 Caroline Perner
Caroline Perner Elke Krüger
Elke Krüger
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 April 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.859703
This article is part of the Research Topic The Role of ER Stress and the UPR in Shaping Immunity: Volume II View all 7 articles
Neuronal cells are specialists for rapid transfer and translation of information. Their electrical properties relay on a precise regulation of ion levels while their communication via neurotransmitters and neuropeptides depends on a high protein and lipid turnover. The endoplasmic Reticulum (ER) is fundamental to provide these necessary requirements for optimal neuronal function. Accumulation of misfolded proteins in the ER lumen, reactive oxygen species and exogenous stimulants like infections, chemical irritants and mechanical harm can induce ER stress, often followed by an ER stress response to reinstate cellular homeostasis. Imbedded between glial-, endothelial-, stromal-, and immune cells neurons are constantly in communication and influenced by their local environment. In this review, we discuss concepts of tissue homeostasis and innate immunity in the central and peripheral nervous system with a focus on its influence on ER stress, the unfolded protein response, and implications for health and disease.
Cell and tissue homeostasis are the foundation of a physiological and healthy organism. Homeostasis means to balance and properly regulate all required components and interactions by actively maintaining certain quantitative characteristics of the system, known as regulated variables, within a desired range (1). This implies a proper osmoregulation, regulation of ion-levels and membrane potentials, pH regulation, glucose and lipid metabolism, protein synthesis, protein folding and degradation on a cellular level, hormone production, thermoregulation, enzyme secretion, transmitter release and host defense on tissue and organismic level. Multicellular organisms have developed a complex machinery, that allows and requires the interaction of multiple cell types to prevent the body from harm, and heal tissue damage. Inflammatory responses represent such a mechanism often required to prevent disruptions in homeostasis and rebuilt it if interruptions occur.
On an organismic level, innate and adaptive immunity are the main drivers for host defense. The innate immune system is evolutionally older and consists of myeloid cells (macrophages, monocytes, microglia, dendritic cells, neutrophils, basophils, eosinophils, mast cells) and NK-cells as well as the complement system. Pathogens invading the organism get recognized through conserved surface structures that are broadly shared by pathogens (glycans, glycoconjugates, lipopolysaccharides (LPS), endotoxins found on cell membranes from gram negative bacteria) and distinguishable from host molecules and therefore called pathogen-associated molecular patterns (PAMPs) that bind to pattern recognition receptors (PRRs) expressed by innate immune cells such as toll-like receptors (TLRs), C-Type lectin receptors (CLRs), NOD-like receptors (NLR & NLRPs) RIG-I-like receptors (RLRs) (2). This allows the cellular innate immune response to start within minutes, reflected by phagocytosis of the invading pathogens or the release of mediators (granzyme, histamine, bradykinin, serotonin, leukotrienes, prostaglandins), cytokines, and chemokines that further promote the elimination of the pathogen by recruiting other immune cells in or directly harm the pathogen (3, 4). Besides the recognition of pathogens, the immune system also recognizes molecules released due to tissue damage (heat-shock proteins, high mobility group protein B 1 (HMGB1), hyaluronan fragments, ATP, uric acid, heparin sulfate, DNA, RNA), called damage associated molecular patterns (DAMPs). DAMPs are endogenous danger signals and can activate innate immune cells trough PRRs to respond to non-infectious “sterile” tissue damage. Cytosolic nucleic acids are sensed by a number of cytosolic nucleic acid receptors such as cyclic-GMP-AMP-synthase (cGAS) and stimulator of interferon (IFN) genes (STING) for DNA or by RIG-like receptors for RNA. These nucleic receptor in turn activate the phosphorylation of transcription factors like IFN regulatory factor 3 (IRF3) by TANK-binding kinase 1 (TBK1) important for the transcription of type I IFN like IFN-α or -β. These proinflammatory cytokines along with a couple of IFN-stimulated molecules can serve as a physical barrier against the spread of the infection but also promote tissue healing (5). But there is also an increasing body of evidence pointing to PRR activation through mitochondrial DNA that will drive IFN-driven inflammation contributing to disease formation (6).
Moreover, we and others discovered recently that there is another cell type involved in the recognition of harmful invaders like bacteria, fungi, and allergens or toxins. Peripheral sensory neurons have a big impact in the initiation of the local inflammation and contribute to the activation of innate and adaptive immune cells (7–10). By sharing many PRRs with innate immune cells and expressing other G-protein coupled receptors (GPCRs) for the recognition of harmful substances like toxins and allergens, they are functioning as a first sensor of tissue damage. Within seconds, these sensory neurons are alarming the host of a potential harm through perception of pain or itch. This is often followed by a neurological reflex arc. Examples are lifting the hand from a hot plate or scratching to remove insects or parasites on the skin, induction of sickness and the initiation of vomiting to remove toxic food intake before it can do more harm to just list some reflexes where the neuronal system acts to defend the host. But besides the neurological implication of the sensed harm by sensory neurons, they also actively participate in initiating inflammation and regulating the immune response. Upon sensing potential harm they react by the immediate release of certain neuropeptides into the tissue where the damage was sensed, but also, and this is the important difference, into the neighboring tissue that gets innervated by other branches of the same sensory neuron (9). This so-called anticipatory immunity allows for an immediate warning of the neighboring tissue and induces inflammation through the release of neuropeptides like calcitonin gene-related peptide (CGRP), Substance P, vasoaktives intestinales peptid (VIP), Pituitary adenylate cyclase-activating polypeptide (PACAP) and Chemokine-like protein TAFA-4 (TAFA-4) (7, 11–13). The release pattern is depended on the pathogen, toxin and tissue damage. Sensory neurons and their neuropeptides are thereby contributing to all aspects of inflammation as defined by Celsus: rubor, calor, tumor, dolor and change of function. Therefore neuropeptides induce vasodilatation, extravasation and function as chemoattractant for immune cells to migrate into the tissue (14). Neuropeptides can induce either a proinflammatory or anti-inflammatory immune response, and promote tissue healing through their respective receptors expressed by immune cells, endothelial cells, and glia cells (15, 16). Thus, small dysregulations in sensory neuron and immune cell function can influence the immune response required to defend properly from a harmful stimulus and rebuild tissue homeostasis as the immune feedback regulation between pro-inflammatory signals for pathogen eradication and anti-inflammation for tissue healing can easily be disrupted.
We discussed above how inflammatory processes are involved in restoring tissue homeostasis in response to infection or injury. On a cellular level, inflammation can occur without infection or damage, but as a response for a disruption in cellular homeostasis (1, 17, 18) Intracellular dysregulation, such as endoplasmic reticulum (ER), mitochondrial or osmotic stress reaching a level that normal homeostatic regulators cannot control, are known to trigger different response mechanism related to inflammation. ER stress is often caused by an increase in misfolded proteins as well as through dysregulations in ER calcium homeostasis or lipid membrane integrity (Figure 1). These stressors are able to induce the unfolded protein response (UPR) that increases transcription of proteins that help with protein processing such as chaperons, the ER-associated degradation system (ERAD), proteins important for lipid metabolism, energy homeostasis and cell proliferation. If cellular homeostasis cannot be rebuilt by the UPR, it can induce apoptosis (21, 22). The UPR stress sensors inositol-requiring protein 1α (IRE1α), protein kinase RNA-like endoplasmic reticulum (ER) kinase (PERK) and activating transcription factor 6 (ATF6) are ER membrane proteins. They transduce information about the protein folding status to the cytosol and nucleus and induce transcription of UPR target genes via splicing of X-box binding protein 1 (XBP1), phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) that can attenuate general protein synthesis and allows translation of ATF4 mRNA to ATF4, which controls the transcription of genes involved in autophagy, apoptosis, amino acid metabolism and antioxidant responses (22). ATF6 is a leucin zipper transcription factor, which is transported to the Golgi apparatus under ER stress conditions where it gets processed via S2P and S1P to release its cytosolic domain fragment and upregulate the gene expression of ERAD and XBP1 components. Thus, the UPR has a major role in preserving correct protein folding, regulate protein degradation, cell metabolism and control of cell organelles thereby protects from cellular death through autophagy (23). Interestingly, by protein quality control of cytokine production, modulation of cytokine transcription factors and cytokine receptor expression, the UPR is also directly involved in inflammation and immunity (24). Activated via TLRs and PRR, ER stress is reported to increase the expression of Interleukine-23 (IL-23) in myeloid cells in response to the intracellular bacterium Chlamydia trachomatis (25). There is further evidence that ER stress and UPR primes cells to respond to innate immune stimuli by activating the IRF3 transcription factor that synergistically augments early type I IFNs and other genes involved in innate immunity (26–29).
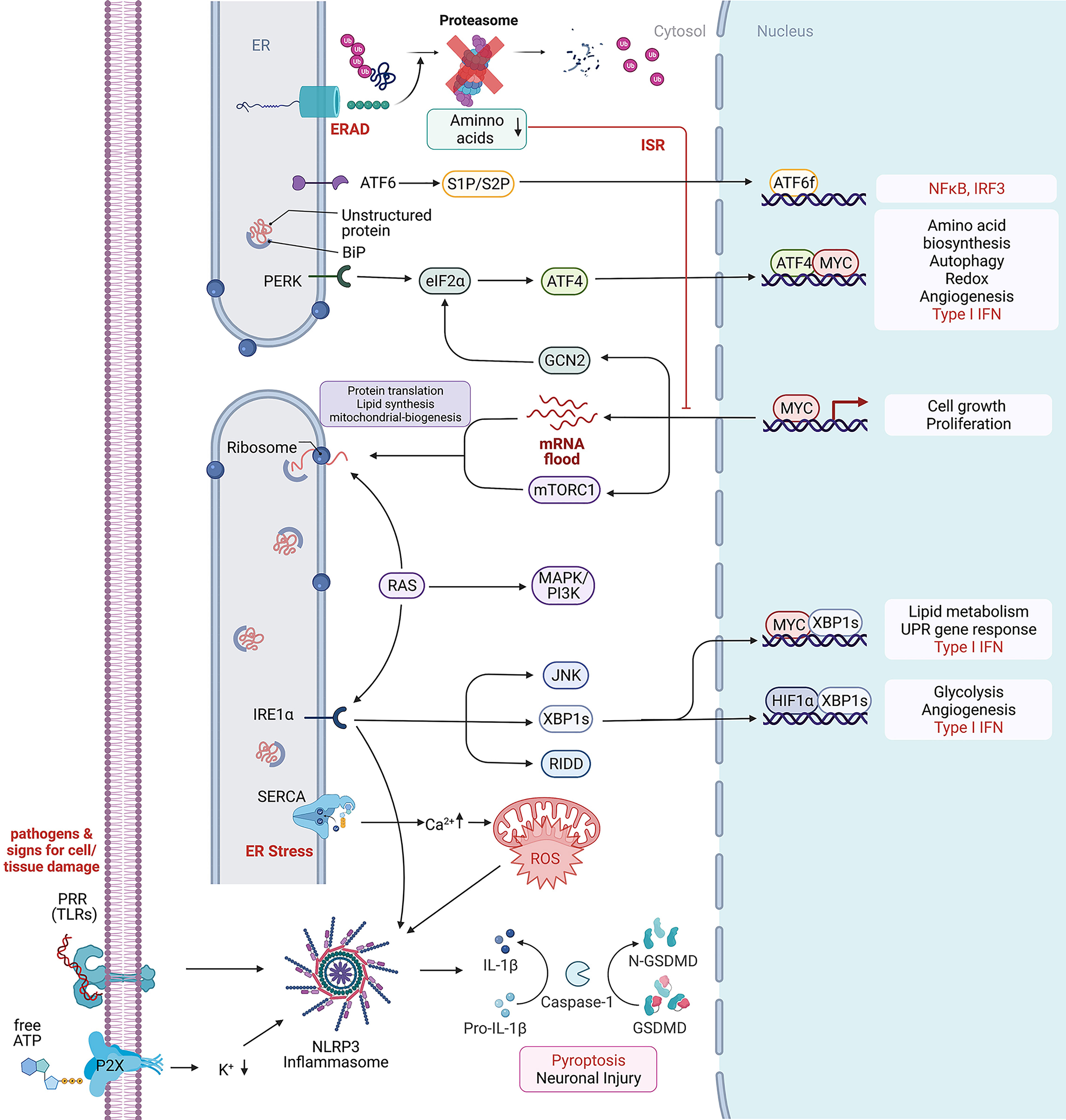
Figure 1 Cellular stress response mechanism. Depicted are intracellular signaling pathways involved in the stress response against extracellular and intracellular stressors. From top to bottom: Endoplasmic-reticulum-associated protein degradation (ERAD) recognizes misfolded proteins of the endoplasmic reticulum (ER) and induces their ubiquitinylation for protein degradation through the proteasome. Low cytosolic amino acid levels can inhibit the proteasomal degradation through the proteasome and further inhibit cell growth and proliferation. Unfolded or misfolded proteins within the ER activate BiP, a major chaperone in the initiation of the unfolded protein response, that further interacts with (ATF6) and the protein kinase RNA-like endoplasmic reticulum kinase (PERK) to induce the UPR, leading to ATF6f and ATF4 dependent transcriptional modification. In rapidly proliferating cells, MYC dependent transcription enriches the cytosol with mRNAs waiting for proper translation on ribosomes. Protein folding mistakes can be recognized by the inositol-requiring enzyme 1 α (IRE1α), that via splicing of X-box binding protein 1 (XBP1) induces transcriptional modification and thereby lipid and glycose metabolism. The RNase domain of IRE1 mediates the cleavage of multiple RNAs in a process known as regulated IRE1-dependent decay (RIDD) (19). c-Jun N-terminal kinase (JNK) activation may be linked to ER stress by IRE1 and autophagy induction after ER stress relies on the IRE1-JNK pathway (20). The ER is highly involved in calcium homeostasis by acting as a calcium storage. Ion-pumps like the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) are actively transporting calcium between the cytosol and ER lumen. High cytosolic calcium concentration can increase the production of reactive oxygen species (ROS) that further are able to promote NLR family pyrin domain containing 3 (NLRP3) inflammasome activation which is usually activated by exogenous pathogens, recognized by their RNA/DNA on pattern recognition receptors (PRR) like Toll-like receptors (TLR) and the involved tissue damage products like ATP that can be recognized by purinergic receptors like P2R. Created with BioRender.com.
Interestingly, there is a big overlap with the diseases associated with ER stress and the UPR (30). Both pathways are important in cellular inflammation and stress response and connected via reactive oxygen species, mitochondrial dysfunction, a disbalance in ion levels, cytokines and Angiotensin II (31). Furthermore, ER stress can independently of the UPR response activate the NLRP3 inflammasome in a Voltage-dependent anion selective channel 1 (VDAC1) dependent manner (32). VDAC1 is located in the outer mitochondrial membrane where it allows ATP to diffuse out of the mitochondria into the cytosol. It is also important for the volume regulation and acts in scaffolding proteins such as hexokinase that are important for metabolic regulation (33). VDAC1 overexpression is therefore known to be associated with Alzheimer´s disease and Parkinson’s disease (34, 35). While inflammasome activation and IL-1β secretion is important in the defense against bacteria, type I IFNs are important for the defense against viral infection sensed through PRRs sensitive to double stranded RNA and DNA. There is increasing evidence for an association of the pathways associated with type I IFNs with neuroinflammation and neurodegenerative diseases just recently reviewed by Fryer A et. al (36). Therefore, the cGAS-STING or RIG-MAVS pathways can also be involved in the initiation of an inflammatory response through the production of type I IFN in the absence of pathogens. Another important cellular response that can be activated through double stranded RNA as well as by IFNs is the integrated stress response (ISR). Similar to the UPR, the ISR is involved in the inhibition of global translation to decrease viral mRNA translation and viral protein assembly, while increasing the translation of selective mRNAs recognized through internal ribosome entry sites (IRESs) or upstream open reading frames (uORFs), such as ATF4. The ISR can be activated through four stress activated kinases, i.e. PERK, protein kinase R (PKR), general control nonderepressible 2 (GCN2), or heme-regulated inhibitor (HRI). Proteotoxic stress by ER stress, misfolded proteins, oxidative stress and proteasome inhibition activate both the ISR and UPR intercrossing via PERK. Viral infection/dsDNA and associated IFNs activate the ISR through PKR. Amino acid deprivation through proteasomal dysregulation, RNA virus infection and UV irradiation are activating the ISR through GCN2, and low heme, heat shock proteins and mitochondrial dysfunction are associated with ISR activation through HRI (37–39). Not surprisingly, there are multiple reports about the ISR to be involved in neurodegeneration as reviewed by Martinez NW et al. (40) and Bond S et al. (37). More recently, proteotoxic stress has been correlated to ER stress and UPR/ISR activation in rare proteasomopathies. This disease spectrum caused by inborn defects of the proteasome includes neuronal manifestation and is linked to dysregulation of type I IFN signaling (41, 42). Mechanistically, PKR serves as an innate immune sensor for proteotoxic stress via cytoplasmic accumulation of IL-24 (43).
UPR and ISR are involved in sensing homeostatic disturbances and are important to restore cellular and tissue homeostasis. As they are intertwined through their stimuli, signaling proteins and through their successful degradation and negative regulation through proteasomal degradation, it is not surprising that they are influencing each other. Disruptions in one of them are likely to induce the other stress response in time and if not regulated properly, they will contribute to cell death rather than restore homeostasis. In this vicious cycle of cell decline due to a dysregulation in cellular stress response, there can be multiple reasons that lead to imbalance, starting a process of a chronic stress response and inflammation. Genetic mutations, metabolic stress due to an unhealthy lifestyle and multiple mechanism associated with aging are known to be highly associated. One rarely discussed cause though, is the accumulation of certain infections during lifetime.
In the last years researchers provided new evidence for a possible causation of viral infection for the development of Alzheimer´s disease (AD) (44–46). In fact, there is evidence that Amyloid beta (Aß) -oligomers bind herpesvirus surface glycoproteins, accelerating ß-amyloid deposition, which is leading to a protective viral entrapment (46). Nevertheless, the accelerated Aß - deposition may induce an increased inflammatory response with a following Aß - pathology and contribute to the continuous and chronic activation of the UPR and inflammasome cellular stress response pathways, initiating the development of neurodegenerative diseases years before the first symptoms occur. Especially now with new RNA-vaccination methods developing for viruses that are known to be difficult to immunize against like severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (47), human immunodeficiency viruses (HIV) (48) and ZIKA virus (49), thinking about vaccinations for viruses likely associated with neurodegenerative diseases like the herpes virus family might be a way to protect from the vicious cycle of UPR, ISR and inflammasome response started through the viral infection. Furthermore, there are more and more clinical trials using inflammasome, ISR and UPR modification as a promising treatment against inflammatory and neurodegenerative diseases (Table 1). In this review, we will further provide an overview about the immune homeostasis (tissue and cellular homeostasis) in central and peripheral neurons, describing the connections to associated neurological diseases and possible treatment strategies.
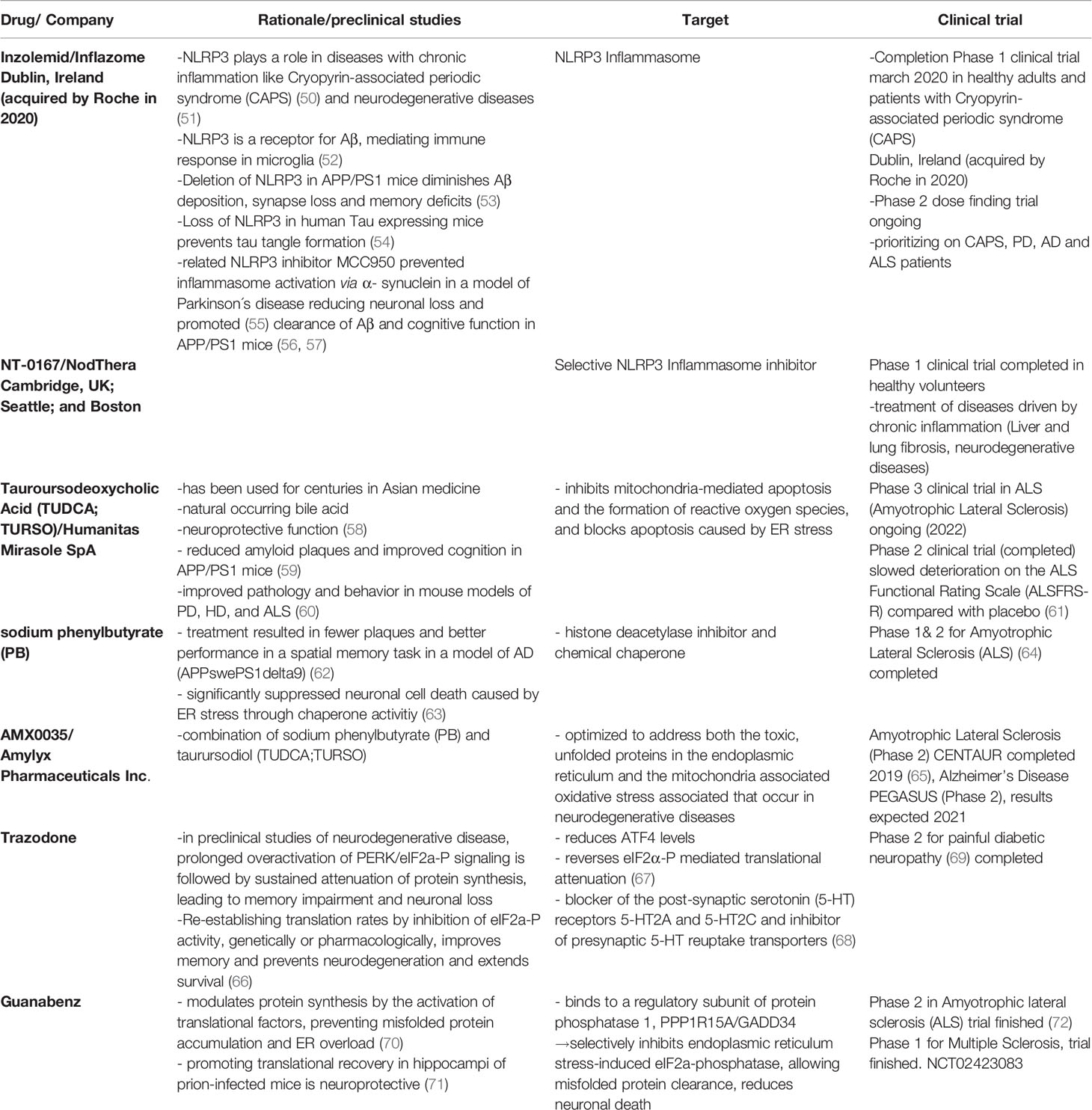
Table 1 Treatments in clinical trials.
The ER is the largest organelle in the cytoplasm of eukaryotic cells. It is composed of a dynamic network of stacked membrane sheets connected with helicoidal motifs (73) with an inner lumen that is connected to the nuclear envelope (Figure 2). The ER that surrounds the nucleus is called rough ER as it contains densely packed ribosomes for protein synthesis on its surface. The proteins are transported from the cytosol into the ER lumen where they get folded and further modified by chaperons and folding enzymes, undergo quality control and get transported to their destination. Proteins that do not achieve a functional conformation within the ER are targeted by the ER quality control and enter the ERAD pathway and thus proteasomal degradation (74). Neurons belong to the biggest cells as their axons can reach over 1m in humans and even more in bigger animals. Therefore, the neuron has to produce a lot of essential proteins. As more protein synthesis is required, as more rough ER is located in the soma and dendrites of the neuron, where it can be seen as Nissl bodies. As the ribosomes are rich in RNA, they can be stained with basic dye. The axon hillock and axon do not contain rough, but smooth ER. The smooth ER with only a few ribosomes consists of connected cylindric tubules and interconnected cisternae. The tubules are involved in lipid biogenesis and glucose metabolism while the cisternae are functioning as calcium storage (75). Phospholipids which are the major lipid component of membranes get biochemically modified and transported together with proteins to an ER region in close juxtaposition to the Golgi apparatus, the ER-Golgi intermediate compartment ERGIC.
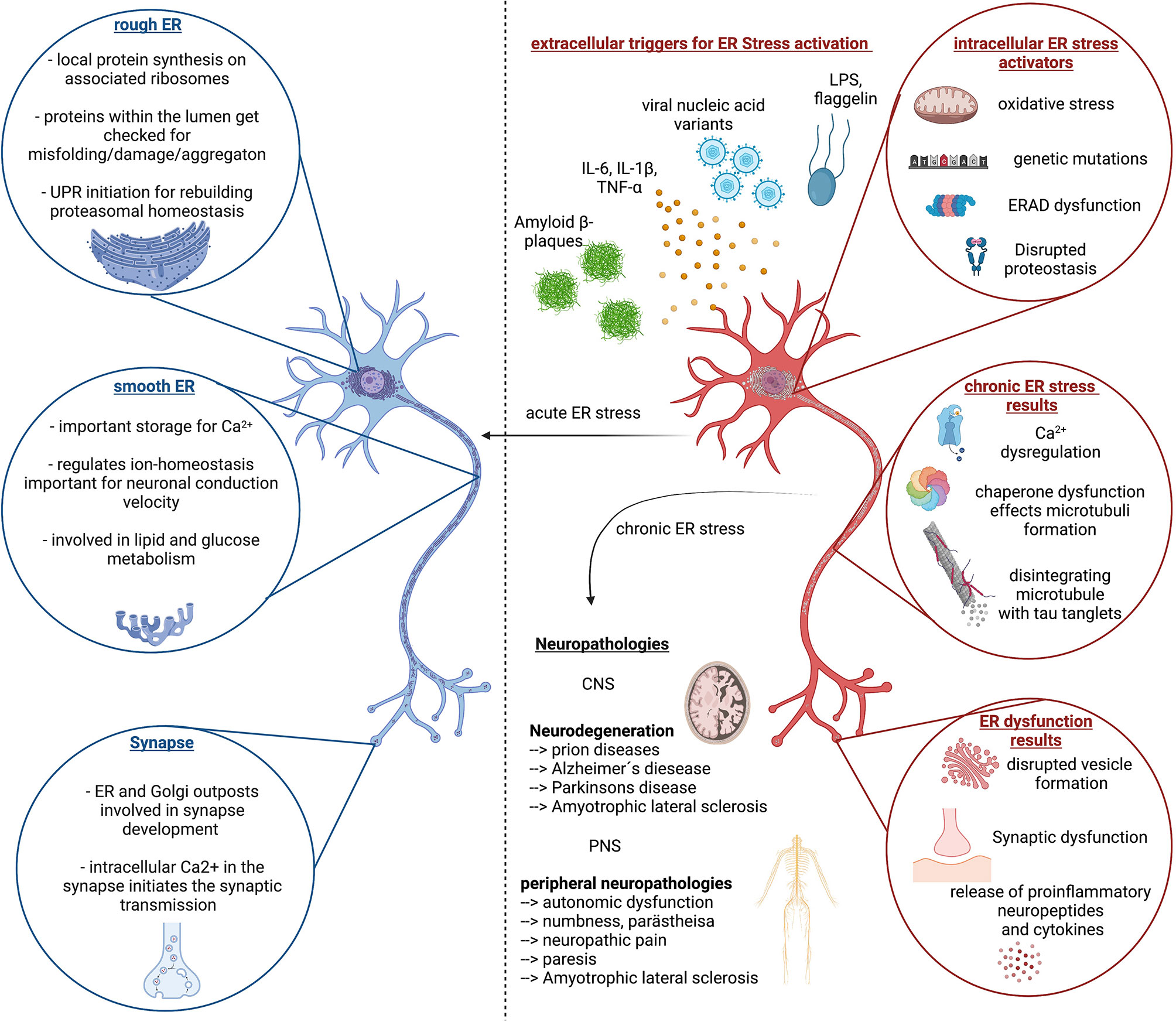
Figure 2 ER stress response in homeostasis and disease. Left (blue color) side depicts important function of the Endoplasmic reticulum (ER) in neuronal cells and highlights the importance of proper ER function for neuronal function, which is reinstated by the unfolded protein response (UPR) and proper regulation of proteasomal degradation and synthesis. The right side (red color) depicts the extra- and intracellular stressors that can lead to chronic ER stress, associated with continues disruption of neuronal cell homeostasis followed by dysfunction and decline. This circulus vicious is a foundation of multiple neurological diseases of the CNS and PNS of which the most common ones are listed here. Created with BioRender.com.
The ER functions as Ca2+ storage, keeping a concentration between 100-800µM. The typical cytosolic Ca2+ concentration is supposed to be around 100nM. In comparison, extracellular space has a Ca2+ concentration of around 2mM. If a cell gets activated through G-protein coupled receptors, phospholipase C (PLC) gets stimulated to cleave phosphatidylinositol 4,5 bisphosphate (PIP2) into diacyl-glycerol (DAG) and IP3, which can then bind to IP3R on the ER membrane leading to Ca2+ release from the ER lumen into the cytosol and transient increase in intracellular Ca2+levels (76). Ca2+ gets pumped back into the ER through the sarcoplasmic reticular calcium ATPases (SERCA) and can additionally enter the cell trough voltage- or ligand gated calcium channels from the extracellular space. The most important function and characteristic of neurons compared to other cell types is the excitability, the transport of information through action potentials as a result of a change in membrane potential. Therefore, it is of utmost importance for the neuron to regulate its ion levels and transfer ions as fast as possible to the right location. In the specific architecture of neurons the ER lumen is connected from the soma, through the whole axon to the synapsis, and thereby allows for fast shifts of ions, but also the transport of required neuropeptides and neurotransmitters for cell communication and signal transduction (77). Signal transmission across synapses can be performed through electric synapses with the transfer of electric signals through gap junctions or chemical synapses where neurotransmitters and neuropeptides get released as messengers. Respective specialized receptors on the post synaptic cell can bind the transmitters and peptides and forward the information through inhibitory or excitatory signals. Reuptake and recycling of neurotransmitters and neuropeptides ensures restorage, enabling the rapid frequency of synaptic transmission if necessary (78).
The ER is therefore a central organelle in neuron physiology. Disruptions of the ER functions are likely to cause dysfunction of neurons through a malfunctioning in protein and lipid biosynthesis for neurotransmitter and neuropeptide vesicles, and a disbalance in calcium levels necessary for the initiation of action potentials and signal transmission through synapses (Figure 2). In sum, ER stress is likely the consequence in different disease mechanism and can contribute positively and negatively to disease pathology for neuropathies and neuronal decline associated with many central and peripheral neurological diseases (79, 80).
The central nervous system (CNS) contains of the brain and the spinal cord. It is surrounded by three layers of meninges, the very thin pia mater that covers the parenchyma and follows supplying arteries, the arachnoid mater, that consist of many spider like columns that build the arachnoid space that is filled with cerebrospinal fluid (CSF) and the outer, very thick and strong dura mater that is connected to the skull and vertebrate bones, builds compartments through the falx cerebri and accommodates the venous sinuses. The meninges offer protection of the CNS parenchyma as does the CSF, that functions as a shock absorber in the arachnoid space and it provides buoyancy, thereby reducing the overall brain weight to protect it from damage. Besides these physical implications for the CNS, the CSF and the meninges are actively contributing to CNS immunity. Other than in the periphery, the CNS is under common, non-inflammatory conditions isolated from blood circulating immune cells and most chemicals through the blood brain barrier (BBB). The BBB is composed of endothelial cells connected through tight junctions, followed by multiple layers of smooth muscle cells and finally large astrocyte (glial) end-feet. Many years there was the prominent concept of the immune-privilege of the brain, pointing to studies, where antigens (pathogens or allografts) implanted only into the brain could not induce a systemic immune response, which was different, if the immune system was pre-primed to the same antigen in the periphery before (81, 82). It was assumed, that this is due to a lack in lymphatics draining the brain from debris and antigens. Indeed, recent studies shed light on the mystery about the lymphatic drainage of the CNS and its impact for CNS immunity and neuronal function which is in detail reviewed by KA Lima, J Rustenhoven and J Kipnis (83). In fact, there are all kinds of innate and adaptive immune cells present in the meninges, especially in locations close to the dural sinuses (Figure 3). Myeloid cells are the dominating cell type and they are recruited directly from the skull bone marrow (84). But multiple studies point especially to a huge impact of cytokines released by lymphocytes in the meninges that can reach the brain parenchyma and directly influence behavior (85, 86). Thereby, meningeal immunity contributes actively through cytokines to CNS homeostasis and neuronal function. Studies with immune compromised mice shows behavioral cognitive deficits which could be rescued through transfer of CD4+ but not CD8+ T cells (87–91). T cells specific to CNS antigens were also shown to contribute to spatial learning, memory and neurogenesis (92). Depletion of CD4+ T cells was therefore shown to reduce hippocampal neurogenesis accompanied by impaired reversal learning in the Morris water maze (93). Indeed, there is evidence that meningeal T cells are supporting learning and memory through the expression of the key cytokine IL-4. Studies revealed a proinflammatory phenotype of meningeal myeloid cells in IL4(-/-) mice that was associated with cognitive impairment and could be reversed through transplantation of wild-type T cells (94, 95). Meningeal γδ T cells expressing high levels of the chemokine receptor CXC chemokine receptor 6 (CXCR6) are seeding meninges shortly after birth. Their release of IL-17a by these cells was shown to be correlated with anxiety-like behavior in mice and was partially dependent on T cell receptor engagement and commensal derived signals. IL-17a receptor is therefore expressed in cortical glutamatergic neurons under steady state and its genetic deletion decreased anxiety-like behavior in mice (85). As cytokines produced by these cells can reach neurons, so can antigens from the brain reach the meninges and their resident immune cells. Therefore, the CSF constantly flows through the brain parenchyma being propelled into the brain via arterial pulsation through aquaporin 4 (AQP4) water channels present in the astrocyte end-feet that form the glial limitans of the BBB. As a result, the CSF flow clears the parenchyma from debris and intestinal solutes along paravenous spaces into the subarachnoid space CSF (96, 97). Moreover, it is shown that the metabolites and proteins (antigens) are traveling through the meninges and via lymphatic vessels into the deep cervical lymph nodes where they might be able to undergo antigen presentation (98). In fact, there is an increasing body of evidence pointing to a contribution of lymphocytes primed to self-antigens in CNS recovery and regeneration after injury and stroke (99–101). In contrast, partial, but not total depletion of Foxp3+ regulatory T cells (Tregs) is neuroprotective in a model of cerebral injury, while boosting Treg function was also shown to be beneficial in a model of stroke (102–104). Examining signatures for T cell activation and residency in aged and young mice revealed no obvious changes in CD4+ T cell phenotypes between young and old dural T cells but revealed IFN-γ production by aged meningeal T cells as a potential source for age related changes in central neuron and glia function (105–107).
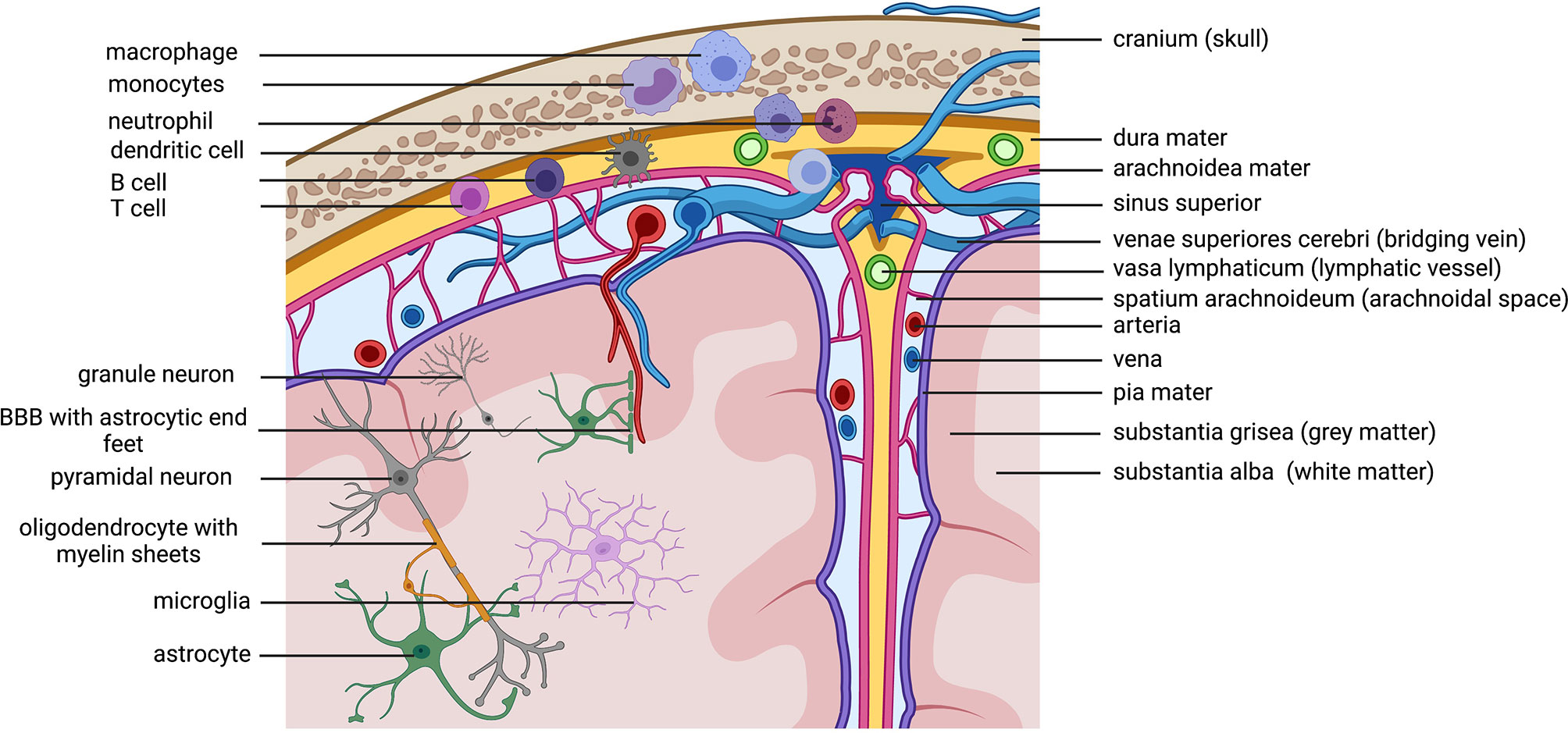
Figure 3 Immune cells patrolling the CNS. Macro anatomical structures important for the brain on the right side The brain is protected by the skull and the meninges. Within the thick dura mater are lymphatic vessels that allow immune cells located within the stroma of the dura mater to drain to the cervical lymphnodes (not shown in the figure). The arachnoidal space is filled with liquor (cerebrospinal fluid= CSF) that flows through the brain and contains and discards cerebral debris. Antigens within the CSF can get presented to patrolling immune cells in the dura mater close to the sinuses. Lymphocytes located in the dura are known to secrete cytokines that can reach the cerebral parenchyma and influence neuronal function. Microanatomical structures /cells on theleft side.Cave! The depiction has no correct scale, the cells are depicted to big. This is only a scheme to annotate involved cell types. Because of the scale the location of cells is not perfectly correct (neuronal cell bodies are always in the grey matter, microglia are also in the grey matter). Important to note is the location of immune cells in the dura mater. Myeloid cells get directly recruited from the bone marrow of the skull. Created with BioRender.com.
Aging research and studies on Alzheimer´s disease also proved an impact on the CSF flow through followed by the lymphatic drainage. Arterial pulsation as well as lymphatic flow are decreasing with age, supporting the cumulation of metabolites and debris in the brain parenchyma (108, 109). Studies could show that decreased lymphatic flow from the meninges to the draining lymph node (LN) is associated with cognitive deficits while aged mice treated with vascular endothelial growth factor (VEGF)-c, a lymphatic endothelial cell mitogen, showed enhanced meningeal lymphatic drainage resulting in improved cognitive function (110). Furthermore, there is new evidence that a reduction in CC chemokine receptor 7 (CCR7) expression by meningeal T cells in old mice is linked to an increase in CD8+ T-betlow effector and CD4+FOXP3+ Treg cells and a reduction in CD4+ T-bethigh IFNγ producing cells. Moreover, a hematopoietic deficiency in CCR7 can induce aging-associated changes in meningeal T cells accompanied by a reduced glymphatic influx and cognitive impairment. Deletion of CCR7 in 5xFAD transgenic mice used as an Alzheimer´s disease model promotes neurovascular and microglial activation and an increase in Aβ deposition in the brain of these mice. Conversely, old mice treated with anti-CD25 antibodies (CD4+FOXP3+Treg cells are mainly CD25high expressing cells and are more responsive to anti-CD25 treatment than CD4+ and CD8+ CD25 low expressing effector T cells) showed a reduced meningeal CD4+FOXP3+ Treg cell response associated with an improved cognitive function (111). It is not shown yet, if the improved cognitive phenotype could also be induced by an associated increase in CD4+ T-bethigh IFN-γ producing cells. Interestingly, these changes due to CCR7 depletion or the reversal through anti-CD25 treatment could only be observed in meningeal T cells and in the respective draining lymph nodes but not in peripheral blood or liver. These studies highlight the therapeutic potential of modulating immune cells in the CNS and meningeal environment to improve brain function in aging and in neurodegenerative diseases.
While meningeal immune cells take part in CNS homeostasis from the outside, microglia function as a CNS resident specialized innate immune cell. Together with glia cells like astrocytes, oligodendrocytes and ependymal cells they all are important to maintain CNS homeostasis from within the BBB. As nicely reviewed by Greenhalgh AD and collogues (112), astrocytes are important for the formation of synapses, tight metabolic coupling with neurons to support their function, rapid neurotransmitter uptake, buffering extracellular potassium, modulation of excitability and plasticity as well as for memory consolidation and generation of circadian rhythm. Microglia, are quite important for neuronal development and learning processes due to synaptic pruning, BDNF production, phagocytosis of cellular and protein debris (e.g. Aβ) and are continuously surveilling the CNS tissue (113). They are highly reactive to changes in the brain through production of signaling molecules that can contribute to homeostasis but also contribute to disease (113). The remaining immune cells besides microglia make about 20% of all immune cells in the brain and consist of neutrophils, monocytes, dendritic cells, innate lymphoid cells and lymphocyte subsets. Under steady state conditions, the CNS environment is mostly anti-inflammatory due to predominant immune-regulatory cytokines like IL-10 and Transforming growth factor beta (TGF-β) (114, 115). This can change rapidly, as CNS immune cells, and astrocytes express PRRs to detect PAMPs and DAMPs recognizing infiltrating pathogens (116, 117) but also misfolded proteins like Aβ (52) followed by a pro-inflammatory cytokine release of IL-1β due to inflammasome activation through NLRs, that is followed by secondary secretion of cytokines like IL-6 and tumor necrosis factor (TNF)-β while IL-18 induces production of IL-17 (118, 119). This group of cellular sensors, belonging to the Nucleotide-binding oligomerization domain (NOD) -like receptor (NLR) and Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing)NLRP PRRs sensing PAMPs, DAMPs and other signs of active inflammation initiate the formation of so called inflammasome multiprotein complexes. There are 6 multi-protein complexes: LRR, NACHT, PYD/CARD domain containing proteins -NLRP1,3,6,12, NLRC4 and AIM2. The most studied and important inflammasome for sensing endogenous stress through signaling events such as potassium ion efflux, changes in calcium signaling and the production of reactive oxygen species by mitochondria, lysosomal leakage and cathepsin B release is the before mentioned NLRP3 (120, 121). Once activated, the NLRP3 builds a multi-protein complex by recruiting caspase 1 via caspase activation and recruitment domain (CARD) assembly to promote caspase enzymatic function in cleaving pro IL-1β and pro IL-18 into their active forms (122). Furthermore, it cleaves the pore forming pro-gasdermin D into its active form that induces inflammatory cell death called pyroptosis (123). The pore formed by gasdermin D promotes the release of IL-18 and IL-1β a proinflammatory cytokine that regulates the function of immune -and neuronal cells (124). Indeed, the majorly important function of inflammasomes is the detection and protection from microbes and the following IL-1β release. Therefore, the cell has to be primed in a first step through a TLR agonist like LPS or inflammatory mediators like TNF, the second signal can be provided by DAMPs or microbe associated virulence factors like toxins. There is not much known about the regulatory signals that can control inflammasome assembly and prevent pyroptosis. It is assumed that adaptive immune cells, namely CD4 T cells and memory T cells are able to suppress NLRP1 and NLRP3 mediated IL-1β secretion and capsase-1 activity, leading to the hypothesis, that the inflammasome is supposed to be active during the first days of innate immune defense against pathogens and be stopped when the adaptive immune cells are at place (125). However, there is a growing body of evidence that chronic inflammasome activity is associated with inflammatory and neurodegenerative diseases (126). High levels of these inflammasome associated cytokines decrease the homeostatic functions of astrocytes (127, 128) and microglia including an increase in glutamate release by astrocytes that is associated with excitotoxicity and microglial activation and state change to a chronic pro-inflammatory state accompanied by inflammasome activation, further promoting neurotoxicity (129–131). Inhibition of the NLRP3 inflammasome is able to convey microglia to a neuroprotective, phagocyting phenotype in a mouse model of Alzheimer´s disease (53). Indeed, a chronic state of immune suppression and anti-inflammation due to aging associated senescence of immune cells is associated with neurodegenerative diseases. There is evidence, that ER stress might contribute to the maintenance of this immune suppressive environment (132). Conversely, there are studies reporting ER stress to be highly associated with neuro-inflammation and neurodegenerative diseases (133–137), while mild ER stress can also be protective and prime CNS immune cells to better cope with infection and tissue damage (138). This paradox occurs due to the fact that the ER stress response, including activation of UPR and ERAD are important in the fight against misfolded and aggregated proteins that are hallmarks in neurodegenerative diseases like Alzheimer’s´ disease and Parkinson´s disease, but the pathways they induce are also supporting inflammation and cell death (139, 140). The clinical outcome of therapeutic targeting ER stress members via suppression or activation can thus both be beneficial (139).
In contrast to the central nervous system, peripheral sensory neurons are not isolated from blood via a BBB, but instead are even required to sense the current metabolites, ions, proteins and chemicals in the blood and in the tissue. This is necessary to initiate adjustments via the autonomic nervous system including sympathetic and parasympathetic neurons but also via the enteric nervous system. Furthermore, sensation via visceral and somatic sensory neurons can be consciously sensed and therefore be important for the initiation of active behavior and responses to stimuli. Unfortunately, injury or endogenous harm of sensory neurons is therefore accompanied by dysesthesia, including paresthesia, numbness and pain. Because of their location throughout the body through innervation of organs, muscles, and skin, sensory neurons are easily exposed to external physical injuries, but they can also be easily harmed by metabolites and chemicals in the blood that have neurotoxic properties. Therefore, the most common cause for peripheral neuropathy is diabetes mellitus (141, 142). In addition, toxicity induced by alcohol consumption or chemotherapy are also common causes (143, 144). Indeed, in the periphery, neurons are imbedded in a tissue environment that is composed of Schwann cells, the peripheral equivalent to oligodendrocytes that are important for the formation of myelin sheets around the neuronal axons for faster conduction velocity, satellite cells that surround the sensory neurons soma, fibroblasts that are important for the formation of the endoneurium, perineurium and epineurium in peripheral nerves, endothelial cells from blood vessels and multiple types of immune cells including macrophages, granulocytes and lymphocytes. Thus, peripheral neuropathies can also be induced by pathologies of the surrounding cells, especially Schwann cells, that are important for the rapid signal transduction in highly myelinated cells including motor and sensory neurons. Auto-immune diseases with auto- antibodies against swan cell structures (like myelin) or structures of neuronal cells can induce acute and chronic peripheral neuropathies (145). There is high evidence, that ER stress activation occurs trough nerve injury where it contributes to neuropathic pain (146–151), while the ER stress response including UPR are required and important for neuronal regeneration and growth (152, 153). Other studies could show the involvement of ER stress in chemotherapy induced neuropathy, for example through treatment with proteasome inhibitors like Bortezomib (154) or through Paclitaxel that induces oscillatory changes in cytosolic calcium by increasing the open probability of the InsP3R and activating calpain, a calcium-dependent protease (155, 156). Neuropathic symptoms could be shown to be prevented or be reversed through treatment with lithium (155) or anti-oxidative plant based compounds like quercetin and lycopene that lead to an increase in neuronal growth factors like brain-derived neurotrophic factor (BDNF) and neural cell adhesion molecule (NCAM) while decreasing the expression of ER stress mediator proteins like ATF6, GRP78, PERK and IRE1 (157, 158). Furthermore, expression of the ER stress marker CCAAT/enhancer-binding protein homologous protein (CHOP) in neuropathy upon treatment with paclitaxel targeting the mitotic spindle could be inhibited via treatment with aucubin, a secondary plant product acting as an anti-inflammatory agent (159). More studies are reporting the contribution of ER stress in the pathology of diabetic neuropathy (160–163) and deliver possible treatment strategies by targeting the ER stress response associated inflammation (164).
Interestingly, there are also positive reports on how the UPR is involved in metabolic fates of tissues innervated by either serotonergic or dopaminergic neurons. Serotonin is thereby inducing chaperone induction and influences protein homeostasis while dopamine is important for metabolic ER-remodeling and lipid depletion (165). Activation and overexpression of the lipophagy component ehbp-1 was further sufficient to remodel ER and promote life span in a model of Caenorhabditis elegans (166). While peripheral neurons are therefore able to influence the ER and UPR in tissues and cells they innervate, ER stress and UPR signaling in neurons and associated Schwann cells have a major impact on neuronal functionality.
As outlined in this review ER stress, UPR and ISR are all necessary to balance cell homeostasis in the central and peripheral nervous system especially upon the defense against pathogens but also to react upon intrinsic cell stressors. Chronic activation and dysregulation of these pathways are contributing to the development of neuroinflammation and neurodegeneration. Possible treatment strategies are focusing on the reduction of stress inducers like anti-oxidative therapy and calcium signaling regulation as mentioned above, but also on restoring the balance of proteasomal degradation and autophagy or proteasome activation and activation or inhibition of translation through phosphorylation/dephosphorylation of eIF2a. Polyamines like spermidine as well as the United States Food and Drug Administration (FDA) approved anti-epileptic treatment carbamazepine are known to induce autophagy and proteasomal activation and could be shown to be neuroprotective in preclinical studies including amyotrophic lateral sclerosis (ALS) and Alzheimer´s disease (167–171). Proteasomal activation is important to provide the cell with the necessary amino acids for protein synthesis and lack of amino acids can thus induce ER stress. Therefore, the regulation of proteasome activation as well as the regulation of translation are key to a balanced protein homeostasis. The eIF2a phosphatase 1 inhibitor guanabenz is one example of how decreased global translation can be neuroprotective in the context of neurodegenerative diseases like prion disease (71) and ALS (72). In Table 1 we list relevant treatments that further target ER stress, UPR, ISR or the Inflammasome and are already investigated in clinical trials for neurological diseases.
The ER stress response including UPR and ERAD signaling are important for the maintenance of neuronal function itself, but also for host defense and tissue immunity (172). Chronic activation of ER associated inflammation in glia cells, immune cells and neurons can turn the valued, protective function into a devastating one. Targeting of ER stress, inflammasome activity and inflammation can therefore be affective through activation and suppression, depending on the disease and progression state (120, 173, 174).
CP and EK conceptualized the work, wrote parts, reviewed and edited. CP additionally wrote major parts of the manuscript and designed the figures, tables, and screened literature. Both authors have read and agreed to the published version of the manuscript.
This work was supported by the German Research Foundation RTG 2719 to EK and DFG PE2864/3-1 to CP. We acknowledge support for the Article Processing Charge by the German Research Foundation and the Open Access Publication Fund of the University of Greifswald.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Meizlish ML, Franklin RA, Zhou X, Medzhitov R. Tissue Homeostasis and Inflammation. Annu Rev Immunol (2021) 39:557–81. doi: 10.1146/annurev-immunol-061020-053734
2. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
3. Palm NW, Rosenstein RK, Medzhitov R. Allergic Host Defences. Nature (2012) 484:465–72. doi: 10.1038/nature11047
4. Tauber AI. Metchnikoff and the Phagocytosis Theory. Nat Rev Mol Cell Biol (2003) 4:897–901. doi: 10.1038/nrm1244
5. Wu YS, Chen SN. Apoptotic Cell: Linkage of Inflammation and Wound Healing. Front Pharmacol (2014) 5:1. Epub 2014/01/31. doi: 10.3389/fphar.2014.00001
6. Lepelley A, Wai T, Crow YJ. Mitochondrial Nucleic Acid as a Driver of Pathogenic Type I Interferon Induction in Mendelian Disease. Front Immunol (2021) 12:729763. doi: 10.3389/fimmu.2021.729763
7. Perner C, Flayer CH, Zhu X, Aderhold PA, Dewan ZNA, Voisin T, et al. Substance P Release by Sensory Neurons Triggers Dendritic Cell Migration and Initiates the Type-2 Immune Response to Allergens. Immunity (2020) 53:1063–77. doi: 10.1016/j.immuni.2020.10.001
8. Lai NY, Musser MA, Pinho-Ribeiro FA, Baral P, Jacobson A, Ma P, et al. Gut-Innervating Nociceptor Neurons Regulate Peyer's Patch Microfold Cells and SFB Levels to Mediate Salmonella Host Defense. Cell (2019) 180(1):33–49. doi: 10.1016/j.cell.2019.11.014
9. Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, et al. Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell (2019) 178(4):919–32. doi: 10.1016/j.cell.2019.06.022
10. Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, et al. Bacteria Activate Sensory Neurons That Modulate Pain and Inflammation. Nature (2013) 501(7465):52–7. doi: 10.1038/nature12479
11. Woodley PK, Min Q, Li Y, Mulvey NF, Parkinson DB, Dun XP, et al. Distinct VIP and PACAP Functions in the Distal Nerve Stump During Peripheral Nerve Regeneration. Front Neurosci (2019) 13:1326. doi: 10.3389/fnins.2019.01326
12. Hou L, Li W, Wang X. Mechanism of Interleukin-1 Beta-Induced Calcitonin Gene-Related Peptide Production From Dorsal Root Ganglion Neurons of Neonatal Rats. J Neurosci Res (2003) 73:188–97. doi: 10.1002/jnr.10651
13. Hoeffel G, Debroas G, Roger A, Rossignol R, Gouilly J, Laprie C, et al. Sensory Neuron-Derived TAFA4 Promotes Macrophage Tissue Repair Functions. Nature (2021) 594:94–9. doi: 10.1038/s41586-021-03563-7
14. Chiu IM, von Hehn CA, Woolf CJ. Neurogenic Inflammation and the Peripheral Nervous System in Host Defense and Immunopathology. Nat Neurosci (2012) 15(8):1063–7. doi: 10.1038/nn.3144
15. Schaffer M, Beiter T, Becker HD, Hunt TK. Neuropeptides: Mediators of Inflammation and Tissue Repair? Arch Surg (1998) 133:1107–16. doi: 10.1001/archsurg.133.10.1107
16. Flayer CH, Perner C, Sokol CL. A Decision Tree Model for Neuroimmune Guidance of Allergic Immunity. Immunol Cell Biol (2021) 454(7203):428–35. doi: 10.1111/imcb.12486
17. Hotamisligil GS. Inflammation and Metabolic Disorders. Nature (2006) 444:860–7. doi: 10.1038/nature05485
18. Medzhitov R. Origin and Physiological Roles of Inflammation. Nature (2008) 454(7203):428–35. doi: 10.1038/nature07201
19. Gomora-Garcia JC, Geronimo-Olvera C, Perez-Martinez X, Massieu L. IRE1alpha RIDD Activity Induced Under ER Stress Drives Neuronal Death by the Degradation of 14-3-3 Theta mRNA in Cortical Neurons During Glucose Deprivation. Cell Death Discov (2021) 7:131. doi: 10.1038/s41420-021-00518-9
20. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science (2000) 287(5453):664–6. doi: 10.1126/science.287.5453.664
21. Rutkowski DT, Hegde RS. Regulation of Basal Cellular Physiology by the Homeostatic Unfolded Protein Response. J Cell Biol (2010) 189:783–94. doi: 10.1083/jcb.201003138
22. Hetz C. The Unfolded Protein Response: Controlling Cell Fate Decisions Under ER Stress and Beyond. Nat Rev Mol Cell Biol (2012) 13:89–102. doi: 10.1038/nrm3270
23. Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J, et al. ER Stress Inhibits Neuronal Death by Promoting Autophagy. Autophagy (2012) 8(6):915–26. doi: 10.4161/auto.19716
24. Smith JA. Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity. Front Immunol (2018) 9:422. doi: 10.3389/fimmu.2018.00422
25. Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, et al. Endoplasmic Reticulum Stress-Induced Transcription Factor, CHOP, is Crucial for Dendritic Cell IL-23 Expression. Proc Natl Acad Sci USA (2010) 107(41):17698–703. doi: 10.1073/pnas.1011736107
26. Liu YP, Zeng L, Tian A, Bomkamp A, Rivera D, Gutman D, et al. Endoplasmic Reticulum Stress Regulates the Innate Immunity Critical Transcription Factor IRF3. J Immunol (2012) 189(9):4630–9. doi: 10.4049/jimmunol.1102737
27. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science (2015) 347(6227):aaa2630. doi: 10.1126/science.aaa2630
28. Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, et al. STING-IRF3 Pathway Links Endoplasmic Reticulum Stress With Hepatocyte Apoptosis in Early Alcoholic Liver Disease. Proc Natl Acad Sci USA (2013) 110(41):16544–9. doi: 10.1073/pnas.1308331110
29. Tanaka Y, Chen ZJ. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci Signal (2012) 5:ra20. doi: 10.1126/scisignal.2002521
30. Ghemrawi R, Khair M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int J Mol Sci (2020) 21(17). doi: 10.3390/ijms21176127
31. Chong WC, Shastri MD, Peterson GM, Patel RP, Pathinayake PS, Dua K, et al. The Complex Interplay Between Endoplasmic Reticulum Stress and the NLRP3 Inflammasome: A Potential Therapeutic Target for Inflammatory Disorders. Clin Transl Immunol (2021) 10(2):e1247. doi: 10.1002/cti2.1247
32. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, et al. ER Stress Activates the NLRP3 Inflammasome via an UPR-Independent Pathway. Cell Death Dis (2012) 3:e261. doi: 10.1038/cddis.2011.132
33. Reina S, Palermo V, Guarnera A, Guarino F, Messina A, Mazzoni C, et al. Swapping of the N-Terminus of VDAC1 With VDAC3 Restores Full Activity of the Channel and Confers Anti-Aging Features to the Cell. FEBS Lett (2010) 584(13):2837–44. doi: 10.1016/j.febslet.2010.04.066
34. Chu Y, Goldman JG, Kelly L, He Y, Waliczek T, Kordower JH. Abnormal Alpha-Synuclein Reduces Nigral Voltage-Dependent Anion Channel 1 in Sporadic and Experimental Parkinson's Disease. Neurobiol Dis (2014) 69:1–14. doi: 10.1016/j.nbd.2014.05.003
35. Smilansky A, Dangoor L, Nakdimon I, Ben-Hail D, Mizrachi D, Shoshan-Barmatz V. The Voltage-Dependent Anion Channel 1 Mediates Amyloid Beta Toxicity and Represents a Potential Target for Alzheimer Disease Therapy. J Biol Chem (2015) 290(52):30670–83. doi: 10.1074/jbc.M115.691493
36. Fryer AL, Abdullah A, Taylor JM, Crack PJ. The Complexity of the cGAS-STING Pathway in CNS Pathologies. Front Neurosci (2021) 15:621501. doi: 10.3389/fnins.2021.621501
37. Bond S, Lopez-Lloreda C, Gannon PJ, Akay-Espinoza C, Jordan-Sciutto KL. The Integrated Stress Response and Phosphorylated Eukaryotic Initiation Factor 2alpha in Neurodegeneration. J Neuropathol Exp Neurol (2020) 79:123–43. doi: 10.1093/jnen/nlz129
38. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The Integrated Stress Response. EMBO Rep (2016) 17(10):1374–95. doi: 10.15252/embr.201642195
39. Suraweera A, Munch C, Hanssum A, Bertolotti A. Failure of Amino Acid Homeostasis Causes Cell Death Following Proteasome Inhibition. Mol Cell (2012) 48:242–53. doi: 10.1016/j.molcel.2012.08.003
40. Martinez NW, Gomez FE, Matus S. The Potential Role of Protein Kinase R as a Regulator of Age-Related Neurodegeneration. Front Aging Neurosci (2021) 13:638208. doi: 10.3389/fnagi.2021.638208
41. Ebstein F, Poli Harlowe MC, Studencka-Turski M, Kruger E. Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front Immunol (2019) 10:2756. doi: 10.3389/fimmu.2019.02756
42. Ebstein F, Kury S, Papendorf JJ, Kruger E. Neurodevelopmental Disorders (NDD) Caused by Genomic Alterations of the Ubiquitin-Proteasome System (UPS): The Possible Contribution of Immune Dysregulation to Disease Pathogenesis. Front Mol Neurosci (2021) 14:733012. doi: 10.3389/fnmol.2021.733012
43. Davidson S, Yu CH, Steiner A, Ebstein F, Baker PJ, Jarur-Chamy V, et al. Protein Kinase R is an Innate Immune Sensor of Proteotoxic Stress via Accumulation of Cytoplasmic IL-24. Sci Immunol (2022) 7(68):eabi6763. doi: 10.1126/sciimmunol.abi6763
44. Haas JG, Lathe R. Microbes and Alzheimer's Disease: New Findings Call for a Paradigm Change. Trends Neurosci (2018) 41(9):555–640. doi: 10.1016/j.tins.2018.07.001
45. Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, et al. Multiscale Analysis of Independent Alzheimer's Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron (2018) 99(1):64–82.e67. doi: 10.1016/j.neuron.2018.05.023
46. Eimer WA, Vijaya Kumar DK, Navalpur, Shanmugam NK, Rodriguez AS, Mitchell T, et al. Alzheimer's Disease-Associated Beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect Against Brain Infection. Neuron (2018) 99(1):56–63.e53. doi: 10.1016/j.neuron.2018.06.030
47. Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med (2020) 383(27):2603–15. doi: 10.1056/NEJMoa2034577
48. Zhang P, Narayanan E, Liu Q, Tsybovsky Y, Boswell K, Ding S, et al. A Multiclade Env-Gag VLP mRNA Vaccine Elicits Tier-2 HIV-1-Neutralizing Antibodies and Reduces the Risk of Heterologous SHIV Infection in Macaques. Nat Med (2021) 27(12):2234–45. doi: 10.1038/s41591-021-01574-5
49. Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, et al. Modified mRNA Vaccines Protect Against Zika Virus Infection. Cell (2017) 168(6):1114–1125.e1110. doi: 10.1016/j.cell.2017.02.017
50. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 Inflammasome Inhibition is Disrupted in a Group of Auto-Inflammatory Disease CAPS Mutations. Nat Immunol (2016) 17:1176–86. doi: 10.1038/ni.3538
51. Heneka MT, McManus RM, Latz E. Inflammasome Signalling in Brain Function and Neurodegenerative Disease. Nat Rev Neurosci (2018) 19(10):610–21. doi: 10.1038/s41583-018-0055-7
52. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 Inflammasome is Involved in the Innate Immune Response to Amyloid-Beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
53. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is Activated in Alzheimer's Disease and Contributes to Pathology in APP/PS1 Mice. Nature (2013) 493(7434):674–8. doi: 10.1038/nature11729
54. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 Inflammasome Activation Drives Tau Pathology. Nature (2019) 575(7784):669–73. doi: 10.1038/s41586-019-1769-z
55. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome Inhibition Prevents Alpha-Synuclein Pathology and Dopaminergic Neurodegeneration in Mice. Sci Transl Med (2018) 10(465). doi: 10.1126/scitranslmed.aah4066
56. Coll RC, et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
57. Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, et al. Inhibiting the NLRP3 Inflammasome With MCC950 Promotes Non-Phlogistic Clearance of Amyloid-Beta and Cognitive Function in APP/PS1 Mice. Brain Behav Immun (2017) 61:306–16. doi: 10.1016/j.bbi.2016.12.014
58. Daruich A, Picard E, Boatright JH, Behar-Cohen F. Review: The Bile Acids Urso- and Tauroursodeoxycholic Acid as Neuroprotective Therapies in Retinal Disease. Mol Vis (2019) 25:610–24.
59. Nunes AF, Amaral JD, Lo AC, Fonseca MB, Viana RJ, Callaerts-Vegh Z, et al. TUDCA, A Bile Acid, Attenuates Amyloid Precursor Protein Processing and Amyloid-Beta Deposition in APP/PS1 Mice. Mol Neurobiol (2012) 45(3):440–54. doi: 10.1007/s12035-012-8256-y
60. Ackerman HD, Gerhard GS. Bile Acids in Neurodegenerative Disorders. Front Aging Neurosci (2016) 8:263. doi: 10.3389/fnagi.2016.00263
61. Elia AE, Lalli S, Monsurro MR, Sagnelli A, Taiello AC, Reggiori B, et al. Tauroursodeoxycholic Acid in the Treatment of Patients With Amyotrophic Lateral Sclerosis. Eur J Neurol (2016) 23(1):45–52. doi: 10.1111/ene.12664
62. Wiley JC, Pettan-Brewer C, Ladiges WC. Phenylbutyric Acid Reduces Amyloid Plaques and Rescues Cognitive Behavior in AD Transgenic Mice. Aging Cell (2011) 10(3):418–28. doi: 10.1111/j.1474-9726.2011.00680.x
63. Mimori S, Okuma Y, Kaneko M, Kawada K, Hosoi T, Ozawa K, et al. Protective Effects of 4-Phenylbutyrate Derivatives on the Neuronal Cell Death and Endoplasmic Reticulum Stress. Biol Pharm Bull (2012) 35(1):84–90. doi: 10.1248/bpb.35.84
64. Cudkowicz ME, Andres PL, Macdonald SA, Bedlack RS, Choudry R, Brown RH Jr, et al. Phase 2 Study of Sodium Phenylbutyrate in ALS. Amyotroph Lateral Scler (2009) 10(2):99–106. doi: 10.1080/17482960802320487
65. Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N Engl J Med (2020) 383(10):919–30. doi: 10.1056/NEJMoa1916945
66. Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne H, et al. Partial Restoration of Protein Synthesis Rates by the Small Molecule ISRIB Prevents Neurodegeneration Without Pancreatic Toxicity. Cell Death Dis (2015) 6:e1672. doi: 10.1038/cddis.2015.49
67. Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC, et al. Repurposed Drugs Targeting Eif2α-P-Mediated Translational Repression Prevent Neurodegeneration in Mice. Brain (2017) 140(6):1768–83. doi: 10.1093/brain/awx074
68. Stahl SM. Mechanism of Action of Trazodone: A Multifunctional Drug. CNS Spectr (2009) 14:536–46. doi: 10.1017/s1092852900024020
69. Lipone P, Ehler E, Nastaj M, Palka-Kisielowska I, Cruccu G, Truini A, et al. Efficacy and Safety of Low Doses of Trazodone in Patients Affected by Painful Diabetic Neuropathy and Treated With Gabapentin: A Randomized Controlled Pilot Study. CNS Drugs (2020) 34(11):1177–89. doi: 10.1007/s40263-020-00760-2
70. Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science (2011) 332:91–4. doi: 10.1126/science.1201396
71. Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, et al. Sustained Translational Repression by Eif2alpha-P Mediates Prion Neurodegeneration. Nature (2012) 485(7399):507–11. doi: 10.1038/nature11058
72. Bella ED, Bersano E, Antonini G, Borghero G, Capasso M, Caponnetto C, et al. The Unfolded Protein Response in Amyotrophic Later Sclerosis: Results of a Phase 2 Trial. Brain (2021) 144(9):2635–47. doi: 10.1093/brain/awab167
73. Terasaki M, Shemesh T, Kasthuri N, Klemm RW, Schalek R, Hayworth KJ, et al. Stacked Endoplasmic Reticulum Sheets are Connected by Helicoidal Membrane Motifs. Cell (2013) 154(2):285–96. doi: 10.1016/j.cell.2013.06.031
74. Ruggiano A, Foresti O, Carvalho P. Quality Control: ER-Associated Degradation: Protein Quality Control and Beyond. J Cell Biol (2014) 204:869–79. doi: 10.1083/jcb.201312042
75. Schwarz DS, Blower MD. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell Mol Life Sci (2016) 73(1):79–94. doi: 10.1007/s00018-015-2052-6
77. Verkhratsky A. Endoplasmic Reticulum Calcium Signaling in Nerve Cells. Biol Res (2004) 37(4):693–9. doi: 10.4067/s0716-97602004000400027
78. Pereda AE. Electrical Synapses and Their Functional Interactions With Chemical Synapses. Nat Rev Neurosci (2014) 15(4):250–63. doi: 10.1038/nrn3708
79. Ozturk Z, O'Kane CJ, Perez-Moreno JJ. Axonal Endoplasmic Reticulum Dynamics and Its Roles in Neurodegeneration. Front Neurosci (2020) 14:48. doi: 10.3389/fnins.2020.00048
80. Shacham T, Sharma N, Lederkremer GZ. Protein Misfolding and ER Stress in Huntington's Disease. Front Mol Biosci (2019) 6:20. doi: 10.3389/fmolb.2019.00020
81. Medawar PB. Immunity to Homologous Grafted Skin; the Fate of Skin Homografts Transplanted to the Brain, to Subcutaneous Tissue, and to the Anterior Chamber of the Eye. Br J Exp Pathol (1948) 29:58–69.
82. Schluter D, Buck C, Reiter S, Meyer T, Hof H, Deckert-Schluter M. Immune Reactions to Listeria Monocytogenes in the Brain. Immunobiology (1999) 201(2):188–95. doi: 10.1016/S0171-2985(99)80058-8
83. Alves de Lima K, Rustenhoven J, Kipnis J. Meningeal Immunity and Its Function in Maintenance of the Central Nervous System in Health and Disease. Annu Rev Immunol (2020) 38:597–620. doi: 10.1146/annurev-immunol-102319-103410
84. Cugurra A, Mamuladze T, Rustenhoven J, Dykstra T, Beroshvili G, Greenberg ZJ, et al. Skull and Vertebral Bone Marrow are Myeloid Cell Reservoirs for the Meninges and CNS Parenchyma. Science (2021) 373. doi: 10.1126/science.abf7844
85. Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal Gammadelta T Cells Regulate Anxiety-Like Behavior via IL-17a Signaling in Neurons. Nat Immunol (2020) 21(11):1421–9. doi: 10.1038/s41590-020-0776-4
86. Salvador AF, de Lima KA, Kipnis J. Neuromodulation by the Immune System: A Focus on Cytokines. Nat Rev Immunol (2021) 21:526–41. doi: 10.1038/s41577-021-00508-z
87. Rattazzi L, Piras G, Ono M, Deacon R, Pariante CM, D'Acquisto F. CD4(+) But Not CD8(+) T Cells Revert the Impaired Emotional Behavior of Immunocompromised RAG-1-Deficient Mice. Transl Psychiatry (2013) 3:e280. doi: 10.1038/tp.2013.54
88. Radjavi A, Smirnov I, Kipnis J. Brain Antigen-Reactive CD4+ T Cells are Sufficient to Support Learning Behavior in Mice With Limited T Cell Repertoire. Brain Behav Immun (2014) 35:58–63. doi: 10.1016/j.bbi.2013.08.013
89. Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T Cell Deficiency Leads to Cognitive Dysfunction: Implications for Therapeutic Vaccination for Schizophrenia and Other Psychiatric Conditions. Proc Natl Acad Sci USA (2004) 101(21):8180–5. doi: 10.1073/pnas.0402268101
90. Ron-Harel N, Segev Y, Lewitus GM, Cardon M, Ziv Y, Netanely D, et al. Age-Dependent Spatial Memory Loss can be Partially Restored by Immune Activation. Rejuvenation Res (2008) 11:903–13. doi: 10.1089/rej.2008.0755
91. Kipnis J, Gadani S, Derecki NC. Pro-Cognitive Properties of T Cells. Nat Rev Immunol (2012) 12:663–9. doi: 10.1038/nri3280
92. Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, et al. Immune Cells Contribute to the Maintenance of Neurogenesis and Spatial Learning Abilities in Adulthood. Nat Neurosci (2006) 9(2):268–75. doi: 10.1038/nn1629
93. Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, et al. CD4-Positive T Lymphocytes Provide a Neuroimmunological Link in the Control of Adult Hippocampal Neurogenesis. J Immunol (2009) 182(7):3979–84. doi: 10.4049/jimmunol.0801218
94. Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, et al. Regulation of Learning and Memory by Meningeal Immunity: A Key Role for IL-4. J Exp Med (2010) 207(5):1067–80. doi: 10.1084/jem.20091419
95. Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-Independent CD4+ T Cells Protect Injured CNS Neurons via IL-4. J Clin Invest (2015) 125(2):699–714. doi: 10.1172/JCI76210
96. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid Beta. Sci Transl Med (2012) 4(147):147ra111. doi: 10.1126/scitranslmed.3003748
97. Preston JE. Ageing Choroid Plexus-Cerebrospinal Fluid System. Microsc Res Tech (2001) 52:31–7. doi: 10.1002/1097-0029(20010101)52:1<31::AID-JEMT5>3.0.CO;2-T
98. Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J. Understanding the Functions and Relationships of the Glymphatic System and Meningeal Lymphatics. J Clin Invest (2017) 127(9):3210–9. doi: 10.1172/JCI90603
99. Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T Cells Protect Neurons From Secondary Degeneration After Central Nervous System Axotomy. Nat Med (1999) 5(1):49–55. doi: 10.1038/4734
100. Moalem G, Yoles E, Leibowitz-Amit R, Muller-Gilor S, Mor F, Cohen IR, et al. Autoimmune T Cells Retard the Loss of Function in Injured Rat Optic Nerves. J Neuroimmunol (2000) 106(1–2):189–97. doi: 10.1016/s0165-5728(00)00240-x
101. Hauben E, Agranov E, Gothilf A, Nevo U, Cohen A, Smirnov I, et al. Posttraumatic Therapeutic Vaccination With Modified Myelin Self-Antigen Prevents Complete Paralysis While Avoiding Autoimmune Disease. J Clin Invest (2001) 108(4):591–9. doi: 10.1172/JCI12837
102. Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective Autoimmunity: Naturally Occurring CD4+CD25+ Regulatory T Cells Suppress the Ability to Withstand Injury to the Central Nervous System. Proc Natl Acad Sci USA (2002) 99(24):15620–5. doi: 10.1073/pnas.232565399
103. Walsh JT, Zheng J, Smirnov I, Lorenz U, Tung K, Kipnis J. Regulatory T Cells in Central Nervous System Injury: A Double-Edged Sword. J Immunol (2014) 193(10):5013–22. doi: 10.4049/jimmunol.1302401
104. Liesz A, Zhou W, Na SY, Hammerling GJ, Garbi N, Karcher S, et al. Boosting Regulatory T Cells Limits Neuroinflammation in Permanent Cortical Stroke. J Neurosci (2013) 33:17350–62. doi: 10.1523/JNEUROSCI.4901-12.2013
105. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. Aging-Induced Type I Interferon Response at the Choroid Plexus Negatively Affects Brain Function. Science (2014) 346(6205):89–93. doi: 10.1126/science.1252945
106. Dulken BW, Buckley MT, Navarro Negredo P, Saligrama N, Cayrol R, Leeman DS, et al. Single-Cell Analysis Reveals T Cell Infiltration in Old Neurogenic Niches. Nature (2019) 571(7764):205–10. doi: 10.1038/s41586-019-1362-5
107. Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected Role of Interferon-Gamma in Regulating Neuronal Connectivity and Social Behaviour. Nature (2016) 535(7612):425–9. doi: 10.1038/nature18626
108. Da Mesquita S, Fu Z, Kipnis J. The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron (2018) 100(2):375–88. doi: 10.1016/j.neuron.2018.09.022
109. Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of Cerebrospinal Fluid is Predominantly Through Lymphatic Vessels and is Reduced in Aged Mice. Nat Commun (2017) 8(1):1434. doi: 10.1038/s41467-017-01484-6
110. Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional Aspects of Meningeal Lymphatics in Ageing and Alzheimer's Disease. Nature (2018) 560:185–91. doi: 10.1038/s41586-018-0368-8
111. Da Mesquita S, Fu Z, Kipnis J. Aging-Associated Deficit in CCR7 is Linked to Worsened Glymphatic Function, Cognition, Neuroinflammation, and Beta-Amyloid Pathology. Sci Adv (2021) 7. doi: 10.1126/sciadv.abe4601
112. Greenhalgh AD, David S, Bennett FC. Immune Cell Regulation of Glia During CNS Injury and Disease. Nat Rev Neurosci (2020) 21:139–52. doi: 10.1038/s41583-020-0263-9
113. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid Cells in the Central Nervous System. Immunity (2017) 46(6):943–56. doi: 10.1016/j.immuni.2017.06.007
114. Irani DN, Lin KI, Griffin DE. Brain-Derived Gangliosides Regulate the Cytokine Production and Proliferation of Activated T Cells. J Immunol (1996) 157:4333–40.
115. Ransohoff RM, Brown MA. Innate Immunity in the Central Nervous System. J Clin Invest (2012) 122(4):1164–71. doi: 10.1172/JCI58644
116. Lehnardt S. Innate Immunity and Neuroinflammation in the CNS: The Role of Microglia in Toll-Like Receptor-Mediated Neuronal Injury. Glia (2010) 58(3):253–63. doi: 10.1002/glia.20928
117. Gorina R, Santalucia T, Petegnief V, Ejarque-Ortiz A, Saura J, Planas AM. Astrocytes are Very Sensitive to Develop Innate Immune Responses to Lipid-Carried Short Interfering RNA. Glia (2009) 57(1):93–107. doi: 10.1002/glia.20738
118. Millward JM, Lobner M, Wheeler RD, Owens T. Inflammation in the Central Nervous System and Th17 Responses are Inhibited by IFN-Gamma-Induced IL-18 Binding Protein. J Immunol (2010) 185(4):2458–66. doi: 10.4049/jimmunol.0902153
119. Van Wagoner NJ, Oh JW, Repovic P, Benveniste EN. Interleukin-6 (IL-6) Production by Astrocytes: Autocrine Regulation by IL-6 and the Soluble IL-6 Receptor. J Neurosci (1999) 19:5236–44. doi: 10.1523/JNEUROSCI.19-13-05236.1999
120. Starobova H, Nadar EI, Vetter I. The NLRP3 Inflammasome: Role and Therapeutic Potential in Pain Treatment. Front Physiol (2020) 11:1016. doi: 10.3389/fphys.2020.01016
121. Zhou R, Yazdi AS, Menu P, Tschopp J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
122. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
123. Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 Inflammasomes Activate Both Apoptotic and Pyroptotic Death Pathways via ASC. Cell Death Differ (2013) 20:1149–60. doi: 10.1038/cdd.2013.37
124. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in Health and Disease. Nature (2012) 481:278–86. doi: 10.1038/nature10759
125. Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, et al. T Cells Dampen Innate Immune Responses Through Inhibition of NLRP1 and NLRP3 Inflammasomes. Nature (2009) 460:269–73. doi: 10.1038/nature08100
126. Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in Neuroinflammatory and Neurodegenerative Diseases. EMBO Mol Med (2019) 11. doi: 10.15252/emmm.201810248
127. Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-Activated Astrocyte Glutamate Release via TNFalpha: Amplification by Microglia Triggers Neurotoxicity. Nat Neurosci (2001) 4:702–10. doi: 10.1038/89490
128. Vesce S, Rossi D, Brambilla L, Volterra A. Glutamate Release From Astrocytes in Physiological Conditions and in Neurodegenerative Disorders Characterized by Neuroinflammation. Int Rev Neurobiol (2007) 82:57–71. doi: 10.1016/S0074-7742(07)82003-4
129. Ho TC, Teresi GI, Segarra JR, Ojha A, Walker JC, Gu M, et al. Higher Levels of Pro-Inflammatory Cytokines Are Associated With Higher Levels of Glutamate in the Anterior Cingulate Cortex in Depressed Adolescents. Front Psychiatry (2021) 12:642976. doi: 10.3389/fpsyt.2021.642976
130. Galic MA, Riazi K, Pittman QJ. Cytokines and Brain Excitability. Front Neuroendocrinol (2012) 33:116–25. doi: 10.1016/j.yfrne.2011.12.002
131. Wang K, Yaghi OK, Spallanzani RG, Chen X, Zemmour D, Lai N, et al. Neuronal, Stromal, and T-Regulatory Cell Crosstalk in Murine Skeletal Muscle. Proc Natl Acad Sci U S A (2020) 117(10):5402–8. doi: 10.1073/pnas.1922559117
132. Salminen A, Kaarniranta K, Kauppinen A. ER Stress Activates Immunosuppressive Network: Implications for Aging and Alzheimer's Disease. J Mol Med (Berl) (2020) 98(5):633–50. doi: 10.1007/s00109-020-01904-z
133. Woodward AM, Di Zazzo A, Bonini S, Argueso P. Endoplasmic Reticulum Stress Promotes Inflammation-Mediated Proteolytic Activity at the Ocular Surface. Sci Rep (2020) 10:2216. doi: 10.1038/s41598-020-59237-3
134. Deslauriers AM, Afkhami-Goli A, Paul AM, Bhat RK, Acharjee S, Ellestad KK, et al. Neuroinflammation and Endoplasmic Reticulum Stress are Coregulated by Crocin to Prevent Demyelination and Neurodegeneration. J Immunol (2011) 187(9):4788–99. doi: 10.4049/jimmunol.1004111
135. Lin L, Liu G, Yang L. Crocin Improves Cognitive Behavior in Rats With Alzheimer's Disease by Regulating Endoplasmic Reticulum Stress and Apoptosis. BioMed Res Int (2019) 2019:9454913. doi: 10.1155/2019/9454913
136. Yang Z, Shao Y, Zhao Y, Li Q, Li R, Xiao H, et al. Endoplasmic Reticulum Stress-Related Neuroinflammation and Neural Stem Cells Decrease in Mice Exposure to Paraquat. Sci Rep (2020) 10(1):17757. doi: 10.1038/s41598-020-74916-x
137. Buchanan H, Mackay M, Palmer K, Tothova K, Katsur M, Platt B, et al. Synaptic Loss, ER Stress and Neuro-Inflammation Emerge Late in the Lateral Temporal Cortex and Associate With Progressive Tau Pathology in Alzheimer's Disease. Mol Neurobiol (2020) 57(8):3258–72. doi: 10.1007/s12035-020-01950-1
138. Wang YW, Zhou Q, Zhang X, Qian QQ, Xu JW, Ni PF, et al. Mild Endoplasmic Reticulum Stress Ameliorates Lipopolysaccharide-Induced Neuroinflammation and Cognitive Impairment via Regulation of Microglial Polarization. J Neuroinflamm (2017) 14:233. doi: 10.1186/s12974-017-1002-7
139. Nomura J, Hosoi T, Kaneko M, Ozawa K, Nishi A, Nomura Y. Neuroprotection by Endoplasmic Reticulum Stress-Induced HRD1 and Chaperones: Possible Therapeutic Targets for Alzheimer's and Parkinson's Disease. Med Sci (Basel) (2016) 4(3). doi: 10.3390/medsci4030014
140. Gerakis Y, Hetz C. Emerging Roles of ER Stress in the Etiology and Pathogenesis of Alzheimer's Disease. FEBS J (2018) 285:995–1011. doi: 10.1111/febs.14332
141. Barrell K, Smith AG. Peripheral Neuropathy. Med Clin North Am (2019) 103:383–97. doi: 10.1016/j.mcna.2018.10.006
142. Feldman EL, Nave KA, Jensen TS, Bennett DLH. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron (2017) 93:1296–313. doi: 10.1016/j.neuron.2017.02.005
143. Staff NP, Windebank AJ. Peripheral Neuropathy Due to Vitamin Deficiency, Toxins, and Medications. Continuum (Minneap Minn) (2014) 20(6):1293–306. doi: 10.1212/01.CON.0000455880.06675.5a
144. Staff NP, Grisold A, Grisold W, Windebank AJ. Chemotherapy-Induced Peripheral Neuropathy: A Current Review. Ann Neurol (2017) 81:772–81. doi: 10.1002/ana.24951
145. Bourque PR, Chardon JW, Massie R. Autoimmune Peripheral Neuropathies. Clin Chim Acta (2015) 449:37–42. doi: 10.1016/j.cca.2015.02.039
146. Yang ES, Bae JY, Kim TH, Kim YS, Suk K, Bae YC. Involvement of Endoplasmic Reticulum Stress Response in Orofacial Inflammatory Pain. Exp Neurobiol (2014) 23:372–80. doi: 10.5607/en.2014.23.4.372
147. Inceoglu B, Bettaieb A, Trindade da Silva CA, Lee KS, Haj FG, Hammock BD, et al. Endoplasmic Reticulum Stress in the Peripheral Nervous System is a Significant Driver of Neuropathic Pain. Proc Natl Acad Sci USA (2015) 112(29):9082–7. doi: 10.1073/pnas.1510137112
148. Ge Y, Jiao Y, Li P, Xiang Z, Li Z, Wang L, et al. Coregulation of Endoplasmic Reticulum Stress and Oxidative Stress in Neuropathic Pain and Disinhibition of the Spinal Nociceptive Circuitry. Pain (2018) 159(5):894–906. doi: 10.1097/j.pain.0000000000001161
149. Yamaguchi Y, Oh-Hashi K, Matsuoka Y, Takemura H, Yamakita S, Matsuda M, et al. Endoplasmic Reticulum Stress in the Dorsal Root Ganglion Contributes to the Development of Pain Hypersensitivity After Nerve Injury. Neuroscience (2018) 394:288–99. doi: 10.1016/j.neuroscience.2018.08.005
150. Mao Y, Wang C, Tian X, Huang Y, Zhang Y, Wu H, et al. Endoplasmic Reticulum Stress Contributes to Nociception via Neuroinflammation in a Murine Bone Cancer Pain Model. Anesthesiology (2020) 132(2):357–72. doi: 10.1097/ALN.0000000000003078
151. Yousuf MS, Samtleben S, Lamothe SM, Friedman TN, Catuneanu A, Thorburn K, et al. Endoplasmic Reticulum Stress in the Dorsal Root Ganglia Regulates Large-Conductance Potassium Channels and Contributes to Pain in a Model of Multiple Sclerosis. FASEB J (2020) 34(9):12577–98. doi: 10.1096/fj.202001163R
152. Onate M, Catenaccio A, Martinez G, Armentano D, Parsons G, Kerr B, et al. Activation of the Unfolded Protein Response Promotes Axonal Regeneration After Peripheral Nerve Injury. Sci Rep (2016) 6:21709. doi: 10.1038/srep21709
153. Ying Z, Zhai R, McLean NA, Johnston JM, Misra V, Verge VM. The Unfolded Protein Response and Cholesterol Biosynthesis Link Luman/CREB3 to Regenerative Axon Growth in Sensory Neurons. J Neurosci (2015) 35(43):14557–70. doi: 10.1523/JNEUROSCI.0012-15.2015
154. Ale A, Bruna J, Navarro X, Udina E. Neurotoxicity Induced by Antineoplastic Proteasome Inhibitors. Neurotoxicology (2014) 43:28–35. doi: 10.1016/j.neuro.2014.02.001
155. Mo M, Erdelyi I, Szigeti-Buck K, Benbow JH, Ehrlich BE. Prevention of Paclitaxel-Induced Peripheral Neuropathy by Lithium Pretreatment. FASEB J (2012) 26:4696–709. doi: 10.1096/fj.12-214643
156. Zheng D, Wang G, Li S, Fan GC, Peng T. Calpain-1 Induces Endoplasmic Reticulum Stress in Promoting Cardiomyocyte Apoptosis Following Hypoxia/Reoxygenation. Biochim Biophys Acta (2015) 1852:882–92. doi: 10.1016/j.bbadis.2015.01.019
157. Celik H, Kucukler S, Ozdemir S, Comakli S, Gur C, Kandemir FM, et al. Lycopene Protects Against Central and Peripheral Neuropathy by Inhibiting Oxaliplatin-Induced ATF-6 Pathway, Apoptosis, Inflammation and Oxidative Stress in Brains and Sciatic Tissues of Rats. Neurotoxicology (2020) 80:29–40. doi: 10.1016/j.neuro.2020.06.005
158. Yardim A, Kandemir FM, Ozdemir S, Kucukler S, Comakli S, Gur C, et al. Quercetin Provides Protection Against the Peripheral Nerve Damage Caused by Vincristine in Rats by Suppressing Caspase 3, NF-Kappab, ATF-6 Pathways and Activating Nrf2, Akt Pathways. Neurotoxicology (2020) 81:137–46. doi: 10.1016/j.neuro.2020.10.001
159. Andoh T, Uta D, Kato M, Toume K, Komatsu K, Kuraishi Y. Prophylactic Administration of Aucubin Inhibits Paclitaxel-Induced Mechanical Allodynia via the Inhibition of Endoplasmic Reticulum Stress in Peripheral Schwann Cells. Biol Pharm Bull (2017) 40(4):473–8. doi: 10.1248/bpb.b16-00899
160. Lupachyk S, Watcho P, Obrosov AA, Stavniichuk R, Obrosova IG. Endoplasmic Reticulum Stress Contributes to Prediabetic Peripheral Neuropathy. Exp Neurol (2013) 247:342–8. doi: 10.1016/j.expneurol.2012.11.001
161. Wu YB, Li HQ, Ren MS, Li WT, Lv XY, Wang L. CHOP/ORP150 Ratio in Endoplasmic Reticulum Stress: A New Mechanism for Diabetic Peripheral Neuropathy. Cell Physiol Biochem (2013) 32:367–79. doi: 10.1159/000354444
162. Kong D, Guo Z, Yang W, Wang Q, Yu Y, Zhang L. Tanshinone II A Affects Diabetic Peripheral Neuropathic Pain via Spinal Dorsal Horn Neuronal Circuitry by Modulating Endoplasmic Reticulum Stress Pathways. Exp Clin Endocrinol Diabetes (2020) 128(1):59–65. doi: 10.1055/a-0919-4614
163. Eftekharpour E, Fernyhough P. Oxidative Stress and Mitochondrial Dysfunction Associated With Peripheral Neuropathy in Type 1 Diabetes. Antioxid Redox Signal (2021). doi: 10.1089/ars.2021.0152
164. Pop-Busui R, Ang L, Holmes C, Gallagher K, Feldman EL. Inflammation as a Therapeutic Target for Diabetic Neuropathies. Curr Diabetes Rep (2016) 16:29. doi: 10.1007/s11892-016-0727-5
165. Higuchi-Sanabria R, Durieux J, Kelet N, Homentcovschi S, de Los Rios Rogers M, Monshietehadi S, et al. Divergent Nodes of Non-Autonomous UPR(ER) Signaling Through Serotonergic and Dopaminergic Neurons. Cell Rep (2020) 33:108489. doi: 10.1016/j.celrep.2020.108489
166. Daniele JR, Higuchi-Sanabria R, Durieux J, Monshietehadi S, Ramachandran V, Tronnes SU, et al. UPR(ER) Promotes Lipophagy Independent of Chaperones to Extend Life Span. Sci Adv (2020) 6:eaaz1441. doi: 10.1126/sciadv.aaz1441
167. Zhang L, Xu J, Gao J, Chen P, Yin M, Zhao W. Decreased Immunoglobulin G in Brain Regions of Elder Female APOE4-TR Mice Accompany With Abeta Accumulation. Immun Ageing (2019) 16:2. doi: 10.1186/s12979-018-0142-7
168. Li L, Zhang S, Zhang X, Li T, Tang Y, Liu H, et al. Autophagy Enhancer Carbamazepine Alleviates Memory Deficits and Cerebral Amyloid-Beta Pathology in a Mouse Model of Alzheimer's Disease. Curr Alzheimer Res (2013) 10(4):433–41. doi: 10.2174/1567205011310040008
169. Mullan LA, Mularczyk EJ, Kung LH, Forouhan M, Wragg JM, Goodacre R, et al. Increased Intracellular Proteolysis Reduces Disease Severity in an ER Stress-Associated Dwarfism. J Clin Invest (2017) 127(10):3861–5. doi: 10.1172/JCI93094
170. Yang Y, Chen S, Zhang Y, Lin X, Song Y, Xue Z, et al. Induction of Autophagy by Spermidine is Neuroprotective via Inhibition of Caspase 3-Mediated Beclin 1 Cleavage. Cell Death Dis (2017) 8(4):e2738. doi: 10.1038/cddis.2017.161
171. Fischer M, Ruhnau J, Schulze J, Obst D, Floel A, Vogelgesang A. Spermine and Spermidine Modulate T-Cell Function in Older Adults With and Without Cognitive Decline Ex Vivo. Aging (Albany NY) (2020) 12(13):13716–39. doi: 10.18632/aging.103527
172. Choi JA, Song CH. Insights Into the Role of Endoplasmic Reticulum Stress in Infectious Diseases. Front Immunol (2019) 10:3147. doi: 10.3389/fimmu.2019.03147
173. Doyle KM, Kennedy D, Gorman AM, Gupta S, Healy SJ, Samali A. Unfolded Proteins and Endoplasmic Reticulum Stress in Neurodegenerative Disorders. J Cell Mol Med (2011) 15(10):2025–39. doi: 10.1111/j.1582-4934.2011.01374.x
Keywords: ER-stress, unfolded and integrated stress response, innate immunity, central nervous system, peripheral nervous system
Citation: Perner C and Krüger E (2022) Endoplasmic Reticulum Stress and Its Role in Homeostasis and Immunity of Central and Peripheral Neurons. Front. Immunol. 13:859703. doi: 10.3389/fimmu.2022.859703
Received: 21 January 2022; Accepted: 28 March 2022;
Published: 27 April 2022.
Edited by:
Hideyuki Takeuchi, Yokohama City University, JapanReviewed by:
Gordon Meares, West Virginia University, United StatesCopyright © 2022 Perner and Krüger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Perner, Y2Fyb2xpbmUucGVybmVyOTFAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.