 Ren-Jun Hsu1,2†
Ren-Jun Hsu1,2† Wei-Chieh Yu3†Guan-Ru Peng3Chih-Hung Ye3SuiYun Hu3Patrick Chun Theng Chong3Kah Yi Yap3Jamie Yu Chieh Lee4Wei-Chen Lin3Shu-Han Yu3*
Wei-Chieh Yu3†Guan-Ru Peng3Chih-Hung Ye3SuiYun Hu3Patrick Chun Theng Chong3Kah Yi Yap3Jamie Yu Chieh Lee4Wei-Chen Lin3Shu-Han Yu3*- 1Cancer Center, Hualien Tzu Chi Hospital, Buddhist Tzuchi Medical Foundation, Hualien, Taiwan
- 2School of Medicine, College of Medicine, Tzu Chi University, Hualien, Taiwan
- 3Institute of Biotechnology, National Taiwan University, Taipei, Taiwan
- 4University of California, Los Angeles, Los Angeles, CA, United States
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has resulted in countless infections and caused millions of deaths since its emergence in 2019. Coronavirus disease 2019 (COVID-19)-associated mortality is caused by uncontrolled inflammation, aberrant immune response, cytokine storm, and an imbalanced hyperactive immune system. The cytokine storm further results in multiple organ failure and lung immunopathology. Therefore, any potential treatments should focus on the direct elimination of viral particles, prevention strategies, and mitigation of the imbalanced (hyperactive) immune system. This review focuses on cytokine secretions of innate and adaptive immune responses against COVID-19, including interleukins, interferons, tumor necrosis factor-alpha, and other chemokines. In addition to the review focus, we discuss potential immunotherapeutic approaches based on relevant pathophysiological features, the systemic immune response against SARS-CoV-2, and data from recent clinical trials and experiments on the COVID-19-associated cytokine storm. Prompt use of these cytokines as diagnostic markers and aggressive prevention and management of the cytokine storm can help determine COVID-19-associated morbidity and mortality. The prophylaxis and rapid management of the cytokine storm appear to significantly improve disease outcomes. For these reasons, this study aims to provide advanced information to facilitate innovative strategies to survive in the COVID-19 pandemic.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and its associated pathology, coronavirus disease 2019 (COVID-19) are of particular concern because of the global pandemic they have unleashed. COVID-19 continues to challenge medical health systems globally, and as the scenario is inevitably worsening (1, 2), developing prophylactic and therapeutic approaches is urgent. The number of cases requiring intensive care has emerged as a critical point in the epidemic. Therefore, it is important to understand in more detail the pathophysiology underlying severe disease, particularly the factors that drive severe lung pathology after infection with the highly pathogenic human coronaviruses (3).
The hyperactive host immune response to SARS-CoV-2 infection leads to an exaggerated inflammatory reaction. Elevated circulating levels of interleukin (IL)-1β, IL-2, IL-6, IL-7, IL-8 (CXCL8), IL-9, IL-10, IL-17, IL-18, IL-22, IL-33, granulocyte-colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon (IFN)-γ, tumor necrosis factor (TNF)-α, chemokine (C-X-C motif) ligand (CXCL)10, monocyte chemoattractant protein 1 (CCL2 or MCP-1), macrophage inflammatory protein 1 (MIP-1)A (CCL3), CX3CL1 and MIP-1B (CCL4) (4, 5) have been reported in patients with COVID-19, particularly in those admitted to the intensive care unit (ICU). The overexpression of cytokines and chemokines triggers the severe cytokine release syndrome (CRS), which increases the severity of the disease even more (6, 7). Moreover, samples of the bronchoalveolar lavage fluid (BALF) of patients with COVID-19 show the accumulation of various immune attractant chemokines, including C-C motif chemokine ligand (CCL)2, CCL3, CCL4, CCL7, CCL8, CCL20, CXCL6, and CXCL11 (4, 8, 9). Many of these chemokines are secreted by monocytes or macrophages (4, 5, 10). Researchers that have analyzed cytokine profiles in patients with COVID-19 suggest that cytokine storms correlate directly with lung injury, multiorgan failure, and unfavorable prognosis in severe COVID-19 (11).
Using available data from experimental and clinical studies, this review aims to provide up-to-date knowledge on cytokine and chemokine secretions induced by host immune responses in COVID-19 as well as on cytokine storm and related pathophysiological features. Identifying the distinct cytokine/chemokine profile(s) and pathophysiological features of COVID-19-induced cytokine storm has practical implications as it can predict clinical deterioration, e.g., the requirement of intubation or mortality. Understanding the nature of the immune response that can lead to recovery from severe COVID-19 remains key to developing effective treatments (12).
Cytokine Secretion Induced by SARS-CoV-2 Infection
SARS-CoV-2 infects cells by attaching itself to the angiotensin-converting enzyme 2 (ACE2) (13) and/or type II transmembrane serine protease (TMPRSS2) receptors (14). Subsequent viral replication and release cause the host cell to undergo pyroptosis and unleash pathogen-associated molecular patterns (PAMPs). In the first few days following infection, innate immune cells, including macrophages, dendritic cells (DCs), neutrophils, and natural killer (NK) cells, are activated when they recognize viral PAMPs (15). Next, PAMPs trigger the production of several proinflammatory cytokines and chemokines, including TNF, IL-6, type-1 IFN, CCL2, CCL3, CCL4, and CXCL10 (IFN-γ-induced protein 10 kDa [IP-10]) (16, 17), which lead to the recruitment of monocytes, macrophages, and T cells to the infection sites. These processes promote further inflammation and recruit NK cells (3) and T cells, which produce more IFN-γ (18). Innate antigen-presenting cells, such as DCs and macrophages, at the infection site also present viral antigens to virus-specific T cells. This leads to the activation of the body’s adaptive immunity, which is mediated by virus-specific B (humoral immunity) and T cells (cellular immunity) (10, 19).
When SARS-CoV-2 binds to the ACE2 receptor to enter the target cell, the renin-angiotensin system (RAS) is activated, and angiotensin II (Ang II) levels increase in circulation (20, 21). Studies have shown that the ACE2-Ang II axis can induce the infiltration of macrophages and secretion of certain cytokines, including IL-6, CCL2, vascular cell adhesion molecule 1 (VCAM-1), and E-selectin, to induce endothelial dysfunction, thrombin formation, and impaired fibrinolysis (20, 22–24). In addition to the ACE2-Ang II axis, viral protein Nsp5 and spike (S) in SARS-CoV-2 can potently induce inflammatory cytokines and chemokines IL-1β, IL-6, TNF-α, IL-2, CXCL1, CXCL2, and CCL2 expressions in monocytes/macrophage through NF-κB signaling pathways (25, 26).
Both aspects of adaptive immunity, i.e., cell-mediated immunity and humoral immunity, have critical roles in COVID-19 (27). Lower absolute numbers of T lymphocytes (CD4+ and CD8+ T cells) occur in both mild and severe infection, with a more notable decrease in severe cases. The decrease in IFN-γ expression by CD4+ T cells is also greater in severe cases than in moderate ones (28). Moreover, pyroptotic cell death can lead to higher serum IL-1β levels and trigger neutrophil migration and T cell activation (29, 30). Indeed, neutrophil cytotoxicity releases leukotrienes and reactive oxygen species, thereby triggering acute lung injury (ALI) and a cytokine storm. Neutrophils may also lead to endothelial injury, which can further promote systemic virus dissemination (17, 30–32). Furthermore, increased serum levels of IL-1β, IFN-γ, CXCL10, and CCL2 strongly point toward the activation of T helper 1 (Th1) cell function (33, 34). Thus, inadequate innate and adaptive antiviral defenses, in addition to high proinflammatory cues, lead to multiorgan damage (10, 35).
To further illustrate the effect of SARS-CoV-2 on cytokine and chemokine production, 27-plex cytokine assay panels and the Bio-Plex 200 system were used to analyze plasma cytokine and chemokine levels during the acute phase of the illness, and available results indicate that the plasma concentrations of specific inflammatory cytokines and chemokines highly correlate with the severity of COVID-19 disease course. In particular, the expression levels of IL-2, IL-7, IL-10, GCS-F, CXCL10, CCL2, CCL3, and TNF-α were significantly higher in ICU patients than in non-ICU patients (33). By contrast, other cytokines, including IL-1β, IL-1RA, IL-7, IL-8, IL-9, IL-10, basic fibroblast growth factors (FGF), GCS-F, GM-CSF, IFN-γ, CXCL10, CCL2, CCL3, CCL4, platelet-derived growth factor subunit B (PDGFB), TNF-α, and vascular endothelial growth factor-A (VEGF-A), showed higher expression in both ICU and non-ICU patients with COVID-19 (33, 36, 37). Of note, the expression of some cytokines, such as IL-5, IL-12, p70, IL-15, CCL5, and eotaxin, were similar in healthy adults and SARS-CoV-2-infected patients (5, 6, 28, 33, 38–40).
Due to the importance of cytokines in COVID-19 disease development, several clinical trials have been focusing on cytokine modulation. According to the ongoing clinical trials, many potential COVID-19 therapeutic drugs, such as anakinra (IL-1 receptor antagonist) (13), siltuximab (IL-6 neutralization) (41), sarilumab (IL-6 receptor antagonist) (41), tocilizumab (IL-6 receptor antagonist) (42), secukinumab (IL-17 neutralization) (43), baricitinib (Jak inhibitor) (44), IFN-β-1α (inducing type I IFN-stimulated genes) (45), recombinant human IFN drugs (46), eculizumab (inhibition of complement activation) (47), G/10 (innate response regulation) (48), CD24F (innate response regulation) (48), anti-GM-CSF monoclonal antibody (49) and NKG2D-ACE2 CAR-NK cells (50) have been applied to COVID-19 patients to boost their immune system, inhibiting viral replication or cytokine storms (42, 44, 48, 51–56). Accordingly, these trials indicated that some cytokines, such as IL-1 (57), IL-6 (58), TNF (57), IFN (59), and GM-CSF (57), could be potential targets to develop as diagnostic markers and personalized drug targets (58). In addition to the potential targets, there are a number of small-molecule drugs under clinical trials to benefit COVID-19 patients. Paxlovid, a ritonavir-boosted protease inhibitor, is the first oral antiviral drug to receive FDA granted EUA in December of 2021 (41). Both remdesivir (60) and molnupiravir (61, 62) can inhibit viral replication by inhibiting RNA-dependent RNA polymerase (RdRp) (60–64).
This section aims to investigate the distinct cytokine profiles associated with COVID-19 immunopathogenesis. They include interleukins (IL), interferon (IFN), tumor necrosis factor (TNF)-α, and chemokines. Changes in immune mechanisms, cytokine, and chemokine secretion caused by SARS-CoV-2 infection, and reported druggable targets are described in the following sections and summarized in Figure 1 and Table 1.
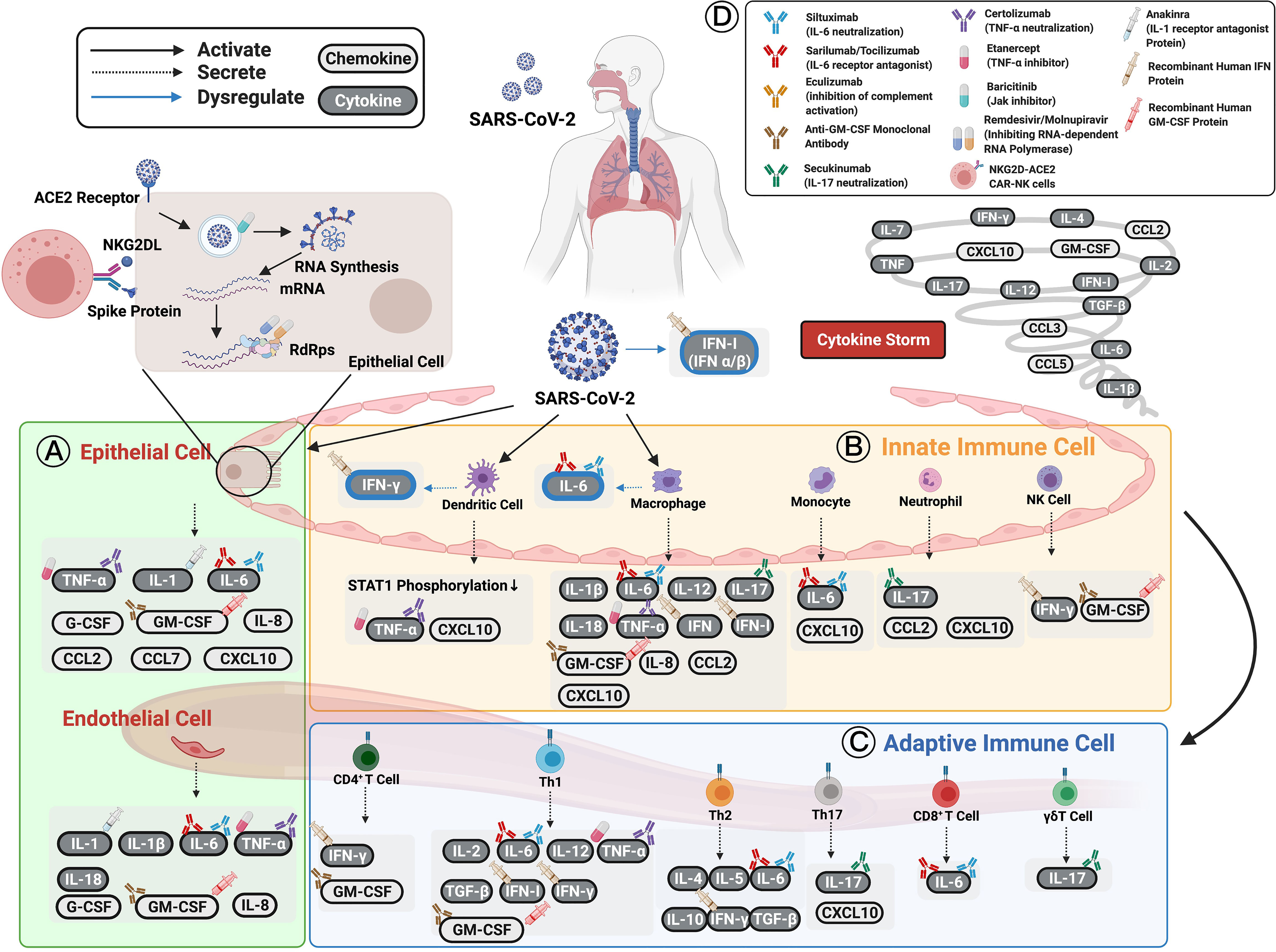
Figure 1 Cytokines and chemokines expression induced by SARS-CoV-2 in patients with COVID-19. (A) Epithelial cells have been shown to express TNF-α, IL-1, IL-6, and other chemokines (e.g., IL-8, CCL2, CCL7, CXCL10, G-CSF, and GM-CSF) as well as initiate immune responses following infection. (B) Innate immune cells, including dendritic cells, macrophages, circulating monocytes, neutrophils, and NK cells, are activated by SARS-CoV-2 infection to secrete various cytokines and chemokines to enhance both innate and adaptive immune cells. (C) Adaptive immune cells consisting of functional CD4+ T cells, Th1 cells, Th2 cells, Th17 cells, CD8+ T cells, and γδT cells defend against SARS-CoV-2 infection. The cytokines and chemokines expressed by adaptive immune cells interact in a positive feedback loop to strengthen innate immune responses. The cellular origin of each cytokine and its regulatory roles are shown in figure and Table 1. The persistent escalation of these responses leads to uncontrolled cytokine and chemokine expression, resulting in a life-threatening systemic inflammatory response syndrome, also known as “cytokine storm”. (D) Selected cytokine/chemokine modulation drugs that have been applied in COVID-19 clinical trials are shown in the upper right of the figure. They include neutralized antibodies (siltuximab, secukinumab, certolizumab); receptor antagonists (anakinra, sarilumab, tocilizumab), human recombinant proteins (IFN-β-1α, IFN-α-1β, IFN-α-2β); small-molecule drugs (baricitinib, remdesivir, molnupiravir, etanercept); and cell therapy (NKG2D-ACE2 CAR-NK cells). The drugs have been applied to COVID-19 patients to boost their immune systems and inhibit viral replication or cytokine storms. Figure created with BiorRender.com.

Table 1 Representative of Histopathological Changes, Cytokine and Chemokine Secretions, and Cellular Origins in SARS-CoV-2 Infection.
Interleukins (ILs)
ILs, a family of cytokines, are involved in immune cell differentiation and activation, directing them to the infection sites, enhancing acute-phase signaling, activating epithelial cells, and mediating secondary cytokine production (113). Both IL-6 and IL-1β are major proinflammatory cytokines released during viral infections (65) to induce acute-phase protein secretion by hepatocytes which activate the complement system. The complement system cascade further increases inflammatory and opsonization. IL-1β may also enhance inflammatory responses in the bronchi and alveoli of patients with lung injury (66, 114). Furthermore, higher IL-2, IL-17, and IL-8 levels have also been noted in patients with COVID-19 (67, 115). Of these cytokines, IL-6 deserves a more extensive discussion with respect to its involvement in the coronavirus-induced cytokine storm (68). However, a previous study showed that compared to the standard care, treatment with IL-6 and IL-1 inhibitors could reduce the neutralizing activity of SARS-CoV-2 antibodies at day 30, which may indicate that patients with cytokine-inhibitor treatments may be at risk for re-infection (116).
IL‐6
IL-6 is critically involved in inflammation owing to its role in regulating the acute-phase protein response (113). It is produced by nearly all stromal cells, B lymphocytes, T lymphocytes, macrophages, monocytes, DCs, mast cells, and other nonlymphocytic cells, such as fibroblasts, endothelial cells, keratinocytes, glomerular mesangial cells, and tumor cells (117). Of note, IL-1β and TNF-α increase IL-6 production (68), and in turn, IL-6 can promote T cell proliferation and resist T cell apoptosis by activating STAT3 (118). Furthermore, TGF-β, along with IL-6 and IL-21, promotes Th17 cell development by enhancing RORγt expression (119). IL-6 can also activate Th2 cytokines and dampen the production of TReg cells via the STAT3 pathway (120–124). Moreover, high IL-6 levels can activate the coagulation system and increase vascular permeability, thereby enabling the rapid spread of inflammation (69). IL-6 dysregulation has also been described to promote cell necrosis and apoptosis (125). Owing to these pleiotropic properties, IL-6 plays an important role in the pathogenesis of the cytokine storm (68).
Recent studies have suggested that the spleen and lymph nodes are infiltrated by macrophages that express ACE2 and that the nucleoprotein antigen is significantly associated with IL-6 production. This suggests that they contribute to excessive inflammation in COVID-19 (6, 70, 107, 126). Further, IL-6 produced by CD14+CD16+ monocytes was significantly higher in ICU-treated patients than in non-ICU patients (6, 127). SARS-CoV-2 can dysregulate host immune responses. For example, the number of Th17 cells could be raised by a virus-driven increase in IL-6 production (68). SARS-CoV-2 may also elevate the levels of IL-10, and TNF-α, which would negatively regulate T cells by increasing exhaustion markers such as PD-1 or Tim-3 (71, 72). Moreover, IL-6 plays a crucial role in the pathology of COVID-19 as it modulates the chemotaxis of neutrophils and lymphocyte exhaustion and necrosis (38). Patients with COVID-19 also display selective induction of the macrophages that produce IL-6, but not TNF-α and IL-1β, and this then directly promotes lymphocyte necrosis (38).
Systematic reviews and meta‐analyses have found higher serum levels of IL-6 in >50% of all patients with COVID-19 (70, 128). Moreover, patients who subsequently developed certain adverse clinical outcomes, including the cytokine storm, ICU admission, acute respiratory distress syndrome (ARDS), or death tended to have higher IL-6 levels (5, 70, 89, 129) than healthy cohorts. The IL-6 levels were elevated nearly threefold in complicated COVID-19 cases, and there were several systemic and extrapulmonary disorders not found in patients with uncomplicated disease (28, 38, 39, 67, 68, 73, 103, 130–133).
In addition to the level association, a combination of multiple cytokines, including IL-6, IL-10, and IL-8, combined into one score may also predict disease severity (74). Thus, serum IL-6 levels (>24 pg/mL) can be used at the initial assessment to predict hypoxemia that requires hospitalization. This may help in the early identification of patients who need hospitalization because it has excellent sensitivity and good specificity (134, 135).
As they do in SARS-CoV, high serum IL-6 levels correlate with shock, respiratory failure, ARDS, and multiorgan dysfunction in severe COVID-19 (58, 73, 75). IL-6 overexpression can induce other complications, including fever, vascular leakage, anemia, cardiomyopathy, acute kidney injury (AKI), interstitial edema, and myocardial dysfunction. In support of this, patients with severe COVID-19 (5) exhibited higher IL-6/IFN-γ ratio and greater lunch damage while a higher risk of hypercytokinemic immune dysregulation (or cytokine syndrome), respiratory failure, and death (76) was characterized by elevated IL-6 levels ≥80 pg/mL. When assessed in parallel, the findings could be associated with a cytokine storm favoring lung damage, than those with moderate COVID-19 (104). Taken together, blocking the IL-6 signaling pathway might inhibit excessive inflammation from SARS-CoV-2 infection (136).
To evaluate the therapeutic efficacy of IL-6 inhibition, clinical trials have tested several monoclonal antibodies, including siltuximab (IL-6 neutralization), sarilumab (IL-6 receptor antagonist), and tocilizumab (IL-6 receptor antagonist) (137). The earliest IL-6-blocking therapies first emerged in China (138), and most of the randomized trials with IL-6 receptor blockers did not have a significant effect on mortality (139–143). Tocilizumab showed no significant improvement in clinical outcome or mortality compared with placebo (140). Nevertheless, recent studies that used a combination of tocilizumab and siltuximab have shown promising results, including clinical and radiographical improvements, as reported by the Randomized, Embedded, Multifactorial Adaptive Platform Trial for Community-Acquired Pneumonia (REMAP-CAP) (144) and other studies (144–147). In the REMAP-CAP study, mortality among patients treated with tocilizumab and sarilumab was 27% compared with 36% in the control group (144). Furthermore, one of the largest initial observational studies in New Jersey has shown reduced mortality owing to tocilizumab, in particular, the mortality rate among 547 ICU patients with COVID-19 treated with tocilizumab was 46% compared with 56% among 134 individuals provided standard care (147). At present, the therapeutic efficacy of IL-6 inhibitors in patients with COVID‐19 appears promising; however, more evidence is needed to establish a definitive benefit (73).
IL-1 Family
Several well-known cytokines in the IL-1 family, including IL-1β and IL-18, have important roles in inflammation, hematopoiesis, and fibrosis (148), and SARS-CoV-2 has been shown to induce the secretion of IL-1 family cytokines, where their levels correlate with virulence (5, 130, 149). Some reports have also indicated that higher levels of IL-1 family cytokines present in the plasma of patients with COVID-19, that levels are higher in severe cases compared with mild cases, and that the levels strongly associate with the Murray score (57, 108, 130, 149, 150). Of note, gene expression analyses concur with these findings, i.e., IL-1-related proinflammatory pathways are highly upregulated in severe cases (151). Therefore, the IL-1 blockade treatment by anakinra (an anti-IL-1 receptor antagonist) may be an effective alternative option for COVID-19 patients (152).
IL-1β
IL-1β is a proinflammatory cytokine that not only responds to infection and inflammation but also plays an important role in acute and chronic autoinflammatory diseases (153). Elevated IL-1β levels have been associated with SARS, hypercoagulation, disseminated intravascular coagulation, and most severe COVID-19 cases (100, 154). Clinical and laboratory parameters indicated that SARS-CoV-2 appears to promote both the activation and maturation of IL-1β and this, in turn, activates other proinflammatory cytokines, including IL-6 and TNF-α. Further, IL-1β dysfunction drives Th1 cell activation (12, 68), macrophage activation syndrome (8, 28, 67, 89, 90), fever (77), ARDS (8, 77, 78), and cytokine storm (100, 155). Using single-cell transcriptomic (156, 157) and flow cytometric analyses (90, 158), some studies have identified that peripheral blood mononuclear cells (PBMCs) from patients with COVID-19 contained IL-1β-associated inflammasome signatures (159).
IL-18
IL-18 is a proinflammatory cytokine produced by macrophages during the very early stages of a viral infection (160). It induces the production of IL-6 and IFN-γ, which are considered important for optimal viral host defense (160, 161). It also drives Th2 cytokine production from CD4+ T cells and NK cells and regulates both Th1 and Th2 responses (93). Additionally, IL-18, along with other cytokines (CCL2, CXCL10, CCL20, IL-18, IL-3, IL-6, G-CSF, GM-CSF, IFN-γ), is suggested to be involved in regulating neutrophil function (162). The activated neutrophils contributed to the formation of neutrophil extracellular traps (163) and increased neutrophil-induced inflammatory response (162). In addition, aberrant production of IL-18 can cause severe tissue injury (160). Patients with severe COVID-19 had higher IL-18 serum concentrations than those with mild symptoms (94, 160), with no difference in IL-18 concentrations between healthy subjects and asymptomatic COVID-19-infected individuals (160). As serum IL-18 concentrations consistently rise with an increase in the severity of pneumonia, significantly higher concentrations were observed in patients who developed macrophage activation syndrome, ARDS (164), or liver damage (77). Further, serum IL-18 levels were higher in males than in females (94, 164). Thus, the serum IL-18 concentration may be used to identify patients who might subsequently need hospitalization (160).
IL-10
IL-10 functions as an anti-inflammatory cytokine, and it is a crucial feedback regulator that exerts immunosuppressive effects on both innate and adaptive inflammation, autoimmune pathologies, and Th1 cell activity (79, 95, 165–173). It is secreted by multiple cell types, including Th1, Th2, Th17 cell subsets, TReg cells, CD8+ T cells, B cells (167, 174–178), mast cells, eosinophils, macrophages, and DCs (179–181). IL-10 is dramatically elevated in COVID-19, and this was believed to be a negative feedback mechanism to suppress inflammation and to induce ARDS (96), acute-phase protein response (77, 97), severe pneumonia, and damage to vital organs (33, 71, 107, 182, 183). Studies have reported higher IL-10 levels in ICU patients than in non-ICU patients (33, 71, 95). In addition, the increased number of IL-10-producing regulatory T cells could be correlated with long-term viral persistence (184). Hence, several lines of clinical evidence suggest that such dramatic and early elevation of IL-10 might exacerbate pathogenesis in determining COVID-19 severity (95). Moreover, the increased IL-10 expression is considered an indicator of poor prognosis in COVID-19 (97, 183). In contrast, other recent studies have demonstrated that IL-10 may directly expand cytotoxic effector CD8+ T cells and contribute to the hyperactivation of adaptive immunity in patients with COVID-19 (95). This observation lent credibility to the idea that both anti-inflammatory and immune-activating roles of IL-10 may occur simultaneously in COVID-19 (68, 95, 185).
IL-22
IL-22 belongs to the IL-10 cytokine family and is produced by γδ T cells, Th1 cells, Th17 cells, Th22 cells, NKT cells, and group 3 innate lymphoid cells (ILC3) (186–188). In general, IL-22 is a pro-and anti-inflammatory cytokine involved in tissue inflammation, immunosurveillance, tissue repair, and homeostasis through the IL-22R activation pathway (186, 189–195). In patients with SARS-CoV-2 infection, IL-22 plasma levels were correlated with disease severity (12, 196), and they were positively correlated with the Th17 population. The increased Th17 proportions and IL-22 were observed in cases with headache, hyperinflammation, respiratory system dysfunction, and ARDS (163, 197). Other studies indicated that IL-22 levels are negatively correlated with the Th22 levels (188, 198). Functional assays revealed that both peripheral blood mononuclear cells (PBMCs) and a human bronchial epithelial cell line (16HBE) can produce IL-22 (188). This suggests that IL-22 could be expressed differently in peripheral blood and tissues.
IL-17
IL-17 is produced by Th17 lymphocytes and other cells, including CD8+ cells and γδT cells, NK cells, and ILC3 (199). IL-17 acts as a neutrophilic inflammation-promoting cytokine (79) whose levels increase during inflammatory processes and autoimmune diseases (200–203). It also plays a role in tissue damage, physiological stress, and infection (100). IL-17, along with IL-22 and TNF-α, induces the production of antimicrobial peptides in the gastrointestinal tract and skin (204, 205). In addition, because both IL-17 and IL-22 originate in Th17 cells, the dysregulation of the above cytokines could contribute to several proinflammatory diseases (206). Therefore, applying Th17-inhibiting therapies such as thiamine to reduce the Th17 mediated pro-inflammatory response (101) may be an effective treatment for decreasing cytokine storms (207). In one study, 16 patients a proinflammatory state from alcohol use disorder were treated with thiamine, and it significantly reduced IL-17 levels and ameliorated the Th17 responses (207).
Upon SARS-CoV-2 infection, IL-17 levels, along with other Th17 related proinflammatory cytokines, such as IL-1, IL-6, IL-15, TNF, and IFN-γ, are increased, which has a positive correlation with disease severity (101, 102, 208). Previous reports indicating the elevation of IL-17 levels in patients with SARS-CoV or MERS (209, 210) as well as elevated IL-17 levels in SARS-CoV-2-induced cytokine storm (33), ALI (102), ARDS (37), viral load have been reported (77, 211). A Janus kinase 2 inhibitor, Fedratinib, has been used to decrease IL-17 expression by inhibiting Th17 cells in murine models (212). By contrast, a recent study illustrated that IL-17 levels were not significantly different between uninfected individuals and patients with COVID-19 with severe versus mild symptoms (115). In addition, secukinumab, a human anti-IL-17 neutralizing monoclonal antibody, also showed no significant difference in adverse effects in COVID-19 patients (43).
IL-33
IL-33 is an alarming and crucial immune modulator released by endothelial and epithelial cells, activated fibroblasts, fibroblast-like cells, and myofibroblasts to maintain the tissue and immune homeostasis and inflammation through IL-33/ST2 signaling (211, 213). IL-33 plays important roles during allergic, fibrotic, infectious, and chronic inflammatory diseases by activating the different immune subgroups, such as Th1, Th2, TReg, CD8+ T cells, mast cells, group 2 innate lymphoid cells (ILC2s), granulocytes, macrophages, DCs, NK cells, iNKT cells, and B cells (209, 210, 213, 214).
In COVID-19 patients, increased IL-33 serum levels were associated with poor outcomes (209, 215–217). IL-33 activation could induce a type-2 immune response by inducing ILC2s (217). These might help differentiate pathogenic γδ T cells (216), thus contributing to pulmonary fibrosis induced by viral infection (216, 218). In addition, transcriptomic analysis of bronchoalveolar lavage fluid (BALF) from COVID-19 patients showed an increased population of IL-33-producing cells with the disease severity (219) and strong upregulation of IL-33 compared to healthy samples (220).
Interferons (IFNs)
IFNs encompass a family of cytokines that play a central role in providing efficient protection against viral infections by activating the antiviral or immunomodulatory properties (221, 222). Human IFNs have been classified into three major types based on signal receptors: IFN type I (IFN-I; IFN-α and IFN-β), IFN type II (IFN-γ), and IFN type III (IFN-λ1) (223). Type I IFNs response can be rapidly triggered when host cells recognize PAMPs such as viral nucleic acids and have been known to combat viruses by inducing the expression of IFN-stimulated genes (ISGs) in epithelial cells, which exert antiviral functions, inhibit viral replication, and indirectly stimulate both innate and adaptive immune responses (3, 68, 224, 225).
While IFN-β-1α and IFN-β recombinant protein have shown great potential in inhibiting SARS-CoV-2 replication in vitro (226). Paralleling the potential, IFN-λ 1/3 has also been induced in early phases of infection and in convalescent COVID-19 patients (227). The results indicated that IFN-β and IFN-λ1 could block virus infection and inhibit the production of SARS-CoV-2 (226, 228). In addition, the dysregulated production of type I IFNs and the exacerbated release of proinflammatory cytokines makes COVID-19 more severe (226, 229). At the same time, the reduced type II IFN was correlated with disease severity in vitro (230), and the IFN-γ plasma levels of COVID-19 ICU patients have been significantly reduced compared to other cohorts (227).
It has been noted that type I IFN, especially IFN-β, can be effective against SARS-CoV-2 to tackle severe COVID-19 and prevent clinical deterioration. Analogously, SARS-CoV-2 may also be sensitive to IFN (231, 232). To evaluate the therapeutic efficacy of IFN-β, several studies in REMAP-CAP and the WHO’s Solidarity Trial administered IFN-β recombinant protein as a potential therapy against COVID-19 (45, 231, 232). A combination of IFN-β-1α, lopinavir/ritonavir, and ribavirin could eliminate the virus from the nasopharyngeal swabs in a phase II clinical trial (45, 233). Another study has shown IFN-β-1α combined with lopinavir/ritonavir or atazanavir/ritonavir and hydroxychloroquine significantly decreased mortality on day 28 in a cohort of 42 severe COVID-19 cases (234). In addition to the combination, pegylated interferon alfa-2β (PEG IFN-α-2β) along with the standard of care (SOC) enabled a faster viral reduction in patients with moderate COVID-19 than with standard care alone (235). The pegylated interferon lambda (PEG IFN-λ) treatment particularly in patients with a high baseline viral load (80), accelerated viral decline and increased the proportion of patients with viral clearance by day 7 has shown a similar role as PEG IFN-α-2β.
IFN-I
IFN-I can be produced by many cell types, including DCs, lymphocytes, macrophages, fibroblasts, endothelial cells, and osteoblasts (236–238). IFN-I can further stimulate both macrophages and NK cells via IRF3/IRF7 antiviral signaling to counter viral infections, and in response to PAMPs, plasmacytoid DCs have been identified as the most potent and natural type I IFN-producing cells (229). Nevertheless, viruses can counteract IFNs by harnessing both structural and nonstructural proteins and can also efficiently suppress IFN induction (239). Of note, such inefficient IFN response may account for progressive virus replication, cytokine storm development, and death in SARS (240).
Many viruses, including SARS-CoV-2, have evolved mechanisms to evade the antiviral effects of IFN, and patients with COVID-19 tend to have suppressed responses to IFN (type I, II, or III) along with impaired monocytes, macrophages, and neutrophils (35, 72, 80). Compared with other respiratory RNA viruses, SARS-CoV-2 is also a poor inducer of IFN-I, both in vitro and in animal models (35, 241). Patients without IFN-α production had poorer outcomes and a higher viral load. Thus, screening patients for IFN production after COVID-19 diagnosis could be crucial in selecting those who could benefit from early intervention with IFN treatment (242). Recent studies suggest that an impaired response of IFN-I in the early stage of the disease may lead to an acute phase and play a major role in creating the cytokine storm (243). Adding to the study, a recent working hypothesis has suggested that the timing of IFN response to SARS-CoV-2 infection varies based on viral load and genetic differences (244) with low viral loads characterized by an IFN response suitable for viral clearance, resulting in mild infection. In contrast, a high viral load can delay IFN response and consequently lead to severe disease (245). Studies have suggested that patients with mild or moderate COVID-19 exhibit greater type I IFN response during days 8-12 compared with patients with severe disease (4, 8). Moreover, some infected patients expressed a low level of IFN-I, but ISGs expression was enhanced in patients’ BALF (240). This suggests that even limited IFN-I production is sufficient to express relevant genes (72, 246).
Despite early reports to the contrary, increasing evidence has highlighted a role for IFN-I responses in severe COVID-19 development (94). A robust type I IFN response has been reported in patients with severe COVID-19, which through diverse mechanisms, exacerbated hyperinflammation (94, 247). Further, a recent longitudinal analysis illustrated that IFN-α levels in peripheral blood remained high in patients with severe COVID-19 and that classical monocytes exhibit both IFN-I and TNF/IL-1β-driven inflammatory responses (94), and approximately 10% of patients with severe COVID-19 had neutralizing immunoglobulin (Ig)G auto-antibodies against type I IFNs (248, 249); hence these patients required plasma exchange (250). Of note, contradictory results among multiple studies on IFN-I responses in patients with COVID-19 might be explained by differences in the definition of disease severity, sampling time points, and/or readout type (for example, IFN-I itself or cellular responses to IFN-I) (247).
TNF-α
TNF-α is a pyrogenic cytokine secreted by macrophages, monocytes, Th1 cells, Th17 cells, CD8+ T cells, and DCs during the acute phase of inflammation or infection (68, 77, 79). It is a central cytokine in viral diseases that increases vascular permeability (251) and is associated with several chronic inflammatory conditions and autoimmune diseases (68). For >20 years now, anti-TNF antibodies have been used for alleviating the severity of some autoimmune inflammatory diseases, including rheumatoid arthritis, inflammatory bowel disease, and ankylosing spondylitis. Further, the United States Food and Drug Administration has approved four off-label indications for anti-TNF therapy, implying that TNF is a valid target in multiple inflammatory diseases (244, 252).
COVID-19 is characterized by unique hyperinflammatory signatures across all types of immune cells, particularly the upregulation of TNF-α-, IL-6-, and IL-1-driven inflammatory responses in severe disease (35). TNF-α presents in the blood and lungs of patients with COVID-19 (244), and in severe COVID-19 cases, high systemic TNF-α levels were associated with respiratory distress syndrome (98, 245, 253, 254), and lower survival along with pulmonary dysfunction [edema, proteinaceous exudates, pneumocyte desquamation, and ARDS (66)] as well as impaired hematopoietic function, disseminated intravascular coagulation, debilitating hyperlipidemia, liver damage, chronic kidney disease, diabetes, and hypertension (8, 33, 77, 135, 255). Thus, high TNF-α levels can be an independent predictor of patient survival (135).
As TNF-α overproduction has been documented in COVID-19, the efficacy and clinical benefits of anti-TNF antibody therapy have been investigated (244). One clinical trial showed that etanercept, a TNF-α inhibitor, can attenuate disease in patients with severe COVID-19 by suppressing their systemic autoinflammatory responses (256), and patients with COVID-19 previously prescribed etanercept treatment did not develop severe COVID-19 (257). Likewise, another anti-TNF-α antibody, certolizumab, may have beneficial effects in patients with COVID-19 (100, 258). A recent study showed that inhibiting both TNF-α and IFN-γ in multiple cytokine storm models protected against death in SARS-CoV-2 infection and other inflammatory syndromes, such as sepsis, hemophagocytic lymphohistiocytosis, and cytokine shock (259). It is still not clear whether a single cytokine blockade (such as TNF-α inhibitors) could be effective in CRS associated with COVID-19. Nevertheless, some results show that anti-TNF therapy should be explored on hospital admission as it may prevent the need for intensive care (244).
Histopathological changes and cytokine and chemokine secretions along with cellular origins in SARS-CoV-2 infection, which are described in the preceding and following sections, are summarized in Table 1.
Chemokines Induced by SARS-CoV-2 Infection
Chemokines, a family of small cytokines, are the crucial mediators of appropriate immune responses (260). However, their excessive release is the primary cause of hyperinflammation, which may be a direct cause of ARDS (261). The exaggerated chemokine response is critical in several viral diseases, such as SARS, MERS, influenza, and SARS-CoV-2 (262). Further, several clinical investigations have revealed that chemokines such as IL-8, CCL2, CCL3, CCL7, CCL8, CXCL2, CXCL16, and CX3CL1 are infiltration signals that mediate the recruitment of mononuclear phagocytes to lungs (6, 240, 261, 263), and they are directly involved in the pathogenesis of severe clinical sequelae in COVID-19, including major complications that cause death in approximately 40% of severe COVID-19 cases (66, 68, 77, 261, 264). We review the role of chemokines in COVID-19 pathogenesis to improve the understanding of the disease immunopathology, which may further aid in developing possible therapeutic targets for COVID-19 (261). Chemokine secretions and their cellular origins in SARS-CoV-2 infection are summarized in Figure 1, Table 1, and described in the following sections.
CXCL10/IP-10
CXCL10, also known as IP-10 or small-inducible cytokine B10, belongs to the C-X-C chemokine family. It binds to the CXCR3 receptor and acts as a chemoattractant for immune cells. That enhances chemotaxis, apoptosis, cell growth, angiostasis, and recruitment of macrophages, Th1 cells, and NK cells (265). Alterations in CXCL10 expression have been associated with inflammatory diseases (including infections), immune dysfunction, and the development of tumors. Thus, CXCL10 is a recognized biomarker of varying progression and severity of disease (265), so it might be used to detect early stages of SARS (68). In COVID-19 cases, levels of CXCL10 serum are highly associated with disease severity, viral load (68, 108), and the Murray score (71, 108). CXCL10 has been found in the BALF from patients with COVID-19 who had higher proinflammatory genes (220). In addition, higher circulation levels of CXCL10 can activate Th1 cell function, and it has been shown to be highly correlated with COVID-19-induced ARDS in clinical and experimental studies (266). Therefore, CXCL10 may also be considered a prognostic and potential therapeutic marker for COVID-19 progression (108).
IL-8/CXCL8
IL-8 or CXCL8 is produced by macrophages and other cell types, such as epithelial cells, airway smooth muscle cells, and endothelial cells (135, 267, 268). Elevated serum IL-8 levels have been observed in many diseases, including severe lung injury, and in patients who developed adverse outcomes in SARS-CoV and MERS-CoV infections (269–272). Upon SARS-CoV-2 infection, increased IL-8 concentrations are associated with ARDS (77) and COVID-19 severity (87, 135, 255, 268, 273–275). Irrespective of demographics or comorbidities (106, 135, 276, 277), lower IL-8 was associated with a shorter duration of illness and in convalescent, recovered, and asymptomatic (276, 278). As a neutrophil chemotactic factor, IL-8 recruits neutrophils to the infection site and activates them (135, 279). Therefore, neutrophilic infiltration, neutrophil-lymphocyte ratio, and increased IL-8 expression are considered realistic prognostic biomarkers of COVID-19 progression and severity (10, 30, 135, 280, 281). However, the correlation between IL-8 levels and disease severity remains controversial (135, 273–275, 281, 282) as the positive correlation is shown only in univariate and not in multivariate analysis (88). Interestingly, the correlation between sex and IL-8 is also unclear in that while some studies have documented no significant differences in serum IL-8 levels (135), others have reported lower IL-8 expression in female SARS-CoV-2 patients than in male patients (275).
GM-CSF
GM-CSF, a proinflammatory cytokine and chemokine also known as CSF2, is a member of the CSF superfamily (112). This monomeric glycoprotein has several cellular origins, including macrophages, T cells, mast cells, NK cells, endothelial cells, and fibroblasts (4, 9, 36). GM-CSF was initially classified as a hematopoietic growth factor, yet it is now believed to play an essential role in communicating between tissue-invading lymphocytes and myeloid cells by stimulating the secretion of proinflammatory cytokines and chemokines (such as IL-1, IL-6, and TNF) (112). Compared with healthy controls, the percentage of GM-CSF-secreting immune cells, such as CD4+ T cells, CD8+ T cells, NK cells, and B cells, was significantly higher in patients with COVID-19 (110, 111). Increased GM-CSF can induce greater acute-phase protein expression (77) and activate Th17 responses (98) to contribute to COVID-19 pathogenesis (33). In addition, to the COVID-19 patients, particularly those admitted in ICU, pathogenic Th1 cells increased rapidly along with GM-CSF and IL-6 secretion (49, 111, 283). Such a proinflammatory environment can further trigger CD14+CD16+ monocytes to secrete more GM-CSF and IL-6. These aberrant and numerous GM-CSF+-IL-6+ cells may enter the lungs and create a hyperinflammatory environment, thereby worsening the cytokine storm in patients with COVID-19 (10, 49, 111). Thus IL-6 and GM-CSF are also considered markers of poor prognosis in the later stages of COVID-19, and these proinflammatory mediators can predispose patients to respiratory failure and eventually ARDS (49).
In current COVID-19 clinical trials, the administration and inhibition of GM-CSF are being therapeutically tested. The inhibition of GM-CSF signaling may function as a potential therapeutic target in patients with COVID-19-associated hyper inflammation and ARDS (49, 284). At present, there are multiple trials with anti-GM-CSF drugs (e.g., Otilimab, Lenzilumab, Namilumab, Gimsilumab, and TJ003234) or GM-CSF receptor antagonists (e.g., Mavrilimumab) (110, 276). On the other hand, the FDA-approved recombinant GM-CSF, Sargramostim (Leukine®), is currently being investigated in a phase IV trial as an adjuvant therapy to restore alveolar anti-inflammatory macrophages to manage acute hypoxic respiratory failure and ARDS (NCT04326920) (57, 110, 276).
CCL2/MCP-1
CCL2, also known as monocyte chemoattractant protein-1 (MCP-1), is produced by many cell types, including endothelial cells, fibroblasts, epithelial cells, smooth muscle cells, mesangial cells, astrocytes, monocytes, and microglia (277, 278, 285–287), serving as a regulator of monocyte/macrophage migration and infiltration. The migration of monocytes through the bloodstream to the vascular endothelium plays a key role in the response to inflammation (286, 288, 289). Both CCL2 and its receptor, CCR2, are known to be activated and involved in several types of inflammatory diseases, such as Alzheimer’s disease, parkinsonism, stroke, ischemic heart disease, arthritis, and COVID-19 (286, 289). With COVID-19, the increased CCL2 is not only associated with respiratory failure but also with extrapulmonary manifestations. The increased CCL2, along with IL-1, IL-6, TNF-α, MMP-8, and ICAM-1, can increase the permeability of the barrier between blood and cerebrospinal fluid, increasing the inflammatory infiltration (290–292). In combination with other inflammatory cytokines, elevated CCL2 can also increase the severity of neurodegeneration, cognitive dysfunction, and stroke (293). Moreover, increased CCL2 levels were reported to be correlated with the development of acute kidney injury in critically ill COVID-19 patients (294), and patients with higher CCL2 expression tend to have detrimental disease progression.
CX3CL1
CX3CL1, also known as fractalkine (FKN), is highly expressed by activated endothelial cells and is known to be involved in endothelial dysfunction, contributing to the development of atherosclerosis and other cardiovascular events (295–297). Additionally, CX3CL1 serves as an infiltration signal that mediates mononuclear phagocyte recruitment to the lungs (261). In COVID-19 patients, increased CX3CL1 is associated with disease severity (298), and patients with neurological syndrome (NS) presented with higher CX3CL1 levels than non-NS patients (299). Studies have shown that CX3CL1 might promote neurological vascular damage and thrombosis during SARS-CoV-2 infection. Spike protein-mediated endocytosis reduces AEC2 in the cell membrane, upregulating the expression of CX3CL1 in the endothelium (300). Increased CX3CL1 can promote a pro-thrombotic environment and enhance immune cell recruitment, leading to more severe COVID-19 and mortality (301). Thus, CX3CL1 may serve as a predictive marker for identifying COVID-19 patients who are at risk of developing thrombotic complications and require more aggressive anti-thrombotic management (301).
Clinical Symptoms
The incubation period of COVID-19 ranges between 1 and 14 days. However, the period is predominantly 3-7 days (302–304). Immune response to SARS-CoV-2 involves both innate and adaptive immunity (3), and although differentiated lymphocytes can take days or weeks to become functional, they have an important role in controlling and shaping host immune responses as they provide multiple immune-related functions and long-lasting protection. In particular, while T cells can either kill an infected cell via cytotoxic CD8+ T cells or balance immune response with the help of CD4+ helper T cells, B cells produce antibodies against pathogenic antigens, which is also known as humoral immunity (305). Both innate and adaptive immune cells are critical for eliminating SARS-CoV-2 infection as they orchestrate the direct clearance and eradication of pathogens and contribute to the generation of long-term adaptive immune responses (10, 80, 306). The hyperactivity of the immune system stimulates the production of several cytokines, including IL-6, IL-8, IL-1β, IL-2, IL-4, IL-7, IL-10, IFN-γ, TNF-α, GM-CSF, CCL2, CCL3, CCL5, and CXCL10 (100) (as described in the previous sections). Cytokine- or chemokine-specific clinical symptoms are as follows: elevated IFN-γ causes fever, headaches, chills, fatigue, malaise, cardiomyopathy, vascular leakage, lung injury, and acute-phase protein production; increased TNF-α causes flu-like symptoms (69, 106); and IL-6 overexpression induces cardiomyopathy and vascular leakage, and it activates the complement pathway, coagulation cascade, diffuse intravascular coagulation, and the cytokine storm (69, 82, 84, 85).
COVID-19 initially presents with “flu”-like symptoms include fever, dry cough, myalgia, fatigue, dyspnea, and anorexia. Atypical presentations are diarrhea and nausea, which can progress to life-threatening systemic inflammation like the CRS (33, 303, 304, 307, 308), which can cause multiple lung immunopathologies, including acute lung injury (ALI), systemic inflammatory response syndrome (SIRS), and ARDS (65, 129). Further, accumulating evidence demonstrates that COVID-19 has an extrapulmonary involvement; this includes the neurological, olfactory, cardiovascular, digestive, hepatobiliary, renal, endocrinological, and dermatological systems, with more than one-third of the patients exhibiting a wide range of neurological symptoms involving the central and/or peripheral nervous systems (10, 308–311). Moreover, in patients with severe COVID-19, cardiovascular complications are accompanied by the accumulation of inflammatory mononuclear cells (e.g., neutrophils) in infected vascular endothelial cells (308). Severe cases also show impaired hepatic function and a higher rate of acute kidney injury (AKI). In patients with prolonged illness, gastrointestinal symptoms also frequently occur. Endocrinological manifestations include exacerbating hyperglycemia, euglycemic ketosis, and diabetic ketoacidosis (312), and thrombosis and visceral embolization can trigger tissue hypoxia and ischemia in patients with COVID-19 (5, 313). Changes in SARS-CoV-2 infection, including pulmonary and extrapulmonary manifestations, are described in the following sections and summarized in Figure 2.
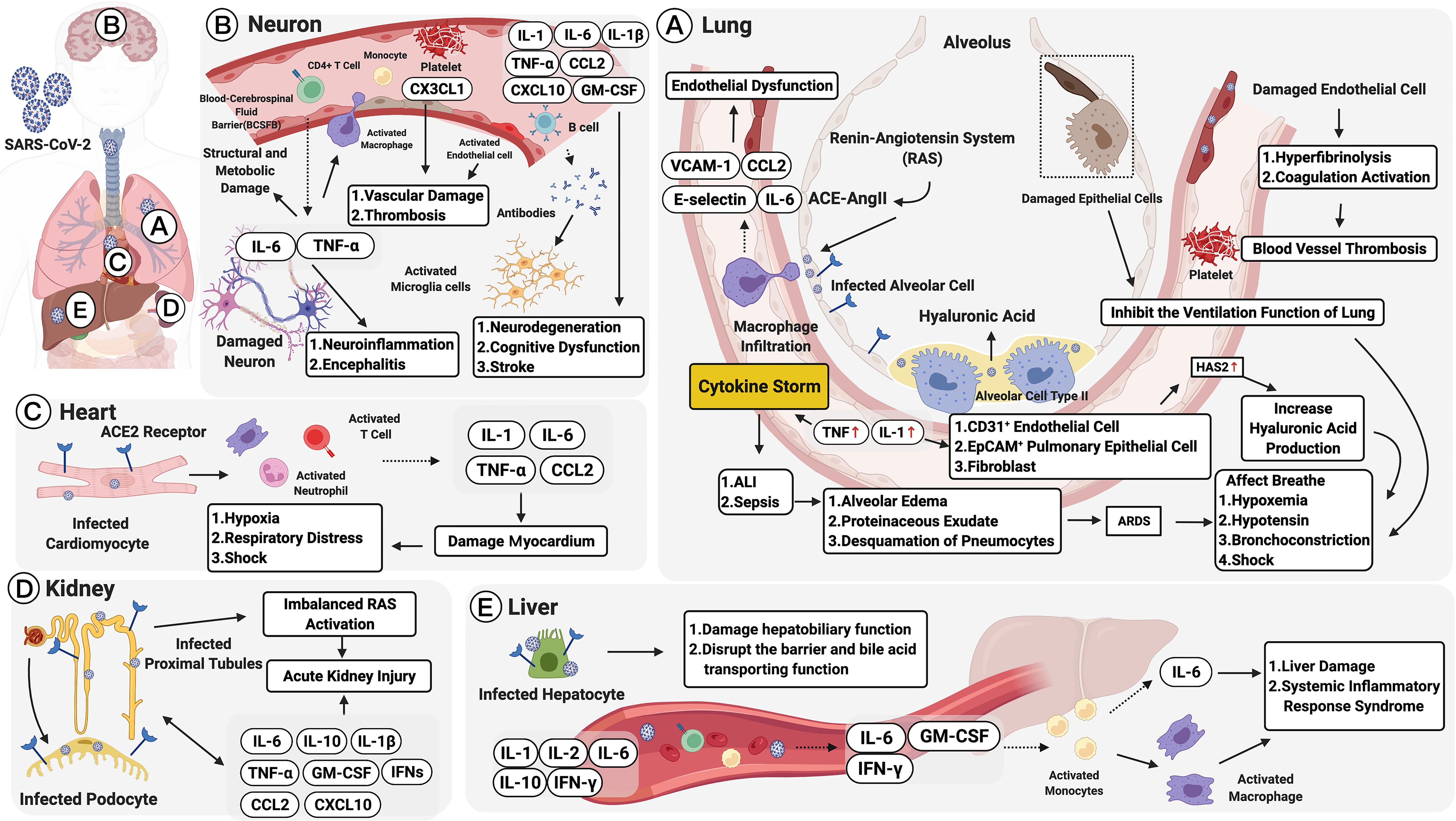
Figure 2 Multiple organ failure in SARS-CoV-2: pulmonary and extrapulmonary manifestations. Pulmonary manifestations: (A) Alveolar cells are infected by the angiotensin-converting enzyme 2 (ACE2) receptor. This leads to acute lung injury (ALI), activation of the renin-angiotensin system (RAS), and macrophage recruitment. Macrophages, activated by the ACE2-Ang II axis, can secrete IL-6, CCL2, VCAM-1, and E-selectin leading to endothelial dysfunction. Damaged endothelial cells may cause hyperfibrinolysis, activate coagulation, and increase inflammatory cytokine levels (IL-1 and TNF), which are strong inducers of hyaluronic acid-synthase-2 (HAS2) in CD31+ endothelium, EpCAM+ lung alveolar epithelial cells, and fibroblasts. The increased hyaluronic acid may cause bronchoconstriction. Extrapulmonary manifestations: (B) In neurons, elevated TNF-α and IL-6 secretions could increase the infiltration of immune cells and the permeability of the blood and cerebrospinal fluid barrier (BCSFB). CX3CL1 could also promote neurological vascular damage and thrombosis. IL-6 along with GM-CSF, IL-1β, TNF-α, CCL2, and CXCL10, could increase the severity of neurodegeneration, cognitive dysfunction, and stroke. (C) In the heart, infected cardiomyocytes would induce elevated secretions of IL-1, IL-6, TNF-α, and CCL2, damaging the myocardium and developing into hypoxia, respiratory distress, and even shock. (D) In the kidney, SARS-CoV-2 infects ACE2 expressing proximal tubules and the podocytes. The infected podocytes promote an imbalanced RAS, which leads to acute kidney injury (AKI). Moreover, IL-6, IL-1β, IL-10, GM-CSF, IFNs, CXCL10, CCL2 and TNF-α promote AKI persistence. (E) In the liver, infected hepatocytes trigger elevated serum IL-1, IL-2, IL-6, IL-10, and IFNγ, as well as pathogenic cytokine secretions mediated by T cells (GM-CSF, IL-6, and IFN-γ). The immune-mediated injury would lead to liver dysfunction and systemic inflammatory response syndrome (SIRS). Figure created with BioRender.com.
Cytokine Storm Syndrome
The terms “cytokine storm” and “CRS” refer to the overproduction of inflammatory cytokines by hyperactivated immune cells. This occurs because of immune dysregulation of varying etiology, and it can cause systemic inflammatory syndromes and life-threatening multiorgan dysfunction (79). During a cytokine storm, elevated levels of circulatory cytokine drive a positive feedback loop in immune cells. This leads to the continuous recruitment of these cells to the sites of inflammation, causing an exponential increase in both inflammation and organ damage (66).
Cytokine storms are associated with various infectious and noninfectious diseases (65), including cancer, autoimmune conditions, and monogenic disorders (79). Despite the variable initial drivers of a cytokine storm, its late-stage clinical manifestations converge and often overlap. As a result, patients exhibit hyperinflammation, coagulopathy, low platelet counts, fatigue, anorexia, headache, rash, diarrhea, arthralgia, myalgia, spontaneous hemorrhage, and neuropsychiatric symptoms. These manifestations are directly caused by cytokine-induced tissue damage, acute-phase physiological changes, or immune cell-mediated responses (79). In addition, many patients develop respiratory symptoms (cough and tachypnea), which can progress to acute respiratory distress syndrome (ARDS) along with hypoxemia that may require mechanical ventilation (314). Fever, renal failure, acute liver injury or cholestasis, and occasionally, stress-related or Takotsubo-like cardiomyopathy are the characteristic manifestations of a severe cytokine storm (315).
In COVID-19, the virus stimulates various infected cells, including lung epithelial cells and alveolar macrophages, to release cytokines and chemokines. The primary cytokines involved in cytokine storms and CRS are ILs, IFNs, TNFs, CSFs, and the chemokine family, among others (100). These, in turn, activate macrophages, DCs, and other immune cells (66) to induce an aggressive inflammatory response and the subsequent release of a large number of proinflammatory cytokines to trigger a cytokine storm (28, 33, 39, 316–318). After which, chemokines recruit additional inflammatory cells, such as monocytes and phagocytes, to the inflammation site resulting in a cascading amplification of infection-induced inflammatory response (66). The abnormal and uncontrolled cytokine storm has been observed in critically ill or severe COVID-19 cases (68), and it triggers a systemic inflammatory response, leading to lung immunopathology and multiple organ failure (65, 66, 129, 319). In fact, the SARS-CoV-2-induced cytokine storm is believed to be the cause of sepsis in 28% of all fatal COVID-19 cases (320). Thus, targeting cytokines could improve survival rates and reduce mortality (11). Any potential treatments should include the direct elimination of coronaviruses, prevention strategies (e.g., vaccine development), and counteracting the imbalance and hyperactivity of the immune system.
Pulmonary Manifestations
Histopathological changes in COVID-19 occur primarily in the lungs (304). Upon first contact with the respiratory mucosa, the virus binds to the surface receptors of ACE2 and TMPRSS2 to enter the alveolar cell (14); this event activates the renin-angiotensin system (20, 21). In the first few days after infection, innate immune cells [including macrophages, DCs, and monocytes (Figure 2)] recognize viral pathogen-associated molecular patterns (PAMPs) via PRRs (10, 15). The activated ACE2-Ang II axis can trigger macrophage infiltration and induce the secretion of several cytokines, including IL-6, CCL2, VCAM-1, and E-selectin, which induce endothelial dysfunction and, subsequently, coagulation (20, 22–24, 65). Therefore, an increase in plasma Ang II levels is considered a marker that correlates with viral load and lung injury in COVID-19 cases (10, 66). IL-1β can enhance inflammatory responses in the bronchi and alveoli in patients with lung injury, and the levels of inflammatory cytokines such as IL-1 and TNF are elevated in the lungs of patients with COVID-19. The increased levels are believed to represent a significant induction of hyaluronic acid-synthase-2 (HAS2) in the CD31+ endothelium, EpCAM+ alveolar epithelial cells, and fibroblasts, which may lead to bronchoconstriction (321).
An unbalanced immune response and elevated proinflammatory cytokines contribute to the major complications of pulmonary coronavirus infection, including sepsis, ARDS, acute lung injury (ALI) (66, 71), SIRS (65), and PF (322, 323); these complications result in respiratory failure and subsequent mortality (324). Nearly 15% of all COVID-19 patients and pneumonia develop ARDS, a common consequence of the cytokine storm in the lung tissue and in systemic circulation (325). In such patients, predominant pulmonary pathologies in the lung tissue, which include low blood oxygen levels, diffuse alveolar damage, alveolar edema, and proteinaceous exudation, alveolar wall thickening, evident pneumocyte desquamation, and hyaline membrane formation (114, 311, 326), provide indicators for ARDS. Damaged alveolar epithelial cells and endothelial cells, and extensive phlegm secretion and exudation can trigger coagulation, hyperfibrinolysis, and small blood vessel thrombosis. They can also inhibit lung ventilation significantly (66), thereby increasing hypoxemia, hypotension and shock, promoting pulmonary embolism, and further increasing the severity of the disease (83, 327–330). Hence, ARDS-induced low oxygen saturation and respiratory failure are major causes of mortality in COVID-19 cases; they are the cause of death in 70% of all fatal COVID-19 cases (328, 330).
The factors that drive severe lung pathology during infection with highly pathogenic human coronaviruses are poorly understood. Computed tomography images revealed that COVID-19-infected lungs accumulate fluid-filled white patches called “ground-glass opacities” (303); however, the nature of the clear jelly remains to be determined. Potential mechanisms include high rates of viral replication that could be responsible for enhanced host cell cytolysis and the strong production of inflammatory cytokines and chemokines by infected epithelial cells and the delayed induction of antiviral IFN responses owing to virus escape mechanisms such as the production of IFN inhibitory proteins that perpetuate viral damage and lead to the excessive accumulation of monocytes, macrophages, and neutrophils (6, 302).
Susceptibility to SARS-CoV-2 infection is not age-dependent (median age at infection is approximately 50 years). However, evidence suggests that clinical severity differs with age (33, 71, 328, 331, 332). In general, compared with young people, children are either asymptomatic or they suffer only from mild pneumonia, whereas older men (>60 years old) with comorbidities are more likely to require intensive treatment, develop severe respiratory disease, or even die (28, 304, 331, 332). Moreover, men with COVID-19 tend to exhibit more severe morbidity and mortality than women (321, 333), and meta-analyses have shown that men with COVID-19 have threefold higher odds of requiring hospitalization and 2.4-fold higher mortality rate (321, 334).
Extrapulmonary Manifestations
Neurological Manifestations
Coronaviruses primarily target the human respiratory system. However, COVID-19 infection has also been reported to trigger neurological manifestations and has neuroinvasive capabilities that permit its spread from the respiratory tract via the olfactory bulb, causing inflammation and demyelination in the central nervous system (CNS) (335, 336). Owing to the ACE2 receptors in olfactory cilial cells, the virus could potentially reach the CSF within seven days (325), while additional studies suggest that the virus could enhance inflammatory cytokines, including TNF-α and IL-6, leading to structural and metabolic damage in the CNS. Moreover, the levels of IL-8, CCL2, CCL3, CCL4, CCL7, CCL12, and CX3CL1 were higher in both the serum and CSF samples of COVID-19 cases with neurological syndrome (NS) (299).
More recent studies demonstrate that the epithelial cells of the blood-cerebrospinal fluid barrier (BCSFB) in the choroid plexus of the ventricles can act as a conduit for SARS-CoV-2 into the CNS (337, 338), leading to inflammation and disruption of the BCSFB integrity (339). The vulnerability to the CNS can result in exposure to complications such as neuroinflammation and encephalitis (340). The inflammation further upregulates the expression of CCL2, IL-1, IL-6, TNF-α, MMP-8, and ICAM-1, increasing the infiltration of inflammatory immune cells, such as monocytes, and enhancing the permeability of the BCSFB (290–292). In addition, CD4+ cells secreting cytokine IL-6, along with mast cells secreting cytokines and chemokines (including IL-1β, TNF-α, CCL2, CXCL10, and GM-CSF) accelerate neurodegeneration, cognitive dysfunction, and stroke (293, 326). As a result, approximately 36% of all patients with COVID-19 have neurological manifestations (312).
Neurological symptoms, involving the central/peripheral nervous system, have been predominantly reported in adults; however, children also experience various neurological insults owing to COVID-19 (341). For example, 28% of pediatric patients in the United States experienced headaches (342), and some developed febrile seizures (335, 343). In adults, neurological manifestations range from mild symptoms, such as headache, dizziness, anorexia, anosmia, myalgia/fatigue, and ageusia (33, 303, 328), to severe manifestations, including acute stroke (344, 345), confusion, or altered consciousness (343, 346), acute inflammatory demyelinating polyneuropathy (Guillain-Barré syndrome) (107, 347), meningoencephalitis, and acute necrotizing encephalopathy of the brainstem and basal ganglia (348, 349). While the cause and correlation of the neurological manifestations of COVID-19 have not been appropriately established, long-term effects on the nervous system deserve further investigation (335). Similar to the respiratory and cardiac manifestations of COVID-19, neurological complications present differently on the basis of age and underlying comorbidities (341).
Cardiovascular Manifestations
Cardiac manifestations have been reported in patients with no clinical features of respiratory disease, and patients with preexisting cardiac-related ailments, such as dyslipidemia, obesity, and diabetes, carry a greater risk of developing severe COVID-19 (350). The severity and lethality of COVID-19-associated cardiac dysfunction are attributed to viral-induced damage to the heart and blood vessels (337). Infection with the SARS-CoV-2 virus directly infects cardiomyocytes to damage the myocardium, and it induces T cell activation. This further increases IL-1, IL-6, and TNF-α secretions (338), and it contributes to COVID-19 post-infective acute myocarditis. In addition, an inclusion can form in the myocardium when SARS-CoV-2 particles are surrounded by pro-inflammatory cells, such as neutrophils, macrophages, and lymphocytes. The inflammatory cells release cytokines such as IL-1β, TNF-α, and CCL2, which are noxious factors to the heart contributing to hypoxia and shock (351). Subsequently, the heightened metabolic rate elevates the demand for myocardial oxygen, whereby hypoxia, respiratory distress, metabolic acidosis, and fluid or electrolyte disturbances are triggered (351, 352). In severe cases, activated neurohumoral systems may trigger cardiac arrest and damage as well as even induce myocarditis, myocardial infarction, and malignant arrhythmias (66, 83, 339, 340, 353–355). Multiple retrospective reports have suggested that in China, cardiovascular disease incidence ranges from 5% to 16%, hypertension incidence ranges from 15% to 31%, and coronary artery disease incidence is 11% (356, 357). Other countries have reported even higher incidence rates of these comorbidities (356).
Renal Manifestations
Acute kidney injury (AKI) is also frequently observed in patients with COVID-19, and its manifestations include hypotension, microvascular damage and contraction, decreased renal perfusion, and hemostasis, and related sepsis (13, 66). In the kidneys, SARS-CoV-2 may infect ACE2-expressing cells, like proximal tubules and podocytes, to accumulate Ang II and further promote an imbalanced RAS. This may result in glomerular dysfunction, fibrosis, vasoconstriction, and inflammation (358). Several studies also illustrated the importance of cytokines, which can interact with kidney-resident cells and induce endothelial and tubular dysfunction. They include IL-6, IL-1β, IL-1RA, IL-7, IL-8, IL-9, IL-10, fibroblast growth factor (FGF), GM-CSF, IFN-γ, G-CSF, CXCL10, CCL2, CCL3, PDGF, TNF-α, and VEGF (33, 89, 123, 359). In particular, elevated IL-6 would cause renal endothelial cells to secrete pro-inflammatory chemokines like CCL14 and CCL2, leading to kidney vascular permeability (360, 361). Moreover, IFNs could cause the loss of podocytes and stimulate glomerulosclerosis to promote AKI persistence (362). One autopsy report has indicated that renal pathology in COVID-19 shows podocyte damage, significant acute proximal tubule injury, and coronavirus particle clusters in podocytes and renal tubular epithelial cells (363).
Hepatobiliary Manifestations
Liver damage in COVID-19 can be directly caused by the viral infection of liver cells, and hepatic injury is, therefore, associated with the degree of disease severity (364). Mechanisms causing liver injury in patients with COVID-19 remain unclear; however, multiple theories have been postulated: (1) ACE2-mediated direct viral infection of gastrointestinal epithelial cells to dysregulate liver functions (351); (2) critical patient status and immune-mediated injuries, such as cytokine storm or pneumonia-associated hypoxia, or IL-6-mediated activation of the complement system and consequent increase in vascular permeability (364); and (3) drug hepatotoxicity (352).
As reported, given the higher ACE2 expression of cholangiocytes as compared to the population of hepatocytes (59.7% vs. 2.6%), SARS-CoV-2 uses ACE2 to gain entry in the cells (351, 365). Cholangiocytes are correlated with liver physiology and adaptive immune response mechanisms. Thus, the impairment of cholangiocyte function would result in hepatobiliary damage and disrupt the barrier and bile-acid-transporting functions (366) to further contribute to SARS-CoV-2-related liver injury (367). Second, an exaggerated hyperinflammatory response from significantly elevated C-reactive protein (CRP), LDH, ferritin, and IL-6 levels contributes to liver injury or even develops into liver failure in critically ill patients (364). After the entry of the virus, pathogenic T cells are activated rapidly, producing GM-CSF, IL-6, and IFN-γ. GM-CSF further induces CD14+CD16+ inflammatory monocytes to produce increased IL-6 (368, 369). This could activate more macrophages and contribute to an inflammatory storm (251, 368, 369). This immune-mediated injury, accompanied by elevated concentrations of IL-1, IL-2, IL-10, and IFNγ, would lead to systemic inflammatory response syndrome (SIRS) (368, 369). As for drug toxicity, it is possible that impairment to the liver is caused by drug hepatotoxicity. This might explain the large variations observed across different cohorts (370).
Multiple reports have indicated that some COVID-19 patients have an abnormal liver function; e.g., the American College of Gastroenterology reported that approximately 20%-30% of patients had elevated liver enzymes, and another study from China suggested that 50.7% of SARS-CoV-2-infected patients had liver abnormalities (371). Of note, elevated liver function results were reported to be associated with moderate-high degree fever and lower T cell function (both CD4+ and CD8+ T cells) (371), and patients with preexisting liver disease (such as hepatitis B infection) carry a higher risk of developing severe disease (372, 373).
Multisystem Inflammatory Syndrome in Children (MIS-C)
Despite initial optimism with respect to children being spared the worst effects of COVID-19, it is now clear that they can also develop severe COVID-19 symptoms and a rare secondary inflammatory syndrome: multisystem inflammatory syndrome in children (MIS-C) (374). MIS-C was first identified in Northern Italy, followed by the UK, and is described as a post-COVID-19 inflammatory syndrome that is approximately 30-fold more common in the COVID-19 cohort than in healthy children (375). MIS-C symptoms range from mild, e.g., fever, rash, and mucocutaneous inflammation, to severe, including vasculitis, cardiac dysfunction, shock, and neurological involvement (376). This post-COVID-19 inflammatory syndrome was originally diagnosed as Kawasaki disease; however, patients with MIS-C display more pronounced lymphopenia, thrombocytopenia, anemia, and elevated serum IFN-γ, IL-1β, IL-6, IL-10, and IL-17 levels (377, 378). However, these effects are not as severe as those seen in severe cytokine storm syndromes, and while increased IL-10 level is correlated with a lower viral load (99), its presence, along with elevated TNF levels, is the optimal marker for distinguishing MIS-C from severe COVID-19 symptoms (379).
Similar to the neuroinvasive capabilities of SARS-CoV-2 and its CNS sequelae observed in adults, children with MIS-C also develop neurological phenomena along with respiratory symptoms and multisystem inflammation (341). Moreover, multiple studies have described neurological symptoms ranging from mild (headache and altered mental status) (380–382) to severe neurological complications, including seizure, coma, encephalitis, demyelinating disorders, encephalopathy, aseptic meningitis dysarthria, dysphagia, cerebellar ataxia, and peripheral neuropathy; the latter leads to global proximal muscle weakness and reduced reflexes (383).
Discussions and Perspectives
The present review describes significant cytokine and chemokine secretions induced by innate and adaptive immune responses in COVID-19 and virus-associated immunopathogenesis. Available data from basic and clinical studies on critical cytokines and chemokines have been presented in this review in addition to a summary of pulmonary and extrapulmonary pathophysiological features of cytokine release syndrome, consequent systemic immune response to SARS-CoV-2, and potential immunotherapeutic approaches. This review suggests that understanding the nature of the COVID-19-induced cytokine/chemokine profile(s) and pathophysiological features of correspondent immune response to control excessive inflammatory response is as important as targeting the virus itself as fatalities are primarily caused by abnormal and exaggerated cytokine storms. Future research should focus on identifying cytokine biomarkers to define the immune correlates of protection and disease severity for the effective triage of patients.
Author Contributions
Conception and design: R-JH and S-HY. Provision of study materials: W-CY, G-RP, C-HY, SH, and PC. Collection and assembly of data: KY, JL, and W-CL. Manuscript writing: All authors. Accountable for all aspects of the work: All authors. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the Young Scholar Fellowship Program by Ministry of Science and Technology (MOST) in Taiwan, under Grant MOST 110-2636-B-002-022 and the Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation in Taiwan, under Grant TCRD111-075.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. WHO. Coronavirus Disease 2019 (Covid-19) Situation Report 127 (2020). Available at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200526-covid-19-sitrep-127.pdf?sfvrsn=7b6655ab_8.
2. Mao R, Liang J, Shen J, Ghosh S, Zhu LR, Yang H, et al. Implications of Covid-19 for Patients With Pre-Existing Digestive Diseases. Lancet Gastroenterol Hepatol (2020) 5(5):425–7. doi: 10.1016/s2468-1253(20)30076-5
3. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The Trinity of Covid-19: Immunity, Inflammation and Intervention. Nat Rev Immunol (2020) 20(6):363–74. doi: 10.1038/s41577-020-0311-8
4. Jafarzadeh A, Chauhan P, Saha B, Jafarzadeh S, Nemati M. Contribution of Monocytes and Macrophages to the Local Tissue Inflammation and Cytokine Storm in Covid-19: Lessons From Sars and Mers, and Potential Therapeutic Interventions. Life Sci (2020) 257:118102. doi: 10.1016/j.lfs.2020.118102
5. Zhou R, To KK, Wong YC, Liu L, Zhou B, Li X, et al. Acute Sars-Cov-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity (2020) 53(4):864–77.e5. doi: 10.1016/j.immuni.2020.07.026
6. Merad M, Martin JC. Pathological Inflammation in Patients With Covid-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol (2020) 20(6):355–62. doi: 10.1038/s41577-020-0331-4
7. Gómez-Rial J, Rivero-Calle I, Salas A, Martinón-Torres F. Role of Monocytes/Macrophages in Covid-19 Pathogenesis: Implications for Therapy. Infect Drug Resist (2020) 13:2485–93. doi: 10.2147/IDR.S258639
8. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should We Stimulate or Suppress Immune Responses in Covid-19? Cytokine and Anti-Cytokine Interventions. Autoimmun Rev (2020) 19(7):102567. doi: 10.1016/j.autrev.2020.102567
9. Pelaia C, Tinello C, Vatrella A, De Sarro G, Pelaia G. Lung Under Attack by Covid-19-Induced Cytokine Storm: Pathogenic Mechanisms and Therapeutic Implications. Ther Adv Respir Dis (2020) 14:1753466620933508. doi: 10.1177/1753466620933508
10. Ye CH, Hsu WL, Peng GR, Yu WC, Lin WC, Hu S, et al. Role of the Immune Microenvironment in Sars-Cov-2 Infection. Cell Transplant (2021) 30:9636897211010632. doi: 10.1177/09636897211010632
11. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The Covid-19 Cytokine Storm; What We Know So Far. Front Immunol (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
12. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal Analyses Reveal Immunological Misfiring in Severe Covid-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
13. Batlle D, Soler MJ, Sparks MA, Hiremath S, South AM, Welling PA, et al. Acute Kidney Injury in Covid-19: Emerging Evidence of a Distinct Pathophysiology. J Am Soc Nephrol (2020) 31(7):1380–3. doi: 10.1681/ASN.2020040419
14. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. Sars-Cov-2 Cell Entry Depends on Ace2 and Tmprss2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80.e8. doi: 10.1016/j.cell.2020.02.052
15. Golonka RM, Saha P, Yeoh BS, Chattopadhyay S, Gewirtz AT, Joe B, et al. Harnessing Innate Immunity to Eliminate Sars-Cov-2 and Ameliorate Covid-19 Disease. Physiol Genomics (2020) 52(5):217–21. doi: 10.1152/physiolgenomics.00033.2020
16. Tu YF, Chien CS, Yarmishyn AA, Lin YY, Luo YH, Lin YT, et al. A Review of Sars-Cov-2 and the Ongoing Clinical Trials. Int J Mol Sci (2020) 21(7):2657–76 doi: 10.3390/ijms21072657
17. Vardhana SA, Wolchok JD. The Many Faces of the Anti-Covid Immune Response. J Exp Med (2020) 217(6):e20200678–88. doi: 10.1084/jem.20200678
18. Aronson JK, DeVito N, Plüddemann A, RE F. Drug Vignettes: Interferons (2020). Available at: https://www.cebm.net/covid-19/drug-vignettes-interferons/.
19. Li X, Geng M, Peng Y, Meng L, Lu S. Molecular Immune Pathogenesis and Diagnosis of Covid-19. J Pharm Anal (2020) 10(2):102–8. doi: 10.1016/j.jpha.2020.03.001
20. Miesbach W. Pathological Role of Angiotensin Ii in Severe Covid-19. TH Open (2020) 4(2):e138–44. doi: 10.1055/s-0040-1713678
21. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and Biochemical Indexes From 2019-Ncov Infected Patients Linked to Viral Loads and Lung Injury. Sci China Life Sci (2020) 63(3):364–74. doi: 10.1007/s11427-020-1643-8
22. Li C, He J, Zhong X, Gan H, Xia Y. Cx3cl1/Cx3cr1 Axis Contributes to Angiotensin Ii-Induced Vascular Smooth Muscle Cell Proliferation and Inflammatory Cytokine Production. Inflammation (2018) 41(3):824–34. doi: 10.1007/s10753-018-0736-4
23. Zhang X, Yang J, Yu X, Cheng S, Gan H, Xia Y. Angiotensin Ii-Induced Early and Late Inflammatory Responses Through Noxs and Mapk Pathways. Inflammation (2017) 40(1):154–65. doi: 10.1007/s10753-016-0464-6
24. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-Converting Enzyme 2 Protects From Severe Acute Lung Failure. Nature (2005) 436(7047):112–6. doi: 10.1038/nature03712
25. Khan S, Shafiei MS, Longoria C, Schoggins JW, Savani RC, Zaki H. Sars-Cov-2 Spike Protein Induces Inflammation Via Tlr2-Dependent Activation of the Nf-Kappab Pathway. Elife (2021) 10:e68563-89. doi: 10.7554/eLife.68563
26. Li W, Qiao J, You Q, Zong S, Peng Q, Liu Y, et al. Sars-Cov-2 Nsp5 Activates Nf-Kappab Pathway by Upregulating Sumoylation of Mavs. Front Immunol (2021) 12:750969. doi: 10.3389/fimmu.2021.750969
27. Wang F, Nie J, Wang H, Zhao Q, Xiong Y, Deng L, et al. Characteristics of Peripheral Lymphocyte Subset Alteration in Covid-19 Pneumonia. J Infect Dis (2020) 221(11):1762–9. doi: 10.1093/infdis/jiaa150
28. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J Clin Invest (2020) 130(5):2620–9. doi: 10.1172/JCI137244
29. Parisi V, Leosco D. Precision Medicine in Covid-19: Il-1beta a Potential Target. JACC Basic Transl Sci (2020) 5(5):543–4. doi: 10.1016/j.jacbts.2020.04.006
30. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting Potential Drivers of Covid-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217(6):e20200652–9. doi: 10.1084/jem.20200652
31. Soy M, Keser G, Atagunduz P, Tabak F, Atagunduz I, Kayhan S. Cytokine Storm in Covid-19: Pathogenesis and Overview of Anti-Inflammatory Agents Used in Treatment. Clin Rheumatol (2020) 39(7):2085–94. doi: 10.1007/s10067-020-05190-5
32. Campana P, Parisi V, Leosco D, Bencivenga D, Della Ragione F, Borriello A. Dendritic Cells and Sars-Cov-2 Infection: Still an Unclarified Connection. Cells (2020) 9(9):2046–69. doi: 10.3390/cells9092046
33. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
34. Mamber SW, Krakowka S, Osborn J, Saberski L, Rhodes RG, Dahlberg AE, et al. Can Unconventional Immunomodulatory Agents Help Alleviate Covid-19 Symptoms and Severity? mSphere (2020) 5(3):e0028820–6. doi: 10.1128/mSphere.00288-20
35. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to Sars-Cov-2 Drives Development of Covid-19. Cell (2020) 181(5):1036–45.e9. doi: 10.1016/j.cell.2020.04.026
36. Nile SH, Nile A, Qiu J, Li L, Jia X, Kai G. Covid-19: Pathogenesis, Cytokine Storm and Therapeutic Potential of Interferons. Cytokine Growth Factor Rev (2020) 53:66–70. doi: 10.1016/j.cytogfr.2020.05.002
37. Mendoza VMM. Interleukin-17: A Potential Therapeutic Target in Covid-19. J Infect (2020) 81(2):e136–e8. doi: 10.1016/j.jinf.2020.05.072
38. Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine Storm in Covid-19: The Current Evidence and Treatment Strategies. Front Immunol (2020) 11:1708. doi: 10.3389/fimmu.2020.01708
39. Chen L, Liu HG, Liu W, Liu J, Liu K, Shang J, et al. [Analysis of Clinical Features of 29 Patients With 2019 Novel Coronavirus Pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi (2020) 43(0):E005. doi: 10.3760/cma.j.issn.1001-0939.2020.0005
40. Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, et al. Sars-Cov-2-Specific T Cell Immunity in Cases of Covid-19 and Sars, and Uninfected Controls. Nature (2020) 584(7821):457–62. doi: 10.1038/s41586-020-2550-z
41. Du P, Geng J, Wang F, Chen X, Huang Z, Wang Y. Role of Il-6 Inhibitor in Treatment of Covid-19-Related Cytokine Release Syndrome. Int J Med Sci (2021) 18(6):1356–62. doi: 10.7150/ijms.53564
42. Tsai A, Diawara O, Nahass RG, Brunetti L. Impact of Tocilizumab Administration on Mortality in Severe Covid-19. Sci Rep (2020) 10(1):19131. doi: 10.1038/s41598-020-76187-y
43. Resende GG, da Cruz Lage R, Lobê SQ, Medeiros AF, Costa e Silva AD, Nogueira Sá AT, et al. Blockade of Interleukin Seventeen (Il-17a) With Secukinumab in Hospitalized Covid-19 Patients – The Bishop Study. medRxiv (2021) 2021.07.21.21260963. doi: 10.1101/2021.07.21.21260963
44. Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, Phelan A, et al. Baricitinib as Potential Treatment for 2019-Ncov Acute Respiratory Disease. Lancet (2020) 395(10223):e30–1. doi: 10.1016/S0140-6736(20)30304-4
45. Jalkanen J, Hollmen M, Jalkanen S. Interferon Beta-1a for Covid-19: Critical Importance of the Administration Route. Crit Care (2020) 24(1):335. doi: 10.1186/s13054-020-03048-5
46. Cohan SL, Hendin BA, Reder AT, Smoot K, Avila R, Mendoza JP, et al. Interferons and Multiple Sclerosis: Lessons From 25 Years of Clinical and Real-World Experience With Intramuscular Interferon Beta-1a (Avonex). CNS Drugs (2021) 35(7):743–67. doi: 10.1007/s40263-021-00822-z
47. Badary OA. Pharmacogenomics and Covid-19: Clinical Implications of Human Genome Interactions With Repurposed Drugs. Pharmacogenomics J (2021) 21(3):275–84. doi: 10.1038/s41397-021-00209-9
48. Napoli C, Benincasa G, Criscuolo C, Faenza M, Liberato C, Rusciano M. Immune Reactivity During Covid-19: Implications for Treatment. Immunol Lett (2021) 231:28–34. doi: 10.1016/j.imlet.2021.01.001
49. Bonaventura A, Vecchie A, Wang TS, Lee E, Cremer PC, Carey B, et al. Targeting Gm-Csf in Covid-19 Pneumonia: Rationale and Strategies. Front Immunol (2020) 11:1625. doi: 10.3389/fimmu.2020.01625
50. Ghasemzadeh M, Ghasemzadeh A, Hosseini E. Exhausted Nk Cells and Cytokine Storms in Covid-19: Whether Nk Cell Therapy Could Be a Therapeutic Choice. Hum Immunol (2022) 83(1):86–98. doi: 10.1016/j.humimm.2021.09.004
51. Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful Treatment of Severe Paediatric Rheumatic Disease-Associated Macrophage Activation Syndrome With Interleukin-1 Inhibition Following Conventional Immunosuppressive Therapy: Case Series With 12 Patients. Rheumatol (Oxf) (2011) 50(2):417–9. doi: 10.1093/rheumatology/keq218
52. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
53. Lounder DT, Bin Q, de Min C, Jordan MB. Treatment of Refractory Hemophagocytic Lymphohistiocytosis With Emapalumab Despite Severe Concurrent Infections. Blood Adv (2019) 3(1):47–50. doi: 10.1182/bloodadvances.2018025858
54. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 Blockade With High-Dose Anakinra in Patients With Covid-19, Acute Respiratory Distress Syndrome, and Hyperinflammation: A Retrospective Cohort Study. Lancet Rheumatol (2020) 2(6):e325–e31. doi: 10.1016/S2665-9913(20)30127-2
55. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in Hospitalized Patients With Covid-19. N Engl J Med (2021) 384(8):693–704. doi: 10.1056/NEJMoa2021436
56. Sterne JAC, Diaz J, Villar J, Murthy S, Slutsky AS, Perner A, et al. Corticosteroid Therapy for Critically Ill Patients With Covid-19: A Structured Summary of a Study Protocol for a Prospective Meta-Analysis of Randomized Trials. Trials (2020) 21(1):734. doi: 10.1186/s13063-020-04641-3
57. Rizk JG, Kalantar-Zadeh K, Mehra MR, Lavie CJ, Rizk Y, Forthal DN. Pharmaco-Immunomodulatory Therapy in Covid-19. Drugs (2020) 80(13):1267–92. doi: 10.1007/s40265-020-01367-z
58. Henry BM, de Oliveira MHS, Benoit S, Plebani M, Lippi G. Hematologic, Biochemical and Immune Biomarker Abnormalities Associated With Severe Illness and Mortality in Coronavirus Disease 2019 (Covid-19): A Meta-Analysis. Clin Chem Lab Med (2020) 58(7):1021–8. doi: 10.1515/cclm-2020-0369
59. Rosenberg HF, Foster PS. Eosinophils and Covid-19: Diagnosis, Prognosis, and Vaccination Strategies. Semin Immunopathol (2021) 43(3):383–92. doi: 10.1007/s00281-021-00850-3
60. Aschenbrenner DS. Remdesivir Approved to Treat Covid-19 Amid Controversy. Am J Nurs (2021) 121(1):22–4. doi: 10.1097/01.NAJ.0000731640.35662.2c
61. Hashemian SMR, Pourhanifeh MH, Hamblin MR, Shahrzad MK, Mirzaei H. Rdrp Inhibitors and Covid-19: Is Molnupiravir a Good Option? BioMed Pharmacother (2021) 146:112517. doi: 10.1016/j.biopha.2021.112517
62. Painter WP, Holman W, Bush JA, Almazedi F, Malik H, Eraut N, et al. Human Safety, Tolerability, and Pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent With Activity Against Sars-Cov-2. Antimicrob Agents Chemother (2021) 65(5):e02428–20. doi: 10.1128/AAC.02428-20
63. Mishra SK, Tripathi T. One Year Update on the Covid-19 Pandemic: Where Are We Now? Acta Trop (2021) 214:105778. doi: 10.1016/j.actatropica.2020.105778
64. Garcia-Lledo A, Gomez-Pavon J, Gonzalez Del Castillo J, Hernandez-Sampelayo T, Martin-Delgado MC, Martin Sanchez FJ, et al. Pharmacological Treatment of Covid-19: An Opinion Paper. Rev Esp Quimioter (2022) 35(2):115–130. doi: 10.37201/req/158.2021
65. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev (2012) 76(1):16–32. doi: 10.1128/mmbr.05015-11
66. Song P, Li W, Xie J, Hou Y, You C. Cytokine Storm Induced by Sars-Cov-2. Clin Chim Acta (2020) 509:280–7. doi: 10.1016/j.cca.2020.06.017
67. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The Role of Cytokines Including Interleukin-6 in Covid-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev (2020) 19(6):102537. doi: 10.1016/j.autrev.2020.102537
68. Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M. The Cytokine Storm in Covid-19: An Overview of the Involvement of the Chemokine/Chemokine-Receptor System. Cytokine Growth Factor Rev (2020) 53:25–32. doi: 10.1016/j.cytogfr.2020.05.003
69. Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic Implications of Il-6 Blockade for Cytokine Storm. Immunotherapy (2016) 8(8):959–70. doi: 10.2217/imt-2016-0020
70. Paces J, Strizova Z, Smrz D, Cerny J. Covid-19 and the Immune System. Physiol Res (2020) 69(3):379–88. doi: 10.33549/physiolres.934492
71. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (Covid-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
72. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe Covid-19 Patients. Science (2020) 369(6504):718–24. doi: 10.1126/science.abc6027
73. Coomes EA, Haghbayan H. Interleukin-6 in Covid-19: A Systematic Review and Meta-Analysis. Rev Med Virol (2020) 30(6):1–9. doi: 10.1002/rmv.2141
74. Nagant C, Ponthieux F, Smet J, Dauby N, Doyen V, Besse-Hammer T, et al. A Score Combining Early Detection of Cytokines Accurately Predicts Covid-19 Severity and Intensive Care Unit Transfer. Int J Infect Dis (2020) 101:342–5. doi: 10.1016/j.ijid.2020.10.003
75. Ulhaq ZS, Soraya GV. Interleukin-6 as a Potential Biomarker of Covid-19 Progression. Med Mal Infect (2020) 50(4):382–3. doi: 10.1016/j.medmal.2020.04.002
76. Chen LYC, Hoiland RL, Stukas S, Wellington CL, Sekhon MS. Confronting the Controversy: Interleukin-6 and the Covid-19 Cytokine Storm Syndrome. Eur Respir J (2020) 56(4):2003006. doi: 10.1183/13993003.03006-2020
77. Sun X, Wang T, Cai D, Hu Z, Chen J, Liao H, et al. Cytokine Storm Intervention in the Early Stages of Covid-19 Pneumonia. Cytokine Growth Factor Rev (2020) 53:38–42. doi: 10.1016/j.cytogfr.2020.04.002
78. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. Covid-19 Infection: The Perspectives on Immune Responses. Cell Death Differ (2020) 27(5):1451–4. doi: 10.1038/s41418-020-0530-3
79. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
80. O’Connell P, Aldhamen YA. Systemic Innate and Adaptive Immune Responses to Sars-Cov-2 as It Relates to Other Coronaviruses. Hum Vaccin Immunother (2020) 16(12):1–12. doi: 10.1080/21645515.2020.1802974
81. Moore JB, June CH. Cytokine Release Syndrome in Severe Covid-19. Science (2020) 368(6490):473–4. doi: 10.1126/science.abb8925
82. Hunter CA, Jones SA. Il-6 as a Keystone Cytokine in Health and Disease. Nat Immunol (2015) 16(5):448–57. doi: 10.1038/ni.3153
83. Geng YJ, Wei ZY, Qian HY, Huang J, Lodato R, Castriotta RJ. Pathophysiological Characteristics and Therapeutic Approaches for Pulmonary Injury and Cardiovascular Complications of Coronavirus Disease 2019. Cardiovasc Pathol (2020) 47:107228. doi: 10.1016/j.carpath.2020.107228
84. Pathan N, Hemingway CA, Alizadeh AA, Stephens AC, Boldrick JC, Oragui EE, et al. Role of Interleukin 6 in Myocardial Dysfunction of Meningococcal Septic Shock. Lancet (2004) 363(9404):203–9. doi: 10.1016/S0140-6736(03)15326-3
85. Hashizume M. Outlook of Il-6 Signaling Blockade for Covid-19 Pneumonia. Inflamm Regener (2020) 40:24. doi: 10.1186/s41232-020-00134-7
86. Ishikawa T. Clinical Preparedness for Cytokine Storm Induced by the Highly Pathogenic H5n1 Influenza Virus. J Pharmacogenom Pharmacoproteomics (2012) 03(06):1. doi: 10.4172/2153-0645.1000e131
87. Ma A, Zhang L, Ye X, Chen J, Yu J, Zhuang L, et al. High Levels of Circulating Il-8 and Soluble Il-2r Are Associated With Prolonged Illness in Patients With Severe Covid-19. Front Immunol (2021) 12:626235. doi: 10.3389/fimmu.2021.626235
88. Zeng Z, Yu H, Chen H, Qi W, Chen L, Chen G, et al. Longitudinal Changes of Inflammatory Parameters and Their Correlation With Disease Severity and Outcomes in Patients With Covid-19 From Wuhan, China. Crit Care (2020) 24(1):525. doi: 10.1186/s13054-020-03255-0
89. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With Covid-19 in Wuhan, China: A Retrospective Cohort Study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/S0140-6736(20)30566-3
90. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex Immune Dysregulation in Covid-19 Patients With Severe Respiratory Failure. Cell Host Microbe (2020) 27(6):992–1000.e3. doi: 10.1016/j.chom.2020.04.009
91. Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC, et al. Attenuated Interferon and Proinflammatory Response in Sars-Cov-2-Infected Human Dendritic Cells Is Associated With Viral Antagonism of Stat1 Phosphorylation. J Infect Dis (2020) 222(5):734–45. doi: 10.1093/infdis/jiaa356
92. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. Covid-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
93. Vecchie A, Bonaventura A, Toldo S, Dagna L, Dinarello CA, Abbate A. Il-18 and Infections: Is There a Role for Targeted Therapies? J Cell Physiol (2021) 236(3):1638–57. doi: 10.1002/jcp.30008
94. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of Covid-19 and Influenza Highlights the Role of Type I Interferons in Development of Severe Covid-19. Sci Immunol (2020) 5(49):eabd1554–70. doi: 10.1126/sciimmunol.abd1554
95. Lu L, Zhang H, Dauphars DJ, He YW. A Potential Role of Interleukin 10 in Covid-19 Pathogenesis. Trends Immunol (2021) 42(1):3–5. doi: 10.1016/j.it.2020.10.012
96. Blot M, Jacquier M, Aho Glele LS, Beltramo G, Nguyen M, Bonniaud P, et al. Cxcl10 Could Drive Longer Duration of Mechanical Ventilation During Covid-19 Ards. Crit Care (2020) 24(1):632. doi: 10.1186/s13054-020-03328-0
97. Han H, Ma Q, Li C, Liu R, Zhao L, Wang W, et al. Profiling Serum Cytokines in Covid-19 Patients Reveals Il-6 and Il-10 Are Disease Severity Predictors. Emerg Microbes Infect (2020) 9(1):1123–30. doi: 10.1080/22221751.2020.1770129
98. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine Storm and Leukocyte Changes in Mild Versus Severe Sars-Cov-2 Infection: Review of 3939 Covid-19 Patients in China and Emerging Pathogenesis and Therapy Concepts. J Leukoc Biol (2020) 108(1):17–41. doi: 10.1002/JLB.3COVR0520-272R
99. Diorio C, Henrickson SE, Vella LA, McNerney KO, Chase J, Burudpakdee C, et al. Multisystem Inflammatory Syndrome in Children and Covid-19 Are Distinct Presentations of Sars-Cov-2. J Clin Invest (2020) 130(11):5967–75. doi: 10.1172/JCI140970
100. Costela-Ruiz VJ, Illescas-Montes R, Puerta-Puerta JM, Ruiz C, Melguizo-Rodriguez L. Sars-Cov-2 Infection: The Role of Cytokines in Covid-19 Disease. Cytokine Growth Factor Rev (2020) 54:62–75. doi: 10.1016/j.cytogfr.2020.06.001
101. Pacha O, Sallman MA, Evans SE. Covid-19: A Case for Inhibiting Il-17? Nat Rev Immunol (2020) 20(6):345–6. doi: 10.1038/s41577-020-0328-z
102. Liu Y, Zhang C, Huang F, Yang Y, Wang F, Yuan J, et al. Novel Coronavirus (2019-Ncov) Infections Trigger an Exaggerated Cytokine Response Aggravating Lung Injury. ChinaXiv (2020). doi: 10.12074/202002.00018
103. Tan M, Liu Y, Zhou R, Deng X, Li F, Liang K, et al. Immunopathological Characteristics of Coronavirus Disease 2019 Cases in Guangzhou, China. Immunology (2020) 160(3):261–8. doi: 10.1111/imm.13223
104. Lagunas-Rangel FA, Chavez-Valencia V. High Il-6/Ifn-Gamma Ratio Could Be Associated With Severe Disease in Covid-19 Patients. J Med Virol (2020) 92(10):1789–90. doi: 10.1002/jmv.25900
105. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional Exhaustion of Antiviral Lymphocytes in Covid-19 Patients. Cell Mol Immunol (2020) 17(5):533–5. doi: 10.1038/s41423-020-0402-2
106. Shimabukuro-Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M, et al. Cytokine Release Syndrome. J Immunother Cancer (2018) 6(1):56. doi: 10.1186/s40425-018-0343-9
107. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With Covid-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
108. Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, et al. Plasma Ip-10 and Mcp-3 Levels Are Highly Associated With Disease Severity and Predict the Progression of Covid-19. J Allergy Clin Immunol (2020) 146(1):119–27 e4. doi: 10.1016/j.jaci.2020.04.027
109. Ghazavi A, Ganji A, Keshavarzian N, Rabiemajd S, Mosayebi G. Cytokine Profile and Disease Severity in Patients With Covid-19. Cytokine (2021) 137:155323. doi: 10.1016/j.cyto.2020.155323
110. Mehta P, Porter JC, Manson JJ, Isaacs JD, Openshaw PJM, McInnes IB, et al. Therapeutic Blockade of Granulocyte Macrophage Colony-Stimulating Factor in Covid-19-Associated Hyperinflammation: Challenges and Opportunities. Lancet Respir Med (2020) 8(8):822–30. doi: 10.1016/S2213-2600(20)30267-8
111. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T-Cells and Inflammatory Monocytes Incite Inflammatory Storms in Severe Covid-19 Patients. Natl Sci Rev (2020) 7(6):998–1002. doi: 10.1093/nsr/nwaa041
112. Becher B, Tugues S, Greter M. Gm-Csf: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity (2016) 45(5):963–73. doi: 10.1016/j.immuni.2016.10.026
113. Brocker C, Thompson D, Matsumoto A, Nebert DW, Vasiliou V. Evolutionary Divergence and Functions of the Human Interleukin (Il) Gene Family. Hum Genomics (2010) 5(1):30–55. doi: 10.1186/1479-7364-5-1-30
114. Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-Mediated Inflammation in Acute Lung Injury. Cytokine Growth Factor Rev (2003) 14(6):523–35. doi: 10.1016/s1359-6101(03)00059-5
115. Wan S, Yi Q, Fan S, Lv J, Zhang X, Guo L, et al. Characteristics of Lymphocyte Subsets and Cytokines in Peripheral Blood of 123 Hospitalized Patients With 2019 Novel Coronavirus Pneumonia (Ncp). medRxiv (2020). doi: 10.1101/2020.02.10.20021832
116. Della-Torre E, Criscuolo E, Lanzillotta M, Locatelli M, Clementi N, Mancini N, et al. Il-1 and Il-6 Inhibition Affects the Neutralising Activity of Anti-Sars-Cov-2 Antibodies in Patients With Covid-19. Lancet Rheumatol (2021) 3(12):e829–e31. doi: 10.1016/S2665-9913(21)00321-0
117. Scheller J, Rose-John S. Interleukin-6 and Its Receptor: From Bench to Bedside. Med Microbiol Immunol (2006) 195(4):173–83. doi: 10.1007/s00430-006-0019-9
118. Fara A, Mitrev Z, Rosalia RA, Assas BM. Cytokine Storm and Covid-19: A Chronicle of Pro-Inflammatory Cytokines. Open Biol (2020) 10(9):200160. doi: 10.1098/rsob.200160
119. Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, et al. Tgf-Beta Promotes Th17 Cell Development Through Inhibition of Socs3. J Immunol (2009) 183(1):97–105. doi: 10.4049/jimmunol.0801986
120. Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, et al. Blockade of Interleukin 6 Trans Signaling Suppresses T-Cell Resistance Against Apoptosis in Chronic Intestinal Inflammation: Evidence in Crohn Disease and Experimental Colitis in Vivo. Nat Med (2000) 6(5):583–8. doi: 10.1038/75068
121. Teague TK, Marrack P, Kappler JW, Vella AT. Il-6 Rescues Resting Mouse T Cells From Apoptosis. J Immunol (1997) 158(12):5791–6.
122. Yoshida H, Hashizume M, Suzuki M, Mihara M. Anti-Il-6 Receptor Antibody Suppressed T Cell Activation by Inhibiting Il-2 Production and Inducing Regulatory T Cells. Eur J Pharmacol (2010) 634(1-3):178–83. doi: 10.1016/j.ejphar.2010.02.026
123. Su H, Lei CT, Zhang C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front Immunol (2017) 8:405. doi: 10.3389/fimmu.2017.00405
124. Kimura A, Kishimoto T. Il-6: Regulator of Treg/Th17 Balance. Eur J Immunol (2010) 40(7):1830–5. doi: 10.1002/eji.201040391
125. Blaser H, Dostert C, Mak TW, Brenner D. Tnf and Ros Crosstalk in Inflammation. Trends Cell Biol (2016) 26(4):249–61. doi: 10.1016/j.tcb.2015.12.002
126. Park MD. Macrophages: A Trojan Horse in Covid-19? Nat Rev Immunol (2020) 20(6):351. doi: 10.1038/s41577-020-0317-2
127. Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, et al. Frontline Science: Covid-19 Infection Induces Readily Detectable Morphologic and Inflammation-Related Phenotypic Changes in Peripheral Blood Monocytes. J Leukocyte Biol (2021) 109(1):13–22. doi: 10.1002/JLB.4HI0720-470R
128. Prompetchara E, Ketloy C, Palaga T. Immune Responses in Covid-19 and Potential Vaccines: Lessons Learned From Sars and Mers Epidemic. Asian Pac J Allergy Immunol (2020) 38(1):1–9. doi: 10.12932/AP-200220-0772
129. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med (2020) 180(7):934–43. doi: 10.1001/jamainternmed.2020.0994
130. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (Covid-19) in Wuhan, China. Clin Infect Dis (2020) 71(15):762–8. doi: 10.1093/cid/ciaa248
131. Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can We Use Interleukin-6 (Il-6) Blockade for Coronavirus Disease 2019 (Covid-19)-Induced Cytokine Release Syndrome (Crs)? J Autoimmun (2020) 111:102452. doi: 10.1016/j.jaut.2020.102452
132. Liu X, Wang H, Shi S, Xiao J. Association Between Il-6 and Severe Disease and Mortality in Covid-19 Disease: A Systematic Review and Meta-Analysis. Postgrad Med J (2021) 0:1–9. doi: 10.1136/postgradmedj-2021-139939
133. Maeda T, Obata R, Rizk DD, Kuno T. The Association of Interleukin-6 Value, Interleukin Inhibitors, and Outcomes of Patients With Covid-19 in New York City. J Med Virol (2021) 93(1):463–71. doi: 10.1002/jmv.26365
134. Sabaka P, Koščálová A, Straka I, Hodosy J, Lipták R, Kmotorková B, et al. Role of Interleukin 6 as a Predictive Factor for a Severe Course of Covid-19: Retrospective Data Analysis of Patients From a Long-Term Care Facility During Covid-19 Outbreak. BMC Infect Dis (2021) 21(1):308. doi: 10.1186/s12879-021-05945-8
135. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An Inflammatory Cytokine Signature Predicts Covid-19 Severity and Survival. Nat Med (2020) 26(10):1636–43. doi: 10.1038/s41591-020-1051-9
136. Hojyo S, Uchida M, Tanaka K, Hasebe R, Tanaka Y, Murakami M, et al. How Covid-19 Induces Cytokine Storm With High Mortality. Inflamm Regener (2020) 40:37. doi: 10.1186/s41232-020-00146-3
137. Khan FA, Stewart I, Fabbri L, Moss S, Robinson K, Smyth AR, et al. Systematic Review and Meta-Analysis of Anakinra, Sarilumab, Siltuximab and Tocilizumab for Covid-19. Thorax (2021) 76:907–19. doi: 10.1136/thoraxjnl-2020-215266
138. Cron RQ. Covid-19 Cytokine Storm: Targeting the Appropriate Cytokine. Lancet Rheumatol (2021) 3(4):e236–7. doi: 10.1016/s2665-9913(21)00011-4
139. Hermine O, Mariette X, Tharaux PL, Resche-Rigon M, Porcher R, Ravaud P, et al. Effect of Tocilizumab Vs Usual Care in Adults Hospitalized With Covid-19 and Moderate or Severe Pneumonia: A Randomized Clinical Trial. JAMA Intern Med (2021) 181(1):32–40. doi: 10.1001/jamainternmed.2020.6820
140. Rosas IO, Brau N, Waters M, Go RC, Hunter BD, Bhagani S, et al. Tocilizumab in Hospitalized Patients With Severe Covid-19 Pneumonia. N Engl J Med (2021) 384(16):1503–16. doi: 10.1056/NEJMoa2028700
141. Salvarani C, Dolci G, Massari M, Merlo DF, Cavuto S, Savoldi L, et al. Effect of Tocilizumab Vs Standard Care on Clinical Worsening in Patients Hospitalized With Covid-19 Pneumonia: A Randomized Clinical Trial. JAMA Intern Med (2021) 181(1):24–31. doi: 10.1001/jamainternmed.2020.6615
142. Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS, et al. Efficacy of Tocilizumab in Patients Hospitalized With Covid-19. N Engl J Med (2020) 383(24):2333–44. doi: 10.1056/NEJMoa2028836
143. Group RC. Tocilizumab in Patients Admitted to Hospital With Covid-19 (Recovery): A Randomised, Controlled, Open-Label, Platform Trial. Lancet (2021) 397(10285):1637–45. doi: 10.1016/S0140-6736(21)00676-0
144. Investigators R-C, Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients With Covid-19. N Engl J Med (2021) 384(16):1491–502. doi: 10.1056/NEJMoa2100433
145. Gritti G, Raimondi F, Ripamonti D, Riva I, Landi F, Alborghetti L, et al. Il-6 Signalling Pathway Inactivation With Siltuximab in Patients With Covid-19 Respiratory Failure: An Observational Cohort Study. medRxiv (2020).
146. Roumier M, Paule R, Groh M, Vallée A, Ackermann F. Interleukin-6 Blockade for Severe Covid-19. medRxiv (2020).
147. Ip A, Berry DA, Hansen E, Goy AH, Pecora AL, Sinclaire BA, et al. Hydroxychloroquine and Tocilizumab Therapy in Covid-19 Patients-An Observational Study. PloS One (2020) 15(8):e0237693. doi: 10.1371/journal.pone.0237693
148. Dinarello CA. Overview of the Il-1 Family in Innate Inflammation and Acquired Immunity. Immunol Rev (2018) 281(1):8–27. doi: 10.1111/imr.12621
149. Tufan A, Avanoglu Guler A, Matucci-Cerinic M. Covid-19, Immune System Response, Hyperinflammation and Repurposing Antirheumatic Drugs. Turk J Med Sci (2020) 50(SI-1):620–32. doi: 10.3906/sag-2004-168
150. Lefebvre AL, McAuliffe L. Targeted Immunomodulatory Therapy: An Overview. R I Med J (2016) 99(12):19–22.
151. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in Sars-Cov-Infected Mice. Cell Host Microbe (2016) 19(2):181–93. doi: 10.1016/j.chom.2016.01.007
152. Figuero-Perez L, Olivares-Hernandez A, Escala-Cornejo RA, Teran-Brage E, Lopez-Gutierrez A, Cruz-Hernandez JJ. Anakinra as a Potential Alternative in the Treatment of Severe Acute Respiratory Infection Associated With Sars-Cov-2 Refractory to Tocilizumab. Reumatol Clin (Engl Ed) (2021) 17(10):559–61. doi: 10.1016/j.reumae.2020.06.008
153. Dinarello CA. Therapeutic Strategies to Reduce Il-1 Activity in Treating Local and Systemic Inflammation. Curr Opin Pharmacol (2004) 4(4):378–85. doi: 10.1016/j.coph.2004.03.010
154. Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, et al. The Use of Anti-Inflammatory Drugs in the Treatment of People With Severe Coronavirus Disease 2019 (Covid-19): The Perspectives of Clinical Immunologists From China. Clin Immunol (2020) 214:108393. doi: 10.1016/j.clim.2020.108393
155. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, et al. Induction of Pro-Inflammatory Cytokines (Il-1 and Il-6) and Lung Inflammation by Coronavirus-19 (Covi-19 or Sars-Cov-2): Anti-Inflammatory Strategies. J Biol Regul Homeost Agents (2020) 34(2):327–31. doi: 10.23812/CONTI-E
156. Guo C, Li B, Ma H, Wang X, Cai P, Yu Q, et al. Single-Cell Analysis of Two Severe Covid-19 Patients Reveals a Monocyte-Associated and Tocilizumab-Responding Cytokine Storm. Nat Commun (2020) 11(1):3924. doi: 10.1038/s41467-020-17834-w
157. Wen W, Su W, Tang H, Le W, Zhang X, Zheng Y, et al. Immune Cell Profiling of Covid-19 Patients in the Recovery Stage by Single-Cell Sequencing. Cell Discovery (2020) 6:31. doi: 10.1038/s41421-020-0168-9
158. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of Covid-19: Current State of the Science. Immunity (2020) 52(6):910–41. doi: 10.1016/j.immuni.2020.05.002
159. Ong EZ, Chan YFZ, Leong WY, Lee NMY, Kalimuddin S, Haja Mohideen SM, et al. A Dynamic Immune Response Shapes Covid-19 Progression. Cell Host Microbe (2020) 27(6):879–82.e2. doi: 10.1016/j.chom.2020.03.021
160. Satis H, Ozger HS, Aysert Yildiz P, Hizel K, Gulbahar O, Erbas G, et al. Prognostic Value of Interleukin-18 and Its Association With Other Inflammatory Markers and Disease Severity in Covid-19. Cytokine (2021) 137:155302. doi: 10.1016/j.cyto.2020.155302
161. Slaats J, Ten Oever J, van de Veerdonk FL, Netea MG. Il-1beta/Il-6/Crp and Il-18/Ferritin: Distinct Inflammatory Programs in Infections. PloS Pathog (2016) 12(12):e1005973. doi: 10.1371/journal.ppat.1005973
162. Borella R. Metabolic Reprograming Shapes Neutrophil Functions in Severe Covid-19. Eur J Immunol (2022) 52(3):484–502. doi: 10.1002/eji.202149481
163. Sadeghi A, Tahmasebi S, Mahmood A, Kuznetsova M, Valizadeh H, Taghizadieh A, et al. Th17 and Treg Cells Function in Sars-Cov2 Patients Compared With Healthy Controls. J Cell Physiol (2021) 236(4):2829–39. doi: 10.1002/jcp.30047
164. Kerget B, Kerget F, Aksakal A, Askin S, Saglam L, Akgun M. Evaluation of Alpha Defensin, Il-1 Receptor Antagonist, and Il-18 Levels in Covid-19 Patients With Macrophage Activation Syndrome and Acute Respiratory Distress Syndrome. J Med Virol (2021) 93(4):2090–98. doi: 10.1002/jmv.26589
165. Saraiva M, O’Garra A. The Regulation of Il-10 Production by Immune Cells. Nat Rev Immunol (2010) 10(3):170–81. doi: 10.1038/nri2711
166. O’Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for Use of Il-10 or Its Antagonists in Human Disease. Immunol Rev (2008) 223:114–31. doi: 10.1111/j.1600-065X.2008.00635.x
167. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the Interleukin-10 Receptor. Annu Rev Immunol (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
168. Hawrylowicz CM, O’Garra A. Potential Role of Interleukin-10-Secreting Regulatory T Cells in Allergy and Asthma. Nat Rev Immunol (2005) 5(4):271–83. doi: 10.1038/nri1589
169. Ouyang W, O’Garra A. Il-10 Family Cytokines Il-10 and Il-22: From Basic Science to Clinical Translation. Immunity (2019) 50(4):871–91. doi: 10.1016/j.immuni.2019.03.020
170. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and Functions of the Il-10 Family of Cytokines in Inflammation and Disease. Annu Rev Immunol (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
171. Gabrysova L, Howes A, Saraiva M, O’Garra A. The Regulation of Il-10 Expression. Curr Top Microbiol Immunol (2014) 380:157–90. doi: 10.1007/978-3-662-43492-5_8
172. Wang X, Wong K, Ouyang W, Rutz S. Targeting Il-10 Family Cytokines for the Treatment of Human Diseases. Cold Spring Harb Perspect Biol. (2019) 11(2):a028548–80. doi: 10.1101/cshperspect.a028548
173. Engelhardt KR, Grimbacher B. Il-10 in Humans: Lessons From the Gut, Il-10/Il-10 Receptor Deficiencies, and Il-10 Polymorphisms. Curr Top Microbiol Immunol (2014) 380:1–18. doi: 10.1007/978-3-662-43492-5_1
174. O’Garra A, Vieira P. T(H)1 Cells Control Themselves by Producing Interleukin-10. Nat Rev Immunol (2007) 7(6):425–8. doi: 10.1038/nri2097
175. Trinchieri G. Interleukin-10 Production by Effector T Cells: Th1 Cells Show Self Control. J Exp Med (2007) 204(2):239–43. doi: 10.1084/jem.20070104
176. Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-Secreting Type 1 Regulatory T Cells in Rodents and Humans. Immunol Rev (2006) 212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x
177. Maynard CL, Weaver CT. Diversity in the Contribution of Interleukin-10 to T-Cell-Mediated Immune Regulation. Immunol Rev (2008) 226:219–33. doi: 10.1111/j.1600-065X.2008.00711.x
178. Maloy KJ, Powrie F. Regulatory T Cells in the Control of Immune Pathology. Nat Immunol (2001) 2(9):816–22. doi: 10.1038/ni0901-816
179. Boonstra A, Rajsbaum R, Holman M, Marques R, Asselin-Paturel C, Pereira JP, et al. Macrophages and Myeloid Dendritic Cells, But Not Plasmacytoid Dendritic Cells, Produce Il-10 in Response to Myd88- and Trif-Dependent Tlr Signals, and Tlr-Independent Signals. J Immunol (2006) 177(11):7551–8. doi: 10.4049/jimmunol.177.11.7551
180. Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, et al. A Toll-Like Receptor 2 Ligand Stimulates Th2 Responses in Vivo, Via Induction of Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase and C-Fos in Dendritic Cells. J Immunol (2004) 172(8):4733–43. doi: 10.4049/jimmunol.172.8.4733
181. Edwards AD, Manickasingham SP, Sporri R, Diebold SS, Schulz O, Sher A, et al. Microbial Recognition Via Toll-Like Receptor-Dependent and -Independent Pathways Determines the Cytokine Response of Murine Dendritic Cell Subsets to Cd40 Triggering. J Immunol (2002) 169(7):3652–60. doi: 10.4049/jimmunol.169.7.3652
182. Wang F, Hou H, Luo Y, Tang G, Wu S, Huang M, et al. The Laboratory Tests and Host Immunity of Covid-19 Patients With Different Severity of Illness. JCI Insight (2020) 5(10):e137799–810. doi: 10.1172/jci.insight.137799
183. Zhao Y, Qin L, Zhang P, Li K, Liang L, Sun J, et al. Longitudinal Covid-19 Profiling Associates Il-1ra and Il-10 With Disease Severity and Rantes With Mild Disease. JCI Insight (2020) 5(13):e139834–45. doi: 10.1172/jci.insight.139834
184. Neumann J, Prezzemolo T, Vanderbeke L, Roca CP, Gerbaux M, Janssens S, et al. Increased Il-10-Producing Regulatory T Cells Are Characteristic of Severe Cases of Covid-19. Clin Transl Immunol (2020) 9(11):e1204. doi: 10.1002/cti2.1204
185. Huang F, Liu X, Sun X, Li Z. Il-10 Served as an Indicator in Severe Covid-19 Patients. J Med Virol (2021) 93(3):1233–5. doi: 10.1002/jmv.26580
186. Sonnenberg GF, Fouser LA, Artis D. Border Patrol: Regulation of Immunity, Inflammation and Tissue Homeostasis at Barrier Surfaces by Il-22. Nat Immunol (2011) 12(5):383–90. doi: 10.1038/ni.2025
187. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of Interleukin 22 But Not Interleukin 17 by a Subset of Human Skin-Homing Memory T Cells. Nat Immunol (2009) 10(8):857–63. doi: 10.1038/ni.1767
188. Gong J, Zhan H, Liang Y, He Q, Cui D. Role of Th22 Cells in Human Viral Diseases. Front Med (Lausanne) (2021) 8:708140. doi: 10.3389/fmed.2021.708140
189. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. Il-22 Increases the Innate Immunity of Tissues. Immunity (2004) 21(2):241–54. doi: 10.1016/j.immuni.2004.07.007
190. Freund E, Stoltz J. [Possibilities of Adjuvant Drug Therapy in Replantation Surgery]. Handchir Mikrochir Plast Chir (1990) 22(4):206–10.
191. Liang SC, Nickerson-Nutter C, Pittman DD, Carrier Y, Goodwin DG, Shields KM, et al. Il-22 Induces an Acute-Phase Response. J Immunol (2010) 185(9):5531–8. doi: 10.4049/jimmunol.0904091
192. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 Mediates Early Host Defense Against Attaching and Effacing Bacterial Pathogens. Nat Med (2008) 14(3):282–9. doi: 10.1038/nm1720
193. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (Il-22) Plays a Protective Role in T Cell-Mediated Murine Hepatitis: Il-22 Is a Survival Factor for Hepatocytes Via Stat3 Activation. Hepatology (2004) 39(5):1332–42. doi: 10.1002/hep.20184
194. Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, et al. Il-22 Regulates the Expression of Genes Responsible for Antimicrobial Defense, Cellular Differentiation, and Mobility in Keratinocytes: A Potential Role in Psoriasis. Eur J Immunol (2006) 36(5):1309–23. doi: 10.1002/eji.200535503
195. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. Il-22 Inhibits Epidermal Differentiation and Induces Proinflammatory Gene Expression and Migration of Human Keratinocytes. J Immunol (2005) 174(6):3695–702. doi: 10.4049/jimmunol.174.6.3695
196. Ellison-Hughes GM, Colley L, O’Brien KA, Roberts KA, Agbaedeng TA, Ross MD. The Role of Msc Therapy in Attenuating the Damaging Effects of the Cytokine Storm Induced by Covid-19 on the Heart and Cardiovascular System. Front Cardiovasc Med (2020) 7:602183. doi: 10.3389/fcvm.2020.602183
197. Tahmasebi S, El-Esawi MA, Mahmoud ZH, Timoshin A, Valizadeh H, Roshangar L, et al. Immunomodulatory Effects of Nanocurcumin on Th17 Cell Responses in Mild and Severe Covid-19 Patients. J Cell Physiol (2021) 236(7):5325–38. doi: 10.1002/jcp.30233
198. Gutierrez-Bautista JF, Rodriguez-Nicolas A, Rosales-Castillo A, Jimenez P, Garrido F, Anderson P, et al. Negative Clinical Evolution in Covid-19 Patients Is Frequently Accompanied With an Increased Proportion of Undifferentiated Th Cells and a Strong Underrepresentation of the Th1 Subset. Front Immunol (2020) 11:596553. doi: 10.3389/fimmu.2020.596553
199. Cua DJ, Tato CM. Innate Il-17-Producing Cells: The Sentinels of the Immune System. Nat Rev Immunol (2010) 10(7):479–89. doi: 10.1038/nri2800
200. McInnes IB, Mease PJ, Ritchlin CT, Rahman P, Gottlieb AB, Kirkham B, et al. Secukinumab Sustains Improvement in Signs and Symptoms of Psoriatic Arthritis: 2 Year Results From the Phase 3 Future 2 Study. Rheumatol (Oxf) (2017) 56(11):1993–2003. doi: 10.1093/rheumatology/kex301
201. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. Il-23 Drives a Pathogenic T Cell Population That Induces Autoimmune Inflammation. J Exp Med (2005) 201(2):233–40. doi: 10.1084/jem.20041257
202. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A Genome-Wide Association Study Identifies Il23r as an Inflammatory Bowel Disease Gene. Science (2006) 314(5804):1461–3. doi: 10.1126/science.1135245
203. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-Producing Cd4+ Effector T Cells Develop Via a Lineage Distinct From the T Helper Type 1 and 2 Lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
204. Isailovic N, Daigo K, Mantovani A, Selmi C. Interleukin-17 and Innate Immunity in Infections and Chronic Inflammation. J Autoimmun (2015) 60:1–11. doi: 10.1016/j.jaut.2015.04.006
205. McGeachy MJ, Cua DJ, Gaffen SL. The Il-17 Family of Cytokines in Health and Disease. Immunity (2019) 50(4):892–906. doi: 10.1016/j.immuni.2019.03.021
206. Eyerich K, Dimartino V, Cavani A. Il-17 and Il-22 in Immunity: Driving Protection and Pathology. Eur J Immunol (2017) 47(4):607–14. doi: 10.1002/eji.201646723
207. Vatsalya V, Li F, Frimodig J, Gala KS, Srivastava S, Kong M, et al. Repurposing Treatment of Wernicke-Korsakoff Syndrome for Th-17 Cell Immune Storm Syndrome and Neurological Symptoms in Covid-19: Thiamine Efficacy and Safety, in-Vitro Evidence and Pharmacokinetic Profile. Front Pharmacol (2020) 11:598128. doi: 10.3389/fphar.2020.598128
208. Mahallawi WH, Khabour OF, Zhang Q, Makhdoum HM, Suliman BA. Mers-Cov Infection in Humans Is Associated With a Pro-Inflammatory Th1 and Th17 Cytokine Profile. Cytokine (2018) 104:8–13. doi: 10.1016/j.cyto.2018.01.025
209. Gaurav R, Anderson DR, Radio SJ, Bailey KL, England BR, Mikuls TR, et al. Il-33 Depletion in Covid-19 Lungs. Chest (2021) 160(5):1656–9. doi: 10.1016/j.chest.2021.06.058
210. Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, et al. Long-Term Il-33-Producing Epithelial Progenitor Cells in Chronic Obstructive Lung Disease. J Clin Invest (2013) 123(9):3967–82. doi: 10.1172/JCI65570
211. Li L, Zhu H, Zuo X. Interleukin-33 in Systemic Sclerosis: Expression and Pathogenesis. Front Immunol (2018) 9:2663. doi: 10.3389/fimmu.2018.02663
212. Wu D, Yang XO. Th17 Responses in Cytokine Storm of Covid-19: An Emerging Target of Jak2 Inhibitor Fedratinib. J Microbiol Immunol Infect (2020) 53(3):368–70. doi: 10.1016/j.jmii.2020.03.005
213. Cayrol C, Girard JP. Interleukin-33 (Il-33): A Nuclear Cytokine From the Il-1 Family. Immunol Rev (2018) 281(1):154–68. doi: 10.1111/imr.12619
214. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in Health and Disease. Nat Rev Immunol (2016) 16(11):676–89. doi: 10.1038/nri.2016.95
215. Burke H, Freeman A, Cellura DC, Stuart BL, Brendish NJ, Poole S, et al. Inflammatory Phenotyping Predicts Clinical Outcome in Covid-19. Respir Res (2020) 21(1):245. doi: 10.1186/s12931-020-01511-z
216. Liang Y, Ge Y, Sun J. Il-33 in Covid-19: Friend or Foe? Cell Mol Immunol (2021) 18(6):1602–4. doi: 10.1038/s41423-021-00685-w
217. Gomez-Cadena A, Spehner L, Kroemer M, Khelil MB, Bouiller K, Verdeil G, et al. Severe Covid-19 Patients Exhibit an Ilc2 Nkg2d(+) Population in Their Impaired Ilc Compartment. Cell Mol Immunol (2021) 18(2):484–6. doi: 10.1038/s41423-020-00596-2
218. Spagnolo P, Balestro E, Aliberti S, Cocconcelli E, Biondini D, Casa GD, et al. Pulmonary Fibrosis Secondary to Covid-19: A Call to Arms? Lancet Respir Med (2020) 8(8):750–2. doi: 10.1016/S2213-2600(20)30222-8
219. Stanczak MA, Sanin DE, Apostolova P, Nerz G, Lampaki D, Hofmann M, et al. Il-33 Expression in Response to Sars-Cov-2 Correlates With Seropositivity in Covid-19 Convalescent Individuals. Nat Commun (2021) 12(1):2133. doi: 10.1038/s41467-021-22449-w
220. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic Characteristics of Bronchoalveolar Lavage Fluid and Peripheral Blood Mononuclear Cells in Covid-19 Patients. Emerg Microbes Infect (2020) 9(1):761–70. doi: 10.1080/22221751.2020.1747363
221. Gresser I. Biologic Effects of Interferons. J Invest Dermatol (1990) 95(6 Suppl):66S–71S. doi: 10.1111/1523-1747.ep12874776
222. Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol (2018) 9:847. doi: 10.3389/fimmu.2018.00847
223. Rojas JM, Alejo A, Martin V, Sevilla N. Viral Pathogen-Induced Mechanisms to Antagonize Mammalian Interferon (Ifn) Signaling Pathway. Cell Mol Life Sci (2021) 78(4):1423–44. doi: 10.1007/s00018-020-03671-z
224. Ivashkiv LB, Donlin LT. Regulation of Type I Interferon Responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
225. Samuel CE. Antiviral Actions of Interferons. Clin Microbiol Rev (2001) 14(4):778–809. doi: 10.1128/CMR.14.4.778-809.2001
226. Clementi N, Ferrarese R, Criscuolo E, Diotti RA, Castelli M, Scagnolari C, et al. Interferon-Beta-1a Inhibition of Severe Acute Respiratory Syndrome-Coronavirus 2 in Vitro When Administered After Virus Infection. J Infect Dis (2020) 222(5):722–5. doi: 10.1093/infdis/jiaa350
227. Kim MH, Salloum S, Wang JY, Wong LP, Regan J, Lefteri K, et al. II, and III Interferon Signatures Correspond to Coronavirus Disease 2019 Severity. J Infect Dis (2021) 224(5):777–82. doi: 10.1093/infdis/jiab288
228. Sohn SY, Hearing J, Mugavero J, Kirillov V, Gorbunova E, Helminiak L, et al. Interferon-Lambda Intranasal Protection and Differential Sex Pathology in a Murine Model of Sars-Cov-2 Infection. mBio (2021) 12(6):e0275621. doi: 10.1128/mBio.02756-21
229. Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay Between Sars-Cov-2 and the Type I Interferon Response. PloS Pathog (2020) 16(7):e1008737. doi: 10.1371/journal.ppat.1008737
230. Ruetsch C, Brglez V, Cremoni M, Zorzi K, Fernandez C, Boyer-Suavet S, et al. Functional Exhaustion of Type I and Ii Interferons Production in Severe Covid-19 Patients. Front Med (Lausanne) (2020) 7:603961. doi: 10.3389/fmed.2020.603961
231. Sallard E, Lescure FX, Yazdanpanah Y, Mentre F, Peiffer-Smadja N. Type 1 Interferons as a Potential Treatment Against Covid-19. Antiviral Res (2020) 178:104791. doi: 10.1016/j.antiviral.2020.104791
232. Gemcioglu E, Davutoglu M, Ozdemir EE, Erden A. Are Type 1 Interferons Treatment in Multiple Sclerosis as a Potential Therapy Against Covid-19? Mult Scler Relat Disord (2020) 42:102196. doi: 10.1016/j.msard.2020.102196
233. Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, et al. Triple Combination of Interferon Beta-1b, Lopinavir-Ritonavir, and Ribavirin in the Treatment of Patients Admitted to Hospital With Covid-19: An Open-Label, Randomised, Phase 2 Trial. Lancet (2020) 395(10238):1695–704. doi: 10.1016/S0140-6736(20)31042-4
234. Davoudi-Monfared E, Rahmani H, Khalili H, Hajiabdolbaghi M, Salehi M, Abbasian L, et al. A Randomized Clinical Trial of the Efficacy and Safety of Interferon Beta-1a in Treatment of Severe Covid-19. Antimicrob Agents Chemother (2020) 64(9):e0106120–34. doi: 10.1128/AAC.01061-20
235. Pandit A, Bhalani N, Bhushan BLS, Koradia P, Gargiya S, Bhomia V, et al. Efficacy and Safety of Pegylated Interferon Alfa-2b in Moderate Covid-19: A Phase Ii, Randomized, Controlled, Open-Label Study. Int J Infect Dis (2021) 105:516–21. doi: 10.1016/j.ijid.2021.03.015
236. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
237. Swiecki M, Colonna M. Type I Interferons: Diversity of Sources, Production Pathways and Effects on Immune Responses. Curr Opin Virol (2011) 1(6):463–75. doi: 10.1016/j.coviro.2011.10.026
238. Ali S, Mann-Nuttel R, Schulze A, Richter L, Alferink J, Scheu S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front Immunol (2019) 10:778. doi: 10.3389/fimmu.2019.00778
239. de Wit E, van Doremalen N, Falzarano D, Munster VJ. Sars and Mers: Recent Insights Into Emerging Coronaviruses. Nat Rev Microbiol (2016) 14(8):523–34. doi: 10.1038/nrmicro.2016.81
240. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened Innate Immune Responses in the Respiratory Tract of Covid-19 Patients. Cell Host Microbe (2020) 27(6):883–90.e2. doi: 10.1016/j.chom.2020.04.017
241. Chu H, Chan JF, Wang Y, Yuen TT, Chai Y, Hou Y, et al. Comparative Replication and Immune Activation Profiles of Sars-Cov-2 and Sars-Cov in Human Lungs: An Ex Vivo Study With Implications for the Pathogenesis of Covid-19. Clin Infect Dis (2020) 71(6):1400–9. doi: 10.1093/cid/ciaa410
242. Trouillet-Assant S, Viel S, Gaymard A, Pons S, Richard JC, Perret M, et al. Type I Ifn Immunoprofiling in Covid-19 Patients. J Allergy Clin Immunol (2020) 146(1):206–8.e2. doi: 10.1016/j.jaci.2020.04.029
243. Kim JS, Lee JY, Yang JW, Lee KH, Effenberger M, Szpirt W, et al. Immunopathogenesis and Treatment of Cytokine Storm in Covid-19. Theranostics (2021) 11(1):316–29. doi: 10.7150/thno.49713
244. Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M, et al. Trials of Anti-Tumour Necrosis Factor Therapy for Covid-19 Are Urgently Needed. Lancet (2020) 395(10234):1407–9. doi: 10.1016/S0140-6736(20)30858-8
245. Li L, Li J, Gao M, Fan H, Wang Y, Xu X, et al. Interleukin-8 as a Biomarker for Disease Prognosis of Coronavirus Disease-2019 Patients. Front Immunol (2020) 11:602395. doi: 10.3389/fimmu.2020.602395
246. Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, et al. Genomic Characterization of the 2019 Novel Human-Pathogenic Coronavirus Isolated From a Patient With Atypical Pneumonia After Visiting Wuhan. Emerg Microbes Infect (2020) 9(1):221–36. doi: 10.1080/22221751.2020.1719902
247. Lee JS, Shin EC. The Type I Interferon Response in Covid-19: Implications for Treatment. Nat Rev Immunol (2020) 20(10):585–6. doi: 10.1038/s41577-020-00429-3
248. Subbian S. The Abstruse Side of Type I Interferon Immunotherapy for Covid-19 Cases With Comorbidities. J Respir (2021) 1(1):49–59. doi: 10.3390/jor1010005
249. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, et al. Autoantibodies Against Type I Ifns in Patients With Life-Threatening Covid-19. Science (2020) 370(6515):eabd4585–98. doi: 10.1126/science.abd4585
250. de Prost N, Bastard P, Arrestier R, Fourati S, Mahevas M, Burrel S, et al. Plasma Exchange to Rescue Patients With Autoantibodies Against Type I Interferons and Life-Threatening Covid-19 Pneumonia. J Clin Immunol (2021) 41(3):536–44. doi: 10.1007/s10875-021-00994-9
251. Anirvan P, Narain S, Hajizadeh N, Aloor FZ, Singh SP, Satapathy SK. Cytokine-Induced Liver Injury in Coronavirus Disease-2019 (Covid-19): Untangling the Knots. Eur J Gastroenterol Hepatol (2021) 33(1S Suppl 1):e42–9. doi: 10.1097/MEG.0000000000002034
252. Steeland S, Libert C, Vandenbroucke RE. A New Venue of Tnf Targeting. Int J Mol Sci (2018) 19(5):1442–97. doi: 10.3390/ijms19051442
253. Gubernatorova EO, Gorshkova EA, Polinova AI, Drutskaya MS. Il-6: Relevance for Immunopathology of Sars-Cov-2. Cytokine Growth Factor Rev (2020) 53:13–24. doi: 10.1016/j.cytogfr.2020.05.009
254. Fu Y, Cheng Y, Wu Y. Understanding Sars-Cov-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol Sin (2020) 35(3):266–71. doi: 10.1007/s12250-020-00207-4
255. Gong J, Dong H, Xia QS, Huang ZY, Wang DK, Zhao Y, et al. Correlation Analysis Between Disease Severity and Inflammation-Related Parameters in Patients With Covid-19: A Retrospective Study. BMC Infect Dis (2020) 20(1):963. doi: 10.1186/s12879-020-05681-5
256. Chen XY, Yan BX, Man XY. Tnfα Inhibitor May Be Effective for Severe Covid-19: Learning From Toxic Epidermal Necrolysis. Ther Adv Respir Dis (2020) 14:1753466620926800. doi: 10.1177/1753466620926800
257. Duret PM, Sebbag E, Mallick A, Gravier S, Spielmann L, Messer L. Recovery From Covid-19 in a Patient With Spondyloarthritis Treated With Tnf-Alpha Inhibitor Etanercept. Ann Rheum Dis (2020) 79(9):1251–2. doi: 10.1136/annrheumdis-2020-217362
258. Zhang R, Wang X, Ni L, Di X, Ma B, Niu S, et al. Covid-19: Melatonin as a Potential Adjuvant Treatment. Life Sci (2020) 250:117583. doi: 10.1016/j.lfs.2020.117583
259. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of Tnf-Alpha and Ifn-Gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in Sars-Cov-2 Infection and Cytokine Shock Syndromes. Cell (2021) 184(1):149–68. doi: 10.1016/j.cell.2020.11.025
260. Mollica Poeta V, Massara M, Capucetti A, Bonecchi R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front Immunol (2019) 10:379. doi: 10.3389/fimmu.2019.00379
261. Khalil BA, Elemam NM, Maghazachi AA. Chemokines and Chemokine Receptors During Covid-19 Infection. Comput Struct Biotechnol J (2021) 19:976–88. doi: 10.1016/j.csbj.2021.01.034
262. Zhang YY, Li BR, Ning BT. The Comparative Immunological Characteristics of Sars-Cov, Mers-Cov, and Sars-Cov-2 Coronavirus Infections. Front Immunol (2020) 11:2033. doi: 10.3389/fimmu.2020.02033
263. Knoll R, Schultze JL, Schulte-Schrepping J. Monocytes and Macrophages in Covid-19. Front Immunol (2021) 12:720109(2952). doi: 10.3389/fimmu.2021.720109
264. Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, et al. Clinical Progression and Viral Load in a Community Outbreak of Coronavirus-Associated Sars Pneumonia: A Prospective Study. Lancet (2003) 361(9371):1767–72. doi: 10.1016/s0140-6736(03)13412-5
265. Liu M, Guo S, Hibbert JM, Jain V, Singh N, Wilson NO, et al. Cxcl10/Ip-10 in Infectious Diseases Pathogenesis and Potential Therapeutic Implications. Cytokine Growth Factor Rev (2011) 22(3):121–30. doi: 10.1016/j.cytogfr.2011.06.001
266. Notz Q, Schmalzing M, Wedekink F, Schlesinger T, Gernert M, Herrmann J, et al. Pro- and Anti-Inflammatory Responses in Severe Covid-19-Induced Acute Respiratory Distress Syndrome-An Observational Pilot Study. Front Immunol (2020) 11:581338. doi: 10.3389/fimmu.2020.581338
267. Shahzad A, Knapp M, Lang I, Kohler G. Interleukin 8 (Il-8) - a Universal Biomarker? Int Arch Med (2010) 3:11. doi: 10.1186/1755-7682-3-11
268. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of Sars-Cov-2 Infected Patients. EBioMedicine (2020) 55:102763. doi: 10.1016/j.ebiom.2020.102763
269. Chien JY, Hsueh PR, Cheng WC, Yu CJ, Yang PC. Temporal Changes in Cytokine/Chemokine Profiles and Pulmonary Involvement in Severe Acute Respiratory Syndrome. Respirology (2006) 11(6):715–22. doi: 10.1111/j.1440-1843.2006.00942.x
270. Chu H, Zhou J, Wong BH, Li C, Cheng ZS, Lin X, et al. Productive Replication of Middle East Respiratory Syndrome Coronavirus in Monocyte-Derived Dendritic Cells Modulates Innate Immune Response. Virology (2014) 454-455:197–205. doi: 10.1016/j.virol.2014.02.018
271. Zhou J, Chu H, Li C, Wong BH, Cheng ZS, Poon VK, et al. Active Replication of Middle East Respiratory Syndrome Coronavirus and Aberrant Induction of Inflammatory Cytokines and Chemokines in Human Macrophages: Implications for Pathogenesis. J Infect Dis (2014) 209(9):1331–42. doi: 10.1093/infdis/jit504
272. Kong SL, Chui P, Lim B, Salto-Tellez M. Elucidating the Molecular Physiopathology of Acute Respiratory Distress Syndrome in Severe Acute Respiratory Syndrome Patients. Virus Res (2009) 145(2):260–9. doi: 10.1016/j.virusres.2009.07.014
273. McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the Inflammatory Response to Severe Covid-19 Illness. Am J Respir Crit Care Med (2020) 202(6):812–21. doi: 10.1164/rccm.202005-1583OC
274. Li S, Jiang L, Li X, Lin F, Wang Y, Li B, et al. Clinical and Pathological Investigation of Patients With Severe Covid-19. JCI Insight (2020) 5(12):e138070–83. doi: 10.1172/jci.insight.138070
275. Chi Y, Ge Y, Wu B, Zhang W, Wu T, Wen T, et al. Serum Cytokine and Chemokine Profile in Relation to the Severity of Coronavirus Disease 2019 in China. J Infect Dis (2020) 222(5):746–54. doi: 10.1093/infdis/jiaa363
276. Lang FM, Lee KM, Teijaro JR, Becher B, Hamilton JA. Gm-Csf-Based Treatments in Covid-19: Reconciling Opposing Therapeutic Approaches. Nat Rev Immunol (2020) 20(8):507–14. doi: 10.1038/s41577-020-0357-7
277. Brown Z, Strieter RM, Neild GH, Thompson RC, Kunkel SL, Westwick J. Il-1 Receptor Antagonist Inhibits Monocyte Chemotactic Peptide 1 Generation by Human Mesangial Cells. Kidney Int (1992) 42(1):95–101. doi: 10.1038/ki.1992.266
278. Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM. Alveolar Macrophage-Derived Cytokines Induce Monocyte Chemoattractant Protein-1 Expression From Human Pulmonary Type Ii-Like Epithelial Cells. J Biol Chem (1991) 266(15):9912–8. doi: 10.1016/S0021-9258(18)92905-4
279. Baggiolini M, Walz A, Kunkel SL. Neutrophil-Activating Peptide-1/Interleukin 8, a Novel Cytokine That Activates Neutrophils. J Clin Invest (1989) 84(4):1045–9. doi: 10.1172/JCI114265
280. Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in Covid-19. Cells (2020) 9(6):1383–91. doi: 10.3390/cells9061383
281. Li H, Zhang J, Fang C, Zhao X, Qian B, Sun Y, et al. The Prognostic Value of Il-8 for the Death of Severe or Critical Patients With Covid-19. Med (Baltimore) (2021) 100(11):e23656. doi: 10.1097/MD.0000000000023656
282. Wilson JG, Simpson LJ, Ferreira AM, Rustagi A, Roque J, Asuni A, et al. Cytokine Profile in Plasma of Severe Covid-19 Does Not Differ From Ards and Sepsis. JCI Insight (2020) 5(17):e140289–95. doi: 10.1172/jci.insight.140289
283. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T-Cells and Inflammatory Monocytes Incite Inflammatory Storms in Severe COVID-19 Patients. Natl Sci Rev (2020) 7(6):998–1002. doi: 10.1093/nsr/nwaa041
284. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. Gm-Csf Inhibition Reduces Cytokine Release Syndrome and Neuroinflammation But Enhances Car-T Cell Function in Xenografts. Blood (2019) 133(7):697–709. doi: 10.1182/blood-2018-10-881722
285. Barna BP, Pettay J, Barnett GH, Zhou P, Iwasaki K, Estes ML. Regulation of Monocyte Chemoattractant Protein-1 Expression in Adult Human Non-Neoplastic Astrocytes Is Sensitive to Tumor Necrosis Factor (Tnf) or Antibody to the 55-Kda Tnf Receptor. J Neuroimmunol (1994) 50(1):101–7. doi: 10.1016/0165-5728(94)90220-8
286. Singh S, Anshita D, Ravichandiran V. Mcp-1: Function, Regulation, and Involvement in Disease. Int Immunopharmacol (2021) 101(Pt B):107598. doi: 10.1016/j.intimp.2021.107598
287. Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, et al. Minimally Modified Low Density Lipoprotein Induces Monocyte Chemotactic Protein 1 in Human Endothelial Cells and Smooth Muscle Cells. Proc Natl Acad Sci USA (1990) 87(13):5134–8. doi: 10.1073/pnas.87.13.5134
288. Ajuebor MN, Flower RJ, Hannon R, Christie M, Bowers K, Verity A, et al. Endogenous Monocyte Chemoattractant Protein-1 Recruits Monocytes in the Zymosan Peritonitis Model. J Leukoc Biol (1998) 63(1):108–16. doi: 10.1002/jlb.63.1.108
289. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte Chemoattractant Protein-1 (Mcp-1): An Overview. J Interferon Cytokine Res (2009) 29(6):313–26. doi: 10.1089/jir.2008.0027
290. Dando SJ, Mackay-Sim A, Norton R, Currie BJ, St John JA, Ekberg JA, et al. Pathogens Penetrating the Central Nervous System: Infection Pathways and the Cellular and Molecular Mechanisms of Invasion. Clin Microbiol Rev (2014) 27(4):691–726. doi: 10.1128/CMR.00118-13
291. Meeker RB, Williams K, Killebrew DA, Hudson LC. Cell Trafficking Through the Choroid Plexus. Cell Adh Migr (2012) 6(5):390–6. doi: 10.4161/cam.21054
292. Gram M, Sveinsdottir S, Cinthio M, Sveinsdottir K, Hansson SR, Morgelin M, et al. Extracellular Hemoglobin - Mediator of Inflammation and Cell Death in the Choroid Plexus Following Preterm Intraventricular Hemorrhage. J Neuroinflamm (2014) 11:200. doi: 10.1186/s12974-014-0200-9
293. Kempuraj D, Selvakumar GP, Ahmed ME, Raikwar SP, Thangavel R, Khan A, et al. Covid-19, Mast Cells, Cytokine Storm, Psychological Stress, and Neuroinflammation. Neuroscientist (2020) 26(5-6):402–14. doi: 10.1177/1073858420941476
294. Bulow Anderberg S, Luther T, Berglund M, Larsson R, Rubertsson S, Lipcsey M, et al. Increased Levels of Plasma Cytokines and Correlations to Organ Failure and 30-Day Mortality in Critically Ill Covid-19 Patients. Cytokine (2021) 138:155389. doi: 10.1016/j.cyto.2020.155389
295. Imaizumi T, Yoshida H, Satoh K. Regulation of Cx3cl1/Fractalkine Expression in Endothelial Cells. J Atheroscler Thromb (2004) 11(1):15–21. doi: 10.5551/jat.11.15
296. Hildemann SK, Schulz C, Fraccarollo D, Schopp C, Flierl U, Wissel K, et al. Fractalkine Promotes Platelet Activation and Vascular Dysfunction in Congestive Heart Failure. Thromb Haemost (2014) 111(4):725–35. doi: 10.1160/TH13-08-0640
297. Flierl U, Bauersachs J, Schafer A. Modulation of Platelet and Monocyte Function by the Chemokine Fractalkine (Cx3 Cl1) in Cardiovascular Disease. Eur J Clin Invest (2015) 45(6):624–33. doi: 10.1111/eci.12443
298. Tong M, Jiang Y, Xia D, Xiong Y, Zheng Q, Chen F, et al. Elevated Expression of Serum Endothelial Cell Adhesion Molecules in Covid-19 Patients. J Infect Dis (2020) 222(6):894–8. doi: 10.1093/infdis/jiaa349
299. Mohammadhosayni M, Sadat Mohammadi F, Ezzatifar F, Mahdavi Gorabi A, Khosrojerdi A, Aslani S, et al. Matrix Metalloproteinases Are Involved in the Development of Neurological Complications in Patients With Coronavirus Disease 2019. Int Immunopharmacol (2021) 100:108076. doi: 10.1016/j.intimp.2021.108076
300. Song B, Zhang ZZ, Zhong JC, Yu XY, Oudit GY, Jin HY, et al. Loss of Angiotensin-Converting Enzyme 2 Exacerbates Myocardial Injury Via Activation of the Ctgf-Fractalkine Signaling Pathway. Circ J (2013) 77(12):2997–3006. doi: 10.1253/circj.cj-13-0805
301. Rivas-Fuentes S, Valdes VJ, Espinosa B, Gorocica-Rosete P, Salgado-Aguayo A. Could Sars-Cov-2 Blocking of Ace2 in Endothelial Cells Result in Upregulation of Cx3cl1, Promoting Thrombosis in Covid-19 Patients? Med Hypotheses (2021) 151:110570. doi: 10.1016/j.mehy.2021.110570
302. Channappanavar R, Perlman S. Pathogenic Human Coronavirus Infections: Causes and Consequences of Cytokine Storm and Immunopathology. Semin Immunopathol (2017) 39(5):529–39. doi: 10.1007/s00281-017-0629-x
303. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA (2020) 323(11):1061–9. doi: 10.1001/jama.2020.1585
304. Hu B, Guo H, Zhou P, Shi ZL. Characteristics of Sars-Cov-2 and Covid-19. Nat Rev Microbiol (2021) 19(3):141–54. doi: 10.1038/s41579-020-00459-7
305. Plüddemann A, Aronson JK. What Is the Role of T Cells in Covid-19 Infection? Why Immunity Is About More Than Antibodies. Cent Evidence-Based Med Evid Serv Support COVID-19 Response (2020).
306. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walters P. Molecular Biology of the Cell. 4th ed. New York: Garland Science (2002).
307. Tang L, Yin Z, Hu Y, Mei H. Controlling Cytokine Storm Is Vital in Covid-19. Front Immunol (2020) 11:570993. doi: 10.3389/fimmu.2020.570993
308. Harrison AG, Lin T, Wang P. Mechanisms of Sars-Cov-2 Transmission and Pathogenesis. Trends Immunol (2020) 41(12):1100–15. doi: 10.1016/j.it.2020.10.004
309. Gavriatopoulou M, Korompoki E, Fotiou D, Ntanasis-Stathopoulos I, Psaltopoulou T, Kastritis E, et al. Organ-Specific Manifestations of Covid-19 Infection. Clin Exp Med (2020) 20(4):493–506. doi: 10.1007/s10238-020-00648-x
310. Catanzaro M, Fagiani F, Racchi M, Corsini E, Govoni S, Lanni C. Immune Response in Covid-19: Addressing a Pharmacological Challenge by Targeting Pathways Triggered by Sars-Cov-2. Signal Transduct Target Ther (2020) 5(1):84. doi: 10.1038/s41392-020-0191-1
311. Robba C, Battaglini D, Pelosi P, Rocco PRM. Multiple Organ Dysfunction in Sars-Cov-2: Mods-Cov-2. Expert Rev Respir Med (2020) 14(9):865–8. doi: 10.1080/17476348.2020.1778470
312. Tsai PH, Lai WY, Lin YY, Luo YH, Lin YT, Chen HK, et al. Clinical Manifestation and Disease Progression in Covid-19 Infection. J Chin Med Assoc (2021) 84(1):3–8. doi: 10.1097/jcma.0000000000000463
313. Rowaiye AB, Okpalefe OA, Onuh Adejoke O, Ogidigo JO, Hannah Oladipo O, Ogu AC, et al. Attenuating the Effects of Novel Covid-19 (Sars-Cov-2) Infection-Induced Cytokine Storm and the Implications. J Inflamm Res (2021) 14:1487–510. doi: 10.2147/JIR.S301784
314. Diamond M, Peniston HL, Sanghavi D, Mahapatra S. Acute Respiratory Distress Syndrome. Treasure Island, FL: Statpearls (2021).
315. Templin C, Ghadri JR, Diekmann J, Napp LC, Bataiosu DR, Jaguszewski M, et al. Clinical Features and Outcomes of Takotsubo (Stress) Cardiomyopathy. N Engl J Med (2015) 373(10):929–38. doi: 10.1056/NEJMoa1406761
316. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical Predictors of Mortality Due to Covid-19 Based on an Analysis of Data of 150 Patients From Wuhan, China. Intensive Care Med (2020) 46(5):846–8. doi: 10.1007/s00134-020-05991-x
317. Gao Y, Li T, Han M, Li X, Wu D, Xu Y, et al. Diagnostic Utility of Clinical Laboratory Data Determinations for Patients With the Severe Covid-19. J Med Virol (2020) 92(7):791–6. doi: 10.1002/jmv.25770
318. Sun D, Li H, Lu XX, Xiao H, Ren J, Zhang FR, et al. Clinical Features of Severe Pediatric Patients With Coronavirus Disease 2019 in Wuhan: A Single Center’s Observational Study. World J Pediatr (2020) 16(3):251–9. doi: 10.1007/s12519-020-00354-4
319. Liu Y, Du X, Chen J, Jin Y, Peng L, Wang HHX, et al. Neutrophil-To-Lymphocyte Ratio as an Independent Risk Factor for Mortality in Hospitalized Patients With Covid-19. J Infect (2020) 81(1):e6–e12. doi: 10.1016/j.jinf.2020.04.002
320. Zhang B, Zhou X, Qiu Y, Song Y, Feng F, Feng J, et al. Clinical Characteristics of 82 Cases of Death From Covid-19. PloS One (2020) 15(7):e0235458. doi: 10.1371/journal.pone.0235458
321. Jin JM, Bai P, He W, Wu F, Liu XF, Han DM, et al. Gender Differences in Patients With Covid-19: Focus on Severity and Mortality. Front Public Health (2020) 8:152. doi: 10.3389/fpubh.2020.00152
322. Vasarmidi E, Tsitoura E, Spandidos DA, Tzanakis N, Antoniou KM. Pulmonary Fibrosis in the Aftermath of the Covid-19 Era (Review). Exp Ther Med (2020) 20(3):2557–60. doi: 10.3892/etm.2020.8980
323. Fraser E. Long Term Respiratory Complications of Covid-19. BMJ (2020) 370:m3001. doi: 10.1136/bmj.m3001
324. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological Findings of Covid-19 Associated With Acute Respiratory Distress Syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
325. Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, et al. Nervous System Involvement After Infection With Covid-19 and Other Coronaviruses. Brain Behav Immun (2020) 87:18–22. doi: 10.1016/j.bbi.2020.03.031
326. Nagu P, Parashar A, Behl T, Mehta V. Cns Implications of Covid-19: A Comprehensive Review. Rev Neurosci (2021) 32(2):219–34. doi: 10.1515/revneuro-2020-0070
327. Tang N, Li D, Wang X, Sun Z. Abnormal Coagulation Parameters Are Associated With Poor Prognosis in Patients With Novel Coronavirus Pneumonia. J Thromb Haemost (2020) 18(4):844–7. doi: 10.1111/jth.14768
328. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet (2020) 395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7
329. Han H, Yang L, Liu R, Liu F, Wu KL, Li J, et al. Prominent Changes in Blood Coagulation of Patients With Sars-Cov-2 Infection. Clin Chem Lab Med (2020) 58(7):1116–20. doi: 10.1515/cclm-2020-0188
330. Zhang B, Zhou X, Qiu Y, Song Y, Feng F, Feng J, et al. Clinical Characteristics of 82 Death Cases With Covid-19. PloS one (2020) 15(7):e0235458–71. doi: 10.1371/journal.pone.0235458
331. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (Covid-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA (2020) 323(13):1239–42. doi: 10.1001/jama.2020.2648
332. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382(18):1708–20. doi: 10.1056/NEJMoa2002032
333. Peckham H, de Gruijter NM, Raine C, Radziszewska A, Ciurtin C, Wedderburn LR, et al. Male Sex Identified by Global Covid-19 Meta-Analysis as a Risk Factor for Death and Itu Admission. Nat Commun (2020) 11(1):6317. doi: 10.1038/s41467-020-19741-6
334. Scully EP, Haverfield J, Ursin RL, Tannenbaum C, Klein SL. Considering How Biological Sex Impacts Immune Responses and Covid-19 Outcomes. Nat Rev Immunol (2020) 20(7):442–7. doi: 10.1038/s41577-020-0348-8
335. Asadi-Pooya AA, Simani L. Central Nervous System Manifestations of Covid-19: A Systematic Review. J Neurol Sci (2020) 413:116832. doi: 10.1016/j.jns.2020.116832
336. Bohmwald K, Galvez NMS, Rios M, Kalergis AM. Neurologic Alterations Due to Respiratory Virus Infections. Front Cell Neurosci (2018) 12:386. doi: 10.3389/fncel.2018.00386
337. Chang WT, Toh HS, Liao CT, Yu WL. Cardiac Involvement of Covid-19: A Comprehensive Review. Am J Med Sci (2021) 361(1):14–22. doi: 10.1016/j.amjms.2020.10.002
338. Bugert CL, Kwiat V, Valera IC, Bugert JJ, Parvatiyar MS. Cardiovascular Injury Due to Sars-Cov-2. Curr Clin Microbiol Rep (2021) 8:167–77. doi: 10.1007/s40588-021-00160-0
339. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (Covid-19). JAMA Cardiol (2020) 5(7):811–8. doi: 10.1001/jamacardio.2020.1017
340. Katsiki N, Banach M, Mikhailidis DP. Lipid-Lowering Therapy and Renin-Angiotensin-Aldosterone System Inhibitors in the Era of the Covid-19 Pandemic. Arch Med Sci (2020) 16(3):485–9. doi: 10.5114/aoms.2020.94503
341. Lin JE, Asfour A, Sewell TB, Hooe B, Pryce P, Earley C, et al. Neurological Issues in Children With Covid-19. Neurosci Lett (2021) 743:135567. doi: 10.1016/j.neulet.2020.135567
342. Team CC-R, Jorden MA, Rudman SL, Villarino E, Hoferka S, Patel MT, et al. Evidence for Limited Early Spread of Covid-19 Within the United States, January-February 2020. MMWR Morb Mortal Wkly Rep (2020) 69(22):680–4. doi: 10.15585/mmwr.mm6922e1
343. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol (2020) 77(6):683–90. doi: 10.1001/jamaneurol.2020.1127
344. Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N Engl J Med (2020) 382(20):e60. doi: 10.1056/NEJMc2009787
345. Yaghi S, Ishida K, Torres J, Mac Grory B, Raz E, Humbert K, et al. Sars-Cov-2 and Stroke in a New York Healthcare System. Stroke (2020) 51(7):2002–11. doi: 10.1161/STROKEAHA.120.030335
346. Pilotto A, Odolini S, Masciocchi S, Comelli A, Volonghi I, Gazzina S, et al. Steroid-Responsive Encephalitis in Coronavirus Disease 2019. Ann Neurol (2020) 88(2):423–7. doi: 10.1002/ana.25783
347. Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, et al. Guillain-Barre Syndrome Associated With Sars-Cov-2. N Engl J Med (2020) 382(26):2574–6. doi: 10.1056/NEJMc2009191
348. Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, et al. A First Case of Meningitis/Encephalitis Associated With Sars-Coronavirus-2. Int J Infect Dis (2020) 94:55–8. doi: 10.1016/j.ijid.2020.03.062
349. Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. Covid-19-Associated Acute Hemorrhagic Necrotizing Encephalopathy: Imaging Features. Radiology (2020) 296(2):E119–E20. doi: 10.1148/radiol.2020201187
350. Bhaskar S, Rastogi A, Chattu VK, Adisesh A, Thomas P, Alvarado N, et al. Key Strategies for Clinical Management and Improvement of Healthcare Services for Cardiovascular Disease and Diabetes Patients in the Coronavirus (Covid-19) Settings: Recommendations From the Reprogram Consortium. Front Cardiovasc Med (2020) 7:112. doi: 10.3389/fcvm.2020.00112
351. Wong SH, Lui RN, Sung JJ. Covid-19 and the Digestive System. J Gastroenterol Hepatol (2020) 35(5):744–8. doi: 10.1111/jgh.15047
352. Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary Manifestations of Covid-19. Nat Med (2020) 26(7):1017–32. doi: 10.1038/s41591-020-0968-3
353. Banach M, Penson PE, Fras Z, Vrablik M, Pella D, Reiner Z, et al. Brief Recommendations on the Management of Adult Patients With Familial Hypercholesterolemia During the Covid-19 Pandemic. Pharmacol Res (2020) 158:104891. doi: 10.1016/j.phrs.2020.104891
354. Doyen D, Moceri P, Ducreux D, Dellamonica J. Myocarditis in a Patient With Covid-19: A Cause of Raised Troponin and Ecg Changes. Lancet (2020) 395(10235):1516. doi: 10.1016/S0140-6736(20)30912-0
355. Bhaskar S, Sinha A, Banach M, Mittoo S, Weissert R, Kass JS, et al. Cytokine Storm in Covid-19-Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The Reprogram Consortium Position Paper. Front Immunol (2020) 11:1648. doi: 10.3389/fimmu.2020.01648
356. Basu-Ray I, Almaddah NK, Adeboye A, Soos MP. Cardiac Manifestations of Coronavirus (Covid-19). In: Statpearls. Treasure Island, FL: StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC (2021).
357. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With Covid-19 in Wuhan, China. JAMA Cardiol (2020) 5(7):802–10. doi: 10.1001/jamacardio.2020.0950
358. Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-Angiotensin-Aldosterone System Inhibitors in Patients With Covid-19. N Engl J Med (2020) 382(17):1653–9. doi: 10.1056/NEJMsr2005760
359. Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, et al. Il-6/Il-6r Axis Plays a Critical Role in Acute Kidney Injury. J Am Soc Nephrol (2008) 19(6):1106–15. doi: 10.1681/ASN.2007070744
360. Desai TR, Leeper NJ, Hynes KL, Gewertz BL. Interleukin-6 Causes Endothelial Barrier Dysfunction Via the Protein Kinase C Pathway. J Surg Res (2002) 104(2):118–23. doi: 10.1006/jsre.2002.6415
361. Taverna G, Di Francesco S, Borroni EM, Yiu D, Toniato E, Milanesi S, et al. The Kidney, Covid-19, and the Chemokine Network: An Intriguing Trio. Int Urol Nephrol (2021) 53(1):97–104. doi: 10.1007/s11255-020-02579-8
362. Legrand M, Bell S, Forni L, Joannidis M, Koyner JL, Liu K, et al. Pathophysiology of Covid-19-Associated Acute Kidney Injury. Nat Rev Nephrol (2021) 17(11):751–64. doi: 10.1038/s41581-021-00452-0
363. Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY, et al. Renal Histopathological Analysis of 26 Postmortem Findings of Patients With Covid-19 in China. Kidney Int (2020) 98(1):219–27. doi: 10.1016/j.kint.2020.04.003
364. Zhang C, Shi L, Wang FS. Liver Injury in Covid-19: Management and Challenges. Lancet Gastroenterol Hepatol (2020) 5(5):428–30. doi: 10.1016/S2468-1253(20)30057-1
365. Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, et al. Specific Ace2 Expression in Cholangiocytes May Cause Liver Damage After 2019-Ncov Infection. bioRxiv (2020):2020.02.03.931766. doi: 10.1101/2020.02.03.931766
366. Alqahtani SA, Schattenberg JM. Liver Injury in Covid-19: The Current Evidence. United Eur Gastroenterol J (2020) 8(5):509–19. doi: 10.1177/2050640620924157
367. Zhao B, Ni C, Gao R, Wang Y, Yang L, Wei J, et al. Recapitulation of Sars-Cov-2 Infection and Cholangiocyte Damage With Human Liver Ductal Organoids. Protein Cell (2020) 11(10):771–5. doi: 10.1007/s13238-020-00718-6
368. Feng G, Zheng KI, Yan QQ, Rios RS, Targher G, Byrne CD, et al. Covid-19 and Liver Dysfunction: Current Insights and Emergent Therapeutic Strategies. J Clin Transl Hepatol (2020) 8(1):18–24. doi: 10.14218/JCTH.2020.00018
369. Portincasa P, Krawczyk M, Machill A, Lammert F, Di Ciaula A. Hepatic Consequences of Covid-19 Infection. Lapping or Biting? Eur J Intern Med (2020) 77:18–24. doi: 10.1016/j.ejim.2020.05.035
370. Ferron PJ, Gicquel T, Megarbane B, Clement B, Fromenty B. Treatments in Covid-19 Patients With Pre-Existing Metabolic Dysfunction-Associated Fatty Liver Disease: A Potential Threat for Drug-Induced Liver Injury? Biochimie (2020) 179:266–74. doi: 10.1016/j.biochi.2020.08.018
371. Fan Z, Chen L, Li J, Cheng X, Yang J, Tian C, et al. Clinical Features of Covid-19-Related Liver Functional Abnormality. Clin Gastroenterol Hepatol (2020) 18(7):1561–6. doi: 10.1016/j.cgh.2020.04.002
372. Kang SH, Cho DH, Choi J, Baik SK, Gwon JG, Kim MY. Association Between Chronic Hepatitis B Infection and Covid-19 Outcomes: A Korean Nationwide Cohort Study. PloS One (2021) 16(10):e0258229. doi: 10.1371/journal.pone.0258229
373. Xu L, Liu J, Lu M, Yang D, Zheng X. Liver Injury During Highly Pathogenic Human Coronavirus Infections. Liver Int (2020) 40(5):998–1004. doi: 10.1111/liv.14435
374. Lingappan K, Karmouty-Quintana H, Davies J, Akkanti B, Harting MT. Understanding the Age Divide in Covid-19: Why Are Children Overwhelmingly Spared? Am J Physiol Lung Cell Mol Physiol (2020) 319(1):L39–44. doi: 10.1152/ajplung.00183.2020
375. Parri N, Lenge M, Buonsenso D. Coronavirus Infection in Pediatric Emergency Departments Research G. Children With Covid-19 in Pediatric Emergency Departments in Italy. N Engl J Med (2020) 383(2):187–90. doi: 10.1056/NEJMc2007617
376. Sperotto F, Friedman KG, Son MBF, VanderPluym CJ, Newburger JW, Dionne A. Cardiac Manifestations in Sars-Cov-2-Associated Multisystem Inflammatory Syndrome in Children: A Comprehensive Review and Proposed Clinical Approach. Eur J Pediatr (2021) 180(2):307–22. doi: 10.1007/s00431-020-03766-6
377. Buszko M, Nita-Lazar A, Park JH, Schwartzberg PL, Verthelyi D, Young HA, et al. Lessons Learned: New Insights on the Role of Cytokines in Covid-19. Nat Immunol (2021) 22(4):404–11. doi: 10.1038/s41590-021-00901-9
378. Henderson LA, Canna SW, Schulert GS, Volpi S, Lee PY, Kernan KF, et al. On the Alert for Cytokine Storm: Immunopathology in Covid-19. Arthritis Rheumatol (2020) 72(7):1059–63. doi: 10.1002/art.41285
379. Consiglio CR, Cotugno N, Sardh F, Pou C, Amodio D, Rodriguez L, et al. The Immunology of Multisystem Inflammatory Syndrome in Children With Covid-19. Cell (2020) 183(4):968–81 e7. doi: 10.1016/j.cell.2020.09.016
380. Cheung EW, Zachariah P, Gorelik M, Boneparth A, Kernie SG, Orange JS, et al. Multisystem Inflammatory Syndrome Related to Covid-19 in Previously Healthy Children and Adolescents in New York City. JAMA (2020) 324(3):294–6. doi: 10.1001/jama.2020.10374
381. Dufort EM, Koumans EH, Chow EJ, Rosenthal EM, Muse A, Rowlands J, et al. Multisystem Inflammatory Syndrome in Children in New York State. N Engl J Med (2020) 383(4):347–58. doi: 10.1056/NEJMoa2021756
382. Abdel-Mannan O, Eyre M, Lobel U, Bamford A, Eltze C, Hameed B, et al. Neurologic and Radiographic Findings Associated With Covid-19 Infection in Children. JAMA Neurol (2020) 77(11):1440–5. doi: 10.1001/jamaneurol.2020.2687
Keywords: COVID-19, cytokines, chemokines, infection, diagnostic markers
Citation: Hsu R-J, Yu W-C, Peng G-R, Ye C-H, Hu S, Chong PCT, Yap KY, Lee JYC, Lin W-C and Yu S-H (2022) The Role of Cytokines and Chemokines in Severe Acute Respiratory Syndrome Coronavirus 2 Infections. Front. Immunol. 13:832394. doi: 10.3389/fimmu.2022.832394
Received: 09 December 2021; Accepted: 24 February 2022;
Published: 07 April 2022.
Edited by:
Rabih Halwani, University of Sharjah, United Arab EmiratesReviewed by:
Nima Taefehshokr, Western University, CanadaBalachandar Selvakumar, University of Sharjah, United Arab Emirates
Francesca Coperchini, University of Pavia, Italy
Copyright © 2022 Hsu, Yu, Peng, Ye, Hu, Chong, Yap, Lee, Lin and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-Han Yu, c2h1aGFueXVAbnR1LmVkdS50dw==
†These authors have contributed equally to this work