 Francesco Fabozzi
Francesco Fabozzi Angela Mastronuzzi
Angela Mastronuzzi Giulia Ceglie
Giulia Ceglie Riccardo Masetti
Riccardo Masetti Davide Leardini
Davide Leardini
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 13 June 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.865773
This article is part of the Research Topic The Role of Transcription Factors in Inborn Errors of Immunity View all 7 articles
GATA2 deficiency is a disease with a broad spectrum of clinical presentation, ranging from lymphedema, deafness, pulmonary dysfunction to miscarriage and urogenital anomalies, but it is mainly recognized as an immune system and bone marrow disorder. It is caused by various heterozygous mutations in the GATA2 gene, encoding for a zinc finger transcription factor with a key role for the development and maintenance of a pool of hematopoietic stem cells; notably, most of these mutations arise de novo. Patients carrying a mutated allele usually develop a loss of some cell populations, such as B-cell, dendritic cell, natural killer cell, and monocytes, and are predisposed to disseminated human papilloma virus and mycobacterial infections. Also, these patients have a predisposition to myeloid neoplasms, including myelodysplastic syndromes, myeloproliferative neoplasms, chronic myelomonocytic leukaemia. The age of symptoms onset can vary greatly even also within the same family, ranging from early childhood to late adulthood; incidence increases by age and most frequently clinical presentation is between the second and third decade of life. Currently, haematopoietic stem cell transplantation represents the only curative treatment, restoring both the hematopoietic and immune system function.
GATA family includes six nuclear transcriptional factors sharing the ability to bind to the consensus DNA sequence T/A (GATA)A/G; of those, GATA4, GATA5, and GATA6 are involved in cardiac formation, GATA3 is expressed in several tissues, whereas GATA1 and GATA2 have a leading role in hematopoiesis (1).
In the past decade, GATA2 deficiency has been recognized to cause previously distinct clinical entities, namely DCML deficiency (dendritic cell, monocyte, B, and NK lymphoid deficiency) (2), MonoMAC syndrome (monocytopenia with Mycobacterium avium complex infection) (3), familial myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML) (4), Emberger syndrome (MDS with lymphedema) (5), and NK deficiency (6, 7). This wide spectrum of clinical manifestations highlights the complex role of this transcription factor: crucial for hematopoietic stem cell (HSC) activity and self-renewal, but also important for vessels and ear development (1, 8). Due to a very high lifetime risk for hematological malignancies, patients with GATA2 deficiency are candidates for hematopoietic stem cell transplantation (HSCT), currently the only current curative treatment. However, given the wide range of disease severity and the not negligible treatment related mortality (TRM), the optimal transplantation timing is still unclear (9). Herein, we briefly review the genetic and clinical features of GATA2 deficiency, focusing on immunological phenotypes. Finally, we summarize the state-of-the-art management options, shedding light on the current main research gaps.
The structure of this important transcription factor consists of two highly conserved zinc finger domains (ZF1 and ZF2), essential for binding to DNA and other proteins, a nuclear localization signal, two transactivation domains, and a negative regulatory domain (1) (Figure 1). While it can also be expressed in endothelial cells, central nervous system, placenta, fetal liver and fetal heart (10, 11), GATA2 plays a leading role in hematopoiesis through a complex network of interactions with other transcription factors such as RUNX1, SCL/TAL1, MYB,GFI1, FLI1,LYL1, or PU.1 (11). In fact, the homozygous knock-out in mouse embryo is lethal, due to failure in the endothelial-hematopoietic transition that produces the first adult HCSs (12). Of note, murine models with homozygous mutations that leave a residual but insufficient protein activity result in embryo death at a later stage, due to endothelial failure instead of the hematopoietic failure, highlightening the importance of gene dose (13). In post-natal life, GATA2 is essential for the maintenance of HSCs pool and for progenitor cell differentiation, for instance, mice carrying heterozygous GATA2 mutations exhibit a reduced amount of HSCs (11). Differently from human GATA2 deficiency phenotype, mouse model with GATA2 haploinsufficiency present normal bone marrow cellularity and peripheral blood cell count without the development of MDS or AML, suggesting a different effect of the mutation (14), probably as a consequence of the shorter mice lifespan (10). Thus, establishing a mouse model to study the mechanisms underlying this disease represents a current research challenge. With the aim of investigating whether further reduced expression of GATA2 could serve as a model for human disease, a hypomorphic mutant mouse line was established by Hoshino and colleagues, in which the expression level of GATA2 was reduced to approximately 20% of normal (15). Despite being apparently healthy at birth, approximately 70% of these mice die early due to bilateral ureterovesical junction obstruction, highlightening the role of gene dose in urogenital organogenesis (15). However, so far no case of death due to fatal urogenital malformations has been reported in humans, although urogenital malformations may occur in a certain proportion of people affected by GATA2 deficiency (16). Interestingly, mice that survive to adulthood are predisposed to develop myeloproliferative neoplasms resembling human chronic myelomonocytic leukemia (17). Nevertheless, distinctive features of GATA2 deficiency, namely cytopenia involving dendritic cells, monocytes, B or NK lymphocytes, were not found in this mouse line, indicating that quantitative deficits of GATA2 do not represent a suitable model of human disease. Recently, a very promising approach to this problem by following the qualitative rather than quantitative deficit way has been published by Hasegawa and colleagues (18). They generated mice that expressed the mutant GATA2 protein in which arginine 398 was substituted for tryptophan, resulting in a dominant-negative effect on the native protein in terms of DNA-binding activity that induces perturbations of the regulation of specific target genes. Mice were found to develop an age-progressive multilineage cytopenia of dendritic cells, monocytes, B and NK lymphocytes, resembling the human phenotype. However, the mice never developed hematopoietic malignancies, indicating that improvements are still needed in establishing a suitable model of GATA2 deficiency.
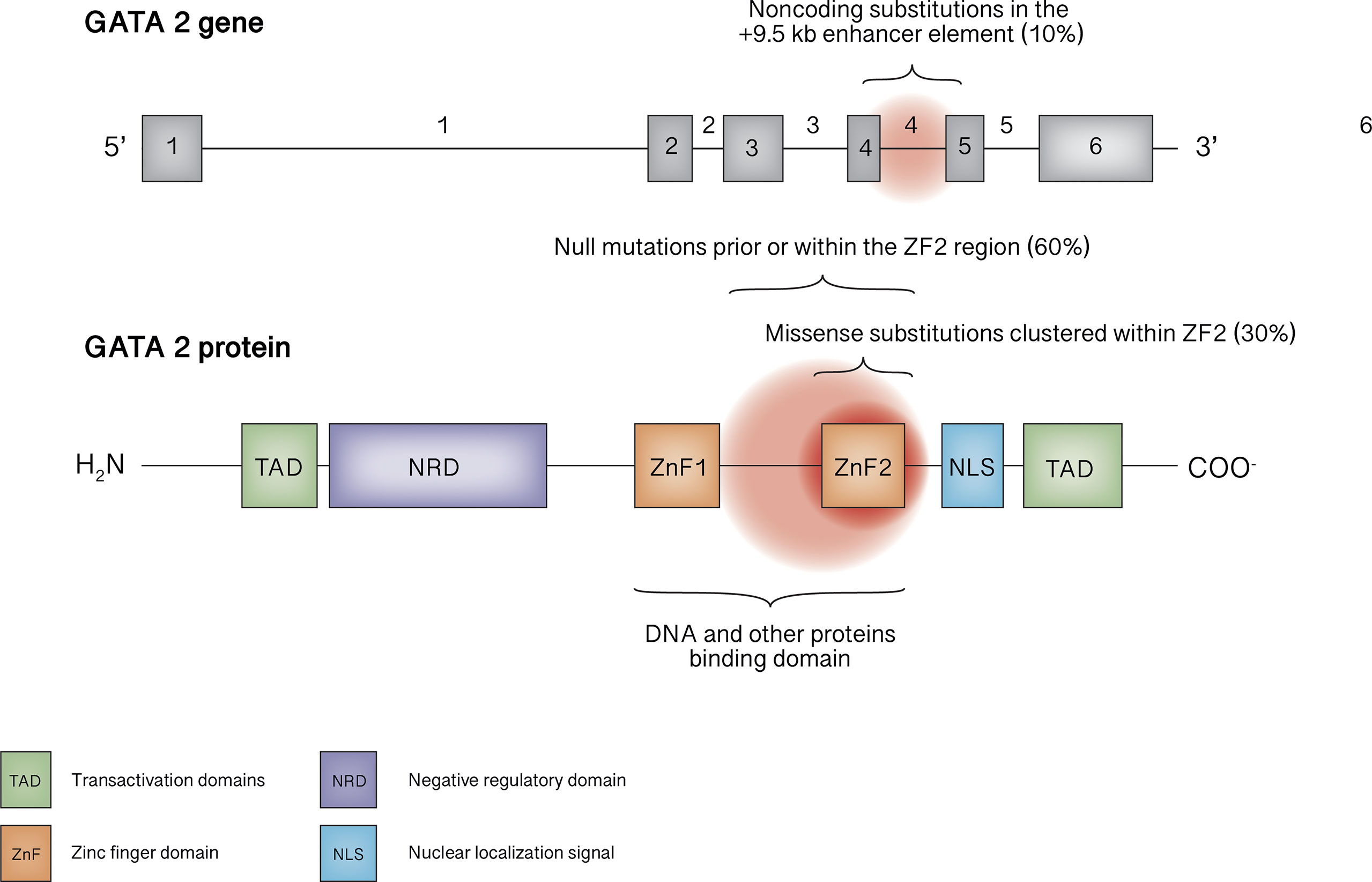
Figure 1 GATA2 gene and protein structure. The mutational spectrum in GATA2 deficiency mainly involves truncating mutations occurring before or within ZF2 or missense mutations within ZF2, as well as noncoding mutations within the +9.5 kb enhancer element.
The GATA2 gene is located on chromosome 3q21.2. To date, more than 150 pathogenic or likely pathogenic germline variants are reported, affecting nearly 500 individuals (11, 16, 19, 20). Null mutations (frameshift, nonsense, splice site and whole gene deletions), located prior or within the ZF2 region, underlie roughly 60% of cases, whereas missense substitutions clustered within ZF2 represent about 30% of mutations. Noncoding substitutions in the +9.5 kb enhancer element (EBOX-GATA-ETS regulatory region in intron 4) are responsible for 4-10% of cases; synonymous mutations introducing new splice donor site, that causes premature translation termination associated with nonsense-mediated decay, account for 8% of cases in the European Working Group of MDS in Childhood (EWOG-MDS) study (21). In addition, tandem GATA2 duplication was recently described in a patient with atypical mycobacterial infection who exhibited an increased transcription of the anti-sense long-non-coding RNA GATA2-AS1, probably responsible for the pathogenetic mechanism (22). Finally, germline mutations involving ZF1 have been described only rarely (23, 24).
Together, all mutations are loss-of-function and result in haploinsufficiency. How reduced levels of GATA2 can cause such a broad spectrum of clinical phenotypes, as well as variable penetrance, even within the same family, is not yet fully elucidated (25). The high variety of mutations could play a role, while epigenetic changes could underlie the reduced penetrance in asymptomatic patients (25–27).
The clinical landscape of GATA2 deficiency is dominated by bone marrow dysfunction, leading to recurrent infection (especially atypical mycobacterial infections and recurrent HPV-related warts) and hematological malignancies. Other characteristic features are pulmonary alveolar proteinosis (PAP) and lymphedema. The median age of clinical onset is 18 years, and the penetrance is estimated at 90% by the age of 60 years (28). At birth, patients show normal cell counts, as demonstrated in carriers recognized by familiar screening. Then, a progressive reduction of CD34+ cells in bone marrow can be demonstrated, inversely correlated with the serum levels of FLT3 ligand, a potential marker for monitoring the evolution of bone marrow failure (29). This leads to the classic peripheral blood cells decrease: monocytopenia, dendritic cell deficiency, NK cell deficiency, B cell deficiency and, less commonly, neutropenia. The severity of manifestations depends on peripheral cell counts: the more severe the cytopenia, the more severe the symptoms (Figure 2).
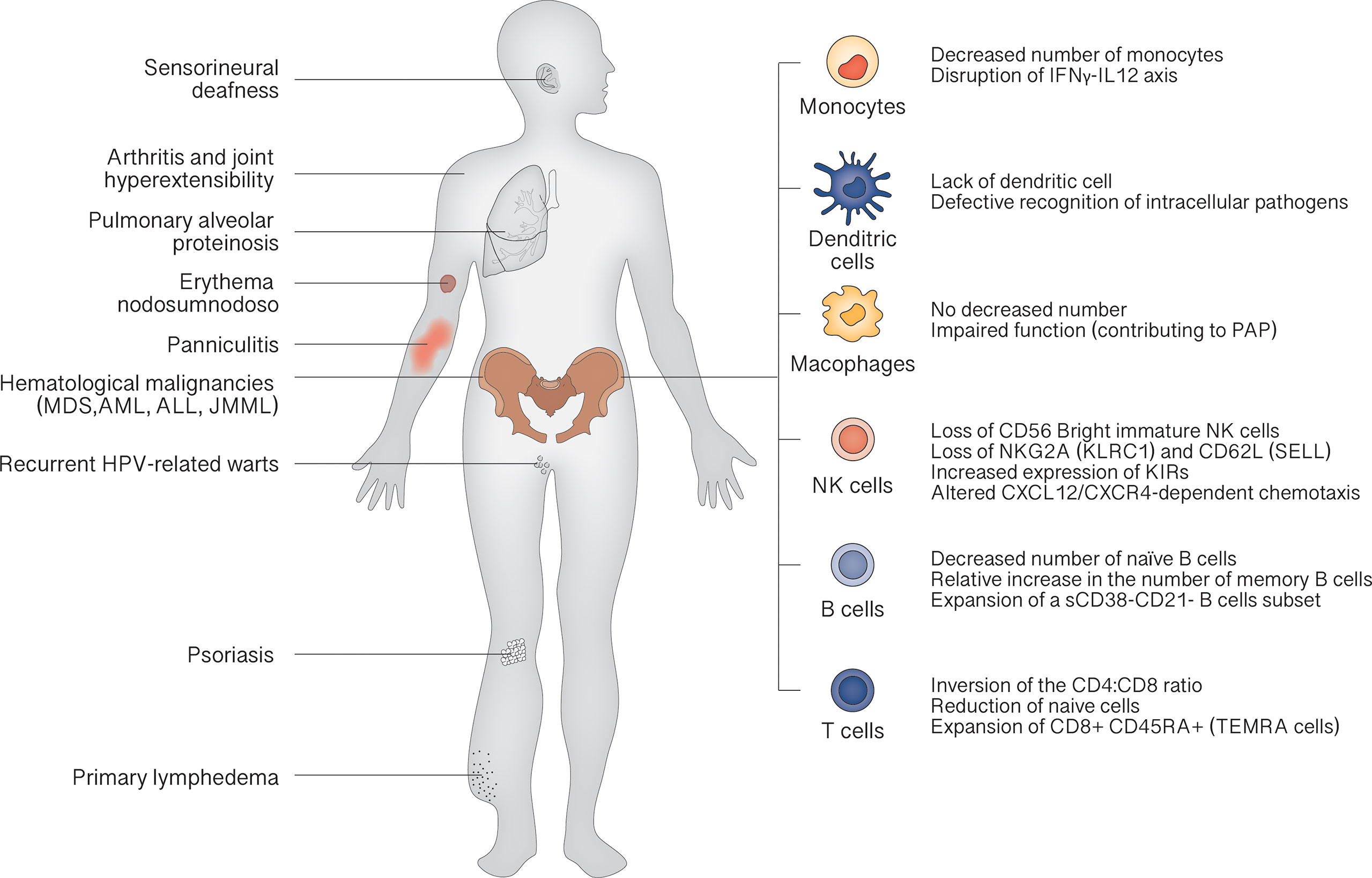
Figure 2 Clinical features of GATA2 deficiency.
According to 2019 Update of the IUIS Phenotypical Classification, GATA2 deficiency is classified among congenital defects of phagocyte, underscoring the role of monocytopenia as a key feature (30). Lack of monocytes results in characteristic susceptibility to non-tuberculous mycobacterial infection, via disruption of IFNγ-IL12 axis that plays a crucial role in resistance to these microorganisms (31). In fact, these infections occur in about half of the cases in the study by Spinner et al. (28). Also lack of DCs contributes to mycobacterial susceptibility by defective recognition of intracellular pathogens. Despite the absence of monocytes, macrophages can be detected in lungs of GATA2 patients, but their function is probably impaired, concurring to PAP trough a not still elucidated mechanism (10, 32).
Once marrow failure is established B and NK precursors are not detectable in the bone marrow of GATA2 patients (33). Loss of CD56 Bright immature NK cells and skewing toward a more highly differentiated phenotype, with loss of NKG2A (KLRC1) and CD62L (SELL) and increased expression of killer cell immunoglobulin-like receptors (KIRs), are typical findings (29). The few circulating NK cells also present an altered CXCL12/CXCR4-dependent chemotaxis (7, 34). A decrease in the number of naïve B cells, with a relative increase in the number of memory B cells and an expansion of a small subset of CD38-CD21- B cells associated with autoimmunity, is constantly found (29).
Although less compromised than other lymphocyte subsets, T cells show inversion of the CD4:CD8 ratio, with a reduction of naive cells and expansion of terminally differentiated CD8+ CD45RA+ effector cells (TEMRA cells). Taken together, the cellular phenotype resembles the pattern of terminal differentiation seen in age-related immunosenescence or in chronic viral infections (35).
The above-described alterations of immune system account for the high rate of infectious diseases in GATA2 patients, HPV and atypical mycobacterial being the most commonly found pathogens. Recurrent warts are the most common presentation of HPV infection, occurring in about half of patients, and are typically refractory to treatment (23, 28). The exact mechanisms of control of HPV infections are unclear, but cellular and cytotoxic immunity may have a crucial role. Both CD4 and CD8 T cells are found in regressing genital warts, and there are increasing evidences about the importance of CD4 T cell responses to E2 and E6 specific HPV proteins in controlling HPV-16 (36). The defective immunosurveillance by NK cells is responsible for a high rate of malignant transformation of HPV lesions. In addition, a significant amount of patients suffers from severe herpes virus infections, occurring in about 1/3 of patients: recurrent stomatitis, esophagitis, genital infections due to HSV 1-2, as well as severe VZV, EBV and CMV infections (28). Reduction of naïve B and T cells, with reduced NK cell-mediated cytotoxicity, promotes severity of EBV infections, as well as neoplastic transformation by the virus; moreover, the ability of the EBV to maintain latency may be disrupted by reduced expression of NOTCH1, TRAF2, and TRAF3 in cells from patients with GATA2 deficiency, resulting in increased virus replication (37). Finally, severe fungal infections (aspergillosis, histoplasmosis and candidiasis) occur in approximately 16% of patients (28).
Recently, rheumatological manifestations were found in 17.8% of patients in a study (38), and a correlation with a lower CD4:CD8 T cells ratio was found. Surprisingly, other than panniculitis, erythema nodosum, psoriasis and arthritis, non-immunologically mediated disorders such as piezogenic pedal papules and joint hyperextensibility were found in a significant number of patients, suggesting a role of GATA2 in collagen architecture. This is a distinguishing feature of GATA2 deficiency, since non-immunologically mediated rheumatologic manifestations have not been described in MDS patients carrying wild type GATA2 (39, 40). On the other hand, autoimmune manifestation can be found also in non-GATA2 mutated MDS with a similar prevalence, although other differences exist between the two phenotypes. In non-GATA2 mutated MDS rheumatologic disorders usually develop months/years after MDS diagnosis, whereas in GATA2 deficiency they can develop independently from hematological manifestations even without any other clinical symptom (40). Moreover, the most prevalent rheumatologic manifestations in the non-GATA2 mutated MDS group are vasculitis, namely polyarteritis nodosa and giant-cell arteritis, differently from GATA2 mutated patients (39, 40).
Although immunodeficiency represents one of the main features of the clinical picture, the high risk of developing myeloid neoplasms is the main sword hanging over the head of patients with GATA2 deficiency, presenting in more than 80% of patients by the age of 40 (41). The mechanism underlying this risk is likely to be found in the continuous stress, due to cytopenias and recurrent infections, to the malfunctioning bone marrow, leading to an accumulation of somatic mutations and selection of a clone with proliferative advantage in a predominantly hypocellular environment; ASXL1 alterations drive most frequently leukemogenic transformation (42, 43). Together with SAMD9/9L mutation, GATA2 germline mutation is one of the most common genetic cause of MDS, causing 15% of advanced forms and 7% of all primary MDS in childhood (44, 45). Mirror wise, MDS is the commonest malignancy among this population, representing roughly 3/4 of neoplasms, followed by AML, that can evolve from MDS or be the initial presentation (23, 41, 44). Lymphoblastic acute leukemia (ALL), juvenile myelomonocytic leukemia (JMML), and aplastic anemia (AA) can also occur (23, 41, 44). Cytogenetic aberrations are frequently identified in GATA2-related MDS: monosomy 7 or der(7; 1q) are the most common, found in about 41% of cases, followed by trisomy 8, identified in roughly 15% of cases among published studies (11, 20); moreover, complex karyotypes are generally very rare, whereas del5q have never been described (11, 20). It should be emphasized that monosomy 7 in pediatric MDS is highly suggestive of a genetic alteration, with a germline mutation of SAMD9/9L or GATA2 found in about half of the cases (45). Notably, a GATA2 mutation is not a negative prognostic factor among MDS (44, 46).
Other clinical features characteristic of GATA2 deficiency are lymphedema and sensorineural deafness, showing the crucial role of GATA2 in the development of both lymphatic vessels and lymphatic valves, as well as of semicircular canals and perilymphatic spaces (47, 48). GATA2 patients also have an increased risk of neoplasms other than hematologic, likely caused by impaired immunosurveillance (10). Miscarriages are frequently reported and are probably due to NK lymphopenia (28, 49).
Given the broad spectrum of clinical manifestations, the diagnosis of GATA2 deficiency can be challenging. A strong clinical suspicion must arise when characteristic features, such lymphedema, deafness, recurrent HPV-related warts, PAP, and atypical mycobacterial infection, are present together with cytopenia including monocytopenia alone. Moreover, all children with high-risk MDS should be considered for GATA2 deficiency, especially those affected by MDS with excess blasts (44, 45). Intrafamilial recurrence of MDS may be another important clue. Once the mutation is found, a baseline bone marrow biopsy should be performed, and all family members should be tested given the variable penetrance. Because of progressive nature of this disease, healthy carriers must be warranted a close follow-up, including complete blood count, lymphocyte subset assessment, bone marrow aspiration (with flow cytometry, cytogenetic and mycobacterial culture), complete dermatologic examination, pulmonary function tests and, in women, gynecologic examination (41, 42, 50). Routine vaccinations should be administered; live virus vaccinations are safe as long as blood counts are normal, while there are no data regarding cytopenic patients (50). Azithromycin prophylaxis could be considered as blood counts decrease, although its use is not supported by consistent evidence (42).
Currently, HSCT represents the only curative treatment, as it has been shown to restore normal hematopoiesis, resolving MDS, clearing underlying infections and improving pulmonary function. However, the great variability in clinical presentation as well as age of onset raises several questions about who and when to transplant. This decision is usually driven by occurrence of malignancies in childhood, and by both malignancies and life-threatening infections in adulthood. Recently, HSCT outcome was demonstrated to be independent of GATA2 germline mutations in a EWOG-MSD study regarding pediatric MDS, suggesting the application of standard MDS algorithms and protocols in this population (46). On the other hand, a better prognosis was found when HSCT is performed early in the disease: in fact, since GATA2 related MDS tends inexorably to progress in more aggressive forms such as leukemia, may be wise considering a preemptive HSCT (46). The excellent HSCT outcome was also confirmed by Nichols-Vinueza et al. in in a different cohort of adult patients (mean age about 28 years) underwent HSCT for life-threatening infections or MDS. The reported overall survival (OS) and event-free survival (EFS) at 4 years were 85% and 82% respectively (51).
Further research is needed to find other effective treatments other than HSCT. Gene editing technologies, such as CRISPR/Cas9 system, representing one of the most intriguing approaches, in the wake of the success achieved in the treatment of other hereditary hematological diseases (52).
Since a heterogeneity of mutations involving different segment of the gene can be found in patients with GATA2 deficiency, the use of homology-direct repair (HDR) as a correction mechanism, may be attempted using large HDR donor templates that cover the whole gene. Unfortunately, the low efficiency of HDR mediated repair combined with the decreased pool of HSCs present in patients, represent one of the major hurdles for gene therapy (53). In addition, the minimum level of chimerism to reverse the phenotype is still not clear. Nevertheless, a case of somatic genetic rescue (SGR) in an asymptomatic individual carrying a pathogenic GATA2 germline mutation was recently described. It was speculated that HSCs carrying the restored GATA2 gene enjoyed a selective advantage over “diseased” cells, restoring normal bone marrow function (54, 55). This observation supports the hypothesis that even a small number of mutation-corrected HSCs might be sufficient to heal or at least mitigate the manifestations of GATA2 deficiency in affected patients.
Germline mutations of GATA2 cause a various and complex clinical phenotype, including immunodeficiency, bone marrow failure, lymphedema, deafness, and pulmonary dysfunction. Despite the emergent findings, the exact pathogenetic mechanism underlying these alterations is still not fully understood. Also, there are still many gaps regarding the optimal management of patients with GATA2 deficiency, highlighting the need for dedicated guidelines.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Crispino JD, Horwitz MS. GATA Factor Mutations in Hematologic Disease. Blood (2017) 129:2103–10. doi: 10.1182/blood-2016-09-687889
2. Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, et al. Mutations in GATA2 Cause Primary Lymphedema Associated With a Predisposition to Acute Myeloid Leukemia (Emberger Syndrome). Nat Genet (2011) 43:929–31. doi: 10.1038/ng.923
3. Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are Associated With the Autosomal Dominant and Sporadic Monocytopenia and Mycobacterial Infection (MonoMAC) Syndrome. Blood (2011) 118:2653–5. doi: 10.1182/blood-2011-05-356352
4. Hahn CN, Chong C-E, Carmichael CL, Wilkins EJ, Brautigan PJ, Li X-C, et al. Heritable GATA2 Mutations Associated With Familial Myelodysplastic Syndrome and Acute Myeloid Leukemia. Nat Genet (2011) 43:1012–7. doi: 10.1038/ng.913
5. Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome Sequencing Identifies GATA-2 Mutation as the Cause of Dendritic Cell, Monocyte, B and NK Lymphoid Deficiency. Blood (2011) 118:2656–8. doi: 10.1182/blood-2011-06-360313
6. Biron CA, Byron KS, Sullivan JL. Severe Herpesvirus Infections in an Adolescent Without Natural Killer Cells. N Engl J Med (1989) 320:1731–5. doi: 10.1056/NEJM198906293202605
7. Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, et al. Mutations in GATA2 Cause Human NK Cell Deficiency With Specific Loss of the CD56bright Subset. Blood (2013) 121:2669–77. doi: 10.1182/blood-2012-09-453969
8. Leubolt G, Redondo Monte E, Greif PA. GATA2 Mutations in Myeloid Malignancies: Two Zinc Fingers in Many Pies. IUBMB Life (2020) 72:151–8. doi: 10.1002/iub.2204
9. Fabozzi F, Strocchio L, Mastronuzzi A, Merli P. GATA2 and Marrow Failure. Best Pract Res Clin Haematol (2021) 34:101278. doi: 10.1016/j.beha.2021.101278
10. Collin M, Dickinson R, Bigley V. Haematopoietic and Immune Defects Associated With GATA2 Mutation. Br J Haematol (2015) 169:173–87. doi: 10.1111/bjh.13317
11. Wlodarski MW, Collin M, Horwitz MS. GATA2 Deficiency and Related Myeloid Neoplasms. Semin Hematol (2017) 54:81–6. doi: 10.1053/j.seminhematol.2017.05.002
12. Tsai FY, Keller G, Kuo FC, Weiss M, Chen J, Rosenblatt M, et al. An Early Haematopoietic Defect in Mice Lacking the Transcription Factor GATA-2. Nature (1994) 371:221–6. doi: 10.1038/371221a0
13. Johnson KD, Hsu AP, Ryu M-J, Wang J, Gao X, Boyer ME, et al. Cis-Element Mutated in GATA2-Dependent Immunodeficiency Governs Hematopoiesis and Vascular Integrity. J Clin Invest (2012) 122:3692–704. doi: 10.1172/JCI61623
14. Rodrigues NP, Janzen V, Forkert R, Dombkowski DM, Boyd AS, Orkin SH, et al. Haploinsufficiency of GATA-2 Perturbs Adult Hematopoietic Stem-Cell Homeostasis. Blood (2005) 106:477–84. doi: 10.1182/blood-2004-08-2989
15. Hoshino T, Shimizu R, Ohmori S, Nagano M, Pan X, Ohneda O, et al. Reduced BMP4 Abundance in Gata2 Hypomorphic Mutant Mice Result in Uropathies Resembling Human CAKUT. Genes Cells (2008) 13:159–70. doi: 10.1111/j.1365-2443.2007.01158.x
16. Shimizu R, Yamamoto M. Quantitative and Qualitative Impairments in GATA2 and Myeloid Neoplasms. IUBMB Life (2020) 72:142–50. doi: 10.1002/iub.2188
17. Harada N, Hasegawa A, Hirano I, Yamamoto M, Shimizu R. GATA2 Hypomorphism Induces Chronic Myelomonocytic Leukemia in Mice. Cancer Sci (2019) 110:1183–93. doi: 10.1111/cas.13959
18. Hasegawa A, Hayasaka Y, Morita M, Takenaka Y, Hosaka Y, Hirano I, et al. Heterozygous Variants in GATA2 Contribute to DCML Deficiency in Mice by Disrupting Tandem Protein Binding. Commun Biol (2022) 5:376. doi: 10.1038/s42003-022-03316-w
19. Homan CC, Venugopal P, Arts P, Shahrin NH, Feurstein S, Rawlings L, et al. GATA2 Deficiency Syndrome: A Decade of Discovery. Hum Mutat (2021) 42:1399–421. doi: 10.1002/humu.24271
20. Sahoo SS, Kozyra EJ, Wlodarski MW. Germline Predisposition in Myeloid Neoplasms: Unique Genetic and Clinical Features of GATA2 Deficiency and SAMD9/SAMD9L Syndromes. Best Pract Res Clin Haematol (2020) 33:101197. doi: 10.1016/j.beha.2020.101197
21. Kozyra EJ, Pastor VB, Lefkopoulos S, Sahoo SS, Busch H, Voss RK, et al. Synonymous GATA2 Mutations Result in Selective Loss of Mutated RNA and are Common in Patients With GATA2 Deficiency. Leukemia (2020) 34:2673–87. doi: 10.1038/s41375-020-0899-5
22. Singh P, Heer M, Resteu A, Mikulasova A, Reza M, Largeaud L, et al. GATA2 Deficiency Phenotype Associated With Tandem Duplication GATA2 and Over-Expression of GATA2-As1. Blood Adv (2021). doi: 10.1182/bloodadvances.2021005217
23. Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, Ouachee M, et al. Natural History of GATA2 Deficiency in a Survey of 79 French and Belgian Patients. Haematologica (2018) 103:1278–87. doi: 10.3324/haematol.2017.181909
24. Shiba N, Funato M, Ohki K, Park M-J, Mizushima Y, Adachi S, et al. Mutations of the GATA2 and CEBPA Genes in Paediatric Acute Myeloid Leukaemia. Br J Haematol (2014) 164:142–5. doi: 10.1111/bjh.12559
25. Bresnick EH, Jung MM, Katsumura KR. Human GATA2 Mutations and Hematologic Disease: How Many Paths to Pathogenesis? Blood Adv (2020) 4:4584–92. doi: 10.1182/bloodadvances.2020002953
26. Al Seraihi AF, Rio-Machin A, Tawana K, Bödör C, Wang J, Nagano A, et al. GATA2 Monoallelic Expression Underlies Reduced Penetrance in Inherited GATA2 -Mutated MDS/AML. Leukemia (2018) 32:2502–7. doi: 10.1038/s41375-018-0134-9
27. Kim N, Choi S, Kim S-M, Lee AC, Im K, Park HS, et al. Monozygotic Twins With Shared De Novo GATA2 Mutation But Dissimilar Phenotypes Due to Differential Promoter Methylation. Leuk Lymphoma (2019) 60:1053–61. doi: 10.1080/10428194.2018.1516039
28. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 Deficiency: A Protean Disorder of Hematopoiesis, Lymphatics, and Immunity. Blood (2014) 123:809–21. doi: 10.1182/blood-2013-07-515528
29. Dickinson RE, Milne P, Jardine L, Zandi S, Swierczek SI, McGovern N, et al. The Evolution of Cellular Deficiency in GATA2 Mutation. Blood (2014) 123:863–74. doi: 10.1182/blood-2013-07-517151
30. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40:66–81. doi: 10.1007/s10875-020-00758-x
31. Wu U-I, Holland SM. Host Susceptibility to Non-Tuberculous Mycobacterial Infections. Lancet Infect Dis (2015) 15:968–80. doi: 10.1016/S1473-3099(15)00089-4
32. Marciano BE, Olivier KN, Folio LR, Zerbe CS, Hsu AP, Freeman AF, et al. Pulmonary Manifestations of GATA2 Deficiency. Chest (2021) 160:1350–9. doi: 10.1016/j.chest.2021.05.046
33. Bigley V, Haniffa M, Doulatov S, Wang X-N, Dickinson R, McGovern N, et al. The Human Syndrome of Dendritic Cell, Monocyte, B and NK Lymphoid Deficiency. J Exp Med (2011) 208:227–34. doi: 10.1084/jem.20101459
34. Maciejewski-Duval A, Meuris F, Bignon A, Aknin M-L, Balabanian K, Faivre L, et al. Altered Chemotactic Response to CXCL12 in Patients Carrying GATA2 Mutations. J Leukoc Biol (2016) 99:1065–76. doi: 10.1189/jlb.5MA0815-388R
35. Strindhall J, Skog M, Ernerudh J, Bengner M, Löfgren S, Matussek A, et al. The Inverted CD4/CD8 Ratio and Associated Parameters in 66-Year-Old Individuals: The Swedish HEXA Immune Study. Age (Dordr) (2013) 35:985–91. doi: 10.1007/s11357-012-9400-3
36. Leiding JW, Holland SM. Warts and All: Human Papillomavirus in Primary Immunodeficiencies. J Allergy Clin Immunol (2012) 130:1030–48. doi: 10.1016/j.jaci.2012.07.049
37. Cohen JI. GATA2 Deficiency and Epstein–Barr Virus Disease. Front Immunol (2017) 8:1869. doi: 10.3389/fimmu.2017.01869
38. Amarnani AA, Poladian KR, Marciano BE, Daub JR, Williams SG, Livinski AA, et al. A Panoply of Rheumatological Manifestations in Patients With GATA2 Deficiency. Sci Rep (2020) 10:8305. doi: 10.1038/s41598-020-64852-1
39. Grignano E, Jachiet V, Fenaux P, Ades L, Fain O, Mekinian A. Autoimmune Manifestations Associated With Myelodysplastic Syndromes. Ann Hematol (2018) 97:2015–23. doi: 10.1007/s00277-018-3472-9
40. Barcellini W, Giannotta JA, Fattizzo B. Autoimmune Complications in Hematologic Neoplasms. Cancers (Basel) (2021) 13:1532. doi: 10.3390/cancers13071532
41. Bruzzese A, Leardini D, Masetti R, Strocchio L, Girardi K, Algeri M, et al. GATA2 Related Conditions and Predisposition to Pediatric Myelodysplastic Syndromes. Cancers (2020) 12:2962. doi: 10.3390/cancers12102962
42. Hsu AP, McReynolds LJ, Holland SM. GATA2 Deficiency. Curr Opin Allergy Clin Immunol (2015) 15:104–9. doi: 10.1097/ACI.0000000000000126
43. West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD. Acquired ASXL1 Mutations are Common in Patients With Inherited GATA2 Mutations and Correlate With Myeloid Transformation. Haematologica (2014) 99:276–81. doi: 10.3324/haematol.2013.090217
44. Wlodarski MW, Hirabayashi S, Pastor V, Starý J, Hasle H, Masetti R, et al. Prevalence, Clinical Characteristics, and Prognosis of GATA2-Related Myelodysplastic Syndromes in Children and Adolescents. Blood (2016) 127:1387–97. doi: 10.1182/blood-2015-09-669937
45. Sahoo SS, Pastor VB, Goodings C, Voss RK, Kozyra EJ, Szvetnik A, et al. Clinical Evolution, Genetic Landscape and Trajectories of Clonal Hematopoiesis in SAMD9/SAMD9L Syndromes. Nat Med (2021) 27:1806–17. doi: 10.1038/s41591-021-01511-6
46. Bortnick R, Wlodarski M, de Haas V, De Moerloose B, Dworzak M, Hasle H, et al. Hematopoietic Stem Cell Transplantation in Children and Adolescents With GATA2-Related Myelodysplastic Syndrome. Bone Marrow Transplant (2021) 56:2732–41. doi: 10.1038/s41409-021-01374-y
47. Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, et al. Loss-Of-Function Germline GATA2 Mutations in Patients With MDS/AML or MonoMAC Syndrome and Primary Lymphedema Reveal a Key Role for GATA2 in the Lymphatic Vasculature. Blood (2012) 119:1283–91. doi: 10.1182/blood-2011-08-374363
48. Haugas M, Lilleväli K, Hakanen J, Salminen M. Gata2 is Required for the Development of Inner Ear Semicircular Ducts and the Surrounding Perilymphatic Space. Dev Dynamics (2010) 239:2452–69. doi: 10.1002/dvdy.22373
49. Fu Y-Y, Ren C-E, Qiao P-Y, Meng Y-H. Uterinenatural Killer Cells and Recurrent Spontaneous Abortion. Am J Reprod Immunol (2021):e13433. doi: 10.1111/aji.13433
50. McReynolds LJ, Calvo KR, Holland SM. Germline GATA2 Mutation and Bone Marrow Failure. Hematology/Oncol Clinics North America (2018) 32:713–28. doi: 10.1016/j.hoc.2018.04.004
51. Nichols-Vinueza DX, Parta M, Shah NN, Cuellar-Rodriguez JM, Bauer TR, West RR, et al. Donor Source and Post-Transplantation Cyclophosphamide Influence Outcome in Allogeneic Stem Cell Transplantation for GATA2 Deficiency. Br J Haematol (2022) 196:169–78. doi: 10.1111/bjh.17840
52. Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med (2021) 384:252–60. doi: 10.1056/NEJMoa2031054
53. Koyunlar C, de Pater E. From Basic Biology to Patient Mutational Spectra of GATA2 Haploinsufficiencies: What Are the Mechanisms, Hurdles, and Prospects of Genome Editing for Treatment. Front Genome Ed (2020) 2:602182. doi: 10.3389/fgeed.2020.602182
54. Catto LFB, Borges G, Pinto AL, Clé DV, Chahud F, Santana BA, et al. Somatic Genetic Rescue in Hematopoietic Cells in GATA2 Deficiency. Blood (2020) 136:1002–5. doi: 10.1182/blood.2020005538
Keywords: GATA2 deficiency, transcription factors, inborn errors of immunity, recurrent infection, autoimmune disease, myelodysplastic syndromes, hematopoietic stem cell transplantation
Citation: Fabozzi F, Mastronuzzi A, Ceglie G, Masetti R and Leardini D (2022) GATA 2 Deficiency: Focus on Immune System Impairment. Front. Immunol. 13:865773. doi: 10.3389/fimmu.2022.865773
Received: 30 January 2022; Accepted: 19 May 2022;
Published: 13 June 2022.
Edited by:
Victoria Bordon, Ghent University, BelgiumReviewed by:
Masayuki Yamamoto, Tohoku University, JapanCopyright © 2022 Fabozzi, Mastronuzzi, Ceglie, Masetti and Leardini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Fabozzi, ZnJhbmNlc2NvLmZhYm96emlAb3BiZy5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.