 Ran Gao
Ran Gao Guo-Ping Shi
Guo-Ping Shi Jing Wang
Jing Wang- 1State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Department of Pathophysiology, Peking Union Medical College, Beijing, China
- 2Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States
Regulatory T cells (Tregs) are a subset of CD4+ T cells with their immunosuppressive activities to block abnormal or excessive immune responses to self and non-autoantigens. Tregs express the transcription factor Foxp3, maintain the immune homeostasis, and prevent the initiation of anti-tumor immune effects in various ways as their mechanisms to modulate tumor development. Recognition of different phenotypes and functions of intratumoral Tregs has offered the possibilities to develop therapeutic strategies by selectively targeting Tregs in cancers with the aim of alleviating their immunosuppressive activities from anti-tumor immune responses. Several Treg-based immunotherapeutic approaches have emerged to target cytotoxic T lymphocyte antigen-4, glucocorticoid-induced tumor necrosis factor receptor, CD25, indoleamine-2, 3-dioxygenase-1, and cytokines. These immunotherapies have yielded encouraging outcomes from preclinical studies and early-phase clinical trials. Further, dual therapy or combined therapy has been approved to be better choices than single immunotherapy, radiotherapy, or chemotherapy. In this short review article, we discuss our current understanding of the immunologic characteristics of Tregs, including Treg differentiation, development, therapeutic efficacy, and future potential of Treg-related therapies among the general cancer therapy.
Introduction
Thymic T cells express T-cell receptor (TCR) at the mature stage when thymus produces immune functional T cells through positive and negative selection to recognize endogenous or exogenous antigens. The purpose of positive selection is to make those T cells carrying TCR that receives survival signals from the major histocompatibility complex-II (MHC-II) molecules on thymic cells, while negative selection mainly leads to the exclusion of T cells with high affinity for MHC-bound antigen peptides. However, some T cells with self-reactive TCR can develop into CD4+CD25+Foxp3+ regulatory T cells (Tregs) after interacting with autoantigen peptides with high affinity (1). Tregs are an immunosuppressive subgroup of the CD4+ T cells. The concept of immunosuppressive T cells was proposed in 1970s (2, 3). A decade later, reports confirmed that Tregs exerted their immunosuppressive effects in mouse tumor models (4, 5). Such immunosuppressive effect of Tregs was further confirmed in 1995 (6). At present, many different types of Tregs have been identified. Naturally developed Tregs account for 5% to 10% of total peripheral CD4+ T cells and are characterized by their high expression of CD25 and low expression of CD45RB (7–9). While the IL-2 receptor CD25 serves as a surface marker of suppressor T cells (10), the forkhead/winged helix transcription factor (Foxp3) is considered a classical combined marker of Tregs (11). The most prominent Treg types include thymus-derived Tregs (tTregs), peripherally generated Tregs (pTregs) from the Foxp3– T conventional (Tconv) cells, and in vitro interleukin-2 (IL-2) and transform growth factor-β (TGF-β)-induced Tregs (iTregs) from Tconv cells (12). tTregs are generated in the thymus through MHC class II-dependent TCR interactions that result in high-avidity selection. Activated polyclonal tTregs modulate T-effector cell trafficking to the target organs, while antigen-specific iTregs inhibit T-effector cell priming by targeting the antigen presenting cells (APCs) (13).
Both tTregs and pTregs are stable in the expression of Foxp3 and other Treg signature genes such as Cd25 and cytotoxic T lymphocyte antigen-4 (Ctla4). These Tregs show sustained immunosuppressive function. The intronic enhancers CNSs (conserved non-coding sequences), also known as TSDRs (Treg-specific demethylation regions), and promoter of the Foxp3 gene play important role in Foxp3 gene stable expression (14, 15). In contrast, the expression of Foxp3 and Treg signature genes in iTregs remains unstable due to incomplete epigenic changes at the TSDRs and these iTregs may become T-effector cells under certain in vivo conditions (16). The stability of Foxp3 expression and immunosuppressive functionality of iTregs relies on the efficient demethylation of the CpG island in the first intron of Foxp3 gene locus CNS2 region (16–19). CNS2 demethylation enhances the recruitment of transcription factors STAT5 (signal transducer and activator of transcription 5), NFAT (nuclear factor of activated T cells), Runx1/Cbfβ, CREB (cAMP-response element binding protein), and Foxp3 itself (20, 21). While reduced demethylation of CNS2 in iTregs leads to impaired Foxp3 expression and iTreg function stability (22), complete demethylation of CNS2 is required for optimal Foxp3 gene expression and iTreg immunosuppression activity (23). In addition to CNS2 demethylation, other key factors determining the development of iTregs include the types of APCs, their differentiation status, and cytokine environment in the activation process.
Tumor infiltrating dendritic cells (DCs), and TGF-β, IL-2, and indoleamine-2, 3-dioxygenase-1 (IDO-1) are all essential cells and molecules that promote CD4+ T-cell differentiation into Tregs (24, 25). Tregs have been among the most extensively studied lymphocytes in oncology for decades. Yet, the successful and precise targeting of Tregs for cancer immunotherapy has been elusive, although these cells may exert different functions depending on their residential tissue types. For example, multiple classes of genes are differentially regulated in Tregs from the visceral adipose tissue (VAT) compared with those in the lymphoid organs, including those encoding the transcription factors, chemokines and cytokines and their receptors, and molecules that are implicated in lipid metabolism to regulate adipose tissue homeostasis and organismal metabolism. These Tregs display much more restricted repertoire of antigen-specific TCRs and stronger dependency on the cytokine IL-33 and its receptors ST2 than those in the lymphoid organs (26, 27). Skeletal muscle Tregs are expanded in response to acute or chronic injury. Like the VAT Tregs, skeletal muscle Tregs also express high levels of transcription factors, chemokines, cytokines, and their receptors (26). The colonic Tregs are developed against microbial antigens. Mice devoid of any microbiota showed much smaller number of colonic Tregs than those in specific pathogen-free (SPF) mice (28). Intestinal Tregs express high levels of ST2 and tissue repair factors. These cells also express inducible costimulator (ICOS), CTLA-4, and ectonucleotidases CD39 and CD73 to regulate Th2- and Th1/Th17-mediated immunity (29). Tregs in the skin are involved in regulating microbial colonization, wound healing, and hair follicle development (29). In tumor microenvironment, Tregs inhibit the antitumor immunity and promote tumor occurrence and development by suppressing the function of immune effector cells via a variety of mechanisms (30) that will be discussed here. Emerging evidence suggests that Tregs demonstrate remarkable adaptability to their local environment and facilitate the immune homeostasis through highly specialized tissue-specific pathways (31). After effective elimination of pathogenic threats, the evolutionarily evolved immune system immediately restores the quiescence and prevents further harm (32).
Regulatory T-Cell Immunosuppressive Function Regulation
Tregs are important mediators of the peripheral tolerance to autoantigens and non-autoantigens, which can be controlled by a variety of inhibitory mechanisms. Treg differentiation, proliferation, and immunosuppression activity vary in response to environmental signals that may alter Treg stability, plasticity, and tissue-specific heterogeneity and shape Treg environmental-dependent immunosuppressive functions (10, 33, 34). These signals include cell-extrinsic factors, such as nutrients, vitamins, and metabolites, and cell-intrinsic metabolic programs. Foxp3 is the major regulatory molecule of Tregs that produces and maintains Treg immunosuppressive activity. Studies have found that lack of Foxp3 in mice increased T-effector cell activity due to Treg depletion (35). Like Treg depletion, mutations of Foxp3 caused fatal immune dysregulation-associated multi-organ autoimmune diseases, such as polyendocrinopathy, enteropathy, and X-linked syndrome (36–38). Transfer of donor CD4+CD25+ Tregs into neonatal Foxp3-deficient mice rescued the lymphoproliferative disorder in recipient mice, suggesting that Foxp3 is a critical regulator of CD4+CD25+ Treg function (11). Yet, both extrinsic and intrinsic factors may enhance or impair Treg differentiation, proliferation, and immunosuppression function, depending on the Treg residential environment and their immune status.
Treg Surface Molecules
Treg surface molecules are among the essential Treg cell intrinsic molecules that respond to the tissue environment and alter Treg stability, plasticity, and tissue-specific heterogeneity. Common surface regulatory molecules include CD25, CTLA-4, TGF-β receptor, CD36, SLC27A1, and glucocorticoid induced tumor necrosis factor receptor (GITR). Expression of these molecules is required for Treg survival and immunosuppression. Foxp3 transfection of CD4+CD25- primitive T cells transformed these cells into CD4+CD25+ Tregs cells that expressed these surface markers (39–41). Tregs regulate T-cell immune response using surface CTLA-4. CTLA-4 or even its truncated form without the cytoplasmic portion regulates the expression of CD80/86 on the surface of APCs during Treg-APC conjugation and immune synapse formation. This process allows CLTA-4 to remove CD80/86 on APC surface via a trogocytosis mechanism, followed by trans-endocytosis for intracellular degradation (40, 42, 43). Decreased CD80/86 expression on APC surface reduces CD28-mediated T-cell stimulation due to cell-extrinsic ligand depletion (40), causes CD80/programed death ligand-1 (PD-L1) heterodimer disruption, and increases free PD-L1 levels from the APCs (43) (Figure 1). Tregs in VAT express CD36 to uptake long-chain fatty acid and contribute to lipid accumulation in obese subjects (44). Short-chain fatty acids also promote iTreg differentiation (45, 46). In brain tumors, high percentages of Tregs express CD36 and SLC27A1 (fatty acid transporters). Inhibition of lipid uptake with sulfo-N-succinimidyl oleate (SSO) or fatty acid oxidation (FAO) with etomoxir prevented Treg immunosuppressive activity under this environment (47). Intratumoral Tregs showed over 15-fold increase of CD36 expression compared with those in the lymphoid organs. CD36 expression supports the suppressive activity of intratumoral Tregs. Disruption of CD36 selectively stimulated intratumoral Treg apoptosis and impaired tumor Treg accumulation and suppressive activity. Therefore, selective depletion of CD36 in Tregs suppressed tumor growth, decreased intratumoral Treg cells, and enhanced the anti-tumor activity in tumor-infiltrating lymphocytes without distrusting the immune homeostasis (48).
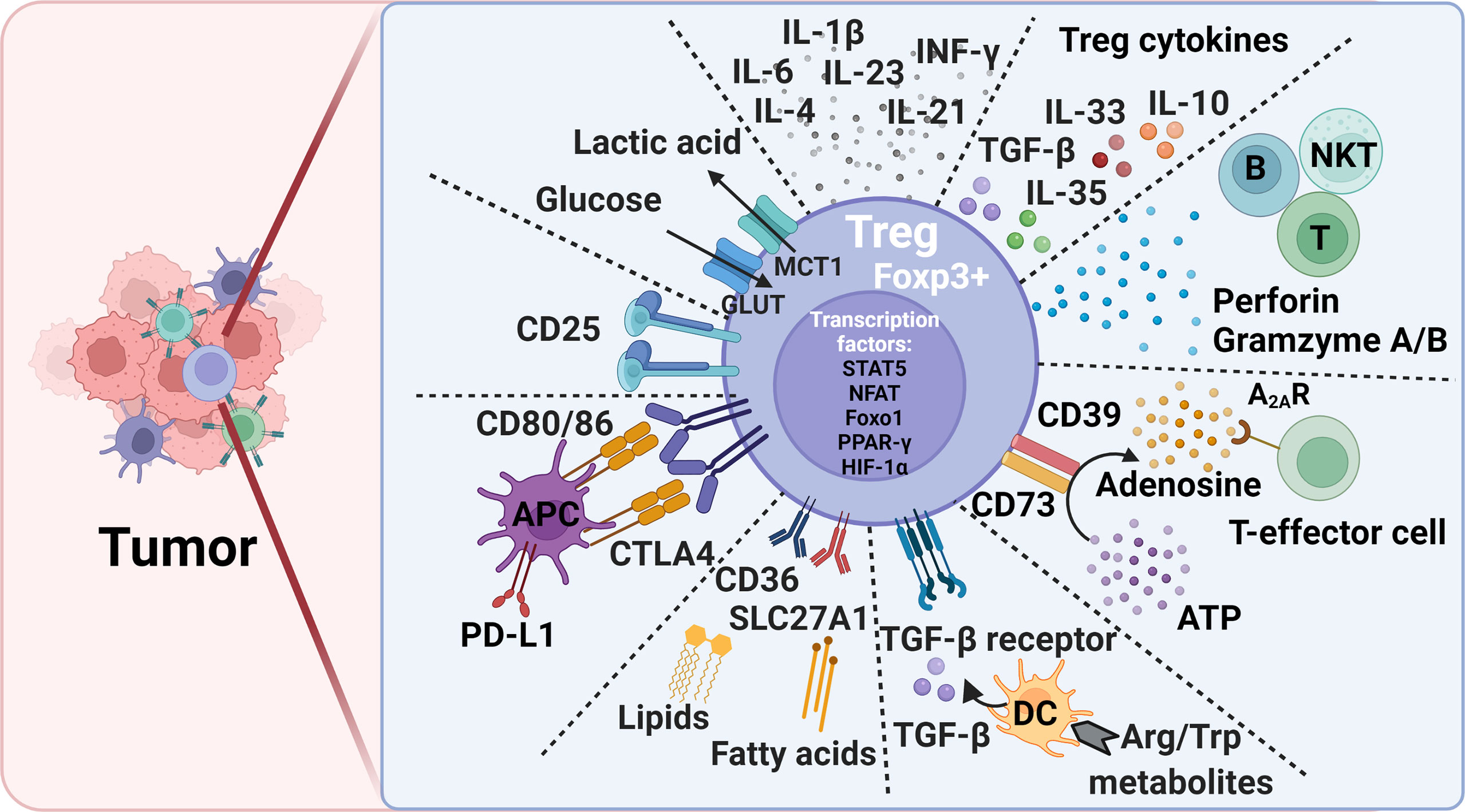
Figure 1 Treg-mediated immunosuppression mechanisms and potential therapeutic targets to alleviate immunosuppression in tumors. Foxp3, forkhead box P3; CTLA4, cytotoxic T lymphocyte antigen-4; PD-L1, programmed death ligand 1; DC, dendritic cell; Trp, L-tryptophan; Arg, L-arginine; ATP, adenosine triphosphate; Glut, Glucose transporters; MCT1, monocarboxylate transporter 1; STAT5, signal transducer and activator of transcription 5; NFAT, Nuclear factor of activated T cells; PPAR-γ, peroxisome proliferator- activated receptor-γ; HIF-1α, hypoxia-inducible factor 1α; Foxo1, Forkhead Box O1.
Secreted Cytokines
Tregs also secrete suppressor cytokines, including TGF-β, IL-10, IL-35, and IL-33 (Figure 1). TGF-β and IL-10 inhibit the function of DCs and CD8+ T-effector cells and promote the transformation of CD4+ T cells into Tregs. TGF-β plays an essential role in Treg differentiation and controls the Treg suppressive activity (42). TGF-β signaling positively regulates Treg-dependent and -independent mechanisms of T-cell development and homeostasis (49). TGF-β signaling is also essential for Treg survival (50). When IL-10 induces M2 macrophages polarization to promote tumor immune escape during tumor progression (51, 52), IL-35 secreted from Tregs induces non-Tregs that inhibit other cells by the “infectious tolerance” mechanism (53, 54). Treg development, maintenance, accumulation, and immnosupression function in VAT and tumor tissues depend upon the expression of TCR, Foxp3, and IL-33. In tumor tissues, IL-33 deficiency attenuated Treg immunosuppressive activity against tumor growth. This activity of IL-33 was IL-33 receptor ST2-independent but depended on the NF-κB-T-bet-dependent IFN-γ production (55). In contrast, Treg development and maintenance in VAT was dependent upon the high expression of ST2 (56). IL-33 treatment increased ST2+ Tregs in VAT in obese mice (57). TCR : MHC-II interactions are also required for VAT Treg development and maintenance (58). In contrast, many extrinsic cytokines play different roles in Treg immunosuppression (Figure 1). Tregs lost their stability or proliferation when cells were exposed to inflammatory cytokines IL-6 and leptin and even IL-4 (19, 59, 60). Tregs became Foxp3- cells that express inflammatory cytokines and failed to mediate immune suppression (61–63). Tregs also express leptin receptor, and leptin decreases Treg proliferation. Leptin is an important environment cue in the adipose tissue to modulate Tregs (60). CD44hi memory T cells release IL-4, IL-21, and IFN-γ that inhibit TGF-β-induced Foxp3 expression (64). IL-6 and downstream STAT3 activation, as well as IL-23, IL-1β, and IL-21 are critical for Treg-to-Th17 conversion (65–67).
Transcription Factors
In the nucleus, besides Foxp3, other transcription factors, such as STAT5, NFAT, Foxo1 (Forkhead Box O1), PPAR-γ (peroxisome proliferator- activated receptor-γ), and HIF-1 (hypoxia-inducible factor 1) control Treg development and maintenance by binding to the Foxp3 gene promoter and regulating Foxp3 gene expression (44, 68–71) (Figure 1). Foxo1 deficiency curtailed Treg development. Tregs from Foxo1-deficient mice were non-functional, did not express CTLA-4, and TGF-β failed to induce Foxp3 expression or to diminish T-bet expression (70). The expression of PPAR-γ is critical for establishing Treg transcriptional program and homeostasis, although this function of PPAR-γ seems mainly in VAT but less so in other organs (44). Hypoxia increases the expression of HIF-1α as a negative regulator of iTreg differentiation that promotes Th17 differentiation (72, 73).
Amino Acid and Nucleic Acid Metabolites
Tumor cells modulate several environmental cues to affect tumor-resident Treg generation and function. Different from lymphoid tissue Tregs, those in tumors are often in activated state with a metabolic signature. Subtle perturbations in metabolic signaling impact tumor-resident Treg cell homeostasis and function (33). Basal amino acid catabolism maintains immune homeostasis, but increased amino acid catabolism enhances immune suppression. L-tryptophan (Trp) and L-arginine (Arg) are probably the most important immune response regulatory amino acids. While TDO (2,3-dioxygenase), IDO-1 and IDO-2 catabolize Trp, iNOS (inducible nitric oxide synthetase), and arginases Arg1 and Arg2 catabolize Arg. Reduction in Treg numbers is associated with reduced Trp and Arg catabolism. DCs express IDO to induce Foxp3+ Treg generation by inhibiting DC IL-6 production (74, 75). In mice, administration of Trp metabolite 3-hydroxyanthranthranilic acid (3-HAA) enhanced TGF-β secretion in DCs, increased Treg cells, and reduced Th1 and Th17 cell conversions (76, 77) (Figure 1). IDO has brought to the attention due to its broad expression in a variety of human tumor types. Under the tumor microenvironment, the immunosuppressive Tregs express lipid phosphatase PTEN (phosphatase and tensin homolog). IDO activates PTEN as a mechanism to maintain Treg immunosuppressive activity (78). Active IDO also maintains Treg immunosuppressive activity by suppressing IL-6 expression from DCs and blocking Treg to Th17 cell conversion. In a mouse melanoma model, IDO inhibitor together with anti-tumor vaccine increased DC IL-6 secretion, Treg to Th17 conversion, and CD8+ T-cell activity and anti-tumor efficacy (74). In addition, suppression of Arg1 and Arg2 activities inhibited Treg proliferation and promoted tumor antigen-specific T-cell tolerance. High levels of Foxp3+ Tregs in the tumor environment and the absence of Arg2 significantly impacted the survival of patients with head and neck squamous cell carcinoma (79, 80).
Extracellular purine metabolites regulate Foxp3 expression via the pro-inflammatory adenosine triphosphate (ATP) and anti-inflammatory adenosine. Tregs are more sensitive to oxidative stress than T-effector cells due to the low level of NRF2 (nuclear factor-erythroid factor 2-related factor 2, a key transcription factor for antioxidant responses) in Tregs. Oxidative stress induces Treg apoptosis, followed by ATP release. Apoptotic Tregs express the ectonucleotidases CD39 and CD73 that convert extracellular ATP into immunosuppressive adenosine to increase Treg suppressive function (53, 81, 82). Adenosine binds to the A2A receptor (A2AR) to inhibit T-effector cell activity (83) (Figure 1). Activation of the adenosine signaling pathway can lead to enhanced Treg function, impaired APC function, and inhibition of NK cell activation (84). While apoptotic Tregs suppressed T-cell activation and tumor necrosis factor-α (TNF-α) and IL-2 expression, pharmacological inhibition of A2A inhibitor blocked these Treg activities (81).
Glucose Uptake and Glycolysis
Tregs use mitochondrial metabolism and oxidative phosphorylation for energy production (85). Treg cell extrinsic nutrients regulate oxidative phosphorylation. High glucose conditions, glucose transporter-mediated glucose uptake, or glucose avidity in Tregs correlate with Treg poor suppressive function and instability (86). In contrast, glucose deprivation drives Foxp3 expression, and shifts the T-cell differentiation from Tconv to iTregs (72, 85, 87). Intratumoral Tregs metabolize the glycolytic by-product lactic acid to support their proliferation and suppressor function. Deletion of the lactate transporter monocarboxylate transporter 1 (MCT1) in Tregs slowed tumor growth and increased responses to immunotherapy (86) (Figure 1). Together, elevated glycolysis can be detrimental to Treg. Accelerated glycolysis in tumor cells enhances glucose consumption and leads to increase of fatty acids. The metabolism of fatty acid then promotes Treg development (88). Increased glycolysis also promotes Th1 cell differentiation by epigenic regulation of the Ifng gene locus (89). Inhibition of glycolysis promotes the induction of Foxp3 expression in response to TGF-β and IL-2 stimulations (72, 90). Glycolytic enzyme enolase-1 also binds to the Foxp3 promoter and represses the expression of a transcription isoform of Foxp3 that is important for Treg suppressive activity (91). T cells use glycolysis during T-effector cell differentiation. While programmed cell death 1 (PD-1) ligation leads to defective T-cell glycolysis, amino acid metabolites and CTLA-4 inhibit T-cell glycolysis (92), supporting a mechanism by which Tregs control T-cell glycolysis and T-effector cell differentiation. Inhibition of the glycolytic pathway with the glucose analogue 2-deoxyglucose blocked Th17 development while promoting Treg generation (72).
The regulation of tissue-specific Treg differentiation, maintenance, and immunosuppression function has been summarized elsewhere (33). In addition to what we discussed above, many other metabolites also affect Treg pathobiology. For example, Vitamin A metabolite all-trans retinoic acid (RA) produced by DC subsets promote Foxp3 expression (93–96). The ten-eleven translocation (TET) family enzymes activate TSDR demethylation (97–99). CNS2 is demethylated in a TET-dependent manner in Foxp3+ iTregs (98). Vitamin C shows its activity to induce TET enzymatic activity in iTregs, thereby providing a new mechanism to stabilize iTregs (98, 100, 101). Tregs also suppress the activities of B cells, natural killer T cells (NKT), and cytotoxic T lymphocyte by secreting perforin and granzymes A and B (102, 103) (Figure 1). Tregs also inhibit the activation of type II innate lymphocytes, including NKT cells, mast cells, basophils, and eosinophils (104).
Regulatory T-Cell Targeting Therapies
Anti-CTLA-4 Monoclonal Antibody
CTLA-4 is an inhibitory receptor on Treg surface and is the most studied and widely used drug target in the clinic. Although Tregs inhibit the immune function in vivo through a variety of ways, deficiency of the CTLA-4 pathway in Tregs makes it difficult for Tregs to maintain their self-tolerance and immune homeostasis even if other inhibitory mechanisms are more active to compensate for the defects. CTLA-4 is an important negative regulator of T-cell responses and a key molecular target for Treg inhibition in physiological and pathological immune responses, including autoimmunity, allergy, and tumor immunity (105). The binding of CTLA-4 with CD80/86 molecule cuts off the CD80/86-CD28 pathway, an important step of T-cell activation. If the CD80/86-CD28 pathway is blocked, T cells become inactive in immune response. It is known that radiotherapy upregulates the expression of CD80 on APC surface. CTLA-4 on Treg surface has higher affinity to CD80, which leads to enhanced immunosuppression after radiotherapy. Therefore, the anti-CTLA-4 antibody therapy can be carried out at the same time of radiotherapy to achieve better anti-tumor immune effect. At present, the anti-CTLA-4 antibody drug mainly refers to iplimumab (IPI). It is the first Treg-targeted drug approved for clinical use, as the first choice for the treatment of advanced malignant melanoma. To enhance the CTLA-4 single drug therapy efficacy, Rudqvist et al. tested and found that combination of radiotherapy and anti-CTLA-4 antibody drug significantly increased the number of infiltrating lymphocytes and improved the survival rate in a mouse breast cancer model (106). Similarly, A phase II clinical trial evaluated the efficacy of local radiotherapy and IPI in patients with metastatic non-small cell lung cancer. The results showed that the objective response rate (ORR) and progression free survival (PFS) were all significantly better in patients received the combined therapy than those from the IPI therapy alone (107). Consistently, in a mouse liver metastasis model, combination of radiotherapy and anti-CD25/CTLA-4 antibody therapy increased tumor CD8+ T-cell accumulation with concurrent decrease of tumor Tregs, suppressed locally irradiated and abscopal unirradiated tumor growth, and improved overall survival rate. Therefore, combined radiotherapy with anti-CTLA-4 antibody reduced liver metastasis (108). Yet, not all studies yielded the same conclusion. A retrospective analysis of 133 tumor samples from patients with metastatic non-small cell lung cancer, melanoma, or renal cell cancer after receiving radiotherapy with or without combined immunotherapy showed that the combination of focal palliative radiation and CTLA-4 and/or PD-1 inhibitors was well tolerated. Patients received the combination therapy experienced more immune-related adverse events than those received either therapy individually (107). These observations might be due to the small patient sample size and many different types of treatments and combinations.
Anti-GITR Monoclonal Antibody
GITR is a member of the TNF receptor protein family. In recent years, GITR has been widely studied as a promising Treg target. Studies have shown that the interaction between GITR and GITR ligand provides costimulatory signals for CD4+ and CD8+ T cells, activates T-effector cells, and suppresses Treg inhibitory activity (109). An agonistic anti-GITR monoclonal antibody or GITR ligands to activate GITR signaling can inhibit CD4+Foxp3+ Treg immunosuppressive activity and induce T-effector cell resistance to CD4+Foxp3+ Treg-mediated immunosuppression (110). The agonist GITR monoclonal antibody enhanced the anti-tumor response by increasing the activity of T-effector cells and reducing the invasion of Tregs (111). TRX-518, a GITR monoclonal antibody, was applied to 40 patients with metastatic solid tumors such as melanoma, non-small cell lung, and colorectal cancers in a phase I study. Single dose of TRX518 up to 8 mg/kg was well tolerated (NCT01239134). A phase I study of AMG228, another GITR agonist, exhibited favorable pharmacokinetics in patients with advanced solid tumors, but there was no evidence of T-cell activation or anti-tumor activity with AMG 228 monotherapy (NCT02437916) (112). In a mouse model of glioma, anti-GITR monoclonal antibody therapy combined with the radiotherapy increased the ratio of CD4+ T cells to Tregs, promoted tumor regression, and significantly improved mouse survival rate (113). Therefore, the effect of GITR on radio-sensitivity is worth to explore further in human trials.
Anti-CD25 Antibody
CD25 antigen, as IL-2 receptor, is a 55-kDa single chain glycoprotein, and is mainly expressed by activated T cells. IL-2 is necessary for the expansion of CD8+ cytotoxic T lymphocyte (CTL) cells. In tumor microenvironment, Tregs express IL-2 receptors with high affinities that surpass the T-effector cells to obtain limited IL-2. Therefore, Tregs gain greater proliferation advantage than the T-effector cells do to promote immunosuppression (114, 115). Oweida et al. showed that anti-CD25 antibody treatment combined with the radiotherapy enhanced the T-cell cytotoxicity and induced the tumor antigen-specific memory response, which cured 57.1% of mouse tumors (116). To confirm further the effect benefit of this combination therapy, Ji et al. proved that radiotherapy combined with anti-CD25 monoclonal antibody therapy reversed the increase of PD-1 expression on CD8+ T cells and CD4+ T cells during the radiotherapy, thereby inhibiting the tumor growth and improving the overall survival rate (108). Yet, few similar studies failed to obtain clinically significant enhancement of Treg-depletion-associated beneficial effects (117–119). One possible reason is that the activated T-effector cells also express CD25. Anti-CD25 antibody treatment may also reduce the activity of activated T-effector cells, thereby weakening the anti-tumor immunity that was enhanced by Treg depletion. Anti-CD25 antibody therapy has also been evaluated in clinical trials. To breast cancer patients, use of an anti-CD25-depleting antibody daclizumab stabilized cancer progression and increased overall survival time in a median follow-up of 22.3 months (120). Meanwhile, the administration time and dose of monoclonal antibody may also be important factors to affect the tumor immunity between Tregs and T-effector cells.
IDO-1 Inhibitor
IDO-1 is a naturally occurred immune regulatory enzyme. Prior studies have found many potential immunosuppressive mechanisms of IDO-1, among which the main mechanism is Trp catabolism in the microenvironment. The relationship between tumor growth and the increase of Trp catabolism becomes more and more recognized. Trp is metabolized into kynurenine by the IDO limiting enzyme, which blocks or inactivates the cell cycle of T-effector cells. This progress plays a direct role in tumor immune escape to promote Treg maturation and activation. IDO inhibitors have been intensively studied in recent years. The radiotherapy could alter IDO-mediated immune activity, and there was a strong correlation between the IDO activity and survival outcomes in patients under a radiotherapy (121). Therefore, the use of IDO inhibitors during radiotherapy can delay the tumor growth. IDO inhibitors in combination with radiotherapy down-regulated the number of CD4+CD25+Foxp3+ Tregs and DC expression of MHC-II in the spleen, along with decreased expression of inhibitory receptors (PD-1 and T cell immunoglobulin domain and mucin domain 3) and ligands (galetin-9 and B- and T-lymphocyte attenuator) to prevent T-cell exhaustion, while DCs and T-effector cells were activated (122). This combination therapy enhanced anti-tumor immunity and inhibited tumor progression.
Immunosuppressive Cytokines
Other therapy related to the inhibition of Treg function is to target the Treg immunosuppressive cytokines, such as TGF-β and IL-10. Hou et al. showed that TGF-β chimeric antigen receptor (CAR) T cells promoted the anti-tumor immune response by alleviating TGF-β-mediated immunosuppression, and reduced Foxp3+ Treg differentiation (123). IL-10 deficiency diminished the Treg-mediated immunosuppression by suppressing neuropillin expression in Treg cells in mouse tumor models (124). Furthermore, pegylated-recombinant human IL-10 has been used in phase I clinical trial (NCT02009449) to decrease TGF-β expression and up-regulate serum levels of IFN-γ and IL-18 in patients with tumors (125). Pegylated-recombinant human IL-10 has also been used to test its efficacy in patients with metastatic pancreatic cancer (phase III clinical trial, NCT02923921) (126).
Dual Therapy
The combination of immunotherapy and radiotherapy enhances the anti-tumor immune response through various mechanisms of immune escape, thereby improving the survival rate more than any single therapy. PD-1 is a type I transmembrane glycoprotein, which was first discovered in 1992 (127). It is mainly expressed on the surface of activated T cells, but its ligand PD-L1 is expressed in many cell types, such as APCs, macrophages, and tumor cells, etc. Under normal circumstances, PD-1 binding of PD-L1 can stop the continuous activation of T cells and prevent the occurrence of autoimmune diseases. Under pathological conditions however, PD-L1 expression from tumor cells was significantly higher than that in other normal cells. The interaction of PD-1 and PD-L1 inhibits the activation of anti-tumor T cells, thereby inducing tumor growth. Many studies have shown that the radiotherapy combined with the anti-PD-1-PD-L1 therapy dramatically suppressed tumor growth. For example, combined use of irradiation and anti-PD-L1 antibody therapy reduced the local accumulation of tumor-infiltrating myeloid-derived suppressor cells with feature of immune suppression (128), which altered the tumor immune microenvironment (129), supporting a close interaction between irradiation, T cells, and the PD-L1/PD-1 axis. Pembrolizumab is a highly selective humanized PD-1 monoclonal antibody. A randomized phase II clinical trial (NCT02492568) of 92 patients of advanced non-small cell lung cancer showed that combined therapy of pembrolizumab after radiotherapy showed much higher overall response rate, progression-free survival rate, and overall survival time than those in patients received pembrolizumab alone (130). A retrospective analysis of 133 patients with metastatic non-small cell lung cancer, melanoma, or renal cell cancer after receiving radiotherapy with or without combined immunotherapy with CTLA-4 and/or PD-1 inhibitors showed that patients were well tolerated with the combination of focal palliative radiation and CTLA-4 and/or PD-1 inhibitors (107). Twyman-Saint Victor et al. put forward the hypothesis that the optimal response in melanoma and other cancer types required radiotherapy together with the anti-CTLA-4 and anti-PD-L1/PD-1 antibodies (Figure 2) (131). They demonstrated that anti-CTLA-4 monoclonal antibody IPI combined with radiotherapy made the tumor substantially subsided in both clinical trials and mouse models of metastatic melanoma, although drug resistance can be a potential concern. Of note, radiotherapy, anti-CTLA-4 antibody, and anti-PD-L1/PD-1 antibody promote tumor immune response with distinct mechanisms. Radiotherapy enhances intratumoral T-cell TCR repertoire diversity, anti-CTLA-4 antibody inhibits Treg and increases CD8+ T cell to Treg ratio, and anti-PD-L1/PD-1 antibody reverses T-cell exhaustion and increases T-cell expansion. Melanoma patients or mice with high expression of PD-L1 on melanoma cells failed to respond to radiotherapy or combined therapy and these patients showed much shorter progression-free survival and overall survival. High expression of PD-L1 on melanoma cells allowed tumors to escape the anti-CTLA-4-based therapy (131). Therefore, the known drug resistance may rely on the PD-L1 upregulation on melanoma cells, leading to T-cell depletion. The results from this clinical study suggest the importance of combined application of anti-CTLA-4 monoclonal antibody, anti-PD-1-PD-L1 monoclonal antibody, and radiotherapy, which activate the anti-tumor responses in multiple mechanisms.
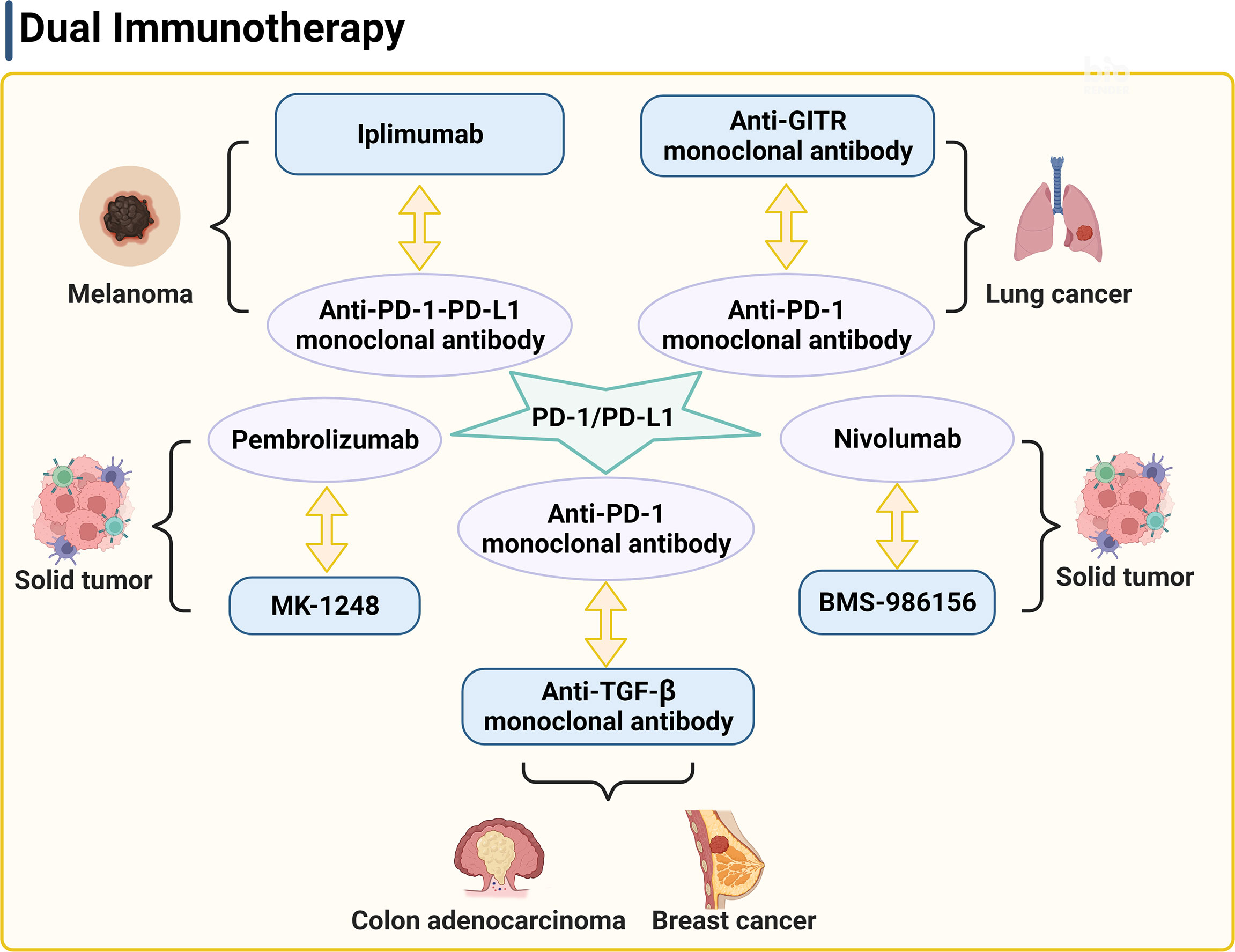
Figure 2 PD-1/PD-L1 is a key target of the dual therapy. The strategy of dual therapy contains anti-PD-1/PD-L1 antibodies to enhance the anti-tumor responses. PD-1, programmed cell death 1; PD-L1, programmed death ligand 1; GITR, glucocorticoid induced tumor necrosis factor receptor.
In a mouse model of anti-PD1 antibody therapy-resistant non-small cell lung adenocarcinoma, combination of anti-GITR monoclonal antibody therapy, anti-PD-1 monoclonal antibody therapy, and radiotherapy also significantly increased the numbers of CD4+ and CD8+ effector memory cells in blood, spleen, and tumor draining lymph node, enhanced tumor-specific immune response, and improved the survival rate (Figure 2) (132). Half of the mice showed no tumors. Furthermore, an anti-GITR antibody (BMS-986156) combined with a PD-1 inhibitor (nivolumab) was well tolerated with low immunogenicity in patients with advanced solid tumors (Figure 2). Antitumor activity was observed with combined use of BMS-986156 and nivolumab at doses predicted to be biologically active (NCT02598960). In a trial of solid tumors, including colorectal cancer, melanoma, and renal cell carcinoma, 17 patients who received a combination therapy of an anti-GITR antibody agonist (MK-1248) and humanized PD-1 monoclonal antibody pembrolizumab reached to an objective response rate of 18% versus no response in 20 patients who received MK-1248 monotherapy (Figure 2) (133). In preclinical experiments, anti-TGF-β monoclonal antibody and anti-PD-1 monoclonal antibody combination with radiotherapy also showed effective antitumor activity (Figure 2) (134, 135). Together, these studies suggest the potential that dual therapy combined with or without radiotherapy overcomes common tumor resistance concerns.
Conclusions
In this short review, we overviewed the origins, functions, and potential clinical applications of Tregs in cancer patients. Tregs participate in the process of immune dysfunction. Based on the mechanism of Tregs, some immunotherapies have demonstrated their clinical efficacy in tumor controls, and candidate drugs have proceeded to clinical trials (Table 1). In particular, the combination of immune inhibitory therapies and radiotherapy has demonstrated its success of inhibitory efficacies in tumor progression, and potential synergistic mechanisms of combination therapy may explain the positive results seen in both clinical trials and experimental models. There are accumulating evidences to support the hypothesis that radiotherapy combined with anti-Treg therapy reverses Treg immunosuppressive activity and enhances the efficacy of radiotherapy. Therefore, the revised Treg immunotherapy has become a potential breakthrough point of cancer therapy. It is necessary to explore more Treg functions and inhibitors in the future to effectively control or reverse the immunosuppressive activities of Tregs and to optimize cancer combinational therapy.

Table 1 Selected immunotherapy targets and clinical trials.
As we discussed above, there are many essential molecules that regulate Treg functions. These include Treg surface molecules, cytokines, transcription factors, amino acid and nucleic acid metabolites, and glucose and glycolytic metabolites. Targeting these molecules and their pathways has led to the development of varies immunotherapy approaches to target the immunosuppressive activity of Tregs against different types of tumors. Yet, while drug efficacy and target selection are of the primary interest of patients and physicians, the development of drug resistance has been the major hurdle in cancer therapy. Here we discussed the benefit of dual therapy by combining the radiotherapy with anti-CTLA-4 and/or anti-PD-1-PD-L1 antibody immunotherapy. While dual therapy may demonstrate synergistic beneficial effects against tumor progression, patients may still develop drug resistance. Immunosuppressive Tregs display adverse effects in tumor growth by down-regulating T-effector cell immune responses. Tregs play protective roles in cardiac, metabolic, autoimmune, and neurological diseases (136–139). We showed that adoptive transfer of Tregs in recipient mice blocked angiotensin II perfusion-induced abdominal aortic aneurysms (140), induced β3-adrenergic receptor agonist-induced adipose tissue thermogenic program (141), and blunted the development of spontaneous systemic lupus erythematosus (142). Therefore, one should consider these beneficial effects of Tregs before treating cancer patients, especially those with cardiovascular, metabolic, and neurological complications. Therefore, discovery of tumor-selective Treg functions and combination therapy to target multiple Treg immunosuppression pathways might be necessary. For example, CD36 expression in intratumoral Tregs was much higher than in those from lymphoid organs (48). CD36 targeting primes tumors to PD-1 blockade and elicited additive anti-tumor responses with the anti-PD1 antibody therapy (48). Tumor resident Tregs express high levels of unique signature genes Ccr8, Tnfrsf8, Cxcr3, and Smasn1 (143, 144), which might serve as valuable targets for tumor immunotherapy. Further, non-lymphoid and tumor tissues differ in their metabolic environments, nutrient supplies, and intracellular metabolic requirements. Consideration of these factors may help develop new generations drugs for cancer patients by selectively targeting the immunosuppressive functions of intratumoral Tregs.
Author Contributions
RG and JW drafted the manuscript. G-PS edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2021-I2M-1-016 to RG), National Natural Science Foundation of China (82100514 to RG), National Key Research and Development Program of China Grants (2019YFA0801804 and 2019YFA0801703 to JW), and grants from the National Heart, Lung, and Blood Institute (HL151627 and HL157073 to G-PS), and the National Institute of Neurological Disorders and Stroke (AG063839 to G-PS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Killebrew JR, Perdue N, Kwan A, Thornton AM, Shevach EM, Campbell DJ. A Self-Reactive TCR Drives the Development of Foxp3+ Regulatory T Cells That Prevent Autoimmune Disease. J Immunol (2011) 187:861–9. doi: 10.4049/jimmunol.1004009
3. Treves AJ, Carnaud C, Trainin N, Feldman M, Cohen IR. Enhancing T Lymphocytes From Tumor-Bearing Mice Suppress Host Resistance to a Syngeneic Tumor. Eur J Immunol (1974) 4:722–7. doi: 10.1002/eji.1830041104
4. North RJ, Bursuker I. Generation and Decay of the Immune Response to a Progressive Fibrosarcoma. I. Ly-1+2- Suppressor T Cells Down-Regulate the Generation of Ly-1-2+ Effector T Cells. J Exp Med (1984) 159:1295–311. doi: 10.1084/jem.159.5.1295
5. Berendt MJ, North RJ. T-Cell-Mediated Suppression of Anti-Tumor Immunity. An Explanation for Progressive Growth of an Immunogenic Tumor. J Exp Med (1980) 151:69–80. doi: 10.1084/jem.151.1.69
6. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155:1151–64.
7. Karkhah A, Javanian M, Ebrahimpour S. The Role of Regulatory T Cells in Immunopathogenesis and Immunotherapy of Viral Infections. Infect Genet Evol (2018) 59:32–7. doi: 10.1016/j.meegid.2018.01.015
8. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol (2016) 37:803–11. doi: 10.1016/j.it.2016.08.012
9. Campbell DJ. Control of Regulatory T Cell Migration, Function, and Homeostasis. J Immunol (2015) 195:2507–13. doi: 10.4049/jimmunol.1500801
10. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
11. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
12. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J Exp Med (2003) 198:1875–86. doi: 10.1084/jem.20030152
13. Shevach EM, Thornton AM. Ttregs, Ptregs, and Itregs: Similarities and Differences. Immunol Rev (2014) 259:88–102. doi: 10.1111/imr.12160
14. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of Conserved Non-Coding DNA Elements in the Foxp3 Gene in Regulatory T-Cell Fate. Nature (2010) 463:808–12. doi: 10.1038/nature08750
15. Schlenner SM, Weigmann B, Ruan Q, Chen Y, von Boehmer H. Smad3 Binding to the Foxp3 Enhancer Is Dispensable for the Development of Regulatory T Cells With the Exception of the Gut. J Exp Med (2012) 209:1529–35. doi: 10.1084/jem.20112646
16. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T Cell Receptor Stimulation-Induced Epigenetic Changes and Foxp3 Expression Are Independent and Complementary Events Required for Treg Cell Development. Immunity (2012) 37:785–99. doi: 10.1016/j.immuni.2012.09.010
17. Li X, Liang Y, LeBlanc M, Benner C, Zheng Y. Function of a Foxp3 Cis-Element in Protecting Regulatory T Cell Identity. Cell (2014) 158:734–48. doi: 10.1016/j.cell.2014.07.030
18. Hilbrands R, Chen Y, Kendal AR, Adams E, Cobbold SP, Waldmann H, et al. Induced Foxp3(+) T Cells Colonizing Tolerated Allografts Exhibit the Hypomethylation Pattern Typical of Mature Regulatory T Cells. Front Immunol (2016) 7:124. doi: 10.3389/fimmu.2016.00124
19. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the Inheritance of Regulatory T Cell Identity by a Cis Element in the Foxp3 Locus. Cell (2014) 158:749–63. doi: 10.1016/j.cell.2014.07.031
20. Kitagawa Y, Ohkura N, Sakaguchi S. Molecular Determinants of Regulatory T Cell Development: The Essential Roles of Epigenetic Changes. Front Immunol (2013) 4:106. doi: 10.3389/fimmu.2013.00106
21. Sekiya T, Nakatsukasa H, Lu Q, Yoshimura A. Roles of Transcription Factors and Epigenetic Modifications in Differentiation and Maintenance of Regulatory T Cells. Microbes Infect (2016) 18:378–86. doi: 10.1016/j.micinf.2016.02.004
22. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA Methylation Controls Foxp3 Gene Expression. Eur J Immunol (2008) 38:1654–63. doi: 10.1002/eji.200838105
23. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic Control of the Foxp3 Locus in Regulatory T Cells. PLoS Biol (2007) 5:e38. doi: 10.1371/journal.pbio.0050038
24. Attias M, Al-Aubodah T, Piccirillo CA. Mechanisms of Human FoxP3(+) Treg Cell Development and Function in Health and Disease. Clin Exp Immunol (2019) 197:36–51. doi: 10.1111/cei.13290
25. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) Regulatory T Cells Through GITR Breaks Immunological Self-Tolerance. Nat Immunol (2002) 3:135–42. doi: 10.1038/ni759
26. Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
27. Li C, Spallanzani RG, Mathis D. Visceral Adipose Tissue Tregs and the Cells That Nurture Them. Immunol Rev (2020) 295:114–25. doi: 10.1111/imr.12850
28. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science (2011) 331:337–41. doi: 10.1126/science.1198469
29. Sharma A, Rudra D. Emerging Functions of Regulatory T Cells in Tissue Homeostasis. Front Immunol (2018) 9:883. doi: 10.3389/fimmu.2018.00883
30. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T Cells in Tumor Microenvironment: New Mechanisms, Potential Therapeutic Strategies and Future Prospects. Mol Cancer (2020) 19:116. doi: 10.1186/s12943-020-01234-1
31. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
32. Chapman NM, Boothby MR, Chi H. Metabolic Coordination of T Cell Quiescence and Activation. Nat Rev Immunol (2020) 20:55–70. doi: 10.1038/s41577-019-0203-y
33. Shi H, Chi H. Metabolic Control of Treg Cell Stability, Plasticity, and Tissue-Specific Heterogeneity. Front Immunol (2019) 10:2716. doi: 10.3389/fimmu.2019.02716
34. Lee W, Lee GR. Transcriptional Regulation and Development of Regulatory T Cells. Exp Mol Med (2018) 50:e456. doi: 10.1038/emm.2017.313
35. Elkord E, Sasidharan NV. T-Regulatory Cells in Health and Disease. J Immunol Res (2018) 2018:5025238. doi: 10.1155/2018/5025238
36. Tanaka A, Sakaguchi S. Targeting Treg Cells in Cancer Immunotherapy. Eur J Immunol (2019) 49:1140–6. doi: 10.1002/eji.201847659
37. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
38. Tanaka A, Sakaguchi S. Regulatory T Cells in Cancer Immunotherapy. Cell Res (2017) 27:109–18. doi: 10.1038/cr.2016.151
39. Furukawa A, Wisel SA, Tang Q. Impact of Immune-Modulatory Drugs on Regulatory T Cell. Transplantation (2016) 100:2288–300. doi: 10.1097/TP.0000000000001379
40. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
41. Seoane J, Gomis RR. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb Perspect Biol (2017) 9:a022277. doi: 10.1101/cshperspect.a022277
42. Li MO, Sanjabi S, Flavell RA. Transforming Growth Factor-Beta Controls Development, Homeostasis, and Tolerance Of T Cells by Regulatory T Cell-Dependent and -Independent Mechanisms. Immunity (2006) 25:455–71. doi: 10.1016/j.immuni.2006.07.011
43. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-Expressed CTLA-4 Depletes CD80/CD86 by Trogocytosis, Releasing Free PD-L1 on Antigen-Presenting Cells. Proc Natl Acad Sci USA (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
44. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-Gamma Is a Major Driver of the Accumulation and Phenotype of Adipose Tissue Treg Cells. Nature (2012) 486:549–53. doi: 10.1038/nature11132
45. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
46. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
47. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. HIF-1alpha Is a Metabolic Switch Between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep (2019) 27:226–37. doi: 10.1016/j.celrep.2019.03.029
48. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-Mediated Metabolic Adaptation Supports Regulatory T Cell Survival and Function in Tumors. Nat Immunol (2020) 21:298–308. doi: 10.1038/s41590-019-0589-5
49. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
50. Liu Q, Yang C, Wang S, Shi D, Wei C, Song J, et al. Wnt5a-Induced M2 Polarization of Tumor-Associated Macrophages via IL-10 Promotes Colorectal Cancer Progression. Cell Commun Signal (2020) 18:51. doi: 10.1186/s12964-020-00557-2
51. Sullivan JA, Tomita Y, Jankowska-Gan E, Lema DA, Arvedson MP, Nair A, et al. Treg-Cell-Derived IL-35-Coated Extracellular Vesicles Promote Infectious Tolerance. Cell Rep (2020) 30:1039–51. doi: 10.1016/j.celrep.2019.12.081
52. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 Limits Anti-Tumor Immunity. Immunity (2016) 44:316–29. doi: 10.1016/j.immuni.2016.01.013
53. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of Ectonucleotidase CD39 by Foxp3+ Treg Cells: Hydrolysis of Extracellular ATP and Immune Suppression. Blood (2007) 110:1225–32. doi: 10.1182/blood-2006-12-064527
54. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T Regulatory and Primed Uncommitted CD4 T Cells Express CD73, Which Suppresses Effector CD4 T Cells by Converting 5'-Adenosine Monophosphate to Adenosine. J Immunol (2006) 177:6780–6. doi: 10.4049/jimmunol.177.10.6780
55. Hatzioannou A, Banos A, Sakelaropoulos T, Fedonidis C, Vidali MS, Köhne M, et al. An Intrinsic Role of IL-33 in Treg Cell-Mediated Tumor Immunoevasion. Nat Immunol (2020) 21:75–85. doi: 10.1038/s41590-019-0555-2
56. Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, et al. Antigen- and Cytokine-Driven Accumulation of Regulatory T Cells in Visceral Adipose Tissue of Lean Mice. Cell Metab (2015) 21:543–57. doi: 10.1016/j.cmet.2015.03.005
57. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The Transcriptional Regulators IRF4, BATF and IL-33 Orchestrate Development and Maintenance of Adipose Tissue-Resident Regulatory T Cells. Nat Immunol (2015) 16:276–85. doi: 10.1038/ni.3085
58. Deng T, Liu J, Deng Y, Minze L, Xiao X, Wright V, et al. Adipocyte Adaptive Immunity Mediates Diet-Induced Adipose Inflammation and Insulin Resistance by Decreasing Adipose Treg Cells. Nat Commun (2017) 8:15725. doi: 10.1038/ncomms15725
59. Kastner L, Dwyer D, Qin FX. Synergistic Effect of IL-6 and IL-4 in Driving Fate Revision of Natural Foxp3+ Regulatory T Cells. J Immunol (2010) 185:5778–86. doi: 10.4049/jimmunol.0901948
60. Galgani M, De Rosa V, La Cava A, Matarese G. Role of Metabolism in the Immunobiology of Regulatory T Cells. J Immunol (2016) 197:2567–75. doi: 10.4049/jimmunol.1600242
61. Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of Natural Foxp3+ T Cells: A Committed Regulatory T-Cell Lineage and an Uncommitted Minor Population Retaining Plasticity. Proc Natl Acad Sci USA (2009) 106:1903–8. doi: 10.1073/pnas.0811556106
62. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the Transcription Factor Foxp3 Leads to the Generation of Pathogenic Memory T Cells In Vivo. Nat Immunol (2009) 10:1000–7. doi: 10.1038/ni.1774
63. Duarte JH, Zelenay S, Bergman ML, Martins AC, Demengeot J. Natural Treg Cells Spontaneously Differentiate Into Pathogenic Helper Cells in Lymphopenic Conditions. Eurj Immunol (2009) 39:948–55. doi: 10.1002/eji.200839196
64. Hill JA, Hall JA, Sun CM, Cai Q, Ghyselinck N, Chambon P, et al. Retinoic Acid Enhances Foxp3 Induction Indirectly by Relieving Inhibition From CD4+CD44hi Cells. Immunity (2008) 29:758–70. doi: 10.1016/j.immuni.2008.09.018
65. Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, et al. DC Activated via Dectin-1 Convert Treg Into IL-17 Producers. Eurj Immunol (2008) 38:3274–81. doi: 10.1002/eji.200838950
66. Li L, Kim J, Boussiotis VA. IL-1beta-Mediated Signals Preferentially Drive Conversion of Regulatory T Cells But Not Conventional T Cells Into IL-17-Producing Cells. J Immunol (2010) 185:4148–53. doi: 10.4049/jimmunol.1001536
67. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity (2008) 29:44–56. doi: 10.1016/j.immuni.2008.05.007
68. Liu B, Salgado OC, Singh S, Hippen KL, Maynard JC, Burlingame AL, et al. The Lineage Stability and Suppressive Program of Regulatory T Cells Require Protein O-GlcNAcylation. Nat Commun (2019) 10:354. doi: 10.1038/s41467-019-08300-3
69. Vaeth M, Schliesser U, Müller G, Reissig S, Satoh K, Tuettenberg A, et al. Dependence on Nuclear Factor of Activated T-Cells (NFAT) Levels Discriminates Conventional T Cells From Foxp3+ Regulatory T Cells. Proc Natl Acad Sci USA (2012) 109:16258–63. doi: 10.1073/pnas.1203870109
70. Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch’en IL, Stockmann C, et al. Foxo Transcription Factors Control Regulatory T Cell Development and Function. Immunity (2010) 33:890–904. doi: 10.1016/j.immuni.2010.12.002
71. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-Inducible Factor-1 Alpha-Dependent Induction of FoxP3 Drives Regulatory T-Cell Abundance and Function During Inflammatory Hypoxia of the Mucosa. Proc Natl Acad Sci USA (2012) 109:E2784–93. doi: 10.1073/pnas.1202366109
72. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J Exp Med (2011) 208:1367–76. doi: 10.1084/jem.20110278
73. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) Balance by Hypoxia-Inducible Factor 1. Cell (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
74. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-Dioxygenase Controls Conversion of Foxp3+ Tregs to TH17-Like Cells in Tumor-Draining Lymph Nodes. Blood (2009) 113:6102–11. doi: 10.1182/blood-2008-12-195354
75. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO Activates Regulatory T Cells and Blocks Their Conversion Into Th17-Like T Cells. J Immunol (2009) 183:2475–83. doi: 10.4049/jimmunol.0900986
76. Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, et al. IDO Upregulates Regulatory T Cells via Tryptophan Catabolite and Suppresses Encephalitogenic T Cell Responses in Experimental Autoimmune Encephalomyelitis. J Immunol (2010) 185:5953–61. doi: 10.4049/jimmunol.1001628
77. Correale J. Immunosuppressive Amino-Acid Catabolizing Enzymes in Multiple Sclerosis. Front Immunol (2020) 11:600428. doi: 10.3389/fimmu.2020.600428
78. Munn DH, Sharma MD, Johnson TS, Rodriguez P. IDO, PTEN-Expressing Tregs and Control of Antigen-Presentation in the Murine Tumor Microenvironment. Cancer Immunol Immunother (2017) 66:1049–58. doi: 10.1007/s00262-017-2010-2
79. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
80. Bron L, Jandus C, Andrejevic-Blant S, Speiser DE, Monnier P, Romero P, et al. Prognostic Value of Arginase-II Expression and Regulatory T-Cell Infiltration in Head and Neck Squamous Cell Carcinoma. Int J Cancer (2013) 132:E85–93. doi: 10.1002/ijc.27728
81. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative Stress Controls Regulatory T Cell Apoptosis and Suppressor Activity and PD-L1-Blockade Resistance in Tumor. Nat Immunol (2017) 18:1332–41. doi: 10.1038/ni.3868
82. Frydrychowicz M, Boruczkowski M, Kolecka-Bednarczyk A, Dworacki G. The Dual Role of Treg in Cancer. Scand J Immunol (2017) 86:436–43. doi: 10.1111/sji.12615
83. Hasanjani RM, Bayani M, Soleimani AS, Mohammadnia-Afrouzi M, Nouri HR, Ebrahimpour S. Evaluation of CD4+ CD25+ FoxP3+ Regulatory T Cells During Treatment of Patients With Brucellosis. J Biol Regul Homeost Agents (2016) 30:675–82.
84. Ohta A, Sitkovsky M. Extracellular Adenosine-Mediated Modulation of Regulatory T Cells. Front Immunol (2014) 5:304. doi: 10.3389/fimmu.2014.00304
85. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
86. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic Support of Tumour-Infiltrating Regulatory T Cells by Lactic Acid. Nature (2021) 591:645–51. doi: 10.1038/s41586-020-03045-2
87. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The Glucose Transporter Glut1 Is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
88. Lv Q, Wang K, Qiao S, Yang L, Xin Y, Dai Y, et al. Norisoboldine, a Natural AhR Agonist, Promotes Treg Differentiation and Attenuates Colitis via Targeting Glycolysis and Subsequent NAD(+)/SIRT1/SUV39H1/H3K9me3 Signaling Pathway. Cell Death Dis (2018) 9:258. doi: 10.1038/s41419-018-0297-3
89. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic Glycolysis Promotes T Helper 1 Cell Differentiation Through an Epigenetic Mechanism. Science (2016) 354:481–4. doi: 10.1126/science.aaf6284
90. Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Antoniadis N, Liakopoulos V, et al. Dichloroacetate at Therapeutic Concentration Alters Glucose Metabolism and Induces Regulatory T-Cell Differentiation in Alloreactive Human Lymphocytes. J Basic Clin Physiol Pharmacol (2013) 24:271–6. doi: 10.1515/jbcpp-2013-0001
91. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis Controls the Induction of Human Regulatory T Cells by Modulating the Expression of FOXP3 Exon 2 Splicing Variants. Nat Immunol (2015) 16:1174–84. doi: 10.1038/ni.3269
92. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
93. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and Regulatory T Cell Differentiation Mediated by Retinoic Acid. Science (2007) 317:256–60. doi: 10.1126/science.1145697
94. Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, et al. Retinoic Acid Increases Foxp3+ Regulatory T Cells and Inhibits Development of Th17 Cells by Enhancing TGF-Beta-Driven Smad3 Signaling and Inhibiting IL-6 and IL-23 Receptor Expression. J Immunol (2008) 181:2277–84. doi: 10.4049/jimmunol.181.4.2277
95. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical Role of All-Trans Retinoic Acid in Stabilizing Human Natural Regulatory T Cells Under Inflammatory Conditions. Proc Natl Acad Sci USA (2014) 111:E3432–40. doi: 10.1073/pnas.1408780111
96. Lu L, Ma J, Li Z, Lan Q, Chen M, Liu Y, et al. All-Trans Retinoic Acid Promotes TGF-Beta-Induced Tregs via Histone Modification But Not DNA Demethylation on Foxp3 Gene Locus. PLoS One (2011) 6:e24590. doi: 10.1371/journal.pone.0024590
97. Toker A, Engelbert D, Garg G, Polansky JK, Floess S, Miyao T, et al. Active Demethylation of the Foxp3 Locus Leads to the Generation of Stable Regulatory T Cells Within the Thymus. J Immunol (2013) 190:3180–8. doi: 10.4049/jimmunol.1203473
98. Cue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, et al. Control of Foxp3 Stability Through Modulation of TET Activity. J Exp Med (2016) 213:377–97. doi: 10.1084/jem.20151438
99. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis. Immunity (2015) 43:251–63. doi: 10.1016/j.immuni.2015.07.017
100. Sasidharan Nair V, Song MH, Oh KI. Vitamin C Facilitates Demethylation of the Foxp3 Enhancer in a Tet-Dependent Manner. J Immunol (2016) 196:2119–31. doi: 10.4049/jimmunol.1502352
101. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, et al. Vitamin C Induces Tet-Dependent DNA Demethylation and a Blastocyst-Like State in ES Cells. Nature (2013) 500:222–6. doi: 10.1038/nature12362
102. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and Perforin Are Important for Regulatory T Cell-Mediated Suppression of Tumor Clearance. Immunity (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
103. Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential Expression of Granzymes A and B in Human Cytotoxic Lymphocyte Subsets and T Regulatory Cells. Blood (2004) 104:2840–8. doi: 10.1182/blood-2004-03-0859
104. Boonpiyathad T, Sozener ZC, Akdis M, Akdis CA. The Role of Treg Cell Subsets in Allergic Disease. Asian Pac J Allergy Immunol (2020) 38:139–49. doi: 10.12932/AP-030220-0754
105. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: A Moving Target in Immunotherapy. Blood (2018) 131:58–67. doi: 10.1182/blood-2017-06-741033
106. Rudqvist NP, Pilones KA, Lhuillier C, Wennerberg E, Sidhom JW, Emerson RO, et al. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol Res (2018) 6:139–50. doi: 10.1158/2326-6066.CIR-17-0134
107. Bang A, Wilhite TJ, Pike L, Cagney DN, Aizer AA, Taylor A, et al. Multicenter Evaluation of the Tolerability of Combined Treatment With PD-1 and CTLA-4 Immune Checkpoint Inhibitors and Palliative Radiation Therapy. Int J Radiat Oncol Biol Phys (2017) 98:344–51. doi: 10.1016/j.ijrobp.2017.02.003
108. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy Induces Responses of Lung Cancer to CTLA-4 Blockade. Nat Med (2018) 24:1845–51. doi: 10.1038/s41591-018-0232-2
109. Narumi K, Miyakawa R, Shibasaki C, Henmi M, Mizoguchi Y, Ueda R, et al. Local Administration of GITR Agonistic Antibody Induces a Stronger Antitumor Immunity Than Systemic Delivery. Sci Rep (2019) 9:5562. doi: 10.1038/s41598-019-41724-x
110. Nishikawa H, Kato T, Hirayama M, Orito Y, Sato E, Harada N, et al. Regulatory T Cell-Resistant CD8+ T Cells Induced by Glucocorticoid-Induced Tumor Necrosis Factor Receptor Signaling. Cancer Res (2008) 68:5948–54. doi: 10.1158/0008-5472.CAN-07-5839
111. Buzzatti G, Dellepiane C, Del ML. New Emerging Targets in Cancer Immunotherapy: The Role of GITR. Esmo Open (2020) 4:e738. doi: 10.1136/esmoopen-2020-000738
112. Tran B, Carvajal RD, Marabelle A, Patel SP, LoRusso PM, Rasmussen E, et al. Dose Escalation Results From a First-in-Human, Phase 1 Study of Glucocorticoid-Induced TNF Receptor-Related Protein Agonist AMG 228 in Patients With Advanced Solid Tumors. J Immunother Cancer (2018) 6:93. doi: 10.1186/s40425-018-0407-x
113. Patel MA, Kim JE, Theodros D, Tam A, Velarde E, Kochel CM, et al. Agonist Anti-GITR Monoclonal Antibody and Stereotactic Radiation Induce Immune-Mediated Survival Advantage in Murine Intracranial Glioma. J Immunother Cancer (2016) 4:28. doi: 10.1186/s40425-016-0132-2
114. Persa E, Balogh A, Safrany G, Lumniczky K. The Effect of Ionizing Radiation on Regulatory T Cells in Health and Disease. Cancer Lett (2015) 368:252–61. doi: 10.1016/j.canlet.2015.03.003
115. Li Y, Strick-Marchand H, Lim AI, Ren J, Masse-Ranson G, Dan L, et al. Regulatory T Cells Control Toxicity in a Humanized Model of IL-2 Therapy. Nat Commun (2017) 8:1762. doi: 10.1038/s41467-017-01570-9
116. Oweida AJ, Darragh L, Phan A, Binder D, Bhatia S, Mueller A, et al. STAT3 Modulation of Regulatory T Cells in Response to Radiation Therapy in Head and Neck Cancer. J Natl Cancer Inst (2019) 111:1339–49. doi: 10.1093/jnci/djz036
117. Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, et al. Dendritic Cell Vaccination in Combination With Anti-CD25 Monoclonal Antibody Treatment: A Phase I/II Study in Metastatic Melanoma Patients. Clin Cancer Res (2010) 16:5067–78. doi: 10.1158/1078-0432.CCR-10-1757
118. Baan CC, Balk AH, van Riemsdijk IC, Vantrimpont PJ, Maat AP, Niesters HG, et al. Anti-CD25 Monoclonal Antibody Therapy Affects the Death Signals of Graft-Infiltrating Cells After Clinical Heart Transplantation. Transplantation (2003) 75:1704–10. doi: 10.1097/01.TP.0000063937.53702.97
119. Wang Z, Xiao L, Shi BY, Qian YY, Bai HW, Chang JY, et al. Short-Term Anti-CD25 Monoclonal Antibody Treatment and Neogenetic CD4(+)CD25(high) Regulatory T Cells in Kidney Transplantation. Transpl Immunol (2008) 19:69–73. doi: 10.1016/j.trim.2008.01.005
120. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, et al. CD25 Blockade Depletes and Selectively Reprograms Regulatory T Cells in Concert With Immunotherapy in Cancer Patients. Sci Transl Med (2012) 4:134r–62r. doi: 10.1126/scitranslmed.3003330
121. Zhu Y, Jiang C, Liu Y, Li Y, Wu H, Feng J, et al. Association Between IDO Activity and Prognosis in Patients With Non-Small Cell Lung Cancer After Radiotherapy. Ann Transl Med (2020) 8:1169. doi: 10.21037/atm-20-5634
122. Liu M, Li Z, Yao W, Zeng X, Wang L, Cheng J, et al. IDO Inhibitor Synergized With Radiotherapy to Delay Tumor Growth by Reversing T Cell Exhaustion. Mol Med Rep (2020) 21:445–53. doi: 10.3892/mmr.2019.10816
123. Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY. TGF-Beta-Responsive CAR-T Cells Promote Anti-Tumor Immune Function. Bioeng Transl Med (2018) 3:75–86. doi: 10.1002/btm2.10097
124. Wang S, Gao X, Shen G, Wang W, Li J, Zhao J, et al. Interleukin-10 Deficiency Impairs Regulatory T Cell-Derived Neuropilin-1 Functions and Promotes Th1 and Th17 Immunity. Sci Rep (2016) 6:24249. doi: 10.1038/srep24249
125. Naing A, Papadopoulos KP, Autio KA, Ott PA, Patel MR, Wong DJ, et al. Safety, Antitumor Activity, and Immune Activation of Pegylated Recombinant Human Interleukin-10 (AM0010) in Patients With Advanced Solid Tumors. J Clin Oncol (2016) 34:3562–9. doi: 10.1200/JCO.2016.68.1106
126. Hecht JR, Lonardi S, Bendell J, Sim HW, Macarulla T, Lopez CD, et al. Randomized Phase III Study of FOLFOX Alone or With Pegilodecakin as Second-Line Therapy in Patients With Metastatic Pancreatic Cancer That Progressed After Gemcitabine (SEQUOIA). J Clin Oncol (2021) 39:1108–18. doi: 10.1200/JCO.20.02232
127. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, Upon Programmed Cell Death. EMBO J (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
128. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
129. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and Anti-PD-L1 Treatment Synergistically Promote Antitumor Immunity in Mice. J Clin Invest (2014) 124:687–95. doi: 10.1172/JCI67313
130. Theelen W, Peulen H, Lalezari F, van der Noort V, de Vries JF, Aerts J, et al. Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced Non-Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial. JAMA Oncol (2019) 5:1276–82. doi: 10.1001/jamaoncol.2019.1478
131. Twyman-Saint VC, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and Dual Checkpoint Blockade Activate Non-Redundant Immune Mechanisms in Cancer. Nature (2015) 520:373–7. doi: 10.1038/nature14292
132. Schoenhals JE, Cushman TR, Barsoumian HB, Li A, Cadena AP, Niknam S, et al. Anti-Glucocorticoid-Induced Tumor Necrosis Factor-Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front Immunol (2018) 9:2170. doi: 10.3389/fimmu.2018.02170
133. Geva R, Voskoboynik M, Dobrenkov K, Mayawala K, Gwo J, Wnek R, et al. First-In-Human Phase 1 Study of MK-1248, an Anti-Glucocorticoid-Induced Tumor Necrosis Factor Receptor Agonist Monoclonal Antibody, as Monotherapy or With Pembrolizumab in Patients With Advanced Solid Tumors. Cancer-Am Cancer Soc (2020) 126:4926–35. doi: 10.1002/cncr.33133
134. Rodriguez-Ruiz ME, Rodriguez I, Mayorga L, Labiano T, Barbes B, Etxeberria I, et al. TGFbeta Blockade Enhances Radiotherapy Abscopal Efficacy Effects in Combination With Anti-PD1 and Anti-CD137 Immunostimulatory Monoclonal Antibodies. Mol Cancer Ther (2019) 18:621–31. doi: 10.1158/1535-7163.MCT-18-0558
135. Vanpouille-Box C, Formenti SC. Dual Transforming Growth Factor-Beta and Programmed Death-1 Blockade: A Strategy for Immune-Excluded Tumors? Trends Immunol (2018) 39:435–7. doi: 10.1016/j.it.2018.03.002
136. Newton R, Priyadharshini B, Turka LA. Immunometabolism of Regulatory T Cells. Nat Immunol (2016) 17:618–25. doi: 10.1038/ni.3466
137. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T Cells in Cardiovascular Diseases. Nat Rev Cardiol (2016) 13:167–79. doi: 10.1038/nrcardio.2015.169
138. Kolios AGA, Tsokos GC, Klatzmann D. Interleukin-2 and Regulatory T Cells in Rheumatic Diseases. Nat Rev Rheumatol (2021) 17:749–66. doi: 10.1038/s41584-021-00707-x
139. Lowther DE, Hafler DA. Regulatory T Cells in the Central Nervous System. Immunol Rev (2012) 248:156–69. doi: 10.1111/j.1600-065X.2012.01130.x
140. Zhou Y, Wu W, Lindholt JS, Sukhova GK, Libby P, Yu X, et al. Regulatory T Cells in Human and Angiotensin II-Induced Mouse Abdominal Aortic Aneurysms. Cardiovasc Res (2015) 107:98–107. doi: 10.1093/cvr/cvv119
141. Fang W, Deng Z, Benadjaoud F, Yang D, Yang C, Shi GP. Regulatory T Cells Promote Adipocyte Beiging in Subcutaneous Adipose Tissue. FASEB J (2020) 34:9755–70. doi: 10.1096/fj.201902518R
142. Zhou Y, Chen H, Liu L, Yu X, Sukhova GK, Yang M, et al. Cathepsin K Deficiency Ameliorates Systemic Lupus Erythematosus-Like Manifestations in Fas(lpr) Mice. J Immuol (2017) 198:1846–54. doi: 10.4049/jimmunol.1501145
143. Magnuson AM, Kiner E, Ergun A, Park JS, Asinovski N, Ortiz-Lopez A, et al. Identification and Validation of a Tumor-Infiltrating Treg Transcriptional Signature Conserved Across Species and Tumor Types. Proc Natl Acad Sci USA (2018) 115:E10672–81. doi: 10.1073/pnas.1810580115
Keywords: regulatory T cell, tumor, immunosuppression, immunotherapy, radiotherapy
Citation: Gao R, Shi G-P and Wang J (2022) Functional Diversities of Regulatory T Cells in the Context of Cancer Immunotherapy. Front. Immunol. 13:833667. doi: 10.3389/fimmu.2022.833667
Received: 12 December 2021; Accepted: 28 February 2022;
Published: 17 March 2022.
Edited by:
Dipayan Rudra, ShanghaiTech University, ChinaReviewed by:
Sung-Min Hwang, Cornell University, United StatesMichal Kuczma, Georgia State University, United States
Copyright © 2022 Gao, Shi and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guo-Ping Shi, Z3NoaUBid2guaGFydmFyZC5lZHU=; Jing Wang, d2FuZ2ppbmdAaWJtcy5wdW1jLmVkdS5jbg==