 Enio Setsuo Arakaki Pacini
Enio Setsuo Arakaki Pacini Naiara Ayako Satori
Naiara Ayako Satori Edwin Kerry Jackson
Edwin Kerry Jackson Rosely Oliveira Godinho
Rosely Oliveira Godinho- 1Division of Cellular Pharmacology, Department of Pharmacology, Universidade Federal de São Paulo, São Paulo, Brazil
- 2Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Adenosine is a purine nucleoside that, via activation of distinct G protein-coupled receptors, modulates inflammation and immune responses. Under pathological conditions and in response to inflammatory stimuli, extracellular ATP is released from damaged cells and is metabolized to extracellular adenosine. However, studies over the past 30 years provide strong evidence for another source of extracellular adenosine, namely the “cAMP-adenosine pathway.” The cAMP-adenosine pathway is a biochemical mechanism mediated by ATP-binding cassette transporters that facilitate cAMP efflux and by specific ectoenzymes that convert cAMP to AMP (ecto-PDEs) and AMP to adenosine (ecto-nucleotidases such as CD73). Importantly, the cAMP-adenosine pathway is operative in many cell types, including those of the airways. In airways, β2-adrenoceptor agonists, which are used as bronchodilators for treatment of asthma and chronic respiratory diseases, stimulate cAMP efflux and thus trigger the extracellular cAMP-adenosine pathway leading to increased concentrations of extracellular adenosine in airways. In the airways, extracellular adenosine exerts pro-inflammatory effects and induces bronchoconstriction in patients with asthma and chronic obstructive pulmonary diseases. These considerations lead to the hypothesis that the cAMP-adenosine pathway attenuates the efficacy of β2-adrenoceptor agonists. Indeed, our recent findings support this view. In this mini-review, we will highlight the potential role of the extracellular cAMP-adenosine pathway in chronic respiratory inflammatory disorders, and we will explore how extracellular cAMP could interfere with the regulatory effects of intracellular cAMP on airway smooth muscle and innate immune cell function. Finally, we will discuss therapeutic possibilities targeting the extracellular cAMP-adenosine pathway for treatment of these respiratory diseases.
Introduction
Adenosine is an endogenous purine nucleoside that, via activation of specific adenosine receptors, modulates inflammation and immune responses (1–3). Adenosine receptors are seven transmembrane G-protein coupled receptors (GPCRs) (4, 5), and consist of a family of four adenosine receptor subtypes called A1, A2A, A2B and A3. These four receptors are encoded by different genes and present high affinities for the α subunit (Gα) of heterotrimeric G-proteins that regulate adenylyl cyclase (AC) activity (6). A1 and A3 receptors are preferentially coupled to the inhibitory Gα (Gi/o) subunit that inhibits AC1, AC5 and AC6, decreasing 3’,5’-cAMP (from here forward referred to as simply cAMP) production, whereas A2A and A2B are strongly coupled to stimulatory Gα (Gs) subunits, which can activate all nine membrane-bound AC isoforms, increasing intracellular cAMP concentrations (7, 8) and triggering one or more cAMP-dependent intracellular signaling pathways (9). cAMP-dependent protein kinase (PKA) is by far the best studied effector of cAMP signaling and regulates many cellular processes including cell proliferation and differentiation, among others (10). By interacting with specific domains in the PKA regulatory subunits, cAMP releases the two catalytic PKA subunits to phosphorylate serine and/or threonine residues in target proteins (11). A2 receptor signaling may also be mediated by cAMP/EPAC (exchange protein directly activated by cAMP) pathways (12). Notably, adenosine receptors can also activate other signaling molecules via canonical GPCR pathways, such as phospholipase C β (PLC β) and Ca2+ via activation of A1, A2A and A3 receptors (4) or by G protein-independent mechanisms mediated by β-arrestin (A1 and A2) (13).
Although cAMP is capable of diffusing throughout the cell, cAMP signaling occurs in microdomains or even nanodomains (14), which are at least in part created by intracellular phosphodiesterases (PDE), metallohydrolases involved in the degradation of cyclic nucleotides and distributed in 11 families (PDE 1–11). While members of PDE 4, 7 and 8 families selectively hydrolyze cAMP, PDE 1-3 and 10-11 are able to hydrolyze both cAMP and 3’,5’-cGMP (15). Intracellular compartmentalization of cAMP signaling also involves A kinase anchoring proteins (AKAPs), which bind to specific domains of PKA regulatory subunits and thereby guide PKA to different subcellular location (16). AKAPs also associate with PDEs, thus influencing the phosphorylation of target proteins and the termination of PKA activation.
Adenosine and Inflammatory/Immune Responses
Several studies associate adenosine with a pro-inflammatory response (17), but many others have linked high concentrations of extracellular adenosine to a protective anti-inflammatory (18–20) and pro-resolving functions (2, 21). It seems that adenosine can limit the progression of inflammatory processes by reducing leukocyte infiltration and the production of inflammatory mediators, as well as by inducing apoptosis of inflammatory cells and promoting tissue repair; for review see (22). In general, these protective effects of adenosine have been associated with A2A receptor activation, since they are drastically reduced in A2A-deficient mice, resulting in increased tissue damage and accumulation of pro-inflammatory cytokines (23, 24).
In fact, because adenosine binds with different affinities to its receptor subtypes (Ki: A1 = ~100 nM; A2A = ~310 nM; A3 = ~290 nM and A2B = ~15,000 nM) (4), its final effect will depend on both the receptor subtype expressed in the target cell and the extracellular concentration of adenosine. Thus, while at early stages of inflammation, neutrophil recruitment, phagocytosis and adhesion to the vascular endothelium are promoted by low concentrations of adenosine via activation of A1 and A3 receptors (25–27), during the healing phase, neutrophil recruitment is inhibited by high concentrations of adenosine via A2A receptors (28).
By activating A2A and A2B receptors, adenosine inhibits inflammatory functions of neutrophils, reducing phagocytosis, degranulation, production of reactive oxygen species (ROS) (25, 29–32) and release of pro-inflammatory mediators (33–35). Via A2A and A2B receptors, adenosine also inhibits monocyte proliferation and differentiation into macrophages, reduces macrophage phagocytosis, attenuates the oxidative burst, and decreases the production of pro-inflammatory mediators such as TNF-α and IL-12 (36). Regarding lymphocytes, activation of A2A receptors by adenosine reduces the production of inflammatory cytokines by T helper cells (37), and inhibits the development and activation of effector cells (38). Finally, adenosine also influences mast cells and eosinophils (17), important players in physiological innate immunity and allergic inflammatory diseases.
Adenosine Signaling and the Regulation of Airway Inflammatory Diseases
Many of adenosine effects on airways involve modulation of intracellular cAMP concentrations on innate and adaptive immune cells (39). Although complex, in general, elevated intracellular cAMP concentrations mediate anti-inflammatory effects and immune suppression; for review see (40). By activating PKA, cAMP inhibits release of pro-inflammatory cytokines from dendritic cells (IL-12 and TNF-α) (41) and macrophages (TNF-α, MIP-1α, and LTB4) (42). cAMP also inhibits T cell chemotaxis (43) and antigen-stimulated B cell proliferation (44).
In the airways, the extracellular adenosine concentrations are relatively low under normal physiological conditions, ranging from 20 to 300 nM (45, 46). However, adenosine concentrations drastically increase in bronchoalveolar lavage fluid and exhaled breath condensate in patients with asthma, COPD and cystic fibrosis (18, 47–50), reaching micromolar to millimolar range. These high concentrations of adenosine are associated with bronchoconstriction and airway smooth muscle hyperresponsiveness (51–54), indicating the involvement of adenosine in the pathophysiology of pulmonary inflammatory diseases. Adenosine induces bronchoconstriction mainly through direct activation of A1 receptors expressed by smooth muscle cells (55). Other studies have associated adenosine-induced bronchoconstriction with release of inflammatory mediators, such as histamine and leukotrienes from mast cells; accordingly, adenosine-induced airway smooth muscle contraction is attenuated by H1‐histamine and CysLT1 leukotriene antagonists (56–58). Moreover, indirect bronchoconstriction induced by adenosine may also involve activation of A1 receptors on vagal afferent neurons. The evidence for this is that vagotomy and inhibitors of cholinergic pathways attenuate adenosine-induced bronchoconstriction (59).
The contribution of adenosine to airway inflammation is multifaceted and involves different receptors and intracellular signaling pathways in dendritic cells, monocytes, macrophages, neutrophils, bronchial smooth muscle and epithelial cells. Pro-inflammatory or anti-inflammatory airway responses are largely dependent on adenosine concentration and disease state over time. Particularly during acute airway inflammation, adenosine induces pro-resolving and tissue-protective actions, while in chronic airway inflammation it promotes deleterious effects such as tissue damage, fibrosis and release of pro-inflammatory cytokines (60, 61). Activation of A2B receptors induces release of pro-inflammatory cytokines such as IL-6 from bronchial smooth muscle cells (62) and lung fibroblasts (63). Adenosine also affects the function of airway epithelial cells (e.g., ciliated cells, mucous cells, secretory cells, and basal cells) (64). While activation of A1 receptors increases production and secretion of mucus (65, 66) and modulates Cl- transport in normal and cystic fibrosis human airway epithelial cells (67, 68), activation of A2A receptors elicits a robust release of pro-inflammatory IL-6, IL-8 and TNF-α from airway epithelia (60, 69, 70). Adenosine also potentiates the recruitment of eosinophils and mast cells (53, 71) and induces airway inflammation by stimulating mast cell degranulation and release of pro-inflammatory mediators such as of IL-5, IL-13 as well as IL-4 (72, 73).
Investigations using adenosine deaminase (ADA)-deficient mice have also provided compelling evidence that accumulation of adenosine in the lung can lead to the development and exacerbation of chronic lung disease (74). ADA-deficient mice exhibit severe pulmonary inflammation and pathological features resembling those seen in asthma and chronic obstructive pulmonary diseases. These features include accumulation of macrophages and eosinophils, enlargement of alveolar spaces, increased production of mucus, higher concentrations of pro-inflammatory cytokines such as IL-5 and IL-13 and increased mast cell degranulation (75–77). Interestingly, all of these changes can be reversed by lowering the adenosine concentrations or by adenosine receptor antagonists (75, 78).
Extracellular cAMP as a Source of Adenosine
The canonical pathway of adenosine formation involves sequential dephosphorylation of ATP into ADP and AMP mediated by intra- or extracellular nucleoside triphosphate diphosphohydrolases (NTPDases), alkaline phosphatases (APs), nucleotide pyrophosphatase/phosphodiesterases (NPPs) and 5’-nucleotidases (5-NTs) (79). A second pathway requires intracellular hydrolysis of S-adenosyl-homocysteine by the enzyme S-adenosyl-L-homocysteine hydrolase (80), and a third mechanism is mediated by the metabolism of extracellular NAD+ to adenosine by a CD38/CD203a/CD73 ectoenzymatic pathway (81).
In addition to the aforementioned pathways of adenosine production, cAMP is now widely recognized as an important source of extracellular adenosine (82). This biochemical route was originally described in the kidney by Jackson in the early 1990s (83) who named this mechanism the “extracellular cAMP-adenosine pathway” (84, 85). This pathway involves the sequential metabolism of extracellular cAMP to 5’-AMP and adenosine by ecto-phosphodiesterases (ecto-PDE) and ecto-5’-nucleotidases (CD73), respectively (86–88) (Figure 1).
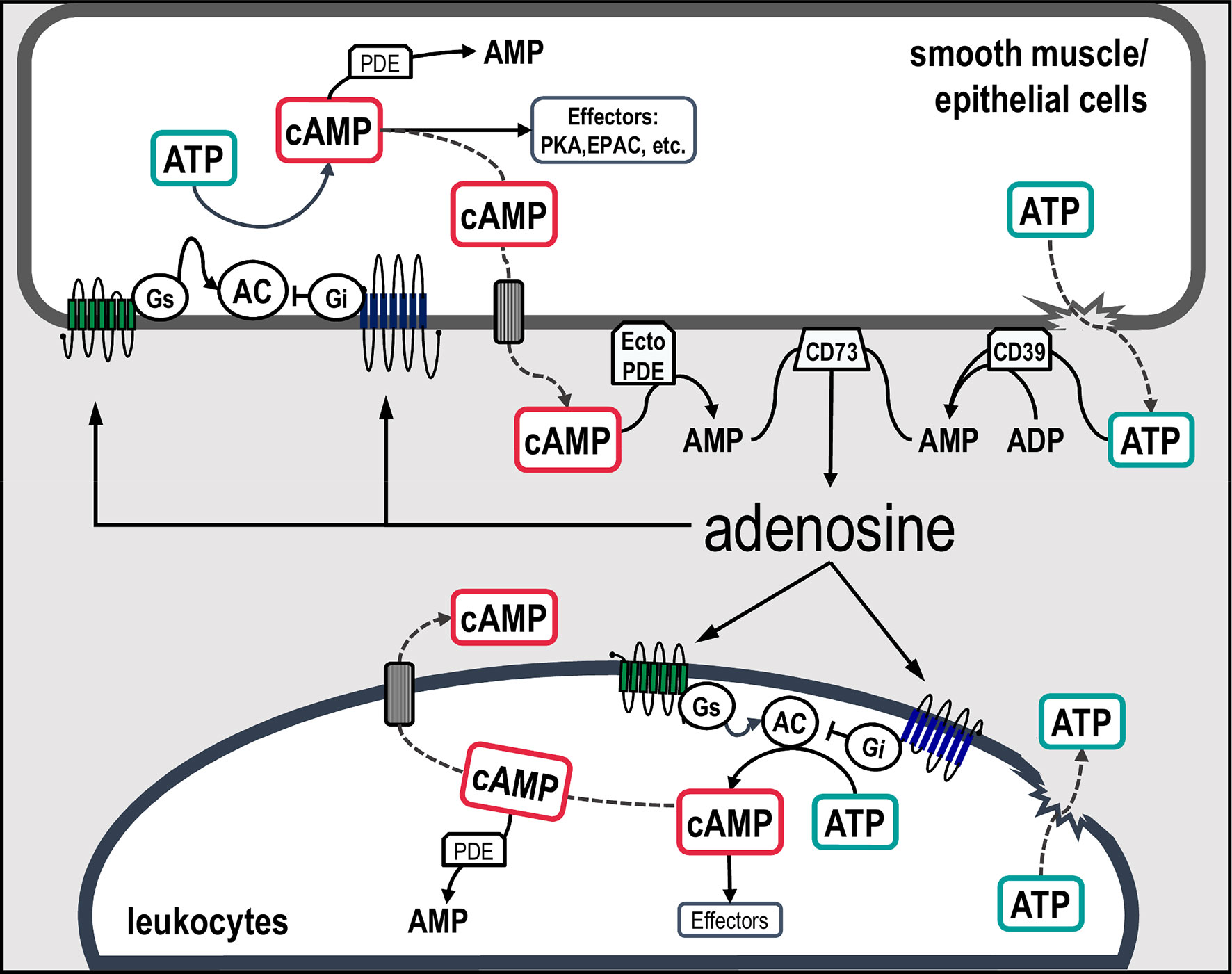
Figure 1 Schematic representation of canonical and non-canonical pathways involved in extracellular adenosine formation in airway cells. Classically, extracellular adenosine is generated mainly via the dephosphorylation of ATP, ADP and AMP by the action of a series of ecto-enzymes. An alternative source of adenosine is the second messenger cAMP. In this scenario, intracellular cAMP synthetized by adenylyl cyclases can be transported to the extracellular compartment and sequentially metabolized into AMP and adenosine by ecto-PDE and CD73, respectively. Increased concentrations of adenosine activate adenosine receptors coupled to Gs or Gi proteins, which in turn regulates the activity of adenylyl cyclases leading to an increase or decrease in cAMP concentrations and stimulation of downstream effectors. AC, adenylyl cyclases; Gs, stimulatory G protein; Gi, inhibitory G protein; PDE, phosphodiesterase; PKA, cAMP-dependent protein kinase; EPAC, exchange protein directly activated by cAMP; CD39, NTPDase-1; CD73, ecto-5’-nucleotidases and Ecto-PDE, ecto-phosphodiesterase.
Ecto-PDE activity has been described in many tissues, including vascular smooth muscle, kidney, intestine, skeletal muscle and airway, and in bovine seminal plasma and epididymal fluid (85, 88–92). Few research groups, however, have attempted to characterized ecto-PDE. Pharmacological studies using selective inhibitors of the intracellular cAMP-specific PDE isoforms demonstrate that ecto-PDEs obtained from the kidney, seminal plasma and vesical fluid present characteristics similar to those of the intracellular PDE8 and PDE10 families (89, 91).
The canonical pathway of extracellular adenosine production requires extracellular ATP as a precursor. In this regard, ATP can be specifically released via vesicular exocytosis and membrane pores upon cell activation or from damaged cells after transient or persistent inflammatory stimuli (93, 94). In contrast, the extracellular cAMP-adenosine pathway requires transport of intracellular cAMP to the extracellular compartment, a process that is mediated by members of ATP-binding cassette (ABC) transporters, subfamily C (ABCC). Due to their ability to extrude various chemotherapeutic agents from tumor cells, ABCC transporters are also referred to as multidrug resistance proteins (MRPs or MDRs). This family of transporters consist of nine members (ABCC1-ABCC9) that use ATP hydrolysis to mediate the efflux of multiple cellular substrates (95). Currently there are four well-defined ABCC proteins (ABCC1/MRP1, ABCC4/MRP4, ABCC5/MRP5 and ABCC11/MRP8) that can transport cAMP out of the cell, with specific kinetic parameters (96, 97); all of these cAMP transporters are expressed in human airway smooth muscle and epithelial cells (98–102). Recent studies analyzing GSE datasets from GEO (Gene Expression Omnibus) show that in airway epithelial cells the rank order of ABCC gene expression is ABCC5 >ABCC1 >ABCC4 >ABCC11 (101), whereas in the airway smooth muscle cells it is ABCC1 >ABCC4 >ABCC5 >ABCC11 (102).
Extracellular cAMP-Adenosine Pathway in Airways and Potential Role in Chronic Inflammatory Diseases
Because of the importance of intracellular cAMP in the physiological modulation of airway smooth muscle tone and as a signaling molecule that mediates the action of bronchodilator agents (e.g., β2-adrenoceptor agonists used for rescue treatment of asthmatic patients suffering acute bronchoconstriction), we have focused our studies on the potential role of extracellular cAMP in the regulation of airway smooth muscle tone and as a source of adenosine. Using isolated rat trachea, we have shown that fenoterol (short-acting) and formoterol (long acting) β2-adrenoceptor agonists induce a time-dependent increase in extracellular cAMP concentrations (54). Interestingly, extracellular cAMP triggers a concentration-dependent contraction of airway smooth muscle that is mimicked by adenosine and increased by drugs that inhibit adenosine degradation (EHNA) and uptake (uridine), indicating that the contracting effects of extracellular cAMP depends on extracellular adenosine formation (54). Indeed, treatment of trachea segments with the adenosine receptor antagonist GCS-15943 increases the potency of fenoterol with regard to inducing smooth muscle relaxation, indicating that the extracellular cAMP-adenosine pathway compromises, via activation of adenosine receptors, the efficacy of β2-adrenoceptor agonists as bronchodilators. Further supporting this idea, using ultraperformance liquid chromatography–tandem mass spectrometry (LC-MS/MS), we found that incubation of isolated rat tracheas with cell impermeable cAMP leads to increases in extracellular concentrations of 5’-AMP, adenosine and inosine (92). The involvement of ecto-phosphodiesterases and ecto-5’-nucleotidase/CD73 in the extracellular metabolism of cAMP is demonstrated by the fact the selective inhibitors of these ecto-enzymes (DPSPX and AMP-CP, respectively) significantly reduce adenosine concentrations (92). Consistent with these observations, studies by Huff et al. (103) demonstrate that cAMP efflux from human airway epithelial cells occurs and is mediated by ABBC4/MRP4 transporters. More recently, Cao and coworkers (102) showed that stimulation of β2-adrenoceptors or AC with formoterol or forskolin, respectively, also promotes extrusion of cAMP from human airway smooth muscle cells in culture. cAMP efflux is markedly reduced by pharmacological inhibition or downregulation of ABCC1 transporters with siRNA, resulting in potentiation of β-agonist induced airway smooth muscle relaxation (102). Collectively, these findings reveal the existence of a functional extracellular cAMP-adenosine pathway in airways.
Increase in plasma concentration of cAMP is observed in physiological and pathological conditions such as during spontaneous or PGE-induced labor (104), insulin-induced hypoglycemia (105) in malignant hyperpyrexia susceptible individuals (106) or following traumatic injury. In fact, according to Cock et al. (107),, there is a positive correlation between the plasma concentrations of cAMP and neutrophil count following injury, which supports the idea that neutrophil mobilization can be activated by extracellular cAMP. Also, cAMP extrusion from neutrophils is stimulated by PGE1, isoproterenol, or forskolin (108), indicating that many cells are able to release cAMP. Regarding the airways, based on the extracellular fluid volume of rat trachea (~1 ml/g of dry tissue weight) (109), we have estimated that after β2-adrenoceptors stimulation, extracellular cAMP can reach micromolar concentrations (54), which are those required to induce smooth muscle contraction.
Studies addressing the expression of transporters and ecto-enzymes responsible for efflux and extracellular degradation of cAMP in the airway cells support a potential role for the extracellular cAMP-adenosine pathway in the pathophysiology of different respiratory diseases. Airway epithelial cells from tobacco smokers and asthmatic patients have increased expression of ABCC1, in comparison with those of healthy individuals (101), which is consistent with the increased concentrations of serum cAMP in asthmatic patients (102). With regard to ecto-enzymes, several studies using human tissues or animal models of respiratory diseases reveal increased expression and enzymatic activity of ecto-5’-nucleotidase/CD73, which could explain the higher level of extracellular adenosine associated with mechanical ventilation-induced lung injury (110), chronic obstructive diseases (111) or long-term cigarette smoking (112). In allergic animal models, airway inflammation and tracheal hyperresponsiveness are largely dependent on CD73 (113). On the other hand, in CD73 deficient mice (CD73-/-) sensitized with ovalbumin, there is an exacerbation of airway inflammation, as reflected by increased formation of mucus and release of pro-inflammatory mediators such like IL-1β, TNFα, IL-4 and IL-5 (114, 115). Other studies have shown that cAMP released from human CD4+ T lymphocytes inhibits T cell proliferation (116) and modulates differentiation of human monocytes into dendritic cells, effects that are mediated via A2A and A2B receptors (117). Likewise, in the experimental autoimmune uveitis mice model, cAMP released from T cells functions as an important source of extracellular adenosine, which in turn contributes to the immunosuppressive function of regulatory T cells (118). Finally, expression of A2B receptors is increased in lungs of patients with COPD or idiopathic pulmonary fibrosis (60). The precise role of the cAMP-adenosine pathway in inflammatory pulmonary diseases is still not clear but it might function as a negative or positive feedback loop limiting or enhancing the output signal initiated by intracellular cAMP.
Currently, there are no clinical trials exploring compounds that target molecules involved in the extracellular cAMP-adenosine pathway (ABCC/MRP, ecto-phosphodiesterases and CD73) for chronic inflammatory airway diseases. However, some Phase I and II studies are testing CD73 inhibitors (e.g., HLX23, LY3475070, IPH5301, AK119, cpi-006, Sym024, IBI325, ORIC-533 and MEDI9447) alone or combined with other agents in different solid tumors, metastatic cancer and COVID-19 (NCT: 04797468, 04148937, 05143970, 04516564, 04572152, 05173792, 03454451, 04672434, 05246995, 05227144, 03381274 and 04668300) with promising results (119, 120). In addition, Phase I and II clinical trials evaluated the safety and efficacy of an MRP-1 inhibitor (sulindac) in patients with advanced melanoma (121)(EUCTR: 2006-006051-12); however, this study was terminated prematurely. Several other clinicals trials (NCR: 00430300, 01640990, 05262218, 03774290, 02635945, 01939587, 04606069) investigated the role of selective adenosine receptor agonists and antagonists (e.g., GW328267X, UK-432097, EPI-2010, CVT-6883, QAF 805, PBF-680 and Regadenoson) in various respiratory diseases, including asthma, COPD, allergic rhinitis, acute lung injury and COVID-19, however, almost all of them were discontinued due to insufficient therapeutic efficacy and/or incidence of side effects (61, 122–124). Since cAMP efflux depends on the preceding increase in intracellular cAMP, future preclinical studies and clinical trials should explore the therapeutic effects of combining treatment with bronchodilator or anti-inflammatory drugs, such as corticosteroids, β2-adrenoceptor agonists, or PDE inhibitors, with inhibitors of either cAMP efflux or the extracellular cAMP-adenosine pathway, to elucidate the potential therapeutic role of this biochemical pathway in inflammatory airway diseases.
Conclusion and Future Perspectives
The discovery of cAMP by Sutherland and colleagues almost 60 years ago provided the basis for the concept of cAMP as an intracellular second messenger (125). Nevertheless, as illustrated in Figure 1, we are now beginning a new phase of understanding the role of cAMP as an autocrine and/or paracrine signaling molecule, in which the extracellular cAMP-adenosine pathway is able to modify cAMP signaling initiated in the intracellular environment (82).
In the airways, cAMP is the main signaling molecule implicated in bronchodilation caused by endogenous catecholamines or beta-adrenergic agents. Even after the identification of membrane transporters capable of exporting cAMP out of cells, the efflux of cAMP from airway epithelial or smooth muscle cells was considered a simple mechanism to reduce intracellular cyclic nucleotide concentrations (102). However, the discovery of an extracellular enzymatic system in airways that converts cAMP into adenosine (92) and can affect the primary cellular response initiated by intracellular cAMP (54) opened new perspectives on how to envision the role of cAMP in airway physiology, highlighting the importance of the mechanisms of cAMP extrusion and its function as an alternative source of extracellular adenosine.
As observed with adenosine, the regulatory effects of extracellular cAMP on the airways will depend on the ectoenzymes and adenosine receptor subtypes expressed in each cell, and on the amount of cAMP released into the extracellular compartment. Although we have revealed bronchoconstrictor effects of the extracellular cAMP-adenosine pathway, the relevance of the extracellular cAMP in inflammatory lung diseases is just beginning to be explored. Considering that increases in intracellular cAMP concentrations are usually followed by efflux of cAMP (88), it is important to explore the impact of the extracellular cAMP-adenosine pathway on the therapeutic effects of bronchodilators and anti-inflammatory drugs used in chronic respiratory diseases, such as glucocorticoids and classical PDE inhibitors, known to increase intracellular cAMP (126–129).
In view of the modulatory effects of adenosine on inflammatory responses and innate immune cell function, there is a potential role for cAMP efflux in airway inflammation/immune responses and bronchial reactivity (Figure 2). The existence of the extracellular cAMP-adenosine pathway in the airways also suggests innovative therapeutic possibilities and new pharmacological targets in airway cells, such as distinct ABCC/MRP transporters and ectoenzymes such ecto-PDEs. Future studies will be needed to decipher the precise role of the extracellular cAMP-adenosine pathway in airway inflammation and immune responses.
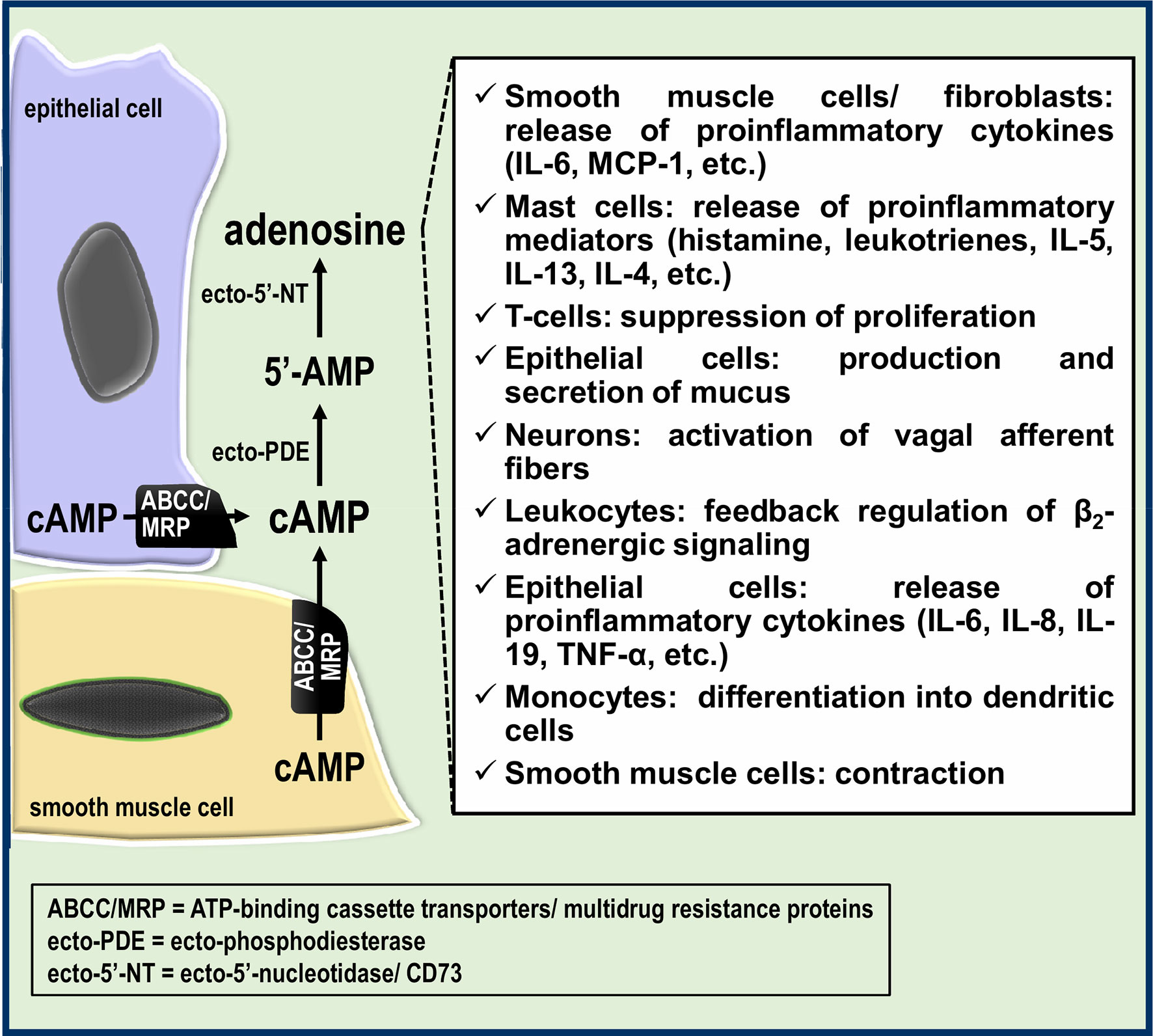
Figure 2 Potential roles of the extracellular cAMP-adenosine pathway in the airways. Part of the cAMP formed in epithelial and smooth muscle cells is transported by ABCC/MRP transporters to the extracellular milieu, giving rise to extracellular adenosine, which in turn can modulate the airway immune response and bronchial reactivity.
Author Contributions
EP and RG contributed to conception and design of the review. RG wrote the first draft of the manuscript. EP, NS, EJ, and RG wrote the sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp, grant #2018/21381-5) and financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brazil (CAPES) - Finance Code 001. RG is a research fellow from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grant # 310498/2019-8. EP is a PNPD postdoctoral fellow and NS is a MSc fellow from Capes, Brazil.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Linden J. New Insights Into the Regulation of Inflammation by Adenosine. J Clin Invest (2006) 116:1835–7. doi: 10.1172/JCI29125
2. Haskó G, Cronstein B. Regulation of Inflammation by Adenosine. Front Immunol (2013) 4:85. doi: 10.3389/fimmu.2013.00085
3. Antonioli L, Fornai M, Blandizzi C, Pacher P, Haskó G. Adenosine Signaling and the Immune System: When a Lot Could be Too Much. Immunol Lett (2019) 205:9–15. doi: 10.1016/j.imlet.2018.04.006
4. Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Muller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors:an Update. Pharmacol Rev (2011) 63:1–34. doi: 10.1124/pr.110.003285
5. Burnstock G, Brouns I, Adriaensen D, Timmermans JP. Purinergic Signaling in the Airways. Pharmacol Rev (2012) 64:834–68. doi: 10.1124/pr.111.005389
6. Franco R, Cordomí A, Llinas Del TC, Lillo A, Serrano-Marín J, Navarro G, et al. Structure and Function of Adenosine Receptor Heteromers. Cell Mol Life Sci (2021) 78:3957–68. doi: 10.1007/s00018-021-03761-6[pii];10.1007/s00018-021-03761-6
7. Sadana R, Dessauer CW. Physiological Roles for G Protein-Regulated Adenylyl Cyclase Isoforms: Insights From Knockout and Overexpression Studies. Neurosignals (2009) 17:5–22. doi: 10.1159/000166277
8. Dessauer CW, Watts VJ, Ostrom RS, Conti M, Dove S, Seifert R. International Union of Basic and Clinical Pharmacology. CI. Structures and Small Molecule Modulators of Mammalian Adenylyl Cyclases. Pharmacol Rev (2017) 69:93–139. doi: 10.1124/pr.116.013078
9. Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol Rev (2018) 98:1591–625. doi: 10.1152/physrev.00049.2017
10. Musheshe N, Schmidt M, Zaccolo M. cAMP: From Long-Range Second Messenger to Nanodomain Signalling. Trends Pharmacol Sci (2018) 39:209–22. doi: 10.1016/j.tips.2017.11.006
11. Billington CK, Ojo OO, Penn RB, Ito S. cAMP Regulation of Airway Smooth Muscle Function. Pulm Pharmacol Ther (2013) 26:112–20. doi: 10.1016/j.pupt.2012.05.007
12. Villarreal F, Epperson SA, Ramirez-Sanchez I, Yamazaki KG, Brunton LL. Regulation of Cardiac Fibroblast Collagen Synthesis by Adenosine: Roles for Epac and PI3K. Am J Physiol Cell Physiol (2009) 296:C1178–84. doi: 10.1152/ajpcell.00291.2008
13. Navarro G, Cordomí A, Brugarolas M, Moreno E, Aguinaga D, Pérez-Benito L, et al. Cross-Communication Between G(I) and G(s) in a G-Protein-Coupled Receptor Heterotetramer Guided by a Receptor C-Terminal Domain. BMC Biol (2018) 16:24. doi: 10.1186/s12915-018-0491-x[pii];491[pii];10.1186/s12915-018-0491-x
14. Agarwal SR, Yang PC, Rice M, Singer CA, Nikolaev VO, Lohse MJ, et al. Role of Membrane Microdomains in Compartmentation of cAMP Signaling. PloS One (2014) 9:e95835. doi: 10.1371/journal.pone.0095835
15. Schmidt M, Cattani-Cavalieri I, Nuñez FJ, Ostrom RS. Phosphodiesterase Isoforms and cAMP Compartments in the Development of New Therapies for Obstructive Pulmonary Diseases. Curr Opin Pharmacol (2020) 51:34–42. doi: 10.1016/j.coph.2020.05.002
16. Omar MH, Scott JD. AKAP Signaling Islands: Venues for Precision Pharmacology. Trends Pharmacol Sci (2020) 41:933–46. doi: 10.1016/j.tips.2020.09.007
17. Spicuzza L, Di MG, Polosa R. Adenosine in the Airways: Implications and Applications. Eur J Pharmacol (2006) 533:77–88. doi: 10.1016/j.ejphar.2005.12.056
18. Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in Bronchoalveolar Lavage Fluid in Asthma. Am Rev Respir Dis (1993) 148:91–7. doi: 10.1164/ajrccm/148.1.91
19. Martin C, Leone M, Viviand X, Ayem ML, Guieu R. High Adenosine Plasma Concentration as a Prognostic Index for Outcome in Patients With Septic Shock. Crit Care Med (2000) 28:3198–202. doi: 10.1097/00003246-200009000-00014
20. Linden J. Molecular Approach to Adenosine Receptors: Receptor-Mediated Mechanisms of Tissue Protection. Annu Rev Pharmacol Toxicol (2001) 41:775–87. doi: 10.1146/annurev.pharmtox.41.1.775
21. Köröskényi K, Duró E, Pallai A, Sarang Z, Kloor D, Ucker DS, et al. Involvement of Adenosine A2A Receptors in Engulfment-Dependent Apoptotic Cell Suppression of Inflammation. J Immunol (2011) 186:7144–55. doi: 10.4049/jimmunol.1002284
22. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Front Immunol (2016) 7:160. doi: 10.3389/fimmu.2016.00160
23. Ohta A, Sitkovsky M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection From Tissue Damage. Nature (2001) 414:916–20. doi: 10.1038/414916a
24. Lukashev D, Ohta A, Apasov S, Chen JF, Sitkovsky M. Cutting Edge: Physiologic Attenuation of Proinflammatory Transcription by the Gs Protein-Coupled A2A Adenosine Receptor In Vivo. J Immunol (2004) 173:21–4. doi: 10.4049/jimmunol.173.1.21
25. Salmon JE, Cronstein BN. Fc Gamma Receptor-Mediated Functions in Neutrophils are Modulated by Adenosine Receptor Occupancy. A1 Receptors are Stimulatory and A2 Receptors are Inhibitory. J Immunol (1990) 145:2235–40.
26. Cronstein BN, Levin RI, Philips M, Hirschhorn R, Abramson SB, Weissmann G. Neutrophil Adherence to Endothelium is Enhanced via Adenosine A1 Receptors and Inhibited via Adenosine A2 Receptors. J Immunol (1992) 148:2201–6.
27. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. Atp Release Guides Neutrophil Chemotaxis. via P2Y2 A3 Receptors. Sci (2006) 314:1792–5. doi: 10.1126/science.1132559
28. Yago T, Tsukamoto H, Liu Z, Wang Y, Thompson LF, McEver RP. Multi-Inhibitory Effects of A2A Adenosine Receptor Signaling on Neutrophil Adhesion Under Flow. J Immunol (2015) 195:3880–9. doi: 10.4049/jimmunol.1500775
29. Bouma MG, Jeunhomme TM, Boyle DL, Dentener MA, Voitenok NN, van den Wildenberg FA, et al. Adenosine Inhibits Neutrophil Degranulation in Activated Human Whole Blood: Involvement of Adenosine A2 and A3 Receptors. J Immunol (1997) 158:5400–8.
30. Gessi S, Varani K, Merighi S, Ongini E, Borea PA. A(2A) Adenosine Receptors in Human Peripheral Blood Cells. Br J Pharmacol (2000) 129:2–11. doi: 10.1038/sj.bjp.0703045
31. van der Hoeven D, Wan TC, Gizewski ET, Kreckler LM, Maas JE, Van OJ, et al. A Role for the Low-Affinity A2B Adenosine Receptor in Regulating Superoxide Generation by Murine Neutrophils. J Pharmacol Exp Ther (2011) 338:1004–12. doi: 10.1124/jpet.111.181792
32. Barletta KE, Cagnina RE, Burdick MD, Linden J, Mehrad B. Adenosine a(2B) Receptor Deficiency Promotes Host Defenses Against Gram-Negative Bacterial Pneumonia. Am J Respir Crit Care Med (2012) 186:1044–50. doi: 10.1164/rccm.201204-0622OC
33. Thiel M, Chouker A. Acting via A2 Receptors, Adenosine Inhibits the Production of Tumor Necrosis Factor-Alpha of Endotoxin-Stimulated Human Polymorphonuclear Leukocytes. J Lab Clin Med (1995) 126:275–82.
34. Flamand N, Boudreault S, Picard S, Austin M, Surette ME, Plante H, et al. Adenosine, a Potent Natural Suppressor of Arachidonic Acid Release and Leukotriene Biosynthesis in Human Neutrophils. Am J Respir Crit Care Med (2000) 161:S88–94. doi: 10.1164/ajrccm.161.supplement_1.ltta-18
35. McColl SR, St-Onge M, Dussault AA, Laflamme C, Bouchard L, Boulanger J, et al. Immunomodulatory Impact of the A2A Adenosine Receptor on the Profile of Chemokines Produced by Neutrophils. FASEB J (2006) 20:187–9. doi: 10.1096/fj.05-4804fje
36. Haskó G, Pacher P, Deitch EA, Vizi ES. Shaping of Monocyte and Macrophage Function by Adenosine Receptors. Pharmacol Ther (2007) 113:264–75. doi: 10.1016/j.pharmthera.2006.08.003
37. Lappas CM, Rieger JM, Linden J. A2A Adenosine Receptor Induction Inhibits IFN-Gamma Production in Murine CD4+ T Cells. J Immunol (2005) 174:1073–80. doi: 10.4049/jimmunol.174.2.1073
38. Linden J, Cekic C. Regulation of Lymphocyte Function by Adenosine. Arterioscler Thromb Vasc Biol (2012) 32:2097–103. doi: 10.1161/ATVBAHA.111.226837
39. Raker VK, Becker C, Steinbrink K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front Immunol (2016) 7:123. doi: 10.3389/fimmu.2016.00123
40. Chinn AM, Insel PA. Cyclic AMP in Dendritic Cells: A Novel Potential Target for Disease-Modifying Agents in Asthma and Other Allergic Disorders. Br J Pharmacol (2020) 177:3363–77. doi: 10.1111/bph.15095
41. Galgani M, De R V, De SS, Leonardi A, D’Oro U, Napolitani G, et al. Cyclic AMP Modulates the Functional Plasticity of Immature Dendritic Cells by Inhibiting Src-Like Kinases Through Protein Kinase a-Mediated Signaling. J Biol Chem (2004) 279:32507–14. doi: 10.1074/jbc.M403355200
42. Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting Edge: Macrophage Inhibition by Cyclic AMP (cAMP): Differential Roles of Protein Kinase a and Exchange Protein Directly Activated by cAMP-1. J Immunol (2005) 174:595–9. doi: 10.4049/jimmunol.174.2.595
43. Hidi R, Timmermans S, Liu E, Schudt C, Dent G, Holgate ST, et al. Phosphodiesterase and Cyclic Adenosine Monophosphate-Dependent Inhibition of T-Lymphocyte Chemotaxis. Eur Respir J (2000) 15:342–9. doi: 10.1034/j.1399-3003.2000.15b21.x
44. Whisler RL, Beiqing L, Grants IS, Newhouse YG. Cyclic AMP Modulation of Human B Cell Proliferative Responses: Role of cAMP-Dependent Protein Kinases in Enhancing B Cell Responses to Phorbol Diesters and Ionomycin. Cell Immunol (1992) 142:398–415. doi: 10.1016/0008-8749(92)90300-e
45. Fredholm BB. Adenosine Receptors as Drug Targets. Exp Cell Res (2010) 316:1284–8. doi: 10.1016/j.yexcr.2010.02.004
46. Chen JF, Eltzschig HK, Fredholm BB. Adenosine Receptors as Drug Targets–What are the Challenges? Nat Rev Drug Discov (2013) 12:265–86. doi: 10.1038/nrd3955
47. Huszar E, Vass G, Vizi E, Csoma Z, Barat E, Molnar VG, et al. Adenosine in Exhaled Breath Condensate in Healthy Volunteers and in Patients With Asthma. Eur Respir J (2002) 20:1393–8. doi: 10.1183/09031936.02.00005002
48. Esther CR Jr., Boysen G, Olsen BM, Collins LB, Ghio AJ, Swenberg JW, et al. Mass Spectrometric Analysis of Biomarkers and Dilution Markers in Exhaled Breath Condensate Reveals Elevated Purines in Asthma and Cystic Fibrosis. Am J Physiol Lung Cell Mol Physiol (2009) 296:L987–93. doi: 10.1152/ajplung.90512.2008
49. Lommatzsch M, Cicko S, Müller T, Lucattelli M, Bratke K, Stoll P, et al. Extracellular Adenosine Triphosphate and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2010) 181:928–34. doi: 10.1164/rccm.200910-1506OC
50. Lazarowski ER, Sesma JI, Seminario L, Esther CR Jr., Kreda SM. Nucleotide Release by Airway Epithelia. Subcell Biochem (2011) 55:1–15. doi: 10.1007/978-94-007-1217-1_1
51. Cushley MJ, Tattersfield AE, Holgate ST. Inhaled Adenosine and Guanosine on Airway Resistance in Normal and Asthmatic Subjects. Br J Clin Pharmacol (1983) 15:161–5. doi: 10.1111/j.1365-2125.1983.tb01481.x
52. Oosterhoff Y, de Jong JW, Jansen MA, Koëter GH, Postma DS. Airway Responsiveness to Adenosine 5’-Monophosphate in Chronic Obstructive Pulmonary Disease is Determined by Smoking. Am Rev Respir Dis (1993) 147:553–8. doi: 10.1164/ajrccm/147.3.553
53. Fan M, Mustafa SJ. Adenosine-Mediated Bronchoconstriction and Lung Inflammation in an Allergic Mouse Model. Pulm Pharmacol Ther (2002) 15:147–55. doi: 10.1006/pupt.2001.0329
54. Pacini ESA, Sanders-Silveira S, Godinho RO. The Extracellular cAMP-Adenosine Pathway in Airway Smooth Muscle. J Pharmacol Exp Ther (2018) 366:75–83. doi: 10.1124/jpet.118.247734
55. Calzetta L, Spina D, Cazzola M, Page CP, Facciolo F, Rendina EA, et al. Pharmacological Characterization of Adenosine Receptors on Isolated Human Bronchi. Am J Respir Cell Mol Biol (2011) 45:1222–31. doi: 10.1165/rcmb.2011-0056OC
56. Phillips GD, Polosa R, Holgate ST. The Effect of Histamine-H1 Receptor Antagonism With Terfenadine on Concentration-Related AMP-Induced Bronchoconstriction in Asthma. Clin Exp Allergy (1989) 19:405–9. doi: 10.1111/j.1365-2222.1989.tb02406.x
57. Rorke S, Jennison S, Jeffs JA, Sampson AP, Arshad H, Holgate ST. Role of Cysteinyl Leukotrienes in Adenosine 5’-Monophosphate Induced Bronchoconstriction in Asthma. Thorax (2002) 57:323–7. doi: 10.1136/thorax.57.4.323
58. Zhong H, Shlykov SG, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, et al. Activation of Murine Lung Mast Cells by the Adenosine A3 Receptor. J Immunol (2003) 171:338–45. doi: 10.4049/jimmunol.171.1.338
59. Hua X, Erikson CJ, Chason KD, Rosebrock CN, Deshpande DA, Penn RB, et al. Involvement of A1 Adenosine Receptors and Neural Pathways in Adenosine-Induced Bronchoconstriction in Mice. Am J Physiol Lung Cell Mol Physiol (2007) 293:L25–32. doi: 10.1152/ajplung.00058.2007
60. Le TT, Berg NK, Harting MT, Li X, Eltzschig HK, Yuan X. Purinergic Signaling in Pulmonary Inflammation. Front Immunol (2019) 10:1633. doi: 10.3389/fimmu.2019.01633
61. Li X, Berg NK, Mills T, Zhang K, Eltzschig HK, Yuan X. Adenosine at the Interphase of Hypoxia and Inflammation in Lung Injury. Front Immunol (2020) 11:604944. doi: 10.3389/fimmu.2020.604944
62. Zhong H, Belardinelli L, Maa T, Feoktistov I, Biaggioni I, Zeng D. A(2B) Adenosine Receptors Increase Cytokine Release by Bronchial Smooth Muscle Cells. Am J Respir Cell Mol Biol (2004) 30:118–25. doi: 10.1165/rcmb.2003-0118OC
63. Zhong H, Belardinelli L, Maa T, Zeng D. Synergy Between A2B Adenosine Receptors and Hypoxia in Activating Human Lung Fibroblasts. Am J Respir Cell Mol Biol (2005) 32:2–8. doi: 10.1165/rcmb.2004-0103OC
64. Young HW, Sun CX, Evans CM, Dickey BF, Blackburn MR. A3 Adenosine Receptor Signaling Contributes to Airway Mucin Secretion After Allergen Challenge. Am J Respir Cell Mol Biol (2006) 35:549–58. doi: 10.1165/rcmb.2006-0060OC
65. McNamara N, Gallup M, Khong A, Sucher A, Maltseva I, Fahy J, et al. Adenosine Up-Regulation of the Mucin Gene, MUC2, in Asthma. FASEB J (2004) 18:1770–2. doi: 10.1096/fj.04-1964fje
66. Lambrecht BN, Hammad H. Allergens and the Airway Epithelium Response: Gateway to Allergic Sensitization. J Allergy Clin Immunol (2014) 134:499–507. doi: 10.1016/j.jaci.2014.06.036
67. McCoy DE, Schwiebert EM, Karlson KH, Spielman WS, Stanton BA. Identification and Function of A1 Adenosine Receptors in Normal and Cystic Fibrosis Human Airway Epithelial Cells. Am J Physiol (1995) 268:C1520–27 doi: 10.1152/ajpcell.1995.268.6.C1520
68. Hentchel-Franks K, Lozano D, Eubanks-Tarn V, Cobb B, Fan L, Oster R, et al. Activation of Airway Cl- Secretion in Human Subjects by Adenosine. Am J Respir Cell Mol Biol (2004) 31:140–6. doi: 10.1165/rcmb.2004-0012OC
69. Sun Y, Wu F, Sun F, Huang P. Adenosine Promotes IL-6 Release in Airway Epithelia. J Immunol (2008) 180:4173–81. doi: 10.4049/jimmunol.180.6.4173
70. Zhong H, Wu Y, Belardinelli L, Zeng D. A2B Adenosine Receptors Induce IL-19 From Bronchial Epithelial Cells, Resulting in TNF-Alpha Increase. Am J Respir Cell Mol Biol (2006) 35:587–92. doi: 10.1165/rcmb.2005-0476OC
71. Spruntulis LM, Broadley KJ. A3 Receptors Mediate Rapid Inflammatory Cell Influx Into the Lungs of Sensitized Guinea-Pigs. Clin Exp Allergy (2001) 31:943–51. doi: 10.1046/j.1365-2222.2001.01087.x
72. Ryzhov S, Goldstein AE, Matafonov A, Zeng D, Biaggioni I, Feoktistov I. Adenosine-Activated Mast Cells Induce Ige Synthesis by B Lymphocytes: An A2B-Mediated Process Involving Th2 Cytokines IL-4 and IL-13 With Implications for Asthma. J Immunol (2004) 172:7726–33. doi: 10.4049/jimmunol.172.12.7726
73. Oldenburg PJ, Mustafa SJ. Involvement of Mast Cells in Adenosine-Mediated Bronchoconstriction and Inflammation in an Allergic Mouse Model. J Pharmacol Exp Ther (2005) 313:319–24. doi: 10.1124/jpet.104.071720
74. Blackburn MR, Kellems RE. Adenosine Deaminase Deficiency: Metabolic Basis of Immune Deficiency and Pulmonary Inflammation. Adv Immunol (2005) 86:1–41. doi: 10.1016/S0065-2776(04)86001-2
75. Blackburn MR, Volmer JB, Thrasher JL, Zhong H, Crosby JR, Lee JJ, et al. Metabolic Consequences of Adenosine Deaminase Deficiency in Mice are Associated With Defects in Alveogenesis, Pulmonary Inflammation, and Airway Obstruction. J Exp Med (2000) 192:159–70. doi: 10.1084/jem.192.2.159
76. Zhong H, Chunn JL, Volmer JB, Fozard JR, Blackburn MR. Adenosine-Mediated Mast Cell Degranulation in Adenosine Deaminase-Deficient Mice. J Pharmacol Exp Ther (2001) 298:433–40.
77. Blackburn MR, Lee CG, Young HW, Zhu Z, Chunn JL, Kang MJ, et al. Adenosine Mediates IL-13-Induced Inflammation and Remodeling in the Lung and Interacts in an IL-13-Adenosine Amplification Pathway. J Clin Invest (2003) 112:332–44. doi: 10.1172/JCI16815
78. Chunn JL, Young HW, Banerjee SK, Colasurdo GN, Blackburn MR. Adenosine-Dependent Airway Inflammation and Hyperresponsiveness in Partially Adenosine Deaminase-Deficient Mice. J Immunol (2001) 167:4676–85. doi: 10.4049/jimmunol.167.8.4676
79. Zimmermann H. History of Ectonucleotidases and Their Role in Purinergic Signaling. Biochem Pharmacol (2020) 187:114322. doi: 10.1016/j.bcp.2020.114322
80. Lloyd HG, Deussen A, Wuppermann H, Schrader J. The Transmethylation Pathway as a Source for Adenosine in the Isolated Guinea-Pig Heart. Biochem J (1988) 252:489–94. doi: 10.1042/bj2520489
81. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 Ectoenzymatic Pathway Independent of CD39 Drives a Novel Adenosinergic Loop in Human T Lymphocytes. Oncoimmunology (2013) 2:e26246. doi: 10.4161/onci.26246
82. Godinho RO, Duarte T, Pacini ES. New Perspectives in Signaling Mediated by Receptors Coupled to Stimulatory G Protein: The Emerging Significance of cAMP Efflux and Extracellular cAMP-Adenosine Pathway. Front Pharmacol (2015) 6:58. doi: 10.3389/fphar.2015.00058
83. Jackson EK. Adenosine: A Physiological Brake on Renin Release. Annu Rev Pharmacol Toxicol (1991) 31:1–35. doi: 10.1146/annurev.pa.31.040191.000245
84. Mi Z, Jackson EK. Metabolism of Exogenous Cyclic AMP to Adenosine in the Rat Kidney. J Pharmacol Exp Ther (1995) 273:728–33.
85. Dubey RK, Mi Z, Gillespie DG, Jackson EK. Cyclic AMP-Adenosine Pathway Inhibits Vascular Smooth Muscle Cell Growth. Hypertension (1996) 28:765–71. doi: 10.1161/01.HYP.28.5.765
86. Cometti B, Dubey RK, Imthurn B, Jackson EK, Rosselli M. Oviduct Cells Express the Cyclic AMP-Adenosine Pathway. Biol Reprod (2003) 69:868–75. doi: 10.1095/biolreprod.103.017962
87. Jackson EK, Raghvendra DK. The Extracellular Cyclic AMP-Adenosine Pathway in Renal Physiology. Annu Rev Physiol (2004) 66:571–99. doi: 10.1146/annurev.physiol.66.032102.111604
88. Chiavegatti T, Costa VL, Araujo MS, Godinho RO. Skeletal Muscle Expresses the Extracellular Cyclic AMP-Adenosine Pathway. Br J Pharmacol (2008) 153:1331–40. doi: 10.1038/sj.bjp.0707648
89. Jackson EK, Ren J, Zacharia LC, Mi Z. Characterization of Renal Ecto-Phosphodiesterase. J Pharmacol Exp Ther (2007) 321:810–5. doi: 10.1124/jpet.106.119057
90. Giron MC, Bin A, Brun P, Etteri S, Bolego C, Florio C, et al. Cyclic AMP in Rat Ileum: Evidence for the Presence of an Extracellular Cyclic AMP-Adenosine Pathway. Gastroenterology (2008) 134:1116–26. doi: 10.1053/j.gastro.2008.01.030
91. Bergeron A, Aragon JP, Guillemette C, Hebert A, Sullivan R, Blondin P, et al. Characterization of cAMP-Phosphodiesterase Activity in Bovine Seminal Plasma. Andrology (2016) 4:1123–30. doi: 10.1111/andr.12267
92. Pacini ESA, Jackson EK, Godinho RO. Extracellular Metabolism of 3’,5’-Cyclic AMP as a Source of Interstitial Adenosine in the Rat Airways. Biochem Pharmacol (2021) 192:114713. doi: 10.1016/j.bcp.2021.114713
93. Faas MM, Sáez T, de Vos P. Extracellular ATP and Adenosine: The Yin and Yang in Immune Responses? Mol Aspects Med (2017) 55:9–19. doi: 10.1016/j.mam.2017.01.002
94. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP Release by Inflammatory Cells. Int J Mol Sci (2018) 19:ijms19041222. doi: 10.3390/ijms19041222
95. Mohammad IS, He W, Yin L. Understanding of Human ATP Binding Cassette Superfamily and Novel Multidrug Resistance Modulators to Overcome MDR. BioMed Pharmacother (2018) 100:335–48. doi: 10.1016/j.biopha.2018.02.038
96. Wielinga PR, van der Heijden I, Reid G, Beijnen JH, Wijnholds J, Borst P. Characterization of the MRP4- and MRP5-Mediated Transport of Cyclic Nucleotides From Intact Cells. J Biol Chem (2003) 278:17664–71. doi: 10.1074/jbc.M212723200
97. Sager G, Ravna AW. Cellular Efflux of cAMP and cGMP - A Question About Selectivity. Mini Rev Med Chem (2009) 9:1009–13. doi: 10.2174/138955709788681654
98. Scheffer GL, Pijnenborg AC, Smit EF, Müller M, Postma DS, Timens W, et al. Multidrug Resistance Related Molecules in Human and Murine Lung. J Clin Pathol (2002) 55:332–9. doi: 10.1136/jcp.55.5.332
99. van der Deen M, de Vries EG, Timens W, Scheper RJ, Timmer-Bosscha H, Postma DS. ATP-Binding Cassette (ABC) Transporters in Normal and Pathological Lung. Respir Res (2005) 6(59). doi: 10.1186/1465-9921-6-59
100. Gold MJ, Hiebert PR, Park HY, Stefanowicz D, Le A, Starkey MR, et al. Mucosal Production of Uric Acid by Airway Epithelial Cells Contributes to Particulate Matter-Induced Allergic Sensitization. Mucosal Immunol (2016) 9:809–20. doi: 10.1038/mi.2015.104
101. Aguiar JA, Tamminga A, Lobb B, Huff RD, Nguyen JP, Kim Y, et al. The Impact of Cigarette Smoke Exposure, COPD, or Asthma Status on ABC Transporter Gene Expression in Human Airway Epithelial Cells. Sci Rep (2019) 9:153. doi: 10.1038/s41598-018-36248-9[pii];36248[pii];10.1038/s41598-018-36248-9
102. Cao G, Lam H, Jude JA, Karmacharya N, Kan M, Jester W, et al. Inhibition of ABCC1 Decreases cAMP Egress and Promotes Human Airway Smooth Muscle Cell Relaxation. Am J Respir Cell Mol Biol (2021) 66:96–106. doi: 10.1165/rcmb.2021-0345OC
103. Huff RD, Rider CF, Yan D, Newton R, Giembycz MA, Carlsten C, et al. Inhibition of ABCC4 Potentiates Combination Beta Agonist and Glucocorticoid Responses in Human Airway Epithelial Cells. J Allergy Clin Immunol (2018) 141:1127–30. doi: 10.1016/j.jaci.2017.10.011
104. Nagata I, Furuya K, Imaizumi E, Seki K, Makimura N, Kato K. Changes in Plasma Cyclic AMP and Cyclic GMP During Spontaneous Labor and Labor Induced by Oxytocin, Prostaglandin F2 Alpha and Prostaglandin E2. Gynecol Obstet Invest (1988) 26:21–8. doi: 10.1159/000293667
105. Brodows RG, Ensinck JW, Campbell RG. Mechanism of Plasma Cyclic AMP Response to Hypoglycemia in Man. Metabolism (1976) 25:659–63. doi: 10.1016/0026-0495(76)90063-9
106. Stanec A, Stefano G. Cyclic AMP in Normal and Malignant Hyperpyrexia Susceptible Individuals Following Exercise. Br J Anaesth (1984) 56:1243–6. doi: 10.1093/bja/56.11.1243
107. Cocks RA, Rainer TH, Chan TY, Maycock PF, Lam NY. Increased Plasma Free Cyclic-AMP Levels Following Major Trauma and Their Relevance to the Immune Response. Resuscitation (2000) 45:105–9. doi: 10.1016/s0300-9572(00)00176-3
108. Harvath L, Robbins JD, Russell AA, Seamon KB. cAMP and Human Neutrophil Chemotaxis. Elevation of cAMP Differentially Affects Chemotactic Responsiveness. J Immunol (1991) 146:224–32.
109. Woie K, Reed RK. Alloxan Diabetes Abolishes the Increased Negativity of Interstitial Fluid Pressure in Rat Trachea Induced by Vagal Nerve Stimulation. Acta Physiol Scand (1997) 161:113–9. doi: 10.1046/j.1365-201X.1997.00194.x
110. Eckle T, Fü llbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, et al. Identification of Ectonucleotidases CD39 and CD73 in Innate Protection During Acute Lung Injury. J Immunol (2007) 178:8127–37. doi: 10.4049/jimmunol.178.12.8127
111. Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR. Alterations in Adenosine Metabolism and Signaling in Patients With Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. PloS One (2010) 5:e9224. doi: 10.1371/journal.pone.0009224
112. Tian Z, Dixon J, Guo X, Deal B, Liao Q, Zhou Y, et al. Co-Inhibition of CD73 and ADORA2B Improves Long-Term Cigarette Smoke Induced Lung Injury. Front Physiol (2021) 12:614330. doi: 10.3389/fphys.2021.614330
113. Schreiber R, Castrop H, Kunzelmann K. Allergen-Induced Airway Hyperresponsiveness is Absent in Ecto-5’-Nucleotidase (CD73)-Deficient Mice. Pflugers Arch (2008) 457:431–40. doi: 10.1007/s00424-008-0543-0
114. Volmer JB, Thompson LF, Blackburn MR. Ecto-5’-Nucleotidase (CD73)-Mediated Adenosine Production is Tissue Protective in a Model of Bleomycin-Induced Lung Injury. J Immunol (2006) 176(7):4449–58. doi: 10.4049/jimmunol.176.7.4449
115. Caiazzo E, Cerqua I, Riemma MA, Turiello R, Ialenti A, Schrader J, et al. Exacerbation of Allergic Airway Inflammation in Mice Lacking ECTO-5’-Nucleotidase (CD73). Front Pharmacol (2020) 11:589343. doi: 10.3389/fphar.2020.589343
116. Vendetti S, Patrizio M, Riccomi A, De Magistris MT. Human CD4+ T Lymphocytes With Increased Intracellular cAMP Levels Exert Regulatory Functions by Releasing Extracellular cAMP. J Leukoc Biol (2006) 80:880–8. doi: 10.1189/jlb.0106072
117. Sciaraffia E, Riccomi A, Lindstedt R, Gesa V, Cirelli E, Patrizio M, et al. Human Monocytes Respond to Extracellular cAMP Through A2A and A2B Adenosine Receptors. J Leukoc Biol (2014) 96:113–22. doi: 10.1189/jlb.3A0513-302RR
118. Su W, Chen X, Zhu W, Yu J, Li W, Li Y, et al. The cAMP-Adenosine Feedback Loop Maintains the Suppressive Function of Regulatory T Cells. J Immunol (2019) 203:1436–46. doi: 10.4049/jimmunol.1801306
119. Allard D, Chrobak P, Allard B, Messaoudi N, Stagg J. Targeting the CD73-Adenosine Axis in Immuno-Oncology. Immunol Lett (2019) 205:31–9. doi: 10.1016/j.imlet.2018.05.001
120. Roh M, Wainwright DA, Wu JD, Wan Y, Zhang B. Targeting CD73 to Augment Cancer Immunotherapy. Curr Opin Pharmacol (2020) 53:66–76. doi: 10.1016/j.coph.2020.07.001
121. O’Connor R, O’Leary M, Ballot J, Collins CD, Kinsella P, Mager DE, et al. A Phase I Clinical and Pharmacokinetic Study of the Multi-Drug Resistance Protein-1 (MRP-1) Inhibitor Sulindac, in Combination With Epirubicin in Patients With Advanced Cancer. Cancer Chemother Pharmacol (2007) 59:79–87. doi: 10.1007/s00280-006-0240-7
122. Polosa R, Blackburn MR. Adenosine Receptors as Targets for Therapeutic Intervention in Asthma and Chronic Obstructive Pulmonary Disease. Trends Pharmacol Sci (2009) 30:528–35. doi: 10.1016/j.tips.2009.07.005
123. Jacobson KA, Tosh DK, Jain S, Gao ZG. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front Cell Neurosci (2019) 13:124. doi: 10.3389/fncel.2019.00124
124. Effendi WI, Nagano T, Kobayashi K, Nishimura Y. Focusing on Adenosine Receptors as a Potential Targeted Therapy in Human Diseases. Cells (2020) 9:cells9030785. doi: 10.3390/cells9030785
125. Davoren PR, Sutherland EW. The Effect of L-Epinephrine and Other Agents on the Synthesis and Release of Adenosine 3´,5´´-Phosphate by Whole Pigeon Erythrocytes. J Biol Chem (1963) 238:3009–15. doi: 10.1016/S0021-9258(18)51859-7
126. Patel BS, Rahman MM, Baehring G, Xenaki D, Tang FS, Oliver BG, et al. Roflumilast N-Oxide in Combination With Formoterol Enhances the Antiinflammatory Effect of Dexamethasone in Airway Smooth Muscle Cells. Am J Respir Cell Mol Biol (2017) 56:532–8. doi: 10.1165/rcmb.2016-0191OC
127. Nuñez FJ, Johnstone TB, Corpuz ML, Kazarian AG, Mohajer NN, Tliba O, et al. Glucocorticoids Rapidly Activate cAMP Production via G( s) to Initiate Non-Genomic Signaling That Contributes to One-Third of Their Canonical Genomic Effects. FASEB J (2020) 34:2882–95. doi: 10.1096/fj.201902521R
128. Koziol-White C, Johnstone TB, Corpuz ML, Cao G, Orfanos S, Parikh V, et al. Budesonide Enhances Agonist-Induced Bronchodilation in Human Small Airways by Increasing cAMP Production in Airway Smooth Muscle. Am J Physiol Lung Cell Mol Physiol (2020) 318:L345–55. doi: 10.1152/ajplung.00393.2019
Keywords: adenosine, adenosine receptors (AR), cyclic AMP (cAMP), airway, chronic inflammatory diseases, inflammation, ecto-phosphodiesterase, ecto-5’nucleotidases
Citation: Pacini ESA, Satori NA, Jackson EK and Godinho RO (2022) Extracellular cAMP-Adenosine Pathway Signaling: A Potential Therapeutic Target in Chronic Inflammatory Airway Diseases. Front. Immunol. 13:866097. doi: 10.3389/fimmu.2022.866097
Received: 30 January 2022; Accepted: 21 March 2022;
Published: 11 April 2022.
Edited by:
Robson Coutinho-Silva, Federal University of Rio de Janeiro, BrazilReviewed by:
Laura Vitiello, IRCCS San Raffaele Roma, ItalyRonald Sluyter, University of Wollongong, Australia
Copyright © 2022 Pacini, Satori, Jackson and Godinho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosely Oliveira Godinho, Z29kaW5ob0B1bmlmZXNwLmJy