57. doi: 10.1126/scisignal.2001811
222. Schmid MC, Khan SQ, Kaneda MM, Pathria P, Shephard R, Louis TL, et al. Integrin CD11b Activation Drives Anti-Tumor Innate Immunity. Nat Commun (2018) 9:5379. doi: 10.1038/s41467-018-07387-4
223. Chiang N, Serhan CN. Structural Elucidation and Physiologic Functions of Specialized Pro-Resolving Mediators and Their Receptors. Mol Aspects Med (2017) 58:114–29. doi: 10.1016/j.mam.2017.03.005
224. Brancaleone V, Mitidieri E, Flower RJ, Cirino G, Perretti M. Annexin A1 Mediates Hydrogen Sulfide Properties in the Control of Inflammation. J Pharmacol Exp Ther (2014) 351:96–104. doi: 10.1124/jpet.114.217034
225. Mustafa M, Zarrough A, Bolstad AI, Lygre H, Mustafa K, Hasturk H, et al. Resolvin D1 Protects Periodontal Ligament. Am J Physiol Cell Physiol (2013) 305:C673–9. doi: 10.1152/ajpcell.00242.2012
226. Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 Regulates Interleukin 23, Interferon-Gamma and Lipoxin A4 to Promote the Resolution of Allergic Inflammation. Nat Immunol (2008) 9:873–9. doi: 10.1038/ni.1627
227. Yazid S, Leoni G, Getting SJ, Cooper D, Solito E, Perretti M, et al. Antiallergic Cromones Inhibit Neutrophil Recruitment Onto Vascular Endothelium via Annexin-A1mobilization. Arterioscler Thromb Vasc Biol (2010) 30:1718–24. doi: 10.1161/ATVBAHA.110.209536
228. Perretti M, D’Acquisto F. Annexin A1 and Glucocorticoids as Effectors of the Resolution of Inflammation. Nat Rev Immunol (2009) 9:62–70. doi: 10.1038/nri2470
229. Yona S, Heinsbroek SE, Peiser L, Gordon S, Perretti M, Flower RJ. Impaired Phagocytic Mechanism in Annexin 1 Null Macrophages. Br J Pharmacol (2006) 148:469–77. doi: 10.1038/sj.bjp.0706730
230. McArthur S, Gobbetti T, Kusters DH, Reutelingsperger CP, Flower RJ, Perretti M. Definition of a Novel Pathway Centered on Lysophosphatidic Acid to Recruit Monocytes During the Resolution Phase of Tissue Inflammation. J Immunol (2015) 195:1139–51. doi: 10.4049/jimmunol.1500733
231. Li Y, Cai L, Wang H, Wu P, Gu W, Chen Y, et al. Pleiotropic Regulation of Macrophage Polarization and Tumorigenesis by Formyl Peptide Receptor-2. Oncogene (2011) 30:3887–99. doi: 10.1038/onc.2011.112
232. McArthur S, Juban G, Gobbetti T, Desgorges T, Theret M, Gondin J, et al. Annexin A1 Drives Macrophage Skewing to Accelerate Muscle Regeneration Through AMPK Activation. J Clin Invest (2020) 130:1156–67. doi: 10.1172/JCI124635
233. Tsai WH, Chien HY, Shih CH, Lai SL, Li IT, Hsu SC, et al. Annexin A1 Mediates the Anti-Inflammatory Effects During the Granulocytic Differentiation Process in All-Trans Retinoic Acid-Treated Acute Promyleocytic Leukemic Cells. J Cell Physiol (2012) 227:3661–9. doi: 10.1002/jcp.24073
234. Leoni G, Neumann PA, Kamaly N, Quiros M, Nishio H, Jones HR, et al. Annexin A1-Containing Extracellular Vesicles and Polymeric Nanoparticles Promote Epithelial Wound Repair. J Clin Invest (2015) 125:1215–27. doi: 10.1172/JCI76693
235. Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, et al. Neutrophil-Derived Microvesicles Enter Cartilage and Protect the Joint in Inflammatory Arthritis. Sci Transl Med (2015) 7:315ra190. doi: 10.1126/scitranslmed.aac5608
236. József L, Zouki C, Petasis NA, Serhan CN, Filep JG. Lipoxin A4 and Aspirin-Triggered 15-Epi-Lipoxin A4 Inhibit Peroxynitrite Formation, NF-κb and AP-1 Activation, and IL-8 Gene Expression in Human Leukocytes. Proc Natl Acad Sci USA (2002) 99:13266–71. doi: 10.1073/pnas.202296999
237. Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, et al. Multi-Pronged Inhibition of Airway Hyper-Responsiveness and Inflammation by Lipoxin A4. Nat Med (2002) 8:1018–23. doi: 10.1038/nm748
238. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting Edge: Lipoxins Rapidly Stimulate Nonphlogistic Phagocytosis of Apoptotic Neutrophils by Monocyte-Derived Macrophages. J Immunol (2000) 164:1663–7. doi: 10.4049/jimmunol.164.4.1663
239. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and Protectin D1 Activate Inflammation-Resolution Programmes. Nature (2007) 447:869–74. doi: 10.1038/nature05877
240. Karp CL, Flick LM, Park KW, Softic S, Greer TM, Keledjian R, et al. Defective Lipoxin-Mediated Anti-Inflammatory Activity in the Cystic Fibrosis Airway. Nat Immunol (2004) 5:388–92. doi: 10.1038/ni1056
241. Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, Iwamoto R, et al. The Lipid Mediator Protectin D1 Inhibits Influenza Virus Replication and Improves Severe Influenza. Cell (2013) 153:112–25. doi: 10.1016/j.cell.2013.02.027
242. Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and Signature Profiles for Pro-Resolving and Inflammatory Lipid Mediators in Human Tissue. Am J Physiol Cell Physiol (2014) 307:C39–54. doi: 10.1152/ajpcell.00024.2014
243. Dalli J, Colas RA, Quintana C, Barragan-Bradford D, Huwitz S, Levy BD, et al. Human Sepsis Eicosanoid and Proresolving Lipid Mediator Temporal Profiles: Correlations With Survival and Clinical Outcomes. Crit Care Med (2017) 45:58–68. doi: 10.1097/CCM.0000000000002014
244. Norling LV, Spite M, Yang R, Flower RJ, Perretti M, Serhan CN. Cutting Edge: Humanized Nano-Proresolving Medicines Mimic Inflammation-Resolution and Enhance Wound Healing. J Immunol (2011) 186:5543–7. doi: 10.4049/jimmunol.1003865
245. The Forsyth Institute. Safety and Preliminary Efficacy of Lipoxin Analog BLXA4-ME Oral Rinse for the Treatment of Gingivitis (Blxa4). Available at: https://clinicaltrials.gov/ct2/show/NCT02342691 (Accessed January 25, 2022).
246. Corminboeuf O, Leroy X. FPR2/ALXR Agonists and the Resolution of Inflammation. J Med Chem (2015) 58:537–59. doi: 10.1021/jm501051x
247. de Gaetano M, Butler E, Gahan K, Zanetti A, Marai M, Chen J, et al. Asymmetric Synthesis and Biological Evaluation of Imidazole- and Oxazole-Containing Synthetic Lipoxin A4 Mimetics (Slxms). Eur J Med Chem (2019) 162:80–108. doi: 10.1016/j.ejmech.2018.10.049
248. Heo SC, Kwon YW, Jang IH, Jeong GO, Lee TW, Yoon JW, et al. Formyl Peptide Receptor 2 is Involved in Cardiac Repair After Myocardial Infarction Through Mobilization of Circulating Angiogenic Cells. Stem Cells (2017) 35:654–65. doi: 10.1002/stem.2535
249. Stalder AK, Lott D, Strasser DS, Cruz HG, Krause A, Groenen PMA, et al. Biomarker-Guided Clinical Development of the First-in-Class Anti-Inflammatory FPR2/ALX Agonist ACT-389949. Br J Clin Pharmacol (2017) 83:476–86. doi: 10.1111/bcp.13149
250. Asahina Y, Wurtz NR, Arakawa K, Carson N, Fujii K, Fukuchi K, et al. Discovery of BMS-986235/LAR-1219: A Potent Formyl Peptide Receptor 2 (FPR2) Selective Agonist for the Prevention of Heart Failure. J Med Chem (2020) 63:9003–19. doi: 10.1021/acs.jmedchem.9b02101
251. Lucas CD, Dorward DA, Tait MA, Fox S, Marwick JA, Allen KC, et al. Downregulation of Mcl-1 has Anti-Inflammatory Pro-Resolution Effects and Enhances Bacterial Clearance From the Lung. Mucosal Immunol (2014) 7:857–68. doi: 10.1038/mi.2013.102
252. Leitch AE, Lucas CD, Marwick JA, Duffin R, Haslett C, Rossi AG. Cyclin-Dependent Kinases 7 and 9 Specifically Regulate Neutrophil Transcription and Their Inhibition Drives Apoptosis to Promote Resolution of Inflammation. Cell Death Differ (2012) 19:1950–61. doi: 10.1038/cdd.2012.80
253. Dorward DA, Felton JM, Robb CT, Craven T, Kipari T, Walsh TS, et al. The Cyclin-Dependent Kinase Inhibitor AT7519 Accelerates Neutrophil Apoptosis in Sepsis-Related Acute Respiratory Distress Syndrome. Thorax (2017) 72:182–5. doi: 10.1136/thoraxjnl-2016-209229
254. Teixeira RAP, Mimura KK, Araujo LP, Greco KV, Oliani SM. The Essential Role of Annexin A1 Mimetic Peptide in the Skin Allograft Survival. J Tissue Eng Regener Med (2016) 10:E44–53. doi: 10.1002/term.1773
255. Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC, et al. Annexin A1 Modulates Natural and Glucocorticoid-Induced Resolution of Inflammation by Enhancing Neutrophil Apoptosis. J Leukoc Biol (2012) 92:249–58. doi: 10.1189/jlb.0112008
256. Guo RF, Ward PA. Role of C5a in Inflammatory Responses. Annu Rev Immunol (2005) 23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835
257. McGrath EE, Marriott HM, Lawrie A, Francis SE, Sabroe I, Renshaw SA, et al. TNF-Related Apoptosis-Inducing Ligand (TRAIL) Regulates Inflammatory Neutrophil Apoptosis and Enhances Resolution of Inflammation. J Leukoc Biol (2011) 90:855–65. doi: 10.1189/jlb.0211062
258. Witko-Sarsat V, Mocek J, Bouayad D, Tamassia N, Ribeil JA, Candalh C, et al. Proliferating Cell Nuclear Antigen Acts as a Cytoplasmic Platform Controlling Human Neutrophil Survival. J Exp Med (2010) 207:2631–45. doi: 10.1084/jem.20092241
259. Martin C, Ohayon D, Alkan M, Mocek J, Pederzoli-Ribeil M, Candalh C, et al. Neutrophil-Expressed P21/Waf1 Favors Inflammation Resolution in Pseudomonas Aeruginosa Infection. Am J Respir Cell Mol Biol (2016) 54:740–50. doi: 10.1165/rcmb.2015-0047OC
260. Arial A, Fredman G, Sun Y-P, Kantarci A, Van Dyke TE, Luster AD, et al. Apoptotic Neutrophils and T Cells Sequester Chemokines During Immune Response Resolution Through Modulation of CCR5 Expression. Nat Immunol (2006) 7:1209–16. doi: 10.1038/ni1392
261. Karbian N, Abutbul A, el-Amore R, Eliaz R, Beeri R, Reicher B, et al. Apoptotic Cell Therapy for Cytokine Storm Associated With Acute Severe Sepsis. Cell Death Dis (2020) 11:535. doi: 10.1038/s41419-020-02748-8
262. Krysko DV, Denecker G, Festjens N, Gabriels S, Parthoens E, D'Herde K, et al. Macrophages Use Different Internalization Mechanisms to Clear Apoptotic and Necrotic Cells. Cell Death Differ (2006) 13:2011–22. doi: 10.1038/sj.cdd.4401900
263. Grégoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F, et al. Impaired Efferocytosis and Neutrophil Extracellular Trap Clearance by Macrophages in ARDS. Eur Respir J (2018) 52:1702590. doi: 10.1183/13993003.02590-2017
264. Bae H-B, Zmijewski JW, Deshane JS, Tadie JM, Chaplin DD, Takashima S, et al. AMP-Activated Protein Kinase Enhances the Phagocytic Ability of Macrophages and Neutrophils. FASEB J (2011) 25:4358–68. doi: 10.1096/fj.11-190587
265. Lee JS, Shin E-C. The Type I Interferon Response in COVID-19: Implications for Treatment. Nat Rev Immunol (2020) 20:585–6. doi: 10.1038/s41577-020-00429-3
266. Patel S, Kumar S, Jyoti A, Srinag BS, Keshari RS, Saluja R, et al. Nitric Oxide Donors Release Extracellular Traps From Human Neutrophils by Augmenting Free Radical Generation. Nitric Oxide (2010) 22:226–34. doi: 10.1016/j.niox.2010.01.001
267. Zheng W, Warner R, Ruggeri R, Su C, Cortes C, Skoura A, et al. PF-1355, a Mechanism-Based Myeloperoxidase Inhibitor, Prevents Immune Complex Vasculitis and Anti-Glomerular-Basement Membrane Glomerulonephritis. J Pharmacol Exp Ther (2015) 353:288–98. doi: 10.1124/jpet.114.221788
268. Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, et al. N-α-Benzoyl-N5-(2-Chloro-1-Iminoethyl)-L-Ornithine Amide, a Protein Arginine Deiminase Inhibitor, Reduces the Severity of Murine Collagen-Induced Arthritis. J Immunol (2011) 186:4396–404. doi: 10.4049/jimmunol.1001620
269. Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, et al. Peptidylarginine Deiminase Inhibition is Immunomodulatory and Vasoprotective in Murine Lupus. J Clin Invest (2012) 123:2981–93. doi: 10.1172/JCI67390
270. Knight JS, Luo W, O'Dell AA, Yalavarthi S, Zhao W, Subramanian V, et al. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ Res (2014) 114:947–56. doi: 10.1161/CIRCRESAHA.114.303312
271. Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, et al. Inhibition of PAD4 Activity Is Sufficient to Disrupt Mouse and Human NET Formation. Nat Chem Biol (2015) 11:189–91. doi: 10.1038/nchembio.1735
272. Cedervall J, Dragomir A, Saupe F, Zhang Y, Ärnlöv J, Larsson E, et al. Pharmacological Targeting of Peptidylarginine Deiminase 4 Prevents Cancer-Associated Kidney Injury in Mice. Oncoimmunology (2017) 6:e1320009. doi: 10.1080/2162402X.2017.1320009
273. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is Essential for Antibacterial Innate Immunity Mediated by Neutrophil Extracellular Traps. J Exp Med (2010) 207:1853–62. doi: 10.1084/jem.20100239
274. Martinod K, Fuchs TA, Zitomersky NL, Wong SL, Demers M, Gallant M, et al. PAD4-Deficiency Does Not Affect Bacteremia in Polymicrobial Sepsis and Ameliorates Endotoxemic Shock. Blood (2015) 125:1948–56. doi: 10.1182/blood-2014-07-587709
275. Cherpokova D, Jouvene CC, Libreros S, DeRoo EP, Chu L, de la Rosa X, et al. Resolvin D4 Attenuates the Severity of Pathological Thrombosis in Mice. Blood (2019) 134:1458–68. doi: 10.1182/blood.2018886317
276. Catz SD, McLeish KR. Therapeutic Targeting of Neutrophil Exocytosis. J Leukoc Biol (2020) 107:393–408. doi: 10.1002/JLB.3RI0120-645R
277. Johnson JL, Ramadass M, He J, Brown SJ, Zhang J, Abgaryan L, et al. Identification of Neutrophil Exocytosis Inhibitors (Nexinhibs), Small Molecule Inhibitors of Neutrophil Exocytosis and Inflammation: Druggability of the Small GTPase Rab27a. J Biol Chem (2016) 291:25965–82. doi: 10.1074/jbc.M116.741884
278. Mejias JC, Forrest OA, Margaroli C, Frey Rubio DA, Viera L, Li J, et al. Neutrophil-Targeted, Protease-Activated Pulmonary Drug Delivery Blocks Airway and Systemic Inflammation. JCI Insight (2019) 4:e131468. doi: 10.1172/jci.insight.131468
279. Uriarte SM, Rane MJ, Merchant ML, Jin S, Lentsch AB, Ward RA, et al. Inhibition of Neutrophil Exocytosis Ameliorates Acute Lung Injury in Rats. Shock (2015) 39:286–92. doi: 10.1097/SHK.0b013e318282c9a1
280. Bai J, Tang L, Lomas-Neira J, Chen Y, McLeish KR, Uriarte SM, et al. TAT-SNAP-23 Treatment Inhibits the Priming of Neutrophil Functions Contributing to Shock and/or Sepsis-Induced Extra-Pulmonary Acute Lung Injury. Innate Immun (2015) 21:42–54. doi: 10.1177/1753425913516524
281. Tremblay GM, Jenelle MF, Bourbonnais Y. Anti-Inflammatory Activity of Neutrophil Elastase Inhibitors. Curr Opin Investig Drugs (2003) 4:556–65.
282. Potey PMD, Rossi AG, Lucas CD, Dorward DA. Neutrophils in the Initiation and Resolution of Acute Pulmonary Inflammation: Understanding Biological Function and Therapeutic Potential. J Pathol (2019) 247:672–85. doi: 10.1002/path.5221
283. Pu S, Wang D, Liu D, Zhao Y, Qi D, He J, et al. Effect of Sivelastat Sodium in Patients With Acute Lung Injury or Acute Respiratory Distress Syndrome: A Meta-Analysis of Randomized Controlled Trials. BMC Pulm Med (2017) 17:148. doi: 10.1186/s12890-017-0498-z
284. Sayah DM, Mallavia B, Liu F, Ortiz-Muñoz G, Caudrillier A, Der Hovanessian A, et al. Neutrophil Extracellular Traps are Pathogenic in Primary Graft Dysfunction After Lung Transplantation. Am J Respir Crit Care Med (2015) 191:455–63. doi: 10.1164/rccm.201406-1086OC
285. Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A, et al. Neutrophil Extracellular Traps Accumulate in Peripheral Blood Vessels and Compromise Organ Function in Tumor-Bearing Animals. Cancer Res (2015) 75:2653–62. doi: 10.1158/0008-5472.CAN-14-3299
286. Macanovic M, Sinicropi D, Shak S, Baughman S, Thiru S, Lachmann PJ. The Treatment of Systemic Lupus Erythematosus (SLE) in NZB/W F1 Hybrid Mice: Studies With Recombinant Murine DNase and With Dexamethasone. Clin Exp Immunol (1996) 106:243–52. doi: 10.1046/j.1365-2249.1996.d01-839.x
287. Pottecher J. Inhaled Dornase Alpha to Reduce Respiratory Failure After Severe Trauma. Available at: https://clinicaltrials.gov/ct2/show/NCT03368092 (Accessed January 20, 2022).
288. Sponsor. Protalix. Safety, Tolerability and Pharmacokinetics Study of AIR DNAse Administered by Inhalation to Healthy Adult Volunteers. Available at: https://clinicaltrials.gov/ct2/show/NCT02605590 (Accessed February 15, 2022).
289. Sponsor. Protalix. Study to Evaluate the Safety Tolerability Pharmacokinetics and Exploratory Efficacy Parameters of AIR DNase in Patients With Cystic Fibrosis Previously Treated With Pulmozyme. Available at: https://clinicaltrials.gov/ct2/show/NCT02722122 (Accessed February 15, 2022).
290. Fuchs TA, Jiménez-Alcázar M, Göbel J, Englert H. US patent application US 2020/0024585 A1 (2019).
Keywords: neutrophil, neutrophil trafficking, apoptosis, neutrophil extracellular trap, pro-resolving mediators, GPCRs, resolution of inflammation
Citation: Filep JG (2022) Targeting Neutrophils for Promoting the Resolution of Inflammation. Front. Immunol. 13:866747. doi: 10.3389/fimmu.2022.866747
Received: 31 January 2022; Accepted: 21 February 2022;
Published: 16 March 2022.
Edited by:
Francesca Granucci, University of Milano-Bicocca, ItalyReviewed by:
Nicola Tamassia, University of Verona, ItalyCopyright © 2022 Filep. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: János G. Filep, amFub3MuZy5maWxlcEB1bW9udHJlYWwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.