 Waltraud C. Schrottmaier
Waltraud C. Schrottmaier Anna Schmuckenschlager
Anna Schmuckenschlager Anita Pirabe
Anita Pirabe Alice Assinger
Alice Assinger
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 March 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.856713
This article is part of the Research TopicPlatelets and Megakaryocytes dysfunctions in Infectious DiseasesView all 8 articles
Viral infections are often associated with platelet activation and haemostatic complications. In line, low platelet counts represent a hallmark for poor prognosis in many infectious diseases. The underlying cause of platelet dysfunction in viral infections is multifaceted and complex. While some viruses directly interact with platelets and/or megakaryocytes to modulate their function, also immune and inflammatory responses directly and indirectly favour platelet activation. Platelet activation results in increased platelet consumption and degradation, which contributes to thrombocytopenia in these patients. The role of platelets is often bi-phasic. Initial platelet hyper-activation is followed by a state of platelet exhaustion and/or hypo-responsiveness, which together with low platelet counts promotes bleeding events. Thereby infectious diseases not only increase the thrombotic but also the bleeding risk or both, which represents a most dreaded clinical complication. Treatment options in these patients are limited and new therapeutic strategies are urgently needed to prevent adverse outcome. This review summarizes the current literature on platelet-virus interactions and their impact on viral pathologies and discusses potential intervention strategies. As pandemics and concomitant haemostatic dysregulations will remain a recurrent threat, understanding the role of platelets in viral infections represents a timely and pivotal challenge.
While the discovery and availability of antibiotics has dampened the fright of bacterial epidemics, viral infections pose a major risk to global health as currently demonstrated by the COVID-19 pandemic. Viral infections can cause a variety of clinical symptoms upon systemic dissemination, frequently including alterations of the haemostatic system, such as increased risk for thrombosis and/or bleeding.
As cellular mediators of haemostasis platelets are prominently involved in many of these haemostatic disturbances. Owing to their evolutionary heritage, which they share with leukocytes, platelets express receptors for various pathogens, enabling them to directly recognize and respond to invading viruses (1, 2). Thereby, haemostatic platelet functions such as maintenance of vascular integrity or thrombus formation are affected by platelet-virus interactions. Activated endothelial cells and leukocytes also modulate platelet functions during viral infections – either via cell-to-cell contacts or indirectly via release of circulating mediators. Increasing evidence emerges that platelets themselves function as immune modulators, thereby signalling back to leukocytes and endothelial cells to alter their effector functions (2–5).
As platelets become activated and/or hyper-responsive during viral infections, they modulate other host responses to interfere with infectious pathogens depending on the local environment, the invading pathogen and the disease state. In re-occurring infections platelets further mediate serological memory via targeted antiviral IgG release at sites of infection (6).
Their highly sensitive nature in combination with their high abundance therefore renders platelets not just crucial mediators of haemostatic functions but also a formidable first line of defence during viral infections. However, some viruses found ways to exploit platelets as a shelter and transport system through the circulation, turning platelets into a guardian as well as a Trojan horse during viral infections.
Platelets express various receptors that mediate virus entry into other cell types and direct interactions between platelets and virus were first described in the late 1990s (7, 8). Today we know that platelets express a diverse repertoire of receptors to directly and indirectly interact with viruses (3, 4, 8, 9) which are summarized in Figures 1 and 2.
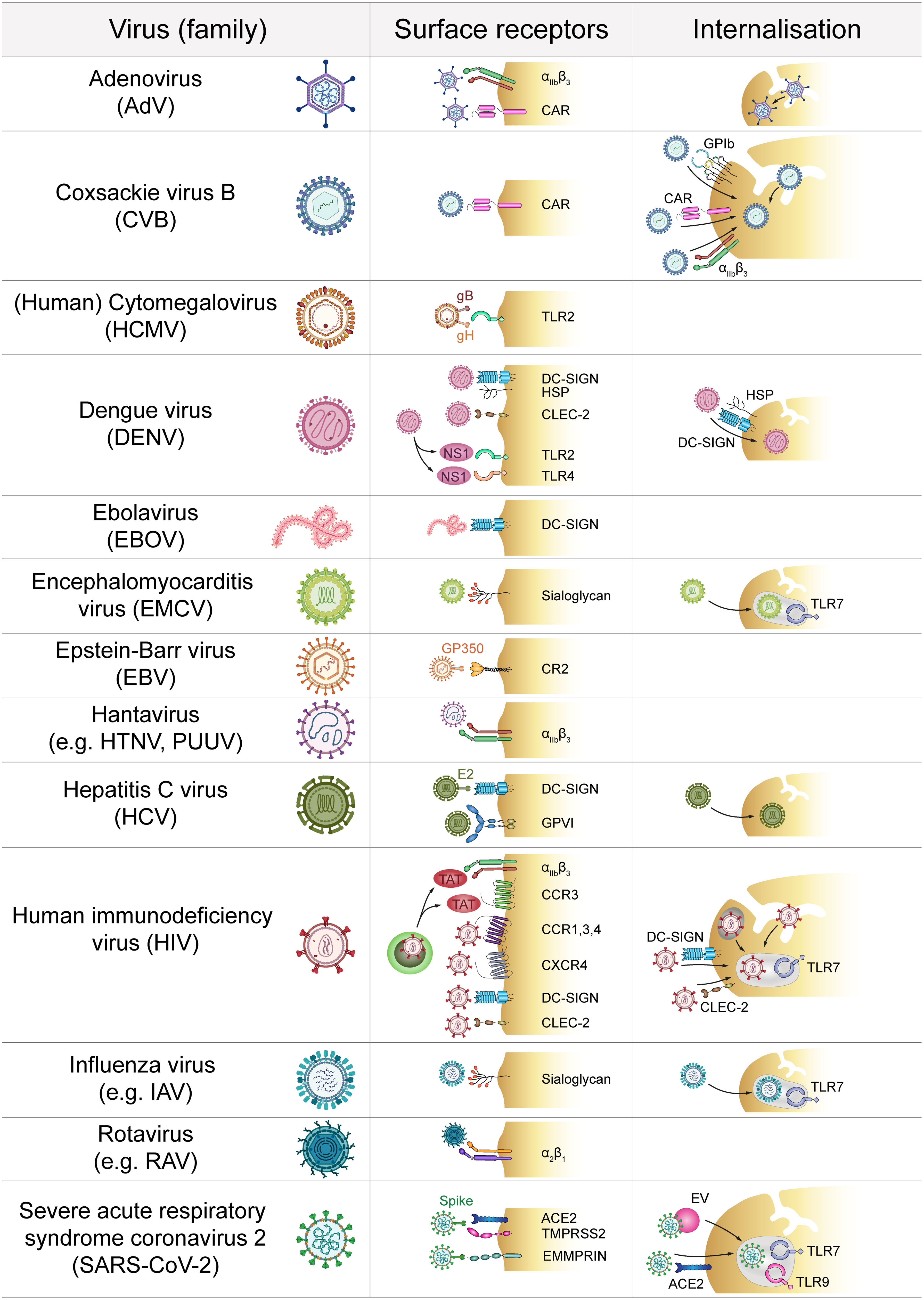
Figure 1 Direct platelet-virus interactions. Platelets express a plethora of surface receptors to directly bind various virus families. Subsequent virus internalisation may occur via the cell surface or the open canalicular system. ACE2, Angiotensin-converting enzyme 2; CAR, Coxsackie and Adenovirus receptor; CCR, C-C chemokine receptor; CLEC-2, C-type lectin receptor 2; CR2, Complement receptor 2; CXCR, C-X-C chemokine receptor; DC-SIGN, Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin; GP, Glycoprotein; HSP, Heparan sulfate proteoglycan; HTNV, Hantaan virus; IAV, Influenza A virus; EMMPRIN, extracellular matrix metalloproteinase inducer; EV, Extracellular vesicle; NS1, Non-structural protein 1; PUUV, Puumala virus; RAV, Rotavirus A; TAT, Trans-activator of transcription; TLR, Toll-like receptor; TMPRSS2, Transmembrane protease serine subtype 2.
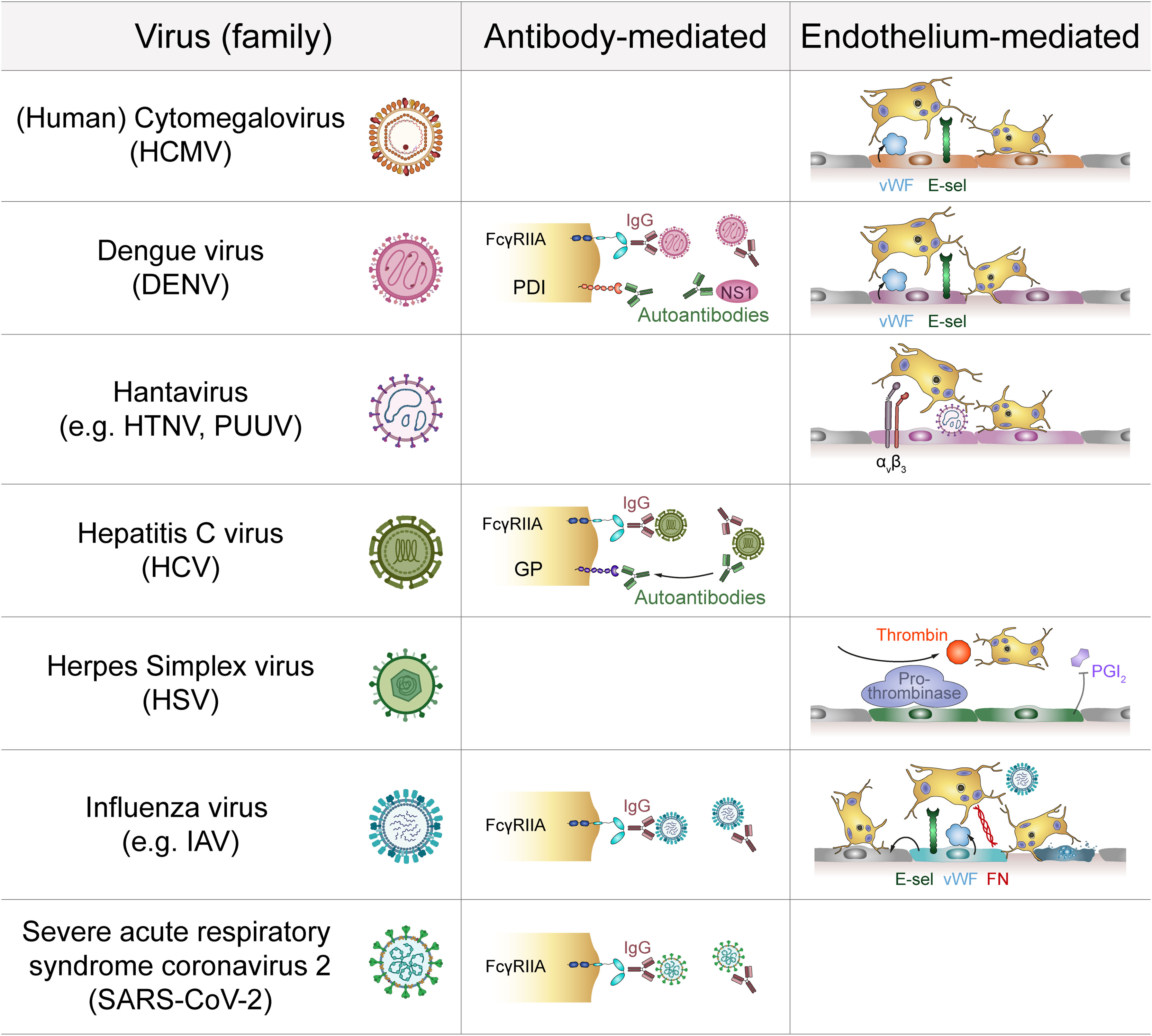
Figure 2 Indirect platelet-virus interactions. Several virus families interact with platelets via the formation of virus-IgG complexes that are recognized by platelet FcγRIIA. Antibodies against viral proteins may also act as autoantibodies by targeting platelet membrane components. Additionally, infection of endothelial cells induces expression of adhesive molecules that promote platelet adhesion. Infected endothelial cells also facilitate pro-thrombinase activity to modulate platelet activation. E-sel, E-selectin; FcγRIIA, Immunoglobulin γ Fc region receptor IIA; FN, Fibronectin; GP, Glycoprotein; HTNV, Hantaan virus; IAV, Influenza A virus; IgG, Immunoglobulin G; NS1, Non-structural protein 1; PDI, Protein disulfide isomerase; PGI2: Prostaglandin I2 (prostacyclin); PUUV, Puumala virus; vWF, von Willebrand factor.
The most rapid interaction between platelets and viruses occurs via direct contact with binding and/or entry receptors (Figure 1). Platelets express a plethora of receptors that specifically allow for interaction with pathogens (2). However, viruses also use receptors that are required for other physiological functions in order to interact with platelets. In this context integrins are of special importance as they are primarily responsible for platelet adhesion but also bind to and might even facilitate entry of specific virus strains (10). Especially, the abundantly expressed β3 integrins are often implicated in binding of viruses, e.g. pathogenic hantaviruses (11). Adenovirus-platelet interaction also involves β3 integrins, such as αIIbβ3 and αVβ3 (12–14), as blocking prominent β3 integrins does not completely abolish virus internalization (15). Other integrins such as α2β1 are involved in viral binding e.g. of rotaviruses (16).
Further, platelets express the Coxsackie and Adenovirus receptor (CAR) (12, 17) which allows for interaction between platelets and Coxsackie virus B (CVB) (18), as well as other viruses (19).
Sialic acid acts as a cellular receptor which interacts with heavily glycosylated glycoproteins (20). As platelets express sialic acid on their surface (21) these sialoyglycans enable interaction with various viruses such as Encephalomyocarditis virus (EMCV) (22) and Influenza virus (23–25).
Also, various cytokine receptors are hijacked by viruses to mediate their interactions with platelets. In that regard, Human immunodeficiency virus (HIV) illustrates how diverse virus-platelet interactions can be. Via expression of C-X-C chemokine receptor (CXCR) type 4 and the required co-receptors C-C chemokine receptor (CCR) type 1, 3 and 4 platelets can directly interact with HIV particles (26, 27). Also, HIV infected cells release the protein Trans-Activator of Transcription (TAT), which activates platelets by binding concurrently to integrin β3 and CCR3 (28). Platelets also bind and internalize HIV via dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) (29–31) and C-type lectin receptor 2 (CLEC-2) (30). DC-SIGN further binds to Ebola virus (EV) (32) which is most likely only captured (33). Hepatitis C virus (HCV) also binds DC-SIGN via viral glycoprotein E2 (34) but also to the collagen receptor glycoprotein (GP) VI (35). For Dengue virus (DENV) a dual receptor recognition involving DC-SIGN and heparan sulfate proteoglycan (HSP) has been suggested to mediate primary platelet binding (36–38). As platelets express no efficient entry receptor, it is not yet fully understood whether binding to DC-SIGN simply mediates virus capture or whether this interaction is also sufficient for cellular entry of HIV and HCV (31, 34).
Apart from DC-SIGN, DENV induced platelet activation also involves CLEC-2 (39). Moreover, DENV non-structural protein 1 (NS1) interacts with Toll-like receptor (TLR) 4 and TLR2, but only TLR4 also mediates platelet activation (40, 41). TLR2 is also important for platelet interaction with Human Cytomegalovirus (HCMV) (42) probably via HCMV glycoproteins B and H (43). Platelets also express complement receptors (CRs) on their surface which allows for the binding of Epstein-Barr virus (EBV) glycoprotein GP350 to platelet CR2 (44, 45).
The interaction of platelets with SARS-CoV-2 is rather controversial. Angiotensin-converting enzyme 2 (ACE2) is proposed as an entry receptor for SARS-CoV-2, which directly interacts with the spike protein (46). However, while some reports demonstrate ACE2 expression on platelets (47), other studies did not find detectable level of ACE2 but suggest mechanisms independent of ACE2 (48), while others found no virus particles within platelets at all (49). Given the low expression of ACE2 on platelets, ACE2-independent platelet activation involving more abundantly expressed receptors such as extracellular matrix metalloproteinase inducer (EMMPRIN/CD147) may be more important for direct platelet dysregulation by SARS-CoV-2 or its spike protein, respectively (50).
Platelets can not only bind but also take up virus particles. However, whether this uptake resembles a true phagocytosis, simple engulfment of virus or a different mechanism remains to be elucidated and might depend on the type of virus and receptors involved. Internalisation of HCV increases virus persistence by sheltering virus from degradation (51–54). DENV particles can be bound and internalized via DC-SIGN and HSP (37). Accordingly, DENV virions were also detected within platelets of infected patients and the presence of negative stranded RNA upon virus uptake suggest that platelets are permissive for virus replication, but this probably does not result in productive infection (55, 56). Other viruses, such as CVB and adenovirus were detected in the surface-connected open canalicular system (OCS) (15, 57), which is involved in internalisation processes. While uptake of CVB is mainly mediated by CAR, the OCS, platelet integrin αIIbβ3 and GPIb might also be involved (19).
Similarly, HIV particles can be engulfed in the OCS but also in vacuoles close to the plasma membrane. Some of these vacuoles resemble or are enclosed by endosome-like structures, suggesting phagocytosis along with simple uptake (27, 58, 59). However, the precise mechanism has not yet been unravelled and might involve HIV co-receptors along with yet unidentified mediators (27, 30). Recently, internalised HIV virions were found to be shuttled from early to late endosomes in an endocytic machinery-dependent manner, providing evidence of true phagocytosis by platelets (29, 59).
Internalisation of viral particles by platelets allows for stimulation of intracellular pattern recognition receptors such as TLRs, which has been observed for HIV and platelet TLR7 (60), but also EMCV and influenza virus are internalised by platelets (61) and their uptake involves TLR7 activation (62). This might represent a general mechanism how single-stranded RNA viruses interact with platelets and subsequently influence their activation. As yet another uptake mechanisms, successful internalisation of SARS-CoV-2 by platelets may be independent of ACE2 expression as virions were found to hitchhike on extracellular vesicles that fuse with the platelet membrane (63).
Platelets not only express several receptors for the recognition of pathogens but are also equipped with a plethora of antimicrobial molecules to fight viral infections. Interaction between platelets and viruses can thus have benefits and risks for the host: while direct interactions can lead to phagocytosis and degradation of viral particles, platelets can also become a hideout for viruses, thereby fostering reproduction and dissemination (Figure 3). Apart from their role in haemostasis, platelets represent relevant regulators of immune responses, e.g. by interacting with leukocytes and subsequently modulating their extravasation and effector functions (5, 64, 65). Thereby, quantification of circulating platelet-leukocyte aggregates (PLAs) is often used to gain insight into immunomodulatory platelet function.
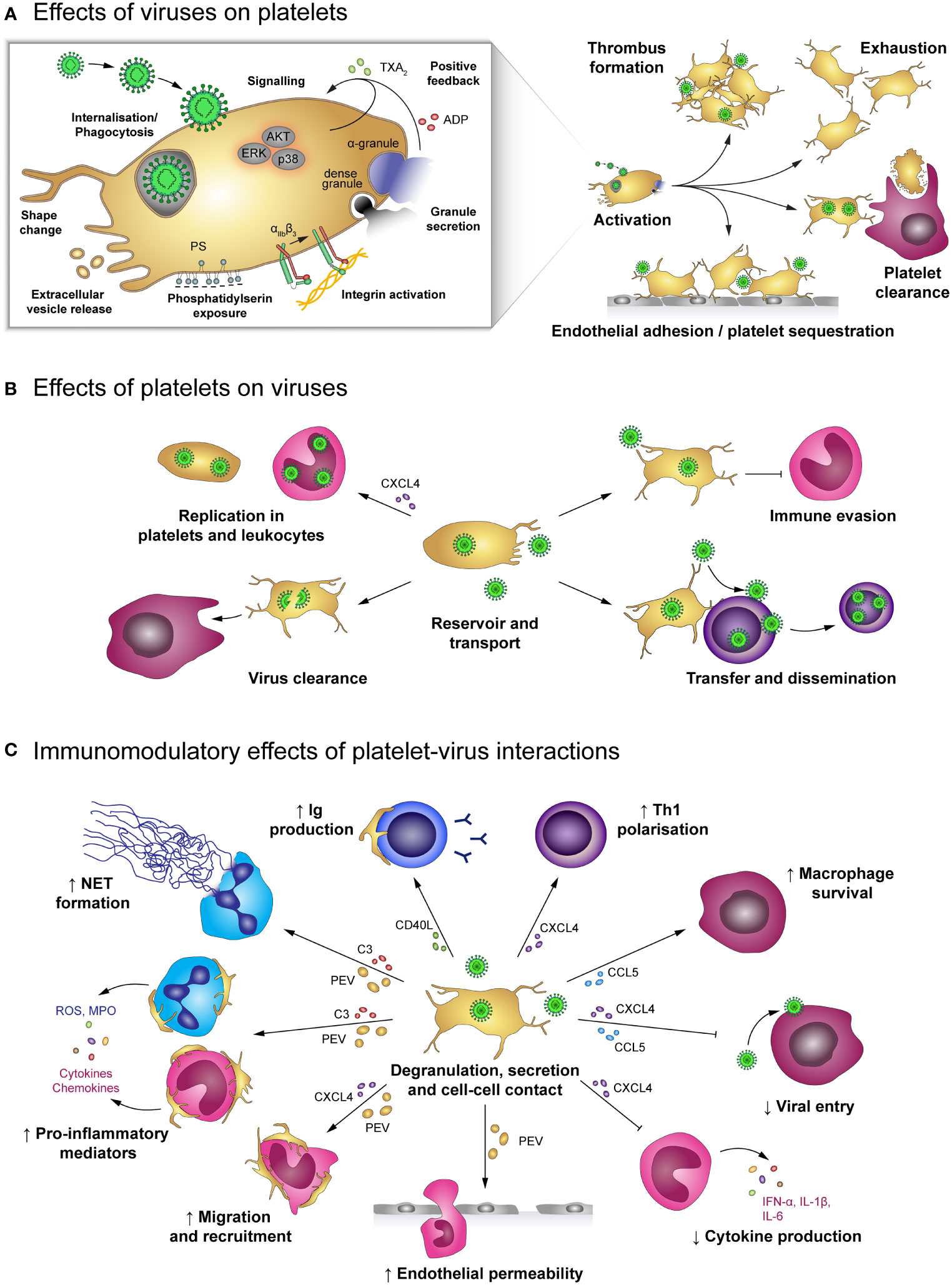
Figure 3 Effects of platelet-virus interaction. (A) Virus binding or internalisation/phagocytosis stimulates platelet activation including degranulation, integrin activation, phosphatidylserin exposure and extracellular vesicle release. Virus-induced platelet activation enhances endothelial adhesion and thrombus formation, but may also lead to platelet exhaustion and clearance from the circulation. (B) Platelets serve as virus reservoir and transport vehicle by supporting viral replication in leukocytes or within platelets themselves and by transferring virus to other host cells, thereby augmenting viral dissemination. Furthermore, platelets shelter intracellular virions from immune-mediated degradation, though degradation of virus-containing platelets contributes to virus clearance. (C) Virus-stimulated platelets modulate immune responses by cell-cell contact and/or soluble mediators. Platelet-derived factors block viral entry into leukocytes and prolong macrophage survival. Platelets also facilitate leukocyte extravasation by augmenting endothelial permeability, leukocyte migration and recruitment. Furthermore, platelets regulate leukocyte effector functions such as IgG production, T cell polarization, NET formation and release of inflammatory mediators, thereby either enhancing or diminishing immune defences. ADP, adenosine diphosphate; AKT, Protein kinase B; C3, complement factor 3; CCL5, chemokine (C-C motif) ligand 5; CD40L, CD40 ligand; CXCL4, chemokine (C-X-C motif) ligand 4; ERK, Extracellular signal-regulated kinase; IFN-α: Interferon α; IgG, immunoglobulin; IL-1β: Interleukin 1β; MPO, myeloperoxidase; NET, neutrophil extracellular trap; p38, p38 mitogen-activated protein kinase; PEV, Platelet-derived extracellular vesicle; PS, phosphatidylserine; ROS, reactive oxygen species; Th1, T helper cell type 1; TXA2, thromboxane A2.
Direct interaction of viruses and platelets is associated with platelet activation indicated by elevated P-selectin (36, 37, 39, 42, 66–68), CD63 (39) and CD40L expression (67), αIIbβ3 activation (66, 68), phosphatidylserine (PS) exposure (36, 68) as well as increased secretion of platelet-derived extracellular vesicles (PEV) (39, 66, 68), platelet factor 4 (PF4/CXCL4) (69), Thromboxane A2 (TXA2) (66) and ADP/ATP (42). In addition, enhanced AKT, p38 and ERK phosphorylation (42) and changes in the platelet morphology (67) support the notion that platelets are stimulated by the direct interaction with viruses (44, 55, 59, 67, 70). As a consequence, platelet activation promotes endothelial adhesion (39) and coagulation (44, 69, 70).
Despite their seemingly simple nature, platelets have been known for decades to bear the ability to kill viral particles by every trick in the book of phagocytosis, including attachment, invagination and formation of phagosome-like structures (7). Viruses are ensnared in endocytic vesicles and killed by antimicrobial peptides when the vesicles fuse with granules. For HIV, this uptake occurs mainly in the OCS, where viruses lose their regular morphology (69) and envelope (29, 59), while phagocytosis of influenza virus is OCS independent (62, 71).
Studies on patient-derived blood showed that platelets rapidly internalise and digest influenza A (H1N1 or H3N2) by fusion of vesicles and granules, suggesting that during viremic influenza infection platelets contribute to viral killing (62, 71). Findings on platelet interaction with HIV are still controversial. While some reports indicate HIV degradation in platelets, others found that platelets can take up HIV particles and keep them in an infectious state. Infectious HIV can then be transmitted from platelets to T cells or dendritic cells, which facilitates viral dissemination (30). This is of special importance, as platelets from HIV-infected individuals on combined antiretroviral drug therapy with low blood CD4+ T cell counts contain replication-competent HIV despite viral suppression (72). Dengue virus particles were also observed in vesicles of patient-derived platelets and Dengue structural protein E was detected inside platelets or in platelet micro-aggregates of in vitro infected platelets (56). However, it is currently unclear whether platelets degrade dengue virions or if the virus enters and/or infects platelets to evade immune responses. While it is clear that platelets directly interact with HCV (51, 73, 74), little is known on internalisation and or degradation of HCV by platelets. But the fact that even after anti-viral therapy HCV RNA was undetectable in serum but still detectable in platelets suggests that platelets serve as a reservoir for HCV and protect them from immune recognition (73).
Intriguingly, although being an anucleated cell, platelets may also serve as host cells for virus replication. Indeed, platelets with internalised Dengue virus particles were shown to enable replication of the viral genome (+ssRNA) of all Dengue virus serotypes (37). During acute infection a high number of Dengue associated copy numbers could be found in Dengue patients’ platelets (68) and isolated platelets contain a higher proportion of Dengue virus-associated RNA than plasma (56), suggesting that Dengue virus infects platelets for reproduction (37, 68). However, while Dengue virus reproduction in platelets themselves is rather inefficient (55), Dengue virus activated platelets enhance viral replication in monocytes and THP-1 cell lines in vitro via secretion of PF4/CXCL4 (75).
Platelet activation in response to viruses can thus indirectly contribute to antiviral effects via modulation of immune responses.
Platelet-virus interaction triggers the release of cytokines and chemokines from platelets including tumour necrosis factor α (TNF-α), interleukin (IL) 6, IL-8, IL-10, monocyte chemoattractant protein 1 (MCP-1/CCL2), transforming growth factor β (TGF-β) and the granulocyte-macrophage colony stimulating factor (GM-CSF) (67). These mediators are important for the initiation of cell migration and immune defence mechanism. In addition, formation of platelet-leukocyte and platelet-lymphocyte aggregates facilitates leukocyte activation, recruitment and reactive oxygen species (ROS) formation (42) as well as B cell isotype switching to immunoglobulin (Ig) G production (76).
In addition to direct platelet-leukocyte interaction, Dengue virus-stimulated platelets also release PEV that enhance vascular permeability, thereby promoting leukocyte migration, and also stimulate the release of pro-inflammatory cytokines such as TNF-α and IL-6 by neutrophils and macrophages. Furthermore, virus-stimulated platelets play a role in formation of neutrophil extracellular traps (NETs) (39), the release of neutrophil-DNA and myeloperoxidase (MPO), which is provoked by platelet-derived complement factor C3 (62).
However, platelet-virus interaction can also have immunosuppressive functions. In acute and chronic EBV infections platelet-derived TGF-β inhibits lung epithelial growth and contributes to disease pathology (44). Also, PF4/CXCL4 diminishes the immune defence against Dengue virus in vitro by inhibiting monocyte secretion of interferon α (IFN-α), IL-1β and IL-6 (75). In addition, activated platelets may influence antiviral immune defences by modulating infection of leukocytes themselves. PF4/CXCL4 reduces HIV infection of T cells and macrophages by binding a subunit of the viral envelope glycoprotein 120 (gp120) and thus blocking viral entry (69, 77, 78).
Dengue patients often present with upregulated platelet apoptosis, indicated by mitochondrial depolarization, elevated PS exposure and high expression of caspase-3 and caspase-9 (36, 79). Moreover, increased platelet levels of annexin V, cyclophilin D and thrombopoietin (TPO) are associated with diminished platelet count as apoptotic platelets get phagocytosed by monocytes via PS recognition (68, 79). Similarly, platelets of dengue patients have impaired mitochondrial membrane potential and constitutively generate ROS, indicating that platelet mitochondria are impaired in viral infections (36).
Thrombocytopenia is a hallmark of viral infections and the underlying causes are multifaceted and complex (2). Thrombocytopenia can be induced by four major mechanisms: (1) direct destruction of circulating platelets, (2) platelet destruction by immunological features such as auto-antibodies, (3) dysregulation of platelet production (megakaryopoiesis and thrombopoiesis) or (4) direct destruction of megakaryocytes (36).
Platelet counts often inversely correlate with viral loads and disease progression, indicating a hallmark in disease pathology, though the underlying mechanisms vary (70, 73, 78). Dengue-associated thrombocytopenia is related to complement-antibody mediated clearance and lysis of activated platelets (36, 37, 68, 75), while platelet activation contributes to thrombocytopenia in HCV infection (73). Influenza-associated thrombocytopenia is, dependent on the influenza strain, induced by viral neuraminidases, which remove sialoglycans after internalisation, thereby stimulating platelet clearance by liver hepatocytes and macrophages (7, 70). In contrast, in vitro Dengue and Junin virus can infect hematopoietic progenitors, which induces production of type I IFN, which in turn attenuates pro-platelet production (80, 81). Interestingly, upregulation of antiviral immune genes e.g. interferon-induced transmembrane protein 3 (IFITM3) in megakaryocytes in response to Dengue infection prevents infection of neighbouring megakaryocytes (81). Of note, patients with an early and sustained anti-viral response may have lower platelet counts during acute infection than patients with no-early response (73). Severe systemic viral infections often result in disseminated intravascular coagulation (DIC), in which excessive and systemic activation of the coagulation system leads to increased consumption of coagulation factors and platelet activation and ultimately results in formation of microthrombi. The subsequent drop in platelet count and concurrent haemorrhages represent a most dreaded clinical complication (82).
Indirect platelet-virus crosstalk on the molecular level may involve additional factors that bridge the contact between platelet receptors and virus particles, but indirect interactions may also be mediated on cellular level when platelets communicate with virus-infected or –activated cells (Figure 2).
Platelets also express FcγRIIA, which is a low-affinity receptor for the constant domain of IgGs. This allows for binding of virus-containing immune complexes that are generated during viral infections (83) and contribute to viral pathogenicity (66). As binding of immune complexes to FcγRIIA activates platelets (66, 84), indirect binding of virions to platelets via bridging IgGs may stimulate platelets even in the absence of virus-specific receptors. Indirect binding of virus-containing immune complexes to platelet FcγRIIA has been described for dengue virus, influenza virus and HCV (35, 68, 85).
Apart from virus-IgG complexes also autoantibodies are generated during infections which are recognized by FcγRIIA. In HCV infection autoantibodies that are cross-reactive towards platelet glycoproteins have been described (86). Autoantibodies against platelets are also common during DENV infection (87, 88). Here, antibodies which initially target DENV NS1 protein are cross-reactive with protein disulfide isomerase (PDI) expressed on the platelet surface (89).
Platelets are primarily known for their role in haemostasis and as such are pivotally involved in preventing and stopping haemorrhages by attaching to the injured endothelium and sealing vessel gaps. This interaction is mediated by various integrins and surface receptors such as αIIbβ3, GPIbα and P-selectin (90), which are also involved in viral interactions. Infected endothelial cells thus also contribute to platelet activation by upregulating adhesion molecules, which influence the binding and interaction with platelets (91). For example, infection with various Hantaviruses e.g. Andes or Hantaan virus induces upregulation of endothelial β3 integrins, which are involved in the recruitment of platelets (11). However, β3 also displays the virus on the endothelial surface, which results in platelet recruitment, as shown for Sin Nombre virus (92). Platelets also bind to endothelial cells infected with Puumala hantavirus (PUUV) (93), which may contribute to the low platelet count observed in patients with PUUV infection.
Increased platelet-endothelium interactions are also observed in DENV (94), Influenza virus (95) and HCMV infection (96). DENV infected endothelial cells express increased E-selectin levels, which in turn promotes platelet adhesion and activation (94). Moreover, DENV-derived NS1 protein triggers degranulation of Weibel-Palade bodies, leading to increased secretion of von Willebrand Factor (vWF) (40), further augmenting platelet adhesion and activation. Similarly, active HCMV replication in endothelial cells increases their expression of vWF and intercellular adhesion molecule 1 (ICAM-1), which mediates platelet-endothelial interactions (97) via platelet GPIb (96). Although vWF and ICAM-1 are upregulated during influenza infection, platelet adhesion to influenza-infected endothelial cells is primarily mediated by other molecules, such as endothelial fibronectin and platelet α5β1. Moreover, paracrine mechanisms also contribute to platelet adhesion in influenza infection, as platelets also attach to cells neighbouring infected ones (95).
In addition to triggering endothelial expression of pro-thrombotic factors and adhesion molecules, virus infection of endothelial cells may also promote endothelial permeability, thereby causing exposure of the subendothelial matrix and subsequent platelet adhesion. Indeed, interaction of DENV with TLR4 was shown to disrupt endothelial integrity (98) and infection of pulmonary microvascular endothelial cells with Influenza virus increases endothelial permeability by inducing apoptosis (99).
Regulation of platelet binding to infected endothelium may have clinical significance. As such, in vitro endothelial cells infected with herpes simplex virus (HSV) display changes in surface composition that facilitate the assembly of the pro-thrombinase complex and thereby accelerate thrombin generation, while at the same time prostacyclin (prostaglandin I2; PGI2) secretion is impaired. Together, these changes in endothelial activation promote platelet binding and induce a pro-thrombotic and pro-coagulant microenvironment (100) that may foster thromboembolic complication.
Platelet interaction with viruses contributes to the pathologies of a plethora of infectious diseases. In the following section we focus on three types of viruses, which are associated with haemostatic imbalances. We summarize the current knowledge on viral haemorrhagic fever (VHF), an infectious disease associated with an increased bleeding risk, influenza and COVID-19, which are primarily pulmonary diseases but also associated with thrombotic complications.
The term viral haemorrhagic fever (VHF) describes an infectious disease associated with an increased bleeding risk. VHF is caused by a distinct group of enveloped RNA viruses that belong to four virus families: Flavivirus (Dengue virus, Yellow fever virus), Bunyaviridae (Hantavirus, Crimean-Congo haemorrhagic fever virus), Arenaviridae (Lassa virus, Junin virus) and Filoviridae (Ebola, Marburg and Sudan virus). An infection with these viruses causes systemic pathogenesis characterized by fever, malaise but also increased vascular permeability and the development of thrombocytopenia, both supporting increased bleeding tendencies (Figure 4). These vascular and haemostatic dysregulations often lead to coagulopathies, which aggravate disease outcome. VHFs are associated with high mortality rates. However, currently no effective therapeutic interventions are known, making them a major global health problem (101). Although, the underlying molecular pathways and mechanism are poorly understood, an involvement of platelets as main players of haemostasis is likely (102).
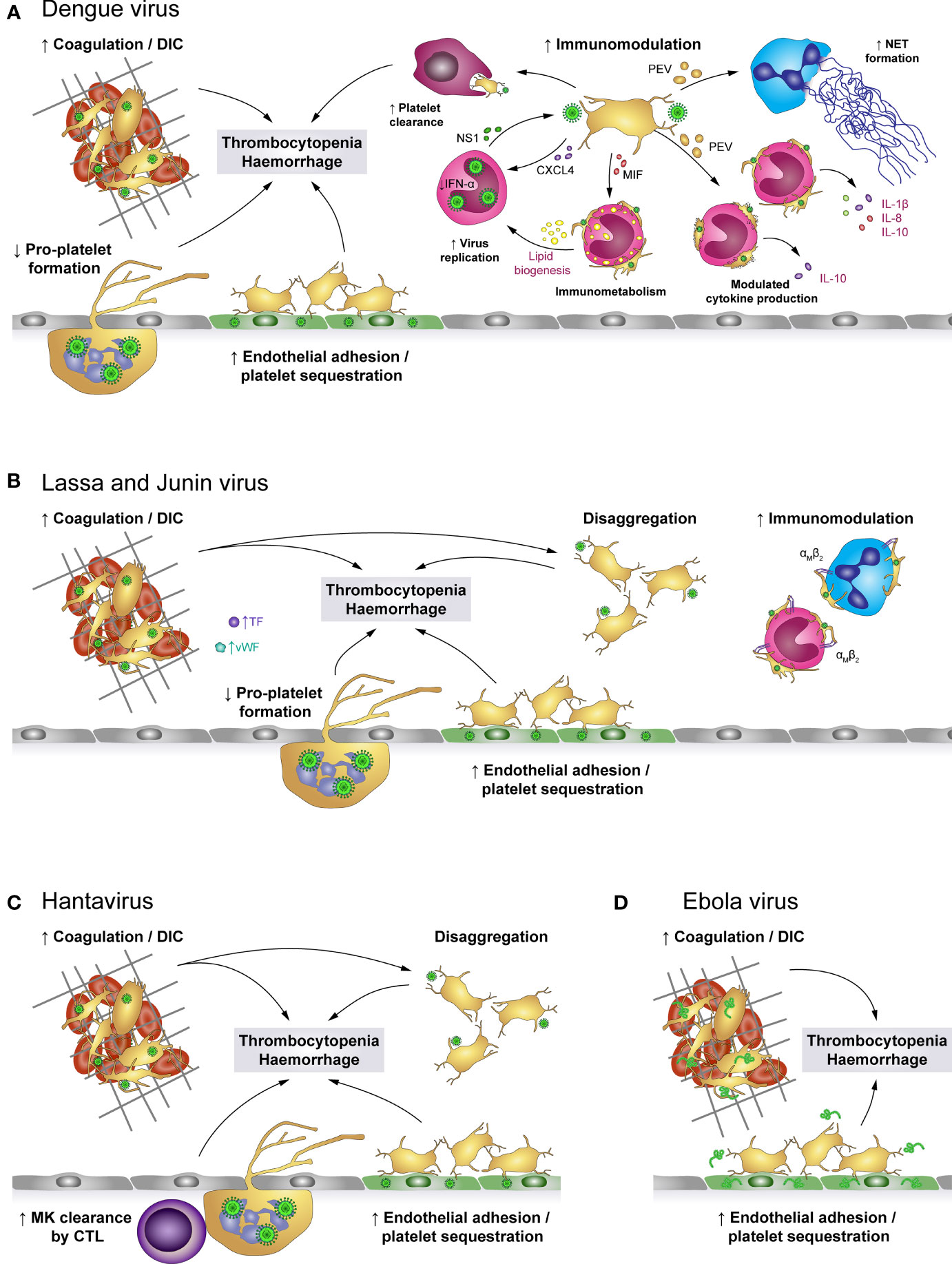
Figure 4 The role of platelets in viral haemorrhagic fevers (VHF). (A) Dengue virus activates platelets and promotes the development of disseminated intravascular coagulation (DIC), leading to platelet consumption. Additionally, activated platelets adhere to infected endothelial cells and infected megakaryocytes (MK) show impaired pro-platelet formation. Together with macrophage-mediated clearance of virus-activated platelets, these mechanisms lead to thrombocytopenia and enhanced haemorrhagic complications. In addition, virus-stimulated platelets bind to leukocytes or release soluble factors to modulate immune responses such as NET formation as well as production of a specific cytokine profile, depending on whether interacting platelets are activated or apoptotic. Platelet-derived factors also promote virus replication in monocytes which is further fostered by modulated immunometabolism and platelet-induced lipid biogenesis. (B) Infection with Lassa or Junin virus is associated with elevated levels of von Willebrand factor (vWF) and tissue factor (TF) which contribute to platelet activation and development of DIC. While activated platelets readily bind to monocytes and neutrophils via their αMβ2 receptor, they are unable to maintain stable platelet-platelet interaction and rapidly disaggregate. Further, virus infection augments endothelial platelet adhesion and sequestration and limits pro-platelet formation of MKs. Thereby, infection with Lassa or Junin virus leads to thrombocytopenia and increases bleeding risk. (C) Hantavirus readily infects endothelial cells which promotes endothelial platelet adhesion and sequestration from the circulation. While infected megakaryocytes display no differentiation dysfunction, they are cleared by cytotoxic T lymphocytes (CTL). Systemic infection triggers a pro-coagulatory state which may exacerbate to DIC. Nevertheless, platelets show impaired capacity to form stable aggregates. Hantavirus thus causes thrombocytopenia and haemorrhagic complications by affecting key mediators of haemostasis. (D) Infection with Ebola virus is associated with systemic coagulation induction and risk for DIC, which leads to platelet consumption. However, platelets may also be sequestrated due to enhanced adhesion to the infected endothelium. Thereby, Ebola virus causes thrombocytopenia and haemorrhagic complications. CTL, Cytotoxic T cell; CXCL4, chemokine (C-X-C motif) ligand 4; DIC, Disseminated intravascular coagulation; IFN-α: Interferon α; IL, Interleukin; MIF, Macrophage migration inhibitory factor; MK, Megakaryocytes; NET, Neutrophil extracellular trap; NS1, Non-structural protein 1; PEV, Platelet-derived extracellular vesicle; TF, Tissue factor; VHF, Viral haemorrhagic fevers; vWF, von Willebrand factor.
Various mechanisms were shown to trigger thrombocytopenia in VHFs (102), though depending on the virus strain different mechanisms dominate. Especially, virus-induced tissue damage and DIC result in excessive platelet activation and hence consumption. DIC is observed during DENV infection (103–105), in Ebola (106, 107) and also in Haemorrhagic Fever with Renal Syndrome (HFRS) caused by PUUV (108). Accordingly, thrombocytopenia along with abnormal coagulation is a known hallmark of pathogenic Hantavirus infection (109, 110), while in Lassa fever DIC seems to play only a minor role (111–113).
Also endothelial cells contribute to haemostatic dysregulations in VHFs. On the one hand infection of endothelial cells induces tissue factor expression, leading to increased thrombin generation and thereby haemostatic disturbances, which increase the risk for clot formation (93). Accordingly, D-dimer levels and prothrombin fragments F1 + 2, are increased in Dengue patients (93), patients with fatal Crimean Congo Haemorrhagic fever (114) and Ebola (106).
On the other hand the infected endothelium contributes to diminished platelet counts via scavenging and/or activation of circulating platelets. Platelet adhesion to DENV-infected endothelium is partially prompted by increased expression of endothelial E-selectin and P-selectin, which further contributes to a drop in circulating platelets (94). Also Ebola patients show elevated E- and P-selectin levels, indicating endothelial cell and platelet activation, which might contribute to diminished circulating platelets due to endothelial scavenging (115). Platelet adhesion to endothelial cells was further observed during PUUV (93) and Hantaan virus (HTNV) infection (11).
DENV-derived NS1 also directly increases platelet adhesion. NS1 activates platelets via TLR-4, thus supporting increased endothelial adhesion but also enhancing platelet aggregation (40). Subsequently, NS1 activated platelets are prompted to perform a phenotypic switch towards inflammation including degranulation, the synthesis but not the release of IL-1β as well as the continued expression and release of NS1 after infection. In turn, released NS1 and granule-stored factors might further enhance platelet activation and aggregation in an autocrine loop (41). Enhanced platelet activation is additionally triggered by DENV-induced synthesis and expression of human leukocyte antigen (HLA) class 1 on platelets which then binds circulating cell-free H2A histones, found in dengue patients’ plasma (116). Enhanced platelet activation and adhesion thus represent a possible link to thrombocytopenia and haemorrhages in dengue fever patients.
Intriguingly, in Lassa fever patients with fatal outcome and during the acute phase of Hantavirus infection dysfunctional platelets with an impaired aggregation potential were found (117). Indeed, normal aggregation is rapidly followed by disaggregation, indicating that Lassa virus (LASV) infection triggers an aggregation inhibition, which is likely due to impaired platelet degranulation as degranulation is essential for sustained platelet aggregation (111, 118, 119). Similarly, infection with Junin virus (JUNV), another virus belonging to the family of Arenaviridae causing Argentine Haemorrhagic Fever, is associated with decreased platelet aggregation, which is mediated by an unidentified plasma component present during acute infection (120). As a consequence, diminished platelet aggregation might contribute to bleeding events that frequently occur in these patients.
Additionally, several proteins important for haemostasis such as TF and vWF are also increased in plasma from Lassa patients, which further implicates pathogenic activation of the coagulation system as well as platelets (111). Similarly, patients suffering from haemorrhagic manifestations during Sudan virus infection also show elevated levels of vWF. This further implicates that VHF viruses contribute to excessive thrombotic events (121).
Phagocytosis of platelets is another mechanism leading to a decrease in circulating platelets. Platelets are cleared via phagocytosis by macrophages upon activation by DENV (40) or severe fever with thrombocytopenia syndrome virus (SFTSV), a member of the Bunyaviridae family like Hantavirus (122). Platelets from dengue patients further shows decreased sialic acid levels, which are associated with increased platelet phagocytosis and hence platelet clearance (123, 124). In line, Dengue fever is associated with increased levels of circulating active vWF, which also induces removal of sialic acids on platelets via neuraminidase mobilization (124). These observations suggest that the underlying mechanisms of thrombocytopenia include platelet clearance.
Not only dysfunctional platelets and/or reduced platelet counts are problematic in VHF, but additional activation of the immune systems e.g. by platelets can further exacerbate disease progression. Platelets also have immunomodulatory functions during VHF. Leukocyte integrin αM (ITGAM, CD11b) is upregulated upon exposure to LASV, which may enhance platelet-leukocyte binding and therefore complicate symptoms (125, 126). During EV infection, a transient increase in sCD40L was measured during the early stages of infection which might be implicated in activating further immune cells (107).
In DENV infection, activated platelets release PMVs, which induce neutrophil activation and subsequent NET formation (39). Additionally, DENV-exposed platelets can also reprogram immunometabolism of uninfected monocytes and amplify potential inflammatory cytokine release. Moreover, by forming aggregates with platelets, monocytes become activated and increase lipid droplet (LD) biogenesis (127). LDs play an important role in the pathogenesis of DENV infection. Viruses are unable to perform lipid synthesis which is a pre-requisite for functional virion production (128) and must therefore exploit cellular mechanisms (129). Indeed, leukocytes from Dengue patients show high LD formation, suggesting that lipid biogenesis might contribute to Dengue pathogenesis (129, 130).
Platelets from dengue patients show increased PS exposure, depolarization of mitochondria as well as the activation of caspase 9 and caspase 3. Thereby DENV-induced platelet apoptosis is not only associated with increased thrombocytopenia but may also enhance monocyte binding (36). Indeed, DENV-activated and apoptotic platelets form aggregates with monocytes and induce secretion of IL-1β, IL-8 and IL-10 or only IL-10, respectively, in a mechanism involving P-selectin-mediated binding as well as recognition of exposed PS (131). All these mechanisms suggest that DENV actually induces cellular changes on many different levels by triggering platelet activation which might aggravate the disease (39).
Lastly, platelets induce the inhibition of IFN-α production in monocytes and enhance DENV replication in monocytes. This effect is at least partially dependent on PF4/CXCL4 and increased plasma levels of PF4/CXCL4 correlate with increased DENV NS1 in monocytes from patients (75).
However, not only platelets but all their progenitors, including both megakaryocytes and haematopoietic stem cells, are affected by VHFs. DENV is able to infect and replicate in megakaryocytes, which leads to diminished megakaryocyte development and platelet production due to diminished pro-platelet formation (132, 133). Similarly, JUNV infection of megakaryocytes also impairs thrombopoiesis by decreasing pro-platelet formation. Reduced pro-platelet formation together with abrogated aggregation responses of circulating platelets contribute to haemorrhages in patients with JUNV infection (80). Also HTNV infects megakaryocytes (134), however no direct effects on megakaryocyte differentiation and survival were observed. Nevertheless, infected megakaryocytes are cleared by cytotoxic T cells, thus also supporting thrombocytopenia (135).
Influenza is an acute respiratory infection caused by negative-strand RNA viruses belonging to the Orthomyxoviridae family that cause seasonal influenza pandemics. While three distinct types of influenza viruses (A, B and C) can infect humans (136, 137), influenza C virus mainly infects children, causing only mild upper respiratory tract infections. Therefore available vaccines target influenza A and B viruses, but not influenza C (138–140). Especially influenza A viruses regularly acquire adaptive mutations by genetic drifts and shifts that create novel influenza A subtypes (136). The virion envelope of influenza is covered with hemagglutinin (HA) and neuraminidase (NA) glycoproteins, which determine the specificity of the virus for a host species and cell type. Each Influenza A subtype is characterized by numbering of HA and NA proteins. The two subtypes H1N1 and H3N2 currently circulate in the human population (139). In contrast, Influenza B virus has only a single subtype with two lineages and is not further sub-categorized (139, 141).
Influenza viruses are primarily transmitted via inhalation of infectious particles when an infected person coughs or sneezes. However, airborne and fomite transmission may also contribute to viral spreading (136). During infection HA binds to the sialic acid (SA)-terminated glycans present at the cell membrane. NA facilitates the release of virus progeny by cleaving SA residues from the cell surface (70) (Figure 5).They primarily target epithelial cells of the upper and lower respiratory tract, causing pneumonia, but also encephalopathy and myocarditis (142, 143). In severe cases pneumonia is often accompanied by acute lung injury (ALI) or even acute respiratory distress syndrome (ARDS), which is characterized by alveolar capillary damage, oedema, parenchymal haemorrhages, pulmonary microvascular thrombosis and hyperinflammatory cytokine responses (142, 144). ALI and ARDS also correlate with organ failure, ICU admission and a high fatality rate (145).
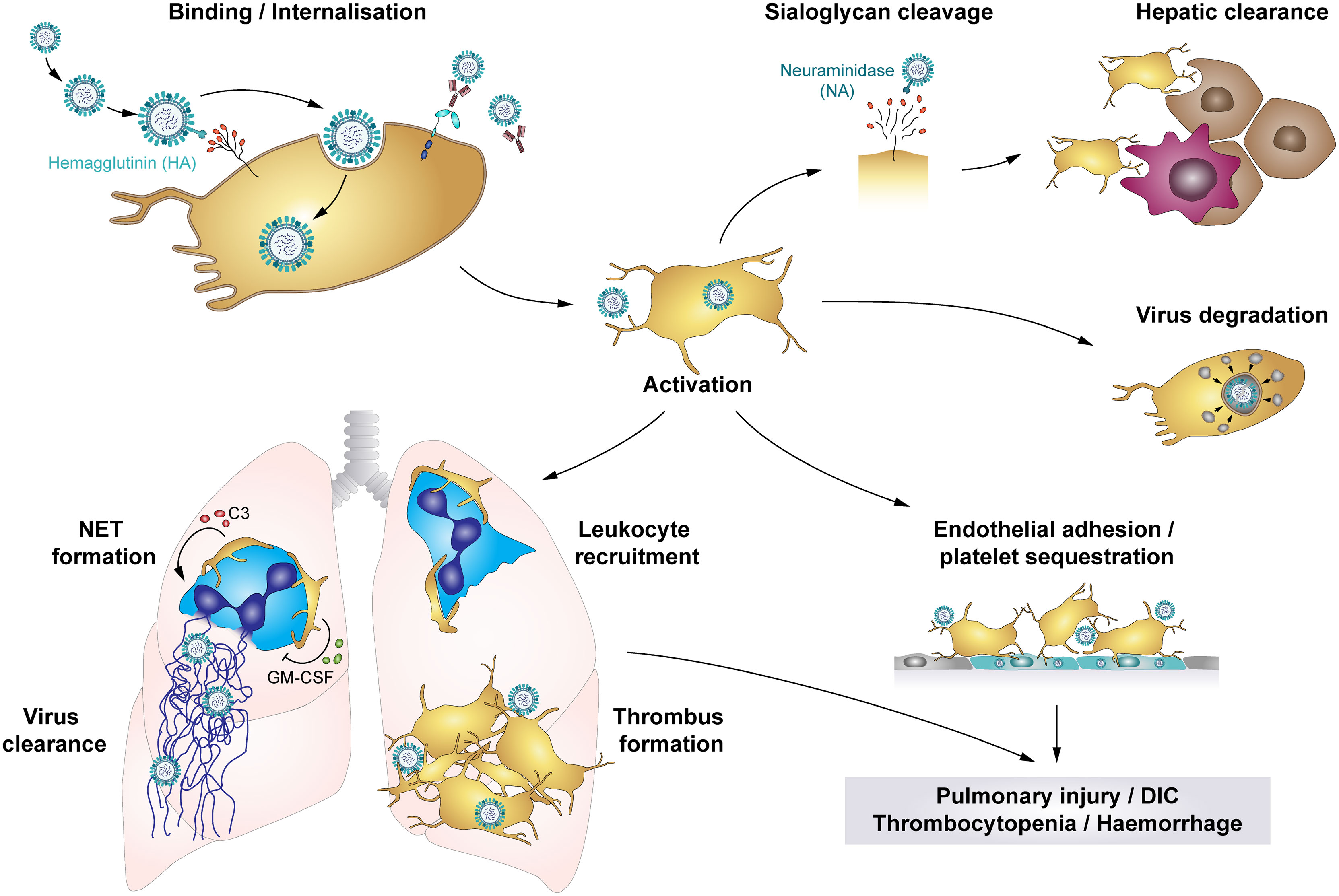
Figure 5 The role of platelets in influenza infections: Platelets bind and internalise influenza virus via the interaction of virus hemagglutinin (HA) proteins and sialic acid (SA)-terminated glycans on the platelet surface, though platelets also bind influenza-containing immune complexes. These interactions result in platelet activation and platelet-mediated immune responses that contribute to influenza-associated pathologies. Cleavage of SA residues by viral neuraminidase (NA) induces platelet clearance by liver hepatocytes and Kupffer cells. However, rapid internalisation of viral particles leads to digestion of influenza virus when vesicles with enclosed virions fuse with platelet granules that contain antimicrobial peptides. Systemically, influenza-infected endothelial cells express pro-thrombotic factors like fibronectin or integrin α5β1, which increases platelet adhesion to the endothelium and thus fosters platelet sequestration. Locally, platelets infiltrate the lung tissue, where thrombus formation may constrict and/or occlude blood vessels or small airways. Furthermore, platelet-leukocyte aggregate formation induces leukocyte recruitment and triggers the formation of NETs to entrap viral particles, regulated by C3 and GM-CSF. However, NETs cause further tissue damage and enhance thrombus formation. Together, these platelet-mediated responses in influenza trigger pulmonary injuries, disseminated intravascular coagulation (DIC), thrombocytopenia and haemorrhages in severe influenza infections. C3, Complement factor C3; DIC, disseminated intravascular coagulation; GM-CSF, Granulocyte-monocyte colony-stimulating factor; HA, hemagglutinin; NA, neuraminidase; NET, neutrophil extracellular traps.
Severe influenza infections can also have major impact on the haemostatic system, with thrombosis and bleedings potentially occurring at the same time. On the one hand, influenza infections are often associated with thrombocytopenia and elevated mean platelet volume (MPV), indicating elevated thrombopoiesis, potentially due to elevated platelet activation and phagocytosis of viral particles (70, 145–148). Accordingly, pulmonary, alveolar and interstitial haemorrhages are frequent complications (70, 137, 149). On the other hand, influenza infection and associated platelet activation increases the risk of thrombus formation.
Influenza-stimulated platelets infiltrate the lungs of infected individuals (137, 149), where they form platelet-platelet and platelet-leukocyte aggregates (137, 147, 149), partially occluding blood vessels or small airways. In addition, neutrophil stimulation triggers NET release, which are highly cytotoxic and thus contribute to tissue damage and induce thrombosis. Interestingly, influenza-mediated platelet activation is not restricted to the lungs, as NET components and platelets co-localize in the heart of influenza-infected mice (144, 149). Also, enhanced deep vein thrombosis (DVT) and pulmonary thromboembolism may occur in influenza patients (142). These data indicate that thrombocytopenia and platelet activation contribute to influenza-associated pulmonary injuries caused by systemic inflammation and local leukocyte infiltrates, thereby fostering pulmonary thrombosis and haemorrhages (71, 147). In addition, in mice anti-platelet therapy [including aspirin, P2Y12 blockage and antagonists of αIIbβ3 or protease-activated receptor 4 (PAR4)] reduces platelet aggregation, leukocyte recruitment and infiltration, viral reproduction and alveolar damage, ameliorating survival and underlining the contribution of platelets to influenza-mediated lung pathologies (137, 149, 150). However, the role of PAR4 remains unclear as one study found a protective effect of PAR4 inhibition on survival in influenza-challenged mice (137), whereas another study reported a detrimental effect of PAR4 deficiency (150).
Recent findings suggest that communication between platelets and neutrophils via CXCL4 is a prerequisite for viral removal and efficient immune response (151). However, dysregulated platelet-neutrophil crosstalk also contributes to influenza-mediated pathologies. Platelet derived complement factor C3 and GM-CSF are regarded as key proteins for platelet-neutrophil communication. Activated platelets form aggregates with neutrophils and secrete C3 from their granules which induces NET formation and thus helps to entrap and kill viruses. In turn, neutrophils trigger platelets to release GM-CSF, which serves as a negative feedback mechanism by reducing C3-mediated NET formation. Dysregulated platelet-neutrophil communication and surplus of C3 and NETs are predictors of acute myocardial infarction and myocardial infarct size and are therefore believed to increase the risk of influenza-associated myocardial infarct (62). Furthermore, platelet aggregation involving integrin αIIb was recently found to play an important role for platelet-leukocyte interplay during influenza as mice deficient for αIIb displayed reduced pulmonary neutrophil influx and NET formation upon challenge with influenza. Similarly, thrombin and PAR4 inhibition also diminished platelet accumulation, neutrophil influx and NET formation in the lungs and reduced pulmonary oedema without affecting viral load, underlining the importance of platelet aggregation for neutrophil-mediated tissue damage (152).
Moreover, platelets contribute to influenza-associated pathologies by interacting with infected endothelial cells. Both in vitro and in vivo platelets adhere to endothelial cells upon infection with influenza A subtypes H1N1 and H3N2, mediated by the interaction of endothelial fibronectin and platelet integrin α5β1. In influenza-infected mice anti-platelet treatment using aspirin thus blocks platelet adhesion to the endothelium, leading to reduced arterial hypoxemia and improved survival (95).
SARS-CoV-2, the causative pathogen of coronavirus-induced disease 2019 (COVID-19), newly emerged in 2019 and led to an ongoing global pandemic.
The COVID-19 pandemic has forced the scientific community to face complex challenges in order to understand the molecular and cellular processes responsible for COVID-19 pathology and improve patient treatment. Although scientific advancements could build on knowledge of the closely related SARS-CoV-1 and Middle East respiratory syndrome coronavirus (MERS-CoV), SARS-CoV-2 shows distinct properties which require in-depth elucidation. Further, the rapid spread of SARS-CoV-2 demanded that new discoveries had to be reported at an unprecedented speed and made broadly available, often curtailing peer review processes and forcing researchers to lower their sights regarding tightly defined cohort composition. In particular, location and time of recruitment may influence clinical studies due to e.g. overtaxed healthcare systems and adapted treatment protocols. As a consequence, despite best efforts studies frequently yield contradictory results that will have to be verified in the future under controlled and comparable settings.
COVID-19 primarily affects the upper and lower respiratory tract, leading to respiratory distress or failure, however complications involving the haemostatic system such as thrombosis, thromboembolism and bleeding events are common, particularly in patients requiring intensive care unit (ICU) treatment (153–155). Additionally, platelet counts are reduced in COVID-19, hinting towards an involvement of primary haemostasis, though counts do not drop to the same extent as during non-COVID-19 pneumonia and clinical thrombocytopenia is rare (156–160). Of note, thrombocytopenia occurs more frequently in severe than non-severe infection (161) and is associated with significant bleeding complications (153).
Despite overall minor reduction of circulating platelets, COVID-19 is associated with increased levels of immature platelets, indicating enhanced platelet turnover (162). Blood smears of COVID-19 patients further revealed the presence of giant platelets, corroborating reports of increased MPV (47, 163, 164). Increased MPV is associated with disease severity (164, 165) and acute kidney failure in COVID-19 (166), but interestingly not with ICU requirement (48).
The primary receptor for SARS-CoV-2 is ACE2 which binds to the viral spike protein and enables cell entry, facilitated by transmembrane protease serine subtype 2 (TMPRSS2) (Figure 6). Both ACE2 and TMPRSS2 were found to be expressed on human and mouse platelets and on the megakaryocytic cell line MEG-01 (47), mediating virus binding and internalisation (47, 158). Additionally, platelet EMMPRIN (CD147) is involved in spike protein-mediated platelet activation and SARS-CoV-2 may hitchhike on extracellular vesicles to enter platelets via membrane fusion, thereby circumventing the need for ACE2 (50, 63). While platelets do not seem to support viral replication, MEG-01 cells can be infected at least temporarily, leading to rising intracellular and shed virions which suggests successful replication (158). However, whether megakaryocyte infection also occurs in vivo and if it affects thrombopoiesis is currently unknown.
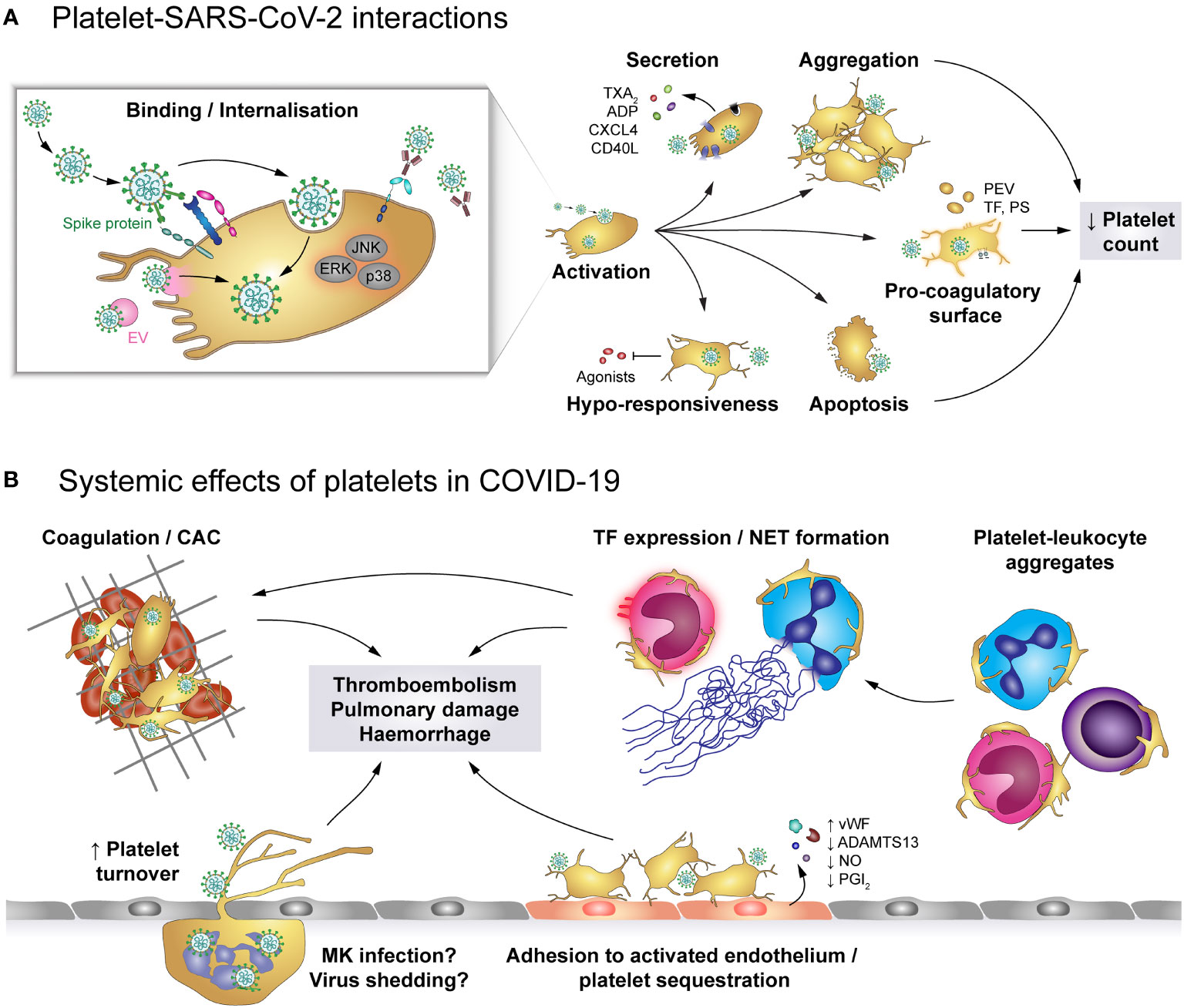
Figure 6 The role of platelets in COVID-19. (A) SARS-CoV-2 may enter platelets upon binding to ACE2/TMPRSS2 or EMMPRIN receptors or by hitchhiking on extracellular vesicles (EV) that fuse with the platelet plasma membrane, though SARS-CoV-2-containing immune complexes are also recognised by FcγRIIA. Binding and/or internalisation of SARS-CoV-2 stimulates platelet activation including degranulation and secretion, integrin activation and aggregation as well as exposure of pro-coagulant surfaces on platelets themselves or platelet-derived extracellular vesicles (PEV). Together with apoptosis of virus-bound platelets, these processes induce a reduction in circulating platelet count. Platelet hyper-activation in COVID-19 is accompanied by hypo-responsiveness to further stimulation. (B) Altered platelet behaviour in COVID-19 has systemic effects on pro-thrombotic and immunomodulatory platelet functions. Hyper-active and pro-coagulant platelets show enhanced adhesion to the inflamed endothelium and foster the development of COVID-19-associated coagulopathy (CAC). Accordingly, platelet turnover is increased in COVID-19. Formation of platelet-leukocyte aggregates triggers monocyte TF expression and NET formation, which add to the pro-coagulant microenvironment. In addition, infected megakaryocytes may shed virions to exacerbate infection. Together, these pathologic alterations increase the risk for thromboembolisms, pulmonary damage and haemorrhages. ACE2, Angiotensin-converting enzyme 2; ADP, adenosine diphosphate; ADAMTS13, A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; CAC, COVID-19-associated coagulopathy; CD40L, CD40 ligand; COVID-19, Coronavirus-induced disease 2019; CXCL4, chemokine (C-X-C motif) ligand 4; EMMPRIN, extracellular matrix metalloproteinase inducer; ERK, Extracellular signal-regulated kinase; FcγRIIA, Immunoglobulin γ Fc region receptor IIA; JNK, c-Jun N-terminal kinase; p38, p38 mitogen-activated protein kinase; MK, Megakaryocyte; EV, Extracellular vesicle; NET, neutrophil extracellular trap; PEV, Platelet-derived extracellular vesicle; NO, Nitric oxide; PGI2, Prostaglandin I2 (prostacyclin); PS, phosphatidylserine; SARS-CoV-2, Severe acute respiratory syndrome coronavirus 2; TF, Tissue factor; TMPRSS2, Transmembrane protease serine subtype 2; TXA2: thromboxane A2; vWF, von Willebrand factor.
SARS-CoV-2 infection is associated with dysregulated platelet functions, affecting all phases of primary haemostasis. Platelets of COVID-19 patients express increased surface levels of CD62P and CD63 relative to either healthy controls or non-COVID-19 pneumonia patients (47, 48, 157, 167–170), demonstrating augmented basal platelet degranulation. In line, α-granule-contained PF4/CXCL4 and dense granule-contained serotonin (5-HT) are reduced within platelets, but increased in plasma (46), along with sCD40L, ADP and TXA2 (164, 167, 169). Of note, surface CD62P, CD63 and plasma TXA2 levels correlate with plasma fibrinogen, D-dimer and C-reactive protein, suggesting a link between enhanced basal platelet degranulation and the hyper-inflammatory and hyper-coagulatory milieu in COVID-19 (167). Additionally, platelets of COVID-19 patients also exhibit enhanced basal αIIbβ3 activation relative to healthy donors or non-COVID-19 pneumonia patients (47, 168, 169).
In vitro and in vivo studies using a humanized mouse model demonstrate that SARS-CoV-2 virions dose-dependently enhance platelet degranulation, αIIbβ3 activation, spreading, aggregation and thrombosis in a mechanism involving spike protein and platelet ACE2 (47), further underlining the link between SARS-CoV-2 infection and platelet hyper-activation. On the other hand, plasma of severely-ill COVID-19 patients also induces platelet CD62P and CD63 expression relative to control plasma (167), providing evidence that platelet activation in COVID-19 is also regulated by plasma components.
While the exact mechanisms of SARS-CoV-2-mediated platelet hyper-activation is still unclear, COVID-19 is associated with enhanced activation of phospholipase A2 (PLA2) and protein kinase Cδ (PKCδ) (46, 48), which are prominently involved in TXA2 generation and platelet degranulation, respectively. Furthermore, janus-activated kinase 3 (JAK3) and mitogen-activated protein kinases such as extracellular signal-regulated kinase (ERK), p38 and c-Jun-N-terminal kinase (JNK) are also triggered in COVID-19 or upon direct exposure to SARS-CoV-2 (47, 48), providing evidence of the complex and diverse dysregulation of the intracellular signalling network that may cause platelet hyper-activation and influence thrombotic risk. Indeed, in a murine thrombosis model SARS-CoV-2 spike protein enhances thrombosis only if mice were transfused with human ACE2-expressing platelets, but not in mice transfused with wildtype platelets (47).
Though reports on elevated basal platelet activation in COVID-19 appear very consistent, associations with disease severity are more variable with some studies showing higher platelet activation in more severe cases (47, 167), whereas other studies did not find any association between disease severity and platelet activation (46, 48).
Interestingly, despite evident platelet hyper-activation in COVID-19, effects on platelet responsiveness are unclear. In fact, initial studies reported facilitated agonist-induced CD62P expression, TXA2 release, adhesion, spreading and aggregation in COVID-19 relative to healthy controls (46, 48). However, accumulating evidence indicates that platelets of COVID-19 patients display a hypo-responsive phenotype that affects degranulation (168, 171), αIIbβ3 activation (48, 157) and aggregation (172), potentially due to alterations in the proteome (173). Further, COVID-19 effects on platelet responsiveness were reported to differ between agonists (169) and to depend on agonist concentration (170) as well as disease stage (174). In line, in vitro thrombus formation under flow is decelerated during the early phase of disease, but comparable to healthy donors in intermediate and late stages, independently of platelet count or severity (175). This provides further evidence of the dynamic alterations of platelet function in COVID-19 and the difficulties in comparing studies of different sampling strategies.
Further, platelets of COVID-19 patients, especially of those with thrombosis, express higher levels of PS than control platelets (48, 176), but agonist-induced upregulation is impaired (177). Together with the occurrence of TF-positive platelets (178) and augmented circulating PMVs (46), these findings provide evidence of the pro-thrombotic and pro-coagulatory microenvironment in COVID-19, which results in thrombotic complications in lungs and other tissues (170). Though, in light of the observed hypo-responsiveness of platelets in aggravated COVID-19, the relative contributions of primary and secondary haemostasis to thromboembolic complications are still incompletely understood.
Hyper-activation of platelets in COVID-19 may not only contribute to elevated thrombotic risk, but could also mediate other disease symptoms such as thrombocytopenia. Aggravating disease severity is associated with increased levels of apoptotic platelets, which in turn negatively correlate with circulating platelet count. To distinguish apoptotic from pro-coagulant platelets, true apoptosis was corroborated by higher mitochondrial inner membrane potential and cytosolic calcium as well as augmented caspase 9 cleavage in addition to PS upregulation. Thereby, elevated platelet apoptosis was identified in COVID-19 patients requiring ICU treatment relative to both healthy controls and to COVID-19 patients in general ward. In vitro this pro-apoptotic phenotype can be reproduced by serum or IgGs derived from ICU patients, suggesting an FcγRIIA-mediated effect (176).
Apart from platelet apoptosis, declining circulating platelets could also be a result of platelet hyper-activation, leading to enhanced adhesion (46, 170) and thus potentially to sequestration. Indeed, platelet count negatively correlates with platelet degranulation markers (47) and associates with mortality (179) and bleeding risk (153, 180), but interestingly not with thrombosis (181). Platelet sequestration may be facilitated by activation of endothelial cells in COVID-19, which show impaired synthesis of nitric oxide and PGI2 (178). In combination with increased levels of vWF and decreased a disintegrin and metalloprotease with thrombospondin type motif 13 (ADAMTS13) in plasma (182), endothelial dysfunction thus generates a pro-thrombotic milieu that fosters the development of microthrombi and sequestration of hyper-active platelets.
Patients with COVID-19 also present with elevated levels of PLAs involving monocytes and neutrophils as well as CD4- and CD8-positive lymphocytes (48, 157, 167, 169). Elevated levels of platelet-monocyte aggregates (PMAs) were found predominantly in severe but not mild COVID-19 (167). Similarly, platelet-neutrophil aggregates (PNAs) also increase with disease severity and worsening blood oxygenation (170). Nevertheless, circulating PLAs are lower in non-survivors than survivors (174). Enhanced PLA formation in COVID-19 may of course be regulated by virus-induced leukocyte activation. However, platelet hyper-activation is also prominently involved as in vitro platelets from COVID-19 patients show higher PMA and PNA formation with naïve leukocytes than platelets from healthy donors (111, 170).
In line with platelet-mediated regulation of leukocyte function, monocytic TF expression in COVID-19 patients is higher on platelet-bound than on solitary monocytes (167) and patient-derived platelets promote NET formation over naïve platelets (170). These findings indicate that platelet activation may promote thromboembolic complications also via augmenting pro-coagulant leukocyte behaviour. Whether platelet hyper-activation and platelet responsiveness in COVID-19 also impact on immune responses and the ability to combat SARS-CoV-2 is currently still unexplored.
Overall, COVID-19 is clearly associated with platelet dysfunction, though its exact characteristics, dynamic changes and underlying mechanisms are still unclear. Current literature supports the idea that platelet dysfunction contributes to (micro)thrombotic events and may affect platelet counts, possibly even immune responses, which in turn could have repercussions for bleeding/thrombotic risk, organ function and ultimately survival.
Hence, the clinically-relevant effects of anti-platelet medication on COVID-19 morbidity and mortality are carefully investigated. Though, fitting to the controversial findings regarding platelet function in COVID-19, studies on the effect of anti-platelet medication also provide variable results.
While dual anti-platelet therapy improves hypoxemia (183) and aspirin has been associated with lowered risk for mechanical ventilation and ICU admission as well as decreased in-hospital mortality without affecting bleeding risk in some studies (184, 185), others found no protective effect of anti-platelet drugs against adverse thrombotic events, severity or mortality (186, 187). An ongoing large randomized controlled trial currently comprising almost 15.000 COVID-19 patients reports no impact of aspirin treatment on the risk for invasive ventilation or mortality (188), which fits to the hypo-reactive platelet phenotype described in various patient cohorts. Future studies will have to determine whether anti-platelet therapy might affect morbidity of COVID-19 patients by impairing other platelet-mediated processes, e.g. vascular surveillance or immunomodulation.
Understanding the underlying mechanisms of platelet dysfunction in viral infectious disease and unravelling the consequences of their interplay with other cellular and non-cellular mediators represents a prerequisite to discover novel and safe therapeutic targets in this complex disease. Current anti-platelet strategies targeting COX-1 and/or P2Y12 to reduce platelet activation and the incidence of arterial thrombosis in patients with cardiovascular diseases. However, they dysregulate the haemostatic balance, leaving patients at risk of systemic side-effects such as bleeding complications.
During chronic and acute inflammatory diseases, including virus infections, distinct mechanisms of platelet activation occur, which are potentially insensitive to classical anti-platelet drugs, e.g. due to alternative pathways of platelet activation that may be of particular importance for immunothrombotic processes during severe infections. The use of thrombin inhibitors or thrombin receptor antagonists which target both primary and secondary haemostasis, represents another available approach, but also bears a risk for haemorrhages. Animal studies on influenza infections could show beneficial effects of the thrombin receptor PAR1 beyond inhibition of platelet activation (189). A recent study suggests that inflammatory and immune-thrombotic mechanisms of platelets can be diminished upon inhibition of PAR4 during influenza infection (152), leading to ameliorated survival (137). However, inhibition of hypo-responsive platelets by these classical platelet agonists might further increase the bleeding risk. Therefore, alternative strategies are necessary to dampen the risk of haemostatic dysregulations in viral diseases.
Recent studies focus on targeting primary platelet activation pathways e.g. via immunoreceptor tyrosine-based activation motif (ITAM)-containing collagen receptor GPVI/FcRγ-chain complex. While GPVI inhibition yielded encouraging results, reducing platelet aggregation without elevating major bleeding complications (190), nothing is yet known on potential beneficial effects in viral diseases. Also targeting immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing receptors could provide an alternative approach for targeted platelet inhibition due to the role of these receptors in the downregulation of platelet ITAM-receptor signalling (191). Again, nothing is known yet on the effect of these inhibitors in viral diseases. However, as some viruses result in GPVI shedding and diminished GPVI responses, like all classical anti-platelet drugs also these inhibitors target platelets behind time. Interfering with platelet-virus interactions would therefore provide another interesting target, though this is likely to also affect immune-defence mechanisms and therefore accelerate the risk of unfavourable outcome.
Another crucial aspect is blocking immune-complex mediated platelet activation via FcγRIIA. The physiologic relevance of platelet FcγRIIA, which may facilitate both direct antimicrobial function of platelets as well as crosstalk with other immune cells, is currently unclear (192). This vicious interaction of immune complexes and platelet FcγRIIA is not only responsible for platelet activation in viral infections but also in a plethora of other diseases – including heparin-induced thrombocytopenia (HIT), autoimmune diseases like systemic lupus erythematosus (SLE) (193) or the more recently discovered vaccine-induced immune thrombotic thrombocytopenia (VITT). To date, no data exist on potential effects of drugs interfering with FcγRIIA signalling in viral infections. Investigations are complicated by the fact that mice do not express FcγRIIA on platelets. However, humanized mouse models do exist and for some viruses a crucial involvement of FcγRIIA for platelet activation and subsequent thrombocytopenia could be unravelled (193). Further studies are warranted to fully understand the impact of platelet inhibitors on viral infections. Beneficial effects of such interventions will likely depend on the virus type, disease stage and co-morbidities of patients. Therefore, experimental models and patient cohorts have to be carefully selected and results cautiously interpreted. Patients are often enrolled at different disease stages and not longitudinally monitored. Moreover, they often suffer from co-infections and/or other co-morbidities, which have to be considered. While some of these obstacles can be overcome by animal models, for some pathogens no appropriate models exist. Also, haemostatic processes as well as platelet surface receptor patterns significantly differ between mice and humans. As pandemics and concomitant haemostatic dysregulations will remain a recurrent threat, understanding the role of platelets in viral infections represents a timely and pivotal challenge.
Conceptualization: AA; Visualization: WS, AA; Funding Acquisition: WS, AA; All authors contributed to Writing, Original Draft Preparation and Writing, Review and Editing. All authors contributed to the article and approved the submitted version.
This work is financially supported by grants of the Austrian National Bank to WS (OENB18450) and of the Austrian Federal Ministry of Education, Science and Research, the Medical-Scientific Fund of the Mayor of Vienna (COVID024) and the Austrian Science Fund (P32064 and P34783) to AA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Manuel Salzmann for his help and provision of resources for figure preparation.
1. Schrottmaier WC, Mussbacher M, Salzmann M, Assinger A. Platelet-Leukocyte Interplay During Vascular Disease. Atherosclerosis (2020) 307:109–20. doi: 10.1016/j.atherosclerosis.2020.04.018
2. Assinger A. Platelets and Infection - an Emerging Role of Platelets in Viral Infection. Front Immunol (2014) 5:649. doi: 10.3389/fimmu.2014.00649
3. Ebermeyer T, Cognasse F, Berthelot P, Mismetti P, Garraud O, Hamzeh-Cognasse H. Platelet Innate Immune Receptors and TLRs: A Double-Edged Sword. Int J Mol Sci (2021) 22(15):7894. doi: 10.3390/ijms22157894
4. Dib PRB, Quirino-Teixeira AC, Merij LB, Pinheiro MBM, Rozini SV, Andrade FB, et al. Innate Immune Receptors in Platelets and Platelet-Leukocyte Interactions. J Leukoc Biol (2020) 108(4):1157–82. doi: 10.1002/JLB.4MR0620-701R
5. Schrottmaier WC, Kral JB, Badrnya S, Assinger A. Aspirin and P2Y12 Inhibitors in Platelet-Mediated Activation of Neutrophils and Monocytes. Thromb Haemost (2015) 114(3):478–89. doi: 10.1160/TH14-11-0943
6. Schrottmaier WC, Salzmann M, Badrnya S, Mussbacher M, Kral-Pointner JB, Morava S, et al. Platelets Mediate Serological Memory to Neutralize Viruses In Vitro and In Vivo. Blood Adv (2020) 4(16):3971–6. doi: 10.1182/bloodadvances.2020001786
7. Terada H, Baldini M, Ebbe S, Madoff MA. Interaction of Influenza Virus With Blood Platelets. Blood (1966) 28(2):213–28. doi: 10.1182/blood.V28.2.213.213
8. Page MJ, Pretorius E. A Champion of Host Defense: A Generic Large-Scale Cause for Platelet Dysfunction and Depletion in Infection. Semin Thromb Hemost (2020) 46(3):302–19. doi: 10.1055/s-0040-1708827
9. Hally K, Fauteux-Daniel S, Hamzeh-Cognasse H, Larsen P, Cognasse F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int J Mol Sci (2020) 21(17):6150. doi: 10.3390/ijms21176150
10. Kasirer-Friede A, Kahn ML, Shattil SJ. Platelet Integrins and Immunoreceptors. Immunol Rev (2007) 218:247–64. doi: 10.1111/j.1600-065X.2007.00532.x
11. Gavrilovskaya IN, Gorbunova EE, Mackow ER. Pathogenic Hantaviruses Direct the Adherence of Quiescent Platelets to Infected Endothelial Cells. J Virol (2010) 84(9):4832–9. doi: 10.1128/JVI.02405-09
12. Gupalo E, Buriachkovskaia L, Othman M. Human Platelets Express CAR With Localization at the Sites of Intercellular Interaction. Virol J (2011) 8:456. doi: 10.1186/1743-422X-8-456
13. Shimony N, Elkin G, Kolodkin-Gal D, Krasny L, Urieli-Shoval S, Haviv YS. Analysis of Adenoviral Attachment to Human Platelets. Virol J (2009) 6:25. doi: 10.1186/1743-422X-6-25
14. Zhang Y, Bergelson JM. Adenovirus Receptors. J Virol (2005) 79(19):12125–31. doi: 10.1128/JVI.79.19.12125-12131.2005
15. Gupalo E, Kuk C, Qadura M, Buriachkovskaia L, Othman M. Platelet-Adenovirus vs. Inert Particles Interaction: Effect on Aggregation and the Role of Platelet Membrane Receptors. Platelets (2013) 24(5):383–91. doi: 10.3109/09537104.2012.703792
16. Fleming FE, Graham KL, Takada Y, Coulson BS. Determinants of the Specificity of Rotavirus Interactions With the Alpha2beta1 Integrin. J Biol Chem (2011) 286(8):6165–74. doi: 10.1074/jbc.M110.142992
17. Othman M, Labelle A, Mazzetti I, Elbatarny HS, Lillicrap D. Adenovirus-Induced Thrombocytopenia: The Role of Von Willebrand Factor and P-Selectin in Mediating Accelerated Platelet Clearance. Blood (2007) 109(7):2832–9. doi: 10.1182/blood-2006-06-032524
18. He Y, Chipman PR, Howitt J, Bator CM, Whitt MA, Baker TS, et al. Interaction of Coxsackievirus B3 With the Full Length Coxsackievirus-Adenovirus Receptor. Nat Struct Biol (2001) 8(10):874–8. doi: 10.1038/nsb1001-874
19. Negrotto S, Jaquenod de Giusti C, Rivadeneyra L, Ure AE, Mena HA, Schattner M, et al. Platelets Interact With Coxsackieviruses B and Have a Critical Role in the Pathogenesis of Virus-Induced Myocarditis. J Thromb Haemost (2015) 13(2):271–82. doi: 10.1111/jth.12782
20. Nilsson EC, Jamshidi F, Johansson SM, Oberste MS, Arnberg N. Sialic Acid is a Cellular Receptor for Coxsackievirus A24 Variant, an Emerging Virus With Pandemic Potential. J Virol (2008) 82(6):3061–8. doi: 10.1128/JVI.02470-07
21. Madoff MA, Ebbe S, Baldini M. Sialic Acid of Human Blood Platelets. J Clin Invest (1964) 43:870–7. doi: 10.1172/JCI104972
22. Guy M, Chilmonczyk S, Cruciere C, Eloit M, Bakkali-Kassimi L. Efficient Infection of Buffalo Rat Liver-Resistant Cells by Encephalomyocarditis Virus Requires Binding to Cell Surface Sialic Acids. J Gen Virol (2009) 90(Pt 1):187–96. doi: 10.1099/vir.0.004655-0
23. Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC. Structure of the Influenza Virus Haemagglutinin Complexed With its Receptor, Sialic Acid. Nature (1988) 333(6172):426–31. doi: 10.1038/333426a0
24. Wagner R, Matrosovich M, Klenk HD. Functional Balance Between Haemagglutinin and Neuraminidase in Influenza Virus Infections. Rev Med Virol (2002) 12(3):159–66. doi: 10.1002/rmv.352
25. Xiong X, Martin SR, Haire LF, Wharton SA, Daniels RS, Bennett MS, et al. Receptor Binding by an H7N9 Influenza Virus From Humans. Nature (2013) 499(7459):496–9. doi: 10.1038/nature12372
26. Clemetson KJ, Clemetson JM, Proudfoot AE, Power CA, Baggiolini M, Wells TN. Functional Expression of CCR1, CCR3, CCR4, and CXCR4 Chemokine Receptors on Human Platelets. Blood (2000) 96(13):4046–54. doi: 10.1182/blood.V96.13.4046.h8004046_4046_4054
27. Simpson SR, Singh MV, Dewhurst S, Schifitto G, Maggirwar SB. Platelets Function as an Acute Viral Reservoir During HIV-1 Infection by Harboring Virus and T-Cell Complex Formation. Blood Adv (2020) 4(18):4512–21. doi: 10.1182/bloodadvances.2020002420
28. Wang J, Zhang W, Nardi MA, Li Z. HIV-1 Tat-Induced Platelet Activation and Release of CD154 Contribute to HIV-1-Associated Autoimmune Thrombocytopenia. J Thromb Haemost (2011) 9(3):562–73. doi: 10.1111/j.1538-7836.2010.04168.x
29. Boukour S, Masse JM, Benit L, Dubart-Kupperschmitt A, Cramer EM. Lentivirus Degradation and DC-SIGN Expression by Human Platelets and Megakaryocytes. J Thromb Haemost (2006) 4(2):426–35. doi: 10.1111/j.1538-7836.2006.01749.x
30. Chaipan C, Soilleux EJ, Simpson P, Hofmann H, Gramberg T, Marzi A, et al. DC-SIGN and CLEC-2 Mediate Human Immunodeficiency Virus Type 1 Capture by Platelets. J Virol (2006) 80(18):8951–60. doi: 10.1128/JVI.00136-06
31. Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, et al. DC-SIGN, a Dendritic Cell-Specific HIV-1-Binding Protein That Enhances Trans-Infection of T Cells. Cell (2000) 100(5):587–97. doi: 10.1016/S0092-8674(00)80694-7
32. Alvarez CP, Lasala F, Carrillo J, Muniz O, Corbi AL, Delgado R. C-Type Lectins DC-SIGN and L-SIGN Mediate Cellular Entry by Ebola Virus in Cis and in Trans. J Virol (2002) 76(13):6841–4. doi: 10.1128/JVI.76.13.6841-6844.2002
33. Simmons G, Reeves JD, Grogan CC, Vandenberghe LH, Baribaud F, Whitbeck JC, et al. DC-SIGN and DC-SIGNR Bind Ebola Glycoproteins and Enhance Infection of Macrophages and Endothelial Cells. Virology (2003) 305(1):115–23. doi: 10.1006/viro.2002.1730
34. Lozach PY, Lortat-Jacob H, de Lacroix de Lavalette A, Staropoli I, Foung S, Amara A, et al. DC-SIGN and L-SIGN are High Affinity Binding Receptors for Hepatitis C Virus Glycoprotein E2. J Biol Chem (2003) 278(22):20358–66. doi: 10.1074/jbc.M301284200
35. Zahn A, Jennings N, Ouwehand WH, Allain JP. Hepatitis C Virus Interacts With Human Platelet Glycoprotein VI. J Gen Virol (2006) 87(Pt 8):2243–51. doi: 10.1099/vir.0.81826-0
36. Hottz ED, Oliveira MF, Nunes PC, Nogueira RM, Valls-de-Souza R, Da Poian AT, et al. Dengue Induces Platelet Activation, Mitochondrial Dysfunction and Cell Death Through Mechanisms That Involve DC-SIGN and Caspases. J Thromb Haemost (2013) 11(5):951–62. doi: 10.1111/jth.12178
37. Simon AY, Sutherland MR, Pryzdial EL. Dengue Virus Binding and Replication by Platelets. Blood (2015) 126(3):378–85. doi: 10.1182/blood-2014-09-598029
38. Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, et al. DC-SIGN (CD209) Mediates Dengue Virus Infection of Human Dendritic Cells. J Exp Med (2003) 197(7):823–9. doi: 10.1084/jem.20021840
39. Sung PS, Huang TF, Hsieh SL. Extracellular Vesicles From CLEC2-Activated Platelets Enhance Dengue Virus-Induced Lethality via CLEC5A/Tlr2. Nat Commun (2019) 10(1):2402. doi: 10.1038/s41467-019-10360-4
40. Chao CH, Wu WC, Lai YC, Tsai PJ, Perng GC, Lin YS, et al. Dengue Virus Nonstructural Protein 1 Activates Platelets via Toll-Like Receptor 4, Leading to Thrombocytopenia and Hemorrhage. PloS Pathog (2019) 15(4):e1007625. doi: 10.1371/journal.ppat.1007625
41. Quirino-Teixeira AC, Rozini SV, Barbosa-Lima G, Coelho DR, Carneiro PH, Mohana-Borges R, et al. Inflammatory Signaling in Dengue-Infected Platelets Requires Translation and Secretion of Nonstructural Protein 1. Blood Adv (2020) 4(9):2018–31. doi: 10.1182/bloodadvances.2019001169
42. Assinger A, Kral JB, Yaiw KC, Schrottmaier WC, Kurzejamska E, Wang Y, et al. Human Cytomegalovirus-Platelet Interaction Triggers Toll-Like Receptor 2-Dependent Proinflammatory and Proangiogenic Responses. Arterioscler Thromb Vasc Biol (2014) 34(4):801–9. doi: 10.1161/ATVBAHA.114.303287
43. Boehme KW, Guerrero M, Compton T. Human Cytomegalovirus Envelope Glycoproteins B and H are Necessary for TLR2 Activation in Permissive Cells. J Immunol (2006) 177(10):7094–102. doi: 10.4049/jimmunol.177.10.7094
44. Ahmad A, Menezes J. Binding of the Epstein-Barr Virus to Human Platelets Causes the Release of Transforming Growth Factor-Beta. J Immunol (1997) 159(8):3984–8.
45. Shannon-Lowe C, Rowe M. Epstein Barr Virus Entry; Kissing and Conjugation. Curr Opin Virol (2014) 4:78–84. doi: 10.1016/j.coviro.2013.12.001
46. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets Can Associate With SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ Res (2020) 127(11):1404–18. doi: 10.1161/CIRCRESAHA.120.317703
47. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J Hematol Oncol (2020) 13(1):120. doi: 10.1186/s13045-020-00954-7
48. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet Gene Expression and Function in Patients With COVID-19. Blood (2020) 136(11):1317–29. doi: 10.1182/blood.2020007214
49. Bury L, Camilloni B, Castronari R, Piselli E, Malvestiti M, Borghi M, et al. Search for SARS-CoV-2 RNA in Platelets From COVID-19 Patients. Platelets (2021) 32(2):284–7. doi: 10.1080/09537104.2020.1859104
50. Maugeri N, De Lorenzo R, Clementi N, Antonia Diotti R, Criscuolo E, Godino C, et al. Unconventional CD147-Dependent Platelet Activation Elicited by SARS-CoV-2 in COVID-19. J Thromb Haemost (2022) 20(2):434–48. doi: 10.1111/jth.15575
51. Hamaia S, Li C, Allain JP. The Dynamics of Hepatitis C Virus Binding to Platelets and 2 Mononuclear Cell Lines. Blood (2001) 98(8):2293–300. doi: 10.1182/blood.V98.8.2293.h8002293_2293_2300
52. Ariede JR, Pardini MI, Silva GF, Grotto RM. Platelets can be a Biological Compartment for the Hepatitis C Virus. Braz J Microbiol (2015) 46(2):627–9. doi: 10.1590/S1517-838246220140553
53. Padovani JL, Corvino SM, Drexler JF, Silva GF, Pardini MI, Grotto RM. In Vitro Detection of Hepatitis C Virus in Platelets From Uninfected Individuals Exposed to the Virus. Rev Soc Bras Med Trop (2013) 46(2):154–5. doi: 10.1590/0037-8682-1627-2013
54. de Almeida AJ, Campos-de-Magalhaes M, Brandao-Mello CE, de Oliveira RV, Yoshida CF, Lampe E. Detection of Hepatitis C Virus in Platelets: Evaluating its Relationship to Viral and Host Factors. Hepatogastroenterology (2007) 54(75):964–8.
55. Kar M, Singla M, Chandele A, Kabra SK, Lodha R, Medigeshi GR. Dengue Virus Entry and Replication Does Not Lead to Productive Infection in Platelets. Open Forum Infect Dis (2017) 4(2):ofx051. doi: 10.1093/ofid/ofx051
56. Noisakran S, Gibbons RV, Songprakhon P, Jairungsri A, Ajariyakhajorn C, Nisalak A, et al. Detection of Dengue Virus in Platelets Isolated From Dengue Patients. Southeast Asian J Trop Med Public Health (2009) 40(2):253–62.
57. Stone D, Liu Y, Shayakhmetov D, Li ZY, Ni S, Lieber A. Adenovirus-Platelet Interaction in Blood Causes Virus Sequestration to the Reticuloendothelial System of the Liver. J Virol (2007) 81(9):4866–71. doi: 10.1128/JVI.02819-06
58. Zucker-Franklin D, Seremetis S, Zheng ZY. Internalization of Human Immunodeficiency Virus Type I and Other Retroviruses by Megakaryocytes and Platelets. Blood (1990) 75(10):1920–3. doi: 10.1182/blood.V75.10.1920.bloodjournal75101920
59. Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM. Host Defense Role of Platelets: Engulfment of HIV and Staphylococcus Aureus Occurs in a Specific Subcellular Compartment and is Enhanced by Platelet Activation. Blood (2002) 99(11):4021–9. doi: 10.1182/blood-2001-12-0191
60. Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, et al. Platelets Endocytose Viral Particles and Are Activated via TLR (Toll-Like Receptor) Signaling. Arterioscler Thromb Vasc Biol (2020) 40(7):1635–50. doi: 10.1161/ATVBAHA.120.314180
61. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 Mediates Host Survival and Platelet Count During Viral Infection in the Absence of Platelet-Dependent Thrombosis. Blood (2014) 124(5):791–802. doi: 10.1182/blood-2013-11-536003
62. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The Role of Platelets in Mediating a Response to Human Influenza Infection. Nat Commun (2019) 10(1):1780. doi: 10.1038/s41467-019-09607-x
63. Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, et al. SARS-CoV-2 Initiates Programmed Cell Death in Platelets. Circ Res (2021) 129(6):631–46. doi: 10.1161/CIRCRESAHA.121.319117
64. Kral JB, Schrottmaier WC, Salzmann M, Assinger A. Platelet Interaction With Innate Immune Cells. Transfus Med Hemother (2016) 43(2):78–88. doi: 10.1159/000444807
65. Finsterbusch M, Schrottmaier WC, Kral-Pointner JB, Salzmann M, Assinger A. Measuring and Interpreting Platelet-Leukocyte Aggregates. Platelets (2018) 29(7):677–85. doi: 10.1080/09537104.2018.1430358
66. Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, et al. Influenza Virus H1N1 Activates Platelets Through FcgammaRIIA Signaling and Thrombin Generation. Blood (2014) 123(18):2854–63. doi: 10.1182/blood-2013-07-515536
67. Nunez-Avellaneda D, Mosso-Pani MA, Sanchez-Torres LE, Castro-Mussot ME, Corona-de la Pena NA, Salazar MI. Dengue Virus Induces the Release of Scd40l and Changes in Levels of Membranal CD42b and CD40L Molecules in Human Platelets. Viruses (2018) 10(7):357. doi: 10.3390/v10070357
68. Ojha A, Nandi D, Batra H, Singhal R, Annarapu GK, Bhattacharyya S, et al. Platelet Activation Determines the Severity of Thrombocytopenia in Dengue Infection. Sci Rep (2017) 7:41697. doi: 10.1038/srep41697
69. Parker ZF, Rux AH, Riblett AM, Lee FH, Rauova L, Cines DB, et al. Platelet Factor 4 Inhibits and Enhances HIV-1 Infection in a Concentration-Dependent Manner by Modulating Viral Attachment. AIDS Res Hum Retroviruses (2016) 32(7):705–17. doi: 10.1089/aid.2015.0344
70. Jansen AJG, Spaan T, Low HZ, Di Iorio D, van den Brand J, Tieke M, et al. Influenza-Induced Thrombocytopenia is Dependent on the Subtype and Sialoglycan Receptor and Increases With Virus Pathogenicity. Blood Adv (2020) 4(13):2967–78. doi: 10.1182/bloodadvances.2020001640
71. Rahbar A, Peredo I, Solberg NW, Taher C, Dzabic M, Xu X, et al. Discordant Humoral and Cellular Immune Responses to Cytomegalovirus (CMV) in Glioblastoma Patients Whose Tumors are Positive for CMV. Oncoimmunology (2015) 4(2):e982391. doi: 10.4161/2162402X.2014.982391
72. Real F, Capron C, Sennepin A, Arrigucci R, Zhu A, Sannier G, et al. Platelets From HIV-Infected Individuals on Antiretroviral Drug Therapy With Poor CD4(+) T Cell Recovery can Harbor Replication-Competent HIV Despite Viral Suppression. Sci Transl Med (2020) 12(535):eaat6263. doi: 10.1126/scitranslmed.aat6263
73. Amer A, Madi MA, Shebl FM, Faridi DA, Alkhinji M, Derbala M. Platelets as a Possible Reservoir of HCV And Predictor of Response to Treatment. J Virol Antiviral Res (2016) 5(3). doi: 10.4172/2324-8955.1000157
74. de Almeida AJ, Campos-de-Magalhaes M, de Melo Marcal OP, Brandao-Mello CE, Okawa MY, de Oliveira RV, et al. Hepatitis C Virus-Associated Thrombocytopenia: A Controlled Prospective, Virological Study. Ann Hematol (2004) 83(7):434–40. doi: 10.1007/s00277-004-0844-0
75. Ojha A, Bhasym A, Mukherjee S, Annarapu GK, Bhakuni T, Akbar I, et al. Platelet Factor 4 Promotes Rapid Replication and Propagation of Dengue and Japanese Encephalitis Viruses. EBioMedicine (2019) 39:332–47. doi: 10.1016/j.ebiom.2018.11.049
76. Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, et al. Platelet-Mediated Modulation of Adaptive Immunity. A Communication Link Between Innate and Adaptive Immune Compartments. Immunity (2003) 19(1):9–19. doi: 10.1016/S1074-7613(03)00177-8
77. Auerbach DJ, Lin Y, Miao H, Cimbro R, Difiore MJ, Gianolini ME, et al. Identification of the Platelet-Derived Chemokine CXCL4/PF-4 as a Broad-Spectrum HIV-1 Inhibitor. Proc Natl Acad Sci USA (2012) 109(24):9569–74. doi: 10.1073/pnas.1207314109
78. Tsegaye TS, Gnirss K, Rahe-Meyer N, Kiene M, Kramer-Kuhl A, Behrens G, et al. Platelet Activation Suppresses HIV-1 Infection of T Cells. Retrovirology (2013) 10(48):48. doi: 10.1186/1742-4690-10-48
79. Alonzo MT, Lacuesta TL, Dimaano EM, Kurosu T, Suarez LA, Mapua CA, et al. Platelet Apoptosis and Apoptotic Platelet Clearance by Macrophages in Secondary Dengue Virus Infections. J Infect Dis (2012) 205(8):1321–9. doi: 10.1093/infdis/jis180
80. Pozner RG, Ure AE, Jaquenod de Giusti C, D'Atri LP, Italiano JE, Torres O, et al. Junin Virus Infection of Human Hematopoietic Progenitors Impairs In Vitro Proplatelet Formation and Platelet Release via a Bystander Effect Involving Type I IFN Signaling. PloS Pathog (2010) 6(4):e1000847. doi: 10.1371/journal.ppat.1000847
81. Campbell RA, Schwertz H, Hottz ED, Rowley JW, Manne BK, Washington AV, et al. Human Megakaryocytes Possess Intrinsic Antiviral Immunity Through Regulated Induction of IFITM3. Blood (2019) 133(19):2013–26. doi: 10.1182/blood-2018-09-873984
82. Laursen MA, Larsen JB, Hvas AM. Platelet Function in Disseminated Intravascular Coagulation: A Systematic Review. Platelets (2018) 29(3):238–48. doi: 10.1080/09537104.2018.1442567
83. Qiao J, Al-Tamimi M, Baker RI, Andrews RK, Gardiner EE. The Platelet Fc Receptor, FcgammaRIIa. Immunol Rev (2015) 268(1):241–52. doi: 10.1111/imr.12370
84. Pollreisz A, Assinger A, Hacker S, Hoetzenecker K, Schmid W, Lang G, et al. Intravenous Immunoglobulins Induce CD32-Mediated Platelet Aggregation In Vitro. Br J Dermatol (2008) 159(3):578–84. doi: 10.1111/j.1365-2133.2008.08700.x
85. Wang TT, Sewatanon J, Memoli MJ, Wrammert J, Bournazos S, Bhaumik SK, et al. IgG Antibodies to Dengue Enhanced for FcgammaRIIIA Binding Determine Disease Severity. Science (2017) 355(6323):395–8. doi: 10.1126/science.aai8128
86. de Almeida AJ, Campos-de-Magalhaes M, Antonietti CL, Brandao-Mello CE, da Silva ML, de Oliveira RV, et al. Autoimmune Thrombocytopenia Related to Chronic Hepatitis C Virus Infection. Hematology (2009) 14(1):49–58. doi: 10.1179/102453309X385106
87. Saito M, Oishi K, Inoue S, Dimaano EM, Alera MT, Robles AM, et al. Association of Increased Platelet-Associated Immunoglobulins With Thrombocytopenia and the Severity of Disease in Secondary Dengue Virus Infections. Clin Exp Immunol (2004) 138(2):299–303. doi: 10.1111/j.1365-2249.2004.02626.x
88. Lin CF, Lei HY, Liu CC, Liu HS, Yeh TM, Wang ST, et al. Generation of IgM Anti-Platelet Autoantibody in Dengue Patients. J Med Virol (2001) 63(2):143–9. doi: 10.1002/1096-9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
89. Cheng HJ, Lei HY, Lin CF, Luo YH, Wan SW, Liu HS, et al. Anti-Dengue Virus Nonstructural Protein 1 Antibodies Recognize Protein Disulfide Isomerase on Platelets and Inhibit Platelet Aggregation. Mol Immunol (2009) 47(2-3):398–406. doi: 10.1016/j.molimm.2009.08.033
90. Hou Y, Carrim N, Wang Y, Gallant RC, Marshall A, Ni H. Platelets in Hemostasis and Thrombosis: Novel Mechanisms of Fibrinogen-Independent Platelet Aggregation and Fibronectin-Mediated Protein Wave of Hemostasis. J BioMed Res (2015) 29:437–44. doi: 10.7555/JBR.29.20150121
91. Fosse JH, Haraldsen G, Falk K, Edelmann R. Endothelial Cells in Emerging Viral Infections. Front Cardiovasc Med (2021) 8:619690. doi: 10.3389/fcvm.2021.619690
92. Goldsmith CS, Elliott LH, Peters CJ, Zaki SR. Ultrastructural Characteristics of Sin Nombre Virus, Causative Agent of Hantavirus Pulmonary Syndrome. Arch Virol (1995) 140(12):2107–22. doi: 10.1007/BF01323234
93. Goeijenbier M, Meijers JC, Anfasa F, Roose JM, van de Weg CA, Bakhtiari K, et al. Effect of Puumala Hantavirus Infection on Human Umbilical Vein Endothelial Cell Hemostatic Function: Platelet Interactions, Increased Tissue Factor Expression and Fibrinolysis Regulator Release. Front Microbiol (2015) 6:220. doi: 10.3389/fmicb.2015.00220
94. Krishnamurti C, Peat RA, Cutting MA, Rothwell SW. Platelet Adhesion to Dengue-2 Virus-Infected Endothelial Cells. Am J Trop Med Hyg (2002) 66(4):435–41. doi: 10.4269/ajtmh.2002.66.435
95. Sugiyama MG, Gamage A, Zyla R, Armstrong SM, Advani S, Advani A, et al. Influenza Virus Infection Induces Platelet-Endothelial Adhesion Which Contributes to Lung Injury. J Virol (2016) 90(4):1812–23. doi: 10.1128/JVI.02599-15
96. Rahbar A, Soderberg-Naucler C. Human Cytomegalovirus Infection of Endothelial Cells Triggers Platelet Adhesion and Aggregation. J Virol (2005) 79(4):2211–20. doi: 10.1128/JVI.79.4.2211-2220.2005
97. van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and Functional Interactions Among Monocytes, Platelets, and Endothelial Cells and Their Relevance for Cardiovascular Diseases. J Leukoc Biol (2009) 85(2):195–204. doi: 10.1189/jlb.0708400
98. Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, et al. Dengue Virus NS1 Protein Activates Cells via Toll-Like Receptor 4 and Disrupts Endothelial Cell Monolayer Integrity. Sci Transl Med (2015) 7(304):304ra142. doi: 10.1126/scitranslmed.aaa3863
99. Armstrong SM, Wang C, Tigdi J, Si X, Dumpit C, Charles S, et al. Influenza Infects Lung Microvascular Endothelium Leading to Microvascular Leak: Role of Apoptosis and Claudin-5. PloS One (2012) 7(10):e47323. doi: 10.1371/journal.pone.0047323
100. Visser MR, Tracy PB, Vercellotti GM, Goodman JL, White JG, Jacob HS, et al. Enhanced Thrombin Generation and Platelet Binding on Herpes Simplex Virus-Infected Endothelium. Proc Natl Acad Sci USA (1988) 85(21):8227–30. doi: 10.1073/pnas.85.21.8227
101. Basler CF. Molecular Pathogenesis of Viral Hemorrhagic Fever. Semin Immunopathol (2017) 39(5):551–61. doi: 10.1007/s00281-017-0637-x
102. Zapata JC, Cox D, Salvato MS. The Role of Platelets in the Pathogenesis of Viral Hemorrhagic Fevers. PloS Negl Trop Dis (2014) 8(6):e2858. doi: 10.1371/journal.pntd.0002858
103. Lee IK, Liu JW, Yang KD. Clinical Characteristics, Risk Factors, and Outcomes in Adults Experiencing Dengue Hemorrhagic Fever Complicated With Acute Renal Failure. Am J Trop Med Hyg (2009) 80(4):651–5. doi: 10.4269/ajtmh.2009.80.651
104. Funahara Y, Sumarmo, Wirawan R. Features of DIC in Dengue Hemorrhagic Fever. Bibl Haematol (1983) 49):201–11. doi: 10.1159/000408460
105. Srichaikul T, Nimmanitaya S, Artchararit N, Siriasawakul T, Sungpeuk P. Fibrinogen Metabolism and Disseminated Intravascular Coagulation in Dengue Hemorrhagic Fever. Am J Trop Med Hyg (1977) 26(3):525–32. doi: 10.4269/ajtmh.1977.26.525
106. Rollin PE, Bausch DG, Sanchez A. Blood Chemistry Measurements and D-Dimer Levels Associated With Fatal and Nonfatal Outcomes in Humans Infected With Sudan Ebola Virus. J Infect Dis (2007) 196 Suppl 2:S364–71. doi: 10.1086/520613
107. Ebihara H, Rockx B, Marzi A, Feldmann F, Haddock E, Brining D, et al. Host Response Dynamics Following Lethal Infection of Rhesus Macaques With Zaire Ebolavirus. J Infect Dis (2011) 204 Suppl 3:S991–9. doi: 10.1093/infdis/jir336
108. Sundberg E, Hultdin J, Nilsson S, Ahlm C. Evidence of Disseminated Intravascular Coagulation in a Hemorrhagic Fever With Renal Syndrome-Scoring Models and Severe Illness. PloS One (2011) 6(6):e21134. doi: 10.1371/journal.pone.0021134
109. Rasche FM, Uhel B, Kruger DH, Karges W, Czock D, Hampl W, et al. Thrombocytopenia and Acute Renal Failure in Puumala Hantavirus Infections. Emerg Infect Dis (2004) 10(8):1420–5. doi: 10.3201/eid1008.031069
110. Koskela SM, Joutsi-Korhonen L, Makela SM, Huhtala H, Vaheri AI, Porsti I, et al. Diminished Coagulation Capacity Assessed by Calibrated Automated Thrombography During Acute Puumala Hantavirus Infection. Blood Coagul Fibrinolysis (2018) 29(1):55–60. doi: 10.1097/MBC.0000000000000667
111. Horton LE, Cross RW, Hartnett JN, Engel EJ, Sakabe S, Goba A, et al. Endotheliopathy and Platelet Dysfunction as Hallmarks of Fatal Lassa Fever. Emerg Infect Dis (2020) 26(11):2625–37. doi: 10.3201/eid2611.191694
112. Peters CJ, Liu CT, Anderson GW Jr., Morrill JC, Jahrling PB. Pathogenesis of Viral Hemorrhagic Fevers: Rift Valley Fever and Lassa Fever Contrasted. Rev Infect Dis (1989) 11 Suppl 4:S743–9. doi: 10.1093/clinids/11.Supplement_4.S743
113. Kunz S. The Role of the Vascular Endothelium in Arenavirus Haemorrhagic Fevers. Thromb Haemost (2009) 102(6):1024–9. doi: 10.1160/TH09-06-0357
114. Ekiz F, Gurbuz Y, Basar O, Aytekin G, Ekiz O, Senturk CS, et al. Mean Platelet Volume in the Diagnosis and Prognosis of Crimean-Congo Hemorrhagic Fever. Clin Appl Thromb Hemost (2013) 19(4):441–4. doi: 10.1177/1076029612440035
115. McElroy AK, Harmon JR, Flietstra TD, Campbell S, Mehta AK, Kraft CS, et al. Kinetic Analysis of Biomarkers in a Cohort of US Patients With Ebola Virus Disease. Clin Infect Dis (2016) 63(4):460–7. doi: 10.1093/cid/ciw334
116. Trugilho MRO, Hottz ED, Brunoro GVF, Teixeira-Ferreira A, Carvalho PC, Salazar GA, et al. Platelet Proteome Reveals Novel Pathways of Platelet Activation and Platelet-Mediated Immunoregulation in Dengue. PloS Pathog (2017) 13(5):e1006385. doi: 10.1371/journal.ppat.1006385
117. Laine O, Joutsi-Korhonen L, Lassila R, Koski T, Huhtala H, Vaheri A, et al. Hantavirus Infection-Induced Thrombocytopenia Triggers Increased Production But Associates With Impaired Aggregation of Platelets Except for Collagen. Thromb Res (2015) 136(6):1126–32. doi: 10.1016/j.thromres.2015.10.003
118. Fisher-Hoch S, McCormick JB, Sasso D, Craven RB. Hematologic Dysfunction in Lassa Fever. J Med Virol (1988) 26(2):127–35. doi: 10.1002/jmv.1890260204
119. Cummins D, Fisher-Hoch SP, Walshe KJ, Mackie IJ, McCormick JB, Bennett D, et al. A Plasma Inhibitor of Platelet Aggregation in Patients With Lassa Fever. Br J Haematol (1989) 72(4):543–8. doi: 10.1111/j.1365-2141.1989.tb04321.x
120. Cummins D, Molinas FC, Lerer G, Maiztegui JI, Faint R, Machin SJ. A Plasma Inhibitor of Platelet Aggregation in Patients With Argentine Hemorrhagic Fever. Am J Trop Med Hyg (1990) 42(5):470–5. doi: 10.4269/ajtmh.1990.42.470
121. McElroy AK, Erickson BR, Flietstra TD, Rollin PE, Towner JS, Nichol ST, et al. Von Willebrand Factor is Elevated in Individuals Infected With Sudan Virus and is Associated With Adverse Clinical Outcomes. Viral Immunol (2015) 28(1):71–3. doi: 10.1089/vim.2014.0072
122. Jin C, Liang M, Ning J, Gu W, Jiang H, Wu W, et al. Pathogenesis of Emerging Severe Fever With Thrombocytopenia Syndrome Virus in C57/BL6 Mouse Model. Proc Natl Acad Sci USA (2012) 109(25):10053–8. doi: 10.1073/pnas.1120246109
123. Sørensen AL, Rumjantseva V, Nayeb-Hashemi S, Clausen H, Hartwig JH, Wandall HH, et al. Role of Sialic Acid for Platelet Life Span: Exposure of β-Galactose Results in the Rapid Clearance of Platelets From the Circulation by Asialoglycoprotein Receptor–Expressing Liver Macrophages and Hepatocytes. Blood (2009) 114(8):1645–54. doi: 10.1182/blood-2009-01-199414
124. Riswari SF, Tunjungputri RN, Kullaya V, Garishah FM, Utari GSR, Farhanah N, et al. Desialylation of Platelets Induced by Von Willebrand Factor is a Novel Mechanism of Platelet Clearance in Dengue. PloS Pathog (2019) 15(3):e1007500. doi: 10.1371/journal.ppat.1007500
125. Zapata JC, Carrion R Jr., Patterson JL, Crasta O, Zhang Y, Mani S, et al. Transcriptome Analysis of Human Peripheral Blood Mononuclear Cells Exposed to Lassa Virus and to the Attenuated Mopeia/Lassa Reassortant 29 (ML29), a Vaccine Candidate. PloS Negl Trop Dis (2013) 7(9):e2406. doi: 10.1371/journal.pntd.0002406
126. Totani L, Evangelista V. Platelet-Leukocyte Interactions in Cardiovascular Disease and Beyond. Arterioscler Thromb Vasc Biol (2010) 30(12):2357–61. doi: 10.1161/ATVBAHA.110.207480
127. Barbosa-Lima G, Hottz ED, de Assis EF, Liechocki S, Souza TML, Zimmerman GA, et al. Dengue Virus-Activated Platelets Modulate Monocyte Immunometabolic Response Through Lipid Droplet Biogenesis and Cytokine Signaling. J Leukoc Biol (2020) 108(4):1293–306. doi: 10.1002/JLB.4MA0620-658R
128. Zhang J, Lan Y, Sanyal S. Modulation of Lipid Droplet Metabolism—A Potential Target for Therapeutic Intervention in Flaviviridae Infections. Front Microbiol (2286) 2017:8. doi: 10.3389/fmicb.2017.02286
129. Samsa MM, Mondotte JA, Iglesias NG, Assuncao-Miranda I, Barbosa-Lima G, Da Poian AT, et al. Dengue Virus Capsid Protein Usurps Lipid Droplets for Viral Particle Formation. PloS Pathog (2009) 5(10):e1000632. doi: 10.1371/journal.ppat.1000632
130. Assuncao-Miranda I, Amaral FA, Bozza FA, Fagundes CT, Sousa LP, Souza DG, et al. Contribution of Macrophage Migration Inhibitory Factor to the Pathogenesis of Dengue Virus Infection. FASEB J (2010) 24(1):218–28. doi: 10.1096/fj.09-139469
131. Hottz ED, Medeiros-de-Moraes IM, Vieira-de-Abreu A, de Assis EF, Vals-de-Souza R, Castro-Faria-Neto HC, et al. Platelet Activation and Apoptosis Modulate Monocyte Inflammatory Responses in Dengue. J Immunol (2014) 193(4):1864–72. doi: 10.4049/jimmunol.1400091
132. Sridharan A, Chen Q, Tang KF, Ooi EE, Hibberd ML, Chen J. Inhibition of Megakaryocyte Development in the Bone Marrow Underlies Dengue Virus-Induced Thrombocytopenia in Humanized Mice. J Virol (2013) 87(21):11648–58. doi: 10.1128/JVI.01156-13
133. Vogt MB, Lahon A, Arya RP, Spencer Clinton JL, Rico-Hesse R. Dengue Viruses Infect Human Megakaryocytes, With Probable Clinical Consequences. PloS Negl Trop Dis (2019) 13(11):e0007837. doi: 10.1371/journal.pntd.0007837
134. Liang KS, Peng LJ, Yin CB, Zhang JL, Xu CG, Liu XD, et al. [Cellular Ultrastructural Changes of Bone Marrow of Patients With Hemorrhagic Fever With Renal Syndrome]. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi (2004) 18(2):165–7.
135. Lutteke N, Raftery MJ, Lalwani P, Lee MH, Giese T, Voigt S, et al. Switch to High-Level Virus Replication and HLA Class I Upregulation in Differentiating Megakaryocytic Cells After Infection With Pathogenic Hantavirus. Virology (2010) 405(1):70–80. doi: 10.1016/j.virol.2010.05.028
136. Gaitonde DY, Moore FC, Morgan MK. Influenza: Diagnosis and Treatment. Am Fam Physician (2019) 100(12):751–8.
137. Le VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, et al. Platelet Activation and Aggregation Promote Lung Inflammation and Influenza Virus Pathogenesis. Am J Respir Crit Care Med (2015) 191(7):804–19. doi: 10.1164/rccm.201406-1031OC
138. Sederdahl BK, Williams JV. Epidemiology and Clinical Characteristics of Influenza C Virus. Viruses (2020) 12(1):89. doi: 10.3390/v12010089
139. Vijaykrishna D, Holmes EC, Joseph U, Fourment M, Su YC, Halpin R, et al. The Contrasting Phylodynamics of Human Influenza B Viruses. Elife (2015) 4:e05055. doi: 10.7554/eLife.05055
140. Wang M, Veit M. Hemagglutinin-Esterase-Fusion (HEF) Protein of Influenza C Virus. Protein Cell (2016) 7(1):28–45. doi: 10.1007/s13238-015-0193-x
141. Webster RG, Govorkova EA. Continuing Challenges in Influenza. Ann N Y Acad Sci (2014) 1323:115–39. doi: 10.1111/nyas.12462
142. Ishiguro T, Matsuo K, Fujii S, Takayanagi N. Acute Thrombotic Vascular Events Complicating Influenza-Associated Pneumonia. Respir Med Case Rep (2019) 28:100884. doi: 10.1016/j.rmcr.2019.100884
143. LeMessurier KS, Tiwary M, Morin NP, Samarasinghe AE. Respiratory Barrier as a Safeguard and Regulator of Defense Against Influenza A Virus and Streptococcus Pneumoniae. Front Immunol (2020) 11:3. doi: 10.3389/fimmu.2020.00003
144. Ashar HK, Mueller NC, Rudd JM, Snider TA, Achanta M, Prasanthi M, et al. The Role of Extracellular Histones in Influenza Virus Pathogenesis. Am J Pathol (2018) 188(1):135–48. doi: 10.1016/j.ajpath.2017.09.014
145. Han Q, Wen X, Wang L, Han X, Shen Y, Cao J, et al. Role of Hematological Parameters in the Diagnosis of Influenza Virus Infection in Patients With Respiratory Tract Infection Symptoms. J Clin Lab Anal (2020) 34(5):e23191. doi: 10.1002/jcla.23191
146. Kazancioglu S, Bastug A, Ozbay BO, Kemirtlek N, Bodur H. The Role of Haematological Parameters in Patients With COVID-19 and Influenza Virus Infection. Epidemiol Infect (2020) 148(e272):1–8. doi: 10.1017/S095026882000271X
147. Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, et al. In Vivo Platelet Activation in Critically Ill Patients With Primary 2009 Influenza a(H1N1). Chest (2012) 141(6):1490–5. doi: 10.1378/chest.11-2860
148. Zhu R, Chen C, Wang Q, Zhang X, Lu C, Sun Y. Routine Blood Parameters are Helpful for Early Identification of Influenza Infection in Children. BMC Infect Dis (2020) 20(1):864. doi: 10.1186/s12879-020-05584-5
149. Pulavendran S, Rudd JM, Maram P, Thomas PG, Akhilesh R, Malayer JR, et al. Combination Therapy Targeting Platelet Activation and Virus Replication Protects Mice Against Lethal Influenza Pneumonia. Am J Respir Cell Mol Biol (2019) 61(6):689–701. doi: 10.1165/rcmb.2018-0196OC
150. Tatsumi K, Schmedes CM, Houston ER, Butler E, Mackman N, Antoniak S. Protease-Activated Receptor 4 Protects Mice From Coxsackievirus B3 and H1N1 Influenza A Virus Infection. Cell Immunol (2019) 344:103949. doi: 10.1016/j.cellimm.2019.103949
151. Guo L, Feng K, Wang YC, Mei JJ, Ning RT, Zheng HW, et al. Critical Role of CXCL4 in the Lung Pathogenesis of Influenza (H1N1) Respiratory Infection. Mucosal Immunol (2017) 10(6):1529–41. doi: 10.1038/mi.2017.1
152. Kim SJ, Carestia A, McDonald B, Zucoloto AZ, Grosjean H, Davis RP, et al. Platelet-Mediated NET Release Amplifies Coagulopathy and Drives Lung Pathology During Severe Influenza Infection. Front Immunol (2021) 12:772859. doi: 10.3389/fimmu.2021.772859
153. Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JCT, Fogerty AE, Waheed A, et al. COVID-19 and Coagulation: Bleeding and Thrombotic Manifestations of SARS-CoV-2 Infection. Blood (2020) 136(4):489–500. doi: 10.1182/blood.2020006520
154. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of Thrombotic Complications in Critically Ill ICU Patients With COVID-19. Thromb Res (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
155. Demelo-Rodríguez P, Cervilla-Munoz E, Ordieres-Ortega L, Parra-Virto A, Toledano-Macias M, Toledo-Samaniego N, et al. Incidence of Asymptomatic Deep Vein Thrombosis in Patients With COVID-19 Pneumonia and Elevated D-Dimer Levels. Thromb Res (2020) 192:23–6. doi: 10.1016/j.thromres.2020.05.018
156. Yin S, Huang M, Li D, Tang N. Difference of Coagulation Features Between Severe Pneumonia Induced by SARS-CoV2 and non-SARS-Cov2. J Thromb Thrombolysis (2021) 51(4):1107–10. doi: 10.1007/s11239-020-02105-8
157. Taus F, Salvagno G, Cane S, Fava C, Mazzaferri F, Carrara E, et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arterioscler Thromb Vasc Biol (2020) 40(12):2975–89. doi: 10.1161/ATVBAHA.120.315175
158. Shen S, Zhang J, Fang Y, Lu S, Wu J, Zheng X, et al. SARS-CoV-2 Interacts With Platelets and Megakaryocytes via ACE2-Independent Mechanism. J Hematol Oncol (2021) 14(1):72. doi: 10.1186/s13045-021-01082-6
159. McCafferty C, Van Den Helm S, Letunica N, Attard C, Karlaftis V, Cai T, et al. Increased Platelet Activation in SARS-CoV-2 Infected non-Hospitalised Children and Adults, and Their Household Contacts. Br J Haematol (2021) 195(1):90–4. doi: 10.1111/bjh.17629
160. Zaid Y, Guessous F, Puhm F, Elhamdani W, Chentoufi L, Morris AC, et al. Platelet Reactivity to Thrombin Differs Between Patients With COVID-19 and Those With ARDS Unrelated to COVID-19. Blood Adv (2021) 5(3):635–9. doi: 10.1182/bloodadvances.2020003513
161. Lippi G, Plebani M, Henry BM. Thrombocytopenia is Associated With Severe Coronavirus Disease 2019 (COVID-19) Infections: A Meta-Analysis. Clin Chim Acta (2020) 506:145–8. doi: 10.1016/j.cca.2020.03.022
162. Cohen A, Harari E, Cipok M, Laish-Farkash A, Bryk G, Yahud E, et al. Immature Platelets in Patients Hospitalized With Covid-19. J Thromb Thrombolysis (2021) 51(3):608–16. doi: 10.1007/s11239-020-02290-6
163. Pezeshki A, Vaezi A, Nematollahi P. Blood Cell Morphology and COVID-19 Clinical Course, Severity, and Outcome. J Hematop (2021) p:1–8. doi: 10.1007/s12308-021-00459-3
164. Comer SP, Cullivan S, Szklanna PB, Weiss L, Cullen S, Kelliher S, et al. COVID-19 Induces a Hyperactive Phenotype in Circulating Platelets. PloS Biol (2021) 19(2):e3001109. doi: 10.1371/journal.pbio.3001109
165. Chen Y, Wang J, Liu C, Su L, Zhang D, Fan J, et al. IP-10 and MCP-1 as Biomarkers Associated With Disease Severity of COVID-19. Mol Med (2020) 26(1):97. doi: 10.1186/s10020-020-00230-x
166. Taha M, Sano D, Hanoudi S, Esber Z, Elahi M, Gabali A, et al. Platelets and Renal Failure in the SARS-CoV-2 Syndrome. Platelets (2021) 32(1):130–7. doi: 10.1080/09537104.2020.1817361
167. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pao CRR, et al. Platelet Activation and Platelet-Monocyte Aggregate Formation Trigger Tissue Factor Expression in Patients With Severe COVID-19. Blood (2020) 136(11):1330–41. doi: 10.1182/blood.2020007252
168. Bongiovanni D, Klug M, Lazareva O, Weidlich S, Biasi M, Ursu S, et al. SARS-CoV-2 Infection is Associated With a Pro-Thrombotic Platelet Phenotype. Cell Death Dis (2021) 12(1):50. doi: 10.1038/s41419-020-03333-9
169. Leopold V, Pereverzeva L, Schuurman AR, Reijnders TDY, Saris A, de Brabander J, et al. Platelets are Hyperactivated But Show Reduced Glycoprotein VI Reactivity in COVID-19 Patients. Thromb Haemost (2021) 121(9):1258–62. doi: 10.1055/a-1347-5555
170. Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation (2020) 142(12):1176–89. doi: 10.1161/CIRCULATIONAHA.120.048488
171. Apostolidis SA, Sarkar A, Giannini HM, Goel RR, Mathew D, Suzuki A, et al. Signaling Through FcgammaRIIA and the C5a-C5aR Pathway Mediates Platelet Hyperactivation in COVID-19. bioRxiv (2021). doi: 10.1101/2021.05.01.442279
172. Ruberto F, Chistolini A, Curreli M, Frati G, Marullo AGM, Biondi-Zoccai G, et al. Von Willebrand Factor With Increased Binding Capacity is Associated With Reduced Platelet Aggregation But Enhanced Agglutination in COVID-19 Patients: Another COVID-19 Paradox? J Thromb Thrombolysis (2021) 52(1):105–10. doi: 10.1007/s11239-020-02339-6
173. Ercan H, Schrottmaier WC, Pirabe A, Schmuckenschlager A, Pereyra D, Santol J, et al. Platelet Phenotype Analysis of COVID-19 Patients Reveals Progressive Changes in the Activation of Integrin Alphaiibbeta3, F13A1, the SARS-CoV-2 Target EIF4A1 and Annexin A5. Front Cardiovasc Med (2021) 8:779073. doi: 10.3389/fcvm.2021.779073
174. Chao Y, Rebetz J, Blackberg A, Hovold G, Sunnerhagen T, Rasmussen M, et al. Distinct Phenotypes of Platelet, Monocyte, and Neutrophil Activation Occur During the Acute and Convalescent Phase of COVID-19. Platelets (2021) p:1–11. doi: 10.1080/09537104.2021.1921721
175. Ghirardello S, Lecchi A, Artoni A, Panigada M, Aliberti S, Scalambrino E, et al. Assessment of Platelet Thrombus Formation Under Flow Conditions in Adult Patients With COVID-19: An Observational Study. Thromb Haemost (2021) 121(8):1087–96. doi: 10.1055/s-0041-1722919
176. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Haberle H, et al. Antibody-Induced Procoagulant Platelets in Severe COVID-19 Infection. Blood (2021) 137(8):1061–71. doi: 10.1182/blood.2020008762
177. Denorme F, Manne BK, Portier I, Petrey AC, Middleton EA, Kile BT, et al. COVID-19 Patients Exhibit Reduced Procoagulant Platelet Responses. J Thromb Haemost (2020) 18(11):3067–73. doi: 10.1111/jth.15107
178. Canzano P, Brambilla M, Porro B, Cosentino N, Tortorici E, Vicini S, et al. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl Sci (2021) 6(3):202–18. doi: 10.1016/j.jacbts.2020.12.009
179. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant Treatment Is Associated With Decreased Mortality in Severe Coronavirus Disease 2019 Patients With Coagulopathy. J Thromb Haemost (2020) 18(5):1094–9. doi: 10.1111/jth.14817
180. Durak K, Kersten A, Grottke O, Zayat R, Dreher M, Autschbach R, et al. Thromboembolic and Bleeding Events in COVID-19 Patients Receiving Extracorporeal Membrane Oxygenation. Thorac Cardiovasc Surg (2021) 69(6):526–36. doi: 10.1055/s-0041-1725180
181. Barrett TJ, Lee AH, Xia Y, Lin LH, Black M, Cotzia P, et al. Platelet and Vascular Biomarkers Associate With Thrombosis and Death in Coronavirus Disease. Circ Res (2020) 127(7):945–7. doi: 10.1161/CIRCRESAHA.120.317803
182. Favaloro EJ, Henry BM, Lippi G. Increased VWF and Decreased ADAMTS-13 in COVID-19: Creating a Milieu for (Micro)Thrombosis. Semin Thromb Hemost (2021) 47(4):400–18. doi: 10.1055/s-0041-1727282
183. Viecca M, Radovanovic D, Forleo GB, Santus P. Enhanced Platelet Inhibition Treatment Improves Hypoxemia in Patients With Severe Covid-19 and Hypercoagulability. A Case Control, Proof of Concept Study. Pharmacol Res (2020) 158:104950. doi: 10.1016/j.phrs.2020.104950
184. Chow JH, Khanna AK, Kethireddy S, Yamane D, Levine A, Jackson AM, et al. Aspirin Use Is Associated With Decreased Mechanical Ventilation, Intensive Care Unit Admission, and In-Hospital Mortality in Hospitalized Patients With Coronavirus Disease 2019. Anesth Analg (2021) 132(4):930–41. doi: 10.1213/ANE.0000000000005292
185. Haji Aghajani M, Moradi O, Amini H, Azhdari Tehrani H, Pourheidar E, Rabiei MM, et al. Decreased in-Hospital Mortality Associated With Aspirin Administration in Hospitalized Patients Due to Severe COVID-19. J Med Virol (2021) 93(9):5390–5. doi: 10.1002/jmv.27053
186. Sahai A, Bhandari R, Godwin M, McIntyre T, Chung MK, Iskandar JP, et al. Effect of Aspirin on Short-Term Outcomes in Hospitalized Patients With COVID-19. Vasc Med (2021) 26(6):626–32. doi: 10.1177/1358863X211012754
187. Wang Y, Ao G, Nasr B, Qi X. Effect of Antiplatelet Treatments on Patients With COVID-19 Infection: A Systematic Review and Meta-Analysis. Am J Emerg Med (2021) 43:27–30. doi: 10.1016/j.ajem.2021.01.016
188. Recovery Collaborative Group. Aspirin in Patients Admitted to Hospital With COVID-19 (RECOVERY): A Randomised, Controlled, Open-Label, Platform Trial. Lancet (2021) 399(10320):143–51. doi: 10.1016/S0140-6736(21)01825-0.
189. Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, et al. PAR1 Contributes to Influenza A Virus Pathogenicity in Mice. J Clin Invest (2013) 123(1):206–14. doi: 10.1172/JCI61667
190. Ungerer M, Rosport K, Bultmann A, Piechatzek R, Uhland K, Schlieper P, et al. Novel Antiplatelet Drug Revacept (Dimeric Glycoprotein VI-Fc) Specifically and Efficiently Inhibited Collagen-Induced Platelet Aggregation Without Affecting General Hemostasis in Humans. Circulation (2011) 123(17):1891–9. doi: 10.1161/CIRCULATIONAHA.110.980623
191. Coxon CH, Geer MJ, Senis YA. ITIM Receptors: More Than Just Inhibitors of Platelet Activation. Blood (2017) 129(26):3407–18. doi: 10.1182/blood-2016-12-720185
192. Arman M, Krauel K. Human Platelet IgG Fc Receptor FcgammaRIIA in Immunity and Thrombosis. J Thromb Haemost (2015) 13(6):893–908. doi: 10.1111/jth.12905
Keywords: platelet, platelet activation, infection, virus, viral haemorrhagic fever, influenza, COVID-19
Citation: Schrottmaier WC, Schmuckenschlager A, Pirabe A and Assinger A (2022) Platelets in Viral Infections – Brave Soldiers or Trojan Horses. Front. Immunol. 13:856713. doi: 10.3389/fimmu.2022.856713
Received: 17 January 2022; Accepted: 03 March 2022;
Published: 28 March 2022.
Edited by:
Amanda M. Jamieson, Brown University, United StatesReviewed by:
Eugenio D. Hottz, Juiz de Fora Federal University, BrazilCopyright © 2022 Schrottmaier, Schmuckenschlager, Pirabe and Assinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alice Assinger, YWxpY2UuYXNzaW5nZXJAbWVkdW5pd2llbi5hYy5hdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.