 Shi-Yue Zheng
Shi-Yue Zheng Jian-Zeng Dong
Jian-Zeng Dong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 April 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.843891
This article is part of the Research Topic Structural Immunology of Molecular Innate Immunity View all 14 articles
Myocarditis is the common cause of sudden cardiac death, dilated cardiomyopathy (DCM) and heart failure (HF) in young adults. The most common type of myocarditis is viral myocarditis (VMC). Toll-like receptors (TLRs) are vital to identify pathogens in vivo. TLRs promote the differentiation of naive CD4+T cells to T helper (Th) cells, activate the immune response, and participate in the pathogenesis of autoimmune and allergic diseases. Although the pathogenesis of VMC is unclear, autoimmune responses have been confirmed to play a significant role; hence, it could be inferred that VMC is closely related to TLRs and Th responses. Some drugs have been found to improve the prognosis of VMC by regulating the immune response through activated TLRs. In this review, we discuss the role of TLRs and Th responses in VMC.
Myocarditis is a myocardial inflammation resulted from infectious, idiopathic, or autoimmune causes, of which the most popular is viral infection brought by enterovirus, Epstein-Barr (EB) virus, or human herpesvirus 6. Besides, myocarditis is the primary cause of dilated cardiomyopathy (DCM) and gradually becomes a cause of sudden cardiovascular death among young people (< 40-year-old) (1). Most patients with myocarditis can recover fully; however, some (up to 20%) develop chronic myocarditis, eventually resulting in DCM and heart failure (HF) (2). Myocarditis is diagnosed by combining clinical presentation, biomarkers, electrocardiogram (ECG), echocardiography, cardiac magnetic resonance imaging (CMRI), and endocardial biopsy (EMB). Tissue taken from EMB should be combined with the results of histology, immunohistochemistry and viral polymerase chain reaction (PCR) for the diagnosis of myocarditis (3). The EMB histology of myocarditis showed a value of leukocytes>14/mm2 with T lymphocytes>7/mm2, while immunohistochemistry showed an increase in the number of CD3+T cells or CD68+macrophages or CD163+M2 macrophages and virus genome could be detected by viral PCR (4, 5). EMB is a non-targeted operation, with low sensitivity as its main shortcoming, which may occur false-negative results when VMC is multifocal, focal, or localized (4). Therefore, the sensitivity of EMB in fulminant myocarditis with extensive inflammatory infiltration is increased, while that in focal myocarditis is relatively low, which may lead to false-negative results. In addition, although the virus can replicate in the myocardium, it does not cause enough myocardial inflammation, and EMB detection of the virus genome may also show false-negative results. The diagnostic criteria of CMRI for myocarditis are based on the ‘Lake-Louise’ criteria (6, 7). Therefore, in order to improve the sensitivity of EMB in diagnosis of myocarditis, we can determine the sampling site by combining CMRI and obtain myocardial tissue from three different sites. Chronic myocarditis also has persistent myocardial inflammation, which is the intermediate stage between acute myocarditis and chronic inflammatory cardiomyopathy. However, there is no detectable inflammation due to myocardial fibrosis in patients with chronic inflammatory cardiomyopathy, which renders diagnosis and treatment rather challenging (8).
Although the pathogenesis of myocarditis is yet unclear, the role of immune response in its process is under intensive focus. Under physiological conditions, a small number of immune cells are detected in the myocardium. After the onset of infections or autoimmune disorders, numerous immune cells and cytokines gather in the myocardial tissue to initiate inflammatory reactions. This process requires the initiation and maintenance of congenital and adaptive immune systems. Toll-like receptors (TLRs) recognize endogenous and exogenous ligands and are expressed on various cells, such as macrophages, neutrophils, dendritic cells (DCs), mast cells, and natural killer (NK) cells (9). They transmit signals to downstream pathways to stimulate innate and adaptive immunity after identifying the ligands involved in the pathogenesis of various autoimmune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), multiple sclerosis (MS), experimental autoimmune encephalitis (EAE), and experimental autoimmune myocarditis (EAM) (10). TLR1-TLR10 mRNA can be detected in normal peripheral blood T cells, but only TLR2-TLR5 and TLR9 expression can be detected at the protein level. TLR1, TLR2, and TLR7 are overexpressed on mRNA level in patients with myocarditis (9, 11). In addition, the inflammatory factors produced after TLRs activation, including interferon-gamma (IFN- γ), interleukin (IL)-6, and tumor growth factor-beta (TGF- β), can also stimulate native CD4+T cells to differentiate into T helper (Th) cells and participate in immune response to aggravate myocarditis. This study reviewed the role of TLRs and Th responses in viral myocarditis (VMC).
VMC is the most common myocarditis caused by various viruses, including enterovirus, adenovirus, influenza virus, EB virus, and parvovirus; the most common is Coxsackievirus B3 (CVB3) that belongs to enterovirus (12). CVB3 may be cleared by innate immune response or stimulate the immune system to produce autoantibodies against the infection. The condition can be cured or progressed to DCM and HF (13). According to the clinical characteristics, VMC can be classified into fulminant, acute, subacute, or chronic myocarditis and localized or diffuse inflammatory infiltration can be observed in myocardial pathology. Fulminant myocarditis is rare and characterized by diffuse inflammatory infiltration in myocardial tissue, which has multiple active lesions and can be completely relieved, die, or progress to chronic myocarditis. Acute or chronic myocarditis progresses latently, resulting in DCM or HF (14). The pathological progression of VMC has three phases at the cellular and tissue level: the acute phase caused by viral entry and replication, the subacute phase characterized by inflammatory cell infiltration, and the chronic phase characterized by cardiac remodeling (15). VMC can be diagnosed by combining biomarkers, ECG, echocardiography, CMRI, and EMB (16). With the continuous update and development of technology, viruses in patients with VMC can be detected by polymerase chain technology, but EMB is still the gold standard for the diagnosis of myocarditis (3, 17, 18). VMC can be divided into eosinophilic, lymphocytic, giant cell, and granulomatous myocarditis based on the histological types observed by EMB. The most common type is lymphocytic myocarditis, wherein the main infiltration is by CD4+T and CD8+T lymphocytes, accompanied by CD68+ macrophages and few B lymphocytes (19, 20). However, only 38% of patients with VMC present viral genomes in their EMB samples (21). Hence, a close correlation is established between virus infection and immune response in the pathogenic process of VMC, while many studies have confirmed that the core of innate immunity and adaptive immunity is related to TLRs (22).
TLRs were first discovered as Drosophila gene and related to the human immune response (23). They are vital receptors on cells to recognize pathogens and belong to the pattern recognition receptor family (PRRs). They can detect pathogen-associated molecular patterns (PAMPs), such as unmethylated cytosine-phosphate-guanine DNA (CpGor TLR3, TLR7, TLR8, and TLR9 transport) and PRAT4A (responsible for TLR1, TLR2, TLR4, and TLR7 transport). These TLRs can only be functional after transport to the internal lysosome (24). Moreover, TLRs can also be heterodimerized, which expands the range of cognitive ligands. Different TLRs correspond to various endogenous ligands that are TLR4 and TLR2 agonists. The abnormal activation of TLRs may lead to unrestricted inflammatory response (25).
Hitherto, 11 TLRs have been found in humans (Table 1) (26). TLRs, such as TLR1, TLR2, TLR4, TLR6, and TLR10, are expressed on the cell surface and can recognize microbial membrane components, such as p-DNA), single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), lipopolysaccharide (LPS), and flagellin and initiate immune response (27). In addition to the above exogenous ligands, TLRs can also recognize endogenous ligands, including high mobility group box 1 (HMGB1), heat shock proteins (HSP), human cardiac myosin (HCM) peptides S2-16, and HCM S2-28 (28, 29). Moreover, TLRs activate various types of cells and are highly expressed in most immune cells, chondrocytes, endothelial cells, and fibroblasts (30). All TLRs consist of an amino-terminal domain and a carboxyl-terminal Toll/interleukin-1 receptor (TIR) domain. The TIR domain interacts with the junction proteins, including myeloid differentiation factor 88 (MyD88), MyD88 adaptor-like (Mal, also known as TIR domain-containing adapter protein (TIRAP)), TIR domain-containing adaptor inducing IFN-β (TRIF), TRIF-related adaptor molecule (TRAM), and sterile a- and armadillo motif-containing protein (SARM), stimulating nuclear factor-kappa B (NF-κB) and the production of various proinflammatory cytokines, thereby initiating an immune response (31). Some TLRs, including TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, are expressed on the cell surface, while others, including TLR3, TLR7, TLR8, and TLR9, are expressed on the intracellular vesicles (32). Intracellular TLRs exist in the endoplasmic reticulum (ER) and are transported by ER resident proteins to the plasma membrane or lysosomes after stimulation: UNC93B (responsible f proteins, lipids, and participate in the recognition of virus proteins (33).
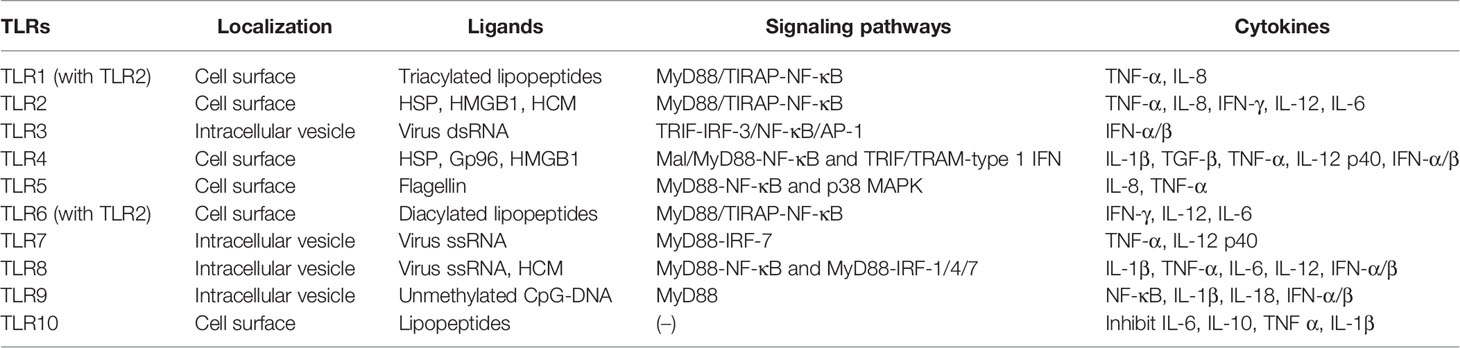
Table 1 Characteristics of TLRs.
TLR2 forms heterodimers with TLR1 or TLR6 and recognizes different TLR ligands, resulting in different functions: dimers combined with TLR1 can recognize triacylated lipopeptides from bacteria while diacylated lipopeptides with TLR6 (34). Both TLR1/2 and TLR2/6 signaling pathways activate downstream inflammatory cytokines, tumor necrosis factor-alpha (TNF-α), (IL-8, IFN-γ, IL-12, and IL-6, through MyD88/Mal-NF-κB signaling pathway (35–37). When recognizing dsRNA, TLR3 transmits signals through TRIF and activates the transcription factor interferon regulatory factor 3 (IRF-3), NF-κB, and AP-1 (the complex of transcription factor 2 and jun), inducing the production of IFN-α/β, cytokines, or chemokines and the maturation of DCs (38, 39). TLR4 is the first molecule identified among TLRs and is mainly expressed in myeloid immune cells and in some non-immune such as endothelial cells (40). TLR4 can recognize heat shock protein, oxidized phospholipid, heparan sulfate, fibrinogen, fibronectin, tendon protein-C, and hyaluronic acid (41). Similar to other TLRs, TLR4 interacts with the intracellular TIR domain responsible for signal transduction (42). It mainly recruits Mal and MyD88 to activate NF-κB and utilizes TRIF and TRAM to activate type 1 IFN to produce proinflammatory factors, such as IL-1β, TGF-β, TNF-α, and IL-12 p40, to eliminate bacteria (43, 44). TLR5 activates the innate immune response against flagella by inducing a MyD88-dependent signaling pathway that stimulates the proinflammatory transcription factor NF-κB in epithelial cells, monocytes, and DCs (45). IL-8 and TNF-α can also be induced by the p38 mitogen-activated protein kinase (MAPK) signaling pathway in response to flagellin infection (46). TLR7 and TLR8 are homologous and located on the X chromosome. Both recognize virus ssRNA and are expressed in various immune cells (47). TLR7 is mainly expressed in plasma-like DCs and B cells, while TLR8 is mainly expressed in monocytes or macrophages, myeloid DCs, and neutrophils (48). Inflammatory factors, such as TNF-α and IL-12 p40 are activated through the MyD88-IRF-7 pathway after TLR7 activation, promoting the innate immune cells to perceive endosomal ssRNA, detecting RNA virus infection (49, 50). However, the overexpression or overactivation of TLR7 promotes the reduction of B cells producing IL-10 in an IFN-γ signal transduction-dependent manner and suppresses the immune response (51). TLR8 induces NF-κB through MyD88 signal transduction and promotes the expression of inflammatory factors, such as IL-1β, TNF-α, IL-6, and IL-12 after recognizing ssRNA. It also induces the production of IFN-α/β through IRAK4, IRAK1, and IRF-7 in response to viral infection (52). TLR9 was first cloned and identified as the receptor of unmethylated CpG-DNA in 2000. It induces the expression of IFN-α/β and proinflammatory cytokines (NF-κB, IL-1β, and IL-18) and activates the immune response only by recruiting MyD88 (53). TLR8 modulates the function of TLR7 on DCs, and TLR9 restrains the response of TLR7 on B cells. TLR7 crosstalk with TLR8 and TLR9 and play a critical role in the immune response of the body (54). Intriguingly, TLR10 is known as an orphan receptor because it lacks classical downstream signaling pathway. It is also an inhibitory receptor, homologous to TLR1 and TLR6, and hence, can form heterodimers with TLR2 and inhibit the production of proinflammatory cytokines, such as IL-6, IL-10, TNF-α, and IL-1β (46). It also inhibits monocyte differentiation, reduces the ability of DCs to stimulate T cells, and suppresses the immune response (55). The human TLR11 gene has no function due to the presence of a stop codon (25).
Naive CD4+T cells can differentiate into different subtypes of CD4+Th cells under the stimulus of cytokines. CD4+Th cells direct the immune response and play key roles in pathogenic infection, chronic inflammation, autoimmune diseases, and cancer. Some studies have found a variety of CD4+Th cells, such as Th1, Th2, Th17, and regulatory T (Treg) cells (Figure 1) (56). Naive CD4+T cells differentiate into Th1 cells post-stimulation of IL-2 and IFN–γ and expression of transcription factors T-bet and secrete IFN-γ, IL-2 and TNF. Th1 cells enhance cell-mediated inflammation and participate in type 1 immune response to intracellular pathogens, such as mycobacteria and viruses (57). Th2 cells are activated by IL-4 and IL-2 and are defined by the expression of transcription factor GATA3, subsequently secreting IL-4, IL-5, IL-6, IL-10, and IL-13. The Th2 cells also participate in type 2 immune response against large extracellular pathogens, such as worms, and play a role in the production of antibodies and allergic reactions (58). Th17 cells, stimulated by TGF-β, IL-6, IL-21, and IL-23 and the expression of transcription factor ROR-γt, produce IL-17, IL-17F, IL-22, and IL-21, which leads to tissue inflammation and promotes participation in type 3 immune response of extracellular pathogens, including bacteria and fungi. Different from other Th cells, Tregs differentiate under the stimulation of IL-10 and TGF-β and the expression of transcription factor Foxp3 to produce anti-inflammatory cytokines, IL-10 and TGF-β (59). Moreover, Tregs inhibit autoimmune diseases and regulate immune response to maintain immune cell homeostasis. Type 1 and 3 immune responses mediate autoimmune diseases, such as SLE, RA, and MES, while type 2 immune responses can lead to allergic diseases, such as asthma (60). Cytokines crosslink each other to maintain Th cells balance. IFN-γ and IL-4 antagonize each other at different levels, and hence the development of Th1 and Th2 cells is mutually exclusive (61). Th17 cells can promote the development of autoimmunity, while Treg cells inhibit autoimmunity; thus, the imbalance of Th17/Treg cells in the body is considered the leading mechanism underlying autoimmune diseases (62).
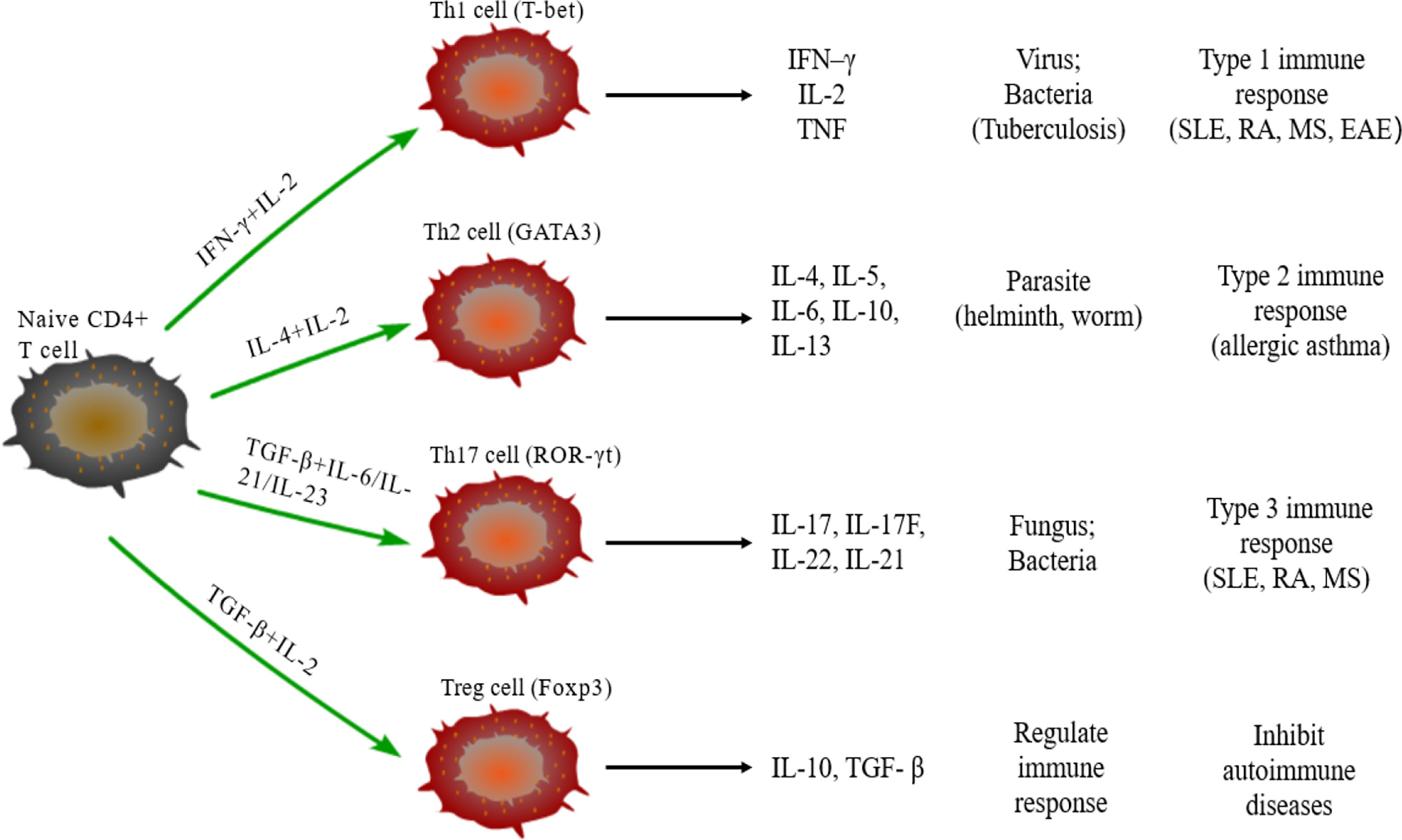
Figure 1 Differentiation of naïve CD4+T cells. Naive CD4+T cells can differentiate into Th1 cells under the stimulus of IFN-γ and IL-2, secrete IFN-γ, IL-2, and TNF, and participate in type 1 immune response. Under the stimulus of IL-2 and IL-4, naive CD4+T cells differentiate into Th2 cells that can secrete IL-4/5/6/10/13 and participate in type 2 immune response. The differentiation of Th17 cells need TGF-β, IL-6, IL-21, and IL-23, and participate in type 3 immune response through secreting IL-17/17F/21/22; TGF-β and IL-2 are required for naïve CD4+T cells to differentiate into Treg cells that secrete IL-10 and TGF-β regulate the immune response.
The activation of TLRs has been shown to bridge innate immunity and acquired immunity. In addition to expression in antigen-presenting cells (DCs and macrophages), TLRs are also expressed in T cells playing a costimulatory role in T cell activation and inducing Th cell differentiation (Figure 2) (63). TLR2 promotes the differentiation of Th17 cells and immune response by disrupting the balance of Th17/Treg cells (64). TLR2/6 ligand is a bacterial lipopeptide that can induce DC tolerance and promote the differentiation of IL-10-producing Tregs through the c-Jun N-terminal kinase (JNK) pathway both in vivo and in vitro. On the other hand, the activation of TLR2/1 promotes the DCs to produce a high level of IL-12 p40 and a low level of IL-10 through p38 MAPK signaling pathway, thereby triggering the differentiation of Th1 or Th 17 cells (65). TLR4 eliminates Th1 response through IRF1 and IFN-α/β receptor-dependent mechanisms. The lack of TLR4 promotes Th1 cell differentiation by enhancing STAT1 pathway, inhibits Th17 cell differentiation by inhibiting STAT3 pathway, and interferes with immune response (66, 67). Bacterial LPS also aggravates allergic inflammation through the production of Th2 cytokines and participates in the immune response of the body post-TLR4 activation (68). Soluble bacterial flagellin stimulates the body to induce Th2 response through TLR5 and inhibits Th1 response to bacterial infection (69). TLR5 promotes DCs in the intestinal tract to differentiate into Th17 cells and respond to pathogen invasion (70). TLR8 induces the expression of IL-12B and IL-23A, promotes the differentiation of IL-23-dependent Th17 cells, and produces immune responses after activating human neutrophils (71). The co-stimulation of TLR7/8 ligands and TLR4 or TLR3 ligands produce IL-12p70 that is the key cytokine to induce Th1 immune response (72). Therefore, ligand co-stimulation is crucial to induce Th1 response. TLR9 is essential in the production of proinflammatory cytokines and other inflammatory responses and to initiate Th1 response and B cell proliferation (73). The interaction between CpG-DNA and TLR9 rapidly activates DCs through the Toll/IL-1 receptor signaling pathway, promoting the differentiation of Th1 cells and the production of cytokines (IL-12 and IL-18) (74). TLR9 ligands also bind to Th cells to promote the proliferation of cells and upregulate the cytokines (75). TLRs-induced immune response is involved in various diseases (9). Presently, several TLR agonists are being tested as adjuvants in the treatment of autoimmune diseases by balancing the immune response.
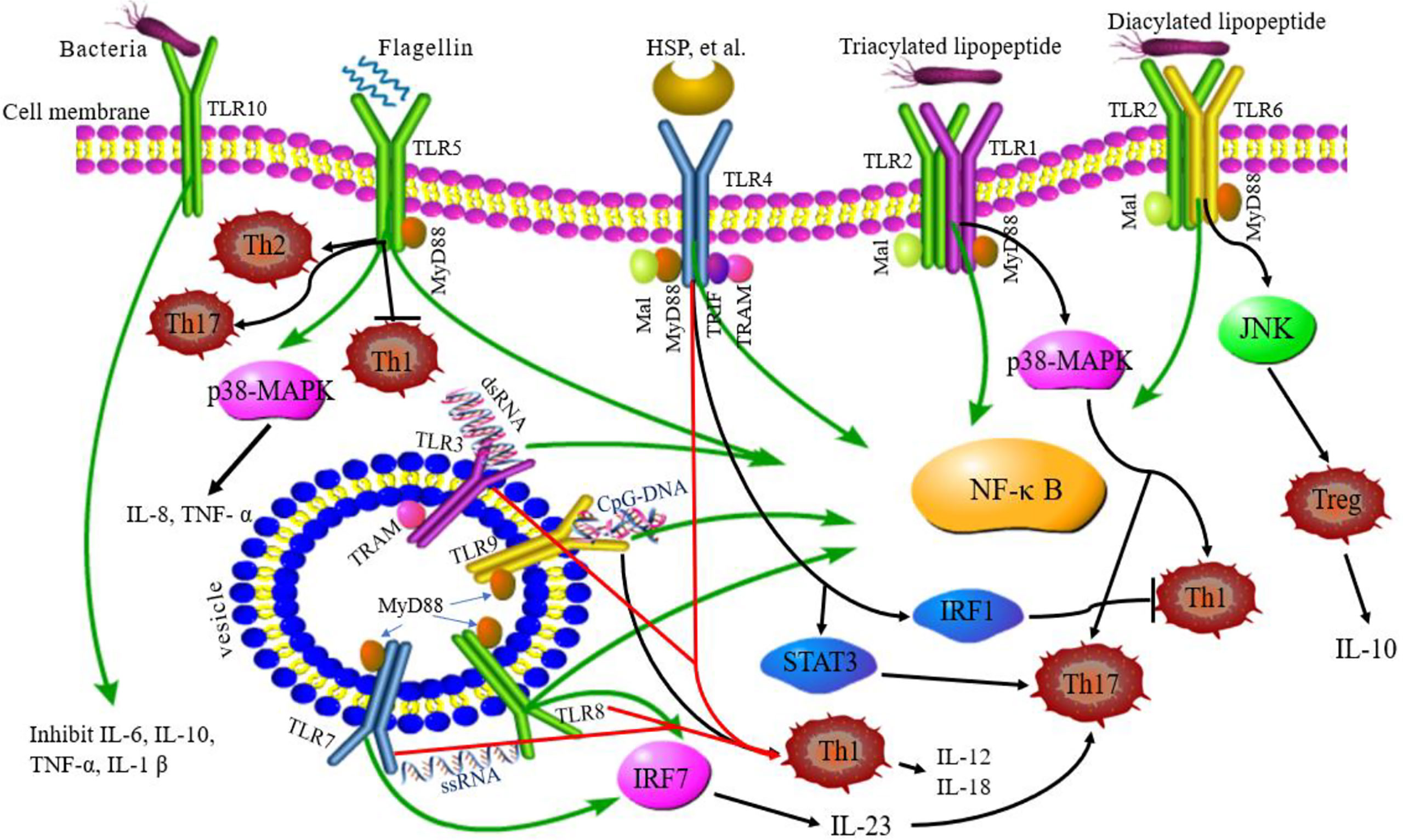
Figure 2 TLRs signaling pathway and related Th responses. TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are expressed on the cell surface, while TLR3, TLR7, TLR8, and TLR9 are expressed on intracellular vesicles. TLRs can recognize different ligands and recruit adapter proteins, MyD88, Mal, TRIF, or TRAM, activating the downstream signaling pathway. TLR10 is an orphan receptor and lacks a classical downstream signaling pathway. Some TLRs promote the differentiation of Th cells. TLR2/6 promotes Treg cell differentiation through the JNK pathway. TLR2/1 promotes Th1 and Th17 cell differentiation through p38 MAPK pathway. TLR4 eliminates the Th1 response through IRF1 and promotes Th17 cell differentiation by STAT3 pathway. TLR5 can stimulate the body to induce Th2 response, inhibit Th1 response, and promote DC differentiation into Th17 cells. TLR8 induces the expression of IL-12B and IL-23A, promotes the differentiation of IL-23-dependent Th17 cells. TLR9 promotes the differentiation of Th1 cells. Green lines: TLR signaling pathways; black lines: TLRs related to Th cell differentiation; red lines: co-stimulation of TLRs induced to Th1 cell.
Th responses play an important role in the pathogenesis of VMC, but different Th responses have different effects on VMC, which may have opposite effects. Besides, the dominant Th responses are different in different stages of VMC.
The imbalance of Th1/Th2 cells can be observed in the process of VMC (Figure 3). Fuse et al. (76) observed the changes in Th1/Th2 ratio of peripheral blood lymphocytes in a patient with acute VMC. In the acute inflammatory phase (day 6), Th1 cells were dominant, while in the recovery phase (days 13 and 20), the proportion of Th2 cells increased. The induction of VMC was related to the dominance of Th1 cells, while the recovery was related to the increased proportion of Th2 cells. However, Th2 immune response induces ventricular remodeling that promotes myocarditis to develop into DCM and HF in the pathogenesis of VMC, while Th1 response alleviates VMC by inhibiting Th2 response and virus replication, but increases acute myocardial inflammation (77). Therefore, when Th2 response begins to be active, the inflammation of VMC decreases, and if Th2 response persists, it will promote myocardial fibrosis and ventricular remodeling. The study also demonstrated that Suramin (a growth factor blocker) inhibits myocardial inflammation in myocarditis by regulating the environment of Th1/Th2 cytokines (78). Therefore, elucidating the Th1/Th2 response might help to understand the activity of VMC. Based on these results, several drugs, such as atorvastatin, tanshinone IIA, apigenin, and cyclooxygenase-2 inhibitors, have been shown to have protective effects on rat model of myocarditis by regulating Th1/Th2 balance (79–82). However, the above drugs can promote Th2 response, which may aggravate the progression of VMC to DMC or HF. Therefore, it is necessary to clarify the therapeutic effect of drugs in the stage from myocarditis to DCM or HF.
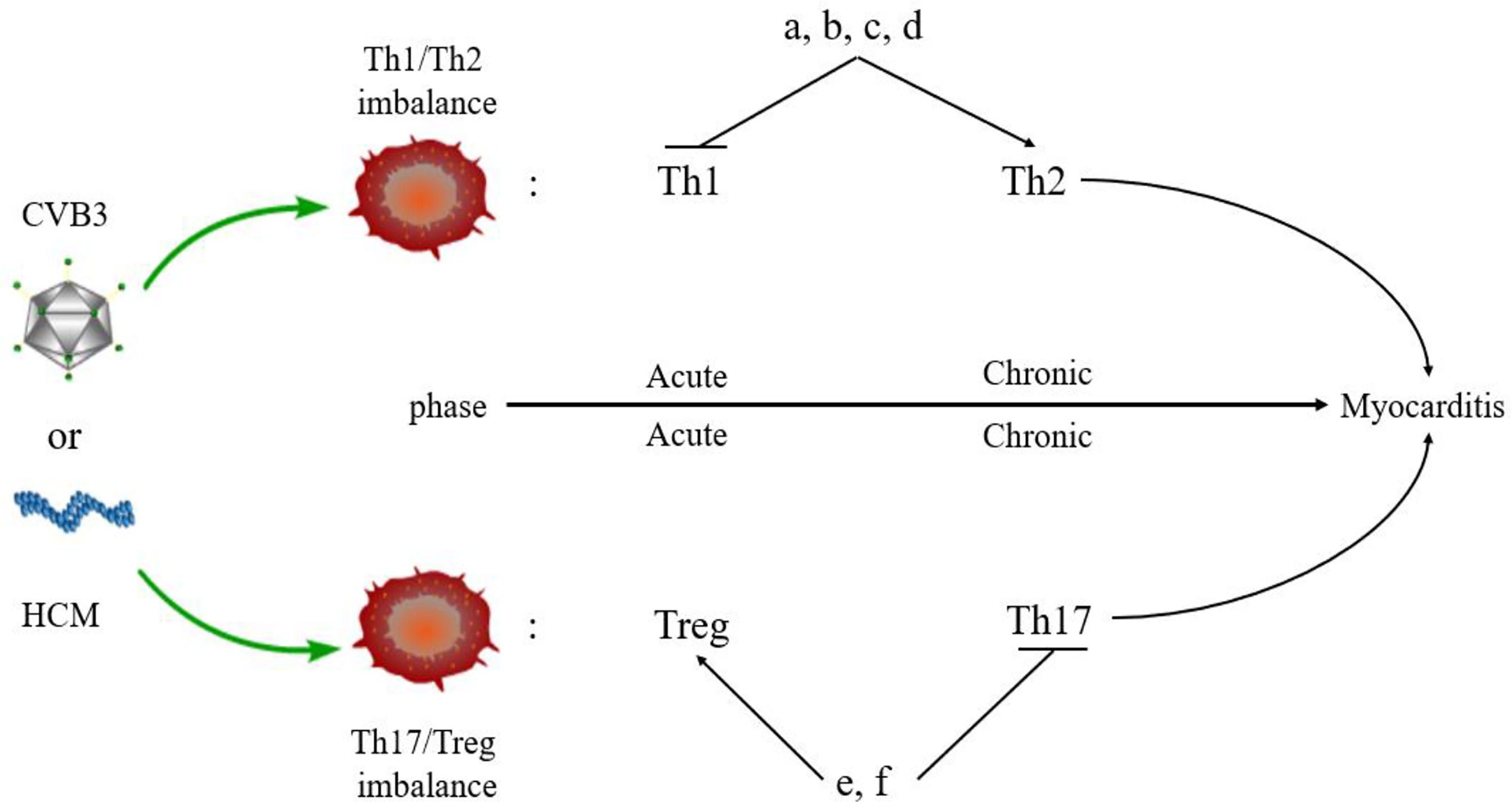
Figure 3 Myocarditis and Th responses. An imbalance of Th cells is observed in the pathogenesis of myocarditis. Th1 and Treg cells are predominant in the acute phase of myocarditis, while Th2 and Th17 cells dominate the chronic phase of myocarditis. Some drugs regulate the balance of Th cells in myocarditis. (A) Atorvastatin; (B) Tanshinone IIA; (C) Apigenin; (D) Cyclooxygenase-2 inhibitors; (E). Valproic acid; (F) Fenofibrate.
Th17 cells secreted IL-17, promoting myocardial fibrosis after myocarditis through protein kinase C β/extracellular signal-regulated kinases 1 and 2/NF-κB pathway, which is an indispensable link in the process of DCM (83). In addition, Tregs can protect mice from CVB3-induced myocarditis progression to cardiomyopathy (84). CVB3 infection stimulates the differentiation of Th17 cells and promotes the secretion of IL-17 by inhibiting the expression of Nucleoporin 98 and aggravating VMC (85). In the acute phase of VMC, Th17 cells stimulate B cells to produce autoantibodies and participate in humoral immune response. The Th2 cells participate in humoral immune response at the late stage of VMC, which is consistent with the above conclusion (86). In the pathogenesis of VMC, the imbalance of Th17/Treg cells plays a critical role in the immune mechanism. MicroRNA-155 (miR-155) is a key regulator of the immune system and promotes the development of myocarditis via differentiation of Th17 cells leading to the imbalance of Th17/Treg cells. The inhibition of miR-155 relieves myocardial injury and the disease (87). Other drugs, such as valproic acid and fenofibrate have also been found to inhibit inflammation, reduce CVB3-induced VMC, and improve prognosis by directly inhibiting the differentiation of Th17 cells (88). Thus, VMC can be treated by promoting the differentiation of Treg cells and regulating the balance of Th17/Treg cells (89). In addition, estrogen inhibits the differentiation of Th17 cells that are mainly induced in males with CVB3 infection but less in females. Thus, Th17 cells show gender bias in myocarditis: the incidence of myocarditis has a male-to-female ratio of 2:1 (90). This indirectly indicates that Th17/Treg cell balance plays a key role in the epidemiological characteristics of myocarditis.
As key members of PRRs, TLRs participate in the upstream signaling pathway that activates innate immune cells and T cells, resulting in the production of proinflammatory cytokines and the activation of T cells. TLRs are considered to be the main factors in the development of autoimmunity, participating in and promoting the occurrence of autoimmune inflammatory diseases (91). The above observations indicate gender differences in the incidence of VMC. Roberts et al. (92) demonstrated that high expression of TLR2 in early infected female mice exerted a protective effect, while that of TLR4 in male mice was lethal. This differential expression between genders resulted in disease resistance in female mice and susceptibility in male mice (Figure 4). Hence, TLRs are deemed to play a critical role in gender difference with respect to myocarditis and understanding the underlying mechanisms would illuminate the epidemiological characteristic of myocarditis (93). TLR3 recognizes dsRNA intermediates produced during CVB3 replication and activates TRIF and TRAF6 to transmit signals to NF-κB (94). TLR3-TRIF signaling pathway helps the host to defend against CVB3 infection. The mechanism might be ascribed to the induction of type II IFN expression, rather than IFN-α/β, and TLR3-TRAF6-III IFN signaling pathway also has antiviral effects (95, 96). The lack of TLR3 increases virus replication and aggravates myocardial inflammation. It also worsens cardiac function and increases the susceptibility to CVB3 (97). The genetic variation of TLR3 affects the host’s susceptibility facing VMC by inhibiting the signal transduction of NF-κB (21). These results proved that TLR3 has a protective effect on the myocardium in the process of virus infection. In addition, neutrophils also interact with and recognize CVB3 through TLR8, activating NF-κB and its downstream factors, resulting in VMC development (98). It also upregulates the expression of TLR4, promotes the expression of NF-κB, and induces myocarditis (99). Based on these results, astragalus polysaccharides have been shown to protect TLR4-induced myocardial injury and inflammation by inhibiting the CVB3-related signaling pathway (100), which provides a potential target to treat myocarditis. TLR7 preferentially promotes the differentiation of Th17 cells and the expression of inflammatory factors, such as IL-17 after CVB3 infection, while TLR8 promotes the production of Th1 cytokines and IFN-α/β response, which are involved in the pathogenesis of myocarditis (101). The potent autoantigen HCM is released from damaged heart during viral infection. HCM peptides S2-16 and S2-28, as an endogenous ligands, can bind to TLR2 and TLR8, and promote the release of pro-inflammatory factors such as IL-8, IL-6, IL-23 and TGF-β, which mainly induce the differentiation of Th17 cells and contribute to DCM or HF (29, 102). Although TLR9 can recognize various DNA viruses unlike the indirect way of recognizing RNA viruses, TLR9-MyD88 signaling pathway mediates myocardial injury in acute phase rather than chronic phase CVB3-induced myocarditis (103). Nonetheless, the mechanisms of other TLRs in myocarditis are yet to be clarified.
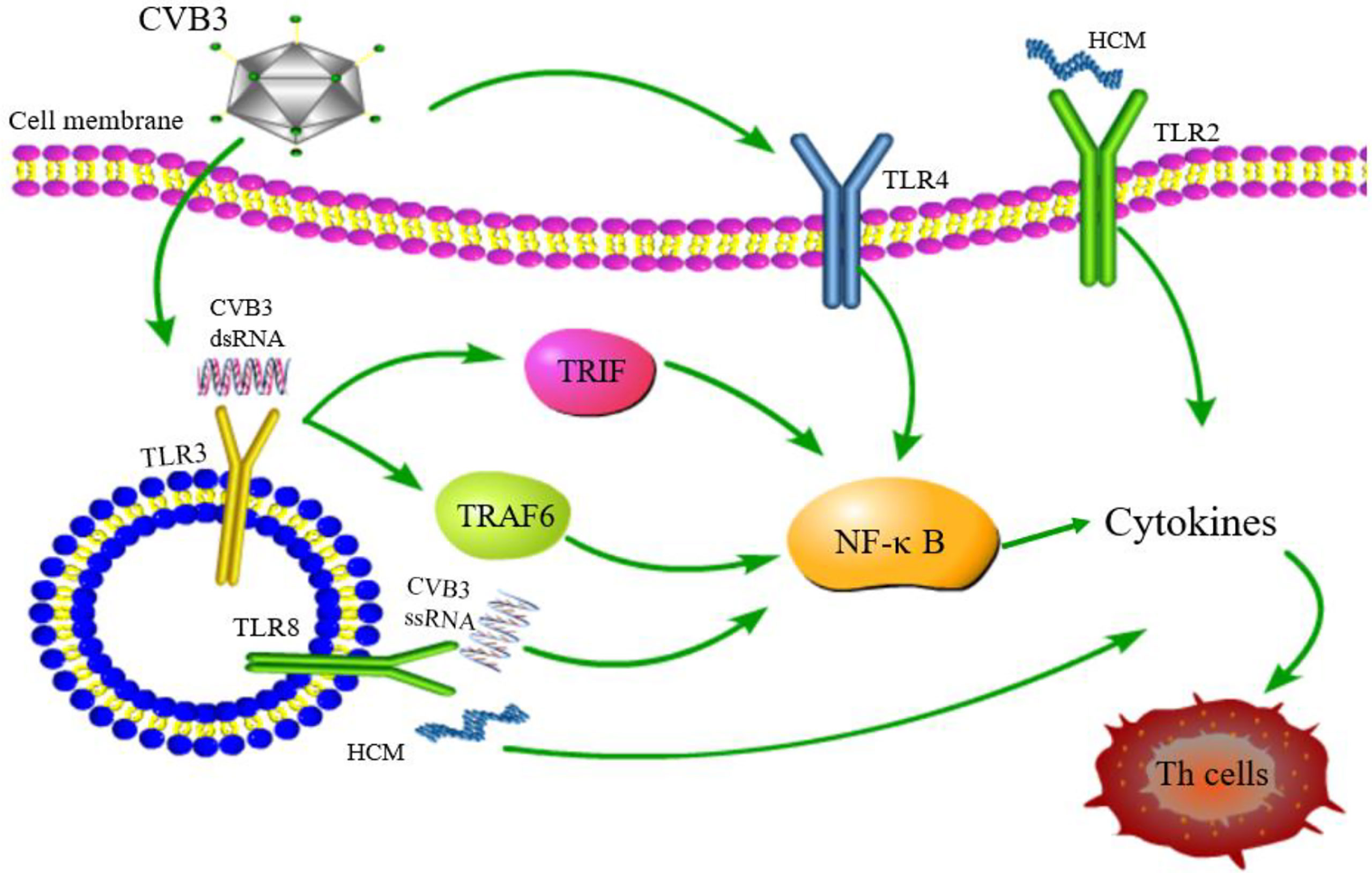
Figure 4 TLR signaling pathway related to CVB3 infection. TLR4/NF-κB is activated after CVB3 infection; TLR3 recognizes CVB3 dsRNA and activates TRIF and TRAF6 to transmit signals to NF-κB; TLR8 recognizes CVB3 ssRNA and activates NF-κB signaling pathway. TLR2 and TLR8 recognizes HCM and promotes the differentiation of Th cells. Although other TLRs may be related to the pathogenesis of myocarditis, whether other TLRs can be activated and its downstream pathway after CVB3 infection has not been reported.
TLRs and Th responses play a critical role in the pathogenesis of VMC and have become the focus of current research. TLRs are a new class of innate immune receptors that mediate CD4+T cell differentiation, induce Th1 and Th2 immune responses, and participate in VMC pathogenesis. Except that CVB3 can directly bind to TLRs to promote Th responses, the release of HCM from damaged heart can also promote DCM or HF through TLRs and Th responses after viral infection. Blocking or activating a single TLR or regulating TLR signaling pathway may affect innate immunity, host resistance, and VMC pathogenesis, indicating that specific TLRs agonists or antagonists comprise new immunotherapy for VMC. In addition, some anti-inflammatory drugs have been found to reduce myocardial injury and improve VMC by interfering with TLR signaling pathways and Th immune responses. However, the role of other TLRs and Th responses in VMC has not yet been reported. Therefore, clarifying the role of TLRs and Th responses in VMC can provide novel ideas for the treatment of VMC.
All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Huber SA. Viral Myocarditis and Dilated Cardiomyopathy: Etiology and Pathogenesis. Curr Pharm Des (2016) 22:408–26. doi: 10.2174/1381612822666151222160500
2. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular Mimicry, Bystander Activation, or Viral Persistence: Infections and Autoimmune Disease. Clin Microbiol Rev (2006) 19:80–94. doi: 10.1128/CMR.19.1.80-94.2006
3. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current State of Knowledge on Aetiology, Diagnosis, Management, and Therapy of Myocarditis: A Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J (2013) 34:2636–48, 48a-48d. doi: 10.1093/eurheartj/eht210
4. Ammirati E, Buono A, Moroni F, Gigli L, Power JR, Ciabatti M, et al. State-Of-the-Art of Endomyocardial Biopsy on Acute Myocarditis and Chronic Inflammatory Cardiomyopathy. Curr Cardiol Rep (2022). doi: 10.1007/s11886-022-01680-x
5. Basso C, Calabrese F, Angelini A, Carturan E, Thiene G. Classification and Histological, Immunohistochemical, and Molecular Diagnosis of Inflammatory Myocardial Disease. Heart Fail Rev (2013) 18:673–81. doi: 10.1007/s10741-012-9355-6
6. Friedrich MG, Sechtem U, Schulz-Menger J, Holmvang G, Alakija P, Cooper LT, et al. Cardiovascular Magnetic Resonance in Myocarditis: A JACC White Paper. J Am Coll Cardiol (2009) 53:1475–87. doi: 10.1016/j.jacc.2009.02.007
7. Luetkens JA, Homsi R, Sprinkart AM, Doerner J, Dabir D, Kuetting DL, et al. Incremental Value of Quantitative CMR Including Parametric Mapping for the Diagnosis of Acute Myocarditis. Eur Heart J Cardiovasc Imaging (2016) 17:154–61. doi: 10.1093/ehjci/jev246
8. Ammirati E, Frigerio M, Adler ED, Basso C, Birnie DH, Brambatti M, et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ Heart Fail (2020) 13:e007405. doi: 10.1161/CIRCHEARTFAILURE.120.007405
9. Vijay K. Toll-Like Receptors in Immunity and Inflammatory Diseases: Past, Present, and Future. Int Immunopharmacol (2018) 59:391–412. doi: 10.1016/j.intimp.2018.03.002
10. Takeda K, Akira S. Toll-Like Receptors. Curr Protoc Immunol (2015) 109:14121–141210. doi: 10.1002/0471142735.im1412s109
11. Heidecker B, Kittleson MM, Kasper EK, Wittstein IS, Champion HC, Russell SD, et al. Transcriptomic Biomarkers for the Accurate Diagnosis of Myocarditis. Circulation (2011) 123:1174–84. doi: 10.1161/CIRCULATIONAHA.110.002857
12. Lasrado N, Reddy J. An Overview of the Immune Mechanisms of Viral Myocarditis. Rev Med Virol (2020) 30:1–14. doi: 10.1002/rmv.2131
13. Narovlyanskaya O, Winokur EJ. Viral Myocarditis. Dimens Crit Care Nurs (2020) 39:75–80. doi: 10.1097/DCC.0000000000000402
14. Hare JM, Baughman KL. Fulminant and Acute Lymphocytic Myocarditis: The Prognostic Value of Clinicopathological Classification. Eur Heart J (2001) 22:269–70. doi: 10.1053/euhj.2000.2272
15. Fung G, Luo H, Qiu Y, Yang D, McManus B. Myocarditis. Circ Res (2016) 118:496–514. doi: 10.1161/CIRCRESAHA.115.306573
16. Olejniczak M, Schwartz M, Webber E, Shaffer A, Perry TE. Viral Myocarditis-Incidence, Diagnosis and Management. J Cardiothorac Vasc Anesth (2020) 34:1591–601. doi: 10.1053/j.jvca.2019.12.052
17. Kociol RD, Cooper LT, Fang JC, Moslehi JJ, Pang PS, Sabe MA, et al. Recognition and Initial Management of Fulminant Myocarditis: A Scientific Statement From the American Heart Association. Circulation (2020) 141:e69–92. doi: 10.1161/CIR.0000000000000745
18. Pollack A, Kontorovich AR, Fuster V, Dec GW. Viral Myocarditis–Diagnosis, Treatment Options, and Current Controversies. Nat Rev Cardiol (2015) 12:670–80. doi: 10.1038/nrcardio.2015.108
19. Imanaka-Yoshida K. Inflammation in Myocardial Disease: From Myocarditis to Dilated Cardiomyopathy. Pathol Int (2020) 70:1–11. doi: 10.1111/pin.12868
20. Seferovic PM, Tsutsui H, McNamara DM, Ristic AD, Basso C, Bozkurt B, et al. Heart Failure Association of the ESC, Heart Failure Society of America and Japanese Heart Failure Society Position Statement on Endomyocardial Biopsy. Eur J Heart Fail (2021) 23:854–71. doi: 10.1002/ejhf.2190
21. Gorbea C, Makar KA, Pauschinger M, Pratt G, Bersola JL, Varela J, et al. A Role for Toll-Like Receptor 3 Variants in Host Susceptibility to Enteroviral Myocarditis and Dilated Cardiomyopathy. J Biol Chem (2010) 285:23208–23. doi: 10.1074/jbc.M109.047464
22. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-Like Receptors: Significance, Ligands, Signaling Pathways, and Functions in Mammals. Int Rev Immunol (2018) 37:20–36. doi: 10.1080/08830185.2017.1380200
23. Muzio M, Polentarutti N, Bosisio D, Manoj Kumar PP, Mantovani A. Toll-Like Receptor Family and Signalling Pathway. Biochem Soc Trans (2000) 28:563–6. doi: 10.1042/bst0280563
24. Lim KH, Staudt LM. Toll-Like Receptor Signaling. Cold Spring Harb Perspect Biol (2013) 5:a011247. doi: 10.1101/cshperspect.a011247
25. Yu L, Wang L, Chen S. Endogenous Toll-Like Receptor Ligands and Their Biological Significance. J Cell Mol Med (2010) 14:2592–603. doi: 10.1111/j.1582-4934.2010.01127.x
26. Kumar V. Toll-Like Receptors in Adaptive Immunity. Handb Exp Pharmacol (2021). doi: 10.1007/164_2021_543
27. Wagner H. Endogenous TLR Ligands and Autoimmunity. Adv Immunol (2006) 91:159–73. doi: 10.1016/S0065-2776(06)91004-9
28. Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-Like Receptors, Endogenous Ligands, and Systemic Autoimmune Disease. Immunol Rev (2005) 204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x
29. Zhang P, Cox CJ, Alvarez KM, Cunningham MW. Cutting Edge: Cardiac Myosin Activates Innate Immune Responses Through TLRs. J Immunol (2009) 183:27–31. doi: 10.4049/jimmunol.0800861
30. Chen JQ, Szodoray P, Zeher M. Toll-Like Receptor Pathways in Autoimmune Diseases. Clin Rev Allergy Immunol (2016) 50:1–17. doi: 10.1007/s12016-015-8473-z
31. McGettrick AF, O'Neill LA. Localisation and Trafficking of Toll-Like Receptors: An Important Mode of Regulation. Curr Opin Immunol (2010) 22:20–7. doi: 10.1016/j.coi.2009.12.002
32. Noreen M, Arshad M. Association of TLR1, TLR2, TLR4, TLR6, and TIRAP Polymorphisms With Disease Susceptibility. Immunol Res (2015) 62:234–52. doi: 10.1007/s12026-015-8640-6
33. Zhou R, Liu L, Wang Y. Viral Proteins Recognized by Different TLRs. J Med Virol (2021) 93:6116–23. doi: 10.1002/jmv.27265
34. Deng Y, Yang J, Qian J, Liu R, Huang E, Wang Y, et al. TLR1/TLR2 Signaling Blocks the Suppression of Monocytic Myeloid-Derived Suppressor Cell by Promoting its Differentiation Into M1-Type Macrophage. Mol Immunol (2019) 112:266–73. doi: 10.1016/j.molimm.2019.06.006
35. Cervantes JL, Weinerman B, Basole C, Salazar JC. TLR8: The Forgotten Relative Revindicated. Cell Mol Immunol (2012) 9:434–8. doi: 10.1038/cmi.2012.38
36. Kim J, Durai P, Jeon D, Jung ID, Lee SJ, Park YM, et al. Phloretin as a Potent Natural TLR2/1 Inhibitor Suppresses TLR2-Induced Inflammation. Nutrients (2018) 10:868. doi: 10.3390/nu10070868
37. Marinho FA, de Paula RR, Mendes AC, de Almeida LA, Gomes MT, Carvalho NB, et al. Toll-Like Receptor 6 Senses Mycobacterium Avium and is Required for Efficient Control of Mycobacterial Infection. Eur J Immunol (2013) 43:2373–85. doi: 10.1002/eji.201243208
38. Matsumoto M, Seya T. TLR3: Interferon Induction by Double-Stranded RNA Including Poly(I:C). Adv Drug Delivery Rev (2008) 60:805–12. doi: 10.1016/j.addr.2007.11.005
39. Mukherjee S, Huda S, Sinha Babu SP. Toll-Like Receptor Polymorphism in Host Immune Response to Infectious Diseases: A Review. Scand J Immunol (2019) 90:e12771. doi: 10.1111/sji.12771
40. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 Trafficking and its Influence on LPS-Induced Pro-Inflammatory Signaling. Cell Mol Life Sci (2021) 78:1233–61. doi: 10.1007/s00018-020-03656-y
41. Lai CY, Su YW, Lin KI, Hsu LC, Chuang TH. Natural Modulators of Endosomal Toll-Like Receptor-Mediated Psoriatic Skin Inflammation. J Immunol Res (2017) 2017:7807313. doi: 10.1155/2017/7807313
42. Kuzmich NN, Sivak KV, Chubarev VN, Porozov YB, Savateeva-Lyubimova TN, Peri F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines (Basel) (2017) 5:34. doi: 10.3390/vaccines5040034
43. Plociennikowska A, Hromada-Judycka A, Borzecka K, Kwiatkowska K. Co-Operation of TLR4 and Raft Proteins in LPS-Induced Pro-Inflammatory Signaling. Cell Mol Life Sci (2015) 72:557–81. doi: 10.1007/s00018-014-1762-5
44. Vaure C, Liu Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front Immunol (2014) 5:316. doi: 10.3389/fimmu.2014.00316
45. Yoon SI, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, et al. Structural Basis of TLR5-Flagellin Recognition and Signaling. Science (2012) 335:859–64. doi: 10.1126/science.1215584
46. Yu Y, Zeng H, Lyons S, Carlson A, Merlin D, Neish AS, et al. TLR5-Mediated Activation of P38 MAPK Regulates Epithelial IL-8 ExpressionVia Posttranscriptional Mechanism. Am J Physiol Gastrointest Liver Physiol (2003) 285:G282–90. doi: 10.1152/ajpgi.00503.2002
47. Wang J, Shao Y, Bennett TA, Shankar RA, Wightman PD, Reddy LG. The Functional Effects of Physical Interactions Among Toll-Like Receptors 7, 8, and 9. J Biol Chem (2006) 281:37427–34. doi: 10.1074/jbc.M605311200
48. Eng HL, Hsu YY, Lin TM. Differences in TLR7/8 Activation Between Monocytes and Macrophages. Biochem Biophys Res Commun (2018) 497:319–25. doi: 10.1016/j.bbrc.2018.02.079
49. Diebold SS, Kaisho T, Hemmi H, Akira S, Sousa CR. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science (2004) 303:1529–31. doi: 10.1126/science.1093616
50. Javmen A, Szmacinski H, Lakowicz JR, Toshchakov VY. Frontline Science: Targeting the TLR7 Signalosome Assembly. J Leukoc Biol (2020) 108:1697–706. doi: 10.1002/JLB.2HI0819-180R
51. Chodisetti SB, Fike AJ, Domeier PP, Choi NM, Soni C, Rahman ZSM. TLR7 Negatively Regulates B10 Cells Predominantly in an IFNgamma Signaling Dependent Manner. Front Immunol (2020) 11:1632. doi: 10.3389/fimmu.2020.01632
52. Saruta M, Michelsen KS, Thomas LS, Yu QT, Landers CJ, Targan SR. TLR8-Mediated Activation of Human Monocytes Inhibits TL1A Expression. Eur J Immunol (2009) 39:2195–202. doi: 10.1002/eji.200939216
53. Muller T, Hamm S, Bauer S. TLR9-Mediated Recognition of DNA. Handb Exp Pharmacol (2008) 183:51–70. doi: 10.1007/978-3-540-72167-3_3
54. Desnues B, Macedo AB, Roussel-Queval A, Bonnardel J, Henri S, Demaria O, et al. TLR8 on Dendritic Cells and TLR9 on B Cells Restrain TLR7-Mediated Spontaneous Autoimmunity in C57BL/6 Mice. Proc Natl Acad Sci USA (2014) 111:1497–502. doi: 10.1073/pnas.1314121111
55. Hess NJ, Felicelli C, Grage J, Tapping RI. TLR10 Suppresses the Activation and Differentiation of Monocytes With Effects on DC-Mediated Adaptive Immune Responses. J Leukoc Biol (2017) 101:1245–52. doi: 10.1189/jlb.3A1116-492R
56. Zhu J. T Helper Cell Differentiation, Heterogeneity, and Plasticity. Cold Spring Harb Perspect Biol (2018) 10:a030338. doi: 10.1101/cshperspect.a030338
57. Gagliani N, Huber S. Basic Aspects of T Helper Cell Differentiation. Methods Mol Biol (2017) 1514:19–30. doi: 10.1007/978-1-4939-6548-9_2
58. Zhu X, Zhu J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int J Mol Sci (2020) 21:8011. doi: 10.3390/ijms21218011
59. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
60. Annunziato F, Romagnani C, Romagnani S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J Allergy Clin Immunol (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
61. Cunningham MW. Cardiac Myosin and the TH1/TH2 Paradigm in Autoimmune Myocarditis. Am J Pathol (2001) 159:5–12. doi: 10.1016/S0002-9440(10)61665-3
62. Lee GR. The Balance of Th17 Versus Treg Cells in Autoimmunity. Int J Mol Sci (2018) 19:730. doi: 10.3390/ijms19030730
63. Jin B, Sun T, Yu XH, Yang YX, Yeo AE. The Effects of TLR Activation on T-Cell Development and Differentiation. Clin Dev Immunol (2012) 2012:836485. doi: 10.1155/2012/836485
64. Nyirenda MH, Sanvito L, Darlington PJ, O'Brien K, Zhang GX, Constantinescu CS, et al. TLR2 Stimulation Drives Human Naive and Effector Regulatory T Cells Into a Th17-Like Phenotype With Reduced Suppressive Function. J Immunol (2011) 187:2278–90. doi: 10.4049/jimmunol.1003715
65. Kamdar K, Nguyen V, DePaolo RW. Toll-Like Receptor Signaling and Regulation of Intestinal Immunity. Virulence (2013) 4:207–12. doi: 10.4161/viru.23354
66. Sacramento LA, Benevides L, Maruyama SR, Tavares L, Fukutani KF, Francozo M, et al. TLR4 Abrogates the Th1 Immune Response Through IRF1 and IFN-Beta to Prevent Immunopathology During L. infantum infection. PloS Pathog (2020) 16:e1008435. doi: 10.1371/journal.ppat.1008435
67. Xu QQ, Zhou Q, Xu LL, Lin H, Wang XJ, Ma WL, et al. Toll-Like Receptor 4 Signaling Inhibits Malignant Pleural Effusion by Altering Th1/Th17 Responses. Cell Biol Int (2015) 39:1120–30. doi: 10.1002/cbin.10485
68. Lee AJ, Ro M, Cho KJ, Kim JH. Lipopolysaccharide/TLR4 Stimulates IL-13 Production Through a MyD88-BLT2-Linked Cascade in Mast Cells, Potentially Contributing to the Allergic Response. J Immunol (2017) 199:409–17. doi: 10.4049/jimmunol.1602062
69. Flores-Langarica A, Bobat S, Marshall JL, Yam-Puc JC, Cook CN, Serre K, et al. Soluble Flagellin Coimmunization Attenuates Th1 Priming to Salmonella and Clearance by Modulating Dendritic Cell Activation and Cytokine Production. Eur J Immunol (2015) 45:2299–311. doi: 10.1002/eji.201545564
70. Liu H, Chen F, Wu W, Cao AT, Xue X, Yao S, et al. TLR5 Mediates CD172alpha(+) Intestinal Lamina Propria Dendritic Cell Induction of Th17 Cells. Sci Rep (2016) 6:22040. doi: 10.1038/srep22040
71. Tamassia N, Arruda-Silva F, Wright HL, Moots RJ, Gardiman E, Bianchetto-Aguilera F, et al. Human Neutrophils Activated via TLR8 Promote Th17 Polarization Through IL-23. J Leukoc Biol (2019) 105:1155–65. doi: 10.1002/JLB.MA0818-308R
72. Bekeredjian-Ding I, Roth SI, Gilles S, Giese T, Ablasser A, Hornung V, et al. T Cell-Independent, TLR-Induced IL-12p70 Production in Primary Human Monocytes. J Immunol (2006) 176:7438–46. doi: 10.4049/jimmunol.176.12.7438
73. Kumagai Y, Takeuchi O, Akira S. TLR9 as a Key Receptor for the Recognition of DNA. Adv Drug Delivery Rev (2008) 60:795–804. doi: 10.1016/j.addr.2007.12.004
74. Wagner H. Interactions Between Bacterial CpG-DNA and TLR9 Bridge Innate and Adaptive Immunity. Curr Opin Microbiol (2002) 5:62–9. doi: 10.1016/s1369-5274(02)00287-4
75. Sharma RK, Sehgal S, Sachdeva N, Kumar R, Gupta A. Direct Engagement of TLR9 Ligand With T Helper Cells Leads to Cell Proliferation & Up-Regulation of Cytokines. Immunol Invest (2019) 48:79–95. doi: 10.1080/08820139.2018.1515223
76. Fuse K, Kodama M, Aizawa Y, Yamaura M, Tanabe Y, Takahashi K, et al. Th1/Th2 Balance Alteration in the Clinical Course of a Patient With Acute Viral Myocarditis. Jpn Circ J (2001) 65:1082–4. doi: 10.1253/jcj.65.1082
77. Fairweather D, Stafford KA, Sung YK. Update on Coxsackievirus B3 Myocarditis. Curr Opin Rheumatol (2012) 24:401–7. doi: 10.1097/BOR.0b013e328353372d
78. Fuse K, Kodama M, Ito M, Okura Y, Kato K, Hanawa H, et al. Polarity of Helper T Cell Subsets Represents Disease Nature and Clinical Course of Experimental Autoimmune Myocarditis in Rats. Clin Exp Immunol (2003) 134:403–8. doi: 10.1111/j.1365-2249.2003.02312.x
79. Guo G, Zhao Q, Wang Q, Li E. Tanshinone IIA Ameliorate Coxsackie Virus B3-Induced Viral Myocarditis Through the Inhibition of Inflammation and Modulation T Helper 1/T Helper 2 Balance in Mice. Pharmacology (2019) 103:136–42. doi: 10.1159/000495755
80. Liu W, Li WM, Gao C, Sun NL. Effects of Atorvastatin on the Th1/Th2 Polarization of Ongoing Experimental Autoimmune Myocarditis in Lewis Rats. J Autoimmun (2005) 25:258–63. doi: 10.1016/j.jaut.2005.06.005
81. Suzuki J, Ogawa M, Futamatsu H, Kosuge H, Tanaka H, Isobe M. A Cyclooxygenase-2 Inhibitor Alters Th1/Th2 Cytokine Balance and Suppresses Autoimmune Myocarditis in Rats. J Mol Cell Cardiol (2006) 40:688–95. doi: 10.1016/j.yjmcc.2006.01.006
82. Zhang S, Liu X, Sun C, Yang J, Wang L, Liu J, et al. Apigenin Attenuates Experimental Autoimmune Myocarditis by Modulating Th1/Th2 Cytokine Balance in Mice. Inflammation (2016) 39:678–86. doi: 10.1007/s10753-015-0294-y
83. Liu Y, Zhu H, Su Z, Sun C, Yin J, Yuan H, et al. IL-17 Contributes to Cardiac Fibrosis Following Experimental Autoimmune Myocarditis by a PKCbeta/Erk1/2/NF-kappaB-Dependent Signaling Pathway. Int Immunol (2012) 24:605–12. doi: 10.1093/intimm/dxs056
84. Vdovenko D, Eriksson U. Regulatory Role of CD4(+) T Cells in Myocarditis. J Immunol Res (2018) 2018:4396351. doi: 10.1155/2018/4396351
85. Long Q, Liao YH, Xie Y, Liang W, Cheng X, Yuan J, et al. Coxsackievirus B3 Directly Induced Th17 Cell Differentiation by Inhibiting Nup98 Expression in Patients With Acute Viral Myocarditis. Front Cell Infect Microbiol (2016) 6:171. doi: 10.3389/fcimb.2016.00171
86. Yuan J, Cao AL, Yu M, Lin QW, Yu X, Zhang JH, et al. Th17 Cells Facilitate the Humoral Immune Response in Patients With Acute Viral Myocarditis. J Clin Immunol (2010) 30:226–34. doi: 10.1007/s10875-009-9355-z
87. Yan L, Hu F, Yan X, Wei Y, Ma W, Wang Y, et al. Inhibition of microRNA-155 Ameliorates Experimental Autoimmune Myocarditis by Modulating Th17/Treg Immune Response. J Mol Med (Berl) (2016) 94:1063–79. doi: 10.1007/s00109-016-1414-3
88. Jin H, Guo X. Valproic Acid Ameliorates Coxsackievirus-B3-Induced Viral Myocarditis by Modulating Th17/Treg Imbalance. Virol J (2016) 13:168. doi: 10.1186/s12985-016-0626-z
89. Cheng H, Xi Y, Chi X, Wu Y, Liu G. Fenofibrate Treatment of Rats With Experimental Autoimmune Myocarditis by Alleviating Treg/Th17 Disorder. Cent Eur J Immunol (2016) 41:64–70. doi: 10.5114/ceji.2016.58817
90. Li Z, Yue Y, Xiong S. Distinct Th17 Inductions Contribute to the Gender Bias in CVB3-Induced Myocarditis. Cardiovasc Pathol (2013) 22:373–82. doi: 10.1016/j.carpath.2013.02.004
91. Mills KH. TLR-Dependent T Cell Activation in Autoimmunity. Nat Rev Immunol (2011) 11:807–22. doi: 10.1038/nri3095
92. Roberts BJ, Dragon JA, Moussawi M, Huber SA. Sex-Specific Signaling Through Toll-Like Receptors 2 and 4 Contributes to Survival Outcome of Coxsackievirus B3 Infection in C57Bl/6 Mice. Biol Sex Differ (2012) 3:25. doi: 10.1186/2042-6410-3-25
93. Roberts BJ, Moussawi M, Huber SA. Sex Differences in TLR2 and TLR4 Expression and Their Effect on Coxsackievirus-Induced Autoimmune Myocarditis. Exp Mol Pathol (2013) 94:58–64. doi: 10.1016/j.yexmp.2012.06.005
94. Marchant D, Si X, Luo H, McManus B, Yang D. The Impact of CVB3 Infection on Host Cell Biology. Curr Top Microbiol Immunol (2008) 323:177–98. doi: 10.1007/978-3-540-75546-3_8
95. Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, et al. A Critical Link Between Toll-Like Receptor 3 and Type II Interferon Signaling Pathways in Antiviral Innate Immunity. Proc Natl Acad Sci USA (2008) 105:20446–51. doi: 10.1073/pnas.0810372105
96. Su R, Shereen MA, Zeng X, Liang Y, Li W, Ruan Z, et al. The TLR3/IRF1/Type III IFN Axis Facilitates Antiviral Responses Against Enterovirus Infections in the Intestine. mBio (2020) 11:e02540–20. doi: 10.1128/mBio.02540-20
97. Abston ED, Coronado MJ, Bucek A, Onyimba JA, Brandt JE, Frisancho JA, et al. TLR3 Deficiency Induces Chronic Inflammatory Cardiomyopathy in Resistant Mice Following Coxsackievirus B3 Infection: Role for IL-4. Am J Physiol Regul Integr Comp Physiol (2013) 304:R267–77. doi: 10.1152/ajpregu.00516.2011
98. Rivadeneyra L, Charo N, Kviatcovsky D, de la Barrera S, Gomez RM, Schattner M. Role of Neutrophils in CVB3 Infection and Viral Myocarditis. J Mol Cell Cardiol (2018) 125:149–61. doi: 10.1016/j.yjmcc.2018.08.029
99. Zhao Z, Cai TZ, Lu Y, Liu WJ, Cheng ML, Ji YQ. Coxsackievirus B3 Induces Viral Myocarditis by Upregulating Toll-Like Receptor 4 Expression. Biochem (Mosc) (2015) 80:455–62. doi: 10.1134/S0006297915040094
100. Liu T, Zhang M, Niu H, Liu J, Ruilian M, Wang Y, et al. Astragalus Polysaccharide From Astragalus Melittin Ameliorates InflammationVia Suppressing the Activation of TLR-4/NF-kappaB P65 Signal Pathway and Protects Mice From CVB3-Induced Virus Myocarditis. Int J Biol Macromol (2019) 126:179–86. doi: 10.1016/j.ijbiomac.2018.12.207
101. de Marcken M, Dhaliwal K, Danielsen AC, Gautron AS, Dominguez-Villar M. TLR7 and TLR8 Activate Distinct Pathways in Monocytes During RNA Virus Infection. Sci Signal (2019) 12:eaaw1347. doi: 10.1126/scisignal.aaw1347
102. Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, et al. Cardiac Myosin-Th17 Responses Promote Heart Failure in Human Myocarditis. JCI Insight (2016) 1:e85851. doi: 10.1172/jci.insight.85851
Keywords: viral myocarditis, TLRs, Th cells, regulatory T cell, immune response
Citation: Zheng SY and Dong JZ (2022) Role of Toll-Like Receptors and Th Responses in Viral Myocarditis. Front. Immunol. 13:843891. doi: 10.3389/fimmu.2022.843891
Received: 04 January 2022; Accepted: 24 March 2022;
Published: 19 April 2022.
Edited by:
Steven O’Reilly, STipe Therapeutics, DenmarkReviewed by:
Sally A. Huber, University of Vermont, United StatesCopyright © 2022 Zheng and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-Zeng Dong, anpkb25nQGNjbXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.