 Erliang Kong
Erliang Kong Yongchang Li1
Yongchang Li1 Tong Hua
Tong Hua Jian Li
Jian Li Hongbin Yuan
Hongbin Yuan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 May 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.861290
This article is part of the Research Topic Orthodox vs Paradox: The Roles of Glycomics, Genetics and Beyond in Immunity, Immune Disorders and Glycomedicine View all 12 articles
Neuropathic pain is characterized by hyperalgesia and allodynia. Inflammatory response is conducive to tissue recovery upon nerve injury, but persistent and exaggerated inflammation is detrimental and participates in neuropathic pain. Synaptic transmission in the nociceptive pathway, and particularly the balance between facilitation and inhibition, could be affected by inflammation, which in turn is regulated by glial cells. Importantly, glycometabolism exerts a vital role in the inflammatory process. Glycometabolism reprogramming of inflammatory cells in neuropathic pain is characterized by impaired oxidative phosphorylation in mitochondria and enhanced glycolysis. These changes induce phenotypic transition of inflammatory cells to promote neural inflammation and oxidative stress in peripheral and central nervous system. Accumulation of lactate in synaptic microenvironment also contributes to synaptic remodeling and central sensitization. Previous studies mainly focused on the glycometabolism reprogramming in peripheral inflammatory cells such as macrophage or lymphocyte, little attention was paid to the regulation effects of glycometabolism reprogramming on the inflammatory responses in glial cells. This review summarizes the evidences for glycometabolism reprogramming in peripheral inflammatory cells, and presents a small quantity of present studies on glycometabolism in glial cells, expecting to promote the exploration in glycometabolism in glial cells of neuropathic pain.
Neuropathic pain is caused by a lesion/disease of the somatosensory system, and is estimated to affect 7%-10% of the general population (1, 2). The pathogenesis of neuropathic pain is complex, and involves the entire nociceptive pathway (primary afferent nerves, spinal cord, brain, and descending pathways) as well as glial cells.
The pathological basis of neuropathic pain is hyperalgesia and allodynia caused by synaptic remodeling in the nociceptive pathway. Chronic nerve injury promotes the release of pro-inflammatory cytokines to activate intracellular signal transduction pathways, and to disturb the balance between facilitation and inhibition in pain signal transduction. A variety of animal models for neuropathic pain (e.g., chronic constriction injury and spinal nerve ligation) have been developed based on persistent nerve injury (3). Recent studies identified abnormal glycometabolism in neurons and the supporting glial cells upon chronic nerve injury (4). Under normal oxygen-rich conditions, pyruvate enters the tricarboxylic acid (TCA) cycle for oxidative phosphorylation into CO2 and NADH in the central nervous system. Under hypoxic conditions, however, pyruvate is converted into lactate and NAD+ through anaerobic glycolysis. Anaerobic glycolysis has low efficiency in energy production than oxidative phosphorylation, but is the preferred metabolic pathway in the active phase of cell proliferation (5).
Glial cells are implicated in a variety of neurophysiological processes, including neuronal development, synaptic remodeling, and neuropathic pain (6). Glial cells have been shown to participate in chronic pain via multiple mechanisms, including regulating glutamate concentration in synaptic cleft through glutamate transporters (7), controlling the release of neurotransmitters (8), altering stability of the synaptic microenvironment (9), and modifying inter-neuronal communications (10). When activated by inflammation, glial cells switch from oxidative phosphorylation to preferentially use glycolysis as energy source, with accompanying changes in pentose phosphate pathway, amino hexanoic acid and glutamine hydrolysis pathway (11). Metabolic intermediates produced in glycometabolism reprogramming also provide substrates for other biosynthetic pathway in cell growth and differentiation, and could regulate a variety of intracellular signaling pathways at both the transcriptional and post-transcriptional levels. To some extent, these effects determine the fate of neurons or glial cells (12).
This review summarizes the neuropathology of neuropathic pain, regulation of phenotypic transition and pain sensitization by glycometabolism reprogramming of glial cells under chronic nerve injury, and the influences of glycometabolism reprogramming on synaptic plasticity and neuronal excitability. The viewpoints are helpful in exploring the crucial roles of glycometabolism reprogramming of glial cells in the development of neuropathic pain and providing potential targets for the intervention of neuropathic pain.
Nociceptive stimuli are converted into electrochemical signals by pain receptors and transmitted to the spinal cord via primary afferent neurons. As the station of signal regulation and integration, the spinal cord sends pain signals to the brain through the upward projection fibers. The upward signal transmission is regulated by downward signals through the spinal cord to effectors via efferent neurons (13). Changes in any part of this nociceptive pathway can lead to allodynia or hyperalgesia.
Chronic nerve injury could produce structural changes in the spinal projection area of afferent neurons. In physiological conditions, peripheral C fibers mainly project to the substantia gelatinosa (lamina II) of spinal cord to transmit chronic pain signals, whereas the Aδ fibers mainly project to lamina I and III to transmit acute pain signals. Tactile information is mainly transmitted by Aβ fibers that project to lamina III and IV. In neuropathic pain, chronic nerve injury induces abnormal projection of the Aβ fibers to neurons in lamina I and III to form additional neural circuits for hyperalgesia and allodynia. Chronic nerve injury also induces actin cytoskeleton remodeling, thus changing the density and length of dendritic spines of the neurons in spinal cord (14, 15). Rho/Rac molecules in the GTPase superfamily could transmit pain signals to intracellular actin cytoskeleton through neurotransmitters, and promote the generation of dendritic spines and ultimately structural communication among neurons. Selective inhibition of Rac1 protein has been shown to attenuate hyperalgesia and reduce the changes in dendritic spines in an animal model of neuropathic pain (16). Chronic nerve injury also promotes autophagy and apoptosis in the inhibitory γ-aminobutyric acid (GABA) interneurons, and reduces the activity of GABA synthase and glutamic acid decarboxylase (GAD), ultimately leading to neuronal disinhibition (17). As the supporting cells of neurons, glial cells are exquisitely sensitive to microenvironment changes. Activated glial cells secrete a variety of substances to promote interneuron sensitization. Intrathecal injection of a microglia activation inhibitor has been shown to attenuate hyperalgesia in a neuropathic pain model by reducing the expression of inflammatory factors in spinal microenvironment (18). Chronic nerve injury also affects a variety of other signaling molecules in central nervous system, including growth factors, neurotransmitters, intracellular second messengers, nuclear transcription factors and membrane receptors. Central sensitization seems to be the result of complex interaction among these mechanisms and disturbed balance between excitatory and inhibitory synapses (19) (Figure 1).
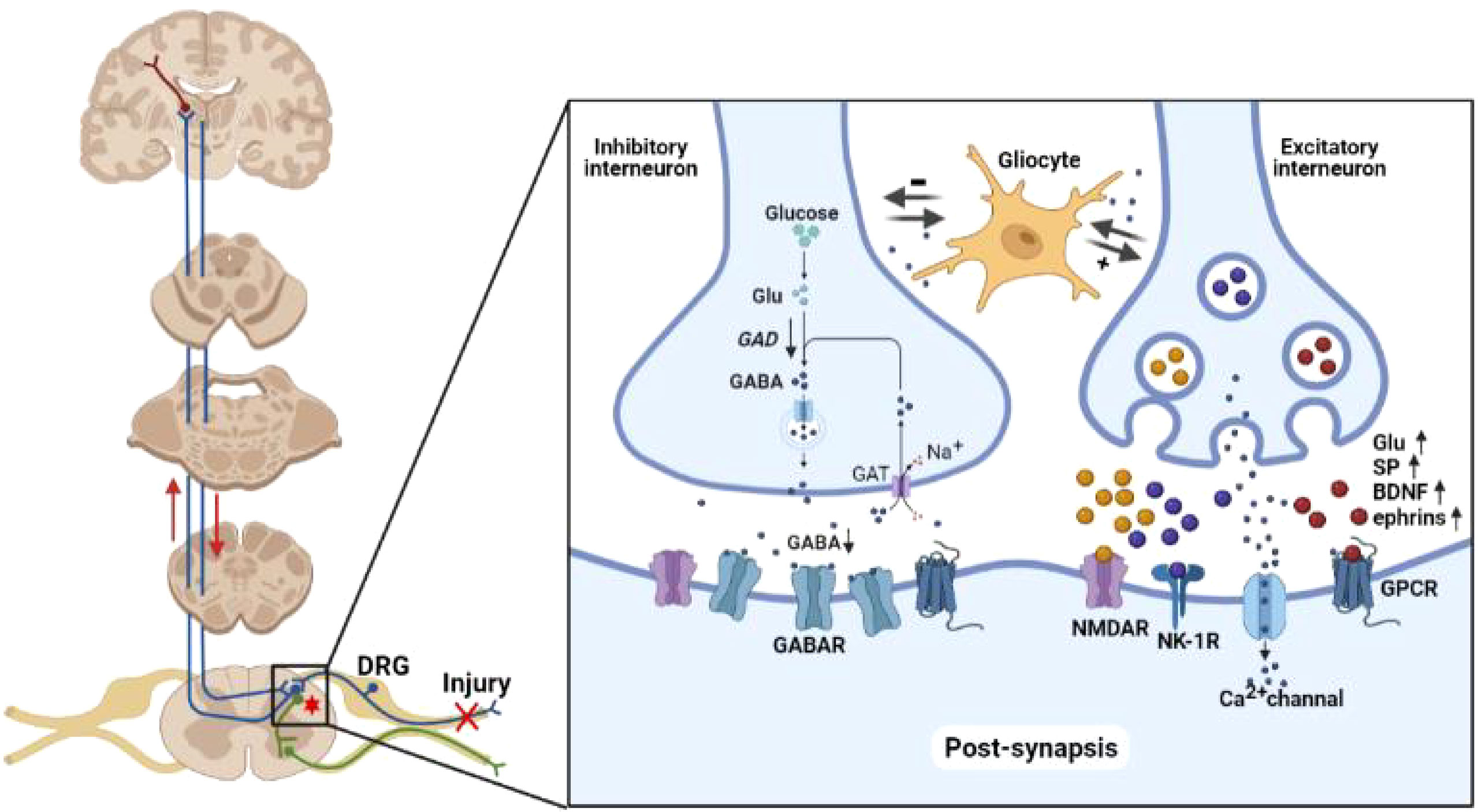
Figure 1 Neuropathology of neuropathic pain. Peripheral nociceptive stimuli are converted into electrochemical signals by pain receptors and transmitted to the brain through the spinal cord via upward projection fibers. The spinal cord also relays downward signal to regulate the nociceptive signal transmission. The imbalance of inhibitory/excitatory interneurons forms the basis of neuropathic pain. In inhibitory interneurons, GABA is synthesized from glutamate by GAD and released into the synaptic cleft. GABA in the synaptic cleft is taken up by interneurons via GABA transporters in a Na+-dependent mechanism. Chronic nerve injury reduces the activity of GABA synthase and GAD, ultimately resulting in disinhibition. Chronic nerve injury enhances the synthesis of glutamate, SP, BDNF and ephrins in excitatory interneurons. These neurotransmitters enhance the spontaneous excitatory postsynaptic currents mediated by AMPAR, NMDAR, NK-1R, and Ca2+ channel on postsynaptic membrane. Glu, glutamate; GAD, glutamic acid decarboxylase; GABA, γ-aminobutyric acid; GAT, GABA transporter; SP, substance P; BDNF, brain derived neurotrophic factor; NMDAR, N-methyl-D-aspartate receptor; NK-1R, neurokinin 1 receptor; GPCR, G protein-coupled receptor.
Synaptic plasticity is implicated in the development and ageing of the central nervous system, as well as the pathophysiology of a number of diseases, including Alzheimer’s disease and neuropathic pain. Synaptic plasticity can be categorized into functional (changes in information transmission) or structural (changes in information storage). Examples of functional synaptic plasticity included long-term potentiation (LTP; strengthening of synaptic connection) and long-term depression (LTD; weakening of synaptic connection). LTP is typically occurs in large synapses and dendritic spines whereas LTD tends to occur in small synapses (20). Upon repeated input of nociceptive signals from peripheral nerves to the spinal neurons, excitatory postsynaptic currents (EPSC) mediated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) on postsynaptic membrane tend to increase over time. This process requires synergistic activation of the N-methyl-D-aspartate (NMDA) receptor, neurokinin 1 (NK1) receptor and low-threshold T-type Ca2+ channel (21). Chronic pain is mainly transmitted to the interneurons in lamina II of spinal cord through C fibers. Chronic depolarizing stimulation removes the conformational block of voltage-dependent Mg2+ channel inside the NMDA receptor of the postsynaptic membrane, and enhance the sensitivity of neurons to subsequent stimulation. Chronic stimulation of C fibers also promotes the platform currents of L-type Ca2+ channel on spinal neurons to further enhance neuronal excitability (22). This complex process involves both excitatory and inhibitory transmitters, as well as neuromodulators such as substance P (SP) and brain derived neurotrophic factor (BDNF), G protein-coupled receptors (GPCR), NK-1 and tyrosine kinase B (TrkB) (23) (Figure 1).
Glucose is the main source of energy in central nervous system. Glucose enters neurons mainly actively through glucose transporter (Glut) on cell membrane (24). Pyruvate, the final product of glycolytic process, enters the TCA cycle in mitochondria under normal oxygen-rich conditions but is converted to lactate under hypoxic conditions. Pyruvate generates 32 ATP molecules through the TCA cycle and electron transport chain in mitochondria and only 2 ATP molecules upon conversion to lactate by lactate dehydrogenase (LDH) (25). Pyruvate dehydrogenase (PDH) is a rate-limiting enzyme in the TCA cycle, and is regulated (inhibited) via phosphorylation by pyruvate dehydrogenase kinase (PDK). When PDH is phosphorylated, pyruvate is shunted from the TCA cycle to anaerobic glycolysis (26). Four human PDK subtypes have been identified. PDK1 is activated in anoxic environment; PDK2 is activated upon acetyl-CoA and NADH accumulation; PDK3 is active in high-ATP environment; PDK4 plays a vital role upon starvation. Increased PDK2 and PDK4 expression has been found in spinal cord neurons in a diabetic model for neuralgia, and double knockout of PDK2 and PDK4 could alleviate hyperalgesia via inhibiting synaptic accumulation of pro-inflammatory factors and lactate and subsequent changes of ion channel permeability in neurons as well as glial cells (27). In primary culture of spinal neurons, exogenous lactate increases the permeability of cell membrane and alters the electrophysiological properties of synapses by facilitating calcium influx through calcium ion channels. The PDK inhibitor dichloroacetate and LDH inhibitor FX11 partially alleviate hyperalgesia in diabetic neuralgia model, providing a potential target for treatment of diabetic neuralgia (28).
Microglia and astrocytes play critical roles in neuroinflammation (29). Microglia are resident immune cells in the central nervous system, and could activate inflammasome, NF-κB and other inflammatory signaling pathways; in contrast, astrocytes are mainly involved in regulating the integrity and permeability of blood-brain barrier (30). In the resting state, microglia primarily rely on oxidative phosphorylation of glucose as energy source. Upon activation, microglia shift from the TCA cycle to anaerobic glycolysis (31) via multiple mechanisms, including increased expression of pro-inflammatory factors and accumulation of advanced glycation end products (AGEs) (32). Microglia activation also promotes autophagy and apoptosis of neurons. Short-term exposure to β amyloid shifts microglia from oxidative phosphorylation to glycolysis via the mammalian target of rapamycin/hypoxia inducible factor-1α (mTOR/HIF-1α) pathway (33). Long-term exposure to β amyloid, however, attenuated both glycolysis and oxidative phosphorylation, and reduced the responsiveness of microglia to noxious stimuli. In a mouse model for Alzheimer’s disease, exogenous interferon-γ (IFN-γ) attenuated neurological deficits by attenuating the stimulation of β amyloid to microglia through promoting glycolysis by activating mTOR pathway (33). In further studies, mice with TREM-2 knockout in Alzheimer’s disease showed decreased mTOR pathway activity, impaired glycolysis and increased neuronal autophagy (34). Enhanced glycolysis in activated microglia has also been noted in patients with multiple sclerosis, an autoimmune disease (35), and has been explored as potential target in the treatment of multiple sclerosis (36).
In addition to increased glycolysis at the expense of TAC cycle, glucometabolic reprogramming in microglia also features enhanced glutamine hydrolysis and pentose phosphate pathway. All together, these metabolic changes lead to the accumulation of a variety of intermediates, including phosphoenolpyruvic acid (PEP), succinate, citric acid, methylene succinic acid, α-ketoglutaric acid, lactate and 2-hydroxyglutaric acid, which in turns alters the acid-base balance in microenvironment, promotes transcription of pro-inflammatory factors and activates inflammatory signaling pathways to change the inflammatory phenotypes of both microglia and peripheral immune cells (37, 38). Upon activation of T cells by chronic nerve injury, PEP accumulation interferes with Ca2+ signaling and promotes the inflammatory cascade (39). When T cell receptor (TCR) is activated by antigen, cell membrane permeability increases and Ca2+ enters the cytoplasm to activate a number of signaling pathways. PEP accumulation by glycolysis inhibits Ca2+ channels in endoplasmic reticulum, thus preventing Ca2+ from entering into the Ca2+ reservoir in endoplasmic reticulum. These changes increase Ca2+ concentration in cytoplasm, further activating inflammatory pathways to maintain the activated state of peripheral immune cells and promoting the transcription of pro-inflammatory factors. PEP accumulation produces similar effects in macrophages, including the induction of M1 polarization and increased expression of pro-inflammatory factors (40). The responses of resident microglia to the glycolysis metabolites are also characterized by a shift towards the inflammatory phenotype (41) (Figure 2).
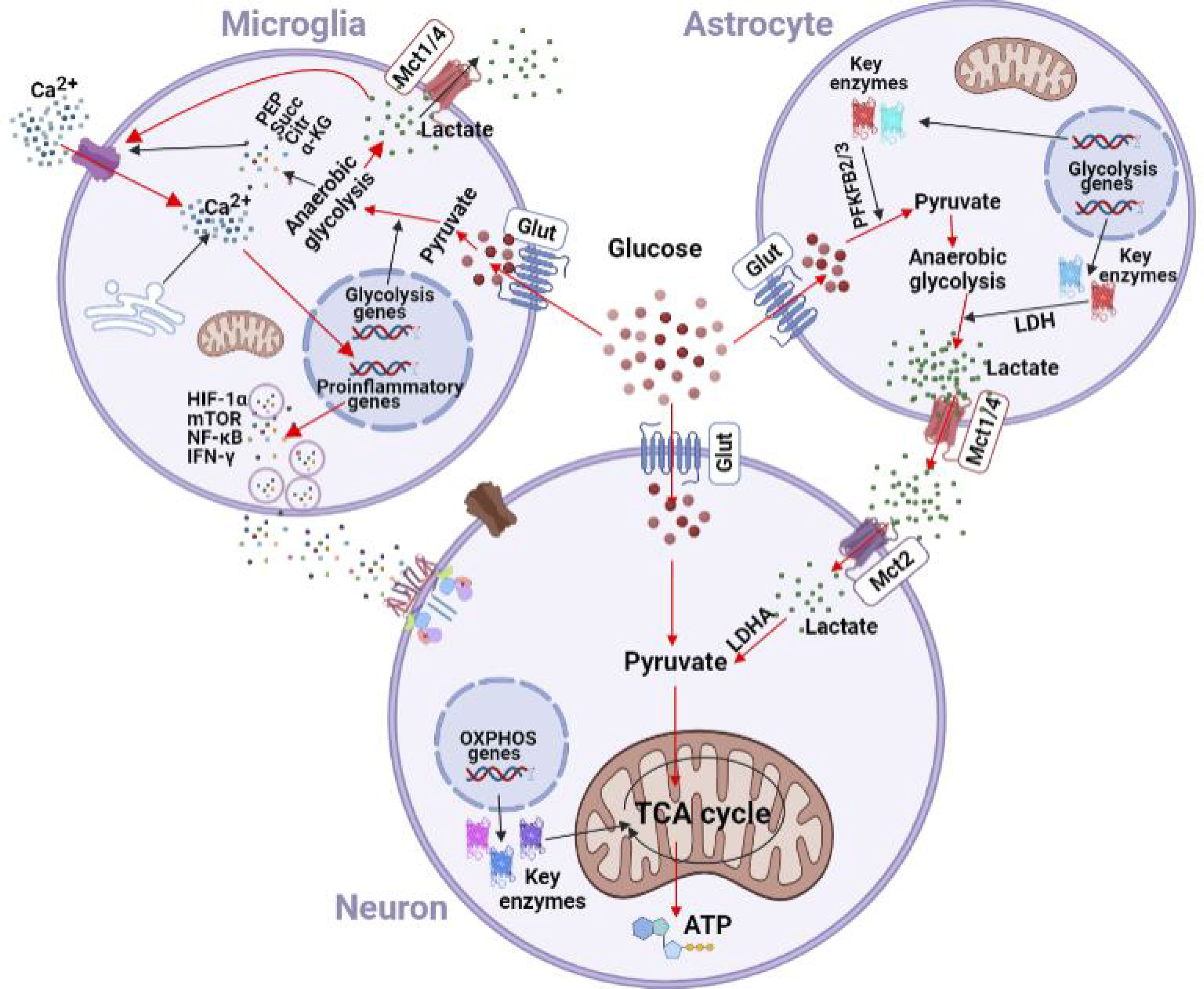
Figure 2 Effects of chronic nerve injury on glycometabolism in microglia and astrocytes. Glycolysis is enhanced in microglia under hypoxic and inflammatory insults. Enhanced glycolysis leads to accumulation of lactate, PEP, succinate, citric acid, and α-KG. Lactate is transferred into synpatic microenvironment by the lactate shuttle to facilitate the Ca2+ channel in cytomembrane and inhibit the Ca2+ channel in the endoplasmic reticulum. The resulting increase of Ca2+ concentration in cytoplasm activates inflammatory pathways (e.g., NF-κB, HIF-1α, mTOR, and IFN-γ) to promote the transcription of pro-inflammatory factors in microglia. Astrocytes express high levels of PFKFB2/3, and could metabolize glucose into pyruvate. Upon chronic nerve injury, glutamate is taken up by astrocytes in a Na+-dependent mechanism, which in turn increases intracellular Na+ concentration and activates Na+-K+-ATPase on the cell membrane to promote glucose uptake and induce anaerobic glycolysis. Lactate is then transferred out of astrocytes via Mct1/4, and enters neurons via Mct2. Glut, glucose transporter; Mct, monocarboxylate transporter; PEP, phosphoenolpyruvic acid; Succ, succinate; Citr, citric acid; α-KG, α-ketoglutarate; HIF-1α, hypoxia inducible factor-1α; mTOR, mammalian target of rapamycin; IFN-γ, interferon-γ; PFKFB, 2, 6-phosphofructo-2-kinase; LDH, lactate dehydrogenase; LDHA, lactate dehydrogenase A; TCA, tricarboxylic acid; OXPHOS, oxidative phosphorylation.
Astrocytes are also part of the resident immune system in the central nervous system, and could release cytokines or chemokines upon activation. Similar to glial cells, astrocytes also undergo glycometabolism reprogramming during the development of neuropathic pain (42). A1 astrocytes are characterized by activation of the classical complement cascade, which in turn promotes neuropathic pain by disrupting the stability and function of synaptic structures. In contrast, A2 astrocytes are characterized by upregulation of neurotrophic factors, which in turn play protective roles during the process of neurodegeneration (43).
Recent in vivo and vitro studies have demonstrated that the astrocyte-neuron lactate shuttle (ANLS) is a crucial additional source of energy for neurons, especially under stress and continuous neuronal stimulation (44, 45). Lactate produced in astrocytes can be transported out of cells and enters neurons by monocarboxylate transporters (MCTs). MCTs of different subtypes have cell-specific distribution. MCT1 and MCT4 have low affinity for lactate and are mainly distributed on glial cells to mediate the outward transport of lactate, whereas the high-affinity MCT2 is mainly distributed on neurons and mediate the uptake of lactate. In comparison to the very low glycogen storage capacity in neurons, astrocytes have high glycogen storage. Upon sensing enhanced energy requirements by surrounding neurons, astrocytes increase lactate production through glycolysis to provide metabolic substrates for neurons (46, 47). In comparison to neurons, astrocytes also express higher levels of 2,6-phosphofructo-2-kinase 2 (PFKFB2) and PFKFB3, and could thus provide energy to neurons via activated glycolysis (44). Accumulated glutamate in synapses upon chronic nerve injury enters astrocytes via glutamate transporters (GLT), increases intracellular Na+ concentration and activates Na+-K+-ATPase on cell membrane of astrocytes. This in turn promotes glucose uptake and induces glycolysis. Anaerobic glycolysis can also produce ATP, which is used for de novo glutamate synthesis, glutamate transfer between astrocytes and neurons, and the maintenance of Na+-K+-ATPase function (48). Dependence of neurons on lactate of astrocyte origin varies among different species, and disruption of glycolysis in astrocytes has been shown to result in loss of neurons in drosophila (49). Metabolic dependence of neurons on lactate of astrocyte origin, and relevance to neuropathic pain are illustrated in Figure 2.
Under normal conditions, lactate concentration is 10 to 50 times higher than pyruvate, and can be released into blood to provide energy for tissues and organs except in the central nervous system. Neurons only use glucose from the blood for energy metabolism in physiological condition, whereas glial cells could provide additional energy source for neurons under stress. Both in vitro and in vivo studies have shown that lactate concentration gradient from astrocytes to neurons plays an important role in neuronal stability, apoptosis and energy metabolism (25, 50). The lactate concentration gradient from astrocytes to neurons could be seen with two-photon microscopy (51). Lactate in astrocytes upregulates learning-related genes and produce intercellular connectivity changes in interneurons (52). Exposure to lipopolysaccharide or interferon increases lactate shuttle between microglia and neurons, indicating that microglia could also provide lactate for neurons under stress (53). Glutamine and glutamate that enter into astrocytes from synaptic cleft could enter the TCA cycle. Since the blood-brain barrier is impervious to lactate, astrocyte-neuron lactate shuttle is critical for neuronal survival, memory and synaptic remodeling under hypoxic conditions or inflammation (54, 55) (Figure 2).
Glycometabolism reprogramming is critical in pain sensitization. Reciprocally, transformation of glycometabolism is subject to epigenetic regulation by intermediates and co-factors, thus forming a feedback regulation system between glycometabolism and pain sensitization (56). Glycometabolism usually affects the expression of pain related genes by regulating substrates required for gene modification. By disrupting the NAD+/NADH balance, glycometabolism transformation can alter the function of histone deacetylase since the catalytic process of acetylase requires a certain concentration of NAD+. Histone deacetylase has been implicated in pain signal transmission and regulation (57). For example, lactate may have opposite roles in different stages of the inflammatory process (58). In the early stage of macrophages activation in peripheral nervous system, lactate accumulation is generated by glycolysis conversion to promote histone acetylation, which in turn upregulate anti-inflammatory genes. In a sense, lactate serves as a feedback signal that switches macrophage to anti-inflammatory phenotype in the development of inflammation. Lactate dehydrogenase A (LDHA), an enzyme that catalyzes the conversion between pyruvate and lactate, regulates the expression of IFN-γ in T cells via acetyl-CoA. In CD4+ T cells, acetyl-CoA is mainly used for histone acetylation of IFN-γ promoters, thereby promoting T cell differentiation into Th1 subsets (59). Reduction of acetyl-CoA levels by ATP-citrate lyase (ACLY) knockout reduces the expression of key enzymes in glycolysis (e.g., hexokinase 2, PFKFB and LDHA), whereas exogenous lactate reduces the effects of ACLY knockout on glycolysis (60). Under low glucose conditions, exogenous lactate enters CD8+ T cells and is converted into acetyl-CoA to increase IFN-γ expression (61). Succinate has been shown to inhibit the DNA methylase TET family genes in Treg cell subsets to alter cell proliferation (62). Changes in the intermediates in glycolysis in macrophages, T cells and other immune cells have been implicated in the abnormal excitability in primary sensory nerve fibers and dorsal root ganglion during pain sensitization and synaptic plasticity. Parallel glycometabolism reprogramming has also been found in spinal cord, where the nociceptive signal is integrated (63).
It is well known that lactate participates in various physiological processes as a signal molecule, and it may exhibit anti-inflammatory effects in some states. Lactate inhibits toll-like receptor induction of inflammasome and production of IL-1β via the GPR81-mediated suppression of innate immunity (64, 65). In dendritic cells, lactate accumulation drives the transformation of inflammatory phenotype by regulating the secretion of interleukin-10 (IL-10) (66). Lactate has been shown to inhibit the migration and cytotoxicity of CD8+ T cells and promote the proliferation of Treg cells via its action on the key enzymes in glycolysis (67). In certain conditions, however, lactate accumulation could enhance the immune response and inflammatory cascade mediated by the NF-κB pathway in Th17 cells (68). Similarly, in endothelial cells, lactate accumulation stimulates the NF-κB/IL-8 pathway and induces the production of reactive oxygen species (ROS), resulting in increased cytomembrane permeability and blood-brain barrier disruption (69). Lactate has also been implicated in ROS production in myogenic cells under stress conditions and could up-regulate the expression of multiple genes related to oxidative stress and pro-inflammatory activities (70). The inflammatory effects and oxidative stress induced by lactate in periphery contribute to pain sensitization. In the brain and spinal cord, lactate accumulated under chronic nerve injury can enter neurons and microglia and serve as a substrate in the TCA cycle to generate both ATP and ROS. Immune regulatory factors and ROS cascades produced by microglia promote apoptosis and autophagy, change the stability and permeability of ion channels, and contribute to pain sensitization by depolarizing synaptic membrane and remodeling (31, 71).
Succinate could be accumulated upon TCA cycle disruption and enhanced glutamine decomposition. Accumulation of succinate in synovial macrophages in response to lipopolysaccharide (LPS) exposure directly inhibits M1 polarization caused by proline hydrolase, thereby promoting stable expression of HIF-1α and IL-1β (72), and inhibiting the transcription of anti-inflammatory cytokine IL-10 (73). Extracellular succinate binds to succinate receptor 1 (SUCNR1) to modulate pro-inflammatory/anti-inflammatory signal pathways in nociceptive neurons. Knockout of the SUCNR1 gene in myeloid cells in adipose tissue has been shown to promote the expression of pro-inflammatory genes in macrophages, adding support to the anti-inflammatory function of succinate (74). Extracellular succinate could produce pro-inflammatory action under different conditions. For example, extracellular succinate has been shown to promote the production of prostaglandin E2 in neural stem cells to exert pro-inflammatory effect (75). Succinate can affect macrophage migration and increase the secretion of pro-inflammatory cytokines, including tumor necrosis factor α (TNF-α) and IL-1β, in dendritic cells (76). Succinate also indirectly enhances IFN-γ and TNF-α production in effector T cells during antigen presentation (77).
Citric acid is generated during the TCA cycle in mitochondria and transported to the cytoplasm by the citrate carrier (CIC) to participate in the synthesis of fatty acids. In the cytoplasm, citric acid is metabolized by ACLY to acetyl-CoA and oxaloacetate. This process also promotes ROS and nitric oxide (NO) synthesis. CIC knockout reduces citric acid level in the cytoplasm and the production of ROS, NO and prostaglandins to prevent transition to the pro-inflammatory phenotype (78). Citric acid is converted into cis-aconitate in mitochondria and transferred into cytoplasm for oxidative dealkylation to methylene succinic acid. Methylene succinic acid accumulates in large quantities in M1 macrophages and acquires anti-bacterial properties when the TCA cycle is disturbed (79). Disruption of methylene succinic acid synthesis using gene knockout techniques has been shown to induce the expression of pro-inflammatory factors (80). In general, methylene succinic acid exerts its anti-inflammatory effects primarily via two signal transduction pathways: one to directly inhibit succinate dehydrogenase to downregulate pro-inflammatory factors, and the other to activate the transcription factors, including nuclear factor E2-related factor 2 (Nrf2) and activating transcription factor 3 (ATF3), in macrophages (81). Through these mechanisms, citric acid participate in synaptic remodeling in pain sensitization.
Several other metabolic intermediates in glycolysis also contribute to pain sensitization. In peripheral monocytes, β-glucan increases cytoplasmic fumaric acid levels and reduce the activity of KDM5 demethylase and promotes cell migration (82). α-Ketoglutarate (α-KG) accumulation induces gene expression related to M2 polarization in macrophages through histone demethylation, promotes the transformation of macrophages to M2 phenotype and enhances their anti-inflammatory activity (83). α-KG seems to produce opposite action in T cells. The IL-2 signal pathway can increase α-KG accumulation in T cells to induce their differentiation into the Th1 subset. α-KG also increases the expression of pro-inflammatory and glycolysis genes through DNA methylation modification, which in turn promotes the glycolysis process in T cells (84). 2-HG, a structural analogue of α-KG, competitively inhibits α-KG-dependent histone demethylase and promotes histone hypermethylation. Increased production of 2-HG under hypoxic conditions increases the hypermethylation of histone and DNA by inhibiting URX and TET2 proteins to increase the expression of CD62L and CD127 and promote T cell differentiation into memory cells to induce synaptic plasticity and pain sensitization (85). Abnormal accumulation of S-adenosylmethionine (SAM) also alters the synaptic microenvironment. LPS stimulation of macrophages promotes histone trimethylation by increasing SAM production and SAM/S-adenosine homocysteine ratio, resulting in the upregulation of pro-inflammatory genes. In vivo and in vitro studies have confirmed that reduced production of SAM caused by methionine deficiency could alter the inflammatory state of primary afferent neurons and dorsal root ganglion neurons by inhibiting T cell proliferation and cytokine production (86). In summary, accumulation of metabolic intermediates in glycolysis could alter the phenotype of immune and glial cells through different pathways to participate in the development of pain sensitization and synaptic plasticity.
Microglia serve as sensors to detect changes in the microenvironment in the central nervous system (87). Through modifying synaptic pruning, microglia regulate experience-dependent plasticity in the barrel cortex and visual cortex after removal of monocular deprivation. Microglia in the resting state plays a vital role learning and memory. Upon infection, injury or stress, microglia migrate to the site of inflammation, assume an amoeboid shape and secrete cytokines, chemokines and ROS (88). Metabolic changes in neurons and glial cells upon injury or stress (e.g., lactate, succinate and citric acid) are important in the formation of synaptic plasticity in pain sensitization and neuropathic pain, as well as a variety of other central nervous system diseases (89).
Microglia are exquisitely sensitive to acidic metabolites. Exposure of microglia to exogenous lactate increases the mRNA of thioredoxin interacting protein (TXNIP) to accelerate neuronal apoptosis and autophagy. This process causes the irreversible changes in synaptic structure and function in the progression of chronic pain, vascular dementia and Alzheimer’s disease (90). H+ could act as a second messenger to regulate the activity of voltage-gated Ca2+ channels, NMDA and GABA receptors. Blocking NMDA receptors could alter the expression of genes related to lactate-induced LTP and LTD, suggesting NMDA receptors are key downstream signal molecules of lactate (91). In addition, lactate and ROS can also promote neuro-inflammation by activating NMDA receptors to mediate Ca2+ influx and the downstream signal cascade. The Src family kinases (SFKs) phosphorylate NMDA receptor subunits to promotes Zn2+ entry into cells through NMDA receptor channels to participate in the activation of the TrKBs/ERK pathway, and ultimately synaptic remodeling and learning/memory (92, 93). A negative correlation between pH and lactate levels in the synaptic microenvironment has been found in patients with schizophrenia or bipolar disorder (94). Overall, the effects of glycometabolism reprogramming in microglia on pH homeostasis in the synaptic microenvironment are important to synaptic plasticity and development of neuropathic pain.
Changes in the NAD+/NADH ratio caused by glycometabolism reprogramming in microglia alter the redox status in synaptic microenvironment. Lactate accumulation increases NADH in both neurons and microglia, and upregulates the expression of a variety of genes, including BDNF, Arc and Zif268 to influence synaptic plasticity (95). Reduced NAD+/NADH ratio activate NMDA receptors and increases Ca2+ influx to trigger downstream inflammatory cascade. Imbalanced NAD+/NADH ratio also affects the levels of transcription factors, deacetylase activity and Ca2+ pathways in microglia (96, 97). The Ca2+ pathway is a critical link in the lactate signal process in glycometabolism reprogramming. Many other key pathways in synaptic plasticity, including the ERK/CREB pathway, dopamine D2 receptors, metabotropic glutamate receptors (mGluRs) and cannabinoid receptors, are regulated by Ca2+ (98). NAD+-dependent sirtuin-1 signal pathway also participate in synaptic plasticity by regulating the expression of plasticity-related genes (e.g., BDNF, Arc and Zif268) and modifying dendritic morphology of neurons. To sum up, cellular redox state and particularly NAD+/NADH ratio are key steps between glycometabolism reprogramming and synaptic plasticity (99) (Figure 3).
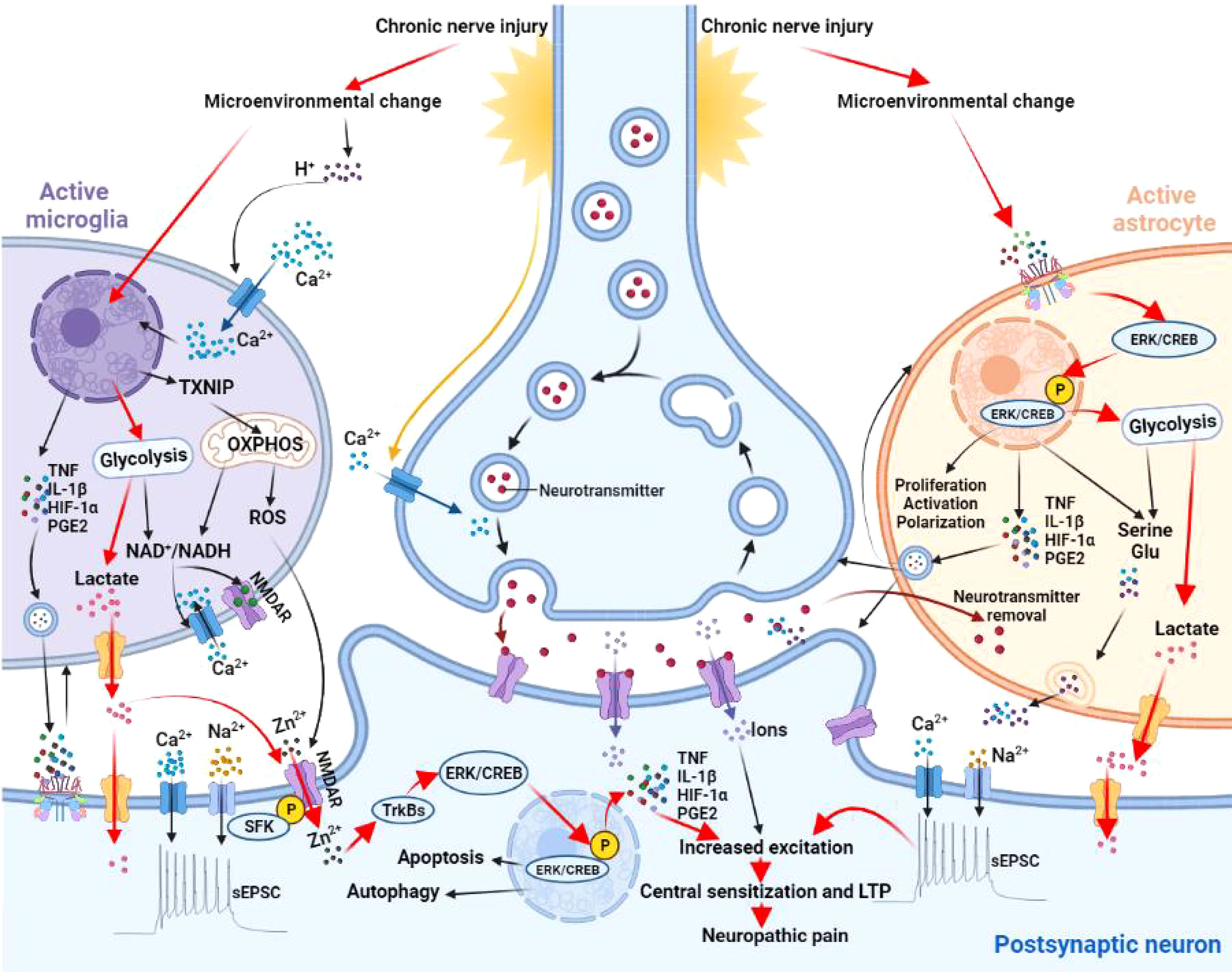
Figure 3 Effects of microglia and astrocyte activation on synaptic plasticity. Microglia sense changes in synaptic microenvironment and H+ accumulation activates a series of stress responses to shift microglia into activated state. These changes promote the transcription and expression of TXNIP, key enzymes of glycolysis and proinflammatory mediators (e.g.,TNF, IL-1β, HIF-1α, and PGE2). TXNIP is closely related to oxidative stress in producing ROS to increase cytomembrane permeability. Increased expression of key enzymes in glycolysis promotes lactate production and disrupts the balance of NAD+/NADH. An imbalanced NAD+/NADH ratio activates the NMDA receptor and increases Ca2+ influx to trigger downstream inflammatory cascade. Increased concentration of proinflammatory mediators in the vicinity of synapses promotes synaptic plasticity and polarization of microglia itself. Activation of NMDA receptors, Ca2+ influx and the downstream signaling cascade are regulated by SFKs and TrKB receptors. SFKs phosphorylates NMDA receptor subunits to promotes Zn2+ entry into postsynaptic neurons through NMDA receptor channels to activate the TrKBs/ERK pathway. Phosphorylation of TrKBs/ERK promotes autophagy, apoptosis, and neuroinflammation. Astrocytes also sense the changes in synaptic microenvironment and activates the ERK/CREB signaling. Upon entering the nucleus, p-ERK activates p-CREB to initiate related gene transcription and promotes proliferation, activation, and polarization. Phosphorylation of ERK/CREB also enhances glycolysis and proinflammatory mediators, leading to the accumulation of lactate, glutamate, and serine. All together, these changes increase the excitability of postsynaptic neurons and contribute to central sensitization in neuropathic pain. TXNIP, thioredoxin interacting protein; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; NMDAR, N-methyl-D-aspartate receptor; HIF-1α, hypoxia inducible factor-1α; TNF, tumor necrosis factor; IL-1β, interleukin-1β; PGE2, prostaglandin E2; SFK, Src family kinases; ERK, extracellular signal regulated kinase; CREB, cyclic AMP response element binding protein; LTP, long-term potentiation; Glu, glutamate; sEPSC, spontaneous excitatory postsynaptic current.
Recent studies indicated that lactate produced through glycogenolysis in astrocytes is critical for long-term memory formation. Disturbing the lactate shuttling between astrocytes and neurons impairs memory formation and consolidation, whereas exogenous lactate attenuates such effects. Astrocyte-neuron lactate shuttle in the spinal cord participates in the formation of abnormal neural circuits in persistent hyperalgesia in neuropathic pain (46). Repeated exposure to noxious stimuli result in substantial changes in astrocyte. Chronic peripheral nerve injury has been shown to promote the proliferation of astrocytes in the spinal cord, as evidenced by increased expression of astrocyte marker glial fibrillary acidic protein (GFAP). Activated astrocytes release a variety of inflammatory mediators and metabolites (e.g., TNF-α, IL-1β, ATP, glutamic acid and serine) to enhance spontaneous excitatory postsynaptic currents and reduce inhibitory interneuron activities, ultimately leading to central sensitization in neuropathic pain (100, 101). Chronic nerve injury has also been shown to increase glutamate concentration in the synaptic cleft via inhibition of glutamate transporters (7).
Synaptic sensitization largely depends on the activation of neuronal receptors and ion channels, intracellular signal transduction pathways and related gene expression. Central sensitization in neuropathic pain shares some common molecular mechanisms with LTP in hippocampus (102). For example, lactate transfer from astrocyte to neuron and subsequent NMDA receptor activation and downstream intracellular signal pathways could alter the synaptic plasticity in context of synaptic plasticity in the spinal cord as well as in LTP in the hippocampus in mice. Disrupting the astrocyte-neuron lactate shuttle has been shown to results in amnesia by impairing LTP in the hippocampus (103).
In the central nervous system, glycogen is stored primarily in astrocytes as a reservoir of energy source upon glucose deprivation. In contrast, glycogen store is practically absent in neurons. In comparison to adult brain, neurons in developing brain (rats less than 3 weeks of age) expresses less glucose transporters but more MCTs, indicating the crucial role of lactate in energy metabolism during development of the central nervous system (104). Intraventricular administration of selective inhibitors of MCT or phosphoenolpyruvate carboxykinase (a key enzyme in lactate metabolism) attenuates the proliferation and differentiation of neuronal precursor cells in newborn mice (105). Lactate in the general circulation can also affect synaptic function. Increased lactate in blood by strenuous exercise has been implicated in synaptic remodeling by stimulating the synthesis of vascular endothelial growth factor (VEGF) via hydroxy-carboxylic acid receptors on vascular endothelial cells (106, 107).
Plantar injection of complete Freund’s adjuvant (CFA) increases lactate release in the anterior cingulate cortex; inhibiting glycolysis in anterior cingulate cortex, in contrast, alleviates CFA-induced chronic inflammatory pain (108). Knockout of MCT genes in neurons of the anterior cingulate cortex also alleviates CFA-induced inflammatory pain, indicating a prominent role of lactate transfer from astrocytes to neurons in central sensitization and neuropathic pain. Numerous studies have demonstrated a critical role of phosphorylated ERK and activation of the transcription factor cyclic AMP response element binding protein (CREB) in synaptic plasticity and central sensitization. Inhibition of glycolysis has been shown to block the ERK/CREB pathway. Specifically, exogenous lactate may participate in synaptic remodeling and central sensitization in neuropathic pain by enhancing the phosphorylation of ERK and CREB (109, 110). Overall, compromised energy supply to neurons due to disruption of the lactate shuttle between astrocytes and neurons contribute significantly to synaptic plasticity and neuropathic pain via multiple mechanisms (111) (Figure 3).
Increasing evidence suggests important role of glycometabolism reprogramming in microglia and astrocytes in neuropathic pain. Glycometabolism reprogramming in microglia promotes the transformation of microglia to the pro-inflammatory phenotype and increases ROS production. The resulting changes in synaptic microenvironment are important pathological basis for pain sensitization. Astrocytes provide energy support to surrounding neurons via astrocyte-neuron lactate shuttle. In addition to a substrate for TAC cycle, lactate that enters the neurons from astrocytes also serves as a signal molecule to promote synaptic plasticity by regulating a variety of signaling pathways. In summary, a plethora of information showed that glycometabolism reprogramming of glial cells contribute to hyperalgesia and allodynia in neuropathic pain and represent potential targets for developing novel treatment for neuropathic pain.
EK, YL, and MD drafted the manuscript; TH, MY, JL, and XF designed and prepared the figures; HY conceived the study. All authors made significant contribution and approved the submitted version.
This work was supported by National Natural Science Foundation of China (81971046 and 82171220) and Medical Science and Technology Research Program of Henan Province (SBGJ202003056, SBGJ202102204 and LHGJ20200781).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic Pain. Nat Rev Dis Primers (2017) 3:17002. doi: 10.1038/nrdp.2017.2
2. Ye J, Ding H, Ren J, Xia Z. The Publication Trend of Neuropathic Pain in the World and China: A 20-Years Bibliometric Analysis. J Headache Pain (2018) 19(1):110. doi: 10.1186/s10194-018-0941-4
3. Finnerup NB, Kuner R, Jensen TS. Neuropathic Pain: From Mechanisms to Treatment. Physiol Rev (2021) 101(1):259–301. doi: 10.1152/physrev.00045.2019
4. Calcutt NA. Diabetic Neuropathy and Neuropathic Pain: A (Con)fusion of Pathogenic Mechanisms. Pain (2020) 161(Suppl 1):S65–86. doi: 10.1097/j.pain.0000000000001922
5. Liu J, Li Y, Lu Z, Gu J, Liang Y, Huang E, et al. Deceleration of Glycometabolism Impedes IgG-Producing B-Cell-Mediated Tumor Elimination by Targeting SATB1. Immunology (2019) 156(1):56–68. doi: 10.1111/imm.12998
6. Murphy-Royal C, Johnston AD, Boyce A, Diaz-Castro B, Institoris A, Peringod G, et al. Stress Gates an Astrocytic Energy Reservoir to Impair Synaptic Plasticity. Nat Commun (2020) 11(1):2014. doi: 10.1038/s41467-020-15778-9
7. Falnikar A, Hala TJ, Poulsen DJ, Lepore AC. GLT1 Overexpression Reverses Established Neuropathic Pain-Related Behavior and Attenuates Chronic Dorsal Horn Neuron Activation Following Cervical Spinal Cord Injury. Glia (2016) 64(3):396–406. doi: 10.1002/glia.22936
8. Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 Receptors Induced in Spinal Microglia Gate Tactile Allodynia After Nerve Injury. Nature (2003) 424(6950):778–83. doi: 10.1038/nature01786
9. Yan X, Yadav R, Gao M, Weng HR. Interleukin-1 Beta Enhances Endocytosis of Glial Glutamate Transporters in the Spinal Dorsal Horn Through Activating Protein Kinase C. Glia (2014) 62(7):1093–109. doi: 10.1002/glia.22665
10. Liu S, Liu YP, Song WB, Song XJ. EphrinB-EphB Receptor Signaling Contributes to Bone Cancer Pain via Toll-Like Receptor and Proinflammatory Cytokines in Rat Spinal Cord. Pain (2013) 154(12):2823–35. doi: 10.1016/j.pain.2013.08.017
11. Wang Z, Guan D, Wang S, Chai L, Xu S, Lam KP. Glycolysis and Oxidative Phosphorylation Play Critical Roles in Natural Killer Cell Receptor-Mediated Natural Killer Cell Functions. Front Immunol (2020) 11:202. doi: 10.3389/fimmu.2020.00202
12. Shan J, Jin H, Xu Y. T Cell Metabolism: A New Perspective on Th17/Treg Cell Imbalance in Systemic Lupus Erythematosus. Front Immunol (2020) 11:1027. doi: 10.3389/fimmu.2020.01027
13. Bannister K, Sachau J, Baron R, Dickenson AH. Neuropathic Pain: Mechanism-Based Therapeutics. Annu Rev Pharmacol Toxicol (2020) 60:257–74. doi: 10.1146/annurev-pharmtox-010818-021524
14. Gao YJ, Ji RR. Chemokines, Neuronal-Glial Interactions, and Central Processing of Neuropathic Pain. Pharmacol Ther (2010) 126(1):56–68. doi: 10.1016/j.pharmthera.2010.01.002
15. Campbell JN, Meyer RA. Mechanisms of Neuropathic Pain. Neuron (2006) 52(1):77–92. doi: 10.1016/j.neuron.2006.09.021
16. Kuner R. Central Mechanisms of Pathological Pain. Nat Med (2010) 16(11):1258–66. doi: 10.1038/nm.2231
17. Sun W, Zhou Q, Ba X, Feng X, Hu X, Cheng X, et al. Oxytocin Relieves Neuropathic Pain Through GABA Release and Presynaptic TRPV1 Inhibition in Spinal Cord. Front Mol Neurosci (2018) 11:248. doi: 10.3389/fnmol.2018.00248
18. Kong EL, Yan H, Wang HQ, Yan Z, Wu F. TLR4 Inhibits Spinal Gabaergic Activities via Microglial Activation in Chronic Constriction Injury Mice. Neuropsychiatry (London) (2018) 08(2):761–71. doi: 10.4172/Neuropsychiatry.1000401
19. Zeilhofer HU, Benke D, Yevenes GE. Chronic Pain States: Pharmacological Strategies to Restore Diminished Inhibitory Spinal Pain Control. Annu Rev Pharmacol Toxicol (2012) 52:111–33. doi: 10.1146/annurev-pharmtox-010611-134636
20. Bliss TV, Collingridge GL, Kaang BK, Zhuo M. Synaptic Plasticity in the Anterior Cingulate Cortex in Acute and Chronic Pain. Nat Rev Neurosci (2016) 17(8):485–96. doi: 10.1038/nrn.2016.68
21. Diering GH, Huganir RL. The AMPA Receptor Code of Synaptic Plasticity. Neuron (2018) 100(2):314–29. doi: 10.1016/j.neuron.2018.10.018
22. Mateos-Aparicio P, Rodríguez-Moreno A. Calcium Dynamics and Synaptic Plasticity. Adv Exp Med Biol (2020) 1131:965–84. doi: 10.1007/978-3-030-12457-1_38
23. Lenz M, Eichler A, Kruse P, Strehl A, Rodriguez-Rozada S, Goren I, et al. Interleukin 10 Restores Lipopolysaccharide-Induced Alterations in Synaptic Plasticity Probed by Repetitive Magnetic Stimulation. Front Immunol (2020) 11:614509. doi: 10.3389/fimmu.2020.614509
24. López-Gambero AJ, Martínez F, Salazar K, Cifuentes M, Nualart F. Brain Glucose-Sensing Mechanism and Energy Homeostasis. Mol Neurobiol (2019) 56(2):769–96. doi: 10.1007/s12035-018-1099-4
25. Yellen G. Fueling Thought: Management of Glycolysis and Oxidative Phosphorylation in Neuronal Metabolism. J Cell Biol (2018) 217(7):2235–46. doi: 10.1083/jcb.201803152
26. Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Zheng Y, et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol Cell (2016) 61(5):705–19. doi: 10.1016/j.molcel.2016.02.009
27. Rahman MH, Bhusal A, Kim JH, Jha MK, Song GJ, Go Y, et al. Astrocytic Pyruvate Dehydrogenase Kinase-2 Is Involved in Hypothalamic Inflammation in Mouse Models of Diabetes. Nat Commun (2020) 11(1):5906. doi: 10.1038/s41467-020-19576-1
28. Park BY, Jeon JH, Go Y, Ham HJ, Kim JE, Yoo EK, et al. PDK4 Deficiency Suppresses Hepatic Glucagon Signaling by Decreasing cAMP Levels. Diabetes (2018) 67(10):2054–68. doi: 10.2337/db17-1529
29. Schain M, Kreisl WC. Neuroinflammation in Neurodegenerative Disorders-A Review. Curr Neurol Neurosci Rep (2017) 17(3):25. doi: 10.1007/s11910-017-0733-2
30. Abbott NJ. Astrocyte-Endothelial Interactions and Blood-Brain Barrier Permeability. J Anat (2002) 200(6):629–38. doi: 10.1046/j.1469-7580.2002.00064.x
31. Devanney NA, Stewart AN, Gensel JC. Microglia and Macrophage Metabolism in CNS Injury and Disease: The Role of Immunometabolism in Neurodegeneration and Neurotrauma. Exp Neurol (2020) 329:113310. doi: 10.1016/j.expneurol.2020.113310
32. Wetzels S, Vanmierlo T, Scheijen J, van Horssen J, Amor S, Somers V, et al. Methylglyoxal-Derived Advanced Glycation Endproducts Accumulate in Multiple Sclerosis Lesions. Front Immunol (2019) 10:855. doi: 10.3389/fimmu.2019.00855
33. Baik SH, Kang S, Lee W, Choi H, Chung S, Kim JI, et al. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer's Disease. Cell Metab (2019) 30(3):493–507.e6. doi: 10.1016/j.cmet.2019.06.005
34. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Cell (2017) 170(4):649–663.e13. doi: 10.1016/j.cell.2017.07.023
35. van der Poel M, Ulas T, Mizee MR, Hsiao CC, Miedema SMS, Adelia, et al. Transcriptional Profiling of Human Microglia Reveals Grey-White Matter Heterogeneity and Multiple Sclerosis-Associated Changes. Nat Commun (2019) 10(1):1139. doi: 10.1038/s41467-019-08976-7
36. Peruzzotti-Jametti L, Pluchino S. Targeting Mitochondrial Metabolism in Neuroinflammation: Towards a Therapy for Progressive Multiple Sclerosis. Trends Mol Med (2018) 24(10):838–55. doi: 10.1016/j.molmed.2018.07.007
37. McBride MA, Owen AM, Stothers CL, Hernandez A, Luan L, Burelbach KR, et al. The Metabolic Basis of Immune Dysfunction Following Sepsis and Trauma. Front Immunol (2020) 11:1043. doi: 10.3389/fimmu.2020.01043
38. Soto-Heredero G, Gómez de Las Heras MM, Gabandé-Rodríguez E, Oller J, Mittelbrunn M. Glycolysis - A Key Player in the Inflammatory Response. FEBS J (2020) 287(16):3350–69. doi: 10.1111/febs.15327
39. Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, et al. Evidence for an Alternative Glycolytic Pathway in Rapidly Proliferating Cells. Science (2010) 329(5998):1492–9. doi: 10.1126/science.1188015
40. Ko CW, Counihan D, Wu J, Hatzoglou M, Puchowicz MA, Croniger CM. Macrophages With a Deletion of the Phosphoenolpyruvate Carboxykinase 1 (Pck1) Gene Have a More Proinflammatory Phenotype. J Biol Chem (2018) 293(9):3399–409. doi: 10.1074/jbc.M117.819136
41. Li M, Lu H, Wang X, Duan C, Zhu X, Zhang Y, et al. Pyruvate Kinase M2 (PKM2) Interacts With Activating Transcription Factor 2 (ATF2) to Bridge Glycolysis and Pyroptosis in Microglia. Mol Immunol (2021) 140:250–66. doi: 10.1016/j.molimm.2021.10.017
42. Lauro C, Limatola C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front Immunol (2020) 11:493. doi: 10.3389/fimmu.2020.00493
43. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic Analysis of Reactive Astrogliosis. J Neurosci (2012) 32(18):6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012
44. Muraleedharan R, Gawali MV, Tiwari D, Sukumaran A, Oatman N, Anderson J, et al. AMPK-Regulated Astrocytic Lactate Shuttle Plays a Non-Cell-Autonomous Role in Neuronal Survival. Cell Rep (2020) 32(9):108092. doi: 10.1016/j.celrep.2020.108092
45. Sun Y, Wang Y, Chen ST, Chen YJ, Shen J, Yao WB, et al. Modulation of the Astrocyte-Neuron Lactate Shuttle System Contributes to Neuroprotective Action of Fibroblast Growth Factor 21. Theranostics (2020) 10(18):8430–45. doi: 10.7150/thno.44370
46. Miyamoto K, Ishikura KI, Kume K, Ohsawa M. Astrocyte-Neuron Lactate Shuttle Sensitizes Nociceptive Transmission in the Spinal Cord. Glia (2019) 67(1):27–36. doi: 10.1002/glia.23474
47. Lindberg D, Ho A, Peyton L, Choi DS. Chronic Ethanol Exposure Disrupts Lactate and Glucose Homeostasis and Induces Dysfunction of the Astrocyte-Neuron Lactate Shuttle in the Brain. Alcohol Clin Exp Res (2019) 43(9):1838–47. doi: 10.1111/acer.14137
48. Estrada-Sánchez AM, Montiel T, Massieu L. Glycolysis Inhibition Decreases the Levels of Glutamate Transporters and Enhances Glutamate Neurotoxicity in the R6/2 Huntington's Disease Mice. Neurochem Res (2010) 35(8):1156–63. doi: 10.1007/s11064-010-0168-5
49. Volkenhoff A, Weiler A, Letzel M, Stehling M, Klämbt C, Schirmeier S. Glial Glycolysis Is Essential for Neuronal Survival in Drosophila. Cell Metab (2015) 22(3):437–47. doi: 10.1016/j.cmet.2015.07.006
50. Ameruoso A, Palomba R, Palange AL, Cervadoro A, Lee A, Di Mascolo D, et al. Ameliorating Amyloid-β Fibrils Triggered Inflammation via Curcumin-Loaded Polymeric Nanoconstructs. Front Immunol (2017) 8:1411. doi: 10.3389/fimmu.2017.01411
51. Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, et al. In Vivo Evidence for a Lactate Gradient From Astrocytes to Neurons. Cell Metab (2016) 23(1):94–102. doi: 10.1016/j.cmet.2015.10.010
52. Descalzi G, Gao V, Steinman MQ, Suzuki A, Alberini CM. Lactate From Astrocytes Fuels Learning-Induced mRNA Translation in Excitatory and Inhibitory Neurons. Commun Biol (2019) 2:247. doi: 10.1038/s42003-019-0495-2
53. Gimeno-Bayón J, López-López A, Rodríguez MJ, Mahy N. Glucose Pathways Adaptation Supports Acquisition of Activated Microglia Phenotype. J Neurosci Res (2014) 92(6):723–31. doi: 10.1002/jnr.23356
54. Veys K, Fan Z, Ghobrial M, Bouché A, García-Caballero M, Vriens K, et al. Role of the GLUT1 Glucose Transporter in Postnatal CNS Angiogenesis and Blood-Brain Barrier Integrity. Circ Res (2020) 127(4):466–82. doi: 10.1161/CIRCRESAHA.119.316463
55. Barros LF. Metabolic Signaling by Lactate in the Brain. Trends Neurosci (2013) 36(7):396–404. doi: 10.1016/j.tins.2013.04.002
56. Chisolm DA, Weinmann AS. Connections Between Metabolism and Epigenetics in Programming Cellular Differentiation. Annu Rev Immunol (2018) 36:221–46. doi: 10.1146/annurev-immunol-042617-053127
57. Verdin E. NAD⁺ in Aging, Metabolism, and Neurodegeneration. Science (2015) 350(6265):1208–13. doi: 10.1126/science.aac4854
58. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
59. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic Glycolysis Promotes T Helper 1 Cell Differentiation Through an Epigenetic Mechanism. Science (2016) 354(6311):481–4. doi: 10.1126/science.aaf6284
60. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science (2009) 324(5930):1076–80. doi: 10.1126/science.1164097
61. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate Promotes T Cell Effector Function During Glucose Restriction. Cell Rep (2019) 27(7):2063–74.e5. doi: 10.1016/j.celrep.2019.04.022
62. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martínez-Reyes I, et al. Mitochondrial Complex III Is Essential for Suppressive Function of Regulatory T Cells. Nature (2019) 565(7740):495–9. doi: 10.1038/s41586-018-0846-z
63. Machelska H, Celik MÖ. Opioid Receptors in Immune and Glial Cells-Implications for Pain Control. Front Immunol (2020) 11:300. doi: 10.3389/fimmu.2020.00300
64. Errea A, Cayet D, Marchetti P, Tang C, Kluza J, Offermanns S, et al. Lactate Inhibits the Pro-Inflammatory Response and Metabolic Reprogramming in Murine Macrophages in a GPR81-Independent Manner. PloS One (2016) 11(11):e0163694. doi: 10.1371/journal.pone.0163694
65. Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate Reduces Liver and Pancreatic Injury in Toll-Like Receptor- and Inflammasome-Mediated Inflammation via GPR81-Mediated Suppression of Innate Immunity. Gastroenterology (2014) 146(7):1763–74. doi: 10.1053/j.gastro.2014.03.014
66. Nasi A, Fekete T, Krishnamurthy A, Snowden S, Rajnavölgyi E, Catrina AI, et al. Dendritic Cell Reprogramming by Endogenously Produced Lactic Acid. J Immunol (2013) 191(6):3090–9. doi: 10.4049/jimmunol.1300772
67. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25(6):1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018
68. Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate Boosts TLR4 Signaling and NF-kappaB Pathway-Mediated Gene Transcription in Macrophages via Monocarboxylate Transporters and MD-2 Up-Regulation. J Immunol (2009) 182(4):2476–84. doi: 10.4049/jimmunol.0802059
69. Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate Influx Through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-κb/IL-8 Pathway That Drives Tumor Angiogenesis. Cancer Res (2011) 71(7):2550–60. doi: 10.1158/0008-5472.CAN-10-2828
70. Hashimoto T, Hussien R, Oommen S, Gohil K, Brooks GA. Lactate Sensitive Transcription Factor Network in L6 Cells: Activation of MCT1 and Mitochondrial Biogenesis. FASEB J (2007) 21(10):2602–12. doi: 10.1096/fj.07-8174com
71. Joshi L, Plastira I, Bernhart E, Reicher H, Koyani CN, Madl T, et al. Lysophosphatidic Acid Induces Aerobic Glycolysis, Lipogenesis, and Increased Amino Acid Uptake in BV-2 Microglia. Int J Mol Sci (2021) 22(4):1–26. doi: 10.3390/ijms22041968
72. Jiang S, Yan W. Succinate in the Cancer-Immune Cycle. Cancer Lett (2017) 390:45–7. doi: 10.1016/j.canlet.2017.01.019
73. Hollander AP, Corke KP, Freemont AJ, Lewis CE. Expression of Hypoxia-Inducible Factor 1alpha by Macrophages in the Rheumatoid Synovium: Implications for Targeting of Therapeutic Genes to the Inflamed Joint. Arthritis Rheum (2001) 44(7):1540–4. doi: 10.1002/1529-0131(200107)44:7<1540::AID-ART277<3.0.CO;2-7
74. Keiran N, Ceperuelo-Mallafré V, Calvo E, Hernández-Alvarez MI, Ejarque M, Núñez-Roa C, et al. SUCNR1 Controls an Anti-Inflammatory Program in Macrophages to Regulate the Metabolic Response to Obesity. Nat Immunol (2019) 20(5):581–92. doi: 10.1038/s41590-019-0372-7
75. Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa A, Kwok CK, Leonardi T, et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell (2018) 22(3):355–368.e13. doi: 10.1016/j.stem.2018.01.020
76. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 Senses Extracellular Succinate Released From Inflammatory Macrophages and Exacerbates Rheumatoid Arthritis. J Exp Med (2016) 213(9):1655–62. doi: 10.1084/jem.20160061
77. Saraiva AL, Veras FP, Peres RS, Talbot J, de Lima KA, Luiz JP, et al. Succinate Receptor Deficiency Attenuates Arthritis by Reducing Dendritic Cell Traffic and Expansion of T(h)17 Cells in the Lymph Nodes. FASEB J (2018) 6(12):1–9, fj201800285. doi: 10.1096/fj.201800285
78. Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, et al. The Mitochondrial Citrate Carrier: A New Player in Inflammation. Biochem J (2011) 438(3):433–6. doi: 10.1042/BJ20111275
79. Williams NC, O Neill L. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front Immunol (2018) 9:141. doi: 10.3389/fimmu.2018.00141
80. Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem (2016) 291(27):14274–84. doi: 10.1074/jbc.M115.685792
81. Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic Properties of Itaconate and Derivatives Regulate the Iκbζ-ATF3 Inflammatory Axis. Nature (2018) 556(7702):501–4. doi: 10.1038/s41586-018-0052-z
82. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab (2016) 24(6):807–19. doi: 10.1016/j.cmet.2016.10.008
83. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-Ketoglutarate Orchestrates Macrophage Activation Through Metabolic and Epigenetic Reprogramming. Nat Immunol (2017) 18(9):985–94. doi: 10.1038/ni.3796
84. Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, León B, Crossman DK, et al. CCCTC-Binding Factor Translates Interleukin 2- and α-Ketoglutarate-Sensitive Metabolic Changes in T Cells Into Context-Dependent Gene Programs. Immunity (2017) 47(2):251–267.e7. doi: 10.1016/j.immuni.2017.07.015
85. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Veliça P, et al. S-2-Hydroxyglutarate Regulates CD8(+) T-Lymphocyte Fate. Nature (2016) 540(7632):236–41. doi: 10.1038/nature20165
86. Yu W, Wang Z, Zhang K, Chi Z, Xu T, Jiang D, et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol Cell (2019) 75(6):1147–1160.e5. doi: 10.1016/j.molcel.2019.06.039
87. Li Q, Barres BA. Microglia and Macrophages in Brain Homeostasis and Disease. Nat Rev Immunol (2018) 18(4):225–42. doi: 10.1038/nri.2017.125
88. Bohlen CJ, Friedman BA, Dejanovic B, Sheng M. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu Rev Genet (2019) 53:263–88. doi: 10.1146/annurev-genet-112618-043515
89. Wu T, He K, Liang X, Wei T, Wang Y, Zou L, et al. The Glycolytic Shift Was Involved in CdTe/ZnS Quantum Dots Inducing Microglial Activation Mediated Through the mTOR Signaling Pathway. J Appl Toxicol (2020) 40(3):388–402. doi: 10.1002/jat.3912
90. Tsubaki H, Tooyama I, Walker DG. Thioredoxin-Interacting Protein (TXNIP) With Focus on Brain and Neurodegenerative Diseases. Int J Mol Sci (2020) 21(24):1–24. doi: 10.3390/ijms21249357
91. Yang J, Ruchti E, Petit JM, Jourdain P, Grenningloh G, Allaman I, et al. Lactate Promotes Plasticity Gene Expression by Potentiating NMDA Signaling in Neurons. Proc Natl Acad Sci U.S.A. (2014) 111(33):12228–33. doi: 10.1073/pnas.1322912111
92. MacDonald JF, Jackson MF, Beazely MA. Hippocampal Long-Term Synaptic Plasticity and Signal Amplification of NMDA Receptors. Crit Rev Neurobiol (2006) 18(1-2):71–84. doi: 10.1615/critrevneurobiol.v18.i1-2.80
93. Liu YN, Yang X, Suo ZW, Xu YM, Hu XD. Fyn Kinase-Regulated NMDA Receptor- and AMPA Receptor-Dependent Pain Sensitization in Spinal Dorsal Horn of Mice. Eur J Pain (2014) 18(8):1120–8. doi: 10.1002/j.1532-2149.2014.00455.x
94. Magistretti PJ, Allaman I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nat Rev Neurosci (2018) 19(4):235–49. doi: 10.1038/nrn.2018.19
95. Patgiri A, Skinner OS, Miyazaki Y, Schleifer G, Marutani E, Shah H, et al. An Engineered Enzyme That Targets Circulating Lactate to Alleviate Intracellular NADH:NAD(+) Imbalance. Nat Biotechnol (2020) 38(3):309–13. doi: 10.1038/s41587-019-0377-7
96. Winkler U, Hirrlinger J. Crosstalk of Signaling and Metabolism Mediated by the NAD(+)/NADH Redox State in Brain Cells. Neurochem Res (2015) 40(12):2394–401. doi: 10.1007/s11064-015-1526-0
97. Requardt RP, Hirrlinger PG, Wilhelm F, Winkler U, Besser S, Hirrlinger J. Ca²⁺ Signals of Astrocytes Are Modulated by the NAD⁺/NADH Redox State. J Neurochem (2012) 120(6):1014–25. doi: 10.1111/j.1471-4159.2012.07645.x
98. Pan B, Zhong P, Sun D, Liu QS. Extracellular Signal-Regulated Kinase Signaling in the Ventral Tegmental Area Mediates Cocaine-Induced Synaptic Plasticity and Rewarding Effects. J Neurosci (2011) 31(31):11244–55. doi: 10.1523/JNEUROSCI.1040-11.2011
99. Singh V, Ubaid S. Role of Silent Information Regulator 1 (SIRT1) in Regulating Oxidative Stress and Inflammation. Inflammation (2020) 43(5):1589–98. doi: 10.1007/s10753-020-01242-9
100. van Deijk AF, Camargo N, Timmerman J, Heistek T, Brouwers JF, Mogavero F, et al. Astrocyte Lipid Metabolism Is Critical for Synapse Development and Function. Vivo Glia (2017) 65(4):670–82. doi: 10.1002/glia.23120
101. Huang Y, Wang L, Ren S, Wu G, Wu J. The Expression of ZnT3 and GFAP Is Potentiated in the Hippocampus of Drug-Resistant Epileptic Rats Induced by Amygdala Kindling. Neuroimmunomodulation (2020) 27(2):104–12. doi: 10.1159/000510399
102. Díaz-Zúñiga J, More J, Melgar-Rodríguez S, Jiménez-Unión M, Villalobos-Orchard F, Muñoz-Manríquez C, et al. Alzheimer's Disease-Like Pathology Triggered by Porphyromonas Gingivalis in Wild Type Rats Is Serotype Dependent. Front Immunol (2020) 11:588036. doi: 10.3389/fimmu.2020.588036
103. Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, et al. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell (2011) 144(5):810–23. doi: 10.1016/j.cell.2011.02.018
104. Vannucci SJ, Simpson IA. Developmental Switch in Brain Nutrient Transporter Expression in the Rat. Am J Physiol Endocrinol Metab (2003) 285(5):E1127–34. doi: 10.1152/ajpendo.00187.2003
105. Álvarez Z, Hyroššová P, Perales JC, Alcántara S. Neuronal Progenitor Maintenance Requires Lactate Metabolism and PEPCK-M-Directed Cataplerosis. Cereb Cortex (2016) 26(3):1046–58. doi: 10.1093/cercor/bhu281
106. Morland C, Andersson KA, Haugen ØP, Hadzic A, Kleppa L, Gille A, et al. Exercise Induces Cerebral VEGF and Angiogenesis via the Lactate Receptor HCAR1. Nat Commun (2017) 8:15557. doi: 10.1038/ncomms15557
107. Zhou J, Liu T, Guo H, Cui H, Li P, Feng D, et al. Lactate Potentiates Angiogenesis and Neurogenesis in Experimental Intracerebral Hemorrhage. Exp Mol Med (2018) 50(7):1–12. doi: 10.1038/s12276-018-0113-2
108. Wang Y, Peng Y, Zhang C, Zhou X. Astrocyte-Neuron Lactate Transport in the ACC Contributes to the Occurrence of Long-Lasting Inflammatory Pain in Male Mice. Neurosci Lett (2021) 764:136205. doi: 10.1016/j.neulet.2021.136205
109. Chang CC, Zhang C, Zhang Q, Sahin O, Wang H, Xu J, et al. Upregulation of Lactate Dehydrogenase a by 14-3-3ζ Leads to Increased Glycolysis Critical for Breast Cancer Initiation and Progression. Oncotarget (2016) 7(23):35270–83. doi: 10.18632/oncotarget.9136
110. Wong CB, Tanaka A, Kuhara T, Xiao JZ. Potential Effects of Indole-3-Lactic Acid, a Metabolite of Human Bifidobacteria, on NGF-Induced Neurite Outgrowth in PC12 Cells. Microorganisms (2020) 8(3):1–14. doi: 10.3390/microorganisms8030398
Keywords: inflammation, glycolysis, microglia, astrocyte, synapse, neuropathic pain, glycometabolism reprogramming
Citation: Kong E, Li Y, Deng M, Hua T, Yang M, Li J, Feng X and Yuan H (2022) Glycometabolism Reprogramming of Glial Cells in Central Nervous System: Novel Target for Neuropathic Pain. Front. Immunol. 13:861290. doi: 10.3389/fimmu.2022.861290
Received: 24 January 2022; Accepted: 26 April 2022;
Published: 20 May 2022.
Edited by:
Gaylia Jean Harry, National Institute of Environmental Health Sciences (NIH), United StatesReviewed by:
Hidetoshi Saitoh, International University of Health and Welfare (IUHW), JapanCopyright © 2022 Kong, Li, Deng, Hua, Yang, Li, Feng and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongbin Yuan, amZqY3p5eUBhbGl5dW4uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.