 Qianqian Ma
Qianqian Ma Jingyun Luan2,3†
Jingyun Luan2,3† Dianwen Ju
Dianwen Ju Hong Xu
Hong Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 March 2022
Sec. Cytokines and Soluble Mediators in Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.851818
Chronic kidney injury has gradually become a worldwide public health problem currently affecting approximately 10% of the population and can eventually progress to chronic end-stage renal disease characteristic by the result of epithelial atrophy. Interleukin-22 (IL-22) is a cytokine produced by activated immune cells, while acting mainly on epithelial cells ranging from innate immune response to tissue regeneration to maintain barrier integrity and promote wound healing. Accumulating data suggests that IL-22 has emerged as a fundamental mediator of epithelial homeostasis in the kidney through promoting tissue repair and regeneration, inhibiting oxidative stress, and producing antimicrobial peptides. Binding of IL-22 to its transmembrane receptor complex triggers janus kinase/tyrosine kinase 2 phosphorylation, which further activates a number of downstream cascades, including signal transducer and activator of transcription 3, MAP kinase, and protein kinase B, and initiates a wide array of downstream effects. However, the activation of the IL-22 signaling pathways promotes the activation of complement systems and enhances the infiltration of chemokines, which does harm to the kidney and may finally result in chronic renal failure of different autoimmune kidney diseases, including lupus nephritis, and IgA nephropathy. This review describes current knowledge of the basic features of IL-22, including structure, cellular origin and associated signaling pathways. Also, we summarize the latest progress in understanding the physiological and pathological effects of IL-22 in the kidney, suggesting the potential strategies for the specific application of this cytokine in the treatment of kidney disease.
The immune system is developed to guard the host against a variety of pathogens while preventing collateral injury and promoting regeneration, wound healing and tissue repair (1). A large proportion of these functions are closely related to the role of cytokines produced by immune cells and functioning through autocrine and paracrine mechanisms (2). Interleukin-22 (IL-22), known as a cytokine, can be produced immediately after injury and initiates an immune response against several types of tissue impairments (3). Studies have exhibited that IL-22 is a critical signaling molecule that exerts multiple biological effects, including antimicrobial immunity, tissue regeneration, inhibition of oxidative stress, activation of complement systems and recruitment of chemokines (3). However, the beneficial and harmful effects of IL-22 may vary among different disease states (4). It is imperative to clarify the specific pathways and mechanisms by which IL-22 transforms between normal physiological processes and pathological states.
The kidney is one of the most complicated organs physiologically, structurally, and metabolically (5). Renal impairment has affected more than 10% of the population and contributes to high morbidity and mortality. Current treatments for kidney disease are mainly immunosuppressive agents, antihypertensives and diuretics, but the effectiveness of which in the clinic is limited, mainly because of their tendency to cause electrolyte disorders, impede growth, microbial infections, drug dependence and other side effects (6). When exploring new treatments, the physiological heterogeneity of the kidneys should be taken into consideration, such as cellular energy metabolism, hypoxia signaling pathways, renal tissue oxygenation and autoimmunity. IL-22 has been reported to be involved in many of these physiological and pathological processes (7). In the pathophysiology of acute kidney injury (AKI), hypoxia-inducible factor (HIF-1α) is expressed in renal tubular cells and can increase the secretion of IL-22 through the activation of CD4+ T cells, thereby regulating glucose metabolism and increasing glycogen storage (8). In addition, IL-22 ameliorates mitochondrial dysfunction and reactive oxygen radical production and inhibits the activation of the NOD-like receptor family containing protein 3 (NLRP3) inflammasome, which consequently reduces inflammatory responses (9, 10). On top of that, IL-22 acts as a regenerative survival factor in renal tissues (11). When the proximal tubules and renal cortical cells in the extrarenal medullary strip are damaged, damage-associated molecular patterns (DAMPs) are released. Dying renal tubular epithelial cells (RTECs) recruit mesenchymal dendritic cells to release IL-22. Toll-like receptor 4 (TLR4)-IL-22 pathway and downstream signaling including signal transducer and activator of transcription 3 (STAT3)/protein kinase B (AKT) are consequently activated to reduce apoptosis and promote regeneration of TECs (12). Furthermore, the effect of IL-22 on the regulation of autoimmune kidney disease is another important area of research. The kidney is acted as a vital objective of pathogenic immune responses against autoantigens or systemic autoimmunity (1). Activation of complement systems and infiltration of chemokines promoted by IL-22 during lupus nephritis (LN) and IgA nephropathy (IgAN) are important causes of kidney damage (13, 14).
Our understanding of the mechanistic link between IL-22 and kidney disease has improved dramatically in recent years. Considerable data has connected the functions of IL-22 to the pathophysiology underlying the most common kidney diseases. This review will introduce the key structural and biological features of IL-22, and focus on the advances in elucidating the biological functions of the IL-22-IL-22 receptor (IL-22-IL-22R) pathway in kidney-associated infection, tissue repair, oxidative stress and autoimmune kidney injury (Figure 1). We will also propose the possibility of targeting IL-22 and its related signaling pathways in the treatment of kidney diseases.
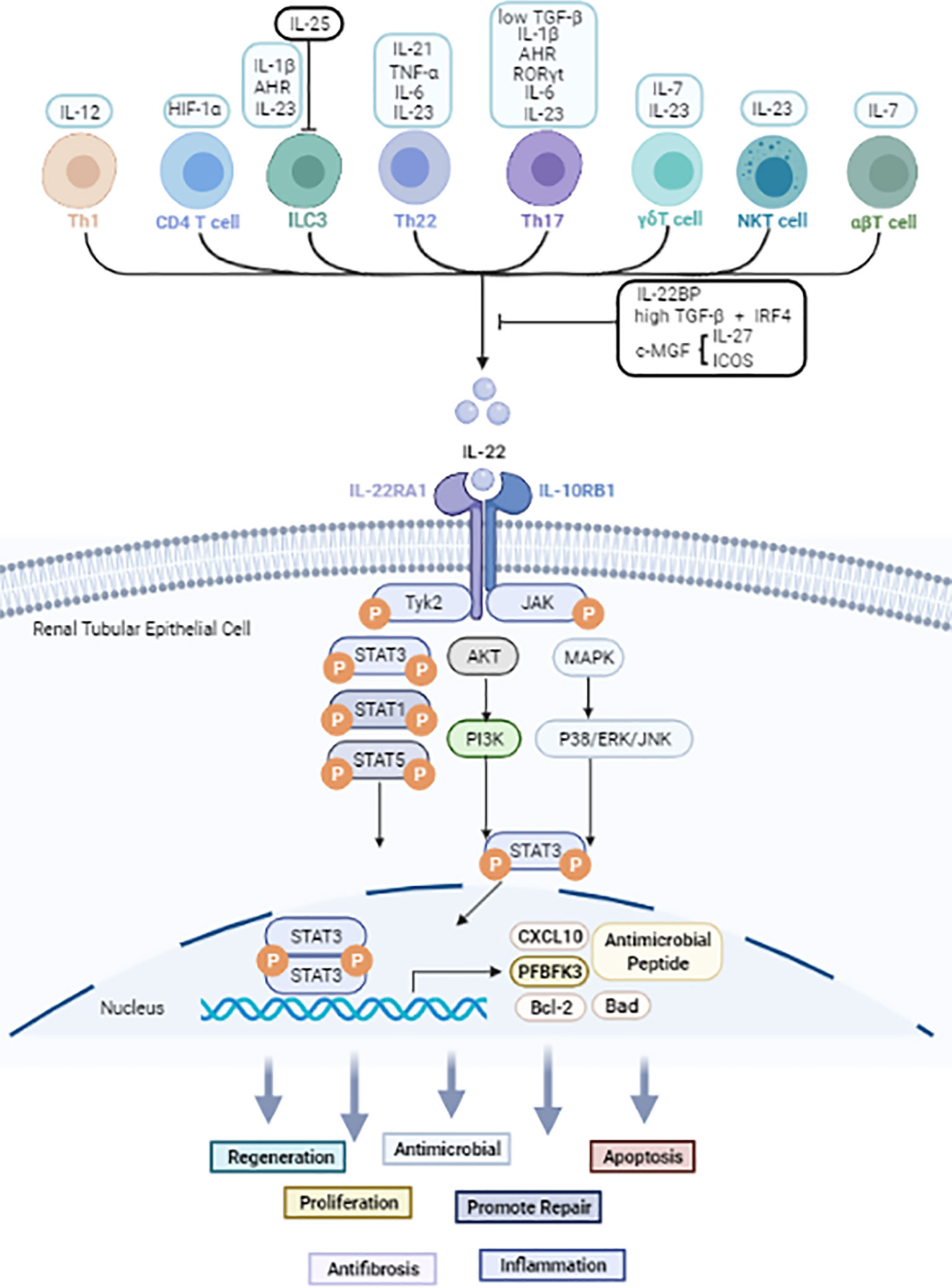
Figure 1 The cellular sources and signals downstream of IL-22 based on current evidence. Black arrow: upregulate. Black line: inhibit. IL-22 is secreted mainly by immune cells. JAK1 and TYK2 molecules are firstly phosphorylated after IL-22 binds to the IL-22R complex. Then further phosphorylate STAT molecules thus causes dimerization and accompanying translocation of STATs to the nucleus. Usually, STAT3 molecules play a pivotal effect, but STAT1 and STAT5 also be involved. Other than JAK/STAT activation, IL-22 also can activate AKT/PI3K and MAPK/P38/ERK/JNK pathways. IL-22 promotes the transcription of proteins such as Bal2, CXCL10 and PFBFK3, and has biological effects including anti-inflammatory, anti-microbial, apoptotic, anti-fibrotic, and proliferative effects in kidney, liver, gastrointestinal tract, and pancreas.
IL-22 was initially cloned from BW5147 T-lymphoma cells stimulated by murine IL-9 (15, 16). According to its biochemical and functional characteristics, IL-22 is classified as a class II cytokine, which belongs to IL-10 family (17). Human IL-22 is 79% homologous to mouse IL-22 and 25% identical to human IL-10, initially referred to as “IL-10-related T-cell inducing factor (IL-TIF)” (15). The human IL-22 gene is located on chromosome 12p15 and contains 5 exons with 5,200 base pairs (18). The secreted human and murine monomeric IL-22 proteins are predicted to be 146 amino acids in length (19). IL-22 is featured with a bundle-like structure consisting of α-helices and connecting loops. There are two disulfide bonds between the N-terminus and the D-E loop, as well as helix C to F forming a stable tertiary structure (19). IL-22 contains three N-glycosylation sites, and the glycosylation may affect the tertiary structure of IL-22 (20). For example, glycosylation of N54 might influence the interaction of IL-22 and IL-10R2 (21). Though the structure change resulting from glycosylation is minor, it should be taken into consideration when using IL-22-specific neutralizing antibodies for treatment (22). Apart from that, the bioactive form of IL-22 appears to be a monomer (23).
In general, IL-22 is primarily produced by lymphoid lineage cells, such as T lymphocytes and innate lymphocytes (ILCs) (24). The production of IL-22 in Th1, Th17, and Th22 cells is mainly induced by cytokines such as IL-6, IL-1β, IL-12, IL-21, IL-23, tumor necrosis factor-α (TNF-α) and other molecules such as RORyt and the aryl hydrocarbon receptor (AHR) ligand FICZ (25, 26). In peripheral blood, naïve T lymphocytes can produce IL-22 after differentiation into Th22 cells (27, 28). γδT cells, αβT cells, and NKT cells are capable of producing IL-22 in the presence of IL-7, IL-23, and HIF-1α (24, 29). Apart from adaptive immune cells, ILC3s and NK cells are also key producers of IL-22 (30–32) under stimulation of IL-1β, IL-23 and AhR (33, 34). In addition, the transcription factor c-Maf, inducible costimulatory (ICOS), IL-25, IL-27 and transforming growth factor β (TGF-β) were classified as negative regulators of IL-22 production (35–38).
IL-22R is a heterodimeric complex consisting of IL-22R1 and IL-10R2 subunits, both of which contain an extracellular moiety, an intracellular moiety, and a transmembrane moiety (39). IL-22 alone has a high affinity for the IL-22R1 (Kd values of 1-20 nM) and cannot bind to IL-10R2. However, IL-10R2 has the capability of binding with IL-22-IL-22R1 complex (Kd values of 7-45 μM) (40, 41). Studies have shown that the interaction between IL-22 and IL-22R is a complicated process. Initially, IL-22 binds to IL-22R1 subunit, which leads to a conformational transformation. Then, the IL-22-IL-22R1 complex binds to the IL-10R2 chain. The integrated structure formed by these three parts is capable of eliciting downstream intracellular signal transduction and a wide range of biological effects (24, 41).
IL-22 binding protein (IL-22BP) is known as the other receptor for IL-22, which is composed of 210 amino acids. Its affinity to IL-22 is 20-1000 times higher than that of IL-22R1 (42, 43). Due to its stable combination with IL-22, IL-22BP acts as a natural antagonist of IL-22 that isolates IL-22 from IL-22R1, thus blocking the activation of downstream signaling (44). IL-22BP has been verified to be predominantly expressed in the lung, skin, lymphoid organs, breast, placenta as well as stomach and intestines (45, 46). In addition to the local inhibitory effect, IL-22BP may support the stabilization and systemic functions of IL-22 to some extent.
Upon binding to its receptor complex, IL-22 can activate JAK and TYK2 phosphorylation, which trigger a series of downstream cascade reactions, including activating STAT1, STAT3, and STAT5 phosphorylation (47). Among these effects, the phosphorylation of STAT3 serine residues is necessary for maximal transcriptional activation. Other molecules, including p38, extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and phosphoinositide 3-kinase (PI3K), are also activated downstream signal pathways mediating the effector function of IL-22 (48–50). IL-22 promotes the transcription of proteins such as Bal2, CXCL10 and PFBFK3, and has biological effects including anti-inflammatory, anti-microbial, apoptotic, anti-fibrotic, and proliferative effects in kidney, liver, gastrointestinal tract, and pancreas.
IL-22 is essential in promoting the regeneration of renal tissue and defending against pathogens, but aberrant activation of IL-22 promotes the pathogenesis of various renal diseases (Table 1). The dual roles of IL-22 including the protective and the pathological effects in kidney-related diseases are described in detail below.
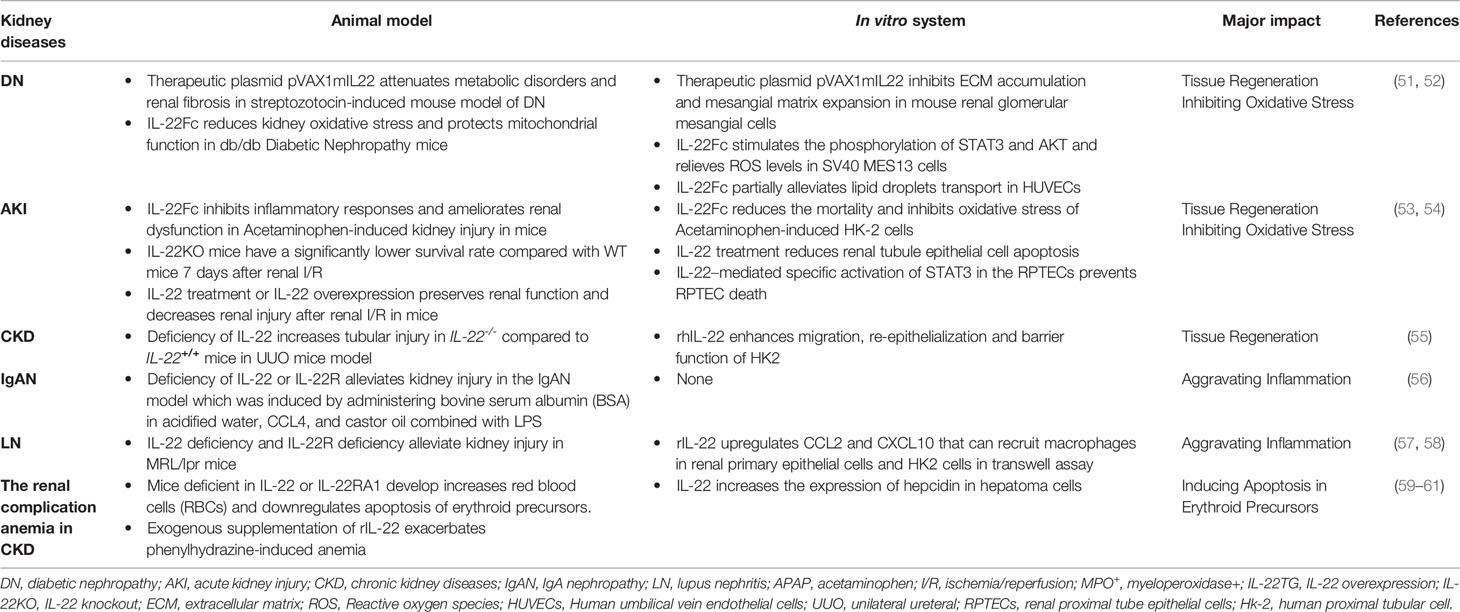
Table 1 Diverse Roles of IL-22 across Different Kidney Diseases.
Cytokines are critical contributors in immune response to external challenges and are also particularly important for epithelial barriers (62). In particular, IL-22 is involved in maintaining microflora homeostasis, kidney epithelial cells and a functional defense system against external threats (63) (Figure 2A).
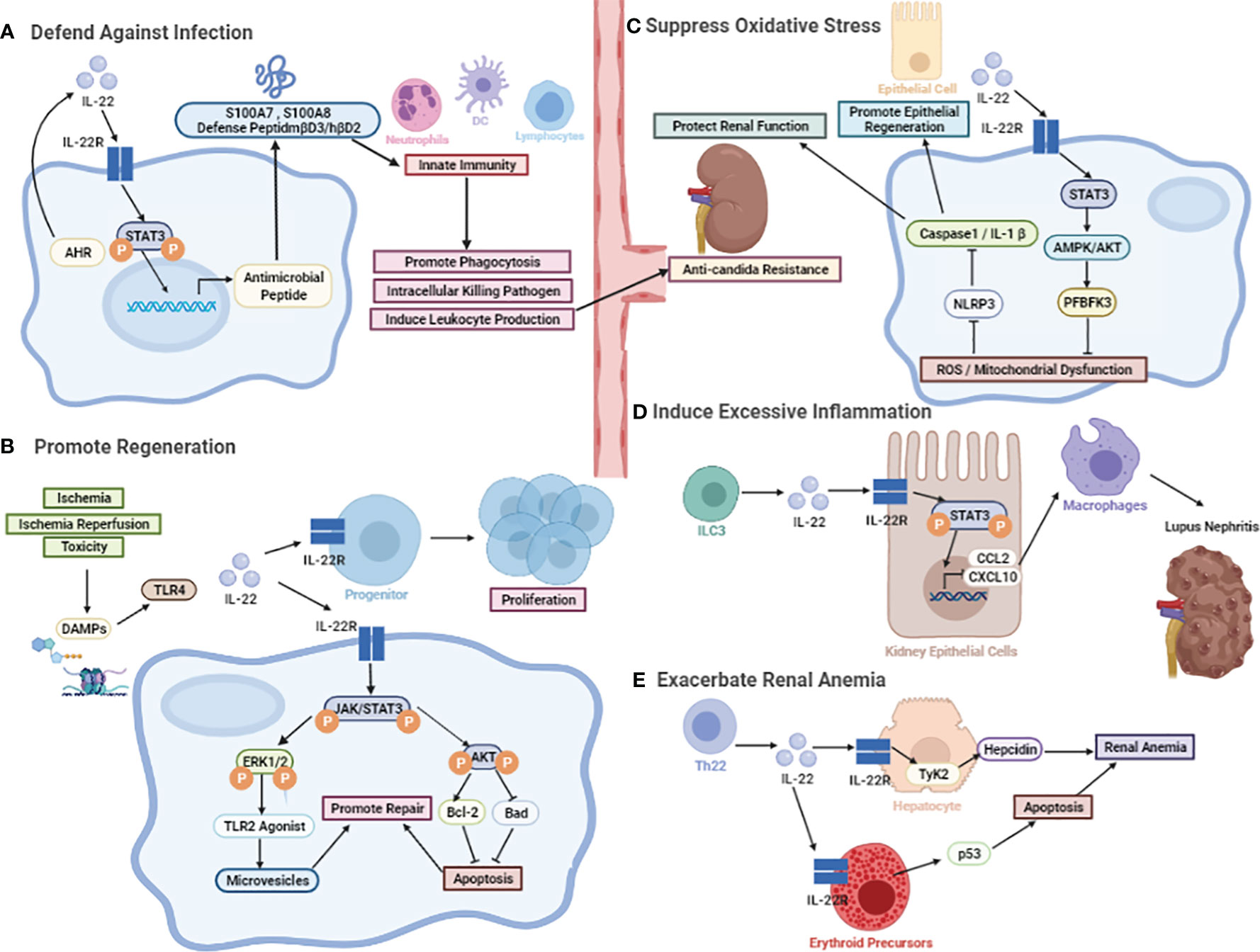
Figure 2 The biological effect of IL-22. (A) The protective role against microorganisms including bacterial and fungal. The induction of various antimicrobial peptides, like psoriasis (S100A7), calgranulin-A (S100A8), calgranulin-B (S100A9), β-defensin-3 (mβD3), and lipocalin-2. (B) The protective role in promoting regeneration. IL-22-mediated regenerative function by activation of JAK-STAT3 and ERK1/2 phosphorylation. After the onset of AKI, TLR4-IL-22 signaling is a specific stimulus for IL-22 induction. Other than that, activation of STAT3 and AKT can be detected after I/R, followed by an increase in Bcl-2 levels and a decrease in Bad levels. (C) The protective role in the inhibition of oxidative stress. It may promote renal repair through the active expression of renal reprogramming PFBFK3 by IL-22 mainly inducing STAT3-AMPK/AKT signaling after injury, slowing down the accumulation of reactive oxygen radical ROS production and mitochondrial dysfunction, which can further protect renal function by inhibiting the NLRP3/Caspase-1/IL-1β signaling pathway. (D) The pathological effects in recruiting chemokines. The involvement of IL-22 in the induction of CCL2 and CXCL10, which promotes the infiltration of macrophages to dampen kidney inflammation. (E) The pathological effects in inducing chronic renal anemia. IL-22 exacerbates anemia through inducing erythropoietin apoptosis and hepcidin production.
The well-documented effect of IL-22 in maintaining microbial homeostasis is mainly achieved by preventing and defending against pathogenic bacterial infections. Production of various antimicrobial peptides, such as psoriasin (S100A7), calgranulin-A (S100A8), calgranulin-B (S100A9), β-defensin-3 (mβD3), and lipocalin-2 serves as a key factor in the IL-22-mediated defense of epithelial cells against pathogenic infection (63). It has been reported that IL-22 plays a major role in protecting skin, gastrointestinal and respiratory tract from pathogenic and commensal bacterial infections through the above mechanisms. Similarly, IL-22 is critical for limiting bacterial replication and hematogenous dissemination in renal infectious diseases, possibly in part by inducing the expression of antimicrobial peptides in epithelial cells on the surface of these barriers. For example, in the case of Staphylococcus aureus (S. aureus) infections, the kidney bacterial burden is the basis for invasive or hematogenous dissemination to distant organs (64). Both renal colony-forming units (CFUs) and activated inflammatory factors correlate with disease severity. IL-22 expression was significantly increased at the abscess expression site in the anti-S. aureus vaccine NDV-3 injection group compared to the control group, accelerating tissue antimicrobial resistance and repair (65, 66). In contrast, the application of cortisol or neutralizing antibodies to inhibit IL-22 significantly promoted microbial proliferation and the rate of abscess progression in the kidney. Mechanistically, IL-22 may induce the production of host defense peptides to fight against bacterial invasion and protect infected tissues and organs. Both mβD3 in mice and β-defensin-2 in humans increase the adaptive immune response (e.g., dendritic cell or lymphocyte function) while recruiting neutrophils and enhancing phagocytosis and the intracellular killing of pathogens (67, 68). In addition to aiming at in-situ infection in the kidney, IL-22 significantly reduces the incidence of hematogenous dissemination after infection, possibly by inducing antimicrobial peptide expression in renal epithelial cells (69). Although little is known whether IL-22 attenuates renal infection through upregulation of related gene expression, IL-22 also acts synergistically with IL-17F or TNF, which stimulates the molecules encoding genes including chemokines CXCL1, CXCL5 and cytokines IL-6 and G-CSF to improve the efficacy of airway, intestinal or skin epithelial cells against bacterial infections (70–72). Furthermore, IL-22 enhances the clearance of abnormal pathogenic microorganisms and regulates flora balance by elevating cytokine and chemokine production in epithelial cells to facilitate leukocyte recruitment and activation at inflammatory sites (73, 74).
IL-22 also exerts a pivotal function in warding off fungal infections of the kidney including primary and hemorrhagic disseminated infections (75). IL-22 is an important natural defender against chronic candidiasis (CMC). As observed in human and mouse models, IL-22 provides the first line of defense against Candida albicans by monitoring fungal growth, especially for bloody disseminated kidney fungal infections (69). Studies have reported that tryptophan metabolites from the microbiota balanced mucosal reactivity resist to fungus through promoting the transcription of IL-22 (76). The highly adaptable Lactobacillus expands and produces an AhR ligand that improves AhR-dependent IL-22 transcription and secretion. IL-22 secreted in this way can regulate the imbalance in the flora, allowing for the survival of commensal communities but resisting fungal colonization (69, 76). The effect of IL-22 in defensing against fungi has been reported in pulmonary Aspergillus fumigatus infection in lungs and Chronic Mucocutaneous Candidiasis. However, more researches are still needed to find the specific mechanisms by which the kidney protects against fungal infection.
In conclusion, IL-22 is involved in the induction of innate defense mechanisms in renal epithelial tissues, thus protecting tissues from injury during microbial infection and recovery of tissue homeostasis.
Apart from strengthening the host barrier defense against microbial infections, IL-22 is able to stimulate downstream signaling pathways and activate cell survival genes. Increasing evidence suggests that IL-22 is associated with both regenerative and antiapoptotic functions in renal tissues, including acute or chronic kidney injury induced by various etiologies (Figure 2B).
AKI involves an aseptic inflammatory response that exacerbates renal tubular cell damage. Necrosis of renal tubular cells after injury can release histones, ATP, and uric acid, which are known as DAMPs (77, 78). Then, DAMPs act on renal parenchymal cells and mesenchymal dendritic cells to induce an array of pattern recognition receptors, such as Toll-like receptors (TLRs) (79, 80). The subsequent innate immune response caused by TLR-4 initiates a variety of immune cells infiltrating into the kidney, promoting an inflammatory response and resulting in tubular cell death (81). After the onset of AKI, TLR4 signaling stimulates the production of IL-22, which is mainly sourced from intrarenal mononuclear phagocytes. IL-22-mediated regenerative functions on renal TECs are followed by the phosphorylation of JAK-STAT3 and ERK1/2 (50). This process increases endogenous TLR2 agonists and induces renal tubular progenitor cells to secrete regenerative proteins and microRNAs and prompt themselves to differentiate into renal TECs. Neutralizing endogenous IL-22 with IL-22 antibodies showed elevated indicators of kidney injury, such as kidney injury molecule (Kim)-1, π-glutathione S-transferase (GST), and α-GST, suggesting impaired tubular epithelial repair in AKI. In addition, IL-22 increased the survival of these epithelial progenitor cells and synthetically achieved renal epithelial regeneration. Overall, IL-22 promotes renal regeneration through epithelial cell proliferation as one of several promising strategies for treating (or avoiding) end-stage renal diseases (12).
Among the multiple causes of AKI, such as renal hypoperfusion, ischemia and toxicity, renal ischemia/reperfusion (I/R) is common, especially in proximal tubular cells (82). Studies have shown that renal dysfunction after I/R is partly attributed to cell apoptosis (83), and IL-22 has anti-apoptotic effects, with a possible molecular mechanism of activating AKT and inhibiting the mitochondrial apoptotic pathway, which is accompanied by an increase in Bcl-2 and a decrease in Bad (53). Compared to wild-type (WT) mice, IL-22-knockout (KO) mice had higher levels of urea nitrogen, and the survival rate at 7 days after injury was significantly lower, suggesting that IL-22 has protective effects on the kidney (53). The results of TUNEL staining displayed that the apoptotic tubule cells in the mIL-22-treated I/R group was significantly reduced; correspondingly, the levels of pAkt473, pSTAT3 and Bcl-2 were increased, and the renal cortex increased significantly, but WT mice had the opposite effects (53). In summary, activation of STAT3 and AKT occurred in the proximal TECs of mice after I/R, followed by an increase in Bcl-2 levels and a decrease in Bad levels, which may play a crucial role in IL-22-mediated regeneration and repair of epithelial cells in response to I/R-induced renal injury.
Apart from AKI, IL-22 is also a regenerative factor that plays an important role in the unilateral ureteral obstruction (UUO) model of chronic progressive obstructive nephropathy (55). In UUO, IL-22+ cells accumulate more abundantly in the renal interstitium than other cells. IL-22-/- mice showed significant tubular cell death and tubular atrophy compared with IL-22+/+ mice. In vitro experiments indicated that IL-22 supplementation promoted the migration, proliferation, metabolic activity, and regeneration of human tubular epithelial cells. Specifically, the regenerative function of IL-22 on RTECs in chronic diseases was linked to the transduction of STAT-AKT-dependent pathways and consequent decrease of Bad protein that is known as a pro-apoptotic mediator. It can be hypothesized that the mechanism by which IL-22 mediates STAT-JAK pathway-induced cell regeneration may be associated with improving apoptotic events. Studies have shown that AKT activation ameliorates renal injury (84, 85). Erlotinib improved animal phenotypes by acting on the AKT signaling pathway and attenuating the activation of thylakoid cells and macrophages in a rat model of CKD (55). Considering the protective role of IL-22 in UUO, modulating IL-22 may be a potential therapeutic strategy in other forms of CKD.
The pathogenesis of AKI is complex and diverse and includes oxidative stress, inflammation, and vascular injury, among which oxidative stress is considered a central exacerbating factor (86). It has been reported that mitochondria are easily damaged during the development of AKI and are the main intracellular source of reactive oxygen species (ROS) (87). ROS are highly reactive products that trigger apoptosis, necrosis, oxidative stress, and even induce chronic inflammation and renal fibrosis (88). Therefore, ROS are critical targets in oxidative stress-mediated AKI treatment, and ROS clearance might reshape the microenvironment of kidney (Figure 2C).
IL-22, a regenerative factor for RTECs, promotes regeneration and repair of renal tissue by inhibiting oxidative stress. In vitro IL-22 can ameliorate the accumulation of dysfunctional mitochondria in TECs after cisplatin injury by activating mitochondrial phagocytosis (89). Meanwhile, compared to the model group, IL-22 treatment protected the mitochondrial health of damaged RTECs, as evidenced by reductions in mitochondrial mass, mitochondrial ROS, and mitochondrial dysfunction. Mechanistically, IL-22 may reprogram PFBFK3 and activate STAT3-AMPK/AKT signaling, alleviating the accumulation of ROS and mitochondrial dysfunction in the kidney (90). Further, the reduction of ROS can ensure metabolic homeostasis of renal cells to some extent by downregulating the NLRP3/Caspase-1/IL-1β pathway, which protects renal function and promotes renal epithelial tissue regeneration and repair (54).
Other than that, once oxidative stress is initiated, persistent stimulation by excess ROS is an important link in the progression of kidney disease to fibrosis. This process recruits immune cells such as Th1, Th2, Th17, and regulatory T (Treg), which can release various growth factors, including IL-13 or TGF-β, promoting the excessive deposition of extracellular matrix and ultimately leading to renal fibrosis. The most common causes of CKD and renal fibrosis are diabetes mellitus (DM) (91–93), adult hypertension and childhood obstructive nephropathy (94). In a mouse model of chronic diabetic nephropathy, IL-22 reduced the abnormal deposition of renal collagen and extracellular matrix and therefore improved kidney fibrosis. The possible mechanism is mainly attributed to the inhibition of renal oxidative stress and mitochondrial dysfunction by IL-22 to improve the inflammatory response (51, 52).
Sustained kidney inflammation is a central player in the development of kidney diseases including IgAN, LN, and psoriatic kidney injury (57, 58, 95–98). Chemokines and their receptors play pivotal roles in inflammation by upregulating the migration and retention of circulating immune cells. Among these chemokines, high expression of CCL2 and CXCL10 was shown to play an important role in exacerbating proteinuria, deteriorating renal pathology and renal dysfunction in patients with LN. In vitro stimulation with rIL-22 alone was sufficient to upregulate the expression of CXCL10 and CCL2 in both primary kidney epithelial cells and a human kidney cell line (HK2), whereas increased macrophage infiltration was observed in the rIL-22-stimulated group. Furthermore, the expression levels of several kinds of chemokines, including CCL2 and CXCL10, have been found to be significantly reduced in rIL-22-deficient and IL-22R-deficient MRL/lpr mice, accompanied by reduced kidney injury in mice. Hu et al. found that IL-22 activated the STAT3 signaling pathway, thus causing deteriorating LN in lupus-prone mice. IL-22 deficiency ameliorated proteinuria, renal function and pathological impairment through downregulating the expression of CCL2 and CXCL10 and reducing the filtration of macrophages into the kidney, ultimately decreasing the systematic disease and the severity of LN (14). These findings indicated that IL-22 might play a role in aggravating inflammatory damage to the kidney and provide a promising novel therapeutic target (14) (Figure 2D).
In addition, IL-22 can also induce the inflammation in the kidney by affecting the complement system. The complement system serves as a vital part of the innate immune system that fights against invasive pathogens; however, uncontrolled activation of complement also promotes excessive inflammation and injury in various kidney diseases, such as LN and IgAN (99, 100). The wide variety of kidney diseases related to complement activation might be attributed to glomerular endothelial cell fenestrations and deficiencies in intrinsic complement regulators in the glomerular basement membrane (101). Indeed, the deposition of complement components in glomeruli has been commonly observed in IgAN and LN patients (102, 103). The association of IL-22 with the complement system was uncovered by analyzing the Gene Expression Omnibus dataset, which showed that treatment with IL-22 positively correlated with increased levels of C3 (56). This finding was further supported by the phenomenon that the mRNA level of C3 was reduced in the liver tissue of IL-22-/- mice after Clostridium difficile infection, and this effect was reversed by systemic administration of IL-22. The mechanism by which IL-22 enhances C3 expression has not been entirely elucidated, but treatment with a STAT3 inhibitor reverses IL-22-induced C3 upregulation, suggesting that the STAT3 signaling pathway may be involved in IL-22-induced upregulation of complement C3 (104). The upregulation of C3 induced by IL-22 has also been demonstrated to exacerbate kidney injury. IL-22 or IL-22R deficiency decreases the deposition of C3 in the kidney but increases serum C3 levels, thus contributing to significant alleviation of kidney injury and prolonged survival rates in the murine model of LN, which is characterized by lower serum C3 levels and higher C3 deposition in the kidney (14). These findings suggested that genetic or pharmaceutical modification to dampen IL-22 signaling may be at least partially alleviate the kidney inflammation in patients with IgAN and LN by downregulating the complement pathway.
Anemia is a common complication of chronic kidney disease (CKD) that can increase the risks of cardiovascular diseases (59). The reasons for anemia were previously thought to be deficient erythropoietin production and poor iron uptake in CKD patients, but therapies intended to increase erythropoietin and iron levels were only effective in a minority of patients (59). Recent progress has been made in identifying a novel mechanism of IL-22 underlying the development of anemia in CKD patients (Figure 2E).
While IL-22 is widely expressed by innate immune cells, the IL-22R was unexpectedly discovered on erythroid precursors, and this receptor was involved in inducing apoptosis in erythroid precursors to exacerbate anemia in CKD patients (60, 61). Mice deficient in IL-22 or IL-22RA1 on a right open reading frame 2 (Riok2)-haploinsufficient background develop excess red blood cells (RBCs) and exhibit downregulated apoptosis in erythroid precursors, while exogenous supplementation with rIL-22 exacerbates phenylhydrazine-induced anemia, as evidenced by upregulated apoptosis and reduced numbers of RBCs. Importantly, rIL-22 treatment failed to worsen phenylhydrazine-induced anemia in mice with erythroid cell-specific knockout of IL-22RA1, suggesting that IL-22RA1 expression on erythroid precursors is absolutely involved in the induction of erythroid precursor apoptosis. Further experiments have identified that p53 is upregulated by IL-22 treatment in vitro and in IL-22RA1+ erythroid precursors from Riok2-haploinsufficient mice. Pharmaceutical inhibitor of p53 suppressed apoptosis in IL-22-free media, indicating that p53 is a critical interrelated molecule by which IL-22 induces apoptosis in erythroid precursors, thus leading to the exacerbation of anemia.
Beyond inducing apoptosis in erythroid progenitors, IL-22 has also been identified to be a positive regulator of hepcidin, which is a central mediator of iron homeostasis. A study showed a marked increase in hepcidin in hepatoma cells treated with IL-22 (105). Mice deficient in IL-22 have decreased transcriptional levels of the hepatic hepcidin 1 and pro-hepcidin gene in response to LPS, while treatment with the IL-22-Fc fusion protein leads to increased levels of hepcidin and iron deficiency in vivo. The mechanism by which IL-22 enhances hepcidin production is not completely understood, but the Tyk2 signaling pathway is likely involved, as IL-22-induced hepcidin upregulation and hemoglobin downregulation are impaired in Tyk2-deficient mice (106). Moreover, plasma levels of IL-22 are significantly elevated in CKD patients with anemia, and this increase correlates with decreased concentrations of hemoglobin (107). These findings highlight the importance of IL-22 in exacerbating anemia by inducing hepcidin production and apoptosis, which may hopefully provide a new target for the treatment of anemia in CKD patients.
Microscopic polyangiitis (MPA) and Granulomatosis with polyangiitis (GPA) are chronic autoimmune inflammatory vascular lesions with unknown etiology, and renal involvement is a typical and potentially life-threatening symptom in these diseases (108). To date, IL-22 has been poorly studied in renal damage associated with these vascular inflammatory diseases. However, high pathogenicity of Th17 to the kidney was observed in GPA patients positive for anti-neutrophil cytoplasmic antibodies (2, 109). The possible mechanism explored was that early infiltration of Th17 cells leads to the upregulation of CXCL9, which subsequently stimulates renal cells to express glutamate-leucine-arginine motif (ELR) chemokines (CXCL1, 2, and 5) that recruit neutrophils to the kidney. And IL-22, the most marked cytokine of Th17 cells, had the possibility of being involved in their pathogenic role in renal disease (110). It was also confirmed that specific IL-22 expression was detected in the renal tissues of GPA patients and diffuse IL-22+ cell infiltration was found in the renal interstitium and local glomeruli medially in renal tissue biopsies, suggesting a possible pathogenic role of IL-22 in GPA-related renal injury (111). Meanwhile, another group found that after 6 months of follow-up of GPA, IL-22 was significantly reduced with the restoration of the kidney and other organs (108). Therefore, the proposed therapeutic strategies targeting Th17 cells, and the corresponding cytokine IL-22 may have promising applications for the treatment of vasculitis-associated nephropathy.
Cytokine-targeted therapy is now a promising therapeutic strategy for various kidney diseases due to the side effects of steroid hormones and other immunosuppressive agents. Currently, no drugs that directly target IL-22 have been approved by the FDA for the treatment of kidney disease. As for other diseases, IL-22-related drugs, including IL-22-Fc, IL-22 specific antibodies and inhibitors, and IL-22R inhibitors, have been in clinical trials for different periods. (Table 2).
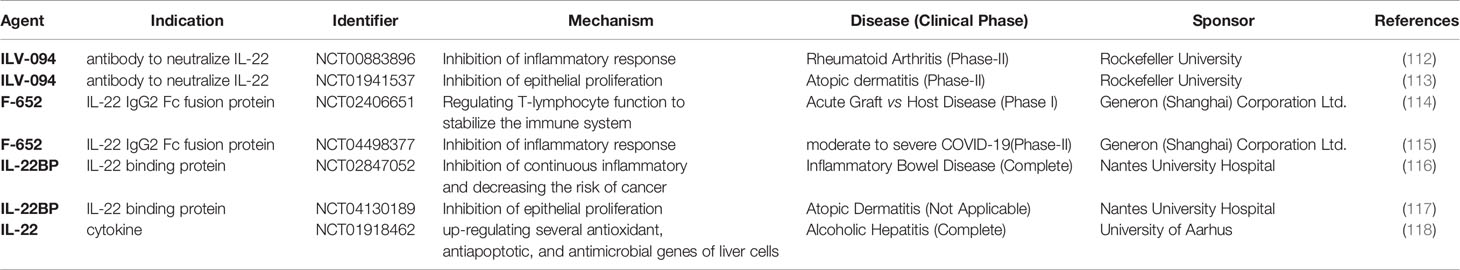
Table 2 Clinical Trials Related to IL-22 Based on Clinicaltrials.gov.
The IL-22-neutralizing antibody Fezakinumab (ILV-094) has currently finished phase I and II clinical trials for rheumatoid arthritis and atopic dermatitis, respectively (4, 112). The only reported adverse reactions with a high incidence were upper respiratory tract infections of different pathogens, including viruses and fungi. F-652, a fusion protein of IL-22 fused to the IgG2 anchoring region, is used its tissue-protective and regenerative effects to treat graft-versus-host disease (aGVHD) and severe COVID-19 (113, 114). After that, existing clinical studies have assessed the expression of IL-22BP in skin and serum to deeply explore the protective role of IL-22BP in atopic dermatitis and inflammatory bowel disease (115, 116). In addition, the IL-22-related signaling pathways PGE2/IL22/IL17 and IL-17/IL-22 are receiving increasing attention in eczema, psoriasis, and chronic obstructive pulmonary disease (117, 119). The production and effects of the cytokine IL-22 in patients with alcoholic hepatitis has been the research interests by certain research teams (120). However, some studies have shown that IL-22 can promote the progression of existing tumors, especially colon cancer, by activating STAT3-related signaling pathways (118, 121). Therefore, the risk of concomitant tumor exacerbation needs to be carefully assessed when applying recombinant IL-22.
In the treatment of renal diseases, the application of enhancing or attenuating IL-22 effects should be determined according to the specific microenvironment. Because IL-22 is able to protect against pathogenic bacterial infections, promote cell proliferation, inhibit apoptosis, and suppress oxidative stress, we propose an attempt to apply IL-22 or its fusion proteins in infectious kidney diseases and AKI. Considering the role of IL-22 in activating the complement system and recruiting chemokines, IL-22-neutralizing antibody and IL-22BP may be used to treat immune-related renal diseases such as IgAN and LN.
In conclusion, IL-22 is a double-edged sword for the kidney. On one hand, studies in diabetic nephropathy, obstructive nephropathy, and acute kidney injury models indicate that IL-22 plays a vital role in tissue protection and regenerative repair. This cytokine is essential not only in host defense against various pathogens but also in restoring immune quiescence and tissue homeostasis after an inflammatory response. Additionally, IL-22 exerts pathogenic effects in certain renal diseases, including systemic lupus erythematosus, IgA nephropathy, and anemia induced by CKD, due to recruitment of chemokines and uncontrolled activation of complement. Upon the protective and pathogenic roles of IL-22 in the kidney, the application of IL-22 in the treatment of renal diseases can be attempted. Further research on IL-22 biology will undoubtedly contribute to a better understanding of many kidney diseases and potential therapeutic approaches, opening the way for the identification of new therapeutic approaches and directions for the application in the clinic.
QM and JL contributed equally to this paper. QM, JL, and CX conceived and drafted the paper. YB and FL prepared the tables and figures. BC was involved in the compilation of the references. DJ and HX instructed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by the National Natural Science Foundation of China under grant 82170793 and The Collaborative Research Projects of Greater Bay Area Institute of Precision Medicine (Guangzhou) in 2021 (IPM2021C003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ouyang W, O'Garra A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity (2019) 50(4):871–91. doi: 10.1016/j.immuni.2019.03.020
2. Kurts C, Panzer U, Anders HJ, Rees AJ. The Immune System and Kidney Disease: Basic Concepts and Clinical Implications. Nat Rev Immunol (2013) 13(10):738–53. doi: 10.1038/nri3523
3. Arshad T, Mansur F, Palek R, Manzoor S, Liska V. A Double Edged Sword Role of Interleukin-22 in Wound Healing and Tissue Regeneration. Front Immunol (2020) 11:2148. doi: 10.3389/fimmu.2020.02148
4. Cobleigh MA, Robek MD. Protective and Pathological Properties of IL-22 in Liver Disease: Implications for Viral Hepatitis. Am J Pathol (2013) 182(1):21–8. doi: 10.1016/j.ajpath.2012.08.043
5. Kimura T, Isaka Y, Yoshimori T. Autophagy and Kidney Inflammation. Autophagy (2017) 13(6):997–1003. doi: 10.1080/15548627.2017.1309485
6. Daehn IS, Duffield JS. The Glomerular Filtration Barrier: A Structural Target for Novel Kidney Therapies. Nat Rev Drug Discovery (2021) 20(10):770–88. doi: 10.1038/s41573-021-00242-0
7. Scholz H, Boivin FJ, Schmidt-Ott KM, Bachmann S, Eckardt KU, Scholl UI, et al. Kidney Physiology and Susceptibility to Acute Kidney Injury: Implications for Renoprotection. Nat Rev Nephrol (2021) 17(5):335–49. doi: 10.1038/s41581-021-00394-7
8. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic Control of Type 1 Regulatory T Cell Differentiation by AHR and HIF1-Alpha. Nat Med (2015) 21(6):638–46. doi: 10.1038/nm.3868
9. Chen W, Luan J, Wei G, Zhang X, Fan J, Zai W, et al. In Vivo Hepatocellular Expression of Interleukin-22 Using Penetratin-Based Hybrid Nanoparticles as Potential Anti-Hepatitis Therapeutics. Biomaterials (2018) 187:66–80. doi: 10.1016/j.biomaterials.2018.09.046
10. Chen Y, Jin S, Teng X, Hu Z, Zhang Z, Qiu X, et al. Hydrogen Sulfide Attenuates LPS-Induced Acute Kidney Injury by Inhibiting Inflammation and Oxidative Stress. Oxid Med Cell Longev (2018) 2018:6717212. doi: 10.1155/2018/6717212
11. Lech M, Anders HJ. Macrophages and Fibrosis: How Resident and Infiltrating Mononuclear Phagocytes Orchestrate All Phases of Tissue Injury and Repair. Biochim Biophys Acta (2013) 1832(7):989–97. doi: 10.1016/j.bbadis.2012.12.001
12. Kulkarni OP, Hartter I, Mulay SR, Hagemann J, Darisipudi MN, Kumar Vr S, et al. Toll-Like Receptor 4-Induced IL-22 Accelerates Kidney Regeneration. J Am Soc Nephrol (2014) 25(5):978–89. doi: 10.1681/ASN.2013050528
13. Xiao C, Zhou Q, Li X, Li H, Meng T, Zhong Y, et al. Differentiation and Recruitment of IL-22-Producing Helper T Cells in lgA Nephropathy. Am J Transl Res (2016) 8(9):3872–82.
14. Hu L, Hu J, Chen L, Zhang Y, Wang Q, Yang X. Interleukin-22 From Type 3 Innate Lymphoid Cells Aggravates Lupus Nephritis by Promoting Macrophage Infiltration in Lupus-Prone Mice. Front Immunol (2021) 12:584414. doi: 10.3389/fimmu.2021.584414
15. Dumoutier L, Van Roost E, Colau D, Renauld JC. Human Interleukin-10-Related T Cell-Derived Inducible Factor: Molecular Cloning and Functional Characterization as an Hepatocyte-Stimulating Factor. Proc Natl Acad Sci USA (2000) 97(18):10144–9. doi: 10.1073/pnas.170291697
16. Dumoutier L, Louahed J, Renauld JC. Cloning and Characterization of IL-10-Related T Cell-Derived Inducible Factor (IL-TIF), a Novel Cytokine Structurally Related to IL-10 and Inducible by IL-9. J Immunol (2000) 164(4):1814–9. doi: 10.4049/jimmunol.164.4.1814
17. Nikoopour E, Bellemore SM, Singh B. IL-22, Cell Regeneration and Autoimmunity. Cytokine (2015) 74(1):35–42. doi: 10.1016/j.cyto.2014.09.007
18. Wolk K, Sabat R. Interleukin-22: A Novel T- and NK-Cell Derived Cytokine That Regulates the Biology of Tissue Cells. Cytokine Growth Factor Rev (2006) 17(5):367–80. doi: 10.1016/j.cytogfr.2006.09.001
19. Nagem RA, Colau D, Dumoutier L, Renauld JC, Ogata C, Polikarpov I. Crystal Structure of Recombinant Human Interleukin-22. Structure (2002) 10(8):1051–62. doi: 10.1016/S0969-2126(02)00797-9
20. Xu T, Logsdon NJ, Walter MR. Crystallization and X-Ray Diffraction Analysis of Insect-Cell-Derived IL-22. Acta Crystallogr D Biol Crystallogr (2004) 60(Pt 7):1295–8. doi: 10.1107/S0907444904010492
21. Logsdon NJ, Jones BC, Allman JC, Izotova L, Schwartz B, Pestka S, et al. The IL-10R2 Binding Hot Spot on IL-22 is Located on the N-Terminal Helix and is Dependent on N-Linked Glycosylation. J Mol Biol (2004) 342(2):503–14. doi: 10.1016/j.jmb.2004.07.069
22. Xu T, Logsdon NJ, Walter MR. Structure of Insect-Cell-Derived IL-22. Acta Crystallogr D Biol Crystallogr (2005) 61(Pt 7):942–50. doi: 10.1107/S0907444905009601
23. de Oliveira Neto M, Ferreira JR Jr., Colau D, Fischer H, Nascimento AS, Craievich AF, et al. Interleukin-22 Forms Dimers That are Recognized by Two Interleukin-22R1 Receptor Chains. Biophys J (2008) 94(5):1754–65. doi: 10.1529/biophysj.107.112664
24. Sabat R, Ouyang W, Wolk K. Therapeutic Opportunities of the IL-22-IL-22R1 System. Nat Rev Drug Discovery (2014) 13(1):21–38. doi: 10.1038/nrd4176
25. Abikhair M, Mitsui H, Yanofsky V, Roudiani N, Ovits C, Bryan T, et al. Cyclosporine A Immunosuppression Drives Catastrophic Squamous Cell Carcinoma Through IL-22. JCI Insight (2016) 1(8):e86434. doi: 10.1172/jci.insight.86434
26. Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, et al. Th22 Cells are an Important Source of IL-22 for Host Protection Against Enteropathogenic Bacteria. Immunity (2012) 37(6):1061–75. doi: 10.1016/j.immuni.2012.08.024
27. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of Interleukin 22 But Not Interleukin 17 by a Subset of Human Skin-Homing Memory T Cells. Nat Immunol (2009) 10(8):857–63. doi: 10.1038/ni.1767
28. Doulabi H, Rastin M, Shabahangh H, Maddah G, Abdollahi A, Nosratabadi R, et al. Analysis of Th22, Th17 and CD4(+)cells Co-Producing IL-17/IL-22 at Different Stages of Human Colon Cancer. BioMed Pharmacother (2018) 103:1101–6. doi: 10.1016/j.biopha.2018.04.147
29. Witte E, Witte K, Warszawska K, Sabat R, Wolk K. Interleukin-22: A Cytokine Produced by T, NK and NKT Cell Subsets, With Importance in the Innate Immune Defense and Tissue Protection. Cytokine Growth Factor Rev (2010) 21(5):365–79. doi: 10.1016/j.cytogfr.2010.08.002
30. Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The Aryl Hydrocarbon Receptor Regulates Gut Immunity Through Modulation of Innate Lymphoid Cells. Immunity (2012) 36(1):92–104. doi: 10.1016/j.immuni.2011.11.011
31. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR Drives the Development of Gut ILC22 Cells and Postnatal Lymphoid Tissues via Pathways Dependent on and Independent of Notch. Nat Immunol (2011) 13(2):144–51. doi: 10.1038/ni.2187
32. Choi SW, Reddy P. Current and Emerging Strategies for the Prevention of Graft-Versus-Host Disease. Nat Rev Clin Oncol (2014) 11(9):536–47. doi: 10.1038/nrclinonc.2014.102
33. Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, et al. Nfil3 is Required for the Development of All Innate Lymphoid Cell Subsets. J Exp Med (2014) 211(9):1733–40. doi: 10.1084/jem.20140145
34. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: Immunobiology and Pathology. Annu Rev Immunol (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
35. Valdez PA, Vithayathil PJ, Janelsins BM, Shaffer AL, Williamson PR, Datta SK. Prostaglandin E2 Suppresses Antifungal Immunity by Inhibiting Interferon Regulatory Factor 4 Function and Interleukin-17 Expression in T Cells. Immunity (2012) 36(4):668–79. doi: 10.1016/j.immuni.2012.02.013
36. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, et al. RORgammat+ Innate Lymphoid Cells Regulate Intestinal Homeostasis by Integrating Negative Signals From the Symbiotic Microbiota. Nat Immunol (2011) 12(4):320–6. doi: 10.1038/ni.2002
37. Wang H, Li Z, Yang B, Yu S, Wu C. IL-27 Suppresses the Production of IL-22 in Human CD4(+) T Cells by Inducing the Expression of SOCS1. Immunol Lett (2013) 152(2):96–103. doi: 10.1016/j.imlet.2013.05.001
38. Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The Inducible Costimulator (ICOS) is Critical for the Development of Human T(H)17 Cells. Sci Transl Med (2010) 2(55):55ra78. doi: 10.1126/scitranslmed.3000448
39. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification of the Functional Interleukin-22 (IL-22) Receptor Complex: The IL-10R2 Chain (IL-10Rbeta ) is a Common Chain of Both the IL-10 and IL-22 (IL-10-Related T Cell-Derived Inducible Factor, IL-TIF) Receptor Complexes. J Biol Chem (2001) 276(4):2725–32. doi: 10.1074/jbc.M007837200
40. Jones BC, Logsdon NJ, Walter MR. Structure of IL-22 Bound to its High-Affinity IL-22R1 Chain. Structure (2008) 16(9):1333–44. doi: 10.1016/j.str.2008.06.005
41. Wang X, Cai J, Lin B, Ma M, Tao Y, Zhou Y, et al. GPR34-Mediated Sensing of Lysophosphatidylserine Released by Apoptotic Neutrophils Activates Type 3 Innate Lymphoid Cells to Mediate Tissue Repair. Immunity (2021) 54(6):1123–36 e8. doi: 10.1016/j.immuni.2021.05.007
42. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and Functions of the IL-10 Family of Cytokines in Inflammation and Disease. Annu Rev Immunol (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
43. Wolk K, Witte E, Hoffmann U, Doecke WD, Endesfelder S, Asadullah K, et al. IL-22 Induces Lipopolysaccharide-Binding Protein in Hepatocytes: A Potential Systemic Role of IL-22 in Crohn's Disease. J Immunol (2007) 178(9):5973–81. doi: 10.4049/jimmunol.178.9.5973
44. Martin JC, Beriou G, Heslan M, Bossard C, Jarry A, Abidi A, et al. IL-22BP is Produced by Eosinophils in Human Gut and Blocks IL-22 Protective Actions During Colitis. Mucosal Immunol (2016) 9(2):539–49. doi: 10.1038/mi.2015.83
45. Dumoutier L, Lejeune D, Colau D, Renauld JC. Cloning and Characterization of IL-22 Binding Protein, a Natural Antagonist of IL-10-Related T Cell-Derived Inducible Factor/IL-22. J Immunol (2001) 166(12):7090–5. doi: 10.4049/jimmunol.166.12.7090
46. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification, Cloning, and Characterization of a Novel Soluble Receptor That Binds IL-22 and Neutralizes its Activity. J Immunol (2001) 166(12):7096–103. doi: 10.4049/jimmunol.166.12.7096
47. Chen P, Schnabl B. Host-Microbiome Interactions in Alcoholic Liver Disease. Gut Liver (2014) 8(3):237–41. doi: 10.5009/gnl.2014.8.3.237
48. Perusina Lanfranca M, Lin Y, Fang J, Zou W, Frankel T. Biological and Pathological Activities of Interleukin-22. J Mol Med (Berl) (2016) 94(5):523–34. doi: 10.1007/s00109-016-1391-6
49. Mitra A, Raychaudhuri SK, Raychaudhuri SP. IL-22 Induced Cell Proliferation is Regulated by PI3K/Akt/mTOR Signaling Cascade. Cytokine (2012) 60(1):38–42. doi: 10.1016/j.cyto.2012.06.316
50. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) Activates the JAK/STAT, ERK, JNK, and P38 MAP Kinase Pathways in a Rat Hepatoma Cell Line. Pathways That are Shared With and Distinct From IL-10. J Biol Chem (2002) 277(37):33676–82. doi: 10.1074/jbc.M204204200
51. Shen Y, Chen W, Han L, Bian Q, Fan J, Cao Z, et al. VEGF-B Antibody and Interleukin-22 Fusion Protein Ameliorates Diabetic Nephropathy Through Inhibiting Lipid Accumulation and Inflammatory Responses. Acta Pharm Sin B (2021) 11(1):127–42. doi: 10.1016/j.apsb.2020.07.002
52. Wang S, Li Y, Fan J, Zhang X, Luan J, Bian Q, et al. Interleukin-22 Ameliorated Renal Injury and Fibrosis in Diabetic Nephropathy Through Inhibition of NLRP3 Inflammasome Activation. Cell Death Dis (2017) 8(7):e2937. doi: 10.1038/cddis.2017.292
53. Xu MJ, Feng D, Wang H, Guan Y, Yan X, Gao B. IL-22 Ameliorates Renal Ischemia-Reperfusion Injury by Targeting Proximal Tubule Epithelium. J Am Soc Nephrol (2014) 25(5):967–77. doi: 10.1681/ASN.2013060611
54. Shen Y, Jin X, Chen W, Gao C, Bian Q, Fan J, et al. Interleukin-22 Ameliorated Acetaminophen-Induced Kidney Injury by Inhibiting Mitochondrial Dysfunction and Inflammatory Responses. Appl Microbiol Biotechnol (2020) 104(13):5889–98. doi: 10.1007/s00253-020-10638-4
55. Weidenbusch M, Song S, Iwakura T, Shi C, Rodler S, Kobold S, et al. IL-22 Sustains Epithelial Integrity in Progressive Kidney Remodeling and Fibrosis. Physiol Rep (2018) 6(16):e13817. doi: 10.14814/phy2.13817
56. Hasegawa M, Yada S, Liu MZ, Kamada N, Munoz-Planillo R, Do N, et al. Interleukin-22 Regulates the Complement System to Promote Resistance Against Pathobionts After Pathogen-Induced Intestinal Damage. Immunity (2014) 41(4):620–32. doi: 10.1016/j.immuni.2014.09.010
57. Liao X, Pirapakaran T, Luo XM. Chemokines and Chemokine Receptors in the Development of Lupus Nephritis. Mediators Inflammation (2016) 2016:6012715. doi: 10.1155/2016/6012715
58. Steinmetz OM, Turner JE, Paust HJ, Lindner M, Peters A, Heiss K, et al. CXCR3 Mediates Renal Th1 and Th17 Immune Response in Murine Lupus Nephritis. J Immunol (2009) 183(7):4693–704. doi: 10.4049/jimmunol.0802626
59. Koury MJ, Haase VH. Anaemia in Kidney Disease: Harnessing Hypoxia Responses for Therapy. Nat Rev Nephrol (2015) 11(7):394–410. doi: 10.1038/nrneph.2015.82
60. Raundhal M, Ghosh S, Myers SA, Cuoco MS, Singer M, Carr SA, et al. Blockade of IL-22 Signaling Reverses Erythroid Dysfunction in Stress-Induced Anemias. Nat Immunol (2021) 22(4):520–9. doi: 10.1038/s41590-021-00895-4
61. Gower M, Foster G, Tikhonova AN. There Won't be Blood. Nat Immunol (2021) 22(4):396–7. doi: 10.1038/s41590-021-00893-6
62. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the Immune System. Nat Rev Immunol (2017) 17(4):219–32. doi: 10.1038/nri.2017.7
63. Sabihi M, Bottcher M, Pelczar P, Huber S. Microbiota-Dependent Effects of IL-22. Cells (2020) 9(10):2205. doi: 10.3390/cells9102205
64. Yeaman MR, Filler SG, Chaili S, Barr K, Wang H, Kupferwasser D, et al. Mechanisms of NDV-3 Vaccine Efficacy in MRSA Skin Versus Invasive Infection. Proc Natl Acad Sci USA (2014) 111(51):E5555–63. doi: 10.1073/pnas.1415610111
65. Singer AJ, Talan DA. Management of Skin Abscesses in the Era of Methicillin-Resistant Staphylococcus Aureus. N Engl J Med (2014) 370(11):1039–47. doi: 10.1056/NEJMra1212788
66. Sugaya M. The Role of Th17-Related Cytokines in Atopic Dermatitis. Int J Mol Sci (2020) 21(4):1314. doi: 10.3390/ijms21041314
67. Tewary P, de la Rosa G, Sharma N, Rodriguez LG, Tarasov SG, Howard OM, et al. Beta-Defensin 2 and 3 Promote the Uptake of Self or CpG DNA, Enhance IFN-Alpha Production by Human Plasmacytoid Dendritic Cells, and Promote Inflammation. J Immunol (2013) 191(2):865–74. doi: 10.4049/jimmunol.1201648
68. Yount NY, Yeaman MR. Emerging Themes and Therapeutic Prospects for Anti-Infective Peptides. Annu Rev Pharmacol Toxicol (2012) 52:337–60. doi: 10.1146/annurev-pharmtox-010611-134535
69. De Luca A, Zelante T, D'Angelo C, Zagarella S, Fallarino F, Spreca A, et al. IL-22 Defines a Novel Immune Pathway of Antifungal Resistance. Mucosal Immunol (2010) 3(4):361–73. doi: 10.1038/mi.2010.22
70. Sonnenberg GF, Fouser LA, Artis D. Border Patrol: Regulation of Immunity, Inflammation and Tissue Homeostasis at Barrier Surfaces by IL-22. Nat Immunol (2011) 12(5):383–90. doi: 10.1038/ni.2025
71. Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological Versus Protective Functions of IL-22 in Airway Inflammation are Regulated by IL-17a. J Exp Med (2010) 207(6):1293–305. doi: 10.1084/jem.20092054
72. Guilloteau K, Paris I, Pedretti N, Boniface K, Juchaux F, Huguier V, et al. Skin Inflammation Induced by the Synergistic Action of IL-17a, IL-22, Oncostatin M, IL-1{Alpha}, and TNF-{Alpha} Recapitulates Some Features of Psoriasis. J Immunol (2010) 184(9):5263–70. doi: 10.4049/jimmunol.0902464
73. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 Inhibits Epidermal Differentiation and Induces Proinflammatory Gene Expression and Migration of Human Keratinocytes. J Immunol (2005) 174(6):3695–702. doi: 10.4049/jimmunol.174.6.3695
74. Rutz S, Wang X, Ouyang W. The IL-20 Subfamily of Cytokines–From Host Defence to Tissue Homeostasis. Nat Rev Immunol (2014) 14(12):783–95. doi: 10.1038/nri3766
75. Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. IL-23 and IL-17A, But Not IL-12 and IL-22, are Required for Optimal Skin Host Defense Against Candida Albicans. J Immunol (2010) 185(9):5453–62. doi: 10.4049/jimmunol.1001153
76. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan Catabolites From Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity (2013) 39(2):372–85. doi: 10.1016/j.immuni.2013.08.003
77. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, et al. Histones From Dying Renal Cells Aggravate Kidney Injury via TLR2 and TLR4. J Am Soc Nephrol (2012) 23(8):1375–88. doi: 10.1681/ASN.2011111077
78. Rabadi MM, Kuo MC, Ghaly T, Rabadi SM, Weber M, Goligorsky MS, et al. Interaction Between Uric Acid and HMGB1 Translocation and Release From Endothelial Cells. Am J Physiol Renal Physiol (2012) 302(6):F730–41. doi: 10.1152/ajprenal.00520.2011
79. Darisipudi MN, Thomasova D, Mulay SR, Brech D, Noessner E, Liapis H, et al. Uromodulin Triggers IL-1beta-Dependent Innate Immunity via the NLRP3 Inflammasome. J Am Soc Nephrol (2012) 23(11):1783–9. doi: 10.1681/ASN.2012040338
80. Anders HJ, Muruve DA. The Inflammasomes in Kidney Disease. J Am Soc Nephrol (2011) 22(6):1007–18. doi: 10.1681/ASN.2010080798
81. Wolk K, Kunz S, Asadullah K, Sabat R. Cutting Edge: Immune Cells as Sources and Targets of the IL-10 Family Members? J Immunol (2002) 168(11):5397–402. doi: 10.4049/jimmunol.168.11.5397
82. Bonventre JV, Yang L. Cellular Pathophysiology of Ischemic Acute Kidney Injury. J Clin Invest (2011) 121(11):4210–21. doi: 10.1172/JCI45161
83. Havasi A, Borkan SC. Apoptosis and Acute Kidney Injury. Kidney Int (2011) 80(1):29–40. doi: 10.1038/ki.2011.120
84. Ghosh S, Sarkar A, Bhattacharyya S, Sil PC. Silymarin Protects Mouse Liver and Kidney From Thioacetamide Induced Toxicity by Scavenging Reactive Oxygen Species and Activating PI3K-Akt Pathway. Front Pharmacol (2016) 7:481. doi: 10.3389/fphar.2016.00481
85. Mohamed AF, Safar MM, Zaki HF, Sayed HM. Telluric Acid Ameliorates Endotoxemic Kidney Injury in Mice: Involvement of TLR4, Nrf2, and PI3K/Akt Signaling Pathways. Inflammation (2017) 40(5):1742–52. doi: 10.1007/s10753-017-0617-2
86. Tanaka S, Tanaka T, Nangaku M. Hypoxia as a Key Player in the AKI-To-CKD Transition. Am J Physiol Renal Physiol (2014) 307(11):F1187–95. doi: 10.1152/ajprenal.00425.2014
87. Plotnikov EY, Kazachenko AV, Vyssokikh MY, Vasileva AK, Tcvirkun DV, Isaev NK, et al. The Role of Mitochondria in Oxidative and Nitrosative Stress During Ischemia/Reperfusion in the Rat Kidney. Kidney Int (2007) 72(12):1493–502. doi: 10.1038/sj.ki.5002568
88. Sahu BD, Kuncha M, Sindhura GJ, Sistla R. Hesperidin Attenuates Cisplatin-Induced Acute Renal Injury by Decreasing Oxidative Stress, Inflammation and DNA Damage. Phytomedicine (2013) 20(5):453–60. doi: 10.1016/j.phymed.2012.12.001
89. Chen W, Shen Y, Fan J, Zeng X, Zhang X, Luan J, et al. IL-22-Mediated Renal Metabolic Reprogramming via PFKFB3 to Treat Kidney Injury. Clin Transl Med (2021) 11(2):e324. doi: 10.1002/ctm2.324
90. Zhao X, Wang LY, Li JM, Peng LM, Tang CY, Zha XJ, et al. Redox-Mediated Artificial Non-Enzymatic Antioxidant MXene Nanoplatforms for Acute Kidney Injury Alleviation. Adv Sci (Weinh) (2021) 8(18):e2101498. doi: 10.1002/advs.202101498
91. Blair P, Flaumenhaft R. Platelet Alpha-Granules: Basic Biology and Clinical Correlates. Blood Rev (2009) 23(4):177–89. doi: 10.1016/j.blre.2009.04.001
92. Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the Proteins Released From Activated Platelets Leads to Localization of Novel Platelet Proteins in Human Atherosclerotic Lesions. Blood (2004) 103(6):2096–104. doi: 10.1182/blood-2003-08-2804
93. De Pascale MR, Sommese L, Casamassimi A, Napoli C. Platelet Derivatives in Regenerative Medicine: An Update. Transfus Med Rev (2015) 29(1):52–61. doi: 10.1016/j.tmrv.2014.11.001
94. Chevalier RL, Thornhill BA, Forbes MS, Kiley SC. Mechanisms of Renal Injury and Progression of Renal Disease in Congenital Obstructive Nephropathy. Pediatr Nephrol (2010) 25(4):687–97. doi: 10.1007/s00467-009-1316-5
95. Xiao C, Xiao P, Li X, Li X, Li H, Chen Y, et al. Cordyceps Sinensis may Inhibit Th22 Cell Chemotaxis to Improve Kidney Function in lgA Nephropathy. Am J Transl Res (2018) 10(3):857–65.
96. Zhong W, Jiang Y, Ma H, Wu J, Jiang Z, Zhao L. Elevated Levels of CCR6(+) T Helper 22 Cells Correlate With Skin and Renal Impairment in Systemic Lupus Erythematosus. Sci Rep (2017) 7(1):12962. doi: 10.1038/s41598-017-13344-w
97. Ren F, Zhang M, Zhang C, Sang H. Psoriasis-Like Inflammation Induced Renal Dysfunction Through the TLR/NF-kappaB Signal Pathway. BioMed Res Int (2020) 2020:3535264. doi: 10.1155/2020/3535264
98. Kassianos AJ, Wang X, Sampangi S, Afrin S, Wilkinson R, Healy H. Fractalkine-CX3CR1-Dependent Recruitment and Retention of Human CD1c+ Myeloid Dendritic Cells by In Vitro-Activated Proximal Tubular Epithelial Cells. Kidney Int (2015) 87(6):1153–63. doi: 10.1038/ki.2014.407
99. Pattrapornpisut P, Avila-Casado C, Reich HN. IgA Nephropathy: Core Curriculum 2021. Am J Kidney Dis (2021) 78(3):429–41. doi: 10.1053/j.ajkd.2021.01.024
100. Bao L, Cunningham PN, Quigg RJ. Complement in Lupus Nephritis: New Perspectives. Kidney Dis (Basel) (2015) 1(2):91–9. doi: 10.1159/000431278
101. Thurman JM. Complement and the Kidney: An Overview. Adv Chronic Kidney Dis (2020) 27(2):86–94. doi: 10.1053/j.ackd.2019.10.003
102. Rizk DV, Maillard N, Julian BA, Knoppova B, Green TJ, Novak J, et al. The Emerging Role of Complement Proteins as a Target for Therapy of IgA Nephropathy. Front Immunol (2019) 10:504. doi: 10.3389/fimmu.2019.00504
103. Dalmasso AP. Complement in the Pathophysiology and Diagnosis of Human Diseases. Crit Rev Clin Lab Sci (1986) 24(2):123–83. doi: 10.3109/10408368609110272
104. Yamamoto H, Kemper C. Complement and IL-22: Partnering Up for Border Patrol. Immunity (2014) 41(4):511–3. doi: 10.1016/j.immuni.2014.10.002
105. Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswoode N, Selvakumar TA, et al. Hepcidin Regulation by Innate Immune and Infectious Stimuli. Blood (2011) 118(15):4129–39. doi: 10.1182/blood-2011-04-351957
106. Smith CL, Arvedson TL, Cooke KS, Dickmann LJ, Forte C, Li H, et al. IL-22 Regulates Iron Availability In Vivo Through the Induction of Hepcidin. J Immunol (2013) 191(4):1845–55. doi: 10.4049/jimmunol.1202716
107. Wallace DF, Subramaniam VN. Analysis of IL-22 Contribution to Hepcidin Induction and Hypoferremia During the Response to LPS In Vivo. Int Immunol (2015) 27(6):281–7. doi: 10.1093/intimm/dxu144
108. Krebs CF, Panzer U. Plasticity and Heterogeneity of Th17 in Immune-Mediated Kidney Diseases. J Autoimmun (2018) 87:61–8. doi: 10.1016/j.jaut.2017.12.005
109. Paust HJ, Turner JE, Steinmetz OM, Peters A, Heymann F, Holscher C, et al. The IL-23/Th17 Axis Contributes to Renal Injury in Experimental Glomerulonephritis. J Am Soc Nephrol (2009) 20(5):969–79. doi: 10.1681/ASN.2008050556
110. Paust HJ, Turner JE, Riedel JH, Disteldorf E, Peters A, Schmidt T, et al. Chemokines Play a Critical Role in the Cross-Regulation of Th1 and Th17 Immune Responses in Murine Crescentic Glomerulonephritis. Kidney Int (2012) 82(1):72–83. doi: 10.1038/ki.2012.101
111. Scrivo R, Peruzzi G, Gattamelata A, Gross CC, Carletti R, Di Gioia C, et al. Multicolor Flow Cytometric Analysis of TLR2 and TLR9 Expression and Function in NK Cells From Patients With ANCA-Associated Vasculitis. Cytometry B Clin Cytom (2018) 94(3):412–22. doi: 10.1002/cyto.b.21586
112. Deleanu D, Nedelea I. Biological Therapies for Atopic Dermatitis: An Update. Exp Ther Med (2019) 17(2):1061–7. doi: 10.3892/etm.2018.6989
113. Justa S, Zhou X, Sarkar S. Endogenous IL-22 Plays a Dual Role in Arthritis: Regulation of Established Arthritis via IFN-Gamma Responses. PloS One (2014) 9(3):e93279. doi: 10.1371/journal.pone.0093279
114. Ahn D, Prince A. Participation of the IL-10rb Related Cytokines, IL-22 and IFN-Lambda in Defense of the Airway Mucosal Barrier. Front Cell Infect Microbiol (2020) 10:300. doi: 10.3389/fcimb.2020.00300
115. Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical Importance of IL-22 Cascade in IBD. J Gastroenterol (2018) 53(4):465–74. doi: 10.1007/s00535-017-1401-7
116. Katoh N. Emerging Treatments for Atopic Dermatitis. J Dermatol (2021) 48(2):152–7. doi: 10.1111/1346-8138.15504
117. Ogawa E, Sato Y, Minagawa A, Okuyama R. Pathogenesis of Psoriasis and Development of Treatment. J Dermatol (2018) 45(3):264–72. doi: 10.1111/1346-8138.14139
118. Sun D, Lin Y, Hong J, Chen H, Nagarsheth N, Peng D, et al. Th22 Cells Control Colon Tumorigenesis Through STAT3 and Polycomb Repression Complex 2 Signaling. Oncoimmunology (2016) 5(8):e1082704. doi: 10.1080/2162402X.2015.1082704
119. Le Rouzic O, Pichavant M, Frealle E, Guillon A, Si-Tahar M, Gosset P. Th17 Cytokines: Novel Potential Therapeutic Targets for COPD Pathogenesis and Exacerbations. Eur Respir J (2017) 50(4):1602434. doi: 10.1183/13993003.02434-2016
120. Lanthier N, Starkel P. Treatment of Severe Alcoholic Hepatitis: Past, Present and Future. Eur J Clin Invest (2017) 47(7):531–9. doi: 10.1111/eci.12767
Keywords: interleukin-22, basic features, biological effects, the protective role, the pathological effect, kidney diseases
Citation: Ma Q, Luan J, Bai Y, Xu C, Liu F, Chen B, Ju D and Xu H (2022) Interleukin-22 in Renal Protection and Its Pathological Role in Kidney Diseases. Front. Immunol. 13:851818. doi: 10.3389/fimmu.2022.851818
Received: 10 January 2022; Accepted: 07 March 2022;
Published: 31 March 2022.
Edited by:
Hai-Feng Pan, Anhui Medical University, ChinaReviewed by:
Kim Maree O’Sullivan, Monash University, AustraliaCopyright © 2022 Ma, Luan, Bai, Xu, Liu, Chen, Ju and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dianwen Ju, ZGlhbndlbmp1QGZ1ZGFuLmVkdS5jbg==; Hong Xu, aHh1QHNobXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.