 Jin Kyung Kim
Jin Kyung Kim Eun-Jin Park
Eun-Jin Park Eun-Kyeong Jo
Eun-Kyeong Jo- 1Department of Microbiology, Chungnam National University College of Medicine, Daejeon, South Korea
- 2Infection Control Convergence Research Center, Chungnam National University College of Medicine, Daejeon, South Korea
Immune metabolic regulation shapes the host-pathogen interaction during infection with Mycobacterium tuberculosis (Mtb), the pathogen of human tuberculosis (TB). Several immunometabolites generated by metabolic remodeling in macrophages are implicated in innate immune protection against Mtb infection by fine-tuning defensive pathways. Itaconate, produced by the mitochondrial enzyme immunoresponsive gene 1 (IRG1), has antimicrobial and anti-inflammatory effects, restricting intracellular mycobacterial growth. L-arginine, a component of the urea cycle, is critical for the synthesis of nitric oxide (NO) and is implicated in M1-mediated antimycobacterial responses in myeloid cells. L-citrulline, a by-product of NO production, contributes to host defense and generates L-arginine in myeloid cells. In arginase 1-expressing cells, L-arginine can be converted into ornithine, a polyamine precursor that enhances autophagy and antimicrobial protection against Mtb in Kupffer cells. Gamma-aminobutyric acid (GABA), a metabolite and neurotransmitter, activate autophagy to induce antimycobacterial host defenses. This review discusses the recent updates of the functions of the three metabolites in host protection against mycobacterial infection. Understanding the mechanisms by which these metabolites promote host defense will facilitate the development of novel host-directed therapeutics against Mtb and drug-resistant bacteria.
Introduction
Metabolites function as innate immune effectors against intracellular bacterial infections, including Mycobacterium tuberculosis (Mtb), the primary pathogen of human tuberculosis (TB). The roles of several metabolites have been determined in host defense against Mtb infection. A significant advance occurred with identifying itaconate, produced by immunoresponsive gene 1 (Irg1) enzymatic activity, in the growth inhibition of mycobacteria possessing isocitrate lyase (1, 2). A recent discovery (3, 4) regarding the anti-inflammatory function of itaconate points to a role in regulating pathological inflammation during Mtb infection. In addition, an earlier study showed that L-arginine (Arg) metabolism is closely related to bacteriostatic activity in macrophages (5). L-Arg consumption, which is accompanied by nitrite/nitrate production, and L-citrulline exert a fungistatic effect in murine macrophages (5). When produced by arginase-expressing macrophages, L-citrulline activates antimycobacterial responses (6). Ornithine, another metabolite of the L-Arg metabolic pathway, is mainly produced by Kupffer cells in the liver and participates in host defense by activating autophagy (7). Furthermore, γ-aminobutyric acid (GABA), a metabolite and neurotransmitter, activates autophagy to induce innate host defenses against intracellular bacteria, including mycobacteria and salmonella (8).
In recent years, several comprehensive studies have indicated the immunoregulatory functions of these metabolites during host-pathogen interactions in Mtb infection. Here, we focus on the roles and mechanisms by which three metabolites—itaconate, Arg, and GABA—enhance host defenses in macrophages during Mtb infection. Understanding the molecular mechanisms by which metabolites modulate host innate immune pathways will provide therapeutic insight into emerging diseases and human TB.
Overview of Immunometabolism in Host-Mtb Infections
Macrophages are the principal host phagocytes for Mtb at sites of infection. After phagocytosis by macrophages, Mtb can reside in the phagosomes and circumvent host immune protection by resisting phagolysosomal acidification (9–12). Several macrophage populations are implicated in innate immune defense and infectious pathogenesis, depending on the context (4, 11, 13). Macrophages initiate intracellular innate immune signaling to activate early inflammatory responses following recognition of Mtb and/or its components via specific pattern-recognition receptors (10, 12, 14). Macrophages can be categorized into two major types, i.e., classically activated (M1) and alternatively activated (M2). M1 macrophages exhibit high microbicidal activity and produce proinflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β, and inducible nitric oxide synthase (iNOS). By contrast, M2 macrophages participate in tissue repair and produce IL-10, tumor growth factor (TGF)-β, and anti-inflammatory cytokines (15, 16). In addition, macrophages have multiple antimicrobial pathways linking innate immune signaling to various effector mechanisms, including cell-autonomous autophagy pathways. Autophagy activation enhances phagosomal maturation to promote host defense against intracellular mycobacterial infection (10, 12, 17–20). Mtb has multiple strategies for manipulating host innate immune responses and escaping from autophagy to survive inside macrophages (10, 12). A deeper understanding of molecular crosstalks between Mtb and host cells would facilitate the development of new therapeutic strategies against human TB, particularly drug-resistant TB.
Macrophages and other immune cells have distinct metabolic and bioenergetics requirements at different stages of infection (21). Studies involving nonhuman primate TB granulomas showed that M1 polarization is related to a favorable disease outcome (22). During infection, TB granulomas are the active sites of host-Mtb interactions, where Mtb develops mechanisms to resist host protective immunity, and the host localizes the condition (23–26). The alveolar macrophages representing bacterial permissiveness are associated with fatty acid β-oxidation, when compared with glycolytically active interstitial macrophages (27). Metabolic rewiring and epigenetic reprogramming are critical for M. bovis bacillus Calmette-Guérin (BCG)-mediated trained immunity, i.e., long-term immune response to infection or vaccination (28–30). Therefore, the orchestrating immunologic and metabolic responses may determine the outcome of Mtb infection. A general overview of immune metabolic remodeling profiles and the underlying mechanisms are reviewed elsewhere (21, 31, 32) and not discussed here.
Role of Itaconate During Mycobacterial Infection
Conversion of isocitrate to succinate and glyoxylate by isocitrate lyase is the first step in the glyoxylate shunt in several bacteria, including Mtb (33–35), and is involved in Mtb survival during chronic infection (1, 36). Michelucci et al. showed that itaconic acid (also known as methylenesuccinic acid), produced by Irg1, restricts the growth of bacteria harboring isocitrate lyase, such as Mtb and Salmonella enterica (1). In addition, itaconate and itaconyl-CoA target methylcitrate lyase in the methylcitrate cycle and B12-dependent methylmalonyl-CoA mutase, respectively, to restrict bacterial growth (1, 37). However, a recent report demonstrates that the Mtb effector Rv2498c, a bifunctional β-hydroxyacyl-CoA lyase, participates in itaconate dissimilation to confer resistance to itaconate (38). Notably, the MtbΔrv2498c strain shows significantly attenuated virulence in a mouse low-dose aerosol infection model (38). Itaconate also has an immunomodulatory function during infection. Irg1, a mitochondrial enzyme for itaconate synthesis, promotes antimicrobial immune responses against Mtb infection by regulating neutrophil-mediated pathological inflammation during infection (39). Indeed, Irg1 suppresses the production of proinflammatory cytokines and reactive oxygen species (ROS). Significantly, treatment of Irg1-/- bone marrow-derived macrophages with a physiologically relevant dose of itaconate (0.25 mM) shifts the transcriptional patterns toward wild-type macrophages (39). Although Irg1-mediated itaconate production is essential for antimycobacterial responses by regulating excessive pulmonary inflammation, it is unclear how Irg1 regulates inflammatory responses in the context of Mtb infection.
Recently, several studies highlighted using the cell-permeable itaconate derivatives to enhance intracellular delivery. Lampropoulou et al. showed that exogenous dimethyl itaconate at physiological doses markedly inhibits S. typhimurium-induced IL-1β, nitric oxide (NO), but not TNF-α, in macrophages (40). The underlying mechanisms are intriguing because both NO and IL-1β are crucial components of antimycobacterial immune responses in murine models (41–44). Itaconate treatment also increases the extracellular acidification rate and inhibits succinate dehydrogenase (Sdh), complex II of the mitochondrial electron transport chain, decreasing mitochondrial respiration (40). However, it is unclear whether itaconate-mediated antimycobacterial responses are associated with increased aerobic glycolysis. Moreover, treatment of human primary macrophages with TNF-α and IL-6 suppresses the intracellular growth of M. avium through by-stander effects via inducing expression of IRG1 (45). Although the endogenous itaconate level is low, direct delivery of itaconate to M. avium phagosomes may contribute to antimicrobial responses (45) (Figure 1).
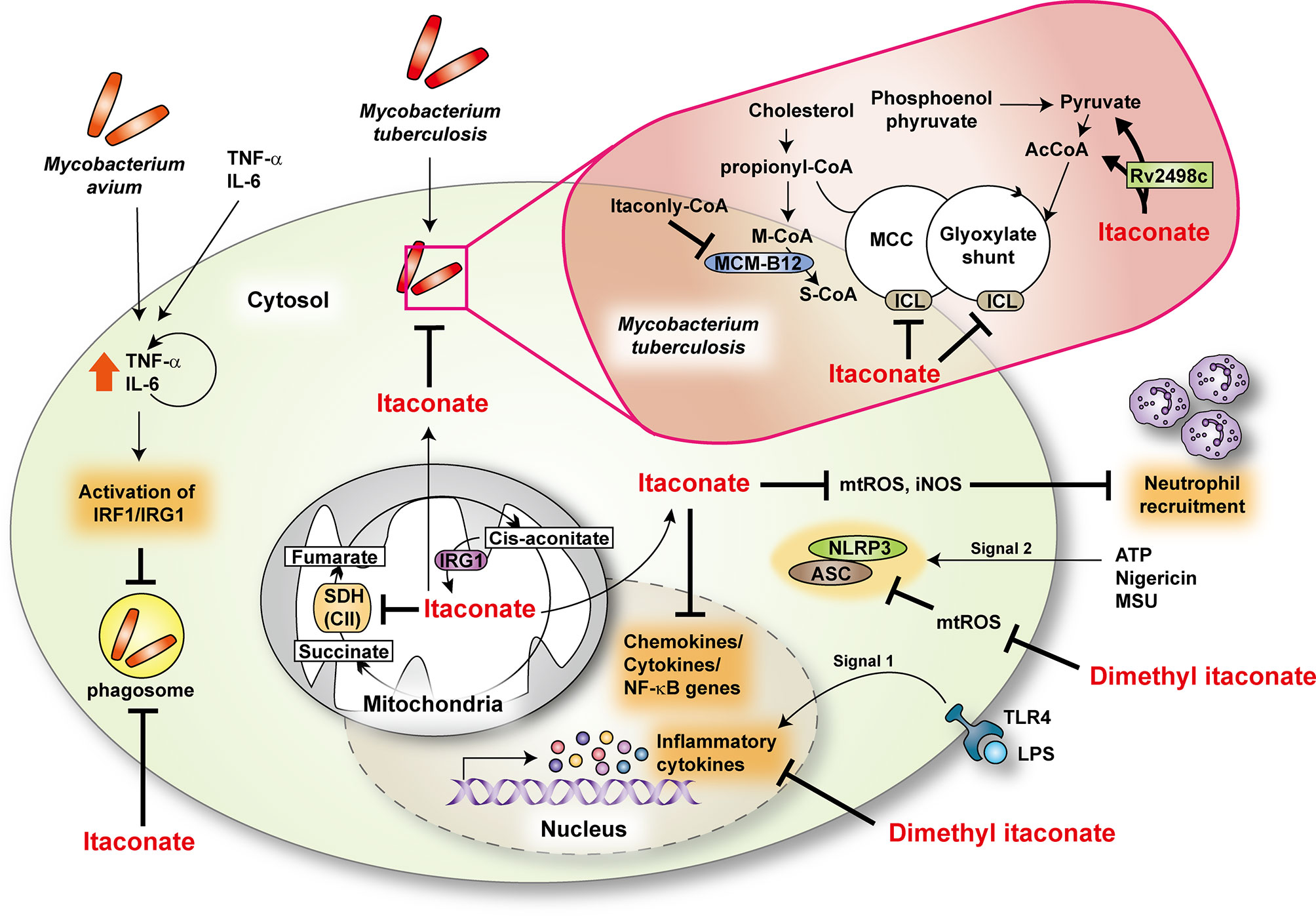
Figure 1 The role of itaconate during mycobacterial infection. Itaconate, produced by immunoresponsive gene 1 (IRG1) from cis-aconitate, modulates TCA cycle by regulating succinate dehydrogenase (SDH, as complex II) activity. Dimethyl itaconate regulates the mRNA expression of inflammatory cytokines in response to LPS. In addition, dimethyl itaconate inhibits SDH and decreases the production of mitochondrial ROS (mtROS) in LPS-treated macrophages. It suppresses the expression of NLRP3 and ASC in NLRP3-activating conditions. During Mtb infection, IRG1 and itaconate downregulate inflammatory responses at the transcriptional level and neutrophil recruitment through inhibiting the production of mtROS and inducible nitric oxide synthase (iNOS). In Mtb, itaconate has an antimicrobial activity for methyl citrate lyase (MCL) in the methyl citrate cycle (MCC) and isocitrate lyase (ICL) in glyoxylate shunt, enzymes that are needed for Mtb survival. Moreover, itaconyl-coenzyme A (CoA) targets B12-dependent methylmalonyl-CoA mutase (MCM-B12) to restrict bacterial growth. Mtb effector Rv2498c participates in itaconate dissimilation to confer resistance to itaconate. During M.avium infection, tumor necrosis factor- α (TNF-α) and interleukin (IL)-6 activate interferon regulatory factor 1 (IRF1)/IRG1 through the autocrine/paracrine signaling pathway. AcCoA, acetyl-coenzyme A; ASC, apoptosis-associated speck-like protein containing a CARD; ATP, adenosine triphosphate; LPS, lipopolysaccharide; M-CoA, methylmalonyl-coenzyme A; MSU, monosodium urate; NLRP3, NLR family pyrin domain containing 3; S-CoA, succinyl-coenzyme A; TLR4, toll-like receptor 4.
To date, the function of itaconate in alveolar macrophages is unclear, although these cells are the first cells that encounter Mtb in the initiation of infection. Future studies are warranted to clarify the role of itaconate in alveolar macrophages during Mtb infection. Recent studies showed that the Irg1 is critically required to control Brucella infection and that dimethyl itaconate has an inhibitory effect against Brucella growth (46). Together, these studies reveal an antimicrobial role for itaconate during bacterial infections, including mycobacteria. However, more studies are warranted to clarify the underlying mechanisms of the itaconate functions of various immune cells, including alveolar macrophages, in mycobacterial diseases.
In an inflammation model, itaconate participates in metabolic remodeling in macrophages toward an anti-inflammatory response by activating nuclear factor erythroid 2–related factor 2 (Nrf2) and inhibiting transcription factor Iκβξ-activating transcription factor 3 inflammatory signaling (47, 48). There are controversial findings upon the function of Nrf2 in the context of mycobacterial infection. Nrf2 is critical for host resistance to pulmonary M. avium complex infection (49, 50); however, it functions in the antioxidant transcriptional responses that delay early Mtb clearance (51). Future studies are needed to address how Nrf2 signaling is associated with itaconate-mediated protection against mycobacterial infection. Moreover, either 4-octyl itaconate or dimethyl itaconate exerts anti-inflammatory effects and inhibits aerobic glycolysis, thus controlling pathologies related to excessive inflammation (4, 46, 52, 53). Therefore these itaconate derivatives may enhance antimycobacterial responses by controlling pathologic inflammation and immunometabolism during infection. More work is needed to define the molecular mechanisms by which endogenous/exogenous itaconate exerts innate host defenses against mycobacterial diseases.
Role of Arg Metabolism During Mycobacterial Infection
Arg metabolism promotes antimicrobial responses in myeloid cells by inducing NO and regulating inflammatory responses (54, 55). M1 and M2 macrophages catabolize Arg via iNOS and ARG1, respectively. In M1 macrophages, NO synthesis promotes proinflammatory and microbicidal activities against intracellular bacteria (54, 56–58). Although NO plays a critical role in antimycobacterial effect in murine macrophages, its role in human macrophages is still debatable (59). In addition to NO, macrophage anti-Mtb activities induced by L-Arg are dependent, in part, on aerobic glycolysis (60). Moreover, L-Arg synthesis from L-citrulline in myeloid cells contributes to host defense against M. bovis BCG and Mtb H37Rv; deficiency of Ass1 or Asl (to eliminate L-Arg synthesis from l-citrulline) increased mycobacterial growth in macrophages and in vivo (61). In M2 macrophages, ARG1 expression is critical for synthesizing ornithine, proline, and polyamines, and contributes to wound healing, defense against parasites, and humoral immunity (54, 57). ARG1 elimination in macrophages reduces the bacterial load in the lung during Mtb infection (62). Additionally, Mtb co-infection with helminths such as Schistosoma mansoni increases ARG1 expression in macrophages to aggravate lung inflammation and impair anti-Mtb T cell responses (63). Overall, both iNOS- and ARG1-dependent pathways in Arg metabolism have opposite roles in host defense against Mtb infection. The Arg-citrulline metabolic axis may enhance host control, whereas ARG1-mediated Arg metabolism leads to inadequate antimicrobial responses during intracellular bacterial infection.
A recent study has identified a novel antimicrobial function of ornithine, an amino acid intermediate produced by ARG1 and ARG2 from Arg metabolism and the urea cycle; and also converts to synthesize proline, polyamines, and citrulline (64, 65). Thandi et al. showed that ornithine is involved in antimycobacterial responses in liver macrophages (7). The authors focused on the liver (7) because of its known role in suppressing Mtb infection (66). Kupffer cells in the liver restrict the growth of Mtb more efficiently than other macrophages, including alveolar macrophages, peritoneal macrophages, and bone marrow-derived macrophages (7). Ornithine promotes Kupffer cell-induced inhibition of intracellular Mtb replication by enhancing autophagy and autolysosome accumulation during Mtb infection (7). Mechanistically, AMP-activated protein kinase (AMPK) is required for ornithine-induced antibacterial autophagy in Kupffer cells during Mtb infection (7). In addition to ornithine, Kupffer cells produce imidazole, which does not induce autophagy but exerts an antimicrobial effect on Mtb by inhibiting mycobacterial cytochrome P450 monooxygenases (7) (Figure 2A). However, the mechanism of how ornithine phosphorylates AMPK in Mtb-infected Kupffer cells is unknown. Also, the roles of ornithine and its metabolism at different stages of mycobacterial infection are unclear. Indeed, several types of tumors exhibit increased polyamines, ornithine-related metabolites, leading to transformation and progression (67, 68). In addition, polyamines suppress the intracellular uptake of fluoroquinolones in M. bovis BCG and Mtb, thus causing phenotypic antibiotic resistance (69). A deeper understanding of the comprehensive molecular mechanisms by which ornithine, citrulline, and polyamines regulate host antimicrobial responses against Mtb infection would facilitate the development of novel strategies to boost host immune defense against human TB. Targeting the L-Arg-related metabolic network may enable the development of novel vaccines and host-directed therapeutics against human TB.
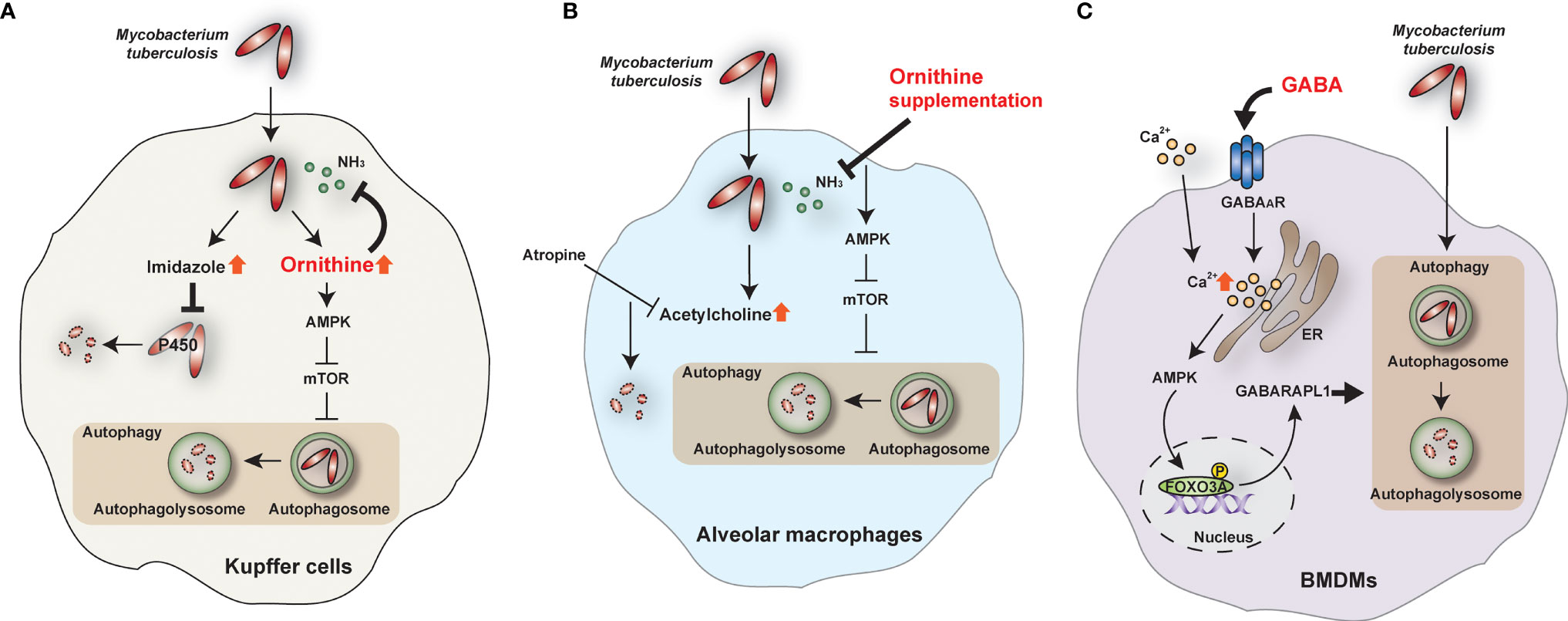
Figure 2 The role of ornithine and GABA during Mtb infection. (A) In Kupffer cells, ornithine and imidazole are top-scoring metabolites during Mtb infection. Ornithine restricts Mtb growth by activating the AMPK-mediated autophagy pathway and inhibiting ammonia (NH3). On the other hand, imidazole kills Mtb directly by inhibiting cytochrome P450 monooxygenases. (B) In alveolar macrophages, ornithine supplementation inhibits NH3 and activates autophagy to restrict Mtb growth. In addition, acetylcholine is increased in Mtb-infected alveolar macrophages, and atropine, an acetylcholine antagonist, inhibits Mtb survival. (C) GABA induces intracellular calcium (Ca2+) influx and activates the autophagy pathway through the AMPK-GABARAPL1 pathway. Autophagy can restrict the intracellular survival of Mtb in bone marrow-derived macrophages (BMDMs). AMPK, AMP-activated protein kinase; FOXO3A, forkhead box O3A; GABAAR, GABA A receptor; GABARAPL1, GABA type A receptor-associated protein-like 1; mTOR, mammalian target of rapamycin.
In contrast to Kupffer cells, alveolar macrophages fail to clear Mtb and facilitate the establishment of Mtb infection through the upregulation of acetylcholine (7). In addition, alveolar macrophages produce higher ammonia , promoting Mtb growth, compared with Kupffer cells (7). Interestingly, supplementation of ornithine, imidazole, and atropine (acetylcholine inhibitor) promotes Mtb clearance in alveolar macrophages (7) (Figure 2B). It is unclear how acetylcholine results in the suppression of Mtb clearance. Given the recent findings that acetylcholine and cholinergic system favor the progression of mycobacterial infection (70), targeting the non-neuronal cholinergic system in the lungs may contribute to the development of new therapeutics against TB.
Role of GABA During Mycobacterial Infection
GABA is an inhibitory neurotransmitter in the central nervous system and is a metabolite synthesized from glutamic acid by glutamic acid decarboxylase (71–73). Also, GABA is produced in peripheral tissues, including the pancreas, pituitary, testes, gastrointestinal tract, ovaries, placenta, uterus, and adrenal medulla (74, 75). Moreover, peripheral immune cells express GABAergic components—such as type A GABA receptors (GABAAR), G-protein-coupled type B receptors (GABABR), and GABA transporters—which modulate GABA biological functions in peripheral tissues and/or cells (76–79). The peripheral GABAergic system plays an essential role in autoimmune and inflammatory diseases like type 1 diabetes, experimental autoimmune encephalomyelitis, collagen-induced arthritis, and dermatitis (77, 80, 81).
GABA signaling activates antimicrobial responses against Mtb and M. bovis BCG (8). GABAergic activation via GABAAR agonists—such as GABA, muscimol, and isoguvacine hydrochloride—induces antibacterial autophagy and phagosomal maturation during Mtb infection. However, GABAergic inhibition suppresses antimicrobial responses during mycobacterial infection. Mechanistically, GABA-induced autophagy activation is mediated by intracellular calcium influx via activation of the AMPK-mediated GABA type A receptor-associated protein-like 1 (GABARAPL1) pathway (8) (Figure 2C). Indeed, GABA treatment activates AMPK-mediated autophagy and GABAAR signaling in intestinal epithelial cells to inhibit enterotoxigenic Escherichia coli-induced excessive apoptosis (82). Certainly, AMPK is a crucial metabolic and autophagic regulator and promotes antimicrobial responses against Mtb infection (83–86). However, it is unclear how GABA triggers intracellular calcium influx in peripheral cells to activate AMPK-autophagy pathways. A recent study suggested a molecular framework for GABA-induced Ca2+ influx in the context of parasitic infection and immune cell migration (87). In myeloid mononuclear phagocytes such as dendritic cells, glutamate-derived GABA is secreted to trigger GABAAR signaling, implicated in Na-K-Cl cotransporters and extracellular Ca2+ influx. The result is phagocyte hypermotility and dissemination of the coccidian parasites Toxoplasma gondii and Neospora caninum (87). Further research is required to identify the role of GABA signaling in different cell types in response to infectious agents.
GABA treatment substantially reduces inflammatory cytokine production in macrophages and lung tissues from infected mice (8). The autocrine or paracrine function of GABA is associated with inhibition of inflammation to ameliorate autoimmune pathologic responses (88, 89). In addition, GABA administration attenuates insulin resistance, obesity-induced adipose tissue macrophage infiltration, and inflammatory responses in subcutaneous inguinal adipose tissues, at least in part via GABABR signaling (90). Therefore, GABA signaling pathways can be therapeutic targets for pathologic inflammation during Mtb infection. By contrast, GABA-mediated mammalian target of rapamycin (mTOR) signaling is required for Th17 cell differentiation in the presence of GABA transporter-2 (SLC6A13) deficiency (91). These data suggest a pleiotropic function for GABA in regulating inflammatory responses via downstream signaling molecules.
Mtb co-opts GABA as an immune-escape strategy to favor intracellular infection. Mtb can adapt to acidic conditions and oxidative stresses via the GABA shunt pathway to reduce NAD+ and proton levels (92). In addition, Mtb uses lactate and pyruvate from host cells via multiple metabolic pathways, including the GABA shunt (35). Given that GABA promotes host protection against intracellular bacterial infection (8), further research should investigate the molecular mechanisms underlying GABAergic defense in host-pathogen interactions during infection. Pavić et al. revealed that MsGabP, a putative GABA transport protein from M. smegmatis and an Mtb homolog, binds GABA and may outcompete the host GABAergic protective system, providing a new target for TB drug development (93). A challenging caveat in developing the immunometabolite-targeted host defensive strategies is the complicated host-mycobacterial relationship to compete for the specific metabolites for their purposes during infection. Further studies are needed to understand better how metabolic communication between Mtb and host cells impact disease outcomes during infection.
Conclusion
The metabolic interaction between host and mycobacteria is a critical determinant of infection outcomes. However, it is unclear how metabolites and their relationships with other metabolic pathways promote the establishment of chronic infection or control of mycobacteria. In addition, it remains unclear how Mtb manipulates host metabolic fluxes and enzymes to escape immune surveillance and survive intracellularly. The complex interactions between host cells and pathogens via metabolic and immune pathways during Mtb infection will be challenging to unravel.
Itaconate and its derivatives exert bactericidal and immuno-modulatory effects critical for antimicrobial defense. Citrulline is a precursor of L-Arg and linked to iNOS-mediated antimycobacterial responses in immune cells; ornithine induces autophagy in Kupffer cells. GABA signaling activates autophagy against intracellular bacteria. Future studies should investigate how these and other immunometabolites influence antimicrobial host defense and pathological inflammation to facilitate a rational design for host-directed therapeutics for mycobacterial infection.
Author Contributions
JK wrote the manuscript and made the illustrations. E-JP was responsible for reviewing and editing the manuscript. E-KJ designed, supervised, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2017R1A5A2015385) and by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No.2019R1A2C1087686).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are indebted to current and past members of our Medical Research Center (i-MRC) for discussions and investigations that contributed to this article. We apologize to colleagues whose publications could not be cited due to space limitations.
References
1. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-Responsive Gene 1 Protein Links Metabolism to Immunity by Catalyzing Itaconic Acid Production. Proc Natl Acad Sci USA (2013) 110:7820–5. doi: 10.1073/pnas.1218599110
2. Zhu X, Guo Y, Liu Z, Yang J, Tang H, Wang Y. Itaconic Acid Exerts Anti-Inflammatory and Antibacterial Effects via Promoting Pentose Phosphate Pathway to Produce ROS. Sci Rep (2021) 11:18173. doi: 10.1038/s41598-021-97352-x
3. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an Anti-Inflammatory Metabolite That Activates Nrf2 via Alkylation of KEAP1. Nature (2018) 556:113–17. doi: 10.1038/nature25986
4. Liao ST, Han C, Xu DQ, Fu XW, Wang JS, Kong LY. 4-Octyl Itaconate Inhibits Aerobic Glycolysis by Targeting GAPDH to Exert Anti-Inflammatory Effects. Nat Commun (2019) 10:5091. doi: 10.1038/s41467-019-13078-5
5. Granger DL, Hibbs JB Jr., Perfect JR, Durack DT. Metabolic Fate of L-Arginine in Relation to Microbiostatic Capability of Murine Macrophages. J Clin Invest (1990) 85:264–73. doi: 10.1172/JCI114422
6. Rapovy SM, Zhao J, Bricker RL, Schmidt SM, Setchell KD, Qualls JE. Differential Requirements for L-Citrulline and L-Arginine During Antimycobacterial Macrophage Activity. J Immunol (2015) 195:3293–300. doi: 10.4049/jimmunol.1500800
7. Sivangala Thandi R, Radhakrishnan RK, Tripathi D, Paidipally P, Azad AK, Schlesinger LS, et al. Ornithine-A Urea Cycle Metabolite Enhances Autophagy and Controls Mycobacterium tuberculosis Infection. Nat Commun (2020) 11:3535. doi: 10.1038/s41467-020-17310-5
8. Kim JK, Kim YS, Lee HM, Jin HS, Neupane C, Kim S, et al. GABAergic Signaling Linked to Autophagy Enhances Host Protection Against Intracellular Bacterial Infections. Nat Commun (2018) 9:4184. doi: 10.1038/s41467-018-06487-5
9. Vergne I, Chua J, Singh SB, Deretic V. Cell Biology of Mycobacterium tuberculosis Phagosome. Annu Rev Cell Dev Biol (2004) 20:367–94. doi: 10.1146/annurev.cellbio.20.010403.114015
10. Dey B, Bishai WR. Crosstalk Between Mycobacterium tuberculosis and the Host Cell. Semin Immunol (2014) 26:486–96. doi: 10.1016/j.smim.2014.09.002
11. Rajaram MV, Ni B, Dodd CE, Schlesinger LS. Macrophage Immunoregulatory Pathways in Tuberculosis. Semin Immunol (2014) 26:471–85. doi: 10.1016/j.smim.2014.09.010
12. Zhai W, Wu F, Zhang Y, Fu Y, Liu Z. The Immune Escape Mechanisms of Mycobacterium tuberculosis. Int J Mol Sci (2019) 20(2):340. doi: 10.3390/ijms20020340
13. Flynn JL, Chan J, Lin PL. Macrophages and Control of Granulomatous Inflammation in Tuberculosis. Mucosal Immunol (2011) 4:271–8. doi: 10.1038/mi.2011.14
14. Corleis B, Dorhoi A. Early Dynamics of Innate Immunity During Pulmonary Tuberculosis. Immunol Lett (2020) 221:56–60. doi: 10.1016/j.imlet.2020.02.010
15. Martinez FO, Gordon S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
16. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
17. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages. Cell (2004) 119:753–66. doi: 10.1016/j.cell.2004.11.038
18. Yuk JM, Yoshimori T, Jo EK. Autophagy and Bacterial Infectious Diseases. Exp Mol Med (2012) 44:99–108. doi: 10.3858/emm.2012.44.2.032
19. Siqueira MDS, Ribeiro RM, Travassos LH. Autophagy and its Interaction With Intracellular Bacterial Pathogens. Front Immunol (2018) 9:935. doi: 10.3389/fimmu.2018.00935
20. Kim YS, Silwal P, Kim SY, Yoshimori T, Jo EK. Autophagy-Activating Strategies to Promote Innate Defense Against Mycobacteria. Exp Mol Med (2019) 51:1–10. doi: 10.1038/s12276-019-0290-7
21. Kumar R, Singh P, Kolloli A, Shi L, Bushkin Y, Tyagi S, et al. Immunometabolism of Phagocytes During Mycobacterium tuberculosis Infection. Front Mol Biosci (2019) 6:105. doi: 10.3389/fmolb.2019.00105
22. Marino S, Cilfone NA, Mattila JT, Linderman JJ, Flynn JL, Kirschner DE. Macrophage Polarization Drives Granuloma Outcome During Mycobacterium tuberculosis Infection. Infect Immun (2015) 83:324–38. doi: 10.1128/IAI.02494-14
23. Saunders BM, Britton WJ. Life and Death in the Granuloma: Immunopathology of Tuberculosis. Immunol Cell Biol (2007) 85:103–11. doi: 10.1038/sj.icb.7100027
24. Mishra A, Surolia A. Mycobacterium tuberculosis: Surviving and Indulging in an Unwelcoming Host. IUBMB Life (2018) 70:917–25. doi: 10.1002/iub.1882
25. Pagan AJ, Ramakrishnan L. The Formation and Function of Granulomas. Annu Rev Immunol (2018) 36:639–65. doi: 10.1146/annurev-immunol-032712-100022
26. Russell DG, VanderVen BC, Lee W, Abramovitch RB, Kim MJ, Homolka S, et al. Mycobacterium tuberculosis Wears What it Eats. Cell Host Microbe (2010) 8:68–76. doi: 10.1016/j.chom.2010.06.002
27. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis In Vivo Segregates With Host Macrophage Metabolism and Ontogeny. J Exp Med (2018) 215:1135–52. doi: 10.1084/jem.20172020
28. Liu Y, Liang S, Ding R, Hou Y, Deng F, Ma X, et al. BCG-Induced Trained Immunity in Macrophage: Reprograming of Glucose Metabolism. Int Rev Immunol (2020) 39:83–96. doi: 10.1080/08830185.2020.1712379
29. Covian C, Fernandez-Fierro A, Retamal-Diaz A, Diaz FE, Vasquez AE, Lay MK, et al. BCG-Induced Cross-Protection and Development of Trained Immunity: Implication for Vaccine Design. Front Immunol (2019) 10:2806. doi: 10.3389/fimmu.2019.02806
30. Arts RJW, Moorlag S, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG Vaccination Protects Against Experimental Viral Infection in Humans Through the Induction of Cytokines Associated With Trained Immunity. Cell Host Microbe (2018) 23:89–100.e5. doi: 10.1016/j.chom.2017.12.010
31. Sheedy FJ, Divangahi M. Targeting Immunometabolism in Host Defence Against Mycobacterium tuberculosis. Immunology (2021) 162:145–59. doi: 10.1111/imm.13276
32. Howard NC, Khader SA. Immunometabolism During Mycobacterium tuberculosis Infection. Trends Microbiol (2020) 28:832–50. doi: 10.1016/j.tim.2020.04.010
33. Moynihan MM, Murkin AS. Cysteine is the General Base That Serves in Catalysis by Isocitrate Lyase and in Mechanism-Based Inhibition by 3-Nitropropionate. Biochemistry (2014) 53:178–87. doi: 10.1021/bi401432t
34. Puckett S, Trujillo C, Wang Z, Eoh H, Ioerger TR, Krieger I, et al. Glyoxylate Detoxification is an Essential Function of Malate Synthase Required for Carbon Assimilation in Mycobacterium tuberculosis. Proc Natl Acad Sci USA (2017) 114:E2225–32. doi: 10.1073/pnas.1617655114
35. Serafini A, Tan L, Horswell S, Howell S, Greenwood DJ, Hunt DM, et al. Mycobacterium tuberculosis Requires Glyoxylate Shunt and Reverse Methylcitrate Cycle for Lactate and Pyruvate Metabolism. Mol Microbiol (2019) 112:1284–307. doi: 10.1111/mmi.14362
36. Kratky M, Vinsova J. Advances in Mycobacterial Isocitrate Lyase Targeting and Inhibitors. Curr Med Chem (2012) 19:6126–37. doi: 10.2174/092986712804485782
37. Ruetz M, Campanello GC, Purchal M, Shen H, McDevitt L, Gouda H, et al. Itaconyl-CoA Forms a Stable Biradical in Methylmalonyl-CoA Mutase and Derails Its Activity and Repair. Science (2019) 366:589–93. doi: 10.1126/science.aay0934
38. Wang H, Fedorov AA, Fedorov EV, Hunt DM, Rodgers A, Douglas HL, et al. An Essential Bifunctional Enzyme in Mycobacterium tuberculosis for Itaconate Dissimilation and Leucine Catabolism. Proc Natl Acad Sci USA (2019) 116:15907–13. doi: 10.1073/pnas.1906606116
39. Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, et al. Irg1 Expression in Myeloid Cells Prevents Immunopathology During M. tuberculosis Infection. J Exp Med (2018) 215:1035–45. doi: 10.1084/jem.20180118
40. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase With Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
41. Yang K, Wu YJ, Xie HP, Li M, Ming SQ, Li LY, et al. Macrophage-Mediated Inflammatory Response Decreases Mycobacterial Survival in Mouse MSCs by Augmenting NO Production. Sci Rep (2016) 6:27326. doi: 10.1038/srep27326
42. Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I. Protective Role of Interleukin-1 in Mycobacterial Infection in IL-1 Alpha/Beta Double-Knockout Mice. Lab Invest (2000) 80:759–67. doi: 10.1038/labinvest.3780079
43. Chao WC, Yen CL, Hsieh CY, Huang YF, Tseng YL, Nigrovic PA, et al. Mycobacterial Infection Induces Higher Interleukin-1beta and Dysregulated Lung Inflammation in Mice With Defective Leukocyte NADPH Oxidase. PLoS One (2017) 12:e0189453. doi: 10.1371/journal.pone.0189453
44. Jamaati H, Mortaz E, Pajouhi Z, Folkerts G, Movassaghi M, Moloudizargari M, et al. Nitric Oxide in the Pathogenesis and Treatment of Tuberculosis. Front Microbiol (2017) 8:2008. doi: 10.3389/fmicb.2017.02008
45. Gidon A, Louet C, Rost LM, Bruheim P, Flo TH. The Tumor Necrosis Factor Alpha and Interleukin 6 Auto-Paracrine Signaling Loop Controls Mycobacterium avium Infection via Induction of IRF1/IRG1 in Human Primary Macrophages. mBio (2021) 12:e0212121. doi: 10.1128/mBio.02121-21
46. Demars A, Vitali A, Comein A, Carlier E, Azouz A, Goriely S, et al. Aconitate Decarboxylase 1 Participates in the Control of Pulmonary Brucella Infection in Mice. PLoS Pathog (2021) 17:e1009887. doi: 10.1371/journal.ppat.1009887
47. Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic Properties of Itaconate and Derivatives Regulate the IkappaBzeta-ATF3 Inflammatory Axis. Nature (2018) 556:501–04. doi: 10.1038/s41586-018-0052-z
48. O’Neill LAJ, Artyomov MN. Itaconate: The Poster Child of Metabolic Reprogramming in Macrophage Function. Nat Rev Immunol (2019) 19:273–81. doi: 10.1038/s41577-019-0128-5
49. Nakajima M, Matsuyama M, Kawaguchi M, Kiwamoto T, Matsuno Y, Morishima Y, et al. Nrf2 Regulates Granuloma Formation and Macrophage Activation During Mycobacterium avium Infection via Mediating Nramp1 and HO-1 Expressions. mBio (2021) 12(1):e01947–20. doi: 10.1128/mBio.01947-20
50. Matsuyama M, Nonaka M, Nakajima M, Morishima Y, Ishii Y, Hizawa N. The Role of NRF2 in Mycobacterial Infection. Antioxidants (2021) 10:1861. doi: 10.3390/antiox10121861
51. Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, et al. Alveolar Macrophages Generate a Noncanonical NRF2-Driven Transcriptional Response to Mycobacterium tuberculosis In Vivo. Sci Immunol (2019) 4(37):eaaw6693. doi: 10.1126/sciimmunol.aaw6693
52. Diskin C, Zotta A, Corcoran SE, Tyrrell VJ, Zaslona Z, O’Donnell VB, et al. 4-Octyl-Itaconate and Dimethyl Fumarate Inhibit COX2 Expression and Prostaglandin Production in Macrophages. J Immunol (2021) 207:2561–69. doi: 10.4049/jimmunol.2100488
53. Nakkala JR, Yao Y, Zhai Z, Duan Y, Zhang D, Mao Z, et al. Dimethyl Itaconate-Loaded Nanofibers Rewrite Macrophage Polarization, Reduce Inflammation, and Enhance Repair of Myocardic Infarction. Small (2021) 17:e2006992. doi: 10.1002/smll.202006992
54. Marti ILAA, Reith W. Arginine-Dependent Immune Responses. Cell Mol Life Sci (2021) 78:5303–24. doi: 10.1007/s00018-021-03828-4
55. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine Metabolism in Myeloid Cells Shapes Innate and Adaptive Immunity. Front Immunol (2017) 8:93. doi: 10.3389/fimmu.2017.00093
56. Das P, Lahiri A, Lahiri A, Chakravortty D. Modulation of the Arginase Pathway in the Context of Microbial Pathogenesis: A Metabolic Enzyme Moonlighting as an Immune Modulator. PLoS Pathog (2010) 6:e1000899. doi: 10.1371/journal.ppat.1000899
57. Munder M. Arginase: An Emerging Key Player in the Mammalian Immune System. Br J Pharmacol (2009) 158:638–51. doi: 10.1111/j.1476-5381.2009.00291.x
58. Morris SM Jr. Arginine: Master and Commander in Innate Immune Responses. Sci Signal (2010) 3:pe27. doi: 10.1126/scisignal.3135pe27
59. Thomas AC, Mattila JT. "Of Mice and Men": Arginine Metabolism in Macrophages. Front Immunol (2014) 5:479. doi: 10.3389/fimmu.2014.00479
60. McKell MC, Crowther RR, Schmidt SM, Robillard MC, Cantrell R, Lehn MA, et al. Promotion of Anti-Tuberculosis Macrophage Activity by L-Arginine in the Absence of Nitric Oxide. Front Immunol (2021) 12:653571. doi: 10.3389/fimmu.2021.653571
61. Lange SM, McKell MC, Schmidt SM, Zhao J, Crowther RR, Green LC, et al. L-Arginine Synthesis From L-Citrulline in Myeloid Cells Drives Host Defense Against Mycobacteria In Vivo. J Immunol (2019) 202:1747–54. doi: 10.4049/jimmunol.1801569
62. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-Like Receptor-Induced Arginase 1 in Macrophages Thwarts Effective Immunity Against Intracellular Pathogens. Nat Immunol (2008) 9:1399–406. doi: 10.1038/ni.1671
63. Monin L, Griffiths KL, Lam WY, Gopal R, Kang DD, Ahmed M, et al. Helminth-Induced Arginase-1 Exacerbates Lung Inflammation and Disease Severity in Tuberculosis. J Clin Invest (2015) 125:4699–713. doi: 10.1172/JCI77378
64. Muthukumaran Sivashanmugam, Jaidev J, Umashankar V, Sulochana KN. Ornithine and Its Role in Metabolic Diseases: An Appraisal. BioMed Pharmacother (2017) 86:185–94. doi: 10.1016/j.biopha.2016.12.024
65. Clemente GS, van Waarde A, Antunes IF, Domling A, Elsinga PH. Arginase as a Potential Biomarker of Disease Progression: A Molecular Imaging Perspective. Int J Mol Sci (2020) 21(15):5291. doi: 10.3390/ijms21155291
66. Chong VH. Hepatobiliary Tuberculosis: A Review of Presentations and Outcomes. South Med J (2008) 101:356–61. doi: 10.1097/SMJ.0b013e318164ddbb
67. Casero RA Jr., Murray Stewart T, Pegg AE. Polyamine Metabolism and Cancer: Treatments, Challenges and Opportunities. Nat Rev Cancer (2018) 18:681–95. doi: 10.1038/s41568-018-0050-3
68. Hesterberg RS, Cleveland JL, Epling-Burnette PK. Role of Polyamines in Immune Cell Functions. Med Sci (Basel) (2018) 6(1):22. doi: 10.3390/medsci6010022
69. Sarathy JP, Lee E, Dartois V. Polyamines Inhibit Porin-Mediated Fluoroquinolone Uptake in Mycobacteria. PLoS One (2013) 8:e65806. doi: 10.1371/journal.pone.0065806
70. Islas-Weinstein L, Marquina-Castillo B, Mata-Espinosa D, Paredes-Gonzalez IS, Chavez J, Balboa L, et al. The Cholinergic System Contributes to the Immunopathological Progression of Experimental Pulmonary Tuberculosis. Front Immunol (2020) 11:581911. doi: 10.3389/fimmu.2020.581911
71. Buddhala C, Hsu CC, Wu JY. A Novel Mechanism for GABA Synthesis and Packaging Into Synaptic Vesicles. Neurochem Int (2009) 55:9–12. doi: 10.1016/j.neuint.2009.01.020
72. Serrano-Regal MP, Bayon-Cordero L, Ordaz RP, Garay E, Limon A, Arellano RO, et al. Expression and Function of GABA Receptors in Myelinating Cells. Front Cell Neurosci (2020) 14:256. doi: 10.3389/fncel.2020.00256
73. Baizabal-Carvallo JF. The Neurological Syndromes Associated With Glutamic Acid Decarboxylase Antibodies. J Autoimmun (2019) 101:35–47. doi: 10.1016/j.jaut.2019.04.007
74. Gladkevich A, Korf J, Hakobyan VP, Melkonyan KV. The Peripheral GABAergic System as a Target in Endocrine Disorders. Auton Neurosci (2006) 124:1–8. doi: 10.1016/j.autneu.2005.11.002
75. Everington EA, Gibbard AG, Swinny JD, Seifi M. Molecular Characterization of GABA-A Receptor Subunit Diversity Within Major Peripheral Organs and Their Plasticity in Response to Early Life Psychosocial Stress. Front Mol Neurosci (2018) 11:18. doi: 10.3389/fnmol.2018.00018
76. Jin Z, Mendu SK, Birnir B. GABA is an Effective Immunomodulatory Molecule. Amino Acids (2013) 45:87–94. doi: 10.1007/s00726-011-1193-7
77. Prud’homme GJ, Glinka Y, Wang Q. Immunological GABAergic Interactions and Therapeutic Applications in Autoimmune Diseases. Autoimmun Rev (2015) 14:1048–56. doi: 10.1016/j.autrev.2015.07.011
78. Wang C, Hao H, He K, An Y, Pu Z, Gamper N, et al. Neuropathic Injury-Induced Plasticity of GABAergic System in Peripheral Sensory Ganglia. Front Pharmacol (2021) 12:702218. doi: 10.3389/fphar.2021.702218
79. Malomouzh A, Ilyin V, Nikolsky E. Components of the GABAergic Signaling in the Peripheral Cholinergic Synapses of Vertebrates: A Review. Amino Acids (2019) 51:1093–102. doi: 10.1007/s00726-019-02754-x
80. Siggs OM, Berger M, Krebs P, Arnold CN, Eidenschenk C, Huber C, et al. A Mutation of Ikbkg Causes Immune Deficiency Without Impairing Degradation of IkappaB Alpha. Proc Natl Acad Sci USA (2010) 107:3046–51. doi: 10.1073/pnas.0915098107
81. Bajic SS, Dokic J, Dinic M, Tomic S, Popovic N, Brdaric E, et al. GABA Potentiate the Immunoregulatory Effects of Lactobacillus Brevis BGZLS10-17 via ATG5-Dependent Autophagy In Vitro. Sci Rep (2020) 10:1347. doi: 10.1038/s41598-020-58177-2
82. Xia Y, Chen S, Zhao Y, Chen S, Huang R, Zhu G, et al. GABA Attenuates ETEC-Induced Intestinal Epithelial Cell Apoptosis Involving GABAAR Signaling and the AMPK-Autophagy Pathway. Food Funct (2019) 10:7509–22. doi: 10.1039/c9fo01863h
83. Paik S, Jo EK. An Interplay Between Autophagy and Immunometabolism for Host Defense Against Mycobacterial Infection. Front Immunol (2020) 11:603951. doi: 10.3389/fimmu.2020.603951
84. Jo EK, Silwal P, Yuk JM. AMPK-Targeted Effector Networks in Mycobacterial Infection. Front Microbiol (2019) 10:520. doi: 10.3389/fmicb.2019.00520
85. Cheng CY, Bohme J, Singhal A. Metabolic Energy Sensors as Targets for Designing Host-Directed Therapies for Tuberculosis. J Leukocyte Biol (2018) 103:215–23. doi: 10.1189/jlb.4MR0617-226R
86. Jia JY, Bissa B, Brecht L, Allers L, Choi SW, Gu YX, et al. AMPK, a Regulator of Metabolism and Autophagy, is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System. Mol Cell (2020) 77:951–69.e9. doi: 10.1016/j.molcel.2019.12.028
87. Bhandage AK, Olivera GC, Kanatani S, Thompson E, Lore K, Varas-Godoy M, et al. A Motogenic GABAergic System of Mononuclear Phagocytes Facilitates Dissemination of Coccidian Parasites. Elife (2020) 9:e60528. doi: 10.7554/eLife.60528
88. Bhat R, Axtell R, Mitra A, Miranda M, Lock C, Tsien RW, et al. Inhibitory Role for GABA in Autoimmune Inflammation. Proc Natl Acad Sci USA (2010) 107:2580–5. doi: 10.1073/pnas.0915139107
89. Nguyen L, Malgrange B, Breuskin I, Bettendorff L, Moonen G, Belachew S, et al. Autocrine/paracrine Activation of the GABA(A) Receptor Inhibits the Proliferation of Neurogenic Polysialylated Neural Cell Adhesion Molecule-Positive (PSA-NCAM+) Precursor Cells From Postnatal Striatum. J Neurosci (2003) 23:3278–94. doi: 10.1523/JNEUROSCI.23-08-03278.2003
90. Hwang I, Jo K, Shin KC, Kim JI, Ji Y, Park YJ, et al. GABA-Stimulated Adipose-Derived Stem Cells Suppress Subcutaneous Adipose Inflammation in Obesity. Proc Natl Acad Sci USA (2019) 116:11936–45. doi: 10.1073/pnas.1822067116
91. Ren W, Liao Y, Ding X, Jiang Y, Yan J, Xia Y, et al. Slc6a13 Deficiency Promotes Th17 Responses During Intestinal Bacterial Infection. Mucosal Immunol (2019) 12:531–44. doi: 10.1038/s41385-018-0111-7
92. Rizvi A, Shankar A, Chatterjee A, More TH, Bose T, Dutta A, et al. Rewiring of Metabolic Network in Mycobacterium tuberculosis During Adaptation to Different Stresses. Front Microbiol (2019) 10:2417. doi: 10.3389/fmicb.2019.02417
Keywords: itaconate, arginine, GABA, host defense, Mycobacterium tuberculosis, innate immunity
Citation: Kim JK, Park E-J and Jo E-K (2022) Itaconate, Arginine, and Gamma-Aminobutyric Acid: A Host Metabolite Triad Protective Against Mycobacterial Infection. Front. Immunol. 13:832015. doi: 10.3389/fimmu.2022.832015
Received: 09 December 2021; Accepted: 13 January 2022;
Published: 04 February 2022.
Edited by:
Danielle Ahn, Columbia University Irving Medical Center, United StatesReviewed by:
Murugesan Rajaram, The Ohio State University, United StatesCopyright © 2022 Kim, Park and Jo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eun-Kyeong Jo, aGF5b3VuZ2pAY251LmFjLmty