 Yanhong Ni
Yanhong Ni Jun T. Low2,3
Jun T. Low2,3 Lorraine A. O’Reilly
Lorraine A. O’Reilly
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 June 2022
Sec. Cytokines and Soluble Mediators in Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.835997
This article is part of the Research Topic Oncoinflammatory Pathways: Focus on Cytokines and Lipid Mediators View all 12 articles
When small proteins such as cytokines bind to their associated receptors on the plasma membrane, they can activate multiple internal signaling cascades allowing information from one cell to affect another. Frequently the signaling cascade leads to a change in gene expression that can affect cell functions such as proliferation, differentiation and homeostasis. The Janus kinase-signal transducer and activator of transcription (JAK-STAT) and the tumor necrosis factor receptor (TNFR) are the pivotal mechanisms employed for such communication. When deregulated, the JAK-STAT and the TNF receptor signaling pathways can induce chronic inflammatory phenotypes by promoting more cytokine production. Furthermore, these signaling pathways can promote replication, survival and metastasis of cancer cells. This review will summarize the essentials of the JAK/STAT and TNF signaling pathways and their regulation and the molecular mechanisms that lead to the dysregulation of the JAK-STAT pathway. The consequences of dysregulation, as ascertained from founding work in haematopoietic malignancies to more recent research in solid oral-gastrointestinal cancers, will also be discussed. Finally, this review will highlight the development and future of therapeutic applications which modulate the JAK-STAT or the TNF signaling pathways in cancers.
Cells can communicate with each other and this is frequently accomplished by molecules, that are produced by one cell, then detected and interpreted by another. One class of these signaling molecules are secreted proteins, including cytokines, interferons and growth factors, otherwise known as ligands. These are detected by discrete receptor molecules which then initiate a cascade of signaling responses in the receptor cell that may result in, for example, small or large, acute or chronic (days to months) and reversible or irreversible changes (live or die) in the receptor cell. It is rare for particular receptor ligand combinations to work in isolation or to signal only one specific outcome, i.e. one signaling event may initiate multiple signaling branches that may in turn intersect, overlap or be affected by other signaling branches occurring concurrently. This makes analysis of signaling challenging but permits cells to respond with a degree of finesse to environmental cues.
The term cytokine encompasses interleukins, chemokines, interferons and tumour necrosis factors and all bind to corresponding receptors to activate cellular signaling pathways, including two related and evolutionary conserved signature signaling entities, the Janus Kinases (JAK: a family of tyrosine kinases) and the Signal Transducer and Activator of Transcription (STAT) protein family, which together constitute the JAK-STAT signaling pathway and has been studied in great detail (1–5). Another well studied pathway activated by cytokines is the NF-κB pathway, a family of inducible transcription factors, required in both innate and adaptive immunity to rapidly activate cellular responses (6, 7). Over 50 cytokines can signal through the JAK-STAT pathway, mediated by JAK-mediated phosphorylation, ultimately regulating a signaling cascade upstream of multiple cellular activities (8–13). The JAK-STAT pathway controls a multitude of biological processes; embryonic and immune-system development, stem cell continuity and innate and adaptive immune responses, inflammation and multiple aspects in the pathway to tumorigenesis (4, 13–15). JAK-STAT signaling therefore serves as a fundamental mechanism for how cells perceive and respond to environmental triggers and how they then further communicate their interpretation of these signals with other cells to control cellular fate outcomes. It is therefore not surprising that this pathway it is tightly regulated at multiple levels (5, 16), with aberrant activation due to dysregulated expression of cytokines or genetic mutations, gene amplifications, or gene polymorphisms resulting in chronic activation of this pathway. This is associated with a variety of human disease states, including; immunodeficiencies, interferonopathies, hematologic malignancies, inflammation, autoimmunity, the cytokine storm associated with COVID-19 mortality (17, 18), predisposition to bacterial and viral infections and, most relevant to this review, solid cancers (8, 19).
At it’s core, JAK-STAT signaling is quite simplistic, with a few elements; a cytokine, a receptor (cytokine receptor), a kinase (JAK kinase) and finally a transcription factor (STAT proteins) (5, 19), coupled with a regulator, the Suppressor Of Cytokine Signaling family of proteins (SOCS) (Figure 1). There are four mammalian members that belong to the JAK family; JAK1, JAK2, JAK3 and Tyk2, each of which selectively binds to different receptor chains, each comprising of seven JAK-homology (JH) domains (20) (Figure 1A). JAKs are capable of interacting with both types of cytokine receptors (I and II) (5), this includes receptors for interleukins, IFNs, and multiple others (21) (Figure 1B). As these receptors have no inherent enzymatic activity, preferential interaction with distinct JAK isoforms is required for their downstream signaling and STAT activation (5, 21, 22) (Figure 1C). The STAT family (STAT1–4, STAT5A, STAT5B, STAT6) is also structurally conserved (Figure 1A), but individual cytokine receptors preferentially activate distinct STATs (Figure 1B) through phosphorylation by the JAK proteins (3, 5, 23). This eventually leads via to exposure of a nuclear localisation signal (NLS) (Figures 1A, B) (1, 24). Within the nucleus, dimerized STATs can bind regulatory sequences such as GAS (Gamma interferon Activation Site) to control transcription of many target genes (8, 25, 26)] (Figure 1C) and integrate inputs from other signaling pathways, such as the NF-κB pathway.
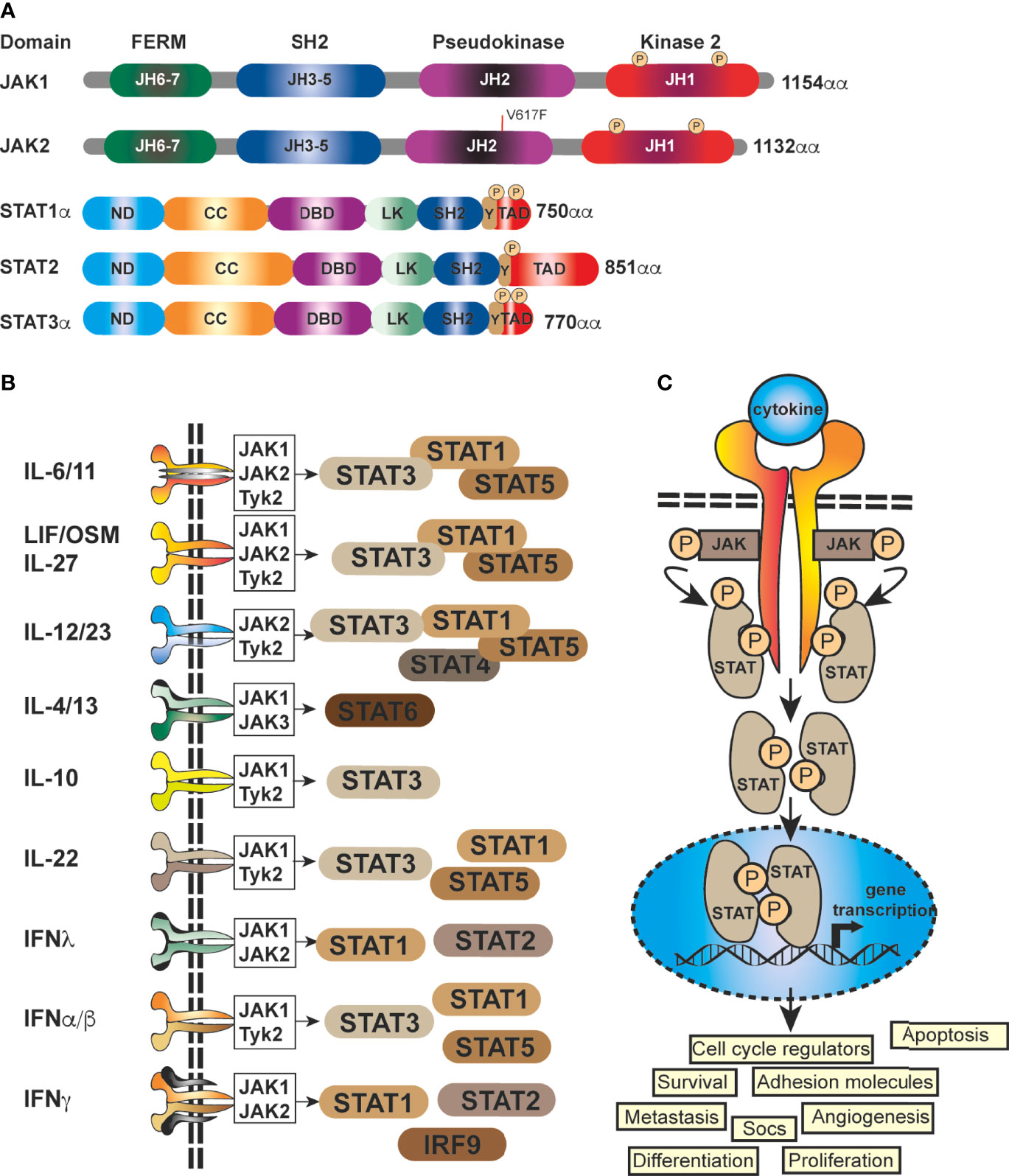
Figure 1 Schematic representation of the generic domain structure of JAKs and STATs. (A) Functional domains of the most important human JAKs and STATs regulating the progression of solid malignancies; ND, N-terminal; CC, coiled-coil; DBD, DNA-binding; LK, linker; SH2, Src-homology; TAD, transcriptional activation domain; JH1, kinase; JH2, pseudo-kinase domain with the ezrin, radixin, moesin (FERM) and SH2 domains forming a JAK receptor-binding module and the PK domain restrains Tyr kinase activity by binding the kinase domain. The common V617F mutation in JAK2 is shown (red line). (B) STATs are activated by a multitude of cytokines and IFNs. The most important ligands, receptors and pairing of JAKs regulating the progression of solid malignancies are shown. Shared receptor subunits are indicated by identical coloring. (C) Simplified schematic representation of the JAK-STAT signaling pathway. JAK activation occurs upon the binding of ligand and receptor multiprotein assembly, at which point two JAKs are brought into close proximity, permitting trans-phosphorylation. Once activated, the JAKs are capable of phosphorylating further targets, firstly the intracellular tails of the receptors on specific tyrosines, which then act as docking sites for their preferred substrates, the STAT proteins. Each STAT contains a conserved tyrosine residue near to the C- terminus transactivation domain (TAD), which is phosphorylated by the JAK proteins. This phospho-tyrosine promotes STAT protein dimerization via binding of an adjacent SRC homology 2, (SH2) domain and leads to exposure of a nuclear localisation signal (NLS), translocation into the nucleus, where they activate the transcription of genes involved in many cellular processes.
Importantly, JAK-STAT signaling is controlled by the SOCS proteins, which function as part of a negative-feedback loop (19, 27, 28) and are rapidly upregulated upon activation of JAK-STAT signaling (29, 30). SOCS1 and SOCS3 are the most mechanistically and functionally defined members of this family and are potent inhibitors of the JAKs, using a range of mechanisms, such as phosphorylation inhibition, blocking STAT recruitment (30, 31) or binding to cytokine receptor complexes via (Figure 2) shutdown off JAK-STAT signaling (21, 27). JAK activity is not only regulated by SOCSs proteins but also by protein tyrosine phosphatases (PTPs) (5, 19). STAT proteins are also controlled at multiple levels, and in addition to ubiquitin-mediated degradation, they can be inhibited by, for example, PIAS (Protein Inhibitors of Activated STAT) in the nucleus (32). In addition to the endogenous control of the JAK-STAT pathway, this signalling cascade can be modulated by various mechanisms, including autocrine/paracrine cytokine production, JAK protein mutation, upstream cancer-causing genes activating STATs, or more rarely STAT mutations themselves, possibly resulting in continuous pathway activation. Mutations and polymorphisms which dysregulate the JAK-STAT pathway can result in a variety of human conditions, such as inflammatory related diseases, an array of leukemias and even solid cancers (8, 15, 33–36).
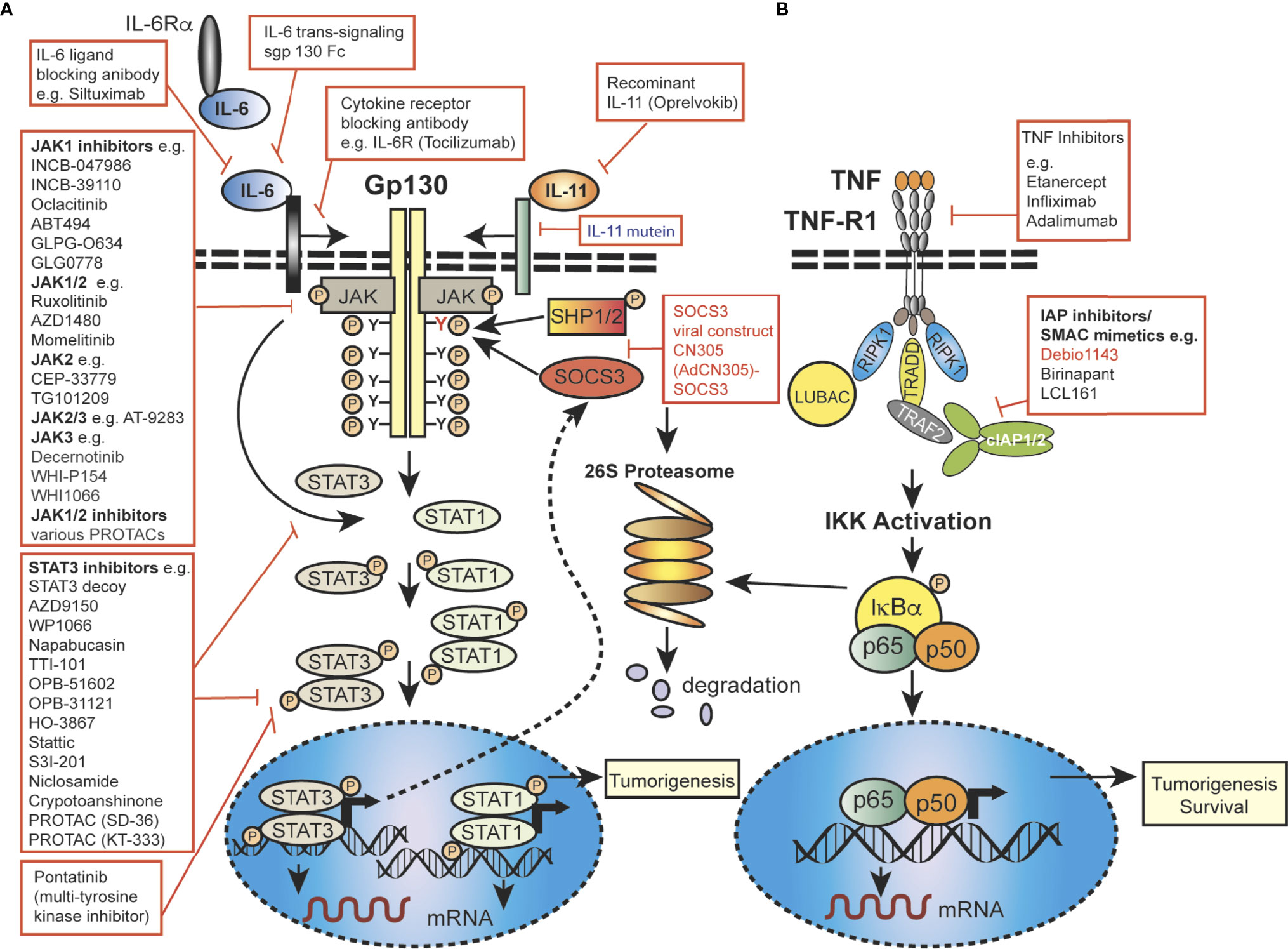
Figure 2 Mechanistic diagram of JAK-STAT and TNFR signalling and modes of inhibition. (A) The gp130 receptor complex and signaling pathways for IL-6 and IL-11 are shown as key activators of STAT transcription factors. The intracellular domain of the gp130 receptor contains a membrane-proximal tyrosine (Y; red), which provides a docking site for the suppressor for SOCSs proteins (e.g. SOCS3) and SHP2, while the membrane-distal tyrosine (Y) sites permits interactions with the SH2 domain of STAT1/3. The membrane proximal Y sites are phosphorylated by JAKs and upon phosphorylation of the distal Y site, STAT1 and STAT3 are recruited, homo-dimerise and are translocated into the nucleus, where they bind to specific target genes to regulate their expression. (B) Simplified schematic diagram of TNF/TNFR1 signaling pathway with downstream interacting proteins indicated. (A, B) Red arrows indicate intervention/inhibition points within each pathway and red boxes list examples of JAK-STAT or cytokine inhibitors/modifying drugs with their target protein.
JAKs are vital components of the JAK-STAT transduction pathway and their influential role in governing cellular survival, proliferation, differentiation and apoptosis is well studied (19, 37). Extensive clinical and genetic studies in human and in murine models have established a role for constitutively activated JAKs in human inflammatory/autoimmune diseases, myeloproliferative diseases and select cancers (8, 12, 19, 34, 38–43). For example, the common V617F mutation (Figure 1A), which results in constitutively activated JAK2, is associated with multiple disease syndromes. including elevated erythrocytes and megakaryocytes. This underlines the importance of JAK signaling in immune cell activities and haematological malignancies and provides the starting point for investigations into solid tumours. The generation of knockout or conditional knockout mice for each JAK family member has also provided an understanding of the interconnectedness between JAKs and their cognate cytokine receptors and the existence of restricted preferential relationships between cytokine receptors and their downstream JAK effectors, paralleling the phenotype of inactivating JAK mutations in humans (reviewed (12, 19, 44–47). However, functional redundancy exists, whereby in the absence of a single JAK, another JAK family member can fulfil the same signaling function (4, 19) (Figure 1B). In addition, the idea of hierarchical role for JAKs, with one JAK family member functioning as the upstream activator of a neighbouring heterotypic JAK family member within a cytokine receptor complex is another factor contributing to their functional complexity [reviewed (19)]. JAK1 and JAK3 GOF mutations occur in human leukemias and multiple myelomas (34, 48) but also in solid cancers, including breast, gastric, colon, lung and hepatitis B associated hepatocellular carcinomas, particularly in the pseudokinase domain and adjoining linker region (Figure 1A) (8, 16, 19, 34, 35). These clinical and genetic studies in human and in murine models have established a role for constitutively activated JAKs in human inflammatory/autoimmune diseases, myeloproliferative diseases and select cancers. Unsurprisingly, this role for activated JAKs in human disease has instigated immense interest by pharmaceutical companies to develop JAK inhibitors (15).
In recent years it has become apparent that the STAT proteins are linked to a variety of normal biological functions and these include cell differentiation, cell cycle control and proliferation, cell death and various aspects of the immune response. Mutations in STAT1, -2, -3, -5B, -6, while rare are associated with multiple inherited conditions, particularly primary immunodeficiencies, autoimmune diseases and even cancers (4, 8). Ultimately, the type of disorder developed is dependent on how STAT signaling is affected and whether the mutation is a GOF or LOF STAT gene defect. Seven inherited disorders are known to be caused by mutations in the STAT family of genes (49), with heterozygous STAT1 GOF mutations linked to increased susceptibility to certain infections, autoimmunity and risk for tumor development, while STAT2 insufficiency is characterized by susceptibility to multiple types of viral infections. Signaling through STAT3 is mediated through multiple receptors (Figure 1B), therefore a range of immunological and phenotypic manifestations is to be anticipated (8, 49). The contribution of specific STAT family members to control of normal cellular processes has also been evaluated by homozygous deletion or conditional knockouts. Probably the most important take home message is the establishment of a non-redundant role for STAT1 protein in IFN signaling (50), while STAT2 and STAT3, homozygous deletion resulted in embryonic lethality (51, 52). Conditional deletion in mice (53) subsequently established that STAT3 plays a crucial role in the signal transduction of various cytokines, including IL-6 (53). Thus, murine STAT proteins, like their human counterparts have diverse effects in response to extracellular signaling proteins, achieved by altering gene transcription in the effector cells.
In terms of human disease, the extended STAT family are also implicated in many aspects of tumorigenesis in humans as well as resistance to chemotherapy treatments. The most habitually mutated gene from this family in haematopoietic cancers is STAT3 [reviewed (4, 54, 55)] and to a lesser extent STAT5B (4, 22, 54). Somatic STAT3 mutation also plays a crucial role in select solid cancers, including skin cancers, GI and neural tumors, while STAT1, -2, -4 and -6 appear to have more limited roles in tumorigenesis (22). The majority of these diseases result from the consequential dysregulation of this signaling pathway, particularly related to JAK family member mutations, but mutation of STATs is not without clinical consequence (56). While mutations in members of the JAK-STAT pathway play a role in the development of a variety of diseases, particularly immunodeficiency syndromes and predisposition to infections, inflammatory processes themselves are one of the major drivers of tumor initiation, progression and metastasis (57, 58). This is in part driven by pro-inflammatory cytokines binding to their cognate receptors resulting in aberrant and often increased activation of JAK-STAT or TNF signaling (Figure 1C), although the exact mechanisms driving the penultimate stages of oncogenic invasion and metastasis are unknown (8). These tightly regulated STAT signaling control mechanisms can be disrupted in cancer cells, altering an otherwise finely-tuned homeostatic balance, which also occurs in GI cancers including Oral Squamous Cell Carcinoma (OSCC) (59), which comes under umbrella term of Head and Neck Squamous Cell Carcinomas (HNSCC) and Gastric Cancer (GC) (37, 60). Dysregulated cytokines levels are a hallmark of many gastrointestinal cancers and this review will focus on the role of JAK-STAT and introduce the TNF signaling pathways. Finally, we will discuss how current therapies and those in development, (eg. cytokine immunotherapy, Smac Mimetics and PROTACs) may be used modulate these signature pathways with the aim to increase curative rates for these low survival cancers.
As outlined above, constitutive triggering of the JAKs and STATs through elevated cytokine levels is linked to many chronic inflammatory diseases (61, reviewed; 57, 62). Such chronic inflammation may assist in promoting the cellular proliferation of nascent tumor cells, transformation and metastasis or conversely constraining the anti-tumor response. Cytokines also act on epithelial cells lining the gastrointestinal tract and other cell types to regulate secretion, proliferation, and differentiation (63). Inappropriate cellular epithelial activation may be one such consequence but may be quite diverse in different solid cancers (15, 16, recently reviewed 57, 64). Cytokines and their receptors are also polymorphic, and subtle genetic variations or single nucleotide polymorphisms (SNPs) predominantly found in the promotor region of genes such as IL-1β, 1-Rα, -2, -6, -10, -12, -13, -16, -18, TNF, IFN-γ, TGF-β are linked to functional changes and correlate with increased susceptibility to infections, autoimmunity, certain cancers and their differing treatment outcomes (65–67). Cytokine production by tumour infiltrating cells (TILs) or stromal cells such as endothelial cells or fibroblasts within solid tumor microenvironments (TME) mediate communication between tumor and TILs (57, 62, 68, 69). For example, chronic STAT3 signaling by cytokines such as IL-6 in transformed cancer cells can induce cell proliferation and activation of MMP’s (matrix metalloproteinases), thereby promoting tumor invasiveness and EMT (epithelia-to-mesenchymal transition) and expression of “master” EMT transcription factors including Twist and Snail (70). In addition, STAT3 becomes phosphorylated (hyperactivated) not only in the tumour cells but also in immune cells and CAFs within the TME (71), which could impact anti-tumor immunity. Chronic STAT3 activation is also associated with the elevated expression of factors promoting cell cycle and cell survival (cyclin D1, survivin and Bcl-xL) (72). Dysregulated cytokine expression levels and JAK-STAT signaling are also hallmarks of oral and gastric cancers (73). Activation of STAT3, STAT5B or JAK2 due to mutational changes are associated with certain heamatopoietic cancers and solid cancers (gastric cancer, breast cancer, lung cancer and oral cancer), while STAT3 and STAT5B are also considered to be bona fide oncogenes, since constitutively-activated forms induce cell transformation and invasion of cancer cells in mice (74–79). However, JAK-STAT signaling is complicated and often has context dependent effects. In certain situations and in certain cell types, it may instead have a tumor suppressive role, as is the case for STAT1 (80) and for STAT5, which may inhibit tumor progression in the liver but also acts as a tumor suppressor in fibroblasts (reviewed 76).
Several cytokines are particularly relevant to oral cancer and GC, such as those having proinflammatory functions; IFNγ, IL-6, IL-11 and other cytokines which are predominantly anti-inflammatory, such as transforming growth factor beta-1 (TGFβ), IL-4, IL-10 and IL-13 which all signal through the JAK-STAT pathway, except TGFβ, which signals through a receptor tyrosine kinase (57, 62). IL-6 mediated JAK-STAT signaling is required for normal homeostatic processes and is kept in check by the restricted pattern of expression of IL-6Rα to a subset of leukocytes and hepatocytes (81). However, trans-signalling through a soluble form of the IL-6Rα released from cells by proteolytic cleavage permits the formation of a IL-6/sIL-6Rα complex, which binds in trans to activate membrane-anchored gp130 (Figure 2A) (81). It is this latter signaling that appears to be involved in the inflammatory response and relevant to this review is involved in inflammation driven tumor response and T cell proliferation, leukocyte recruitment and activation of stromal cells (57, 82). Within the tumor microenvironment IL-6 is produced by a multitude of cell types including tumor cells, immune cells, and stromal cells. Elevated triggering of STAT3 by dysregulated IL-6 combined with additional oncogenic driver mutations, such as in KRAS or TP53, can drive tumor development in the oral cavity (83) and stomach (84).
In addition to cytokines which signal through the JAK-STAT pathway, initiation and development of inflammation driven cancers can also be driven by the master proinflammatory cytokine Tumor Necrosis Factor (TNF), an important regulator of the immune response in both the steady state and in disease processes and a critical mediator of carcinogenesis, as regulator of cellular proliferation, invasiveness and metastasis of a multitude of cancers (85, 86). Macrophages and T-cells are major sources of TNF, but other cells such as B-cells, endothelial cells and neutrophils also produce it (86, 87). TNF binding to its receptors TNFR1 and TNFR2, can cause a dizzying range of effects facilitating TIL invasion of tumors and promoting angiogenesis and tumour cell migration and invasion, but also promoting cell survival or cell death (88, 89). TNF is expressed as a type II transmembrane protein but is also liberated into a soluble form by cleavage by a membrane associated metalloproteinase, ADAM17 also known as TNF Converting enzyme (TACE) which also, incidentally, liberates IL6R for trans-signalling (90). Both forms of TNF interact with TNFR1 and TNFR2 but while membrane TNF stimulates both TNF receptors, soluble TNF largely fails to stimulate TNFR2 despite high-affinity binding (91). TNFR1 is widely expressed on most cell types and stimulates transcription of a host of inflammatory mediators including other cytokines by activating transcription and stabilising mRNA in a Nuclear factor of kappa B (NF-κB) (Figure 2B) and MAP kinase dependent manner (91, 92). However, in certain circumstances, particularly where transcription or signalling is interfered with, TNF can also induce cell death. In general, TNFR2 has a more restricted expression pattern and is predominantly expressed in myeloid cells and regulatory T-cells. TNFR2 has no intrinsic cell death inducing mechanism but stimulates NF-κB signaling and activation of various kinases and, while it cannot directly induce cell death it can switch TNFR1 signalling to cell death (91, 93). Activation of the TNF/TNFR2 pathway has been established as a critical bio-marker of several cancers including oral (94) and gastric cancers (63, 95).
Oral squamous cell carcinoma (OSCC) is one of the most common human malignancies and a leading cause of morbidity and mortality world-wide, constituting 4% of all systemic malignant tumors (96–98) OSCC also constitutes a major subcategory of HNSCC. These cancers originate in the oral and oropharyngeal subsites (tongue, lips, gingiva and retromolar trigone) and account for approximately 90% of all oral cancers (96). The oral cavity is the first station of digestive tract to be exposed to a multitude of environmental stimuli, including viral and bacterial infections and continuous chemical irritation. The main risk factors for OSCC are entirely predictable and include; tobacco smoking, alcohol consumption, continuous mastication of Areca-nut/betel-leaf/tobacco-quid (particularly in Southeast Asia), infection with human papillomavirus (HPV) and the inflammatory autoimmune condition Oral Lichen Planus (OLP, especially the erosive form), a possible precursor lesion of OSCC (96, 99, 100). These carcinogens and inflammatory agents and conditions contribute to a progressive influx of inflammatory cells (CD4 and CD8 T cells, B cells, NK cells, neutrophils, eosinophils, macrophages and plasma cells (101)), resulting in the accumulation of genetic and epigenetic lesions throughout the oral mucosa, affecting cell cycle, DNA repair mechanisms, cell differentiation and apoptosis and resulting in the transformation of normal keratinocytes to hyperkeratosis, oral dysplasia, the development of carcinoma in situ, then invasive cancer (102) (Figure 3A). Indeed, the oral cavity may be carpeted with pre-cancerous lesions with a high risk for malignant transformation (96). During the preceding period, chronic inflammation, driven in part by dysregulated salivary cytokines, a hallmark of OSCC, drives and maintains neoplastic transformation; a highly complex multifactorial process occurring in epithelial cells through the premalignant lesions, leukoplakia and erythroplakia to epithelial-to-mesenchymal transition EMT and eventually invasive cancer (Figure 3A) (94, 96, 100, 103). A direct link between inflammation in the oral cavity and promotion of tumor invasion has been established (1, 94, 104), in which elevated cytokines (94) and dysregulation of the JAK-STAT signaling pathway have been implicated (105, 106).
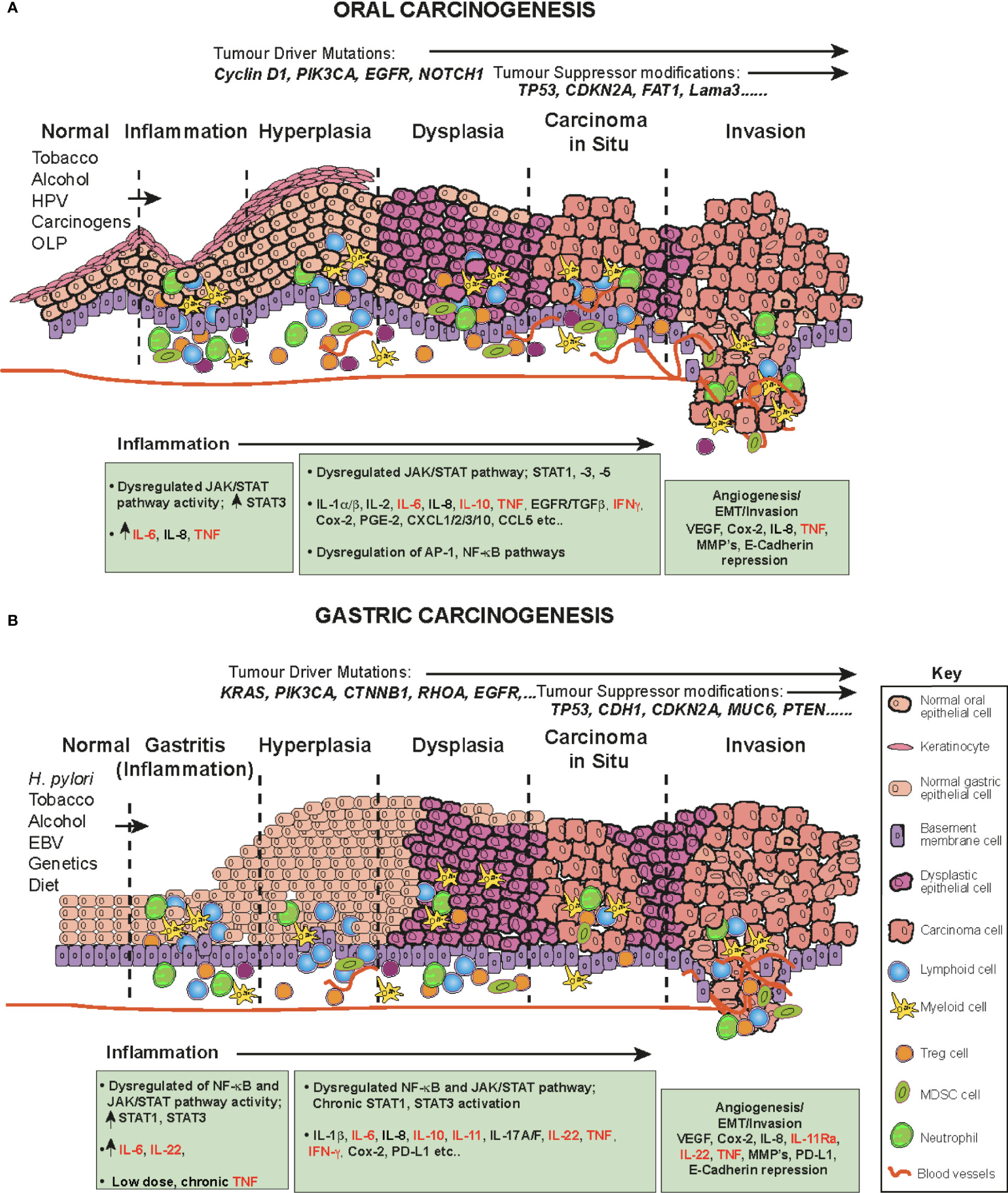
Figure 3 The role of cytokines and JAK/STAT in OSCC and GC in a step-wise model. The normal epithelium (left) progresses to (A) OSCC or (B) or GC, through a series of histopathological precursors. During early disease stage the immune system is activated by inflammatory stimuli, resulting in both pro-tumor and anti-tumor effects mediated by inflammatory cells. The cytokine and JAK-STAT pathway are dysregulated within the cells of the inflammatory microenvironment and target genes serve as fuel to control both apoptosis and inflammation. Pro-inflammatory cytokine mediators promote neoplastic growth and development, proliferation, tissue remodeling (EMT), angiogenesis and metastases. During this progression, several alterations in key genes also accumulate (e.g. KRAS, TP53) to progress tumorigenesis.
Certain syndromes such as Fanconi anemia or Dyskeratosis congenita can predispose to cancer of the oral cavity, however, these conditions are rare and usually OSCC occurs spontaneously in aged individuals (107). The associated accumulation of genetic and epigenetic alterations due to chronic inflammation results in oncogenic activation and inactivation or loss of tumor suppressor genes. These include frequent DNA copy number gains at chromosomes 3q, 5p and 8q and copy number losses on 3p and 8p (108). In addition, several driver/tumor suppressor modifications have been identified in OSCC, including; TP53, NOTCH1, EGFR (epidermal growth factor receptor), CDKN2A (cyclin-dependent kinase inhibitor 2a), Cyclin D1, Rb (retinoblastoma), FAT1 and Lama3 (105, 108–113) (Figure 3A).
Despite recent advances in clinical diagnosis and improvements in systemic therapeutics (cisplatin, 5FU (5-fluorouracil), docetaxel) and radiotherapy, the 5-year survival for HNSCC has remained at ~50% for the last 30 years for regional and more disseminated disease (99, 104). In addition, both the cancer and its treatment cause considerable morbidity with acute and long-term toxicity impacting speech, swallowing, nutrition and appearance (114, 115). The difficulty with early diagnosis and the high potential for neck lymph node metastasis with invasive properties continues to account for the high mortality (103, 116).
Potential novel biomarkers to identify high risk disease individuals, such as activating JAK mutations have been detected in some solid tumors, such as gastric (77) and for both JAKs and STATs in haematological malignancies (4, 8). However, these haven’t been reported to be associated with OSCC (117). Tests of predicted functional polymorphisms in HNSCC for SOCS3, have been conflicting; rs2280148 located at the 3′-untranslated region, indicated a predicted increase in the risk for this cancer, while rs8064821 located in the promoter region was associated with an decreased risk (118). Functional polymorphisms affecting gene expression of IL-4,-6,-8,-10 and TNF have been shown to strongly associate with increased risk for oral cancer (119). Single nucleotide polymorphisms (SNPs) in the promoter region of the TNF gene are linked to HNSCC susceptibility. For example, the TNF-308 position (AA/GA haplotype) SNP was shown to increase TNF expression and contribute to greater malignant clinical aggressiveness and reduced overall survival (120). More recently, patients with the CC genotype of TNF–1211, were shown to have higher plasma TNF associated with more severe oral mucositis complications and reduced survival when administered with chemoradiotherapy (121). On the other hand, patients with the TNF−238 A/G SNP homozygous recessive allelic variant had a reduced risk of oral precancer (122). Overall, these genetic association studies addressing the predisposition to oral cancer, highlight susceptibility differences at the population level, possible predictable treatment outcomes of these cancers and novel target avenues, which may be tested in animal models and are discussed later in this review.
Cells of the upper aerodigestive tract epithelium, are regulated, in part, by multiple cellular growth factors and OSCC demonstrates inappropriate activation of JAK-STAT signaling, particularly STAT3 but also STAT-1A, 5A and -5B (102, 123–125) as discussed in a number of recent comprehensive reviews (72, 117). However, the mechanistic links of the JAK-STAT pathway in OSCC are still being investigated. Inflammatory cytokines and chemokines, which activate the JAK-STAT pathway have been identified as early bio-markers and mediators of OSCC [recently reviewed (94)]. Gene expression microarray analysis of OSCC cell lines with high NF-κB activity and OSCC patient samples identified dysregulation of genes involved in inflammation, wound healing, angiogenesis and growth regulation and upregulated levels of IL-8, CCL5, STAT1, and VEGF in OSCC (102). In this review we primarily summarize the role of the cytokine IL-6, which signals through JAK1/JAK2 to activate STAT-1,-3,-5 (Figures 1B, 2A) and TNF (Figure 2B), since these cytokines play major roles in OSCC initiation and development of OSCC (94) (Figure 3A).
In general, the early phase of OSCC is asymptomatic, accounting for both generalized late detection and diagnosis (97). The identification of early OSCC biomarkers is of great importance to improving survival rates (126), because early detection dramatically improves survival but unfortunately it is exceptionally difficult and lesions are often overlooked (127). To address this problem, non-invasive salivary secretion diagnostics, termed a “liquid biopsies”, are being explored. TNF has been shown to have role in the progression of OLP and meta-analysis of multiple studies has shown higher levels of TNF in salivary secretions (128). Initial studies indicated that it was possible to distinguish cancer from non-malignant lesions. IFN, IL-1β, IL-2, IL-6, TNF and IL-8 were amongst the most commonly studied saliva cytokines, with elevated levels possible predictors of early malignancy [reviewed (129–131)]. However, differentiating between inflammatory conditions, such as periodontitis and OSCC is difficult (130). More recently several systemic and qualitative reviews from meta-analyses of OSCC case-controlled studies identified IL-2, IL-6, IL-8, TNF, IL1β and IL-10 as being significantly upregulated in OSCC patient saliva by ELISA (129, 132, 133). Except for IL-10, these cytokines were also higher in OSCC patients and oral, potentially malignant, disorders such as leukoplakia when this latter group were compared to healthy controls (132). Overall, the most promising and reliable diagnostic cytokines for OSCC, significantly elevated from other oral potentially malignant disorders from these studies are IL-6, IL-8 and TNF (101, 132, 133) (Figure 3A). While this early screening approach is promising, there are still many issues to be tackled before it can be adopted clinically for early diagnosis screening of high-risk populations.
Saliva cytokine levels of IL-1β, IL-2, IL-6 and TNF are not only associated with oral inflammation but also with the severity of oral mucosal damage in cancer patients. For example, a literature analysis by Uz et al. suggested that overexpression and elevated serum and/or saliva IL-6 concentrations in patients with HNSCC are related to poor survival and increased tumorigenicity (134). TNF also plays additional roles in promoting tumorigenesis and is overexpressed in human oral cancer tissues (135). It is also associated with pain, albeit in murine studies (136). However, HNSCC are a group of cancers associated with a high prevalence of pain (137) and pre-treatment for pain may serve as a prognostic factor for patient survival (138). In vitro studies also suggest that TNF enhances the invasion and metastasis ability of OSCC cell lines via the NF-κB signaling pathway (139). In addition, while it is believed that HPV infection alone may be insufficient for the oncogenic transformation of normal epithelial cells recent studies suggest that chronic exposure of TNF to HPV-infected oral keratinocyte cell lines increases cancer stem cell-like populations and stemness (140). This would suggest that HPV and TNF may act in concert to promote oral malignant conversion and that mechanisms to inhibit TNF may be of benefit for OSCC.
The 2015 TCGA study of head and neck cancers (constituting 62% oral cancers) identified amplifications in several oncogenes including the JAK-STAT linked receptor tyrosine kinases, EGFR and IGF1R (105). Nuclear and cytoplasmic interactions between EGFR and STAT3, increase the expression of several EMT promotors (iNOS, cyclin D1, c-fos) through direct binding of the EGFR/STAT3 complex to their promoters [reviewed (141)] resulting in consistent JAK-STAT triggering in HNSCC (4) (Figure 3A). IGF1R is also an upstream signal for JAK-STAT activation, particularly STAT3 (141).
Both HPV+ and HPV- HNSCCs demonstrate aberrant regulation of JAK-STAT signaling, with upregulation of STAT3 and its many gene targets contributing to malignancy and therapy resistance via cell cycle, cell growth and inhibition of cell death mechanisms (72, 73, 117, 142) (Figure 3A). Inhibition of aberrant STAT3 activity has been shown to impede OSCC growth both in vitro and in vivo, (72), and, coupled with the association of STAT3 hyperactivation with poor prognosis, resistance to standard therapies, and immune escape, this makes it a potential therapeutic target for OSCC (72, 102). IL-6 is one of the key upstream cytokines implicated in poor clinical outcomes in OSCC patients (143). STAT3 activity is controlled in part by the activity of the prototypical pro-inflammatory IL-6/gp130 signaling cascade (144) (Figure 2A). This STAT3 activation can in turn lead to elevated IL-6 expression creating a positive feedback loop that fuels the creation of an inflammatory and a pro-tumorigenic milieu (144). Multiple STAT3-controlled signal transduction pathways are associated with OSCC/HNSCC. In addition growth factors including EGF, TGF-α and platelet-derived growth factor can activate members of the STAT family, including STAT1 and STAT3 (145). Overexpression of EGFR and its ligand TGF have been detected in tumors and cell lines established from OSCC patients and EGFR protein is required to sustain OSCC cells in vitro (146). Stimulation of TGF/EGFR activates both STAT1 and STAT3, however, this TGF-α/EGFR mediated autocrine growth of transformed OSCC epithelial cells appears to be reliant on activation of STAT3 but not STAT1 (123).
In solid tumors, STAT1 is generally considered as a tumor suppressor but there is growing evidence that STAT1 also has a pro-tumorigenic function, perhaps in a cell-type specific context (80). The role of STAT1 in OSCC is controversial (117). Previous studies have shown that in chemotherapy treated OSCC patients, increased levels of p-STAT1 had a positive prognostic association, with a predictive increase in overall survival (147, 148). However, another more recent study showed the opposite correlation, with higher intra-tumoral p-STAT1 associated with worse survival (149). Despite these contradictory studies, microarray analysis of OSCC transcriptomes recently suggested that STAT1 is an important contributor to OSCC development, and that STAT1 might therefore serve as a potential diagnostic biomarker (150). One issue is that the role of STAT1 in OSCC may not actually correlate with expression levels within the tumor itself, but rather with the immune cells constituting the inflammatory tumor milieu. For example, in pre-malignant OSCC, CD163+ TAMs (tumor associated macrophages) are the main cells that express STAT1 and p-STAT1 (151). Relevant to both OSCC and EBV+-gastric cancer, JAK2/STAT1 signaling has been shown to mediate respectively EGFR- or IRF-1, IFNγ-induced upregulation of the programmed cell death-ligand 1 (PD-L1), an inhibitor of T-cell-mediated tumor cytotoxicity (152, 153) as discussed later in this review.
STAT3 transforms human epithelial cells and has been defined as an OSCC oncogene and identified as a negative prognostic factor in human OSCC (154). A constitutively activated JAK-STAT pathway, particularly STAT3 is an early event in OSCC (124) and shown to be mediated by the autocrine/paracrine stimulation of the IL-6/gp130 cytokine/receptor (155). In addition to being a core early event in OSCC, abnormal STAT3 activation represents a potential risk factor for poor prognosis in early-stage patients (106, 124) and in later disease stages correlates with poor tumor differentiation, lymph node metastasis and reduced survival (16, 106). Expression of a dominant-negative mutant STAT3 in HNSCC/OSCC cell lines has been shown to prevent proliferation, trigger apoptosis, and inhibit the downstream pathways associated with STAT activation (124). Thus, STAT3 activation is a fundamental underlying factor in a multitude of malignant behaviours in OSCC, contributing to cell proliferation, differentiation and apoptosis resulting in neovascularization, establishment of a pro-inflammatory state. Hyperactivation of STAT3 is also implicated in both treatment resistance and immune escape within the oral cavity (72, 123, 124, 156). In addition, LOF protein tyrosine phosphatase receptor type T (PTPRT) by somatic mutation or promoter hypermethylation (31-60% of HNSCC), increases STAT3 activation and sensitivity to STAT3 inhibition (72, 157). Tumor suppressors that dephosphorylate STAT3, may also lead to prolonged phosphorylation and activation of STAT3 (72, 105, 157). Dysregulation of STAT3 and enhanced expression of the active phospho-form not only within the tumor cells themselves but also within the TILs, including fibroblasts constituting the tumor microenvironment can support solid tumor growth (158). Therefore, STAT3 activation most likely engages the communication between these types in the OSCC microenvironment promoting tumor progression. In addition to serving as an oncogene in OSCC, STAT3 is a resistance mechanism for standard chemotherapeutics and radiation, the current treatment modalities for this cancer (99).
Cancer-associated fibroblasts (CAFs), are major cellular components of the OSCC stroma and communicate with tumor cells to stimulate cancer cell growth, survival, and invasion and associated with poorer survival outcomes. Epiregulin (EREG) a member of the Epithelial Growth Factor (EGF) family promotes tumor development, migration and invasion (159). We recently showed that EREG is upregulated in CAFs from OSCC patients, with elevated expression correlating with the tumor severity and predicted shorter overall survival (160). Mechanistically, EREG appeared to be essential for normal fibroblast to CAF transformation and essential for the induction of tumor cell EMT in a JAK2-STAT3- and IL-6-dependent manner (160). This recent finding further underlines the importance of the JAK-STAT pathway in the progression of OSCC by activating genes important in the initiation of tumor metastasis.
Compared to STAT3, STAT5 activation plays a relatively minor role in OSCC/HNSSC but STAT5 can contribute to the development of tumorigenesis and increased expression of STAT5 proteins and phospho-STAT5 has been demonstrated in these tumors (161). SOCS2 protein is significantly downregulated in OSCC patients, and its levels are inversely correlated with miR-424-5p expression, through a newly described IL-8/miR-424-5p/STAT5 pathway in OSCC (162). This pathway involves the pro-inflammatory cytokine IL-8 activating STAT5, which then induces SOCS2 (a STAT5 inhibitor) and miR-424-5p. MiR-424-5p expression however suppressed SOCS2 activity and led to constitutive STAT5 expression. Such elevated STAT5 expression correlated with increased tumor cell migration and invasion through elevated matrix metalloproteinase activity in OSCC cancer cells. Copy number variations were increased in OSCC when compared with normal or pre-malignant oral lichen planus samples. In 7/15 samples there was an increase in chromosome 9 sequences in a region which encompass JAK2, as well as 38 other genes (163).
Clinical and descriptive studies of biopsy samples and analysis of cell lines from patient OSCC, are informative for later disease stages but rarely provide insight into disease development. Animal models of chemical induced oral carcinogenesis and transgenic animals are useful in this regard and also to assess therapeutic approaches and the impact of the immune system (103). The most frequently used chemicals are DMBA (9,10-dimethyl-1,2-benzanthracene) and 4NQO (4-nitroquinoline-1 oxide). DMBA is highly irritating and produces an inflammatory response and necrosis. 4NQO, acts similarly but is more efficacious in inducing tumorigenesis and has been shown to mimic the process of oral carcinogenesis in humans (113, 124, 164, 165). These murine cancers, share pathologic and biochemical features with tobacco-related human OSCC, including epidermal growth factor receptor (EGFR) overexpression downregulation of p16 (164), elevated STAT1 (166), STAT3 (164, 165), overexpression of SOCS1, -3 (167), elevated pro-inflammatory cytokines (IL-1β, IL-6, TNF) and MMPs (136, 168). Furthermore, as we have shown, loss of NF-κB signalling accelerates tumorigenesis (169). Regulatory T cell (Tregs) enrichment and function also appears to be modulated by STAT3 in response to radiation therapy in a murine oral orthotopic model with DMBA induced HNSCC/OSCC (170). Since radioresistance is a major issue with human HNSCC, this suggests that STAT3 inhibition may benefit patients receiving this type of therapy.
Moreover, the mutational spectrum of human OSCC was shown recently to be replicated in the 4NQO mouse model, including mutations in Tp53, PIK3ca, Notch1, Fat1 and Lama3 with gene ontogeny analysis identifying cytokine signalling as a major biological process linking the human and murine disease (113). In addition, a cross-species genomic comparison of DMBA carcinogen-induced murine and human OSCCs with indolent or metastatic growth using next gen sequencing also revealed conservation of the human driver pathway mutations in mouse OSCC; Tp53, Mapks, Pi3k, Notch, Fat1–4 and the Jak-Stat pathway (171). There is an opportunity to delve more deeply into the roles of these factors in initiation and development of oral cancer because not many studies have used genetically modified mouse models deficient in cytokines or elements of the JAK-STAT pathway to explore their roles in these chemical induced OSCC models (169).
The stomach is the next station in the alimentary tract down from the oral cavity and aids in the adsorption of nutrients by secreting hydrochloric acid and enzymes. As with the oral epithelium, the gastric epithelium is exposed to multiple exogenous stimuli, which can result in chronic inflammation. In most human GCs, this inflammation is initiated by infectious agents, such as Helicobacter pylori (H. pylori) or Epstein-Barr virus (EBV) infection or prolonged exposure to gastric irritants, such as a diet high in salt and nitrated foods (172) (Figure 3B). These agents promote chronic inflammatory gastritis, subsequently leading to pre-cancerous alterations in the stomach epithelial lining, which combined with aging may have an additive role in the promotion of GC (63, 172–174) (Figure 3B). Gastric cancer (GC) is the 5th most common human cancer that imposes the 4th highest cause of cancer mortality world-wide (98). This is in part due to the frequently asymptomatic nature of this disease which often results in a late-stage diagnosis with locally advanced or metastatic disease and limited curative opportunities (172). More than 90% of gastric cancers (GCs) are adenocarcinomas, which originate from epithelial cells in the chronically inflamed gastric mucosa. A pathology based, classification of GC identified diffuse, intestinal or mixed sub-types (175), which have been subsequently divided into four molecular sub-types; genomically stable (GS), microsatellite instability (MSI), EBV+, and a Chromosomal instable type (CIN) (77). A small proportion of chronic gastritis patients develop gastric cancer, suggesting additional risk for genetic and environmental factors in GC development (172). DNA polymorphisms that increase GC risk have mapped to genes encoding cytokines (63). Many cytokines secreted by immune cells and epithelial cells during chronic gastritis have been linked to poor patient outcomes (176).
GC usually occurs spontaneously with only about 8-10% of cases due to inherited mutations, such as in E-cadherin (CDH1) resulting in an autosomal dominant GC predisposition (177). Gastrointestinal polyposis syndromes can also predispose to GC (178). Multiple studies including the Cancer Genome Atlas Research Network (77), in addition to the risk factors described above, have revealed certain genetic alterations and/or mutations can contribute to GC pathogenesis including; PIK3CA, TP53, KRAS, APC, STK11, CTNNB1, CDKN2A, ARID1A, ERBB2, FGFR1, FGFR2, EGFR and MET and (Figure 3B) (77). A novel recurrent amplification at 9p24.1, the locus containing JAK2, CD274 (PD-L1) and PDCD1LG2 (PD-L2) were also described in the EBV+ GC subtype (77, 179, 180). CD274 (PD-L1) and PDCD1LG2 (PD-L2) are involved in immunosuppression by tumor cells to evade cytotoxic T cell mediated killing and are associated predominantly with the EBV+ sub-type (77) in a STAT1 driven manner as discussed later in this review (153).
Hopes that anti-inflammatory drugs and H. pylori eradication during early disease stages may prevent disease progression, have not been entirely realised (181) but may reduce risk (182). However, current treatments (chemotherapy, radiation, surgical resection) are often given with palliative intent. High rates of relapse for GC are indicative of the failure to address the chronic inflammation that is believed to one of the critical disease drivers. Both the NF-κB and JAK-STAT pathways are known to promote inflammation-associated tumorigenesis within the GI tract (7, 60) and deregulation of these pro-inflammatory signaling pathways, results in gastric intestinal dysplasia and eventually, over decades results in invasive adenocarcinoma (173). Significant improvements in overall survival rates of patients, which currently stand at 30% 5 years post diagnosis have not, to date, been achieved (98).
H. pylori is the leading risk factor for GC, infecting the stomach lumen. In most cases the immune response is unable to clear the infection and the inflammation becomes chronic, creating molecular and cellular changes favouring the transition to tumorigenesis (174). However, this response is variable and is dependent upon both host genetics and H. pylori strain (183). Activation of inflammatory genes and elevated pro-inflammatory cytokines including IL-1β, IL-1Rα and the neutrophil attracting cytokine IL-8, IL-10, -11, -17A, -17F, -22 and TNF (reviewed (63, 84, 184) increase the risk for atrophic gastritis and GC (174) (Figure 3B). Many of these cytokines activate the JAK-STAT and NF-κB pathways, resulting in their activation and the formation of an inflammatory microenvironment containing a complex combination of cytokines and chemokines, which accelerates GC development and progression (174). Our discussion will centre on cytokines, since the role of chemokines in GC has been recently excellently reviewed (185).
A number of DNA polymorphisms in TNF and STAT controlled cytokine genes have been mapped, including IL-1β, IL-1R, -8, -6, -17A, 17F, -22, that may mediate differences in response to chronic H. pylori infection and therefore risk for gastric cancer (186–191) and reviewed (183). Specifically, a recent meta-analysis study of 46 publications strengthened the idea that several TNF polymorphisms may be associated with GC risk, particularly TNF-857 in Caucasians and TNF-1031 in Asian populations (192). As a pro-inflammatory cytokine, low-dose chronic TNF secreted by inflammatory cells sustains inflammation, chemokine expression, whilst promoting angiogenesis and inflammation-associated neoplastic progression (193, 194). A recent analysis of patient GC RNA transcript levels has shown that many TNF responsive cell survival genes, (e. g. TRAF2, C-FLIP) are up-regulated favoring a pro-tumoral effect, while pro-apoptotic genes as caspase-3 and TNFR1 are down-regulated during GC development (Figure 2B). This suggests that a disequilibrium between the cell death and proliferative processes occurs in GC. Whilst TNF is elevated in H. pylori–infected patients (188) its role in GC still remains to be rigorously investigated.
The IL-6 family of cytokines mediates their inflammatory or additional pro-tumor effects through the JAK-STAT signaling pathway by binding to specific transmembrane signal transducers (Figure 1B, 2A). Signalling through Gp130, a shared receptor element of many of these transducers, is often dysregulated in GC due to elevated levels of IL-6 and IL-11 (84), which activates STAT3 in GC and GC stem cells and is associated with poorer outcomes (195, 196). IL-11, rather than IL-6, appears to show a greater correlation with elevated STAT3 activation in both human and murine GC (197, 198). Another inflammatory cytokine IL-22, which is produced by CAFs, has been shown to enhance the invasive capacity of human GC cells in vitro by activating both STAT3 and ERK signaling (184). Elevated IL-11Ra expression is also associated with lymphatic invasion and blood vessel infiltration (199).
Dysregulation of the JAK-STAT pathway more generally has also been documented to contribute to gastric tumorigenesis (60, 200). The role of STAT1 in GC is complex as discussed below and in GC tissues it is regulated in an IFNγ-JAK-STAT-dependent manner (153). Chronic STAT3 activation appears to be pivotal in GC induction (200) and STAT3 has been shown to control the production of many pro-inflammatory cytokines (TNF, IL-1β, IL-6 and IL-22). These factors are known to control the cellular function of immune cells, resulting in a pro-inflammatory state and can also impact multiple functions contributing to nascent tumorigenesis in epithelial cells (201, 202). Active STAT3 is expressed in several established GC cell lines (203) and its inhibition has been shown to mediate their apoptosis. STAT3 is also known to promote the formation of new blood vessels by increasing expression levels of VEGF in GC (60). A recent study also highlighted the role of H. pylori in the epigenetic silencing of SOCS1 in GC through hypermethylation of the promotor region, which in addition to inflammatory cytokines, further amplifies JAK-STAT signaling in this cancer (204). Collectively, multiple human GC studies have intimated a role for JAK-STAT signaling, in particular STAT3 in multiple cell types associated with tumorigenic phenomena, such as inflammation, EMT transition and metastasis (60).
Transgenic and gene knockout technologies have been utilised to develop murine models of inflammation induced GC, which have been useful to study the influence of cytokines on the progression to tumorigenesis, without the influence of Helicobacter (63, 205). Many of these models include mice carrying mutations that result in the overexpression of the JAK-STAT transcription factors, cytokines or modulation of the NF-κB pathway impacting cytokine and STAT protein expression (206–209). Constitutive STAT3 activation promotes gastric tumorigenesis not only in human adenocarcinomas and but also mouse models (200) (Figure 3B). The common gp130 subunit heterodimerizes with several IL-6 family co-receptors (Figure 2A) (210). One of the best characterized murine models to investigate the role of these cytokines is the gp130Y757F/Y757F (gp130F/F) mouse, which carries a homozygous mutation (Y757 residue) in the gp130 receptor chain (206, 209). This mutation disrupts the SHP2 and SOCS3 binding site preventing them from shutting down signalling and resulting in the hyperactivation of STAT3, and to a lesser extent STAT1. This in turn leads to upregulation of many STAT3 target genes, including pro-inflammatory cytokines (19, 27, 28). The gp130F/F mice develop spontaneous gastric adenomas, akin to human intestinal GC (209). Similar to human GC, in this model IL-11 has a more prominent role compared to IL-6 during the progression to GC and a stronger correlation with elevated STAT3 activation. This suggests that for GC, targeting IL-11 rather than IL-6 may be a more beneficial option (198, 211). Excessive STAT3 activation, fuelled and maintained by tumor-associated IL-11 expression, appears to be sufficient to trigger neoplastic behaviour of gastric epithelium without the requirement for additional predisposing genetic alterations (212). This IL-6/IL-11-dependent increase of STAT3 expression also contributes to the development and progression of H.pylori-associated GC (213), while loss of STAT3 or STAT1 prevented disease (211). Overall, these animal studies have provided valuable insights into how STAT3-driven inflammation drives GC tumorigenesis and identify IL-11 as a crucial cytokine promoting chronic gastric inflammation and associated tumorigenesis mediated by excessive activation of STAT3 and STAT1 (197, 212).
H. pylori or EBV infection in addition to activating STAT3 can result in sustained activation of NF-κB. We have recently shown that mice lacking NF-κB1 (Nfkb1-/-), a member of the canonical NF-κB signaling pathway develop inflammation-driven invasive gastric adenocarcinoma (208). GC in Nfkb1-/- mice mimics the histopathology of the human disease and is associated with elevated STAT1, and to a lesser extent STAT3, in the gastric mucosa (208), recapitulating the pathogenic loss-of-function polymorphisms in NF-κB1 associated with human GC (207, 214). Similar to the human disease, GC in this mouse model is associated with elevated pro-inflammatory cytokines and chemokines such as TNF, IL-6 and IFNγ. Consistent with the foregoing discussion there is an earlier onset of GC in Nfkb1-/-/gp130+/F mice (207, 208).
As discussed for oral cancer, while STAT1, exerts tumor suppressive activities, by integrating the anti-proliferative and pro-cell death signals elicited by interferons, it can also drive tumor promoting activities in stromal cells. These activities include inducing an immunosuppressive tumor environment by regulating immune checkpoint inhibitor expression, such PD-L1 and PD-L2 the ligands for PD-1, which can be induced in tumors by IFNs, resulting in immune evasion (80). We established a direct link between NF-κB1 loss-of-function and upregulation of STAT1 pro-tumorigenic functions and the immune checkpoint PD-L1 (208). Pertinently, JAK2 is also amplified in human EBV+ GC (77) and cellular RNA expression of Jak2 is abnormally elevated in the gastric mucosa of Nfkb1-/- mice before overt GC (208). Moreover, the mechanism underlying the regulation of PD1 and PD-L1 in human the EBV+ GC subtype has recently been shown to be controlled by the JAK-STAT1-IRF3 signaling axis (153). Therefore, the immune evasion of EBV+GC cells could be regulated by this signaling pathway, which may be further investigated using these murine models (215). More recently we showed that IL-6, and IL-22 and the receptor for IL-11 (IL-11Rα) are dispensable for the development of GC in Nfkb1-/- mice (207). However, the loss of IL-11Rα significantly reduced invasive GC disease and loss of TNF inhibited GC development in Nfkb1-/- mice but to a lesser extent than complete loss of STAT1 (207). Notably, the loss of either TNF or STAT1 reduced gastric inflammation and PD-L1 expression in the stomach (207). Insights from this model reveal a role for TNF in GC development, identify a role for IL-11Rα in invasive GC disease and uncover a link between elevated TNF levels and aberrant STAT1 activation shaping the gastric immune microenvironment. These findings suggest that inhibition of IL-11/IL-11Rα signaling for example by using IL11-Mutein might have clinical benefit (197, 198, 216). While inhibitors of TNF are readily available compared to STAT1 (currently do not exist), these potential therapies may extend to a broad range of GCs, not only those with NFKB1 gene polymorphisms, a topic we will discuss in the final section of this review.
Surgery and chemoradiotherapy/radiotherapy (CRT) are the mainstays for advanced OSCC/HNSCC, because effective and targeted therapies which are still wanting (142, 217). Similarly, the prognosis for advanced GC remains abject due to the poor response to current therapies (chemotherapy, surgery, chemo/radiotherapy) (60, 96, 218, 219). Due to a deficit of targeted therapies, additional or adjunct therapies are warranted to advance treatments for these cancers. Blocking JAK-STAT signaling may be a solution, either as a single therapy, or in combination with other anti-cancer agents. There are multiple potential intervention points for targeting JAK-STAT signaling, including various cytokines, their receptors, STATs, JAKs, SOCS (Figure 3) as well as other cytokine signalling pathways, such as TNF. Inhibitors abrogating the JAK-STAT pathway tested in pre-clinical and clinical studies have been recently extensively reviewed (4, 8, 15, 56, 71, 82, 220, 221) and so we will here focus on HNSCC/OSCC and GC.
Therapeutic targeting of STAT transcription factors is not without challenges. Transcription factors have been deemed “undruggable” due to their lack of catalytic function (82, 222). While there are many studies using conventional small molecule drugs, unfortunately, both off-target adverse effects and on-target toxicity have tarnished the progression of these compounds and few have transited to clinical trials for solid cancers (4, 56, 60, 71, 72, 82, 220, 221) and even more limited for HNSCC/OSCC and GC (Table 1).
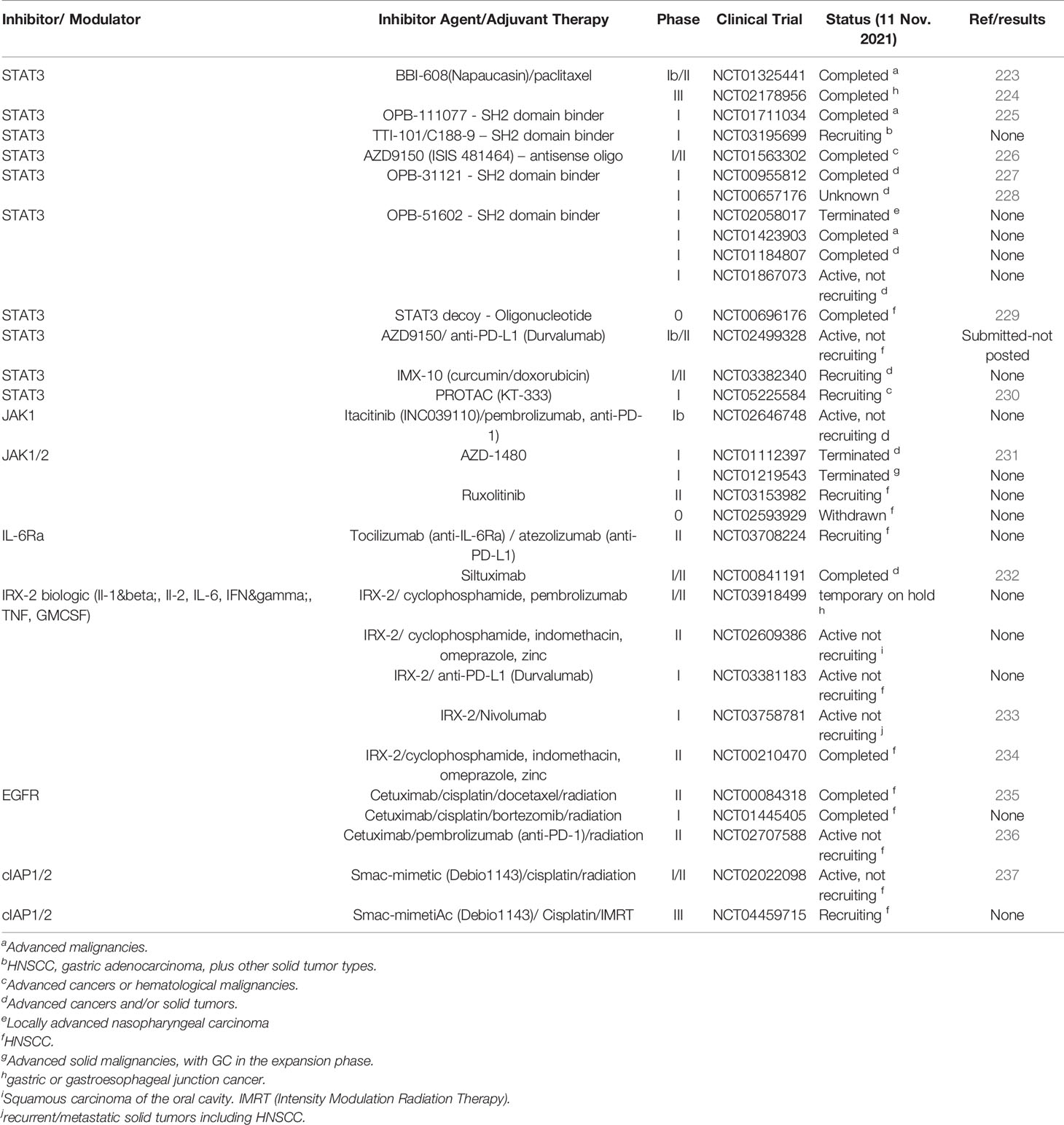
Table 1 Clinical Trials for JAK-STAT/TNF Pathway modifiers in HNSCC or GC.
STAT1 inhibitors are still under development using comparative virtual screening and docking validation (238). An important point to bear in mind is that STAT1 takes part in IFN signaling and thus plays important roles in barrier function and host defence against infections, therefore its inhibition may not be of optimal benefit for translation to the clinic (4, 8, 220).
Because of its strong pro-oncogenic function, most STAT inhibition studies have focused on this transcription factor with the aim of blocking phosphorylation and/or STAT dimerization (221). However, inhibition of STAT3 is also problematic, since it can be activated by several different upstream kinases and development of an inhibitor that specifically targets STAT3 rather than STAT1 to improve therapeutic efficacy remains challenging (15, 56, 239). While a number of STAT3-inhibiting compounds have been developed (4, 71, 221) (Figure 2A), they generally have low potency, poor specificity and inappropriate pharmacology constraining their progression into the clinic and approval (71, 82, 239). Newer compounds are being synthesized and evaluated by computational methods to improve the understanding of the STAT3 functional mechanism and aid in the design of STAT3 inhibitors as anti-cancer drugs (239, 240). These consist of direct STAT3 inhibitors (peptides, small molecules, oligonucleotides), indirect inhibitors (JAKs, IL-6, EGFR) or those that can be combined with immunotherapy (e.g. immune checkpoint inhibitors, CAR-T cell therapy, dendritic cell based cancer vaccine and immunostimulatory Toll Ligand Receptor (TLR) agonists) (71).
STAT3 is also a major therapeutic target under investigation for HNSCC/OSCC/GC [reviewed (71, 72, 82)] and while studies are predominantly at the preclinical or early clinical stage, they hold some promise. For example, the STAT3 decoy (double-stranded DNA containing STAT3-binding site) that sequesters dimeric STAT3 away from endogenous targets has been shown to increase apoptotic death and reduce tumor growth in laryngeal squamous cell carcinoma (PCI-37A) (241). This STAT3 decoy is being tested in the clinic, where the expression levels of STAT3 target genes were shown to decrease in HNSCC/OSCC following STAT3 decoy injection (229) (Table 1). STAT3 signaling activity can also be attenuated by Stattic, a small molecule STAT inhibitor, which targets the SH2 domain, resulting in the modulation of invasion and migration of OSCC cell lines (242). In nasopharyngeal carcinoma cell lines Stattic has been shown to inhibit cell viability and proliferation, induce apoptosis and enhanced chemo/radio sensitivity (243).
The most successful STAT3 inhibitor to date is napabucasin/BBI-608 which inhibits JAK2 and STAT3 phosphorylation and transcription of target genes (244) (Figure 2A; Table 1). It received orphan designation from the FDA (2016) for gastric cancer due to early positive results for the phase Ib/II trial (223), however the phase III (BRIGHTER trial) as a second-line treatment in combination with paclitaxel in patients with gastric and pre-treated advanced gastric and gastroesophageal junction (GEJ) cancer showed no improvement in overall or progression free survival (224). Subgroup analysis is pending, and this may provide some insight for better patient selection for napabucasin in the future.
The Otsuka Pharmaceutical Co. have developed a number of non-peptide STAT3-SH2 domain inhibitors (OPB-31121, OPB‐111077, OPB‐51602), which have been documented in many phase I trials (selected; Table 1). OPB-31121 has been shown to reduce proliferation of gastric cancer cells and in a xenograft model, where it was shown to synergize with 5-fluorouracil (5-FU) and cisplatin (245). This inhibitor also interacts with STAT5 and showed antitumor activity in various hematopoietic malignancies (246). However, phase I trials for advanced malignancies were terminated before many participants had been enrolled (Table 1). On the other hand, it has undergone successful phase I/II trials for advanced hepatocellular carcinoma (ClinicalTrials.gov #NCT01406574) and for solid cancers (Table 1), (227) and is currently in a phase III trial in combination with 5-FU, Leucovorin and Irinotecan (FOLFIRI) for metastatic colorectal cancer (ClinicalTrials.gov #NCT03522649) and in a completed but yet to be reported trial with nab-paclitaxel and gemcitabine for metastatic pancreatic ductal carcinoma (CanStem11P trial, ClinicalTrials.gov #NCT02993731). OPB‐111077, another novel inhibitor of STAT3, also exhibits promising anti-cancer activity in patients with diffuse large B‐cell lymphoma (DLBCL) and modest efficacy was observed against other tumors, including GC, when given as a monotherapy (225). Phase I studies with OPB‐51602, have been disappointing in hematological malignancies (247) and locally advanced nasopharyngeal carcinoma but are currently in trial for solid cancers (Table 1).
The small-molecule competitive STAT3 inhibitor TTI-101 (formerly C188-9) developed by Tvardi, targets the pY-peptide binding site within the SH2 domain (Figure 2A) to prevent phosphorylation, homodimerization, nuclear translocation and transcriptional activation (221). TTI-101 has been shown to inhibit cytokine-stimulated pSTAT3 and reduce constitutive pSTAT3 activity in multiple HNSCC cell lines, including the radioresistant HNSCC cell line UM-SCC-17B in a xenograft model. In this system, TTI-101 prevented tumor growth by modulating many STAT3-regulated genes affecting oncogenesis and radio-resistance, as well as radio-resistance genes regulated by STAT1, due to its potent activity against not only STAT3 but also STAT1 (248). More recent studies have shown that TTI-101 can be given orally and without toxicity (249) and this STAT3 inhibitor is currently being trialled as a monotherapy for solid tumors including HNSCC and GC (Table 1).
AZD9150 is a second generation antisense oligonucleotide targetting the 3’ untranslated region of STAT3. It inhibits mRNA translation and has shown efficacy in pre-clinical models (226), where it inhibited tumor growth and expression of STAT3 downstream target genes. It has also shown efficacy in phase I/Ib clinical trials for both lung cancer and lymphoma (142, 226, 250). A Phase I/II dose-expansion study for the treatment of patients with advanced cancers, DLBCL and other advanced lymphomas (ClinicalTrials.gov NCT01563302) has been completed but no results are available. Another phase II trial using AZD9150 in advanced solid tumors including metastatic HNSCC as a monotherapy or combined with MED14736/Durvalumab immunotherapy (anti-PD-L1) is currently underway (ClinicalTrials.gov #NCT02499328) (Table 1).
Another method to target STAT3, involves the use of double-stranded “decoy” oligonucleotides, corresponding to STAT3 response elements, such as those present in the c-fos promoter. An analogous STAT3 decoy has been shown to block binding of STAT3 and when used to treat HNCSS cell lines inhibited proliferation and reduced STAT3-mediated gene expression (241). A phase 0 trial of this STAT3 decoy (ClinicalTrials.gov #NCT00696176, Table 1) determined that intra-tumoral administration in HNSCC patients inhibited STAT3 driven gene expression. In addition, a kinetic study involving a xenograft model showed that administration of the STAT3 decoy, but not the mutant control decoy, decreased expression of STAT3 target genes (Bcl-xL and/or Cyclin D1) (229).
Finally, YHO-1701 a novel quinoline- carboxamide derivative of STX-0119 and a non-peptide SH2 domain STAT3 inhibitor has been shown to inhibit the SH2 binding to a p-Tyr peptide more potently than the original STX-0119 in biochemical assays (251). YHO-1701 also exhibited strong activity in abrogating STAT3 signaling in the human OSCC cell line SAS, by inhibiting STAT3 dimerization and also suppressing STAT3 promoter activity. In addition, YHO-1701 showed anti-tumor effects in SAS xenograft models in combination with sorafenib producing an anti-proliferative and synergistic effect in SAS OSCC cells, justifying future clinical follow-up (252). Overall, the studies evaluating STAT3 inhibitors for OSCC/HNSCC and GC at pre-clinical and early clinical trial stages suggest that it is unlikely that they will be used as a monotherapy. However, there is still the possibility of using them as adjunct therapy, which will be discussed later in this section.
The cornerstone of JAK-STAT signaling inhibition are the JAK family members (Figure 1) and these have been extensively studied for inflammatory diseases such as rheumatoid arthritis (253). A variety of JAK inhibitor compounds are available at the clinical or pre-clinical stage as orally delivered small molecules targeting the ATP-binding site of the JAK protein, preventing their phosphorylation and the subsequent phosphorylation of STATs thus diminishing JAK activity and nuclear signaling (59, 82). Since JAKs are upstream of STATs and therefore might also interfere with other mechanisms involved in cancer progression this could explain a perceived improved efficacy of JAK inhibitors and the popularity of Tofacitinib (JAK1/JAK3 inhibitor) and Baricitinib (JAK1/JAK2 inhibitor) for the treatment of inflammatory conditions (4, 8, 59, 60, 82, 253, 254). As an interesting aside, a relatively selective JAK1 inhibitor, Oclacitinib, is used to treat dermatitis in dogs and lacks the side effects that most JAK inhibitors have in humans (255). Ruxolitinib, a JAK1/JAK2 inhibitor is FDA approved for the bone marrow cancer myelofibrosis, polycythemia vera and topically for atopic dermatitis (59, 71, 82, 254) and has also shown efficacy in solid cancers, for example in HNSCC cell lines (256). However, an early phase 0/I trial with ruxolitinib as a monotherapy administered prior to surgical resection for operable HNSCC was withdrawn due to adverse events (ClinicalTrials.gov #NCT02593929), but a phase II trial is currently recruiting (ClinicalTrials.gov #NCT03153982) (Table 1). Trials have also recently commenced for a range of other JAK inhibitors for solid malignancies, but with limited success (4, 59, 71, 254). For example, AZD140 (JAK1/2 inhibitor) abrogated STAT3 activation and HNSCC tumor growth in a patient-derived xenograft preclinical model (257), but a study to assess the safety and tolerability of AZD140 as an oral monotherapy in patients with solid tumors (ClinicalTrials.gov #NCT01112397) (231) and a phase I study including GC in the expansion phase (ClinicalTrials.gov #NCT01219543) have both been terminated due to a decision to stop development of this JAK inhibitor (Table 1). It is noteworthy that JAK inhibitors are not specific for each JAK and there are issues with off-target effects (253), thus many clinical trials for solid tumors have been terminated (59) (Table 1). It is clear that additional JAK inhibitor specificity is necessary to optimize future therapeutic applications. In addition, adverse events associated with the wide-ranging immunological effects of JAKs (258), have been reported and thus the future of JAK monotherapy applications may be limited (15, 253). However, novel PROTACs targeting the Janus kinase family (JAK1, JAK2, JAK3 and TYK2) have been recently described which may overcome these shortfalls (259, 260).
Since SOCS proteins negatively regulate JAK-STAT cytokine signaling, they can play major roles in limiting the evolution and subsequent progression of tumorigenesis. Atypical SOCS1 and SOCS3 expression in established tumor cell lines and also at advanced clinical stages of cancer is considerably variable (261). However, SOCS1 mediated negative signaling feedback has been shown to be important for inflammation reduction (262) and also to limit nascent tumor growth (263). Similarly, epigenetic inactivation due to CpG methylation in SOCS1 is frequently linked to several human solid cancers including GC and may be involved in its development, progression and even metastasis, since reduced expression of SOCS1 was associated with lymph node metastasis and more severe GC tumor stage (264). In addition, SOCS1 and SOCS3 can control the development and activation status of immune cells in tumorigenesis but their exact roles in this process are still unclear (261, 265, 266). Thus, SOCS-based targeting is in its infancy (267).
Since SOCS seem to inhibit tumor progression different strategies have been used to either increase or mimic SOCS activity. For example, SOCS1 and SOCS3 expressing oncolytic adenovirus (CN305 (AdCN305)-SOCS3) or recombinant, cell-permeable proteins has been shown to efficiently control aberrant STAT signaling in hepatocellular carcinoma (268). A SOCS1 peptide mimetic (containing the KIR domain) which acts as a pseudosubstrate for JAK1, JAK2, TYK2 and JAK3 and is able to activate the endogenous SOCS1 protein has also been pursued for therapeutic applications. Their safety and efficacy particularly in comparison to JAK inhibitors is yet to be fully evaluated (263).
An emerging opportunity for cancer therapy to circumvent the hazards of targeting intracellular signaling molecules such JAKs, or STATs is through pharmacologic inhibition of proteins that activate them, such as pro-inflammatory cytokines (71, 73, 198, 269). Since cytokines are major drivers of several autoimmune/inflammatory diseases, it is not surprising that cytokine inhibition has revolutionized therapy for these disorders, particularly with monoclonal antibodies targeting TNF, IL-1, IL-2 and IL-6 (116). Cytokine-based immunotherapy for cancer treatment is encouraging, since cytokines are able to regulate the host immune response to directly induce cancer cell death (270). Recently this has led to an interest in the efficacy of cytokine-based drugs, as single agents or in a combinative approach with other immunotherapy drugs to improve the anti-tumor properties of cytokines. Newer second-generation drugs aimed at improving cytokine activity in the tumour microenvironment or towards the desired effector immune cells, however, are yet to reach the clinic (270). In this review we will focus on modulation of IL-6 and IL-11 which signal through the JAK-STAT pathway (Figures 1, 2A) and TNF, which signals through the TNF family (Figure 2B). In addition, we discuss how antagonizing the activity of these cytokines has merit in overcoming toxicity to improve tolerance of cancer immunotherapy, including for OSCC and GC.
Many therapies target cytokines or their receptors (Figure 2) and to date, much focus has been placed on antagonizing the activity of IL-6, since elevated levels of this cytokine can mediate hyperstimulation of JAK-STAT signaling through the gp130 receptor (197, 269, 271). Antagonistic monoclonal antibodies such as tocilizumab, against IL-6R, and siltuximab against IL-6 (Figure 2A), were originally developed to treat inflammatory diseases, and tocilizumab was granted an emergency use authorization in the US for COVID. Other IL-6 targeting agents are also in development: sarilumab, ALX-0061, olokizumab, sirukinumab and clazakinumab (82, 269). Inhibitors of IL-6 and IL-6Rs are now undergoing clinical trials for solid cancers: ovarian, renal, prostate, lung, melanoma, pancreatic and breast (71, 82, 272, 273). However, the IL-6 ligand antibody (siltuximab/CNTO-28) trial for prostate cancer, did not improve clinical outcomes (274).
A phase I/II trail with siltuximab for select solid advanced solid tumors, including HNSCC with KRAS mutations, while well tolerated was without clinical activity (232) (Table 1). Our preclinical studies in a GC cancer model driven by the absence of NF-κB1, have shown that genetic deletion of IL-6 is dispensable for the development of GC, with a minor role for IL-6 at the early stages of gastric dysplasia (207). Studies antagonizing the activity of IL-6 for HNSCC and GC are not yet forthcoming. However, as indicated earlier in this review, IL-6 trans-signalling through a soluble form of the IL-6Rα (81) is involved in inflammation driven tumor response. This signalling can be selectively inhibited using a soluble form of gp130 (sgp130) fused to an IgG Fc fragment, as sgp130Fc (olamkicept/FE 999301/TJ301) (Figure 2A). This first-in-class decoy protein exclusively blocks IL-6 proinflammatory trans-signaling and has shown clinical efficacy in early phase IIa trials for ulcerative colitis/inflammatory bowel disease (IBD) without immunosuppression but with p-STAT3 reduction in the inflamed IBD mucosa [ClinicalTrials.gov #NCT03235752), (275). However, sgp130Fc is yet to be trialled in solid tumors. Other designer mutants to modulate IL-6 signaling are in early development (197)].
Another IL-6 family cytokine, IL-11 also signals through gp130 to activate JAKs/STAT3 (Figure 2A). IL-11 is known to be a pleiotropic in character and to play a role in hematopoiesis, adipogenesis and platelet maturation (197). In this capacity, IL-11 predominantly acts as an anti-inflammatory cytokine, with potential to increase platelet counts in chronic myelogenous leukemia. More recently its role in several inflammation driven cancers, such as gastrointestinal cancers has been identified (197, 276), perhaps providing a link between inflammation and cancer. IL-11 can increase the oncogenic properties of cells, including, cancer cell proliferation and survival (197, 277). For example, elevated expression has been linked to several cancers, including GC, where IL-11 levels are elevated in preclinical models of GC (277). These findings support a role for IL-11/IL-11Rα signaling inhibition as an emerging therapeutic opportunity for multiple cancers, including GC, perhaps through implementation of IL11-Mutein, an antagonist of IL11Rα (216). However, our understanding of how IL-11 impacts the initiation and progression cancers is still limited and no agent inhibiting IL-11 is currently approved for the treatment of solid cancers (197).
The master proinflammatory cytokine TNF can serve as either therapeutic target or agent. As a therapeutic target, it can be inhibited with well established anti-TNF biologics, including etanercept, infliximab, adalimumab, golimumab, and certolizumab pegol (278) for inflammatory diseases (116). As discussed in this review, TNF is also important at many stages in OSCC (94, 136) and GC (63), therefore targeting the TNFR signaling pathway may be an effective preventive or therapeutic strategy for solid cancers. Indeed, the use of anti-TNF drugs to treat cancer has a long and interesting history (58, 85). Some early preclinical studies were encouraging in this regard; for example blocking TNF with golimumab reduced tumor growth, angiogenesis and metastasis of OSCC in a murine model of orthotopic human OSCC (279), and antagonizing TNF reduced oral cancer proliferation and cytokine production in mice with 4NQO induced oral cancer (135). TNF/TNFR1 signaling also been shown to promote gastric tumorigenesis in the Gan mouse model, in which transgenesis activates both canonical Wnt signaling and the COX-2/PGE2 pathway (280). Clinical studies with anti-TNF therapy are yet to be published for OSCC or GC (273).
Another approach to enlist TNF signaling are the smac-mimetics, a drug class that antagonises Inhibitor of APoptosis proteins (IAPs: XIAP, cIAP1, and cIAP2). Several small molecule smac-mimetic compounds have been developed and many have been or are currently in clinical trials, including Debio 1143/AT-406/SM-406, LCL161, APG1387, BI 891065 and ASTX660 (281, 282). These promote the proteasomal degradation of the cIAPs, resulting in TNF production in a subset of ‘responsive ‘ cells (283–285). IAP overexpression is associated with tumorigenesis, chemoresistance and poor prognosis (281, 286, 287) and the IAPs also positively (canonical) and negatively (non-canonical) regulate NF-κB signalling pathway (281) (Figure 2B). We, and others, have shown that smac-mimetics function by instigating auto-ubiquitylation and proteasomal degradation of cIAP1 and cIAP2 (284, 285, 288, 289), which in the presence of a stimulus such as TNF, chemotherapy or TLR ligand, results in the formation of a RIPK1/caspase-8 complex, followed by caspase-8 cleavage and the induction of tumor cell death (281, 285, 290, 291). A number of smac-mimetic compounds have been developed and due to their limited anti-cancer activities when used as single agents are now being tested in combination with immunotherapy and/or chemotherapy (6, 281, 282). For example, a trial reported in 2020, using Debio 1143 in combination with chemo-radiotherapy was the first drug combination in more than three decades to significantly improve overall survival in high risk HNSCC (includes OSCC) patients (ClinicalTrials.gov NCT02022098) (237) and a phase III trial is currently recruiting (ClinicalTrials.gov NCT04459715) (Table 1).
Immune checkpoint dysregulation to escape from immune system surveillance is used by a number of cancers including HNSCC/OSCC and GC to allow their development. Immune checkpoint inhibitor therapy (anti- CTLA-4, PD-1 or PD-L1 antibodies) boosts the cytotoxic activity of tumour antigen-specific T cells and has revolutionized treatment and increased survival for a subset of cancers (292, 293). Boosting the immune response with anti-PD-1 for both HNSCC/OSCC (96, 294) and GC (215, 295), while proven for subsets of patients is not without challenges, including patient selection, bio-markers and adverse events (296, 297). Therapy for OSCC and GC is changing, and pre-clinical studies suggest that JAK-STAT signaling could play a role in regulating the activity of Immune Checkpoint Inhibitor (ICI) therapies (71, 152, 153, 295). The FDA has given approval for the use of ICI therapies for the MSI sub-type of GC (with EBV+ also a subtype which might benefit) (77). These subtypes are generally more immunogenic due to high mutational burden and ICI exploits this leading to generally better outcomes (215). However, since IFNγ is a major driver of JAK-STAT1 associated PD-L1 expression (ligand for the immune-checkpoint PD-1), JAK-STAT therapies, which have the potential to block IFNγ signalling, may result in reduced PD-L1 expression, thereby diminishing the effects of ICI. However, it may be possible to circumvent this since it was recently reported that JAK2 inhibitors in combination with ICI had better therapeutic responses, by blocking JAK signaling and repressing the effects on PD-L1 (73). These considerations are starting to focus attention on the immunological context of GC and particularly STAT1 expression as an immunotherapy biomarker (56, 73, 215, 298).
These new advances are exemplified by a new era of personalized therapy in the IMMUNOGAST phase II Trial (ClinicalTrials.gov #NCT04739202) for recurrent GC based on TCGA sub-type and trialling immunotherapy (anti-PD-L1, atezolizumab) plus ipatasertib (AKT inhibitor) for EBV+ GC, which exhibits amplified JAK2, ErbB2, PD-L1 and PD-L2 (Table 1). Similarly, for advanced solid cancers, including HNSCC, an active phase 1b trial combines pembrolizumab (anti-PD1) with the JAK1 inhibitor itacitinib (INCB039110) (ClinicalTrials.gov #NCT02646748) (Table 1; Figure 2A). APROPOS, a phase II clinical trial for atezolizumab (anti-PD-L1) in combination with tocilizumab to modulate T cell infiltration as a pre-surgery immunotherapy (ClinicalTrials.gov identifier NCT03708224) is currently recruiting for HNSCC (Table 1). Recent studies also highlight the potential for smac-mimetics specifically in combination with ICI and CAR-T-cell therapies (299–301).
Cytokine therapy is challenging because of their high degree of pleiotropism, resulting in many adverse effects. However, their real value in cancer therapy, particularly for oral and gastric cancers may be in enhancing the safety of immunotherapies. Thus, ICI or CAR-T cell therapy can result in inflammatory toxicities, such as immune-related adverse events (irAEs) or cytokine release syndrome (CRS). These are life threatening systemic diseases affecting multiple organs (302, 303). By perturbing the inhibitory control processes of the immune system, ipilimumab (anti-CTLA-4), atezolizumab (anti-PD-L1), or nivolumab/pembrolizumab (anti-PD-1) (293) unleash immune cells potentially resulting in the destruction of healthy tissues, limiting therapy duration and otherwise durable remission (302). Serious irAEs manifest with increased mononuclear cells and elevated levels of inflammatory cytokines, including IL-6 and TNF (302). In this context, treatment of advanced GC with nivolumab has been associated with liver injury accompanied with elevated TNF levels (14), highlighting an opportunity for anti-TNF to be used in conjunction with ICIT (193). A recent review on the topic suggests that short course TNF inhibitors are safe irAE treatment for cancer patients undergoing ICI therapy (304). However, the safety profile for long-term TNF inhibitor use for irAEs is lacking and further clinical studies that directly assess the effect of TNF inhibitor treatment on ICI efficacy are required. Data from preclinical studies hint that TNF neutralization in combination with ICIs reduces clinical irAE and improves antitumor efficacy in tumor models (305).
Improvements in selectivity and prevention of drug resistance in cancer treatment may be achievable with a novel approach using Proteolysis Targeting Chimeric (PROTAC) technology developed by the Crews and Deshaies labs (306, 307). In one approach, the ubiquitin-proteasome system (UPS) is hijacked for targeted protein degradation through hetero-functional molecules linking the target protein ligand to an E3 ubiquitin ligase (E3) by a functional linker, resulting in the continuous and rapid deletion of the target protein (181, 308–312). PROTACs have been developed targeting proteins important in cancer growth such as BTK (Bruton’s tyrosine kinase), FAK (Focal adhesion kinase), CDK’s (Cyclin-dependent kinases), Bromodomain-containing protein 4 (BRD4), Mcl-1, MDM2 (312–314) for haematological malignancies (315) and solid cancers, including a photo-controlled BRD4 PROTAC for tongue squamous carcinoma (314).
To date only two orally available PROTACs have reached phase II trials using CRBN (cereblon) as the E3 ligase (312). These PROTACs are showing promising results for prostate (ARV-110) and breast (ARV-471) cancers, targeting the androgen or oestrogen receptors respectively (312, 313, 316). PROTACs are also in clinical development for Bcl-xL (ClinicalTrials.gov. NCT04886622) using an alternative E3 ligase, VHL (von Hippel-Lindau) for both hematologic and solid tumours (312). The success of PROTACs for targeted protein degradation has also progressed from proteasome strategy to harness additional cellular degradation pathways (317). Pertinently, newly emerging degradation technologies are also expanding the scope of PROTACs (312, 318, 319) and their development looks likely to upend the viewpoint that transcription factors are “undruggable” and bring proteins such as those within the NF-κB (230) and JAK-STAT (318) signaling pathways into the druggable orbit.
Despite being considered as a therapeutic target for cancers, attempts to target STAT3 as we have discussed have not met with much success, due to poor target specificity and ineffectiveness in blocking the transcriptional activity of the monomeric form (82, 222, 310, 320). However, STAT3 is an example of the application of PROTAC technology towards the development of small molecule STAT3 inhibitors (306, 310, 320, 321). SD-36, the first described STAT3 PROTAC was designed and developed by the Wang lab using the cell permeable STAT3 SH2 domain and transcriptional inhibitor, SI-109, attached to an E3 ligase analogue of the CRBN ligand lenalidomide (320, 322, 323). SD-36 selectively degraded STAT3 (Figure 2), including mutated STAT3 proteins over other STAT proteins, even though they share a conserved SH2 domain (323) in various leukemia, lymphoma (323) and glioma cell lines (324). Functionally in certain leukemia and lymphoma cell lines SD-36 inhibited STAT3 dimerization, DNA binding and target gene activity (323). Prolonged tumor regression in mice xenografted with leukemia, lymphoma cell lines in vivo was also achieved with SD-36 and was well tolerated, bringing this PROTAC into the realm of possible therapeutic utility (323). While, SD-36 is yet to enter clinical trials, excitingly the STAT3 targeting PROTAC developed by Kymera (KT-333, Figure 2) (321) is recruiting for a first in human phase I trial (Feb 04, 2022, Clinical Trials.gov. NCT05225584) for leukemia, lymphoma and non-specified solid tumors (Table 1). Similar to SD-36, KT-333 showed selective degradation of STAT3 over other STAT family members, apoptosis upon treatment of tumor cells, depletion of STAT3 and down-regulation of STAT3 target proteins (321). In pre-clinical in vivo xenograft studies KT-333, similar to SD-36 showed a dose dependent growth suppression of hematologic tumors and was well tolerated (321). The development and early clinical trials of PROTACs is an encouraging next phase in the search for STAT3 inhibitors that may yet provide clinical benefit for STAT3-driven cancers such as gastric and oral cancers.
Specific inhibition of select JAK family members is a therapeutic challenge, due to the sequence similarity within pivotal catalytic domains such as the ATP binding site. PROTAC drugs, which destroy their target, are not necessarily constrained to these conserved domains and therefore open up the possibility of specifically targeting JAK family members. Efforts are already underway to exploit this with the design of the first of several series of heterobifunctional PROTAC compounds targeting the JAK family (JAK1, JAK2, JAK3 and TYK2), of proximal membrane bound proteins (259, 260, 325). Solving the structure of JAK inhibitors (e.g. Pyrimidine 1, Quinoxaline 2 or modification of ruxolitinib or baricitinib) bound to the JAK2 tyrosine kinase domain enabled the design and optimization of a series JAK PROTACS t capable of inducing proteosomal degradation of JAK1 and JAK2 (260, 325). The PROTACs capable of removing all possible functions of JAK signaling, rather than just the catalytic activity traditionally associated with JAK inhibitors themselves (259, 325). The most advanced of the JAK inhibitors are series of cereblon (CRBN)-directed JAK PROTACs linked to derivatives of the JAK inhibitors ruxolitinib or baricitinib tested in a panel of leukemia/lymphoma cell lines and xenograft models of kinase-driven acute lymphoblastic leukemia (325). These early studies highlight the potential of JAK-directed protein degradation as a therapeutic approach in JAK-STAT mediated diseases, however they are yet to be tested in vivo for solid cancer models or clinically for any disease. We speculate that PROTAC development may extend to the targeting of other JAK-STAT related proteins, such as cytokines (Figure 2) and that they may be beneficial for the treatment of not only hematologic malignancies but also extend to solid cancers including oral and gastric cancer, diseases yet to be explored both in pre-clinical models and in the clinic. In conclusion, PROTACS are likely to become a welcome addition to the already available armoury of other targeted drugs and chem-radio therapy available for solid cancers.
Tumor-induced immune suppression is common in patients with advanced malignancies, including HNSCC and GC. Strategies for relieving immunosuppression in HNSCC to restore anti-tumor immune functions, include the neoadjuvant IRX-2, derived from stimulating human PBMCs with phytohemagglutinin (326). IRX-2 contains a cocktail of cytokines including IL-2, IL-1β IL-6, IL-8, TNFα, GM-CSF, and IFNγ (Table 1). In vitro studies have shown that IRX-2, enhances DC maturation, T cell activation, and NK cell stimulation, to overcome tumor-mediated immunosuppression by activating the tumor environment (326). In clinical settings, IRX-2 administered with chemotherapy promotes anticancer immune responses (234). For example, in a phase IIa trial, patients with previously untreated HNSCC, IRX-2 increased tumor infiltration of T cells, B cells, and DCs and was associated with tumor reduction and prolonged patient survival (Table 1). Follow-up and data analysis are under way in a multi-center, randomized, phase IIbINSPIRE trial evaluating the IRX-2 regimen as a stand-alone therapy for activating the immune system to recognize and specifically destroy OSCC tumors (ClinicalTrials.gov #NCT02609386). The IRX-2 regime combined with immune checkpoint therapy is also being trialled for HNSCC and other solid tumors (ClinicalTrials.gov #NCT03758781) (233) (Table 1).
One of the few natural compounds tested for treating tumorigenesis is the polyphenol curcumin (diferuloylmethane), the yellow pigment found in turmeric. This natural compound targets many signaling pathways and has been shown to have multiple anti-inflammatory and anti-tumor qualities including anti-tumor metastatic activities through a variety of mechanisms (327). High dose curcumin (30-100mg/kg per day) reduced the tumor inducing potential of the carcinogen 4NQO when administered over the same period in a rat model of OSCC, which was associated with downregulation of STAT3 (327). Oral cancer patients given the curcumin containing drug APG-157, which also contains several other polyphenolic compounds, showed a reduction of the inflammatory cytokines IL-1β, IL-6, and IL-8 in salivary fluid, accompanied with elevated immune T cells in the tumor tissue, suggesting its potential use in combination with immunotherapy (328). Further studies in GC have suggested that curcumin can prevent GC cancer cell proliferation, invasion, metastasis, and angiogenesis, reviewed in (329). However, it is difficult to extrapolate from these studies into curcumin consumption due to its various effects on multiple cellular and molecular mechanisms. In addition, it is evident from the current and expanding treatment strategies discussed for both oral (142) and gastric cancer that an appreciation of patient/tumor genetics, the tumor microenvironment and possibly the microbiome also need to be considered for further optimized treatment strategies.
JAK-STAT/cytokine signaling pathways have been comprehensively studied due to their pivotal roles in both inflammation and many disease conditions including cancer. The core JAK unit activating the transcription factor STAT element is triggered by a variety of receptors and activates a wide range of downstream targets. Deregulated JAK-STAT signalling and particularly dysregulation of JAK2, STAT3, Il-6 and TNF are commonly associated with driving inflammation and carcinogenesis, including both OSCC and GC. Therefore, the targeting of these signaling pathways with inhibitors holds potential as clinical interventions for these cancers. There are a number of specific inhibitors of the JAK-STAT/cytokine cascade that have already been approved by the FDA for inflammatory conditions and which are being investigated for efficacy in clinical trials for haematopoietic malignancies and a few select solid tumors. However, despite promising clinical outcomes in inflammatory immune diseases and in tumor animal studies, toxicity issues have hindered adoption of these therapeutic approaches in the clinic. Investigating other avenues, such as the addition of cytokine blockade or smac-mimetics may provide synergy for more efficacious inhibition of OSCC and GC tumorigenic progression. More recent advances such as blocking JAK-STAT signaling with small molecules such as PROTACs may provide even better therapeutic precision in conjunction with other anti-cancer agents already in the clinic such as immunotherapy.
LO’R wrote the original review and YN, JL and JS reviewed edited and assisted with figures. All authors have read and agreed to the published version of the manuscript.
This review was in part funded by a Cancer Council Victoria Grant-in-Aid project, grant number 1161400 (to LO’R), a Cancer Australia and a Cancer Council New South Wales project grant number G20-13 (to LO’R), the Victorian State Government (OIS grant), NHMRC fellowships to (to JS) NHMRC 1107149 & 1195038 and National Natural Science Foundation of China Grant Nos. 81772880, 82173159 (to YN), a Key Research and Development Project (Jiangsu Province) No.BE2020628 (to YN).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT Signaling Pathway. J Cell Sci (2004) 117:1281–3. doi: 10.1242/jcs.00963
2. Darnell JEJ, Kerr IM, Stark GR. Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins. Science (1994) 264:1415–21. doi: 10.1126/science.8197455
3. Stark GR, Darnell JE Jr. The JAK-STAT Pathway at Twenty. Immunity (2012) 36:503–14. doi: 10.1016/j.immuni.2012.03.013
4. Bousoik E, Montazeri Aliabadi H. "Do We Know Jack" About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front Oncol (2018) 8:287.
5. Morris R, Kershaw NJ, Babon JJ. The Molecular Details of Cytokine Signaling via the JAK/STAT Pathway. Protein Sci Publ Protein Soc (2018) 27:1984–2009. doi: 10.1002/pro.3519
6. Silke J, O’Reilly LA. NF-κb and Pancreatic Cancer; Chapter and Verse. Cancers (2021) 13:4510. doi: 10.3390/cancers13184510
7. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-Kappab: A Blossoming of Relevance to Human Pathobiology. Cell (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
8. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu Rev Med (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
9. Aaronson DS, Horvath CM. A Road Map for Those Who Don't Know JAK-STAT. Science (2002) 296(5573):1653–5. doi: 10.1126/science.1071545
10. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-Type Cytokine Signalling Through the Gp130/Jak/STAT Pathway. Biochem J (1998) 334(Pt 2):297–314. doi: 10.1042/bj3340297
11. Moresi V, Adamo S, Berghella L. The JAK/STAT Pathway in Skeletal Muscle Pathophysiology. Front Physiol (2019) 10:500. doi: 10.3389/fphys.2019.00500
12. O'Shea JJ, Plenge R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity (2012) 36:542–50. doi: 10.1016/j.immuni.2012.03.014
13. Schindler C, Shuai K, Prezioso VR, Darnell JE Jr. Interferon-Dependent Tyrosine Phosphorylation of a Latent Cytoplasmic Transcription Factor. Science (1992) 257:809–13. doi: 10.1126/science.1496401
14. Oda H, Ishihara M, Miyahara Y, Nakamura J, Kozuka Y, Iwasa M, et al. First Case of Cytokine Release Syndrome After Nivolumab for Gastric Cancer. Case Rep Oncol (2019) 12:147–56. doi: 10.1159/000496933
15. Groner B, von Manstein V. Jak Stat Signaling and Cancer: Opportunities, Benefits and Side Effects of Targeted Inhibition. Mol Cell Endocrinol (2017) 451:1–14. doi: 10.1016/j.mce.2017.05.033
16. Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. The Role of JAK/STAT Signalling in the Pathogenesis, Prognosis and Treatment of Solid Tumours. Br J Cancer (2015) 113:365–71. doi: 10.1038/bjc.2015.233
17. Elkon KB. Cell Death, Nucleic Acids, and Immunity: Inflammation Beyond the Grave. Arthritis Rheumatol (Hoboken NJ). (2018) 70:805–16. doi: 10.1002/art.40452
18. Satarker S, Tom AA, Shaji RA, Alosious A, Luvis M, Nampoothiri M. JAK-STAT Pathway Inhibition and Their Implications in COVID-19 Therapy. Postgrad. Med (2021) 133:489–507.
19. Babon JJ, Lucet IS, Murphy JM, Nicola NA, Varghese LN. The Molecular Regulation of Janus Kinase (JAK) Activation. Biochem J (2014) 462:1–13. doi: 10.1042/BJ20140712
20. Haan C, Kreis S, Margue C, Behrmann I. Jaks and Cytokine Receptors–an Intimate Relationship. Biochem Pharmacol (2006) 72:1538–46. doi: 10.1016/j.bcp.2006.04.013
21. Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol Sci (2019) 40:298–308. doi: 10.1016/j.tips.2019.03.001
22. Shan Y, Gnanasambandan K, Ungureanu D, Kim ET, Hammaren H, Yamashita K, et al. Molecular Basis for Pseudokinase-Dependent Autoinhibition of JAK2 Tyrosine Kinase. Nat Struct Mol Biol (2014) 21:579–84. doi: 10.1038/nsmb.2849
23. Villarino AV, Kanno Y, Ferdinand JR, O'Shea JJ. Mechanisms of Jak/STAT Signaling in Immunity and Disease. J Immunol (2015) 194:21–7. doi: 10.4049/jimmunol.1401867
24. Levy DE, Darnell JE Jr. Stats: Transcriptional Control and Biological Impact. Nat Rev Mol Cell Biol (2002) 3:651–62. doi: 10.1038/nrm909
25. Huang G, Wu X, Li S, Xu X, Zhu H, Chen X. The Long Noncoding RNA CASC2 Functions as a Competing Endogenous RNA by Sponging miR-18a in Colorectal Cancer. Sci Rep (2016) 6:26524. doi: 10.1038/srep26524
26. Pencik J, Pham HT, Schmoellerl J, Javaheri T, Schlederer M, Culig Z, et al. JAK-STAT Signaling in Cancer: From Cytokines to non-Coding Genome. Cytokine (2016) 87:26–36. doi: 10.1016/j.cyto.2016.06.017
27. Croker BA, Kiu H, Nicholson SE. SOCS Regulation of the JAK/STAT Signalling Pathway. Semin Cell Dev Biol (2008) 19:414–22. doi: 10.1016/j.semcdb.2008.07.010
28. Larsen L, Ropke C. Suppressors of Cytokine Signalling: SOCS. APMIS (2002) 110:833–44. doi: 10.1034/j.1600-0463.2002.1101201.x
29. Kubo M, Hanada T, Yoshimura A. Suppressors of Cytokine Signaling and Immunity. Nat Immunol (2003) 4:1169–76. doi: 10.1038/ni1012
30. Linossi EM, Nicholson SE. Kinase Inhibition, Competitive Binding and Proteasomal Degradation: Resolving the Molecular Function of the Suppressor of Cytokine Signaling (SOCS) Proteins. Immunol Rev (2015) 266:123–33. doi: 10.1111/imr.12305
31. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The Molecular Basis of JAK/STAT Inhibition by SOCS1. Nat Commun (2018) 9:1558. doi: 10.1038/s41467-018-04013-1
32. Shuai K, Liu B. Regulation of JAK-STAT Signalling in the Immune System. Nat Rev Immunol (2003) 3:900–11. doi: 10.1038/nri1226
33. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. Gp130-Mediated Stat3 Activation in Enterocytes Regulates Cell Survival and Cell-Cycle Progression During Colitis-Associated Tumorigenesis. Cancer Cell (2009) 15:91–102. doi: 10.1016/j.ccr.2009.01.002
34. Hammaren HM, Virtanen AT, Raivola J, Silvennoinen O. The Regulation of JAKs in Cytokine Signaling and its Breakdown in Disease. Cytokine (2019) 118:48–63. doi: 10.1016/j.cyto.2018.03.041
35. Jeong EG, Kim MS, Nam HK, Min CK, Lee S, Chung YJ, et al. Somatic Mutations of JAK1 and JAK3 in Acute Leukemias and Solid Cancers. Clin Cancer Res (2008) 14:3716–21. doi: 10.1158/1078-0432.CCR-07-4839
36. O'Shea JJ, Holland SM, Staudt LM. JAKs and STATs in Immunity, Immunodeficiency, and Cancer. N. Engl J Med (2013) 368:161–70. doi: 10.1056/NEJMra1202117
37. Rane SG, Reddy EP. Janus Kinases: Components of Multiple Signaling Pathways. Oncogene (2000) 19:5662–79. doi: 10.1038/sj.onc.1203925
38. Lotz DR, Knutsen AP. Janus Kinase 3 Missense Mutation in a Child With Jacobsen Syndrome. Ann Allergy Asthma Immunol (2010) 104:536–7. doi: 10.1016/j.anai.2010.03.016
39. Skoda RC, Duek A, Grisouard J. Pathogenesis of Myeloproliferative Neoplasms. Exp Hematol (2015) 43:599–608. doi: 10.1016/j.exphem.2015.06.007
40. Vainchenker W, Constantinescu SN. JAK/STAT Signaling in Hematological Malignancies. Oncogene (2013) 32:2601–13. doi: 10.1038/onc.2012.347
41. Walters DK, Mercher T, Gu TL, O'Hare T, Tyner JW, Loriaux M, et al. Activating Alleles of JAK3 in Acute Megakaryoblastic Leukemia. Cancer Cell (2006) 10:65–75. doi: 10.1016/j.ccr.2006.06.002
42. Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, et al. A TEL-JAK2 Fusion Protein With Constitutive Kinase Activity in Human Leukemia. Science (1997) 278:1309–12. doi: 10.1126/science.278.5341.1309
43. Tefferi A. JAK and MPL Mutations in Myeloid Malignancies. Leuk. Lymphoma. (2008) 49:388–97. doi: 10.1080/10428190801895360
44. Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 Deficiency Defines an Essential Developmental Checkpoint in Definitive Hematopoiesis. Cell (1998) 93:397–409. doi: 10.1016/S0092-8674(00)81168-X
45. Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell (1998) 93:373–83. doi: 10.1016/S0092-8674(00)81166-6
46. Baird AM, Thomis DC, Berg LJ. T Cell Development and Activation in Jak3-Deficient Mice. J Leukoc Biol (1998) 63:669–77. doi: 10.1002/jlb.63.6.669
47. Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, et al. Partial Impairment of Cytokine Responses in Tyk2-Deficient Mice. Immunity (2000) 13:549–60. doi: 10.1016/S1074-7613(00)00054-6
48. Nielsen C, Birgens HS, Nordestgaard BG, Kjaer L, Bojesen SE. The JAK2 V617F Somatic Mutation, Mortality and Cancer Risk in the General Population. Haematologica (2011) 96:450–3. doi: 10.3324/haematol.2010.033191
49. Lorenzini T., Dotta L., Giacomelli M., Vairo Badolato D. R. STAT Mutations as Program Switchers: Turning Primary Immunodeficiencies Into Autoimmune Diseases. J Leukoc Biol (2017) 101:29–38. doi: 10.1189/jlb.5RI0516-237RR
50. Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted Disruption of the Mouse Stat1 Gene Results in Compromised Innate Immunity to Viral Disease. Cell (1996) 844:43–450. doi: 10.1016/S0092-8674(00)81289-1
51. Kimura T, Kadokawa Y, Harada H, Matsumoto M, Sato M, Kashiwazaki Y, et al. Essential and non-Redundant Roles of P48 (ISGF3 Gamma) and IRF-1 in Both Type I and Type II Interferon Responses, as Revealed by Gene Targeting Studies. Genes Cells (1996) 1:115–24. doi: 10.1046/j.1365-2443.1996.08008.x
52. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted Disruption of the Mouse Stat3 Gene Leads to Early Embryonic Lethality. Proc Natl Acad Sci U S A (1997) 94:3801–4. doi: 10.1073/pnas.94.8.3801
53. Sano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, Miura H, et al. Keratinocyte-Specific Ablation of Stat3 Exhibits Impaired Skin Remodeling, But Does Not Affect Skin Morphogenesis. EMBO J (1999) 18:4657–68. doi: 10.1093/emboj/18.17.4657
54. Couronne L, Scourzic L, Pilati C, Della Valle V, Duffourd Y, Solary E, et al. STAT3 Mutations Identified in Human Hematologic Neoplasms Induce Myeloid Malignancies in a Mouse Bone Marrow Transplantation Model. Haematologica (2013). 11:1748–1752. doi: 10.3324/haematol.2013.085068
55. Shahmarvand N, Nagy A, Shahryari J, Ohgami RS. Mutations in the Signal Transducer and Activator of Transcription Family of Genes in Cancer. Cancer Sci (2018) 109:926–33. doi: 10.1111/cas.13525
56. Verhoeven Y, Tilborghs S, Jacobs J, De Waele J, Quatannens D, Deben C, et al. The Potential and Controversy of Targeting STAT Family Members in Cancer. Semin Cancer Biol (2019) 60:41–56. doi: 10.1016/j.semcancer.2019.10.002
57. Briukhovetska D, Dörr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in Cancer: From Biology to Therapy. Nat Rev Cancer. (2021) 21:481–99. doi: 10.1038/s41568-021-00363-z
58. Crusz SM, Balkwill FR. Inflammation and Cancer: Advances and New Agents. Nat Rev Clin Oncol (2015) 12:584–96. doi: 10.1038/nrclinonc.2015.105
59. Qureshy Z, Johnson DE, Grandis JR. Targeting the JAK/STAT Pathway in Solid Tumors. J Cancer meta. Treat (2020) 6:27.
60. Khanna P, Chua PJ, Bay BH, Baeg GH. The JAK/STAT Signaling Cascade in Gastric Carcinoma (Review). Int J Oncol (2015) 47:1617–26. doi: 10.3892/ijo.2015.3160
61. Leonard WJ, O'Shea JJ. Jaks and STATs: Biological Implications. Annu Rev Immunol (1998) 16:293–322. doi: 10.1146/annurev.immunol.16.1.293
62. Lan T, Chen L, Wei X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells (2021) 10:100. doi: 10.3390/cells10010100
63. Bockerstett KA, DiPaolo RJ. Regulation of Gastric Carcinogenesis by Inflammatory Cytokines. Cell Mol Gastroenterol Hepatol. (2017) 4:47–53. doi: 10.1016/j.jcmgh.2017.03.005
64. Leaman DW, Leung S, Li X, Stark GR. Regulation of STAT-Dependent Pathways by Growth Factors and Cytokines. FASEB J (1996) 10:1578–88. doi: 10.1096/fasebj.10.14.9002549
65. Norhalifah HK, Mat NFC, Edinur HA. Cytokine Gene Polymorphisms in Cancer and Inflammatory Disorders. Curr Immunol Rev (2018) 14:81–93. doi: 10.2174/1573395514666180724121419
66. Smith AJP, Humphries SE. Cytokine and Cytokine Receptor Gene Polymorphisms and Their Functionality. Cytokine Growth Factor Rev (2009) 20:43–59. doi: 10.1016/j.cytogfr.2008.11.006
67. Jin P, Panelli MC, Marincola FM, Wang E. Cytokine Polymorphism and its Possible Impact on Cancer. Immunologic Res (2004) 30:181–90. doi: 10.1385/IR:30:2:181
68. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
69. Landskron G, de la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J Immunol Res (2014) 149185:2014. doi: 10.1155/2014/149185
70. Wendt MK, Balanis N, Carlin CR, Schiemann WP. STAT3 and Epithelial-Mesenchymal Transitions in Carcinomas. JAK-STAT (2014) 3:e28975. doi: 10.4161/jkst.28975
71. Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in Cancer Immunotherapy. Mol Cancer (2020) 19:145. doi: 10.1186/s12943-020-01258-7
72. Geiger JL, Grandis JR, Bauman JE. The STAT3 Pathway as a Therapeutic Target in Head and Neck Cancer: Barriers and Innovations. Oral Oncol (2016) 56:84–92. doi: 10.1016/j.oraloncology.2015.11.022
73. Vaddi K. Kerr D, Johnson R, editors. Immunotherapy for Gastrointestinal Cancer. (Switerland: Springer International Publishing) (2017).
74. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an Oncogene. Cell (1999) 98:295–303. doi: 10.1016/S0092-8674(00)81959-5
75. Bowman T, Garcia R, Turkson J, Jove R. STATs in Oncogenesis. Oncog (2000) 19:2474–88. doi: 10.1038/sj.onc.1203527
76. Ferbeyre G, Moriggl R. The Role of Stat5 Transcription Factors as Tumor Suppressors or Oncogenes. Biochim Biophys Acta (2011) 1815(1):104–14. doi: 10.1016/j.bbcan.2010.10.004
77. Cancer Genome Atlas Research N. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature (2014) 513:202–9. doi: 10.1038/nature13480
78. Ashrafizadeh M, Zarrabi A, Orouei S, Zarrin V, Rahmani Moghadam E, Zabolian A, et al. STAT3 Pathway in Gastric Cancer: Signaling, Therapeutic Targeting and Future Prospects. Biol (Basel). (2020) 9(6):126. doi: 10.3390/biology9060126
79. de Araujo ED, Erdogan F, Neubauer HA, Meneksedag-Erol D, Manaswiyoungkul P, Eram MS, et al. Structural and Functional Consequences of the STAT5BN642H Driver Mutation. Nat Commun (2019) 10(1):2517. doi: 10.1038/s41467-019-10422-7
80. Meissl K, Macho-Maschler S, Muller M, Strobl B. The Good and the Bad Faces of STAT1 in Solid Tumours. Cytokine (2017) 89:12–20. doi: 10.1016/j.cyto.2015.11.011
81. Fisher DT, Appenheimer MM, Evans SS. The Two Faces of IL-6 in the Tumor Microenvironment. Semin Immunol (2014) 26:38–47. doi: 10.1016/j.smim.2014.01.008
82. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 Signalling Axis in Cancer. Nat Rev Clin Oncol (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8
83. Gasche JA, Hoffmann J, Boland CR, Goel A. Interleukin-6 Promotes Tumorigenesis by Altering DNA Methylation in Oral Cancer Cells. Int J Cancer. (2011) 129:1053–63. doi: 10.1002/ijc.25764
84. Necula LG, Chivu-Economescu M, Stanciulescu EL, Bleotu C, Dima SO, Alexiu I, et al. IL-6 and IL-11 as Markers for Tumor Aggressiveness and Prognosis in Gastric Adenocarcinoma Patients Without Mutations in Gp130 Subunits. J gastrointestin. liver Dis (2012) 21:23–9.
85. Balkwill F. Tumour Necrosis Factor and Cancer. Nat Rev Cancer. (2009) 9:361–71. doi: 10.1038/nrc2628
86. Brenner D, Blaser H, Mak TW. Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nat Rev Immunol (2015) 15:362–74. doi: 10.1038/nri3834
87. Zelová H, Hošek J. TNF-α Signalling and Inflammation: Interactions Between Old Acquaintances. Inflammation Res (2013) 62:641–51. doi: 10.1007/s00011-013-0633-0
88. Holbrook J, Lara-Reyna S, Jarosz-Griffiths H, McDermott M. Tumour Necrosis Factor Signalling in Health and Disease. F1000Research (2019) 8:F1000 Faculty Rev–1111. doi: 10.12688/f1000research.17023.1
89. Webster JD, Vucic D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front Cell Dev Biol (2020) 8:365. doi: 10.3389/fcell.2020.00365
90. Horiuchi K. A Brief History of Tumor Necrosis Factor α–Converting Enzyme: An Overview of Ectodomain Shedding. Keio J Med (2013) 62:29–36. doi: 10.2302/kjm.2012-0003-RE
91. Wajant H, Siegmund D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front Cell Dev Biol (2019) 7:91. doi: 10.3389/fcell.2019.00091
92. Wajant H, Scheurich P. TNFR1-Induced Activation of the Classical NF-kappaB Pathway. FEBS J (2011) 278:862–76. doi: 10.1111/j.1742-4658.2011.08015.x
93. Grell M, Zimmermann G, Gottfried E, Chen CM, Grünwald U, Huang DC, et al. Induction of Cell Death by Tumour Necrosis Factor (TNF) Receptor 2, CD40 and CD30: A Role for TNF-R1 Activation by Endogenous Membrane-Anchored TNF. EMBO J (1999) 18:3034–43. doi: 10.1093/emboj/18.11.3034
94. Niklander SE. Inflammatory Mediators in Oral Cancer: Pathogenic Mechanisms and Diagnostic Potential. Front Oral Health (2021) 2:2. doi: 10.3389/froh.2021.642238
95. Quan X, Ding Y, Feng R, Zhu X, Zhang Q. Expression Profile of Cytokines in Gastric Cancer Patients Using Proteomic Antibody Microarray. Oncol Lett (2017) 14:7360–6. doi: 10.3892/ol.2017.7104
96. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and Neck Squamous Cell Carcinoma. Nat Rev Dis Primers (2020) 6(1):92. doi: 10.1038/s41572-020-00224-3
97. Siegel RL, Miller KD, Jemal A. Cancer Statistic. CA Cancer J Clin (2019) 69:7–34. doi: 10.3322/caac.21551
98. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
99. Kumar M, Nanavati R, Modi TG, Dobariya C. Oral Cancer: Etiology and Risk Factors: A Review. J Cancer Res Ther (2016) 12:458–63. doi: 10.4103/0973-1482.186696
101. Goertzen C, Mahdi H, Laliberte C, Meirson T, Eymael D, Gil-Henn H, et al. Oral Inflammation Promotes Oral Squamous Cell Carcinoma Invasion. Oncotarget (2018) 9:25540. doi: 10.18632/oncotarget.25540
102. Rao SK, Pavicevic Z, Du Z, Kim JG, Fan M, Jiao Y, et al. Pro-Inflammatory Genes as Biomarkers and Therapeutic Targets in Oral Squamous Cell Carcinoma. J Biol Chem (2010) 285:32512–21. doi: 10.1074/jbc.M110.150490
103. Tanaka T, Ishigamori R. Understanding Carcinogenesis for Fighting Oral Cancer. J Oncol (2011) 2011:603740. doi: 10.1155/2011/603740
104. Elmusrati A, Wang J, Wang CY. Tumor Microenvironment and Immune Evasion in Head and Neck Squamous Cell Carcinoma. Int J Oral Sci (2021) 13:24. doi: 10.1038/s41368-021-00131-7
105. Cancer Genome Atlas N. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature (2015) 517:576–82. doi: 10.1038/nature14129
106. Shah NG, Trivedi TI, Tankshali RA, Goswami JA, Jetly DH, Kobawala TP, et al. Stat3 Expression in Oral Squamous Cell Carcinoma: Association With Clinicopathological Parameters and Survival. Int J Biol Mark. (2006) 21:175–83. doi: 10.1177/172460080602100307
107. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
108. Walter V, Yin X, Wilkerson MD, Cabanski CR, Zhao N, Du Y, et al. Molecular Subtypes in Head and Neck Cancer Exhibit Distinct Patterns of Chromosomal Gain and Loss of Canonical Cancer Genes. PloS One (2013) 8:e56823. doi: 10.1371/journal.pone.0056823
109. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome Sequencing of Head and Neck Squamous Cell Carcinoma Reveals Inactivating Mutations in NOTCH1. Science (2011) 333(6046):1154–7. doi: 10.1126/science.1206923
110. Callender T, el-Naggar AK, Lee MS, Frankenthaler R, Luna MA, Batsakis JG. PRAD-1 (CCND1)/cyclin D1 Oncogene Amplification in Primary Head and Neck Squamous Cell Carcinoma. Cancer (1994) 74:152–8. doi: 10.1002/1097-0142(19940701)74:1<152::AID-CNCR2820740124>3.0.CO;2-K
111. Hammerman PS, Hayes DN, Grandis JR. Therapeutic Insights From Genomic Studies of Head and Neck Squamous Cell Carcinomas. . Cancer discov (2015) 5:239–44. doi: 10.1158/2159-8290.CD-14-1205
112. Smeets SJ, Brakenhoff RH, Ylstra B, van Wieringen WN, van de Wiel MA, Leemans CR, et al. Genetic Classification of Oral and Oropharyngeal Carcinomas Identifies Subgroups With a Different Prognosis. Cell Oncol (2009) 31:291–300. doi: 10.1155/2009/267928
113. Sequeira I, Rashid M, Tomás IM, Williams MJ, Graham TA, Adams DJ, et al. Genomic Landscape and Clonal Architecture of Mouse Oral Squamous Cell Carcinomas Dictate Tumour Ecology. Nat Commun (2020) 11:5671. doi: 10.1038/s41467-020-19401-9
114. Hutcheson KA, Warneke CL, Yao C, Zaveri J, Elgohari BE, Goepfert R, et al. Dysphagia After Primary Transoral Robotic Surgery With Neck Dissection vs Nonsurgical Therapy in Patients With Low- to Intermediate-Risk Oropharyngeal Cancer. JAMA Otolaryngol Head Neck Surg (2019) 145:1053–63. doi: 10.1001/jamaoto.2019.2725
115. Machtay M, Moughan J, Trotti A, Garden AS, Weber RS, Cooper JS, et al. Factors Associated With Severe Late Toxicity After Concurrent Chemoradiation for Locally Advanced Head and Neck Cancer: An RTOG Analysis. J Clin Oncol (2008) 26:3582–9. doi: 10.1200/JCO.2007.14.8841
116. Kyttaris VC. Targeting Cytokines to Treat Autoimmunity. Clin Immunol (2019) 206:108251. doi: 10.1016/j.clim.2019.108251
117. Cedars EJDE, Grandis JR. Jak/STAT Signaling in Head and Neck Cancer. In: Molecular Determinants of Head and Neck Cancer Current Cancer Research. Spinger (2018). p. 155–84.
118. Hang D, Yin Y, Wang L, Yuan H, Du J, Zhu M, et al. Effects of Potentially Functional Polymorphisms in Suppressor of Cytokine Signaling 3 (SOCS3) on the Risk of Head and Neck Squamous Cancer. J Oral Pathol Med (2017) 46:598–602. doi: 10.1111/jop.12539
119. Serefoglou Z, Yapijakis C, Nkenke E, Vairaktaris E. Genetic Association of Cytokine DNA Polymorphisms With Head and Neck Cancer. Oral Oncol (2008) 44:1093–9. doi: 10.1016/j.oraloncology.2008.02.012
120. Corrêa GT, Bandeira GA, Cavalcanti BG, de Carvalho Fraga CA, dos Santos EP, Silva TF, et al. Association of -308 TNF-α Promoter Polymorphism With Clinical Aggressiveness in Patients With Head and Neck Squamous Cell Carcinoma. Oral Oncol (2011) 47(9):888–94. doi: 10.1016/j.oraloncology.2011.07.001
121. Mlak R, Powrózek T, Brzozowska A, Homa-Mlak I, Mazurek M, Gołębiowski P, et al. The Relationship Between TNF-α Gene Promoter Polymorphism (- 1211 T > C), the Plasma Concentration of TNF-α, and Risk of Oral Mucositis and Shortening of Overall Survival in Patients Subjected to Intensity-Modulated Radiation Therapy Due to Head and Neck Cancer. Support. Care Cancer (2020) 28:531–40. doi: 10.1007/s00520-019-04838-6
122. Erdei E, Luo L, Sheng H, Maestas E, White KAM, Mackey A. Cytokines and Tumor Metastasis Gene Variants in Oral Cancer and Precancer in Puerto Rico. PloS One (2013) 8:e79187. doi: 10.1371/journal.pone.0079187
123. Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, et al. Requirement of Stat3 But Not Stat1 Activation for Epidermal Growth Factor Receptor- Mediated Cell Growth In Vitro. J Clin Invest (1998) 102:1385–92. doi: 10.1172/JCI3785
124. Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, et al. Constitutive Activation of Stat3 Signaling Abrogates Apoptosis in Squamous Cell Carcinogenesis In Vivo. Proc Natl Acad Sci U S A (2000) 97:4227–32. doi: 10.1073/pnas.97.8.4227
125. Leong PL, Xi S, Drenning SD, Dyer KF, Wentzel AL, Lerner EC, et al. Differential Function of STAT5 Isoforms in Head and Neck Cancer Growth Control. Oncogene (2002) 21:2846–53. doi: 10.1038/sj.onc.1205385
126. Song X, Yang X, Narayanan R, Shankar V, Ethiraj S, Wang X, et al. Oral Squamous Cell Carcinoma Diagnosed From Saliva Metabolic Profiling. Proc Natl Acad Sci USA (2020) 117:16167–73. doi: 10.1073/pnas.2001395117
127. Ganesh D, Sreenivasan P, Öhman J, Wallström M, Braz-Silva PH, Giglio D, et al. Potentially Malignant Oral Disorders and Cancer Transformation. Anticancer Res (2018) 38:3223–9. doi: 10.21873/anticanres.12587
128. Mozaffari HR, Ramezani M, Mahmoudiahmadabadi M, Omidpanah N, Sadeghi M. Salivary and Serum Levels of Tumor Necrosis Factor-Alpha in Oral Lichen Planus: A Systematic Review and Meta-Analysis Study. Oral surg. Oral med. Oral Pathol Oral Radiol (2017) 124:e183–9. doi: 10.1016/j.oooo.2017.06.117
129. Diesch T, Filippi C, Fritschi N, Filippi A, Ritz N. Cytokines in Saliva as Biomarkers of Oral and Systemic Oncological or Infectious Diseases: A Systematic Review. Cytokine (2021) 143:155506. doi: 10.1016/j.cyto.2021.155506
130. Prasad G, McCullough M. Chemokines and Cytokines as Salivary Biomarkers for the Early Diagnosis of Oral Cancer. Int J Dentist. (2013) 2013:813756. doi: 10.1155/2013/813756
131. Sahibzada HA, Khurshid Z, Khan RS, Naseem M, Siddique KM, Mali M, et al. Salivary IL-8, IL-6 and TNF-α as Potential Diagnostic Biomarkers for Oral Cancer. Diagnostic (Basel Switzerland) (2017) 7:21. doi: 10.3390/diagnostics7020021
132. Chiamulera MMA, Zancan CB, Remor AP, Corderio MF, Gleber-Netto FO, Baptistella AR. Salivary Cytokines as Biomarkers of Oral Cancer: A Systematic Review and Meta-Analysis. BMC Cancer. (2021) 21:205. doi: 10.1186/s12885-021-07932-3
133. Ferrari E, Pezzi ME, Cassi D, Pertinhez TA, Spisni A, Meleti M. Salivary Cytokines as Biomarkers for Oral Squamous Cell Carcinoma: A Systematic Review. Int J Mol Sci (2021) 22:6795. doi: 10.3390/ijms22136795
134. Uz U, Eskiizmir G. Association Between Interleukin-6 and Head and Neck Squamous Cell Carcinoma: A Systematic Review. Clin Exp otorhinolaryngol. (2021) 14:50–60. doi: 10.21053/ceo.2019.00906
135. Salvo E, Tu NH, Scheff NN, Dubeykovskaya ZA, Chavan SA, Aouizerat BE, et al. Tnfα Promotes Oral Cancer Growth, Pain, and Schwann Cell Activation. Sci Rep (2021) 202111:1840. doi: 10.1038/s41598-021-81500-4
136. Scheff NN, Ye Y, Bhattacharya A, MacRae J, Hickman DN, Sharma AK, et al. Umor Necrosis Factor Alpha Secreted From Oral Squamous Cell Carcinoma Contributes to Cancer Pain and Associated Inflammation. Pain (2017) 158:2396–409. doi: 10.1097/j.pain.0000000000001044
137. van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, Schouten HC, van Kleef M, Patijn J. Prevalence of Pain in Patients With Cancer: A Systematic Review of the Past 40 Years. Ann Oncol (2007) 18:1437–49. doi: 10.1093/annonc/mdm056
138. Reyes-Gibby CC, Anderson KO, Merriman KW, Todd KH, Shete SS, Hanna EY. Survival Patterns in Squamous Cell Carcinoma of the Head and Neck: Pain as an Independent Prognostic Factor for Survival. J Pain (2014) 15:1015–22. doi: 10.1016/j.jpain.2014.07.003
139. Tang D, Tao D, Fang Y, Deng C, Xu Q, Zhou J. TNF-Alpha Promotes Invasion and Metastasis via NF-Kappa B Pathway in Oral Squamous Cell Carcinoma. Med Sci monit. basic Res (2017) 23:141–9. doi: 10.12659/msmbr.903910
140. Hong HS, Akhavan J, Lee SH, Kim RH, Kang MK, Park NH, et al. Proinflammatory Cytokine Tnfα Promotes HPV-Associated Oral Carcinogenesis by Increasing Cancer Stemness. Int J Oral Sci (2020) 12:3. doi: 10.1038/s41368-019-0069-7
141. Jin W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells (2020) 9:217. doi: 10.3390/cells9010217
142. Alsahafi E, Begg K, Amelio I, Raulf N, Lucarelli P, Sauter T, et al. Clinical Update on Head and Neck Cancer: Molecular Biology and Ongoing Challenges. Cell Death Dis (2019) 10:540. doi: 10.1038/s41419-019-1769-9
143. Choudhary MM, France TJ, Teknos TN, Kumar P. Interleukin-6 Role in Head and Neck Squamous Cell Carcinoma Progression. World J Otorhinolaryngol. Head Neck Surg (2016) 2:90–7. doi: 10.1016/j.wjorl.2016.05.002
144. Ernst M, Thiem S, Nguyen PM, Eissmann M, Putoczki TL. Epithelial Gp130/Stat3 Functions: An Intestinal Signaling Node in Health and Disease. Semin Immunol (2014) 26:29–37. doi: 10.1016/j.smim.2013.12.006
145. Park OK, Schaefer TS, Nathans D. In Vitro Activation of Stat3 by Epidermal Growth Factor Receptor Kinase. Proc Natl Acad Sci U S A (1996) 93:13704–8. doi: 10.1073/pnas.93.24.13704
146. He Y, Zeng Q, Drenning SD, Melhem MF, Tweardy DJ, Huang L, et al. Inhibition of Human Squamous Cell Carcinoma Growth In Vivo by Epidermal Growth Factor Receptor Antisense RNA Transcribed From the U6 Promoter. J Natl Cancer Inst (1998) 90:1080–7. doi: 10.1093/jnci/90.14.1080
147. Laimer K, Spizzo G, Obrist P, Gastl G, Brunhuber T, Schafer G, et al. STAT1 Activation in Squamous Cell Cancer of the Oral Cavity: A Potential Predictive Marker of Response to Adjuvant Chemotherapy. Cancer (2007) 110:326–33. doi: 10.1002/cncr.22813
148. Pappa E, Nikitakis N, Vlachodimitropoulos D, Avgoustidis D, Oktseloglou V, Papadogeorgakis N. Phosphorylated Signal Transducer and Activator of Transcription-1 Immunohistochemical Expression Is Associated With Improved Survival in Patients With Oral Squamous Cell Carcinoma. J Oral Maxillofac. Surg (2014) 72:211–21. doi: 10.1016/j.joms.2013.06.198
149. Schmitt NC, Trivedi S, Ferris RL. STAT1 Activation Is Enhanced by Cisplatin and Variably Affected by EGFR Inhibition in HNSCC Cells. Mol Cancer Ther (2015) 14:2103–11. doi: 10.1158/1535-7163.MCT-15-0305
150. Sun Y, Sang Z, Jiang Q, Ding X, Yu Y. Transcriptomic Characterization of Differential Gene Expression in Oral Squamous Cell Carcinoma: A Meta-Analysis of Publicly Available Microarray Data Sets. Tumour Biol (2016) 37. doi: 10.1007/s13277-016-5439-6
151. Mori K, Haraguchi S, Hiori M, Shimada J, Ohmori Y. Tumor-Associated Macrophages in Oral Premalignant Lesions Coexpress CD163 and STAT1 in a Th1-Dominated Microenvironment. BMC Cancer. (2015) 15:573. doi: 10.1186/s12885-015-1587-0
152. Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, et al. Identification of the Cell-Intrinsic and -Extrinsic Pathways Downstream of EGFR and IFNgamma That Induce PD-L1 Expression in Head and Neck Cancer. Cancer Res (2016) 76:1031–43. doi: 10.1158/0008-5472.CAN-15-2001
153. Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, et al. IFNgamma Induces PD-L1 Overexpression by JAK2/STAT1/IRF-1 Signaling in EBV-Positive Gastric Carcinoma. Sci Rep (2017) 7:17810. doi: 10.1038/s41598-017-18132-0
154. Shah NG, Trivedi TI, Tankshali RA, Goswami JV, Jetly DH, Shukla SN, et al. Prognostic Significance of Molecular Markers in Oral Squamous Cell Carcinoma: A Multivariate Analysis. Head Neck. (2009) 31:1544–56. doi: 10.1002/hed.21126
155. Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS. Epidermal Growth Factor Receptor-Independent Constitutive Activation of STAT3 in Head and Neck Squamous Cell Carcinoma is Mediated by the Autocrine/Paracrine Stimulation of the Interleukin 6/Gp130 Cytokine System. Cancer Res (2003) 63:2948–56.
156. Cedars EJDE, Grandis JR. Jak/STAT Signaling in Head and Neck Cancer. In: Molecular Determinants of Head and Neck Cancer. Cham: Humana Press (2018).
157. Peyser ND, Freilino M, Wang L, Zeng Y, Li H, Johnson DE, et al. Frequent Promoter Hypermethylation of PTPRT Increases STAT3 Activation and Sensitivity to STAT3 Inhibition in Head and Neck Cancer. Oncogene (2016) 35:1163–9. doi: 10.1038/onc.2015.171
158. Yu H, Kortylewski M, Pardoll D. Crosstalk Between Cancer and Immune Cells: Role of STAT3 in the Tumour Microenvironment. Nat Rev Immunol (2007) 7:41–51. doi: 10.1038/nri1995
159. Bauer AK, Velmurugan K, Xiong KN, Alexander CM, Xiong J, Brooks R. Epiregulin is Required for Lung Tumor Promotion in a Murine Two-Stage Carcinogenesis Model. Mol carcinogen. (2017) 56:94–105. doi: 10.1002/mc.22475
160. Wang Y, Jing Y, Ding L, Zhang X, Song Y, Chen S, et al. Epiregulin Reprograms Cancer-Associated Fibroblasts and Facilitates Oral Squamous Cell Carcinoma Invasion via JAK2-STAT3 Pathway. J Exp Clin Cancer Res (2019) 38:274. doi: 10.1186/s13046-019-1277-x
161. Xi S, Zhang Q, Gooding WE, Smithgall TE, Grandis JR. Constitutive Activation of Stat5b Contributes to Carcinogenesis In Vivo. Cancer Res (2003) 63:6763–71.
162. Peng HY, Jiang SS, Hsiao JR, Hsiao M, Hsu YM, Wu GH, et al. IL-8 Induces miR-424-5p Expression and Modulates SOCS2/STAT5 Signaling Pathway in Oral Squamous Cell Carcinoma. Mol Oncol (2016) 10:895–909. doi: 10.1016/j.molonc.2016.03.001
163. Nemeth CG, Rocken C, Siebert R, Wiltfang J, Ammerpohl O, Gassling V. Recurrent Chromosomal and Epigenetic Alterations in Oral Squamous Cell Carcinoma and its Putative Premalignant Condition Oral Lichen Planus. PLoS One (2019) 14:e0215055. doi: 10.1371/journal.pone.0215055
164. Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral Cavity and Esophageal Carcinogenesis Modeled in Carcinogen-Treated Mice. Clin Cancer Res (2004) 10:301–13. doi: 10.1158/1078-0432.CCR-0999-3
165. Vitale-Cross L, Czerninski R, Amornphimoltham P, Patel V, Molinolo AA, Gutkind JS. Chemical Carcinogenesis Models for Evaluating Molecular-Targeted Prevention and Treatment of Oral Cancer. Cancer Prev Res (Phila) (2009) 2:419–22. doi: 10.1158/1940-6207.CAPR-09-0058
166. Chao JL, Korzinkin M, Zhavoronkov A, Ozerov IV, Walker MT, Higgins K, et al. Effector T Cell Responses Unleashed by Regulatory T Cell Ablation Exacerbate Oral Squamous Cell Carcinoma. Cell Rep Med (2021) 2:100399. doi: 10.1016/j.xcrm.2021.100399
167. Cabrera Ortega AA, Goncalves Vde P, Guimaraes MR, Rossa Junior C, Spolidorio LC. Overexpression of Bcl-2, SOCS 1, 3 and Cdh 1, 2 are Associated With the Early Neoplasic Changes in Modified 4-Nitroquinoline 1-Oxide-Induced Murine Oral Cancer Model. J Oral Pathol Med (2016) 45:573–80. doi: 10.1111/jop.12413
168. Oghumu S, Knobloch TJ, Terrazas C, Varikuti S, Ahn-Jarvis J, Bollinger CE, et al. Deletion of Macrophage Migration Inhibitory Factor Inhibits Murine Oral Carcinogenesis: Potential Role for Chronic Pro-Inflammatory Immune Mediators. Int J Cancer (2016) 139:1379–90. doi: 10.1002/ijc.30177
169. Ni Y, Yap T, Silke N, Silke J, McCullough MJ, Celentano A, et al. Loss of NF-Kb1 and C-Rel Accelerates Oral Carcinogenesis in Mice. Oral Dis (2020) 27:168–72. doi: 10.1111/odi.13508
170. Oweida AJ, Darragh L, Phan A, Binder D, Bhatia S, Mueller A, et al. STAT3 Modulation of Regulatory T Cells in Response to Radiation Therapy in Head and Neck Cancer. J Natl Cancer Institut. (2019) 111:1339–49. doi: 10.1093/jnci/djz036
171. Onken MD, Winkler AE, Kanchi KL, Chalivendra V, Law JH, Rickert CG, et al. A Surprising Cross-Species Conservation in the Genomic Landscape of Mouse and Human Oral Cancer Identifies a Transcriptional Signature Predicting Metastatic Disease. Clin Cancer Res (2014) 20:2873–84. doi: 10.1158/1078-0432.CCR-14-0205
172. Sitarz R, Skierucha M, Mielko J, Offerhaus G, Maciejewski R, Polkowski WP. Gastric Cancer: Epidemiology, Prevention, Classification, and Treatment. Cancer Manage Res (2018) 10:239–48. doi: 10.2147/CMAR.S149619
173. Correa P. Human Gastric Carcinogenesis: A Multistep and Multifactorial Process–First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res (1992) 52:6735–40.
174. Fox JG, Wang TC. Inflammation, Atrophy, and Gastric Cancer. J Clin Invest. (2007) 117:60–9. doi: 10.1172/JCI30111
175. Lauren P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol Microbiol Scand (1965) 64:31–49. doi: 10.1111/apm.1965.64.1.31
176. Zou Z, Zhao L, Su S, Liu Q, Yu L, Wei J, et al. The Plasma Levels of 12 Cytokines and Growth Factors in Patients With Gastric Cancer. Medicine (2018) 97:e0413. doi: 10.1097/MD.0000000000010413
177. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-Cadherin Germline Mutations in Familial Gastric Cancer. Nature (1998) 392(6674):402–5. doi: 10.1038/32918
178. Hamilton SR, Aaltonen LA. Pathology and Genetics of Tumours of the Digestive System. Lyon: IARC Press (2000).
179. Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, et al. Molecular Analysis of Gastric Cancer Identifies Subtypes Associated With Distinct Clinical Outcomes. Nat Med (2015) 21:449–56. doi: 10.1038/nm.3850
180. Wong SS, Kim KM, Ting JC, Yu K, Fu J, Liu S, et al. Genomic Landscape and Genetic Heterogeneity in Gastric Adenocarcinoma Revealed by Whole-Genome Sequencing. Nat Commun (2014) 5:5477. doi: 10.1038/ncomms6477
181. Li B, Cheung KS, Wong IY, Leung WK, Law S. Nonaspirin Nonsteroidal Anti-Inflammatory Drugs and Gastric Cancer Risk After Helicobacter Pylori Eradication: A Territory-Wide Study. Cancer (2021) 127:1805–15. doi: 10.1002/cncr.33412
182. Epplein M, Nomura AM, Wilkens LR, Henderson BE, Kolonel LN. Nonsteroidal Antiinflammatory Drugs and Risk of Gastric Adenocarcinoma: The Multiethnic Cohort Study. Am J Epidemiol (2009) 170(4):507–14. doi: 10.1093/aje/kwp162
183. Negovan A, Iancu M, Fülöp E, Bănescu C. Helicobacter Pylori and Cytokine Gene Variants as Predictors of Premalignant Gastric Lesions. World J Gastroenterol (2019) 25:4105–24. doi: 10.3748/wjg.v25.i30.4105
184. Fukui H, Zhang X, Sun C, Hara K, Kikuchi S, Yamasaki T, et al. IL-22 Produced by Cancer-Associated Fibroblasts Promotes Gastric Cancer Cell Invasion via STAT3 and ERK Signaling. Br J Cancer (2014) 111:763–71. doi: 10.1038/bjc.2014.336
185. Pawluczuk E, Łukaszewicz-Zając M, Mroczko B. The Role of Chemokines in the Development of Gastric Cancer - Diagnostic and Therapeutic Implications. Int J Mol Sci (2020) 21:8456. doi: 10.3390/ijms21228456
186. Qin SY, Yang XW, Luo W, Chen M, Liu ZL, Su SB, et al. Association of Interleukin 22 Polymorphisms With Gastric Cancer Risk. Tumour Biol (2015) 36:2033–9. doi: 10.1007/s13277-014-2810-3
187. Ruzzo A, Catalano V, Canestrari E, Giacomini E, Santini D, Tonini G, et al. Genetic Modulation of the Interleukin 6 (IL-6) System in Patients With Advanced Gastric Cancer: A Background for an Alternative Target Therapy. BMC Cancer (2014) 14:357. doi: 10.1186/1471-2407-14-357
188. Sugimoto M, Furuta T, Shirai N, Nakamura A, Xiao F, Kajimura M, et al. Different Effects of Polymorphisms of Tumor Necrosis Factor-Alpha and Interleukin-1 Beta on Development of Peptic Ulcer and Gastric Cancer. J Gastroenterol hepatol. (2007) 22:51–9. doi: 10.1111/j.1440-1746.2006.04442.x
189. Sugimoto M, Yamaoka Y, Furuta T. Influence of Interleukin Polymorphisms on Development of Gastric Cancer and Peptic Ulcer. World J Gastroenterol (2010) 16:1188–200. doi: 10.3748/wjg.v16.i10.1188
190. Xu T, Kong Z, Zhao H. Relationship Between Tumor Necrosis Factor-α Rs361525 Polymorphism and Gastric Cancer Risk: A Meta-Analysis. Front Physiol (2018) 9:469. doi: 10.3389/fphys.2018.00469
191. Zheng W, Zhang S, Zhang S, Min L, Wang Y, Xie J, et al. The Relationship Between Tumor Necrosis Factor-α Polymorphisms and Gastric Cancer Risk: An Updated Meta-Analysis. Biomed Rep (2017) 7:133–42. doi: 10.3892/br.2017.934
192. Gholamalizadeh M, Mirzaei Dahka S, Sedigh Ebrahim-Saraie H, Akbari ME, Pourtaheri A, Rastgoo S, et al. The Role of Tumor Necrosis Factor-α (TNF-α) Polymorphisms in Gastric Cancer: A Meta-Analysis. J Gastrointestin. Cancer (2021). doi: 10.1007/s12029-021-00688-w
193. Montfort A, Dufau C, Colacios C, Andrieu-Abadie N, Levade T, Filleron T, et al. Anti-TNF, a Magic Bullet in Cancer Immunotherapy? J ImmunoTher. Cancer (2019) 7:303. doi: 10.1186/s40425-019-0802-y
194. Wang X, Lin Y. Tumor Necrosis Factor and Cancer, Buddies or Foes? Acta pharmacol. Sin (2008) 29:1275–88. doi: 10.1111/j.1745-7254.2008.00889.x
195. Hajimoradi M, Mohammad Hassan Z, Ebrahimi M, Soleimani M, Bakhshi M, Firouzi J, et al. STAT3 is Overactivated in Gastric Cancer Stem-Like Cells. Cell J (2016) 17:617–28. doi: 10.22074/cellj.2016.3834
196. Kim DY, Cha ST, Ahn DH, Kang HY, Kwon CI, Ko KH, et al. STAT3 Expression in Gastric Cancer Indicates a Poor Prognosis. J Gastroenterol Hepatol (2009) 24:646–51. doi: 10.1111/j.1440-1746.2008.05671.x
197. Metcalfe RD, Putoczki TL, Griffin M. Sructural Understanding of Interleukin 6 Family Cytokine Signaling and Targeted Therapies: Focus on Interleukin 11. Front Immunol (2020) 11:1424. doi: 10.3389/fimmu.2020.01424
198. Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, Ziegler PK, et al. Interleukin-11 is the Dominant IL-6 Family Cytokine During Gastrointestinal Tumorigenesis and Can Be Targeted Therapeutically. Cancer Cell (2013) 24:257–71. doi: 10.1016/j.ccr.2013.06.017
199. Nakayama T, Yoshizaki A, Izumida S, Suehiro T, Miura S, Uemura T, et al. Expression of Interleukin-11 (IL-11) and IL-11 Receptor Alpha in Human Gastric Carcinoma and IL-11 Upregulates the Invasive Activity of Human Gastric Carcinoma Cells. Int J Oncol (2007) 30:825–33.
200. Giraud AS, Menheniott TR, Judd LM. Targeting STAT3 in Gastric Cancer. Expert Opin Ther Targets (2012) 16:889–901. doi: 10.1517/14728222.2012.709238
201. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell (2009) 15:103–13. doi: 10.1016/j.ccr.2009.01.001
202. Grivennikov SI, Karin M. Dangerous Liaisons: STAT3 and NF-kappaB Collaboration and Crosstalk in Cancer. Cytokine Growth Factor Rev (2010) 21:11–9. doi: 10.1016/j.cytogfr.2009.11.005
203. Wang Z, Si X, Xu A, Meng X, Gao S, Qi Y, et al. Activation of STAT3 in Human Gastric Cancer Cells via Interleukin (IL)-6-Type Cytokine Signaling Correlates With Clinical Implications. PLoS One (2013) 8:e75788. doi: 10.1371/journal.pone.0075788
204. Jan I, Rather RA, Mushtaq I, Malik AA, Besina S, Baba AB, et al. Helicobacter Pylori Subdues Cytokine Signaling to Alter Mucosal Inflammation via Hypermethylation of Suppressor of Cytokine Signaling 1 Gene During Gastric Carcinogenesis. Front Oncol (2021) 10:604747. doi: 10.3389/fonc.2020.604747
205. Poh AR, O'Donoghue RJ, Ernst M, Putoczki TL. Mouse Models for Gastric Cancer: Matching Models to Biological Questions. J Gastroenterol Hepatol (2016) 31:1257–72. doi: 10.1111/jgh.13297
206. Jenkins BJ, Grail D, Nheu T, Najdovska M, Wang B, Waring P, et al. Hyperactivation of Stat3 in Gp130 Mutant Mice Promotes Gastric Hyperproliferation and Desensitizes TGF-Beta Signaling. Nat Med (2005) 11:845–52. doi: 10.1038/nm1282
207. Low J, Christie M, Ernst M, Dumoutier L, Preaudet A, Ni Y, et al. Loss of NFKB1 Results in Expression of Tumor Necrosis Factor and Activation of Signal Transducer and Activator of Transcription 1 to Promote Gastric Tumorigenesis in Mice. Gastroenterology (2020) 159:144–1458. doi: 10.1053/j.gastro.2020.06.039
208. O'Reilly LA, Putoczki TL, Mielke LA, Low JT, Lin A, Preaudet A, et al. Loss of NF-Kappab1 Causes Gastric Cancer With Aberrant Inflammation and Expression of Immune Checkpoint Regulators in a STAT-1-Dependent Manner. Immunity (2018) 48:570–583.e578. doi: 10.1016/j.immuni.2018.03.003
209. Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, et al. Reciprocal Regulation of Gastrointestinal Homeostasis by SHP2 and STAT-Mediated Trefoil Gene Activation in Gp130 Mutant Mice. Nat Med (2002) 8:1089–97. doi: 10.1038/nm763
210. Silver JS, Hunter CA. Gp130 at the Nexus of Inflammation, Autoimmunity, and Cancer. J Leukocyte Biol (2010) 88:1145–56. doi: 10.1189/jlb.0410217
211. Ernst M, Najdovska M, Grail D, Lundgren-May T, Buchert M, Tye H, et al. STAT3 and STAT1 Mediate IL-11-Dependent and Inflammation-Associated Gastric Tumorigenesis in Gp130 Receptor Mutant Mice. J Clin Invest (2008) 118:1727–38. doi: 10.1172/JCI34944
212. Ernst M, Putoczki TL. Stat3: Linking Inflammation to (Gastrointestinal) Tumourigenesis. Clin Exp Pharmacol Physiol (2012) 39:711–8. doi: 10.1111/j.1440-1681.2011.05659.x
213. Jackson CB, Giraud AS. STAT3 as a Prognostic Marker in Human Gastric Cancer. J Gastroenterol Hepatol (2009) 24:505–7. doi: 10.1111/j.1440-1746.2009.05822.x
214. Concetti J, Wilson CL. NFKB1 and Cancer: Friend or Foe? Cells (2018) 133:7. doi: 10.3390/cells7090133
215. Coutzac C, Pernot S, Chaput N, Zaanan A. Immunotherapy in Advanced Gastric Cancer, Is It the Future? Crit Rev Oncol Hematol (2019) 133:25–32. doi: 10.1016/j.critrevonc.2018.10.007
216. Nguyen PM, Abdirahman SM, Putoczki TL. Emerging Roles for Interleukin-11 in Disease. Growth Factors (Chur Switzerland) (2019) 37:1–11. doi: 10.1080/08977194.2019.1620227
217. Anderson G, Ebadi M, Vo K, Novak J, Govindarajan A, Amini A. An Updated Review on Head and Neck Cancer Treatment With Radiation Therapy. Cancers (2021) 13:4912. doi: 10.3390/cancers13194912
218. Proserpio I, Rausei S, Barzaghi S, Frattini F, Galli F, Iovino D, et al. Multimodal Treatment of Gastric Cancer. World J Gastrointestin Surg (2014) 6:55–8. doi: 10.4240/wjgs.v6.i4.55
219. Siebenhüner AR, De Dosso S, Helbling D, Astaras C, Szturz P, Moosmann P, et al. Advanced Gastric Cancer: Current Treatment Landscape and a Future Outlook for Sequential and Personalized Guide: Swiss Expert Statement Article. Oncol Res Treat (2021) 44:485–94. doi: 10.1159/000518107
220. Owen KL, Brockwell NK, Parker BS. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers (2019) 11:2002. doi: 10.3390/cancers11122002
221. Tolomeo M, Cascio A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. Int J Mol Sci (2021) 22:603. doi: 10.3390/ijms22020603
222. Beebe JD, Liu JY, Zhang JT. Two Decades of Research in Discovery of Anticancer Drugs Targeting STAT3, How Close Are We? Pharmacol Ther (2018) 191:74–91. doi: 10.1016/j.pharmthera.2018.06.006
223. Becerra C, Stephenson J, Jonker DJ, Cohn AL, Asmis TR, Bekaii-Saab TS, et al. Phase Ib/II Study of Cancer Stem Cell (CSC) Inhibitor BBI608 Combined With Paclitaxel in Advanced Gastric and Gastroesophageal Junction (GEJ) Adenocarcinoma. J Clin Oncol (2015) 33:4069–9. doi: 10.1200/jco.2015.33.15_suppl.4069
224. Shah MA, Shitara K, Lordick F, Bang YJ, Tebbutt N, Metges JP, et al. The BRIGHTER Trial: A Phase 3 Randomized Double-Blind Study of Napabucasin (NAPA) Plus Paclitaxel (PTX) Versus Placebo (PBO) Plus PTX in Patients (Pts) With Pretreated Advanced Gastric and Gastroesophageal Junction (GEJ) Adenocarcinoma. J Clin Oncol (2018) 36:4010. doi: 10.1200/JCO.2018.36.15_suppl.4010
225. Tolcher A, Flaherty K, Shapiro GI, Berlin J, Witzig T, Habermann T, et al. A First-In-Human Phase I Study of OPB-111077, a Small-Molecule STAT3 and Oxidative Phosphorylation Inhibitor, in Patients With Advanced Cancers. Oncol. (2018) 23:658–e672. doi: 10.1634/theoncologist.2017-0325
226. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a Next-Generation Antisense Oligonucleotide Inhibitor of STAT3 With Early Evidence of Clinical Activity in Lymphoma and Lung Cancer. Sci Trans Med (2015) 7:314ra185. doi: 10.1126/scitranslmed.aac5272
227. Bendell JC, Hong DS, Burris HA, Naing A, Jones SF, Falchook G, et al. Phase 1, Open-Label, Dose-Escalation, and Pharmacokinetic Study of STAT3 Inhibitor OPB-31121 in Subjects With Advanced Solid Tumors. Cancer chemother. Pharmacol (2014) 74:125–30. doi: 10.1007/s00280-014-2480-2
228. Oh DY, Lee SH, Han SW, Kim MJ, Kim TM, Kim TY, et al. Phase I Study of OPB-31121, an Oral STAT3 Inhibitor, in Patients With Advanced Solid Tumors. Cancer Res Treat (2015) 47:607–15. doi: 10.4143/crt.2014.249
229. Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT, et al. First-In-Human Trial of a STAT3 Decoy Oligonucleotide in Head and Neck Tumors: Implications for Cancer Therapy. Can Discov (2012) 2:694–705. doi: 10.1158/2159-8290.CD-12-0191
230. Liu J, Chen H, Kaniskan H, Xie L, Chen X, Jin J, et al. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J Am Chem Soc (2021) 143(23):8902–10. doi: 10.1021/jacs.1c03852
231. Plimack ER, Lorusso PM, McCoon P, Tang W, Krebs AD, Curt G, et al. AZD1480: A Phase I Study of a Novel JAK2 Inhibitor in Solid Tumors. oncol. (2013) 18:819–20. doi: 10.1634/theoncologist.2013-0198
232. Angevin E, Tabernero J, Elez E, Cohen SJ, Bahleda R, van Laethem JL, et al. A Phase I/II, Multiple-Dose, Dose-Escalation Study of Siltuximab, an Anti-Interleukin-6 Monoclonal Antibody, in Patients With Advanced Solid Tumors. Clin Cancer Res (2014) 20:2192–204. doi: 10.1158/1078-0432.CCR-13-2200
233. Jain RK, Zhang J, Eroglu Z, Creelan BC, Brohl AS, Shafique MR, et al. The IRX-2 Regimen Combined With Nivolumab in Recurrent/Metastatic Solid Tumors: A Phase 1b Study to Evaluate the Safety, Determine Recommended Phase 2 Dose (RP2D), and Investigate the Biologic and Clinical Activity. J Clin Oncol (2019) 37:TPS2662–TPS2662. doi: 10.1200/JCO.2019.37.15_suppl.TPS2662
234. Wolf GT, Moyer JS, Kaplan MJ, Newman JG, Egan JE, Berinstein NL, et al. IRX-2 Natural Cytokine Biologic for Immunotherapy in Patients With Head and Neck Cancers. OncoTargets Ther (2018) 11:3731–46. doi: 10.2147/OTT.S165411
235. Harari PM, Harris J, Kies MS, Myers JN, Jordan RC, Gillison ML, et al. Postoperative Chemoradiotherapy and Cetuximab for High-Risk Squamous Cell Carcinoma of the Head and Neck: Radiation Therapy Oncology Group RTOG-0234. J Clin Oncol (2014) 32:2486–95. doi: 10.1200/JCO.2013.53.9163
236. Bourhis J, Sire C, Tao Y, Martin L, Alfonsi M, Prevost JB, et al. LBA38 Pembrolizumab Versus Cetuximab, Concomitant With Radiotherapy (RT) in Locally Advanced Head and Neck Squamous Cell Carcinoma (LA-HNSCC): Results of the GORTEC 2015-01 “PembroRad” Randomized Trial. Annal Oncol (2020) 31:S1168. doi: 10.1016/j.annonc.2020.08.2268
237. Sun XS, Tao Y, Le Tourneau C, Pointreau Y, Sire C, Kaminsky MC, et al. Debio 1143 and High-Dose Cisplatin Chemoradiotherapy in High-Risk Locoregionally Advanced Squamous Cell Carcinoma of the Head and Neck: A Double-Blind, Multicentre, Randomised, Phase 2 Study. Lancet (2020) 21:1173–87. doi: 10.1016/S1470-2045(20)30327-2
238. Szelag M, Czerwoniec A, Wesoly J, Bluyssen HA. Identification of STAT1 and STAT3 Specific Inhibitors Using Comparative Virtual Screening and Docking Validation. PLoS One (2015) 10:e0116688. doi: 10.1371/journal.pone.0116688
239. Kolosenko I, Yu Y, Busker S, Dyczynski M, Liu J, Haraldsson M, et al. Identification of Novel Small Molecules That Inhibit STAT3-Dependent Transcription and Function. PLoS One (2017) 12:e0178844. doi: 10.1371/journal.pone.0178844
240. Sgrignani J, Garofalo M, Matkovic M, Merulla J, Catapano CV, Cavalli A. Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development. Int J Mol Sci (2018) 19:1591. doi: 10.3390/ijms19061591
241. Leong PL AG, Andrews GA, Johnson DE, Dyer KF, SXi S, Mai JC, et al. Targeted Inhibition of Stat3 With a Decoy Oligonucleotide Abrogates Head and Neck Cancer Cell Growth. Proc Natl Acad Sci USA (2003) 100:4138–43. doi: 10.1073/pnas.0534764100
242. Wang Y, Guo W, Li Z, Wu Y, Jing C, Ren Y, et al. Role of the EZH2/miR-200 Axis in STAT3-Mediated OSCC Invasion. Int J Oncol (2018) 52:1149–64. doi: 10.3892/ijo.2018.4293
243. Pan Y, Zhou F, Zhang R, Claret FX. Stat3 Inhibitor Stattic Exhibits Potent Antitumor Activity and Induces Chemo- and Radio-Sensitivity in Nasopharyngeal Carcinoma. PLoS One (2013) 8:e54565. doi: 10.1371/journal.pone.0054565
244. Sonbol MB, Ahn DH, Bekaii-Saab TS. Therapeutic Targeting Strategies of Cancer Stem Cells in Gastrointestinal Malignancies. Biomedicines. Biomedicines (2019) 7:17. doi: 10.3390/biomedicines7010017
245. Kim MJ, Nam HJ, Kim HP, Han SW, Im SA, Kim TY, et al. OPB-31121, a Novel Small Molecular Inhibitor, Disrupts the JAK2/STAT3 Pathway and Exhibits an Antitumor Activity in Gastric Cancer Cells. Cancer Lett (2013) 335:145–52. doi: 10.1016/j.canlet.2013.02.010
246. Hayakawa F, Sugimoto K, Harada Y, Hashimoto N, Ohi N, Kurahashi S, et al. A Novel STAT Inhibitor, OPB-31121, has a Significant Antitumor Effect on Leukemia With STAT-Addictive Oncokinases. Blood Cancer J (2013) 3:e166. doi: 10.1038/bcj.2013.63
247. Ogura M, Uchida T, Terui Y, Hayakawa F, Kobayashi Y, Taniwaki M, et al. Phase I Study of OPB-51602, an Oral Inhibitor of Signal Transducer and Activator of Transcription 3, in Patients With Relapsed/Refractory Hematological Malignancies. Cancer Sci (2015) 106:896–901. doi: 10.1111/cas.12683
248. Bharadwaj U, Eckols TK, Xu X, Kasembeli MM, Chen Y, Adachi M, et al. Small-Molecule Inhibition of STAT3 in Radioresistant Head and Neck Squamous Cell Carcinoma. Oncotarget (2016) 7:26307–30. doi: 10.18632/oncotarget.8368
249. Kasembeli MM, Singhmar P, Ma J, Edralin J, Tang Y, Adams C, et al. TTI-101: A Competitive Inhibitor of STAT3 That Spares Oxidative Phosphorylation and Reverses Mechanical Allodynia in Mouse Models of Neuropathic Pain. Biochem Pharmacol (2021) 192:114688. doi: 10.1016/j.bcp.2021.114688
250. Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y, et al. STAT3 Antisense Oligonucleotide AZD9150 in a Subset of Patients With Heavily Pretreated Lymphoma: Results of a Phase 1b Trial. J Immunother. Can (2018) 6:119. doi: 10.1186/s40425-018-0436-5
251. Taniguchi K, Konishi H, Yoshinaga A. Efficacy of Combination Treatment Using YHO-1701, an Orally Active STAT3 Inhibitor, With Molecular-Targeted Agents on Cancer Cell Lines. Sci Rep (2021) 11:6685. doi: 10.1038/s41598-021-86021-8
252. Nishisaka F, Taniguchi K, Tsugane M, Hirata G, Takagi A, Asakawa N, et al. Antitumor Activity of a Novel Oral Signal Transducer and Activator of Transcription 3 Inhibitor YHO-1701. Cancer Sci (2020) 111:1774–84. doi: 10.1111/cas.14369
253. Fragoulis GE, McInnes IB, Siebert S. JAK-Inhibitors. New Players in the Field of Immune-Mediated Diseases, Beyond Rheumatoid Arthritis. Rheumatol (Oxford) (2019) 58:i43–54. doi: 10.1093/rheumatology/key276
254. Buchert M, Burns CJ, Ernst M. Targeting JAK Kinase in Solid Tumors: Emerging Opportunities and Challenges. Oncogene (2016) 35:939–51. doi: 10.1038/onc.2015.150
255. Marsella R, Ahrens K, Wilkes R, Trujillo A, Dorr M. Comparison of Various Treatment Options for Canine Atopic Dermatitis: A Blinded, Randomized, Controlled Study in a Colony of Research Atopic Beagle Dogs. Vet. Dermatol (2020) 31:284–e269. doi: 10.1111/vde.12849
256. Vallath S, Sage EK, Kolluri KK, Lourenco SN, Teixeira VS, Chimalapati S, et al. CADM1 Inhibits Squamous Cell Carcinoma Progression by Reducing STAT3 Activity. Sci Rep (2016) 6:24006. doi: 10.1038/srep24006
257. Sen M, Pollock NI, Black J, DeGrave KA, Wheeler S, Freilino ML, et al. JAK Kinase Inhibition Abrogates STAT3 Activation and Head and Neck Squamous Cell Carcinoma Tumor Growth. Neoplasia (2015) 17:256–64. doi: 10.1016/j.neo.2015.01.003
258. Dunn GP, Koebel CM, Schreiber RD. Interferons, Immunity and Cancer Immunoediting. Nat Rev Immunol (2006) 6:836–48. doi: 10.1038/nri1961
259. Kargbo RB. PROTAC-Mediated Degradation of Janus Kinase as a Therapeutic Strategy for Cancer and Rheumatoid Arthritis. ACS medicinal Chem letters. (2021) 12(6):945–6. doi: 10.1021/acsmedchemlett.1c00245
260. Shah RR, Redmond JM, Mihut A, Menon M, Evans JP, Murphy JA, et al. Hi-JAK-Ing the Ubiquitin System: The Design and Physicochemical Optimisation of JAK PROTACs. Bioorganic. Medicinal Chem (2020) 28(5):115326. doi: 10.1016/j.bmc.2020.115326
261. Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, Inflammation, and Cancer. JAKSTAT (2013) 2:e24053. doi: 10.4161/jkst.24053
262. Collins EL, Jager LD, Dabelic R, Benitez P, Holdstein K, Lau K, et al. Inhibition of SOCS1-/- Lethal Autoinflammatory Disease Correlated to Enhanced Peripheral Foxp3+ Regulatory T Cell Homeostasis. J Immunol (2011) 187:2666–76. doi: 10.4049/jimmunol.1003819
263. Sharma J, Larkin J 3rd. Therapeutic Implication of SOCS1 Modulation in the Treatment of Autoimmunity and Cancer. Front Pharmacol (2019) 10:324. doi: 10.3389/fphar.2019.00324
264. Oshimo Y, Kuraoka K, Nakayama H, Kitadai Y, Yoshida K, Chayama K, et al. Epigenetic Inactivation of SOCS-1 by CpG Island Hypermethylation in Human Gastric Carcinoma. Int J Cancer (2004) 112:1003–9. doi: 10.1002/ijc.20521
265. Jo D, Liu D, Yao S, Collins RD, Hawiger J. Intracellular Protein Therapy With SOCS3 Inhibits Inflammation and Apoptosis. Nat Med (2005) 11:892–8. doi: 10.1038/nm1269
266. Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 Binds Specific Receptor-JAK Complexes to Control Cytokine Signaling by Direct Kinase Inhibition. Nat Struct Mol Biol (2013) 20:469–76. doi: 10.1038/nsmb.2519
267. Jiang M, Zhang WW, Liu P, Yu W, Liu T, Yu J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front Immunol (2017) 8:70. doi: 10.3389/fimmu.2017.00070
268. Liu L, Li W, Wei X, Cui Q, Lou W, Wang G, et al. Potent Antitumor Activity of Oncolytic Adenovirus-Mediated SOCS1 for Hepatocellular Carcinoma. Gene Ther (2013) 20:84–92. doi: 10.1038/gt.2012.4
269. Schwartz DM, Bonelli M, Gadina M, O'Shea JJ. Type I/II Cytokines, JAKs, and New Strategies for Treating Autoimmune Diseases. Nat Rev Rheumatol (2016) 12:25–36. doi: 10.1038/nrrheum.2015.167
270. Berraondo P, Sanmamed MF, Ochoa MC. Cytokines in Clinical Cancer Immunotherapy. Br J Cancer (2019) 120:6–16. doi: 10.1038/s41416-018-0328-y
271. Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting Interleukin-6 Signaling in Clinic. Immunity (2019) 50(4):1007–23. doi: 10.1016/j.immuni.2019.03.026
272. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 Signaling Pathway in Targeted Therapy for Cancer. Cancer Treat Rev (2012) 38:904–10. doi: 10.1016/j.ctrv.2012.04.007
273. Qiu Y, Su M, Liu L, Tang Y, Pan YM, Sun J. Clinical Application of Cytokines in Cancer Immunotherapy. Drug Des Devel Ther (2021) 15:2269–87. doi: 10.2147/DDDT.S308578
274. Fizazi K, De Bono JS, Flechon A, Heidenreich A, Voog E, Davis NB, et al. Randomised Phase II Study of Siltuximab (CNTO 328), an Anti-IL-6 Monoclonal Antibody, in Combination With Mitoxantrone/Prednisone Versus Mitoxantrone/Prednisone Alone in Metastatic Castration-Resistant Prostate Cancer. Eur J Cancer (2012) 48:85–93. doi: 10.1016/j.ejca.2011.10.014
275. Schreiber S, Aden K, Bernardes JP, Conrad C, Tran F, Höper H, et al. Therapeutic Interleukin-6 Trans-Signaling Inhibition by Olamkicept (Sgp130fc) in Patients With Active Inflammatory Bowel Disease. Gastroenterology (2021) 160:2354–66.e2311. doi: 10.1053/j.gastro.2021.02.062
276. Xu DH, Zhu Z, Wakefield MR, Xiao H, Bai Q, Fang Y. The Role of IL-11 in Immunity and Cancer. Cancer Lett (2016) 373:156–63. doi: 10.1016/j.canlet.2016.01.004
277. Putoczki TL, Ernst M. IL-11 Signaling as a Therapeutic Target for Cancer. Immunotherapy (2015) 7:441–53. doi: 10.2217/imt.15.17
278. Li P, Zheng Y, Chen X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front Pharmacol (2017) 8:460. doi: 10.3389/fphar.2017.00460
279. Lai KC, Liu CJ, Lin TJ, Mar AC, Wang HH, Chen CW, et al. Blocking TNF-α Inhibits Angiogenesis and Growth of IFIT2-Depleted Metastatic Oral Squamous Cell Carcinoma Cells. Cancer Lett (2016) 370:207–15. doi: 10.1016/j.canlet.2015.10.016
280. Oshima H, Ishikawa T, Yoshida GJ, Naoi K, Maeda Y, Naka K, et al. TNF-α/TNFR1 Signaling Promotes Gastric Tumorigenesis Through Induction of Noxo1 and Gna14 in Tumor Cells. Oncogene (2014) 33:3820–9. doi: 10.1038/onc.2013.356
281. Morrish E, Brumatti G, Silke J. Future Therapeutic Directions for Smac-Mimetics. Cells (2020) 9:406. doi: 10.3390/cells9020406
282. Zhao XY, Wang XY, Wei QY, Xu YM, Lau ATY. Potency and Selectivity of SMAC/DIABLO Mimetics in Solid Tumor Therapy. Cells (2020) 9:1012. doi: 10.3390/cells9041012
283. Gaither A, Porter D, Yao Y, Borawski J, Yang G, Donovan J, et al. Smac Mimetic Rescue Screen Reveals Roles for Inhibitor of Apoptosis Proteins in Tumor Necrosis Factor-Alpha Signaling. Cancer Res Treat (2007) 67:11493–8. doi: 10.1158/0008-5472.CAN-07-5173
284. Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP Antagonists Induce Autoubiquitination of C-IAPs, NF-kappaB Activation, and TNFalpha-Dependent Apoptosis. Cell (2007) 131:669–81. doi: 10.1016/j.cell.2007.10.030
285. Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP Antagonists Target Ciap1 to Induce TNFalpha-Dependent Apoptosis. Cell (2007) 131:682–93. doi: 10.1016/j.cell.2007.10.037
286. Condon SM, Mitsuuchi Y, Deng Y, LaPorte MG, Rippin SR, Haimowitz T, et al. Birinapant, a Smac-Mimetic With Improved Tolerability for the Treatment of Solid Tumors and Hematological Malignancies. J Med Chem (2014) 57:3666–77. doi: 10.1021/jm500176w
287. Silke J, Vucic D. IAP Family of Cell Death and Signaling Regulators. Methods Enzymology (2014) 545:35–65. doi: 10.1016/B978-0-12-801430-1.00002-0
288. Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, et al. Antagonists Induce a Conformational Change in Ciap1 That Promotes Autoubiquitination. Science (2011) 334:376–80. doi: 10.1126/science.1207862
289. Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, Condon SM, et al. Smac Mimetics Activate the E3 Ligase Activity of Ciap1 Protein by Promoting RING Domain Dimerization. J Biol Chem (2011) 286:17015–28. doi: 10.1074/jbc.M111.222919
290. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs Block Ripoptosome Formation, a RIP1/caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol Cell (2011) 43:449–63. doi: 10.1016/j.molcel.2011.06.011
291. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a Signaling Platform That Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol Cell (2011) 43:432–48. doi: 10.1016/j.molcel.2011.06.006
292. Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune Modulation in Cancer With Antibodies. Annu Rev Med (2014) 65:185–202. doi: 10.1146/annurev-med-092012-112807
293. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
294. Mohan SP, Bhaskaran MK, George AL, Thirutheri A, Somasundaran M, Pavithran A. Immunotherapy in Oral Cancer. J Pharm Bioallied Sci (2019) 11:S107–11. doi: 10.4103/JPBS.JPBS_31_19
295. Dolcetti R, De Re V, Canzonieri V. Immunotherapy for Gastric Cancer: Time for a Personalized Approach? Int J Mol Sci (2018) 19:1602. doi: 10.3390/ijms19061602
296. Almangush A, Leivo I, Mäkitie AA. Biomarkers for Immunotherapy of Oral Squamous Cell Carcinoma: Current Status and Challenges. Front Oncol (2021) 11:616629. doi: 10.3389/fonc.2021.616629
297. Xie T, Zhang Z, Zhang X, Qi C, Shen L, Peng Z. Appropriate PD-L1 Cutoff Value for Gastric Cancer Immunotherapy: A Systematic Review and Meta-Analysis. Front Oncol (2021) 11:646355. doi: 10.3389/fonc.2021.646355
299. Kearney CJ, Lalaoui N, Freeman AJ, Ramsbottom KM, Silke J, Oliaro J. PD-L1 and IAPs Co-Operate to Protect Tumors From Cytotoxic Lymphocyte-Derived TNF. Cell Death Differ (2017) 24:1705–16. doi: 10.1038/cdd.2017.94
300. Michie J, Beavis PA, Freeman AJ, Vervoort SJ, Ramsbottom KM, Narasimhan V, et al. Antagonism of IAPs Enhances CAR T-Cell Efficacy. Cancer Immunol Res (2019) 7:183–92. doi: 10.1158/2326-6066.CIR-18-0428
301. Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell (2019) 178:585–99. doi: 10.1016/j.cell.2019.06.014
302. Dougan M, Luoma AM, Dougan SK, Wucherpfennig KW. Understanding and Treating the Inflammatory Adverse Events of Cancer Immunotherapy. Cell (2020) 184:1575–88. doi: 10.1016/j.cell.2021.02.011
303. Huarte E, O'Connor RS, Peel MT, Nunez-Cruz S, Leferovich J, Juvekar A, et al. Itacitinib (INCB039110), A JAK1 Inhibitor, Reduces Cytokines Associated With Cytokine Release Syndrome Induced by CAR T-Cell Therapy. Clin Cancer Res (2020) 26:6299–309. doi: 10.1158/1078-0432.CCR-20-1739
304. Chen AY, Wolchok JD, Bass AR. TNF in the Era of Immune Checkpoint Inhibitors: Friend or Foe? Nat Rev Rheumatol (2021) 17:213–23. doi: 10.1038/s41584-021-00584-4
305. Jacoberger-Foissac C, Blake SJ, Liu J, McDonald E, Triscott H, Nakamura K, et al. Concomitant or Delayed Anti-TNF Differentially Impact on Immune-Related Adverse Events and Antitumor Efficacy After Anti-CD40 Therapy. J immunotherapy Cancer (2020) 8:e001687. doi: 10.1136/jitc-2020-001687
306. Bond MJ, Crews CM. Proteolysis Targeting Chimeras (PROTACs) Come of Age: Entering the Third Decade of Targeted Protein Degradation. RSC Chem Biol (2021) 2(3):725–42. doi: 10.1039/D1CB00011J
307. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc Natl Acad Sci USA (2001) 98(15):8554–9. doi: 10.1073/pnas.141230798
308. Toure M, Crews CM. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angewandte Chemie (International Ed English) (2016) 55(6):1966–73. doi: 10.1002/anie.201507978
309. Nalawansha DA, Crews CM. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem Biol (2020) 27(8):998–1014. doi: 10.1016/j.chembiol.2020.07.020
310. Shiah JV, Grandis JR, Johnson DE. Targeting STAT3 With Proteolysis Targeting Chimeras and Next-Generation Antisense Oligonucleotides. Mol Cancer Ther (2021) 20(2):219–28. doi: 10.1158/1535-7163.MCT-20-0599
311. Hu Z, Crews CM. Recent Developments in PROTAC-Mediated Protein Degradation: From Bench to Clinic. Chembiochem Eur J Chem Biol (2022) 23(2):e202100270. doi: 10.1002/cbic.202100270
312. Békés M, Langley DR, Crew CM. PROTAC Targeted Protein Degraders: The Past is Prologue. Nat Rev Drug Discovery (2022). doi: 10.1038/s41573-021-00371-6
313. Qi SM Dong J, Xu ZY, Cheng XD, Zhang WD, Qin JJ. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front Pharmacol (2021) 12:692574. doi: 10.3389/fphar.2021.692574
314. Li Z, Ma S, Yang X, Zhang L, Liang D, Dong G, et al. Development of Photocontrolled BRD4 PROTACs for Tongue Squamous Cell Carcinoma (TSCC). Eur J Medicinal Chem (2021) 222:113608. doi: 10.1016/j.ejmech.2021.113608
315. He Y, Khan S, Huo Z, Lv D, Zhang X, Liu X, et al. Proteolysis Targeting Chimeras (PROTACs) are Emerging Therapeutics for Hematologic Malignancies. J Hematol Oncol (2020) 13(1):103. doi: 10.1186/s13045-020-00924-z
316. Lin X., Xiang H., Luo G. Targeting Estrogen Receptor α for Degradation With PROTACs: A Promising Approach to Overcome Endocrine Resistance. Europ J Medicinal Chem (2020) 206: 112689. doi: 10.1016/j.ejmech.2020.112689
317. Alabi SB, Crews CM. Major Advances in Targeted Protein Degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem (2021) 296:100647. doi: 10.1016/j.jbc.2021.100647
318. Li H, Dong J, Cai M, Xu Z, Cheng XD, Qin JJ. Protein Degradation Technology: A Strategic Paradigm Shift in Drug Discovery. J Hematol Oncol (2021) 14(1):138. doi: 10.1186/s13045-021-01146-7
319. Farnaby W, Koegl M, McConnell DB, Ciulli A. Transforming Targeted Cancer Therapy With PROTACs: A Forward-Looking Perspective. Curr Opin Pharmacol (2021) 57:175–83. doi: 10.1016/j.coph.2021.02.009
320. Heppler LN, Frank DA. Inhibit Versus Destroy: Are PROTAC Degraders the Solution to Targeting Stat3? Cancer Cell (2019) 36(5):459–61. doi: 10.1016/j.ccell.2019.10.010
321. Liu PCC, Dixit V, Mayo M, Dey J, Yuan K, Karnik R, et al. A First-In-Class STAT3 Degrader KT-333 in Development for Treatment of Hematologic Cancers. 138(S1):1865.
322. Zhou H, Bai L, Xu R, Zhao Y, Chen J, McEachern D, et al. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J Medicinal Chem (2019) 62(24):11280–300. doi: 10.1021/acs.jmedchem.9b01530
323. Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell (2019) 36(5):498–511.e17. doi: 10.1016/j.ccell.2019.10.002
324. Kong S, Ge X, Li X, Liu Z, Zhang R, Yang M, et al. SD-36 Promotes Growth Inhibition and Induces Apoptosis via Suppression of Mcl-1 in Glioma. J Cell Mol Med (2021) 25(17):8261–70. doi: 10.1111/jcmm.16754
325. Chang Y, Min J, Jarusiewicz JA, Actis M, Yu-Chen Bradford S, Mayasundari A, et al. Degradation of Janus Kinases in CRLF2-Rearranged Acute Lymphoblastic Leukemia. Blood (2021) 138(23):2313–26. doi: 10.1182/blood.2020006846
326. Egan JE, Quadrini KJ, Santiago-Schwarz F, Hadden JW, Brandwein HJ, Signorelli KL. IRX-2, A Novel In Vivo Immunotherapeutic, Induces Maturation and Activation of Human Dendritic Cells In Vitro. J Immunother. (2007) 30:624–33. doi: 10.1097/CJI.0b013e3180691593
327. Goncalves Vde P, Ortega AA, Guimaraes MR, Curylofo FA, Rossa Junior C, Ribeiro DA, et al. Chemopreventive Activity of Systemically Administered Curcumin on Oral Cancer in the 4-Nitroquinoline 1-Oxide Model. J Cell Biochem (2015) 116:787–96. doi: 10.1002/jcb.25035
328. Basak SK, Bera A, Yoon AJ, Morselli M, Jeong C, Tosevska A, et al. A Randomized, Phase 1, Placebo-Controlled Trial of APG-157 in Oral Cancer Demonstrates Systemic Absorption and an Inhibitory Effect on Cytokines and Tumor-Associated Microbes. Cancer (2020) 126:1668–82. doi: 10.1002/cncr.32644
Keywords: cytokines, JAK, STAT, oral, gastric, cancer, PROTAC (proteolysis-targeting chimeric molecule), TNF
Citation: Ni Y, Low JT, Silke J and O’Reilly LA (2022) Digesting the Role of JAK-STAT and Cytokine Signaling in Oral and Gastric Cancers. Front. Immunol. 13:835997. doi: 10.3389/fimmu.2022.835997
Received: 15 December 2021; Accepted: 16 May 2022;
Published: 29 June 2022.
Edited by:
Fabiola Castro, University of São Paulo, BrazilReviewed by:
Robert Welner, University of Alabama at Birmingham, United StatesCopyright © 2022 Ni, Low, Silke and O’Reilly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorraine A. O’Reilly, b3JlaWxseUB3ZWhpLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.