
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 13 April 2022
Sec. Alloimmunity and Transplantation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.845301
 Gesa Tiller1
Gesa Tiller1 Rosa G. M. Lammerts1,2
Rosa G. M. Lammerts1,2 Jessy J. Karijosemito1
Jessy J. Karijosemito1 Firas F. Alkaff1,3
Firas F. Alkaff1,3 Arjan Diepstra4
Arjan Diepstra4 Robert A. Pol5Anita H. Meter-Arkema1Marc. A. Seelen1Marius C. van den Heuvel4
Robert A. Pol5Anita H. Meter-Arkema1Marc. A. Seelen1Marius C. van den Heuvel4 Bouke G. Hepkema2
Bouke G. Hepkema2 Mohamed R. Daha6
Mohamed R. Daha6 Jacob van den Born1
Jacob van den Born1 Stefan P. Berger1* on behalf of the COMBAT Consortium
Stefan P. Berger1* on behalf of the COMBAT ConsortiumBackground: The role of the complement system in antibody-mediated rejection (ABMR) is insufficiently understood. We aimed to investigate the role of local and systemic complement activation in active (aABMR). We quantified complement activation markers, C3, C3d, and C5b-9 in plasma of aABMR, and acute T-cell mediated rejection (aTCMR), and non-rejection kidney transplant recipients. Intra-renal complement markers were analyzed as C4d, C3d, C5b-9, and CD59 deposition. We examined in vitro complement activation and CD59 expression on renal endothelial cells upon incubation with human leukocyte antigen antibodies.
Methods: We included 50 kidney transplant recipients, who we histopathologically classified as aABMR (n=17), aTCMR (n=18), and non-rejection patients (n=15).
Results: Complement activation in plasma did not differ across groups. C3d and C4d deposition were discriminative for aABMR diagnosis. Particularly, C3d deposition was stronger in glomerular (P<0,01), and peritubular capillaries (P<0,05) comparing aABMR to aTCMR rejection and non-rejection biopsies. In contrast to C3d, C5b-9 was only mildly expressed across all groups. For C5b-9, no significant difference between aABMR and non-rejection biopsies regarding peritubular and glomerular C5b-9 deposition was evident. We replicated these findings in vitro using renal endothelial cells and found complement pathway activation with C4d and C3d, but without terminal C5b-9 deposition. Complement regulator CD59 was variably present in biopsies and constitutively expressed on renal endothelial cells in vitro.
Conclusion: Our results indicate that terminal complement might only play a minor role in late aABMR, possibly indicating the need to re-evaluate the applicability of terminal complement inhibitors as treatment for aABMR.
Antibody-mediated rejection (ABMR) is a major reason for impaired function and reduced longevity of the kidney allograft, accounting for about 60% of chronic graft failure in kidney transplant recipients (KTR) (1–5). Understanding the underlying pathoimmunological processes associated with ABMR is crucial for accurate diagnosis and implementation of effective treatment strategies. Former Banff classifications identified ABMR based on the presence of donor-specific HLA-antibodies (DSA), C4d complement deposition, microvascular inflammation, and/or transplant glomerulopathy in renal specimens (6).
From a traditional perspective, ABMR pathomechanism was thought to be based on the activation of the classical complement pathway, initiated by HLA-Antibodies (HLA-Abs), subsequent binding of C1q, C3-convertase formation, and final cellular cytotoxic damage through C5b-9 (2, 7–11). C4d, a split product of the classical and lectin pathway, is relatively stable and binds covalently to renal capillaries. Therefore, C4d was introduced and is still utilized as a diagnostic marker for complement pathway activation (8, 12).
However, recent Banff classifications recognize ABMR subtypes, including C4d-negative ABMR and C4d-positive ABMR without DSA, thereby questioning the universal pathogenic role of the classical complement pathway in ABMR (4, 13–15). These Banff updates indicate that ABMR encompasses a more heterogeneous group, which is possibly linked to diverse pathomechanisms. Moreover, studies evaluating the efficacy of terminal complement inhibitors as a potential treatment for ABMR, showed that terminal complement inhibition was insufficient in preventing ABMR occurrence in the long term (13, 16, 17).
A possible reason for the lack of C4d deposition in the biopsies of some ABMR patients and the insufficient efficacy of complement inhibitors might be that the classical complement pathway does not serve as a universal pathogenetic model for ABMR. Coherently, a recent study by Mező et al. (2019) showed that complement factors in plasma failed to predict ABMR outcome (18).
A more nuanced view on the role of the complement system in ABMR is necessary to precisely define the role of the complement system in its development. However, evaluation of the full complement orchestra with analysis of the consecutive steps of the classical complement pathway activation in ABMR is still missing. We aimed to analyze the role of the classical complement pathway in ABMR, specifically active ABMR, by quantification of representative complement activation products, both in plasma, in kidney transplant biopsies, and upon HLA-Ab binding to renal endothelial cells in vitro.
The study population encompassed a total of 227 KTR of whom blood samples for direct complement measurement were taken and who had either an indication or protocol kidney biopsy at the University Medical Center Groningen (UMCG), the Netherlands, between 2010 and 2020. KTR were recruited with informed consent in the Transplantlines biobank cohort study, following the Declarations of Helsinki and Istanbul, with NCT02811835 as ClinicalTrials.gov identifier. The research was approved by the UMCG institutional review board (METc 2008/186) (19).
KTRs were retrospectively included and classified as aABMR, acute T-cell mediated rejection (aTCMR), and non-rejection (NR) patients. All NR patients had a protocol biopsy taken without signs of rejection. Inclusion and exclusion criteria are depicted in Figure 1. 50 KTR were included, of whom 17 were classified as aABMR, 18 as aTCMR patients, and 15 as NR patients. As included patients were diagnosed based on different versions of the Banff guideline depending on the year of consultation, standardized re-evaluation of the diagnosis by the most recent Banff’19 guideline was performed. Re-evaluation according to Banff’19 classification considered presence of HLA-Abs, signs of microvascular inflammation in the biopsy, and C4d deposition (15). In case antibody screening in serum revealed no HLA-Abs in pre-categorized cases of aABMR, we only included these HLA-Abs-negative patients as aABMR patients, if otherwise clear histopathological signs of microvascular inflammation were present. Coherently, C4d-negative patients were only included as aABMR patients, if histopathological microvascular inflammation was unambiguously identified and serum samples were positive for either HLA-Abs class I or II or both (15).
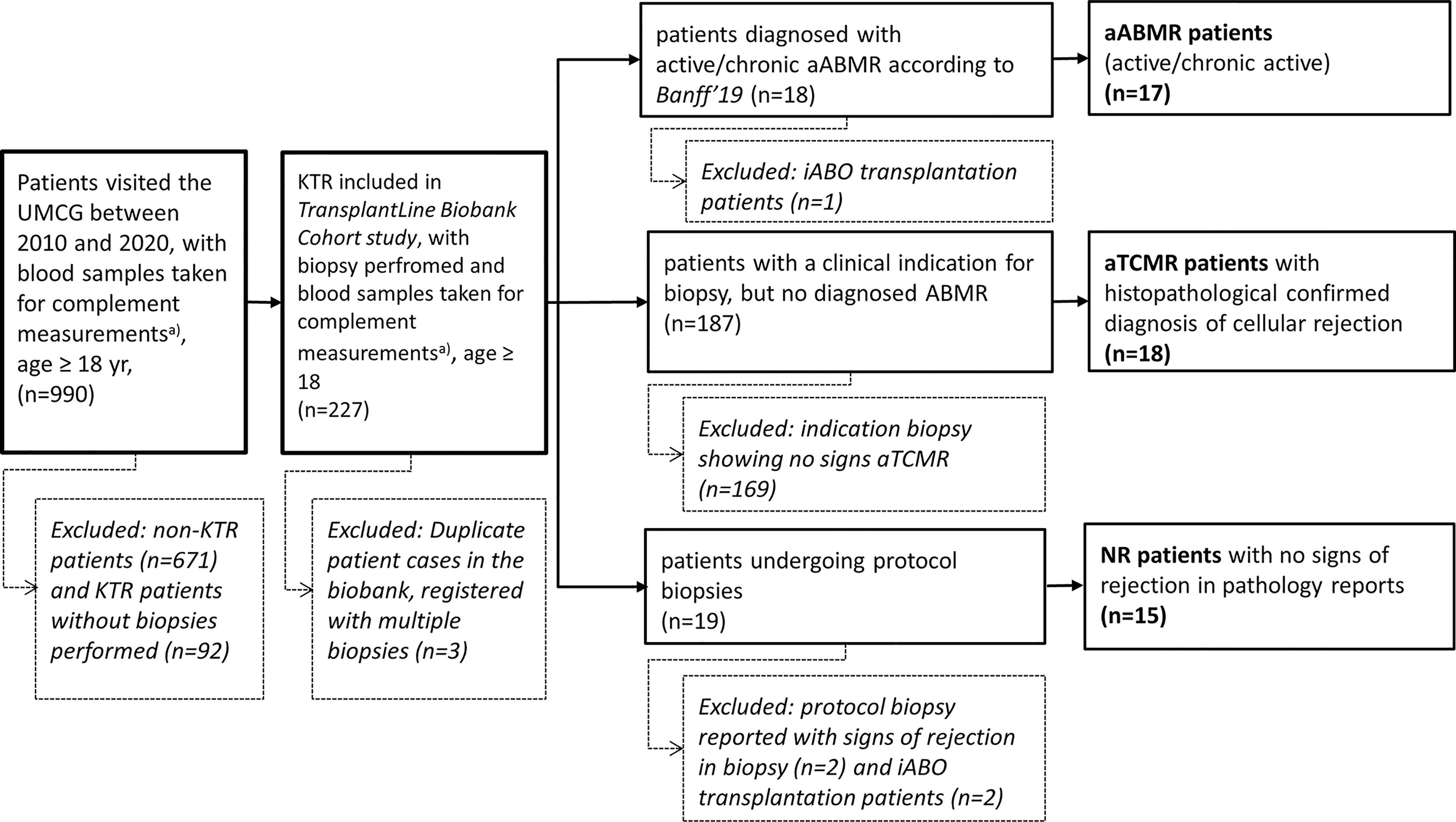
Figure 1 Patient Selection with Inclusion and Exclusion Criteria. Flow chart representing selection of patients based on exclusion criteria. Total amount of patients excluded and included are depicted as (n). Exclusion of patients is presented in box with dotted lines. KTR, Kidney Transplant Recipients; UMCG, University Medical Center; yr, years; non-KTR, patients without a renal transplant; aABMR, active antibody-mediated rejection; iABO, blood group incompatible; aTCMR, acute T-cell mediated rejection; NR, non-rejection. a) with blood samples directly placed on ice.
Plasma samples taken closest to the date of biopsy were immediately stored at minus 80°C and only thawn directly prior to complement measurements. Clinical data regarding patient demographics, underlying renal disease, transplantation, immunological status, and kidney function around the time of sampling were derived from the hospital electronic patient file (Epic Systems Cooperation, EPIC, Wisconsin, United States). Kidney function was assessed by estimating glomerular filtration rate (eGFR) by applying the Chronic Kidney Disease Epidemiology Collaboration equation (20). Protein excretion of ≥ 0.5 g per day was defined as proteinuria. Renal biopsy samples were taken based on clinical indication or in the context of protocol biopsies, fixed in formalin, embedded, and stored in paraffin.
The presence of HLA-Abs class I and II n serum was evaluated prior to anti-rejection therapy using the Life screen the Luxe (LsdL), in accordance with the manufacturer’s protocol (Immucor Transplant Diagnostics, Norcross, Georgia). Serum samples of aABMR and aTCMR patients, which were tested positive in LsdL class I and/or II were subsequently tested with Lifecodes Single Antigen Bead (LSA;Immucor Transplant Diagnostics) class I and/or II assay respectively. In light of the HLA-donor type, LSA test results were evaluated regarding donor-specificity and mean fluorescence intensity (MFI) by B.G.H. and R.G.L. (Department of Transplant Immunology, Groningen).
C3 quantification in patients’ plasma was performed using radial immunodiffusion assay (RID) measurement according to Mancini et al. (21). C3d and soluble C5b-9 were quantified by enzyme-linked immunosorbent assay (ELISA) (22, 23). C3d measurements were preceded by Polyethylene Glycol precipitation for fractionation of C3d (22). For accurate quantification, standard curves were derived from a serial dilution with a standard sample kindly provided by the University of Leiden for C3d; and by serial dilution of zymosan-activated normal human serum in PTB [PBS (Phosphate Buffered Saline) with 0.05% Tween and 1% Bovin Serum Albumin (BSA)] of defined concentration for C5b-9. The C3d/C3 ratio was determined by dividing the C3d values in ng/mL by the C3 concentration in ng/mL. Details on antibodies and conjugates used for quantification of respective complement factors are provided in Table 1.

Table 1 Antibodies for ELISA on plasma samples.
Paraffin-embedded renal specimens were stained with rabbit anti-human C4d (clone SP91, Ventana, Benchmark automated immunostainer) according to the manufacturer’s protocol, as part of the standardized pathological evaluation of biopsies. C3d, C5b-9, and CD59 staining were performed using in-house stainings. Details on C4d, C3d, C5b-9, and CD59 staining in renal biopsies are given in Table 2. Solutions were prepared with BSA (A9647, Sigma, St Louis, United States) and PBS (17-512Q, Lonza, Wijchen, The Netherlands) with specific dilutions provided in Table 2. Between every incubation step, slides were washed three times with PBS (17-512Q, Lonza, Wijchen, The Netherlands).
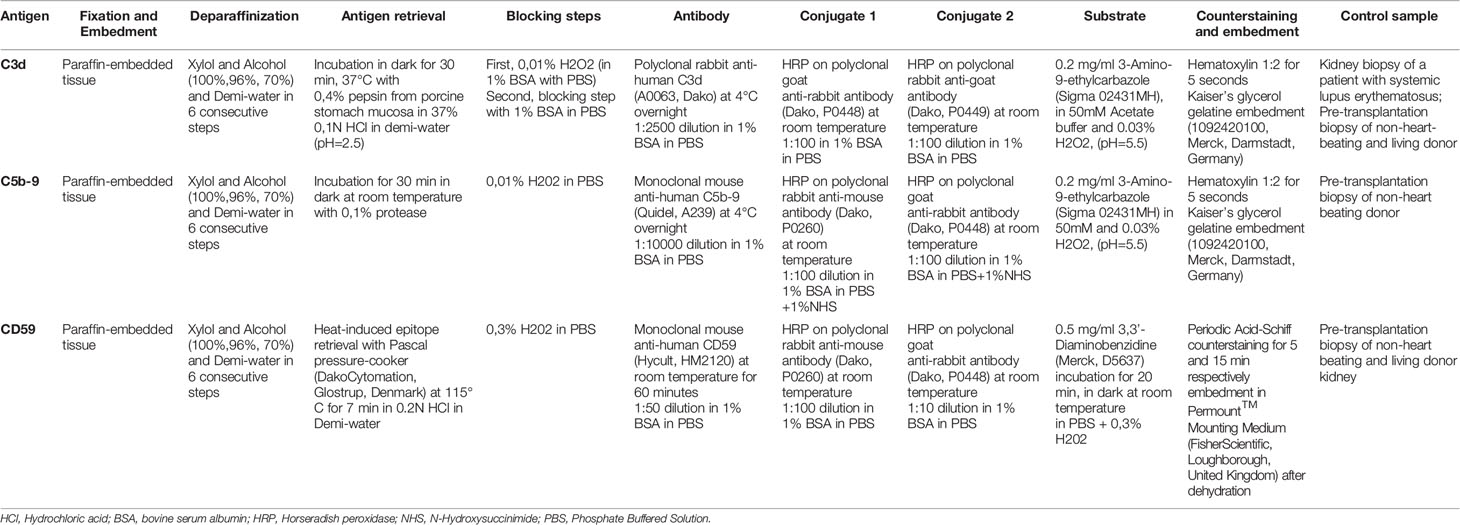
Table 2 Complement factor staining in biopsies from kidney transplant recipients.
A renal pathologist (M. vd H.) blindly scored the presence of peritubular capillary C4d according to the recent Banff`19 classification standard (15). For inter-group statistical analysis, semiquantitative scores of the pathologist were categorized as ‘C4d positive’ or ‘C4d negative’.
Slides stained for C3d and C5b-9 were digitalized by the Department of Pathology & Medical Biology of the UMCG, using a Hamamatsu slide scanner (Hamamatsu Photonics, Hamamatsu, Japan). Stained slides were blindly graded by M. vd H. in the Aperio ImageScope software (Leica Biosystems, Nussloch, Germany). Complement factors C3d and C5b-9 were scored with a semiquantitative scoring system previously described by Bobka et al. (2018) as a method of analyzing complement deposition in biopsies (24). Glomerular and peritubular semiquantitative scores ranged from 0 to 4, indicating lack of any deposit, weak (≤25%), moderate (≤50%), substantial (≤75%), and intense deposition.
To evaluate whether HLA-Abs can activate the complement cascade up to C5b-9 on renal endothelial cells, in vitro experiments using conditionally immortalized glomerular endothelial cells (CiGEnCs) were conducted (25). In addition, the presence of terminal complement regulator CD59 was analyzed. Details on handling and characterization of the CiGenCs cell-line are provided in the Supplementary Material 2A. Depending on the incubation condition of the complement activation assay, cells were incubated with either human monoclonal HLA-antibodies (A2/A28 IgG1, provided by Dr. F. Claas, Leiden University Medical Center, The Netherlands), or 25% human serum containing HLA-A2 antibodies directed against HLA antigens expressed on the CiGEnC cell membrane, or a pan-HLA-class I antibody (clone W6/32). 25% heat-inactivated blood group compatible serum was used as a control. The following washing steps are described in Supplementary Material 2B. For detection of activated C3, C4d, C5b-9, and CD59, cells were incubated with primary and secondary antibodies, as depicted in Table 3. To exclude apoptotic and necrotic cells, propidium iodide 1 μg/ml (Molecular Probes, Leiden, The Netherlands) was added just before measuring. Activated C3, C4d, and neoantigen C9 deposition on viable non-apoptotic cells were analyzed in a FACSCalibur™ (FACSCalibur, Becton Dickinson, New Jersey, USA). Results were derived from three independent experiments.

Table 3 Antibodies for flow cytometric analysis on CiGEnCs.
Statistical tests were conducted for the baseline characteristics and complement component measurements. Normality was examined by the Kolmogorov Smirnov test for each variable. Categorical parameters are represented as n (%). For continuous data, normally distributed variables are expressed as mean ± standard deviation (SD) and skewed data as median and interquartile range (IQR). All three groups were analyzed by Kruskal-Wallis for non-parametric variables and by one-way ANOVA for parametric variables. Analyses between subgroups were additionally performed for complement measures in serum and biopsy, comparing aABMR with aTCMR patients, and aABMR with NR patients. These two-group analyses were conducted by Mann-Whitney U test for non-parametric and by independent unpaired t-test for parametric data. Correlations between plasma and local complement factors were derived from Spearman’s correlation analyses. Computations were performed by SPSS (IBM SPSS Statistics, Chicago, United States) version 23, and GraphPad software 8.0 (GraphPad Software, San Diego, United States) was used for graphical visualization.
Among 50 KTR, 17 were identified as aABMR patients according to the Banff ‘19 classification, 18 as aTCMR, and 15 as NR (Figure 1) (2, 4). Mean age at biopsy was 46.8 ± 12.5 years in the ABMR group, 48.1 ± 16.8 years in the aTCMR group, and 51.1 ± 15.3 years in the NR group. No significant differences were found in demographic characteristics between all groups (Table 4). We included ABMR patients based on biopsy specimens with clear histopathological evidence for ongoing rejection, and only considered cases of active or chronic active ABMR, following the Banff’19 criteria (15). Thus, KTR with chronic, inactive ABMR were excluded (Figure 1 and Table 5). Within the aABMR group, 3 patients showed acute active ABMR, with the diagnosis made within the first 30 days after transplantation (Table 5). Based on inclusion criteria, all transplants were ABO-compatible.
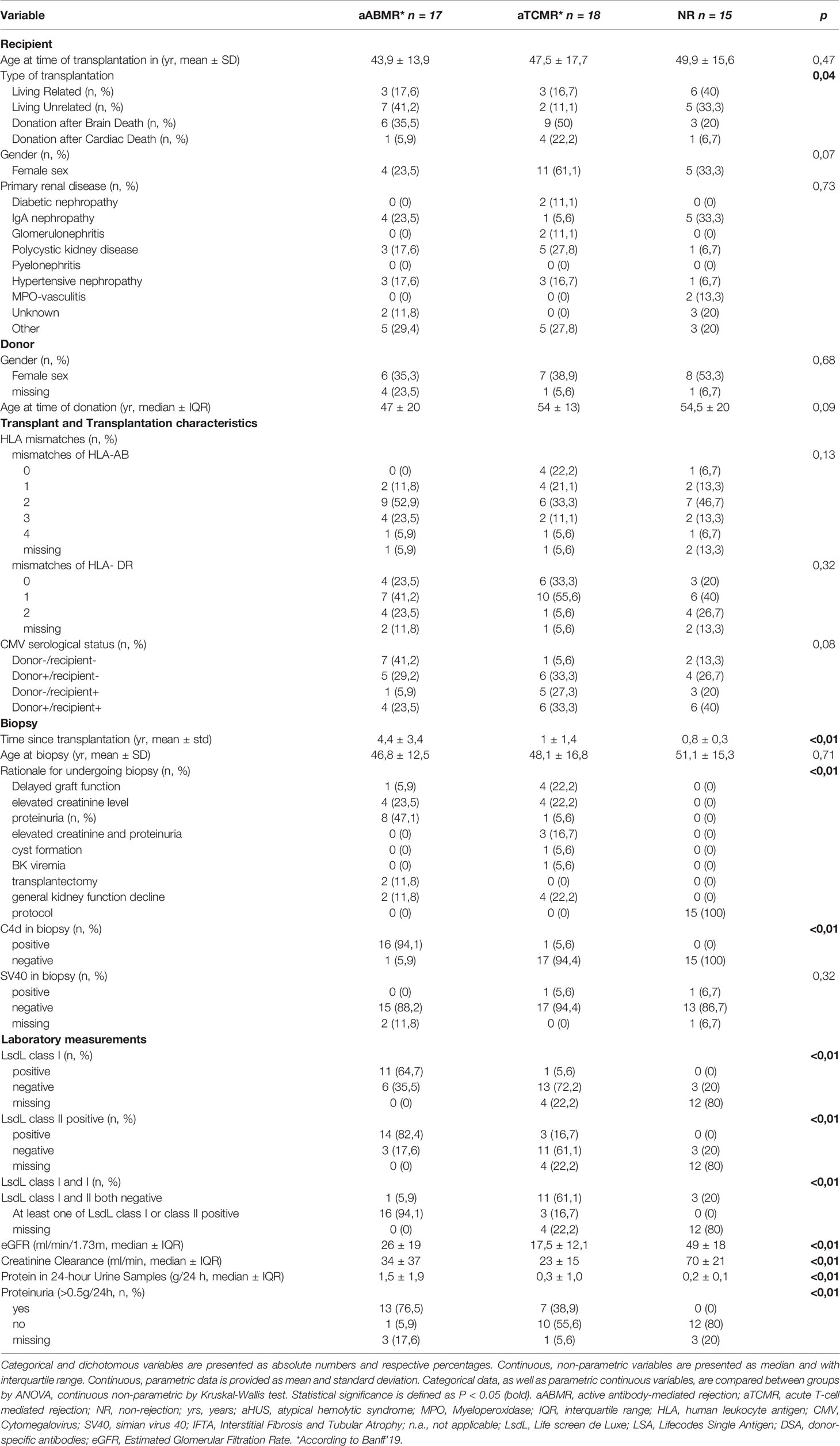
Table 4 Baseline characteristics of aABMR, aTCMR, and NR patients.
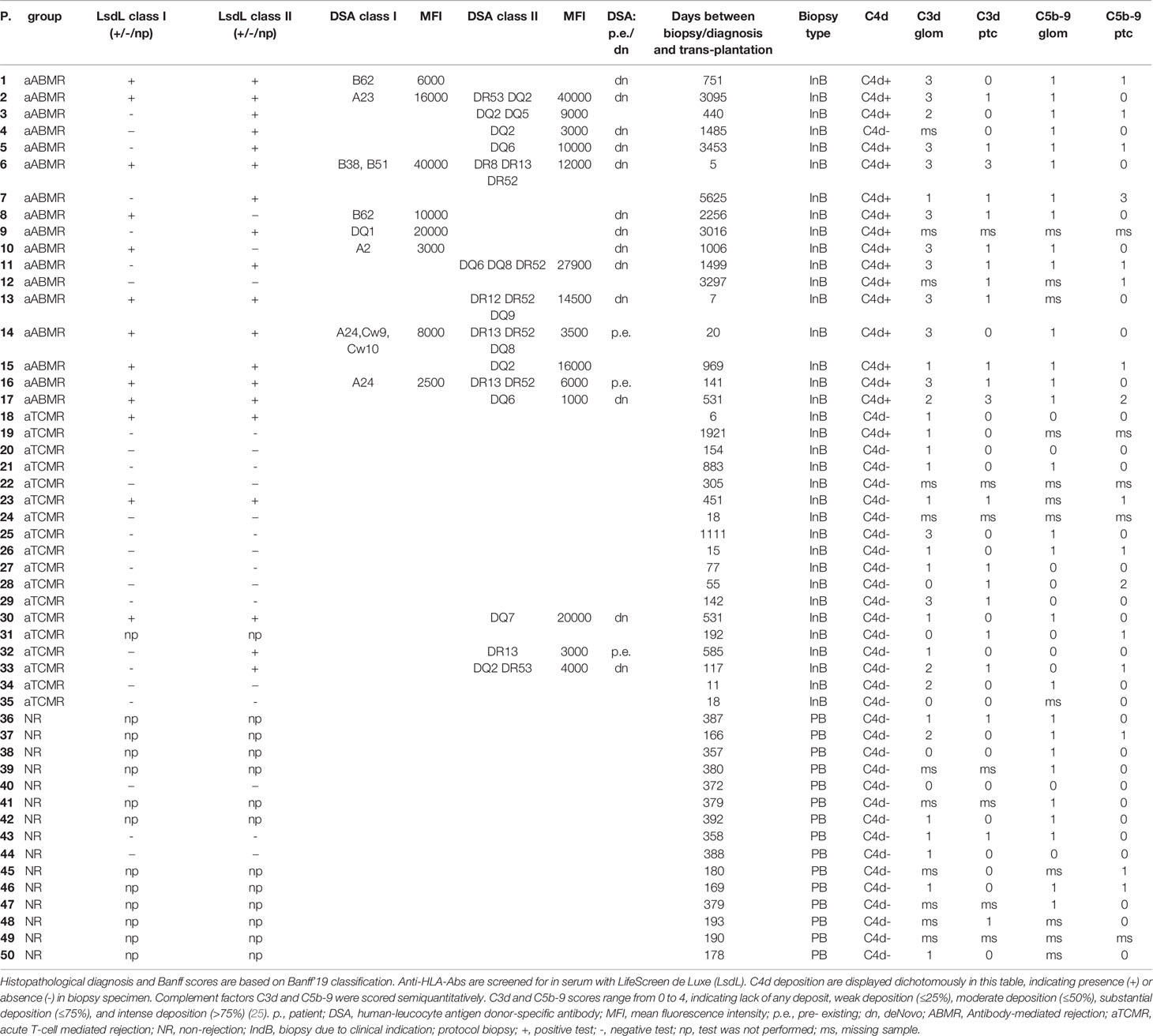
Table 5 Detailed immunological, histopathological and clinical information of individual patients included.
LsdL-tests were performed on serum samples, which were taken at the same timepoints as plasma samples for aforementioned complement measurements. LsdL-tests for HLA-Abs class I and class II in serum were more commonly positive in aABMR patients when compared to TCMR and NR patients (P<0,01 respectively) (Table 4). Specifically, LsdL tests showed that 52,9% of ABMR patients were positive for HLA-Abs class I and 82,4% positive for HLA class II (Table 4).
HLA-Abs were identified as donor-specific in 70,6% of tested aABMR patients and in 16,7% of tested aTCMR patients. Respective mean MFIs of HLA class I of 13187,5 ± 12421 for aABMR and of HLA class II of 129991 ± 11711. For aTCMR with DSA class II, mean MFI was 90000± 9539. DSA-negative aABMR patients showed otherwise clear histopathological evidence for aABMR diagnosis according to the Banff’19 classification (Tables 4, 5).
Quantification of plasma complement activation markers C3d, and C5b-9 did not show significant differences across groups. C3d/C3 ratio was evaluated as an indication for complement catabolism (26). Both C3 and C3d/C3 did not differ when comparing aABMR with aTCMR (P=0,7; P=0,4) and when comparing aABMR with NR (P=0,1; P=0,4) (Figure 2). Similarly, plasma C5b-9 did not differ when comparing ABMR with aTCMR (P=0,5) and with NR KTR (P=0,9). Plasma levels of C3d and C5b-9 complement factors, as well as C3d/C3 ratio, did not correlate with the intensity of complement deposition in renal biopsies of ABMR patients.
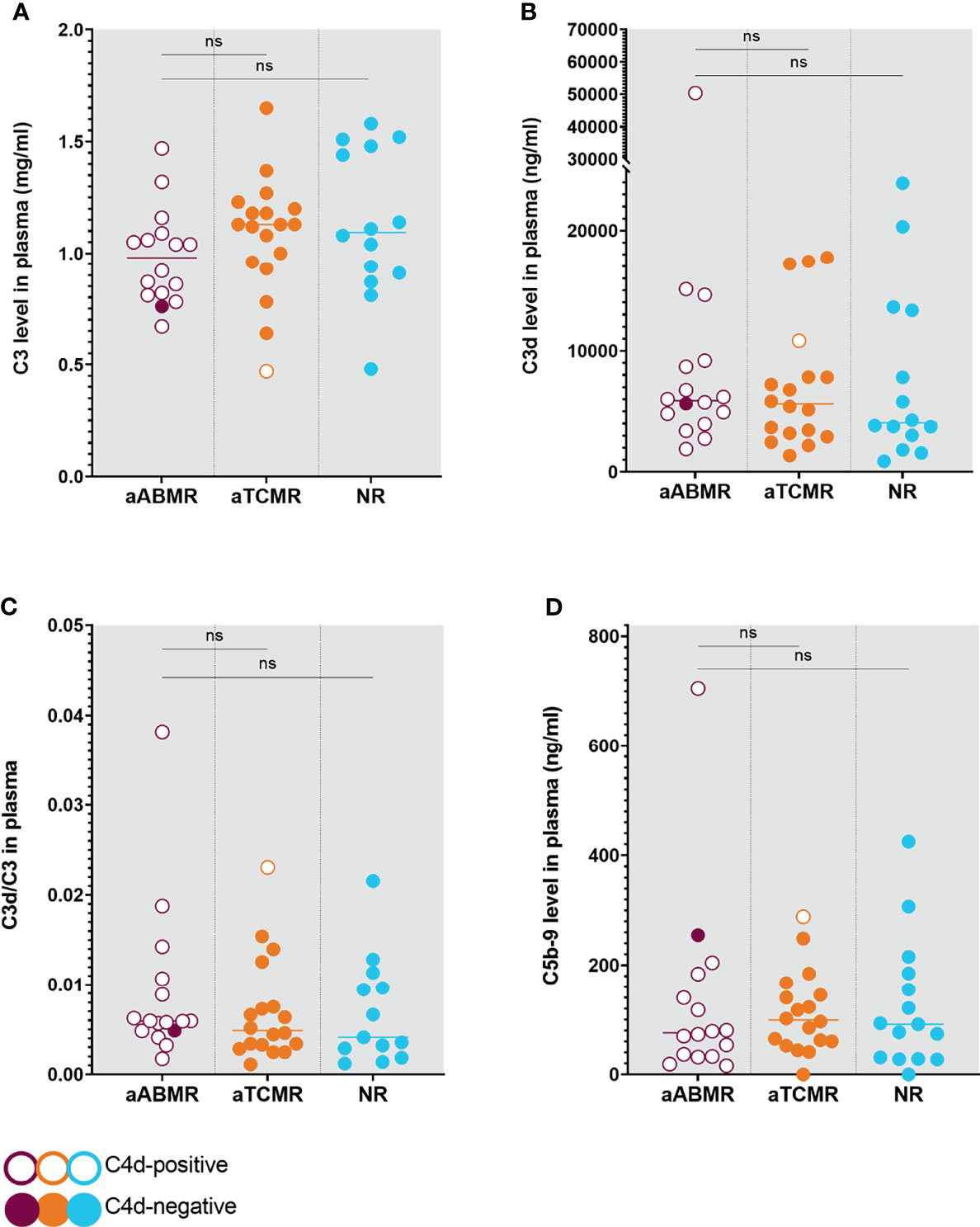
Figure 2 Complement Levels in Plasma of aABMR, aTCMR, and NR patients. Plasma levels were measured for C3 (A), C3d (B), and C5b-9 (D). Ratios were calculated from respective C3 and C3d levels (C), indicating systemic complement consumption. Circles represent individual patients with open circles indicating C4d-positive patients and filled circles symbolizing C4d-negative patients. P values depict results from Mann-Whitney U statistical analysis. Statistical significance is defined as P < 0.05. aABMR, active antibody-mediated Rejection; aTCMR, acute T-cell mediated rejection; NR, non-rejection; ns, not significant.
Biopsy samples were chosen with the closest temporal proximity to plasma samples, with a mean time difference of 3,9 ± 44 days. In a number of cases, complement deposition could not be determined due to missing biopsy material or due to missing glomeruli in available specimens (Tables 3, 5).
C4d, as the standard clinicopathological marker, was scored according to Banff’19 (2, 15). C4d deposition was clearly linked to ABMR diagnosis, with 16 of 17 C4d-positive patients identified as aABMR patients (Tables 3, 5).
The median score for glomerular C3d deposition in the aABMR group was 3, and in the aTCMR and NR group 1. Glomerular C3d deposition was significantly higher in the aABMR patients compared to aTCMR patients (P<0,01) and NR (P<0,01) (Figure 3A). The median score for C3d in the peritubular capillaries in the AMR group was 1, and in the aTCMR and NR group 0. Expression of C3d in peritubular capillaries was higher in aABMR compared to aTCMR (P=0,04) and NR patients (P=0,03) (Figure 3B). C3d expression appeared more intense in glomerular than in peritubular structures across all three groups (Figure 3).
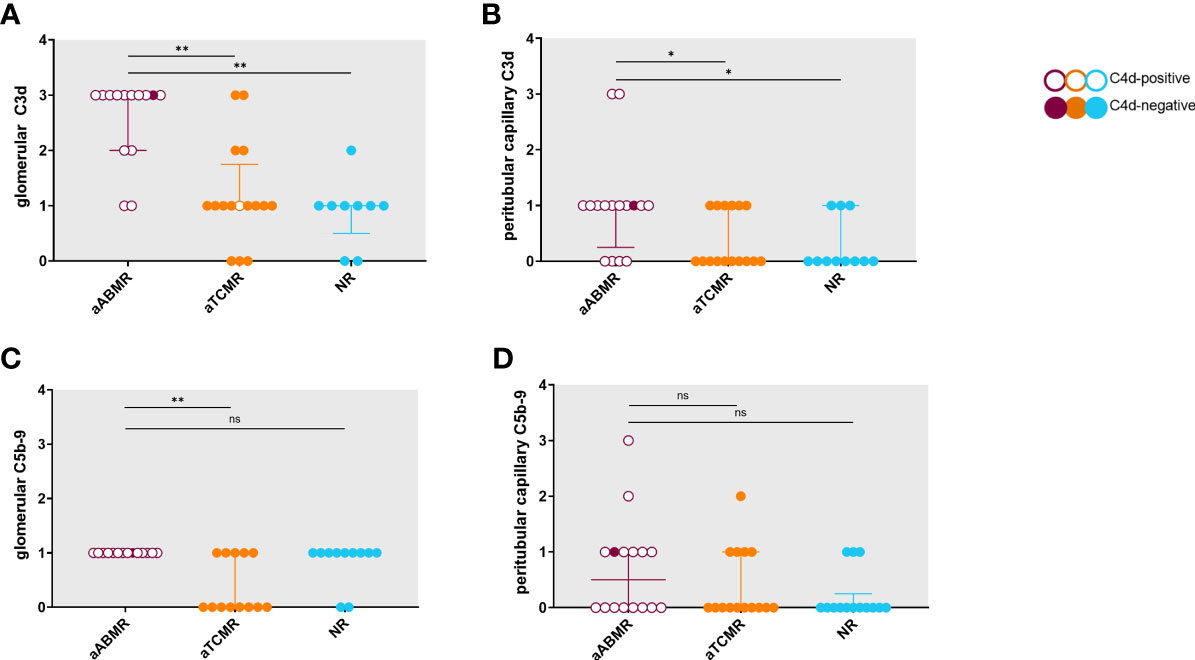
Figure 3 Semiquantitative scores for C3d and C5b-9 deposition in renal biopsy specimens of aABMR, aTCMR, and NR patients. Semiquantitative scores range from 0 to 4, with higher scores indicating increased positivity in biopsy. Groups are compared regarding glomerular C3d deposition (A), peritubular capillary C3d (B), glomerular C5b-9 (C), and peritubular capillary C5b-9 (D) deposition. Data represent staining results of individual patients. Error bars represent median with interquartile range. Unfilled circles symbolize biopsies of C4d-positive patients, filled circles C4d-negative patients. P-values are derived from Mann-Whitney U tests with statistical significance defined as *P < 0.05, **P < 0.001; aABMR, active antibody-mediated rejection; aTCMR, acute T-cell mediated rejection; NR, non-rejection; ns, not significant.
Median scores for glomerular C5b-9 showed mild intensity (1) for aABMR and NR patients and negativity (0) for aTCMR patients. While glomerular C5b-9 deposition was not significantly increased in aABMR compared to NR patients (P=0,5), glomerular C5b-9 staining was stronger in aABMR compared to aTCMR patients (P<0,01) (Figure 3C). Peritubular capillary deposition of C5b-9 was similar across groups with median semiquantitative scores of 0 in all groups (Figure 3D).
Figure 4 exemplifies the expression pattern in a C4d-positive aABMR patient, who presented with strong C3d deposition in the glomerulus, but only mild C3d in peritubular capillaries and scarce glomerular and no peritubular C5b-9. In comparison, in aTCMR and NR patients, C3d and C5b-9 were absent or only mildly expressed (Figures 5, 6).
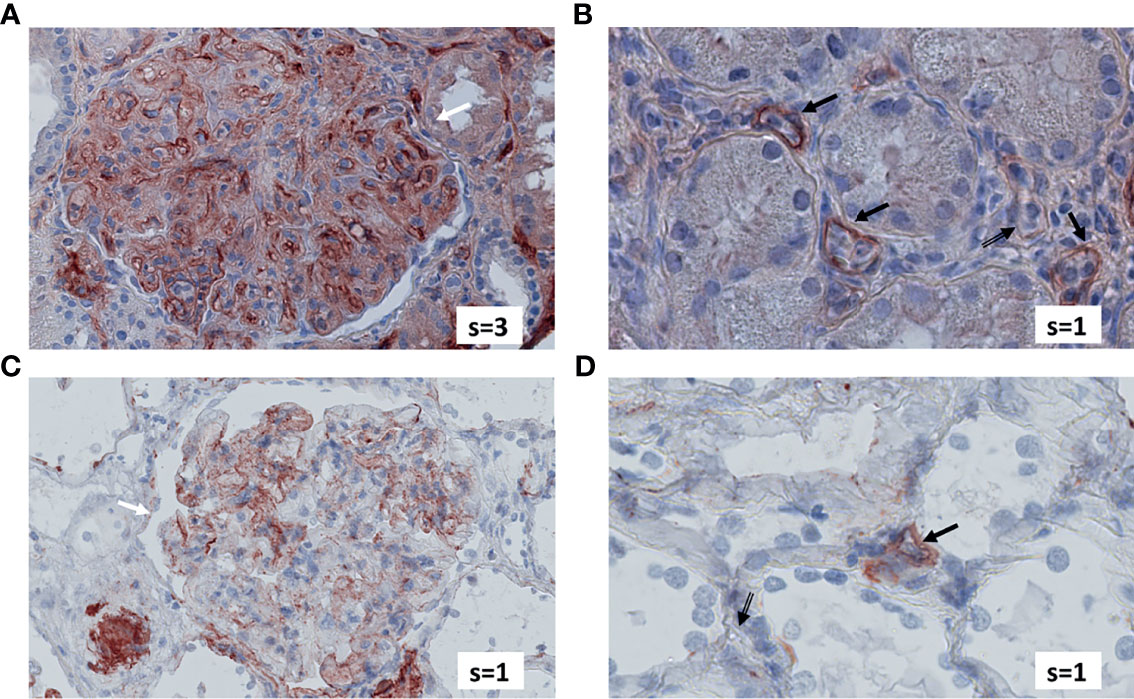
Figure 4 Renal biopsy staining for complement factors in a KTR diagnosed with C4d-positive, aABMR. Selected section of biopsy slide stained for C3d in glomeruli (A) and in peritubular capillaries (B). Selected section of biopsy slide stained for C5b-9 in glomeruli (C) and in peritubular capillaries (D). White arrows indicate glomeruli and black arrows indicate peritubular capillaries. Double-compound arrows indicate C5b-9 negative peritubular capillaries (D). Semi-quantitative scores (0-4) are indicated in the lower right corner of each picture. KTR, kidney transplant recipients; aABMR, active antibody-mediated rejection; s, semi-quantitative score.
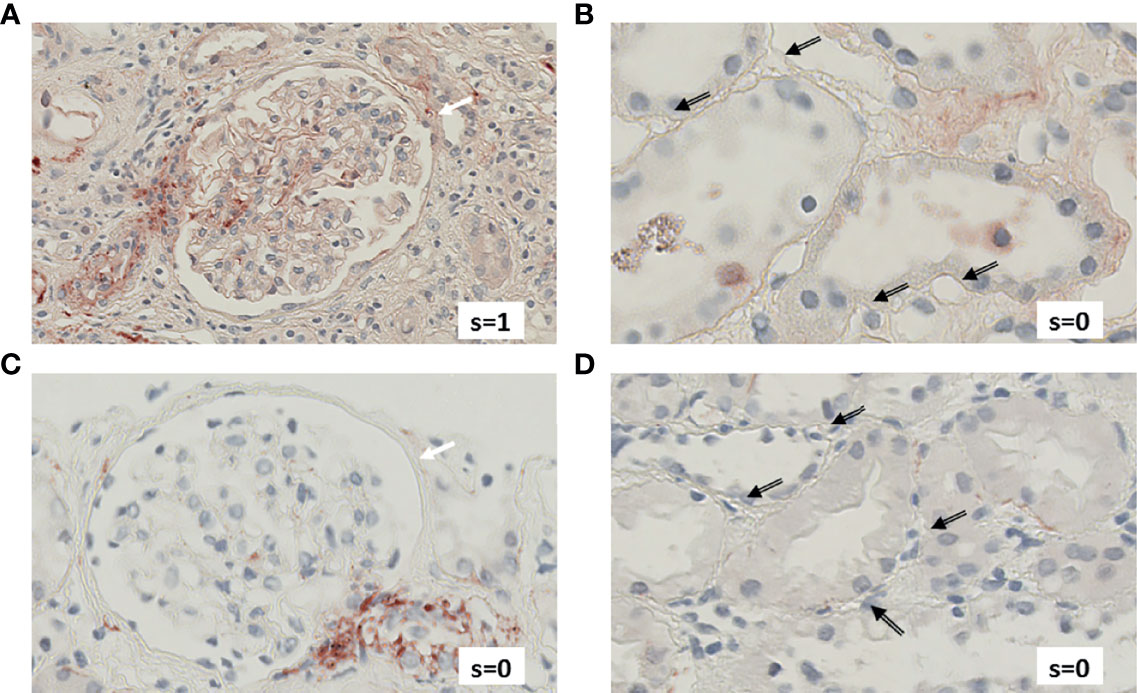
Figure 5 Renal biopsy staining for complement factors in a KTR diagnosed with aTCMR. Selected section of biopsy slide stained for C3d in glomeruli (A) and in peritubular capillaries (B). Selected section of biopsy slide stained for C5b-9 in glomeruli (C) and in peritubular capillaries (D). White arrows indicate glomeruli and black arrows indicate peritubular capillaries. Semi-quantitative scores (0-4) are indicated in the lower right corner of each picture. KTR, kidney transplant recipients; aTCMR, acute T-cell-mediated rejection; s, semi-quantitative score.
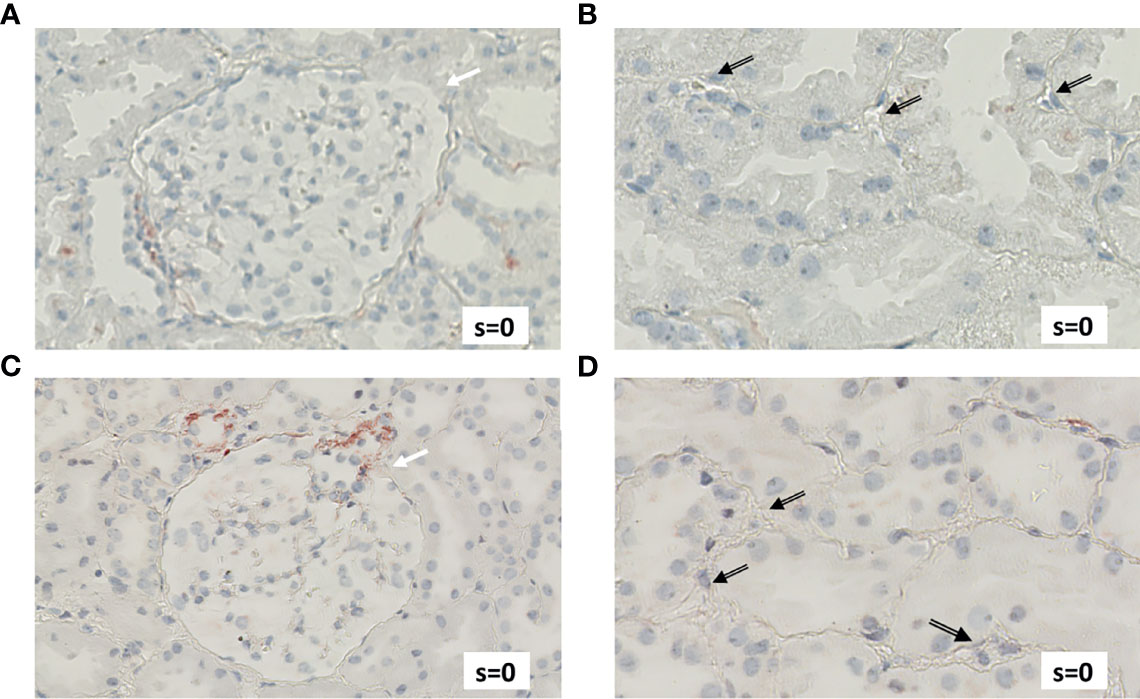
Figure 6 Renal biopsy staining for complement factors in a KTR without indication for undergoing biopsy and without signs of rejection in biopsy (NR patient). Selected section of biopsy slide stained for C3d in glomeruli (A) and in peritubular capillaries (B). Selected section of biopsy slide stained for C5b-9 in glomeruli (C) and in peritubular capillaries (D). White arrows indicate glomeruli and black arrows indicate peritubular capillaries. Semi-quantitative scores (0-4) are indicated in the lower right corner of each picture. KTR, kidney transplant recipients; s, semi-quantitative score.
Complement regulator CD59 in human renal tissue derived from a pre-transplantation biopsy is depicted in Figures 7A, B. CD59 was clearly expressed in both glomeruli and peritubular capillaries, with stronger deposition in peritubular structures (Figure 7B). CD59 staining in 5 aABMR patients showed a tendency of lower CD59 expression (Figures 7C, D) as compared to pre-transplantation control biopsy (Figures 7A, B).
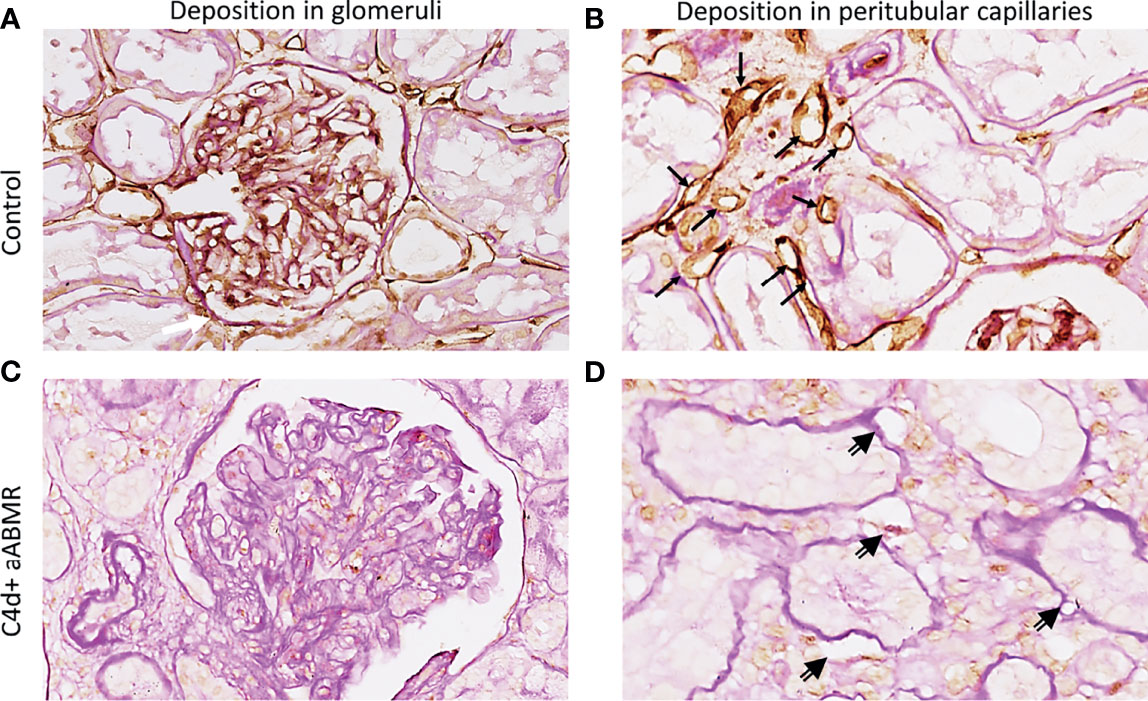
Figure 7 CD59 staining in vivo. Renal biopsy staining for complement regulator CD59 in glomeruli (A, C) and peritubular capillaries (B, D). Staining was performed on human kidney before transplantation as control (A, B), on biopsy specimen from a C4d-positive (C4d+) aABMR (C, D). Black arrows point to CD59-positive peritubular capillaries, double-compound arrows to CD59-negative peritubular capillaries. aABMR, active antibody-mediated rejection.
Quantification of complement factors on CiGEnCs showed strong expression of C3d and C4d, but only slight or no deposition of C5b-9 after incubation with HLA-Abs. This expression pattern varied across the different incubation conditions but revealed a general trend of strong C3d and C4d with minor or absent C5b-9 expression. Notably, incubation with IgG HLA-A2 antibodies resulted in expression of complement factors C3d and C4d without simultaneous C5b-9 deposition on the cell surface. In the pan-HLA antibody incubation condition, C5b-9 expression was highest when compared to the other incubation conditions (Figure 8A). Complement regulator, CD59, was constitutively expressed on CiGEnCs, independent of the incubation condition (Figure 8B).
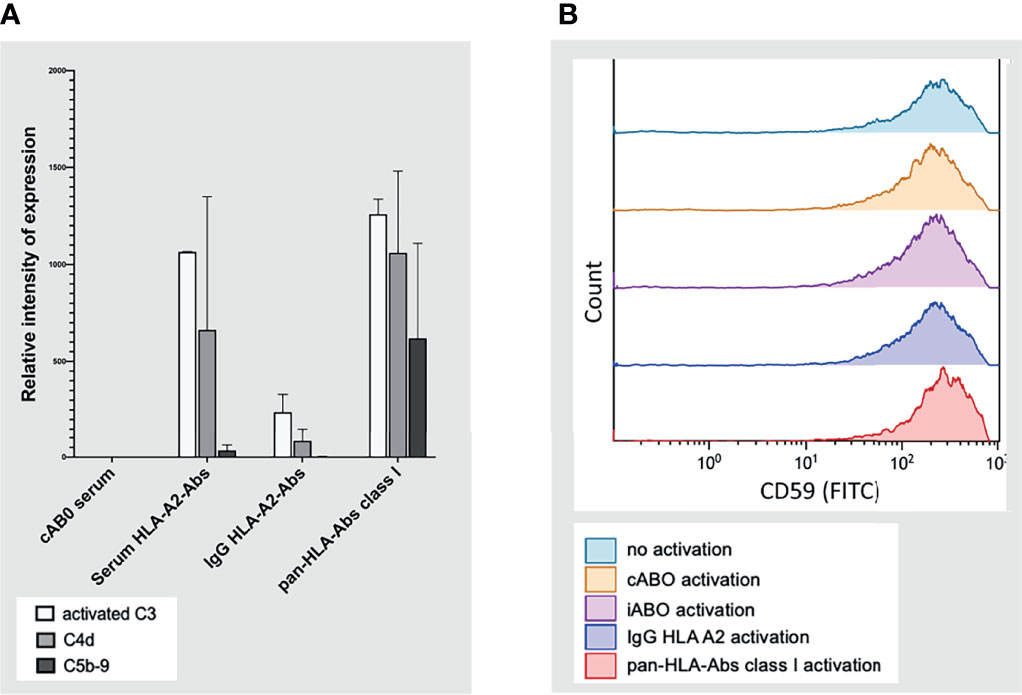
Figure 8 Complement system activation on conditionally immortalized glomerular endothelial cells in vitro in flow cytometric analysis. Complement factors C3 (activated), C4d, and C5b-9 on conditionally immortalized glomerular endothelial cells in vitro in flow cytometric analysis are depicted in (A) with the four different incubation conditions plotted on the y-axis. Deposition of complement regulator, CD59, was measured in flow-cytometry under five different incubation conditions (B). cABO, ABO-compatible; HLA, Human Leucocyte Antigen; Abs, antibodies; IgG, Immunoglobulin G; FITC, Fluorescein isothiocyanate; iABO, ABO-incompatible.
In this study, we show robust proximal complement activation in aABMR renal biopsies compared to controls, without or only mild, concomitant C5b-9 deposition. The absence of co-expression of proximal and terminal complement factors might imply that terminal complement is less involved in aABMR pathogenesis. In this line of reasoning, the minor role of terminal complement could explain the disappointing efficacy of terminal complement inhibition as treatment for aABMR, particularly for late, aABMR (27–30).
Earliest research on immunohistopathology of ABMR by Feucht et al. revealed similar results. Next to their primary findings on the role of C4d deposition and its diagnostic value, Feucht et al. demonstrated the absence of C5b-9 deposits in C4d-positive ABMR patients (8).
Terminal complement regulation could account for the absence of C5b-9 deposits in aABMR patients, who did otherwise stain positive for C3d and C4d (Figure 4) (10, 31). Other authors indicated that complement regulation and degradation by CD59 could explain why C5b-9 deposition is volatile, discounting C5b-9 as a diagnostic marker in ABMR (10, 32, 33). Stronger protection against terminal complement by CD59 compared to the proximal regulators CD46 and CD55 might further explain why C3d and C4d but not C5b-9 was pronounced in aABMR (34).
We confirmed this assumed influence of CD59 in vitro, as it was continuously expressed on CiGEnCs, independent of the antibody stimulation condition. Similarly, we revealed clear endothelial CD59 expression in vivo, in pre-transplantation kidney biopsies (Figures 7A, B). Interaction of CD59 and C5b-9 might cause internalization and/or degradation of the CD59/C9 complex, possibly explaining the absence of both C5b-9 and CD59 in ABMR biopsies. Particularly, Cai et al. showed that binding to the complement-binding side of CD59 can cause internalization of the complex in cancer cells (35). Despite these findings, literature on CD59 actions, degradation, and recycling processes is scarce. Thus, interpretations of CD59 expression in our aABMR samples remain speculative. Regarding the sustained CD59 positivity after complement activation in vitro, we assume that down-modulation by internalization of the CD59/C9 complex takes more time than the 45-minute time frame used in the in vitro complement activation experiments. In short, while the kidney endothelium seems to be highly protected against C5b-9 activation, this protection does not prevent aABMR. The absence of systemic and local C5b-9 fundamentally questions the relevance of terminal complement in aABMR pathogenesis (11, 31).
A broad body of evidence proved the efficacy of terminal complement inhibition in complement-mediated diseases like atypical hemolytic uremic syndrome or paroxysmal sleep hemoglobinuria (36, 37). In contrast, studies on the efficacy of terminal complement inhibition for ABMR presented with more equivocal results (27–30, 38, 39). Particularly, these studies were unable to show universal and enduring effectiveness of the terminal complement inhibitor, eculizumab (27–30). Interestingly, two case studies demonstrated substantial effectiveness of eculizumab in a pre-sensitized child (39) and a young adult (38), who showed clear deposition of immunoglobulins, C4d, and C5b-9 in renal biopsies. Complement deposits fully resolved within days after eculizumab administration in both case studies. Case studies in which eculizumab was effective as ABMR treatment, showed C5b-9 positivity, stressing the importance of C5b-9 involvement for eculizumab treatment (38, 39). Thus, we do not deny the potential relevance of identifying C5b-9-positive ABMR as an immunophenotypic subtype (40), although we showed that only a minority of our ABMR patients were C5b-9-positive.
Our in vitro results provide some indication for why C5b-9 deposition occurs in particular ABMR subgroups. Specifically, they suggest that a particularly high level of antibody-antigen interaction on the endothelial surface, as in the pan-HLA incubation condition, might be able to overwhelm any local complement regulatory barrier on endothelial cells. Overwhelming this regulation barrier possibly explains the mild expression of C5b-9 on endothelial cells after incubation with pan-HLA-Abs. Thus, C5b-9 might be involved in pathogenesis in specific ABMR patients, with early-onset rejection and/or with high titers of HLA-antibodies, in whom terminal complement inhibition might be relevant (41–43). In our cohort, a minority of aABMR patients presented with early acute rejection, partially because early acute ABMR is a rare event in recipients without pre-transplant HLA sensitization. Therefore, our findings are of particular relevance for the majority of ABMR cases in the clinical setting (41).
In C5b-9 negative aABMR, complement cascade split products like C4a, C3dg, iC3b, and C5a arising proximal to ultimate C5b-9 assembly might be sufficient to cause ABMR by binding to respective receptors on endothelium, creating a pro-inflammatory environment, or recruiting other immunogenic agents (33, 44–46).
Besides the complement system, alternative immunopathogenic pathways could be involved in aABMR, accounting for aABMR in the absence of C5b-9 in biopsies. A broad body of evidence emphasizes complement-independent mechanisms in ABMR pathogenesis, possibly mediated via Natural Killer cells (47–51). Others emphasized that DSA-binding could directly cause complement-independent endothelial cell activation (52–55).
To evaluate systemic evidence of complement activation in aABMR, complement factors in plasma were quantified, and no differences were found regarding C3, C3d, and C5b-9 levels between groups. This is in line with recently published findings revealing that ABMR and TCMR with circulating DSAs could not be distinguished by plasma complement factors (18)
Notably, aABMR patients showed increased expression of C3d in glomerular and peritubular structures compared to aTCMR and NR patients. C4d and C3d are considered stable split products that deposit on vascular structures after cleavage of C4 and C3 (11, 31). Pronounced C3d deposition in C4d-positive aABMR patients suggests that HLA-Abs cause classical pathway activation at least up to the level of C3 convertase in aABMR.
Other studies indicate that renal, C3d, and not, C4d deposition, could serve as a prognostic factor for graft functioning and survival in KTR with acute rejection (56, 57).
Our study has several strengths. First, we provide a sophisticated level of comparison for complement activation by including a well-defined, biopsy-proven cohort of aABMR, NR, and aTCMR patients. The aABMR group encompasses a relatively homogenous and clinically relevant group, with the majority of ABMR patients showing active or chronic active ABMR, and classical C4d- and DSA-positivity. Second, we provide a holistic picture of complement system activation, with factor quantification on local and systemic levels. This integrative complement analysis is crucial, considering that local and systemic complement system activation did not mirror each other. Third, we strengthened our findings in vivo by reproducing similar complement deposition patterns on glomerular, endothelial cells in vitro.
Some limitations of our study have to be noted. Our study was based on a small, single-center cohort, with some patients missing biopsy material. Future research should be performed on a larger group of ABMR patients to reflect and address the immunopathogenic heterogeneity of ABMR subgroups. Notably, we mainly included late, chronic active and active ABMR patients, whereas patients with chronic inactive ABMR, early ABMR and DSA-/C4d-negative ABMR are missing or underrepresented in our study. This underrepresentation limits any implications for these particular ABMR subgroups. Chronic active and active ABMR might differ in terms of the underlying complement-related pathogenesis, particularly as chronic active ABMR was more commonly C4d-negative (58). The possible influence of chronicity on complement activation cannot fully be addressed by our study due to the small sample size. However, as 16 of 17 included aABMR patients were C4d-positive, we do not expect that the chronicity component had a significant effect on complement analyses in our study. In addition, the Banff classification has its limitations and may not always represent the true underlying pathophysiology. For example, one patient classified as aTCMR on the basis of tubulitis and interstitial inflammation. This patient showed C4d-positivity in biopsy, but no other histological evidence for ABMR to fulfil the diagnostic criteria for aABMR. Within the boundaries of the Banff classification, the origin of C4d-deposition in this patient remains unexplained and a potential influence of undetected (non-)HLA-Abs cannot fully be excluded. With regard to the CD59 staining, it should be noted that it was only performed for 5 aABMR patients and preliminary results need to be validated in a larger aABMR population, as well as in aTCMR patients.
In conclusion, local C3d, and C4d are predominant hallmarks in the majority of aABMR patients, indicating upstream complement cascade activation of the classical pathway, which is not reflected by systemic complement activation. Terminal complement regulation, especially in peritubular capillaries, might explain this absence of C5b-9 despite C3d and C4d deposits. Thus, cytotoxicity is probably not mediated through C5b-9 in a significant proportion of aABMR patients. Together with our findings in vitro, our results provide sufficient justification to hypothesize that proximal complement factors or Fc-receptor mediated pathways could be more efficient pharmacological targets than terminal complement treatment.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by University Medical Center Groningen (Groningen, The Netherlands) institutional review board (METc 2008/186). The patients/participants provided their written informed consent to participate in this study.
SB and JB were mainly responsible for the design of the study, as well as the supervision of the design of experiments, analyses, and writing processes. GT, RL, JK, AM-A, and FA carried out the experiments. MH conducted classifications and semi-quantitative scoring of biopsies. BH and RL were mainly responsible for quantification of HLA-antibodies in serum of patients. AD provided histopathological support for initial biopsy sample collection and selection of patients. RP provided advice for manuscript writing and the collection of pathological biopsy material. MD and MS provided advice in study design, analyses and writing of the manuscript. GT and RL were mainly responsible for writing, analysing, and designing figures and tables. All authors approved the final version of the manuscript.
This study was supported by the Dutch Kidney foundation (grant number:13O CA27).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Dr. T. F. Claas from the University, Leiden (Groningen, the Netherlands) for kindly providing the HLA-antibodies for in vitro experiments; H. Burgerhoff for his advice regarding analyses and graphical display (Figure 3) and A.W. Gomez for his initial help in designing the database and requesting samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.845301/full#supplementary-material
1. Schinstock CA, Mannon RB, Budde K, Chong AS, Haas M, Knechtle S, et al. Recommended Treatment for Antibody-Mediated Rejection After Kidney Transplantation: The 2019 Expert Consensus From the Transplantion Society Working Group. Transplantation (2020) 104(5):911–22. doi: 10.1097/TP.0000000000003095
2. Roufosse C, Simmonds N, Clahsen-van Groningen M, Haas M, Henriksen KJ, Horsfield C, et al. A 2018 Reference Guide to the Banff Classification of Renal Allograft Pathology. Transplantation (2018) 102(11):1795–814. doi: 10.1097/TP.0000000000002366
3. Loupy A, Aubert O, Orandi BJ, Naesens M, Bouatou Y, Raynaud M, et al. Prediction System for Risk of Allograft Loss in Patients Receiving Kidney Transplants: International Derivation and Validation Study. BMJ (2019) 366:l4923. doi: 10.1136/bmj.l4923
4. Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D, et al. The Banff 2017 Kidney Meeting Report: Revised Diagnostic Criteria for Chronic Active T Cell–Mediated Rejection, Antibody-Mediated Rejection, and Prospects for Integrative Endpoints for Next-Generation Clinical Trials. Am J Transplant (2018) 18(2):293–307. doi: 10.1111/ajt.14625
5. Sellarés J, De Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the Causes of Kidney Transplant Failure: The Dominant Role of Antibody-Mediated Rejection and Nonadherence. Am J Transplant (2012) 12(2):388–99. doi: 10.1111/j.1600-6143.2011.03840.x
6. Solez K, Colvin RB, Racusen LC, Sis B, Halloran PF, Birk PE, et al. Banff ‘05 Meeting Report: Differential Diagnosis of Chronic Allograft Injury and Elimination of Chronic Allograft Nephropathy (‘Can’). Am J Transplant (2007) 7:518–26. doi: 10.1111/j.1600-6143.2006.01688.x
7. Collins AB, Schneeberger EE, Pascual MA, Saidman SL, Williams WW, Tolkoff-Rubin N, et al. Complement Activation in Acute Humoral Renal Allograft Rejection: Diagnostic Significance of C4d Deposits in Peritubular Capillaries. J Am Soc Nephrol (1999) 10:2208–14. doi: 10.1681/ASN.V10102208
8. Feucht HE, Schneeberger H, Hillebrand G, Burkhardt K, Weiss M, Riethmuller G, et al. Capillary Deposition of C4d Complement Fragment and Early Renal Graft Loss. Kidney Int (1993) 43(6):1333–8. doi: 10.1038/ki.1993.187
9. Biglarnia AR, Huber-Lang M, Mohlin C, Ekdahl KN, Nilsson B. The Multifaceted Role of Complement in Kidney Transplantation. Nat Rev Nephrol (2018) 14(12):767–81. doi: 10.1038/s41581-018-0071-x
10. Nishi S, Imai N, Ito Y, Ueno M, Fukase S, Mori H, et al. Pathological Study on the Relationship Between C4d, CD59 and C5b-9 in Acute Renal Allograft Rejection. Clin Transplantation Suppl (2004) 18(11):18–23. doi: 10.1111/j.1399-0012.2004.00242
11. Thurman JM, Panzer SE, Le Quintrec M. The Role of Complement in Antibody Mediated Transplant Rejection. Mol Immunol (2019) 112:240–6. doi: 10.1016/j.molimm.2019.06.002
12. Murata K, Baldwin WM III. Mechanisms of Complement Activation, C4d Deposition, and Their Contribution to the Pathogenesis of Antibody-Mediated Rejection ☆. Transplant Rev (2009) 23:139–50. doi: 10.1016/j.trre.2009.02.005
13. Montgomery RA, Loupy A, Segev DL. Antibody-Mediated Rejection: New Approaches in Prevention and Management. Am J Transplant (2018) 18:3–17. doi: 10.1111/ajt.14584
14. Haas M. Pathology of C4d-Negative Antibody-Mediated Rejection in Renal Allografts. Curr Opin Organ Transplant (2013) 18(3):319–26. doi: 10.1097/MOT.0b013e32835d4daf
15. Loupy A, Haas M, Roufosse C, Naesens M, Adam B, Afrouzian M, et al. The Banff 2019 Kidney Meeting Report (I): Updates on and Clarification of Criteria for T Cell– and Antibody-Mediated Rejection. Am J Transplant (2020) 20(9):2318–31. doi: 10.1111/ajt.15898
16. Eskandary F, Wahrmann M, Mühlbacher J, Böhmig GA. Complement Inhibition as Potential New Therapy for Antibody-Mediated Rejection. Transpl Int (2016) 29(4):392–402. doi: 10.1111/tri.12706
17. Stegall MD, Gloor JM. Deciphering Antibody-Mediated Rejection: New Insights Into Mechanisms and Treatment. Curr Opin Organ Transplant (2010) 15(1):8–10. doi: 10.1097/MOT.0b013e3283342712
18. Mező B, Heilos A, Böhmig GA, Eskandary F, Wahrmann M, Bond G, et al. Complement Markers in Blood and Urine: No Diagnostic Value in Late Silent Antibody-Mediated Rejection. Transplant Direct (2019) 5(7):1–11. doi: 10.1097/TXD.0000000000000915
19. Eisenga MF, Gomes-Neto AW, Van Londen M, Ziengs AL, Douwes RM, Stam SP, et al. Rationale and Design of Transplantlines: A Prospective Cohort Study and Biobank of Solid Organ Transplant Recipients. BMJ Open (2018) 8:24502. doi: 10.1136/bmjopen-2018-024502
20. Levey AS, Stevens LA, Schmid CH, Zhang Y, Castro AF, Feldman HI, et al. A New Equation to Estimate Glomerular Filtration Rate. Ann Intern Med (2009) 150(9):604–12. doi: 10.7326/0003-4819-150-9-200905050-00006
21. Mancini G, Carbonara AO, Heremans JF. Immunochemical Quantitation of Antigens by Single Radial Immunodiffusion. Immunochemistry (1965) 2(3):235–54. doi: 10.1016/0019-2791(65)90004-2
22. Peakman M, Lobo-Yeo A, Senaldi G, Nilsson M, Tee DEH, Vergani D. Quantification of C3d in Biological Fluids by an Enzyme-Linked Immunosorbent Assay. J Immunol Methods (1987) 104(1-2):51–6. doi: 10.1016/0022-1759(87)90486-8
23. Damman J, Seelen MA, Moers C, Daha MR, Rahmel A, Leuvenink HG, et al. Systemic Complement Activation in Deceased Donors Is Associated With Acute Rejection After Renal Transplantation in the Recipient. Transplantation (2011) 92(2):163–9. doi: 10.1097/TP.0b013e318222c9a0
24. Bobka S, Ebert N, Koertvely E, Jacobi J, Wiesener M, Büttner-Herold M, et al. Is Early Complement Activation in Renal Transplantation Associated With Later Graft Outcome? Kidney Blood Press Res (2018) 43:1488–504. doi: 10.1159/000494014
25. Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, Von Ruhland CJ, et al. Conditionally Immortalized Human Glomerular Endothelial Cells Expressing Fenestrations in Response to VEGF. Kidney Int (2006) 69(9):1633–40. doi: 10.1038/sj.ki.5000277
26. Ristau T, Paun C, Ersoy L, Hahn M, Lechanteur Y, Hoyng C, et al. Impact of the Common Genetic Associations of Age-Related Macular Degeneration Upon Systemic Complement Component C3d Levels. PloS One (2014) 9(3):e93459. doi: 10.1371/journal.pone.0093459
27. Cornell LD, Schinstock CA, Gandhi MJ, Kremers WK, Stegall MD. Positive Crossmatch Kidney Transplant Recipients Treated With Eculizumab: Outcomes Beyond 1 Year. Am J Transplant (2015) 15(5):1293–302. doi: 10.1111/ajt.13168
28. Marks WH, Mamode N, Montgomery RA, Stegall MD, Ratner LE, Cornell LD, et al. Safety and Efficacy of Eculizumab in the Prevention of Antibody-Mediated Rejection in Living-Donor Kidney Transplant Recipients Requiring Desensitization Therapy: A Randomized Trial. Am J Transplant (2019) 19(10):2876–88. doi: 10.1111/ajt.15364
29. Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, et al. Terminal Complement Inhibition Decreases Antibody-Mediated Rejection in Sensitized Renal Transplant Recipients. Am J Transplant (2011) 11(11):2405–13. doi: 10.1111/j.1600-6143.2011.03757.x
30. Orandi BJ, Zachary AA, Dagher NN, Bagnasco SM, Garonzik-Wang JM, Van Arendonk KJ, et al. Eculizumab and Splenectomy as Salvage Therapy for Severe Antibody-Mediated Rejection After HLA-Incompatible Kidney Transplantation. Transplantation (2014) 98(8):857–63. doi: 10.1097/TP.0000000000000298
31. Stites E, Le Quintrec M, Thurman JM. Antibody-Mediated Transplant Rejection the Complement System and. J Immunol Ref (2015) 195:5525–31. doi: 10.4049/jimmunol.1501686
32. Koopman JJE, Van Essen MF, Rennke HG, De Vries APJ, Van Kooten C. Deposition of the Membrane Attack Complex in Healthy and Diseased Human Kidneys. Front Immunol (2021) 11:599974. doi: 10.3389/fimmu.2020.599974
33. González-Molina M, Ruiz-Esteban P, Caballero A, Burgos D, Cabello M, Leon M, et al. Immune Response and Histology of Humoral Rejection in Kidney Transplantation. Nefrologia (2016) 36(4):354–67. doi: 10.1016/j.nefro.2016.03.023
34. Ichida S, Yuzawa Y, Okada H, Yoshioka K, Matsuo S. Localization of the Complement Regulatory Proteins in the Normal Human Kidney. Kidney Int (1994) 46(1):89–96. doi: 10.1038/ki.1994.247
35. Cai B, Xie S, Liu F, Simone LC, Caplan S, Qin X, et al. Rapid Degradation of the Complement Regulator, CD59, by a Novel Inhibitor. J Biol Chem (2014) 289(17):12109–25. doi: 10.1074/jbc.M113.547083
36. Keating GM. Eculizumab: A Review of its Use in Atypical Haemolytic Uraemic Syndrome. Drugs (2013) 73:2053–66. doi: 10.1007/s40265-013-0147-7
37. Zhou S, Dong X, Chen C, Ma L, Wu Y, Zhou Y, et al. Efficacy and Safety of Eculizumab for Paroxysmal Nocturnal Hemoglobinuria: A Systematic Review and Meta-Analysis. J Pediatr Hematol Oncol (2021) 43(6):203–10. doi: 10.1097/MPH.0000000000002178
38. Locke JE, Magro CM, Singer AL, Segev DL, Haas M, Hillel AT, et al. The Use of Antibody to Complement Protein C5 for Salvage Treatment of Severe Antibody-Mediated Rejection. Am J Transplant (2009) 9(1):231–5. doi: 10.1111/j.1600-6143.2008.02451.x
39. Chehade H, Rotman S, Matter M, Girardin E, Aubert V, Pascual M. Eculizumab to Treat Antibody-Mediated Rejection in a 7-Year-Old Kidney Transplant Recipient. Pediatrics (2015) 135(2):e551–5. doi: 10.1542/peds.2014-2275
40. Goutaudier V, Perrochia H, Mucha S, Bonnet M, Delmas S, Garo F, et al. C5B9 Deposition in Glomerular Capillaries Is Associated With Poor Kidney Allograft Survival in Antibody-Mediated Rejection. Front Immunol (2019) 10:235. doi: 10.3389/fimmu.2019.00235
41. Cornell LD. Histopathologic Features of Antibody Mediated Rejection: The Banff Classification and Beyond. Front Immunol (2021) 12:718122. doi: 10.3389/fimmu.2021.718122
42. Tan EK, Bentall A, Dean PG, Shaheen MF, Stegall MD, Schinstock CA. Use of Eculizumab for Active Antibody-Mediated Rejection That Occurs Early Post-Kidney Transplantation: A Consecutive Series of 15 Cases. Transplantation (2019) 103(11):2397–404. doi: 10.1097/TP.0000000000002639
43. Haas M. Eculizumab as Primary Therapy for Active Antibody-Mediated Rejection of Renal Allografts: A Matter of Timing, Severity, and Donor-Specific Antibodies. Transplantation (2019) 103(11):2219–20. doi: 10.1097/TP.0000000000002640
44. Min X, Liu C, Wei Y, Wang N, Yuan G, Liu D, et al. Expression and Regulation of Complement Receptors by Human Natural Killer Cells. Immunobiology (2014) 219(9):671–9. doi: 10.1016/j.imbio.2014.03.018
45. Noris M, Remuzzi G. Overview of Complement Activation and Regulation. Semin Nephrol (2013) 33(6):479–92. doi: 10.1016/j.semnephrol.2013.08.001
46. Grafals M, Thurman JM. The Role of Complement in Organ Transplantation. Front Immunol (2019) 10:2380. doi: 10.3389/fimmu.2019.02380
47. Koenig A, Chen CC, Marçais A, Barba T, Mathias V, Sicard A, et al. Missing Self Triggers NK Cell-Mediated Chronic Vascular Rejection of Solid Organ Transplants. Nat Commun (2019) 10(1):5350. doi: 10.1038/s41467-019-13113-5
48. Yazdani S, Callemeyn J, Gazut S, Lerut E, de Loor H, Wevers M, et al. Natural Killer Cell Infiltration Is Discriminative for Antibody-Mediated Rejection and Predicts Outcome After Kidney Transplantation. Kidney Int (2019) 95(1):188–98. doi: 10.1016/j.kint.2018.08.027
49. Yagisawa T, Tanaka T, Miyairi S, Tanabe K, Dvorina N, Yokoyama WM, et al. In the Absence of Natural Killer Cell Activation Donor-Specific Antibody Mediates Chronic, But Not Acute, Kidney Allograft Rejection. Kidney Int (2019) 95(2):350–62. doi: 10.1016/j.kint.2018.08.041
50. Koenig A, Mezaache S, Callemeyn J, Barba T, Mathias V, Sicard A, et al. Missing Self-Induced Activation of NK Cells Combines With Non-Complement-Fixing Donor-Specific Antibodies to Accelerate Kidney Transplant Loss in Chronic Antibody-Mediated Rejection. J Am Soc Nephrol (2021) 32:479–94. doi: 10.1681/ASN.2020040433
51. O’Neill MA, Hidalgo LG. NK Cells in Antibody-Mediated Rejection – Key Effector Cells in Microvascular Graft Damage. Int J Immunogenet (2021) 48(2):110–9. doi: 10.1111/iji.12532
52. Penack O, Dressel R, Coppage M, Mooney N, Cross AR, Glotz D. The Role of the Endothelium During Antibody-Mediated Rejection: From Victim to Accomplice. Front Immunol (2018) 9:29. doi: 10.3389/fimmu.2018.00106
53. Cross AR, Lion J, Poussin K, Assayag M, Taupin JL, Glotz D, et al. HLA-DQ Alloantibodies Directly Activate the Endothelium and Compromise Differentiation of Foxp3high Regulatory T Lymphocytes. Kidney Int (2019) 96(3):689–98. doi: 10.1016/j.kint.2019.04.023
54. Taflin C, Favier B, Baudhuin J, Savenay A, Hemon P, Bensussan A, et al. Human Endothelial Cells Generate Th17 and Regulatory T Cells Under Inflammatory Conditions. Proc Natl Acad Sci USA (2011) 108(7):2891–6. doi: 10.1073/pnas.1011811108
55. Lion J, Taflin C, Cross AR, Robledo-Sarmiento M, Mariotto E, Savenay A, et al. HLA Class II Antibody Activation of Endothelial Cells Promotes Th17 and Disrupts Regulatory T Lymphocyte Expansion. Am J Transplant (2016) 16(5):1408–20. doi: 10.1111/ajt.13644
56. Kuypers DRJ, Lerut E, Evenepoel P, Maes B, Vanrenterghem Y, Van Damme B. C3d Deposition in Peritubular Capillaries Indicates a Variant of Acute Renal Allograft Rejection Characterized by a Worse Clinical Outcome. Transplantation (2003) 76(1):102–8. doi: 10.1097/01.TP.0000069040.16457.06
57. Lv R, Zhang W, Han F, Liu G, Xie W, Chen J. Capillary Deposition of Complement C4d and C3d in Chinese Renal Allograft Biopsies. Dis Markers (2015), 2015. doi: 10.1155/2015/397613
Keywords: antibody-mediated rejection, kidney transplantation, complement system, membrane-attack complex, C5b-9
Citation: Tiller G, Lammerts RGM, Karijosemito JJ, Alkaff FF, Diepstra A, Pol RA, Meter-Arkema AH, Seelen MA, van den Heuvel MC, Hepkema BG, Daha MR, van den Born J and Berger SP (2022) Weak Expression of Terminal Complement in Active Antibody-Mediated Rejection of the Kidney. Front. Immunol. 13:845301. doi: 10.3389/fimmu.2022.845301
Received: 29 December 2021; Accepted: 14 March 2022;
Published: 13 April 2022.
Edited by:
Oriol Bestard, Vall d’Hebron University Hospital, SpainReviewed by:
Francesc J. Moreso, Vall d’Hebron University Hospital, SpainCopyright © 2022 Tiller, Lammerts, Karijosemito, Alkaff, Diepstra, Pol, Meter-Arkema, Seelen, van den Heuvel, Hepkema, Daha, van den Born and Berger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan P. Berger, cy5wLmJlcmdlckB1bWNnLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.