 Marie-Sophie Meuleman
Marie-Sophie Meuleman Anna Duval
Anna Duval Véronique Fremeaux-Bacchi
Véronique Fremeaux-Bacchi Lubka T. Roumenina
Lubka T. Roumenina Sophie Chauvet
Sophie Chauvet
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 April 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.860689
This article is part of the Research TopicCurrent Challenges in Complement DiagnosticsView all 27 articles
As part of the innate immune system, the complement system plays a key role in defense against pathogens and in host cell homeostasis. This enzymatic cascade is rapidly triggered in the presence of activating surfaces. Physiologically, it is tightly regulated on host cells to avoid uncontrolled activation and self-damage. In cases of abnormal complement dysregulation/overactivation, the endothelium is one of the primary targets. Complement has gained momentum as a research interest in the last decade because its dysregulation has been implicated in the pathophysiology of many human diseases. Thus, it appears to be a promising candidate for therapeutic intervention. However, detecting abnormal complement activation is challenging. In many pathological conditions, complement activation occurs locally in tissues. Standard routine exploration of the plasma concentration of the complement components shows values in the normal range. The available tests to demonstrate such dysregulation with diagnostic, prognostic, and therapeutic implications are limited. There is a real need to develop tools to demonstrate the implications of complement in diseases and to explore the complex interplay between complement activation and regulation on human cells. The analysis of complement deposits on cultured endothelial cells incubated with pathologic human serum holds promise as a reference assay. This ex vivo assay most closely resembles the physiological context. It has been used to explore complement activation from sera of patients with atypical hemolytic uremic syndrome, malignant hypertension, elevated liver enzymes low platelet syndrome, sickle cell disease, pre-eclampsia, and others. In some cases, it is used to adjust the therapeutic regimen with a complement-blocking drug. Nevertheless, an international standard is lacking, and the mechanism by which complement is activated in this assay is not fully understood. Moreover, primary cell culture remains difficult to perform, which probably explains why no standardized or commercialized assay has been proposed. Here, we review the diseases for which endothelial assays have been applied. We also compare this test with others currently available to explore complement overactivation. Finally, we discuss the unanswered questions and challenges to overcome for validating the assays as a tool in routine clinical practice.
As part of the complex innate immune surveillance system, the complement system plays a key role in defense against pathogens and in host homeostasis. This enzymatic cascade is rapidly triggered in the presence of activating surfaces, such as bacteria or apoptotic necrotic cells. However, the cascade is highly physiologically regulated on host cells to avoid self-aggression. The endothelium is one of the primary targets of complement dysregulation. There is increasing evidence of complement implications in the pathophysiology of many human diseases. Many complement-blocking therapeutics are under development, and some are already available in clinical practice. Nevertheless, detection of abnormal functioning complement is challenging, because in many pathological conditions C3 and C4 plasma levels, the two main biomarkers of complement activation, remain within normal ranges. The available tests to demonstrate such overactivation with diagnostic, prognostic, and therapeutic implications are limited. Methods are poorly standardized, and only a few have functional value. Therefore, there is a need to develop a robust and standardized tool for identifying infraclinical complement activation.
The final objective is to allow better pathophysiologically based therapeutic management of patients. The analysis of complement deposits on cultured endothelial cells (EC) incubated with patient serum holds promise as a reference assay. This approach has been used to explore complement activation in the sera of patients with atypical hemolytic uremic syndrome (aHUS), malignant hypertension, hemolysis, elevated liver enzymes, and low platelet (HELLP) syndrome, sickle cell disease (SCD), and pre-eclampsia. In some cases, adjusting the complement-blocking drugs has been considered. Nevertheless, the international standard for this test is lacking, and the mechanism by which complement is activated in this assay is not fully understood.
After a brief summary of the complement cascade, we present the mechanisms of complement activation and how they contribute to cell damage in several human diseases. We then provide an overview of the tests currently available to explore complement overactivation in routine practice. Finally, through a comparative analysis of the available endothelial assays for complement exploration, we discuss the unanswered questions and challenges to overcome to validate the study of complement deposition on cultured EC as a tool in routine clinical practice.
The complement system plays a key role in cell homeostasis, inflammation, and defense against pathogens. It is the first line of defense. The system comprises more than 30 soluble and membrane-bound proteins. Three different pathways lead to complement activation: the classical (CP), lectin (LP), and alternative (AP) pathways. When activated, these serine protease cascades converge to the formation of two enzymes, C3 convertase and C5 convertase, allowing the generation of the main effectors of this system: anaphylatoxins (C3a and C5a), opsonin (C3b/iC3b), and the membrane attack complex (MAC) (C5b-9). CP and LP are initiated by the recognition of pathogen-associated molecular patterns or damage-associated molecular patterns by pattern-recognition molecules (C1q and mannose-binding lectin). Conversely, AP is constantly activated at a low level in the fluid phase, generating a small quantity of C3b. In the presence of an activating surface (apopto-necrotic or bacterial), C3b covalently binds to the surface, and thus, initiates cell surface C3 convertase formation (C3bBb) and the AP amplification loop. To avoid self-aggression, AP is highly regulated in the fluid phase and on the host cell surface by soluble (factor H (FH), factor I (FI)) and membrane-bound regulators (membrane cofactor protein (MCP) or CD46, complement receptor 1 (CR1) or CD35, decay accelerating factor or CD55, and CD59). In humans, deficiencies in complement regulatory proteins are associated with rare diseases, such as aHUS, C3 glomerulopathy (C3G), and paroxysmal nocturnal hemoglobinuria (PNH). However, complement activation triggered by different pathophysiological processes that overwhelm the capacity of regulation has been increasingly described in a wide spectrum of diseases.
While AP overactivation is the central mechanism of cell and tissue injury in complementopathies (aHUS, C3G, and PNH), complement is crucial to tissue injury in a wide variety of diseases. These include age-related macular degeneration (AMD), antibody-mediated rejection (ABMR), cryoglobulinemic vasculitis (CV), IgA nephropathy (IgAN), systemic lupus erythematosus (SLE), anti-phospholipid syndrome (APS), ANCA-associated vasculitis (AAV), rheumatoid arthritis (RA), HELLP syndrome, pre-eclampsia, myasthenia gravis (MG), neuromyelitis optica spectrum disorder (NMOSD), SCD, and rhabdomyolysis-induced acute kidney injury (RIAKI). To a lesser extent, complement seems to be involved in an increasing spectrum of human pathological conditions, such as inflammatory disorders, ischemia/reperfusion, cancer, degenerative disorders (e.g., Alzheimer’s disease, atherosclerosis), and more recently, viral infections that include COVID-19 (1) (Figure 1).
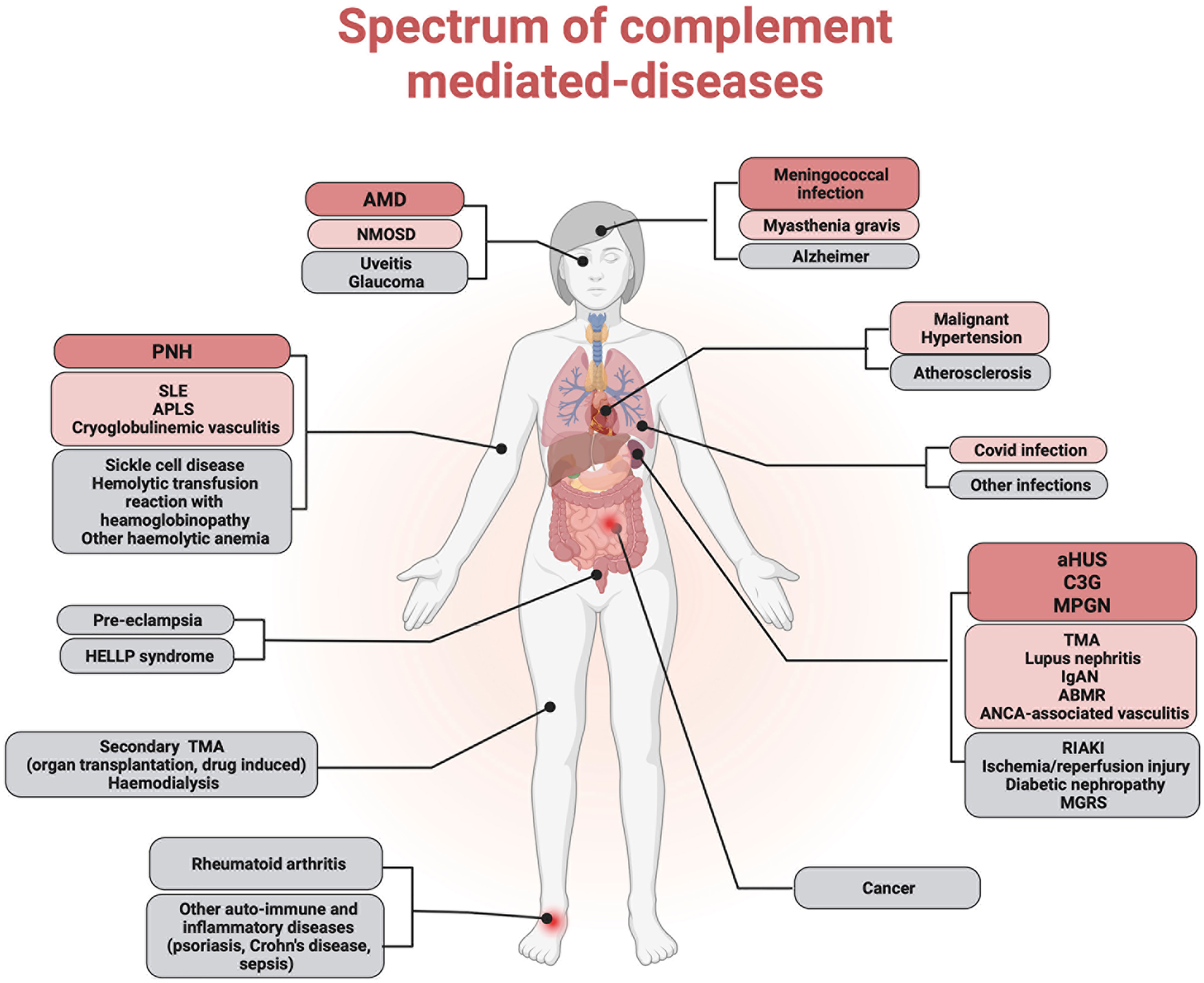
Figure 1 Complement implication in human diseases. Complement dysregulation has been implicated in the pathophysiology of several human diseases. Complementopathies in which alternative pathway dysregulation is the central mechanism of cell and tissue injury are represented in red. Conditions in which the complement system has been demonstrated to contribute significantly to tissue injury are represented in pink. Other diseases in which complement plays an accessory role are represented in gray.
Complementopathies are characterized by a specific cell target of AP-mediated damage. In aHUS and PNH, AP dysregulation occurs on the cell membrane, EC surface or platelets (2) and erythrocyte surface (3). In C3G, overactivation of C3 and C5 convertases may occur in the fluid phase or locally within the glomeruli, where the targeted surface remains to be determined (suggestions include glomerular EC and mesangial cells). AP dysregulation is a central pathophysiological mechanism in these diseases. It can be related to innate or acquired abnormalities in complement components, mainly regulators (FH, FI, or MCP) or C3 convertase components (C3 or FB) (4–15).
In diseases with major complement contributions, complement activation can be triggered by one or another pathway. In CV (16) and ABMR (17), activation occurs through CP in the presence of immune complexes (IC). In cryoglobulinemia (type II), IC are composed of IgM with rheumatoid factor activity associated with polyclonal IgG. In ABMR, IC are composed of IgG and donor HLA molecules. Conversely, despite the disease being triggered by the presence of IC, AP appears to be essential for disease development in mouse models of RA (18, 19) and SLE (20–22). This activation can be enhanced by apoptotic and necrotic cells due to prior damage (23) or by proteins of the extracellular matrix (ECM) from damaged cartilage in RA (24). In IgAN, AP (25), and LP (26) activation is mediated by polymeric IgA. In vitro, a correlation was found between C3 cleavage products (iC3b, C3c, C3dg) and IgA-A-IgG IC levels, suggesting that IC-containing IgA may act as a surface for soluble AP activation (27). In AAV, AP may be activated by neutrophil extracellular traps, thus amplifying complement activation and damage of EC (28). Finally, a disease-specific soluble factor has been implicated in complement activation. Free heme renders EC more sensitive to complement activation in SCD (29), aHUS (30), and RIAKI (31). In vitro, thrombin induces C5 cleavage in C5a in APS (32).
Complement activation does not arise from a unique mechanism but can be triggered in several ways according to the disease pathophysiology. Identification of the precise mechanisms of complement activation will help determine different potential therapeutic targets within the cascade.
Complement activation contributes to cell and tissue injuries in different ways. First, it promotes inflammatory cell recruitment mainly in CV (33), ABMR (34), AMD (35), SLE (36)and RA. C5a and its receptor C5aR are involved in neutrophil recruitment (37–39) and endothelial activation (40) in AAV. Complement activation can promote specific disease processes. Thus, MAC can directly affect collagenase production by synovial fibroblasts in RA (41). In IgAN, mesangial cells exposed to complement activation and C3 deposition promote phenotypic conversion to a more synthetic and proliferative state (42). In AMD, C3a and C5a promote choroidal and C5a induces vascular endothelial growth factor secretion by retinal pigment epithelium (35). In pre-eclampsia, it has been suggested that the binding of C5a to C5aR expressed on trophoblasts contributes to the acquisition of their anti-angiogenic phenotype (43).
The complement system can also act as an amplifier for other molecules involved in injury. The C5a/C5aR axis participates in neutrophil recruitment and activation, which in turn can induce complement activation in AAV (38). C5a induces tissue factor expression by neutrophils, leading to factor X activation and thrombin generation, which in turn cleaves C5 into C5a in APS (32).
Ultimately, several triggers of complement activation and effectors may contribute to cell and tissue damage in heterogeneous human diseases. The identification of specific triggers of complement activation and fine pathophysiological mechanisms resulting in cell and tissue complement-mediated injury is needed to determine the best therapeutic target within the cascade. Complement inhibitor anti-C5 monoclonal antibody (eculizumab, and more recently its long-acting form, ravulizumab) is the gold standard in two complementopathies, aHUS and PNH, and has obtained Food and Drug Administration (FDA) approval for MG and NMOSD. Avacopan is a C5aR1 antagonist that has also been approved by the FDA for patients with AAV, another disease with a major complement contribution. Understanding the detailed mechanisms of complement activation and complement-mediated damage is necessary to guide the prescription of new complement inhibitors.
Currently available tests mainly consist in quantification of individual complement components or activation products.
- For the quantification of individual complement proteins in plasma, various types of immunoassays are used to determine the concentration of individual complement components. The most common is nephelometry. Polyclonal antibodies to component are added in excess of the sample and bind to their target. Quantification is performed by passing a light beam through the sample, which is distorted by the IC that have formed (44).
- Quantification of complement activation products corresponding to cleavage fragments or complement protein complexes (C3a, C3dg, C4a, C4d, Ba, Bb, C5a, C3bBbP, MASP2, and sC5b-9) is possible. Several assays have been described, mostly based on the recognition of a neoepitope of the complement component in an enzyme-linked immunosorbent assay (ELISA) format. Thus, C4a and C4d reflect CP/LP activation, Ba, Bb, and C3bBbP reflect AP activation, MASP2 is a key enzyme in LP activation (45) and increasing soluble C5b-9 reflects TP activation (46). C3a and C5a are common to the three activation pathways.
- Detection of auto-Abs (anti-FH, FB, C3b, C3bBb, and C1q) targeting complement proteins can be performed using ELISA (14).
• Quantification of complement function is used to explore the activity of a pathway or the entire cascade.
- In hemolytic assays, CP activation can be assessed by incubating patient sera with sheep erythrocytes coated with rabbit anti-sheep red blood cell antibodies (47). In this assay, termed the CH50 assay, C1q binds to immunoglobulins, initiates the formation of CP C3 convertase, and leads to MAC assembly and erythrocyte lysis. Hemoglobin release is determined to calculate the number of hemolytic sites per cell. Activation through AP can be assessed using rabbit or guinea pig erythrocytes, which are activators of human AP, incubated with patient serum added to ethylene glycol-bis(β-aminoethyl ether) (EGTA), which chelates Ca2+ and inhibits activation via CP and LP (48). This hemolytic assay is termed the AP50 assay.
- Liposomes coated with an activator can be used in a similar manner to CH50 assays (49). The main difference is the readout, which consists of the unquenching of a fluorescent dye and not the lysis of erythrocytes.
- Assays based on ELISA method can also be used to explore the function of the three pathways. Microtiter plate wells are coated with recognition structures specific to each pathway (IgM for CP, mannan or acetylated bovine serum albumin for LP, and LPS for AP). Patient serum is added and incubated under conditions in which only one pathway is operative at any given time; the other two pathways are blocked. Finally, activation capacity is detected through the formation of the C5b-9 complex by monoclonal antibodies targeting a neo-epitope in complex-bound C9 (50).
• Different hemolytic assays have been developed to explore specific steps of the AP.
- Sanchez-Corral et al. (51) developed a hemolytic assay to study FH functional defects in aHUS. The assay relies on the knowledge that sheep erythrocytes are highly sialylated and favor FH binding, whereas their membrane complement regulators are incompatible with human complement proteins. Therefore, they are protected from complement lysis due to the binding of human FH to their surface. In the assay, sheep erythrocytes are incubated with human plasma in Mg-EGTA buffer, allowing activation of AP only. Normal plasma does not induce lysis, whereas aHUS plasma with FH functional defects (mutations or autoantibodies) induces lysis under these conditions (51).
- Hemolytic assays can also be used to study the stabilization of cell-bound AP convertases (52). This assay has been used to detect C3Nef in C3G cells. Sheep erythrocytes bearing C3 convertase C3bBb (generated by exposure of sheep erythrocytes bearing C3b to FB and FD) were incubated with patient IgG. C3Nef activity correlates with residual C3bBb hemolytic sites, and lysis is developed by the addition of rat serum.
• Staining of tissue sections for the deposition of complement activation products can provide information about local complement activation in tissue. This can be performed by immunohistochemistry or immunofluorescence (53). For example, this technique has been used to study C5b-9 deposition in the skin of patients with aHUS (54).
These tests allow only the characterization of a specific molecule or step of the complement cascade. To reproduce human pathological conditions and their complexity, several authors have proposed the use of an ex vivo endothelial assay. The assay detects and quantifies complement component deposition on the EC surface after incubation with human serum. The EC surface is used as the regulating surface. The objective is to detect abnormal complement deposition that could result from either complement overactivation exceeding the capacity of regulation, or from a defect in complement regulation in fluid or on the EC surface. The ex vivo endothelial assay is presented in Figure 2.
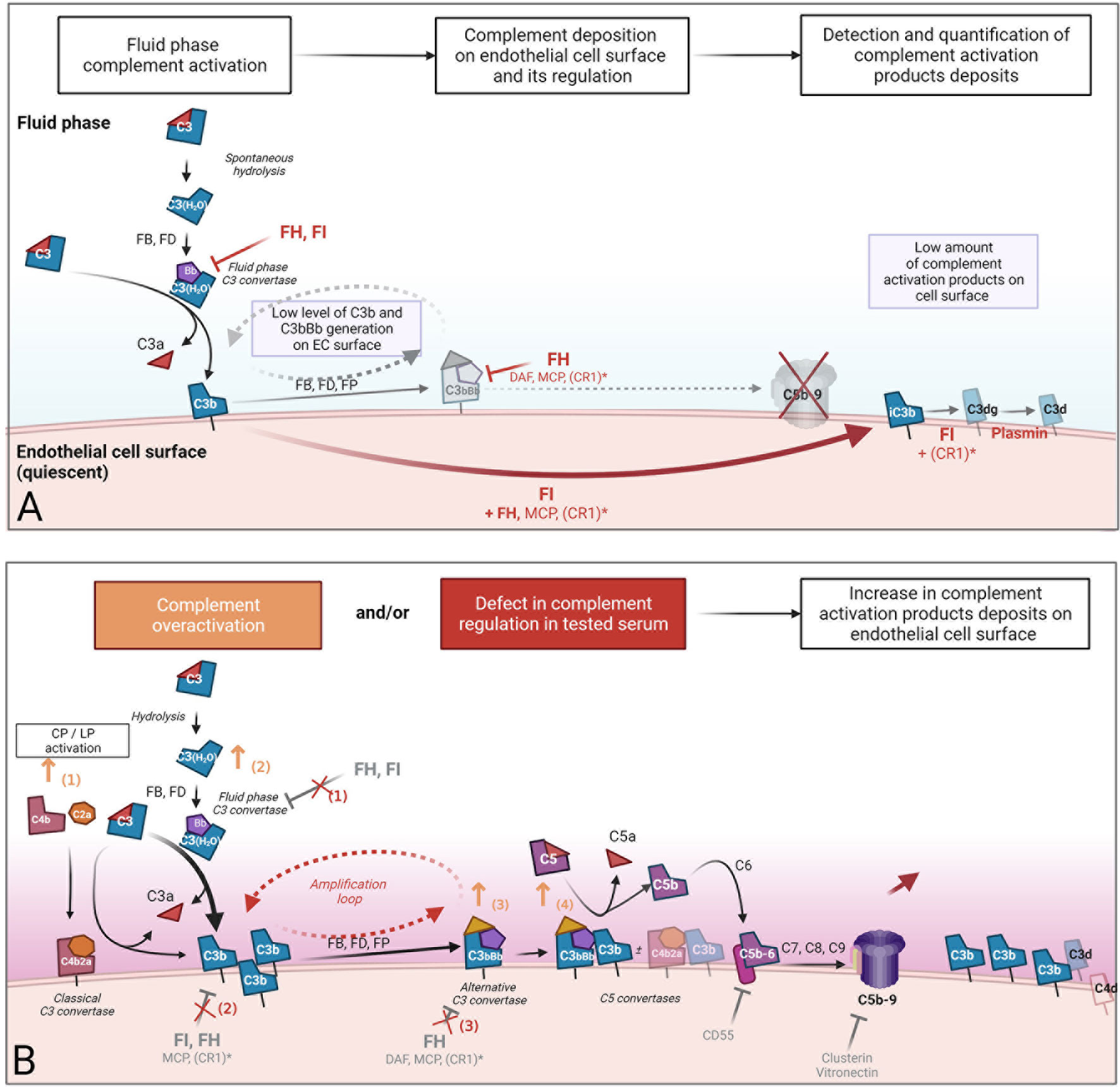
Figure 2 Concept of ex vivo complement deposition on endothelial cells. An ex vivo endothelial assay was developed to reproduce human pathological conditions and their complexity. The assay consists of the detection and quantification of complement component deposition on the cultured endothelial cells (EC) surface after incubation with human serum. The EC surface was used as the regulatory surface. (A) In serum from healthy individuals, the alternative pathway is active at low levels but tightly regulated in the fluid phase by regulators, resulting in a very low level of complement activation product deposition on the EC surface. The detection of an increased complement deposition when incubation is performed with pathological serum (B) could result in either i) complement overactivation that overwhelms EC capacity of regulation (orange) or ii) defect in complement regulation in fluid or solid phase. Both are induced by tested human serum incubated with EC. Orange arrows represent some mechanisms involved in complement overactivation in serum (1): the participation of a coactivation of classical/alternative pathway due to pathological immunoglobins, immune complexes, or lectin pathway activation by polymeric IgA in IgA nephropathy (2), an increase in the formation of fluid phase C3 convertases in the presence of heme or fluid phase activating surface, and the stabilization of C3 (3) or C5 (4) convertases by pathological immunoglobulins, such as C3 and C5 nephritic factors. Red crosses represent potential defects in alternative complement pathway regulation in the fluid phase (1) and on the cell surface (2, 3). These defects in complement regulation could be the consequence of inhibition of the main alternative pathway regulator FH due to anti-factor H antibodies (such as in aHUS), a lack of function, or a quantitative deficiency of FH and FI due to pathological genetic variants. *CR1: weak expression of CR1 on endothelial cells. CR1, complement receptor 1 (CD35); FB, factor B; FD, factor D; FH, factor H; FI, factor I; FP, properdin; MCP, membrane cofactor protein.
We next discuss the advantages and limits of this functional approach.
EC line blood vessels and constitute an active regulatory organ that has been implicated in vascular homeostasis, permeability regulation, vasomotor tone, angiogenesis, and diapedesis of immune cells (55). As first barrier between the blood and interstitium it is in constant equilibrium with the environment. Thus, heterogeneity in the structure and function of EC is a core property of the endothelium, allowing diverse vascular functions and regional specificity (56, 57). This diversity can be partially explained by a distinct transcriptional profile (58) in relation to neighboring cells (59). Hence, EC from different blood vessels have distinct and dynamic expression profiles of complement components and regulators, which may explain the different susceptibility and specific organ tropism observed in some complement-mediated diseases (60).
At a steady state, EC can produce most complement components and express high levels of complement regulators on their membranes (Table S1). Under inflammatory conditions, complement component production and regulatory protein expression are modified (Table 1). In addition to the steady state, the modulation of complement protein expression under inflammatory conditions differs according to the EC type and probably contributes to a specific damage mediated by AP and the different organ tropisms observed in complement-mediated diseases. Sartain et al. demonstrated that resting or tumor necrosis factor (TNF)-stimulated brain microvascular EC expressed higher levels of regulatory molecules (FH, FI, CD46, CD55, and THBD), generated lower levels of C3a and C4a, and enhanced lower degree AP activation (measured by lower Ba generation) than human renal glomerular EC (HRGEC) (61). The authors also demonstrated a slight increase in CD46 expression, decrease in thrombomoduline (TM), and increase in C3 and FB transcription in HRGEC exposed to TNF (62). These results agree with the prior demonstrations of an increase in C3 and FB production by human umbilical vein EC (HUVEC) exposed to TNF (63), increased FH transcription and production by HUVEC exposed to interferon (INF) gamma (64), increased C2, FH, FB, and C1inh transcription, and decreased C3 production by HUVEC exposed to INF gamma (65). May et al. compared the properties of four EC types (HRGEC, glomerular EC (GEnC), human microvascular EC (HMEC), and HUVEC) in the resting state and after overnight exposure to heme (66). While there was no difference in expression of regulatory factors (MCP, CD55, TM) at resting state, after overnight heme exposure, C3 deposits on glomerular EC were greater than on other EC. This was associated with, and possibly explained by, weaker FH binding and TM upregulation and lower upregulation of heme-oxygenase 1 (cytoprotective heme-degrading enzyme) compared to HUVEC. Moreover, HUVEC, but not EC, of glomerular origin were protected from complement deposition after re-challenge with heme (Table S2).

Table 1 Production of distinct complement components and expression of regulators according to endothelial cell type after stimulation.
EC used for ex vivo experiments comprise two types: conditionally immortalized EC (CI-EC) and primary EC (Table 2). Primary EC can be difficult to isolate and maintain in culture, and have a limited lifespan. Moreover, differences in the genetic background of individual donors can lead to interexperimental variability. In particular, inter-individual heterogeneity in complement regulator expression at the EC surface cannot be excluded. CI-EC has been developed to overcome these difficulties. HMEC-1 and CI-GEnC are HMEC and GEnCs, respectively, that have been transfected with SV40 large T antigen (67, 68). EA.hy926 cells were obtained by fusing HUVEC with A549 cells obtained from human lung carcinoma (69). The EA.hy926 cells were used to generate glycosylphosphatidylinositol-anchored complement regulatory protein-deficient cells when treated with phosphatidylinositol-specific phospholipase C. These cells have been used along with the PIGA-mutant TF-1 to study complement deposits by confocal microscopy and flow cytometry after incubation with serum from patients with thrombotic microangiopathy (TMA), this test was called the modified Ham test (70). After incubation with serum from aHUS patients, cell surface C5b-9 deposits were reportedly higher than after incubation with thrombotic thrombocytopenic purpura (TTP) serum. Therefore, this test has been considered a tool to distinguish aHUS from TTP. It is important to note that sC5b-9, reflecting terminal pathway activation and regulation, is elevated under both aHUS and TTP plasma conditions (71, 72). One possibility is that both conditions are associated with complement activation. However, in aHUS, complement overactivation exceeds alternative and terminal pathway regulation, leading to C5b-9 deposits. In contrast, in TTP, complement activation is counterbalanced by complement regulation, leading to sC5b-9 release, but not C5b-9 deposits in the modified Ham test.
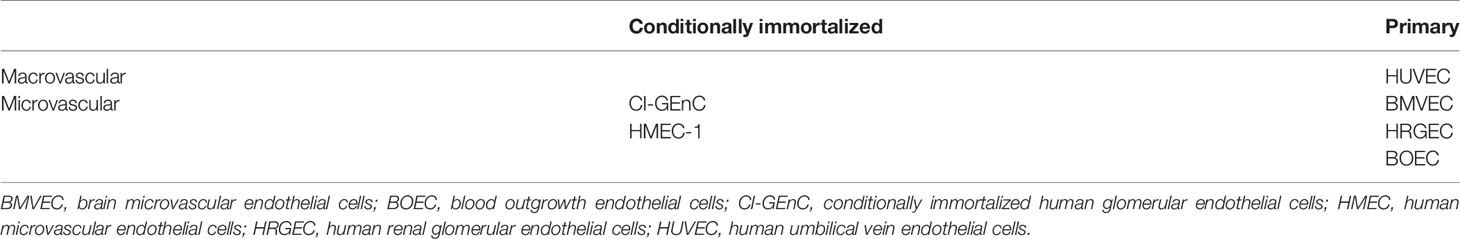
Table 2 Endothelial cells used for ex vivo experiments.
Micro- or macrovascular origin of the EC tissue lineages also needs to be considered. Complement-mediated EC injury demonstrates specific cell tropism according to pathophysiological processes. In HUS and TTP, microvascular EC of dermal, renal, and cerebral origin are more sensitive to apoptosis, whereas microvascular EC of pulmonary and hepatic origin and macrovascular EC are resistant (73). Distinct sensitivity of EC to complement attack has also been explored in aHUS and heme exposure. The demonstration of a distinct EC response in terms of complement regulator expression after a trigger (here heme) was proposed to partially explain the kidney tropism in this disease (66).
HUVEC are primary macrovascular EC isolated from human umbilical cords. These are the most frequently used cells for ex vivo assays (74). If tissue specificity is required, HRGEC (75) or GEnCs (76), which are both isolated from human glomeruli, can be used. More recently, the use of blood outgrowth EC obtained from the differentiation of circulating marrow-derived endothelial progenitor cells isolated from peripheral blood has been proposed (77).
These tests consist of the quantification of complement activation products (C3 activation fragments and C5b-9) deposits on EC by immunofluorescence (IF) measured by confocal microscopy or flow cytometry (fluorescence-activated cell sorting, FACS) after incubation with a serum sample of interest. Different protocols have been proposed to study complement activation on the EC surface in several pathological conditions, including aHUS (30, 78–88), TMA of other etiologies (89, 90), HELLP syndrome and pre-eclampsia (91), C3G (14, 92), lupus nephritis (LN) (93, 94), APS (95, 96), SCD (29), hemolytic anemia (97) and hyperhemolytic transfusion reaction without hemoglobinopathy (98).
The general procedure of the ex vivo assay and the different protocols are presented in Figure 3.
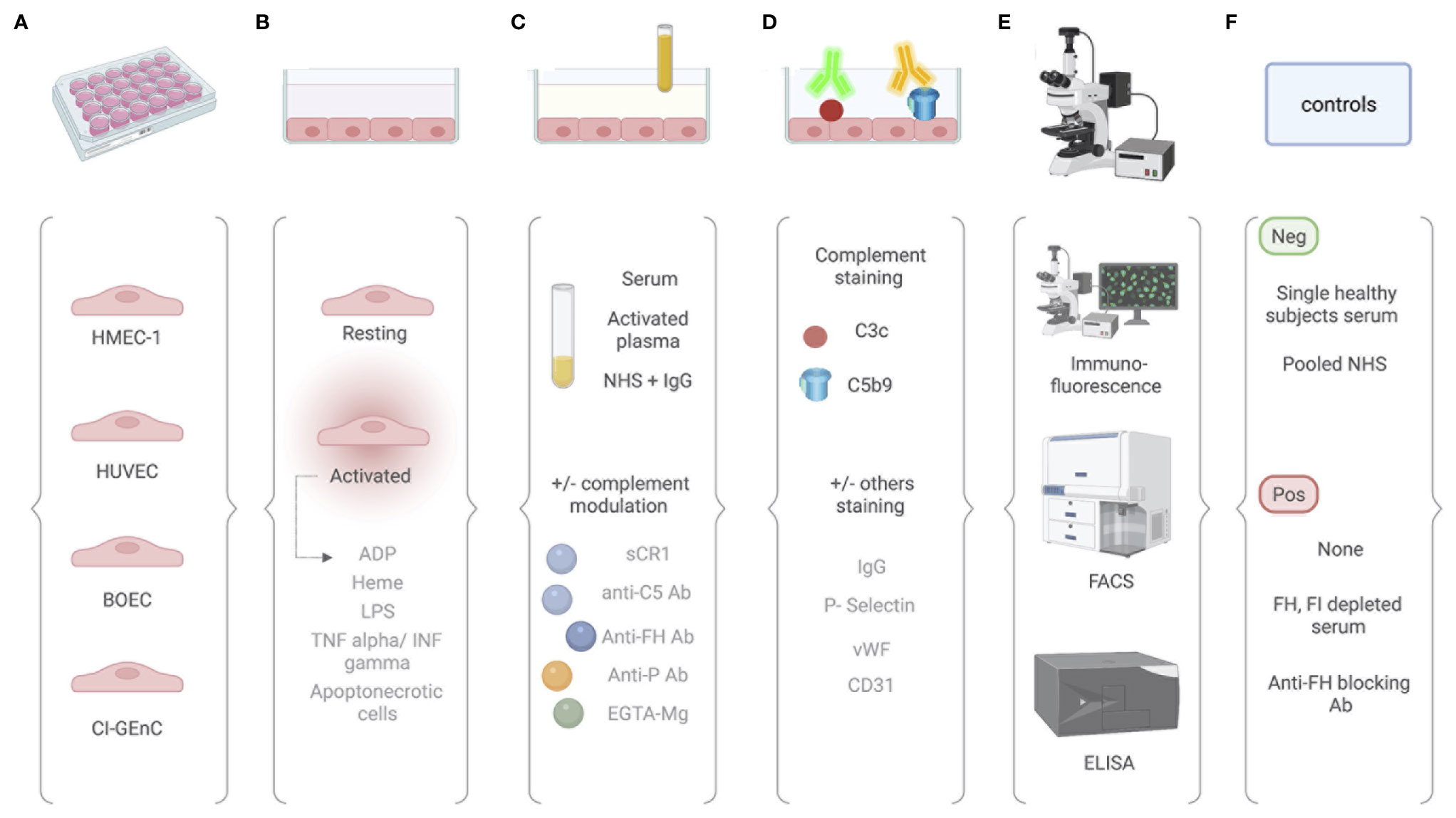
Figure 3 Comparative analysis of different protocols used for the ex vivo complement activation test with endothelial cells. (A) The ex vivo test for measuring complement attack on endothelial cells can be performed on different endothelial cells, including human dermal microvascular endothelial cells (HMEC-1), human umbilical vein endothelial cells (HUVEC), blood outgrowth endothelial cells (BOEC), and glomerular endothelial cells (GEnC). (B) Cultured EC are then used at their resting state or after an activation by either ADP, heme, LPS, TNF/INF gamma, or apoptonecrotic cells. (C) EC are incubated with sample of interest. Either serum or activated plasma (consisting of patient citrated plasma mixed 1:1 with control serum pool) or normal human serum with addition of the protein of interest (e.g., IgG). Complement activation can be modulated in by addition of sCR1, anti-C5 antibody, anti-FH antibody, anti-properdin antibody, or EGTA-Mg buffer. (D) Complement activation products are then revealed by fluorescent tagged antibody. Antibody directed again C3c or C5b9 can be used. According to the context, staining for other molecules have been proposed and include IgG, P-selectin, vWF, and CD31. (E) Quantification is then performed using immunofluorescence scanning, flow cytometry, or ELISA. (F) Controls are required and vary according to the protocols.
Resting EC or EC pre-activated by cytokines, ADP, or heme can be used (Table S3) to provide additional information.
When resting HUVEC were incubated with aHUS FB mutants added to FB-depleted normal human serum (NHS), enhanced C3b/iC3b-fragment deposition as measured by an anti-C3c-reacting antibody was observed (78). The same result was obtained for some cases when aHUS patient serum was incubated with resting HUVEC (30, 79). Nevertheless, incubation with NHS depleted in FB and reconstituted with other aHUS FB variants (80) or incubation with aHUS serum from patients carrying some C3 or FH variants (30, 79) may be insufficient to induce C3 or C5b-9 deposits. When quiescent HMEC-1 were incubated with aHUS serum from patients carrying mutations in FH, FI, C3, or FH/CFHR1 hybrid, enhanced C3c or C5b-9 deposition was reported only if serum was collected during the acute phases of the disease and not after reaching remission (82, 89). Furthermore, deposits on quiescent HMEC-1 are better correlated with relapse risk during the tapering or discontinuation of eculizumab (85).
To increase test sensitivity, the authors proposed pre-activating EC. Modifications in surface-bound protein expression enable complement activation. This was achieved in the case of P-selectin expression on HMEC-1 pre-activated with ADP, LPS, or thrombin (82) or P-selectin expression on HUVEC or GEnC pre-activated with heme (30, 80, 99), which could allow C3b binding and C3 convertase formation. Enhanced formation of C3 fragments by TNF/IFN pre-activation and C5b-9 deposition by ADP pre-activation on HUVEC or HMEC-1 cells was described after incubation of these cells with serum from asymptomatic carriers of mutations in AP regulatory proteins or C3 (79, 82). The normal range was established when pre-activated EC were incubated with sera from healthy donors. In addition, serum from healthy family members without the mutation was within the normal range in this assay (79).
Serum has been used as the source of complement proteins in the vast majority of the tests described above. One limitation of these tests, particularly when deposits are detected by IF, is the variation in the results, reportedly from 30% to 52% when activated HMEC-1 were incubated with serum collected at the acute phase of aHUS (91). To reduce this variation, Palomo et al. proposed the use of activated plasma, which refers to citrated plasma mixed 1:1 with a control serum pool. Using this approach, the authors derived a coefficient of variation of 9% to 18% (91). C3 consumption by the patient or loss of C3 activity during the pre-analytical phase are also potential factors responsible for this variation (4). Finally, for all complement assays and to avoid in vitro complement activation, proper blood collection and processing must be achieved (100). Processing of plasma or serum sample must be performed within a few hours of collection, with storage at –80°C and defrosting immediately before use to avoid repeated freezing and thawing.
To explore the functional consequences of autoantibodies against C3 and properdin in SLE, Vasilev et al. and Radanova et al. incubated HUVEC with NHS supplemented with purified IgG from patients positive for such autoantibodies (93, 94). Using this strategy, complement deposition on EC can be directly ascribed to the addition of autoantibodies to NHS. The same approach was applied for anti-C3b/FB autoantibodies in patients with C3G (14). To understand the mechanism behind complement deposits on EC from patients with SCD, microvesicles from normal or patient-derived erythrocytes were added to normal serum to model the disease condition. Enhanced binding of the C3 activation products was demonstrated (29, 101).
Most often, NHS is used as a negative control (14, 29, 30, 78–83) (Table S4). An important aspect to consider is the inter-individual variability in deposits induced by normal sera. FACS analysis has revealed that this variability was relatively low when sera from 50 healthy donors were tested (79). However, this is a concern, particularly when deposits are detected by IF. This has not been directly reported, but has been suggested by the use of pooled sera in more recent papers (85, 91) and our own experience. Aiello et al. reported that C3 and C5b-9 deposits obtained after a single healthy subject serum (N=12) incubation range from 0.5 to 1.5 fold increase of stained surface area compared to pooled serum (from 10 healthy donors) run in parallel (87).
Several authors did not use any positive controls for their experiments (82, 83, 85, 90, 91). The comparison was only made with the deposits obtained with negative controls. It might be interesting to position the results on a scale. Positive controls with published data are FH or FI depleted NHS (14, 79, 81) or normal serum supplemented with blocking anti-FH antibodies targeting the N-terminus or C-terminus (30, 80) or with FH19-20, corresponding to the two last domains of FH, able to compete with the full FH protein for cell surface binding (97).
The main issues with this type of assay are the lack of validated international standards as well as standardized positive and negative controls. The variability of the results in samples from healthy donors needs to be studied extensively to determine the appropriate cutoff. In addition, the impact of C3 or other complement protein consumption in the patient and the influence of the pre-analytical phase must be determined to avoid false positive and false negative results.
The objective of these tests is to demonstrate and explore complement overactivation or dysregulation on the EC surface after incubation with blood samples of interest. This is enabled by quantification of the deposition of complement component products resulting from activation or regulation. C3c (a common epitope to C3, C3(H2O), C3b, and iC3b) (which reflects C3 convertase activity and the early phase of the complement cascade) can be detected by polyclonal anti-C3c antibody. Antibody targeting C5b-9 reveals the final step of the cascade. When a signal is detected on the cell membrane, it can be assumed that the detected fragment is C3b or iC3b covalently attached to the surface. Nevertheless, heme-activated EC and likely ADP-activated EC (102, 103) express P-selectin, which recruits C3b, C3(H2O), and a C3(H2O)-like form of C3 generated after contact with heme (30, 99). Properdin also binds to heme-exposed or stressed EC, promoting complement activation in a similar manner without covalent C3b binding (97). This is an additional mechanism for amplification of complement activation on the EC surface. C5b-9 deposits may be more relevant in identifying dysregulation at any step. Nevertheless, early dysregulation can induce C3 activation fragment deposits without C5b-9 formation because of TP regulation. C5b-9 is readily detectable by IF but is much more difficult to detect by FACS because of the weak shifts of the peaks. To test for CP participation, the presence of C4d-positive deposits was also investigated (82, 89). Staining can also be performed under the same conditions for von Willebrand Factor, C5aR1, P-selectin, and others (87).
The test can be modified to assess which complement pathway is activated in given pathological settings. The test can be performed under different conditions to avoid CP and LP contributions, which include C2 (30) or C1q (104) depleted NHS, addition of SCR1 (87) or Mg-EGTA buffer (30, 78, 93, 94). EGTA chelates Ca2+, which is crucial for CP and LP activation, whereas AP depends on Mg2+. If AP has to be inhibited, FB-depleted NHS can be used. These reagents are applicable for test conditions, where the activating factor is added externally to the serum (i.e., IgG, heme, microvesicles, etc.). When patient samples are used directly, the same effect can be achieved by inhibiting C1q, C4, FB, or properdin with blocking antibodies, protein constructs, or small molecules, if available (97). Quantification of complement activation products (split fragments generated by cleavage of complement components or protein complexes when activated components bind their target (i.e., C3a, C4a, Ba, Bb, C5a, and sC5b-9) in the supernatant might be an additional element to study complement cascade activation.
The two main detection techniques commonly used are FACS and IF. HUVEC pre-activated with heme and then incubated with NHS or aHUS serum showed results similar by FACS or IF detection (30). IF directly analyzes deposits on EC grown on slides. FACS requires a cell detachment step before staining, with the potential risk of losing a part of the deposit signal. In contrast, as mentioned by Gavriilaki et al., obtaining quantitative data by IF requires confocal microscopy and further analysis using specialized software (70). When IF is used, the area occupied by fluorescent staining in fields systematically digitized along the surface is quantified. The quantified results expressed as the mean of the square number of pixels per field are compared with the negative control (82, 83, 85, 89, 91). Considering the number of EC on which fluorescence has been measured and the staining intensity might appear relevant. In contrast, FACS allows the rapid and objective quantification of deposits.
If enhancement of complement fragment deposits on EC is interpreted as pathogenic, the functional consequences of such deposits must be questioned. Lactate dehydrogenase release from EC reflects cell damage. This release can be measured in the cell culture supernatant (104). Analysis of complement deposits can also be associated with a cell viability assay, corresponding to a colorimetric assay based on cleavage of the WST-1 tetrazolium salt by mitochondrial dehydrogenases in viable cells (70). Cellular integrity can be verified by May-Grunwald Giemsa staining (88). Direct cell death rarely occurs under these experimental conditions. Experiments testing cell activation status by complement overactivation have not been reported in the literature and are needed to further understand the impact of complement on endothelial injury. Analysis of transcriptomic modifications in EC exposed to complement deposits under several conditions could also be of interest.
The ex vivo EC assay, consisting in the quantification of complement activation products (C3 activation fragments or C5b-9) deposits on EC (by IF measured on confocal microscopy or FACS), after incubation with a serum sample of interest, was first used for specific characterization of complement component abnormalities (78–81, 83) or exploration of mechanisms implicated in EC injury (30) in the main complementopathy, aHUS. The assay was then used to demonstrate and explore complement activation and participation in the pathophysiology of several diseases, including C3G (14, 92), HELLP syndrome and pre-eclampsia (91, 105), TMA associated with severe hypertension (89, 106), drug-induced TMA (90), SCD (29), hemolytic anemia (97, 98), SLE (93, 94), and APS (96). Demonstration of increasing complement deposits on EC incubated with pathological sera is not sufficient to determine what is responsible for complement activation at the EC surface. Modulation of the test conditions can help in detailing complement activation. This was the case when complement activation was inhibited by the addition of hemopexin to the sera of patients with SCD (29).
Noris et al. and Galbusera et al. also proposed the use of this ex vivo EC test to monitor eculizumab therapy in patients with aHUS (82, 85). During eculizumab tapering or discontinuation, disease relapse preceded or was associated with an increase in C5b-9 deposits on resting HMEC-1 in all patients. In contrast, only one patient without relapse showed increased deposits (85). In clinical practice, CH50 is the only routine test used to monitor eculizumab therapy. CH50 is reportedly strongly suppressed in patients receiving eculizumab according to the standard protocol. However, CH50 does not allow monitoring of eculizumab dosage tapering or discontinuation, as it is not well correlated with relapse risk (82, 85). Eculizumab therapy monitoring using the Wieslab® complement system screen (107) or the modified Ham test (108) has also been proposed. Thus, the ex vivo EC assay could represent a test of interest to a better personalized complement-blocking therapy, but first needs to be more standardized.
This test can also be used to better classify and assess the prognosis of specific diseases. This is the case for hypertensive TMA, as Timmermans et al. demonstrated in a cohort of hypertension emergencies associated with TMA (106). The authors demonstrated a statistical association between increased C5b-9 deposition in the EC ex vivo test and kidney survival. Moreover, they reported an improvement in renal function for those with increased deposits treated with eculizumab. The authors proposed a classification of TMA-hypertensive emergency based on the EC ex vivo test (106).
Finally, many new anti-complement drugs targeting specific steps of the cascade have been under development in recent years (109). A standardized and validated assay to study complement activation could be a useful tool in their development.
The increasing demonstration of complement involvement in the pathophysiology of many human diseases has mandated the development of tools to finely explore complement activation. Complement is a complex enzymatic cascade that is highly regulated in constant interplay with its environment. The current arsenal for complement exploration does not provide functional characterization and does not report on the complex interplay between complement and its environment, particularly the cell surface.
The development of tests with these capabilities could allow for a deeper exploration of the mechanisms of complement activation in several diseases. This information could inform the development of a complement blocking therapeutic strategy based on pathophysiological mechanisms.
Ex vivo complement activation on EC represents a promising tool for demonstrating and exploring complement activation (Figure 4). It not only recapitulates complex complement cascade regulation in vivo, but also allows modification of several steps of the experimental procedure to characterize complement activation mechanisms.
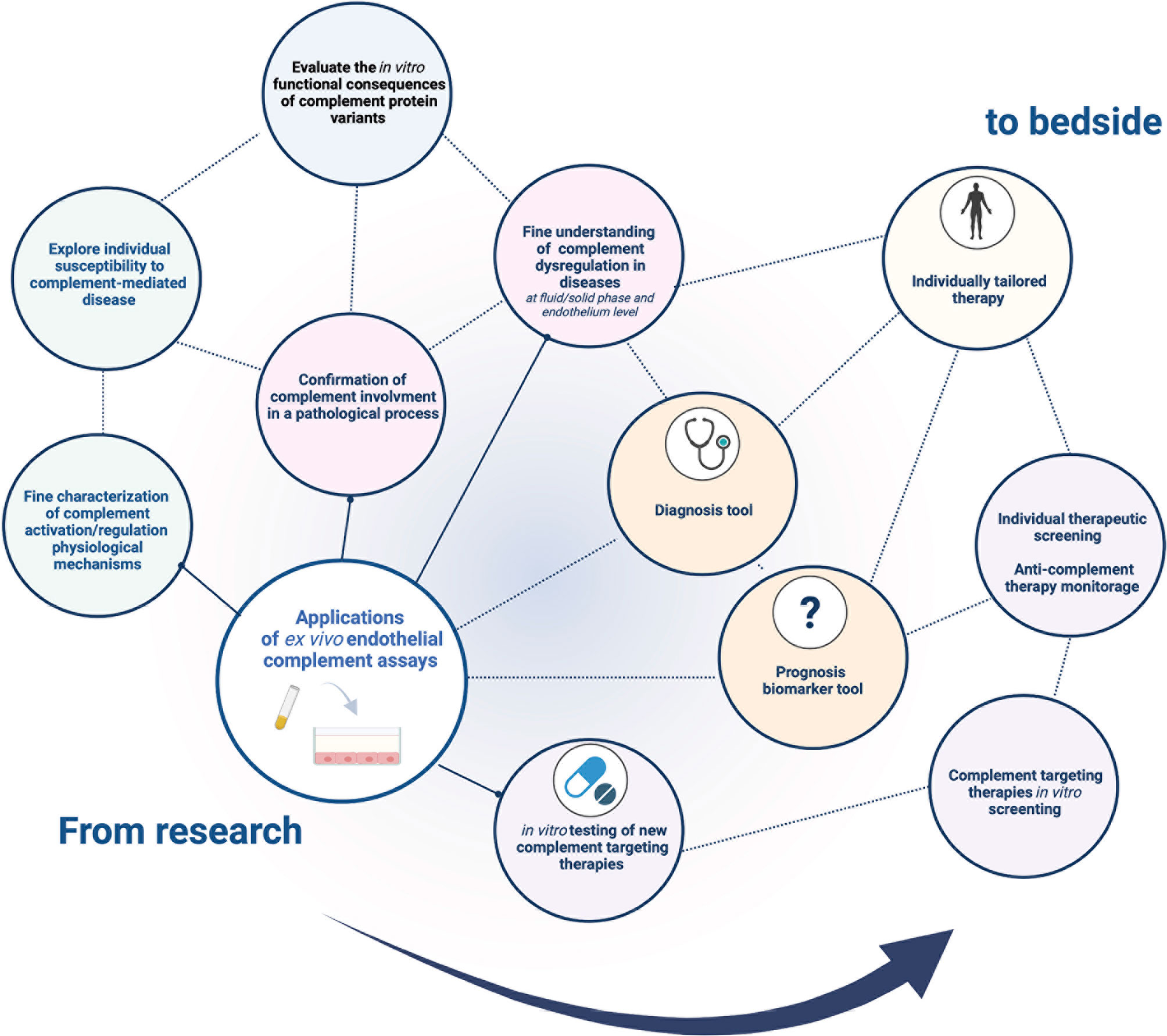
Figure 4 Current and future application fields of the ex vivo complement activation test on endothelial cells. There is a wide range of potential applications of ex vivo complement activation tests in endothelial cells. Currently used to decipher in vitro complement pathophysiology in research, a standardized test would represent a promising tool in clinical and therapeutic fields, paving the way for tailored medicine in complementopathies.
However, there are still unanswered questions hindering broad used. The first is the variability in the results and the inter-individual variability in deposits induced by normal sera. Comprehension of the precise mechanism responsible for complement deposition in this assay would improve its better use. The second issue is to standardize the main steps of the procedure to improve the interexperimental comparison.
The use of such a test could be multiple, including molecular functional characterization, disease pathophysiology exploration, prognosis classification, complement targeting drug development, and complement therapeutic monitoring. The use of standardized conditions will expand the field of this promising tool.
M-SM, SC, and LR conceptualized and conceived the manuscript. M-SM drafted the manuscript, including the literature search, reading, and writing. SC, VF-B, AD, and LR edited and critically evaluated the manuscript. All authors have contributed to the manuscript and approved the submitted version.
This research was funded by ANR JCJC 2020 COMSIGN.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.860689/full#supplementary-material
1. Magro C, Mulvey JJ, Laurence J, Seshan S, Crowson AN, Dannenberg AJ, et al. Docked SARS CoV-2 Proteins Within the Cutaneous and Subcutaneous Microvasculature and Their Role in the Pathogenesis of Severe COVID-19. Hum Pathol (2020) 106:106–16. doi: 10.1016/j.humpath.2020.10.002
2. Fakhouri F, Zuber J, Frémeaux-Bacchi V, Loirat C. Haemolytic Uraemic Syndrome. Lancet (2017) 390:681–96. doi: 10.1016/S0140-6736(17)30062-4
3. Brodsky RA. Paroxysmal Nocturnal Hemoglobinuria. Blood (2014) 124:2804–11. doi: 10.1182/blood-2014-02-522128
4. Roumenina LT, Loirat C, Dragon-Durey M-A, Halbwachs-Mecarelli L, Sautes-Fridman C, Fremeaux-Bacchi V. Alternative Complement Pathway Assessment in Patients With Atypical HUS. J Immunol Methods (2011) 365:8–26. doi: 10.1016/j.jim.2010.12.020
5. Heinen S, Józsi M, Hartmann A, Noris M, Remuzzi G, Skerka C, et al. Hemolytic Uremic Syndrome: A Factor H Mutation (E1172Stop) Causes Defective Complement Control at the Surface of Endothelial Cells. J Am Soc Nephrol (2007) 18:506–14. doi: 10.1681/ASN.2006091069
6. Dragon-Durey M-A. Heterozygous and Homozygous Factor H Deficiencies Associated With Hemolytic Uremic Syndrome or Membranoproliferative Glomerulonephritis: Report and Genetic Analysis of 16 Cases. J Am Soc Nephrol (2004) 15:787–95. doi: 10.1097/01.ASN.0000115702.28859.A7
7. Lehtinen MJ, Rops AL, Isenman DE, van der Vlag J, Jokiranta TS. Mutations of Factor H Impair Regulation of Surface-Bound C3b by Three Mechanisms in Atypical Hemolytic Uremic Syndrome. J Biol Chem (2009) 284:15650–8. doi: 10.1074/jbc.M900814200
8. Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, et al. Mutations in Factor H Reduce Binding Affinity to C3b and Heparin and Surface Attachment to Endothelial Cells in Hemolytic Uremic Syndrome. J Clin Invest (2003) 111:1181–90. doi: 10.1172/JCI16651
9. Kavanagh D, Richards A, Noris M, Hauhart R, Liszewski MK, Karpman D, et al. Characterization of Mutations in Complement Factor I (CFI) Associated With Hemolytic Uremic Syndrome. Mol Immunol (2008) 45:95–105. doi: 10.1016/j.molimm.2007.05.004
10. Bienaime F, Dragon-Durey M-A, Regnier CH, Nilsson SC, Kwan WH, Blouin J, et al. Mutations in Components of Complement Influence the Outcome of Factor I-Associated Atypical Hemolytic Uremic Syndrome. Kidney Int (2010) 77:339–49. doi: 10.1038/ki.2009.472
11. Fremeaux-Bacchi V, Moulton EA, Kavanagh D, Dragon-Durey M-A, Blouin J, Caudy A, et al. Genetic and Functional Analyses of Membrane Cofactor Protein (CD46) Mutations in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol (2006) 17:2017–25. doi: 10.1681/ASN.2005101051
12. Dragon-Durey M-A, Loirat C, Cloarec S, Macher M-A, Blouin J, Nivet H, et al. Anti–Factor H Autoantibodies Associated With Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol (2005) 16:555–63. doi: 10.1681/ASN.2004050380
13. Blanc C, Togarsimalemath SK, Chauvet S, Le Quintrec M, Moulin B, Buchler M, et al. Anti–Factor H Autoantibodies in C3 Glomerulopathies and in Atypical Hemolytic Uremic Syndrome: One Target, Two Diseases. J Immunol (2015) 194:5129–38. doi: 10.4049/jimmunol.1402770
14. Marinozzi MC, Roumenina LT, Chauvet S, Hertig A, Bertrand D, Olagne J, et al. Anti-Factor B and Anti-C3b Autoantibodies in C3 Glomerulopathy and Ig-Associated Membranoproliferative Gn. J Am Soc Nephrol (2017) 28:1603–13. doi: 10.1681/ASN.2016030343
15. Iatropoulos P, Noris M, Mele C, Piras R, Valoti E, Bresin E, et al. Complement Gene Variants Determine the Risk of Immunoglobulin-Associated MPGN and C3 Glomerulopathy and Predict Long-Term Renal Outcome. Mol Immunol (2016) 71:131–42. doi: 10.1016/j.molimm.2016.01.010
16. Sansonno D, Gesualdo L, Manno C, Schena FP, Dammacco F. Hepatitis C Virus-Related Proteins in Kidney Tissue From Hepatitis C Virus-Infected Patients With Cryoglobulinemic Membranoproliferative Glomerulonephritis. Hepatology (1997) 25:1237–44. doi: 10.1002/hep.510250529
17. Stegall MD, Chedid MF, Cornell LD. The Role of Complement in Antibody-Mediated Rejection in Kidney Transplantation. Nat Rev Nephrol (2012) 8:670–8. doi: 10.1038/nrneph.2012.212
18. Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic Complement Activation in Collagen Antibody- Induced Arthritis in Mice Requires Amplification by the Alternative Pathway. J Immunol (2007) 179:4101–9. doi: 10.4049/jimmunol.179.6.4101
19. Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, et al. Alternative Complement Pathway Activation Is Essential for Inflammation and Joint Destruction in the Passive Transfer Model of Collagen-Induced Arthritis. J Immunol (2006) 177:1904–12. doi: 10.4049/jimmunol.177.3.1904
20. Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, et al. Modulation of Renal Disease in MRL/ Lpr Mice Genetically Deficient in the Alternative Complement Pathway Factor B. J Immunol (2000) 164:786–94. doi: 10.4049/jimmunol.164.2.786
21. Elliott MK, Jarmi T, Ruiz P, Xu Y, Holers VM, Gilkeson GS. Effects of Complement Factor D Deficiency on the Renal Disease of MRL/lpr Mice. Kidney Int (2004) 65:129–38. doi: 10.1111/j.1523-1755.2004.00371.x
22. Sekine H, Kinser TTH, Qiao F, Martinez E, Paulling E, Ruiz P, et al. The Benefit of Targeted and Selective Inhibition of the Alternative Complement Pathway for Modulating Autoimmunity and Renal Disease in MRL/lpr Mice. Arthritis Rheum (2011) 63:1076–85. doi: 10.1002/art.30222
23. Nauta AJ, Trouw LA, Daha MR, Tijsma O, Nieuwland R, Schwaeble WJ, et al. Direct Binding of C1q to Apoptotic Cells and Cell Blebs Induces Complement Activation. Eur J Immunol (2002) 32:1726. doi: 10.1002/1521-4141(200206)32:6<1726::AID-IMMU1726>3.0.CO;2-R
24. Sjöberg AP, Manderson GA, Mörgelin M, Day AJ, Heinegård D, Blom AM. Short Leucine-Rich Glycoproteins of the Extracellular Matrix Display Diverse Patterns of Complement Interaction and Activation. Mol Immunol (2009) 46:830–9. doi: 10.1016/j.molimm.2008.09.018
25. Russell MW, Mansa B. Complement-Fixing Properties of Human IgA Antibodies Alternative Pathway Complement Activation by Plastic-Bound, But Not Specific Antigen-Bound, IgA. Scand J Immunol (1989) 30:175–83. doi: 10.1111/j.1365-3083.1989.tb01199.x
26. Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA Activates the Complement System Via the Mannan-Binding Lectin Pathway. J Immunol (2001) 167:2861–8. doi: 10.4049/jimmunol.167.5.2861
27. Maillard N, Boerma LJ, Hall S, Huang ZQ, Mrug M, Moldoveanu Z, et al. L’analyse Protéomique De Complexes Immuns Artificiels IgA1-IgG Révèle Une Association Avec Des Formes Activées De C3. Néphrologie Thérapeutique (2014) 10:279. doi: 10.1016/j.nephro.2014.07.347
28. Schreiber A, Rousselle A, Becker JU, von Mässenhausen A, Linkermann A, Kettritz R. Necroptosis Controls NET Generation and Mediates Complement Activation, Endothelial Damage, and Autoimmune Vasculitis. Proc Natl Acad Sci (2017) 114:E9618–25. doi: 10.1073/pnas.1708247114
29. Roumenina LT, Chadebech P, Bodivit G, Vieira-Martins P, Grunenwald A, Boudhabhay I, et al. Complement Activation in Sickle Cell Disease: Dependence on Cell Density, Hemolysis and Modulation by Hydroxyurea Therapy. Am J Hematol (2020) 95:456–64. doi: 10.1002/ajh.25742
30. Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, et al. Complement Activation by Heme as a Secondary Hit for Atypical Hemolytic Uremic Syndrome. Blood (2013) 122:282–92. doi: 10.1182/blood-2013-03-489245
31. Boudhabhay I, Poillerat V, Grunenwald A, Torset C, Leon J, Daugan MV, et al. Complement Activation is a Crucial Driver of Acute Kidney Injury in Rhabdomyolysis. Kidney Int (2020) 99(3):581–97. doi: 10.1016/j.kint.2020.09.033
32. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A Novel C5a Receptor-Tissue Factor Cross-Talk in Neutrophils Links Innate Immunity to Coagulation Pathways. J Immunol (2006) 177:4794–802. doi: 10.4049/jimmunol.177.7.4794
33. Sansonno D, Dammacco F. Hepatitis C Virus, Cryoglobulinaemia, and Vasculitis: Immune Complex Relations. Lancet Infect Dis (2005) 5:227–36. doi: 10.1016/S1473-3099(05)70053-0
34. Loupy A, Lefaucheur C. Antibody-Mediated Rejection of Solid-Organ Allografts. N Engl J Med (2018) 379:1150–60. doi: 10.1056/NEJMra1802677
35. Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, et al. Drusen Complement Components C3a and C5a Promote Choroidal Neovascularization. Proc Natl Acad Sci (2006) 103:2328–33. doi: 10.1073/pnas.0408835103
36. Botto M, Dell’ Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q Deficiency Causes Glomerulonephritis Associated With Multiple Apoptotic Bodies. Nat Genet (1998) 19:56–9. doi: 10.1038/ng0598-56
37. Dick J, Gan P-Y, Ford SL, Odobasic D, Alikhan MA, Loosen SH, et al. C5a Receptor 1 Promotes Autoimmunity, Neutrophil Dysfunction and Injury in Experimental Anti-Myeloperoxidase Glomerulonephritis. Kidney Int (2018) 93:615–25. doi: 10.1016/j.kint.2017.09.018
38. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a Receptor Mediates Neutrophil Activation and ANCA-Induced Glomerulonephritis. J Am Soc Nephrol (2009) 20:289–98. doi: 10.1681/ASN.2008050497
39. Camous L, Roumenina L, Bigot S, Brachemi S, Frémeaux-Bacchi V, Lesavre P, et al. Complement Alternative Pathway Acts as a Positive Feedback Amplification of Neutrophil Activation. Blood (2011) 117:1340–9. doi: 10.1182/blood-2010-05-283564
40. Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, et al. C5a-Induced Expression of P-Selectin in Endothelial Cells. J Clin Invest (1994) 94:1147–55. doi: 10.1172/JCI117430
41. Jahn B, Von Kempis J, Krämer KL, Filsinger S, Hänsch GM. Interaction of the Terminal Complement Components C5b-9 With Synovial Fibroblasts: Binding to the Membrane Surface Leads to Increased Levels in Collagenase-Specific mRNA. Immunology (1993) 78:329–34.
42. Wan J-X, Fukuda N, Endo M, Tahira Y, Yao E-H, Matsuda H, et al. Complement 3 is Involved in Changing the Phenotype of Human Glomerular Mesangial Cells. J Cell Physiol (2007) 213:495–501. doi: 10.1002/jcp.21129
43. Ma Y, Kong L-R, Ge Q, Lu Y-Y, Hong M-N, Zhang Y, et al. Complement 5a-Mediated Trophoblasts Dysfunction is Involved in the Development of Pre-Eclampsia. J Cell Mol Med (2017) 22(2):1034–46. doi: 10.1111/jcmm.13466
44. Ekdahl KN, Persson B, Mohlin C, Sandholm K, Skattum L, Nilsson B. Interpretation of Serological Complement Biomarkers in Disease. Front Immunol (2018) 9:2237. doi: 10.3389/fimmu.2018.02237
45. Elhadad S, Chapin J, Copertino D, Van Besien K, Ahamed J, Laurence J. MASP2 Levels Are Elevated in Thrombotic Microangiopathies: Association With Microvascular Endothelial Cell Injury and Suppression by Anti-MASP2 Antibody Narsoplimab. Clin Exp Immunol (2021) 203:96–104. doi: 10.1111/cei.13497
46. Mollnes TE, Lea T, Froland SS, Harboe M. Quantification of the Terminal Complement Complex in Human Plasma by an Enzyme-Linked Immunosorbent Assay Based on Monoclonal Antibodies Against a Neoantigen of the Complex. Scand J Immunol (1985) 22:197–202. doi: 10.1111/j.1365-3083.1985.tb01871.x
47. Mayer MM. On the Destruction of Erythrocytes and Other Cells by Antibody and Complement. Cancer Res (1961) 21:1262–9.
48. Platts-Mills TA, Ishizaka K. Activation of the Alternate Pathway of Human Complements by Rabbit Cells. J Immunol Baltim Md 1950 (1974) 113:348–58.
49. Yamamoto S, Kubotsu K, Kida M, Kondo K, Matsuura S, Uchiyama S, et al. Automated Homogeneous Liposome-Based Assay System for Total Complement Activity. Clin Chem (1995) 41:586–90. doi: 10.1093/clinchem/41.4.586
50. Seelen MA, Roos A, Wieslander J, Mollnes TE, Sjöholm AG, Wurzner R, et al. Functional Analysis of the Classical, Alternative, and MBL Pathways of the Complement System: Standardization and Validation of a Simple ELISA. J Immunol Methods (2005) 296:187–98. doi: 10.1016/j.jim.2004.11.016
51. Sanchezcorral P. Functional Analysis in Serum From Atypical Hemolytic Uremic Syndrome Patients Reveals Impaired Protection of Host Cells Associated With Mutations in Factor H. Mol Immunol (2004) 41:81–4. doi: 10.1016/j.molimm.2004.01.003
52. Daha MR, Fearon DT, Austen KF. C3 Nephritic Factor (C3NeF): Stabilization of Fluid Phase and Cell-Bound Alternative Pathway Convertase. J Immunol Baltim Md 1950 (1976) 116:1–7.
53. Mori H, Cardiff RD. “Methods of Immunohistochemistry and Immunofluorescence: Converting Invisible to Visible.,”. In: Ursini-Siegel J, Beauchemin N, editors. The Tumor Microenvironment. New York, NY: Springer (2016). p. 1–12. doi: 10.1007/978-1-4939-3801-8_1
54. Magro CM, Momtahen S, Mulvey JJ, Yassin AH, Kaplan RB, Laurence JC. Role of the Skin Biopsy in the Diagnosis of Atypical Hemolytic Uremic Syndrome. Am J Dermatopathol (2015) 37:349–56. doi: 10.1097/DAD.0000000000000234
55. Sturtzel C. “Endothelial Cells”. In: Sattler S, Kennedy-Lydon T, editors. The Immunology of Cardiovascular Homeostasis and Pathology. Cham: Springer International Publishing (2017). p. 71–91. doi: 10.1007/978-3-319-57613-8_4
56. Aird WC. Endothelial Cell Heterogeneity. Cold Spring Harb Perspect Med (2012) 2:a006429–a006429. doi: 10.1101/cshperspect.a006429
57. Dumas SJ, Meta E, Borri M, Luo Y, Li X, Rabelink TJ, et al. Phenotypic Diversity and Metabolic Specialization of Renal Endothelial Cells. Nat Rev Nephrol (2021) 17:441–64. doi: 10.1038/s41581-021-00411-9
58. Chi J-T, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, et al. Endothelial Cell Diversity Revealed by Global Expression Profiling. Proc Natl Acad Sci (2003) 100:10623–8. doi: 10.1073/pnas.1434429100
59. Jambusaria A, Hong Z, Zhang L, Srivastava S, Jana A, Toth PT, et al. Endothelial Heterogeneity Across Distinct Vascular Beds During Homeostasis and Inflammation. eLife (2020) 9:e51413. doi: 10.7554/eLife.51413
60. Roumenina LT, Rayes J, Frimat M, Fremeaux-Bacchi V. Endothelial Cells: Source, Barrier, and Target of Defensive Mediators. Immunol Rev (2016) 274:307–29. doi: 10.1111/imr.12479
61. Sartain SE, Turner NA, Moake JL. Brain Microvascular Endothelial Cells Exhibit Lower Activation of the Alternative Complement Pathway Than Glomerular Microvascular Endothelial Cells. J Biol Chem (2018) 293:7195–208. doi: 10.1074/jbc.RA118.002639
62. Sartain SE, Turner NA, Moake JL. TNF Regulates Essential Alternative Complement Pathway Components and Impairs Activation of Protein C in Human Glomerular Endothelial Cells. J Immunol (2016) 196:832–45. doi: 10.4049/jimmunol.1500960
63. Kawakami Y, Watanabe Y, Yamaguchi M, Sakaguchi H, Kono I, Ueki A. TNF-α Stimulates the Biosynthesis of Complement C3 and Factor B by Human Umbilical Cord Vein Endothelial Cells. Cancer Lett (1997) 116:21–6. doi: 10.1016/S0304-3835(97)04737-X
64. Brooimans RA, van der Ark AA, Buurman WA, van Es LA, Daha MR. Differential Regulation of Complement Factor H and C3 Production in Human Umbilical Vein Endothelial Cells by IFN-Gamma and IL-1. J Immunol Baltim Md 1950 (1990) 144:3835–40.
65. Lappin DF, Guc D, Hill A, McShane T, Whaley K. Effect of Interferon-γ on Complement Gene Expression in Different Cell Types. Biochem J (1992) 281:437–42. doi: 10.1042/bj2810437
66. May O, Merle NS, Grunenwald A, Gnemmi V, Leon J, Payet C, et al. Heme Drives Susceptibility of Glomerular Endothelium to Complement Overactivation Due to Inefficient Upregulation of Heme Oxygenase-1. Front Immunol (2018) 9:3008. doi: 10.3389/fimmu.2018.03008
67. Ades EW, Candal FJ, Swerlick RA, George VG, Susan S, DC B, et al. HMEC-1: Establishment of an Immortalized Human Microvascular Endothelial Cell Line. J Invest Dermatol (1992) 99:683–90. doi: 10.1111/1523-1747.ep12613748
68. Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, et al. Conditionally Immortalized Human Glomerular Endothelial Cells Expressing Fenestrations in Response to VEGF. Kidney Int (2006) 69:1633–40. doi: 10.1038/sj.ki.5000277
69. Edgell CJ, McDonald CC, Graham JB. Permanent Cell Line Expressing Human Factor VIII-Related Antigen Established by Hybridization. Proc Natl Acad Sci (1983) 80:3734–7. doi: 10.1073/pnas.80.12.3734
70. Gavriilaki E, Yuan X, Ye Z, Ambinder AJ, Shanbhag SP, Streiff MB, et al. Modified Ham Test for Atypical Hemolytic Uremic Syndrome. Blood (2015) 125:3637–46. doi: 10.1182/blood-2015-02-629683
71. Réti M, Farkas P, Csuka D, Rázsó K, Schlammadinger Á, Udvardy ML, et al. Complement Activation in Thrombotic Thrombocytopenic Purpura. J Thromb Haemost JTH (2012) 10:791–8. doi: 10.1111/j.1538-7836.2012.04674.x
72. Bettoni S, Galbusera M, Gastoldi S, Donadelli R, Tentori C, Spartà G, et al. Interaction Between Multimeric Von Willebrand Factor and Complement: A Fresh Look to the Pathophysiology of Microvascular Thrombosis. J Immunol (2017) 199:1021–40. doi: 10.4049/jimmunol.1601121
73. Mitra D, Jaffe EA, Weksler B, Hajjar KA, Soderland C, Laurence J. Thrombotic Thrombocytopenic Purpura and Sporadic Hemolytic-Uremic Syndrome Plasmas Induce Apoptosis in Restricted Lineages of Human Microvascular Endothelial Cells. Blood (1997) 89:1224–34. doi: 10.1182/blood.V89.4.1224
74. Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of Human Endothelial Cells Derived From Umbilical Veins. Identification By Morphologic And Immunologic Criteria. J Clin Invest (1973) 52:2745–56. doi: 10.1172/JCI107470
75. Mcginn S, Poronnik P, Gallery ED, Pollock CA. A Method for the Isolation of Glomerular and Tubulointerstitial Endothelial Cells and a Comparison of Characteristics With the Human Umbilical Vein Endothelial Cell Model. Nephrology (2004) 9:229–37. doi: 10.1111/j.1440-1797.2004.00254.x
76. van Setten PA, van Hinsbergh VWM, van der Velden TJAN, van de Kar NCAJ, Vermeer M, Mahan JD, et al. Effects of Tnfα on Verocytotoxin Cytotoxicity in Purified Human Glomerular Microvascular Endothelial Cells. Kidney Int (1997) 51:1245–56. doi: 10.1038/ki.1997.170
77. Martin-Ramirez J, Hofman M, van den Biggelaar M, Hebbel RP, Voorberg J. Establishment of Outgrowth Endothelial Cells From Peripheral Blood. Nat Protoc (2012) 7:1709–15. doi: 10.1038/nprot.2012.093
78. Roumenina LT, Jablonski M, Hue C, Blouin J, Dimitrov JD, Dragon-Durey M-A, et al. Hyperfunctional C3 Convertase Leads to Complement Deposition on Endothelial Cells and Contributes to Atypical Hemolytic Uremic Syndrome. Blood (2009) 114:2837–45. doi: 10.1182/blood-2009-01-197640
79. Roumenina LT, Frimat M, Miller EC, Provot F, Dragon-Durey M-A, Bordereau P, et al. A Prevalent C3 Mutation in aHUS Patients Causes a Direct C3 Convertase Gain of Function. Blood (2012) 119:4182–91. doi: 10.1182/blood-2011-10-383281
80. Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, et al. Complement Factor B Mutations in Atypical Hemolytic Uremic Syndrome—Disease-Relevant or Benign? J Am Soc Nephrol (2014) 25:2053–65. doi: 10.1681/ASN.2013070796
81. Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira-Martins P, Hue C, et al. Mapping Interactions Between Complement C3 and Regulators Using Mutations in Atypical Hemolytic Uremic Syndrome. Blood (2015) 125:2359–69. doi: 10.1182/blood-2014-10-609073
82. Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, et al. Dynamics of Complement Activation in aHUS and How to Monitor Eculizumab Therapy. Blood (2014) 124:1715–26. doi: 10.1182/blood-2014-02-558296
83. Valoti E, Alberti M, Tortajada A, Garcia-Fernandez J, Gastoldi S, Besso L, et al. A Novel Atypical Hemolytic Uremic Syndrome–Associated Hybrid CFHR1/CFH Gene Encoding a Fusion Protein That Antagonizes Factor H–Dependent Complement Regulation. J Am Soc Nephrol (2015) 26:209–19. doi: 10.1681/ASN.2013121339
84. Rigothier C, Delmas Y, Roumenina LT, Contin-Bordes C, Lepreux S, Bridoux F, et al. Distal Angiopathy and Atypical Hemolytic Uremic Syndrome: Clinical and Functional Properties of an Anti–Factor H Igaλ Antibody. Am J Kidney Dis (2015) 66:331–6. doi: 10.1053/j.ajkd.2015.03.039
85. Galbusera M, Noris M, Gastoldi S, Bresin E, Mele C, Breno M, et al. An Ex Vivo Test of Complement Activation on Endothelium for Individualized Eculizumab Therapy in Hemolytic Uremic Syndrome. Am J Kidney Dis (2019) 74:56–72. doi: 10.1053/j.ajkd.2018.11.012
86. Madden I, Roumenina LT, Langlois-Meurinne H, Guichoux J, Llanas B, Frémeaux-Bacchi V, et al. Hemolytic Uremic Syndrome Associated With Bordetella Pertussis Infection in a 2-Month-Old Infant Carrying a Pathogenic Variant in Complement Factor H. Pediatr Nephrol (2019) 34:533–7. doi: 10.1007/s00467-018-4174-1
87. Aiello S, Gastoldi S, Galbusera M, Ruggenenti PL, Portalupi V, Rota S, et al. C5a and C5aR1 are Key Drivers of Microvascular Platelet Aggregation in Clinical Entities Spanning From aHUS to COVID-19. Blood Adv (2021) 6(3):866–81. doi: 10.1182/bloodadvances.2021005246
88. Piras R, Iatropoulos P, Bresin E, Todeschini M, Gastoldi S, Valoti E, et al. Molecular Studies and an Ex Vivo Complement Assay on Endothelium Highlight the Genetic Complexity of Atypical Hemolytic Uremic Syndrome: The Case of a Pedigree With a Null CD46 Variant. Front Med (2020) 7:579418. doi: 10.3389/fmed.2020.579418
89. Timmermans SAMEG, Abdul-Hamid MA, Potjewijd J, Theunissen ROMFIH, Damoiseaux JGMC, Reutelingsperger CP, et al. C5b9 Formation on Endothelial Cells Reflects Complement Defects Among Patients With Renal Thrombotic Microangiopathy and Severe Hypertension. J Am Soc Nephrol (2018) 29:2234–43. doi: 10.1681/ASN.2018020184
90. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, Garcia-Herrera A, Rosiñol L, Castro P, et al. Complement as the Enabler of Carfilzomib-Induced Thrombotic Microangiopathy. Br J Haematol (2020) 193(1):181–7. doi: 10.1111/bjh.16796
91. Palomo M, Blasco M, Molina P, Lozano M, Praga M, Torramade-Moix S, et al. Complement Activation and Thrombotic Microangiopathies. Clin J Am Soc Nephrol (2019) 14:1719–32. doi: 10.2215/CJN.05830519
92. Chauvet S, Roumenina LT, Bruneau S, Marinozzi MC, Rybkine T, Schramm EC, et al. A Familial C3GN Secondary to Defective C3 Regulation by Complement Receptor 1 and Complement Factor H. J Am Soc Nephrol (2016) 27:1665–77. doi: 10.1681/ASN.2015040348
93. Vasilev VV, Noe R, Dragon-Durey M-A, Chauvet S, Lazarov VJ, Deliyska BP, et al. Functional Characterization of Autoantibodies Against Complement Component C3 in Patients With Lupus Nephritis. J Biol Chem (2015) 290:25343–55. doi: 10.1074/jbc.M115.647008
94. Radanova M, Mihaylova G, Ivanova D, Daugan M, Lazarov V, Roumenina L, et al. Clinical and Functional Consequences of Anti-Properdin Autoantibodies in Patients With Lupus Nephritis. Clin Exp Immunol (2020) 201(2):135–44. doi: 10.1111/cei.13443
95. Chaturvedi S, Braunstein EM, Yuan X, Yu J, Alexander A, Chen H, et al. Complement Activity and Complement Regulatory Gene Mutations are Associated With Thrombosis in APS and CAPS. Blood (2020) 135:239–51. doi: 10.1182/blood.2019003863
96. Timmermans S, Damoiseaux J, Reutelingsperger C, van Paassen P. More About Complement in the Antiphospholipid Syndrome. Blood (2020) 136(12):1456–9. doi: 10.1182/blood.2020005171
97. Chen JY, Galwankar NS, Emch HN, Menon SS, Cortes C, Thurman JM, et al. Properdin Is a Key Player in Lysis of Red Blood Cells and Complement Activation on Endothelial Cells in Hemolytic Anemias Caused by Complement Dysregulation. Front Immunol (2020) 11:1460. doi: 10.3389/fimmu.2020.01460
98. Cid J, Fernández J, Palomo M, Blasco M, Bailó N, Diaz-Ricart M, et al. Hyperhemolytic Transfusion Reaction in Non-Hemoglobinopathy Patients and Terminal Complement Pathway Activation: Case Series and Review of the Literature. Transfus Med Rev (2020) 34(3):172–7. doi: 10.1016/j.tmrv.2020.06.002
99. Merle NS, Paule R, Leon J, Daugan M, Robe-Rybkine T, Poillerat V, et al. P-Selectin Drives Complement Attack on Endothelium During Intravascular Hemolysis in TLR-4/Heme-Dependent Manner. Proc Natl Acad Sci (2019) 116:6280–5. doi: 10.1073/pnas.1814797116
100. Mollnes TE, Garred P, Bergseth G. Effect of Time, Temperature and Anticoagulants on In Vitro Complement Activation: Consequences for Collection and Preservation of Samples to be Examined for Complement Activation. Clin Exp Immunol (1988) 73:484–8.
101. Merle NS, Grunenwald A, Rajaratnam H, Gnemmi V, Frimat M, Figueres M-L, et al. Intravascular Hemolysis Activates Complement via Cell-Free Heme and Heme-Loaded Microvesicles. JCI Insight (2018) 3:e96910. doi: 10.1172/jci.insight.96910
102. del Conde I, Crúz MA, Zhang H, López JA, Afshar-Kharghan V. Platelet Activation Leads to Activation and Propagation of the Complement System. J Exp Med (2005) 201:871–9. doi: 10.1084/jem.20041497
103. Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, et al. Alternative Pathway Activation of Complement by Shiga Toxin Promotes Exuberant C3a Formation That Triggers Microvascular Thrombosis. J Immunol (2011) 187:172–80. doi: 10.4049/jimmunol.1100491
104. Noone DG, Riedl M, Pluthero FG, Bowman ML, Liszewski MK, Lu L, et al. Von Willebrand Factor Regulates Complement on Endothelial Cells. Kidney Int (2016) 90:123–34. doi: 10.1016/j.kint.2016.03.023
105. Youssef L, Miranda J, Blasco M, Paules C, Crovetto F, Palomo M, et al. Complement and Coagulation Cascades Activation Is the Main Pathophysiological Pathway in Early-Onset Severe Preeclampsia Revealed by Maternal Proteomics. Sci Rep (2021) 11:3048. doi: 10.1038/s41598-021-82733-z
106. Timmermans SAMEG, Wérion A, Damoiseaux JGMC, Morelle J, Reutelingsperger CP, van Paassen P. Diagnostic and Risk Factors for Complement Defects in Hypertensive Emergency and Thrombotic Microangiopathy. Hypertension (2020) 75:422–30. doi: 10.1161/HYPERTENSIONAHA.119.13714
107. Volokhina EB, van de Kar NCAJ, Bergseth G, van der Velden TJAM, Westra D, Wetzels JFM, et al. Sensitive, Reliable and Easy-Performed Laboratory Monitoring of Eculizumab Therapy in Atypical Hemolytic Uremic Syndrome. Clin Immunol Orlando Fla (2015) 160:237–43. doi: 10.1016/j.clim.2015.05.018
108. Merrill SA, Brittingham ZD, Yuan X, Moliterno AR, Sperati CJ, Brodsky RA. Eculizumab Cessation in Atypical Hemolytic Uremic Syndrome. Blood (2017) 130:368–72. doi: 10.1182/blood-2017-02-770214
Keywords: complement, endothelial cells, diagnostics, prognostics, therapeutics, kidney injury, nephrology, explorations
Citation: Meuleman M-S, Duval A, Fremeaux-Bacchi V, Roumenina LT and Chauvet S (2022) Ex Vivo Test for Measuring Complement Attack on Endothelial Cells: From Research to Bedside. Front. Immunol. 13:860689. doi: 10.3389/fimmu.2022.860689
Received: 23 January 2022; Accepted: 21 March 2022;
Published: 12 April 2022.
Edited by:
Erik J. M. Toonen, Hycult Biotech, NetherlandsReviewed by:
Jeffrey Laurence, Cornell University, United StatesCopyright © 2022 Meuleman, Duval, Fremeaux-Bacchi, Roumenina and Chauvet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie-Sophie Meuleman, bWFyaWUtc29waGllLm1ldWxlbWFuQGluc2VybS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.