 Xiaoyue Wen
Xiaoyue Wen Bing Xie
Bing Xie Shiying Yuan
Shiying Yuan Jiancheng Zhang
Jiancheng Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 29 April 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.833479
Sepsis is a life-threatening organ dysfunction caused by the host’s malfunctioning response to infection. Due to its high mortality rate and medical cost, sepsis remains one of the world’s most intractable diseases. In the early stage of sepsis, the over-activated immune system and a cascade of inflammation are usually accompanied by immunosuppression. The core pathogenesis of sepsis is the maladjustment of the host’s innate and adaptive immune response. Many immune cells are involved in this process, including neutrophils, mononuclear/macrophages and lymphocytes. The immune cells recognize pathogens, devour pathogens and release cytokines to recruit or activate other cells in direct or indirect manner. Pyroptosis, immune cell-extracellular traps formation and autophagy are several novel forms of cell death that are different from apoptosis, which play essential roles in the progress of sepsis. Immune cells can initiate “self-sacrifice” through the above three forms of cell death to protect or kill pathogens. However, the exact roles and mechanisms of the self-sacrifice in the immune cells in sepsis are not fully elucidated. This paper mainly analyzes the self-sacrifice of several representative immune cells in the forms of pyroptosis, immune cell-extracellular traps formation and autophagy to reveal the specific roles they play in the occurrence and progression of sepsis, also to provide inspiration and references for further investigation of the roles and mechanisms of self-sacrifice of immune cells in the sepsis in the future, meanwhile, through this work, we hope to bring inspiration to clinical work.
Sepsis is a systemic inflammatory response syndrome (SIRS) induced by infection, which can cause multiple organ dysfunction in severe cases. Sepsis is accompanied by a release of a variety of inflammatory cytokines secreted from immune cells, including neutrophils, monocytes/macrophages, lymphocytes and microglia (1–3). With the rapid progression of the severity of sepsis and in the critical condition with severe sepsis and septic shock, the average mortality rate of sepsis is 25% to 30%, and even 40%–50% in septic shock (4). Previous studies have demonstrated that the pathological process of sepsis is very complex, which still poses a big challenge in critical care medicine at present (5, 6).
In recent years, the pyroptosis and autophagy, as well as immune cells extracellular traps, including neutrophil extracellular traps (NETs), macrophages extracellular traps (METs), mast cells extracellular traps (MCETs), eosinophils extracellular traps (EETs), basophil extracellular traps (BETs) and dendritic cells extracellular traps (DCETs), are defined as unique forms of programmed cell death, which is different from necrosis and apoptosis (Table 1). These are programmed deaths, but none of them are genetically regulated under physiological conditions, they are caused by pathological stimuli or changes in the external environment, then further release of substances by cell death continue to perform essential roles on the body. The death of immune cells to protect or damage the body is undoubtedly similar to a kind of self-sacrificing death of cells, however, they have not been comprehensively evaluated.
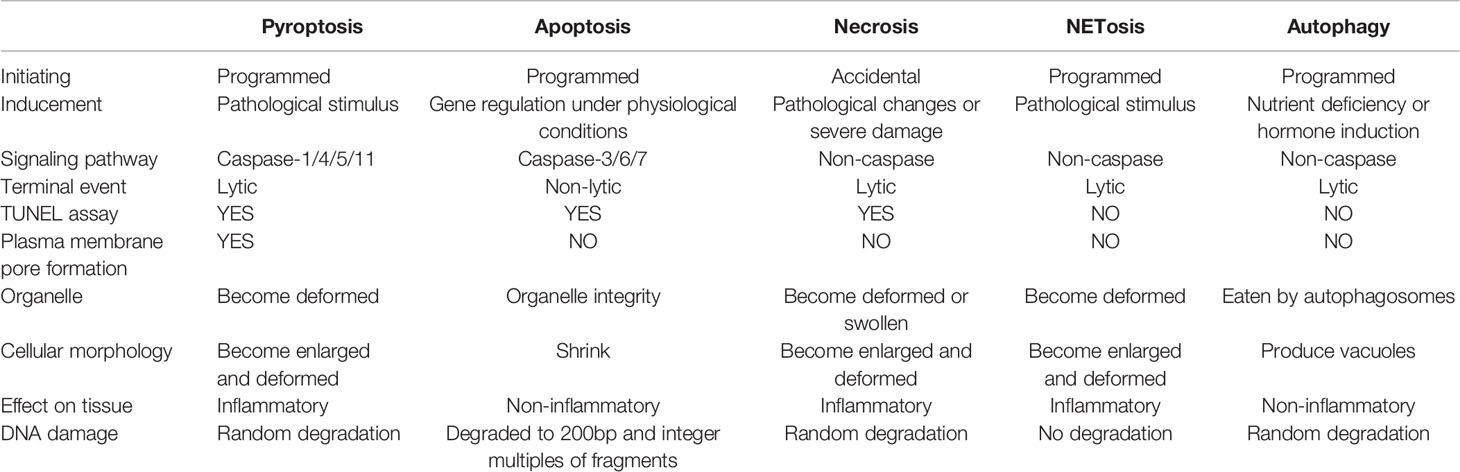
Table 1 Comparison of different cell death modes.
Researches have confirmed that the pyroptosis is an inflammatory form of programmed cell death dependent on inflammatory caspase-1, caspase-4, caspase-5 and caspase-11, as well as the release of a large number of pro-inflammatory cytokines (3, 7). The pyroptosis mainly relies on inflammasome to activate caspase family and then cleave and activate gasdermin protein, which is transferred to the membrane to form holes and thus leads to cell membrane rupture (8). The role of immune cell pyroptosis in sepsis is by far the most controversial. This self-sacrificing way of death was initially thought to play a protective role in sepsis, but now more and more studies have proved the disadvantages of excessive pyroptosis (8–10).
Immune cells extracellular traps are relatively new as research topics, among which NETs has made significant advances in oncology, autoimmune disease and COVID-19 in recent years (11–13). Neutrophils activate trap and kill pathogens by releasing substances composed of depolymerized chromatin and intracellular granular proteins. Neutrophils would die during the formation of NETs, which is called NETosis (14). With further study, more types of immune cells have been found that they can launch extracellular traps formation with disrupted cell membrane releasing DNA and histone to protect or damage the host tissue in programmed cell death. NETs plays an important role in the clearance of pathogens in sepsis and improves the survival rate of patients with sepsis. Neutrophils release histone, histamine and other inflammatory mediators into the blood after forming NETs, which could damage the tissue and promote the apoptosis of macrophages, aggravating the inflammatory response in sepsis (15). They can activate platelets to start the coagulation process and induce the formation of thrombosis as well, further aggravating the disease (16–18). Therefore, the detailed mechanism of NETs in the pathophysiological process of sepsis deserves further investigation.
The autophagy is a process enveloping bacteria and viruses escaped from the phagosomes or damaged mitochondria into vesicles, fusing with lysosomes to form autophagosomes and degrading their encapsulated contents. The relatively broad term “autophagy” itself has been utilized with rather variable and sometimes misleading connotations. The material in the cytoplasm and lysosomal degradation are acknowledged to be involved in the autophagy (19). In the early stage of sepsis, autophagy is induced in many important organs such as heart, brain, lung, liver and kidney, and it plays a protective role in the body. With the suppression of immune cell function, the body enters a period of continuous immunosuppression, with the development of sepsis, autophagy activity decreased (20). In the progression of sepsis, autophagy of immune cells plays an important role. Many studies have proved that macrophages, lymphocytes and neutrophils play a role in actively removing microorganisms and participating in inflammatory reactions (21–23).
The pyroptosis, autophagy, and immune cells extracellular traps formations can all play significant roles in sepsis in a way that resembles self-sacrifice. Much evidence has shown that pyroptosis, immune cells, extracellular traps formations and autophagy are widely involved in the occurrence and development of infectious diseases, neurological diseases, atherosclerotic diseases, and endocrine disease (14, 19, 24, 25). The immune cells through the self-sacrifice to achieve their goal is very interesting, so far, there are a lot of studies on these three ways of death in sepsis, but whether these three ways play a protective or damaging role in the process of sepsis is controversial at present, and no consistent answers have been obtained in cell experiments or clinical trials. Therefore, an in-depth study of this “self-sacrifice” in sepsis is important and necessary. This review focuses on the roles they play in the occurrence and progression of sepsis. The inspiration and reference could also be provided to us with immune cells’ self-sacrifice in sepsis, which could provide a deeper understanding of the mechanism of sepsis, and new treatment strategies for sepsis.
The phenomenon of pyroptosis was first discovered by Zychlinsky et al. in 1992. They observed a novel pathogenic mechanism of cell death in Shigella flexneri-infected macrophages, they found that this process mainly depended on caspase-1 rather than the traditional form of caspase-3-dependent cell death (26). In 1998, Hubert Hilbi claimed that caspase-1 played an essential role in the specific type of cell death. Pro-interleukin(IL)-1β would be cleaved after the activation of caspase-1, which results in inflammation, and this mode of cell death is distinct from apoptosis. Besides, they proposed that caspase-1 had dual effects of promoting apoptosis and inflammation (27). In 2001, Boise LH and Collins CM first discussed this new type of programmed cell death and used “Pyroptosis” to name the cell death mode dependent on caspase-1 (28, 29). In 2011, Kayagaki et al. highlighted the importance of caspase-11 in pyroptosis and pointed it as a non-classical pathway. Moreover, they demonstrated in vivo, as opposed to current reasoning, that caspase-11 rather than caspase-1 might be the basic effector of deleterious inflammatory responses. Their discoveries likewise feature the need to return to the job of caspase-1 versus caspase-11 in various mouse infection models, as so far, all investigations have utilized Casp1/11 twofold knockout mice (30). In 2018, Kambara et al. raised an essential point to explain the pathological mechanisms of sepsis. They reported that gasdermin D (GSDMD), a pro-inflammatory factor, could exert anti-inflammatory effects by promoting neutrophil death, and GSDMD would be a potential therapeutic target in the future (31).
In 2004, Volker Brinkmann firstly used IL-8, phosphor myristate (PMA) and lipopolysaccharide (LPS) to stimulate neutrophils. They found that activated neutrophils became flat and formed prominent extracellular structures called NETs, and this process was called NETosis. The fibrous structure of NETs could not only kill bacteria efficiently, but also serve as a physical barrier to prevent the further spread of bacteria. Whereas they observed that the excessive exposure of extracellular histone complexes could cause damage to the immune system, which indicated that NETosis was a double-edged sword to the immune system (32). In 2007, further investigation by Dr. Brinkmann’s group concluded that NETosis was a novel form of cell death pathway. They stressed that NETosis was mainly produced by neutrophils and described the way it killed bacteria effectively postmortem (33). In 2008, for the first time, von Köckritz-Blickwede et al. proposed MECTs as a novel cell death pathway in leukocytes (34). In 2012, Yipp et al. provided evidence that NETosis might lead to tissue damage besides its bactericidal properties (35). In 2015, Boe et al. described extracellular traps in macrophages and called it METosis (36).
Ashford TP and Porter KR first proposed the term “autophagy” in 1962 after discovering the phenomenon of “self-eating” in cells (37). It referred to the double layer of membrane shedding from the ribosomal region of the rough endoplasmic reticulum. Autophagosome was formed by the degraded organelles, proteins and other components, which fused with lysosomes to create autophagosome and degraded the encapsulated contents to meet the metabolic needs of the cell itself and the renewal of some organelles (38). In 1997, Matsuura et al. from the Ohsumi Ryoshinori Laboratory discovered that APG1 encodes a new type of serine/threonine protein kinase, and its kinase activity is necessary for autophagy (39). As the molecular mechanism of autophagy has slowly been elucidated, the relationship between autophagy and disease such as oncogenesis, immune system diseases, cardiovascular and neurological diseases has constantly being discovered and understood (40). In 2013, Choi et al. proposed the possible mechanism of autophagy and relevant therapeutic strategies by pharmacological means. Provided for the focal part about AKT-mechanistic target of rapamycin (MTOR) to directing autophagy, they evaluated fasting-induced autophagic reactions and found that they were impeded for hearts for these mice. Moreover, These discoveries showed that the signal defect of impair autophagy caused by LMNA gene mutation is one of the pathogenesis of the disease (41). In 2016, the Nobel Prize in Physiology or Medicine was awarded to Tokyo Institute of Technology researcher Yoshinori Ohsumi due to his discovery of the early identification and characterization mechanisms of the autophagy machinery.
According to the cleavage of inflammatory molecules upstream of GSDMD, pyroptosis could be divided into caspase-1-dependent classical pathway and caspase-4/5/11-dependent non-classical pathway (9, 42). In the classical pathway, under the stimulation of pathogens, bacteria and other signals, intracellular Nod-like receptor (NLR) family recognizes these signals and activates casepase-1 by binding with pro-caspase-1 through adaptor protein ASC. On the one hand, activated caspase-1 could cut GSDMD. GSDMD is divided into N-terminal and C-terminal, the N-terminal of GSDMD and phosphatidylinositol on cell membrane are combined to form holes which called “gasdermin channel”, then releasing contents and inducing inflammation (43). On the other hand, activated caspase-1 cleaves pro-IL-1β and pro-IL-18 to transform into IL-1β and IL-18, which is released to extracellular and lead to inflammation as well. In the non-classical pathway, taking LPS as an example, the activation of caspase-4/5/11 does not require the involvement of ASC, LPS could directly enter the cells without the receptors. The lipid A portion of LPS binds to the N-terminal of caspase activation and recruitment domain of caspase-4/5/11 and induces its oligomerization. After caspase-4/5/11 is activated, then the activated caspase-4/5/11 could cleave GSDMD and induce pyroptosis, meanwhile inducing the caspase-1 activation to cleave pro-IL-1β and pro-IL-18, the expression of IL-1β causes a febrile reaction, dilates blood vessels, causes hypotension, promotes immune cells (polymorphonuclear neutrophils, monocytes, macrophages, and dendritic cells) infiltration, and ultimately damages tissues (44). Meanwhile, IL-18 is a cytokine with multiple pro-inflammatory functions, it could activate Th1 cells and produces interferon-γ (IFN-γ). The overproduction of IL-18 may policy the inflammatory response and cause tissue damage (45) resulting in an inflammatory response (43, 46). It is worth noting that pannexin-1and P2X7 downstream of caspase-11 play a significant role for pyroptosis and susceptibility to sepsis induced by the non-classical pathway (47). Then we will focus on exploring how this complex process plays a role in immune cells in sepsis (Figure 1).
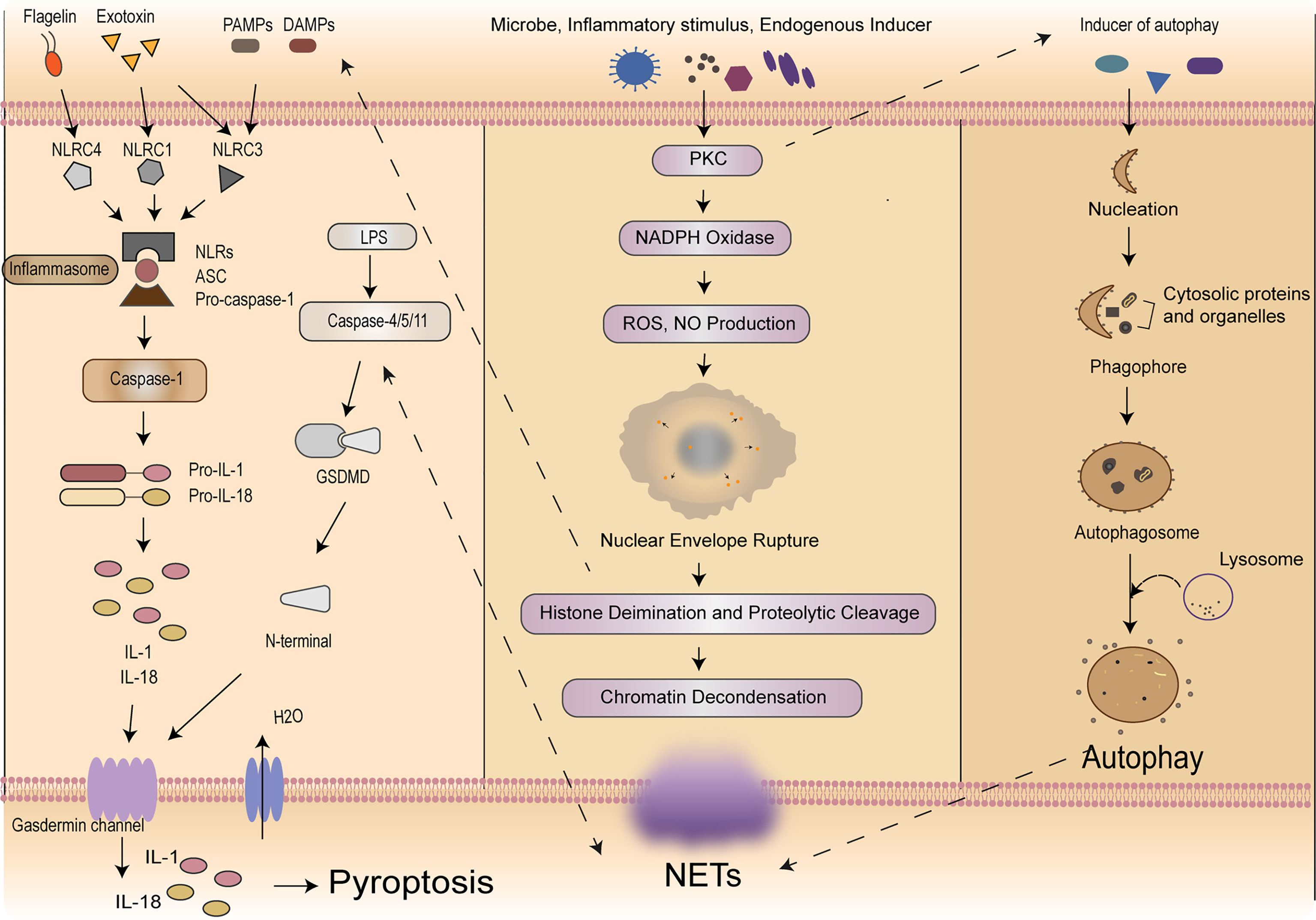
Figure 1 Mechanisms of self-sacrifice in neutrophils. Pyroptosis mainly relies on inflammasome to activate caspase family, caspase family could cleave and activate gasdermin protein, which is transferred to the membrane to form holes and thus leads to cell membrane rupture. NETs components DNA and histones can act as DAMPs molecules to directly initiate or amplify inflammatory responses. NETosis induced by LPS in neutrophils can elicit caspase-11 and GSDMD-dependent histone H3 citrullination. NETosis is a multi-step process involving the destruction of neutrophil nuclear and cytoplasmic granular membranes, chromatin relaxation, chromatin interaction with granular proteins, and chromatin release from the cells. A variety of stimulants, such as PMA, microflora, LPS, eubacteria, activated blood plates, IL-8 and immune complexes, can induce the formation of NETs through the PKC signaling pathway. Autophagy is a process enveloping bacteria and viruses escaped from the phagosomes or damaged mitochondria into vesicles, fusing with lysosomes to form autophagosomes and degrading their encapsulated contents. Neutrophil autophagy could subsequently activate NETs. NETs, neutrophil extracellular traps; DAMPs, damage‐associated molecular patterns; LPS, lipopolysaccharide; GSDMD, gasdermin D; PMA, phosphor myristate; IL, interleukin; PKC, protein kinase C.
Pyroptosis of neutrophils is a pro-inflammatory programmed cell death process performed by the protein GSDMD. From the point of view of protection, the intracellular replicative niche of the bacteria is cleared through pyroptosis of neutrophils, immune factors released during pyroptosis active the effector cells to kill the pathogen (10). In contrast, Mei Yang et al. suggested that neutrophils could secrete IL-1β through caspase-1-dependent classical pathway and then lead to a lower survival rate in mice. Compared to those in controls, the expression of caspase-1, NLRP-1, IL-1β, and IL-18 decreased in mice with AC-YVAD-CMK (caspase-1-inhibitor) treatment, additionally, with AC-YVAD-CMK treatment, they observed that the accumulation of neutrophils and macrophages were suppressed, serum creatinine and blood urea nitrogen level were decreased, and the expression of GSDMD reduced (48). In 2014, Chen et al. confirmed that neutrophils could express multiple Nod Like Receptors (NLRs) and are significant source of IL-1β during acute Salmonella infection. In addition, the neutrophil NLRC4 inflammasome drove caspase-1 and IL-1β activation (49). In 2018, Chen et al. clarified that due to the low expression of caspase-1 in neutrophils and the classical inflammasome signal transduction aptamer protein ASC, GSDMD could not be cleaved effectively, resulting in focal death independent of the classical pathway in neutrophils. However, IL-1β could be released during this process, which in turn recruits more neutrophils to accumulate (50).
In caspase-4/5/11-dependent non-classical pathway, it is worth emphasizing that Kumari et al. revealed that the release of caspase-11 in neutrophils was crucial and indispensable for LPS-induced mortality after sepsis, but it does not play primary roles in other cells, such as intestinal epithelial cells (51).
Analogously, during polymicrobial sepsis, Gentile et al. evidenced that the level of inflammatory cytokines and neutrophils’ phagocytosis could increase, the survival rate of wild-type mice and bacterial colonization could decrease after ablation of caspase-1/11, moreover, they emphasize that caspase-1/11 activity, not only could accelerate the magnitude of the inflammatory response, but also suppress protective immunity (52). In contrast, Cheng et al. used the mature neutrophils extracted from mouse bone marrow, and found neutrophils was sensitive to caspase-11-dependent non-classical pathway, in which could effectively cleave the GSDMD in neutrophils, conditional deletion of caspase-11 in lung injury caused by bacterial sepsis could reduce neutrophils accumulation and pyroptosis, indirectly proving that caspase-11-dependent non-classical pathway might be involved in the lung injury after sepsis, moreover, they evidenced that caspase-11 and GSDMD contributed to host defense and the decrease of bacterial load though driving the NETosis (53). Peptidylarginine deiminases (PADs) are a family of calcium-dependent enzymes. In 2020, Yuzi Tian et al. testified that PAD2 protein is increased in sepsis patients and CLP-induced mouse models of sepsis. Through the using of PAD2-specific inhibitors, they demonstrate that it could decrease NETosis and macrophage’casepase-11-dependent pyroptosis, and inhibition of caspase-11 activation could lead to reduce the release of IL-1α and high mobility group box 1 (HMGB1) in the peritoneal cavity, which increases the number of macrophages, significantly reduces bacterial load and inflammation in the blood, and ultimately increases survival and organ function after sepsis (54).
At present, there is no sufficient evidence showing that pyroptosis of lymphocytes can directly aggravate the occurrence of sepsis. Whereas Sarkar et al. used caspase-1 knockout, IL-1 knockout, IL-1/IL-18 double knockout and their respective wild-type mice to analyze the survival rate in 2006, they found that mice with caspase-1 knockout had a higher survival rate and they proposed that caspase-1-dependent pyroptosis might cause delayed apoptosis of B lymphocytes, which further affected the phenotype of macrophages and thus improved the survival rate, of note, this process is not regulated by cytokines, finally they proved that caspase-1 was essential to surviving live E. coli-induced septic shock (55). Moreover, studies have shown that Group 2 Innate lymphoid Cells (ILC2) can inhibit the activation of caspase-1 by secreting IL-9, thus inhibiting pyroptosis of vascular endothelial cells, additionally, granzyme released by cytotoxic lymphocytes can trigger pyroptosis in target cells by cleaving GSDMB (56). Kader et al. manifested that in Ehrlichia-induced sepsis mice, caspase-11-mediated HMGB1 cytosolic translocation and extracellular secretion are linked to the induction of pyroptosis, the IFNAR signaling involve in this process, which played an essential role in bacterial replication and NKT cells, CD8+ cells and neutrophils expanded during this process, resulting the damage of liver in sepsis (57). In a word, the pyroptosis in lymphocytes does have a relationship with sepsis, but the mechanism of their interaction is still worth further exploration.
Sepsis-associated disseminated intravascular coagulation (DIC) could inhibit macrophage pyroptosis through platelet endothelial cell adhesion molecule-1 (PECAM-1), thus restoring vascular barrier integrity (58). PECAM-1 could significantly increase or decrease the expression of caspase-11 through the up-regulation or down-regulation of the expression of Sphingosine-1-phosphate receptors-2 (S1PR2) in macrophages. Many sphingosine-1-phosphate receptors are expressed in macrophages, which could promote caspase-11-dependent pyroptosis of macrophages. Loss of S1PR2 could reduce the pyroptosis of macrophages and improve the prognosis of sepsis caused by E. coli infection. They also proved that a RhoA inhibitor significantly reduced caspase-11 activation (59). In 2018, Wang et al. found that the level of the caspase-1, the percentage of caspase-1-induced peripheral blood monocyte pyroptosis, and the level of IL-18 were significantly increased in patients with post-traumatic sepsis compared with healthy subjects, and the percentage of peripheral blood mononuclear cells pyroptosis could predict the development and the occurrence of post-traumatic sepsis (60). In the same year, Chen et al. proved that the activity of bone marrow-derived macrophages in sepsis mice was inhibited after treatment with HMGB1, leading to the increased release of IL-1β and IL-18 through caspase-11-dependent pyroptosis. They demonstrated that HMGB1 interacted with extracellular LPS to intervene caspase-11-dependent pyroptosis in fatal sepsis, which is a physical and specific bond interaction, and in this process RAGE-dependent internalization was a decisive pathway by disrupting the membrane of the acidic lysosome, allowing LPS to enter the cytoplasm. More importantly, they found that HMGB1 and caspase-11 was upregulated 24 hours after the onset of sepsis, providing the basis for targeted therapy for sepsis (61). Unlike caspase-11-dependent pyroptossis in neutrophils, recently studies presented that caspase-11-dependent-pyroptosis in macrophages mainly plays a damaging role in the process of sepsis. Salvamoser et al. showed that the levels of caspase-1 and caspase-11, and the expression of caspase-1 in macrophages were increased in mice after LPS-induced sepsis. Besides, the deficiency of caspase-1 and caspase-11 in mice resulted in increased tolerance to septic shock and reduced mortality in mice (62). Similarly, Kang et al. confirmed that regulating caspase-11-mediated GSDMD cleavage in macrophages improved immune hyperactivation in sepsis, indicating that genetic or pharmacologic suppression of excessive pyroptosis in macrophages could enhance the survival rate of mice in sepsis (63).
Microglia and peripheral macrophages belong to mononuclear macrophages of hematopoietic origin and microglia is the only resident macrophages of the brain parenchyma acting as innate immune effector cells in the central nervous system (64). Microglia activation and neuroinflammation are the main features of neuropathology (65, 66). In 2019, Xu et al. confirmed that caspase-1 inhibitors could suppress the expression of GSDMD and its cleavage from GSDMD-NT, then inhibit the pyroptosis of microglia on day 1 and 7. In addition, it could also reduce the detrimental effects of IL-1β, monocyte chemoattractant protein-1 (MCP-1), and TNF-α in the serum and brain of septic mice and thus alleviate brain injury. Therefore, caspase-1 inhibitors can protect against brain injury caused by sepsis to some extent, mainly by blocking the microglial pyroptosis pathway and reducing the release of pro-inflammatory cytokines (67). Furthermore, Xu et al. further found that that TREM-1 was a key regulator of inflammation. Microglia further increased N-terminal of GSDMD and the formation of GSDMD pores through upregulation of TREM-1, leading to pyroptosis of microglia, then resulting the damage in nerve. TREM-1-induced spleen tyrosine kinase (SYK) mobilization is responsible for microglial pyroptosis through CARD9/NF-κB and NLRP3/caspase-1 pathways both in vivo and in vitro. It is worth mentioning that such inflammation is not directly equivalent to sepsis, according to the definition of sepsis and their role in sepsis remains to be studied (68). In the same year, Liang Li et al. illustrated that microglial with Cylindromatosis deficiency could exacerbate LPS-induced pyroptosis in septic mice (69). In 2020, a study in a rat model of cardiac arrest found that selective inhibition of NLRP3 and caspase-1 with MCC950 and Ac-YVAD-cmk could significantly prevent microglial pyroptosis (70).
NETotic cell death was originally characterized by neutrophils, of note, similar structures have been found in macrophages, eosinophils, basophils, and DC cells by now. This designation has so far been controversial and not widely advocated (71). At present, NETs is mainly through NADPH oxidase (NOX) dependent and NOX independent pathways. A variety of stimulants, such as PMA, microflora, LPS, eubacteria, activated blood plates, IL-8 and immune complexes (IC), could induce the formation of NETs through the NOX-dependent pathway (72–75), protein kinase C (PKC) or rapidly accelerated fibrosarcoma (RAF)-mitogen-activated protein kinase (MEK)-extracellular signal-regulated kinase (ERK) signaling pathway (75, 76). After the activation of neutrophils, NETs can be formed in the NOX-dependent cell death process (77). The NOX-independent pathway occurs without lysis of neutrophils, and ROS generation is not involved in. NETs are transported mainly through nuclear membrane blister and subsequent vesicles. Activated platelets may be the inducer of this pathway. The specific mechanism remains unclear and needs further study. The NETotic cell death is largely unknown, both in terms of naming and mechanism, but its role in sepsis is still being explored.
NETosis is a multi-step process involving the destruction of neutrophil nuclear and cytoplasmic granular membranes, chromatin relaxation, chromatin interaction with granular proteins, and chromatin release from the cells (14). In 2013, Yipp BG and Kubes P divided NETosis into suicidal NETosis and vital NETosis, and the two categories differ in whether cells are cleaved. Vital NETosis primarily provides extracellular antibacterial action while neutrophils remain mobile and phagocytic (15). Khan et al. added one more point and elucidated that those increasing concentrations of LPS was a critical switch that suicidal NETosis could be turned on (78).
Suicidal NETosis is shown to be able to promote the inflammatory response, induces coagulation disorder, and damages the tissue directly (14, 79). There is growing evidence showing that NETs and their components are cytotoxic and can directly kill endothelial and epithelial cells in vitro. Moreover, excessive accumulation of NETs in vivo leads to epithelial and endothelial tissue damage (80). Endothelial cell activation is an integral part of sepsis pathogenesis. In the study of Gupta et al. in 2010, activated endothelial cells induced NETs through IL-8 resulting in severe tissue damage (81). What’s more, Villanueva et al. demonstrated that “low-density granulocytes” had a greater capacity to produce NETs. By creating a higher proportion of NETs, these cells might drive disease pathogenesis (82). Xu et al. concluded that Extracellular histones from NETs are cytotoxic in vitro and have lethal effect on mice, in vivo, histone administration might result in vacuolated endothelium and thrombosis. Besides, After infusing of APC (activated protein C) with E. coli in baboons, they detected that histone in the circulation of baboons decreased and observed it could prevent lethality in mice, implying that APC could cleavage the histone (mainly H3 and H4), and the overactivation of NETs led to extracellular histones accumulation, thus contributed to the death of mice in sepsis (83). Inversely, Schauer et al. in 2014 ascertained that the mice with NETosis-deficient could exacerbate disease that can be decreased by adoptive by aggregated NETs’ transformation. Besides, the hydrolytic protease adhered to NETs interfered with neutrophil recruitment and activation as well as degraded cytokines and chemokines, thereby alleviating local inflammation (84). The above studies show that the specific effects of NETs are being explored and dialectically.
Coagulation disorder is a typical pathological feature of sepsis, and the thrombogenic mechanisms of NETs are varied. First, during the formation of NETs, tissue factors can be produced and released, which act as the trigger for the exogenous clotting pathway, thereby activating the coagulation cascade and simultaneously stimulating platelet activation, leading to platelet aggregation and ultimately thrombosis (85). NETs can then activate endogenous pathways by activating clotting factor XII in the blood, resulting in deep vein thrombosis (86).
Furthermore, pyroptosis and NETosis are not two independent processes in neutrophils. They are mechanistically linked with each other and have mutual effects. One study has shown that NETosis induced by LPS in neutrophils can elicit caspase-11 and GSDMD-dependent histone H3 citrullination. It revealed that neutrophils used an inflammasome and GSDMD-dependent mechanism to activate NETosis as a defense response against cytosolic bacteria. But whether their cooperation is beneficial for removing pathogens still needs more evidence to study. This kind of cooperation is not limited to neutrophils but also in other immune cells.
NETs components DNA and histones can act as damage‐associated molecular patterns (DAMPs) molecules to directly initiate or amplify inflammatory responses (Figure 2). Szatmary et al. described the direct damage in three ways, including thrombosis, tissue hypoperfusion and organ damage (80). Tsourouktsoglou et al. found that NETs induced the up-regulation of IL-6 and pro-IL-1β transcription levels in macrophages by TLR2 or TLR4 (87). Similarly, Song et al. in China recently found that NETs could induce the pro-inflammatory M1-type polarization of lung tissue macrophages, thus aggravating lung injury (88). More interestingly, Chen et al. reported that NETs could facilitate macrophage pyroptosis in sepsis through RAGE and dynamin-dependent signaling, in which histone H3 plays an important role (89). At this point, how pyroptosis and NETs cooperate and constrain with each other in neutrophils has been discovered sustainedly in sepsis. Linson Chen further emphasized the connection between NETs and macrophage pyroptosis in sepsis. He stressed out that NETs could promote pyroptosis of macrophages, which could exacerbate the inflammatory response of sepsis (89).
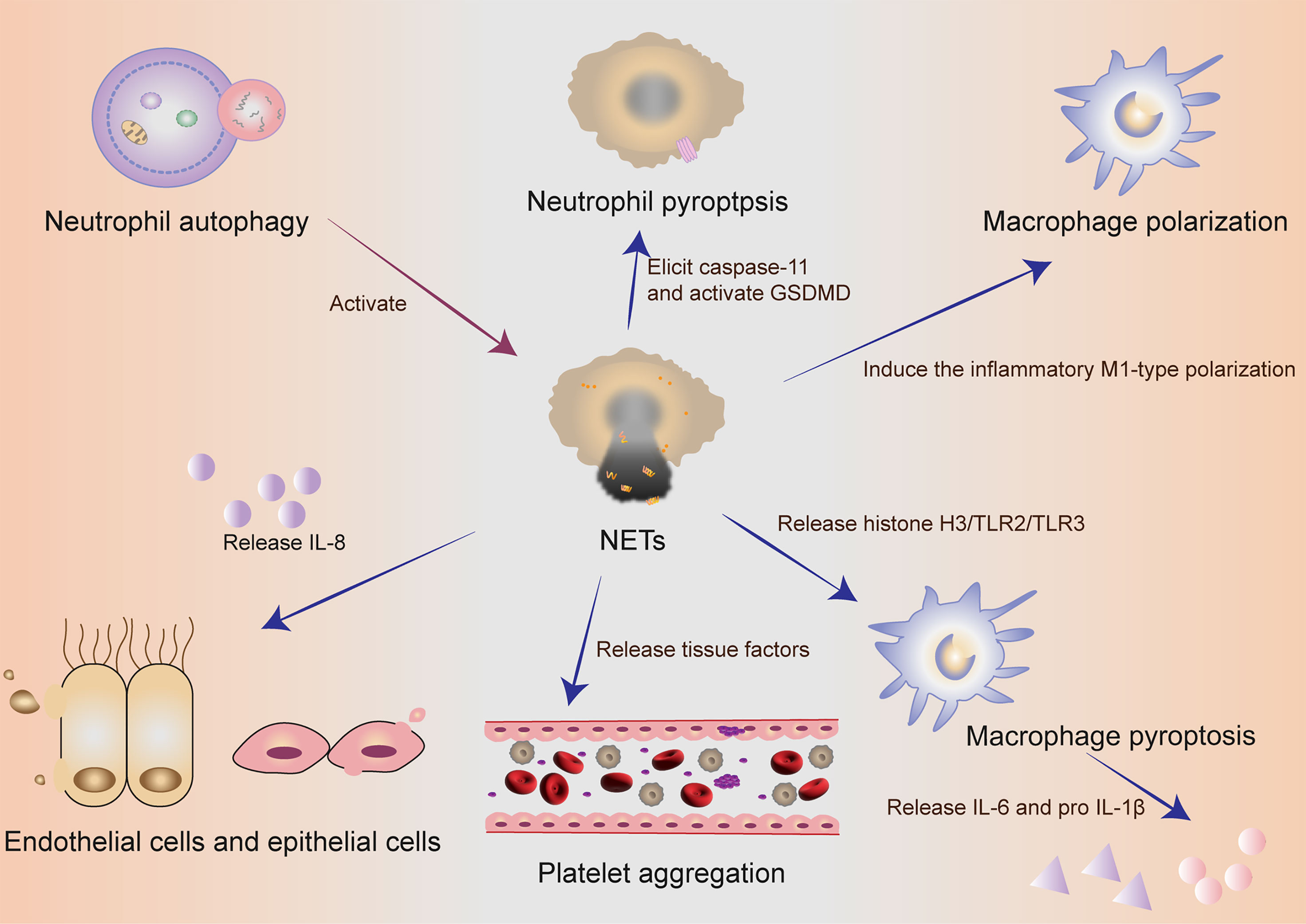
Figure 2 The collaboration of different cells in self-sacrifice. Neutrophil autophagy could subsequently activate NETs. Neutrophils used an inflammasome and GSDMD-dependent mechanism to activate NETosis. NETs could induce the pro-inflammatory M1-type polarization of lung tissue macrophages, thus aggravating lung injury. NETs could promote pyroptosis of macrophages, which could exacerbate the inflammatory response of sepsis. Activated endothelial cells induced NETs through IL-8 resulting in severe tissue damage. NETs and their components are cytotoxic and can directly kill endothelial and epithelial cells. During the formation of NETs, tissue factors can be produced and released, thereby activating the coagulation cascade and simultaneously stimulating platelet activation, leading to platelet aggregation and ultimately thrombosis. NETs, neutrophil extracellular traps; GSDMD, gasdermin D; IL, interleukin.
Studies have suggested that T lymphocyte depletion is the main feature of immunosuppression in sepsis (90, 91). Immunosuppression of T cells in sepsis could be through cytokines secretion, including the release of IL-10, down-regulation of IL-7, and up-regulation of T cell proliferation by programmed death 1 (PD-1)/PD-1 ligand (PD-L1) (91, 92).
Unfortunately, whether lymphocytes could have extracellular traps’ nets as neutrophils is on doubt and there is no research reported. If lymphocytes also had such structure, we would suppose that the primary function might be as follows (1): a physical barrier and a signal-bearer (2); capturing and killing the bacteria through specific enzyme secretion from extracellular nets (3); killing themselves in programmed cell death, which is caused by excessive activation of lymphocytes, thus giving rise to immunosuppression and exacerbation of sepsis.
Extracellular traps exit not only in neutrophils but also in Macrophages. Macrophages extracellular traps (METs) are thought to be composed of cellular DNA, histones and MPO (myeloperoxidase), cellular proteins, which have similar structures with neutrophils. King et al. and Aulik et al. clarified that the increase of ROS might be involved in the pathways (93, 94). Liu et al. proposed a ROS-independent mechanism of METs in the defensive effects against microbes (95). However, whether elastase and myeloperoxidase could contribute to METosis need to be further elucidated (96–98). What is even more interesting is that METs was thought to be capable of killing microorganisms at first defense. However, a recent study noted that the METs induced by a rapid growing mycobacterium release actually provided a scaffold to enhance bacterial growth, and thus facilitated the bacteria’s survival in the disease, intriguingly Sungmo et al. emphasized that this process depended on calcium influx instead of NAPHP oxidase activity, in addition, they found histone, MPO and elastase made up of microbicidal proteins (93). We need to emphasize two points here: firstly, bacterial infection and sepsis are not identical, and further verification is needed to determine whether this mechanism has the same effect in the sepsis models, moreover, PMA-Differentiated THP-1 cells and mycobacterium were used in this trial, which are unique.
Like NETs, mast cells have appeared to secret their atomic DNA and shape mast cell-extracellular traps (MCETs), which can entangle and eliminate different organisms (34). MCETs mainly consists of histones, tryptase and LL-37 (99). LL-37 an 18-kDa precursor protein, which could defend against bacterial due to its pore-forming activity and induce mast cells to release nucleic acids to kill bacteria (100, 101). MCETs could also recruit neutrophils by storing and releasing TNF-α to further promoting inflammation (102). However, this uncontrolled mast cell degranulation will enhance the nearby immune reaction in this manner, exaggerating and maintaining tissue damage (103–105). Here, we need to point out that the relationship between MCETs and sepsis has not been discussed at present, and the discussion about MCETs mainly focuses on the antibacterial effects (34, 103), and chemical inducers of MCET formation, like listeria monocytogenes and mycobacterium tuberculosis (106–108). Therefore, we expect further studies to point out the close connection between sepsis and MCETs.
Different from NETs, eosinophils release their mitochondrial DNA and granule proteins to form eosinophils extracellular traps (EETs) activated by LPS, IL-5, C5a and eotaxin after response to the physical antibacterial mechanism in the inflammation (109, 110). Notably, its hyperactivation can also be amplified to induce a self-sacrifice attack. Eosinophils can produce some toxins, which are involved in coagulation disorders (111). It is interesting to note that the infiltration of eosinophils to lungs under the infection of A. fumigates leads to the degranulation of eosinophils, which provides a protective effect against lethal respiratory virus infections (112). As for basophils, basophils extracellular traps (BETs) has been reported to possess antimicrobial activity by exerting its extracellular traps (104).
Dendritic cells (DCs) is specialized antigen presenting cells. DCs play an indirect role mainly by regulating T cells in the progression of sepsis (113). Loures et al. found that plasmacytoid DCs (pDCs) could form pDC extracellular traps (pETs) containing DNA and citrullinated histone H3, which had similar structures with NETs. Infection of A. fumigatus hyphae could stimulate the formation of pETs via Dectin-2. It has been shown that pETs acted mainly as an antibacterial agent (114). Besides, a loss of DC autophagy slowed sepsis by increasing lifespan and decreasing IFNα (115).
In sepsis, there are multiple targets that could induce autophagy. LPS, as an important pathogenic factor in sepsis, has been shown to induce autophagy through toll-like receptor 4 (TLR4) dependent pathway. TLR4 signaling pathways can be divided into two categories: myeloid differentiation factor 88 (MyD88) dependent pathway and MyD88-independent pathway. LPS activates a downstream P62-dependent aggregation-like inducible structure (ALIS) of selective autophagy through the MyD88-TLR4-dependent pathway (21). In addition, LPS could activate the autophagy by affecting NF-κB in sepsis model (116).
At present, there are a majority of studies on the relationship between autophagy and sepsis, and much evidence concludes that autophagy brings a more positive impact on immediate clearance of pathogens (117), neutralization of microbial toxins (118), regulation of cytokine release, and reduction of apoptosis (119). Who has an agreement with the above points is that, Hsieh, C. H has emphasized that in CLP-induced-sepsis mice, the inhibition of autophagy can damage relevant organs, they observed that in the septic hearts the reveal of the autophagosomes could increase dramatically, but few autolysosomes were detected, suggesting that incomplete autophagy might be one of the reasons of tissue damage caused by autophagy (120). On the contrary, a few research results have the opposite conclusion. Autophagy is usually induced in the early phase of sepsis, and its activity decreases in the late stage of sepsis. Studies have also underlined that autophagy was involved in mitochondrial damage caused by sepsis and had toxic effects on the human body, Unuma Kana concluded that LPS stimulated cells to release the exocytosis of autolysosomes, resulting in multi-organ injuries due to its release into the circulation (121).
Autophagy can also promote the formation of NETs (3, 8). In 2017, an analysis of neutrophil autophagy markers in 44 patients with sepsis in South Korea found that neutrophil autophagy increased in patients with sepsis survivors and the autophagy of healthy neutrophil could subsequently activate NETs, which was confirmed in vivo in a subsequent study performed in mice, neutrophils isolated from patients who died of sepsis could induced dysregulation of autophagy (72).
Additionally, different from the two-sidedness of pyroptosis and NETosis on immune cells, the primary effect of lymphocyte autophagy activation is to improve the severe inflammatory response of sepsis. In 2017, Oami et al. demonstrated that the lack of autophagy on CD4+ T cells will increase the mortality of mice, compared with the control mice, a blockade of autophagy could increase the expression of Bcl-2-like 11, lead to mitochondrial accumulation and accelerate programmed cell death in T cells. And the autophagy of T cells will protect mice from immunosuppression from a control experiment (23). Similarly, in a TLR7-mediated model of autoimmunity, Weindel et al. certified the loss of autophagy in B cells and DCs could lead to excessive tissue inflammation and cytokines related to inflammasome (115). In cecal ligation and puncture (CLP)-induced sepsis model in T-cell-specific autophagy related protein 7 (Atg7)-knockout mice and control mice, the mice with T-cell autophagy deficiency had higher mortality via suppressing bacterial clearance in the spleen, they finally proposed that autophagy might inhibit sepsis-induced apoptosis and immunosuppression in T lymphocytes (122). Ge et al. proposed that IL-36β reduced the immunosuppressive activity of CD4+ CD25+ Tregs by initiating the autophagy, subsequently improving to progress of the host immune reaction in sepsis and reducing the mortality rate in mice. Mechanistic studies uncovered that IL-36β set off autophagy of CD4+ CD25+ Tregs. These effects were obviously receded under the precondition of the autophagy inhibitor 3-methyladenine or Beclin1 knockdown (123).
In sepsis, pattern recognition molecules (PRMs), such as LPS from the pathogen, can bind to pattern recognition receptors (PRRs) on the macrophage membrane, and activate downstream inflammatory pathways in macrophages. Activated macrophages express many pro-inflammatory cytokines such as IL-6 and TNF-α, which could promote the inflammatory response of sepsis (128).
In addition to pyroptosis and METs, autophagy also plays a vital role in modulating macrophages in sepsis. Autophagy mainly affects the progress of sepsis by influencing the senescence and phagocytosis of macrophages, regulating the death, polarization and activation of macrophages, and the release of inflammatory cytokines from macrophage (127, 129, 130). The effects of autophagic macrophages on the pathophysiological process of diseases are now debated (131). The excessive activation of macrophages can result in many inflammatory and autoimmune diseases, including sepsis. It has been elucidated that autophagy induced by rapamycin can negatively regulate the abnormal activation of macrophages and reduce the inflammatory response (126).
Autophagy and pyroptosis are not two separate processes in macrophages, they can interact with each other. In a mouse model of Pseudomonas aeruginosa-induced sepsis, the deficiency of Atg7 gene, an indispensable regulatory factor in inducing autophagy (132) through phagophore initiation, expansion, transition and fusion (130, 133), could enhance the activity of inflammasomes in macrophages via elevating blood levels of IL-1β and IL-18 and increasing macrophage pyroptosis (134). A further study proved that in Pseudomonas aeruginosa–induced sepsis, flagellin is an effective activator of the inflammasome, and loss of Atg7 led to increased pyroptosis (135). Consequently, the regulation mechanisms of autophagy in pyroptosis might be a future direction.
The immune cells die in such a self-sacrificing way in sepsis, which undoubtedly has a huge impact on the procession of sepsis. Based on the above studies, how to control the process of these self-sacrificing immune cells towards the direction of protecting the body is one goal of our clinical struggle. Though the research in this area is extremely limited, there are still several research, which might provide some reference and intelligence for the future clinical research.
Based on the above injury mechanism, treatments for sepsis have been extensively developed now. For instance, nitrosonisoldipine, a photodegradation product of calcium channel inhibitor nisoldipine, can serve as a selective inhibitor of inflammatory caspase to block caspase-1-depenpent classical pyroptosis way and caspase-11-depenpent non-classical way, which ultimately contributes to the improvement of the survival time in LPS- and CLP-induced septic models (124). Similarly, Ethyl pyruvate, is a simple aliphatic ester, which has been proved to inhibit the connection of LPS to caspase-11-depenpent non-classical pyroptosis in macrophages, then positively affects the antibacterial ability of the body in sepsis (136). An Fc-modified HIT-like monoclonal antibody was invented to bind to PF4-NET complexes, further enhance DNase resistance. Treatment with this antibody has the capability of decreasing bacterial dissemination and increasing survival in mouse sepsis models, evidencing a novel NET-targeting approach to improve outcomes in sepsis (125). The same antibody-based strategy was reported in 2020, tetranectin could bind with HGBM-1 to inhibit the recruitment of lactate dehydrogenase and caspase in macrophage, this interaction could further reduce inflammatory damage and play a positive role in lethal sepsis (137).
In 2019, a study showed that Dexmedetomidine could reduce pyroptosis and histone release of astrocytes by reducing NLRP3 and caspase-1 recruitment in vivo and vitro, which might protect comprehensively nerve cells from damage in sepsis (138). What’ more, there are exploration on treating sepsis through magnesium ions, chemical destruction and of GSDMD, cathelicidin peptide LL-37 (139–141). Although many studies have suggested that caspase and NADPH are potential clinical therapeutic targets in sepsis, unfortunately, they have not been widely practiced and recommended. In 2015, David Nobuhiro Douda proposed a possible therapeutic approach to combat with NETs, which is mediated by ROS and a calcium activated small conductance potassium channel (142).
Although the pathways of pyroptosis, NETS and have attracted a lot of attention from researchers, and their stimulants, pathways, receptors, and effects are increasingly being discovered (143). However, the progress of clinical research and drugs development targeting immune cells is not satisfying. Although targeted drugs have been developed for NETs and pyroptosis like FDA-approved disulfiram and so on (124, 144–148), their roles in immune cells in sepsis remains unclear. In 2020, Stiel et al. collected statistics of complete blood count, C-reactive protein (CRP), IL-6, levels of cell-free DNA (cfDNA), neutrophil elastase (NE) and myeloperoxidase (MPO) in umbilical cord blood to try to predict the early-onset sepsis. Unfortunately, the markers of NETosis they testified could not predict the happening of sepsis (149).
Immune cells in previous studies on sepsis mainly revolved around the protective effects of recruiting and activating immune cells on the host and the damage to the body by immunosuppression. In the battle between immune cells and pathogens, if immune cells fail, they will face unprogrammed death, which means necrosis, or apoptosis, but if immune cells could initiate a self-sacrifice programmed death before the failure, for example, T cells and B cells release tissue factors and cytokines to kill bacteria by launching autophagy, and neutrophils and macrophages’ pyroptosis directly or indirectly to improve immune suppression, in this way immune cells will protect the host through sacrificing themselves. Therefore, we separately discussed the function of three different death modes in sepsis immune cells, and this self-sacrificing death mode is intelligent for immune cells. However, not all immune cells can play an active role in protecting the host in this process, as well as neutrophils, macrophages, and microglia, if their pyroptosis and extracellular traps are over-activated, their damage to the organism will be greater than the protective effects in sepsis. (Tables 2, 3) What is worth paying attention to in the future is how to amplify the advantages of self-sacrifice of immune cells, while reducing the disadvantages of self-sacrifice of immune cells. With the continuous discovery of the mechanisms of these self-sacrifice, the corresponding treatments related to sepsis will be the direction of future exploration.
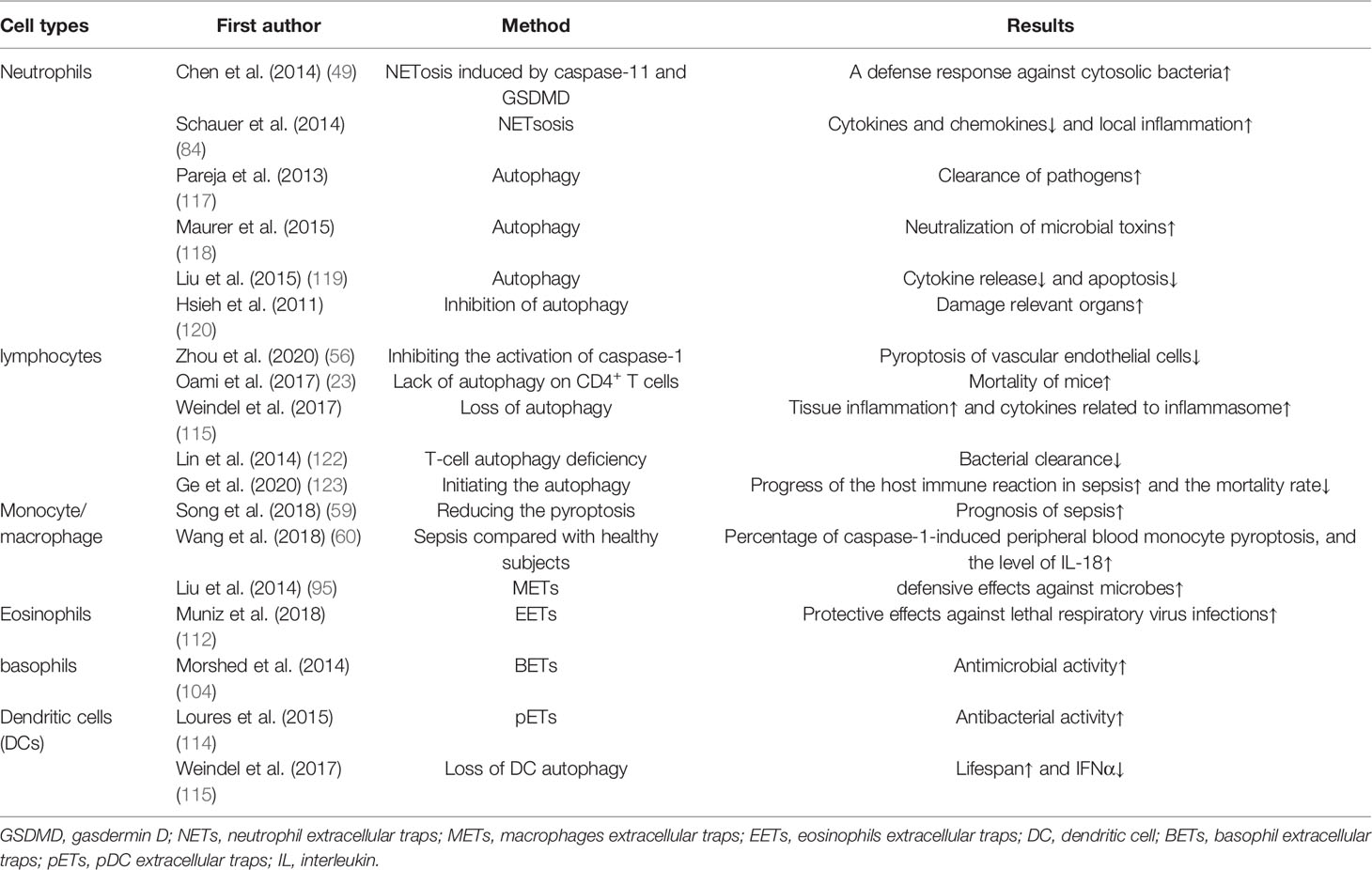
Table 2 The pros of various self-sacrifice of immune cells.
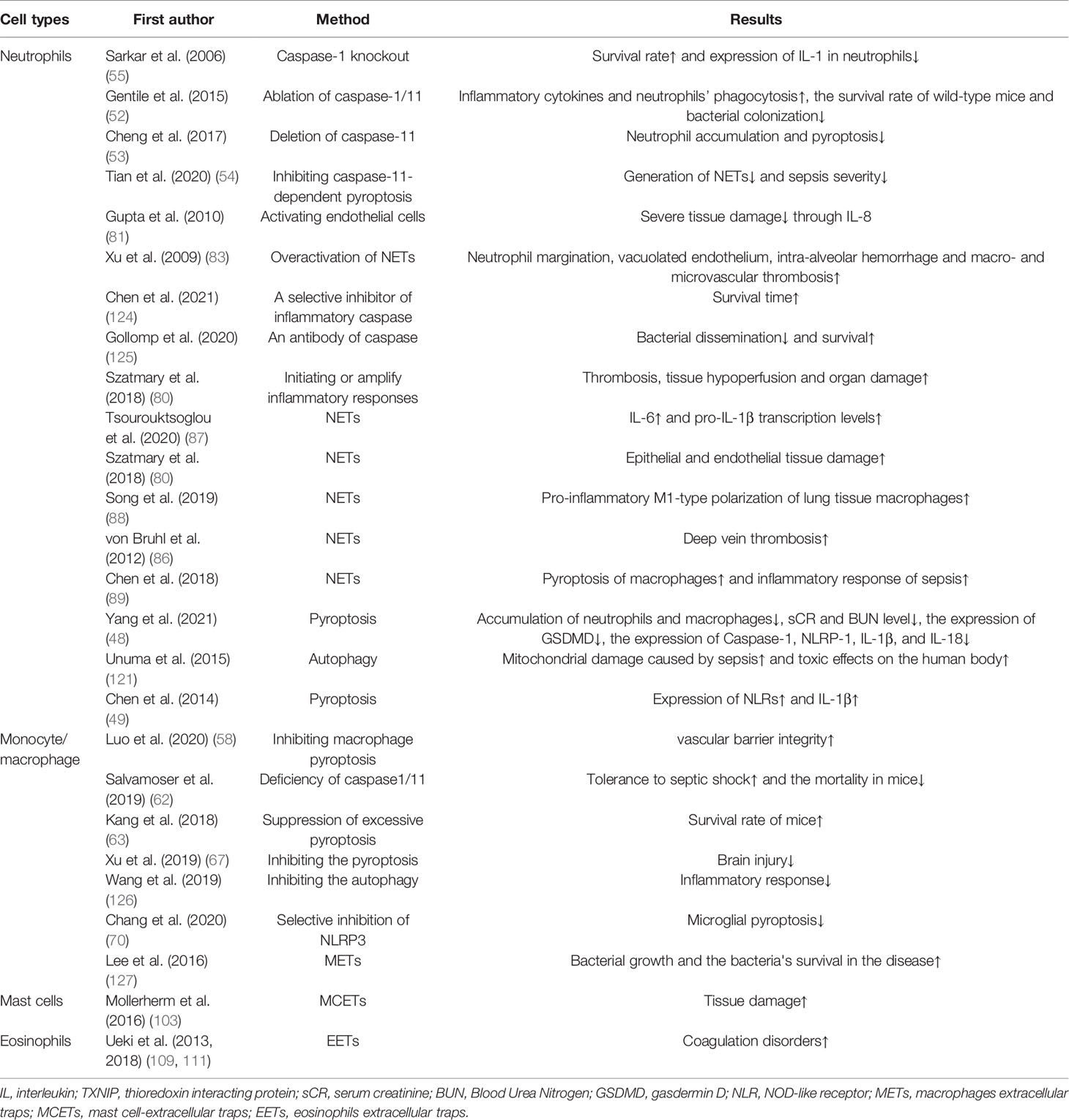
Table 3 The cons of various self-sacrifice of immune cells.
What we want to emphasize here is that not all immune cells can produce extracellular traps structures. Eosinophils, neutrophils, basophils, mast cells, macrophages and microglia have been proved to be capable of forming extracellular traps (104, 150). However, some immune cells can only produce extracellular traps in some specific and certain stimulating conditions. Whether the extracellular traps in the immune cells exert positive or negative effects depends on different physiopathology condition, including the different diseases, location of immune cells, immune cell types, and type and intensity of stimulus. But according to the current studies, the extracellular traps of the immune cell are almost a double-edged sword. Whether it is protective or detrimental depends on the pathophysiology of the disease. We propose for the first time here that the structures of histone, enzyme and chromatin DNA secreted by immune cells after specific stimulation are collectively called immune cell-extracellular traps (ICETs). They all contain DNA, histones and granular proteins involved in the innate immune response and die in a programmed way in which their cell membrane is dissolved.
In sepsis, the phenomenon of self-sacrifice of these immune cells is very interesting and notable, which provides us with a novel understanding of the roles and mechanisms of immune cell in the pathogenesis of sepsis. Nevertheless, our discussion is limited in three ways of the self-sacrifice attack in which immune cells die without discussing other ways of self-sacrifice attack. Whether the self-sacrifice attack is elf or devil is still under further study. There have been evidence demonstrating that from the point of view of self-sacrifice, neutrophils do more harm than protection, which means that we could focus on this characteristic in the treatment of sepsis in the future clinical studies. Unfortunately, there is no conclusive evidence proving that the other immune cells do more harm than good through pyroptosis, ICETs or autophagy. The exploration and exploitation of the initiating and regulating pathways, mechanisms of action and potential therapeutic drugs are on the way. On the other hand, how to magnify the protective effects and lower the detrimental effects of “ self-sacrifice “ of immune cells is worth exploring in the future. When to intervene the “ self-sacrifice “ of immune cells is also needed to be clarified. Moreover, the interplay and relationship between pyroptosis, ICETs and autophagy are also an important challenge for future research. Finally, it is worth mentioning that it is regrettable that we have not seen an attempt based entirely on this principle in clinical practice, but it is gratifying to see that there is an upper pathway in the use of a few drugs, and we believe that this path will be gradually explored.
In addition to the immune cells, pyroptosis, extracellular traps or autophagy has also been observed in tissue cells, including cardiomyocytes and endothelial cells. Meanwhile, our review does not cover all immune cells, only several relatively important immune cells are chosen as represents to be discussed.
XW and JZ performed the study design, data collection, data analysis, data interpretation, and writing. XW and BX performed the data collection and data analysis. SY and JZ performed the study design, data interpretation, and writing. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant No. 82071480 [to JZ]).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Salomao R, Ferreira BL, Salomao MC, Santos SS, Azevedo LCP, Brunialti MKC. Sepsis: Evolving Concepts and Challenges. Braz J Med Biol Res (2019) 52(4):e8595. doi: 10.1590/1414-431X20198595
2. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
3. Zheng X, Chen W, Gong F, Chen Y, Chen E. The Role and Mechanism of Pyroptosis and Potential Therapeutic Targets in Sepsis: A Review. Front Immunol (2021) 12:711939. doi: 10.3389/fimmu.2021.711939
4. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and Septic Shock. Lancet (2018) 392(10141):75–87. doi: 10.1016/S0140-6736(18)30696-2
5. Gul F, Arslantas MK, Cinel I, Kumar A. Changing Definitions of Sepsis. Turk J Anaesthesiol Reanim (2017) 45(3):129–38. doi: 10.1016/S0140-6736(18)30696-2
6. Banerjee D, Levy MM. Sepsis Definitions. Providence, RI, USA: Springer International Publishing (2017).
7. Fu Q, Wu J, Zhou XY, Ji MH, Mao QH, Li Q, et al. NLRP3/Caspase-1 Pathway-Induced Pyroptosis Mediated Cognitive Deficits in a Mouse Model of Sepsis-Associated Encephalopathy. Inflammation (2019) 42(1):306–18. doi: 10.1007/s10753-018-0894-4
8. Gao YL, Zhai JH, Chai YF. Recent Advances in the Molecular Mechanisms Underlying Pyroptosis in Sepsis. Mediators Inflamm (2018) 2018:5823823. doi: 10.1155/2018/5823823
9. Liu L, Sun B. Neutrophil Pyroptosis: New Perspectives on Sepsis. Cell Mol Life Sci (2019) 76(11):2031–42. doi: 10.1007/s00018-019-03060-1
10. Jorgensen I, Miao EA. Pyroptotic Cell Death Defends Against Intracellular Pathogens. Immunol Rev (2015) 265(1):130–42. doi: 10.1111/imr.12287
11. Janiuk K, Jabłońska E, Garley M. Significance of NETs Formation in COVID-19. Cells (2021) 10(1):151. doi: 10.3390/cells10010151
12. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front Immunol (2020) 11:1749. doi: 10.3389/fimmu.2020.01749
13. Fousert E, Toes R, Desai J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells (2020) 9(4):915. doi: 10.3390/cells9040915
15. Yipp BG, Kubes P. NETosis: How Vital is it? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
16. Shi CX, Wang Y, Chen Q, Jiao FZ, Pei MH, Gong ZJ. Extracellular Histone H3 Induces Pyroptosis During Sepsis and May Act Through NOD2 and VSIG4/NLRP3 Pathways. Front Cell Infect Microbiol (2020) 10:196. doi: 10.3389/fcimb.2020.00196
17. Sorensen OE, Borregaard N. Neutrophil Extracellular Traps - the Dark Side of Neutrophils. J Clin Invest (2016) 126(5):1612–20. doi: 10.1172/JCI84538
18. Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil Extracellular Traps Induce Organ Damage During Experimental and Clinical Sepsis. PloS One (2016) 11(2):e0148142. doi: 10.1371/journal.pone.0148142
19. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular Definitions of Autophagy and Related Processes. EMBO J (2017) 36(13):1811–36. doi: 10.15252/embj.201796697
20. Daviaud F, Grimaldi D, Dechartres A, Charpentier J, Geri G, Marin N, et al. Timing and Causes of Death in Septic Shock. Ann Intensive Care (2015) 5(1):16. doi: 10.1186/s13613-015-0058-8
21. Qiu P, Liu Y, Zhang J. Review: The Role and Mechanisms of Macrophage Autophagy in Sepsis. Inflammation (2019) 42(1):6–19. doi: 10.1007/s10753-018-0890-8
22. Skibsted S, Bhasin MK, Henning DJ, Jaminet SC, Lewandowski J, Kirkegaard H, et al. Leukocyte Transcriptional Response in Sepsis. Shock (2019) 52(2):166–73. doi: 10.1097/SHK.0000000000001258
23. Oami T, Watanabe E, Hatano M, Sunahara S, Fujimura L, Sakamoto A, et al. Suppression of T Cell Autophagy Results in Decreased Viability and Function of T Cells Through Accelerated Apoptosis in a Murine Sepsis Model. Crit Care Med (2017) 45(1):e77–85. doi: 10.1097/CCM.0000000000002016
24. Croker BA, O’Donnell JA, Gerlic M. Pyroptotic Death Storms and Cytopenia. Curr Opin Immunol (2014) 26:128–37. doi: 10.1016/j.coi.2013.12.002
25. Roshanravan N, Alamdari NM, Jafarabadi MA, Mohammadi A, Shabestari BR, Nasirzadeh N, et al. Effects of Oral Butyrate and Inulin Supplementation on Inflammation-Induced Pyroptosis Pathway in Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. Cytokine (2020) 131:155101. doi: 10.1016/j.cyto.2020.155101
26. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella Flexneri Induces Apoptosis in Infected Macrophages. Nature (1992) 358(6382):167–9. doi: 10.1038/358167a0
27. Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. Shigella-Induced Apoptosis is Dependent on Caspase-1 Which Binds to IpaB. J Biol Chem (1998) 273(49):32895–900. doi: 10.1074/jbc.273.49.32895
28. Boise LH, Collins CM. Salmonella-Induced Cell Death: Apoptosis, Necrosis or Programmed Cell Death? Trends Microbiol (2001) 9(2):64–7. doi: 10.1016/s0966-842x(00)01937-5
29. da Silva FP, Nizet V. Cell Death During Sepsis: Integration of Disintegration in the Inflammatory Response to Overwhelming Infection. Apoptosis (2009) 14(4):509–21. doi: 10.1007/s10495-009-0320-3
30. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
31. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D Exerts Anti-Inflammatory Effects by Promoting Neutrophil Death. Cell Rep (2018) 22(11):2924–36. doi: 10.1016/j.celrep.2018.02.067
32. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
33. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J Cell Biol (2007) 176(2):231–41. doi: 10.1083/jcb.200606027
34. von Köckritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-Independent Antimicrobial Activity of Mast Cells by Means of Extracellular Trap Formation. Blood (2008) 111(6):3070–80. doi: 10.1182/blood-2007-07-104018
35. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-Induced NETosis is a Dynamic Process Involving Neutrophil Multitasking In Vivo. Nat Med (2012) 18(9):1386–93. doi: 10.1038/nm.2847
36. Boe DM, Curtis BJ, Chen MM, Ippolito JA, Kovacs EJ. Extracellular Traps and Macrophages: New Roles for the Versatile Phagocyte. J Leukoc Biol (2015) 97(6):1023–35. doi: 10.1189/jlb.4RI1014-521R
37. Ashford TP, Porter KR. Cytoplasmic Components in Hepatic Cell Lysosomes. J Cell Biol (1962) 12(1):198–202. doi: 10.1083/jcb.12.1.198
38. Ho J, Yu J, Wong SH, Zhang L, Liu X, Wong WT, et al. Autophagy in Sepsis: Degradation Into Exhaustion? Autophagy (2016) 12(7):1073–82. doi: 10.1080/15548627.2016.1179410
39. Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a Novel Protein Kinase Required for the Autophagic Process in Saccharomyces Cerevisiae. Gene (1997) 192(2):245–50. doi: 10.1016/s0378-1119(97)00084-x
40. Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and Cellular Immune Responses. Immunity (2013) 39(2):211–27. doi: 10.1016/j.immuni.2013.07.017
41. Choi JC, Worman HJ. Reactivation of Autophagy Ameliorates LMNA Cardiomyopathy. Autophagy (2013) 9(1):110–1. doi: 10.4161/auto.22403
42. Aziz M, Jacob A, Wang P. Revisiting Caspases in Sepsis. Cell Death Dis (2014) 5:e1526. doi: 10.1038/cddis.2014.488
43. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
44. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci (2019) 20(13):3328. doi: 10.3390/ijms20133328
45. Vecchie A, Bonaventura A, Toldo S, Dagna L, Dinarello CA, Abbate A. IL-18 and Infections: Is There a Role for Targeted Therapies? J Cell Physiol (2021) 236(3):1638–57. doi: 10.1002/jcp.30008
46. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for non-Canonical Inflammasome Signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
47. Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity (2015) 43(5):923–32. doi: 10.1016/j.immuni.2015.10.009
48. Yang M, Fang JT, Zhang NS, Qin LJ, Zhuang YY, Wang WW, et al. Caspase-1-Inhibitor AC-YVAD-CMK Inhibits Pyroptosis and Ameliorates Acute Kidney Injury in a Model of Sepsis. BioMed Res Int (2021) 2021:6636621. doi: 10.1155/2021/6636621
49. Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1beta Maturation Without Pyroptosis During Acute Salmonella Challenge. Cell Rep (2014) 8(2):570–82. doi: 10.1016/j.celrep.2014.06.028
50. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6676. doi: 10.1126/sciimmunol.aar6676
51. Kumari P, Russo AJ, Wright SS, Muthupalani S, Rathinam VA. Hierarchical Cell-Type-Specific Functions of Caspase-11 in LPS Shock and Antibacterial Host Defense. Cell Rep (2021) 35(3):109012. doi: 10.1016/j.celrep.2021.109012
52. Gentile LF, Cuenca AL, Cuenca AG, Nacionales DC, Ungaro R, Efron PA, et al. Improved Emergency Myelopoiesis and Survival in Neonatal Sepsis by Caspase-1/11 Ablation. Immunology (2015) 145(2):300–11. doi: 10.1111/imm.12450
53. Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, et al. Caspase-11-Mediated Endothelial Pyroptosis Underlies Endotoxemia-Induced Lung Injury. J Clin Invest (2017) 127(11):4124–35. doi: 10.1172/JCI94495
54. Tian Y, Qu S, Alam HB, Williams AM, Wu Z, Deng Q, et al. Peptidylarginine Deiminase 2 has Potential as Both a Biomarker and Therapeutic Target of Sepsis. JCI Insight (2020) 5(20):e138873. doi: 10.1172/jci.insight.138873
55. Sarkar A, Hall MW, Exline M, Hart J, Knatz N, Gatson NT, et al. Caspase-1 Regulates Escherichia Coli Sepsis and Splenic B Cell Apoptosis Independently of Interleukin-1beta and Interleukin-18. Am J Respir Crit Care Med (2006) 174(9):1003–10. doi: 10.1164/rccm.200604-546OC
56. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A From Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
57. Kader M, El Andaloussi A, Vorhaour J, Tamama K, Nieto N, Scott MJ, et al. Interferon Type I Regulates Inflammasome Activation and High Mobility Group Box 1 Translocation in Hepatocytes During Ehrlichia-Induced Acute Liver Injury. Hepatol Commun (2021) 5(1):33–51. doi: 10.1002/hep4.1608
58. Luo L, Xu M, Liao D, Deng J, Mei H, Hu Y. PECAM-1 Protects Against DIC by Dampening Inflammatory Responses via Inhibiting Macrophage Pyroptosis and Restoring Vascular Barrier Integrity. Transl Res (2020) 222:1–16. doi: 10.1016/j.trsl.2020.04.005
59. Song F, Hou J, Chen Z, Cheng B, Lei R, Cui P, et al. Sphingosine-1-Phosphate Receptor 2 Signaling Promotes Caspase-11-Dependent Macrophage Pyroptosis and Worsens Escherichia Coli Sepsis Outcome. Anesthesiology (2018) 129(2):311–20. doi: 10.1097/ALN.0000000000002196
60. Wang YC, Liu QX, Liu T, Xu XE, Gao W, Bai XJ, et al. Caspase-1-Dependent Pyroptosis of Peripheral Blood Mononuclear Cells Predicts the Development of Sepsis in Severe Trauma Patients: A Prospective Observational Study. Med (Baltimore) (2018) 97(8):e9859. doi: 10.1097/MD.0000000000009859
61. Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity (2018) 49(4):740–53 e7. doi: 10.1016/j.immuni.2018.08.016
62. Salvamoser R, Brinkmann K, O’Reilly LA, Whitehead L, Strasser A, Herold MJ. Characterisation of Mice Lacking the Inflammatory Caspases-1/11/12 Reveals No Contribution of Caspase-12 to Cell Death and Sepsis. Cell Death Differ (2019) 26(6):1124–37. doi: 10.1038/s41418-018-0188-2
63. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe (2018) 24(1):97–108. e4. doi: 10.1016/j.chom.2018.05.009
64. Baaklini CS, Rawji KS, Duncan GJ, Ho MFS, Plemel JR. Central Nervous System Remyelination: Roles of Glia and Innate Immune Cells. Front Mol Neurosci (2019) 12:225. doi: 10.3389/fnmol.2019.00225
65. Refolo V, Stefanova N. Neuroinflammation and Glial Phenotypic Changes in Alpha-Synucleinopathies. Front Cell Neurosci (2019) 13:263. doi: 10.3389/fncel.2019.00263
66. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell (2018) 173(5):1073–81. doi: 10.1016/j.cell.2018.05.003
67. Xu XE, Liu L, Wang YC, Wang CT, Zheng Q, Liu QX, et al. Caspase-1 Inhibitor Exerts Brain-Protective Effects Against Sepsis-Associated Encephalopathy and Cognitive Impairments in a Mouse Model of Sepsis. Brain Behav Immun (2019) 80:859–70. doi: 10.1016/j.bbi.2019.05.038
68. Xu P, Zhang X, Liu Q, Xie Y, Shi X, Chen J, et al. Microglial TREM-1 Receptor Mediates Neuroinflammatory Injury via Interaction With SYK in Experimental Ischemic Stroke. Cell Death Dis (2019) 10(8):555. doi: 10.1038/s41419-019-1777-9
69. Li L, Shu MQ, Chen J. CYLD Deficiency Exacerbates Lipopolysaccharide (LPS)-Induced Pyroptosis in Astrocytes of Mice With Sepsis. Biochem Biophys Res Commun (2019) 514(4):1066–73. doi: 10.1016/j.bbrc.2019.05.033
70. Chang Y, Zhu J, Wang D, Li H, He Y, Liu K, et al. NLRP3 Inflammasome-Mediated Microglial Pyroptosis is Critically Involved in the Development of Post-Cardiac Arrest Brain Injury. J Neuroinflamm (2020) 17(1):219. doi: 10.1186/s12974-020-01879-1
71. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
72. Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, et al. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation During Sepsis. Am J Respir Crit Care Med (2017) 196(5):577–89. doi: 10.1164/rccm.201603-0596OC
73. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and Molecular Pathways Involved in the Regulation of Neutrophil Extracellular Trap Formation Mediated by Activated Platelets. J Leukoc Biol (2016) 99(1):153–62. doi: 10.1189/jlb.3A0415-161R
74. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-Selectin Promotes Neutrophil Extracellular Trap Formation in Mice. Blood (2015) 126(2):242–6. doi: 10.1182/blood-2015-01-624023
75. Murao A, Arif A, Brenner M, Denning NL, Jin H, Takizawa S, et al. Extracellular CIRP and TREM-1 Axis Promotes ICAM-1-Rho-Mediated NETosis in Sepsis. FASEB J (2020) 34(7):9771–86. doi: 10.1096/fj.202000482R
76. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK Pathway is Required for Neutrophil Extracellular Trap Formation. Nat Chem Biol (2011) 7(2):75–7. doi: 10.1038/nchembio.496
77. Spel L, Martinon F. Gasdermin D Opens the Way for NETs. Nat Rev Rheumatol (2018) 14(12):690–2. doi: 10.1038/s41584-018-0124-3
78. Khan MA, Farahvash A, Douda DN, Licht JC, Grasemann H, Sweezey N, et al. JNK Activation Turns on LPS- and Gram-Negative Bacteria-Induced NADPH Oxidase-Dependent Suicidal NETosis. Sci Rep (2017) 7(1):3409. doi: 10.1038/s41598-017-03257-z
79. Yu X, Tan J, Diamond SL. Hemodynamic Force Triggers Rapid NETosis Within Sterile Thrombotic Occlusions. J Thromb Haemost (2018) 16(2):316–29. doi: 10.1111/jth.13907
80. Szatmary P, Huang W, Criddle D, Tepikin A, Sutton R. Biology, Role and Therapeutic Potential of Circulating Histones in Acute Inflammatory Disorders. J Cell Mol Med (2018) 22(10):4617–29. doi: 10.1111/jcmm.13797
81. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated Endothelial Cells Induce Neutrophil Extracellular Traps and are Susceptible to NETosis-Mediated Cell Death. FEBS Lett (2010) 584(14):3193–7. doi: 10.1016/j.febslet.2010.06.006
82. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (2011) 187(1):538–52. doi: 10.4049/jimmunol.1100450
83. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular Histones are Major Mediators of Death in Sepsis. Nat Med (2009) 15(11):1318–21. doi: 10.1038/nm.2053
84. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, et al. Aggregated Neutrophil Extracellular Traps Limit Inflammation by Degrading Cytokines and Chemokines. Nat Med (2014) 20(5):511–7. doi: 10.1038/nm.3547
85. Wolberg AS, Aleman MM, Leiderman K, Machlus KR. Procoagulant Activity in Hemostasis and Thrombosis: Virchow’s Triad Revisited. Anesth Analg (2012) 114(2):275–85. doi: 10.1213/ANE.0b013e31823a088c
86. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, Neutrophils, and Platelets Cooperate to Initiate and Propagate Venous Thrombosis in Mice In Vivo. J Exp Med (2012) 209(4):819–35. doi: 10.1084/jem.20112322
87. Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep (2020) 31(5):107602. doi: 10.1016/j.celrep.2020.107602
88. Song C, Li H, Li Y, Dai M, Zhang L, Liu S, et al. NETs Promote ALI/ARDS Inflammation by Regulating Alveolar Macrophage Polarization. Exp Cell Res (2019) 382(2):111486. doi: 10.1016/j.yexcr.2019.06.031
89. Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, et al. Neutrophil Extracellular Traps Promote Macrophage Pyroptosis in Sepsis. Cell Death Dis (2018) 9(6):597. doi: 10.1038/s41419-018-0538-5
91. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Nivolumab. Intensive Care Med (2019) 45(10):1360–71. doi: 10.1007/s00134-019-05704-z
92. Andreu-Ballester JC, Cuellar C, Garcia-Ballesteros C, Perez-Griera J, Amigo V, Peiro-Gomez A, et al. Deficit of Interleukin 7 in Septic Patients. Int Immunopharmacol (2014) 23(1):73–6. doi: 10.1016/j.intimp.2014.08.015
93. Je S, Quan H, Yoon Y, Na Y, Kim BJ, Seok SH. Mycobacterium Massiliense Induces Macrophage Extracellular Traps With Facilitating Bacterial Growth. PloS One (2016) 11(5):e0155685. doi: 10.1371/journal.pone.0155685
94. King PT, Sharma R, O’Sullivan K, Selemidis S, Lim S, Radhakrishna N, et al. Nontypeable Haemophilus Influenzae Induces Sustained Lung Oxidative Stress and Protease Expression. PloS One (2015) 10(3):e0120371. doi: 10.1371/journal.pone.0120371
95. Liu P, Wu X, Liao C, Liu X, Du J, Shi H, et al. Escherichia Coli and Candida Albicans Induced Macrophage Extracellular Trap-Like Structures With Limited Microbicidal Activity. PloS One (2014) 9(2):e90042. doi: 10.1371/journal.pone.0090042
96. Mohanan S, Horibata S, McElwee JL, Dannenberg AJ, Coonrod SA. Identification of Macrophage Extracellular Trap-Like Structures in Mammary Gland Adipose Tissue: A Preliminary Study. Front Immunol (2013) 4:67. doi: 10.3389/fimmu.2013.00067
97. Munoz-Caro T, Silva LM, Ritter C, Taubert A, Hermosilla C. Besnoitia Besnoiti Tachyzoites Induce Monocyte Extracellular Trap Formation. Parasitol Res (2014) 113(11):4189–97. doi: 10.1007/s00436-014-4094-3
98. Wong KW, Jacobs WR Jr. Mycobacterium Tuberculosis Exploits Human Interferon Gamma to Stimulate Macrophage Extracellular Trap Formation and Necrosis. J Infect Dis (2013) 208(1):109–19. doi: 10.1093/infdis/jit097
99. Elieh Ali Komi D, Kuebler WM. Significance of Mast Cell Formed Extracellular Traps in Microbial Defense. Clin Rev Allergy Immunol (2022) 62(1):160–79. doi: 10.1007/s12016-021-08861-6
100. Dahl S, Anders E, Gidlöf O, Svensson D, Nilsson BO. The Host Defense Peptide LL-37 Triggers Release of Nucleic Acids From Human Mast Cells. Peptides (2018) 109:39–45. doi: 10.1016/j.peptides.2018.10.001
101. Clark M, Kim J, Etesami N, Shimamoto J, Whalen RV, Martin G, et al. Group A Streptococcus Prevents Mast Cell Degranulation to Promote Extracellular Trap Formation. Front Immunol (2018) 9:327. doi: 10.3389/fimmu.2018.00327
102. Kikuchi-Ueda T, Kamoshida G, Ubagai T, Nakano R, Nakano A, Akuta T, et al. The TNF-α of Mast Cells Induces Pro-Inflammatory Responses During Infection With Acinetobacter Baumannii. Immunobiology (2017) 222(11):1025–34. doi: 10.1016/j.imbio.2017.05.015
103. Möllerherm H, von Köckritz-Blickwede M, Branitzki-Heinemann K. Antimicrobial Activity of Mast Cells: Role and Relevance of Extracellular DNA Traps. Front Immunol (2016) 7:265. doi: 10.3389/fimmu.2016.00265
104. Morshed M, Hlushchuk R, Simon D, Walls AF, Obata-Ninomiya K, Karasuyama H, et al. NADPH Oxidase-Independent Formation of Extracellular DNA Traps by Basophils. J Immunol (2014) 192(11):5314–23. doi: 10.4049/jimmunol.1303418
105. Undem BJ, Taylor-Clark T. Mechanisms Underlying the Neuronal-Based Symptoms of Allergy. J Allergy Clin Immunol (2014) 133(6):1521–34. doi: 10.1016/j.jaci.2013.11.027
106. Naqvi N, Srivastava R, Naskar P, Puri N. Mast Cells Modulate Early Responses to Mycobacterium Bovis Bacillus Calmette-Guerin by Phagocytosis and Formation of Extracellular Traps. Cell Immunol (2021) 365:104380. doi: 10.1016/j.cellimm.2021.104380
107. Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, Rodríguez-López GM, Soria-Castro R, García-Pérez BE, et al. Mycobacterium Tuberculosis Catalase Inhibits the Formation of Mast Cell Extracellular Traps. Front Immunol (2018) 9:1161. doi: 10.3389/fimmu.2018.01161
108. Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, González-Jiménez M, Paredes-Vivas Y, Cerbulo-Vázquez A, et al. Listeria Monocytogenes Induces Mast Cell Extracellular Traps. Immunobiology (2017) 222(2):432–9. doi: 10.1016/j.imbio.2016.08.006
109. Ueki S, Tokunaga T, Melo RCN, Saito H, Honda K, Fukuchi M, et al. Charcot-Leyden Crystal Formation is Closely Associated With Eosinophil Extracellular Trap Cell Death. Blood (2018) 132(20):2183–7. doi: 10.1182/blood-2018-04-842260
110. Silveira JS, Antunes GL, Gassen RB, Breda RV, Stein RT, Pitrez PM, et al. Respiratory Syncytial Virus Increases Eosinophil Extracellular Traps in a Murine Model of Asthma. Asia Pacific Allergy (2019) 9(4):e32. doi: 10.5415/apallergy.2019.9.e32
111. Ueki S, Melo RC, Ghiran I, Spencer LA, Dvorak AM, Weller PF. Eosinophil Extracellular DNA Trap Cell Death Mediates Lytic Release of Free Secretion-Competent Eosinophil Granules in Humans. Blood (2013) 121(11):2074–83. doi: 10.1182/blood-2012-05-432088
112. Muniz VS, Silva JC, Braga YAV, Melo RCN, Ueki S, Takeda M, et al. Eosinophils Release Extracellular DNA Traps in Response to Aspergillus Fumigatus. J Allergy Clin Immunol (2018) 141(2):571–85.e7. doi: 10.1016/j.jaci.2017.07.048
113. Kumar V. Dendritic Cells in Sepsis: Potential Immunoregulatory Cells With Therapeutic Potential. Mol Immunol (2018) 101:615–26. doi: 10.1016/j.molimm.2018.07.007
114. Loures FV, Rohm M, Lee CK, Santos E, Wang JP, Specht CA, et al. Recognition of Aspergillus Fumigatus Hyphae by Human Plasmacytoid Dendritic Cells is Mediated by Dectin-2 and Results in Formation of Extracellular Traps. PloS Pathog (2015) 11(2):e1004643. doi: 10.1371/journal.ppat.1004643
115. Weindel CG, Richey LJ, Mehta AJ, Shah M, Huber BT. Autophagy in Dendritic Cells and B Cells Is Critical for the Inflammatory State of TLR7-Mediated Autoimmunity. J Immunol (2017) 198(3):1081–92. doi: 10.4049/jimmunol.1601307
116. Su Y, Qu Y, Zhao F, Li H, Mu D, Li X. Regulation of Autophagy by the Nuclear Factor kappaB Signaling Pathway in the Hippocampus of Rats With Sepsis. J Neuroinflamm (2015) 12:116. doi: 10.1186/s12974-015-0336-2
117. Pareja ME, Colombo MI. Autophagic Clearance of Bacterial Pathogens: Molecular Recognition of Intracellular Microorganisms. Front Cell Infect Microbiol (2013) 3:54. doi: 10.3389/fcimb.2013.00054
118. Maurer K, Reyes-Robles T, Alonzo F 3rd, Durbin J, Torres VJ, Cadwell K. Autophagy Mediates Tolerance to Staphylococcus Aureus Alpha-Toxin. Cell Host Microbe (2015) 17(4):429–40. doi: 10.1016/j.chom.2015.03.001
119. Liu Y, Levine B. Autosis and Autophagic Cell Death: The Dark Side of Autophagy. Cell Death Differ (2015) 22(3):367–76. doi: 10.1038/cdd.2014.143
120. Hsieh CH, Pai PY, Hsueh HW, Yuan SS, Hsieh YC. Complete Induction of Autophagy is Essential for Cardioprotection in Sepsis. Ann Surg (2011) 253(6):1190–200. doi: 10.1097/SLA.0b013e318214b67e
121. Unuma K, Aki T, Funakoshi T, Hashimoto K, Uemura K. Extrusion of Mitochondrial Contents From Lipopolysaccharide-Stimulated Cells: Involvement of Autophagy. Autophagy (2015) 11(9):1520–36. doi: 10.1080/15548627.2015.1063765
122. Lin CW, Lo S, Hsu C, Hsieh CH, Chang YF, Hou BS, et al. T-Cell Autophagy Deficiency Increases Mortality and Suppresses Immune Responses After Sepsis. PloS One (2014) 9(7):e102066. doi: 10.1371/journal.pone.0102066
123. Ge Y, Huang M, Dong N, Yao YM. Effect of Interleukin-36β on Activating Autophagy of CD4+CD25+ Regulatory T Cells and Its Immune Regulation in Sepsis. J Infect Dis (2020) 222(9):1517–30. doi: 10.1093/infdis/jiaa258
124. Chen Q, Zheng J, Wang D, Liu Q, Kang L, Gao X, et al. Nitrosonisoldipine is a Selective Inhibitor of Inflammatory Caspases and Protects Against Pyroptosis and Related Septic Shock. Eur J Immunol (2021) 51(5):1234–45. doi: 10.1002/eji.202048937
125. Gollomp K, Sarkar A, Harikumar S, Seeholzer SH, Arepally GM, Hudock K, et al. Fc-Modified HIT-Like Monoclonal Antibody as a Novel Treatment for Sepsis. Blood (2020) 135(10):743–54. doi: 10.1182/blood.2019002329
126. Wang Z, Li Y, Yang X, Zhang L, Shen H, Xu W, et al. Protective Effects of Rapamycin Induced Autophagy on CLP Septic Mice. Comp immunol Microbiol Infect Dis (2019) 64:47–52. doi: 10.1016/j.cimid.2019.01.009
127. Lee JP, Foote A, Fan H, de Castro CP, Lang T, Jones SA, et al. Loss of Autophagy Enhances MIF/macrophage Migration Inhibitory Factor Release by Macrophages. Autophagy (2016) 12(6):907–16. doi: 10.1080/15548627.2016.1164358
128. Winkler MS, Rissiek A, Priefler M, Schwedhelm E, Robbe L, Bauer A, et al. Human Leucocyte Antigen (HLA-DR) Gene Expression is Reduced in Sepsis and Correlates With Impaired TNFalpha Response: A Diagnostic Tool for Immunosuppression? PloS One (2017) 12(8):e0182427. doi: 10.1371/journal.pone.0182427
129. Obba S, Hizir Z, Boyer L, Selimoglu-Buet D, Pfeifer A, Michel G, et al. The PRKAA1/AMPKalpha1 Pathway Triggers Autophagy During CSF1-Induced Human Monocyte Differentiation and is a Potential Target in CMML. Autophagy (2015) 11(7):1114–29. doi: 10.1080/15548627.2015.1034406
130. Ko JH, Yoon SO, Lee HJ, Oh JY. Rapamycin Regulates Macrophage Activation by Inhibiting NLRP3 Inflammasome-P38 MAPK-Nfκb Pathways in Autophagy- and P62-Dependent Manners. Oncotarget (2017) 8(25):40817–31. doi: 10.18632/oncotarget.17256
131. Ceylan-Isik AF, Zhao P, Zhang B, Xiao X, Su G, Ren J. Cardiac Overexpression of Metallothionein Rescues Cardiac Contractile Dysfunction and Endoplasmic Reticulum Stress But Not Autophagy in Sepsis. J Mol Cell Cardiol (2010) 48(2):367–78. doi: 10.1016/j.yjmcc.2009.11.003
132. Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T. Autophagy and Autophagy-Related Proteins in the Immune System. Nat Immunol (2015) 16(10):1014–24. doi: 10.1038/ni.3273
133. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature (2008) 456(7219):264–8. doi: 10.1038/nature07383
134. Dikic I, Elazar Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat Rev Mol Cell Biol (2018) 19(6):349–64. doi: 10.1038/s41580-018-0003-4
135. Pu Q, Gan C, Li R, Li Y, Tan S, Li X, et al. Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis in Pseudomonas Sepsis. J Immunol (2017) 198(8):3205–13. doi: 10.4049/jimmunol.1601196
136. Qiu X, Cheng X, Zhang J, Yuan C, Zhao M, Yang X. Ethyl Pyruvate Confers Protection Against Endotoxemia and Sepsis by Inhibiting Caspase-11-Dependent Cell Pyroptosis. Int Immunopharmacol (2020) 78:106016. doi: 10.1016/j.intimp.2019.106016
137. Chen W, Qiang X, Wang Y, Zhu S, Li J, Babaev A, et al. Identification of Tetranectin-Targeting Monoclonal Antibodies to Treat Potentially Lethal Sepsis. Sci Transl Med (2020) 12(539):eaaz3833. doi: 10.1126/scitranslmed.aaz3833
138. Sun YB, Zhao H, Mu DL, Zhang W, Cui J, Wu L, et al. Dexmedetomidine Inhibits Astrocyte Pyroptosis and Subsequently Protects the Brain in In Vitro and In Vivo Models of Sepsis. Cell Death Dis (2019) 10(3):167. doi: 10.1038/s41419-019-1416-5
139. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical Disruption of the Pyroptotic Pore-Forming Protein Gasdermin D Inhibits Inflammatory Cell Death and Sepsis. Sci Immunol (2018) 3(26):eaat2738. doi: 10.1126/sciimmunol.aat2738
140. Wang D, Zheng J, Hu Q, Zhao C, Chen Q, Shi P, et al. Magnesium Protects Against Sepsis by Blocking Gasdermin D N-Terminal-Induced Pyroptosis. Cell Death Differ (2020) 27(2):466–81. doi: 10.1038/s41418-019-0366-x
141. Hu Z, Murakami T, Suzuki K, Tamura H, Kuwahara-Arai K, Iba T, et al. Antimicrobial Cathelicidin Peptide LL-37 Inhibits the LPS/ATP-Induced Pyroptosis of Macrophages by Dual Mechanism. PloS One (2014) 9(1):e85765. doi: 10.1371/journal.pone.0085765
142. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in Sepsis: A Novel Understanding of the Disorder and a New Therapeutic Approach. Lancet Infect Dis (2013) 13(3):260–8. doi: 10.1016/S1473-3099(13)70001-X
143. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 Channel and Mitochondrial ROS Mediate NADPH Oxidase-Independent NETosis Induced by Calcium Influx. Proc Natl Acad Sci USA (2015) 112(9):2817–22. doi: 10.1073/pnas.1414055112
144. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
145. Li Z, Jia Y, Feng Y, Cui R, Miao R, Zhang X, et al. Methane Alleviates Sepsis-Induced Injury by Inhibiting Pyroptosis and Apoptosis: In Vivo and In Vitro Experiments. Aging (Albany NY) (2019) 11(4):1226–39. doi: 10.18632/aging.101831
146. Pfalzgraff A, Heinbockel L, Su Q, Brandenburg K, Weindl G. Synthetic Anti-Endotoxin Peptides Inhibit Cytoplasmic LPS-Mediated Responses. Biochem Pharmacol (2017) 140:64–72. doi: 10.1016/j.bcp.2017.05.015
147. Pfalzgraff A, Weindl G. Intracellular Lipopolysaccharide Sensing as a Potential Therapeutic Target for Sepsis. Trends Pharmacol Sci (2019) 40(3):187–97. doi: 10.1016/j.tips.2019.01.001
148. Zhou J, Zhang F, Lin H, Quan M, Yang Y, Lv Y, et al. The Protein Kinase R Inhibitor C16 Alleviates Sepsis-Induced Acute Kidney Injury Through Modulation of the NF-κb and NLR Family Pyrin Domain-Containing 3 (NLPR3) Pyroptosis Signal Pathways. Med Sci Monit (2020) 26:e926254. doi: 10.12659/MSM.926254
149. Stiel CU, Ebenebe CU, Trochimiuk M, Pagarols Raluy L, Vincent D, Singer D, et al. Markers of NETosis Do Not Predict Neonatal Early Onset Sepsis: A Pilot Study. Front Pediatr (2019) 7:555. doi: 10.3389/fped.2019.00555
Keywords: sepsis, pyroptosis, NETosis, immune cells, self-sacrifice
Citation: Wen X, Xie B, Yuan S and Zhang J (2022) The “Self-Sacrifice” of ImmuneCells in Sepsis. Front. Immunol. 13:833479. doi: 10.3389/fimmu.2022.833479
Received: 11 December 2021; Accepted: 05 April 2022;
Published: 29 April 2022.
Edited by:
Rksubbarao Malireddi, St. Jude Children’s Research Hospital, United StatesCopyright © 2022 Wen, Xie, Yuan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiancheng Zhang, emhqY2hlbmcxQDEyNi5jb20=; Shiying Yuan, eXVhbl9zaGl5aW5nQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.