 Gaëtan Caravello
Gaëtan Caravello- UPR9022, University of Strasbourg, Institut de Biologie Moléculaire et Cellulaire (IBMC), Modèles Insectes D’Immunité Innée (M3I) Unité Propre Recherche (UPR) 9022 du Centre National de la Recherche Scientifique (CNRS), Strasbourg, France
Microsporidia are obligate intracellular parasites able to infest specifically a large range of species, including insects. The knowledge about the biology of microsporidial infections remains confined to mostly descriptive studies, including molecular approaches such as transcriptomics or proteomics. Thus, functional data to understand insect host defenses are currently lacking. Here, we have undertaken a genetic analysis of known host defenses of the Drosophila melanogaster using an infection model whereby Tubulinosema ratisbonensis spores are directly injected in this insect. We find that phagocytosis does confer some protection in this infection model. In contrast, the systemic immune response, extracellular reactive oxygen species, thioester proteins, xenophagy, and intracellular antiviral response pathways do not appear to be involved in the resistance against this parasite. Unexpectedly, several genes such as PGRP-LE seem to promote this infection. The prophenol oxidases that mediate melanization have different functions; PPO1 presents a phenotype similar to that of PGRP-LE whereas that of PPO2 suggests a function in the resilience to infection. Similarly, eiger and Unpaired3, which encode two cytokines secreted by hemocytes display a resilience phenotype with a strong susceptibility to T. ratisbonensis.
Introduction
Microsporidia are a group of more than 1,400 species of obligate intracellular parasites placed at the root of the fungal kingdom that are able to infect a large array of animal hosts, vertebrates and invertebrates (1). These pathogens are well adapted to their hosts, as attested by their reduced genomically-encoded metabolism and in some cases highly compacted genomes (2). Indeed, these parasites have lost numerous metabolic pathways, such as de novo synthesis pathways for amino acids, nucleotides; importantly, they lack mitochondria (2–5). Microsporidia infect their hosts by delivering the spore content directly or indirectly into the cytoplasm of the host cell through a polar tube (6). Next, the sporoplasm forms meronts in the host cell cytoplasm that multiply until filling the cell prior to differentiating into sporonts, sporoblasts, and then into mature spores. However, the signals triggering the eversion of the polar tube from the spore are generally scarcely known although several stimuli such as hydration or ultraviolet light have been described for some microsporidia species (7–9).
In humans, microsporidia essentially behave as opportunistic parasites. Human infections have increased at the turn of the century due to increased rates of immunodeficient AIDS patients, and microsporidia infections mostly affected the gut and/or the brain (10). Other microsporidia species like Nosema cerenae may contribute to honeybee colony collapse disorder alongside other factors (11). Indeed, it was reported that honeybees succumb even faster when also exposed to sub-lethal doses of pesticides, e.g., the phenyl pyrazole fipronil (12). Some species are also inducing the formation of giant specialized structures called xenomas, syncytia resulting from the fusion of infected host cells that become parasite-producing factories (13, 14).
In vertebrates, both innate and adaptive immune responses have a role in controlling microsporidia infections. Innate immune cells such as, γδT cells, natural killer cells (NKs), macrophages and dendritic cells (DCs) are able to partially eliminate and/or present the spores to lymphocytes. However, the host requires the adaptive immune responses including cytototoxic T lymphocytes (CTL) and humoral immune responses to completely eliminate the parasite (15). Classically activated (M1) macrophages were shown to reduce Encephalitozoon infection (16–18) through a process that involves reactive nitrogen species as well as reactive oxygen species (ROS) (17, 19, 20). Macrophages can also secrete chemokines in response to microsporidia, which relies on the TLR2-NF-κB signaling pathway (21). Reports also highlight the important roles of IFN-γ and IL-12 in DCs response upon infection (22, 23), and the role of NKs (24). Moreover, several antimicrobial peptides were shown to have an effect on spore germination while reducing the infection of enterocytes (25). Finally, T-cell mediated immunity was shown to confer a critical protection against microsporidia (26).
In Caenorhabditis elegans, the parasites successfully invade epithelial cells, suggesting that defenses from these cells are not sufficient to control the infection (27). Nevertheless, ubiquitylation components, the proteasome, and autophagy have been shown to limit Nematocida parisii infection (28) while the p38 Mitogen-Associated Protein Kinases (MAPKs) and insulin/insulin-like growth factor (IGF) signaling pathways do not have roles in the resistance against microsporidia. More recently, a transcriptional immune/stress response called the intracellular pathogen response (IPR) has been shown to be triggered by microsporidia or viral infections as well as by proteotoxic stress and mutations in a purine nucleoside phosphorylase enzyme. These distinct activation pathways converge on the ZIP-1 transcription factor, which likely acts through the gene pals-5 in the intestine (28–31). While these studies constitute major advances in our understanding of defenses against microsporidia in protostomes, the IPR may be specific to nematode worms. Indeed, most genes involved in the IPR do not appear to have homologues in the D. melanogaster genome.
Some host defenses such as the Toll pathway-mediated systemic immune response, phagocytosis, melanization or autophagy are believed to fight microsporidia infection in other invertebrates. Nosema bombycis is a pathogen of the silkworm Bombyx mori that contributed to “pébrine”, which caused important economic losses to the silkworm industry in the 19th century. Transcriptomic studies revealed that potential host defenses including autophagy, oxidative stress, Toll, JAK-STAT and antimicrobial peptides (AMPs) are induced upon the infection while melanization was suppressed, suggesting that these pathways might be involved in the fight against these intracellular parasites in the silkworm (32–34). In Aedes aegypti, a study showed that antimicrobial defensins are upregulated upon Vavraia culiculis infection (35). Moreover, infected honeybees exhibited an increase in midgut oxidative stress (36–38). However, the parasites are able to suppress honeybee immunity as shown by the downregulation of genes coding for serine proteases, glucose dehydrogenase, lysozyme, GMC oxidoreductase, AMPs, dopa decarboxylase and catalases (36, 39). In Drosophila, one transcriptomic study showed that genes potentially involved in the host defenses, such as lysozyme and a scavenger receptor from the CD36 family coding genes, were induced upon ingestion of Octosporea (40). The biology of the intestinal Octosporea infection has not been described, thus limiting the interpretation of these data.
Tubulinosema ratisbonensis has been identified in a laboratory colony of Drosophila melanogaster (41). In vitro, the parasite is also able to infect insect and human cells (42). Previously, we described that T. ratisbonensis hijacks a specific metabolite playing a key role in the biosynthesis of triglycerides, phosphatidic acid, and thereby enhances its proliferation (43). In our infection model, we inject a controlled dose of spores directly into the fly hemolymph, perhaps inducing several immune pathways before intracellular infections of host tissues including the fat body occur. Few intracellular responses are known in Drosophila and have been essentially described for some bacterial and viral infections. Of note, adult Drosophila flies do not appear to be infected per os by Tubulinosema species, in contrast to larvae (44).
Drosophila melanogaster is often used as a model to study host-pathogen interactions in the framework of bacterial, viral, or fungal infections. Indeed, the fruit fly is able to fight most of infections through a variety of immune defenses including the systemic immune response (Toll, IMD, JAK-STAT & JNK pathways), the local immune response (AMP expression & ROS production by barrier epithelia) and the cellular immune response (phagocytosis, opsonization, encapsulation, coagulation and melanization) (45). In addition, some intracellular defenses have been documented in vivo such as Peptidoglycan Recognition Protein-LE (PGRP-LE)-mediated xenophagy of Listeria monocytogenes (46) and the Dicer2-Ago2 RNAi (47) as well as the Sting-Relish pathways as antiviral defenses (48–50). However, how Drosophila or insects reacts to microsporidia infection remains poorly explored (51). Furthermore, almost no infection models implicating a eukaryotic intracellular pathogen are described in this model insect. As the impact of microsporidia on economically important invertebrates (e.g., silkworm, shrimps) and broadly on insect populations (e.g., honeybees, mosquitoes) has been increasing over the years, using Drosophila melanogaster and its genetic tools and knowledge will be helpful to investigate insect host-defenses to the parasite. Indeed, a current limitation of the study of these defenses is that they rely essentially on descriptive transcriptomic and proteomic studies (51, 52) but are rarely followed up by experimental characterization. In Drosophila, it is easier to directly test the functional relevance of specific host defenses by genetic loss-of-function approaches. For instance, it is possible to silence gene expression in a specific tissue or cell-type by transgenic RNA interference (RNAi) (53). Alternatively, classical genetic mutants can be used when available and not affecting essential genes. We shall generically refer to RNAi or classical mutants simply as mutants. If a gene is specifically involved in resistance to T. ratisbonensis infection, the expectation is that the cognate mutant line will display a higher susceptibility to this infection in survival experiments and also an increased parasitic burden due to the heightened proliferation of the pathogen in immunodeficient flies. In contrast, if a gene is required in disease tolerance or resilience (54–56), homeostatic processes that help the host cope with damages inflicted during infection, its corresponding mutant will also display an increased sensitivity to the parasite but without any clear-cut impact on the microsporidial load (57). Alternatively, if the gene product is used by the parasite to enhance its infectivity, survival and microbial loads opposite to those of resistance mutants are expected.
Here, we functionally test using a genetic approach the known facets of the adult Drosophila melanogaster host defense to infections by its natural intracellular parasite T. ratisbonensis in a spore injection model. Unexpectedly, we found that most of the immune defenses including Toll, IMD, JAK-STAT, JNK, xenophagy, RNAi, STING, melanization & complement-related thioester-containing proteins (TEPs) are not required to control the parasite. The exception is phagocytosis that is effective to some degree against the parasite. We however did not identify receptors involved in the specific recognition of the parasite in Drosophila. Some signal transduction pathways yielded rather ambiguous phenotypes and we have observed in several instances an uncoupling between the survival phenotype and the microsporidial burden. Finally, some host defenses such as melanization are paradoxically required for the parasite to proliferate.
Materials and Methods
Parasite Culture
The microsporidia T. ratisbonensis was propagated and harvested as described (42, 58). The human lung fibroblast (MRC-5) cells used for this purpose (a gift from Thomas Baumert) were grown in DMEM + GlutaMAX (Gibco), supplemented with 10% (v/v) FCS and 1% (v/v) PenStrep (Invitrogen) in a tissue culture incubator under 5% CO2 at 37°C. The cell culture was intermittently tested for the presence of mycoplasma. The MRC-5 cell line was not authenticated and does not appear in the databases of commonly misidentified cell lines maintained by ICLAC and NCBI Biosample.
Fly Strains
Fly lines were raised at 25°C with 60% humidity on a standard medium composed of 25 L of sterile water containing 1.2 kg cornmeal (Primeíal), 1.2 kg glucose (Tereos Syral), 1.5 kg yeast (Bio Springer), 90 g nipagin (VWR Chemicals) diluted into 350 ml ethanol (Sigma-Aldrich), and 120 g of agar-agar (Sobigel). Female flies were used in all experiments.
For experiments using mutant flies, wA5001, w1118, yw, or Canton S flies were used as wild-type controls as needed. MyD88c03881, Tep3 and Tep4 mutants were isogenized in the wA5001 background (59). To silence gene expression ubiquitously or specifically in the fat body or in hemocytes, Ubi-Gal4-Gal80ts, Yolk-Gal4, or Hml-Gal4-Gal80ts virgin females were respectively crossed to males carrying relevant UAS-RNAi transgenes from the Vienna Drosophila RNAi Center (VDRC) or from the Transgenic RNAi Project (TRiP) at Harvard Medical School (Boston, MA, USA) (53, 60). For the VDRC RNAi flies, control w1118 (no. 60000) were used for the GD construct and control w1118 (no. 60200) were used for shRNA lines. For TRiP RNAi flies, the control flies were mCherry VALIUM20 (no.35785). To check autophagy flux, we crossed Ubi-Gal4-Gal80ts virgins females with males carrying UAS-GFP-mCherry-Atg8a; the fusion of autophagosomes with lysosomes is quenching GFP fluorescence by the acidic hydrolases, resulting in red autolysosomes (61). Trans heterozygous Atg7d14/Atg7d77 mutant flies were generated as described (62). To generate hemoless flies, virgin females Hml-Gal4-UAS-GFP were crossed with males carrying both UAS-rpr and UAS-hid transgenes. Crosses done with Ubi-Gal4-Gal80ts or Hml-Gal4-Gal80ts were launched at 18°C and the progeny was harvested and kept at 29°C for 7 days before performing the experiment. Crosses done with Yolk-Gal4 were launched at 25°C. For the generation of hemoless flies, crosses were launched and kept at 29°C during all developmental stages. Efficiency of hemocytes ablation was controlled by checking Hemocytes-GFP signal under the fluorescent microscope (Figure S5A). The effectiveness of autophagy inhibition was checked by performing starvation experiments (in mutant or knockdown flies). We decided to work mostly on Atg7 lines as they were the ones displaying the strongest phenotype in starvation experiments. Using the proper wild-type control in all experiments was important as the fly genetic background impacts the fly resistance to the spores. All fly lines used in this study and their origins are described in Table 1.

Table 1 Summary of fly lines used in this study.
Microsporidia Infection
For microsporidia infection, spores were stored in PBS at 4°C. Microsporidia spores were injected into the thorax, precisely into the mesopleuron on adult flies at a concentration of 2,000 spores (unless indicated otherwise) in 9.2 nl PBS containing 0.01% Tween20 using a microcapillary connected to a Nanoject II Auto-Nanoliter Injector (Drummond). The same volume of PBS-0.01% Tween20 was injected for control experiments. Experiments were performed at 25°C or 29°C depending on fly strains used.
For the feeding experiments, spores were resuspended in 100 mM sucrose solution to obtain a solution of 2.105 spores/mL. Flies were exposed to 200 µL of solution that was added in Eppendorf caps, which were placed at the bottom of medium-size vials (3.5 cm diameter). A 100 mM sucrose solution was used as a control. Experiments were performed at 29°C and flies were switched back to food after one day of exposure.
Survival Tests
Survival tests were performed using 10 to 20 flies per vial in biological triplicates per experiment. Adult flies used for survival tests were 5–7-days old from 25°C stock. For survival tests using RNAi-silencing genes, flies were kept for 7 days more at 29°C to express the RNAi prior to the experiment. Flies were counted every day. The number of independent experiments is specified in each figure legend.
Parasite Quantification
Parasite quantification was determined using five adult flies per condition. Flies were transferred into 2-mL microtubes (Starstedt) containing five 1.4-mm ceramic beads (Dominique Dutcher) in 200 μl proteinase K solution and crushed using the Precellys 24 Tissue homogenizer (Bertin Technologies). Total genomic DNA was extracted using the NucleoSpin 96 Tissue kit (Macherey-Nagel) according to the manufacturer’s instructions. Samples of genomic DNA were diluted to 1/10 into milliQ water. Quantification was performed by qPCR using SYBR Green (Bio-Rad) as described previously (58). Only primer couples with over 90% efficiency were used. Data were normalized 1) using the Drosophila ubiquitous gene RpL32 (encoding ribosomal protein 49) 2) using flies frozen on the day of injection and 3) using wild-type controls. Primers used for RpL32 were the forward 5’-GACGCTTCAAGGGACAGTATCTG-3’ and the reverse 5’-AAACGCGGTTCTGCATGAG-3’. Primers used for T. ratisbonensis were the forward 5’- TCTCACAGTAGTGGCGAATG-3’ and the reverse 5’-AACACCGTATTGGAATACAG-3’.
Antibody Production
Polyclonal rabbit antiserum raised against T. ratisbonensis spores was produced as described (74).
Triacylglyceride Quantification
Triglycerides (TAGs) were quantified on samples of five adult flies per condition. Flies were transferred into 2 mL Micro tube (Starstedt) containing five 14 mm ceramic beads (Dominique Dutcher) and mixed using the Precellys 24 Tissue homogenizer (Bertin Technologies). Total TAGs were extracted using the Triglyceride Colorimetric Assay Kit (Cayman Chemicals, #10010303) according to the instructions of the manufacturer.
Stainings
Fat body dissections were performed on flies expressing GFP-mCherry-Atg8a. Flies were cut transversally with a scalpel on a petri dish cleaned with 70% ethanol to observe the fat bodies. Fat bodies were fixed with PFA 8% and mounted on diagnostic microscope slides (Thermo Fisher Scientific) in Vectashield with DAPI (Vector Laboratories).
To observe hemocytes, wild-type larvae injected with untreated or heat killed spores (treatment at 100°C for 15 minutes) were opened 6 hours after injection in a drop of 1x PBS directly on diagnostic microscope slides. After dissection, samples were left for 30 minutes to settle the cells on the slides. Hemocytes were fixed with 8% PFA, permeabilized for 15 min with 1x PBS and 0.1% Triton X-100. Samples were blocked for 2h in 1x PBS, 0.1% Triton X-100 and 2% BSA (PTB). Hemocytes were incubated in PTB plus the primary rabbit antibody anti-Tr spores (1/500) and 10 μM of FITC phalloidin (Sigma-Aldrich #P5282). Samples were washed for 15 min with 1x PBS and 0.1% Triton X-100. Cells were incubated for 2h on PTB plus the secondary goat antibody anti-rabbit coupled to Cy3 (1/500) (Invitrogen #A10522). Hemocytes were washed for 15 min with 1x PBS and 0.1% Triton X-100 and mounted in Vectashield with DAPI (Vector Laboratories). All samples were observed using a LSM 780 confocal microscope (Zeiss).
Fluorescence Quantification
Fluorescence was quantified using the ImageJ program. Channels were separated and analyzed by measuring the fluorescent signal intensity for each channel. For autophagy quantifications, a ratio of GFP/mCherry signal intensity was measured.
Injection of GPIs
1 mM of chemically synthesized GPIs structures (GIcN-IP or Man4GIcN-IP) (75) or 104 T. ratisbonensis spores were injected into pDipt-LacZ reporter flies.
Latex Beads Injection
Adult wA5001 flies were injected with 69 nL of latex beads solution to saturate hemocytes as described (76). Flies were also injected with 69 nL of 1x PBS as a control. We checked the efficiency of phagocytosis blockage by injecting pHrodo-labeled Escherichia coli (Figure S5B).
Cytochalasin D Injection
Cytochalasin D (Sigma-Aldrich #PC8273) was resuspended in DMSO to achieve a 1 mg/mL stock solution and eventually diluted in 1x PBS to get a working solution at 20 µg/mL. Cytochalasins are metabolites obtained from fungi, which act as mycotoxins by blocking actin polymerization. We inhibited hemocytes activity by injecting 69 nL of cytochalasin D solution and mock-injected flies were injected with a PBS solution containing 2% DMSO.
pHrodo Injection
Adult flies were injected with killed E. coli coupled to pHrodo Red (Thermo Fisher Scientific) as described (77). Hemocytes were stimulated by injecting 69 nL of pHrodo Red coupled E. coli. The fluorescence of pHrodo Red increases as pH decreases indicating when pHrodo is located into the mature phagosome with its acidic pH. At neutral or basic pH, pHrodo Red is non-fluorescent. The same volume of 1x PBS was injected as a control.
H2O2 Measurements
H2O2 measurements were performed on 4x5 flies using the hydrogen peroxide assay kit (Sigma-Aldrich #MAK165) following the instructions provided by the supplier.
Statistical Analyses
All graphs and statistical tests were performed using GraphPad Prism. The statistical test used for the survival curves was Log-rank. For parasite load experiments, Mann-Whitney, unpaired t-test or one-way ANOVA were used. For autophagy, H2O2 and TAGs quantifications experiments Kruskall-Wallis or one-way ANOVA test were performed. When performing parametric unpaired t-tests, a Gaussian distribution of data was checked using either D’Agostino-Pearson omnibus or Shapiro-Wilk normality tests. The number of stars (*) represents the P values P≥0.05 (ns), P<0.05 (*), P<0.01 (**), P<0.001 (***) and P<0.0001 (****).
Results
The Systemic Immune Response Toll & IMD Pathways Are Not Effective to Restrict T. ratisbonensis Infection
The injection of microorganisms within the body cavity usually triggers one or both NF-κB-type pathways; Toll or IMD. We have previously reported that we did not observe any consistent induction of these pathways by measuring the expression of AMP genes by RTqPCR, in keeping with the absence of peptidoglycan or ß-glucans in microsporidia (43, 78). However, we occasionally observed signals in some fat body lobules with a pDipt-LacZ reporter transgene but not with a pDrosomycin-GFP reporter (Figure S1A). We therefore wondered whether other constituents of the spore cell wall might be detected, albeit weakly, by the innate immune system. A class of compounds present on the surface of parasites detected by the vertebrate innate immune system are glycosylphosphatidyl-inositols (GPIs) that anchor some surface proteins to the cytoplasmic membrane. We therefore wondered whether GPI anchors might also elicit the Drosophila systemic immune response. To identify the type of GPIs potentially present on the surface of T. ratisbonensis spores, we have incubated a T. ratisbonensis-specific antiserum on a chip that displays some synthetic GPIs (Figure S1B). Whereas sera directed against the human-infecting microsporidia species yielded a weak positive signal with EtN-Man4GlcN-PI, we observed a strong signal with two epitopes, GlcN-PI and Man4GlcN-IP as well as milder signals with some other GPIs (Figure S1C). The injection of the two synthetic GPIs that yielded a strong signal did not however induce a consistent expression of the pDipt-LacZ reporter transgene as observed after an Escherichia coli challenge (Figures S1A, D).
We next examined the survival and parasite titers of Toll pathway mutant MyD88c03881 flies following the injection of T. ratisbonensis. MyD88c03881 infected flies were not more sensitive to the infection compared to wild-type flies (Figure 1A) and both wild-type and mutant flies exhibited a similar parasite load (Figure 1B). The other major NF-κB pathway in Drosophila, Immune deficiency (IMD), also did not appear to be involved in the host defense against T. ratisbonensis since kenny mutant and wild-type flies displayed similar survival curves and parasite burden (Figures 1C, D). Additionally, we decided to test a mutant fly line in which all major known AMP genes have been deleted (65). The survival and parasite load of these ΔAMPs mutant infected flies were similar to those of wild-type flies (Figures 1E, F).
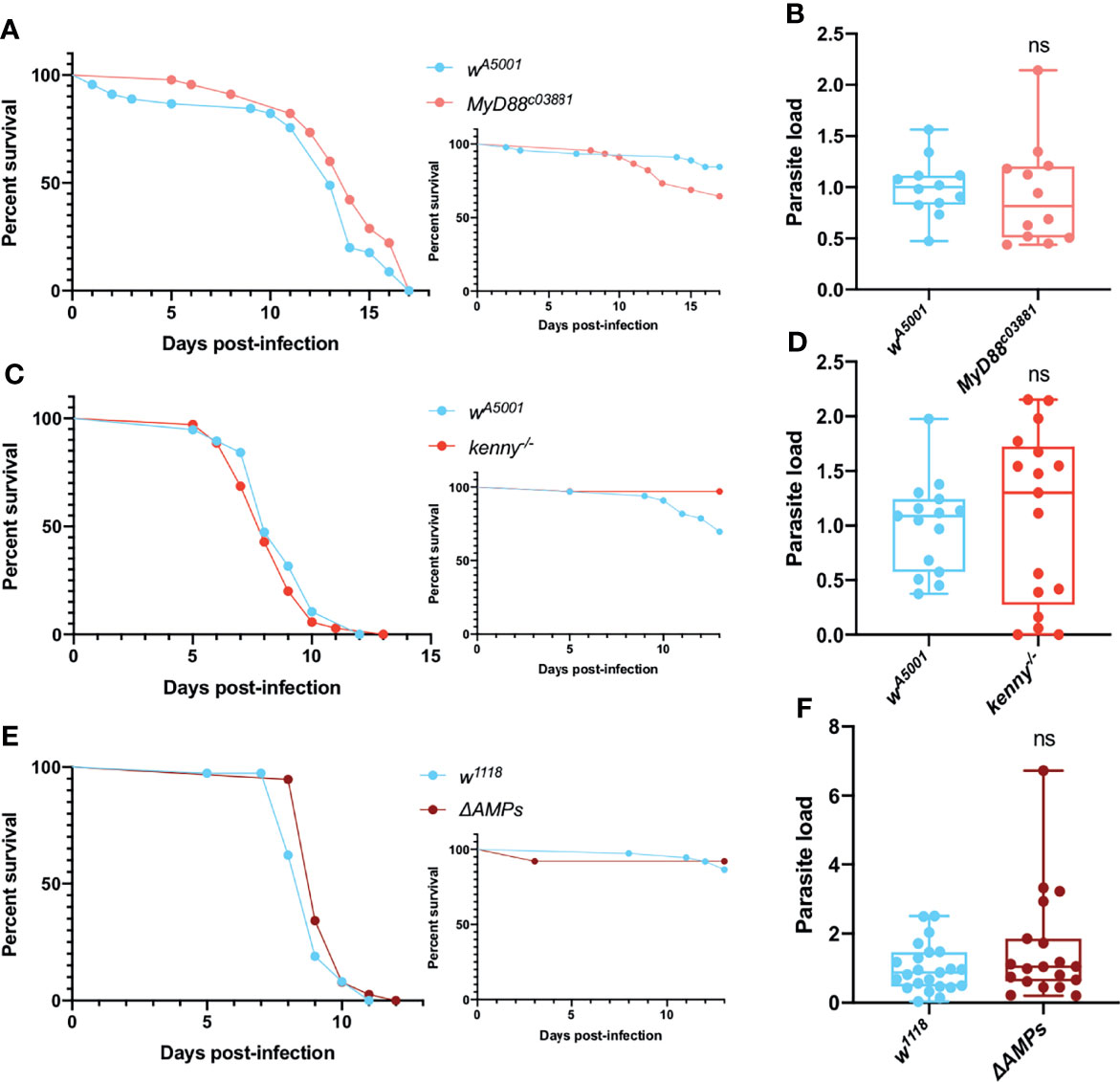
Figure 1 Toll and IMD pathways are not involved in parasite control. (A, B) Survival curve and relative parasite load measured by qPCR at 9 days post-infection of isogenized Myd88c03881 mutant flies infected by T. ratisbonensis. (C, D) Survival curve and relative parasite load measured by qPCR at 6 days post-infection of kenny mutant flies infected by T. ratisbonensis. (E, F) Survival curve and relative parasite load measured by qPCR at 6 days post-infection of ΔAMPs mutant flies infected by T. ratisbonensis. The silencing of genes involved in the Toll & IMD pathways was not impacting either fly survival or parasite loads, which was confirmed by testing the ΔAMPs mutant. Experiments were performed at 25°C (A, B) or 29°C (C-F) on initially 5-7 day-old female flies. The inset graphs display survivals of control noninfected flies injected with PBS. Each survival graph is representative of at least three independent experiments. Parasite load graphs represent the pooled data of at least three independent experiments. Survival data were analyzed using the log-rank statistical test. qPCR data were analyzed using an unpaired t-test. ns, not significant.
Taken together, these data suggest that the Toll and IMD pathways do not significantly contribute to Drosophila host defense against T. ratisbonensis.
Ambiguous Roles of Oxidative Stress and Stress Response Pathways in Host Defense Against T. ratisbonensis
Physical or biological stresses such as exposure to ROS or infections are also known to activate MAPKs pathways in Drosophila. We first silenced by RNAi the Jun kinase (JNK) gene basket in the fat body. Even though the silenced flies behaved as control flies in survival assays (Figure 2A), they consistently exhibited a decreased parasitic burden (Figure 2B). In contrast, when ubiquitously knocking-down the apoptotic signal-regulating kinase 1 Ask1 or the MAPK p38b, variable survival curves were observed, from increased sensitivity to improved resistance against T. ratisbonensis infection (Ask1) or increased sensitivity to no phenotype (p38b) (Figure 2C and Figures S2A, B). The parasite load was equal in p38b knockdown flies whereas it was somewhat increased after Ask1 knockdown (Figure 2D).
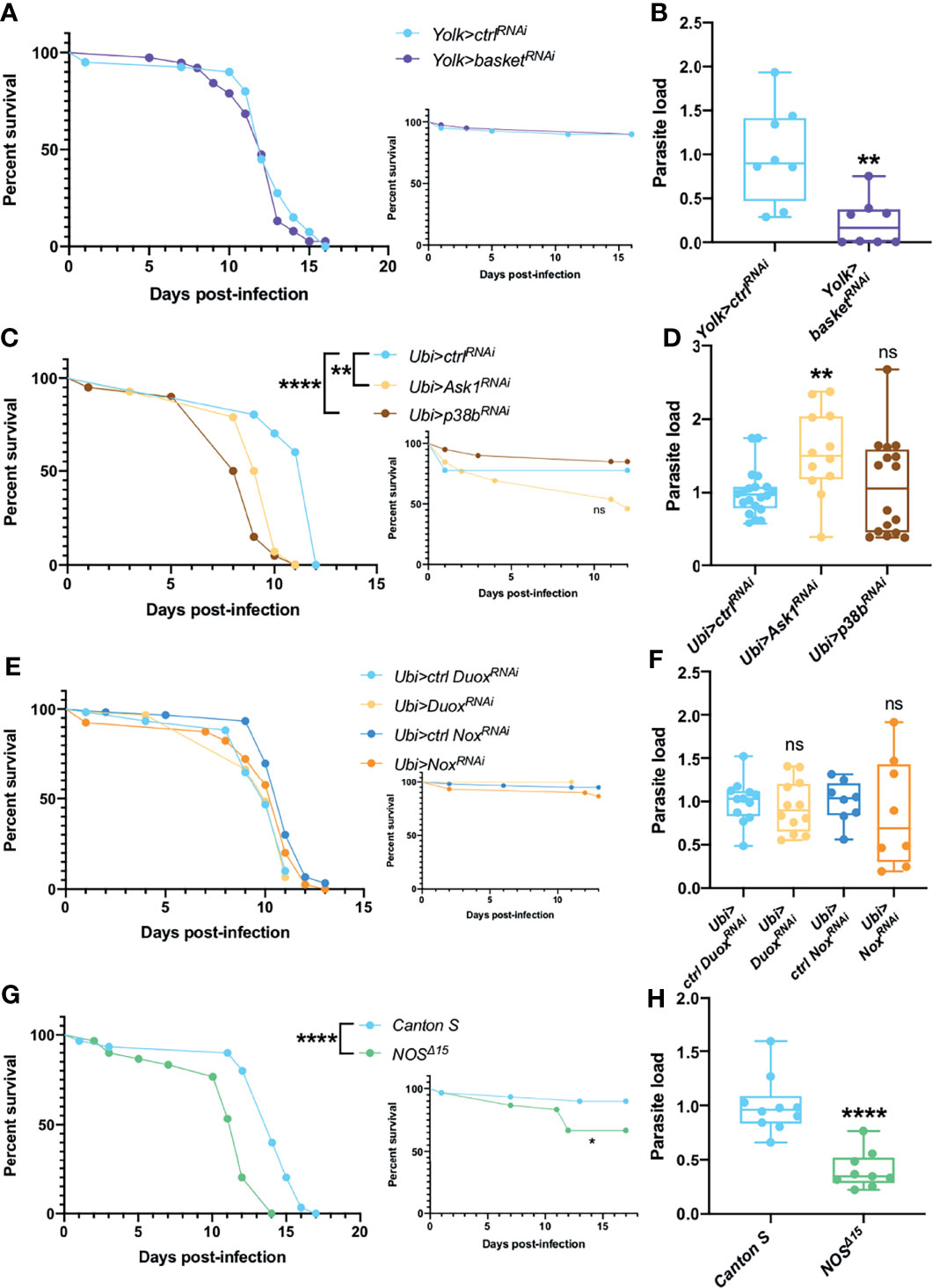
Figure 2 JNK, ROS, NOS & NOX are not involved in parasite control. (A, B) Survival test and relative parasite load measured by qPCR at 6 days post-infection of flies infected with T. ratisbonensis after basket RNAi knockdown driven in the fat body using a Yolk-Gal4 driver. Downregulating the expression of basket did not affect fly survival, but decreased the parasite load. (C, D) Survival test and relative parasite load measured by qPCR at 6 days post-infection of flies infected with T. ratisbonensis after Ask1 or p38b RNAi knockdown in the whole fly using a Ubi-Gal4-Gal80ts driver. Downregulating the expression of Ask1 or p38b negatively affected fly survival but only the RNAi knockdown of Ask1 increased the parasite load. (E, F) Survival test and relative parasite load measured by qPCR 6 days post-infection of flies infected with T. ratisbonensis after Duox or NOX knockdown using a Ubi-Gal4-Gal80ts driver. (G, H) Survival test and relative parasite load measured by qPCR 9 days post-infection of NOSΔ15 mutant flies infected with T. ratisbonensis. Silencing of Duox or NOX did not reveal any immune defense phenotype but NOSΔ15 mutant flies exhibited higher sensitivity in survival (4/4 experiments) and lower loads. Experiments were performed at 29°C (A-F), except for experiments shown in G-H that were done at 25°C, on initially 5-7 day-old female flies. The inset graphs display survivals of control non-infected flies injected with PBS. Each survival graph is representative of at least two to three independent experiments, except for Ask1 and p38b, as survivals were highly variable (Figures S2A, B). Parasite load graphs represent the pooled data of at least two independent experiments. Survival data were analyzed using the log-rank statistical test. qPCR data were analyzed using an unpaired t-test. The number of stars (*) represents the P values P≥0.05 (ns), (**), and P<0.0001 (****). ns, not significant.
We have also tested whether enzymes that generate ROS or Reactive Nitrogen Species such as the Nitric Oxide Synthase NOS are involved in the host defense against T. ratisbonensis. The ubiquitous silencing of NADPH-oxidase genes Duox or Nox in the whole fly had no significant impact on fly survival and parasite load (Figures 2E, F) and quantifying H2O2 levels upon infection did not present any phenotype (Figure S2C). However, NOS null mutants were more susceptible to the infection (Figure 2G). Unexpectedly, the parasite load was significantly decreased (Figure 2H). Taken together, these data suggest that the ROS response is not involved in parasite control. In contrast, there is an uncoupling between the phenotypes of survival and parasite loads when the basket, Ask1-p38 or the NOS genes are affected.
Xenophagy Does Not Control T. ratisbonensis Infection
Autophagy is involved in the elimination of damaged endogenous components such as defective mitochondria as well as the removal of exogenous material in a process called xenophagy when dealing with invading pathogens. In Drosophila, it has been shown that L. monocytogenes is inducing xenophagy through PGRP-LE recognition (46). As microsporidia are intracellular parasites and proliferate mostly in fat body cells by stealing lipids (43), autophagy might be involved in parasite control either directly by xenophagy or indirectly by regulating the access to lipid stores by lipophagy.
To study the role of PGRP-LE we used two different strategies. We silenced PGRP-LE expression in the whole fly and we used PGRP-LE112 null mutant flies. For both type of mutations, we observed an increased resistance to infection that correlated well with a decreased parasitic burden (Figures 3A–D). As T. ratisbonensis proliferates by preying onto host lipids, we also checked triacylglyceride (TAG) levels. The TAG reserves were intact in uninfected PGRP-LE silenced flies. Unexpectedly, whereas control flies effectively exhibited depleted TAG stores upon T. ratisbonensis infection, this was no longer the case when PGRP-LE was silenced (Figure 3E). One interpretation of these data is that PGRP-LE is required by the parasite to hijack host lipid reserves.
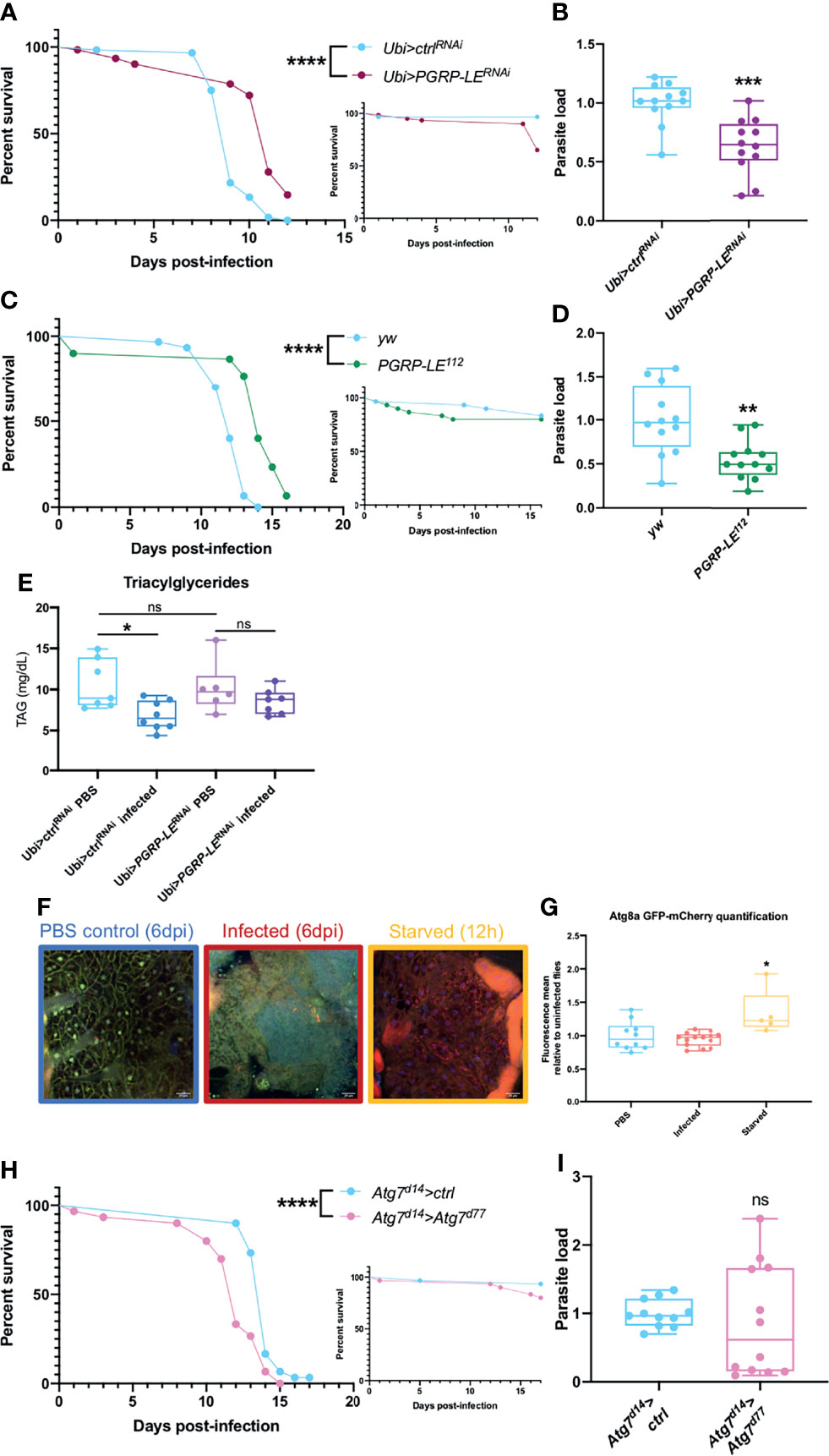
Figure 3 Xenophagy is not involved in parasite control. (A, B) Survival test and relative parasite load measured by qPCR 6 days post-infection of flies infected with T. ratisbonensis after PGRP-LE knockdown using a Ubi-Gal4-Gal80ts driver. (C, D) Survival test and relative parasite load measured by qPCR 9 days post-infection of PGRP-LE112 mutant flies infected with T. ratisbonensis. Performing a knockdown of PGRP-LE or using a mutant line improved fly survival and reduced parasite loads. (E) TAGs quantification on PGRP-LE knockdown infected flies. (F) Confocal pictures of GFP-mCherry-Atg8a uninfected flies 6 days post-injection (left), infected with T. ratisbonensis 6 days post-infection (middle), or starved flies after 12 hours on water (right). Blue = nuclei; yellow = autophagosomes; red = autolysosomes (G) Quantification of the ratio of mCherry fluorescence over GFP fluorescence (flies from the experiment shown in F). (H, I) Survival and relative parasite load measured by qPCR at 7 days post-infection of Atg7 trans heterozygous mutant flies infected by T. ratisbonensis. Autophagy was not induced upon T. ratisbonensis infection and flies lacking functional autophagy were more sensitive to the infection, which was not correlated with a higher load. Experiments were performed at 29°C except for experiments shown in (C, D) that were done at 25°C, on initially 5-7 days old female flies. The inset graphs display survivals of control non-infected flies injected with PBS. Each survival graph is representative of at least three independent experiments. Parasite load graphs represent the pooled data of at least three independent experiments. Autophagy flux quantification graph is a merge of two independent experiments. TAG graph represents the pooled data of two independent experiments. Survival data were analyzed using the log-rank statistical test. qPCR data were analyzed using an unpaired t-test. Autophagy quantification was analyzed using a Kruskall-Wallis test. TAG graph was analyzed using one-way ANOVA statistical test. The number of stars (*) represents the P values P≥0.05 (ns), P<0.05 (*), P<0.01 (**), P<0.001 (***) and P<0.0001 (****). ns, not significant.
As PGRP-LE is connected to autophagy and as autophagy has been reported to be an intracellular host defense against microsporidia in C. elegans (28, 79), we next used a fly reporter GFP-mCherry-Atg8a line (61) to assess whether any autophagic vacuoles form upon invasion by T. ratisbonensis. We did not observe any autophagy induction in microsporidia infected flies compared to uninfected control (Figures 3F, G). To further exclude a role for autophagy during infection, we also tested an Atg7d14/Atg7d77 transheterozygous mutant line and performed survival and parasite quantification. Even though these mutant flies were more sensitive to starvation (Figure S3A), as expected, and to T. ratisbonensis infection (Figure 3H), Atg7 mutant flies displayed an unaltered parasite burden (Figure 3I). To exclude a potential developmental effect in the Atg7d14/Atg7d77 (62), we silenced Atg7 expression solely in the adult fat body. The survival and parasite load of silenced flies was not impaired upon infection compared to control flies (Figures S3C, D), yet, these silenced flies were more sensitive to starvation (Figure S3B).
These results allow us to conclude that T. ratisbonensis proliferation is not controlled by xenophagy in Drosophila. Surprisingly, the parasite needs PGRP-LE to proliferate within the fat body cells, potentially by allowing an efficient depletion of lipidic stores.
Antiviral Pathways Are Not Able to Control Microsporidia
Like viruses, microsporidia are obligate intracellular pathogens. We therefore checked for a potential involvement of antiviral pathways. One of the major antiviral defense in Drosophila is RNAi (47) mediated by Dicer-2. A Dicer-2 null mutant line appeared to be more sensitive to a T. ratisbonensis challenge than a control rescued line (Figure S4A); yet, this may reflect a sensitivity to wounding as PBS-injected flies displayed a similar behavior (Figure S4A, inset). Indeed, the parasite load was equal in the Dicer-2 mutant flies compared to rescued flies (Figure S4B). We conclude that RNA interference is not involved in parasite control.
The cGAS-STING pathway is also involved in the control of intracellular pathogens such as virus or bacteria, through the recognition of nucleic acids. Recently, the antiviral function of the Drosophila ortholog of STING against picorna-like viruses in Drosophila has been shown (48–50). dSTING null mutant flies were not more sensitive to infection compared to wild-type flies and their parasite burden was unaltered (Figures S4C, D). Altogether, we failed to obtain any evidence for an involvement of intracellular antiviral pathways in the host defense against T. ratisbonensis.
Pro-Phenoloxidases (PPOs) Are Required for Parasite Proliferation
Melanization is one of the major host immune response in insects (80). This pathway is dependent on key enzymes: the phenoloxidases (POs), catalyzing the oxidation of phenols to quinones and ultimately polymerizing into melanin at the wounding site. POs are first synthesized as inactive zymogens called proPOs (PPOs) prior to being cleaved to generate active POs. The survival phenotypes of PPO1 and PPO2 mutant flies were opposite, with the former being more resistant and the latter more sensitive to T. ratisbonensis infection (Figure 4B). We note that both mutants exhibit a late, mild sensitivity to injury (Figure 4A). In contrast, the PPO1-PPO2 double mutant was highly sensitive to “clean” wounds in this series of experiments precluding an interpretation of its survival curve when infected (Figures 4A, B). Unexpectedly, the microsporidial burden was lower than in wild-type for all PPO mutants (Figure 4C).
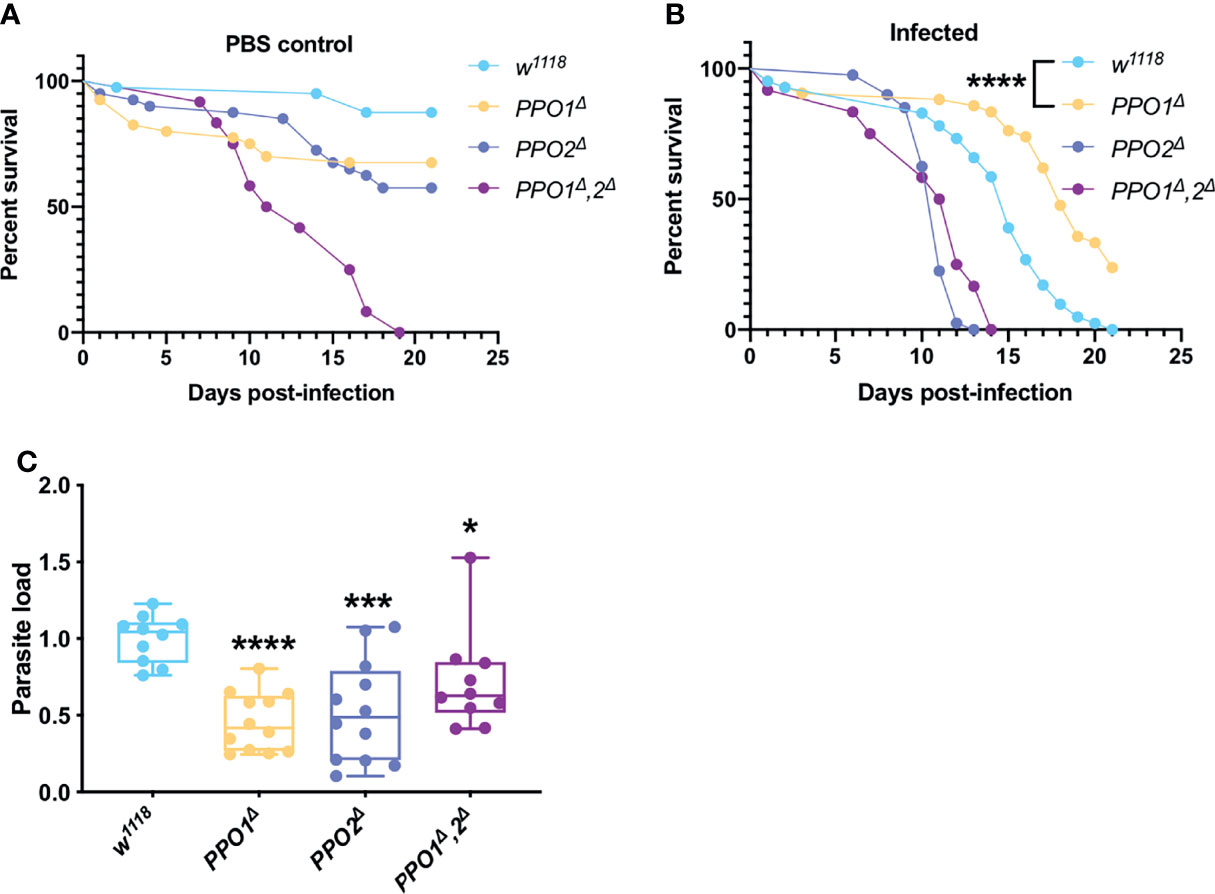
Figure 4 PPOs are required for parasite growth. (A) Survival test of PPO1Δ, PPO2Δ, and PPO1Δ-PPO2Δ mutant flies injected with PBS. (B ,C) Survival tests and relative parasite loads measured qPCR of PPO1Δ, PPO2Δ, and PPO1Δ-PPO2Δ mutant flies infected by T. ratisbonensis. PPO mutant flies were slowly succumbing from PBS injection; however, PPO1Δ mutant flies were more resistant to the infection and this was correlated with a lower parasitic burden, a phenotype found for all PPO mutants. All experiments were performed at 25°C on initially 5-7 day-old female flies. Parasite loads were performed at 9 days post infection. Each survival graph is representative of at least three independent experiments. Parasite load graphs represent the pooled data from three independent experiments. All experiments were also performed using another wild-type control and yielded similar results. Survival data were analyzed using the log-rank statistical test. qPCR data were analyzed using an unpaired t-test. The number of stars (*) represents the P values P≥0.05 (ns), P<0.05 (*), P<0.001 (***) and P<0.0001 (****).
Thus, even though PPO2 appeared to be required in host resilience against T. ratisbonensis, the lack of enhanced parasite load is not consistent with such a conclusion. In contrast, PPO1 is required for an efficient T. ratisbonensis infection, a puzzling inference.
Phagocytosis Is an Essential Defense to Control Microsporidia
In adults, the cellular immune response mainly eliminates invading pathogens through phagocytosis. We first checked that plasmatocytes are able to ingest lived and killed injected T. ratisbonensis spores using confocal microscopy on hemocytes retrieved from injected larvae (Figure 5A). We next used several functional approaches to inactivate phagocytosis in adults. First, we depleted hemocytes in adults by inducing their apoptosis throughout development (Figure S5A). These “hemoless” flies were somewhat more sensitive to infection and displayed an increased microsporidial titer (Figures 5B, C; Figure S5B). Similar results were obtained upon the saturation of the phagocytic apparatus by the prior injection of nondegradable latex beads that are phagocytosed by plasmatocytes (Figures 5D, E). The injection of cytochalasin also blocks phagocytosis and in keeping with the previous results led to an enhanced sensitivity to T. ratisbonensis infection (Figure S5C).
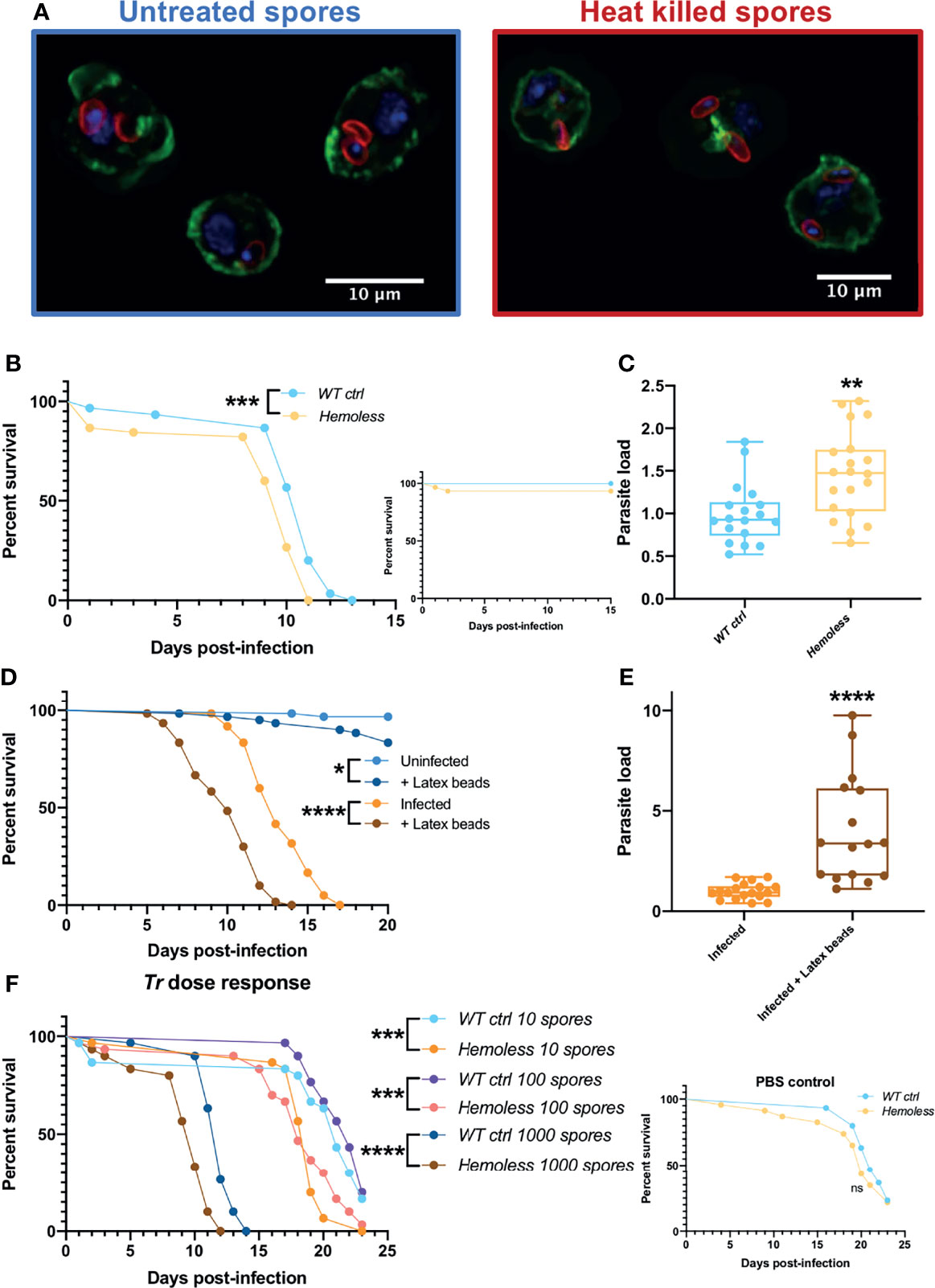
Figure 5 Phagocytosis provides a degree of protection against T. ratisbonensis infections. (A) Confocal pictures of hemocytes extracted from infected larvae with T. ratisbonensis untreated spores (left) or heat killed (right). Blue = nuclei; green = actin; red = spores (B, C), Survival test and relative parasite load measured by qPCR of T. ratisbonensis-infected flies in which apoptosis of hemocytes was induced by crossing an Hml-Gal4-UAS-GFP driver with a UAS-rpr; UAS-hid line (hemoless flies). (D, E) Survival curves and relative parasite load measured by qPCR of flies injected with latex beads to saturate the phagocytic apparatus. (F) Survival curves of hemoless flies injected with low doses of spores (10 to 1000 spores). Blocking phagocytosis, genetically or mechanically, always impaired fly survival and correlated with a higher parasitic burden, indicating a role in resistance to T. ratisbonensis infection. All experiments were performed at 29°C on initially 5-7 day-old female flies. The inset graphs display survivals of control non-infected flies injected with PBS. Parasite loads were performed 6 days post-infection. Each survival graph is representative of at least three independent experiments. Parasite load graphs represent the pooled data of at least three independent experiments. Survival data were analyzed using the log-rank statistical test. qPCR data were analyzed using an unpaired t-test. The number of stars (*) represents the P values P≥0.05 (ns), P<0.05 (*), P<0.01 (**), P<0.001 (***) and P<0.0001 (****).
Wild-type flies do succumb within 10-15 days to the injection of 2000 spores as documented in all experiments shown so far. They also succumb at the same rate to the injection of 1000 spores (Figure 5F). However, when they were injected with lower doses such as 10 or 100 spores on average, they succumbed at the same rate as PBS-injected controls (Figure 5F and inset). In contrast, “hemoless” flies injected with low doses were killed at a significantly faster pace (Figure 5F). These data indicate that the cellular immune response can control low intensity infections but becomes overwhelmed when exposed to higher inocula.
The Uptake of Microsporidia Does Not Require Most Known Phagocytic Receptors
Several potential phagocytosis receptors that mediate the uptake of microbes or apoptotic bodies have been identified in Drosophila (68, 81–85).
We first tested mutant lines for eater, NimA, and Draper. The survival and parasitic loads of eater and NimA mutants were comparable to wild-type controls infected with T. ratisbonensis spores (Figures S6A–D). Unexpectedly, the survival of DrprHP37013 mutant infected flies was improved compared to wild-type controls (Figure S6E) and parasite load was decreased (Figure S6F), which suggests that Draper promotes T. ratisbonensis infection.
We relied on RNA interference induced only at the adult stage to test other potential phagocytosis receptors (or opsonins). Several lines did not display an altered survival to a T. ratisbonensis challenge, those targeting NimB1, NimB2, croquemort (crq), and one NimB4 RNAi line (Figure S6G). Three lines appeared to be more resistant to T. ratisbonensis infection, i.e., those affecting peste (pes), NimB5, and NimB4 (a second RNAi line). No conclusion can be drawn as regards NimC1 as mock-infected controls died as rapidly as T. ratisbonensis-infected RNAi flies. With respect to the microsporidial burden, a trend toward a reduced T. ratisbonensis titer was observed for NimB5, NimB4, and crq (Figure S6H), although the survival of the latter two RNAi lines was not affected. For most other RNAi lines, the measured load was variable from experiment to experiment, as exemplified by the observed bimodal distribution for NimB1, NimB2, NimB4 (the sensitive line), NimC1, and pes (Figure S6H). Thus, the results are difficult to interpret reliably, even though the titers for the pes RNAi line was statistically significant but not correlating to their survival phenotypes (no enhanced sensitivity or protection).
Thioester-containing proteins (TEPs) belong to the superfamily of complement-like factors and have been shown in some instances to act as opsonins in insects (86, 87). We have tested Drosophila null mutant lines affecting either individual Tep genes or removing all of them, except Tep6, which has been shown to function in intestinal epithelium barrier function (88, 89). The individual Tep2, Tep3, and Tep4 lines were isogenized in the wA5001 genetic background while the Tep1 and compound deletion mutant Tep-qΔ were isogenized in the Drosdel w1118 background. All the mutant lines displayed a survival and parasitic burden that were similar to those of their respective controls, except for Tep3 that was slightly more resistant to T. ratisbonensis infection (Figure S7).
In conclusion, we have not identified so far the receptors or putative opsonins that may be required for the uptake of microsporidia by plasmatocytes.
Hemocyte Signaling Does Not Impact Parasite Proliferation But May Improve Fly Resilience to T. ratisbonensis Infection
We cannot formally exclude that the approaches that we have used to block phagocytosis may also impede other functions of hemocytes such as cytokine signaling.
A recent study has documented that Drosophila pupal macrophages cross the blood-brain barrier upon receiving a PDGF-like factor, Pvf2, signal from glial cells that is induced by infection (90). Such signals have also been described for the developmental migration of hemocytes and their invasion of the embryonic epithelium (91, 92). We therefore ubiquitously silenced pvf2 in adult flies and tested their survival and microsporidial burden upon T. ratisbonensis infection and did not observe any difference when compared to control flies (Figures 6A, B).
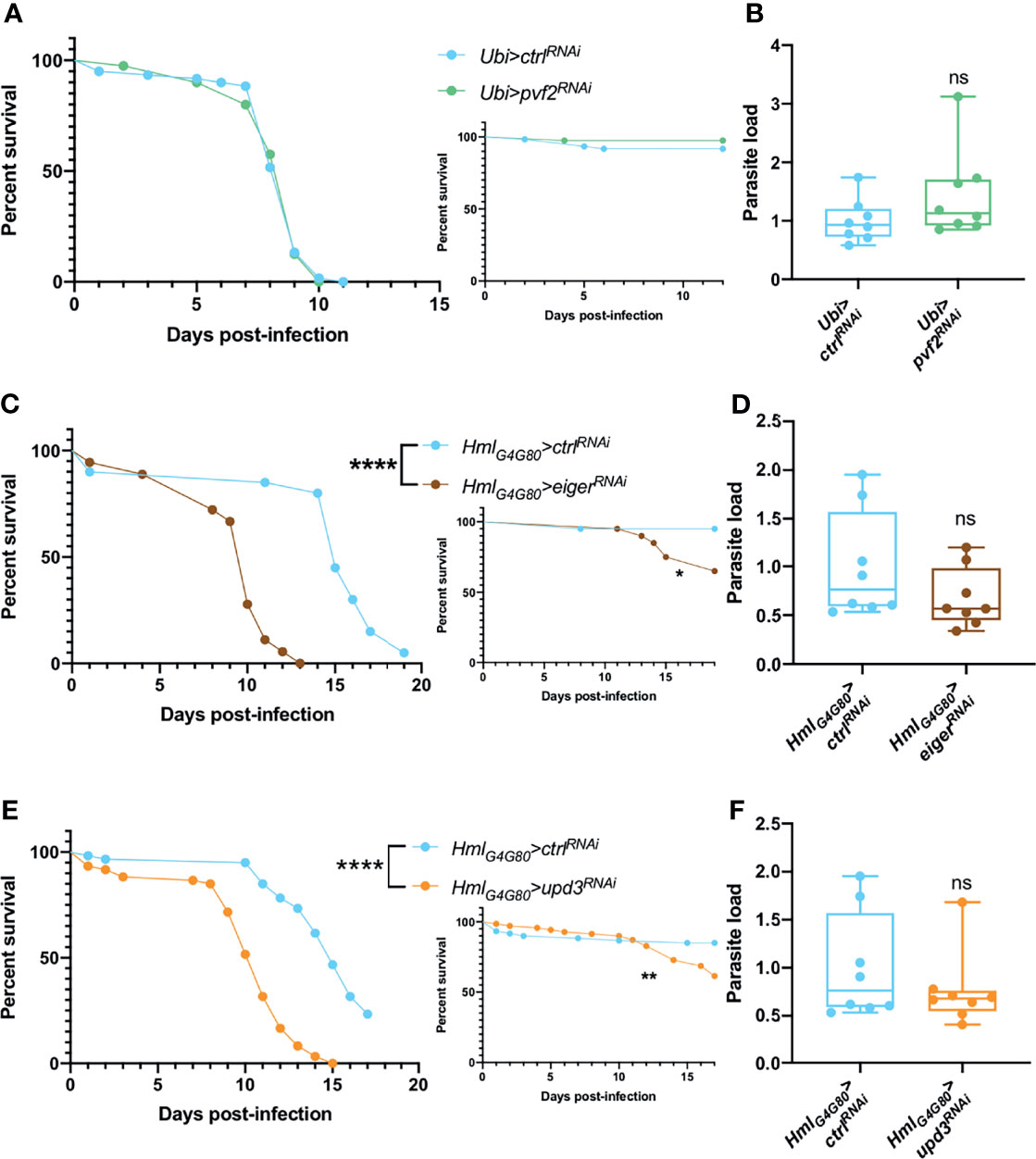
Figure 6 Hemocyte signaling might be involved in fly resilience upon T. ratisbonensis infection. (A, B) Survival test and relative parasite load measured by qPCR of flies infected with T. ratisbonensis after pvf2 knockdown using a Ubi-Gal4-Gal80ts driver. (C, D) Survival test and relative parasite load measured by qPCR of flies infected with T. ratisbonensis after eiger knockdown using a Hml-Gal4-Gal80ts driver. (E, F) Survival test and relative parasite load measured by qPCR of flies infected with T. ratisbonensis after upd3 knockdown using a Hml-Gal4-Gal80ts driver. Survivals and loads performed after the knockdown of pvf2 did not shown any phenotype; however, downregulating eiger or upd3 expression strongly impaired the survival of infected flies compared to wild-type and non-infected controls, without impacting the parasite titer. All experiments were performed at 29°C on initially 5-7 day-old female flies. The inset graphs display survivals of control non-infected flies injected with PBS. Parasite loads were performed at 6 days post infection. Each survival graph is representative of two independent experiments. Parasite load graphs represent the pooled data of two independent experiments. Survival data were analyzed using the log-rank statistical test. qPCR data were analyzed using an unpaired t-test. The number of stars (*) represents the P values P≥0.05 (ns), P<0.05 (*), P<0.01 (**), and P<0.0001 (****).
We next tested the roles of Eiger, a ligand that activates the JNK pathway (93, 94) and UPD3, a cytokine involved in JAK-STAT activation and highly expressed in hemocytes upon septic injury (95). Upon inhibiting eiger or upd3 expression specifically in hemocytes by RNAi, we observed a strongly enhanced susceptibility to T. ratisbonensis infection (Figures 6C, E) that however did not correlate with an increased parasitic load (Figures 6D, F).
Thus, the silencing of either eiger or upd3 in hemocytes yielded the strongest phenotype observed so far in survival experiments, which underscores their importance in the host defense against T. ratisbonensis. The observation that the microsporidial burden is not affected in these mutants argues that these genes are required in resilience and not resistance to T. ratisbonensis infection.
Discussion
We have here systematically tested in a T. ratisbonensis systemic infection model the known Drosophila antimicrobial host defenses using a genetic approach. Contrary to our expectations, we found that only phagocytosis is able to confer a degree of protection against this microsporidial infection (Table 2). Several other host defenses were either not relevant or unexpectedly promoted the infection by the parasite (Table 2). Our work therefore provides a novel perspective on insect host defenses against one obligate intracellular parasite that differs from the picture gained through descriptive transcriptomic or proteomic analyses (51, 52).
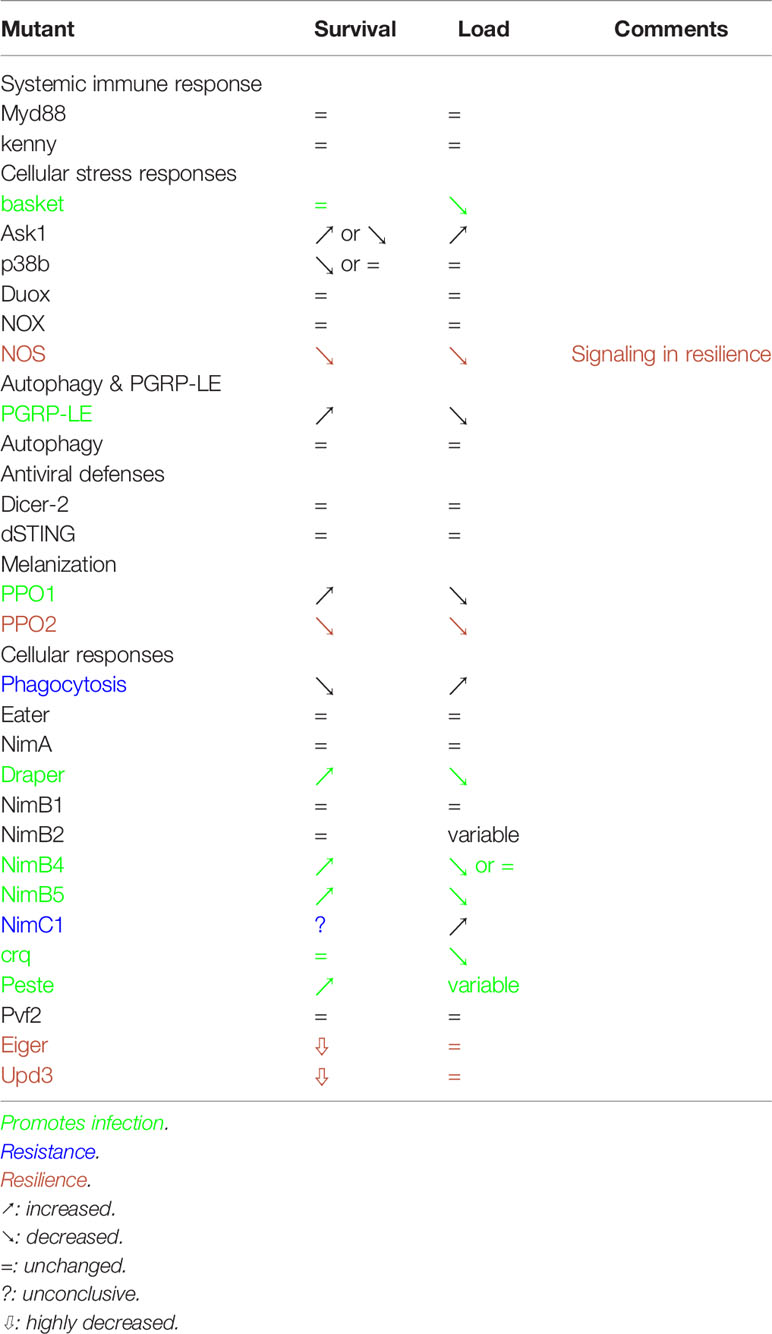
Table 2 Summary of the phenotypes obtained in this study.
Several host defenses do not appear to be required or to be efficient in the Drosophila host defense against T. ratisbonensis infection. This is the case for the systemic humoral immune response mediated by the Toll and IMD pathways that jointly regulate the expression of AMP genes (96). These pathways do not appear to be consistently induced and did not present any altered phenotype to a T. ratisbonensis challenge. Although ROS have been proposed to be important defenses against microsporidial infections, e.g., infections of the intestinal epithelium of honeybees by Nosema ceranae (12), we did not find that the two major enzymes known to secrete ROS extracellularly, NOX and Duox, appeared to play a role against injected T. ratisbonensis spores. In this respect, it is important to note that by using an injection model, we bypass local epithelial barrier defenses that may very well be highly relevant in other infection models. A more definitive answer would be provided by developing a consistent larval stage infection model. The insect complement system plays a primary role against extracellular eukaryotic parasites in insects, for instance against Plasmodium infections in mosquitoes (97–99). Even though injected T. ratisbonensis spores are initially found in the hemolymph, TEP proteins do not appear to confer any protection, possibly because of the original infection mode used by microsporidia in which a polar tube is everted within seconds near or inside target cells, a time scale that may be too rapid for an effective response. In addition, the injected sporoplasm would be shielded by the polar tube from the action of such factors. Our results do not support the possibility that xenophagy contributes to Drosophila host defense against T. ratisbonensis, in as much as several other mutant lines affecting other autophagy genes failed to yield reproducible phenotypes consistent with this possibility (Figures S8A–D). Finally, known antiviral defenses acting intracellularly also do not appear to be involved in the protection against this parasite.
Several genes involved in various aspects of host defense displayed a mutant phenotype consistent with a proactive role in infection and not in host defense. The mutants displayed an enhanced survival rate coupled to a decreased microsporidial load. In none of the cases is it fully clear whether these gene products are actively repurposed by the parasite, for instance through secreted virulence factors, or play a passive role in a process hijacked by the parasite. While it had originally been proposed to act extracellularly, PGRP-LE appears mostly to function as an intracellular sensor of diaminopimelic type of peptidoglycan found in the cell wall of bacilli and Gram-negative bacteria that triggers IMD pathway activation (100–102). It was therefore unexpected to find a phenotype for PGRP-LE since peptidoglycan is not synthesized by microsporidia. A role for the IMD pathway has been ruled out (see above) and we did not find any evidence for a requirement for autophagy, a second function of PGRP-LE against intracellular DAP-type peptidoglycan containing L. monocytogenes (46). Our data suggest a potential role for PGRP-LE in lipid metabolism, a critical resource for parasite growth. We note that PGRP-LE did not display any TAG store alteration in mock-infected flies, thereby excluding a basal role for PGRP-LE in the regulation of lipidic reserves. We cannot however formally exclude that the lower depletion of host lipidic stores results from the lower parasitic burden in PGRP-LE mutants due to independent causes. One open possibility would be that microsporidia systemic infection alter the Drosophila microbiota and that such changes are detected through PGRP-LE, to the parasite’s advantage by an as yet unidentified process.
Several studies have reported that melanization might play a role during infection as PPO genes appeared to be induced by microsporidial infections (33, 103). However, the fact that a gene is induced during microsporidia invasion does not necessarily mean that it plays a role in host defense. One should keep in mind that the induction of a given gene may not necessarily reflect the induction of a host defense but a manipulation by the parasite. This might be the case for PPO1 which appears to promote T. ratisbonensis infection. PPO1 is produced by crystal cells in larvae and released in the hemolymph upon the rupture of the cytoplasmic membranes after a septic injury (104). How PPO1 promotes T. ratisbonensis infection is unclear at present. A first step would be to determine whether its function is required within hemocytes or once PPO1 is secreted in the hemolymph. It does not appear to compete with PPO2, which displays an opposite survival phenotype, because the double mutant also harbors a decreased titer of the parasite.
Draper has first been shown to be involved in the phagocytosis of apoptotic bodies during development (105). One hypothesis is that Draper might be involved in scavenging cell debris resulting from lyzed cells and therefore providing building materials for the parasite. It will be interesting to test whether its signaling function within the Src42A-Shark-Rac1 axis is required as well as determining whether receptors such as Six-microns-under that are involved in efferocytosis, the disposal of apoptotic bodies (106), are also displaying a phenotype similar to that of Draper. In this respect, NimB4 has recently been reported to also participate in efferocytosis (107) and its Draper-like phenotype in our study reinforces this interpretation (Table 2). crq has initially been shown to function in efferocytosis; crq mutants displayed a strongly decreased T. ratisbonensis load, yet their survival was unaltered. The scavenger receptor Peste might play a related role but its phenotype remains uncertain despite its enhanced resistance because of a variability in its T. ratisbonensis burden. NimB5 also promotes the proliferation of the parasite as it displays a mutant phenotype of enhanced resistance to the microsporidial infection that correlates well with a decreased titer of T. ratisbonensis. Although it belongs to the Nimrod superfamily, it is a secreted protein, like NimB4, that has been described to function as an adipokine in starving larvae. It then inhibits the peripheral proliferation of hemocytes as well as their adhesion. How it functions in adults remains to be determined. It is unlikely to regulate the proliferation of hemocytes in adults, which has so far not been convincingly demonstrated (108). It might be released by fat body cells that undergo a starvation-like experience as the parasite depletes its metabolic stores. Its absence in the mutant might be construed to lead to an increased adherence of hemocytes to tissues, a hypothesis difficult to support since most hemocytes are sessile in the adult. In any case, an increased adherence is expected to lower the phagocytic function of plasmatocytes. As described below, interfering with phagocytosis yielded a phenotype opposite to that of NimB5. It will therefore be important to determine the function of NimB5 in the adult, especially during T. ratisbonensis infections.
The one process providing a degree of protection against T. ratisbonensis infection is phagocytosis, which only delays the fatal issue when challenged with some one thousand spores. It however appears to be able to control lower doses. One interesting idea is that it may control parasites that have crossed the intestinal barrier, much like it does for ingested Serratia marcescens (76) or Pseudomonas aeruginosa (109) bacteria. This might explain why intestinal infections are not successful in adults (Figures S9A, B). Our experiments did not support this possibility (Figure S9C). Our analysis of potential phagocytosis receptors such as Eater, NimA or Peste was unsuccessful; however, an increased microsporidial burden in NimC1 mutants might point out to redundant function with Eater (81). Although the different methods we have used to probe cellular defenses all affect the phagocytosis function of plasmatocytes, they might at the same time affect other functions of hemocytes such as the secretion of cytokines (110–112). In this respect, mutants affecting upd3 or eiger exhibited a greatly enhanced susceptibility to T. ratisbonensis in survival experiments, even though the microsporidial burden was not changed. This phenotype is consistent with a resilience function of these cytokine genes. Further studies will allow determining in which tissues the corresponding cytokine receptors are required and thus provide some insight into the homeostatic process that allow the fly to better survive to this intracellular parasite.
The phenotypes of NOS and PPO2 may also suggest a role in resilience, except that the mutants undergo a significantly reduced parasite burden. NOS might lead indirectly to the formation of reactive nitrogen species that are thought to be noxious to pathogens. This possibility is however not compatible with the decreased T. ratisbonensis titer measured in these mutants. Thus, it is likely that NO generated by NOS may diffuse in the organism and fulfill a signaling function as described in mammalian immune systems (113). The function of PPO2 remains enigmatic at this stage. In larvae, it forms the crystal found in crystal cells (67). The ablation of PPO2-expressing hemocytes might reveal whether these cells are required for this resistance-independent defense function of PPO2. The classical view of PPO-mediated melanization killing injected parasites is not relevant in the case of T. ratisbonensis infection. Indeed, we have failed to observe any melanin deposition on the surface of spores.
In conclusion, this study provides a largely unexpected view of classical antimicrobial host defenses, which are irrelevant or possibly diverted by the parasite for its own purpose, at least in this infection model in which T. ratisbonensis spores are injected directly within the hemocoel. In the long term, it would be interesting to reproduce this study in a larval infection model, which at present is not available due to the limited control of the spore load upon ingestion and the large quantity of parasites that would be required. It is an open possibility that other unknown defense mechanisms may be also at work and might be revealed upon using -omics studies. Nevertheless, the parasite appears to win the competition with the host as we are not aware of the existence of Drosophila lines refractory to microsporidia infections. The finding that hemocytes are required to protect the flies from such infections, at least to a degree, establish that injected spores do elicit an immune response that is not systemic.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://figshare.com/, https://figshare.com/s/566ee89ed278eb6b34f3.
Author Contributions
GC performed most of the experimental work described in this study. AF and SN initiated this study and performed early experiments. GC and DF designed the experiments, analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to J. Nguyen and M. Yamba for expert technical assistance. We thank Bruno Lemaitre, Patick O’Farrell, Akira Goto and Shigeo Hayashi for sharing fly lines used in this study. Stocks obtained from VDRC (www.vdrc.at) and the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. We are indebted to Nahid Azzouz and Peter Seeberger (MPI for Colloids and Interfaces, Berlin) for the kind gift of GPIs microarrays and chemically synthesized GPIs structures used in this work. This work has been founded by CNRS, University of Strasbourg, Fondation Simone and Cino Del Duca, Institut d’Etude Avancée de l’Université de Strasbourg, Fondation pour la Recherche Médicale (Equipe FRM to DF), and Agence Nationale de la Recherche (BEELOSS, ANR-11-EQPX-0022, LIPIDIC_PURGE).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.858360/full#supplementary-material
References
1. Weiss LM, Becnel JJ. Microsporidia: Pathogens of Opportunity: First Edition. Wiley-Blackwell (2014). doi: 10.1002/9781118395264
2. Katinka MD, Duprat S, Cornillot E, Méténier G, Thomarat F, Prensier G, et al. Genome Sequence and Gene Compaction of the Eukaryote Parasite Encephalitozoon Cuniculi. Nature (2001) 414:450–3. doi: 10.1038/35106579
3. Goldberg AV, Molik S, Tsaousis AD, Neumann K, Kuhnke G, Delbac F, et al. Localization and Functionality of Microsporidian Iron-Sulphur Cluster Assembly Proteins. Nature (2008) 452:624–8. doi: 10.1038/nature06606
4. Keeling PJ, Corradi N, Morrison HG, Haag KL, Ebert D, Weiss LM, et al. The Reduced Genome of the Parasitic Microsporidian Enterocytozoon Bieneusi Lacks Genes for Core Carbon Metabolism. Genome Biol Evol (2010) 2:304–9. doi: 10.1093/gbe/evq022
5. Nakjang S, Williams TA, Heinz E, Watson AK, Foster PG, Sendra KM, et al. Reduction and Expansion in Microsporidian Genome Evolution: New Insights From Comparative Genomics. Genome Biol Evol (2013) 5:2285–303. doi: 10.1093/gbe/evt184
6. Han B, Weiss LM. Microsporidia: Obligate Intracellular Pathogens Within the Fungal Kingdom. Microbiol Spectr (2017) 5:2. doi: 10.1128/microbiolspec.FUNK-0018-2016
7. Undeen AH, Epsky ND. In Vitro and In Vivo Germination of Nosema Locustae (Microspora: Nosematidae) Spores. J Invertebr Pathol (1990) 56:371–9. doi: 10.1016/0022-2011(90)90124-O
8. Undeen AH, Vander Meer RK. The Effect of Ultraviolet Radiation on the Germination of Nosema Algerae Vávra and Undeen (Microsporida: Nosematidae) Spores. J Protozool (1990) 37:194–9. doi: 10.1111/j.1550-7408.1990.tb01127.x
9. Whitlock VH, Johnson S. Stimuli for the In Vitro Germination and Inhibition of Nosema Locusta (Microspora: Nosematidae) Spores. J Invertebr Pathol (1990) 56:57–62. doi: 10.1016/0022-2011(90)90144-U
10. Franzen C, Müller A. Microsporidiosis: Human Diseases and Diagnosis. Microbes Infect (2001) 3:389–400. doi: 10.1016/S1286-4579(01)01395-8
11. Higes M, Meana A, Bartolomé C, Botías C, Martín-Hernández R. Nosema Ceranae (Microsporidia), a Controversial 21st Century Honey Bee Pathogen. Environ Microbiol Rep (2013) 5:17–29. doi: 10.1111/1758-2229.12024
12. Vidau C, Diogon M, Aufauvre J, Fontbonne R, Viguès B, Brunet J-L, et al. Exposure to Sublethal Doses of Fipronil and Thiacloprid Highly Increases Mortality of Honeybees Previously Infected by Nosema Ceranae. PloS One (2011) 6:e21550. doi: 10.1371/journal.pone.0021550
13. Balla KM, Luallen RJ, Bakowski MA, Troemel ER. Cell-To-Cell Spread of Microsporidia Causes Caenorhabditis Elegans Organs to Form Syncytia. Nat Microbiol (2016) 1:16144. doi: 10.1038/nmicrobiol.2016.144
14. Lom J, Dyková I. Microsporidian Xenomas in Fish Seen in Wider Perspective. Folia Parasitol (2005) 52:69–81. doi: 10.14411/fp.2005.010
15. Han Y, Gao H, Xu J, Luo J, Han B, Bao J, et al. Innate and Adaptive Immune Responses Against Microsporidia Infection in Mammals. Front Microbiol (2020) 11:1468. doi: 10.3389/fmicb.2020.01468
16. Didier ES, Shadduck JA. IFN-Gamma and LPS Induce Murine Macrophages to Kill Encephalitozoon Cuniculi In Vitro. J Eukaryot Microbiol (1994) 41:34S.
17. Didier ES. Reactive Nitrogen Intermediates Implicated in the Inhibition of Encephalitozoon Cuniculi (Phylum Microspora) Replication in Murine Peritoneal Macrophages. Parasite Immunol (1995) 17:405–12. doi: 10.1111/j.1365-3024.1995.tb00908.x
18. Fischer J, Tran D, Juneau R, Hale-Donze H. Kinetics of Encephalitozoon Spp. Infection of Human Macrophages. J Parasitol (2008) 94:169–75. doi: 10.1645/GE-1303.1
19. Didier ES, Bowers LC, Martin AD, Kuroda MJ, Khan IA, Didier PJ. Reactive Nitrogen and Oxygen Species, and Iron Sequestration Contribute to Macrophage-Mediated Control of Encephalitozoon Cuniculi (Phylum Microsporidia) Infection In Vitro and In Vivo. Microbes Infect (2010) 12:1244–51. doi: 10.1016/j.micinf.2010.09.010
20. Franzen C, Hartmann P, Salzberger B. Cytokine and Nitric Oxide Responses of Monocyte-Derived Human Macrophages to Microsporidian Spores. Exp Parasitol (2005) 109:1–6. doi: 10.1016/j.exppara.2004.10.001
21. Fischer J, Suire C, Hale-Donze H. Toll-Like Receptor 2 Recognition of the Microsporidia Encephalitozoon Spp. Induces Nuclear Translocation of NF-κb and Subsequent Inflammatory Responses. Infect Immun (2008) 76:4737–44. doi: 10.1128/IAI.00733-08
22. Moretto MM, Weiss LM, Combe CL, Khan IA. IFN-γ-Producing Dendritic Cells Are Important for Priming of Gut Intraepithelial Lymphocyte Response Against Intracellular Parasitic Infection. J Immunol (2007) 179:2485–92. doi: 10.4049/jimmunol.179.4.2485
23. Moretto MM, Khan IA, Weiss LM. Gastrointestinal Cell Mediated Immunity and the Microsporidia. PloS Pathog (2012) 8:e1002775. doi: 10.1371/journal.ppat.1002775
24. Niederkorn JY, Brieland JK, Mayhew E. Enhanced Natural Killer Cell Activity in Experimental Murine Encephalitozoonosis. Infect Immun (1983) 41:302–7. doi: 10.1128/iai.41.1.302-307.1983
25. Leitch GJ, Ceballos C. A Role for Antimicrobial Peptides in Intestinal Microsporidiosis. Parasitology (2009) 136:175–81. doi: 10.1017/S0031182008005313
26. Valencakova A, Halanova M. Immune Response to Encephalitozoon Infection Review. Comp Immunol Microbiol Infect Dis (2012) 35:1–7. doi: 10.1016/j.cimid.2011.11.004
27. Troemel ER, Félix M-A, Whiteman NK, Barrière A, Ausubel FM. Microsporidia are Natural Intracellular Parasites of the Nematode Caenorhabditis Elegans. PloS Biol (2008) 6:2736–52. doi: 10.1371/journal.pbio.0060309
28. Bakowski MA, Desjardins CA, Smelkinson MG, Dunbar TL, Dunbar TA, Lopez-Moyado IF, et al. Ubiquitin-Mediated Response to Microsporidia and Virus Infection in C. Elegans. PloS Pathog (2014) 10:e1004200. doi: 10.1371/journal.ppat.1004200
29. Reddy KC, Dror T, Sowa JN, Panek J, Chen K, Lim ES, et al. An Intracellular Pathogen Response Pathway Promotes Proteostasis in C. Elegans. Curr Biol (2017) 27:3544–3553.e5. doi: 10.1016/j.cub.2017.10.009
30. Reddy KC, Dror T, Underwood RS, Osman GA, Elder CR, Desjardins CA, et al. Antagonistic Paralogs Control a Switch Between Growth and Pathogen Resistance in C. Elegans. PloS Pathog (2019) 15:e1007528. doi: 10.1371/journal.ppat.1007528
31. Lažetić V, Wu F, Cohen LB, Reddy KC, Chang Y-T, Gang SS, et al. The Transcription Factor ZIP-1 Promotes Resistance to Intracellular Infection in Caenorhabditis Elegans. Nat Commun (2022) 13:17. doi: 10.1038/s41467-021-27621-w
32. Yue Y-J, Tang X-D, Xu L, Yan W, Li Q-L, Xiao S-Y, et al. Early Responses of Silkworm Midgut to Microsporidium Infection–A Digital Gene Expression Analysis. J Invertebr Pathol (2015) 124:6–14. doi: 10.1016/j.jip.2014.10.003
33. Ma Z, Li C, Pan G, Li Z, Han B, Xu J, et al. Genome-Wide Transcriptional Response of Silkworm (Bombyx Mori) to Infection by the Microsporidian Nosema Bombycis. PloS One (2013) 8:e84137. doi: 10.1371/journal.pone.0084137
34. He X, He X, Liu H, Li M, Cai S, Fu Z, et al. Proteomic Analysis of BmN Cells (Bombyx Mori) in Response to Infection With Nosema Bombycis. Acta Biochim Biophys Sin (2014) 46:982–90. doi: 10.1093/abbs/gmu092
35. Desjardins CA, Sanscrainte ND, Goldberg JM, Heiman D, Young S, Zeng Q, et al. Contrasting Host–Pathogen Interactions and Genome Evolution in Two Generalist and Specialist Microsporidian Pathogens of Mosquitoes. Nat Commun (2015) 6:7121. doi: 10.1038/ncomms8121
36. Aufauvre J, Misme-Aucouturier B, Viguès B, Texier C, Delbac F, Blot N. Transcriptome Analyses of the Honeybee Response to Nosema Ceranae and Insecticides. PloS One (2014) 9:e91686. doi: 10.1371/journal.pone.0091686
37. Kurze C, Dosselli R, Grassl J, Le Conte Y, Kryger P, Baer B, et al. Differential Proteomics Reveals Novel Insights Into Nosema–honey Bee Interactions. Insect Biochem Mol Biol (2016) 79:42–9. doi: 10.1016/j.ibmb.2016.10.005
38. Vidau C, Panek J, Texier C, Biron DG, Belzunces LP, Le Gall M, et al. Differential Proteomic Analysis of Midguts From Nosema Ceranae-Infected Honeybees Reveals Manipulation of Key Host Functions. J Invertebr Pathol (2014) 121:89–96. doi: 10.1016/j.jip.2014.07.002
39. Chaimanee V, Chantawannakul P, Chen Y, Evans JD, Pettis JS. Differential Expression of Immune Genes of Adult Honey Bee (Apis Mellifera) After Inoculated by Nosema Ceranae. J Insect Physiol (2012) 58:1090–5. doi: 10.1016/j.jinsphys.2012.04.016
40. Roxström-Lindquist K, Terenius O, Faye I. Parasite-Specific Immune Response in Adult Drosophila Melanogaster: A Genomic Study. EMBO Rep (2004) 5:207–12. doi: 10.1038/sj.embor.7400073
41. Franzen C, Fischer S, Schroeder J, Schölmerich J, Schneuwly S. Morphological and Molecular Investigations of Tubulinosema Ratisbonensis Gen. Nov., Sp. Nov. (Microsporidia: Tubulinosematidae Fam. Nov.), a Parasite Infecting a Laboratory Colony of Drosophila Melanogaster (Diptera: Drosophilidae). J Eukaryot Microbiol (2005) 52:141–52. doi: 10.1111/j.1550-7408.2005.04-3324.x
42. Franzen C, Fischer S, Schroeder J, Bleiss W, Schneuwly S, Schölmerich J, et al. In Vitro Cultivation of an Insect Microsporidian Tubulinosema Ratisbonensis in Mammalian Cells. J Eukaryot Microbiol (2005) 52:349–55. doi: 10.1111/j.1550-7408.2005.00043x
43. Franchet A, Niehus S, Caravello G, Ferrandon D. Phosphatidic Acid as a Limiting Host Metabolite for the Proliferation of the Microsporidium Tubulinosema Ratisbonensis in Drosophila Flies. Nat Microbiol (2019) 4:645–55. doi: 10.1038/s41564-018-0344-y
44. Vijendravarma RK, Godfray HCJ, Kraaijeveld AR. Infection of Drosophila Melanogaster by Tubulinosema Kingi: Stage-Specific Susceptibility and Within-Host Proliferation. J Invertebr Pathol (2008) 99:239–41. doi: 10.1016/j.jip.2008.02.014
45. Lemaitre B, Hoffmann J. The Host Defense of Drosophila Melanogaster. Annu Rev Immunol (2007) 25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615
46. Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, et al. Autophagic Control of Listeria Through Intracellular Innate Immune Recognition in Drosophila. Nat Immunol (2008) 9:908–16. doi: 10.1038/ni.1634
47. Mussabekova A, Daeffler L, Imler J-L. Innate and Intrinsic Antiviral Immunity in Drosophila. Cell Mol Life Sci (2017) 74:2039–54. doi: 10.1007/s00018-017-2453-9
48. Goto A, Okado K, Martins N, Cai H, Barbier V, Lamiable O, et al. The Kinase Ikkβ Regulates a STING- and NF-κb-Dependent Antiviral Response Pathway in Drosophila. Immunity (2018) 49:225–234.e4. doi: 10.1016/j.immuni.2018.07.013
49. Holleufer A, Winther KG, Gad HH, Ai X, Chen Y, Li L, et al. Two cGAS-Like Receptors Induce Antiviral Immunity in Drosophila. Nature (2021) 597:114–8. doi: 10.1038/s41586-021-03800-z
50. Slavik KM, Morehouse BR, Ragucci AE, Zhou W, Ai X, Chen Y, et al. cGAS-Like Receptors Sense RNA and Control 3’2’-cGAMP Signalling in Drosophila. Nature (2021) 597:109–13. doi: 10.1038/s41586-021-03743-5
51. Pan G, Bao J, Ma Z, Song Y, Han B, Ran M, et al. Invertebrate Host Responses to Microsporidia Infections. Dev Comp Immunol (2018) 83:104–13. doi: 10.1016/j.dci.2018.02.004
52. Houdelet C, Arafah K, Bocquet M, Bulet P. Molecular Histoproteomy by MALDI Mass Spectrometry Imaging to Uncover Markers of the Impact of Nosema On Apis Mellifera. Proteomics (2022):e2100224. doi: 10.1002/pmic.202100224
53. Dietzl G, Chen D, Schnorrer F, Su K-C, Barinova Y, Fellner M, et al. A Genome-Wide Transgenic RNAi Library for Conditional Gene Inactivation in Drosophila. Nature (2007) 448:151–6. doi: 10.1038/nature05954
54. Soares MP, Teixeira L, Moita LF. Disease Tolerance and Immunity in Host Protection Against Infection. Nat Rev Immunol (2017) 17:83–96. doi: 10.1038/nri.2016.136
55. Ferrandon D. The Complementary Facets of Epithelial Host Defenses in the Genetic Model Organism Drosophila Melanogaster: From Resistance to Resilience. Curr Opin Immunol (2013) 25:59–70. doi: 10.1016/j.coi.2012.11.008
56. Medzhitov R, Schneider DS, Soares MP. Disease Tolerance as a Defense Strategy. Science (2012) 335:936–41. doi: 10.1126/science.1214935
57. Duneau D, Ferdy J-B, Revah J, Kondolf H, Ortiz GA, Lazzaro BP, et al. Stochastic Variation in the Initial Phase of Bacterial Infection Predicts the Probability of Survival in D. Melanogaster. Elife (2017) 6:e28298. doi: 10.7554/eLife.28298
58. Niehus S, Giammarinaro P, Liégeois S, Quintin J, Ferrandon D. Fly Culture Collapse Disorder: Detection, Prophylaxis and Eradication of the Microsporidian Parasite Tubulinosema Ratisbonensis Infecting Drosophila Melanogaster. Fly (Austin) (2012) 6:193–204. doi: 10.4161/fly.20896
59. Thibault ST, Singer MA, Miyazaki WY, Milash B, Dompe NA, Singh CM, et al. A Complementary Transposon Tool Kit for Drosophila Melanogaster Using P and Piggybac. Nat Genet (2004) 36:283–7. doi: 10.1038/ng1314
60. Ni J-Q, Zhou R, Czech B, Liu L-P, Holderbaum L, Yang-Zhou D, et al. A Genome-Scale shRNA Resource for Transgenic RNAi in Drosophila. Nat Methods (2011) 8:405–7. doi: 10.1038/nmeth.1592
61. DeVorkin L, Gorski SM. Monitoring Autophagy in Drosophila Using Fluorescent Reporters in the UAS-GAL4 System. Cold Spring Harb Protoc (2014) 2014:pdb.prot080341. doi: 10.1101/pdb.prot080341
62. Juhász G, Érdi B, Sass M, Neufeld TP. Atg7-Dependent Autophagy Promotes Neuronal Health, Stress Tolerance, and Longevity But is Dispensable for Metamorphosis in Drosophila. Genes Dev (2007) 21:3061–6. doi: 10.1101/gad.1600707
63. Tauszig-Delamasure S, Bilak H, Capovilla M, Hoffmann JA, Imler J-L. Drosophila MyD88 is Required for the Response to Fungal and Gram-Positive Bacterial Infections. Nat Immunol (2002) 3:91–7. doi: 10.1038/ni747
64. Rutschmann S, Jung AC, Zhou R, Silverman N, Hoffmann JA, Ferrandon D. Role of Drosophila Ikkγ in a Toll-Independent Antibacterial Immune Response. Nat Immunol (2000) 1:342–7. doi: 10.1038/79801
65. Hanson MA, Dostálová A, Ceroni C, Poidevin M, Kondo S, Lemaitre B. Synergy and Remarkable Specificity of Antimicrobial Peptides In Vivo Using a Systematic Knockout Approach. Elife (2019) 8:e44341. doi: 10.7554/eLife.44341
66. Donelick HM, Talide L, Bellet M, Aruscavage PJ, Lauret E, Aguiar ERGR, et al. In Vitro Studies Provide Insight Into Effects of Dicer-2 Helicase Mutations in Drosophila Melanogaster. RNA (2020) 26:1847–61. doi: 10.1261/rna.077289.120
67. Binggeli O, Neyen C, Poidevin M, Lemaitre B. Prophenoloxidase Activation Is Required for Survival to Microbial Infections in Drosophila. PloS Pathog (2014) 10:e1004067. doi: 10.1371/journal.ppat.1004067
68. Kocks C, Cho JH, Nehme N, Ulvila J, Pearson AM, Meister M, et al. Eater, a Transmembrane Protein Mediating Phagocytosis of Bacterial Pathogens in Drosophila. Cell (2005) 123:335–46. doi: 10.1016/j.cell.2005.08.034
69. Aoun RB, Hetru C, Troxler L, Doucet D, Ferrandon D, Matt N. Analysis of Thioester-Containing Proteins During the Innate Immune Response of Drosophila Melanogaster. J Innate Immun (2011) 3:52–64. doi: 10.1159/000321554
70. Dostálová A, Rommelaere S, Poidevin M, Lemaitre B. Thioester-Containing Proteins Regulate the Toll Pathway and Play a Role in Drosophila Defence Against Microbial Pathogens and Parasitoid Wasps. BMC Biol (2017) 15:79. doi: 10.1186/s12915-017-0408-0
71. Yakubovich N, Silva EA, O’Farrell PH. Nitric Oxide Synthase is Not Essential for Drosophila Development. Curr Biol (2010) 20:R141–2. doi: 10.1016/j.cub.2009.12.011
72. Reichhart JM, Meister M, Dimarcq JL, Zachary D, Hoffmann D, Ruiz C, et al. Insect Immunity: Developmental and Inducible Activity of the Drosophila Diptericin Promoter. EMBO J (1992) 11:1469–77. doi: 10.1002/j.1460-2075.1992.tb05191.x
73. Ferrandon D, Jung AC, Criqui M, Lemaitre B, Uttenweiler-Joseph S, Michaut L, et al. A Drosomycin-GFP Reporter Transgene Reveals a Local Immune Response in Drosophila That is Not Dependent on the Toll Pathway. EMBO J (1998) 17:1217–27. doi: 10.1093/emboj/17.5.1217
74. Belkorchia A, Biderre C, Militon C, Polonais V, Wincker P, Jubin C, et al. In Vitro Propagation of the Microsporidian Pathogen Brachiola Algerae and Studies of its Chromosome and Ribosomal DNA Organization in the Context of the Complete Genome Sequencing Project. Parasitol Int (2008) 57:62–71. doi: 10.1016/j.parint.2007.09.002
75. Kamena F, Tamborrini M, Liu X, Kwon Y-U, Thompson F, Pluschke G, et al. Synthetic GPI Array to Study Antitoxic Malaria Response. Nat Chem Biol (2008) 4:238–40. doi: 10.1038/nchembio.75
76. Nehme NT, Liégeois S, Kele B, Giammarinaro P, Pradel E, Hoffmann JA, et al. A Model of Bacterial Intestinal Infections in Drosophila Melanogaster. PloS Pathog (2007) 3:e173. doi: 10.1371/journal.ppat.0030173
77. Cuttell L, Vaughan A, Silva E, Escaron CJ, Lavine M, Van Goethem E, et al. Undertaker, a Drosophila Junctophilin, Links Draper-Mediated Phagocytosis and Calcium Homeostasis. Cell (2008) 135:524–34. doi: 10.1016/j.cell.2008.08.033
78. Taupin V, Garenaux E, Mazet M, Maes E, Denise H, Prensier G, et al. Major O-Glycans in the Spores of Two Microsporidian Parasites are Represented by Unbranched Manno-Oligosaccharides Containing Alpha-1,2 Linkages. Glycobiology (2007) 17:56–67. doi: 10.1093/glycob/cwl050
79. Balla KM, Lažetić V, Troemel ER. Natural Variation in the Roles of C. Elegans Autophagy Components During Microsporidia Infection. PloS One (2019) 14:e0216011. doi: 10.1371/journal.pone.0216011
80. Cerenius L, Lee BL, Söderhäll K. The proPO-System: Pros and Cons for its Role in Invertebrate Immunity. Trends Immunol (2008) 29:263–71. doi: 10.1016/j.it.2008.02.009
81. Melcarne C, Ramond E, Dudzic J, Bretscher AJ, Kurucz É, Andó I, et al. Two Nimrod Receptors, NimC1 and Eater, Synergistically Contribute to Bacterial Phagocytosis in Drosophila Melanogaster. FEBS J (2019) 286:2670–91. doi: 10.1111/febs.14857
82. Rämet M, Manfruelli P, Pearson A, Mathey-Prevot B, Ezekowitz RAB. Functional Genomic Analysis of Phagocytosis and Identification of a Drosophila Receptor for E. coli. Nature (2002) 416:644–8. doi: 10.1038/nature735
83. Rämet M, Pearson A, Manfruelli P, Li X, Koziel H, Göbel V, et al. Drosophila Scavenger Receptor CI Is a Pattern Recognition Receptor for Bacteria. Immunity (2001) 15:1027–38. doi: 10.1016/S1074-7613(01)00249-7
84. Hashimoto Y, Tabuchi Y, Sakurai K, Kutsuna M, Kurokawa K, Awasaki T, et al. Identification of Lipoteichoic Acid as a Ligand for Draper in the Phagocytosis of Staphylococcus Aureus by Drosophila Hemocytes. J Immunol (2009) 183:7451–60. doi: 10.4049/jimmunol.0901032
85. Ramond E, Petrignani B, Dudzic JP, Boquete J-P, Poidevin M, Kondo S, et al. The Adipokine NimrodB5 Regulates Peripheral Hematopoiesis in Drosophila. FEBS J (2020) 287:3399–426. doi: 10.1111/febs.15237
86. Haller S, Franchet A, Hakkim A, Chen J, Drenkard E, Yu S, et al. Quorum-Sensing Regulator RhlR But Not its Autoinducer RhlI Enables Pseudomonas to Evade Opsonization. EMBO Rep (2018) 19:6. doi: 10.15252/embr.201744880
87. Levashina EA, Moita LF, Blandin S, Vriend G, Lagueux M, Kafatos FC. Conserved Role of a Complement-Like Protein in Phagocytosis Revealed by dsRNA Knockout in Cultured Cells of the Mosquito, Anopheles Gambiae. Cell (2001) 104:709–18. doi: 10.1016/s0092-8674(01)00267-7
88. Bätz T, Förster D, Luschnig S. The Transmembrane Protein Macroglobulin Complement-Related is Essential for Septate Junction Formation and Epithelial Barrier Function in Drosophila. Development (2014) 141:899–908. doi: 10.1242/dev.102160
89. Hall S, Bone C, Oshima K, Zhang L, McGraw M, Lucas B, et al. Macroglobulin Complement-Related Encodes a Protein Required for Septate Junction Organization and Paracellular Barrier Function in Drosophila. Development (2014) 141:889–98. doi: 10.1242/dev.102152
90. Winkler B, Funke D, Benmimoun B, Spéder P, Rey S, Logan MA, et al. Brain Inflammation Triggers Macrophage Invasion Across the Blood-Brain Barrier in Drosophila During Pupal Stages. Sci Adv (2021) 7:eabh0050. doi: 10.1126/sciadv.abh0050
91. Cho NK, Keyes L, Johnson E, Heller J, Ryner L, Karim F, et al. Developmental Control of Blood Cell Migration by the Drosophila VEGF Pathway. Cell (2002) 108:865–76. doi: 10.1016/S0092-8674(02)00676-1
92. Parsons B, Foley E. The Drosophila Platelet-Derived Growth Factor and Vascular Endothelial Growth Factor-Receptor Related (Pvr) Protein Ligands Pvf2 and Pvf3 Control Hemocyte Viability and Invasive Migration *. J Biol Chem (2013) 288:20173–83. doi: 10.1074/jbc.M113.483818
93. Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, et al. Eiger, a TNF Superfamily Ligand That Triggers the Drosophila JNK Pathway. EMBO J (2002) 21:3009–18. doi: 10.1093/emboj/cdf306
94. Fogarty CE, Diwanji N, Lindblad JL, Tare M, Amcheslavsky A, Makhijani K, et al. Extracellular Reactive Oxygen Species Drive Apoptosis-Induced Proliferation via Drosophila Macrophages. Curr Biol (2016) 26:575–84. doi: 10.1016/j.cub.2015.12.064
95. Agaisse H, Petersen UM, Boutros M, Mathey-Prevot B, Perrimon N. Signaling Role of Hemocytes in Drosophila JAK/STAT-Dependent Response to Septic Injury. Dev Cell (2003) 5:441–50. doi: 10.1016/s1534-5807(03)00244-2
96. De Gregorio E, Spellman PT, Tzou P, Rubin GM, Lemaitre B. The Toll and Imd Pathways are the Major Regulators of the Immune Response in Drosophila. EMBO J (2002) 21:2568–79. doi: 10.1093/emboj/21.11.2568
97. Castillo JC, Ferreira ABB, Trisnadi N, Barillas-Mury C. Activation of Mosquito Complement Antiplasmodial Response Requires Cellular Immunity. Sci Immunol (2017) 2:eaal1505. doi: 10.1126/sciimmunol.aal1505
98. Blandin SA, Wang-Sattler R, Lamacchia M, Gagneur J, Lycett G, Ning Y, et al. Dissecting the Genetic Basis of Resistance to Malaria Parasites in Anopheles Gambiae. Science (2009) 326:147–50. doi: 10.1126/science.1175241
99. Fraiture M, Baxter RHG, Steinert S, Chelliah Y, Frolet C, Quispe-Tintaya W, et al. Two Mosquito LRR Proteins Function as Complement Control Factors in the TEP1-Mediated Killing of Plasmodium. Cell Host Microbe (2009) 5:273–84. doi: 10.1016/j.chom.2009.01.005
100. Takehana A, Katsuyama T, Yano T, Oshima Y, Takada H, Aigaki T, et al. Overexpression of a Pattern-Recognition Receptor, Peptidoglycan-Recognition Protein-LE, Activates Imd/Relish-Mediated Antibacterial Defense and the Prophenoloxidase Cascade in Drosophila Larvae. Proc Natl Acad Sci U S A (2002) 99:13705–10. doi: 10.1073/pnas.212301199
101. Kaneko T, Yano T, Aggarwal K, Lim J-H, Ueda K, Oshima Y, et al. PGRP-LC and PGRP-LE Have Essential Yet Distinct Functions in the Drosophila Immune Response to Monomeric DAP-Type Peptidoglycan. Nat Immunol (2006) 7:715–23. doi: 10.1038/ni1356
102. Neyen C, Poidevin M, Roussel A, Lemaitre B. Tissue- and Ligand-Specific Sensing of Gram-Negative Infection in Drosophila by PGRP-LC Isoforms and PGRP-Le. J Immunol (2012) 189:1886–97. doi: 10.4049/jimmunol.1201022
103. Bargielowski I, Koella JC. A Possible Mechanism for the Suppression of Plasmodium Berghei Development in the Mosquito Anopheles Gambiae by the Microsporidian Vavraia Culicis. PloS One (2009) 4:e4676. doi: 10.1371/journal.pone.0004676
104. Bidla G, Dushay MS, Theopold U. Crystal Cell Rupture After Injury in Drosophila Requires the JNK Pathway, Small GTPases and the TNF Homolog Eiger. J Cell Sci (2007) 120:1209–15. doi: 10.1242/jcs.03420
105. Melcarne C, Lemaitre B, Kurant E. Phagocytosis in Drosophila: From Molecules and Cellular Machinery to Physiology. Insect Biochem Mol Biol (2019) 109:1–12. doi: 10.1016/j.ibmb.2019.04.002
106. Kurant E, Axelrod S, Leaman D, Gaul U. Six-Microns-Under Acts Upstream of Draper in the Glial Phagocytosis of Apoptotic Neurons. Cell (2008) 133:498–509. doi: 10.1016/j.cell.2008.02.052
107. Petrignani B, Rommelaere S, Hakim-Mishnaevski K, Masson F, Ramond E, Hilu-Dadia R, et al. A Secreted Factor NimrodB4 Promotes the Elimination of Apoptotic Corpses by Phagocytes in Drosophila. EMBO Rep (2021) 22:e52262. doi: 10.15252/embr.202052262
108. Sanchez Bosch P, Makhijani K, Herboso L, Gold KS, Baginsky R, Woodcock KJ, et al. Adult Drosophila Lack Hematopoiesis But Rely on a Blood Cell Reservoir at the Respiratory Epithelia to Relay Infection Signals to Surrounding Tissues. Dev Cell (2019) 51:787–803.e5. doi: 10.1016/j.devcel.2019.10.017
109. Limmer S, Haller S, Drenkard E, Lee J, Yu S, Kocks C, et al. Pseudomonas Aeruginosa RhlR is Required to Neutralize the Cellular Immune Response in a Drosophila Melanogaster Oral Infection Model. Proc Natl Acad Sci U S A (2011) 108:17378–83. doi: 10.1073/pnas.1114907108
110. Chakrabarti S, Dudzic JP, Li X, Collas EJ, Boquete J-P, Lemaitre B. Remote Control of Intestinal Stem Cell Activity by Haemocytes in Drosophila. PloS Genet (2016) 12:e1006089. doi: 10.1371/journal.pgen.1006089
111. Chakrabarti S, Visweswariah SS. Intramacrophage ROS Primes the Innate Immune System via JAK/STAT and Toll Activation. Cell Rep (2020) 33:108368. doi: 10.1016/j.celrep.2020.108368
112. Schneider DS, Ayres JS, Brandt SM, Costa A, Dionne MS, Gordon MD, et al. Drosophila Eiger Mutants are Sensitive to Extracellular Pathogens. PloS Pathog (2007) 3:e41. doi: 10.1371/journal.ppat.0030041
Keywords: Tubulinosema ratisbonensis, phagocytosis, Drosophila, intracellular parasite, immunity, mutant analysis
Citation: Caravello G, Franchet A, Niehus S and Ferrandon D (2022) Phagocytosis Is the Sole Arm of Drosophila melanogaster Known Host Defenses That Provides Some Protection Against Microsporidia Infection. Front. Immunol. 13:858360. doi: 10.3389/fimmu.2022.858360
Received: 19 January 2022; Accepted: 18 March 2022;
Published: 13 April 2022.
Edited by:
Akira Goto, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Aaron Reinke, University of Toronto, CanadaJorge Contreras-Garduño, National Autonomous University of Mexico, Mexico
Copyright © 2022 Caravello, Franchet, Niehus and Ferrandon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominique Ferrandon, D.Ferrandon@ibmc-cnrs.unistra.fr
†Present address: Adrien Franchet, The Francis Crick Institute, London, UK