 Ruoxi Zhang
Ruoxi Zhang Rui Kang
Rui Kang Daolin Tang
Daolin Tang- Department of Surgery, University of Texas (UT) Southwestern Medical Center, Dallas, TX, United States
Stimulator of interferon response cGAMP interactor 1 (STING1), also known as TMEM173, is an immune adaptor protein that governs signal crosstalk that is implicated in many physiological and pathological processes. Although it has been established that STING1 traffics from the endoplasmic reticulum (ER) to Golgi apparatus (Golgi) upon DNA-triggered activation, emerging evidence reveals that STING1 can be transported to different organelles, which dictate its immune-dependent (e.g., the production of type I interferons and pro-inflammatory cytokines) and -independent (e.g., the activation of autophagy and cell death) functions. In this brief review, we outline the roles of STING1 in different organelles (including the ER, ER-Golgi intermediate compartment, Golgi, mitochondria, endosomes, lysosomes, and nucleus) and discuss the potential relevance of these roles to diseases and pharmacological interventions.
Introduction
Stimulator of interferon response cGAMP interactor 1 (STING1, also known as STING or TMEM173), an evolutionarily conserved transmembrane protein, is expressed in various endothelial and epithelial cell types as well as in T cells, B cells, and myeloid cells (e.g., macrophages and monocytes) and mainly localized on endoplasmic reticulum (ER). It was originally described as an adaptor protein that mediated the production of type I interferons (IFNs) in DNA-induced immune responses (1–4). Although STING1-mediated innate immunity plays significant roles in shaping host defense against microbe invasion and tumor growth (5), aberrative activation of STING1 can also disturb immune balance, thereby leading to pathological conditions and human diseases, such as STING-associated vasculopathy with onset in infancy (SAVI), Aicardi-Goutieres syndrome, systemic lupus erythematosus (SLE), and sepsis (6, 7), as well as neurodegenerative diseases (8), and metabolic diseases (9). Therefore, STING1 is an emerging therapeutic target in translational research.
Accumulating evidence demonstrates that the subcellular distribution of STING1 is not restricted to an ER-to-Golgi apparatus (Golgi) membranous network. Under different circumstances, the localization on other organelles enables some immune-independent functions of STING1, contributing to autophagy (10), regulated cell death (11), ER stress (12), lipid metabolism (13), and DNA damage response (DDR) (14). The participation and crosstalk of these organelles determine the function of STING1 in diseases by controlling its location, binding partners, and signaling recognition. In this mini-review, we highlight recent scientific advances regarding STING1 in different subcellular structures, and will draw parallels and differences across functions and diseases.
Roles of STING1 in Organelles
ER
STING1 possesses an N-terminal transmembrane (TM) domain that spans the ER membrane four times, a cytosolic cyclic dinucleotide (CDN) binding domain (CBD), and a C-terminal tail (CTT) (15). Under steady-state conditions, STING1 is retained in the ER by its ER-binding partners, with its CBD face to the cytosol, which facilitates the detection of second messengers. The second messenger is the small molecule and ion that transmits the signal received by the cell surface receptor to the effector protein. The cytosolic DNA sensor cyclic GMP-AMP synthase (CGAS) recognizes various DNA driven from pathogens or hosts, then generates the second messenger’s cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). The cGAMP or bacteria-produced CDNs bind to STING1, causing STING1 to activate through conformational changes and oligomerization. This protein secondary structural change of STING1 subsequently promotes the translocation of STING1 from ER to Golgi, where the CTT of STING1 binds and phosphorylates the TANK binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3). Finally, activated IRF3 promotes gene transcription of type I IFNs (Figure 1). Alternatively, STING1-mediated nuclear factor kappa B (NF-κB) pathway activation favors the production of proinflammatory cytokines, such as tumor necrosis factor (TNF) and interleukin 6 (IL6). This supports the hypothesis that STING1 is a mediator of inflammatory disease (16).
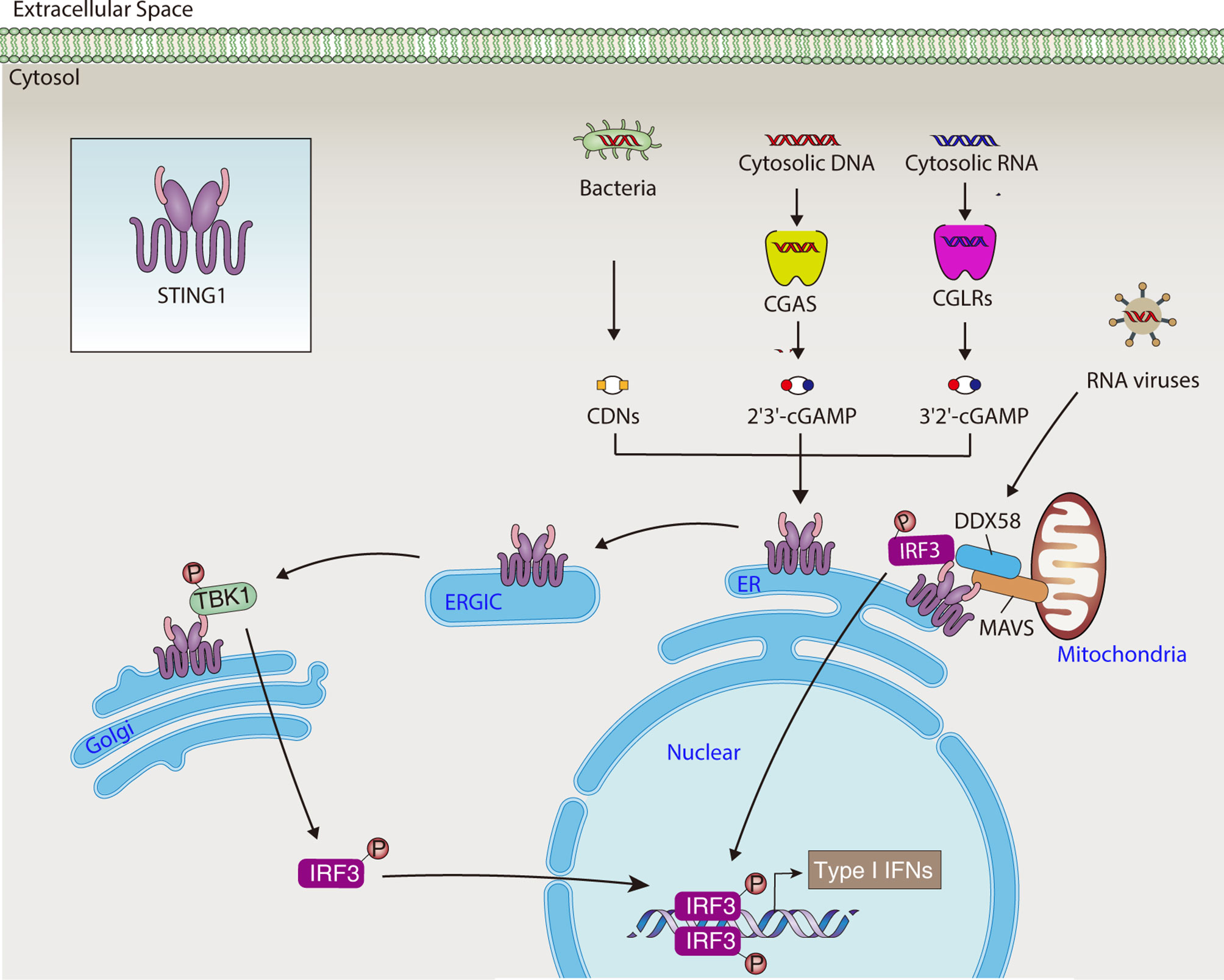
Figure 1 STING1-mediated type I IFNs production. In response to DNA or RNA from pathogens and hosts, STING1 activates the TBK1-IRF3 pathway, leading to the production of type I IFNs. CDN, cytosolic cyclic dinucleotide; cGAMP, cyclic GMP-AMP; CGAS, cyclic GMP-AMP synthase; CGLR, CGAS-like receptor; DDX58, DExD/H-box helicase 58; ER, endoplasmic reticulum; ERGIC, endoplasmic reticulum-Golgi intermediate compartment; IRF3, interferon regulatory factor 3; MAVS, mitochondrial antiviral signaling protein; STING1, Stimulator of interferon response cGAMP interactor 1; TBK1, TANK binding kinase 1; type I IFNs, type I interferons.
Although there is no clear consensus on the ER targeting signal sequence of STING1, several binding partners of STING1 regulate the ER retention of STING1. The ER calcium sensor, stromal interaction molecule 1 (STIM1), physically interacts with and retains STING1 in ER, inhibiting downstream immune responses and ER stress in mouse embryonic fibroblasts (MEFs) and human embryonic kidney 293 (HEK293) cells (17). In contrast, transmembrane protein 203 (TMEM203) competes with STIM1 to bind STING1, promoting cGAMP-induced STING1 activation in human macrophages (18). In addition, toll interacting protein (TOLLIP), an ubiquitin-binding protein that interacts with several components of the toll-like receptor, can stabilize STING1 on the ER and reduce cGAMP-induced lysosomal degradation of STING1 in MEFs (19). These findings reinforce the notion that STING1 is an adaptor protein with strong activity in binding multiple proteins. Different STING1 protein complexes not only affect the location of STING1 in the ER, but also regulate its degradation and activity.
The exit of STING1 from the ER is a kinetic process involving interaction with STING1 ER exit protein 1 (STEEP1) to recruit phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), leading to phosphatidylinositol-3-phosphate (PtdIns3P) synthesis and membrane curvature on the ER, and finally packaging STING1 into vesicles that bud from the ER (20) (Figure 2). STING1 mutations in the TM domain cause constitutive ER exit and subsequent autoimmunity in SAVI patients, the process of which is independent of cGAMP binding. Brefeldin A, an inhibitor of protein trafficking between the ER and Golgi, blocks STING1 trafficking regardless of its mutation status (21). These data indicate that the membrane topology of STING1 is attributed to its transmembrane domain and PtdIns3P synthesis. To support this, virus-mediated cell fusion, including that of DNA viruses (e.g., herpes simplex virus type 1 [HSV-1]) and RNA viruses (e.g., influenza A virus), can activate membrane-proximal signaling phospholipase C-γ (PLC-γ) and phosphatidylinositol-3 kinase (PI3K) pathways, triggering STING1-dependent immune responses independent of nucleotide recognition (22, 23). Overall, these data suggest that PtdIns3P acts as a key signal to promote ER exit of STING1. Given that STING1 has two calcium (Ca2+) binding sites and PtdIns3P triggers Ca2+ efflux from ER to induce STING1 activation (24, 25), Ca2+ may be another ER exit signal for STING1.
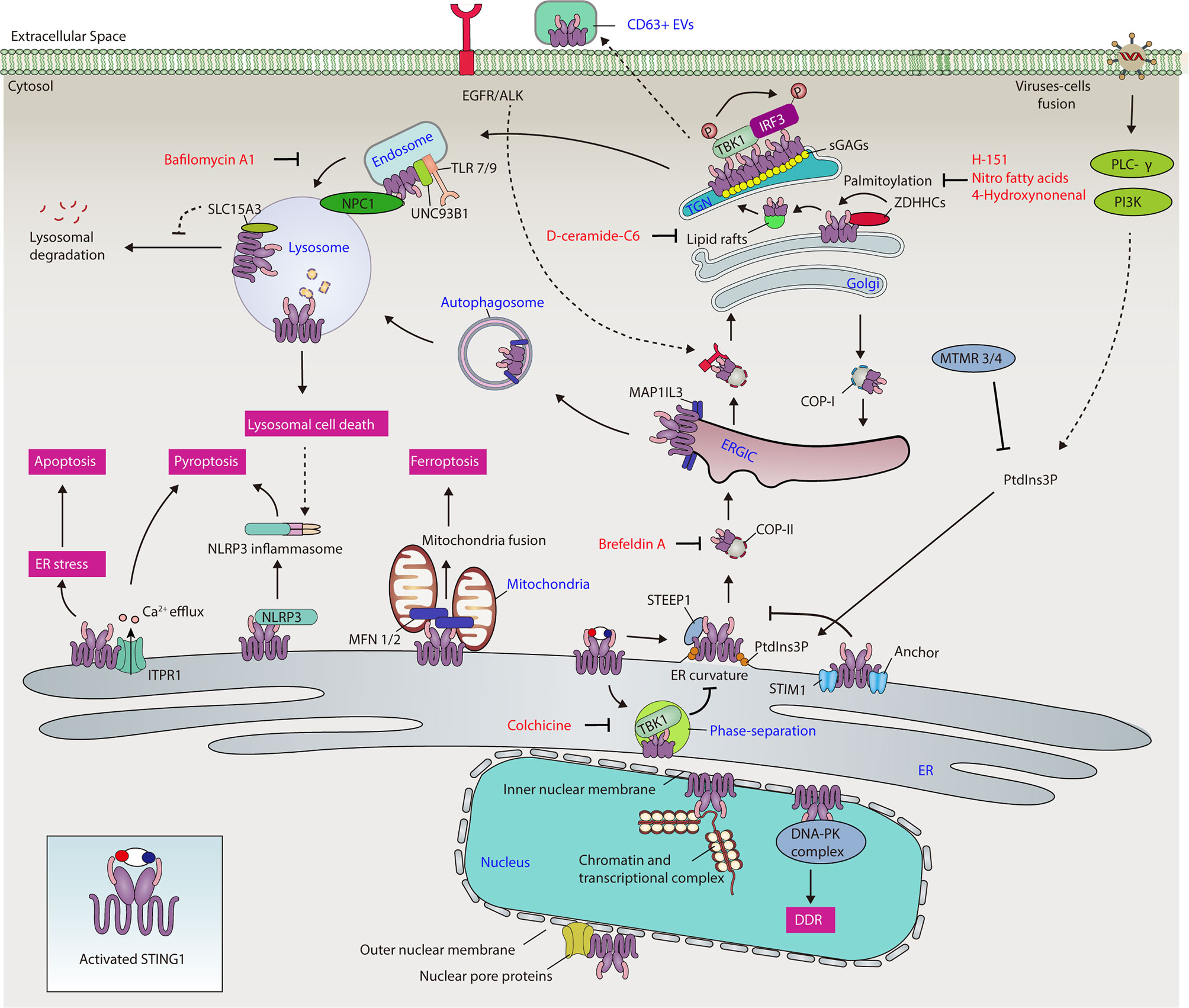
Figure 2 The network of STING1 in different organelles. Activated STING1 traffics to the ER, ERGIC, Golgi, mitochondria, endosomes, lysosomes, and nucleus, contributing to different cellular processes, including immune response, ER stress, cell death, autophagy, gene transcription, and DNA damage response. ALK, ALK receptor tyrosine kinase; Ca2+, calcium; COP-I, coat protein complex I; COP-II, coat protein complex II; DDR, DNA damage response; DNA-PK, DNA-dependent protein kinase; EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; ERGIC, endoplasmic reticulum-Golgi intermediate compartment; EV, extracellular vesicle; IRF3, interferon regulatory factor 3; ITPR1, inositol 1,4,5-trisphosphate receptor type 1; MAP1LC3, microtubule-associated protein 1 light chain 3; MFN 1/2, mitofusin 1/2; MTMR3/4, myotubularin-related protein 3/4; NLRP3, NLR family pyrin domain containing 3; NPC1, Niemann-Pick type C1; PI3K, phosphatidylinositol-3 kinase; PLC-γ, phospholipase C-γ; PtdIns3P, phosphatidylinositol-3-phosphate; sGAG, sulfated glycosaminoglycan; SLC15A3, solute carrier family 15 member 3; STING1, Stimulator of interferon response CGAMP interactor 1; STIM1, stromal interaction molecule 1; STEEP1, STING1 ER exit protein 1; TBK1, TANK binding kinase 1; TGN, trans-Golgi network; TLR, toll-like receptor; type I IFN, type I interferon; UNC93B1, unc-93 homolog B1, TLR signaling regulator; ZDHHC, zinc finger dhhc-type palmitoyltransferase.
Phase separation, the process of spontaneous separation of a mixed solution of macromolecules (such as a protein or nucleic acid) into two phases, is an alternative location for STING1 after ER exit. Functionally, to prevent STING1 overactivation, a high cGAMP level induces STING1 condensation on the ER and the formation of a puzzle-like droplet that separates STING1 and TBK1 from downstream IRF3 (26) (Figure 2). However, the constitutive activating mutants in STING1 (including N154S or V155M mutations) reduce STING1 phase separators. The lack of this sponge-based negative feedback mechanism contributes to the autoimmunity of patients with SAVI. The microtubule inhibitor colchicine (but not actin polymerization inhibitors or brefeldin A) inhibits STING1 condensation, indicating that the ER exit of STING1 is also microtubule-dependent.
Many recent studies have focused on how ER-associated STING1 modulates ER stress and cell death (Figure 2). For example, during HSV-1 infection, STING1 inhibits the ubiquitin degradation of NLR family pyrin domain 3 (NLRP3) in macrophages through direct protein-protein interaction in the ER, causing inflammasome activation and subsequent pyroptosis (27). In bacterial sepsis, the interaction of STING1 with ER Ca2+ channel inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) increases Ca2+ release from ER to cytoplasm in macrophages, leading to GSDMD-dependent pyroptosis, the release of coagulation factor III, and subsequent activation of systemic coagulation in septic mice (25, 28). The constitutive activation of STING1 can also trigger apoptosis in CD8+ T cells by disrupting Ca2+ homeostasis and activating the unfolded protein response and ER stress through its CBD (12). In contrast, the interaction of notch intracellular signaling domain (NICD) with STING1 CBD limits STING1-mediated apoptosis in CD4+ T cells, preventing immunosuppression in the late stage of sepsis (29). Together, these findings establish STING1-dependent Ca2+ signaling in the control of regulated cell death in immune cells.
ER STING1 also links the lipid metabolism and immune response through the insulin signaling pathway (30–33). On one hand, STING1-mediated type I IFN signaling limits cholesterol biosynthetic flux in MEFs. On the other hand, insulin-induced gene 1 (INSIG1), sterol regulatory element binding transcription factor 1 (SREBP), and SREBF chaperone (SCAP) show strong activity to bind STING1, enhancing STING1-mediated cytokine production. These results raise the concern of whether lifestyle factors (such as a high-fat diet) affect STING1 signaling and thereby affect the outcome of metabolic diseases.
ER-Golgi Intermediate Compartment
The ER-Golgi intermediate compartment (ERGIC) is a pre-Golgi intermediate composed of vesicular tubular clusters that deliver secretory cargo from ER exit sites to the Golgi (34). Upon activation, STING1 exits from ER and relocates to the Golgi via the ERGIC in a coat protein complex II (COP-II)-dependent manner (10, 35). Pharmacological or genetic inhibition of ER-to-ERGIC trafficking significantly suppresses STING1 activation (21, 36). On the other hand, coat protein complex I (COP-I) mediates the retrograde transport of STING1 to the ERGIC from Golgi, while deficiency in COP-I transport causes failure of Golgi-to-ER STING1 retrieval and ligand-independent activation of STING1, thus contributing to COPA syndrome (37–40) (Figure 2). COPA syndrome is a rare inherited autoimmune disease caused by mutations in the coat protein complex subunit alpha (COPA) gene. Studies highlight the importance of the ER-ERGIC/Golgi axis in the control of STING1 activation, demonstrate a “tug-of-war” between the ER and the ERGIC/Golgi for STING1, and suggest therapeutic strategies for inflammatory and autoimmune diseases (37–40). However, whether STING1 can be directly activated on ERGIC needs to be further clarified by inhibition of “ERGIC-to-Golgi” membrane trafficking.
In addition to its role in immune response, ERGIC is also involved in STING1-dependent autophagy, a process that relies on the formation of various membrane structures to degrade cytoplasmic cargo. ERGIC could serve as a membrane source for the formation of autophagosomes through STING1 activation-induced microtubule-associated protein 1 light chain 3 (MAP1LC3) lipidation. This STING1-mediated autophagy initiation is further enhanced by WD repeat domain, phosphoinositide interacting 2 (WIPI2) (10) or autophagy-related 16-like 1 (ATG16L1) (41). Given that STING1 directly interacts with MAP1LC3 upon activation, MAP1LC3 lipidation-mediated transportation may provide more trafficking routes for STING1 on intracellular organelles. Nevertheless, autophagy-mediated STING1 degradation limits the immune activity of STING1.
Golgi
The Golgi connects the ER, mitochondria, endosomes, and other organelles through membranous networks for vesicular trafficking and protein/lipid secretion. It also serves as a platform for signaling transduction connecting multiple innate immune pathways (42). Consistently, activated and polymerized STING1 traffics to the Golgi and recruits TBK1 for the phosphorylation of IRF3, inducing a set of downstream immune and non-immune events, such as IRF3-mediated type I IFN production, apoptosis, and necroptosis (11). As a part of Golgi membranous networks, activated Golgi STING1 is further transported to endosomes for degradation or transported to ER for recycling (43).
The Golgi membranous network contains a tubular reticular network of membranes facing the ER, namely the cis-Golgi network (CGN, including ERGIC) and another tubular reticular network of membranes facing plasma membrane and compartments of the endocytic pathway, called the trans-Golgi network (TGN) (44). The regulation of ER-to-Golgi trafficking of STING1 is context-dependent. For example, phosphatidylinositol 3 phosphatases myotubularin-related protein 3 (MTMR3) and myotubularin-related protein 4 (MTMR4), which inhibits PtdIns3P production, thereby suppressing STING1 trafficking to Golgi and subsequent STING1-mediated innate immune response in mouse macrophage RAW264.7 cells (45). Epidermal growth factor receptor (EGFR)-mediated phosphorylation of STING1 on Tyr 245 contributes to STING1 trafficking to Golgi and subsequent activation in RAW264.7 cells, whereas EGFR deficiency leads to autophagosome localization of activated STING1, instead of transport to the Golgi (46). In line with this, the activation of EGFR and ALK receptor tyrosine kinase can mediate STING1 activation in macrophages caused by CDNs (47). STING couples with the downstream kinases of EGFR and PI3K to control the accumulation of F-actin during the activation of B-cell receptor (BCR) (48), highlighting an interplay between STING1 and cytoskeleton in B cells. Overall, these data indicate that a phosphoinositide-dependent cytoskeleton transporting system is highly associated with ER-to-Golgi trafficking of STING1, and this process is fine-tuned through the EGFR signal pathway (Figure 2).
Evidence indicates the potential role of TGN in mediating STING1 activation. First, the active form of TBK1 is only localized at the TGN, but not at the rest of the Golgi domains (49). Second, the lipid molecule D-ceramide-C6 disrupts lipid rafts at the Golgi and TGN location of proteins, and inhibits STING1-dependent phosphorylation of TBK1 and IRF3 without affecting the translocation of STING1 to the Golgi (49). Third, sphingomyelin and cholesterol are enriched in the TGN, which contribute to the phosphorylation of STING1 by TBK1 (50). Understanding the active mechanism of STING1 from TGN is therefore important in innate immunity. Whether CGN plays the opposite role in STING1 activation is not fully understood.
STING1 activation is highly dependent on palmitoylation, which is implicated in the clustering of many proteins into cholesterol- and sphingomyelin-enriched lipid rafts and TGN (51). Upon activation, STING1 interacts with several palmitoyl transferases on Golgi, such as zinc finger dhhc-type palmitoyltransferase (ZDHHC) (43, 52) (Figure 2). The palmitoylation of two membrane-proximal Cys residues (C88/91) of STING1 is crucial for STING1 activation, even for gain-of-function STING1 mutants in SAVI patients, which makes STING1 constitutively expressed on Golgi (49, 53). Accordingly, nitro fatty acids directly modify STING1 palmitoylation by nitro alkylation, leading to the inhibition of type I IFN production in fibroblasts derived from SAVI patients (54). In addition, 4-hydroxynonenal (4HNE), one of end products of lipid peroxidation, inhibits STING1 translocation to Golgi and activation by the carbonylation of STING1 at C88 in mouse primary peritoneal macrophages (55). This 4HNE-mediated STING1 inhibition is enhanced by the depletion of glutathione peroxidase 4 (GPX4), which is known for specifically catalysing the reduction of lipid peroxides. However, excessive lipid peroxidation can lead to cell death (especially ferroptosis), which may trigger damage-associated molecular pattern (DAMP)-mediated inflammation and immune responses.
In addition, the sulfated glycosaminoglycans (sGAGs) can bind the TM domain of STING1 and promote STING1 clustering in Golgi and subsequent activation in cancer cells (THP1, HeLa, and HT1080 cells) (56) (Figure 2). A mutant of STING1 lacking a TM domain and residues C88/91 could form a self-clustered tetrameric structure and effectively trigger the TBK1 and IRF3 activation by cGAMP under physiological conditions (57). Therefore, clustering of STING1 may be a prerequisite for the interaction of STING1 and TBK1, although it is unclear whether this change is different under physiological and pathological conditions.
Mitochondria
The ER and mitochondria share a close relationship via multiple molecular interaction-bridged mitochondria-associated membranes (MAMs), which enable signal messenger movement between the two organelles, regulating mitochondrial fusion and fission, immune response, metabolic homeostasis, and cell death (58). The location of STING1 on mitochondria including MAMs has recently been documented (59). During some RNA virus infections, STING1 serves as an immune adaptor on the outer membrane of mitochondria by interacting with mitochondrial antiviral signaling protein (MAVS). Viral RNA sensor DExD/H-box helicase 58 (DDX58, best known as RIG-I) is recruited to the STING1-MAVS complex on the MAM (4, 60). STING1 then links IRF3 and TBK1 to MAVS, thus transmitting DDX58-MAVS–mediated signals (4, 60). Recently, CGAS-like receptors (CGLRs) were identified as a double-stranded RNA sensors in Drosophila, which can produce a novel second messenger, cG[3’-5’]pA[2’-5’]p (3’2’-cGAMP), for activating a STING1-dependent immune response (61). These data imply a crosstalk between CGAS/STING1- and DDX58/MAVS-dependent innate immune response pathways on MAMs (Figure 1).
Mitochondria are the central regulator of cell death. STING1 is accumulated in mitochondria in human ferroptotic pancreatic ductal adenocarcinoma cells (62). Although reactive oxygen species (ROS) is closely related to various types of regulated cell death, the specific factors driving STING1 mitochondrial accumulation in ferroptosis are still elusive. Consequently, mitochondrial STING1 promotes ferroptosis sensitivity through binding to mitochondria fusion regulators, namely mitofusins (including mitofusin 1 [MFN1] and mitofusin 2 [MFN2]), to increase mitochondria fusion-mediated ROS production and lipid peroxidation (Figure 2). Although these findings establish a direct role of STING1 in mitochondrial dynamics and cell death, the mitochondria fission mediator dynamin 1-like (DNM1L) can function as a negative regulator of STING1-dependent IFNB/IFN-β production (63, 64). The dynamic relationship between mitochondrial DNA damage, cell death, and the STING1 pathway in the control of sterile inflammation and tissue damage needs further investigation.
Endosomes
Endosomes are essential cellular stations where endocytic and secretory trafficking routes converge. The proteins transiting at endosomes are degraded by lysosomes or recycled to the other organelles (65). STING1 can be translocated to acidified endosomes and finally degraded by lysosomes at the late stage of STING1 activation (66). In contrast, inhibition of this process by the lysosomal inhibitor bafilomycin A1 that targets the fusion step of endosomes and lysosomes could enhance STING1-mediated antitumor immune response (Figure 2). Many endosomal interactors with STING1 have been identified at the late activation stage in RAW264.7 cells, providing an integrated mechanism to control STING1 trafficking (43). Unc-93 homolog B1, TLR signaling regulator (UNC93B1) is a chaperone of endosomal toll-like receptors (TLRs) and acts as a repressor of STING1 activation by anchoring STING1 in the endosomes of human and mouse fibroblasts (67). Endosomal TLRs play a role in the activation of B cells in the context of autoimmune diseases (68). STING1-deficient mice had significantly less arthritic joint inflammation, but these mice still produced autoantibodies (69). In contrast, depletion of the chaperone protein UNC93B required for endosomal localization of TLR7 and TLR9 limits autoantibody production in experimental arthritis models (69). Notably, a recent study showed that the STING1 pathway is not essential for the development of experimental SLE in mice, suggesting that other DNA-sensing pathways have alternative roles in mediating autoimmune pathology (70). STING1 can also be exocytosed in CD63+ extracellular vesicles, generating an antiviral effect in recipient cells during HSV-1 infection (71–74) (Figure 2). The function of extracellular STING1 in the setting of disease remains elusive, but it may represent a mechanism to shape immune response.
Lysosomes
As mentioned above, after the activation of the CGAS-STING1 pathway, STING1 can be transported to the lysosome through autophagosomes and endosomes for degradation. Lysosomal disorder is associated with increased STING1-mediated immune response, highlighting that lysosomal damage and substance accumulation are the activation signals of the STING1 pathway. Although the mechanism still depends on the context, several proteins, including small GTPase RAB7A (10, 66), autophagy receptor sequestosome 1 (SQSTM1) (75), and lysosomal membrane protein Niemann-Pick type C1 (NPC1) (33), contribute to STING1 translocation to lysosomes for autophagic degradation in various disease conditions (Figure 2). This plasticity may explain why the degradation of STING1 needs different stimulation signals and mediators.
The most well-understood autophagy pathway involving ubiquitin is selective autophagy. There is a complex relationship between STING1 and autophagy receptors during stress. SQSTM1 is required for STING1-dependent autophagy to inhibit M. tuberculosis infection in macrophages, but SQSTM1 is dispensable for STING1-mediated autophagy in HeLa cancer cells (76). STING1 is degraded in a SQSTM1-dependent manner correlating with its K63-linked ubiquitination in monocyte cells (75, 77). However, in HeLa cells, the autophagy receptor coiled-coil domain containing 50 (CCDC50) binds to and targets K63-polyubiquitinated STING1 for autophagic degradation. Studies reveal that CCDC50 mainly mediates the delivery of STING1 to MAP1LC3-positive autophagosomes, rather than directly promoting STING1 degradation in lysosomes (78). Therefore, each step of selective autophagy degradation of STING1 may require different autophagy receptors to mediate.
The inhibition of STING1 degradation in lysosomes promotes downstream immune responses. A lysosomal protein solute carrier family 15 member 3 (SLC15A3) can interact with STING1 and elevate STING1-dependent type I IFN response, thus protecting against HSV-1 infection in human primary monocytes (79) (Figure 2). However, whether and how SLC15A3 mediates the protein stability of STING1 in lysosomes remains obscure.
In addition to immune response regulation, lysosomal STING1 can affect the function of lysosomes in cell death and pH homeostasis (Figure 2). In primary monocytes, activated STING1 traffics to the lysosomes, triggering lysosomal membrane permeabilization and subsequently lysosome-dependent cell death (80). Lysosome STING1 also participates in lysosomal rupture, cathepsin B release, and lysosome-dependent cell death in rat neuronal cells after hypoxia ischemia (81). In addition, circulating mitochondrial DNA-mediated robust STING1 activation in macrophages induces STING1 accumulation in lysosomes, which blocks lysosomal acidification and subsequently autophagic clearance of DAMPs in lethal sepsis models (82), suggesting a different anti-autophagy role of lysosomal STING1. Given these contradictory observations, further research is needed to reveal whether the dual role of STING1 in autophagy really depends on its subcellular location. Whether STING1 can act as an autophagy receptor is still unknown.
The Nucleus
CGAS is present in the nucleus and regulates genomic stability and cell cycle (83–85), raising the possibility that STING1 may have a role in the nucleus. In fact, the nuclear STING1 was originally identified by a proteomics study of the nuclear envelope, and this finding is supported by two subsequent studies, which show a role for nuclear STING1 in promoting chromatin compaction through epigenetic modifications (86–88). STING1 can bind the DNA-dependent protein kinase (DNA-PK) complex in the inner nuclear membrane, which protects breast cancer cells from DNA instability by promoting DDR in a CGAS-independent manner (14). Nuclear STING1 also interacts with various nucleotide-binding proteins, which regulate gene transcription of type I IFNs (89). STING1 redistributes from the nucleus to the ER, increasing both dsDNA- and dsRNA-triggered immune responses. In addition, subcellular localization analysis and the nuclear interactome show that STING1 co-localizes with the lamina, which serves as an anchoring point for chromatin and transcription factors and interacts with transcriptional activators and co-activators (14, 89), indicating that nuclear STING1 may be directly involved in the regulation of gene transcriptional activity (Figure 2).
The precise nuclear location and function of STING1 is still controversial. By using structured illumination super-resolution microscopy, STING1 has been found redistributed into different nuclear envelope locations (89). Given that the ER is adjacent to the nuclear envelope, STING1 may diffuse to the contiguous nuclear membrane after its initial insertion into ER membranes (90). Thus, it is not surprising that STING1 localizes on outer and inner nuclear membranes. STING1 can also co-localize with spectrin repeat containing nuclear envelope protein 1 (SYNE1) at the nuclear lamina, and mediates the docking of capsid protein of human herpes viruses to nuclear pore complex proteins (91) (Figure 2). These findings provide a framework to explain the import of viral genomes into the nucleus of susceptible cells in the early stages of infection (91). Additional studies are required to fully elucidate the mechanism of ER-located STING1 trafficking into the nucleus and other nuclear functions of STING1, particularly in regulating genome transcription and chromosome stabilization.
Conclusions and Perspectives
STING1 is a multifaceted protein, probably known best as an adaptor protein of DNA sensor pathways to activate innate immunity. Under normal conditions, most of STING1 is in the ER and acts as a regulator of ER homeostasis. Upon challenge with a range of environmental stresses, STING1 translocates to other organelles, such as Golgi, mitochondria, endosomes, lysosomes, and nuclei, and plays an immune-dependent and -independent role in infection, cell death, and gene expression. Although the subcellular localization sequence of STING1 is unclear, the transport of STING1 in subcellular organelles is regulated by the cytoskeleton, binding proteins, and posttranslational modifications.
This complexity of location and modulation may increase the side effects should STING1 be indiscriminately targeted in diseases. In contrast, using small molecules to target STING1 translocation may be a precision treatment strategy (Figure 2). Further studies on the effectors that alter STING1 trafficking and localization may provide novel therapeutic targets for enhancing STING1-dependent immune response or reducing STING1-mediated hyperinflammatory and autoimmune responses. In the meantime, we should uncover the function of cell and tissue type-specific STING1 in health and disease using advanced conditional knockout strategies.
Author Contributions
RZ wrote the manuscript. RK and DT edited the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the National Institutes of Health of USA (R01CA160417, R01CA211070, and R01GM127791).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript.
References
1. Ishikawa H, Barber GN. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
2. Ishikawa H, Ma Z, Barber GN. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature (2009) 461:788–92. doi: 10.1038/nature08476
3. Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an Endoplasmic Reticulum IFN Stimulator, Activates Innate Immune Signaling Through Dimerization. Proc Natl Acad Sci USA (2009) 106:8653–8. doi: 10.1073/pnas.0900850106
4. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity (2008) 29:538–50. doi: 10.1016/j.immuni.2008.09.003
5. Barber GN. STING: Infection, Inflammation and Cancer. Nat Rev Immunol (2015) 15:760–70. doi: 10.1038/nri3921
6. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
7. Zhang RX, Kang R, Tang DL. STING1 in Sepsis: Mechanisms, Functions, and Implications. Chin J Traumatol (2022) 25:1–10. doi: 10.1016/j.cjtee.2021.07.009
8. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms Underlying Inflammation in Neurodegeneration. Cell (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
9. Bai J, Liu F. cGASSTING Signaling and Function in Metabolism and Kidney Diseases. J Mol Cell Biol (2021) 13:728–38. doi: 10.1093/jmcb/mjab066
10. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy Induction via STING Trafficking Is a Primordial Function of the cGAS Pathway. Nature (2019) 567:262–66. doi: 10.1038/s41586-019-1006-9
11. Zhang R, Kang R, Tang D. The STING1 Network Regulates Autophagy and Cell Death. Signal Transduct Target Ther (2021) 6:208. doi: 10.1038/s41392-021-00613-4
12. Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-Mediated Disruption of Calcium Homeostasis Chronically Activates ER Stress and Primes T Cell Death. J Exp Med (2019) 216:867–83. doi: 10.1084/jem.20182192
13. Akhmetova K, Balasov M, Chesnokov I. Drosophila STING Protein has a Role in Lipid Metabolism. Elife (2021) 10:e67358. doi: 10.7554/eLife.67358
14. Cheradame L, Guerrera IC, Gaston J, Schmitt A, Jung V, Goudin N, et al. STING Protects Breast Cancer Cells From Intrinsic and Genotoxic-Induced DNA Instability via a non-Canonical, Cell-Autonomous Pathway. Oncogene (2021) 40:6627–40. doi: 10.1038/s41388-021-02037-4
15. Zhang X, Bai XC, Chen ZJ. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity (2020) 53:43–53. doi: 10.1016/j.immuni.2020.05.013
16. Hopfner KP, Hornung V. Molecular Mechanisms and Cellular Functions of cGAS-STING Signalling. Nat Rev Mol Cell Biol (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
17. Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca(2+) Sensor STIM1 Regulates the Type I Interferon Response by Retaining the Signaling Adaptor STING at the Endoplasmic Reticulum. Nat Immunol (2019) 20:152–62. doi: 10.1038/s41590-018-0287-8
18. Li Y, James SJ, Wyllie DH, Wynne C, Czibula A, Bukhari A, et al. TMEM203 Is a Binding Partner and Regulator of STING-Mediated Inflammatory Signaling in Macrophages. Proc Natl Acad Sci USA (2019) 116:16479–88. doi: 10.1073/pnas.1901090116
19. Pokatayev V, Yang K, Tu X, Dobbs N, Wu J, Kalb RG, et al. Homeostatic Regulation of STING Protein at the Resting State by Stabilizer TOLLIP. Nat Immunol (2020) 21:158–67. doi: 10.1038/s41590-019-0569-9
20. Zhang BC, Nandakumar R, Reinert LS, Huang J, Laustsen A, Gao ZL, et al. STEEP Mediates STING ER Exit and Activation of Signaling. Nat Immunol (2020) 21:868–79. doi: 10.1038/s41590-020-0730-5
21. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING Activation by Translocation From the ER Is Associated With Infection and Autoinflammatory Disease. Cell Host Microbe (2015) 18:157–68. doi: 10.1016/j.chom.2015.07.001
22. Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, et al. Virus-Cell Fusion as a Trigger of Innate Immunity Dependent on the Adaptor STING. Nat Immunol (2012) 13:737–43. doi: 10.1038/ni.2350
23. Holm CK, Rahbek SH, Gad HH, Bak RO, Jakobsen MR, Jiang Z, et al. Influenza A Virus Targets a cGAS-Independent STING Pathway That Controls Enveloped RNA Viruses. Nat Commun (2016) 7:10680. doi: 10.1038/ncomms10680
24. Shu C, Yi G, Watts T, Kao CC, Li P. Structure of STING Bound to Cyclic Di-GMP Reveals the Mechanism of Cyclic Dinucleotide Recognition by the Immune System. Nat Struct Mol Biol (2012) 19:722–4. doi: 10.1038/nsmb.2331
25. Zhang R, Kang R, Klionsky DJ, Tang D. Ion Channels and Transporters in Autophagy. Autophagy (2022) 18:4–23. doi: 10.1080/15548627.2021.1885147
26. Yu X, Zhang L, Shen J, Zhai Y, Jiang Q, Yi M, et al. The STING Phase-Separator Suppresses Innate Immune Signalling. Nat Cell Biol (2021) 23:330–40. doi: 10.1038/s41556-021-00659-0
27. Wang W, Hu D, Wu C, Feng Y, Li A, Liu W, et al. STING Promotes NLRP3 Localization in ER and Facilitates NLRP3 Deubiquitination to Activate the Inflammasome Upon HSV-1 Infection. PloS Pathog (2020) 16:e1008335. doi: 10.1371/journal.ppat.1008335
28. Zhang H, Zeng L, Xie M, Liu J, Zhou B, Wu R, et al. TMEM173 Drives Lethal Coagulation in Sepsis. Cell Host Microbe (2020) 27:556–70 e6. doi: 10.1016/j.chom.2020.02.004
29. Long J, Yang C, Zheng Y, Loughran P, Guang F, Li Y, et al. Notch Signaling Protects CD4 T Cells From STING-Mediated Apoptosis During Acute Systemic Inflammation. Sci Adv (2020) 6:eabc5447. doi: 10.1126/sciadv.abc5447
30. Wang Q, Liu X, Cui Y, Tang Y, Chen W, Li S, et al. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity (2014) 41:919–33. doi: 10.1016/j.immuni.2014.11.011
31. Chen W, Li S, Yu H, Liu X, Huang L, Wang Q, et al. ER Adaptor SCAP Translocates and Recruits IRF3 to Perinuclear Microsome Induced by Cytosolic Microbial DNAs. PloS Pathog (2016) 12:e1005462. doi: 10.1371/journal.ppat.1005462
32. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell (2015) 163:1716–29. doi: 10.1016/j.cell.2015.11.045
33. Chu TT, Tu X, Yang K, Wu J, Repa JJ, Yan N. Tonic Prime-Boost of STING Signalling Mediates Niemann-Pick Disease Type C. Nature (2021) 596:570–75. doi: 10.1038/s41586-021-03762-2
34. Appenzeller-Herzog C, Hauri HP. The ER-Golgi Intermediate Compartment (ERGIC): In Search of Its Identity and Function. J Cell Sci (2006) 119:2173–83. doi: 10.1242/jcs.03019
35. Ran Y, Xiong MG, Xu ZS, Luo WW, Wang SY, Wang YY. YIPF5 Is Essential for Innate Immunity to DNA Virus and Facilitates COPII-Dependent STING Trafficking. J Immunol (2019) 203:1560–70. doi: 10.4049/jimmunol.1900387
36. Ogawa E, Mukai K, Saito K, Arai H, Taguchi T. The Binding of TBK1 to STING Requires Exocytic Membrane Traffic From the ER. Biochem Biophys Res Commun (2018) 503:138–45. doi: 10.1016/j.bbrc.2018.05.199
37. Mukai K, Ogawa E, Uematsu R, Kuchitsu Y, Kiku F, Uemura T, et al. Homeostatic Regulation of STING by Retrograde Membrane Traffic to the ER. Nat Commun (2021) 12:61. doi: 10.1038/s41467-020-20234-9
38. Lepelley A, Martin-Niclos MJ, Le Bihan M, Marsh JA, Uggenti C, Rice GI, et al. Mutations in COPA Lead to Abnormal Trafficking of STING to the Golgi and Interferon Signaling. J Exp Med (2020) 217:e20200600. doi: 10.1084/jem.20200600
39. Kato T, Yamamoto M, Honda Y, Orimo T, Sasaki I, Murakami K, et al. Augmentation of Stimulator of Interferon Genes-Induced Type I Interferon Production in COPA Syndrome. Arthritis Rheumatol (2021) 73:2105–15. doi: 10.1002/art.41790
40. Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A Defect in COPI-Mediated Transport of STING Causes Immune Dysregulation in COPA Syndrome. J Exp Med (2020) 217:e20201045. doi: 10.1084/jem.20201045
41. Fischer TD, Wang C, Padman BS, Lazarou M, Youle RJ. STING Induces LC3B Lipidation Onto Single-Membrane Vesicles via the V-ATPase and ATG16L1-WD40 Domain. J Cell Biol (2020) 219:e202009128. doi: 10.1083/jcb.202009128
42. Tao Y, Yang Y, Zhou R, Gong T. Golgi Apparatus: An Emerging Platform for Innate Immunity. Trends Cell Biol (2020) 30:467–77. doi: 10.1016/j.tcb.2020.02.008
43. Motani K, Kosako H. BioID Screening of Biotinylation Sites Using the Avidin-Like Protein Tamavidin 2-REV Identifies Global Interactors of Stimulator of Interferon Genes (STING). J Biol Chem (2020) 295:11174–83. doi: 10.1074/jbc.RA120.014323
44. Gu F, Crump CM, Thomas G. Trans-Golgi Network Sorting. Cell Mol Life Sci (2001) 58:1067–84. doi: 10.1007/PL00000922
45. Dewi Pamungkas Putri D, Kawasaki T, Murase M, Sueyoshi T, Deguchi T, Ori D, et al. PtdIns3P Phosphatases MTMR3 and MTMR4 Negatively Regulate Innate Immune Responses to DNA Through Modulating STING Trafficking. J Biol Chem (2019) 294:8412–23. doi: 10.1074/jbc.RA118.005731
46. Wang C, Wang X, Veleeparambil M, Kessler PM, Willard B, Chattopadhyay S, et al. EGFR-Mediated Tyrosine Phosphorylation of STING Determines its Trafficking Route and Cellular Innate Immunity Functions. EMBO J (2020) 39:e104106. doi: 10.15252/embj.2019104106
47. Zeng L, Kang R, Zhu S, Wang X, Cao L, Wang H, et al. ALK Is a Therapeutic Target for Lethal Sepsis. Sci Transl Med (2017) 9:eaan5689. doi: 10.1126/scitranslmed.aan5689
48. Jing Y, Dai X, Yang L, Kang D, Jiang P, Li N, et al. STING Couples With PI3K to Regulate Actin Reorganization During BCR Activation. Sci Adv (2020) 6:eaax9455. doi: 10.1126/sciadv.aax9455
49. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING Requires Palmitoylation at the Golgi. Nat Commun (2016) 7:11932. doi: 10.1038/ncomms11932
50. Takahashi K, Niki T, Ogawa E, Fumika K, Nishioka Y, Sawa M, et al. A Cell-Free Assay Implicates a Role of Sphingomyelin and Cholesterol in STING Phosphorylation. Sci Rep (2021) 11:11996. doi: 10.1038/s41598-021-91562-z
51. Linder ME, Deschenes RJ. Palmitoylation: Policing Protein Stability and Traffic. Nat Rev Mol Cell Biol (2007) 8:74–84. doi: 10.1038/nrm2084
52. Zhou Q, Lin H, Wang S, Wang S, Ran Y, Liu Y, et al. The ER-Associated Protein ZDHHC1 Is a Positive Regulator of DNA Virus-Triggered, MITA/STING-Dependent Innate Immune Signaling. Cell Host Microbe (2014) 16:450–61. doi: 10.1016/j.chom.2014.09.006
53. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING With Covalent Small-Molecule Inhibitors. Nature (2018) 559:269–73. doi: 10.1038/s41586-018-0287-8
54. Hansen AL, Buchan GJ, Ruhl M, Mukai K, Salvatore SR, Ogawa E, et al. Nitro-Fatty Acids Are Formed in Response to Virus Infection and are Potent Inhibitors of STING Palmitoylation and Signaling. Proc Natl Acad Sci USA (2018) 115:E7768–E75. doi: 10.1073/pnas.1806239115
55. Jia M, Qin D, Zhao C, Chai L, Yu Z, Wang W, et al. Redox Homeostasis Maintained by GPX4 Facilitates STING Activation. Nat Immunol (2020) 21:727–35. doi: 10.1038/s41590-020-0699-0
56. Fang R, Jiang Q, Guan Y, Gao P, Zhang R, Zhao Z, et al. Golgi Apparatus-Synthesized Sulfated Glycosaminoglycans Mediate Polymerization and Activation of the cGAMP Sensor STING. Immunity (2021) 54:962–75 e8. doi: 10.1016/j.immuni.2021.03.011
57. He Y, Hong C, Yan EZ, Fletcher SJ, Zhu G, Yang M, et al. Self-Assembled cGAMP-STINGDeltaTM Signaling Complex as a Bioinspired Platform for cGAMP Delivery. Sci Adv (2020) 6:eaba7589. doi: 10.1126/sciadv.aba7589
58. Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: More Than Just a Housekeeper. Trends Cell Biol (2009) 19:81–8. doi: 10.1016/j.tcb.2008.12.002
59. Zevini A, Olagnier D, Hiscott J. Crosstalk Between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol (2017) 38:194–205. doi: 10.1016/j.it.2016.12.004
60. Nazmi A, Mukhopadhyay R, Dutta K, Basu A. STING Mediates Neuronal Innate Immune Response Following Japanese Encephalitis Virus Infection. Sci Rep (2012) 2:347. doi: 10.1038/srep00347
61. Slavik KM, Morehouse BR, Ragucci AE, Zhou W, Ai X, Chen Y, et al. cGAS-Like Receptors Sense RNA and Control 3’2’-cGAMP Signalling in Drosophila. Nature (2021) 597:109–13. doi: 10.1038/s41586-021-03743-5
62. Li C, Liu J, Hou W, Kang R, Tang D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front Cell Dev Biol (2021) 9:698679. doi: 10.3389/fcell.2021.698679
63. Kwon D, Sesaki H, Kang SJ. Intracellular Calcium is a Rheostat for the STING Signaling Pathway. Biochem Biophys Res Commun (2018) 500:497–503. doi: 10.1016/j.bbrc.2018.04.117
64. Kwon D, Park E, Sesaki H, Kang SJ. Carbonyl Cyanide 3-Chlorophenylhydrazone (CCCP) Suppresses STING-Mediated DNA Sensing Pathway Through Inducing Mitochondrial Fission. Biochem Biophys Res Commun (2017) 493:737–43. doi: 10.1016/j.bbrc.2017.08.121
65. Yong X, Mao L, Shen X, Zhang Z, Billadeau DD, Jia D. Targeting Endosomal Recycling Pathways by Bacterial and Viral Pathogens. Front Cell Dev Biol (2021) 9:648024. doi: 10.3389/fcell.2021.648024
66. Gonugunta VK, Sakai T, Pokatayev V, Yang K, Wu J, Dobbs N, et al. Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-Tumor Response. Cell Rep (2017) 21:3234–42. doi: 10.1016/j.celrep.2017.11.061
67. He Z, Ye S, Xing Y, Jiu Y, Zhong J. UNC93B1 Curbs Cytosolic DNA Signaling by Promoting STING Degradation. Eur J Immunol (2021) 51:1672–85. doi: 10.1002/eji.202048901
68. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-Like Receptor 7 and TLR9 Dictate Autoantibody Specificity and Have Opposing Inflammatory and Regulatory Roles in a Murine Model of Lupus. Immunity (2006) 25:417–28. doi: 10.1016/j.immuni.2006.07.013
69. Baum R, Sharma S, Carpenter S, Li QZ, Busto P, Fitzgerald KA, et al. Cutting Edge: AIM2 and Endosomal TLRs Differentially Regulate Arthritis and Autoantibody Production in DNase II-Deficient Mice. J Immunol (2015) 194:873–7. doi: 10.4049/jimmunol.1402573
70. Motwani M, McGowan J, Antonovitch J, Gao KM, Jiang Z, Sharma S, et al. cGAS-STING Pathway Does Not Promote Autoimmunity in Murine Models of SLE. Front Immunol (2021) 12:605930. doi: 10.3389/fimmu.2021.605930
71. Dogrammatzis C, Saleh S, Deighan C, Kalamvoki M. Diverse Populations of Extracellular Vesicles With Opposite Functions During Herpes Simplex Virus 1 Infection. J Virol (2021) 95:e02357–20. doi: 10.1128/JVI.02357-20
72. Deschamps T, Kalamvoki M. Extracellular Vesicles Released by Herpes Simplex Virus 1-Infected Cells Block Virus Replication in Recipient Cells in a STING-Dependent Manner. J Virol (2018) 92:e01102–18. doi: 10.1128/JVI.01102-18
73. Kalamvoki M, Du T, Roizman B. Cells Infected With Herpes Simplex Virus 1 Export to Uninfected Cells Exosomes Containing STING, Viral mRNAs, and microRNAs. Proc Natl Acad Sci USA (2014) 111:E4991–6. doi: 10.1073/pnas.1419338111
74. Kalamvoki M, Deschamps T. Extracellular Vesicles During Herpes Simplex Virus Type 1 Infection: An Inquire. Virol J (2016) 13:63. doi: 10.1186/s12985-016-0518-2
75. Prabakaran T, Bodda C, Krapp C, Zhang BC, Christensen MH, Sun C, et al. Attenuation of cGAS-STING Signaling Is Mediated by a P62/SQSTM1-Dependent Autophagy Pathway Activated by TBK1. EMBO J (2018) 37:e97858. doi: 10.15252/embj.201797858
76. Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING Directly Activates Autophagy to Tune the Innate Immune Response. Cell Death Differ (2019) 26:1735–49. doi: 10.1038/s41418-018-0251-z
77. Watson RO, Manzanillo PS, Cox JS. Extracellular M. Tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
78. Hou P, Lin Y, Li Z, Lu R, Wang Y, Tian T, et al. Autophagy Receptor CCDC50 Tunes the STING-Mediated Interferon Response in Viral Infections and Autoimmune Diseases. Cell Mol Immunol (2021) 18:2358–71. doi: 10.1038/s41423-021-00758-w
79. He L, Wang B, Li Y, Zhu L, Li P, Zou F, et al. The Solute Carrier Transporter SLC15A3 Participates in Antiviral Innate Immune Responses Against Herpes Simplex Virus-1. J Immunol Res (2018) 2018:5214187. doi: 10.1155/2018/5214187
80. Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell (2017) 171:1110–24 e18. doi: 10.1016/j.cell.2017.09.039
81. Gamdzyk M, Doycheva DM, Araujo C, Ocak U, Luo Y, Tang J, et al. cGAS/STING Pathway Activation Contributes to Delayed Neurodegeneration in Neonatal Hypoxia-Ischemia Rat Model: Possible Involvement of LINE-1. Mol Neurobiol (2020) 57:2600–19. doi: 10.1007/s12035-020-01904-7
82. Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y, et al. Circulating Mitochondrial DNA-Triggered Autophagy Dysfunction via STING Underlies Sepsis-Related Acute Lung Injury. Cell Death Dis (2021) 12:673. doi: 10.1038/s41419-021-03961-9
83. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature (2018) 563:131–36. doi: 10.1038/s41586-018-0629-6
84. Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, et al. Chromatin-Bound cGAS is an Inhibitor of DNA Repair and Hence Accelerates Genome Destabilization and Cell Death. EMBO J (2019) 38:e102718. doi: 10.15252/embj.2019102718
85. Chen H, Chen H, Zhang J, Wang Y, Simoneau A, Yang H, et al. cGAS Suppresses Genomic Instability as a Decelerator of Replication Forks. Sci Adv (2020) 6:eabb8941. doi: 10.1126/sciadv.abb8941
86. Schirmer EC, Florens L, Guan T, Yates JR 3rd, Gerace L. Nuclear Membrane Proteins With Potential Disease Links Found by Subtractive Proteomics. Science (2003) 301:1380–2. doi: 10.1126/science.1088176
87. Malik P, Zuleger N, de las Heras JI, Saiz-Ros N, Makarov AA, Lazou V, et al. NET23/STING Promotes Chromatin Compaction From the Nuclear Envelope. PloS One (2014) 9:e111851. doi: 10.1371/journal.pone.0111851
88. Malik P, Korfali N, Srsen V, Lazou V, Batrakou DG, Zuleger N, et al. Cell-Specific and Lamin-Dependent Targeting of Novel Transmembrane Proteins in the Nuclear Envelope. Cell Mol Life Sci (2010) 67:1353–69. doi: 10.1007/s00018-010-0257-2
89. Dixon CR, Malik P, de Las Heras JI, Saiz-Ros N, de Lima Alves F, Tingey M, et al. STING Nuclear Partners Contribute to Innate Immune Signaling Responses. iScience (2021) 24:103055. doi: 10.1016/j.isci.2021.103055
90. Katta SS, Smoyer CJ, Jaspersen SL. Destination: Inner Nuclear Membrane. Trends Cell Biol (2014) 24:221–9. doi: 10.1016/j.tcb.2013.10.006
Keywords: adaptor protein, autophagy, cell death, immunity, organelle, STING1
Citation: Zhang R, Kang R and Tang D (2022) STING1 in Different Organelles: Location Dictates Function. Front. Immunol. 13:842489. doi: 10.3389/fimmu.2022.842489
Received: 23 December 2021; Accepted: 28 February 2022;
Published: 17 March 2022.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Jacquelyn Gorman, Oklahoma Medical Research Foundation, United StatesCopyright © 2022 Zhang, Kang and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daolin Tang, ZGFvbGluLnRhbmdAdXRzb3V0aHdlc3Rlcm4uZWR1