 Anna J. Jasinska
Anna J. Jasinska Ivona Pandrea
Ivona Pandrea Cristian Apetrei
Cristian Apetrei
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 January 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.835994
This article is part of the Research Topic CCR5: A Receptor at the Center Stage in Infection View all 12 articles
CCR5, a chemokine receptor central for orchestrating lymphocyte/cell migration to the sites of inflammation and to the immunosurveillance, is involved in the pathogenesis of a wide spectrum of health conditions, including inflammatory diseases, viral infections, cancers and autoimmune diseases. CCR5 is also the primary coreceptor for the human immunodeficiency viruses (HIVs), supporting its entry into CD4+ T lymphocytes upon transmission and in the early stages of infection in humans. A natural loss-of-function mutation CCR5-Δ32, preventing the mutated protein expression on the cell surface, renders homozygous carriers of the null allele resistant to HIV-1 infection. This phenomenon was leveraged in the development of therapies and cure strategies for AIDS. Meanwhile, over 40 African nonhuman primate species are long-term hosts of simian immunodeficiency virus (SIV), an ancestral family of viruses that give rise to the pandemic CCR5 (R5)-tropic HIV-1. Many natural hosts typically do not progress to immunodeficiency upon the SIV infection. They have developed various strategies to minimize the SIV-related pathogenesis and disease progression, including an array of mechanisms employing modulation of the CCR5 receptor activity: (i) deletion mutations abrogating the CCR5 surface expression and conferring resistance to infection in null homozygotes; (ii) downregulation of CCR5 expression on CD4+ T cells, particularly memory cells and cells at the mucosal sites, preventing SIV from infecting and killing cells important for the maintenance of immune homeostasis, (iii) delayed onset of CCR5 expression on the CD4+ T cells during ontogenetic development that protects the offspring from vertical transmission of the virus. These host adaptations, aimed at lowering the availability of target CCR5+ CD4+ T cells through CCR5 downregulation, were countered by SIV, which evolved to alter the entry coreceptor usage toward infecting different CD4+ T-cell subpopulations that support viral replication yet without disruption of host immune homeostasis. These natural strategies against SIV/HIV-1 infection, involving control of CCR5 function, inspired therapeutic approaches against HIV-1 disease, employing CCR5 coreceptor blocking as well as gene editing and silencing of CCR5. Given the pleiotropic role of CCR5 in health beyond immune disease, the precision as well as costs and benefits of such interventions needs to be carefully considered.
CCR5, a C-C chemokine receptor 5 (formerly known as CC-CKR-5 or CKR5), is primarily involved in immune surveillance, inflammatory response, tumor formation and metastasis (1–3), pathogenesis of inflammatory diseases (4–6), asthma (7, 8), and cancer (2, 3). It plays a key role in the recruitment of the immune cells to inflammation sites by directing immune cell migration (chemotaxis) along the chemokine gradient (9, 10). CCR5 regulates trafficking and effector functions of memory/effector T lymphocytes, macrophages, and immature dendritic cells (11). Beyond its direct involvement in mediation of the immune processes, it acts as a suppressor of learning, memories and synaptic connections in the brain (12).
CCR5 is a seven-transmembrane, G protein-coupled receptor (GPCR), a member of the family of class A GCPRs. As a GPCR, CCR5 comprises of seven transmembrane α-helices, three extracellular loops, three intracellular loops, an amino-terminal domain and a carboxyl-terminal domain (13).
The natural ligands for CCR5 include chemokines (small chemoattractant cytokines) involved in innate immunity, which are natural suppressors of HIV-1 infection (14–17): macrophage inflammatory proteins CCL3 (MIP-1 α) and CCL4 (MIP-1 β), CCL5 (RANTES - regulated on activation, normal T-cell expressed and secreted) and CCL3L1, the most potent among the agonists of CCR5 and HIV-1-suppressant (18). CCL7 (MCP-3) is, on the other hand, the main antagonist ligand of the CCR5 receptor (19). Activation of the CCR5 receptor by its agonist ligands stimulates cell migration and mediates inflammatory responses.
CCR5 activation occurs upon binding its agonist ligands and leads to stimulation of pertussis toxin-sensitive heterotrimeric αβγ G protein by catalysing the exchange of GTP for GDP in the Gα subunit that triggers intracellular pathways involved in chemotaxis and activation of leukocytes (20). Upon ligand binding, CCR5 receptor undergoes rapid phosphorylation in the carboxy-terminal region that promotes desensitization and internalization regulated by β-arrestin, an adaptor protein causing sequestration of the receptor to clathrin-coated pits. Upon clathrin-mediated endocytosis, CCR5 receptor moves to endosomes and Golgi network, and then is recycled back to plasma membrane (21–23). The conformation of the CCR5 receptor is dynamically impacted through this process and dependent on cellular localization (24).
CCR5 is expressed on a wide array of bone-marrow-derived cells, including lymphocytes, monocyte/macrophages, granulocytes, T cells, and specialized immune cells including natural killer (NK) cells and regulatory T (Treg) cells, located in primary and secondary lymphoid organs, including thymus and spleen, nonhematopoietic peripheral tissues, such as epithelium, endothelium, vascular smooth muscles, fibroblasts, and in central nervous system in neurons, astrocytes, microglia (15, 25–29). In the normal adult brain, CCR5 is highly expressed in microglia, yet it is undetectable in neurons (30).
Increased levels of CCR5 expression on mononuclear cells is characteristic to chronically inflamed tissues, suggesting that CCR5+ cells are recruited to the inflammatory sites (25). In addition to the lymphoid tissues, CCR5 expression is induced in the cortical neurons and transiently lowered in microglia/macrophages in response to stroke (30).
While CCR5 appears an essential player in various aspects of immune health, knockout alleles of this gene, leading to the loss of function of the CCR5 coreceptor, are present in different primate species at high frequency and with occurrence of null homozygous genotypes (Figure 1).
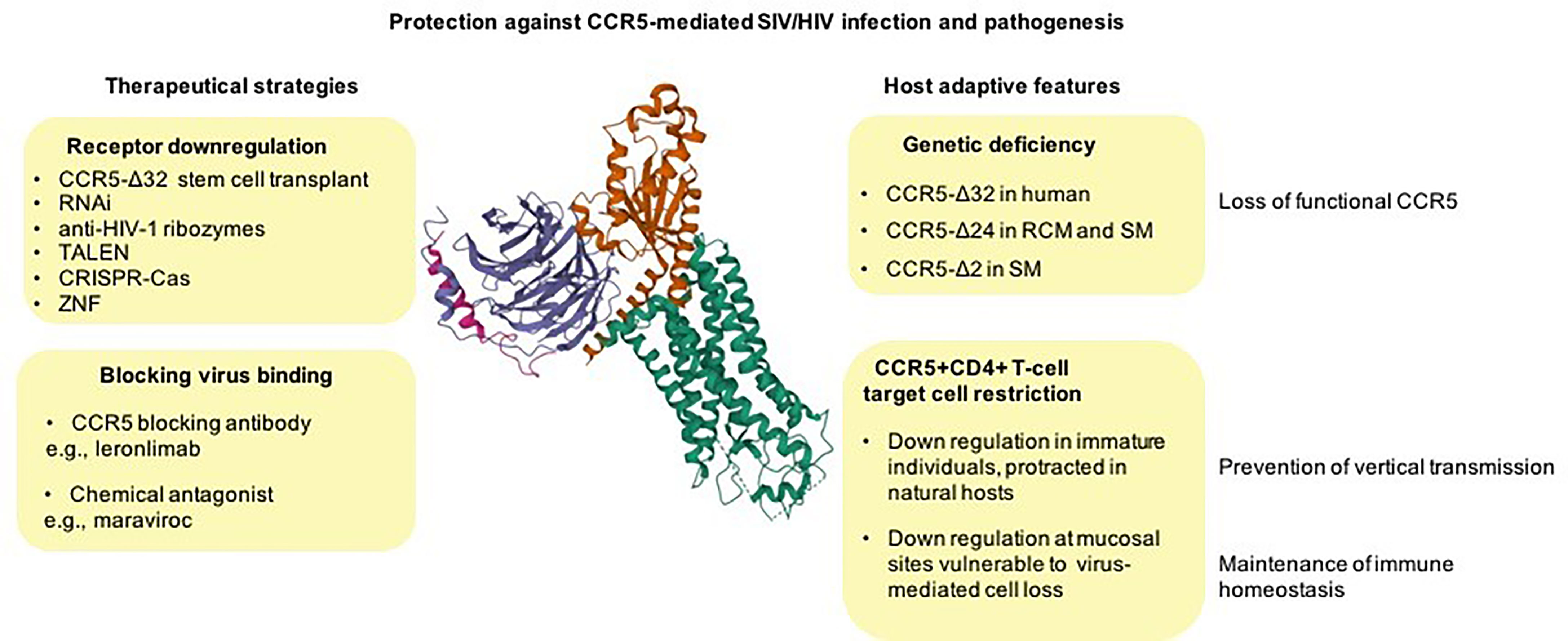
Figure 1 From natural control of CCR5 activity to therapeutic approaches against HIV disease. CCR5 null alleles, preventing CCR5-mediated virus entry, naturally emerged and raised to high frequency in different primate species most likely as a result of host adaptation to lethal pathogens (unknown ancient pathogen in humans and SIV in natural hosts) (top right). CCR5 downregulation of CCR5 on CD4+ T-cells may be an adaptive feature of natural hosts protecting against the vertical transmission of the virus (via breast feeding), and it may also represent an evolutionary adaptation to spare essential CD4+ T-cell subsets from killing by the virus (bottom right). Examples of approaches for therapeutic disruption of CCR5 expression include natural CCR5 gene knockouts (resulting in functional cure), programmable nucleases, and gene silencing (top left) and approaches to blocking the virus fusion with cell membrane via chemical CCR5 receptor antagonists or antibodies (bottom left). Cryo-EM structure of the chemokine receptor CCR5 (green) in complex with RANTES and guanine nucleotide-binding protein Gi subunits alpha-1 (red), beta-1 (blue), and gamma-2 (magenta) was acquired from https://www.rcsb.org/3d-view/7F1R/1 wwPDB: Worldwide Protein Data Bank (13, 31, 32).
CCR5 coreceptor expression on the cell surface can be prevented by a natural genetic variant, a 32-bp deletion (Δ32) observed in human populations. This mutation is localized in the region encoding the second extracellular loop of the receptor and results in a frameshift in the protein coding sequence leading to premature truncation of the normal CCR5 protein and abrogating its availability on the cell surface. The loss of function of the CCR5 gene modulates the risk for HIV transmission and counteract the pathogenesis of HIV infection (33–35).
The CCR5-Δ32 allele is primarily observed in populations of European descent, where its average frequency is ~10%, while being virtually absent in SubSaharan Africans, Asians and Native Americans (34–37). Migrations have likely contributed to the global distribution of the CCR5-Δ32 allele, as, despite its predominant presence in Europe, high frequencies of CCR5-Δ32 were also observed in specific populations of European ancestry outside Europe, e.g., in South Africa (13%) and Chile (12%) (38). The CCR5-Δ32 is also present in African Americans, yet at low incidence (2%) (35) and in some Jewish populations, with the highest frequency in the Ashkenazis (11-20%) (39), where it probably emerged through admixture with people of European descent. The presence of this variant mainly in Euroasian populations suggests that the mutation occurred after their separation from the founders/ancestors of African, Asian, and Native American populations (40).
In Europe, the distribution of the CCR5-Δ32 variant shows marked clines, North-South and East-West. The highest frequency of the CCR5-Δ32 variant was observed among Northern Europeans, for example, in Finland and Russia (16%), Iceland (15%), Sweden (14%), Denmark (13%), Northern France (14%), and Norway (10%) (36, 37, 41, 42), while its lowest frequency was seen in Southern European and Mediterranean populations, such as Spain (7%), Italy (5.6%), Portugal (5.2%), Sardinia (4%), with the lowest prevalence being observed in Corsica (0.9%) (36, 37, 41).
The spatial distribution of the CCR5-Δ32 mutation, with the highest frequency in the Nordic countries suggests a Northern European origin of this variant (37, 41) and subsequent spreading out from the Scandinavian peninsula across Europe by the Vikings through their raids in the 8th-10th centuries (41). While the role of the long-range dispersal consistent with the Viking mediated spread was demonstrated, other models raise the possibility that the allele arose outside of Scandinavia and moved into the region via dispersers from the South (43) or from the Finno-Ugrian tribes of Russia, where the mutation is frequent nowadays (37).
Several features of the CCR5-Δ32 mutation suggest that it had a unitary origin, subsequently becoming a subject to positive selection, as supported by: (a) its high frequency, (b) its virtual absence in other populations than those of European descent, (c) a striking gradient of this variant across Europe, as well as (d) a long-range disequilibrium in this CCR5 locus (36, 37, 44). Age estimates of the ancestral CCR5-Δ32 variant based on microsatellite analysis suggested that the deletion arose relatively recently, yet in a time frame that varies widely: about 700 years ago with an estimated range of 275-1,875 yrs (based on haplotype coalescence) (36); 3,400 years ago (based on the recombination frequency near the CCR5 locus); 1,400 years ago (based on the microsatellite mutation rate); or 2,000-2,200 years ago (based on mutation and crossover events) (37). Other studies suggested that the original single mutation occurred 2,500 years ago (44).
The relatively recent origin of the mutation, ranging between the Neolithic period (37) and Middle Ages (36), suggests that a positive selection or a selection acting on heterozygotes associated with selective advantage, rather than random drifts, could have driven the allele to the currently high frequency in the populations of European descent (37, 41). In comparison to the calculations based on the linkage disequilibrium, mutation rate, and the spatial allele distribution, which indicated the age of CCR5-Δ32 variant between several hundred to several thousand years, the age estimate for this variant was 127,500 years (i.e., two orders of magnitude higher), when its frequency was determined assuming neutral drift (36). This discrepancy points to intense natural selection, rather than random drift, as a force shaping the frequency of CCR5-Δ32 allele, suggesting an existence of local environmental factors, such as major pathogens, that have exerted a marked selective pressure on this locus in the historical time (43, 45).
Pathogens and infectious diseases represent major selective forces that shape the frequencies of alleles involved in protective immune mechanisms. Nowadays, the CCR5-Δ32 variant plays an important role against the HIV transmission in the human populations. An ancestor of HIV-1 originated from a cross-species transmission from chimpanzee to humans at the beginning of the 20th century (46) and then it spread out in the European populations only in the 1980s (47, 48). Meanwhile, several cross-species transmissions of the SIVsmm that naturally infect the sooty mangabeys are at the origin of HIV-2 (49–52). These cross-species transmissions also occurred during the 20th century (53). Since HIVs were passed to humans only recently, they have not had sufficient time to exert such profound selective effect on the allele frequency. Instead, several other pathogens and resultant diseases were proposed to drive the CCR5-Δ32 mutation to the contemporarily high frequencies.
The first proposed selective factor was the plague, which had a high mortality rate and was confined to Europe, where it persisted for ~300 years, from 1347 to 1670 (36). The plague hypothesis, which still remains one of the most “popular” concepts, despite some contradicting observations, proposed that macrophages infected with Yersinia pestis (54) were introduced in the human bloodstream by bites by fleas travelling on black rats in the Middle Ages (36), spreading the disease that killed ~40% of the population of Europe during epidemics, such as the Black Death of 1348–1350. In vitro studies showed, however, that although the Ccr5-deficient macrophages have a drastically reduced uptake of Yersinia pestis (an isolate from a fatal human case of plague), they experience a similar mortality with the wild-type Ccr5-expressing macrophages (55), suggesting that, if this model is representative to humans, the CCR5 deficiency did not have a protective effect against plague in people (56). The plague hypothesis was also challenged by the observation that plague may have not generated sufficient selective pressure for increasing the CCR5-Δ32 allele to contemporary frequencies (57). Instead, the selective raise of the CCR5-Δ32 allele was proposed to be attributed to smallpox (Variola major) caused by the poxvirus, based on population genetic analysis, which considered the temporal pattern and age-dependent nature of the diseases (57). The smallpox hypothesis, on the other hand, was opposed by the argument that the lethal form of smallpox emerged only recently (in England ~1628), not long before the introduction of variolation ~1750 and vaccination ~1800 (44) that gave the pathogen a narrow time window to push the mutant allele frequency to the current level (less than estimated 600 years needed for that) (57). As a selecting factor, other models considered recurrent epidemics of viral hemorrhagic fevers (“hemorrhagic plague”) affecting the eastern Mediterranean region since at least 1500 BC (44) or a pathogen spread during the time of the expansion of Roman Empire (58). A potential influence of other microbes, such as Shigella, Salmonella, and Mycobacterium tuberculosis, on the frequency of CCR5-Δ32 variant was also proposed (36).
Sabetti et al. reported that the variation of the CCR5-Δ32 was consistent with the pattern of neutral selection and estimated that the ancestral haplotype carrying CCR5-Δ32 variant might have arisen more than 5,000 years ago, with a certain probability that some selection has occurred thereafter (40).
The case of CCR5-Δ32 allele in humans resembles deletions in the CCR5 gene present in the African monkeys of the genus Cercocebus, preventing CCR5 coreceptor-mediated SIV entry.
West African natural host species of SIVs, particularly the Cercocebus species, such as the RCM and the SM often carry a 24-bp deletion mutation in the CCR5 gene (CCR5-Δ24) that causes an in-frame deletion of eight aminoacids in the fourth transmembrane region, abrogating the cell surface expression and coreceptor function of CCR5 for SIV entry (59, 60). The CCR5-Δ24 mutation in RCMs has a high frequency (86.6%) (60), exceeding that of the CCR5-Δ32 allelic variant in the human populations (44). It was observed in geographically distant RCM populations, from Gabon and Nigeria (and mangabeys in the US zoos), which demonstrates that its frequency is not due to local founder effects, but rather attributable to an old age and selective advantage of the variant (60, 61). The CCR5-Δ24 allele was also observed in SMs, yet at lower frequencies (4.1%) (60). The geographic ranges of RCMs and SMs are not overlapping at present days, as RCMs inhabit the swamps, mangroves and riverine forests along the Gulf of Guinea shore of Nigeria, Cameroon, Equatorial Guinea, Gabon, and the Gabon-Congo border (62), while the habitat of the SMs ranges in the forests at the Atlantic coast from Senegal to the Ivory Coast (63). The exact time of origin of the CCR5-Δ24 mutation in these two Cercocebus species is not known. The timing of divergence for these species is estimated at 2.29 MYA (64), suggesting either an ancient age of mutation before the split of these species, or its emergence following recurrent events. Unlike HIVs, which on the pandemic scale have been in the human populations for ~60 years, SIVs have been present in African NHPs for a much longer evolutionary time scale (65, 66) and potentially could have been a selective factor behind the high frequency of null CCR5 alleles. However, this hypothesis has yet to be confirmed.
In addition to the CCR5-Δ24 allele (that they carry at a frequency of 3%), SMs are also frequently (26%) carrying a 2-bp deletion in the CCR5 gene (CCR5-Δ2), which, like the CCR5-Δ24 allele, encodes a truncated molecule that is not expressed at the cell surface (59).
The presence of these common deletion alleles of CCR5 in different primate species (CCR5-Δ32 in humans, CCR5-Δ24 in RCMs and SM, and CCR5-Δ2 in SM) suggests that the emergence and high frequencies of these alleles may represent a convergent evolution, yet it remains unclear what pathogens were driving these adaptations, most likely different for humans and African NHPs.
To infect CD4+ T cells in humans, HIV-1 utilizes CCR5 (mediating entry of R5 viruses) or CXCR4 (mediating entry of X4 viruses), or both entry coreceptors (67). CCR5 coreceptor is mostly expressed on memory CD4+ T cells, while CXCR4 is expressed on both memory and naive cells. The change in coreceptor usage towards CXCR4-tropism during the later stage of HIV-1 infection may contribute to accelerated disease progression (68–70). Meanwhile, in addition to CCR-5, HIV-2 uses GPR15 (BOB) and CXCR6 (BONZO) (71).
CCR5 density on the surface of CD4+ T cells is a key regulator of cell infectability and virus production, and a critical determinant of the HIV-1 disease progression (72). The extent of cell death correlates with the virus replication, and the capacity of HIV to induce cell death depends on the level of CCR5 expression on the surface of the CD4+ T cells (73). The density of CCR5 receptors on target cells is logarithmically correlated with HIV-1 viremia (72) and disease progression (74). In vitro studies suggested a dual role of CCR5 in determining HIV-1 production: as an entry coreceptor, it acts as a critical factor for infection, yet exerts only a moderate influence on the magnitude of viral loads, while as a postentry regulator of the HIV-1 life cycle, particularly at reverse transcription stage, it accounts for the logarithmic relation between the viremia and CCR5 density (75).
HIV entry via CCR5 receptor occurs through a series of processes, depending on the conformational state of both viral envelope protein and cellular receptor (24). CCR5 stabilizes the CD4-induced conformation of Env protein and anchors the virus near the cell surface (76). Chemokines that are native CCR5 ligands naturally restrict HIV-1 infection sterically, by masking the viral envelope glycoprotein gp120 binding sites and promoting CCR5 endocytosis, reducing the CCR5 cell surface level (77, 78). The second extracellular loop and amino-terminal domain of CCR5 receptor are critical for interacting with HIV Env protein and binding natural chemokine ligands, such as CCL4 and RANTES. While these molecules bind different regions, they both compete with the virus for the binding site (79, 80).
Biological activity of CCR5 depends on its conformations (81), which influences interaction with HIV gp120 and native chemokines (82). Some receptors have low binding affinity for native CCR5 chemokines and therefore chemokines are weak inducers of CCR5 endocytosis (82).
The CCR5-Δ32 variant generates a nonfunctional entry coreceptor for HIV that does not support fusion between the virus and the target-cell membrane, thus preventing infection and pathogenesis. The homozygous CCR5-Δ32bp genotype (Δ32/Δ32) carriers (about 1% of Europeans) are highly protected from HIV-1 infection (33–35, 83–85), yet this protection is not complete, as rare cases of HIV infection were reported in the homozygotes (86–88). The Δ32bp knock-out of the CCR5 gene was observed in cohorts of multiple HIV-exposed seronegative (HESN) individuals (33, 35), and resistance of circulating cells to HIV infection in vitro was reported (34).
The WT/Δ32 heterozygotes exhibited a reduced ability to support HIV-1 replication compared to the wild type homozygotes (WT/WT) (33–35), had reduced viral loads (33, 34, 36, 84), a slower rate of CD4+ T-cell depletion (84), resulting in a 2-3 years delayed progression to AIDS (33–35, 84, 85, 89), and improved virological response to antiretroviral therapy (90). CCR5-Δ32bp heterozygosity also appeared to be associated with reduced susceptibility to HIV-1 infection (91), yet this observation was not universally confirmed (92). The CCR5 WT/Δ32 genotype was also associated with protection from AIDS-related lymphoma, a non-Hodgkin’s B cell malignancy that is common in patients with AIDS (93, 94).
An increased prevalence of heterozygotes for the CCR5-Δ32 mutation was found in some, but not all, cohorts of HIV long-term nonprogressors (LTNP, i.e., HIV infected individuals with little or no clinical signs of progression), but it does not appear either essential or sufficient for protection against disease progression (87, 95). In elite controllers (ECs, which spontaneously control HIV replication to undetectable viral loads and maintain stable CD4+ T-cell counts), the prevalence of CCR5-Δ32 heterozygotes appears somewhat elevated compared to the general population, yet this difference is not striking (96).
When compared to homozygotes with both normal copies of CCR5 WT/WT, CCR5 heterozygotes WT/Δ32 associate a >50% reduction in cell surface expression of CCR5, and display a lower infectability of blood cells by the M-tropic HIV-1 in vitro (97, 98). The abrogation of cell surface expression of CCR5 coreceptor is caused by the interruption of CCR5 transport to the cell membrane. While normal CCR5 protein can undergo both post-translational phosphorylation and/or cotranslational multimerization, the mutant CCR5-Δ32 can only form multimers and is incapable of being phosphorylated. In the CCR5 heterozygotes, the heterodimers between the normal CCR5 and mutant CCR5-Δ32 proteins are retained in the endoplasmic reticulum causing reduced cell surface expression of the functional CCR5 coreceptor (98).
The delayed progression to HIV disease was associated with other types of genetic variation in the CCR5 locus. For example, variants located within the CCR5 promoter (99–102) showed regulation of CCR5 cell surface expression and of CD4+ T-cell apoptosis, as well as a correlation with HIV disease progression (103). The CCR5 promoter variant 59029 G/A reduced the activity of the CCR5 promoter by ~45% and resulted in ~4 years delayed progression to AIDS in the carriers of this mutation (104). Downregulation of active transcription of CCR5, paralleled with reduced cell surface expression of CCR5, was observed in a subset of elite and viremic controllers with an R5-resistance phenotype (105). The transcriptomic downregulation of CCR5 (9-fold) was associated with downregulation of multiple genes, including CCR2, in the 500 kb block around the CCR2-CCR5 locus on the chromosome 3p21 (105).
Also, genetic variation in genes coding ligands of CCR5 may influence the functionality of this receptor. For example, CCL3L, a HIV-1 suppressive chemokine, shows a copy number variation that is associated in a dosage dependent manner with susceptibility to HIV infection; lower number of copies of CCL3L are associated with an increased risk of HIV (106).
The effect of the CCR5 deficiency (Δ32/Δ32) in conferring nearly complete prevention of HIV-1 infection, was achieved through experimental manipulation that blocked HIV-1 entry into cells with an anti-CCR5 reagent (97). Given that the genetic variation lowering the CCR5 expression has an advantageous effect on taming HIV pathogenesis and did not seem to be associated with a deleterious phenotype in humans (33), interventions blocking or reducing the CCR5 expression emerged as promising approaches to the prevention and treatment of the HIV disease (107–115). However, while the initial studies suggested that the loss of function due to the CCR5-Δ32 does not bear marked impact on health, there is increasing evidence that CCR5 plays a complex role in organism homeostasis, and is not completely dispensable (45, 116–118).
Beyond SIV/HIV infection, CCR5 plays multiple roles in viral diseases (119), bacterial and parasitic infections (120). It is anticipated that CCR5-deficiency may exert several different, some mutually opposing, effects: (a) prevent infection with CCR5-tropic pathogens, (b) weaken the immune response to some pathogens, leading to increased susceptibility to infection, and (c) reduce CCR5-mediated inflammation, which can either hamper protective inflammatory response, or reduce problems related to excessive inflammation.
CCR5 is a key protective factor against some pathogens. For example, it promotes survival during infection with the West Nile virus (WNV), which can cause fatal encephalitis, by promoting leukocyte trafficking to the brain during the infection (116). However, genetic deficiency of CCR5 (Δ32/Δ32) confers a strong risk of symptomatic WNV infection associated with a fatal outcome (117, 121). In influenza patients, CCR5 deficiency causes a four-fold increased mortality (122). These findings warrant a question regarding the safety of some HIV therapies employing null CCR5 alleles and motivates a development of strategies blocking virus binding CCR5 while preserving the functionality of CCR5 as a chemokine receptor (118).
On the other hand, CCR5 is implicated in infections with CCR5-tropic pathogens, such as Dengue virus (123) and Staphylococcus aureus (124). CCR5-Δ32 mutation showed a protective effect against community acquired pneumonia caused by Streptococcus pneumoniae (125) and against a severe form of COVID-19 (126).
The CCR5-Δ32 variant plays a complex pro- and antimicrobial role in Mycoplasma pneumoniae infection, showing an association with development of chronic infection, yet also with a reduced risk of asthma development in infected children, when compared to children with a nondeleted version of CCR5 (127). Analogically, CCR5 null allele plays a protective effect against toxoplasmosis (Toxoplasma gondii) infection (128, 129), while the functional CCR5 receptor is an essential regulator of the inflammatory response following this parasitic infection (10, 130).
CCR5-Δ32 mutation was implicated as a factor modulating the risk of neurodegenerative dementias (131, 132) and recovery after stroke and traumatic brain injury (30). CCR5 link with Alzheimer disease was suggested by several studies, yet contradicting association results were reported in the others (131). In the human populations, the CCR5-Δ32 allele was not significantly associated with neurodegenerative dementias, however, an earlier age of onset of neurodegenerative disease was observed in carriers of the CCR5-Δ32 allele, suggesting that the deletion may have a detrimental effect in the context of neurodegeneration (132). Humans that carry CCR5-Δ32 have better outcomes after stroke, with an enhanced motor recovery and reduced cognitive deficits (30). Based on that observation, CCR5 was proposed as “a translational target for neural repair in stroke and traumatic brain injury” (30). This is consistent with the observation in a mouse model that inhibition of CCR5 signaling enhanced neuroplasticity processes, learning and memory, while overexpression of CCR5 led to learning and memory deficits (12).
Many African NHP species (e.g., SM, RCM, African green monkey-AGM, mandrill-MND) carry species-specific SIVs, a family of viruses from which HIV evolved. Yet, in contrast to the progressing hosts, such as humans and macaques, the African species do not typically develop immunodeficiency despite many years of infection and high levels of viral replication (133–135).
This lack of disease progression in the natural hosts is attributed to the long-term host-pathogen coevolution spanning between hundreds of thousands to possibly millions of years (65, 66), which allowed for the development of protective mechanisms, including lower levels of immune activation upon infection (136–142). Such nonpathogenic SIV infections in natural hosts, some of which utilize specific CCR5 regulations to minimize pathogenesis, provide an insight into adaptive mechanisms protective against the disease (143).
SIV infection in its respective natural hosts is usually nonprogressive and presents the following main features: a) only a transient depletion of peripheral CD4+ T cell, b) absence of intestinal dysfunction and its deleterious consequences, allowing the maintenance of integrity of gut barrier, and c) resolution of immune activation after acute infection. These features are in stark contrast to the pathogenic SIV/HIV infection in a non-natural host, which is characterized by a) progressive CD4+ T-cell loss, b) disruption of the intestinal barrier leading to severe gut dysfunction, and c) chronic inflammation and immune activation (144).
Despite the fact that progressing and nonprogressing hosts display stark differences in the course of infection and pathogenesis, they share several common features of the lentiviral infection, such as the high virus replication rates and fast turnover of infected cells (145–147). Natural SIV infections are therefore different from HIV-1 long-term nonprogressors and SIV-infected RMs, in which the deleterious impact of HIV/SIV infection is minimized through a control of viral replication. Instead, it resembles more to the viremic HIV controllers, a small fraction of HIV-infected individuals that control disease progression by keeping at bay chronic inflammation and T-cell immune activation, in the context of a very active viral replication (148).
The SIV’s usage of CCR5 coreceptor to infect its target cells (149) renders the cells coexpressing CCR5 and CD4 (T-cells, the monocytes/macrophages, dendritic cells) the main targets for SIV/HIV infection.
The natural hosts of SIV, both uninfected and SIV-infected, are characterized by markedly lowered abundance of CCR5+ CD4+ T cells at the mucosal sites, as well as in peripheral blood, lymph nodes and in bone marrow compared to pathogenic hosts, human and macaques (134). In addition to the CD4+ T cells, CCR5 expression on monocytes is lower in the natural host than in humans and macaques, yet to a lesser extent (134). Intermediate levels of CCR5 expression on the CD4+ T cells were observed in the chimpanzee, a non-natural host, which acquired its species-specific SIVcpz more recently than the African monkey hosts. Still, chimpanzees were infected in the wild for considerably longer periods than humans and macaques. It was therefore postulated that chimpanzees did not have sufficient evolutionary time to adapt well to the virus and thus remain vulnerable to its pathogenic effects (149). Indeed, studies in wild chimpanzees reported that they can progress to AIDS-like disease and develop CD4+ T cell depletion, also their mortality rate was 10-16-fold increase compared to uninfected chimpanzees (150, 151).
In nonprogressing hosts, downregulation of CCR5 expression on CD4+ T cells is associated with lower levels of infection than in non-natural hosts (e.g., SMs vs RMs) (152). In SMs, CD4+ T cells, in particular central memory cells, did not upregulate CCR5 in response to in vitro stimulation, and the low CCR5 expression on central memory cells was associated with reduced susceptibility to infection (152). This specific regulation of CCR5 expression on different cell types may protect from SIV infection (and subsequent death) the CD4+ T cell subsets critical to a mild course of infection, while the virus replicates in less dispensable cells (152). It was postulated that long-standing selective pressure of SIV has led to the adaptive shift toward immune functions less dependent on the CD4+ CCR5+ T cells in natural hosts (134).
The CD4+ T cells expressing CCR5 on their surface are the main targets for HIV/SIV infection in both natural and non-natural hosts. The CCR5 surface expression shows a distinctive ontogenetic pattern characterized by an increase of CCR5 expression with the host maturation; as a result, availability of CCR5+ CD4+ target T cells increases with age. In general, the levels of target cells are very low in newborns compared to adults, in both natural hosts of SIV (i.e., AGMs) and in non-natural vulnerable hosts, i.e., macaques and humans (153–156). This pattern, however, markedly varies between progressing and nonprogressing hosts with respect to the timing. The target cell maturation is programmed distinctively among different primate species: (a) rapid in RMs, reaching CCR5+ CD4+ T-cell levels comparable to those in adults by the age of 9 months i.e., at the end of lactation (154, 155, 157); (b) intermediate in humans, with a gradual increase, reaching the adult level by 5-6 years i.e., long after weaning, yet still during childhood (158); (c) slow in natural hosts, in which at the end of the lactation period, the levels of target cells are not significantly different from newborns and much lower than in adults, and an increased CCR5 expression on CD4+ T cells only occurs at sexual maturity (144, 153, 159).
Vertical transmission of HIV and SIV can occur in utero, intrapartum, and postnatally, through breastfeeding (BFT) (160). Humans and NHPs show striking species-specific differences in the timing of maturation of the CCR5 expression on CD4+ T cells (i.e., onset of CD4+ CCR5+ T target cells) in immature individuals: early in RM, intermediate in humans, and later in AGMs. The increasing age of target cells availability was paralleled by the rates of BFT: 60% in RMs (161), 29% in humans (162), and 0-5% in AGMs (66, 153, 159) suggesting that the delayed maturation of the SIV target cells in natural hosts compared to pathogenic hosts may be the factor behind the lack of/low SIV BFT (144).
These observations are concordant with the age-related increase of SIV transmission in natural populations of AGMs in West Africa (sabaeus) and South Africa (pygerythrus/vervet) (66, 159) and ontogenetic changes in the abundance of CCR5+ CD4+ T cells in the blood of sabaeus AGMs. Among the uninfected monkeys, the levels of CCR5+ on circulating CD4+ T cells are low in infants and juveniles, and markedly increased in adults, that usually become infected at the age of sexual maturity (159). While there is no significant difference in the mean levels of target cells between SIV infected and uninfected adults, in immature individuals the availability of target cells is positively associated with the SIV infection status (159). A convergent observation was made in infants of rhesus macaques experimentally exposed to SIV treated with maraviroc - the maraviroc treatment prevented vertical SIV transmission only in individuals with naturally low levels of CCR5 on the CD4+ T cells prior to the treatment (163). The susceptibility to infection is proportional to the target cell availability at mucosal sites (164) and therefore the natural restriction of CCR5 expression in young individuals may represent a strategy to protect target cells from infection.
In comparison to non-natural hosts, natural host show much lower expression of CCR5 on CD4+ T cells (but not CD8+ T cells) at mucosal sites, in particular in the gut, that leads to overall low numbers of CCR5+ CD4+ T target cell (134, 153), especially memory cells, at these locations (134). These adaptive roles of CCR5+ CD4+ target cell restriction suggest that interventions mimicking the natural phenomenon of limited abundance of target cells at critical developmental periods and tissues may protect against transmission and excessive cell death, respectively.
Despite the difference in availability of target cells supporting viral replication, both natural and non-natural hosts show high viremia. It brings up a question, if not CD4+ CCR5+ T cells, which other cells facilitate virus replication. A plausible explanation is that SIVs can infect cells in natural hosts through other coreceptors than CCR5, and in this way protect target cells and specific anatomical sites from SIV pathogenesis, and prevent vertical SIV transmission (153).
SIVs predominantly use the canonical CCR5-mediated cell entry pathway (165), yet they can also use other coreceptors, usually in addition to CCR5: CXCR4, CXCR6 (AKA BONZO, STRL33), orphan receptors GPR1 and GPR15, as demonstrated through in vitro studies (165–169). A unique alternative coreceptor usage, CCR2, was developed by SIVrcm and SIVmm (59, 61, 170, 171).
As a result of the high frequency of deletion alleles, some RCMs and SMs are homozygous for nonfunctional CCR5 variants and therefore completely lack the functional CCR5 receptor on the cell surface. The frequency of null homozygotes in RCMs exceeds 70% (60). However, the CCR5 null genotype in RCMs and SMs is not sufficient to protect them from SIV infection in vivo (59–61, 172–174) in contrast to homozygotes of CCR5-Δ32 in humans that are nearly completely protected from HIV infection. This suggests that SIVs infecting these species bypassed the inactivated CCR5-mediated entry pathway by developing the ability to use alternative receptors (Figure 2).
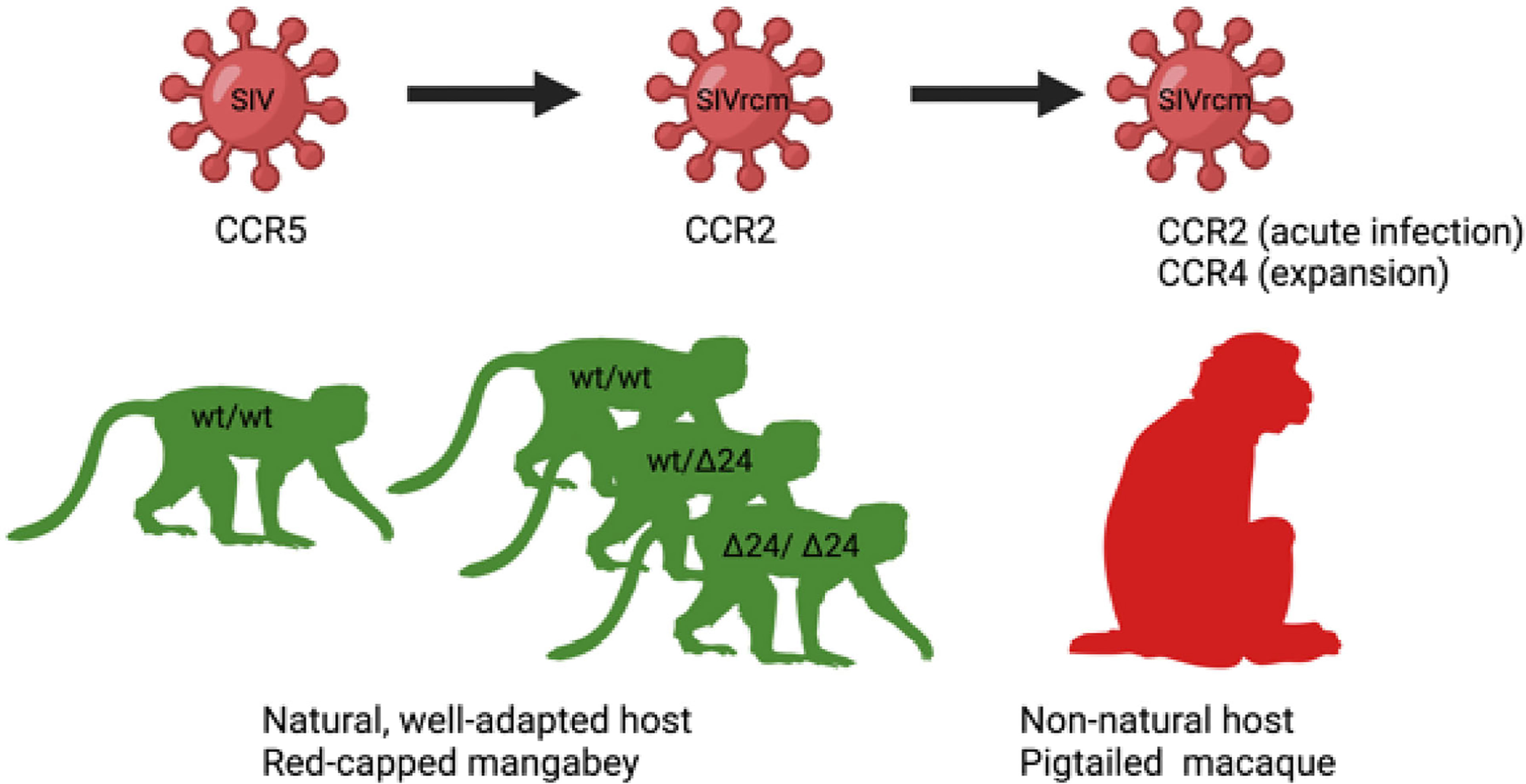
Figure 2 Host-pathogen co-adaptations in a natural (left) and non-natural host (right). On the left, a putative origin of a unique among SIVs CCR2-tropic SIVrcm infecting red-capped mangabey RCM: A high frequency of CCR5-Δ24 mutation, disrupting CCR5 function and protecting from infection with CCR5-tropic SIV, led to the virus adaptation via changing the usage of CCR5 to CCR2b for cell entry (60). On the right, SIVrcm adaptation of coreceptor usage to a new host: experimental SIVrcm infection of pigtailed macaque, a natural host, showed a CCR2 usage in early infection and expansion of coreceptor usage to CCR4 demonstrating that lentiviral adaptation may occur rapidly through strain selection (170).
In spite of being frequently CCR5-Δ24 homozygous and thus widely protected against infection with a CCR5-tropic SIV, the RCMs are naturally infected with a species-specific SIVrcm. The SIVrcm strains collected from naturally-infected RCMs from distant geographic locations, Gabon and Nigeria, are members of the same lineage of SIV (60, 61). SIV prevalence in RCMs is significant, about 22% (175, 176). The explanation for this successful SIVrcm spread in the RCM populations is that the virus utilizes CCR2b as the main coreceptor for virus entry, unlike the vast majority of SIV and HIV strains that use CCR5 (60, 177). The high allelic frequency of the CCR5-Δ24 in RCMs paralleled with the unique among primate SIVs R2b-tropism of SIVrcm suggest that the CCR2 coreceptor usage may have been acquired by SIVrcm as an adaptation to CCR5 genetic defects in its host.
CCR2 is mainly expressed on monocytes (long lived cells) and nearly absent on T lymphocytes (short lived cells), and therefore it could be anticipated that, upon experimental infection, SIVrcm would show in vivo tropism toward monocytes (170). Yet, such an experimental infection of pigtailed macaques led to a surprising pattern of viral replication characteristic for short lived cells and a significant CD4+ T cell loss in the intestine and blood, in particular effector memory CD4+ T cells, and only a minimal monocyte depletion (170). These pathogenic features were explained by an in vivo expansion of the SIVrcm tropism upon infection of the pigtailed macaques upon expanded coreceptor use to CCR4. This coreceptor use expansion led to a selective depletion of CCR4-expressing memory CD4+ T cells (170). CCR4 was indeed reported to be expressed mainly on lymphocytes and only at very low levels on monocytes (178).
Blocking of CCR5 coreceptor in vitro did not prevent SIV infection in circulating lymphocytes of SMs or AGMs (171) suggesting an existence of alternative entry pathways. In both cases, in addition to the CCR5 coreceptor, CXCR6 was an efficient entry pathway of SIV in in vitro experiments. Thus, SIVagm and SIVsmm utilize, in addition to CCR5, CXCR6 and GPR15 (171, 179). Additionally, SIVsmm utilizes GPR1 less frequently (59). In vitro, CCR5 appears nonessential for SIVsmm infection in SMs as SIVsmm glycoprotein can interact with GPR15 and CXCR6 supporting a similar level of infection as that mediated via CCR5 (59).
Alternative pathways are exclusively responsible for SIVsmm replication in animals that genetically lack functional CCR5, while both CCR5 and alternative coreceptors may be used in hosts where both CCR5 and alternative pathways are available (180). It was postulated that the usage of alternative non-CCR5-mediated pathways in natural hosts may be a counter measure to minimize pathogenicity of infection, yet still maintain high virus replication levels by directing the virus to different cell subsets, less critical to the maintenance of immune homeostasis (59).
Note, however, that these in vitro experiments were carried out using SIV clones, some of them derived after passage through pathogenic hosts, which might have impacted their in vitro tropism. In vivo, SIVsab was shown to preferentially infect and deplete central and effector memory cells (181), being thus possible that, in vivo, the transmitted-founder viruses preferentially infect CCR5-expressing CD4+ T cells, thus recapitulating the pathogenesis of HIV-1 transmission, when transmitted founder HIV strains infect exclusively CCR5-expressing cells and display specific phenotypes (182–184). It is very likely that, as infection progresses, SIVs confronted with a low availability of the CCR5+-expressing cells, expand their in vivo tropism towards the other coreceptors described above (59, 171, 179, 180).
This idea is also supported by reports of coreceptor expansion for SIVsmm to efficiently infect naïve cells. While typically SM do not experience chronic CD4+ T-cell loss or clinical signs of disease, a small subset of SMs showed a profound CD4+ T-cell depletion associated with carrying SIVsmm variants with an expanded use of SM-derived coreceptos (180) and human coreceptors, including CXCR4 (180, 185, 186), and generally expanded tropism. These coreceptors may support virus replication in SMs that have restricted CCR5 expression and lack functional CCR5 due to loss of function mutations.
SIVcpz infecting chimpanzees and HIV-1 infecting humans are members of the same virus lineage and are both pathogenic (Figure 3). Both can use CCR5 as a principal entry coreceptor, but cannot use CXCR6, which was a coreceptor used for the entry of the ancestor of HIV-1 originated from cross-species transmission of SIVcpz infecting chimpanzees (46). In chimps, SIVcpz emerged from a Cercopithecus lineage of SIV (188), which env gene has a recombinant origin SIVgsn/mus/mon from greater spot-nosed monkey (Cercopithecus nictitans), mustached monkey (C. cephus), and mona monkey (C. mona) (189, 190). Contemporary SIVmus, similarly to SIVsmm and SIVagm, uses both CCR5 and CXCR6 for cell entry (187), suggesting the CXCR6 usage as a major coreceptor is attributable to SIVs with nonpathogenic course of infection in their respective hosts MUS, SM, and AGM. In the SIVcpz and HIV-1 lineage, the ability to infect via CXCR6 was lost and the viruses shifted their tropism exclusively towards CCR5. CXCR6 is expressed on CD4+ effector memory T-cells, yet on a subpopulation distinct from those with expression CCR5 (187) and therefore the switch in coreceptor use resulted in the change of target cells, probably to more vulnerable cell subsets what can lead to pathogenesis (187).
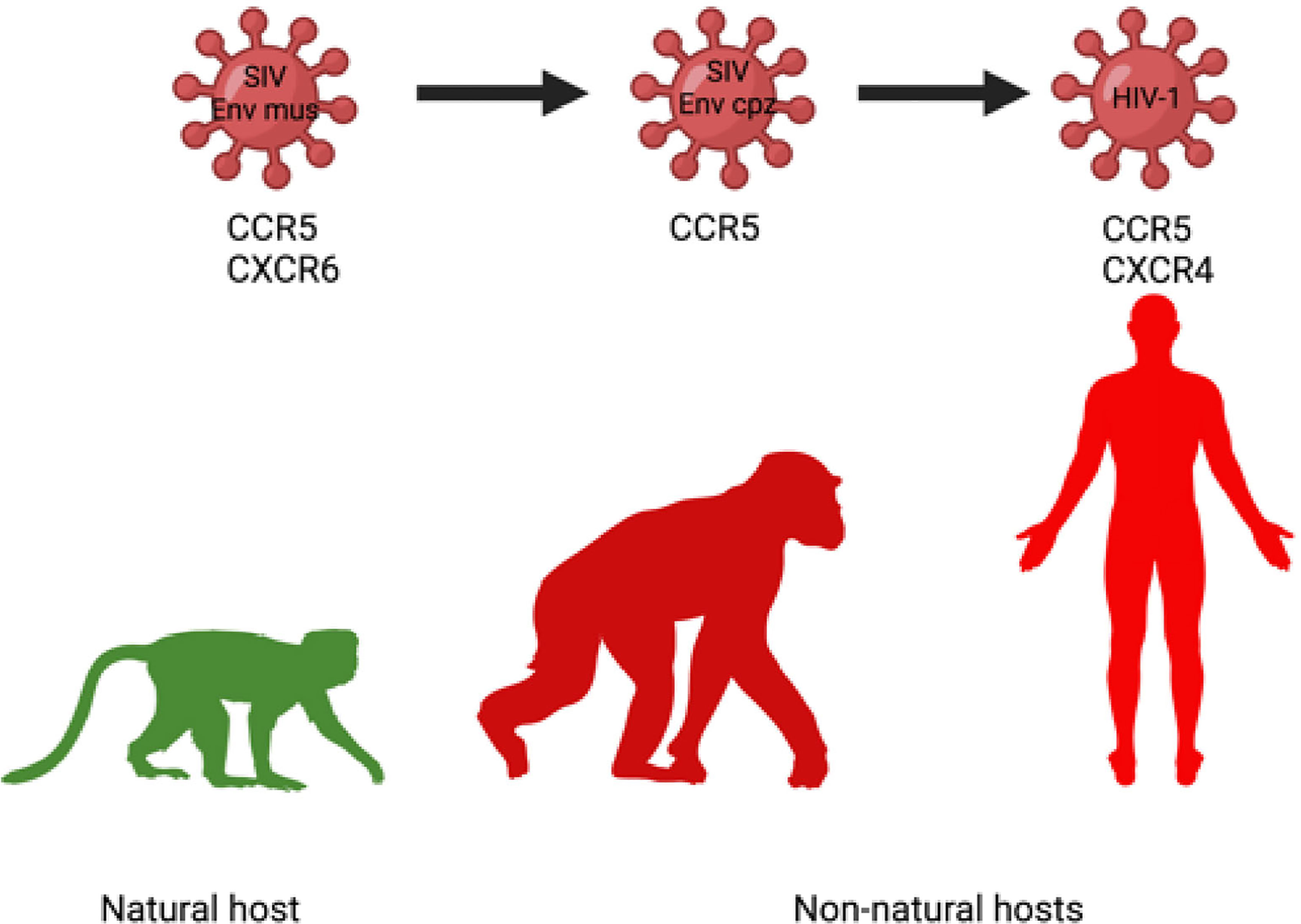
Figure 3 Change of coreceptor tropism (loss of CXCR6 usage) in the SIVcpz/HIV-1 lineage may contribute to increased use of CCR5 as an entry coreceptor, more widespread infection of target cells and enhanced pathogenicity of SIVcpz/HIV-1 in chimpanzee and human, respectively (187).
Note, however, that the main limitation of this hypothesis that SIV pathogenicity in different species is dependent on the coreceptor usage is that SIVagm and SIVsmm can actually be highly pathogenic in macaques upon direct cross-species transmission (137, 191). In macaques, both SIVsmm and SIVsab target preferentially memory cells and not naïve cells (137, 191, 192). Therefore, in order to confirm this theory, comparative pathogenesis studies in the AGMs/SMs versus macaques should be carried out to show that the viruses restrict coreceptor usage to exclusively CCR5 upon cross-species transmission.
Thus, the SIV coreceptor expansion may enable the viruses to circumvent the lowered CCR5 expression in the natural hosts, making SIV infection in natural hosts less dependent on the use of a CCR5 coreceptor (193). In more recent hosts, high and unaltered CCR5 expression appears indispensable for HIV-1 infection in humans and SIVmac infection in RMs (33–35, 84). Differential cell targeting between natural and non-natural hosts could contribute to different outcomes between infection in natural and non-natural hosts (194), but in vivo studies are needed to confirm these in vitro observations and infection of naïve cells in natural hosts of SIVs.
A more promiscuous coreceptor usage allows the virus to expand their spectrum of target cells. These new target cell subsets are less essential, with short life span, and their infection has minimal impact on immune health. This can explain why low levels of CCR5 expression on CD4+ T cells in natural hosts does not reduce the infection of the CD4+ T cells, permit high viremia, and result in infection of cells with short lifespan in natural host in comparison to non-natural host. Restricted expression of CCR5 coreceptor may thus protect essential cells from infection (in particular memory CD4+ T-cells) and preserve immune homeostasis. Note, however, that, in pathogenic HIV infections, high pathogenicity and disease progression is associated with an expansion in coreceptor usage rather than a more restrictive coreceptor use.
The impact of natural CCR5 loss-of-function mutations and the phenomenon of target cell restriction via downregulating cell surface CCR5 expression on preventing infection or minimizing SIV/HIV pathogenesis pointed to the central role of CCR5 in the process of natural protection against SIV/HIV. It led to development of therapeutic approaches to inactivate or block the function of CCR5 gene or protein, such as chemical or antibody-based blocking of CCR5 receptors, or generation of CCR5 cells which are deficient or downregulate CCR5.
CCR5 blockers mimic the effects of naturally occurring CCR5-Δ24 mutation, at least in part, with respect to inhibiting HIV-1 utilization of cell surface CCR5 for cell entry.
Maraviroc (MVC), a nonpeptidic small molecule, causing a pharmacological blockade of CCR5 signaling, was the first CCR5 antagonist approved by FDA for the treatment of patients infected with R5-using HIV-1 virus (195). MVC blocks binding of viral envelope, gp120, to CCR5 to prevent the membrane fusion and viral entry, through allosteric inhibition (i.e. without occupying the binding site for chemokines and the HIV envelope glycoprotein gp120) and without affecting CCR5 cell surface levels or associated intracellular signaling (196–198). Maraviroc administration, originally devised for HIV treatment, may have applications beyond HIV/AIDS. Rodent models either treated with maraviroc or with CCR5 knockout showed an induced recovery after traumatic brain injury (30), however, maraviroc blockade of infant macaques only marginally impacted the rate of oral SIVmac transmission (163).
Leronlimab, a CCR5-blocking monoclonal antibody, binds to the external domains of CCR5 and through a competitive mechanism prevents HIV and SIV from binding to the CCR5 coreceptor, entering the cell and replication (199, 200). Beside the CCR5 masking from SIV/HIV, leronlimab CCR5 binding does not downregulate CCR5 expression or deplete CCR5-expressing cells (199), but prevents CCL-5-induced activation and migration of inflammatory CCR5-expressing monocytes and T lymphocytes along a chemical gradient (199). In this context, leronlimab appears a an excellent prospect for treatment of diseases, in which the CCL5-CCR5 pathway is involved in the pathogenesis. Given the role of CCR5 in immune cell migration and inflammation, CCR5 blockade with leronlimab was applied in critical COVID-19 patients, and led to the reduction of the IL-6 levels, restoration of CD4/CD8 ratio, and resolution of SARS-CoV-2 burden, thus implicating CCR5 as a potential therapeutic target for COVID-19 (201).
Antibody conjugates (ACs) comprising of an antibody carrier and small molecule CCR-5 antagonist were developed to enhance the CCR5-dependent therapies, specifically, to increase their clinical effects, reduce off-target effect and toxicity, and extend the pharmacokinetic profile of the attached molecule (202). To increase the potency of CCR5 targeted therapies, anti-CCR5 monoclonal antibodies were conjugated with a CCR5 small molecule antagonist, targeting nonoverlapping epitopes (203) and with a fusion inhibitor (204). The neutralization activity of CCR5 antagonists, such as MVC, can be further extended by formulating them with long-lived carriers - chemically programmed antibodies (cpAbs) and PEGylated derivatives. Such compounds containing MVC had significantly extended pharmacokinetic profiles (205).
Sustained remission of HIV infection was achieved using stem-cell transplantation from donors homozygous for CCR5 null allele Δ32/Δ32, lacking functional expression of the CCR5 coreceptor and showing HIV resistance in two patients treated for acute myelogenous leukemia (Berlin patient) and refractory Hodgkin lymphoma (London patient) (206, 207). The Berlin patient received two rounds of total body irradiation and allogeneic hematopoietic stem-cell transplantation (allo-HSCT) and the London patient underwent one HSCT procedure.
The Berlin patient achieved long-term post treatment control of HIV (206, 208, 209) and the London Patient has been in HIV-1 remission for at least 30 months with no detectable replication-competent virus in blood, CSF, intestinal tissue, or lymphoid tissue. Both these cases potentially represent cases of HIV-1 cure (207, 210–212).
While the allogenic transplantation of CCR5 deficient cells demonstrated a feasibility of cure, finding HLA-matched donors with naturally occurring homozygous CCR5 deletions is a limiting factor of this approach. Therefore, various genomic manipulations have been attempted to disrupt CCR5 function.
Gene editing and silencing technologies have been implemented to block the CCR5 function, including (i) modifications of naturally existing anti-HIV-1 ribozymes (108), (ii) gene silencing using RNA interference to suppress CCR5 (110), (iii) programmable nucleases, such as zinc-finger transcription activator-like effector nucleases (TALENs) and (iv) clustered regularly interspaced short palindromic repeat (CRISPR)-Cas (113), and engineered zinc-finger nucleases (ZFNs) (107, 112, 213, 214).
Editing the CCR5 gene via CRISPR-Cas9 technology was also applied to genetically modify Mauritian cynomolgus macaque embryos as the foundation for developing a model system of SIV resistance for studying SIV/HIV disease and development of therapies. Through this technology, a disrupted gene CCR5, containing homozygous deletions in CCR5 (including a 24-bp deletion region, which does not occur spontaneously in macaques) was introduced into macaque embryos and edited cells (115).
Germline editing using CRISPR-Cas9 technology was also used to introduce a null genotype of CCR5 in human embryos from an HIV discordant couple, which is similar, yet not identical to CCR5-Δ32. Twin girls with this genetic alteration were born in 2018. While the intention behind the germline inactivation of CCR5 in the human embryo appears to be protection from HIV infection in later life, this intervention evoked questions regarding the necessity of such permanent gene inactivation, while other preventive methods are available. It also evoked discussion about readiness of germline editing technology (its safety and control of target off effect) and still limited knowledge of pleiotropic function of immune genes in health, and therefore difficulty to precisely predict the effects of the introduced alterations (215).
The pleiotropic role of CCR5, which makes this chemokine receptor a promising target for therapies of various diseases, needs to be closely studied in relation to potential undesirable effects of CCR5-targeted therapies (the receptor blockage or disruption). While they can prevent the cell-to-cell spread of HIV/SIV and reduce chronic T-cell immune activation and inflammation, the inactivation of natural CCR5 function may have various unintended consequences. For example, there is increasing evidence on the prominent role of CCR5 in the differentiation, activation and migration of the CD8+ T cells to the sites of inflammation (216, 217), and these processes are impaired by CCR5 deficiency or blockade (218, 219). CCR5 is also highly expressed in virus-specific CD8+ T cells during various viral infections, including HIV-1, suggesting a role of CCR5 in the CD8+ T cell responses to viral infections (216, 220–222). CCR5-expressing CD8+ T cells display an effector memory phenotype, age-related increase in rhesus macaques, and a marked reduction during the progression of SIV disease (223). MVC treatment reduced the in vitro activation of CD8+ T-cells from SIV-infected macaques (223). This effect could be beneficial as it may reduce disease-related chronic immune inflammation; on the other hand, it may limit the CD8+ T-cell responses to the virus, and potentially increase a risk of the virus latency (223). The complex role of CCR5 in immune health highlights the need for studies of the CCR5-directed therapies on CD8+ T cell and immune health in general.
CCR5 is central to HIV pathogenesis. Targeting this receptor was successfully used as an antiretroviral therapy and could be successfully expanded to either other infections or to medical areas which are unrelated to infectious diseases. Natural hosts of SIVs adapted, over an evolutionary history of millennia, to counter SIV infection by limiting the expression of the CCR5 receptor on the target cells. Meanwhile, viruses naturally infecting natural hosts of SIVs found escape routes to counter replication restrictions due to low CCR5 expression. As such, the CCR5-SIV relation represents a perfect illustration of the red queen principle which proposes that species must constantly adapt, evolve and proliferate in order to survive in contact with the opposed species (224). “Now, here, you see, it takes all the running you can do, to keep in the same place.” (225). This remarkable ability of the SIVs and their species-specific hosts to continuously adapt calls for a careful evaluation of the cure approaches targeting CCR5 expression.
AJJ, IP, and CA designed, wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
IP and CA are supported by grants from the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases/National Heart, Lung and Blood Institute/National Institute of Allergy and Infectious Diseases: RO1 HL117715 (IP), R01 HL123096 (IP), R01 HL154862 (IP), R01 DK130481 (IP), R01 DK113919 (IP/CA), R01 DK119936 (CA), R01 DK131476 (CA), R01 AI119346 (CA). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Figures 2 and 3 were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Viola A, Luster AD. Chemokines and Their Receptors: Drug Targets in Immunity and Inflammation. Annu Rev Pharmacol Toxicol (2008) 48:171–97. doi: 10.1146/annurev.pharmtox.48.121806.154841
2. Jiao X, Velasco-Velázquez MA, Wang M, Li Z, Rui H, Peck AR, et al. CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion. Cancer Res (2018) 78:1657–71. doi: 10.1158/0008-5472.CAN-17-0915
3. Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D, et al. Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res (2019) 79:4801–7. doi: 10.1158/0008-5472.can-19-1167
4. Eri R, Jonsson JR, Pandeya N, Purdie DM, Clouston AD, Martin N, et al. CCR5-Δ32 Mutation Is Strongly Associated With Primary Sclerosing Cholangitis. Genes Immun (2004) 5:444–50. doi: 10.1038/sj.gene.6364113
5. Pokorny V, McQueen F, Yeoman S, Merriman M, Merriman A, Harrison A, et al. Evidence for Negative Association of the Chemokine Receptor CCR5 D32 Polymorphism With Rheumatoid Arthritis. Ann Rheum Dis (2005) 64:487–90. doi: 10.1136/ard.2004.023333
6. Prahalad S. Negative Association Between the Chemokine Receptor CCR5-Δ32 Polymorphism and Rheumatoid Arthritis: A Meta-Analysis. Genes Immun (2006) 7:264–8. doi: 10.1038/sj.gene.6364298
7. Hall IP, Wheatley A, Christie G, McDougall C, Hubbard R, Helms PJ. Association of CCR5 ▵32 With Reduced Risk of Asthma. Lancet (1999) 354:1264–5. doi: 10.1016/s0140-6736(99)03425-x
8. Berce V, Repnik K, Potocnik U. Association of CCR5-Delta32 Mutation With Reduced Risk of Nonatopic Asthma in Slovenian Children. J Asthma (2008) 45:780–4. doi: 10.1080/02770900802386024
9. Moreira AP, Cavassani KA, Massafera Tristão FS, Campanelli AP, Martinez R, Rossi MA, et al. CCR5-Dependent Regulatory T Cell Migration Mediates Fungal Survival and Severe Immunosuppression. J Immunol (2008) 180:3049–56. doi: 10.4049/jimmunol.180.5.3049
10. Luangsay S, Kasper LH, Rachinel N, Minns LA, Mennechet FJD, Vandewalle A, et al. CCR5 Mediates Specific Migration of Toxoplasma Gondii—Primed CD8+ Lymphocytes to Inflammatory Intestinal Epithelial Cells. Gastroenterology (2003) 125:491–500. doi: 10.1016/S0016-5085(03)00903-X
11. Oppermann M. Chemokine Receptor CCR5: Insights Into Structure, Function, and Regulation. Cell Signal (2004) 16:1201–10. doi: 10.1016/j.cellsig.2004.04.007
12. Zhou M, Greenhill S, Huang S, Silva TK, Sano Y, Wu S, et al. CCR5 Is a Suppressor for Cortical Plasticity and Hippocampal Learning and Memory. Elife (2016) 5. doi: 10.7554/eLife.20985
13. Zhang H, Chen K, Tan Q, Shao Q, Han S, Zhang C, et al. Structural Basis for Chemokine Recognition and Receptor Activation of Chemokine Receptor CCR5. Nat Commun (2021) 12:4151. doi: 10.1038/s41467-021-24438-5
14. Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Molecular Cloning and Functional Expression of a New Human CC-Chemokine Receptor Gene. Biochemistry (1996) 35:3362–7. doi: 10.1021/bi952950g
15. Raport CJ, Gosling J, Schweickart VL, Gray PW, Charo IF. Molecular Cloning and Functional Characterization of a Novel Human CC Chemokine Receptor (CCR5) for RANTES, MIP-1beta, and MIP-1alpha. J Biol Chem (1996) 271:17161–6. doi: 10.1074/jbc.271.29.17161
16. Combadiere C, Ahuja SK, Tiffany HL, Murphy PM. Cloning and Functional Expression of CC CKR5, a Human Monocyte CC Chemokine Receptor Selective for MIP-1(Alpha), MIP-1(Beta), and RANTES. J Leukoc Biol (1996) 60:147–52. doi: 10.1002/jlb.60.1.147
17. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 Entry Cofactor: Functional cDNA Cloning of a Seven-Transmembrane, G Protein-Coupled Receptor. Science (1996) 272:872–7. doi: 10.1126/science.272.5263.872
18. Miyakawa T, Obaru K, Maeda K, Harada S, Mitsuya H. Identification of Amino Acid Residues Critical for LD78β, a Variant of Human Macrophage Inflammatory Protein-1α, Binding to CCR5 and Inhibition of R5 Human Immunodeficiency Virus Type 1 Replication. J Biol Chem (2002) 277:4649–55. doi: 10.1074/jbc.m109198200
19. Blanpain C, Migeotte I, Lee B, Vakili J, Doranz BJ, Govaerts C, et al. CCR5 Binds Multiple CC-Chemokines: MCP-3 Acts as a Natural Antagonist. Blood (1999) 94:1899–905. doi: 10.1182/blood.V94.6.1899.418k31_1899_1905
20. Zhao J, Ma L, Wu YL, Wang P, Hu W, Pei G. Chemokine Receptor CCR5 Functionally Couples to Inhibitory G Proteins and Undergoes Desensitization. J Cell Biochem (1998) 71:36–45. doi: 10.1002/(SICI)1097-4644(19981001)71:1<36::AID-JCB4>3.0.CO;2-2
21. Mack M, Luckow B, Nelson PJ, Cihak J, Simmons G, Clapham PR, et al. Aminooxypentane-RANTES Induces CCR5 Internalization But Inhibits Recycling: A Novel Inhibitory Mechanism of HIV Infectivity. J Exp Med (1998) 187:1215–24. doi: 10.1084/jem.187.8.1215
22. Venuti A, Pastori C, Siracusano G, Pennisi R, Riva A, Tommasino M, et al. The Abrogation of Phosphorylation Plays a Relevant Role in the CCR5 Signalosome Formation With Natural Antibodies to CCR5. Viruses (2017) 10(1):9. doi: 10.3390/v10010009
23. Signoret N, Pelchen-Matthews A, Mack M, Proudfoot AE, Marsh M. Endocytosis and Recycling of the HIV Coreceptor CCR5. J Cell Biol (2000) 151:1281–94. doi: 10.1083/jcb.151.6.1281
24. Flegler AJ, Cianci GC, Hope TJ. CCR5 Conformations Are Dynamic and Modulated by Localization, Trafficking and G Protein Association. PloS One (2014) 9:e89056. doi: 10.1371/journal.pone.0089056
25. Rottman JB, Ganley KP, Williams K, Wu L, Mackay CR, Ringler DJ. Cellular Localization of the Chemokine Receptor CCR5. Correlation to Cellular Targets of HIV-1 Infection. Am J Pathol (1997) 151:1341–51.
26. Khan IA, Thomas SY, Moretto MM, Lee FS, Islam SA, Combe C, et al. CCR5 Is Essential for NK Cell Trafficking and Host Survival Following Toxoplasma Gondii Infection. PloS Pathog (2006) 2:e49. doi: 10.1371/journal.ppat.0020049
27. Weiss ID, Shoham H, Wald O, Wald H, Beider K, Abraham M, et al. Ccr5 Deficiency Regulates the Proliferation and Trafficking of Natural Killer Cells Under Physiological Conditions. Cytokine (2011) 54:249–57. doi: 10.1016/j.cyto.2011.01.011
28. Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 Signaling Suppresses Inflammation and Reduces Adverse Remodeling of the Infarcted Heart, Mediating Recruitment of Regulatory T Cells. Am J Pathol (2010) 176:2177–87. doi: 10.2353/ajpath.2010.090759
29. Tan MCB, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-Dependent Homing of Regulatory T Cells Inhibits Tumor Growth in a Murine Model of Pancreatic Cancer. J Immunol (2009) 182:1746–55. doi: 10.4049/jimmunol.182.3.1746
30. Joy MT, Ben Assayag E, Shabashov-Stone D, Liraz-Zaltsman S, Mazzitelli J, Arenas M, et al. CCR5 Is a Therapeutic Target for Recovery After Stroke and Traumatic Brain Injury. Cell (2019) 176(5):1143–57.e13. doi: 10.1016/j.cell.2019.01.044
31. Sehnal D, Bittrich S, Deshpande M, Svobodová R, Berka K, Bazgier V, et al. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res (2021) 49:W431–7. doi: 10.1093/nar/gkab314
32. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Research (2000) 28(1):235–42. doi: 10.1093/nar/28.1.235
33. Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, et al. Homozygous Defect in HIV-1 Coreceptor Accounts for Resistance of Some Multiply-Exposed Individuals to HIV-1 Infection. Cell (1996) 86:367–77. doi: 10.1016/S0092-8674(00)80110-5
34. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 Infection in Caucasian Individuals Bearing Mutant Alleles of the CCR-5 Chemokine Receptor Gene. Nature (1996) 382:722–5. doi: 10.1038/382722a0
35. Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, et al. Genetic Restriction of HIV-1 Infection and Progression to AIDS by a Deletion Allele of the CKR5 Structural Gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science (1996) 273:1856–62. doi: 10.1126/science.273.5283.1856
36. Stephens JC, Reich DE, Goldstein DB, Shin HD, Smith MW, Carrington M, et al. Dating the Origin of the CCR5-Δ32 AIDS-Resistance Allele by the Coalescence of Haplotypes. Am J Hum Genet (1998) 62:1507–15. doi: 10.1086/301867
37. Libert F. The Deltaccr5 Mutation Conferring Protection Against HIV-1 in Caucasian Populations Has a Single and Recent Origin in Northeastern Europe. Hum Mol Genet (1998) 7:399–406. doi: 10.1093/hmg/7.3.399
38. Solloch UV, Lang K, Lange V, Böhme I, Schmidt AH, Sauter J. Frequencies of Gene Variant CCR5-Δ32 in 87 Countries Based on Next-Generation Sequencing of 1.3 Million Individuals Sampled From 3 National DKMS Donor Centers. Hum Immunol (2017) 78:710–7. doi: 10.1016/j.humimm.2017.10.001
39. Klitz W, Brautbar C, Schito AM, Barcellos LF, Oksenberg JR. Evolution of the CCR5 δ32 Mutation Based on Haplotype Variation in Jewish and Northern European Population Samples. Hum Immunol (2001) 62:530–8. doi: 10.1016/S0198-8859(01)00239-7
40. Sabeti PC, Walsh E, Schaffner SF, Varilly P, Fry B, Hutcheson HB, et al. he Case for Selection at CCR5-Δ32. Plos Biol (2005) 3(11):e378. doi: 10.1371/journal.pbio.0030378
41. Lucotte G, Mercier G. Distribution of the CCR5 Gene 32-Bp Deletion in Europe. J Acquir Immune Defic Syndr Hum Retrovirol (1998) 19:174–7. doi: 10.1097/00042560-199810010-00011
42. Martinson JJ, Chapman NH, Rees DC, Liu YT, Clegg JB. Global Distribution of the CCR5 Gene 32-Basepair Deletion. Nat Genet (1997) 16:100–3. doi: 10.1038/ng0597-100
43. Novembre J, Galvani AP, Slatkin M. The Geographic Spread of the CCR5 Δ32 HIV-Resistance Allele. PloS Biol (2005) 3:e339. doi: 10.1371/journal.pbio.0030339
44. Duncan SR, Scott S, Duncan CJ. Reappraisal of the Historical Selective Pressures for the CCR5-Δ32 Mutation. J Med Genet (2005) 42:205–8. doi: 10.1136/jmg.2004.025346
45. Galvani AP, Novembre J. The Evolutionary History of the CCR5-Delta32 HIV-Resistance Mutation. Microbes Infect (2005) 7:302–9. doi: 10.1016/j.micinf.2004.12.006
46. Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, et al. Chimpanzee Reservoirs of Pandemic and Nonpandemic HIV-1. Science (2006) 313:523–6. doi: 10.1126/science.1126531
47. CDC. Pneumocystis Pneumonia Los Angeles. MMWR {CENTERS FOR Dis CONTROL} (1981) 30:250–2. Available at: https://www.cdc.gov/mmwr/preview/mmwrhtml/june_5.htm.
48. WHO. HIV/AIDS in Europe: Moving From Death Sentence to Chronic Disease Management. Geneva: WHO {World Health Organization. Regional Office for Europe} (2006).
49. Gao F, Yue L, Robertson DL, Hill SC, Hui H, Biggar RJ, et al. Genetic Diversity of Human Immunodeficiency Virus Type 2: Evidence for Distinct Sequence Subtypes With Differences in Virus Biology. J Virol (1994) 68:7433–47. doi: 10.1128/jvi.68.11.7433-7447.1994
50. Chen Z, Gettie A, Ho DD, Marx PA. Primary SIVsm Isolates Use the CCR5 Coreceptor From Sooty Mangabeys Naturally Infected in West Africa: A Comparison of Coreceptor Usage of Primary SIVsm, HIV-2, and SIVmac. Virology (1998) 246:113–24. doi: 10.1006/viro.1998.9174
51. Damond F, Apetrei C, Robertson DL, Souquière S, Leprêtre A, Matheron S, et al. Variability of Human Immunodeficiency Virus Type 2 (Hiv-2) Infecting Patients Living in France. Virology (2001) 280:19–30. doi: 10.1006/viro.2000.0685
52. Damond F, Worobey M, Campa P, Farfara I, Colin G, Matheron S, et al. Identification of a Highly Divergent HIV Type 2 and Proposal for a Change in HIV Type 2 Classification. AIDS Res Hum Retroviruses (2004) 20:666–72. doi: 10.1089/0889222041217392
53. Lemey P, Pybus OG, Wang B, Saksena NK, Salemi M, Vandamme A-M. Tracing the Origin and History of the HIV-2 Epidemic. Proc Natl Acad Sci USA (2003) 100:6588–92. doi: 10.1073/pnas.0936469100
54. Bi Y, Du Z, Han Y, Guo Z, Tan Y, Zhu Z, et al. Yersinia Pestis and Host Macrophages: Immunodeficiency of Mouse Macrophages Induced by YscW. Immunology (2009) 128:e406–17. doi: 10.1111/j.1365-2567.2008.02990.x
55. Elvin SJ, Williamson ED, Scott JC, Smith JN, de Lema GP, Chilla S, et al. Ambiguous Role of CCR5 in Y. Pestis Infection. Nature (2004) 430:418–8. doi: 10.1038/nature02822
56. Mecsas J, Franklin G, Kuziel WA, Brubaker RR, Falkow S, Mosier DE. Evolutionary Genetics: CCR5 Mutation and Plague Protection. Nature (2004) 427:606. doi: 10.1038/427606a
57. Galvani AP, Slatkin M. Evaluating Plague and Smallpox as Historical Selective Pressures for the CCR5-Δ32 HIV-Resistance Allele. Proc Natl Acad Sci USA (2003) 100:15276–9. doi: 10.1073/pnas.2435085100
58. Faure E, Royer-Carenzi M. Is the European Spatial Distribution of the HIV-1-Resistant CCR5-Δ32 Allele Formed by a Breakdown of the Pathocenosis Due to the Historical Roman Expansion? Infect Genet Evol (2008) 8:864–74. doi: 10.1016/j.meegid.2008.08.007
59. Riddick NE, Hermann EA, Loftin LM, Elliott ST, Wey WC, Cervasi B, et al. A Novel CCR5 Mutation Common in Sooty Mangabeys Reveals SIVsmm Infection of CCR5-Null Natural Hosts and Efficient Alternative Coreceptor Use In Vivo. PloS Pathog (2010) 6:e1001064. doi: 10.1371/journal.ppat.1001064
60. Chen Z, Kwon D, Jin Z, Monard S, Telfer P, Jones MS, et al. Natural Infection of a Homozygous Delta24 CCR5 Red-Capped Mangabey With an R2b-Tropic Simian Immunodeficiency Virus. J Exp Med (1998) 188:2057–65. doi: 10.1084/jem.188.11.2057
61. Beer BE, Foley BT, Kuiken CL, Tooze Z, Goeken RM, Brown CR, et al. Characterization of Novel Simian Immunodeficiency Viruses From Red-Capped Mangabeys From Nigeria (SIVrcmNG409 and -NG411). J Virol (2001) 75:12014–27. doi: 10.1128/jvi.75.24.12014-12027.2001
62. Maisels F, Oates JF, Linder J, Ikemeh R, Imong I, Etiendem D. Cercocebus Torquatus, IUCN. IUCN Red List of Threatened Species. (2019). doi: 10.2305/iucn.uk.2019-1.rlts.t4201a17955626.en
63. Koné I, McGraw S, Gonedelé Bi S, Barrie A. Cercocebus Atys, IUCN. IUCN Red List of Threatened Species. (2019). doi: 10.2305/iucn.uk.2020-2.rlts.t136933a92248451.en
64. Kumar S, Stecher G, Suleski M, Hedges SB. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol (2017) 34:1812–9. doi: 10.1093/molbev/msx116
65. Worobey M, Telfer P, Souquière S, Hunter M, Coleman CA, Metzger MJ, et al. Island Biogeography Reveals the Deep History of SIV. Science (2010) 329:1487. doi: 10.1126/science.1193550
66. Ma D, Jasinska A, Kristoff J, Grobler JP, Turner T, Jung Y, et al. SIVagm Infection in Wild African Green Monkeys From South Africa: Epidemiology, Natural History, and Evolutionary Considerations. PloS Pathog (2013) 9:e1003011. doi: 10.1371/journal.ppat.1003011
67. Zhang YJ, Moore JP. Will Multiple Coreceptors Need to be Targeted by Inhibitors of Human Immunodeficiency Virus Type 1 Entry? J Virol (1999) 73:3443–8. doi: 10.1128/JVI.73.4.3443-3448.1999
68. Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in Coreceptor Use Correlates With Disease Progression in HIV-1–Infected Individuals. J Exp Med (1997) 185:621–8. doi: 10.1084/jem.185.4.621
69. Scarlatti G, Tresoldi E, Björndal A, Fredriksson R, Colognesi C, Deng HK, et al. In Vivo Evolution of HIV-1 Co-Receptor Usage and Sensitivity to Chemokine-Mediated Suppression. Nat Med (1997) 3:1259–65. doi: 10.1038/nm1197-1259
70. van Rij RP, Blaak H, Visser JA, Brouwer M, Rientsma R, Broersen S, et al. Differential Coreceptor Expression Allows for Independent Evolution of Non-Syncytium-Inducing and Syncytium-Inducing HIV-1. J Clin Invest (2000) 106:1039–52. doi: 10.1172/JCI7953
71. Blaak H, Boers PHM, Gruters RA, Schuitemaker H, van der Ende ME, Osterhaus ADME. CCR5, GPR15, and CXCR6 Are Major Coreceptors of Human Immunodeficiency Virus Type 2 Variants Isolated From Individuals With and Without Plasma Viremia. J Virol (2005) 79:1686–700. doi: 10.1128/JVI.79.3.1686-1700.2005
72. Reynes J, Portales P, Segondy M, Baillat V, André P, Réant B, et al. CD4+ T Cell Surface CCR5 Density as a Determining Factor of Virus Load in Persons Infected With Human Immunodeficiency Virus Type 1. J Infect Dis (2000) 181:927–32. doi: 10.1086/315315
73. Lelièvre J-D, Petit F, Perrin L, Mammano F, Arnoult D, Ameisen J-C, et al. The Density of Coreceptors at the Surface of CD4+ T Cells Contributes to the Extent of Human Immunodeficiency Virus Type 1 Viral Replication-Mediated T Cell Death. AIDS Res Hum Retroviruses (2004) 20:1230–43. doi: 10.1089/0889222042545045
74. Reynes J, Portales P, Segondy M, Baillat V, André P, Avinens O, et al. CD4 T Cell Surface CCR5 Density as a Host Factor in HIV-1 Disease Progression. AIDS (2001) 15:1627–34. doi: 10.1097/00002030-200109070-00004
75. Lin Y-L, Mettling C, Portales P, Reynes J, Clot J, Corbeau P. Cell Surface CCR5 Density Determines the Postentry Efficiency of R5 HIV-1 Infection. Proc Natl Acad Sci USA (2002) 99:15590–5. doi: 10.1073/pnas.242134499
76. Shaik MM, Peng H, Lu J, Rits-Volloch S, Xu C, Liao M, et al. Structural Basis of Coreceptor Recognition by HIV-1 Envelope Spike. Nature (2019) 565:318–23. doi: 10.1038/s41586-018-0804-9
77. Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 Alpha, and MIP-1 Beta as the Major HIV-Suppressive Factors Produced by CD8+ T Cells. Science (1995) 270:1811–5. doi: 10.1126/science.270.5243.1811
78. Gong W, Howard OM, Turpin JA, Grimm MC, Ueda H, Gray PW, et al. Monocyte Chemotactic Protein-2 Activates CCR5 and Blocks CD4/CCR5-Mediated HIV-1 Entry/Replication. J Biol Chem (1998) 273:4289–92. doi: 10.1074/jbc.273.8.4289
79. Blanpain C, Doranz BJ, Bondue A, Govaerts C, De Leener A, Vassart G, et al. The Core Domain of Chemokines Binds CCR5 Extracellular Domains While Their Amino Terminus Interacts With the Transmembrane Helix Bundle. J Biol Chem (2003) 278:5179–87. doi: 10.1074/jbc.M205684200
80. Lopalco L. CCR5: From Natural Resistance to a New Anti-HIV Strategy. Viruses (2010) 2:574–600. doi: 10.3390/v2020574
81. Blanpain C, Vanderwinden J-M, Cihak J, Wittamer V, Le Poul E, Issafras H, et al. Multiple Active States and Oligomerization of CCR5 Revealed by Functional Properties of Monoclonal Antibodies. Mol Biol Cell (2002) 13:723–37. doi: 10.1091/mbc.01-03-0129
82. Colin P, Bénureau Y, Staropoli I, Wang Y, Gonzalez N, Alcami J, et al. HIV-1 Exploits CCR5 Conformational Heterogeneity to Escape Inhibition by Chemokines. Proc Natl Acad Sci USA (2013) 110:9475–80. doi: 10.1073/pnas.1222205110
83. Ansari-Lari MA, Ali Ansari-Lari M, Liu X-M, Metzker ML, Rut AR, Gibbs RA. The Extent of Genetic Variation in the CCR5 Gene. Nat Genet (1997) 16:221–2. doi: 10.1038/ng0797-221
84. Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T, et al. The Role of a Mutant CCR5 Allele in HIV–1 Transmission and Disease Progression. Nat Med (1996) 2:1240–3. doi: 10.1038/nm1196-1240
85. Michael NL, Louie LG, Rohrbaugh AL, Schultz KA, Dayhoff DE, Wang CE, et al. The Role of CCR5 and CCR2 Polymorphisms in HIV-1 Transmission and Disease Progression. Nat Med (1997) 3:1160–2. doi: 10.1038/nm1097-1160
86. O’Brien TR, Winkler C, Dean M, Nelson JAE, Carrington M, Michael NL, et al. HIV-1 Infection in a Man Homozygous for CCR5▵ 32. Lancet (1997) 349:1219. doi: 10.1016/S0140-6736(97)24017-1
87. Biti R, Ffrench R, Young J, Bennetts B, Stewart G, Liang T. HIV-1 Infection in an Individual Homozygous for the CCR5 Deletion Allele. Nat Med (1997) 3:252–3. doi: 10.1038/nm0397-252
88. Theodorou L, Meyer L, Magierowska M, Katlama C, Rouzioux C. HIV-1 Infection in an Individual Homozygous for CCR5▵32. Lancet (1997) 349:1219–20. doi: 10.1016/s0140-6736(05)62411-7
89. Eugen-Olsen J, Iversen AK, Garred P, Koppelhus U, Pedersen C, Benfield TL, et al. Heterozygosity for a Deletion in the CKR-5 Gene Leads to Prolonged AIDS-Free Survival and Slower CD4 T-Cell Decline in a Cohort of HIV-Seropositive Individuals. AIDS (1997) 11:305–10. doi: 10.1097/00002030-199703110-00007
90. Laurichesse JJ, Persoz A, Theodorou I, Rouzioux C, Delfraissy JF, Meyer L. Improved Virological Response to Highly Active Antiretroviral Therapy in HIV-1-Infected Patients Carrying the CCR5 Delta32 Deletion. HIV Med (2007) 8:213–9. doi: 10.1111/j.1468-1293.2007.00455.x
91. Marmor M, Sheppard HW, Donnell D, Bozeman S, Celum C, Buchbinder S, et al. Homozygous and Heterozygous CCR5-Δ32 Genotypes Are Associated With Resistance to HIV Infection. J Acquired Immune Deficiency Syndromes (2001) 27:472–81. doi: 10.1097/00042560-200108150-00009
92. Liu S, Kong C, Wu J, Ying H, Zhu H. Effect of CCR5-Δ32 Heterozygosity on HIV-1 Susceptibility: A Meta-Analysis. PloS One (2012) 7:e35020. doi: 10.1371/journal.pone.0035020
93. Rabkin CS, Yang Q-E, Goedert JJ, Nguyen G, Mitsuya H, Sei S. Chemokine and Chemokine Receptor Gene Variants and Risk of Non-Hodgkin’s Lymphoma in Human Immunodeficiency Virus-1–Infected Individuals. Blood J Am Soc Hematol (1999) 93:1838–42. doi: 10.1182/blood.V93.6.1838.406k39_1838_1842
94. Dean M, Jacobson LP, McFarlane G, Margolick JB, Jenkins FJ, Zack Howard OM, et al. Reduced Risk of AIDS Lymphoma in Individuals Heterozygous for the CCR5-Δ32 Mutation. Cancer Res (1999) 59:3561–4.
95. Morawetz RA, Rizzardi GP, Glauser D, Rutschmann O, Hirschel B, Perrin L, et al. Genetic Polymorphism of CCR5 Gene and HIV Disease: The Heterozygous (CCR5/delta Ccr5) Genotype Is Neither Essential Nor Sufficient for Protection Against Disease Progression. Swiss HIV Cohort. Eur J Immunol (1997) 27:3223–7. doi: 10.1002/eji.1830271220
96. International HIV Controllers Study, Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PIW, et al. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science (2010) 330:1551–7. doi: 10.1126/science.1195271
97. Wu L, Paxton WA, Kassam N, Ruffing N, Rottman JB, Sullivan N, et al. CCR5 Levels and Expression Pattern Correlate With Infectability by Macrophage-Tropic HIV-1. Vitro J Exp Med (1997) 185:1681–91. doi: 10.1084/jem.185.9.1681
98. Benkirane M, Jin D-Y, Chun RF, Koup RA, Jeang K-T. Mechanism of Transdominant Inhibition of CCR5-Mediated HIV-1 Infection by Ccr5Δ32. J Biol Chem (1997) 272:30603–6. doi: 10.1074/jbc.272.49.30603
99. Martin MP, Dean M, Smith MW, Winkler C, Gerrard B, Michael NL, et al. Genetic Acceleration of AIDS Progression by a Promoter Variant of CCR5. Science (1998) 282:1907–11. doi: 10.1126/science.282.5395.1907
100. Catano G, Chykarenko ZA, Mangano A, Anaya J-M, He W, Smith A, et al. Concordance of CCR5 Genotypes That Influence Cell-Mediated Immunity and HIV-1 Disease Progression Rates. J Infect Dis (2011) 203:263–72. doi: 10.1093/infdis/jiq023
101. Mummidi S, Bamshad M, Ahuja SS, Gonzalez E, Feuillet PM, Begum K, et al. Evolution of Human and Non-Human Primate CC Chemokine Receptor 5 Gene and mRNA. Potential Roles for Haplotype and mRNA Diversity, Differential Haplotype-Specific Transcriptional Activity, and Altered Transcription Factor Binding to Polymorphic Nucleotides in the Pathogenesis of HIV-1 and Simian Immunodeficiency Virus. J Biol Chem (2000) 275:18946–61. doi: 10.1074/jbc.M000169200
102. Gonzalez E, Bamshad M, Sato N, Mummidi S, Dhanda R, Catano G, et al. Race-Specific HIV-1 Disease-Modifying Effects Associated With CCR5 Haplotypes. Proc Natl Acad Sci USA (1999) 96:12004–9. doi: 10.1073/pnas.96.21.12004
103. Joshi A, Punke EB, Sedano M, Beauchamp B, Patel R, Hossenlopp C, et al. CCR5 Promoter Activity Correlates With HIV Disease Progression by Regulating CCR5 Cell Surface Expression and CD4 T Cell Apoptosis. Sci Rep (2017) 7:232. doi: 10.1038/s41598-017-00192-x
104. McDermott DH, Zimmerman PA, Guignard F, Kleeberger CA, Leitman SF, Murphy PM. CCR5 Promoter Polymorphism and HIV-1 Disease Progression. Lancet (1998) 352:866–70. doi: 10.1016/S0140-6736(98)04158-0
105. Gonzalo-Gil E, Rapuano PB, Ikediobi U, Leibowitz R, Mehta S, Coskun AK, et al. Transcriptional Down-Regulation of Ccr5 in a Subset of HIV+ Controllers and Their Family Members. Elife (2019) 8. doi: 10.7554/eLife.44360
106. Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, et al. The Influence of CCL3L1 Gene-Containing Segmental Duplications on HIV-1/AIDS Susceptibility. Science (2005) 307:1434–40. doi: 10.1126/science.1101160
107. Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 Resistance in CD4+ T Cells by Genome Editing Using Zinc-Finger Nucleases. Nat Biotechnol (2008) 26:808–16. doi: 10.1038/nbt1410
108. Scarborough RJ, Gatignol A. HIV and Ribozymes. Adv Exp Med Biol (2015) 848:97–116. doi: 10.1007/978-1-4939-2432-5_5
109. Cornu TI, Mussolino C, Bloom K, Cathomen T. Editing CCR5: A Novel Approach to HIV Gene Therapy. Adv Exp Med Biol (2015) 848:117–30. doi: 10.1007/978-1-4939-2432-5_6
110. Swamy MN, Wu H, Shankar P. Recent Advances in RNAi-Based Strategies for Therapy and Prevention of HIV-1/AIDS. Adv Drug Delivery Rev (2016) 103:174–86. doi: 10.1016/j.addr.2016.03.005
111. Wang CX, Cannon PM. The Clinical Applications of Genome Editing in HIV. Blood (2016) 127:2546–52. doi: 10.1182/blood-2016-01-678144
112. Ando D, Meyer K. Gene Editing: Regulatory and Translation to Clinic. Hematol Oncol Clin North Am (2017) 31:797–808. doi: 10.1016/j.hoc.2017.06.002
113. Cornu TI, Mussolino C, Cathomen T. Refining Strategies to Translate Genome Editing to the Clinic. Nat Med (2017) 23:415–23. doi: 10.1038/nm.4313
114. Vangelista L, Vento S. The Expanding Therapeutic Perspective of CCR5 Blockade. Front Immunol (2017) 8:1981. doi: 10.3389/fimmu.2017.01981
115. Schmidt JK, Strelchenko N, Park MA, Kim YH, Mean KD, Schotzko ML, et al. Genome Editing of CCR5 by CRISPR-Cas9 in Mauritian Cynomolgus Macaque Embryos. Sci Rep (2020) 10:18457. doi: 10.1038/s41598-020-75295-z
116. Glass WG, Lim JK, Cholera R, Pletnev AG, Gao J-L, Murphy PM. Chemokine Receptor CCR5 Promotes Leukocyte Trafficking to the Brain and Survival in West Nile Virus Infection. J Exp Med (2005) 202:1087–98. doi: 10.1084/jem.20042530
117. Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, et al. CCR5 Deficiency Increases Risk of Symptomatic West Nile Virus Infection. J Exp Med (2006) 203:35–40. doi: 10.1084/jem.20051970
118. Ahuja SK, He W. Double-Edged Genetic Swords and Immunity: Lesson From CCR5 and Beyond. J Infect Dis (2010) 201:171–4. doi: 10.1086/649427
119. Ellwanger JH, Kulmann-Leal B, Kaminski V de L, Rodrigues AG, Bragatte MA de S, Chies JAB. Beyond HIV Infection: Neglected and Varied Impacts of CCR5 and CCR5Δ32 on Viral Diseases. Virus Res (2020) 286:198040. doi: 10.1016/j.virusres.2020.198040
120. Ellwanger JH, Kaminski V de L, Rodrigues AG, Kulmann-Leal B, Chies JAB. CCR5 and CCR5Δ32 in Bacterial and Parasitic Infections: Thinking Chemokine Receptors Outside the HIV Box. Int J Immunogenet (2020) 47:261–85. doi: 10.1111/iji.12485
121. Lim JK, Louie CY, Glaser C, Jean C, Johnson B, Johnson H, et al. Genetic Deficiency of Chemokine Receptor CCR5 Is a Strong Risk Factor for Symptomatic West Nile Virus Infection: A Meta-Analysis of 4 Cohorts in the US Epidemic. J Infect Dis (2008) 197:262–5. doi: 10.1086/524691
122. Falcon A, Cuevas MT, Rodriguez-Frandsen A, Reyes N, Pozo F, Moreno S, et al. CCR5 Deficiency Predisposes to Fatal Outcome in Influenza Virus Infection. J Gen Virol (2015) 96:2074–8. doi: 10.1099/vir.0.000165
123. Marques RE, Guabiraba R, Del Sarto JL, Rocha RF, Queiroz AL, Cisalpino D, et al. Dengue Virus Requires the CC-Chemokine Receptor CCR5 for Replication and Infection Development. Immunology (2015) 145:583–96. doi: 10.1111/imm.12476
124. Alonzo F 3rd, Kozhaya L, Rawlings SA, Reyes-Robles T, DuMont AL, Myszka DG, et al. CCR5 Is a Receptor for Staphylococcus Aureus Leukotoxin ED. Nature (2013) 493:51–5. doi: 10.1038/nature11724
125. Salnikova LE, Smelaya TV, Moroz VV, Golubev AM, Rubanovich AV. Host Genetic Risk Factors for Community-Acquired Pneumonia. Gene (2013) 518:449–56. doi: 10.1016/j.gene.2012.10.027
126. Hubacek JA, Dusek L, Majek O, Adamek V, Cervinkova T, Dlouha D, et al. CCR5Delta32 Deletion as a Protective Factor in Czech First-Wave COVID-19 Subjects. Physiol Res (2021) 70:111–5. doi: 10.33549/physiolres.934647
127. Ungvári I, Tölgyesi G, Semsei AF, Nagy A, Radosits K, Keszei M, et al. CCR5 Delta 32 Mutation, Mycoplasma Pneumoniae Infection, and Asthma. J Allergy Clin Immunol (2007) 119:1545–7. doi: 10.1016/j.jaci.2007.02.038
128. Meyer L, Magierowska M, Hubert JB. Others. CCR5 A32 Deletion and Reduced Risk of Toxoplasmosis in HIV-1 Infected Patients. J Infect Dis (1999) 180:920–4. doi: 10.1086/314933
129. Ashton LJ, Stewart GJ, Biti R, Law M, Cooper DA, Kaldor JM. Heterozygosity for CCR5-Delta32 But Not CCR2b-64I Protects Against Certain Intracellular Pathogens. HIV Med (2002) 3:91–6. doi: 10.1046/j.1468-1293.2002.00106.x
130. Bonfá G, Benevides L, Souza M do C, Fonseca DM, Mineo TWP, Rossi MA, et al. CCR5 Controls Immune and Metabolic Functions During Toxoplasma Gondii Infection. PloS One (2014) 9:e104736. doi: 10.1371/journal.pone.0104736
131. Li T, Zhu J. Entanglement of CCR5 and Alzheimer’s Disease. Front Aging Neurosci (2019) 11:209. doi: 10.3389/fnagi.2019.00209
132. Wojta KJ, Ayer AH, Ramos EM, Nguyen PD, Karydas AM, Yokoyama JS, et al. Lack of Association Between the CCR5-Delta32 Polymorphism and Neurodegenerative Disorders. Alzheimer Dis Assoc Disord (2020) 34:244–7. doi: 10.1097/WAD.0000000000000367
133. Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV Hosts: Showing AIDS the Door. Science (2012) 335:1188–93. doi: 10.1126/science.1217550
134. Pandrea I, Apetrei C, Gordon S, Barbercheck J, Dufour J, Bohm R, et al. Paucity of CD4+CCR5+ T Cells Is a Typical Feature of Natural SIV Hosts. Blood (2007) 109:1069–76. doi: 10.1182/blood-2006-05-024364
135. Peeters M, Ma D, Liegeois F, Apetrei C. Chapter 3 - Simian Immunodeficiency Virus Infections in the Wild. In: Ansari AA, Silvestri G, editors. Natural Hosts of SIV. (2014) Amsterdam: Elsevier. p. 37–67.
136. Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, McClure HM, et al. Nonpathogenic SIV Infection of Sooty Mangabeys Is Characterized by Limited Bystander Immunopathology Despite Chronic High-Level Viremia. Immunity (2003) 18:441–52. doi: 10.1016/S1074-7613(03)00060-8
137. Silvestri G, Fedanov A, Germon S, Kozyr N, Kaiser WJ, Garber DA, et al. Divergent Host Responses During Primary Simian Immunodeficiency Virus SIVsm Infection of Natural Sooty Mangabey and Nonnatural Rhesus Macaque Hosts. J Virol (2005) 79:4043–54. doi: 10.1128/JVI.79.7.4043-4054.2005
138. Kaur A, Grant RM, Means RE, McClure H, Feinberg M, Johnson RP. Diverse Host Responses and Outcomes Following Simian Immunodeficiency Virus SIVmac239 Infection in Sooty Mangabeys and Rhesus Macaques. J Virol (1998) 72:9597–611. doi: 10.1128/JVI.72.12.9597-9611.1998
139. Broussard SR, Staprans SI, White R, Whitehead EM, Feinberg MB, Allan JS. Simian Immunodeficiency Virus Replicates to High Levels in Naturally Infected African Green Monkeys Without Inducing Immunologic or Neurologic Disease. J Virol (2001) 75:2262–75. doi: 10.1128/JVI.75.5.2262-2275.2001
140. Kornfeld C, Ploquin MJ-Y, Pandrea I, Faye A, Onanga R, Apetrei C, et al. Antiinflammatory Profiles During Primary SIV Infection in African Green Monkeys Are Associated With Protection Against AIDS. J Clin Invest (2005) 115:1082–91. doi: 10.1172/JCI23006
141. Onanga R, Souquière S, Makuwa M, Mouinga-Ondeme A, Simon F, Apetrei C, et al. Primary Simian Immunodeficiency Virus SIVmnd-2 Infection in Mandrills (Mandrillus Sphinx). J Virol (2006) 80:3301–9. doi: 10.1128/JVI.80.7.3301-3309.2006
142. Pandrea I, Onanga R, Kornfeld C, Rouquet P, Bourry O, Clifford S, et al. High Levels of SIVmnd-1 Replication in Chronically Infected Mandrillus Sphinx. Virology (2003) 317:119–27. doi: 10.1016/j.virol.2003.08.015
143. Svardal H, Jasinska AJ, Apetrei C, Coppola G, Huang Y, Schmitt CA, et al. Ancient Hybridization and Strong Adaptation to Viruses Across African Vervet Monkey Populations. Nat Genet (2017) 49:1705–13. doi: 10.1038/ng.3980
144. Raehtz K, Pandrea I, Apetrei C. The Well-Tempered SIV Infection: Pathogenesis of SIV Infection in Natural Hosts in the Wild, With Emphasis on Virus Transmission and Early Events Post-Infection That May Contribute to Protection From Disease Progression. Infect Genet Evol (2016) 46:308–23. doi: 10.1016/j.meegid.2016.07.006
145. Pandrea I, Silvestri G, Onanga R, Veazey RS, Marx PA, Hirsch V, et al. Simian Immunodeficiency Viruses Replication Dynamics in African Non-Human Primate Hosts: Common Patterns and Species-Specific Differences. J Med Primatol (2006) 35:194–201. doi: 10.1111/j.1600-0684.2006.00168.x
146. Gordon SN, Dunham RM, Engram JC, Estes J, Wang Z, Klatt NR, et al. Short-Lived Infected Cells Support Virus Replication in Sooty Mangabeys Naturally Infected With Simian Immunodeficiency Virus: Implications for AIDS Pathogenesis. J Virol (2008) 82:3725–35. doi: 10.1128/JVI.02408-07
147. Pandrea I, Gaufin T, Brenchley JM, Gautam R, Monjure C, Gautam A, et al. Cutting Edge: Experimentally Induced Immune Activation in Natural Hosts of Simian Immunodeficiency Virus Induces Significant Increases in Viral Replication and CD4+ T Cell Depletion. J Immunol (2008) 181:6687–91. doi: 10.4049/jimmunol.181.10.6687
148. Choudhary SK, Vrisekoop N, Jansen CA, Otto SA, Schuitemaker H, Miedema F, et al. Low Immune Activation Despite High Levels of Pathogenic Human Immunodeficiency Virus Type 1 Results in Long-Term Asymptomatic Disease. J Virol (2007) 81:8838–42. doi: 10.1128/JVI.02663-06
149. VandeWoude S, Apetrei C. Going Wild: Lessons From Naturally Occurring T-Lymphotropic Lentiviruses. Clin Microbiol Rev (2006) 19:728–62. doi: 10.1128/CMR.00009-06
150. Keele BF, Jones JH, Terio KA, Estes JD, Rudicell RS, Wilson ML, et al. Increased Mortality and AIDS-Like Immunopathology in Wild Chimpanzees Infected With SIVcpz. Nature (2009) 460:515–9. doi: 10.1038/nature08200
151. Terio KA, Kinsel MJ, Raphael J, Mlengeya T, Lipende I, Kirchhoff CA, et al. Pathologic Lesions in Chimpanzees (Pan Trogylodytes Schweinfurthii) From Gombe National Park, Tanzania, 2004–2010. J Zoo Wildlife Med (2011) 42:597–607. doi: 10.1638/2010-0237.1
152. Paiardini M, Cervasi B, Reyes-Aviles E, Micci L, Ortiz AM, Chahroudi A, et al. Low Levels of SIV Infection in Sooty Mangabey Central Memory CD4+ T Cells Are Associated With Limited CCR5 Expression. Nat Med (2011) 17:830–6. doi: 10.1038/nm.2395
153. Pandrea I, Onanga R, Souquiere S, Mouinga-Ondéme A, Bourry O, Makuwa M, et al. Paucity of CD4+ CCR5+ T Cells May Prevent Transmission of Simian Immunodeficiency Virus in Natural Nonhuman Primate Hosts by Breast-Feeding. J Virol (2008) 82:5501–9. doi: 10.1128/JVI.02555-07
154. Wang X, Das A, Lackner AA, Veazey RS, Pahar B. Intestinal Double-Positive CD4+CD8+ T Cells of Neonatal Rhesus Macaques Are Proliferating, Activated Memory Cells and Primary Targets for SIVMAC251 Infection. Blood (2008) 112:4981–90. doi: 10.1182/blood-2008-05-160077
155. Wang X, Xu H, Pahar B, Alvarez X, Green LC, Dufour J, et al. Simian Immunodeficiency Virus Selectively Infects Proliferating CD4+ T Cells in Neonatal Rhesus Macaques. Blood (2010) 116:4168–74. doi: 10.1182/blood-2010-03-273482
156. Winters MA, Van Rompay KKA, Kashuba ADM, Shulman NS, Holodniy M. Maternal-Fetal Pharmacokinetics and Dynamics of a Single Intrapartum Dose of Maraviroc in Rhesus Macaques. Antimicrob Agents Chemother (2010) 54:4059–63. doi: 10.1128/AAC.00747-10
157. Wang X, Rasmussen T, Pahar B, Poonia B, Alvarez X, Lackner AA, et al. Massive Infection and Loss of CD4+ T Cells Occurs in the Intestinal Tract of Neonatal Rhesus Macaques in Acute SIV Infection. Blood (2007) 109:1174–81. doi: 10.1182/blood-2006-04-015172
158. Aldrovandi GM, Kuhn L. What Infants and Breasts Can Teach Us About Natural Protection From HIV Infection. J Infect Dis (2010) 202 Suppl 3:S366–70. doi: 10.1086/655972
159. Ma D, Jasinska AJ, Feyertag F, Wijewardana V, Kristoff J, He T, et al. Factors Associated With Siman Immunodeficiency Virus Transmission in a Natural African Nonhuman Primate Host in the Wild. J Virol (2014) 88:5687–705. doi: 10.1128/JVI.03606-13
160. Peckham C, Gibb D. Mother-To-Child Transmission of the Human Immunodeficiency Virus. N Engl J Med (1995) 333:298–302. doi: 10.1056/NEJM199508033330507
161. Amedee AM, Rychert J, Lacour N, Fresh L, Ratterree M. Viral and Immunological Factors Associated With Breast Milk Transmission of SIV in Rhesus Macaques. Retrovirology (2004) 1:17. doi: 10.1186/1742-4690-1-17
162. Nagelkerke NJ, Moses S, Embree JE, Jenniskens F, Plummer FA. The Duration of Breastfeeding by HIV-1-Infected Mothers in Developing Countries: Balancing Benefits and Risks. J Acquir Immune Defic Syndr Hum Retrovirol (1995) 8:176–81. doi: 10.1097/00042560-199502000-00008
163. Brocca-Cofano E, Xu C, Wetzel KS, Cottrell ML, Policicchio BB, Raehtz KD, et al. Marginal Effects of Systemic CCR5 Blockade With Maraviroc on Oral Simian Immunodeficiency Virus Transmission to Infant Macaques. J Virol (2018) 92(17). doi: 10.1128/JVI.00576-18
164. Pandrea I, Parrish NF, Raehtz K, Gaufin T, Barbian HJ, Ma D, et al. Mucosal Simian Immunodeficiency Virus Transmission in African Green Monkeys: Susceptibility to Infection is Proportional to Target Cell Availability at Mucosal Sites. J Virol (2012) 86:4158–68. doi: 10.1128/JVI.07141-11
165. Deng HK, Unutmaz D, KewalRamani VN, Littman DR. Expression Cloning of New Receptors Used by Simian and Human Immunodeficiency Viruses. Nature (1997) 388:296–300. doi: 10.1038/40894
166. Rucker J, Edinger AL, Sharron M, Samson M, Lee B, Berson JF, et al. Utilization of Chemokine Receptors, Orphan Receptors, and Herpesvirus-Encoded Receptors by Diverse Human and Simian Immunodeficiency Viruses. J Virol (1997) 71:8999–9007. doi: 10.1128/jvi.71.12.8999-9007.1997
167. Farzan M, Choe H, Martin K, Marcon L, Hofmann W, Karlsson G, et al. Two Orphan Seven-Transmembrane Segment Receptors Which Are Expressed in CD4-Positive Cells Support Simian Immunodeficiency Virus Infection. J Exp Med (1997) 186:405–11. doi: 10.1084/jem.186.3.405
168. Schols D, De Clercq E. The Simian Immunodeficiency Virus Mnd(GB-1) Strain Uses CXCR4, Not CCR5, as Coreceptor for Entry in Human Cells. J Gen Virol (1998) 79( Pt 9):2203–5. doi: 10.1099/0022-1317-79-9-2203
169. Owen SM, Masciotra S, Novembre F, Yee J, Switzer WM, Ostyula M, et al. Simian Immunodeficiency Viruses of Diverse Origin Can Use CXCR4 as a Coreceptor for Entry Into Human Cells. J Virol (2000) 74:5702–8. doi: 10.1128/JVI.74.12.5702-5708.2000
170. Gautam R, Gaufin T, Butler I, Gautam A, Barnes M, Mandell D, et al. Simian Immunodeficiency Virus SIVrcm, a Unique CCR2-Tropic Virus, Selectively Depletes Memory CD4+ T Cells in Pigtailed Macaques Through Expanded Coreceptor Usage. In Vivo J Virol (2009) 83:7894–908. doi: 10.1128/JVI.00444-09
171. Wetzel KS, Yi Y, Elliott STC, Romero D, Jacquelin B, Hahn BH, et al. CXCR6-Mediated Simian Immunodeficiency Virus SIVagmSab Entry Into Sabaeus African Green Monkey Lymphocytes Implicates Widespread Use of Non-CCR5 Pathways in Natural Host Infections. J Virol (2017) 91(4). doi: 10.1128/JVI.01626-16
172. Georges-Courbot MC, Lu CY, Makuwa M, Telfer P, Onanga R, Dubreuil G, et al. Natural Infection of a Household Pet Red-Capped Mangabey (Cercocebus Torquatus Torquatus ) With a New Simian Immunodeficiency Virus. J Virol (1998) 72:600–8. doi: 10.1128/jvi.72.1.600-608.1998
173. Smith SM, Makuwa M, Lee F, Gettie A, Russo C, Marx PA. SIVrcm Infection of Macaques. J Med Primatol (1998) 27:94–8. doi: 10.1111/j.1600-0684.1998.tb00232.x
174. Peeters M, Courgnaud V, Abela B, Auzel P, Pourrut X, Bibollet-Ruche F, et al. Risk to Human Health From a Plethora of Simian Immunodeficiency Viruses in Primate Bushmeat. Emerg Infect Dis (2002) 8:451–7. doi: 10.3201/eid0805.010522
175. Simon F, Souquière S, Damond F, Kfutwah A, Makuwa M, Leroy E, et al. Synthetic Peptide Strategy for the Detection of and Discrimination Among Highly Divergent Primate Lentiviruses. AIDS Res Hum Retroviruses (2001) 17:937–52. doi: 10.1089/088922201750290050
176. Apetrei C, Robertson DL, Marx PA. The History of SIVS and AIDS: Epidemiology, Phylogeny and Biology of Isolates From Naturally SIV Infected Non-Human Primates (NHP) in Africa. Front Biosci (2004) 9:225–54. doi: 10.2741/1154
177. Zhang Y, Lou B, Lal RB, Gettie A, Marx PA, Moore JP. Use of Inhibitors to Evaluate Coreceptor Usage by Simian and Simian/Human Immunodeficiency Viruses and Human Immunodeficiency Virus Type 2 in Primary Cells. J Virol (2000) 74:6893–910. doi: 10.1128/JVI.74.15.6893-6910.2000
178. Simmons G, Reeves JD, McKnight A, Dejucq N, Hibbitts S, Power CA, et al. CXCR4 as a Functional Coreceptor for Human Immunodeficiency Virus Type 1 Infection of Primary Macrophages. J Virol (1998) 72:8453–7. doi: 10.1128/JVI.72.10.8453-8457.1998
179. Riddick NE, Wu F, Matsuda K, Whitted S, Ourmanov I, Goldstein S, et al. Simian Immunodeficiency Virus SIVagm Efficiently Utilizes Non-CCR5 Entry Pathways in African Green Monkey Lymphocytes: Potential Role for GPR15 and CXCR6 as Viral Coreceptors. J Virol (2015) 90:2316–31. doi: 10.1128/JVI.02529-15
180. Elliott STC, Riddick NE, Francella N, Paiardini M, Vanderford TH, Li B, et al. Cloning and Analysis of Sooty Mangabey Alternative Coreceptors That Support SIVsmm Entry Independently of CCR5. J Virol (2011) 86(2):898–908. doi: 10.1128/JVI.06415-11
181. Pandrea IV, Gautam R, Ribeiro RM, Brenchley JM, Butler IF, Pattison M, et al. Acute Loss of Intestinal CD4+ T Cells Is Not Predictive of Simian Immunodeficiency Virus Virulence. J Immunol (2007) 179:3035–46. doi: 10.4049/jimmunol.179.5.3035
182. Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, et al. Identification and Characterization of Transmitted and Early Founder Virus Envelopes in Primary HIV-1 Infection. Proc Natl Acad Sci (2008) 105:7552–7. doi: 10.1073/pnas.0802203105
183. Parker ZF, Iyer SS, Wilen CB, Parrish NF, Chikere KC, Lee F-H, et al. Transmitted/founder and Chronic HIV-1 Envelope Proteins Are Distinguished by Differential Utilization of CCR5. J Virol (2013) 87:2401–11. doi: 10.1128/JVI.02964-12
184. Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, et al. Phenotypic Properties of Transmitted Founder HIV-1. Proc Natl Acad Sci USA (2013) 110:6626–33. doi: 10.1073/pnas.1304288110
185. Milush JM, Mir KD, Sundaravaradan V, Gordon SN, Engram J, Cano CA, et al. Lack of Clinical AIDS in SIV-Infected Sooty Mangabeys With Significant CD4+ T Cell Loss Is Associated With Double-Negative T Cells. J Clin Invest (2011) 121(3):1102–10. doi: 10.1172/JCI44876
186. Milush JM, Reeves JD, Gordon SN, Zhou D, Muthukumar A, Kosub DA, et al. Virally Induced CD4+ T Cell Depletion Is Not Sufficient to Induce AIDS in a Natural Host. J Immunol (2007) 179:3047–56. doi: 10.4049/jimmunol.179.5.3047
187. Wetzel KS, Yi Y, Yadav A, Bauer AM, Bello EA, Romero DC, et al. Loss of CXCR6 Coreceptor Usage Characterizes Pathogenic Lentiviruses. PloS Pathog (2018) 14:e1007003. doi: 10.1371/journal.ppat.1007003
188. Bailes E, Gao F, Bibollet-Ruche F, Courgnaud V, Peeters M, Marx PA, et al. Hybrid Origin of SIV in Chimpanzees. Science (2003) 300:1713. doi: 10.1126/science.1080657
189. Courgnaud V, Salemi M, Pourrut X, Mpoudi-Ngole E, Abela B, Auzel P, et al. Characterization of a Novel Simian Immunodeficiency Virus With a Vpu Gene From Greater Spot-Nosed Monkeys (Cercopithecus Nictitans) Provides New Insights Into Simian/Human Immunodeficiency Virus Phylogeny. J Virol (2002) 76:8298–309. doi: 10.1128/JVI.76.16.8298-8309.2002
190. Courgnaud V, Abela B, Pourrut X, Mpoudi-Ngole E, Loul S, Delaporte E, et al. Identification of a New Simian Immunodeficiency Virus Lineage With a Vpu Gene Present Among Different Cercopithecus Monkeys (C. Mona, C. Cephus and C. Nictitans) From Cameroon. J Virol (2003) 77:12523–34. doi: 10.1128/JVI.77.23.12523-12534.2003
191. Mandell DT, Kristoff J, Gaufin T, Gautam R, Ma D, Sandler N, et al. Pathogenic Features Associated With Increased Virulence Upon Simian Immunodeficiency Virus Cross-Species Transmission From Natural Hosts. J Virol (2014) 88:6778–92. doi: 10.1128/JVI.03785-13
192. Pandrea I, Gaufin T, Gautam R, Kristoff J, Mandell D, Montefiori D, et al. Functional Cure of SIVagm Infection in Rhesus Macaques Results in Complete Recovery of CD4+ T Cells and Is Reverted by CD8+ Cell Depletion. Plos Pathogen (2011) 7(8):e1002170. doi: 10.1371/journal.ppat.1002170
193. Veazey RS, Klasse PJ, Ketas TJ, Reeves JD, Piatak M, Kunstman K, et al. Use of a Small Molecule CCR5 Inhibitor in Macaques to Treat Simian Immunodeficiency Virus Infection or Prevent Simian–Human Immunodeficiency Virus Infection. J Exp Med (2003) 198:1551–62. doi: 10.1084/jem.20031266
194. Wetzel KS, Elliott STC, Collman RG. SIV Coreceptor Specificity in Natural and Non-Natural Host Infection: Implications for Cell Targeting and Differential Outcomes From Infection. Curr HIV Res (2018) 16:41–51. doi: 10.2174/1570162X15666171124121805
195. Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, et al. Maraviroc for Previously Treated Patients With R5 HIV-1 Infection. New Engl J Med (2008) 359:1429–41. doi: 10.1056/nejmoa0803152
196. Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, et al. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science (2013) 341:1387–90. doi: 10.1126/science.1241475
197. Garcia-Perez J, Rueda P, Alcami J, Rognan D, Arenzana-Seisdedos F, Lagane B, et al. Allosteric Model of Maraviroc Binding to CC Chemokine Receptor 5 (CCR5). J Biol Chem (2011) 286:33409–21. doi: 10.1074/jbc.M111.279596
198. Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, et al. Maraviroc (UK-427,857), a Potent, Orally Bioavailable, and Selective Small-Molecule Inhibitor of Chemokine Receptor CCR5 With Broad-Spectrum Anti-Human Immunodeficiency Virus Type 1 Activity. Antimicrob Agents Chemother (2005) 49:4721–32. doi: 10.1128/AAC.49.11.4721-4732.2005
199. Olson WC, Rabut GE, Nagashima KA, Tran DN, Anselma DJ, Monard SP, et al. Differential Inhibition of Human Immunodeficiency Virus Type 1 Fusion, Gp120 Binding, and CC-Chemokine Activity by Monoclonal Antibodies to CCR5. J Virol (1999) 73:4145–55. doi: 10.1128/JVI.73.5.4145-4155.1999
200. Chang XL, Wu HL, Webb GM, Tiwary M, Hughes C, Reed JS, et al. CCR5 Receptor Occupancy Analysis Reveals Increased Peripheral Blood CCR5+CD4+ T Cells Following Treatment With the Anti-CCR5 Antibody Leronlimab. Front Immunol (2021) 12:794638. doi: 10.3389/fimmu.2021.794638
201. Patterson BK, Seethamraju H, Dhody K, Corley MJ, Kazempour K, Lalezari J, et al. CCR5 Inhibition in Critical COVID-19 Patients Decreases Inflammatory Cytokines, Increases CD8 T-Cells, and Decreases SARS-CoV2 RNA in Plasma by Day 14. Int J Infect Dis (2021) 103:25–32. doi: 10.1016/j.ijid.2020.10.101
202. Umotoy JC, de Taeye SW. Antibody Conjugates for Targeted Therapy Against HIV-1 as an Emerging Tool for HIV-1 Cure. Front Immunol (2021) 12:708806. doi: 10.3389/fimmu.2021.708806
203. Ji C, Zhang J, Dioszegi M, Chiu S, Rao E, Derosier A, et al. CCR5 Small-Molecule Antagonists and Monoclonal Antibodies Exert Potent Synergistic Antiviral Effects by Cobinding to the Receptor. Mol Pharmacol (2007) 72:18–28. doi: 10.1124/mol.107.035055
204. Kopetzki E, Jekle A, Ji C, Rao E, Zhang J, Fischer S, et al. Closing Two Doors of Viral Entry: Intramolecular Combination of a Coreceptor- and Fusion Inhibitor of HIV-1. Virol J (2008) 5:56. doi: 10.1186/1743-422X-5-56
205. Asano S, Gavrilyuk J, Burton DR. Barbas CF 3rd. Preparation and Activities of Macromolecule Conjugates of the CCR5 Antagonist Maraviroc. ACS Med Chem Lett (2014) 5:133–7. doi: 10.1021/ml400370w
206. Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. N Engl J Med (2009) 360:692–8. doi: 10.1056/NEJMoa0802905
207. Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, et al. HIV-1 Remission Following CCR5Δ32/Δ32 Haematopoietic Stem-Cell Transplantation. Nature (2019) 568:244–8. doi: 10.1038/s41586-019-1027-4
208. Hütter G, Thiel E. Allogeneic Transplantation of CCR5-Deficient Progenitor Cells in a Patient With HIV Infection: An Update After 3 Years and the Search for Patient No. 2. AIDS (2011) 25:273–4. doi: 10.1097/qad.0b013e328340fe28
209. Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the Cure of HIV Infection by CCR5Δ32/Δ32 Stem Cell Transplantation. Blood (2011) 117:2791–9. doi: 10.1182/blood-2010-09-309591
210. Lisziewicz J, Rosenberg E, Lieberman J, Jessen H, Lopalco L, Siliciano R, et al. Control of HIV Despite the Discontinuation of Antiretroviral Therapy. N Engl J Med (1999) 340:1683–4. doi: 10.1056/NEJM199905273402114
211. Jessen H, Allen TM, Streeck H. How a Single Patient Influenced HIV Research–15-Year Follow-Up. N Engl J Med (2014) 370:682–3. doi: 10.1056/NEJMc1308413
212. Gupta RK, Peppa D, Hill AL, Gálvez C, Salgado M, Pace M, et al. Evidence for HIV-1 Cure After CCR5Δ32/Δ32 Allogeneic Haemopoietic Stem-Cell Transplantation 30 Months Post Analytical Treatment Interruption: A Case Report. Lancet HIV (2020) 7:e340–7. doi: 10.1016/S2352-3018(20)30069-2
213. Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, et al. Human Hematopoietic Stem/Progenitor Cells Modified by Zinc-Finger Nucleases Targeted to CCR5 Control HIV-1. Vivo Nat Biotechnol (2010) 28:839–47. doi: 10.1038/nbt.1663
214. DiGiusto DL, Cannon PM, Holmes MC, Li L, Rao A, Wang J, et al. Preclinical Development and Qualification of ZFN-Mediated CCR5 Disruption in Human Hematopoietic Stem/Progenitor Cells. Mol Ther Methods Clin Dev (2016) 3:16067. doi: 10.1038/mtm.2016.67
215. Xu M. CCR5-Δ32 Biology, Gene Editing, and Warnings for the Future of CRISPR-Cas9 as a Human and Humane Gene Editing Tool. Cell Biosci (2020) 10:48. doi: 10.1186/s13578-020-00410-6
216. Palendira U, Chinn R, Raza W, Piper K, Pratt G, Machado L, et al. Selective Accumulation of Virus-Specific CD8+ T Cells With Unique Homing Phenotype Within the Human Bone Marrow. Blood (2008) 112:3293–302. doi: 10.1182/blood-2008-02-138040
217. Kohlmeier JE, Reiley WW, Perona-Wright G. Inflammatory Chemokine Receptors Regulate CD8+ T Cell Contraction and Memory Generation Following Infection. J Exp Med (2011) 208(8):1621–34. doi: 10.1084/jem.20102110
218. Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, et al. The Chemokine Receptor CCR5 Plays a Key Role in the Early Memory CD8+ T Cell Response to Respiratory Virus Infections. Immunity (2008) 29:101–13. doi: 10.1016/j.immuni.2008.05.011
219. Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines Enhance Immunity by Guiding Naive CD8+ T Cells to Sites of CD4+ T Cell–Dendritic Cell Interaction. Nature (2006) 440:890–5. doi: 10.1038/nature04651
220. Fukada K, Sobao Y, Tomiyama H, Oka S, Takiguchi M. Functional Expression of the Chemokine Receptor CCR5 on Virus Epitope-Specific Memory and Effector CD8+ T Cells. J Immunol (2002) 168:2225–32. doi: 10.4049/jimmunol.168.5.2225
221. Glass WG, Lane TE. Functional Analysis of the CC Chemokine Receptor 5 (CCR5) on Virus-Specific CD8+ T Cells Following Coronavirus Infection of the Central Nervous System. Virology (2003) 312:407–14. doi: 10.1016/S0042-6822(03)00237-X
222. Shacklett BL, Cox CA, Sandberg JK, Stollman NH, Jacobson MA, Nixon DF. Trafficking of Human Immunodeficiency Virus Type 1-Specific CD8+ T Cells to Gut-Associated Lymphoid Tissue During Chronic Infection. J Virol (2003) 77:5621–31. doi: 10.1128/JVI.77.10.5621-5631.2003
223. Wang X, Russell-Lodrigue KE, Ratterree MS, Veazey RS, Xu H. Chemokine Receptor CCR5 Correlates With Functional CD8 T Cells in SIV-Infected Macaques and the Potential Effects of Maraviroc on T-Cell Activation. FASEB J (2019) 33:8905–12. doi: 10.1096/fj.201802703r
224. Clarke DK, Duarte EA, Elena SF, Moya A, Domingo E, Holland J. The Red Queen Reigns in the Kingdom of RNA Viruses. Proc Natl Acad Sci USA (1994) 91:4821–4. doi: 10.1073/pnas.91.11.4821
225. Charles Dodgson ALC. Through the Looking-Glass, And What Alice Found There. The Project Gutenberg Ebook. Available at: https://www.gutenberg.org/files/12/12-h/12-h.htm (Accessed December 14, 2021).
Keywords: CCR5, human immunodeficiency virus, simian immunodeficiency virus, delta 32, red-capped mangabey, sooty mangabey, African green monkey, virus transmission
Citation: Jasinska AJ, Pandrea I and Apetrei C (2022) CCR5 as a Coreceptor for Human Immunodeficiency Virus and Simian Immunodeficiency Viruses: A Prototypic Love-Hate Affair. Front. Immunol. 13:835994. doi: 10.3389/fimmu.2022.835994
Received: 15 December 2021; Accepted: 10 January 2022;
Published: 27 January 2022.
Edited by:
Joel Henrique Ellwanger, Federal University of Rio Grande do Sul, BrazilReviewed by:
Gianluca Veggiani, University of Toronto, CanadaCopyright © 2022 Jasinska, Pandrea and Apetrei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristian Apetrei, YXBldHJlaWNAcGl0dC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.