 Paul Shafer
Paul Shafer Lauren M. Kelly
Lauren M. Kelly Valentina Hoyos
Valentina Hoyos
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 March 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.835762
This article is part of the Research TopicNew Frontiers in Gene-Modified T Cell TechnologyView all 16 articles
To redirect T cells against tumor cells, T cells can be engineered ex vivo to express cancer-antigen specific T cell receptors (TCRs), generating products known as TCR-engineered T cells (TCR T). Unlike chimeric antigen receptors (CARs), TCRs recognize HLA-presented peptides derived from proteins of all cellular compartments. The use of TCR T cells for adoptive cellular therapies (ACT) has gained increased attention, especially as efforts to treat solid cancers with ACTs have intensified. In this review, we describe the differing mechanisms of T cell antigen recognition and signal transduction mediated through CARs and TCRs. We describe the classes of cancer antigens recognized by current TCR T therapies and discuss both classical and emerging pre-clinical strategies for antigen-specific TCR discovery, enhancement, and validation. Finally, we review the current landscape of clinical trials for TCR T therapy and discuss what these current results indicate for the development of future engineered TCR approaches.
The past decades have seen rapid advancements in our understanding of the mechanisms underlying the antitumor function of immune cells, and as such adoptive cell therapy (ACT) strategies have emerged as a major platform of cancer therapeutics. A milestone in ACT was the success of tumor infiltrating lymphocyte (TIL) therapy for metastatic melanoma beginning in the 1980s (1). While TIL therapy remains an important ACT modality, the manufacture of TIL products is logistically challenging. ACT efforts have thus largely transitioned towards strategies to engineer peripheral blood T cells with receptors that confer desired antigen specificity. These predominantly include chimeric antigen receptor T cell (CAR T) and T cell receptor engineered T cell (TCR T) therapies. Due to the remarkable efficacy of CAR T therapies in treating B cell malignancies (2), interest in CAR T therapy has eclipsed that of TCR T therapy. However, TCR T therapy is gaining interest as CAR T trials have so far failed to elicit satisfactory responses in the treatment of solid cancers (2), and many believe TCRs may be better suited for the treatment of solid cancers (3). Indeed, exciting clinical results are now emerging that demonstrate safety and efficacy of TCR T therapies in both hematological and solid cancers. In this review we describe the biology of TCRs and tumor antigen targets and discuss state of the art techniques for TCR discovery and preclinical assessment. Finally, we describe the current landscape of TCR T trials and the challenges that remain.
Conventional T cells recognize MHC-presented antigens through their T cell receptor (TCR), a disulfide-linked heterodimer comprised of an α and β chain. To form a functional receptor, TCR α/β heterodimers further complex with CD3ϵ/γ/δ/ζ subunits (4–7). TCRs recognize enzymatically cleaved peptides that are presented at the cell surface by MHC molecules (pMHC). In humans, antigen-presenting MHC alleles are broadly classified as HLA class I (A, B, or C) or HLA class II (DR, DP, or DQ), which predominantly present cytosolic or extracellular derived peptides, respectively (4). The coreceptors CD8 and CD4 enhance TCR antigen sensitivity through interaction with MHC class I or II molecules, respectively (8). TCR binding to cognate pMHC leads to the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) in intracellular regions of the CD3 subunits (5, 6), which results in T cell activation and initiation of effector functions including proliferation, cytokine secretion, and cytolysis via secretion of perforin and granzyme (Figure 1). In TCR T therapy, T cells are edited to express TCR α and β chains that confer a desired specificity. Here, introduced TCR α and β chains dimerize and complex with endogenous CD3 components to form a functional TCR that redirects T cell specificity towards an antigen of interest.
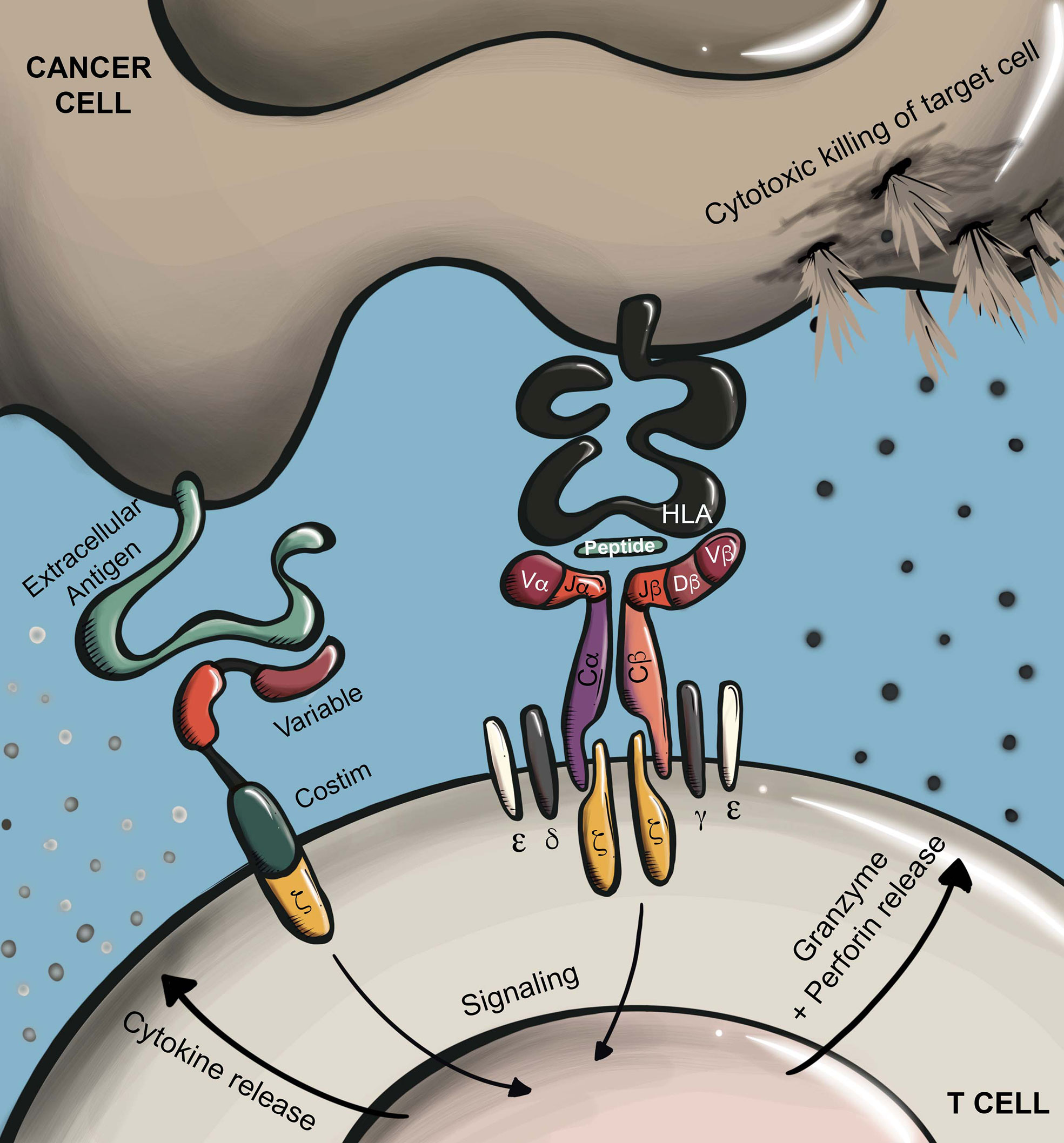
Figure 1 Antigen recognition by CARs and TCRs. CARs recognize surface proteins typically through an antibody-derived scFv recognition domain. Antigen recognition leads to T cell activation via phosphorylation of ITAMs in a conjugated intracellular CD3ζ domain. In the case of later generation CARs, ligand binding also leads to additional stimulation of conjugated costimulatory receptors (e.g. CD28, 4-1BB). TCRs recognize HLA-presented peptides which may be derived from any cellular compartment. Antigen recognition by TCRs leads to T cell activation through phosphorylation of ITAMs in the associated CD3ϵ/γ/δ/ζ subunits. Depending on T cell subtype, T cell activation through either receptor type will trigger effector functions including proliferation, cytokine secretion, and target cell killing through directed secretion of perforin and granzyme.
Another common method for redirecting T cell specificity is through the genetic transfer of chimeric antigen receptors (CARs), which are broadly comprised of an extracellular-facing antigen-binding domain linked to an intracellular immune cell activation signaling domain. Most often, CARs recognize antigen through the single-chain variable fragment (scFv) of an antibody. In a typical CAR design, the antigen-binding scFv is linked via a hinge, or spacer, region to a transmembrane domain that is further conjugated to an intracellular CD3ζ signaling domain. In this manner, antigen binding by the scFv drives CD3ζ phosphorylation and downstream T cell activation. Later generation CARs include the addition of intracellular costimulatory domains such as CD28 and 4-1BB, which further improve CAR T function and persistence (Figure 1) (9).
While TCRs recognize antigens in the context of HLA presentation, CARs recognize natively folded proteins at the cell surface. Therefore, CARs overcome clinical limitations imposed by the HLA-restriction of TCRs. HLA encoding genes are the most polymorphic in the human genome, with over 20,000 HLA-class I alleles identified to date (10). Therefore, unlike CAR T therapy, patients selected for TCR T therapy must express not only the targeted antigen, but also the corresponding antigen-restricting HLA allele. For this reason, TCR T therapies typically utilize TCRs that are restricted to relatively common HLA alleles, such as HLA-A*02:01, which is present in about 47.8% and 16.8% of Caucasian and African American populations in the United States, respectively (11).
As CARs and TCRs utilize differing signaling mechanisms, they exhibit several important differences in their functional response to antigen stimulation. While TCRs can elicit a cytotoxic response to as few as a single pMHC molecule, CARs typically require thousands of target surface molecules to mediate an effective response (12–14). A consequence of the reduced antigen sensitivity of CARs is seen in patients with B cell malignancies who initially respond to CAR T therapy but subsequently relapse with progression of antigen-low cancer cells (15). Upon stimulation, CARs mediate supraphysiologic T cell activation, leading to enhanced cytokine secretion. For this reason, CAR T cells are more likely to cause cytokine release syndrome (CRS) in patients as compared to TCR T cells; however, recent advancements in treatment have made CRS generally clinically manageable (16, 17).
Tumor antigens recognized by CARs must be located at the surface of cancer cells. Conversely, TCRs recognize HLA presented peptides that may be derived from any cellular compartment. As transmembrane proteins constitute only an estimated 14-26% of the proteome (18–20), CAR-targetable antigens are considerably more limited. However, the repertoire of CAR-targetable antigens is extended to some degree by the ability of CARs to recognize not only protein antigens, but also other molecules like glycoproteins and glycolipids (21). This difference in repertoire of potential targetable antigens has significant implications for CAR T and TCR T therapies, which must aggressively target tumor cells while avoiding toxicity directed towards healthy tissue. Indeed, expression of target antigen on normal cells can lead to T cell-mediated destruction of healthy tissue, known as ‘on-target off-tumor’ toxicity (22). Therefore, the degree to which a target antigen is exclusively expressed by cancer cells is an important factor. So far, the primary success of CAR T therapy has been in the treatment of B cell malignancies targeting CD19, an antigen expressed ubiquitously on malignant and healthy B cells. While CD19-directed CAR T therapy leads to ablation of both malignant and healthy B cells, such on-target off-tumor toxicity is clinically manageable through replacement antibody therapy (22). However, in the case of many other types of cancer, including almost all solid cancers, such T cell-mediated ablation of healthy organ tissue is not clinically manageable, and thus target antigens with exclusivity of expression in cancer cells are best. Currently described tumor antigens with the greatest specificity of expression in cancer cells are predominantly intracellular derived antigens, accessible to TCRs but not CARs (23). Therefore, TCR T therapies may have an advantage over CAR T therapies in the ability to aggressively target cancer cells while minimizing toxicity.
Significant progress has been made in identifying the precise cancer antigens that mediate immune rejection. While the nomenclature describing tumor antigens varies, widely studied classes of tumor antigens include tumor-associated antigens (TAAs), cancer-germline antigens (CGAs), and tumor-specific antigens (TSAs) (24–26) (Figure 2).
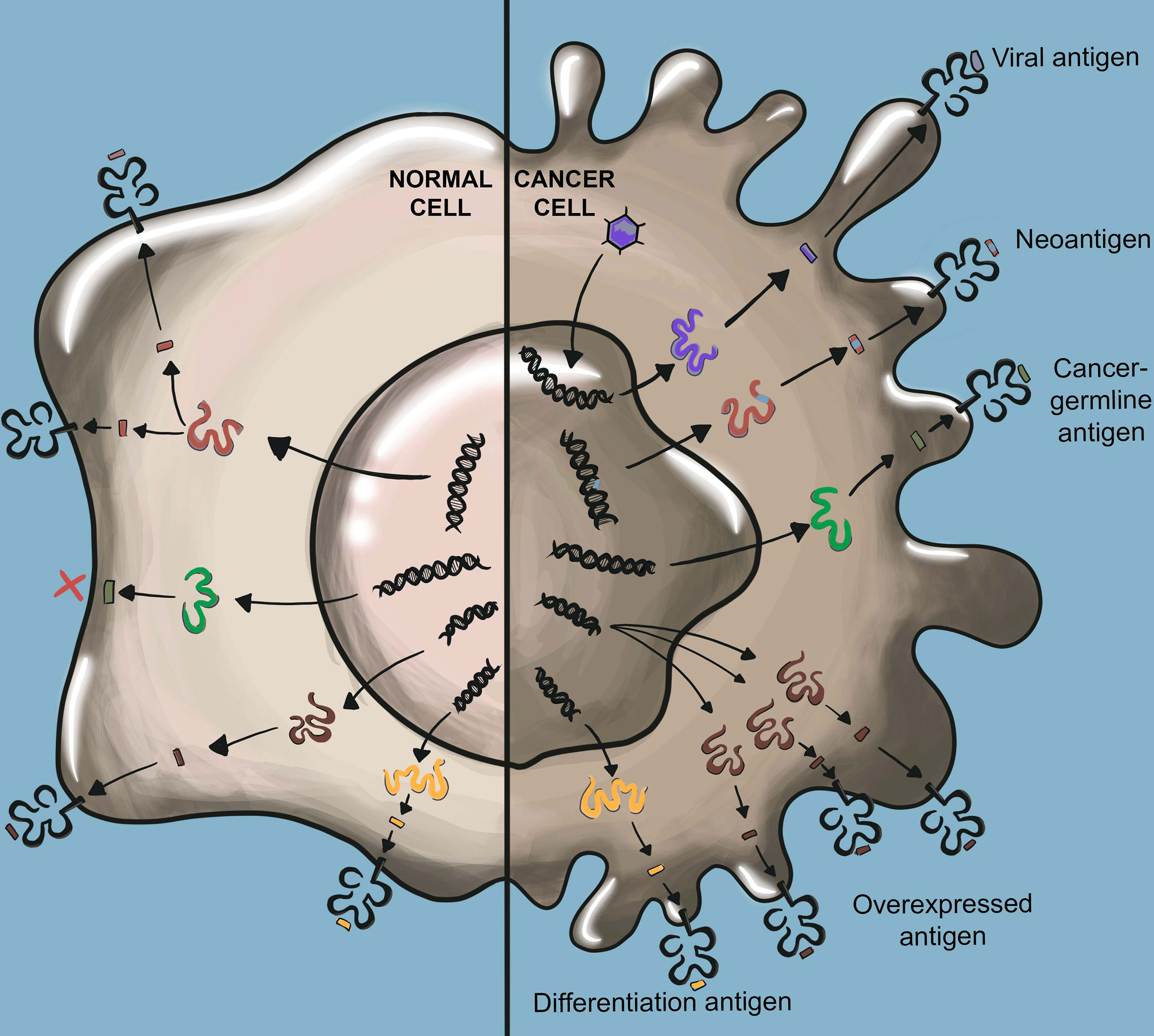
Figure 2 TCR-recognized tumor antigens. Viral antigens result from viral oncogenes which are not present in normal cells. Neoantigens arise from somatic mutations not found in normal cells. Viral antigens and neoantigens are collectively referred to as tumor-specific antigens (TSAs). Cancer germline antigens (CGAs) are derived from proteins that are normally only expressed in germ cells such as testis which lack HLA class I expression. Overexpressed antigens arise from proteins highly overexpressed in cancer tissue as compared to normal tissue. Cancer differentiation antigens are expressed by cancer cells and their expression is otherwise limited to only the normal cells of the same tissue origin as the cancer. Overexpressed antigens and cancer differentiation antigens are collectively referred to as tumor-associated antigens (TAAs).
TAAs are expressed by tumor cells but are also expressed in at least some healthy tissue. As a result, therapies targeting TAAs must contend with potential T cell mediated on-target off-tumor toxicity. TAAs are further classified as differentiation antigens or overexpressed antigens.
Differentiation antigens are expressed by cancer cells as well as normal cells of the same tissue origin. Melanoma differentiation antigens were among the first discovered tumor antigens and include the widely studied melanoma-associated antigen recognized by T cells (MART-1) (27) and glycoprotein 100 (gp100) (28). Among tumor antigens, differentiation antigens typically pose the greatest risk for on-target off-tumor toxicity. As discussed in further detail in a later section, clinical experience has demonstrated that targeting differentiation antigens is likely only clinically appropriate when antigen expression is restricted to dispensable healthy tissue, such as CD19-expressing B cells. Likely for this reason, only one TCR T clinical trial targeting a solid cancer differentiation antigen has been initiated since 2012 (Supplementary Table 1).
Overexpressed antigens are expressed at high levels in cancer cells but are minimally expressed in healthy cells. While targeting such antigens continues to pose a risk for on-target off-tumor toxicity, the differential expression between cancer and normal cells allows for the possibility of achieving a therapeutic window by which adoptively transferred T cells may destroy high-antigen expressing cancer cells with minimal destruction of low-antigen expressing healthy tissue. An example of a widely studied overexpressed antigen is Wilms’ Tumor Antigen 1 (WT1), a transcription factor with 10- to 1000- fold higher expression in leukemic cells as compared to normal cells (29, 30).
CGAs are aberrantly expressed in cancer cells while their expression in normal tissue is restricted to germline cells, such as those of testis, which lack HLA-class I expression, thus greatly reducing the risk for on-target off-tumor toxicity. As such, CGAs are currently among the most aggressively pursued targets for TCR-based immunotherapies. Examples of CGAs with high clinical significance include NY-ESO-1 and MAGE-A4, which are detected with high levels of expression in various solid and hematological cancers (31–35). However, several studies have reported that CGAs are heterogeneously expressed within tumors, which could limit potential therapeutic efficacy when targeting a single CGA (36).
TSAs are genetically encoded in cancer cells but are not present in the genome of any normal cells. TSAs are further classified as viral antigens or neoantigens.
Many human cancers are caused by viral infections, such as human papillomavirus (HPV) (37), hepatitis B virus (HBV) (38), and Epstein-Barr virus (EBV) (39, 40). In many cases, virus-driven cancers are mediated by the expression of viral oncogenes that drive cellular transformation and cancer progression (41–43). As viral oncogenes are often homogenously expressed in virus-driven cancers, and their expression is nearly absent in normal cells, they represent highly attractive tumor antigen targets. Specific examples of clinically relevant viral antigens include the HPV viral oncogenes E6 and E7, which are expressed in several types of epithelial carcinoma (37, 44, 45), and the EBV viral oncogenes LMP1 and LMP2, which are expressed in several solid and hematological cancers (40, 46–49).
Genomic instability is a cardinal feature of cancer (50), which results in the accumulation of many tumor-specific mutations. Some of these mutations will give rise to new proteins, or neoantigens. As neoantigens are expressed exclusively by cancer cells, these serve as attractive targets for ACT that would pose essentially no risk for on-target off-tumor toxicity. However, a challenge is that the vast majority of cancer mutations are so-called bystander mutations, which do not enhance the fitness of the cancer cell. Such random, non-selected mutations are typically heterogeneously expressed and are unlikely to be shared across patients, rendering them ineffective antigen targets. Conversely, a small fraction of cancer mutations improve cellular fitness and directly promote cancer progression, which are known as driver mutations. These mutations may be expressed homogenously by cancer cells and shared among patients within particular cancer types (51–54). If immunogenic and restricted to a common HLA, such driver mutations give rise to so called ‘public neoantigens’ (55–57). While few public neoantigens have been discovered so far, these highly selective targets are of significant clinical interest. Examples of currently described public neoantigens include KRAS G12D/G12V, collectively found in 60-70% of pancreatic adenocarcinomas and 20-30% of colorectal cancers (58), and PIK3CA H1047L, detected in about 5% of metastatic breast cancers (59, 60).
V(D)J recombination of TCRs during thymic development results in a tremendous diversity of TCR sequences within the human T cell repertoire. It is estimated that in an average adult human, there are approximately 4 x 1011 total circulating T cells and an estimated 1010 unique T cell clonotypes (61). Thus, for the vast majority of T cell clones with specificity towards non-viral antigens, the clonal frequency in peripheral blood is far below what is needed to perform the various manipulations required to isolate antigen-specific TCRs given current technologies. Therefore, TCR isolation efforts generally begin with a method that allows for enrichment of T cells with the desired antigen specificity. The following section describes several common T cell enrichment methods employed for TCR discovery.
In certain types of solid cancers there is often a large presence of tumor infiltrating lymphocytes (TILs) (62). Compared to peripheral blood T cells, T cells within the tumor tissue are often enriched in clones with tumor-antigen specificity. Several groups have used expanded TILs as sources for discovery of tumor specific TCRs (63–67). Of note, for decades ex vivo expanded TILs have in themselves served as an effective ATC for several types of solid cancer (66–70). The TIL therapy lifileucel from Iovance has demonstrated strong efficacy in clinical trials and the company plans to submit for FDA approval (71).
T cells with specificity towards antigens of interest can be selectively expanded in vivo through vaccination strategies. A common approach is to vaccinate human HLA transgenic mice with an antigen of interest, which can result in robust enrichment of antigen-specific T cells harvested from the lymph nodes and spleen (46, 58, 72). In select cases, the peripheral blood of patients participating in cancer vaccine trials has been used as a source of antigen-enriched T cells for TCR discovery (73, 74).
Several strategies have been developed to stimulate peripheral blood T cells in vitro in an antigen-specific manner, driving the selective expansion of T cells with a desired specificity. Early pioneering work in this regard performed in vitro stimulations of peripheral blood T cells to preferentially expand virus-specific T cells (75–78). These stimulation methods have since been used to expand T cells enriched in specificity for TAAs (79–81) and neoantigens (60, 82, 83). These approaches typically stimulate T cells via autologous antigen presenting cells (APCs), usually dendritic cells (DCs), pulsed with the antigens of interest in the form of exogenous peptide or through cDNA/RNA delivery (60, 80, 82–84). In the case of patient-derived peripheral blood, several studies have shown that initial selection of PD-1+ and/or antigen-experienced (CD45RO+CD62L+, CD45RO+CD62L-, or CD45RO-CD62L-) T cells can further enhance in vitro enrichment of tumor-specific T cells (82, 83, 85–87).
To overcome the requirements of generating autologous mature DCs for antigen stimulation, several groups have developed so called artificial antigen presenting cells (aAPCs). One common aAPC system uses the myelogenous leukemia cell line K562, which is negative for HLA-A, B, and DR. This cell line serves as a modular aAPC through the stable transduction of various HLA alleles and costimulatory molecules. Other cell-free aAPC systems have been developed that conjugate HLA and costimulatory molecules onto beads and nanoparticles (88–90).
After obtaining polyclonal T cell products that are enriched for T cells with specificities of interest, it is necessary to isolate the antigen-specific T cells from the bulk T cell population.
Approaches in this regard typically involve stimulating T cells with the cognate antigen of interest, and then isolating antigen-responsive T cells based on increased expression of known T cell activation-associated molecules. This includes antibody staining of transmembrane proteins that are transiently upregulated following T cell stimulation (e.g., 4-1BB and OX40 in CD8+ and CD4+ T cells, respectively), allowing for isolation of these cells by FAC sorting or magnetic bead separation (82, 83). Another approach is IFN-γ-capture, whereby antigen stimulated T cells are identified and captured based on production of IFN-γ, which is rapidly secreted by antigen-stimulated CD8+ and Th1 CD4+ T cells. In certain cases, staining with peptide-HLA multimers followed by FAC sorting or magnetic bead separation is an efficient method to identify and isolate antigen specific T cells (63). However, this requires upfront knowledge of an antigen restricting HLA and minimal epitope. Although the repertoire of HLA multimer reagents is expanding, these reagents remain limited to relatively common HLA alleles.
TCR α and β chains of T cells of interest are then cloned from cDNA through PCR amplification. However, a unique challenge is that their 5’ regions are highly variable. To overcome this, one of two PCR variations are typically employed, 5’ RACE or multiplex PCR (66, 67, 73, 79, 82, 91). The fact that TCR specificity is encoded by regions of two separate genes imposes a unique challenge for determining functional TCR sequences from a population of T cells; that is, once T cells are lysed for RNA extraction, the TCR α and β transcripts from each T cell clone intermix, making it ambiguous as to which TCR α sequence pairs with which TCR β sequence. Therefore, prior to sequencing the functional TCR α and β chain transcripts of antigen-specific T cells, it is typically required to first separate individual T cell clones. While by no means exhaustive, we outline here several classical and emerging strategies to isolate T cell clones for TCR sequencing.
A classical method for obtaining T cell clones is the outgrowth of T cell clones in individual wells. In the limiting dilution method, T cells are diluted to obtain a cell concentration allowing for approximately one cell to be deposited into each well of a 96-well plate. An alternative method is to FACs sort the T cell population to deliver a single cell into each well. The goal is to obtain expanded clonal populations of the T cells of interest, which can then be additionally screened for antigen-specificity and sequenced via Sanger sequencing (27, 46, 72, 73, 84, 92–97).
Several studies have obviated the need to expand T cell clones following antigen-specific T cell separation by instead performing single cell RT-PCR to amplify TCR α and β chains. In such methods, single T cells are FAC sorted into wells containing RT-PCR reaction buffer, and from a single cell RT-PCR is performed and the TCR α and β chains are PCR amplified (66, 79, 82, 83, 91, 98). This method reduces the time and labor required for expansion of individual T cell clones; however, a downside to this approach is that confirmatory assays to assess antigen specificity cannot be performed on the T cell clones prior to sequencing.
Single-cell RNA sequencing (scRNAseq) is a rapidly advancing technology that has emerged as a uniquely effective platform for TCR discovery, as it allows for single-cell assessment of cellular gene expression as well as the sequence of gene transcripts. As such, several recent studies have successfully used this platform for TCR discovery by stimulating T cells with antigens of interest and then performing scRNAseq. This allowed the researchers to identify antigen specific T cells through their increased expression of effector cytokines such as IFN-γ, TNF-α, and/or IL-2, and from this same data set the researchers then obtained the sequences of transcripts for the TCR α and β chains from the activated cells (60, 65, 99).
TCR discovery efforts often yield sequences of several TCRs with a desired antigen specificity. How does one select an optimal TCR from this list of candidates? And once a lead TCR is selected, what preclinical evaluations can be performed that may predict the likelihood of clinical success? The following section describes several TCR features that are commonly evaluated in preclinical TCR studies.
Often the first characteristic of an isolated TCR that must be determined is its HLA restriction (Figure 3). Not only is understanding the HLA restriction of a TCR necessary to identify patients that may respond to a TCR T therapy, it is also needed to perform many of the experiments for preclinical assessment. In the case of several TCR discovery approaches, knowledge of HLA restriction is already incorporated into the pipeline, such as in the case of vaccination of HLA-transgenic mice or the use of HLA tetramers to select T cells, in which case the HLA restriction of resulting TCRs will be near certain. However, in TCR discovery approaches that do not incorporate a priori knowledge of HLA restriction, such as through stimulating with autologous DCs, HLA restriction must be determined experimentally. A commonly used approach is to deliver individual cloned HLA alleles into the non-human primate COS-7 cell line, which possesses antigen processing and presentation capabilities but does not express potentially confounding endogenous human HLA alleles. The COS-7 cells are then induced to express one of the HLA candidates and the antigen of interest through delivery of cDNA/RNA or peptide loading, and cocultured with TCR T cells. Here, the antigen restricting HLA is evident as the HLA that elicits a TCR T cell response, observed through functional responses such as cytokine secretion and/or 4-1BB/OX40 upregulation (58, 66, 82, 83, 87, 93, 100–104).
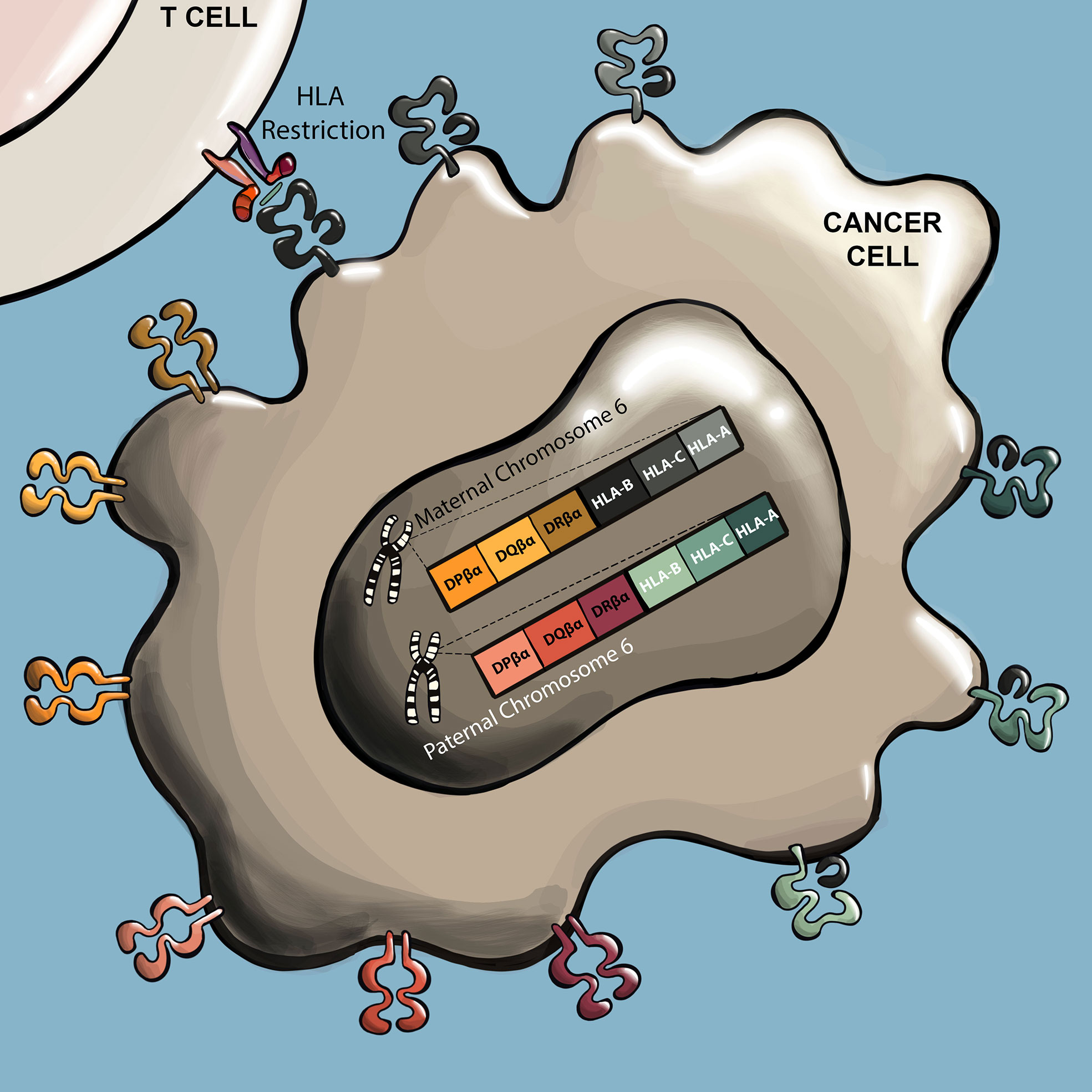
Figure 3 TCRs recognize antigens presented by specific HLA alleles. TCR antigens are predominantly presented by six HLA genes. These include genes for HLA class I (A, B, and C), and class II (DR, DP, and DQ). These HLA genes are highly polymorphic, with many allele variants in the human population. Humans inherit one set of each gene from each parent, and human cells can therefore express up to twelve different HLA presenting alleles. For a given TCR, the specific HLA allele that presents the cognate peptide is referred to as the ‘restricting HLA’ of the TCR.
The successful interaction between a TCR and the appropriate pMHC complex is a critical component of effective antitumor immune responses. TCR affinity and avidity describe the binding and kinetic interactions between the TCR and the pMHC (105). Affinity plays a central role in TCR sensitivity and specificity, and refers to the physical strength of the interaction (105–107). Affinity is quantified via surface plasmon resonance (SPR), a 3D interaction that measures binding in terms of an association rate (Kon) and a dissociation rate (Koff) (105, 107, 108). Together, Koff and Kon make up the binding constant (KD), where KD = koff/kon (105, 107). High affinity TCRs recognize lower levels of antigen, do not require the CD8 coreceptor, and can enable CD4+ T cells to recognize and lyse tumor cells in an MHC class I-dependent manner (106, 109).
TCR avidity usually correlates with affinity, and refers to the combined effect of multiple TCR-pMHC interactions, coreceptors (CD8), TCR density, and T cell functional status (105, 110). Different aspects of avidity (i.e. structural or TCR avidity) can be measured via staining with pMHC monomers or multimers with defined valency (105). TCR affinity, avidity, and the various kinetic constants all contribute directly/indirectly to functional avidity, which describes how well T cells expressing a specific TCR respond to decreasing abundance of peptide, and is sometimes referred to as antigen sensitivity (105, 108, 111). Assessments of TCR functional avidity typically include measurement of TCR T cell cytokine secretion or cytolytic function in response to target cells that are pulsed with titering concentrations of peptide.
HLA class I epitopes are peptide fragments, typically 8-12 amino acids in length (112, 113), generated through processing of ubiquitinated proteins by the proteasome. The proteasome is a large protein complex responsible for the degradation of endogenous proteins that have been damaged or are not needed by the cell and have been tagged by ubiquitin conjugation. The subunits β1, β2, and β5 of the proteasome’s 20S catalytic core are associated with the three major catalytic activities of the proteasome. While proteasomes that incorporate subunits β1, β2, and β5 are referred to as the ‘standard proteasome’, hematopoietic cells and cells stimulated with certain inflammatory cytokines (e.g., INF-γ, IFN-α, IFN-β, and TNF-α) alternatively express β1i, β2i, and β5i subunits that displace β1, β2, and β5 subunits in the proteasome, forming an isoform termed the ‘immunoproteasome’. The immunoproteasome displays several biochemical differences that influence peptide cleavage activity. This results in the immunoproteasome producing peptide products with enhanced immunogenicity compared to the standard proteasome, as these immunoproteasome-generated peptides are more likely to contain C-terminal hydrophobic residues, which are associated with more efficient HLA-class I binding (24, 114). In addition, there are ‘intermediate proteasomes’ that contain a mixture of standard and immunoproteasome subunits, specifically substituting only β5i or β1i plus β5i and result in a peptide repertoire similar to that produced by the immunoproteasome, but includes additional unique peptide products (24, 115).
Several tumor antigens have now been characterized as being produced by the standard, intermediate, and/or immunoproteasomes (101, 115–118). Because the dendritic cells used to enrich tumor specific T cells express predominantly intermediate and immunoproteasomes (115), it is likely that most preclinical TCRs will recognize antigens produced by immunoproteasome and/or intermediate proteasome. These cognate peptides may or may not be additionally produced by the standard proteasome. The proteasomal requirements of a cognate peptide can be determined in several ways. Many cell lines predominantly express the standard proteasome, but will upregulate immunoproteasome subunits in response to IFN-γ. Therefore, the response of TCR T cells can be compared against antigen expressing cell lines with or without pretreatment with IFN-γ (73, 101, 116, 117). Peptide proteasomal requirements have also been determined with further resolution by testing T cell responses against antigen expressing 293 cells with or without overexpression of specific inducible proteasomal β subunits (101, 115–117, 119). There is a growing appreciation of the importance of proteasomal processing dynamics in immunotherapies such as immune checkpoint blockade (120) and TCR T therapy (121). In cases in which a therapeutic TCR recognizes a peptide processed exclusively by the immunoproteasome, it may be useful to select patients whose tumors have confirmed expression of immunoproteasome subunits. As downregulation of immunoproteasome subunits has now been observed in some cancer types (122, 123), it is likely ideal for a therapeutic TCR to recognize an antigen that is generated by both the standard and immunoproteasome.
Of major importance is the identification of potential safety concerns of lead TCRs, as previous clinical trials have observed severe cases of both on-target off-tumor toxicities (124–126) and off-target toxicities (127, 128). This section describes state-of-the-art techniques to assess the safety of preclinical TCRs and their cognate-antigen targets.
In cases in which a candidate TCR targets a novel or putative TAA or CGA epitope, it is imperative to preclinically assess its pattern of expression and HLA-presentation in healthy tissue. The importance of such validation was made clear by the fatal neurotoxicity that occurred in two patients following administration of T cells expressing an affinity enhanced TCR recognizing an epitope shared by MAGE-A3 and MAGE-A12. Autopsy performed on these patients revealed infiltration of CD3+CD8+ T cells in the brain. Further investigation identified unexpected expression of MAGE-A12 in a subset of neurons in the human brain (129). Several strategies are now available for preclinical assessment of expression profiles of putative CGA or TAA targets that should be employed. Kunert et al. provide suggested strategies based on their experience of assessing the expression profile of a MAGE-C2 derived epitope that is now being targeted clinically (NCT04729543). As an early step in the assessment of putative TAAs or CGAs, the authors suggest consulting online databases such as The Human Protein Atlas (proteinatlas.org) and the CTdatabase (cta.lncc.br) (130), which compile extensive data throughout literature concerning RNA and protein expression of many genes in both healthy and cancerous tissue (131). An additional tool that has recently emerged for the validation and/or discovery of CGAs and TAAs is the HLA Ligand Atlas (hla-ligand-atlas.org), an open-source, community resource of comprehensive human HLA ligandome data collected originally from 29 distinct non-malignant tissues derived from 21 individuals (113). To experimentally and independently evaluate antigen expression in healthy tissue, Kunert et al. suggest performing qPCR on commercially available cDNA libraries derived from a wide array of healthy tissues types, and if possible, further evaluating protein expression by performing IHC on a panel of healthy tissue types (131).
In addition to establishing the safety of the cognate-antigen target, it is of critical importance to investigate potential off-target reactivities of candidate TCRs. The clinical importance of such investigation was highlighted by the deaths of two patients resulting from off-target reactivity of an affinity-enhanced MAGE-A3-specific TCR towards a Titin-derived epitope expressed in cardiomyocytes (127, 128). Several preclinical strategies are now commonly employed by investigators to identify possible off-target reactivities of candidate TCRs. Several groups have identified specific amino acids within the cognate peptide that are necessary for TCR recognition. This is accomplished by mutating each residue within the cognate peptide and identifying the mutant versions unable to elicit a T cell response. The investigators then searched for all other human peptides containing an identical or similar amino acid motif through the use of webtools such as BLAST and ScanProsite, and then assessed whether these structurally similar peptides elicited a response by the candidate TCR (60, 63, 131–134). In cases in which one or several off-target peptides were identified, the researchers further investigated the immunogenicity of these peptides by determining TCR T cell response at titering concentrations (63, 131, 134) or determining if the off-target peptide is actually capable of being naturally processed (132, 133). While this approach is highly valuable for identifying cross-reactive peptides that are structurally similar to the cognate peptide, it would not identify structurally dissimilar peptides that mediate cross-reactivity (135).
Several groups have also assessed potential alloreactivity of therapeutic TCRs by performing functional assays in which TCR T cells are cultured with many different lymphoblastoid cell lines (LCLs) expressing various HLA alleles (131–133). The utility of this approach is highlighted by Sanderson et al., who identified that a lead HLA-A*02:01 restricted MAGE-A4 specific TCR mediated an alloresponse to HLA-A*02:05, indicating that patients that express HLA-A*02:05 should be excluded from treatments using this TCR (133).
TCR α/β chains form heterodimers largely through interactions within TCR constant regions. A challenge facing TCR T applications is that the endogenous TCR α/β chains expressed by conventional T cells can pair with introduced TCR α/β chains. Several consequences arise from such TCR mispairing. Firstly, TCR mispairing reduces the surface expression of introduced TCRs, as a significant fraction of the introduced TCR α/β chains will participate in non-productive mispairings with endogenous TCR α/β chains. Furthermore, mispaired TCRs compete with the engineered TCR heterodimer for association with limiting CD3 components (136). A second consequence of TCR mispairing is the production of brand new TCRs that have not undergone thymic selection, and which may have unexpected specificity for autoantigens. Indeed, TCR mispairing was shown to cause lethal graft-versus-host-disease (GVHD) in mice (137), and led to the formation of alloreactive and autoreactive human T cells in vitro (138). However, incidence of GVHD has not been observed in human TCR T clinical trials to date, including early TCR T trials utilizing unmodified human TCRs (139). To avoid issues associated with TCR mispairing, the vast majority of TCR T applications now use at least one strategy to reduce TCR mispairing (Figure 4).
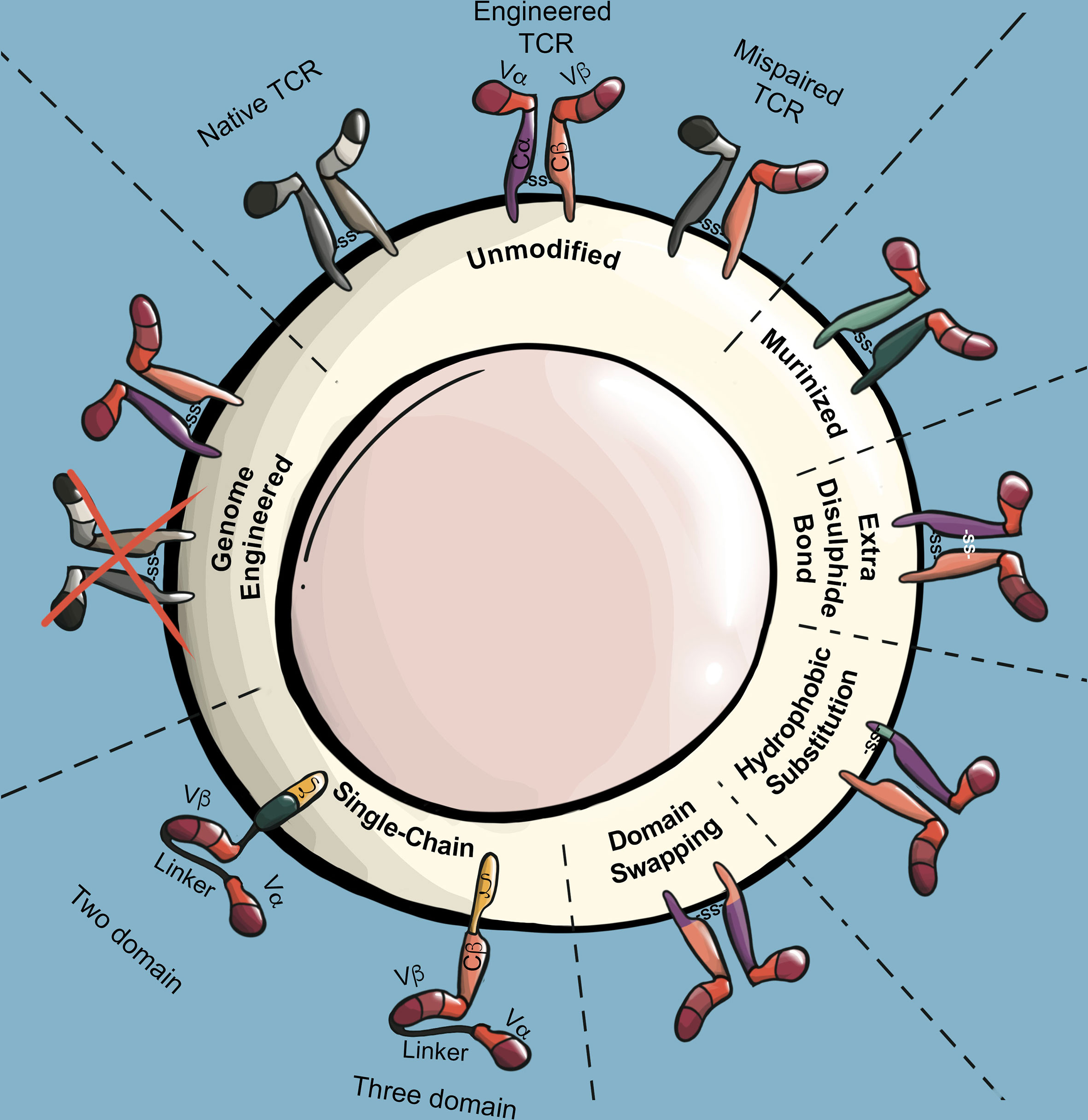
Figure 4 TCR modifications to prevent mispairing and maximize surface expression. Illustration of mispairing between endogenous TCR and engineered TCR. Murinized TCRs replace the human TCR constant regions with those of a mouse TCR constant region. The addition of an extra disulfide bond in the TCR constant region through cysteine substitutions stabilizes interchain binding affinity of engineered TCR α/β chains while reducing their binding affinity with endogenous TCR α/β chains. Stability of the engineered TCR α chain can be increased through select hydrophobic substitutions in its transmembrane region. Domain swapped TCRs invert large or specific segments of the engineered TCR α/β constant regions, which reduces propensity of engineered TCRs to mispair. Single-chain TCRs (scTCR) encode TCR antigen recognition and signaling domains into a single chain. Three-domain and two-domain scTCRs differ by the inclusion or absence of the TCR β constant region, respectively. Genome engineering strategies utilize RNA interference or endonuclease technologies to reduce or ablate endogenous TCR expression.
Extending on the serendipitous observation that human T cells exhibited greater biological activity when engineered with a murine-derived TCR as compared to human-derived TCRs, Cohen et al. demonstrated that murine TCR α/β chains preferentially dimerize with each other in the presence of endogenous human TCR α/β chains. The investigators further demonstrated that preferential pairing of introduced TCRs is also achieved when the constant regions of human TCRs are replaced with murine constant regions (140). A concern of using engineered TCRs with murine constant regions is that the foreign murine sequences may elicit an immune response in patients, as has been observed in other cell therapy trials utilizing foreign proteins such as green fluorescent protein (141, 142) and the HyTK suicide gene (143). One study identified anti-murine TCR antibodies in the post-treatment sera of 6/26 patients treated with TCR T cells expressing fully murine TCRs. However, epitope mapping revealed that the antibodies were specific for the variable regions of the TCRs, not the constant regions (144). In a separate TCR T trail utilizing a human TCR with murinized constant regions, anti-TCR serum antibodies were not detected in any of the 11 patients screened (44). Together, these clinical findings suggest that murine TCR constant regions have low or negligible immunogenicity. Nonetheless, strategies have also been developed to partially murinize TCRs by substituting specific murine amino acid sequences (145, 146).
Endogenous TCR α/β chains form a disulfide bond between TCR α constant region (Cα) residue 94 and TCR β constant region (Cβ) residue 130 (147). The proper pairing of introduced TCRs can be improved by introducing a second stabilizing disulfide bond through cysteine substitutions at Cα residue 48 and Cβ residue 57, which increases interchain binding affinity of introduced TCR α/β chains while decreasing binding affinity with endogenous TCR α/β chains (148, 149).
The endogenous TCR α chain has a relatively low stability, which can be increased by substituting leucine and valine residues within the Cα transmembrane region. TCR α chains containing these stabilizing mutations, termed α-LVL, demonstrate increased TCR surface expression and biological activity. While this strategy promotes pairing of an introduced TCR by stabilizing the TCR α chain, the TCR β chain remains unmodified and thus susceptible to mispairing. However, this can be addressed by incorporating the α-LVL substitutions into murinized TCRs, the combination of which can synergistically enhance TCR expression and biological activity (150).
Through TCR crystal structure analysis, Voss et al. identified several amino acids mediating TCR α/β dimerization. By swapping two such interacting residues, a Cα glycine and Cβ arginine, the authors generated mutant TCR α/β chains with a similar propensity for dimerizing with each other, but with a significantly reduced propensity to bind with unmodified endogenous TCR α/β chains (151). Bethune et al. employed a similar strategy when they designed TCRs with large regions of Cα and Cβ segments exchanged, referred to as dsTCRc. Interestingly, mispairing of introduced dsTCRc α/β chains with endogenous α/β chains was completely undetectable (152). Other examples of TCR domain swapping/conjugation strategies include swapping with γδ TCR constant regions (152), replacing regions with CD3ζ (153, 154) or CD28/CD3ϵ (155), or conjugation to leucine zipper dimerization motifs (156, 157).
To combine the antigen recognition properties of a TCR α/β heterodimer into a single chain, several groups have developed so-called three-domain single-chain TCRs (scTCR), which are composed of Vα/Vβ regions fused by a short peptide linker and conjugated to a Cβ domain (153, 158, 159). To mediate signal transduction, three-domain scTCRs are typically further conjugated to CD3ζ (153, 158, 160). Zhang et al. compared the function of nine three-domain scTCR constructs conjugated to CD3ζ with or without conjugation to the additional stimulatory domains CD28 and Lck. Although the addition of both CD28 and Lck improved scTCR function, none of the scTCR constructs performed as well as native TCRs in terms of functional avidity (160). scTCR constructs utilizing CD3ζ transmembrane and signaling domains function independently of the CD3 complex (160), which theoretically allows for higher surface expression to be achieved with scTCRs than with native TCRs, as scTCRs are not limited by the abundance of CD3 components. The CD3-independence of scTCRs may also be beneficial in applications where it is desirable to maintain levels of endogenous TCR expression. However, scTCRs utilize signaling mechanisms distinct from those of CD3-dependent native TCRs, which may partially explain the reduced functional avidity of scTCRs (160). To generate scTCRs that preserve CD3-dependence, Voss et al. designed a system whereby three-domain TCRs without CD3ζ conjugation were coexpressed with a Cα domain. In this manner, the three domain scTCR dimerizes with the coexpressed Cα domain, presenting at the cell surface in a four-domain structure similar to that of a native TCR heterodimer. Intriguingly, these scTCR/Cα constructs have similar functional avidities as native TCRs (161). However, Aggen et al. demonstrated that three-domain scTCRs continue to mispair to some extent with endogenous TCR α chains due to the presence of the Cβ domain. To generate a scTCR system that completely eliminates mispairing, the group generated two-domain scTCRs, which utilize stabilizing Vα/Vβ mutations to obviate the need for a Cβ domain (162). To mediate signaling, two-domain scTCRs are conjugated to intracellular signaling domains such as CD3ζ and CD28. Intriguingly, CD3ζ/CD28 containing two-domain scTCRs are essentially CARs that utilize a Vα/Vβ antigen recognition domain. As such, two-domain scTCRs display features typical of CARs including CD3-independent signaling and decreased sensitivity to low antigen density. However, unlike typical CARs, two-domain scTCRs are still dependent on HLA presentation (14).
Rather than modifying the introduced TCR, other strategies address mispairing through knock-down or knock-out of the endogenous TCR. Provasi et al. combined the use of zinc-finger nucleases (ZFN) to knock-out endogenous TRAC and TRBC genes with lenti-viral delivery of a WT-1 specific TCR. Here, the authors described an elegant, although relatively extensive manufacture system utilizing sequential rounds of TRAC/TRBC disruption, magnetic bead separation, and TCR α/β chain delivery. This resulted in a TCR T product with enhanced expression of the introduced TCR and a complete absence of endogenous TCR α/β chains (163). In a recent first-in-human trial, Stadtmauer et al. employed multiplexed CRISPR/Cas9 editing to disrupt T cell TRAC, TRBC, and PDCD1 genes in combination with lenti-viral delivery of a NY-ESO-1 specific TCR. In the four patient-derived products described in this report, disruption of TRAC and TRBC was achieved in an average of 45% and 15% of cells, respectively. However, as TRAC/TRBC edited T cells were not selected prior to lenti-viral transduction with the NY-ESO-1 TCR, a significant fraction of the TCR-engineered T cells likely continued to express endogenous TCR α/β chains (164). Several groups have also developed virus-free systems to deliver TCRs and/or disrupt endogenous TRAC/TRBC genes, which may also aid in improving TCR T clinical cost and feasibility. Davo et al. electroporated T cells with Dicer-substrate small interfering RNAs (DsiRNA) targeting the endogenous TRAC and TRBC loci, achieving an approximately 6-fold and 3-fold reduction in expression of TRAC and TRBC, respectively. The authors then electroporated the T cells with a codon optimized WT1-specific TCR that isn’t recognized by the DsiRNA. This resulted in T cell products with relatively high engineered TCR expression (60.2% tetramer+) with no observable TCR mispairing. However, as transgene expression in this system is transient, this would likely necessitate multiple infusions in a clinical setting (165). Roth et al. used CRISPR/Cas9 editing to mediate targeted insertion of TCR α and β variable regions into the first exon of the TRAC and TRBC loci, respectively. This mediated the combined effect of disrupting the endogenous TCR α/β chains, while placing expression of the introduced TCR under physiologic control. A potential challenge of this approach is the relatively low editing efficiency, with about 3% of cells expressing the introduced TCR, which could therefore necessitate sorting and/or extended selective expansion (166).
Given that affinity plays a central role in TCR function, the manufacturing of high-affinity TCRs is an attractive method to improve the efficacy of TCR T therapies. Naturally occurring TCRs, including those that recognize self/tumor antigens, have relatively low affinities as a result of negative selection (105, 167). Methods to improve TCR affinity focus on the introduction of amino acid sequence (AAS) variations into the TCR complementarity-determining regions (CDRs). For example, single and dual AAS substitutions that enhanced the functions of TCRs specific for NY-ESO-1 (1G4) and MART-1 (DMF4, DMF5) were generated through overlapping PCR (168). These TCRs had affinities in the low μM and even nM range, which surpass the affinity of most naturally occurring TCRs, which range from 1-100 μM (106, 110). Yeast and bacteriophage display are additional powerful, high-throughput tools that can generate TCRs with affinities in the pM range (109, 167, 169). While these techniques are effective at identifying high affinity TCR variants, higher affinity has been associated with increased cross-reactivity (106, 109, 168). TCRs with affinities greater than the normal range (1-100 μM) are more likely to demonstrate cross-reactivity to similar or completely different peptides (109, 135, 170). In various studies of high affinity TCRs, increasing the affinity within the nM and pM range resulted in recognition of control antigen and antigen negative target cells (109, 168). Efforts to improve the affinity of TCRs should thus proceed with caution and thorough evaluation as these high affinity TCRs could have detrimental effects when used as patient therapies.
Recent work in TCR affinity maturation has focused on incorporating a more thorough assessment of the structure of the TCR and how it interacts with the target pMHC. Hellman et al. utilized a structure-guided design that incorporated both positive and negative designs (106, 170). In other words, they utilized mutations that either enhanced or weakened the interaction of the TCR with the MHC protein. These mutations redistributed the binding free energy in a way that forces the TCR-pMHC interaction to rely more on the presence of the correct target peptide, leaving less flexibility for off-target peptides. In the MART-1 specific DMF5 TCR, these structure-guided modifications decreased cross-reactivity to MART-1 homologs and eliminated cross-recognition of a selection of divergent peptides. Structure-guided approaches, therefore, have the potential to improve ACT while minimizing the risk of off-target toxicities.
As of October 3rd, 2021, the search term “TCR” (and synonyms “T Cell Receptor” and “T Cell Antigen Receptor”) in clinicaltrials.gov yielded 538 interventional trials. Through manual inspection of these trials, 119 were identified to include the adoptive transfer of TCR T cells. One TCR T trial that did not include these search terms (NCT04044768) was also identified and included in this analysis (Supplementary Table 1). The first TCR T trial was initiated in 2004 by Steven Rosenberg at the National Institutes of Health (NIH) targeting the melanoma differentiation antigen gp100 (NCT00085462). Since then, the number of new TCR T trials initiated has steadily increased, with a particular acceleration between the years of 2017 – 2019, in which 51 new TCR T trials were initiated (Figure 5A). Of the TCR T trials to-date, 53% have been in phase I, 24% in phase I/II, and 22% in phase II. To date, no phase III TCR T trials have been initiated (Supplementary Table 1). The status of these TCR T trials was assessed as of November 3rd, 2021 (Figure 5B). There were 118 antigens targeted in the 116 TCR T trials with specified targets. Of the targeted antigens, the majority are CGAs (47%), followed by viral antigens (24%), tumor-associated antigens (21%), neoantigens (7%), and fetal oncogenes (3%) (Figure 5C). The CGA NY-ESO-1 is by far the most targeted antigen in TCR T trials to-date (36 trials). Although not first targeted until 2014, HPV now constitutes the second most common TCR T target (10 trials). While the melanoma differentiation antigen MART-1 is the third most targeted antigen in TCR T trials (7 trials), these largely constitute early TCR T trials, as MART-1 targeted-trials have not been initiated since 2012 (Figure 5D). The majority of the TCR T trials to date have been for the treatment of solid cancers (85%), followed by hematological malignancies (9%), and trials targeting both solid and hematological cancers (2%). A small subset of TCR T trials have been for the treatment of HIV, CMV, or EBV infections (4%) (Figure 5E). More information about the precise disease targets of these TCR T trials can be found in Supplementary Table 1. Of the 100 trials with specified HLA-restrictions, 111 restricting-HLA alleles were listed, as some trials included multiple antigen targets and HLA restrictions. HLA-A*02 was by far the most common restricting HLA allele (80%), followed by HLA-A*11 (7%) and HLA-A*24 (5%). HLA class II restricted antigens were targeted in 3 trials, all of which were restricted by HLA-DP*04 (3%) (Figure 5F). The majority of TCR T trial locations were in the United States (56%), followed by China (18%), and the United Kingdom (6%) (Figure 5G). Among the 80 TCR T trials occurring in the United States, 44% were sponsored by the NIH, 28% by academic institutions, and 29% by industry. The support of industry in TCR T, which accelerated around 2017 (Supplementary Table 1), will likely aid in the development of later phase TCR T trials. The NCI has been by far the most active individual institution in United States TCR T trials, sponsoring 31 trials to date. Among academic institutions, the Fred Hutchingson Cancer Research Center has sponsored the most trials (7 trials), followed by the Johnson Comprehensive Cancer Center (6 trials). Among TCR T trials sponsored by pharmaceutical companies, Adaptimmune and GlaxoSmithKline have sponsored the most trials (8 trials each) (Figure 5H).

Figure 5 Trends in TCR T trials initiated thus far. TCR T trials registered in clicaltrials.gov were assessed as of October 3rd 2021. (A) The number of new TCR T trials initiated each year and the cumulative number of registered TCR T trials by year. (B) Clinical status of the 120 TCR T trials. (C) Classifications of 118 tumor antigen targets in 116 TCR T trials with specified target antigens. (D) Ten most common targets in TCR T trials. (E) Diseases targeted in TCR T trials. (F) Frequency of 111 target antigen-restricting HLAs in 100 TCR T trials that specified HLA restriction. (G) Locations where TCR T trials have been conducted by country. (H) Primary sponsors of the 80 TCR T trials conducted in the United States.
There is a quickly expanding body of literature detailing the results of TCR T trials. Clinical results encompassing over twenty-five TCR T trials are detailed in Table 1. This section discusses broad findings that have emerged from these early phase trials, particularly relating to TCR T safety and efficacy. Of note, many TCR T clinical protocols include lymphodepleting regimens prior to TCR T infusion, which has been demonstrated in early ACT trials to improve T cell engraftment and persistence. Many studies also include systemic administration of IL-2 following TCR T infusion to support T cell activity and persistence. Both interventions consistently induce various toxicities that, while undesirable, are generally clinically manageable. Detailed description of the impact of these interventions is beyond the scope of this review, but is extensively reviewed elsewhere (189–191). As such, discussion of toxicities observed in TCR T trials will focus on those mediated directly by the infused T cells. Finally, it is worth noting that to date all TCR T trials have been early phase and almost exclusively treating patients with highly advanced, treatment refractory disease.
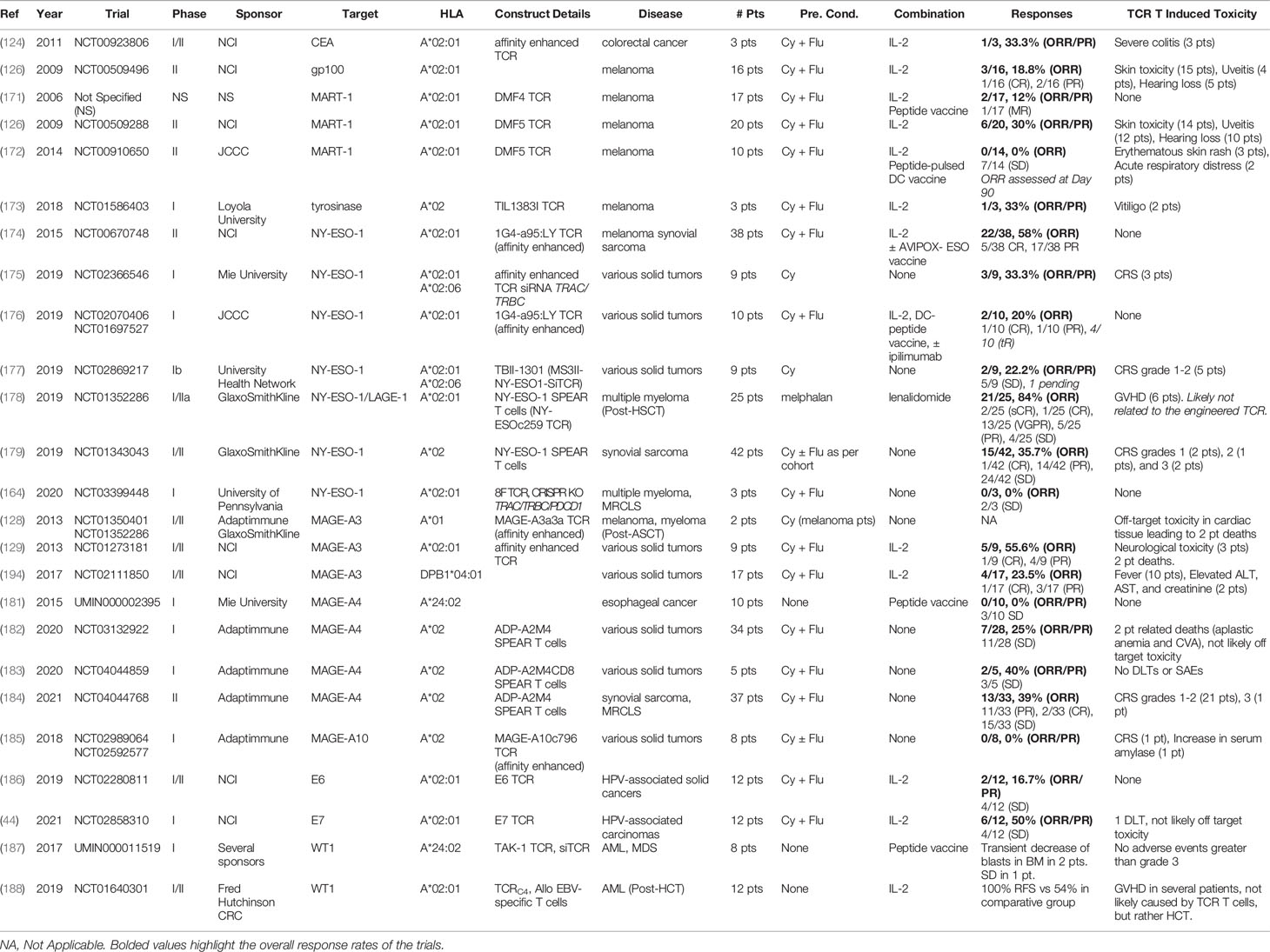
Table 1 Available TCR T trial results.
In one of the earliest TCR T trials, patients with metastatic melanoma were treated with autologous T cells transduced with a TCR (DMF4) recognizing the melanoma differentiation antigen MART-1. The objective response rate in these patients was relatively modest, with 2/17 (12%) patients achieving durable partial responses. No TCR T induced toxicity was observed (171). In a later related study, patients were treated with TCR T cells that expressed a higher affinity MART-1 specific TCR (DMF5). Compared to the prior trial using the lower affinity DMF4 receptor, this trial observed enhanced efficacy, with objective responses observed in 6/20 (30%) patients. However, the increased biological activity mediated by the DMF5 TCR was also associated with the emergence of on-target off-tumor destruction of melanocytes, leading to widespread erythematous skin rash (14 pts), uveitis (12 pts), and hearing loss (10 pts). Similar results were observed in patients treated with T cells expressing a high affinity mouse-derived TCR targeting the melanoma differentiation antigen gp100 (126). In a later study, three patients with metastatic colorectal cancer were treated with an affinity-enhanced TCR recognizing the cancer differentiation antigen carcinoembryonic antigen (CEA). While a partial response was observed in 1/3 (33%) patients, all three patients presented with severe transient colitis as a result of on-target off-tumor destruction of CEA-expressing colonic mucosa (124). Ultimately, these studies demonstrated that while TCR T therapies targeting cancer differentiation antigens can mediate objective clinical responses, they are often associated with potentially dangerous on-target off-tumor toxicities. Likely owing to this, few TCR T trials targeting cancer differentiation antigens have been initiated in the past decade (Supplementary Table 1).
The CGA NY-ESO-1 has been the most widely targeted antigen in TCR T trials, and several groups have now published promising results from trials targeting this antigen. An early landmark trial treating patients with refractory melanoma or synovial sarcoma with TCR T cells expressing an affinity-enhanced NY-ESO-1 specific TCR reported objective responses in 11/20 (55%) melanoma patients, including four durable complete responses, and 11/18 (61%) synovial sarcoma patients, including one durable complete response. No TCR T associated toxicities were observed (174). Later, a large study of synovial sarcoma patients treated with TCR T cells expressing an affinity enhanced NY-ESO-1 specific TCR (SPEAR T cells) observed clinical responses in 15/42 patients (36%), including one complete response, and 24/42 patients presenting with stable disease. These patients were also divided into treatment cohorts based on magnitude of NY-ESO-1 tumor expression and the lymphodepleting regimen they received. The greatest clinical responses were observed in a cohort of twelve patients (cohort 1) whose tumor expressed +2 or +3 NY-ESO-1 staining by immunohistochemistry in ≥50% of cells and who received a relatively intensive lymphodepleting regimen. Here, clinical responses were observed in 6/12 (50%) patients, including one complete response, and a median duration of response of 30.9 weeks (179). Of these twelve patients, five experienced cytokine release syndrome of grades 1 (2 pts), 2 (1 pt), or 3 (2 pts) (192). Several other studies reporting TCR T trials targeting NY-ESO-1 to treat various cancer types have also observed clinical responses without instances of on- or off- target toxicity attributed to TCR T cells (164, 176–178, 193). Together, these studies demonstrate that NY-ESO-1 targeting TCR T therapies are safe and capable of eliciting potent antitumor responses.
The MAGE-A family of proteins have also served as highly attractive CGA targets, and results from several TCR T trials targeting members of this family are now available. Unfortunately, two early reports of trials targeting MAGE-A3 described fatal TCR T mediated toxicity. In one study, two patients treated with TCR T cells expressing an affinity enhanced MAGE-A3 specific TCR died of cardiac toxicity following TCR T infusion. Post hoc investigation into the cause of these fatalities revealed cross-reactivity of the affinity enhanced TCR towards Titin expressed in cardiomyocytes (127, 128). This study emphasized the need for extensive preclinical investigation into potential off-target reactivities of lead TCRs. In a study published the same year, nine patients with metastatic cancer were treated with TCR T cells expressing an affinity enhanced TCR recognizing a similar MAGE-A3 and MAGE-A12 epitope. Clinical responses were achieved in 5/9 (56%) of patients, including a durable complete response in one patient. However, three patients experienced severe neurotoxicity following TCR T cell infusion, with two patients dying as a result. Post hoc analysis identified unexpected expression of MAGE-A12 in a subset of neurons in the brain, and the observed toxicity was thus presumed to be due to on-target off-tumor recognition of MAGE-A12 the brain (129). This study demonstrated the need for extensive preclinical characterization of cognate antigen targets in healthy tissue. A later study targeted MAGE-A3 with a high affinity HLA-DPB1*04:01 restricted TCR derived from a T regulatory cell. Objective responses were achieved in 4/17 (24%) of patients, with one patient achieving a durable complete response. Following TCR T treatment, one patient experienced grade 4 toxicities including increased ALT, AST, and creatinine, and eventually developed respiratory failure requiring hospitalization. A second patient experienced grade 3 toxicities of increased ALT, AST, and creatinine lasting two days. The cause of these toxicities was not described (194). Two recent reports of phase I trials of TCR T cells expressing an affinity-enhanced TCR specific for MAGE-A4 (ADP-A2M4 SPEAR T cells) to treat various solid cancers observed antitumor responses without evidence of serious TCR T mediated toxicity (182, 183). This year, Adaptimmune reported results of a phase II trial of ADP-A2M4 SPEAR T cells (now afamitresgene autoleucel) to treat patients with synovial sarcoma or myxoid/round cell liposarcoma (MRCLS). Here, objective responses were observed in 13/33 (39.4%) patients, including two durable complete responses, and disease control was achieved in 28/33 (84.8%) patients. Grades 1-2 CRS were observed in 21 patients and grade 3 CRS was observed in one patient. Based on this data, the company plans to file for afamitresgene autoleucel approval next year (184). In summary, results of early TCR T trials targeting MAGE-A3 demonstrated the need for more extensive preclinical testing of both on-target and off-target effects of lead TCRs and enhanced preclinical safety assessment strategies have emerged as a result of these studies. However, recent trials of Adaptimmune’s MAGE-A4-targeting afamitresgene autoleucel have demonstrated strong efficacy treating solid cancers in the absence of major TCR T cell mediated toxicity and may be nearing FDA approval.
Recent results have demonstrated efficacy of TCR T cells targeting HPV antigens in patients with various HPV-associated solid cancers. Relatively modest efficacy was achieved in a study treating twelve patients with TCR T cells expressing an E6-specific TCR, with objective responses observed in 2/12 (17%) patients, with no dose-limiting toxicities (186). In a related study using a higher affinity TCR recognizing E7, objective responses were achieved in 6/12 (50%) patients, with one dose-limiting toxicity that was presumably unrelated to TCR T mediated toxicity (44). However, the duration of clinical responses observed in both trials were relatively short (2 – 9 months). Recent studies have also demonstrated safety and efficacy of TCR T trials targeting the cancer overexpression antigen WT1. In one study, twelve AML patients at high risk for relapse following hematopoietic cell transplant (HCT) were treated with allogenic EBV-specific T cells engineered to express a WT1 specific TCR. Relapse free survival was achieved in 100% of the TCR T treated patients, as compared to 54% relapse free survival post-HCT in a comparative group of eighty-eight AML patients at similar risk of relapse. Nine patients exhibited grade 1-2 GVHD following TCR T infusion, with one patient developing grade 3 acute GVHD. However, the GVDH was determined to be most likely caused by the use of allogenic T cells rather than the introduced TCR (188).
Recent years have seen the emergence of clinical trials reporting on the safety and efficacy of TCR T therapeutics in the context of both hematological malignancies and solid tumors (Table 1). Several TCR T trials focused on the latter have achieved improved therapeutic outcomes as compared to those achieved in CAR T trials (195), and it now appears that TCR T therapeutics are closer to receiving FDA approval for the treatment of solid cancers. A possible explanation for this disparity lies in the biological differences between TCRs and CARs (Figure 1), including 1) the ability of TCRs to recognize HLA-presented antigens derived from any cellular compartment including high specificity antigens such as CGAs and viral antigens, 2) TCRs are considerably more sensitive to low concentrations of a target antigen as compared to CARs, particularly in the case of affinity-enhanced TCRs (12–14), and 3) unlike CAR T cells, engineered TCRs do not drive ligand-independent tonic signaling (196, 197), making them better equipped to maintain function in vivo. Ultimately, the aforementioned factors are speculated to play a role in the comparatively improved performance of TCR T therapeutics targeting solid cancers, however, both fields are evolving rapidly with new antigen discoveries and further genetic engineering of the T cells, which should lead to improved efficacy in the upcoming solid tumor CAR T trials.
Intense efforts have been made to understand and address factors that limit the efficacy and clinical applicability of T cell-based ACTs, including TCR T therapy. This section describes select challenges currently facing TCR T therapy and the strategies available to meet them.
TCR T products typically require 1-3 weeks of manufacture and involve several relatively complex processing steps that must be performed in highly regulated good manufacturing process (GMP) facilities. Despite the relative complexity, current TCR T manufacturing processes generally result in high rates of successful product manufacture. However, the cost of these T cell products is high. This is demonstrated by the high costs of FDA approved CAR T therapies, which utilize similar manufacturing processes. For example, single infusions of axicabtagene ciloleucel or tisagenlecleucel cost $373,000 and $475,000, respectively (198). In response to this, a strategy under development by many groups is the use allogeneic, or off-the-shelf, TCR T and CAR T products. However, a major challenge facing this approach is the likelihood of GVHD mediated by allogeneic T cells. Strategies to mediate GVHD of allogeneic T cell products include the use of oligoclonal virus-specific T cells with tightly restricted antigen specificities (188, 199), invariant T cell subsets such as γδ T cells (200) and iNKT cells (201), or TRAC/TRBC disrupted T cells (202). Another strategy to reduce the manufacturing cost of both TCR T and CAR T products is the use of non-viral gene delivery methods such as RNA electroporation (203, 204), transposons (205–207), and CRISPR/Cas9 (166).
The ability of infused T cells to persist within the patient is an important factor mediating ACT antitumor efficacy (208). As such, intense efforts have been made to develop strategies that improve T cell persistence. Perhaps the most common strategy is to administer non-myeloablative lymphodepleting chemotherapy prior to T cell infusion, which alleviates competition of infused T cells with endogenous T cells for homeostatic cytokines such as IL-7 and IL-15, among other likely mechanisms (189). Several studies in mice have demonstrated that less differentiated T cell subsets (e.g., central memory and stem cell memory), have improved in vivo engraftment and persistence compared to highly differentiated T cells subsets (e.g., effector memory and terminally differentiated effectors) (209–211). As such, several strategies are now commonly employed to preserve less differentiated T cell subsets during ACT manufacture, including reduced expansion times and use of less differentiation-inducing cytokines (181, 212, 213). Some TCR T trials have further extended this approach by selecting specific T cell differentiation subsets prior to infusion (NCT02062359, NCT02408016). Another factor that limits T cell persistence is the paucity of costimulatory ligands within the tumor microenvironment. Several early TCR T trials performed coadministration of vaccines in efforts to provide TCR stimulation and costimulation in vivo. However, several of these studies failed to observe increased TCR T efficacy mediated by combination with vaccination (174, 176, 180). Genetic strategies to improve T cell costimulation include modified TCRs incorporating costimulatory domains (155) and coexpression of costim-only CARs (214) or domain swapped inhibitory receptors (215). Finally, several genetic strategies are also being explored to maintain T cell homeostatic cytokine signaling, including auto-secreting IL-15 or IL-12 elements (216–219) and a constitutively active IL-7 receptor (220).
Another obstacle faced by adoptively transferred T cells is the immunosuppressive tumor microenvironment (TME). The TME supports tumor survival by recruiting immunosuppressive cell types, including myeloid derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages (TAMs) (221). Several pre-clinical studies of ACT are working to incorporate strategies to enable T cells to function in the hostile TME, including cotreatment with immune checkpoint blockade (222, 223), and genetic incorporation of inverted cytokine receptors (224) or MDSC targeting costimulatory receptors (214). A thorough understanding of the TME and its effect on T cells is necessary for the future success of ACT for solid cancers.
TCR T efficacy is influenced by the heterogeneity of cognate-antigen expression by tumor cells. This is especially the case when targeting CGAs, such as NY-ESO-1 and MAGE-A family proteins, which are often heterogeneously expressed by tumors (36). Nonetheless, complete responses have been observed in patients treated with TCR T cells targeting NY-ESO-1 and MAGE-A family proteins (Table 1). This may be partially explained by a phenomenon known as epitope spreading, whereby the immune response mounted by the infused TCR T cells leads to priming and activation of endogenous T cells to other non-cognate tumor antigens. Indeed, epitope spreading has been observed in humans following vaccination (225) and ACT (226, 227). Several genetic engineering strategies have now been developed to promote epitope spreading, including constitutively expressing inflammatory cytokines IL-12 (217, 218) and IL-18 (228), CD40L (229), or the DC growth factor FLT3 (230). Another approach to address antigen heterogeneity is to genetically encode specificity towards multiple antigens. While several multitargeting approaches have been developed in the CAR T realm (231), far less progress has been made in the development of multitargeting TCR T therapies to date.
TCR T therapies are also susceptible to tumor cell escape through perturbations in APM pathways. Downregulation of HLA-class I and other APM components such as TAP1 has been observed in many cancer types (232, 233). The impact of these escape mechanisms has been clearly demonstrated in TCR T and TIL trials where mutations in tumor APM components resulted in tumor escape and cancer progression (44, 67). These studies highlight an urgent need for strategies to address such tumor intrinsic mechanisms of escape, especially as we see further improvements of the antitumor efficacy of TCR T therapeutics. In cases of tumor downregulation of APM pathways, expression of these components may be recovered through administration of interferon (234) or epigenetic modifying drugs (235). Addressing cases where APM components are lost through hardwired genetic mutations is considerably more challenging. One strategy that has been investigated in mice is in situ gene delivery of β2M with an adenoviral vector (236). However, this approach delivered adenovirus into relatively small tumors (7 – 10 mm in diameter) by intratumoral injection, therefore it is unclear if this approach will be effective in patients whose tumors are large, inaccessible, and/or dispersed. Therefore, patients with hardwired loss of APM components will likely need to be treated with HLA-independent therapies such as CAR T. Interestingly, HLA-independent TCRs are also currently under development. These TCRs bind natively folded surface proteins similar to CARs, but possess binding affinities within the range of pMHC-TCR interactions (237).
Immunotherapies have swiftly risen to become one of the major pillars in cancer treatment. Among them, TCR-engineered T cell therapies are a rapidly growing, active, and evolving field. Since the first report of redirected T cell specificity through TCR transfer in 1986 (238), tremendous progress has been made in TCR T therapies and their applications. Emerging technologies and enhanced strategies have made TCR discovery efforts considerably more time and cost effective. This will allow for more groups to become involved in TCR discovery and will ultimately lead to an increase in TCRs targeting new tumor antigens and restricted to a broader range of HLAs. Several clinical findings from early TCR T trials have shaped the past decade of TCR T development. Toxicities observed in early trials have led to improved preclinical safety assessments of TCRs and a transition towards antigen targets with increased tumor specificity. Since 2015 there has been an influx of results of TCR T trials treating various solid cancers and hematological malignancies. Several of these trials have demonstrated impressive clinical responses in the absence of serious toxicities, and it now seems that the first approval of TCR T therapies for solid cancer may be around the corner. These early results give reason for optimism in the continued development of TCR T therapies for cancer.
PS and LK contributed to the development, writing, and review of the article. VH contributed to the figures and review of the article. All authors contributed to the article and approved the submitted version.
Cancer Prevention and Research Institute of Texas (CPRIT) Grant ID: RR170024 and the Susan Komen Career Catalyst Research grant # CCR19606699.
PS and VH have submitted a patent application for TCR discovery.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Malcolm Brenner for editing the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.835762/full#supplementary-material
1. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients With Metastatic Melanoma. A Preliminary Report. N Engl J Med (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
2. Weber EW, Maus MV, Mackall CL. The Emerging Landscape of Immune Cell Therapies. Cell (2020) 181:46–62. doi: 10.1016/j.cell.2020.03.001
3. Garber K. Driving T-Cell Immunotherapy to Solid Tumors. Nat Biotechnol (2018) 36:215–9. doi: 10.1038/nbt.4090
4. Rudolph MG, Stanfield RL, Wilson IA. How TCRs Bind MHCs, Peptides, and Coreceptors. Annu Rev Immunol (2006) 24:419–66. doi: 10.1146/annurev.immunol.23.021704.115658
5. Huang Y, Wange RL. T Cell Receptor Signaling: Beyond Complex Complexes. J Biol Chem (2004) 279:28827–30. doi: 10.1074/JBC.R400012200
6. Anton van der Merwe P, Dushek O. Mechanisms for T Cell Receptor Triggering. Nat Rev Immunol (2011) 11:47–55. doi: 10.1038/nri2887
7. Davis MM. T Cell Receptor Gene Diversity and Selection. Annu Rev Biochem (1990) 59:475–96. doi: 10.1146/annurev.bi.59.070190.002355
8. Mørch AM, Bálint Š, Santos AM, Davis SJ, Dustin ML. Coreceptors and TCR Signaling – the Strong and the Weak of it. Front Cell Dev Biol (2020) 8:597627. doi: 10.3389/fcell.2020.597627
9. Abate-Daga D, Davila ML. CAR Models: Next-Generation CAR Modifications for Enhanced T-Cell Function. Mol Ther - Oncolytics (2016) 3:16014. doi: 10.1038/MTO.2016.14
10. Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA Database: Allele Variant Databases. Nucleic Acids Res (2015) 43:D423–31. doi: 10.1093/NAR/GKU1161
11. Gonzalez-Galarza FF, McCabe A, Melo dos Santos EJ, Jones J, Takeshita L, Ortega-Rivera ND, et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acids Res (2020) 48:D783–8. doi: 10.1093/NAR/GKZ1029
12. Chandran SS, Klebanoff CA. T Cell Receptor-Based Cancer Immunotherapy: Emerging Efficacy and Pathways of Resistance. Immunol Rev (2019) 290:127–47. doi: 10.1111/IMR.12772
13. Salter AI, Rajan A, Kennedy JJ, Ivey RG, Shelby SA, Leung I, et al. Comparative Analysis of TCR and CAR Signaling Informs CAR Designs With Superior Antigen Sensitivity and In Vivo Function. Sci Signal (2021) 14:eabe2606. doi: 10.1126/scisignal.abe2606
14. Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J Immunol (2018) 200:1088–100. doi: 10.4049/JIMMUNOL.1700236
15. Majzner RG, Mackall CL. Tumor Antigen Escape From CAR T-Cell Therapy. Cancer Discovery (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
16. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1–Specific TCR–engineered T Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat Med (2015) 21:914–21. doi: 10.1038/nm.3910
17. Frey N, Porter D. Cytokine Release Syndrome With Chimeric Antigen Receptor T Cell Therapy. Biol Blood Marrow Transplant (2019) 25:e123–7. doi: 10.1016/J.BBMT.2018.12.756
18. Fagerberg L, Jonasson K, Von Heijne G, Uhlén M, Berglund L. Prediction of the Human Membrane Proteome. Proteomics (2010) 10:1141–9. doi: 10.1002/PMIC.200900258
19. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-Based Map of the Human Proteome. Science (2015) 347:1260419. doi: 10.1126/science.1260419
20. Bausch-Fluck D, Goldmann U, Müller S, van Oostrum M, Müller M, Schubert OT, et al. The in Silico Human Surfaceome. Proc Natl Acad Sci USA (2018) 115:E10988–97. doi: 10.1073/pnas.1808790115
21. Ponterio E, De Maria R, Haas TL. Identification of Targets to Redirect CAR T Cells in Glioblastoma and Colorectal Cancer: An Arduous Venture. Front Immunol (2020) 11:565631. doi: 10.3389/fimmu.2020.565631
22. Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell (2017) 168:724–40. doi: 10.1016/j.cell.2017.01.016
23. Watanabe K, Kuramitsu S, Posey ADJ, June CH. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front Immunol (2018) 9:2486. doi: 10.3389/FIMMU.2018.02486
24. Vigneron N. Human Tumor Antigens and Cancer Immunotherapy. BioMed Res Int (2015) 2015:948501. doi: 10.1155/2015/948501
25. Ilyas S, Yang JC. Landscape of Tumor Antigens in T Cell Immunotherapy. J Immunol (2015) 195:5117–22. doi: 10.4049/jimmunol.1501657
26. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour Antigens Recognized by T Lymphocytes: At the Core of Cancer Immunotherapy. Nat Rev Cancer (2014) 14:135–46. doi: 10.1038/nrc3670
27. Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, et al. Cloning of the Gene Coding for a Shared Human Melanoma Antigen Recognized by Autologous T Cells Infiltrating Into Tumor. Proc Natl Acad Sci USA (1994) 91:3515–9. doi: 10.1073/pnas.91.9.3515
28. Bakker ABH, Schreurs MWJ, De Boer AJ, Kawakami Y, Rosenberg SA, Adema GJ, et al. Melanocyte Lineage-Specific Antigen Gp100 Is Recognized by Melanoma-Derived Tumor-Infiltrating Lymphocytes. J Exp Med (1994) 179:1005–9. doi: 10.1084/jem.179.3.1005
29. Cilloni D, Gottardi E, De Micheli D, Serra A, Volpe G, Messa F, et al. Quantitative Assessment of WT1 Expression by Real Time Quantitative PCR may be a Useful Tool for Monitoring Minimal Residual Disease in Acute Leukemia Patients. Leukemia (2002) 16:2115–21. doi: 10.1038/sj.leu.2402675
30. Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, et al. Aberrant Overexpression of the Wilms Tumor Gene (WT1) in Human Leukemia. Blood (1997) 89:1405–12. doi: 10.1182/BLOOD.V89.4.1405
31. Chen Y-T, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, et al. A Testicular Antigen Aberrantly Expressed in Human Cancers Detected by Autologous Antibody Screening. Proc Natl Acad Sci U.S.A. (1997) 94:1914–8. doi: 10.1073/pnas.94.5.1914
32. Dhodapkar MV, Osman K, Teruya-Feldstein J, Filippa D, Hedvat CV, Iversen K, et al. Expression of Cancer/Testis (CT) Antigens MAGE-A1, MAGE-A3, MAGE-A4, CT-7, and NY-ESO-1 in Malignant Gammopathies is Heterogeneous and Correlates With Site, Stage and Risk Status of Disease. Cancer Immun Arch (2003) 3:9.
33. Bergeron A, Picard V, LaRue H, Harel F, Hovington H, Lacombe L, et al. High Frequency of MAGE-A4 and MAGE-A9 Expression in High-Risk Bladder Cancer. Int J Cancer (2009) 125:1365–71. doi: 10.1002/IJC.24503
34. Duffour M-T, Chaux P, Lurquin C, Cornelis G, Boon T, van der Bruggen P. A MAGE-A4 Peptide Presented by HLA-A2 is Recognized by Cytolytic T Lymphocytes. Eur J Immunol (1999) 29:3329–37. doi: 10.1002/(SICI)1521-4141(199910)29:10<3329::AID-IMMU3329>3.0.CO;2-7
35. Yakirevich E, Sabo E, Lavie O, Mazareb S, Spagnoli GC, Resnick MB. Expression of the MAGE-A4 and NY-ESO-1 Cancer-Testis Antigens in Serous Ovarian Neoplasms. Clin Cancer Res (2003) 9:6453–60.
36. Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen Y-T. Cancer/testis Antigens: An Expanding Family of Targets for Cancer. Immunol Rev (2002) 188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x
37. de Martel C, Plummer M, Vignat J, Franceschi S. Worldwide Burden of Cancer Attributable to HPV by Site, Country and HPV Type. Int J Cancer (2017) 141:664–70. doi: 10.1002/IJC.30716
38. Perz JF, Armstrong GL, Farrington LA, Hutin YJF, Bell BP. The Contributions of Hepatitis B Virus and Hepatitis C Virus Infections to Cirrhosis and Primary Liver Cancer Worldwide. J Hepatol (2006) 45:529–38. doi: 10.1016/J.JHEP.2006.05.013
39. Thompson MP, Kurzrock R. Epstein-Barr Virus and Cancer. Clin Cancer Res (2004) 10:803–21. doi: 10.1158/1078-0432.CCR-0670-3
40. Khan G, Hashim MJ. Global Burden of Deaths From Epstein-Barr Virus Attributable Malignancies 1990-2010. Infect Agent Cancer (2014) 9:38. doi: 10.1186/1750-9378-9-38
41. Pal A, Kundu R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front Microbiol (2020) 10:3116. doi: 10.3389/FMICB.2019.03116
42. Yim E-K, Park J-S. The Role of HPV E6 and E7 Oncoproteins in HPV-Associated Cervical Carcinogenesis. Cancer Res Treat (2005) 37:319–24. doi: 10.4143/CRT.2005.37.6.319
43. Dawson CW, Port RJ, Young LS. The Role of the EBV-Encoded Latent Membrane Proteins LMP1 and LMP2 in the Pathogenesis of Nasopharyngeal Carcinoma (NPC). Semin Cancer Biol (2012) 22:144–53. doi: 10.1016/J.SEMCANCER.2012.01.004
44. Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, et al. TCR-Engineered T Cells Targeting E7 for Patients With Metastatic HPV-Associated Epithelial Cancers. Nat Med (2021) 27:419–25. doi: 10.1038/s41591-020-01225-1
45. Draper LM, Kwong ML, Gros A, Stevanović S, Tran E, Kerkar S, et al. Targeting of HPV-16+ Epithelial Cancer Cells by TCR Gene Engineered T Cells Directed Against E6. Clin Cancer Res (2015) 21:4431–9. doi: 10.1158/1078-0432.CCR-14-3341
46. Cho H-I, Kim U-H, Shin A-R, Won J-N, Lee H-J, Sohn H-J, et al. A Novel Epstein–Barr Virus-Latent Membrane Protein-1-Specific T-Cell Receptor for TCR Gene Therapy. Br J Cancer (2018) 118:534–45. doi: 10.1038/BJC.2017.475
47. Gottschalk S, Heslop H, Rooney C. Adoptive Immunotherapy for EBV-Associated Malignancies. Leuk Lymphoma (2005) 46:1–10. doi: 10.1080/10428190400002202
48. Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained Complete Responses in Patients With Lymphoma Receiving Autologous Cytotoxic T Lymphocytes Targeting Epstein-Barr Virus Latent Membrane Proteins. J Clin Oncol (2014) 32:798–808. doi: 10.1200/JCO.2013.51.5304
49. Cho SG, Kim N, Sohn HJ, Lee SK, Oh ST, Lee HJ, et al. Long-Term Outcome of Extranodal Nk/T Cell Lymphoma Patients Treated With Postremission Therapy Using EBV LMP1 and LMP2a-Specific CTLs. Mol Ther (2015) 23:1401–9. doi: 10.1038/MT.2015.91
50. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic Instability — an Evolving Hallmark of Cancer. Nat Rev Mol Cell Biol (2010) 11:220–8. doi: 10.1038/nrm2858
51. Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell (2018) 173:371–85. doi: 10.1016/j.cell.2018.02.060
52. Stratton MR, Campbell PJ, Futreal PA. The Cancer Genome. Nature (2009) 458:719–24. doi: 10.1038/NATURE07943
53. Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, et al. A Compendium of Mutational Cancer Driver Genes. Nat Rev Cancer (2020) 20:555–72. doi: 10.1038/s41568-020-0290-x
54. Reiter JG, Makohon-Moore AP, Gerold JM, Heyde A, Attiyeh MA, Kohutek ZA, et al. Minimal Functional Driver Gene Heterogeneity Among Untreated Metastases. Science (2018) 361:1033–7. doi: 10.1126/SCIENCE.AAT7171
55. Armistead PM. Cellular Therapy Against Public Neoantigens. J Clin Invest (2019) 129:506–8. doi: 10.1172/JCI126116
56. Pearlman AH, Hwang MS, Konig MF, Hsiue EH-C, Douglass J, DiNapoli SR, et al. Targeting Public Neoantigens for Cancer Immunotherapy. Nat Cancer (2021) 2:487–97. doi: 10.1038/s43018-021-00210-y
57. Klebanoff CA, Wolchok JD. Shared Cancer Neoantigens: Making Private Matters Public. J Exp Med (2018) 215:5–7. doi: 10.1084/JEM.20172188
58. Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-Cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol Res (2016) 4:204–14. doi: 10.1158/2326-6066.CIR-15-0188
59. Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and Clinical Significance of ESR1 Mutations in ER-Positive Metastatic Breast Cancer Patients Receiving Fulvestrant. Nat Commun (2016) 7:1–10. doi: 10.1038/ncomms11579
60. Chandran SS, Ma J, Klatt MG, Dundar F, Bandlamudi C, Razavi P, et al. Immunogenicity of a Public Neoantigen Derived From Mutated PIK3CA. bioRxiv (2021). doi: 10.1101/2021.04.08.439061
61. Lythe G, Callard RE, Hoare RL, Molina-París C. How Many TCR Clonotypes Does a Body Maintain? J Theor Biol (2016) 389:214–24. doi: 10.1016/j.jtbi.2015.10.016
62. Balch CM, Riley LB, Bae YJ, Salmeron MA, Platsoucas CD, von EA, et al. Patterns of Human Tumor-Infiltrating Lymphocytes in 120 Human Cancers. Arch Surg (1990) 125:200–5. doi: 10.1001/ARCHSURG.1990.01410140078012
63. Jin BY, Campbell TE, Draper LM, Stevanović S, Weissbrich B, Yu Z, et al. Engineered T Cells Targeting E7 Mediate Regression of Human Papillomavirus Cancers in a Murine Model. JCI Insight (2018) 3:e99499. doi: 10.1172/jci.insight.99488
64. Lu YC, Zheng Z, Robbins PF, Tran E, Prickett TD, Gartner JJ, et al. An Efficient Single-Cell RNA-Seq Approach to Identify Neoantigen-Specific T Cell Receptors. Mol Ther (2018) 26:379–89. doi: 10.1016/J.YMTHE.2017.10.018
65. Lu Y-C, Zheng Z, Lowery FJ, Gartner JJ, Prickett TD, Robbins PF, et al. Direct Identification of Neoantigen-Specific TCRs From Tumor Specimens by High-Throughput Single-Cell Sequencing. J Immunother Cancer (2021) 9:e002595. doi: 10.1136/jitc-2021-002595
66. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune Recognition of Somatic Mutations Leading to Complete Durable Regression in Metastatic Breast Cancer. Nat Med (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
67. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
68. Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, et al. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. JNCI J Natl Cancer Inst (1994) 86:1159–66. doi: 10.1093/JNCI/86.15.1159
69. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive Cell Transfer: A Clinical Path to Effective Cancer Immunotherapy. Nat Rev Cancer (2008) 8:299–308. doi: 10.1038/NRC2355
70. Wang S, Sun J, Chen K, Ma P, Lei Q, Xing S, et al. Perspectives of Tumor-Infiltrating Lymphocyte Treatment in Solid Tumors. BMC Med (2021) 19:1–7. doi: 10.1186/S12916-021-02006-4
71. Lee S, Psyrri A, Sukari A, Thomas S, Wenham R, Gogas H, et al. Phase 2 Efficacy and Safety of Autologous Tumor-Infiltrating Lymphocyte (TIL) Cell Therapy in Combination With Pembrolizumab in Immune Checkpoint Inhibitor-Naïve Patients With Advanced Cancers [Conference Presentation]. In: SITC. Washington, D.C., United States: J ImmunoTher Cancer (2021). doi: 10.1136/jitc-2021-SITC2021.492
72. Chinnasamy N, Wargo JA, Yu Z, Rao M, Frankel TL, Riley JP, et al. A TCR Targeting the HLA-A*0201–Restricted Epitope of MAGE-A3 Recognizes Multiple Epitopes of the MAGE-A Antigen Superfamily in Several Types of Cancer. J Immunol (2011) 186:685–96. doi: 10.4049/JIMMUNOL.1001775
73. Kunert A, van Brakel M, van Steenbergen-Langeveld S, da Silva M, Coulie PG, Lamers C, et al. MAGE-C2–Specific TCRs Combined With Epigenetic Drug-Enhanced Antigenicity Yield Robust and Tumor-Selective T Cell Responses. J Immunol (2016) 197:2541–52. doi: 10.4049/JIMMUNOL.1502024
74. Abrams SI, Khleif SN, Bergmann-Leitner ES, Kantor JA, Chung Y, Hamilton JM, et al. Generation of Stable CD4+ and CD8+ T Cell Lines From Patients Immunized With Ras Oncogene-Derived Peptides Reflecting Codon 12 Mutations. Cell Immunol (1997) 182:137–51. doi: 10.1006/CIMM.1997.1224
75. Heslop PIHE, Brenner MK, Rooney C, Krance C-IRA, Roberts WM, Rochester R, et al. Administration of Neomycin Resistance Gene Marked EBV Specific Cytotoxic T Lymphocytes to Recipients of Mismatched-Related or Phenotypically Similar Unrelated Donor Marrow Grafts. Hum Gene Ther (1994) 5:381–97. doi: 10.1089/HUM.1994.5.3-381
76. Greenberg SR, Riddell M, Rabin AP, Geballe WJ, Britt PD. Class I MHC-Restricted Cytotoxic T Lymphocyte Recognition of Cells Infected With Human Cytomegalovirus Does Not Require Endogenous Viral Gene Expression(1991). Available at: http://www.jimmunol.org/content/146/8/2795 (Accessed October 19, 2021).
77. Reusser P, Riddell S, Meyers J, Greenberg P. Cytotoxic T-Lymphocyte Response to Cytomegalovirus After Human Allogeneic Bone Marrow Transplantation: Pattern of Recovery and Correlation With Cytomegalovirus Infection and Disease. Blood (1991) 78:1373–80. doi: 10.1182/BLOOD.V78.5.1373.1373
78. Laubscher A, Bluestein HG, Spector SA, Zvaifler NJ. Generation of Human Cytomegalovirus-Specific Cytotoxic T-Lymphocytes in a Short-Term Culture. J Immunol Methods (1988) 110:69–77. doi: 10.1016/0022-1759(88)90084-1
79. Bethune MT, Li X-H, Yu J, McLaughlin J, Cheng D, Mathis C, et al. Isolation and Characterization of NY-ESO-1-Specific T Cell Receptors Restricted on Various MHC Molecules. Proc Natl Acad Sci USA (2018) 115:E10702–11. doi: 10.1073/pnas.1810653115
80. Gerdemann U, Katari U, Christin AS, Cruz CR, Tripic T, Rousseau A, et al. Cytotoxic T Lymphocytes Simultaneously Targeting Multiple Tumor-Associated Antigens to Treat EBV Negative Lymphoma. Mol Ther (2011) 19:2258–68. doi: 10.1038/mt.2011.167
81. Leung WK, Workineh A, Mukhi S, Tzannou I, Brenner D, Watanabe N, et al. Evaluation of Cyclin A1-Specific T Cells as a Potential Treatment for Acute Myeloid Leukemia. Blood Adv (2020) 4:387–97. doi: 10.1182/bloodadvances.2019000715
82. Cafri G, Yossef R, Pasetto A, Deniger DC, Lu YC, Parkhurst M, et al. Memory T Cells Targeting Oncogenic Mutations Detected in Peripheral Blood of Epithelial Cancer Patients. Nat Commun (2019) 10:1–9. doi: 10.1038/s41467-019-08304-z
83. Malekzadeh P, Yossef R, Cafri G, Paria BC, Lowery FJ, Jafferji M, et al. Antigen Experienced T Cells From Peripheral Blood Recognize P53 Neoantigens. Clin Cancer Res (2020) 26:1267–76. doi: 10.1158/1078-0432.ccr-19-1874
84. Arber C, Feng X, Abhyankar H, Romero E, Wu M-F, Heslop HH, et al. Survivin-Specific T Cell Receptor Targets Tumor But Not T Cells. J Clin Invest (2015) 125:157–68. doi: 10.1172/JCI75876
85. Inozume T, Hanada K, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, et al. Selection of CD8+PD-1+ Lymphocytes in Fresh Human Melanomas Enriches for Tumor-Reactive T-Cells. J Immunother (2010) 33:956–64. doi: 10.1097/CJI.0B013E3181FAD2B0
86. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective Identification of Neoantigen-Specific Lymphocytes in the Peripheral Blood of Melanoma Patients. Nat Med (2016) 22:433–8. doi: 10.1038/NM.4051
87. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 Identifies the Patient-Specific CD8+ Tumor-Reactive Repertoire Infiltrating Human Tumors. J Clin Invest (2014) 124:2246–59. doi: 10.1172/JCI73639
88. Butler MO, Hirano N. Human Cell-Based Artificial Antigen-Presenting Cells for Cancer Immunotherapy. Immunol Rev (2014) 257:191–209. doi: 10.1111/IMR.12129
89. Kim JV, Latouche J-B, Rivière I, Sadelain M. The ABCs of Artificial Antigen Presentation. Nat Biotechnol (2004) 22:403–10. doi: 10.1038/nbt955
90. Neal LR, Bailey SR, Wyatt MM, Bowers JS, Majchrzak K, Nelson MH, et al. The Basics of Artificial Antigen Presenting Cells in T Cell-Based Cancer Immunotherapies. J Immunol Res Ther (2017) 2:68–79.
91. Sprouse ML, Blahnik G, Lee T, Tully N, Benarjee P, James EA, et al. Streamlined Single Cell TCR Isolation and Generation of Retroviral Vectors for In Vitro and In Vivo Expression of Human TCRs. JoVE (J Vis Exp (2017) 127:e55379. doi: 10.3791/55379
92. Rosati SF, Parkhurst MR, Hong Y, Zheng Z, Feldman SA, Rao M, et al. A Novel Murine T-Cell Receptor Targeting NY-ESO-1. J Immunother (2014) 37:135–46. doi: 10.1097/CJI.0000000000000019
93. Van Der Bruggen P, Russo E, Schultz GR, Cornelis T, Boon V, Stroobant V, et al. Identification of Five MAGE-A1 Epitopes Recognized by Cytolytic T Lymphocytes Obtained by In Vitro Stimulation With Dendritic Cells Transduced With MAGE-A1(1999). Available at: http://www.jimmunol.org/content/163/5/2928.full (Accessed October 17, 2021).
94. Mariotti S, Nisini R. Generation of Human T Cell Clones. TC ell Protoc Second Ed (2009) 514:65–93. doi: 10.1007/978-1-60327-527-9_6
95. Verhoef A. Production of Human T-Cell Clones. Methods Mol Med Allergy Methods Protoc (2008) 138:43–50. doi: 10.1007/978-1-59745-366-0_4
96. Thompson T, Boon PD, Greenberg C, Yee MJ, Gilbert SR, Riddell VG, et al. Isolation of Tyrosinase-Specific CD8+ and CD4+ T Cell Clones From the Peripheral Blood of Melanoma Patients Following In Vitro Stimulation With Recombinant Vaccinia Virus(1996). Available at: http://www.jimmunol.org/content/157/9/4079 (Accessed November 4, 2020).
97. Ho WY, Nguyen HN, Wolfl M, Kuball J, Greenberg PD. In Vitro Methods for Generating CD8+ T-Cell Clones for Immunotherapy From the Naïve Repertoire. J Immunol Methods (2006) 310:40–52. doi: 10.1016/j.jim.2005.11.023
98. Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, et al. Tumor- and Neoantigen-Reactive T-Cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol Res (2016) 4:734–43. doi: 10.1158/2326-6066.CIR-16-0001
99. Lulla PD, Tzannou I, Vasileiou S, Carrum G, Ramos CA, Kamble R, et al. The Safety and Clinical Effects of Administering a Multiantigen-Targeted T Cell Therapy to Patients With Multiple Myeloma. Sci Transl Med (2020) 12:eaaz3339. doi: 10.1126/SCITRANSLMED.AAZ3339
100. Parkhurst M, Gros A, Pasetto A, Prickett T, Crystal JS, Robbins P, et al. Isolation of T-Cell Receptors Specifically Reactive With Mutated Tumor-Associated Antigens From Tumor-Infiltrating Lymphocytes Based on CD137 Expression. Clin Cancer Res (2017) 23:2491–505. doi: 10.1158/1078-0432.CCR-16-2680
101. Ma W, Vigneron N, Chapiro J, Stroobant V, Germeau C, Boon T, et al. A MAGE-C2 Antigenic Peptide Processed by the Immunoproteasome is Recognized by Cytolytic T Cells Isolated From a Melanoma Patient After Successful Immunotherapy. Int J Cancer (2011) 129:2427–34. doi: 10.1002/IJC.25911
102. Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, et al. Identification of a Human Melanoma Antigen Recognized by Tumor-Infiltrating Lymphocytes Associated With In Vivo Tumor Rejection. Proc Natl Acad Sci USA (1994) 91:6458. doi: 10.1073/PNAS.91.14.6458
103. Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of Somatic Mutations in Human Gastrointestinal Cancers. Science (2015) 350:1387–90. doi: 10.1126/science.aad1253
104. Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient Identification of Mutated Cancer Antigens Recognized by T Cells Associated With Durable Tumor Regressions. Clin Cancer Res (2014) 20:3401–10. doi: 10.1158/1078-0432.CCR-14-0433
105. Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N. Identifying Individual T Cell Receptors of Optimal Avidity for Tumor Antigens. Front Immunol (2015) 6:582. doi: 10.3389/FIMMU.2015.00582
106. Spear TT, Evavold BD, Baker BM, Nishimura MI. Understanding TCR Affinity, Antigen Specificity, and Cross-Reactivity to Improve TCR Gene-Modified T Cells for Cancer Immunotherapy. Cancer Immunol Immunother (2019) 68:1881–9. doi: 10.1007/s00262-019-02401-0
107. Stone JD, Chervin AS, Kranz DM. T-Cell Receptor Binding Affinities and Kinetics: Impact on T-Cell Activity and Specificity. Immunology (2009) 126:165. doi: 10.1111/J.1365-2567.2008.03015.X
108. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria J-CF, et al. Evidence for a TCR Affinity Threshold Delimiting Maximal CD8 T Cell Function. J Immunol (2010) 184:4936–46. doi: 10.4049/jimmunol.1000173
109. Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LYL, et al. High-Affinity TCRs Generated by Phage Display Provide CD4 + T Cells With the Ability to Recognize and Kill Tumor Cell Lines. J Immunol (2007) 179:5845–54. doi: 10.4049/jimmunol.179.9.5845
110. Zhong S, Malecek K, Johnson LA, Yu Z, de Miera EV-S, Darvishian F, et al. T-Cell Receptor Affinity and Avidity Defines Antitumor Response and Autoimmunity in T-Cell Immunotherapy. Proc Natl Acad Sci (2013) 110:6973–8. doi: 10.1073/PNAS.1221609110
111. Hu Z, Zhu L, Wang J, Wan Y, Yuan S, Chen J, et al. Immune Signature of Enhanced Functional Avidity CD8+ T Cells In Vivo Induced by Vaccinia Vectored Vaccine. Sci Rep (2017) 7:1–12. doi: 10.1038/srep41558
112. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielson M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol (2017) 199:3360–8. doi: 10.4049/JIMMUNOL.1700893
113. Marcu A, Bichmann L, Kuchenbecker L, Kowalewski DJ, Freudenmann LK, Backert L, et al. HLA Ligand Atlas: A Benign Reference of HLA-Presented Peptides to Improve T-Cell-Based Cancer Immunotherapy. J Immunother Cancer (2021) 9:e002071. doi: 10.1136/JITC-2020-002071
114. Tanaka K. The Proteasome: Overview of Structure and Functions. Proc Jpn Acad (2009) 85:12–36. doi: 10.2183/PJAB.85.12
115. Guillaume B, Chapiro J, Stroobant V, Colau D, Van HB, Parvizi G, et al. Two Abundant Proteasome Subtypes That Uniquely Process Some Antigens Presented by HLA Class I Molecules. Proc Natl Acad Sci (2010) 107:18599–604. doi: 10.1073/PNAS.1009778107
116. Schultz E, Chapiro J, Lurquin C, Claverol S, Burlet-Schiltz O, Warnier G, et al. The Production of a New MAGE-3 Peptide Presented to Cytolytic T Lymphocytes by HLA-B40 Requires the Immunoproteasome. J Exp Med (2002) 195:391–9. doi: 10.1084/JEM.20011974
117. Guillaume B, Stroobant V, Bousquet-Dubouch M-P, Colau D, Chapiro J, Parmentier N, et al. Analysis of the Processing of Seven Human Tumor Antigens by Intermediate Proteasomes. J Immunol (2012) 189:3538–47. doi: 10.4049/JIMMUNOL.1103213
118. Morel S, Lévy F, Burlet-Schiltz O, Brasseur F, Probst-Kepper M, Peitrequin A, et al. Processing of Some Antigens by the Standard Proteasome But Not by the Immunoproteasome Results in Poor Presentation by Dendritic Cells. Immunity (2000) 12:107–17. doi: 10.1016/S1074-7613(00)80163-6
119. Chapiro J, Claverol S, Piette F, Ma W, Stroobant V, Guillaume B, et al. Destructive Cleavage of Antigenic Peptides Either by the Immunoproteasome or by the Standard Proteasome Results in Differential Antigen Presentation. J Immunol (2006) 176:1053–61. doi: 10.4049/JIMMUNOL.176.2.1053
120. Kalaora S, Lee JS, Barnea E, Levy R, Greenberg P, Alon M, et al. Immunoproteasome Expression is Associated With Better Prognosis and Response to Checkpoint Therapies in Melanoma. Nat Commun (2020) 11:1–12. doi: 10.1038/s41467-020-14639-9
121. Greenberg PD. Selecting and Validating Targets, Isolating High Affinity TCRs, and Engineering T Cells That Can Be Effective in Therapy, in: 2nd NCI Workshop on Cell-Based Immunotherapy for Solid Tumors . Available at: https://nci.rev.vbrick.com/ (Accessed October 20, 2021).
122. Liu Q, Hao C, Su P, Shi J. Down-Regulation of HLA Class I Antigen-Processing Machinery Components in Esophageal Squamous Cell Carcinomas: Association With Disease Progression. Scand J Gastroenterol (2009) 44:960–9. doi: 10.1080/00365520902998679
123. Hasim A, Abudula M, Aimiduo R, Ma J-Q, Jiao Z. Post-Transcriptional and Epigenetic Regulation of Antigen Processing Machinery (APM) Components and HLA-I in Cervical Cancers From Uighur Women. PloS One (2012) 7:e44952. doi: 10.1371/journal.pone.0044952
124. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-AN, Feldman SA, et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer But Induce Severe Transient Colitis. Mol Ther (2011) 19:620–6. doi: 10.1038/MT.2010.272
125. Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, et al. Melanocyte Destruction After Antigen-Specific Immunotherapy of Melanoma: Direct Evidence of T Cell–mediated Vitiligo. J Exp Med (2000) 192:1637–44. doi: 10.1084/JEM.192.11.1637
126. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene Therapy With Human and Mouse T-Cell Receptors Mediates Cancer Regression and Targets Normal Tissues Expressing Cognate Antigen. Blood (2009) 114:535–46. doi: 10.1182/BLOOD-2009-03-211714
127. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-Derived HLA-A1–Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3–Directed T Cells. Sci Transl Med (2013) 5:197ra103. doi: 10.1126/SCITRANSLMED.3006034
128. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular Toxicity and Titin Cross-Reactivity of Affinity-Enhanced T Cells in Myeloma and Melanoma. Blood (2013) 122:863–71. doi: 10.1182/BLOOD-2013-03-490565
129. Morgan R, Chinnasamy N, Abate-Daga D, Gros A, Robbins P, Zheng Z, et al. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J Immunother (2013) 36:133–51. doi: 10.1097/CJI.0B013E3182829903
130. Almeida LG, Sakabe NJ, de Oliveira AR, Silva MCC, Mundstein AS, Cohen T, et al. CTdatabase: A Knowledge-Base of High-Throughput and Curated Data on Cancer-Testis Antigens. Nucleic Acids Res (2009) 37:D816–9. doi: 10.1093/NAR/GKN673
131. Kunert A, Obenaus M, Lamers C, Blankenstein T, Debets R. T-Cell Receptors for Clinical Therapy: In Vitro Assessment of Toxicity Risk. Clin Cancer Res (2017) 23:6012–20. doi: 10.1158/1078-0432.CCR-17-1012
132. Luo X, Cui H, Cai L, Zhu W, Yang W, Patrick M, et al. Selection of a Clinical Lead TCR Targeting Alpha-Fetoprotein-Positive Liver Cancer Based on a Balance of Risk and Benefit. Front Immunol (2020) 11:623. doi: 10.3389/FIMMU.2020.00623
133. Sanderson J, Crowley D, Wiedermann G, Quinn L, Crossland K, Tunbridge H, et al. Preclinical Evaluation of an Affinity-Enhanced MAGE-A4-Specific T-Cell Receptor for Adoptive T-Cell Therapy. Oncoimmunology (2019) 9:e1682381. doi: 10.1080/2162402X.2019.1682381
134. Cai L, Caraballo Galva LD, Peng Y, Luo X, Zhu W, Yao Y, et al. Preclinical Studies of the Off-Target Reactivity of AFP158-Specific TCR Engineered T Cells. Front Immunol (2020) 11:607. doi: 10.3389/FIMMU.2020.00607
135. Stone JD, Kranz DM. Role of T Cell Receptor Affinity in the Efficacy and Specificity of Adoptive T Cell Therapies. Front Immunol (2013) 4:244. doi: 10.3389/FIMMU.2013.00244
136. Minami Y, Weissman AM, Samelson LE, Klausner RD. Building a Multichain Receptor: Synthesis, Degradation, and Assembly of the T-Cell Antigen Receptor. Biochemistry (1987) 84:2688–92. doi: 10.1073/pnas.84.9.2688
137. Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal Graft-Versus-Host Disease in Mouse Models of T Cell Receptor Gene Therapy. Nat Med (2010) 16:565–70. doi: 10.1038/nm.2128
138. van Loenen MM, de BR, AL A, RS H, GL V, Willemze R, et al. Mixed T Cell Receptor Dimers Harbor Potentially Harmful Neoreactivity. Proc Natl Acad Sci (2010) 107:10972–7. doi: 10.1073/PNAS.1005802107
139. Rosenberg SA. Of Mice, Not Men: No Evidence for Graft-Versus-Host Disease in Humans Receiving T-Cell Receptor–Transduced Autologous T Cells. Mol Ther (2010) 18:1744–5. doi: 10.1038/MT.2010.195
140. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced Antitumor Activity of Murine-Human Hybrid T-Cell Receptor (TCR) in Human Lymphocytes Is Associated With Improved Pairing and TCR/CD3 Stability. Cancer Res (2006) 66:8878–86. doi: 10.1158/0008-5472.CAN-06-1450
141. Stripecke R, Villacres C, Skelton DC, Satake N, Halene S, Kohn DB. Immune Response to Green Fluorescent Protein: Implications for Gene Therapy. Gene Ther (1999) 6:1305–12. doi: 10.1038/sj.gt.3300951
142. Rosenzweig M, Connole M, Glickman R, Yue S-PS, Noren B, DeMaria M, et al. Induction of Cytotoxic T Lymphocyte and Antibody Responses to Enhanced Green Fluorescent Protein Following Transplantation of Transduced CD34+ Hematopoietic Cells. Blood (2001) 97:1951–9. doi: 10.1182/BLOOD.V97.7.1951
143. Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, et al. T-Cell Mediated Rejection of Gene-Modified HIV-Specific Cytotoxic T Lymphocytes in HIV-Infected Patients. Nat Med (1996) 2:216–23. doi: 10.1038/nm0296-216
144. Davis J, Theoret M, Zheng Z, Lamers C, Rosenberg S, Morgan R. Development of Human Anti-Murine T-Cell Receptor Antibodies in Both Responding and Nonresponding Patients Enrolled in TCR Gene Therapy Trials. Clin Cancer Res (2010) 16:5852–61. doi: 10.1158/1078-0432.CCR-10-1280
145. Bialer G, Horovitz-Fried M, Ya’acobi S, Morgan RA, Cohen CJ. Selected Murine Residues Endow Human TCR With Enhanced Tumor Recognition. J Immunol (2010) 184:6232–41. doi: 10.4049/JIMMUNOL.0902047
146. Sommermeyer D, Uckert W. Minimal Amino Acid Exchange in Human TCR Constant Regions Fosters Improved Function of TCR Gene-Modified T Cells. J Immunol (2010) 184:6223–31. doi: 10.4049/JIMMUNOL.0902055
147. Krshnan L, Park S, Im W, Call MJ, Call ME. A Conserved αβ Transmembrane Interface Forms the Core of a Compact T-Cell Receptor–CD3 Structure Within the Membrane. Proc Natl Acad Sci USA (2016) 113:E6649. doi: 10.1073/PNAS.1611445113
148. Kuball J, Dossett ML, Wolfl M, Ho WY, Voss R-H, Fowler C, et al. Facilitating Matched Pairing and Expression of TCR Chains Introduced Into Human T Cells. Blood (2007) 109:2331–8. doi: 10.1182/BLOOD-2006-05-023069
149. Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors With a Second Disulfide Bond. Cancer Res (2007) 67:3898–903. doi: 10.1158/0008-5472.CAN-06-3986
150. Haga-Friedman A, Horovitz-Fried M, Cohen CJ. Incorporation of Transmembrane Hydrophobic Mutations in the TCR Enhance Its Surface Expression and T Cell Functional Avidity. J Immunol (2012) 188:5538–46. doi: 10.4049/JIMMUNOL.1103020
151. Voss R-H, Willemsen RA, Kuball J, Grabowski M, Engel R, Intan RS, et al. Molecular Design of the Cαβ Interface Favors Specific Pairing of Introduced Tcrαβ in Human T Cells. J Immunol (2008) 180:391–401. doi: 10.4049/JIMMUNOL.180.1.391
152. Bethune MT, Gee MH, Bunse M, Lee MS, Gschweng EH, Pagadala MS, et al. Domain-Swapped T Cell Receptors Improve the Safety of TCR Gene Therapy. Elife (2016) 5:e19095. doi: 10.7554/ELIFE.19095
153. Willemsen RA, Weijtens MEM, Ronteltap C, Eshhar Z, Gratama JW, Chames P, et al. Grafting Primary Human T Lymphocytes With Cancer-Specific Chimeric Single Chain and Two Chain TCR. Gene Ther (2000) 7:1369–77. doi: 10.1038/sj.gt.3301253
154. Sebestyén Z, Schooten E, Sals T, Zaldivar I, José ES, Alarcón B, et al. Human TCR That Incorporate Cd3ζ Induce Highly Preferred Pairing Between Tcrα and β Chains Following Gene Transfer. J Immunol (2008) 180:7736–46. doi: 10.4049/JIMMUNOL.180.11.7736
155. Govers C, Sebestyén Z, Roszik J, van Brakel M, Berrevoets C, Szöőr Á, et al. TCRs Genetically Linked to CD28 and CD3ϵ Do Not Mispair With Endogenous TCR Chains and Mediate Enhanced T Cell Persistence and Anti-Melanoma Activity. J Immunol (2014) 193:5315–26. doi: 10.4049/jimmunol.1302074
156. Willcoj BE, Gao GF, Wyer JR, O’callaghan CA, Jakobsen BK, Boulter JM, et al. Production of Soluble αβ T-Cell Receptor Heterodimers Suitable for Biophysical Analysis of Ligand Binding. Protein Sci (2008) 8:2418–23. doi: 10.1110/PS.8.11.2418
157. Foley KC, Spear TT, Murray DC, Nagato K, Garrett-Mayer E, Nishimura MI. HCV T Cell Receptor Chain Modifications to Enhance Expression, Pairing, and Antigen Recognition in T Cells for Adoptive Transfer. Mol Ther - Oncolytics (2017) 5:105–15. doi: 10.1016/J.OMTO.2017.05.004
158. Chung S, Wucherpfennig KW, Friedman SM, Hafler DA, Strominger JL. Functional Three-Domain Single-Chain T-Cell Receptors. Proc Natl Acad Sci U.S.A. (1994) 91:12654–8. doi: 10.1073/PNAS.91.26.12654
159. Plaksin D, Polakova K, McPhie P, Margulies DH. A Three-Domain T Cell Receptor is Biologically Active and Specifically Stains Cell Surface MHC/peptide Complexes. J Immunol (1997) 158:2218–27.
160. Zhang T, He X, Tsang TC, Harris DT. Transgenic TCR Expression: Comparison of Single Chain With Full-Length Receptor Constructs for T-Cell Function. Cancer Gene Ther (2004) 11:487–96. doi: 10.1038/sj.cgt.7700703
161. Voss R-H, Thomas S, Pfirschke C, Hauptrock B, Klobuch S, Kuball J, et al. Coexpression of the T-Cell Receptor Constant α Domain Triggers Tumor Reactivity of Single-Chain TCR-Transduced Human T Cells. Blood (2010) 115:5154–63. doi: 10.1182/BLOOD-2009-11-254078
162. Aggen DH, Chervin AS, Schmitt TM, Engels B, Stone JD, Richman SA, et al. Single-Chain Vαvβ T-Cell Receptors Function Without Mispairing With Endogenous TCR Chains. Gene Ther (2011) 19:365–74. doi: 10.1038/gt.2011.104
163. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, et al. Editing T Cell Specificity Towards Leukemia by Zinc-Finger Nucleases and Lentiviral Gene Transfer. Nat Med (2012) 18:807. doi: 10.1038/NM.2700
164. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-Engineered T Cells in Patients With Refractory Cancer. Science (2020) 367:eaba7365. doi: 10.1126/SCIENCE.ABA7365
165. Campillo-Davo D, Fujiki F, Van den Bergh JMJ, De Reu H, Smits ELJM, Goossens H, et al. Efficient and Non-Genotoxic RNA-Based Engineering of Human T Cells Using Tumor-Specific T Cell Receptors With Minimal TCR Mispairing. Front Immunol (2018) 9:2503. doi: 10.3389/FIMMU.2018.02503
166. Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming Human T Cell Function and Specificity With Non-Viral Genome Targeting. Nature (2018) 559:405. doi: 10.1038/S41586-018-0326-5
167. Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed Evolution of Human T-Cell Receptors With Picomolar Affinities by Phage Display. Nat Biotechnol (2005) 23:349–54. doi: 10.1038/nbt1070
168. Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and Dual Amino Acid Substitutions in TCR CDRs Can Enhance Antigen-Specific T Cell Functions. J Immunol (2008) 180:6116–31. doi: 10.4049/jimmunol.180.9.6116
169. Holler PD, Holman PO, Shusta EV, O’Herrin S, Wittrup KD, Kranz DM. In Vitro Evolution of a T Cell Receptor With High Affinity for Peptide/MHC. Proc Natl Acad Sci USA (2000) 97:5387–92. doi: 10.1073/pnas.080078297
170. Hellman LM, Foley KC, Singh NK, Alonso JA, Riley TP, Devlin JR, et al. Improving T Cell Receptor On-Target Specificity via Structure-Guided Design. Mol Ther (2019) 27:300–13. doi: 10.1016/j.ymthe.2018.12.010
171. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science (2006) 314:126–9. doi: 10.1126/SCIENCE.1129003
172. Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive Transfer of MART-1 T-Cell Receptor Transgenic Lymphocytes and Dendritic Cell Vaccination in Patients With Metastatic Melanoma. Clin Cancer Res (2014) 20:2457–65. doi: 10.1158/1078-0432.CCR-13-3017
173. Moore T, Wagner C, Scurti G, Hutchens K, Godellas C, Clark A, et al. Clinical and Immunologic evaluation of Three Metastatic Melanoma Patients Treated With Autologous Melanoma-Reactive TCR-Transduced T Cells. Cancer Immunol Immunother (2018) 67:311–25. doi: 10.1007/S00262-017-2073-0
174. Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A Pilot Trial Using Lymphocytes Genetically Engineered With an NY-ESO-1–Reactive T-Cell Receptor: Long-Term Follow-Up and Correlates With Response. Clin Cancer Res (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
175. Ishihara M, Kitano S, Hattori H, Miyahara Y, Kato H, Mishima H, et al. Tumor Responses and Early Onset Cytokine Release Syndrome in Synovial Sarcoma Patients Treated With a Novel Affinity-Enhanced NY-ESO-1-Targeting TCR-Redirected T Cell Transfer [Conference presentation]. Am Soc Clin Oncol (2019) 530. doi: 10.1200/JCO.2019.37.15_SUPPL.2530
176. Nowicki T, Berent-Maoz B, Cheung-Lau G, Huang R, Wang X, Tsoi J, et al. A Pilot Trial of the Combination of Transgenic NY-ESO-1-Reactive Adoptive Cellular Therapy With Dendritic Cell Vaccination With or Without Ipilimumab. Clin Cancer Res (2019) 25:2096–108. doi: 10.1158/1078-0432.CCR-18-3496
177. Butler MO, Sotov V, Saibil S, Bonilla L, Boross-Harmer S, Fyrsta M, et al. Adoptive T Cell Therapy With TBI-1301 Results in Gene-Engineered T Cell Persistence and Anti-Tumour Responses in Patients With NY-ESO-1 Expressing Solid Tumours. Ann Oncol (2019) 30:v481. doi: 10.1093/ANNONC/MDZ253.009
178. Stadtmauer E, Faitg T, Lowther D, Badros A, Chagin K, Dengel K, et al. Long-Term Safety and Activity of NY-ESO-1 SPEAR T Cells After Autologous Stem Cell Transplant for Myeloma. Blood Adv (2019) 3:2022–34. doi: 10.1182/BLOODADVANCES.2019000194
179. Ramachandran I, Lowther D, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, et al. Systemic and Local Immunity Following Adoptive Transfer of NY-ESO-1 SPEAR T Cells in Synovial Sarcoma. J Immunother Cancer (2019) 7:276. doi: 10.1186/S40425-019-0762-2
180. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive Transfer of MAGE-A4 T-Cell Receptor Gene-Transduced Lymphocytes in Patients With Recurrent Esophageal Cancer. Clin Cancer Res (2015) 21:2268–77. doi: 10.1158/1078-0432.CCR-14-1559
181. Berard M, Brandt K, Bulfone-Paus S, Tough DF. IL-15 Promotes the Survival of Naive and Memory Phenotype CD8 + T Cells. J Immunol (2003) 170:5018–26. doi: 10.4049/jimmunol.170.10.5018
182. Hong DS, Van Tine BA, Olszanski AJ, Johnson ML, Liebner DA, Trivedi T, et al. Phase I Dose Escalation and Expansion Trial to Assess the Safety and Efficacy of ADP-A2M4 SPEAR T Cells in Advanced Solid Tumors [Conference Presentation]. In: 2020 ASCO Annual Meeting (American Society of Clinical Oncology). Journal of Clinical Immunology (2020) p. 102. doi: 10.1200/JCO.2020.38.15_SUPPL.102
183. Hong D, Clarke J, Johanns T, Kebriaei P, Heymach J, Galal A, et al. Initial Safety, Efficacy, and Product Attributes From the SURPASS Trial With ADP-A2M4CD8, a SPEAR T-Cell Therapy Incorporating an Affinity Optimized TCR Targeting MAGE-A4 and a CD8α Co-Receptor [Conference Presentation]. In: SITC 2020 (BMJ Specialist Journals). Journal for ImmunoTherapy of Cancer (2020) p. A231. doi: 10.1136/JITC-2020-SITC2020.0379
184. D’Angelo SP, Van Tine BA, Attia S, Blay J-Y, Strauss SJ, Morales CMV, et al. SPEARHEAD-1: A Phase 2 Trial of Afamitresgene Autoleucel (Formerly ADP-A2M4) in Patients With Advanced Synovial Sarcoma or Myxoid/Round Cell Liposarcoma [Conference Presentation]. In: 2021 ASCO Annual Meeting (Wolters Kluwer Health). Journal of Clinical Immunology (2020) p. 11504–4. doi: 10.1200/JCO.2021.39.15_SUPPL.11504
185. Lam VK, Hong DS, Heymach J, Blumenschein GR, Creelan BC, Bradbury PA, et al. Initial Safety Assessment of MAGE-A10c796TCR T-Cells in Two Clinical Trials [Conference presentation]. Am Soc Clin Oncol (2018) 3056. doi: 10.1200/JCO.2018.36.15_SUPPL.3056
186. Doran SL, Stevanović S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, et al. T-Cell Receptor Gene Therapy for Human Papillomavirus–Associated Epithelial Cancers: A First-In-Human, Phase I/II Study. J Clin Oncol (2019) 37:2759–68. doi: 10.1200/JCO.18.02424
187. Tawara I, Kageyama S, Miyahara Y, Fujiwara H, Nishida T, Akatsuka Y, et al. Safety and Persistence of WT1-Specific T-Cell Receptor Gene–Transduced Lymphocytes in Patients With AML and MDS. Blood (2017) 130:1985–94. doi: 10.1182/BLOOD-2017-06-791202
188. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T Cell Receptor Gene Therapy Targeting WT1 Prevents Acute Myeloid Leukemia Relapse Post-Transplant. Nat Med (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
189. Bechman N, Maher J. Lymphodepletion Strategies to Potentiate Adoptive T-Cell Immunotherapy–What are We Doing; Where are We Going? Expert Opin Biol Ther (2021) 21:627–37. doi: 10.1080/14712598.2021.1857361
190. Jiang T, Zhou C, Ren S. Role of IL-2 in Cancer Immunotherapy. Oncoimmunology (2016) 5:e1163462. doi: 10.1080/2162402X.2016.1163462
191. Rosenberg SA. IL-2: The First Effective Immunotherapy for Human Cancer. J Immunol (2014) 192:5451–8. doi: 10.4049/JIMMUNOL.1490019
192. D’Angelo S, Melchiori L, Merchant M, Bernstein D, Glod J, Kaplan R, et al. Antitumor Activity Associated With Prolonged Persistence of Adoptively Transferred NY-ESO-1 C259 T Cells in Synovial Sarcoma. Cancer Discov (2018) 8:944–57. doi: 10.1158/2159-8290.CD-17-1417
193. Ishihara M, Kitano S, Hattori H, Miyahara Y, Kato H, Mishima H, et al. Tumor Responses and Early Onset Cytokine Release Syndrome in Synovial Sarcoma Patients Treated With a Novel Affinity-Enhanced NY-ESO-1-Targeting TCR-Redirected T Cell Transfer [Conference Presentation]. In: ASCO Annual Meeting (American Society of Clinical Oncology). (2019). p. 2530. doi: 10.1200/JCO.2019.37.15_SUPPL.2530
194. Lu Y, Parker L, Lu T, Zheng Z, Toomey M, White D, et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol (2017) 35:3322–9. doi: 10.1200/JCO.2017.74.5463
195. Wagner J, Wickman E, Derenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther (2020) 28:2320–39. doi: 10.1016/j.ymthe.2020.09.015
196. Calderon H, Mamonkin M, Guedan S. Analysis of CAR-Mediated Tonic Signaling. Methods Mol Biol (2020) 2086:223–36. doi: 10.1007/978-1-0716-0146-4_17
197. Ajina A, Maher J. Strategies to Address Chimeric Antigen Receptor Tonic Signalling. Mol Cancer Ther (2018) 17:1795–815. doi: 10.1158/1535-7163.MCT-17-1097
198. Fiorenza S, Ritchie DS, Ramsey SD, Turtle CJ, Roth JA. Value and Affordability of CAR T-Cell Therapy in the United States. Bone Marrow Transplant (2020) 55:1706–15. doi: 10.1038/s41409-020-0956-8
199. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-The-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections After Allogeneic Hematopoietic Stem-Cell Transplantation. J Clin Oncol (2017) 35:3547–57. doi: 10.1200/JCO.2017.73.0655
200. Nussbaumer O, Koslowski M. The Emerging Role of γδ T Cells in Cancer Immunotherapy. Immuno-Oncol Technol (2019) 1:3–10. doi: 10.1016/j.iotech.2019.06.002
201. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT Cells With Chimeric Antigen Receptor Provide a Novel Platform for Safe and Effective Cancer Immunotherapy. Blood (2014) 124:2824. doi: 10.1182/BLOOD-2013-11-541235
202. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300
203. Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, et al. High-Efficiency Transfection of Primary Human and Mouse T Lymphocytes Using RNA Electroporation. Mol Ther (2006) 13:151–9. doi: 10.1016/J.YMTHE.2005.07.688
204. Birkholz K, Hombach A, Krug C, Reuter S, Kershaw M, Kämpgen E, et al. Transfer of mRNA Encoding Recombinant Immunoreceptors Reprograms CD4+ and CD8+ T Cells for Use in the Adoptive Immunotherapy of Cancer. Gene Ther (2009) 16:596–604. doi: 10.1038/gt.2008.189
205. Nakazawa Y, Huye LE, Dotti G, Foster AE, Vera JF, Manuri PR, et al. Optimization of the PiggyBac Transposon System for the Sustained Genetic Modification of Human T-Lymphocytes. J Immunother (2009) 32:826–36. doi: 10.1097/CJI.0B013E3181AD762B
206. Deniger DC, Pasetto A, Tran E, Parkhurst MR, Cohen CJ, Robbins PF, et al. Stable, Nonviral Expression of Mutated Tumor Neoantigen-Specific T-Cell Receptors Using the Sleeping Beauty Transposon/Transposase System. Mol Ther (2016) 24:1078–89. doi: 10.1038/MT.2016.51
207. Morita D, Nishio N, Saito S, Tanaka M, Kawashima N, Okuno Y, et al. Enhanced Expression of Anti-CD19 Chimeric Antigen Receptor in Piggybac Transposon-Engineered T Cells. Mol Ther - Methods Clin Dev (2018) 8:131–40. doi: 10.1016/J.OMTM.2017.12.003
208. Chan JD, Lai J, Slaney CY, Kallies A, Beavis PA, Darcy PK. Cellular Networks Controlling T Cell Persistence in Adoptive Cell Therapy. Nat Rev Immunol (2021) 21:769–84. doi: 10.1038/s41577-021-00539-6
209. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of Full Effector Function In Vitro Paradoxically Impairs the In Vivo Antitumor Efficacy of Adoptively Transferred CD8+ T Cells. J Clin Invest (2005) 115:1616–26. doi: 10.1172/JCI24480
210. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central Memory Self/Tumor-Reactive CD8+ T Cells Confer Superior Antitumor Immunity Compared With Effector Memory T Cells. Proc Natl Acad Sci (2005) 102:9571–6. doi: 10.1073/PNAS.0503726102
211. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A Human Memory T-Cell Subset With Stem Cell-Like Properties. Nat Med (2011) 17:1290–7. doi: 10.1038/NM.2446
212. Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, et al. IL-15 Enhances the In Vivo Antitumor Activity of Tumor-Reactive CD8+ T Cells. Proc Natl Acad Sci USA (2004) 101:1969–74. doi: 10.1073/PNAS.0307298101
213. Mo F, Yu Z, Li P, Oh J, Spolski R, Zhao L, et al. An Engineered IL-2 Partial Agonist Promotes CD8+ T Cell Stemness. Nature (2021) 597:544–8. doi: 10.1038/s41586-021-03861-0
214. Nalawade S, Shafer P, Bajgain P, McKenna K, Ali A, Kelly L, et al. Selectively Targeting Myeloid-Derived Suppressor Cells Through TRAIL Receptor 2 to Enhance the Efficacy of CAR T Cell Therapy for Treatment of Breast Cancer. J Immunother Cancer (2021) 9:e003237. doi: 10.1136/jitc-2021-003237
215. Oda S, Daman A, Garcia N, Wagener F, Schmitt T, Tan X, et al. A CD200R-CD28 Fusion Protein Appropriates an Inhibitory Signal to Enhance T-Cell Function and Therapy of Murine Leukemia. Blood (2017) 130:2410–9. doi: 10.1182/BLOOD-2017-04-777052
216. Feng J, Xu H, Cinquina A, Wu Z, Chen Q, Zhang P, et al. Treatment of Aggressive T Cell Lymphoblastic Lymphoma/leukemia Using Anti-CD5 CAR T Cells. Stem Cell Rev Rep (2021) 17:652–61. doi: 10.1007/S12015-020-10092-9
217. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-Targeted T Cells Modified to Secrete IL-12 Eradicate Systemic Tumors Without Need for Prior Conditioning. Blood (2012) 119:4133–41. doi: 10.1182/BLOOD-2011-12-400044
218. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors In Vivo. Oncoimmunology (2015) 4:e994446. doi: 10.4161/2162402X.2014.994446
219. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-Specific T Lymphocytes With Interleukin-15 and a Suicide Gene to Enhance Their Anti-Lymphoma/Leukemia Effects and Safety. Leukemia (2010) 24:1160–70. doi: 10.1038/LEU.2010.75
220. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive Signaling From an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
221. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current Progress in Car-T Cell Therapy for Solid Tumors. Int J Biol Sci (2019) 15:2548–60. doi: 10.7150/ijbs.34213
222. Irving M, de Silly RV, Scholten K, Dilek N, Coukos G. Engineering Chimeric Antigen Receptor T-Cells for Racing in Solid Tumors: Don’t Forget the Fuel. Front Immunol (2017) 8:267. doi: 10.3389/fimmu.2017.00267
223. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
224. Bajgain P, Tawinwung S, D’Elia L, Sukumaran S, Watanabe N, Hoyos V, et al. CAR T Cell Therapy for Breast Cancer: Harnessing the Tumor Milieu to Drive T Cell Activation. J Immunother Cancer (2018) 6:1–13. doi: 10.1186/s40425-018-0347-5
225. Corbière V, Chapiro J, Stroobant V, Ma W, Lurquin C, Lethé B, et al. Antigen Spreading Contributes to MAGE Vaccination-Induced Regression of Melanoma Metastases. Cancer Res (2011) 71:1253–62. doi: 10.1158/0008-5472.CAN-10-2693
226. Chapuis AG, Roberts IM, Thompson JA, Margolin KA, Bhatia S, Lee SM, et al. T-Cell Therapy Using Interleukin-21–Primed Cytotoxic T-Cell Lymphocytes Combined With Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression. J Clin Oncol (2016) 34:3787–95. doi: 10.1200/JCO.2015.65.5142
227. Vasileiou S, Lulla PD, Tzannou I, Watanabe A, Kuvalekar M, Callejas WL, et al. T-Cell Therapy for Lymphoma Using Nonengineered Multiantigen-Targeted T Cells Is Safe and Produces Durable Clinical Effects. J Clin Oncol (2021) 39:1415–25. doi: 10.1200/JCO.20.02224
228. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and Through Activation of the Endogenous Immune System. Cell Rep (2018) 23:2130–41. doi: 10.1016/J.CELREP.2018.04.051
229. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, Van Leeuwen DG, et al. Enhancing Antitumor Efficacy of Chimeric Antigen Receptor T Cells Through Constitutive CD40L Expression. Mol Ther (2015) 23:769–78. doi: 10.1038/MT.2015.4
230. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive Cellular Therapy With T Cells Expressing the Dendritic Cell Growth Factor Flt3L Drives Epitope Spreading and Antitumor Immunity. Nat Immunol (2020) 21:914–26. doi: 10.1038/s41590-020-0676-7
231. Shah NN, Maatman T, Hari P, Johnson B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front Oncol (2019) 9:146. doi: 10.3389/FONC.2019.00146
232. Jhunjhunwala S, Hammer C, Delamarre L. Antigen Presentation in Cancer: Insights Into Tumour Immunogenicity and Immune Evasion. Nat Rev Cancer (2021) 21:298–312. doi: 10.1038/s41568-021-00339-z
233. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
234. Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, et al. Low-Dose IFN-γ Induces Tumor MHC Expression in Metastatic Malignant Melanoma. Clin Cancer Res (2003) 9:84–92.
235. Vlková V, Štěpánek I, Hrušková V, Šenigl F, Mayerová V, Šrámek M, et al. Epigenetic Regulations in the Ifnγ Signalling Pathway: Ifnγ-Mediated MHC Class I Upregulation on Tumour Cells Is Associated With DNA Demethylation of Antigen-Presenting Machinery Genes. Oncotarget (2014) 5:6923. doi: 10.18632/ONCOTARGET.2222
236. Del Campo AB, Aptsiauri N, Méndez R, Zinchenko S, Vales A, Paschen A, et al. Efficient Recovery of HLA Class I Expression in Human Tumor Cells After Beta2-Microglobulin Gene Transfer Using Adenoviral Vector: Implications for Cancer Immunotherapy. Scand J Immunol (2009) 70:125–35. doi: 10.1111/J.1365-3083.2009.02276.X
237. Hiscox MJ, Wasmuth A, Williams CL, Foot JN, Wiedermann GE, Fadda V, et al. In Vitro Selection and Engineering of a Human Leukocyte Antigen-Independent T-Cell Receptor Recognizing Human Mesothelin [Conference Presentation]. In: ASGCT 2021. (2021).
Keywords: T cell receptor, TCR, chimeric antigen receptor, CAR, TCR-engineered T cells, TCR T, adoptive cell therapy
Citation: Shafer P, Kelly LM and Hoyos V (2022) Cancer Therapy With TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 13:835762. doi: 10.3389/fimmu.2022.835762
Received: 14 December 2021; Accepted: 10 February 2022;
Published: 03 March 2022.
Edited by:
Paul G. Schlegel, University Children’s Hospital Würzburg, GermanyReviewed by:
Matthias Eyrich, University Hospital Würzburg, GermanyCopyright © 2022 Shafer, Kelly and Hoyos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Shafer, cGF1bC5zaGFmZXJAYmNtLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.