
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 16 May 2022
Sec. B Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.828734
 Melisa Gorosito Serrán1
Melisa Gorosito Serrán1 Facundo Fiocca Vernengo1Laura Almada1†Cristian G. Beccaria1†
Facundo Fiocca Vernengo1Laura Almada1†Cristian G. Beccaria1† Yamila Gazzoni1
Yamila Gazzoni1 Pablo F. Canete2
Pablo F. Canete2 Jonathan A. Roco2
Jonathan A. Roco2 Jimena Tosello Boari1Maria Cecilia Ramello1Ellen Wehrens3Yeping Cai2Elina I. Zuniga3
Jimena Tosello Boari1Maria Cecilia Ramello1Ellen Wehrens3Yeping Cai2Elina I. Zuniga3 Carolina L. Montes1
Carolina L. Montes1 Ian A. Cockburn2Eva V. Acosta Rodriguez1Carola G. Vinuesa2,4
Ian A. Cockburn2Eva V. Acosta Rodriguez1Carola G. Vinuesa2,4 Adriana Gruppi1*
Adriana Gruppi1*During infections with protozoan parasites or some viruses, T cell immunosuppression is generated simultaneously with a high B cell activation. It has been described that, as well as producing antibodies, plasmablasts, the differentiation product of activated B cells, can condition the development of protective immunity in infections. Here, we show that, in T. cruzi infection, all the plasmablasts detected during the acute phase of the infection had higher surface expression of PD-L1 than other mononuclear cells. PD-L1hi plasmablasts were induced in vivo in a BCR-specific manner and required help from Bcl-6+CD4+T cells. PD-L1hi expression was not a characteristic of all antibody-secreting cells since plasma cells found during the chronic phase of infection expressed PD-L1 but at lower levels. PD-L1hi plasmablasts were also present in mice infected with Plasmodium or with lymphocytic choriomeningitis virus, but not in mice with autoimmune disorders or immunized with T cell-dependent antigens. In vitro experiments showed that PD-L1hi plasmablasts suppressed the T cell response, partially via PD-L1. Thus, this study reveals that extrafollicular PD-L1hi plasmablasts, whose peaks of response precede the peak of germinal center response, may have a modulatory function in infections, thus influencing T cell response.
Pathogens develop different strategies to settle in the host. We identified a plasmablast population that expressed high levels of PD-L1, induced by pathogens in acute infections which influences T cell response.
Persistent replicating parasites and viruses can overcome the initial immune response by suppressing the innate and T cell response to allow the establishment of chronic infection. Immune suppression in chronic infections serves to prevent pathogenic immune-mediated inflammation and to suppress protective effector immune responses (1, 2). As such, Trypanosoma cruzi infection, also known as Chagas disease, has been an important model for understanding the delicate balance between protective T cell immunity, immune suppression, and parasite establishment.
Chagas disease presents an acute phase both in mice and humans, characterized by a state of immunosuppression in which T. cruzi replicates extensively and induces immunomodulatory molecules that delay parasite-specific T cell effector responses (3–6). It has been reported that during this phase of T. cruzi infection the expression of PD-1, one of the members of the CD28/CTLA4 family with inhibitory capacity, increases on the myocardium infiltrating CD4+ and CD8+ T cells (7). The cross-linking of PD-1 with any of its two ligands, PD-L1 (8) or PD-L2 (9), inhibits the activation of T cells and the production of IL-2 and IFNγ.
Many studies have shown that other protozoan parasites, such as Plasmodium (10) as well as viruses (11) and bacteria (12), also benefit from the PD-1/PD-L1 pathway to suppress and evade the host’s adaptive immunity. The activation of the PD-1/PD-L1 pathway might enable the establishment of microorganisms and chronicity (13–15). Accordingly, the blocking of PD-1/PD-L1 signals, in combination with therapeutic immunizations or therapies with cytokines, has been proposed as a strategy to improve the efficacy of vaccinations (16) and to revitalize exhausted T cells (17).
During infection with T. cruzi, blocking PD-1 and PD-L1 or deletion of the PD-1 gene result in a reduction in parasitemia and tissue parasitism, but also in increased mortality due to an augmented cardiac inflammatory response (7). These results reveal that the signaling pathway of PD-1/PD-L1 has an important role in the control of inflammation induced by T. cruzi and provide another perspective on the mechanisms of regulation in the pathogenesis of Chagas disease. However, PD-L1-expressing regulatory cells have not been fully identified in this context.
It has been reported that PD-L1+ regulatory plasmablasts have a critical role in suppressing immune T cell responses in cancer models (18); however, their role in chronic infection has not been described. Recently, we reported that Blimp-1fl/flCD23Cre mice, which are deficient in plasmablasts, had a significantly higher number of trypomastigotes in blood and splenic TNF+CD4+ T cells than infected CD23Cre mice after T. cruzi infection (19), suggesting that plasmablasts and plasma cells are important for parasite replication control and also for the regulation of TNF-producing cells. Considering that the absence of plasmablasts improves effector T cell responses, we explored whether PD-L1+ plasmablasts are present in T. cruzi-infected mice and in other parasitic and chronic infections. Moreover, given the proximity of extrafollicular plasmablasts to T cell zones (20), we asked whether PD-L1+ plasmablasts can regulate the T cell response via the PD-1/PD-L1 pathway.
Here we show that, in T. cruzi infection, all the plasmablasts detected had higher surface expression of PD-L1 than other B-lymphocyte lineage cells or other mononuclear cells. These plasmablasts were also present in other infections which involve high inflammatory conditions. This study reveals that extrafollicular PD-L1hi plasmablasts in vitro can operate as suppressor cells on PD-1 expressing T cells, providing new insights that may be harnessed for cell targeting immune therapies.
To understand the mechanisms by which plasmablasts regulate the CD4+ T cell response in experimental Chagas disease, we first investigated the kinetics of the plasmablast response. Compared with uninfected mice, flow cytometry analysis revealed that the number of splenic plasmablasts (CD3-B220lowIgD-CD138+) increased prior to the GC reaction and peaked at 15-18 days post infection (Dpi) with T. cruzi (Figure 1A). The number of plasmablasts declined at 20 Dpi, whereas the number of GC B cells (CD19+CD3-CD138-B220+CD38lowBcl6+) remained high until 25 Dpi and decreased later. The peak of the total CD4+ T cell response occurred after the frequency and number of plasmablasts had decreased (Figure 1A). This cellular kinetics may be related to the pathogen load since the parasitemia peaks at 10-12 Dpi (19).
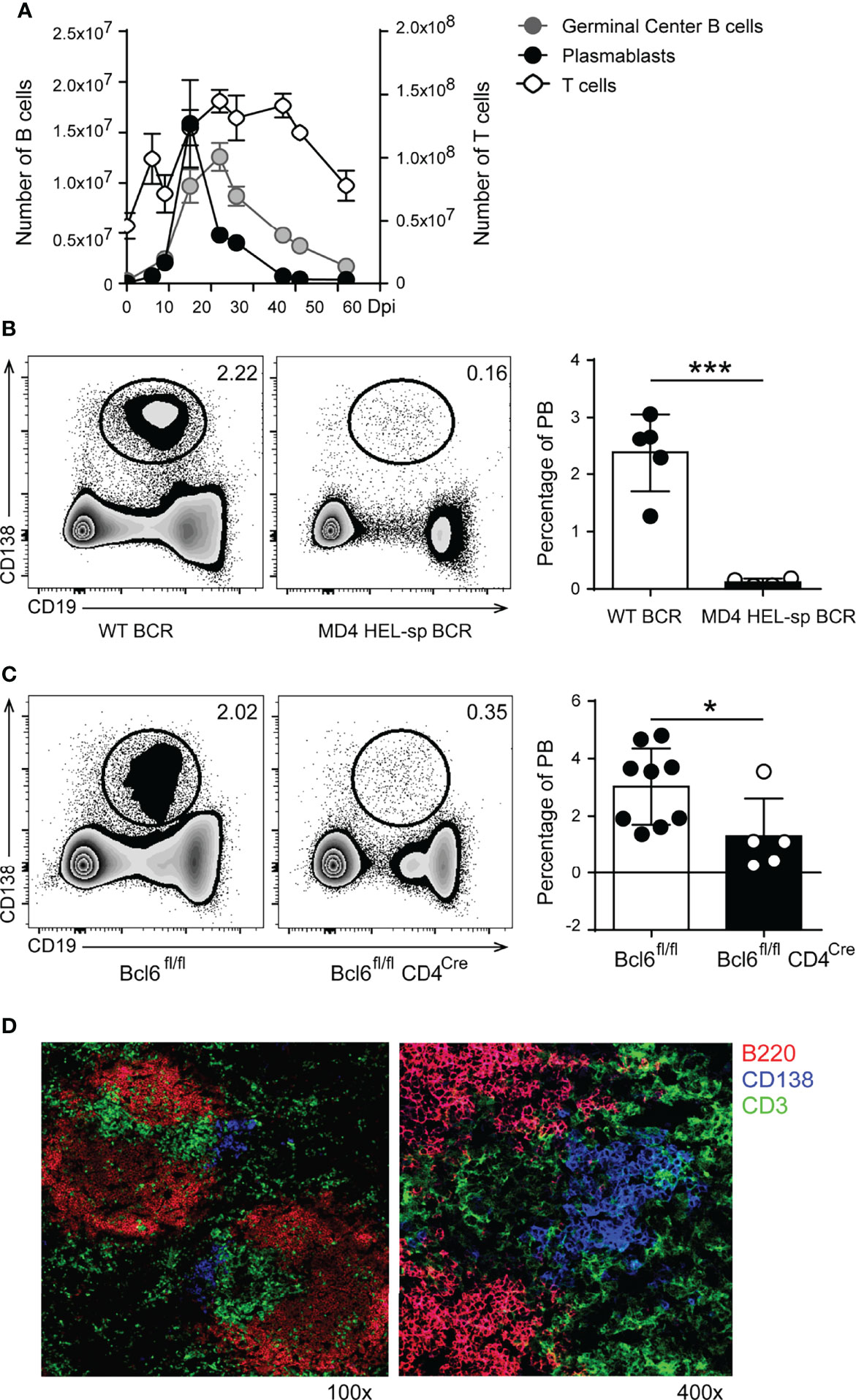
Figure 1 In T. cruzi infection, the plasmablast response peaks before GC reaction, was generated in a BCR specific manner and required Bcl-6+CD4+T cell collaboration. (A) Kinetics of plasmablasts (PB, CD19intB220lowCD138hi), Germinal Center (GC) B cells (CD19+CD3-CD138- B220+CD38lowBcl6+) and CD3+CD4+ T cells number in the spleen of T. cruzi infected mice. (B) Representative flow cytometry plots and statistical analysis of the percentage of splenic PB (CD138+CD19int/low) from WT (WT BCR, n = 5) and MD4 (HEL-sp BCR, n = 8) mice at 14 Dpi with T. cruzi (***p < 0.001, two tailed t Test). (C) Representative flow cytometry plots and statistical analysis of the percentage of splenic PB from Cre negative littermates Bcl6fl/fl (n = 9) and Bcl6fl/flCD4Cre (Bcl-6negCD4+ T cells, n = 5) mice at 14 Dpi with T. cruzi. (*p < 0.05, two tailed t Test) (D) Representative Immunofluorescence of spleen sections (7um) from T. cruzi-infected C57BL/6 mice obtained at 14 Dpi, stained with anti-B220 (red), anti-CD3 (green) and anti-CD138 (blue). Magnification: x100 (left) and x400 (right) (n = 9). Results shown in (A, D) are representative of five independent experiments. Data in (B, C) are representative of two independent experiments.
To understand in more depth the origin and characteristics of plasmablasts in T. cruzi infection, we infected MD4 mice (whose B cells express a transgenic BCRs specific for a T. cruzi non-related protein, HEL) and their non-transgenic controls (WT-BCR, whose B cells express a wild-type BCR repertoire). Figure 1B illustrates a representative staining of CD19 and CD138 on lymphocytes from the spleen of T. cruzi-infected WT-BCR and MD4 mice. Infection of MD4 mice showed that plasmablast induction depends on BCR signaling, since plasmablasts were almost absent from infected MD4 mice compared to WT mice around the peak of the plasmablast responses at 14 Dpi (Figure 1B, left and right graphs).
Since extrafollicular (EF) antibody responses require Bcl-6 expression by T cells (21), we investigated whether plasmablast induction and/or survival required the collaboration of Bcl-6+CD4+T cells. For this, we took advantage of Bcl6fl/flCD4Cre mice that lack Tfh and CXCR5negBcl-6+CD4+ T cells. The dot plots (Figure 1C) show representative data illustrating the identification of CD19int/lowCD138+ cells among lymphocytes from the spleen of T. cruzi Bcl6fl/fl and Bcl6fl/flCD4Cre infected mice. While Cre-negative littermates develop a normal Bcl-6+CD4+T cell response (22), T. cruzi-infected Bcl6fl/flCD4Cre mice had a significant decrease in the frequency of plasmablasts (Figure 1C, left and right graph), indicating that the plasmablast response is critically dependent on Bcl-6+CD4+ T cells.
Immunofluorescence analysis of the spleen of T. cruzi-infected mice (Figure 1D) identified CD138+ cells extending beyond the classical sites of extrafollicular plasmablast growth (Figure 1D, CD138+ cells, blue) into the T zone areas (Figure 1D, CD3+ cells, green), a finding consistent with our previous reports (20, 23). The results suggest that, in T. cruzi infection, plasmablasts were induced in a BCR-dependent manner, required Bcl-6+CD4+ T cell collaboration, and were located at extrafollicular sites in close proximity to T cells.
To identify if plasmablasts express inhibitory molecules able to modulate the T cell response, we evaluated the expression of PD-L1 and PD-L2 (24) on plasmablasts from T. cruzi-infected mice. In comparison with B and non-B cells, plasmablasts from T. cruzi-infected mice, evaluated at different Dpi, expressed the highest levels of PD-L1 (determined as MFI, Figure 2A). Most of the plasmablasts (nearly 97% of them) detected during the acute phase of T. cruzi infection expressed high levels of PD-L1 (Figures 2A, B). As a consequence of the infection, B cells and plasmablasts also expressed PD-L2, but at similar levels (Figure 2A, see histograms and bar graph). PD-L1hi plasmablasts were absent in the spleen of infected mice during the chronic phase of the infection (130 Dpi, data not shown). The frequency of CD138+ Blimp-1+ cells in the spleen and bone marrow of infected mice, obtained at 15 and 130 Dpi, is illustrated in the dot plots of Figure 2B. As a control, the frequency of CD138+ Blimp-1+ cells in the bone marrow of age-matched non-infected mice is also shown (Figure 2B). CD138+ Blimp-1+ cells in the bone marrow of 15 Dpi-infected mice were few, and the frequency of these cells was significantly higher in the bone marrow of 130 Dpi-infected mice than in age-matched non-infected mice or 15 Dpi-infected mice (Figure 2B). While most plasmablasts analyzed during the acute phase were PD-L1+ (Figure 2C, grey histogram), only ~ 50% of the bone marrow plasma cells detected in the chronic phase (130 Dpi) expressed PD-L1 (Figure 2C, black histograms). PD-L1 expression on plasma cells detected in chronic infection was significantly lower (Figure 2C, bar graph, black bar) than PD-L1 expression on plasmablasts present at 15 Dpi (Figure 2C, bar graph, white bar).
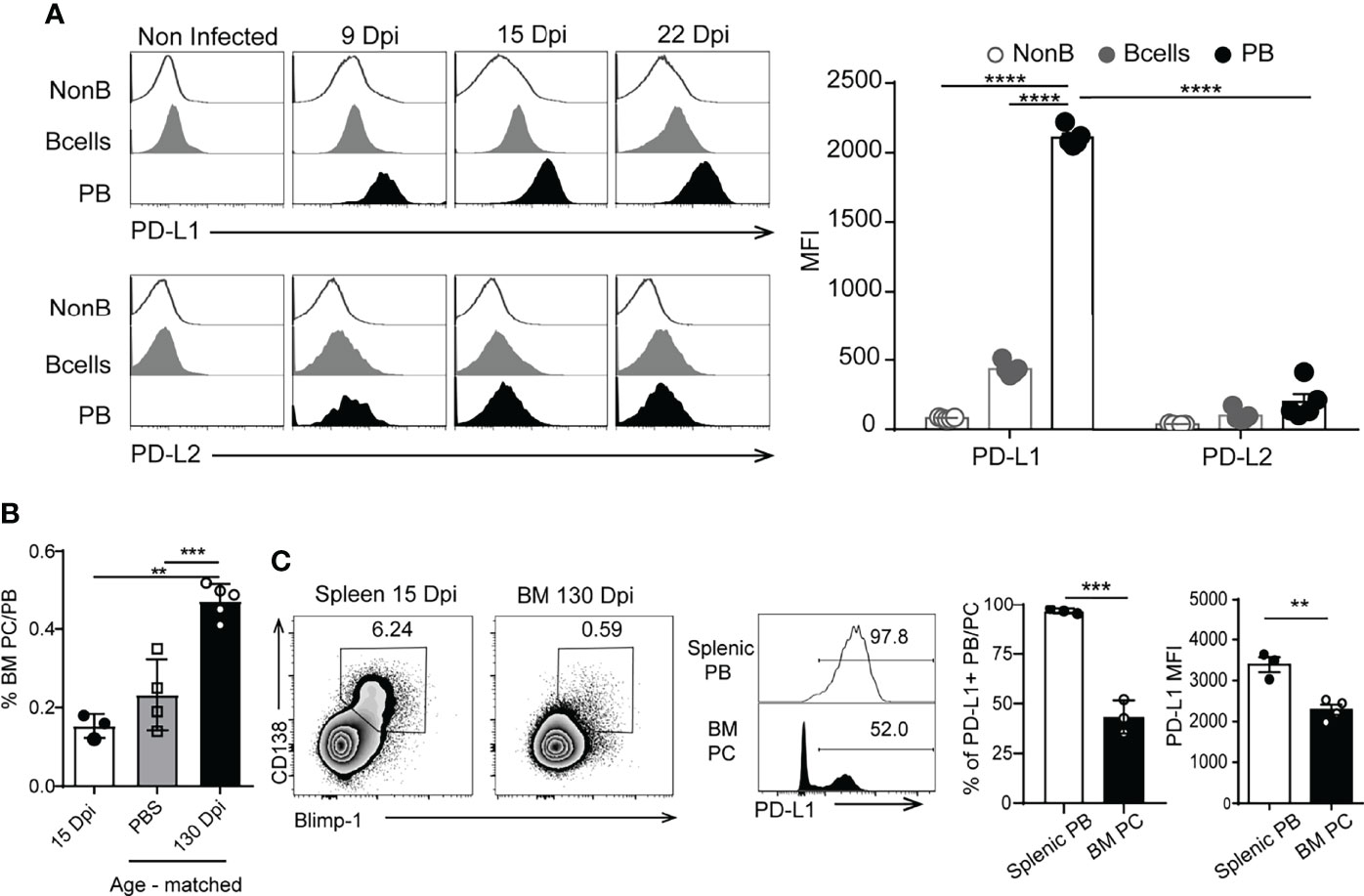
Figure 2 Plasmablasts from T. cruzi-infected mice expressed high levels of PD-L1. (A) Representative histograms of PD-L1 or PD-L2 expression on splenic non-B cells (CD19-CD138-B220-), B cells (CD19+CD3-CD138-B220+) from non-infected mice (n=5) and T. cruzi-infected C57BL/6 mice (n=5) and on plasmablasts (PB) (CD19intB220lowCD138hi) from T. cruzi-infected C57BL/6 mice evaluated at 9, 15 and 22 Dpi. Statistical analysis of mean fluorescence intensity (MFI) of PD-L1 or PD-L2 on the mentioned cells analyzed at 15 Dpi. ****p < 0.0001 one-way ANOVA, Bonferroni post hoc test. (B) Statistical analysis of the frequency of PB/PC (plasma cell) CD138+Blimp-1+ of live single lymphocytes (gated on IgD-IgM-CD11b-CD24-) from bone marrow (BM) obtained at 15 Dpi (white bar) and at 130 Dpi with T. cruzi (black bar). Cells from BM from age-matched non-infected mice, injected with PBS, were used as controls (gray bar). (C) Representative plots of CD138 vs Blimp-1 of live single lymphocytes (gated on IgD-IgM-CD11b-CD24-) from spleen and bone marrow (BM) obtained at 15 Dpi and 130 Dpi with T. cruzi, respectively. Representative histograms of PD-L1 on gated CD138+Blimp-1+B220+ cells. Numbers in histograms indicate the percentage of PD-L1+ cells. Bar graph shows statistical analysis of the frequency of PD-L1+ PB/PC and MFI of PD-L1+ on splenic PB from 15 Dpi-Tcruzi infected mice (white bar) and 130 Dpi-BM plasma cells (PC). **p < 0.01, one-way ANOVA, Bonferroni post hoc test. Data in (A, B) are representative of three independent experiments. ***p < 0.001.
We also investigated whether PD-L1hi plasmablasts are unique to T. cruzi-infected mice or if they are present in other persistent infections. We also analyzed the expression of PD-L1 on other B lineage cells. We evaluated the presence of PD-L1hi plasmablasts in mice infected with the Armstrong strain of LCMV, which causes acute infection, or with the LCMV clone 13, which causes chronic infection (25). Analysis of the spleens of mice infected with LCMV (either Armstrong –ARM- or clone 13 –Cl13- isolates) or with P. chabaudi analyzed after 9 and 10 Dpi, respectively, showed similar frequencies of PD-L1+plasmablasts (Figure S1), and that almost all the plasmablasts were PD-L1+ (Figure S1 and Figure 3A). In a similar way to our findings with T. cruzi, the plasmablasts from P. chabaudi and from ARM- or Cl13-LCMV-infected mice also expressed high levels of PD-L1 in comparison to naive and GC B cells and T cells from the same mice (Figure 3A).
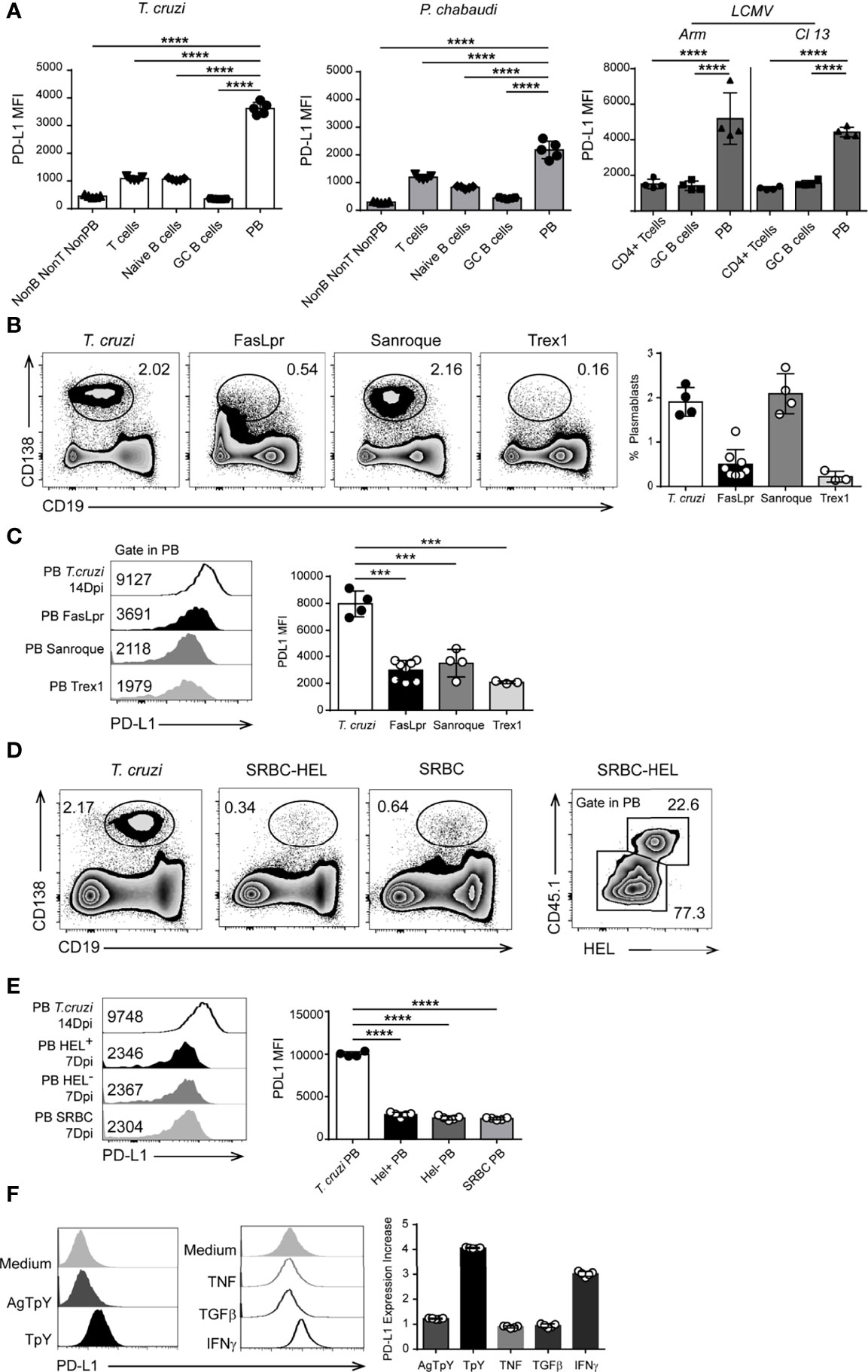
Figure 3 PD-L1hi plasmablasts were present in infected but not in autoimmune mice. (A) Statistical analysis of MFI of PD-L1 on Non-B non-T non-PB cells (CD3-CD19-B220-CD138-), T cells (CD3+CD19-B220-), naïve B cells (CD19+CD3-CD138-B220+IgD+CD38+Bcl6-), GC B cells (CD19+CD3-CD138-B220+CD38lowBcl6+) and plasmablasts (PB, CD19 intCD138+) from the spleen of T. cruzi-infected mice (n=5) obtained at 14 Dpi, or from the spleen of P. chabaudi-infected mice obtained at 10 Dpi (n=5); or CD4+ T cells, GC B cells, and PB from the spleen of Arm- or Cl13 LCMV-infected mice obtained at 9 Dpi (n = 4 for each LCMV infection). ****p < 0.0001, one-way ANOVA, Bonferroni post hoc test. (B) Representative flow cytometry plots and statistical analysis of the percentage of splenic plasmablasts from T. cruzi-infected- and from FasLpr, Sanroque and Trex1 mice. C57BL6 mice (n = 4) were infected with T. cruzi Y strain, and the spleens were obtained at 14 Dpi. Spleens from FasLpr (n = 7), Sanroque (n = 4) and Trex1 (n = 3) mice were obtained at 10-12 weeks of age. Splenocytes were stained with anti-B220, anti-CD138 and anti-PD-L1. (C) Representative histograms and statistical analysis of PD-L1 expression (MFI) on plasmablasts (PB) from the spleen of T cruzi-infected mice and FasLpr, Sanroque and Trex1 mice. (D) Representative flow cytometry plots of the percentage of plasmablasts from 14 Dpi-T. cruzi-infected mice and from mice immunized with 2x108 SRBC or with SRBC conjugated with HEL. SWHEL-specific B cells identified as CD45.1+HEL+ cells and the percentage of SWHEL-specific plasmablasts CD138+CD19low in CD45.2 mice transferred with 30.000 HEL-specific B cells and immunized with 2x108 SRBC conjugated with HEL. (E) Representative flow cytometry histograms and statistical analysis of PD-L1 expression (MFI) on plasmablasts from T. cruzi-infected mice (n = 4), SWHEL-specific (PB HEL+) and -non-specific (PB HEL-) plasmablasts from CD45.2 mice transferred with 30,000 HEL-specific B cells and immunized with 2x108 SRBC conjugated with HEL (n = 5), and on plasmablasts from mice immunized with SRBC (7 DPI) (PB SRBC, n = 5). (F) Representative histograms of PD-L1 expression on B cells cultured with medium, T. cruzi trypomastigote antigens (AgTpY), live T. cruzi trypomastigotes (TpY), TNF, TGFβ, and IFNγ. Bar graph shows statistical analysis of the PD-L1 expression increase, described as a fold change, on B cells cultured with the mentioned stimulus respect to medium (n = 5). ***p < 0.001, ****p < 0.0001 one-way ANOVA, Bonferroni post hoc test. Data in (A) are representative of three independent experiments and in B–F) are representative of two independent experiments.
We next investigated whether PD-L1hi plasmablasts occur in autoimmune disease or after immunization with non-microbial protein antigens. Mice known to develop spontaneous autoimmunity, such as Faslpr, Sanroque and Trex1-deficient mice, had varying frequencies of splenic plasmablasts (Figure 3B, representative dot plots and bar graphs). Regardless of the frequency of plasmablasts present in the autoimmune mice, PD-L1 expression on splenic plasmablasts determined as MFI from autoimmune mice was significantly lower than on plasmablasts in mice infected with T. cruzi (Figure 3C, representative histograms and bar graphs), although they remained positive compared to the isotype control.
To test whether the protein antigen-immunization that induces EF plasmablasts triggers PD-L1 expression on plasmablasts, we used the SwHEL cell transfer model (26). HEL-specific B cells from SwHEL mice (CD45.1+, which contain a large population of B cells with BCR that recognize HEL, hen egg lysozyme) were adoptively transferred into C57BL/6 mice, which were then immunized iv with sheep red blood cells (SRBC) alone (immunization control) or SRBC conjugated to HEL2x (26). PD-L1 expression was analyzed on CD19lowCD138+ cells gated in CD45.1+HEL+ cells (Figure 3D). PD-L1 expression at the peak of the EF HEL-specific plasmablast response (day 6) was significantly lower than that from plasmablasts at the peak of acute T. cruzi infection (14 Dpi) (Figure 3E, histograms - PB T cruzi MFI: 9748 vs PB HEL+ MFI: 2346). Importantly, HEL-specific plasmablasts had a PD-L1 expression similar to that of non-HEL binding plasmablasts and that of plasmablasts generated by SRBC alone (Figure 3E). Overall, the results suggest that BCR binding on B cells was not sufficient to induce high PD-L1 expression on plasmablasts.
Since PD-L1hi expression on plasmablasts was observed in infected mice, we hypothesize that a strong inflammatory environment or the pathogens favor the high expression of PD-L1 on plasmablasts. To test this, purified B cells from uninfected C57BL/6 mice were cultured in the presence or absence of T. cruzi antigens, live trypomastigotes, or cytokines, and PD-L1 expression was evaluated after 24 h. IFNγ, TNF and TGFβ were the selected cytokines and these were tested as PD-L1-inducers on B cells because they have been shown to increase PD-L1 expression on other cell types (27–29), and they are present at high levels during the acute phase of T. cruzi infection, when PD-Lhi plasmablasts are present (19, 30).
In comparison to B cells cultured with medium alone, PD-L1 upregulation was observed when B cells were cultured with live trypomastigotes or with recombinant IFNγ but not with T. cruzi antigens, TNF, or TGFβ (Figure 3F). However, PD-L1hi plasmablasts were present in IFNγ-/- mice infected with T. cruzi (Figure S2), indicating that this cytokine may be redundant with other cytokines for mediating PD-L1 upregulation on plasmablasts in vivo. Together, these results suggest that live parasites are required to induce maximal PD-L1 expression, possibly through inducing a combination of pro-inflammatory cytokines.
To test the role of PD-L1 on TNF- and IFNγ-producing cells from T. cruzi-infected mice, splenocytes stimulated with T. cruzi antigens were cultured with medium or with anti-PD-1, anti-PD-L1, or anti-PD-L2 and media, with isotypes as controls, and the concentration of cytokines was evaluated in the culture supernatant. The blockade of PD-1 and PD-L1 signaling significantly increased TNF and IFNγ concentration in the culture supernatants (Figure 4A). As expected, no significant changes were detected in the concentration of cytokines in the supernatant of splenocytes cultured with anti-PD-L2 compared to splenocytes cultured with medium (Figure 4A), indicating, as expected, that the PD-L2 pathway was not involved in the in vitro regulation of cytokine-producing splenic cells from T. cruzi-infected mice.
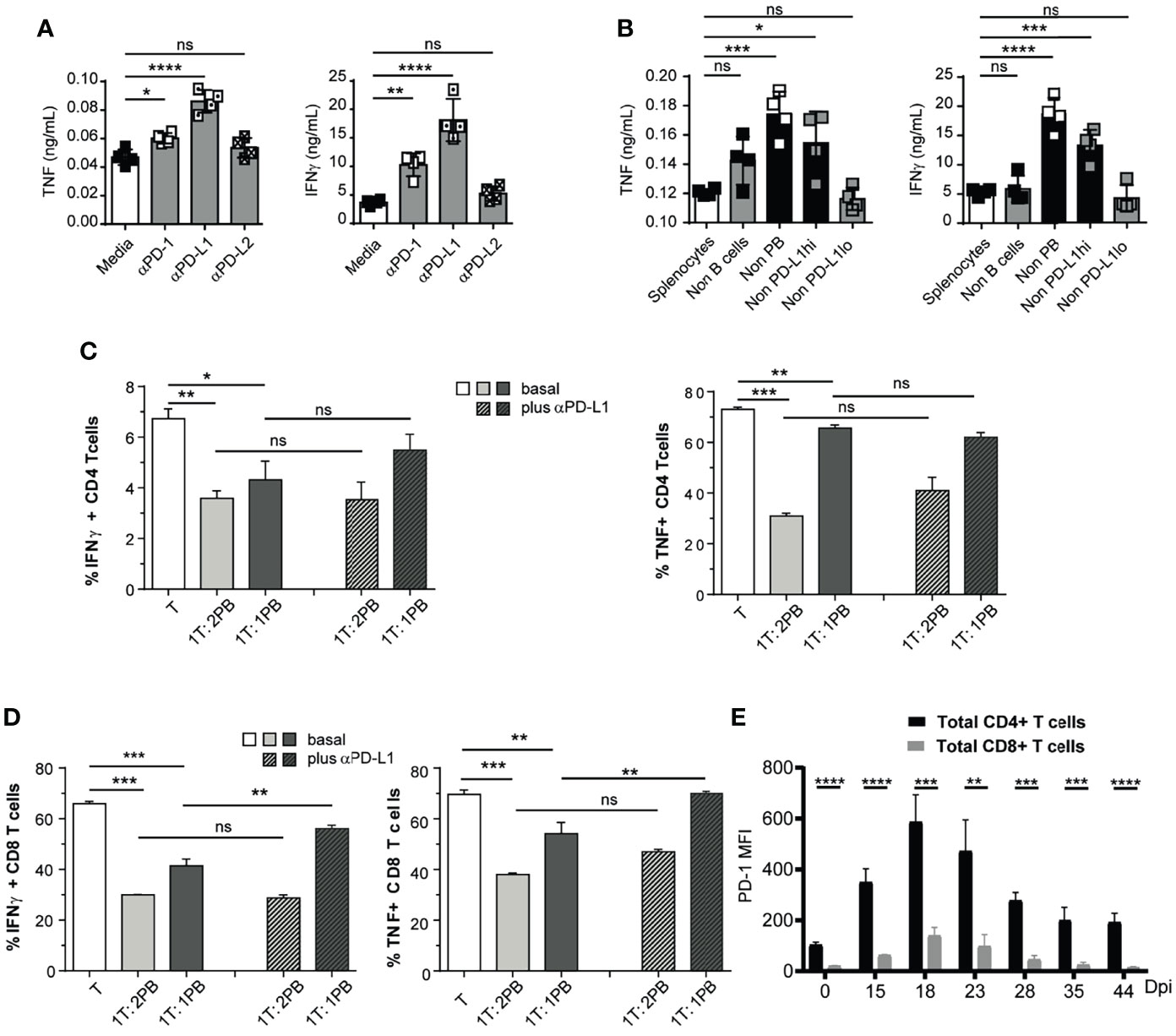
Figure 4 Plasmablasts from T. cruzi-infected mice suppressed CD8+ T cell response via PD-L1. (A) 1 x 105 Splenocytes from T. cruzi-infected mice obtained at 14 Dpi were incubated with T. cruzi antigens and cultured for 48h with media, anti-PD-1, anti-PD-L1, or anti-PD-L2. TNF and IFNγ concentrations were determined by Elisa in the culture supernatant. (B) 1x 105 Non-B (CD19negCD138neg) cells in a suspension of total splenocytes (non-depleted) from T. cruzi infected mice obtained at 14 Dpi or depleted of B cells (Non-B cells), plasmablasts (Non-PB), PD-L1hi cells (Non-PD-L1hi) or PD-L1low (Non-PD-L1lo) cells were cultured with T. cruzi antigens for 48h. TNF and IFNγ concentrations were determined by Elisa in the culture supernatant. (C) Purified CD4+ and (D) CD8+ (responder T cell from non-infected mice activated with anti-CD3 plus anti-CD28 were co-cultured with sorted plasmablast from T. cruzi-infected mice obtained at 14 Dpi in 1:1 or 1:2 ratio with for 3 days, in the presence of control antibody (basal) or anti-PD-L1. Bars show the frequency of TNF+ and IFNγ+ CD4+ or CD8+ T cells which were determined by flow cytometry. (E) PD-1 expression, determined as MFI by flow cytometry, of splenic CD4+ (black) and CD8+ (grey) T cells obtained from T. cruzi-infected mice at different Dpi. Data in (A–D) are representative of two independent experiments. ta are shown as mean ± SD of triplicates cultures. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 two-tailed t Test). ns, non significant.
To identify the B-lineage cell population responsible for limiting cytokine-producing T cells, splenocytes from T. cruzi-infected mice obtained at 14 Dpi were depleted of plasmablasts or B cells by cell sorting (Figure S3A–C). Later, the cells were cultured with parasite antigens, and cytokine concentration was determined in culture supernatants. Only plasmablast depletion significantly increased the concentration of TNF and IFNγ in the culture supernatant (Figure 4B). Depletion of PD-L1hi cells, but not of PD-L1low cells, from splenocytes also led to an increase in the concentration of cytokines in the supernatant (Figure 4B). Given that depletion of PD-L1hi cells concomitantly leads to plasmablast elimination (Figure S3D, E), these data suggest that plasmablasts with high PD-L1 expression inhibit cytokine-producing cells in vitro.
Next, we investigated the regulatory capacity of PD-L1hi plasmablasts on CD4+ and CD8+ T cell cytokine production. For this, sorted plasmablast from 14 Dpi-T. cruzi-infected mice were incubated with purified splenic CD4+ or CD8+ T cells obtained from the spleen of non-infected control mice in a plate coated with anti-CD3/CD28. Three days after co-culture, the intracellular cytokine content of T cells was assessed by FACS. When T cells were co-cultured with plasmablasts, the frequency of TNF+ and IFNγ+ CD4+ or CD8+ T cells significantly decreased (Figures 4C, D, plain light and dark gray bars).
To explore the role of PD-L1, we performed blocking experiments by adding an anti-PD-L1 mAb from the beginning of the co-culture. PD-L1 blockade increased the frequency of IFNγ- and TNF-producing CD8+ T cells. In contrast to CD8+ T cells, the effect of anti-PD-L1 blockade in co-culture with CD4+ T cells was less pronounced or absent (Figure 4C, plain gray bars vs striped gray bars). Next, we evaluated the expression of PD-1, the target of PD-L1, on T cells from T. cruzi-infected mice at different times post infection. Figure 4E shows PD-1 expression of splenic CD4+ and CD8+ T cells, determined as MFI by flow cytometry, obtained at different Dpi. We observed that splenic CD4+ T cells from T. cruzi-infected mice had significantly higher expression of PD-1 than CD8+ T cells. Thus, regardless of PD-1 expression, CD8+ T cells were more susceptible to PD-L1 expressing plasmablast suppression.
Our current understanding of humoral responses has been derived largely from immunization with model antigens. In the last decade, murine models of infections that also present a significant inflammatory component have enabled new cell populations to be identified with fundamental roles in the induction of the B cell response (31, 32), the discovery of novel sites where B cells are activated (33) and the identification of distinct regulatory B cell functions (23, 34). Here, we describe plasmablasts with a very high expression of PD-L1 that were detected in infected mice but were absent in mice with autoimmunity or immunized with T-dependent model antigens. Our findings provide the first evidence that most of the plasmablasts in T. cruzi, Plasmodium or LCMV infections express high levels of PD-L1, in comparison to naïve and GC B cells, T cells or other lymphoid cells. Interestingly, CD138+B220lowBlimp-1hi cells present in bone marrow at the chronic phase of T. cruzi infection did not exhibit high expression of PD-L1, suggesting that PD-L1hi expression is not a characteristic of all antibody-secreting cells. PD-L1 expression was highest in one antibody-secreting cell population, the plasmablasts, present in the early acute phase of the infection, when there is a strong inflammation, and the effector response of T cells begins to develop (35).
Most of the plasmablasts detected during the acute phase of T. cruzi infection are PD-L1hi and are extrafollicular since the CD138+B220low cells, located outside the follicle, are the only antibody-secreting cells expressing CD138 at this time of infection (20) and Figure 1). The peak of PD-L1hi plasmablasts precedes the peak of GC reaction and, as previously reported for the extrafollicular response (21), Bcl-6 expression in T cells is essential for their optimal induction/survival. Consistent with this, very few PD-L1hiB220low cells were detected in the spleens of infected mice lacking Bcl-6 expression in CD4+T cells. Additionally, despite the prevailing view that, during the acute phase of T. cruzi infection most B cells lack parasite specificity, suggesting non-antigen specific B cell activation (36), PD-L1hi plasmablasts required parasite-specific BCRs. Indeed, mice whose B cells expressed BCRs non-specific for T. cruzi exhibited a very low number of plasmablasts at 14 Dpi.
High PD-L1 expression on antibody-secreting cells was previously reported on intestinal lamina propria IgA+ plasma cells, which are the major PD-L1 expressing cells in that tissue. PD-L1 expression levels on intestinal IgA plasma cells are higher than those on IgG plasma cells in peripheral lymphoid tissues (37). Probably, the high expression of PD-L1 in intestinal IgA plasma cells is triggered by microbiota, as we observed that microorganisms (in this report, T. cruzi) per se and/or inflammatory cytokines can induce high PD-L1 expression on B cells. PD-L1 expression on plasmablasts was also described in different mouse prostate cancer models under Oxaliplatin treatment (18). This therapy induces tumor-infiltrating plasmacytes that express IgA, IL-10 and PD-L1. Elimination of these cells, which also infiltrate human-therapy-resistant prostate cancer, allows cytotoxic T cell-dependent eradication of Oxaliplatin-treated tumors (18), suggesting a suppressive function in this population. Considering that PD-L1 plays a major role in suppressing the adaptive arm of the immune system, PD-L1hi plasmablasts emerge as a population with regulatory capacity.
The majority of studies on Breg cells have focused on the predisposition of immature and mature B cells to produce IL-10 and to suppress a range of autoimmune or allergic conditions in both mice and humans (38). Little information is available about the regulatory function and mechanisms of action of antibody-secreting cells. In this study, we found that plasmablasts from T. cruzi-infected mice exert a regulatory function through PD-L1 in vitro on T cells. When splenocytes from T. cruzi-infected mice were depleted from PD-L1hi cells or plasmablasts and later cultured with T. cruzi antigens, we observed a greater increase in TNF and IFNγ levels in the culture supernatant than in the culture of total splenocytes. We observed that T. cruzi plasmablasts suppressed both the CD4+ and the CD8+ T cell response. Analyzing possible targets of PD-L1hi plasmablasts, we identified CD8+ T cells as targets of PD-L1–expressing plasmablasts. It should be noted that, although there was a tendency of anti-PD-L1 to revert the suppression of CD4+ T cells triggered by plasmablasts, this was not significant, suggesting that plasmablasts may suppress the CD4+ T cell response using other mechanisms.
The immunomodulatory effect exerted by plasmablasts may involve more than one mediator. It has been described that IL-10-producing plasmablasts exert regulatory functions in autoimmunity (39) and we observed that plasmablasts from T. cruzi-infected mice produce IL-10 (data not shown). Additionally, it was reported that a high consumption of glutamine by plasmablasts can deprive other populations of substrate, conditioning their response (40). Whether these mechanisms operate in T. cruzi infection needs to be determined.
Consistent with our previous report on B cells from patients with rheumatoid arthritis (41), this study reveals that extrafollicular PD-L1hi plasmablasts, which precede the GC response, can operate in vitro as modulatory cells on PD-1 expressing CD8+ T cells, providing new insights that could be harnessed for cell targeting immune therapies. It has been described that human and mouse glioblastoma-associated B cells that overexpress PD-L1 also have immunosuppressive activity directed towards activated CD8+ T cells (42). B cells located in the tumor microenvironment, but not circulating or from lymph nodes, express high levels of PD-L1, but this expression apparently does not depend on the inflammatory environment but rather tumor myeloid-derived suppressive cells deliver microvesicles transporting PD-L1, which is taken up by tumoral B cells.
Plasmablasts also express PD-L2, another molecule that interacts with PD-1 and suppresses T cell proliferation and cytokine release (9). However, we observed that PD-L2 was not mediating effector cell suppression, since PD-L2 blockade in cell culture did not affect cytokine production.
The data obtained in this work allow us to partially explain why, during infections with protozoan parasites such as T. cruzi and Plasmodium, a delay is generated in T cell response simultaneously with high B cell activation. The acute phase of Chagas disease in mice and humans is marked by a state of immunosuppression, which coexists with polyclonal B cell activation, in which T. cruzi replicates extensively and induces immunomodulatory molecules that delay parasite-specific responses mediated by effector T cells (43). Also, it has been reported that, in the acute phase of infection with P. chabaudi, the composition of the B cell compartment is altered, with vigorous extrafollicular growth of plasmablasts and GC formation. In Plasmodium infection, extrafollicular foci of plasmablasts are visible from day 4, initiating a very strong plasma cell response. By day 10, plasma cells are localized in the periarteriolar region of the white pulp and form clusters, occupying part of the area normally filled by T cells. During this phase of infection, as in T. cruzi infection, there is a delayed GC response and a significant reduction of the humoral response to T-dependent antigens (44).
Finally, we observed that mice infected with LCMV ARM or Cl13 exhibited high PD-L1 expression on plasmablasts at 9 Dpi. Infection of mice with LCMV ARM and with Cl13 has served as a powerful model to study immune responses during acute and chronic viral infections, respectively (2). In particular, LCMV Cl13, which produces chronic infection, induces higher and more sustained PD-1 upregulation in T cells, and PD-1/PD-L1 interaction significantly contributes to T cell suppression and viral persistence (45). Interestingly, LCMV infection also triggers polyclonal B cell activation (46, 47). Given that virus-specific CD4+ and CD8+ T cells from LCMV Cl13- (but not ARM-)-infected mice express high levels of PD-1 (45), it is tempting to speculate that PD-L1hi plasmablasts may also contribute to suppressing antiviral T cells during chronic LCMV infection. It should be noted that, although PD-L1hi plasmablasts were also induced after acute LCMV ARM infection, they are unlikely to exert a significant regulatory function in this setting, as virus-specific T cells from ARM-infected mice exhibit low levels of PD-1 expression (45).
It may be important to point out that plasmablasts may have more than one function. Plasmablasts produce cytokines (19, 48) and, therefore, can influence the response of other cell populations independent of PD-L1 expression. Plasmablasts secrete antibodies which could act directly on the parasite, controlling parasite replication (19). Therefore, we hypothesize that plasmablasts may act according to the context and the characteristics of the target cells.
In summary, our results revealed a new function of extrafollicular plasmablasts in the context of infections, which could provide a new target for the rational design of new therapeutic treatments aimed at enhancing protective T cell responses during Chagas disease, malaria and potentially other chronic infections.
C57BL/6, and B6.129S7-Ifngtm1Ts/J (IFNγKO) mice were initially obtained from The Jackson Laboratories (USA). These mice were housed and bred in the Animal Facility of the CIBICI-CONICET, FCQ-UNC.
Mice from the Australian National University Bioscience Services were maintained in specific pathogen-free conditions and had access to food and water ad libitum. The mice used in the experiments conducted at the John Curtin School of Medical Research (JCSMR) were:
HEL-IgM-BCR transgenic mice (MD4, knock in for a transgenic BCR specific for HEL, a T. cruzi non-related protein). As controls of the MD4 mice, we used their littermates whose genotype was negative for the knock in and therefore their B cells expressed a complete BCR repertoire.
SWHEL CD45.1 mice, which carry a Vk10k light chain transgene and a knocked in VH10 Ig heavy chain in place of the JH segments of the endogenous IgH gene that encode a high-affinity antibody for HEL (hen egg lysozyme), were obtained from Robert Brink’s laboratory (Garvan Institute, Sydney, New South Wales, Australia). The BCR of all mature B cells recognize HEL.
Bcl6fl/fl mice were mated to CD4-cre mice to generate Bcl6fl/fl CD4Cre mice (21). Mice expressing Cre lack Tfh, while Cre-negative T lymphocyte littermates will present a functional Bcl6 molecule and develop a normal Tfh response.
Mice experiments conducted at the CIBICI were approved by, and performed in accordance with the guidelines of, the Institutional Animal Care and Use Committee of the FCQ-UNC (Approval Number HCD 1525/14). Mice experiments conducted at JCSMR were approved by the Animal Experimentation Ethics Committee (Australian National University protocol number 2016/17). Male and female mice were age-matched (8-12 weeks-old) and housed with a 12-h light-dark cycle.
Mice experiments conducted at the Division of Molecular Biology, Department of Biological Sciences, University of California, San Diego, were handled in accordance with the requirements of the National Institutes of Health and the Institutional Animal Care and Use Guidelines of University of California, San Diego.
T. cruzi infection: mice were infected intraperitoneally with 1 × 104 trypomastigotes of T. cruzi Y-Br strain (49) diluted in a sterile solution of 1% glucose in PBS (23). Uninfected littermates were injected with 1% glucose in PBS and processed in parallel. Spleens were collected at different Dpi for immune response analysis. Bone marrow cells of T. cruzi-infected mice were isolated at 15 and 130 Dpi by flushing mice femurs and tibias with RPMI-1640.
Plasmodium chabaudi infection: mice were infected intravenously (iv) with 10000 parasites of P. chabaudi (strain AS) obtained from a mouse infected 8 days previously from a frozen stock. The spleens of P chabaudi-infected mice were obtained at 10 Dpi.
LCMV infection: mice were infected with 2 × 106 plaque-forming units (pfu) of LCMV Arm (ARM) or LCMV clone 13 (Cl13) iv via tail vein. The Armstrong strain of LCMV causes acute infection in which the virus is cleared 8 days post infection, while the LCMV clone 13 causes chronic infection, with the virus persisting in the brain and kidneys for >3 mo (25).
Viruses were propagated on BHK cells and quantified by plaque assay performed on Vero cells (50). The spleens of LCMV-infected mice were obtained at 9 Dpi.
T. cruzi parasites (Y-Br strain) were cultured in NIH3T3 mouse fibroblasts and were collected as described (49). Trypomastigotes were washed twice in sterile PBS (37 °C) and were resuspended in 500 ul of PBS (4 °C) and sonicated in ice-cold water for 10 minutes. Aliquots were stored at -80 °C until use and thawed only once. Protein quantification was carried out after thawing in a Synergy HTX Multi-Mode Reader (Biotek).
To evaluate PD-L1 expression on plasmablasts generated by TD-antigen immunization, two different schedules were performed: one group of C57BL/6 mice were immunized intravenously with 2 × 108 SRBCs (Applied Biological Products Management, Australia) and the plasmablast phenotype was analyzed at 7 days post immunization (DPI). The other group of C57BL/6 mice were transferred, by iv injection, with splenocytes from CD45.1 SWHEL mice (Het/Het) containing 30.000 CD19+HEL+ cells simultaneously with 2x108 Hel-conjugated SRBCs. The frequency of plasmablasts and plasmablast phenotypes was analyzed at 7 Dpi.
Spleens were obtained and tissues were homogenized through a 0.70um cell strainer. Erythrocytes in splenic or bone marrow cell suspensions were lysed for 5 min in Tris–ammonium chloride buffer. Viable cell numbers were determined by trypan blue exclusion using a hemocytometer.
For surface staining, single-cell suspensions were washed first in ice-cold PBS and incubated for ten minutes with a Live/Dead Staining. Cells were washed in PBS and then incubated with fluorochrome-labeled Abs for 30 min at 4°C (surface staining). For intracellular cytokine staining, cells were cultured for 5 h with 50 ng/ml PMA (phorbol 12-myristate 13-acetate) (Sigma), 1,000 ng/ml ionomycin (Sigma) and Brefeldin A (eBioscience) or Monensin (eBioscience). Cells were fixed and permeabilized with BD Cytofix/Cytoperm and Perm/Wash (BD Biosciences) according to the manufacturer’s instructions. Data were collected on a BD FACSCanto II, BD LSR II, or BD Fortessa X20, and were analyzed using the FlowJo software (TreeStar). The specific cell populations and gating strategy in each case are described in Figure S4.
The following anti-mouse antibodies were used for FACS: CD19-PE (6D5), CD19-BV605 (6D5), CD3-AlexaFluor700 (17A2), CD3-APCCY7 (17A2), CD4-APC-Cy7 (GK1.5), CD8-PerCP-Cy5.5 (53-6.7), CD8-FITC (5H10-1), Streptavidin BV605, PD-1-BV421 (29F.1A12), PDL-1-Pecy7 (10F.9G2), Bcl6-PE-Dazzle594 (7D1), ICOS-AlexaFluor488 (C398.4A), CD45.1- BV605 (A20), B220-APCCy7 (RA3-6B2), Live Dead Aqua 430 were from Biolegend; CD19-AlexaFluor700 (1D3), CD19-FITC (1D3), IgD-FITC (11-26c), CD8-PECy7 (53-6.7), TNF-APC (MP6-XT22), PD-L1-PE (MIH5) and were from eBioscience; CD138-BV605 (281-2), CD138-APC (281-2), CD138-PE (281-2), CD138- Biotin, CD38-BV421 (90/CD38), CXCR5-Biotin (2G8), IFN-γ-FITC (XMG1.2), Ki-67-AlexaFluor647 (B56), Blimp-1-PE-CF594 (5E7), and CD8-BUV805 (53-6.7) were from BD Biosciences.
The following anti-mouse antibodies were used for tissue immunofluorescence: B220-PE (RA3-6B2) and CD3 FITC anti-CD3 (145-2C11) from Biolegend and CD138-APC (281-2) from BD Biosciences.
HEL was conjugated with AlexaFluor647 with a protein labeling kit (Invitrogen).
For tissue immunofluorescence, spleens from infected WT mice were collected and frozen over liquid nitrogen. Frozen sections, 7 μm thick, were cut, fixed for 10 min in cold acetone, left to dry at 25 °C and stored at -80 °C until use. Slides were hydrated in TRIS buffer and blocked for 30 min at 25 °C with 10% normal mouse serum in TRIS buffer. After blockade, slides were incubated for 50 min at 25 °C with the different Abs diluted in TRIS Buffer. Slices were mounted with FluorSave (Merck Millipore). Images were collected with an Olympus microscope (FV1000) and those recorded in the far-red channel were pseudo-colored blue.
Splenic cells from infected mice were obtained at 14 Dpi and splenocytes were stained with fluorochrome-conjugated anti-CD19, anti-CD138 and anti-PD-L1. The elimination of plasmablasts, PD-L1hi, PD-L1low and non-B cells from the splenocyte population was carried out by cell sorting in a FACS Aria II (BD Biosciences), using different gate strategies (see Figure S3).
Total splenocytes from infected mice or splenocytes depleted from plasmablasts, non-B cells, PD-L1hi and PD-L1low cells were incubated with AgTpY (5ug/mL) plus anti-PD-1, anti-PD-L1, anti-PD-L2 (5ug/mL) or control antibodies (5ug/mL). After 48 h of culture, TNF and IFNγ were quantified by ELISA in the culture supernatant according to the manufacturer’s instructions (eBioscience).
CD4+ and CD8+ T cells were purified from a pool of the spleens of 2-3 non-infected C57BL/6 mice using EasySep Mouse CD4 or CD8 negative Selection Kit II (StemCell). CD19+ CD138+ plasmablasts were obtained from the spleens of T. cruzi-infected mice at 14 Dpi by cell-sorting using a FACS Aria IIb Cell Sorter (BD Biosciences). For sorting experiments, the spleens of at least 3 mice were pooled.
5x104 CD4+ or CD8+ T cells were cultured alone or in combination with plasmablasts in a plate previously coated with anti-CD3 (1μg/mL, eBioscience) and anti-CD28 (0.25μg/mL, BD Biosciences) for 3 days in a 1:1 (T cell:plasmablast) or 1:2 ratio. When indicated, anti-PD-L1 or isotype control (both 5μg/mL) was added at the beginning of the cell culture. Next, cells were centrifuged and re-stimulated for 5 hours with PMA and ionomycin, and the frequency of TNF+ and IFNγ+ T cells was determined by flow cytometry.
Statistical significance through comparison of mean values was assessed by two-tailed Student’s t-test, one-way or two-way ANOVA, followed by the Bonferroni post hoc test using GraphPad software.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Institutional animal Care and Use Committee Facultad de Ciencias Químicas - UNC.
MGS performed and designed most of the experiments, analyzed data and prepared figures and manuscript. FFV, CGB, LA, YG, MCR, and JTB collaborated with performing experiments. EIZ supervised design, analysis, and interpretation of the LCMV experiments. EW designed, performed, analyzed, and interpreted the LCMV experiments. IAC provided P. chabaudi parasites and contributed to the design of the Plasmodium experiments. IAC and YC performed the Plasmodium experiments. CLM and EVAR contributed to the study design and analysis. AG and CGV conceived, designed, and supervised the study and wrote the manuscript. All authors reviewed the manuscript before submission.
This work was supported by grants from the Agencia Nacional de Promoción Cientı́fica y Técnica (PICT 2015-0645 and PICT 2018-01494), CONICET (PIP 201511220150100560CO) and R01AI116432 and R01AI110340 from NIH (USA). EVAR, CLM, and AG are Researchers from CONICET. MGS, FFV, LA, CB, YG, MR, and JTB, thank CONICET for the fellowship awarded. CGV was supported by Australian NH&MRC program, project, and fellowship grants. MGS was recipient of a Endeavour Scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the staff from Cell Sorter and Cytometry Core and Animal facility from CIBICI-CONICET and A Cook and J Cappelo from JCSMR for their excellent technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.828734/full#supplementary-material
1. Geiger A, Bossard G, Sereno D, Pissarra J, Lemesre J-L, Vincendeau P, et al. Escaping Deleterious Immune Response in Their Hosts: Lessons From Trypanosomatids. Front Immunol (2016) 7:212/abstract. doi: 10.3389/fimmu.2016.00212/abstract
2. Zuniga EI, Macal M, Lewis GM, Harker JA. Innate and Adaptive Immune Regulation During Chronic Viral Infections. Annu Rev Virol (2015) 2(1):573–97. doi: 10.1146/annurev-virology-100114-055226
3. Arocena AR, Onofrio LI, Pellegrini AV, Carrera Silva AE, Paroli A, Cano RC, et al. Myeloid-Derived Suppressor Cells are Key Players in the Resolution of Inflammation During a Model of Acute Infection. Eur J Immunol (2014) 44(1):184–94. doi: 10.1002/eji.201343606
4. Morán-Utrera Y, López-Monteon A, Rosales-Encina JL, Méndez-Bolaina E, Ramos-Ligonio A. Trypanosoma Cruzi SSP4 Amastigote Protein Induces Expression of Immunoregulatory and Immunosuppressive Molecules in Peripheral Blood Mononuclear Cells. J Trop Med (2012) 2012:1–10. doi: 10.1155/2012/829139
5. Poncini CV, Giménez G, Pontillo CA, Alba-Soto CD, de Isola ELD, Piazzón I, et al. Central Role of Extracellular Signal-Regulated Kinase and Toll-Like Receptor 4 in IL-10 Production in Regulatory Dendritic Cells Induced by Trypanosoma Cruzi. Mol Immunol (2010) 47(11–12):1981–8. doi: 10.1016/j.molimm.2010.04.016
6. Souza PEA, Rocha MOC, Menezes CAS, Coelho JS, Chaves ACL, Gollob KJ, et al. Trypanosoma Cruzi Infection Induces Differential Modulation of Costimulatory Molecules and Cytokines by Monocytes and T Cells From Patients With Indeterminate and Cardiac Chagas’ Disease. Infect Immun (2007) 75(4):1886–94. doi: 10.1128/IAI.01931-06
7. Gutierrez FRS, Mariano FS, Oliveira CJF, Pavanelli WR, Guedes PMM, Silva GK, et al. Regulation of Trypanosoma Cruzi-Induced Myocarditis by Programmed Death Cell Receptor 1. In: Urban JF, editor. Infect Immun, vol. 79 (2011). p. 1873–81. doi: 10.1128/IAI.01047-10
8. Dong H, Zhu G, Tamada K, Chen L. B7-H1, A Third Member of the B7 Family, Co-Stimulates T-Cell Proliferation and Interleukin-10 Secretion. Nat Med (1999) 5(12):1365–9. doi: 10.1038/70932
9. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a Second Ligand for PD-1 and Inhibits T Cell Activation. Nat Immunol (2001) 2(3):261–8. doi: 10.1038/85330
10. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, et al. Therapeutic Blockade of PD-L1 and LAG-3 Rapidly Clears Established Blood-Stage Plasmodium Infection. Nat Immunol (2012) 13(2):188–95. doi: 10.1038/ni.2180
11. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 Expression on HIV-Specific T Cells is Associated With T-Cell Exhaustion and Disease Progression. Nature (2006) 443(7109):350–4. doi: 10.1038/nature05115
12. Patil NK, Luan L, Bohannon JK, Hernandez A, Guo Y, Sherwood ER. Frontline Science: Anti-PD-L1 Protects Against Infection With Common Bacterial Pathogens After Burn Injury. J Leukoc Biol (2018) 103(1):23–33. doi: 10.1002/JLB.5HI0917-360R
13. Akhmetzyanova I, Drabczyk M, Neff CP, Gibbert K, Dietze KK, Werner T, et al. PD-L1 Expression on Retrovirus-Infected Cells Mediates Immune Escape From CD8+ T Cell Killing. In: Wherry EJ, editor. PLoS Pathog, vol. 11 (2015). p. e1005224. doi: 10.1371/journal.ppat.1005224
14. Dyck L, Mills KHG. Immune Checkpoints and Their Inhibition in Cancer and Infectious Diseases. Eur J Immunol (2017) 47(5):765–79. doi: 10.1002/eji.201646875
15. López-Medina M, Carrillo-Martín I, Leyva-Rangel J, Alpuche-Aranda C, Ortiz-Navarrete V. Salmonella Impairs CD8 T Cell Response Through PD-1: PD-L Axis. Immunobiology (2015) 220(12):1369–80. doi: 10.1016/j.imbio.2015.07.005
16. Ha S-J, Mueller SN, Wherry EJ, Barber DL, Aubert RD, Sharpe AH, et al. Enhancing Therapeutic Vaccination by Blocking PD-1–Mediated Inhibitory Signals During Chronic Infection. J Exp Med (2008) 205(3):543–55. doi: 10.1084/jem.20071949
17. West EE, Jin H-T, Rasheed A-U, Penaloza-MacMaster P, Ha S-J, Tan WG, et al. PD-L1 Blockade Synergizes With IL-2 Therapy in Reinvigorating Exhausted T Cells. J Clin Invest (2013) 123(6):2604–15. doi: 10.1172/JCI67008
18. Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive Plasma Cells Impede T-Cell-Dependent Immunogenic Chemotherapy. Nature (2015) 521(7550):94–8. doi: 10.1038/nature14395
19. Gorosito Serrán M, Tosello Boari J, Fiocca Vernengo F, Beccaría CG, Ramello MC, Bermejo DA, et al. Unconventional Pro-Inflammatory CD4+ T Cell Response in B Cell-Deficient Mice Infected With Trypanosoma Cruzi. Front Immunol (2017) 8:1548. doi: 10.3389/fimmu.2017.01548
20. Bermejo DA, Amezcua Vesely MC, Khan M, Acosta Rodríguez EV, Montes CL, Merino MC, et al. Trypanosoma Cruzi Infection Induces a Massive Extrafollicular and Follicular Splenic B-Cell Response Which is a High Source of non-Parasite-Specific Antibodies: B-Cell Response to T. Cruzi Immunol (2011) 132(1):123–33. doi: 10.1111/j.1365-2567.2010.03347.x
21. Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B Cell Priming for Extrafollicular Antibody Responses Requires Bcl-6 Expression by T Cells. J Exp Med (2011) 208(7):1377–88. doi: 10.1084/jem.20102065
22. Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, et al. Insights Into the Role of Bcl6 in Follicular Th Cells Using a New Conditional Mutant Mouse Model. JI (2013) 191(7):3705–11. doi: 10.4049/jimmunol.1300378
23. Bermejo DA, Jackson SW, Gorosito-Serran M, Acosta-Rodriguez EV, Amezcua-Vesely MC, Sather BD, et al. Trypanosoma Cruzi Trans-Sialidase Initiates a Program Independent of the Transcription Factors Rorγt and Ahr That Leads to IL-17 Production by Activated B Cells. Nat Immunol (2013) 14(5):514–22. doi: 10.1038/ni.2569
24. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and Its Ligands in Tolerance and Immunity. Annu Rev Immunol (2008) 26(1):677–704. doi: 10.1146/annurev.immunol.26.021607.090331
25. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J Virol (2003) 77(8):4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003
26. Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen Recognition Strength Regulates the Choice Between Extrafollicular Plasma Cell and Germinal Center B Cell Differentiation. J Exp Med (2006) 203(4):1081–91. doi: 10.1084/jem.20060087
27. Song S, Yuan P, Wu H, Chen J, Fu J, Li P, et al. Dendritic Cells With an Increased PD-L1 by TGF-β Induce T Cell Anergy for the Cytotoxicity of Hepatocellular Carcinoma Cells. Int Immunopharmacol (2014) 20(1):117–23. doi: 10.1016/j.intimp.2014.02.027
28. Funaki S, Shintani Y, Kawamura T, Kanzaki R, Minami M, Okumura M. Chemotherapy Enhances Programmed Cell Death 1/Ligand 1 Expression via TGF-β Induced Epithelial Mesenchymal Transition in non-Small Cell Lung Cancer. Oncol Rep (2017) 38(4):2277–84. doi: 10.3892/or.2017.5894
29. Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory Cytokines IL-17 and TNF-α Up-Regulate PD-L1 Expression in Human Prostate and Colon Cancer Cells. Immunol Lett (2017) 184:7–14. doi: 10.1016/j.imlet.2017.02.006
30. Waghabi MC, de Souza EM, de Oliveira GM, Keramidas M, Feige J-J, Araújo-Jorge TC, et al. Pharmacological Inhibition of Transforming Growth Factor β Signaling Decreases Infection and Prevents Heart Damage in Acute Chagas’ Disease. Antimicrob Agents Chemother (2009) 53(11):4694–701. doi: 10.1128/AAC.00580-09
31. Gaya M, Barral P, Burbage M, Aggarwal S, Montaner B, Warren Navia A, et al. Initiation of Antiviral B Cell Immunity Relies on Innate Signals From Spatially Positioned NKT Cells. Cell (2018) 172(3):517–533.e20. doi: 10.1016/j.cell.2017.11.036
32. Merluzzi S, Frossi B, Gri G, Parusso S, Tripodo C, Pucillo C. Mast Cells Enhance Proliferation of B Lymphocytes and Drive Their Differentiation Toward IgA-Secreting Plasma Cells. Blood (2010) 115(14):2810–7. doi: 10.1182/blood-2009-10-250126
33. Trivedi N, Weisel F, Smita S, Joachim S, Kader M, Radhakrishnan A, et al. Liver Is a Generative Site for the B Cell Response to Ehrlichia Muris. Immunity (2019) 51(6):1088–1101.e5. doi: 10.1016/j.immuni.2019.10.004
34. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-Producing B Cells are Critical Regulators of Immunity During Autoimmune and Infectious Diseases. Nature (2014) 507(7492):366–70. doi: 10.1038/nature12979
35. Cardillo F, Pinho RT, Antas PRZ, Mengel J. Immunity and Immune Modulation in Trypanosoma Cruzi Infection. Pathog Dis (2015) 73(9):ftv082. doi: 10.1093/femspd/ftv082
36. Minoprio P, Burlen O, Pereira P, Guilbert B, Andrade L, Hontebeyrie-Joskowicz M, et al. Most B Cells in Acute Trypanosoma Cruzi Infection Lack Parasite Specificity. Scand J Immunol (1988) 28(5):553–61. doi: 10.1111/j.1365-3083.1988.tb01487.x
37. Doi T, Kanai T, Mikami Y, Sujino T, Jun L, Ono Y, et al. IgA Plasma Cells Express the Negative Regulatory Co-Stimulatory Molecule Programmed Cell Death 1 Ligand and Have a Potential Tolerogenic Role in the Intestine. Biochem Biophys Res Commun (2012) 425(4):918–23. doi: 10.1016/j.bbrc.2012.08.010
38. Mauri C, Bosma A. Immune Regulatory Function of B Cells. Annu Rev Immunol (2012) 30(1):221–41. doi: 10.1146/annurev-immunol-020711-074934
39. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-Producing Plasmablasts Exert Regulatory Function in Autoimmune Inflammation. Immunity (2014) 41(6):1040–51. doi: 10.1016/j.immuni.2014.10.016
40. Vijay R, Guthmiller JJ, Sturtz AJ, Surette FA, Rogers KJ, Sompallae RR, et al. Infection-Induced Plasmablasts Are a Nutrient Sink That Impairs Humoral Immunity to Malaria. Nat Immunol (2020) 21(7):790–801. doi: 10.1038/s41590-020-0678-5
41. Zacca ER, Onofrio LI, Acosta CDV, Ferrero PV, Alonso SM, Ramello MC, et al. PD-L1+ Regulatory B Cells Are Significantly Decreased in Rheumatoid Arthritis Patients and Increase After Successful Treatment. Front Immunol (2018) 9:2241. doi: 10.3389/fimmu.2018.02241
42. Lee-Chang C, Rashidi A, Miska J, Zhang P, Pituch KC, Hou D, et al. Myeloid-Derived Suppressive Cells Promote B Cell–Mediated Immunosuppression via Transfer of PD-L1 in Glioblastoma. Cancer Immunol Res (2019) 7(12):1928–43. doi: 10.1158/2326-6066.CIR-19-0240
43. DosReis GA. Evasion of Immune Responses by Trypanosoma Cruzi, the Etiological Agent of Chagas Disease. Braz J Med Biol Res (2011) 44(2):84–90. doi: 10.1590/S0100-879X2011007500005
44. Wilmore JR, Maue AC, Lefebvre JS, Haynes L, Rochford R. Acute Plasmodium Chabaudi Infection Dampens Humoral Responses to a Secondary T-Dependent Antigen But Enhances Responses to a Secondary T-Independent Antigen. JI (2013) 191(9):4731–9. doi: 10.4049/jimmunol.1301450
45. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring Function in Exhausted CD8 T Cells During Chronic Viral Infection. Nature (2006) 439(7077):682–7. doi: 10.1038/nature04444
46. Greczmiel U, Kräutler NJ, Borsa M, Pedrioli A, Bartsch I, Richter K, et al. LCMV-Specific CD4 T Cell Dependent Polyclonal B-Cell Activation Upon Persistent Viral Infection is Short Lived and Extrafollicular. Eur J Immunol (2020) 50(3):396–403. doi: 10.1002/eji.201948286
47. Jellison ER, Guay HM, Szomolanyi-Tsuda E, Welsh RM. Dynamics and Magnitude of Virus-Induced Polyclonal B Cell Activation Mediated by BCR-Independent Presentation of Viral Antigen. Eur J Immunol (2007) 37(1):119–28. doi: 10.1002/eji.200636516
48. Fiocca Vernengo F, Beccaria CG, Araujo Furlan CL, Tosello Boari J, Almada L, Gorosito Serrán M, et al. CD8 + T Cell Immunity Is Compromised by Anti-CD20 Treatment and Rescued by Interleukin-17a. In: Kaufmann SHE, Boothroyd JC, editors. Mbio, vol. 11 (2020). p. e00447–20. doi: 10.1128/mBio.00447-20
49. Norris KA, Schrimpf JE, Szabo MJ. Identification of the Gene Family Encoding the 160-Kilodalton Trypanosoma Cruzi Complement Regulatory Protein. Infect Immun (1997) 65(2):349–57. doi: 10.1128/iai.65.2.349-357.1997
50. Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of Genetic Variants of Lymphocytic Choriomeningitis Virus in Spleens of Persistently Infected Mice. Role in Suppression of Cytotoxic T Lymphocyte Response and Viral Persistence. J Exp Med (1984) 160(2):521–40. doi: 10.1084/jem.160.2.521
Keywords: plasmablasts/plasma cells, B cell, PD-L1, Trypanosoma cruzi, Plasmodium, LCMV, immunosuppression, microorganism evasion
Citation: Serrán MG, Vernengo FF, Almada L, Beccaria CG, Gazzoni Y, Canete PF, Roco JA, Boari JT, Ramello MC, Wehrens E, Cai Y, Zuniga EI, Montes CL, Cockburn IA, Rodriguez EVA, Vinuesa CG and Gruppi A (2022) Extrafollicular Plasmablasts Present in the Acute Phase of Infections Express High Levels of PD-L1 and Are Able to Limit T Cell Response. Front. Immunol. 13:828734. doi: 10.3389/fimmu.2022.828734
Received: 03 December 2021; Accepted: 11 April 2022;
Published: 16 May 2022.
Edited by:
Pablo Engel, University of Barcelona, SpainReviewed by:
Luisa María Villar, Ramón y Cajal University Hospital, SpainCopyright © 2022 Serrán, Vernengo, Almada, Beccaria, Gazzoni, Canete, Roco, Boari, Ramello, Wehrens, Cai, Zuniga, Montes, Cockburn, Rodriguez, Vinuesa and Gruppi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adriana Gruppi, YWdydXBwaUBmY3EudW5jLmVkdS5hcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.