 Hua Zhu
Hua Zhu Siping Hu3†
Siping Hu3† Yuntao Li
Yuntao Li Yao Sun
Yao Sun Xiaoxing Xiong
Xiaoxing Xiong- 1Department of Neurosurgery, The Affiliated Huzhou Hospital, Zhejiang University School of Medicine (Huzhou Central Hospital), Huzhou, China
- 2Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, China
- 3Department of Anesthesiology, The Affiliated Huzhou Hospital, Zhejiang University School of Medicine (Huzhou Central Hospital), Huzhou, China
- 4Department of General Surgery, The Affiliated Huzhou Hospital, Zhejiang University School of Medicine (Huzhou Central Hospital), Huzhou, China
Ischemic stroke after cerebral artery occlusion is one of the major causes of chronic disability worldwide. Interleukins (ILs) play a bidirectional role in ischemic stroke through information transmission, activation and regulation of immune cells, mediating the activation, multiplication and differentiation of T and B cells and in the inflammatory reaction. Crosstalk between different ILs in different immune cells also impact the outcome of ischemic stroke. This overview is aimed to roughly discuss the multiple roles of ILs after ischemic stroke. The roles of IL-1, IL-2, IL-4, IL-5, IL-6, IL-8, IL-9, IL-10, IL-12, IL-13, IL-15, IL-16, IL-17, IL-18, IL-19, IL-21, IL-22, IL-23, IL-32, IL-33, IL-34, IL-37, and IL-38 in ischemic stroke were discussed in this review.
1 Introduction
Ischemic stroke after cerebral artery occlusion is one of the major causes of chronic disability worldwide, and there is still a lack of effective methods to improve functional recovery after cerebral stroke (1). After ischemic stroke, a severe shortage of blood supply to the brain leads to the insufficient oxygen supply to the brain, which in turn leads to neuronal death. Inflammatory responses at the blood-endothelial interface of brain capillaries are the basis of ischemic tissue damage. Furthermore, inflammatory interactions at the blood-endothelial interface, including adhesion molecules, cytokines, chemokines and white blood cells, are crucial for the pathogenesis of tissue injury in cerebral infarction (2). Pathophysiological changes after ischemic stroke include ion imbalance, neuroinflammation, and abnormal activation of immune cells, can lead to neuronal death. However, despite extensive research work have been made, the exact mechanisms of stroke damage are not fully understood. It is clear that ILs play a major role in the progression of ischemic stroke disease.
IL, refers to a lymphocyte medium that interacts between white blood cells or immune cells. It is a cytokine in the same category as blood cell growth factor. Both IL and hemocyte growth factor belong to cytokines, and they coordinate and interact with each other to complete hematopoiesis and immune regulation functions together. IL plays a crucial role in information transmission, activation and regulation of immune cells, mediating the activation, multiplication and differentiation of T and B cells and in the inflammatory reaction (3). There is a close relationship between IL and the pathogenesis of ischemic stroke. This review is to discuss the inflammatory effects of IL in the pathogenesis of stroke, the interactions between different IL-mediated pathways, the cell-type dependent effects of different mediators and how different ILs regulate complex inflammatory cascades. The role of IL-1, IL-4, IL-6, and IL-10 were discussed in more detail.
2 IL-1 Family
IL-1, described earlier as a fever-causing protein called human leukocyte pyrogen, is one of the pro-inflammatory cytokines produced by monocytes, macrophages, and epithelial cells. The IL-1 family consists of IL-1α, IL-1β, and specific receptor antagonist (IL-1RN) (4). The IL-1 gene complex is located on chromosome-14 and consists of three linked genes, namely IL-1α, IL-1β and IL-β (5). IL-1α and IL-1β was regarded as pro-inflammatory cytokines. The sequence homology of IL-1α and IL-1β is not high, but they bind to the same receptor complex and have similar biological activity. IL-1RN is a 16-18 kD protein that binds competitively with IL-1 and its receptor to become an important anti-inflammatory cytokine. There are five alleles for IL-1RN, and IL-1RN*1 is the most common genotype, followed by IL-1RN*2. The incidence of the remaining alleles (IL-1RN*3, IL-1RN*4 and IL-1RN*5) is less than 1% (6). Among them, IL-1RN*2 polymorphism is considered to be a genetic risk factor for coronary artery disease and atherosclerosis, which is closely related to ischemic stroke.
2.1 Mechanism of Pleiotropic Effects of IL-1 on Ischemic Stroke
IL-1 is a multifactorial cytokine with multiple biological effects in many cell types, many of which are associated with stroke risk and outcome. Downstream effects of IL-1 include increased expression of cytokines, chemokines, and growth factors, activation of matrix metalloproteinases, upregulation of adhesion molecules, increased leukocyte infiltration, activation of platelets, alteration of blood flow, increased angiogenesis, decreased neurogenesis, and numerous other effects. We have discussed some of all these effects and related mechanisms in detail, and the rest can be found in the reviews (7, 8).
Stroke-related comorbidities and risk factors are associated with elevated systemic inflammation, mediated in part by IL-1. As shown in Figure 1, in acute phase, the increase of IL-1 in the brain after stroke mediates the harmful the inflammatory process, including up-regulation of IL-6, TNF-α, MMP-9 and chemokines in astrocytes; inhibition of neurogenesis (9); increase of adhesion molecules and neutrophil infiltration, decrease of BBB integrity and blood flow by acting on endothelial cells, leading to worse outcomes. Moreover, IL-1 stimulates the proliferation and activation of astrocytes, leading to astrocyte hyperplasia, which is a typical response to brain injury. Data reported in many studies confirm that IL-1 upregulates a large number of genes in astrocytes, which encode neurotoxic factors including MMPs, chemokines, pro-inflammatory cytokines such as IL-6 and TNFα, but also survival-promoting mediators such as NGF (nerve growth factor) (10). IL-1 also exerts its effects on cerebrovascular endothelial cells to increase the production of chemoattractant and adhesion molecules such as CCL2 (CC chemokine ligand-2), ICAM-1 (intercellular adhesion molecule-1), and E- and P-selectin, and even promotes the breakdown of BBB, events that are associated with recruitment of leukocytes (11). IL-1 can act directly on neurons through an alternative signaling mechanism involving ceramide production and activation of Src kinase that phosphorylates the NMDAR [NMDA (N-methyl-D-aspartate) receptor] subunit 2B, leading to enhanced calcium entry and increased vulnerability to additional injury (12). IL-1 may also induce neurotoxicity indirectly through its action on the vascular endothelium to promote the recruitment of leukocytes, especially neutrophils that damage the neurovascular unit through the release of MMPs and reactive oxygen species (ROS) (11).
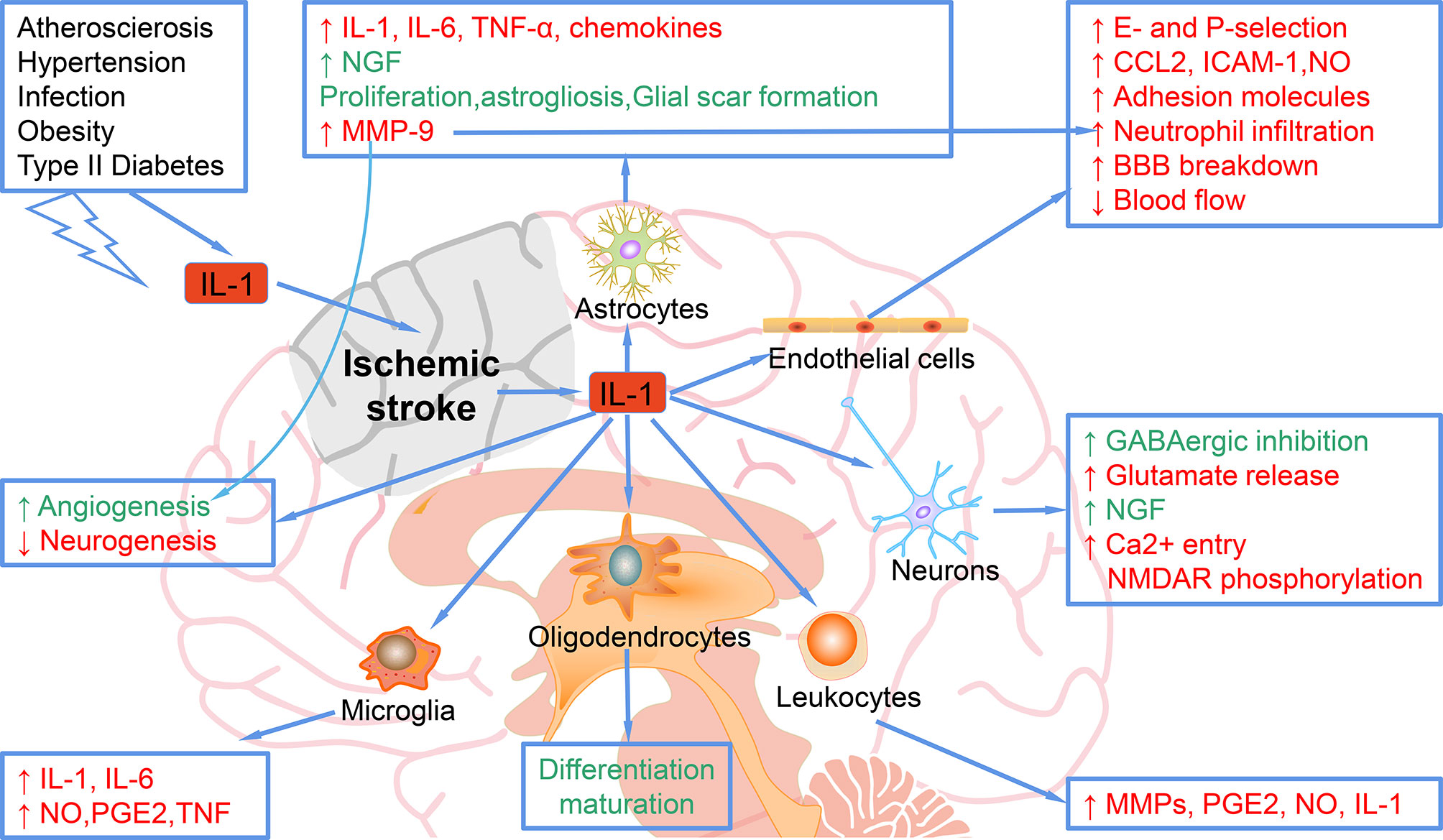
Figure 1 Mechanisms of the role of IL-1 in ischemic stroke. IL-1, elevated after ischemic stroke or other risk factors, can target astrocytes and up-regulate the expression of IL-1, IL-6, TNF-α, MMP-9, and chemokines. IL-1 target astrocytes to promote the NGF, proliferation, astrogliosis, and glial scar formation, which ameliorate ischemic injury. IL-1 targets endothelial cells and increases the E- and P-selection, CCL-2, ICAM-1, NO, adhesion molecules, neutrophil infiltration, blood flow decrease and BBB breakdown, which exacerbate ischemic stroke. When targets neurons, IL-1 increases the GABAergic inhibition and NGF, which rescue ischemic injury, while IL-1 also increases glutamate release and Ca2+ entry NMDAR phosphorylation, which cause more damage in ischemic stroke. Leukocytes can be regulated by IL-1 to secret more MMPs, PGE2, NO, and IL-1. In addition, microglia can be regulated by IL-1 to elevate the expression of IL-1, IL-6, NO, PGE2 and TNF. Oligodendrocytes can also be regulated by IL-1 for differentiation maturation.
However, in the subacute and chronic phases post-stroke, some of the effects of IL-1 may be beneficial. For example, IL-1 promotes the glial scar formation and enhances angiogenesis, thereby promoting ischemic stroke recovery (13). In addition, IL-1 is not toxic to pure neurons in culture and can even promote survival through enhancement of synaptic GABA(γ-aminobutyric acid)ergic inhibition or production of NGF (14, 15).
2.2 The Mechanisms of IL-1β-Induced Brain Damage in Ischemic Stroke
In acute ischemic stroke, blood perfusion to the brain is reduced. In cerebral infarction, when blood flow is 10%-25% lower than normal, nerve cells will suffer irreversible damage or even death, and inflammatory cells in the tissue will release inflammatory factors. As one of the most powerful pro-inflammatory cytokines, IL-1β exerts an essential role in ischemic stroke mainly through the following mechanisms.
2.2.1 IL-1β Aggravates the BBB Dysfunction
After ischemic stroke, increased secretion of IL-1β activates phospholipase A2 to degrade arachidonic acid and destroy the phospholipid bilayer (16). Moreover, the metabolites, prostaglandin and leukotriene, can promote the increase of microvascular permeability, resulting in blood-brain barrier (BBB) dysfunction and the formation of vasogenic brain edema (17). Meanwhile, after ischemic stroke, the reduction of glucose and oxygen supply, insufficient ATP production, and enhanced glycolysis, lead to the occurrence of cytotoxic brain edema. The interaction between the vasogenic brain edema and cytotoxic brain edema causes cranial pressure increase, secondary injury of brain tissue, and the possible occurrence of cerebral hernia in severe cases, endangering the life of the patient. In addition, IL-1β also aggravates ischemic injury by promoting the expression of adhesion molecules between endothelial cells, inducing leukocyte migration to the ischemic area to trigger inflammatory response (18).
2.2.2 IL-1β Mediates the Inflammatory Response in Ischemic Stroke
IL-1β stimulates the activation of microglia, which, as the main effector cells in the neuroinflammatory response, aggravates the inflammatory response and leads to secondary brain damage by secreting and releasing a series of potential neurotoxic substances, such as TNF-α and iNOS. IL-1β activates IκB kinase through the IRAK pathway, resulting in the phosphorylation and ubiquitination of IL-1β-mediated IκB-α, which ultimately upregulates the expression of NF-kB in the cell nucleus and induces the increase of the transcription of target genes such as IL-8 and TNF-α (19). Study has demonstrated that IL-1β regulates the PI3K/AKT pathway to stimulate IL-6 and other cytokines, which synergistically act on ischemic areas and aggravate the damage effect (20). Other studies have shown that up-regulation of IL-6 and other pro-inflammatory cytokines can promote the phosphorylation of JAK2/STAT3 (21, 22). After P-STAT3 enters the nucleus, it will bind to the DNA sequence characteristic of the promoter region of target genes and up-regulate the transcription of IL-1β, IL-6 and TNF-α genes (22). This vicious cycle leads to persistent inflammation, and damaged brain cells fail to recover.
2.2.3 IL-1β Promotes Apoptosis After Ischemic Stroke
After ischemic stroke, a large number of potentially salvageable neurons exist in the ischemic penumbra. However, with the prolongation of ischemia time, IL-1β promotes the apoptosis of the damaged cells by activating the apoptotic molecular mechanism, leading to the original ischemic penumbra gradually becoming the area of cerebral infarction, and finally the aggravated brain damage. Studies have verified that IL-1β plays a crucial role in the process of apoptosis of injured cells. This effect is mainly through the following two aspects: (1) activation of the excitatory toxicity mediated by glutamate (23); (2) activating the apoptotic cascade to activate the JNK/AP-1 pathway (24). Recently, it has been reported that the AIM2 inflammasome-derived IL-1β production activated triggers the expression of FasL in the spleen monocytes which evokes the apoptosis of Fas-dependent extrinsic T cells, causing an increased risk of infection by bacteria after ischemic stroke (25). Therefore, IL-1β may be involved in the signaling cascade activated by AIM2 inflammasome, causing immune suppression and secondary infection after stroke injury.
3 IL-2
Il-2, also known as cell growth factor, is an immunomodulatory lymphocyte secreted by T lymphocytes after being stimulated by antigen. In addition to maintaining the long-term multiplication and differentiation of T cells in vitro, IL also has important biological functions such as enhancing the killing activity of NK cells, promoting the proliferation and differentiation of B cells, and inducing the production of lymphokine-activated killer cells.
3.1 IL-2 Expression Decreased After Ischemic Stroke
Clinical trials showed that the level of serum IL-2 in patients with acute cerebral apoplexy was significantly lower than that in normal control group (26). The mechanism may be related to the following two factors: (1) In acute stroke, the body stress response, the immune stability in vivo is destroyed, especially the function of T cell is affected, so that the blood level of IL-2 is significantly decreased; (2) In acute stroke, the brain tissue cells are damaged, and local ischemia and hypoxia reduce the synthesis of IL-2 in the brain.
3.2 The Role of IL-2 in Ischemic Stroke
The IL-2/IL-2 antibody complex (IL-2/IL-2Ab) may improve the prognosis of ischemic stroke by regulating the amount of regulatory T cells (Tregs) in the body (27). Tregs are known to prevent ischemic stroke. However, the small amount of Tregs limits their clinical efficacy.
Previous research has showed that IL-2/IL-2Ab treatment selectively increases the amount of Tregs in the lymph nodes, spleen, and blood, significantly reduces the infarct volume, inhibits neuroinflammation, and improves sensorimotor function compared to stroke mice treated with isotype IgG (27). IL-2mAb has been reported to reduce demyelination after ischemic stroke by suppressing CD8 + T cells (28). The depletion of Tregs by diphtheria toxin eliminated neuroprotective effect provided by IL-2/IL-2Ab. IL-2/IL-2Ab promotes the expression of CD39 and CD73 in expanded Tregs, the deficiency of which may reduce the protective action of Tregs stimulated by IL-2/IL-2Ab in ischemic stroke mice (27). After stroke, increasing Treg cell numbers by delivering IL-2:IL-2 antibody complexes can improve white matter integrity and rescue neurological functions over the long term (29). In addition, Zhao et al. has found that ischemic stroke patients with poor functional outcomes at 3 months have significantly higher levels of IL-2 receptor α (sIL-2Rα) and lower levels of IL-2 than patients with good outcomes. Higher sIL-2Rα and IL-2 levels were associated with an increased and reduced risk of unfavorable outcomes, respectively (30), indicating that increased plasma sIL-2Rα and IL-2 levels manifested opposite correlations with functional outcome, illustrating the importance of IL-2/IL-2R autocrine loops in ischemic stroke.
4 IL-4
IL-4 regulates various immune responses, including the differentiation of T cells and nonspecific transformation of B cells (31). It is also the most characteristic M2 macrophage polarization promoter to date. Numerous evidences suggest that IL-4 plays a critical role in brain function under physiological and pathological conditions. For example, T-cell-derived IL-4 is essential for learning and memory in the normal brain. Levels of IL-4 in the brain tissue decrease with age, which may contribute to cognitive decline in older people and also increase the risk of Alzheimer’s disease (32). After ischemic stroke, IL-4 treatment has been shown to enhance white matter integrity (33).
4.1 IL-4 Promotes the M2 Polarization of Microglia/Macrophages
Recent animal and clinical researches have demonstrated the importance of IL-4 in the acute phase of stroke (34, 35). Several hours after the onset of stroke, the level of IL-4 in serum was observably increased (36). In addition, 24 hours after transient middle cerebral artery occlusion (MCAO), IL-4 deficiency resulted in brain injury and neurological dysfunction (37). IL-4 plays an important role in the M2 polarization and long-term recovery of microglia/macrophages after ischemic stroke. Mice lacking IL-4 have more M1-polarized microglia/macrophages, larger infarcts and more severe functional deficits after cerebral ischemia, while recombinant IL-4 can eliminate these effects (38). IL-4-polarized microglia cells may alleviate the ischemic stroke injury by promoting angiogenesis through the secretion of exosomes containing miRNA-26a (39). There is a direct salutary effect of IL-4 on oligodendrocyte differentiation that is mediated by the peroxisome proliferator-activated receptor gamma (PPARγ) axis. Additionally, PPARγ is essential for IL-4-induced oligodendrocyte progenitor cell differentiation and long-term functional improvements after stroke (33).
4.1.1 Inhibition Pro-Inflammatory Cytokines
The neuroprotective effect of IL-4 may be achieved by stimulating IL-4/STAT6 signal transduction and inhibiting pro-inflammatory cytokines. Previous study has showed that IL-4 knockout mice produce more pro-inflammatory cytokines, including IL-1β and TNF-α (40). The loss of IL-4 in mice also increases sensitivity to mechanical pain.
4.1.2 IL-4 Is Essential for Sex Differences in Vulnerability to Stroke
IL-4 protects against cerebral ischemia in male mice. However, female mice generally exhibit less damage in response to the same challenge of cerebral ischemia. Infarct volume in WT female mice in proestrus and estrus phases is markedly smaller than in males. IL-4 is required for female neuroprotection during the estrus phase of the estrus cycle (38). In protected female WT mice, microglia/macrophages were dominated by M2 polarization and inflammatory infiltration was reduced (40). Therefore, increasing macrophage M2 polarization, with or without added inhibition of inflammatory infiltration, may be a novel approach for stroke treatment.
4.1.3 IL-4 Affects Neuronal Excitability
Chen et al. have shown that cortical pyramidal and stellate neurons common for ischemic penumbra after cerebral ischemia-reperfusion injury exhibit intrinsic hyperexcitability and enhanced excitatory synaptic transmissions in IL-4 knockout mice. In addition, upregulation of Nav1.1 channel, and downregulations of KCa3.1 channel and α6 subunit of GABAA receptors are observed in the cortical tissues and primary cortical neurons in IL-4 knockout mice (34), indicating that IL-4 deficiency results in neural hyperexcitability and aggravates cerebral ischemia-reperfusion injury.
5 IL-6
IL-6 is a glycoprotein with a molecular mass of 20 to 30 kDa, depending on the cellular source and preparation, and is a cytokine with pleiotropic, playing a role in central host defense (41). The IL-6 family of cytokines recruits gp130 for signaling. For IL-6 specifically, a hexamer forms (two IL-6, two IL-6R and two gp130) that can activate intracellular tyrosin-kinases such as JAK and, to a lesser extent, TYK, which, in turn, activate a number of proteins including the STAT family of transcription factors, or the RAS-RAF-MAPK pathway, PI3K, or IRS (insulin receptor substrate) (42). IL-6, mainly produced by monocyte macrophages, lymphoid cells, T cells, B cells, granulocytes, mast cells and endothelial cells, is a kind of multi-effector cytokine. IL-6 has critical effect in immune reactions, acute phase response and hematopoiesis regulation, mainly in autocrine or paracrine ways. By activating target genes, IL-6 not only serves as a differentiation and growth factor of hematopoietic cells, B cells, T cells, osteoclasts and endothelial cells, but also plays an important role in the growth, differentiation, regeneration and degradation of peripheral and central nervous system nerve cells. IL-6 activates and recruits neutrophils and monocytes, stimulates vascular endothelial cells to secrete adhesion molecules and other inflammatory transmitters, and enhances local inflammatory response (43). Circulating and local IL-6 production will lead to the state of pre-thrombosis, which can induce the production of platelet derived growth factor, fibroblast growth factor, TNF, macrophage colony stimulating factor, and promote the proliferation of smooth muscle cells (44).
5.1 Dual Role of IL-6 in Ischemic Stroke
The dysregulation of IL-6 is closely related to the occurrence and outcome of many clinical diseases, including coronary heart disease, leukemia, hypertension, ischemic stroke and so on (45). Su et al. demonstrated that elevated IL-6 induced by ischemia and hypoxia, oxidative stress, vascular occlusion and inflammation, partly leads to the production of the acute phase protein in the liver, thereby stimulating leukocyte recruitment and thrombosis, ultimately causing multiple cardio-cerebrovascular diseases including ischemic stroke (46). Elevated serum IL-6 levels are implicated in a higher risk of incident stroke and mediate the racial disparity in stroke via inflammatory effects of risk factors (47). Elevated plasma IL-6 has been reported to be a signatures of post-stroke delirium (48). Additionally, high IL-6 levels at 24 hours are associated with futile reperfusion in patients with acute ischemic stroke with large vessel occlusion treated with mechanical thrombectomy (49). Moreover, a lower admission level of IL-6 is positively correlated with the first-pass effect, which is defined as a complete or near-complete reperfusion achieved after a single thrombectomy pass is predictive of favorable outcome in acute ischemic stroke patients (50). These findings indicate that IL-6 may be a predictor of the prognosis of ischemic stroke patients.
IL-6 is a marker of inflammation after stroke, and elevated IL-6 is mainly secreted from neurons, microglia, astrocytes, and endothelia cells in the ischemic hemisphere, traditionally regarding as an adverse prognostic factor (51) (Figure 2). In the ischemic brain, IL-6 protein is mainly localized in the neurons of the cerebral cortex. The neuronal expression of IL-6 starts 3.5 h after ischemia, peaks after 24 h of reperfusion, and remains for 7 days. The immunoreactivity of IL-6 was most upregulated in ischemic penumbra. IL-6 released into the cerebrospinal fluid after stroke may lead to impaired cerebrovascular autoregulation and increased histopathology. In addition, IL-6 is related to the inflammation, which contributes to both damage and recovery process after ischemic stroke (52). The high levels of serum IL-6 have been reported to be related to the body temperature, early neurologic deterioration, infarct volume, and a long-term poor outcome. It has been identified that after stroke brain is the main source of IL-6 (53). In addition, inflammatory biomarkers, including C-reactive protein, fibrinogen, IL-1 receptor antagonist, and TNF-α are also elevated in parallel with IL-6 (54).
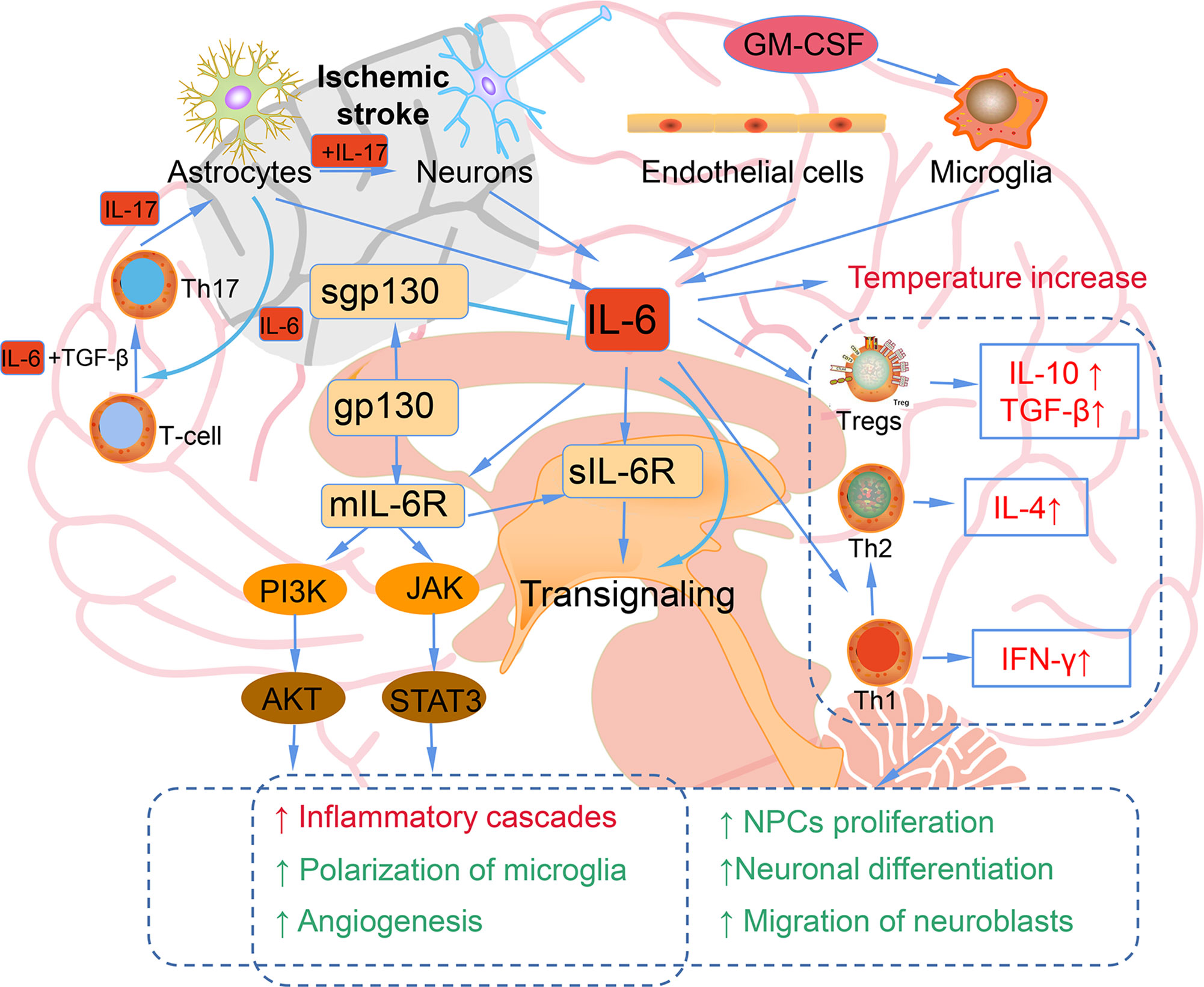
Figure 2 The bidirectional role of IL-6 in ischemic stroke. Astrocytes, neurons, endothelial cells, and microglia can produce IL-6 after ischemic stroke. Elevated IL-6 leads to the temperature increase; inflammatory cascades, microglia polarization, and angiogenesis mediated by JAK/STAT3 and PI3K/AKT pathways; increased IL-10, TGF-β induced by Tregs; Th1 polarizing to Th2; NPCs proliferation; neuronal differentiation; migration of neuroblasts.
However, IL-6 is also a neurotrophic cytokines that shares a common receptor subunit, gp130, with other neurotrophic cytokines, such as leukemia inhibitory factor (LIF) and ciliary neurotrophic factor (55). The IL-6 expression is mainly observed in neuronal cells in the ischemic penumbra region, and the expression of LIF shows a similar pattern. The direct injection of these cytokines into the brain tissue after ischemic stroke can reduce cerebral ischemic damage. The main downstream signaling pathway of IL-6 is JAK-STAT, and the activation of STAT3 occurs primarily in neuronal cells after ischemic reperfusion. Since the role of STAT3 in stroke is also diverse and controversial, further studies are needed to explore the accurate action of STAT3 signaling in neuroprotective effect (54). IL-6 secreted from astrocytes promotes Th1 polarize into Th2 to mediate immunosuppressive microenvironment and contribute to the neurogenesis and angiogenesis and neuronal differentiation (51). IL-6 stimulates the phosphorylation of STAT3 and the early transcriptional activation of angiogenesis-related genes, thereby leading to the enhanced angiogenesis and elevated cerebral blood flow in the delayed period after ischemic stroke. IL-6R simultaneously activates the PI3K/AKT and JAK-STAT pathways, which play vital roles in angiogenesis after ischemic stroke (56). Additionally, IL-6 also been reported to facilitate post-traumatic healing in the CNS through repair of endothelial cells, which also demonstrates that IL-6 may enhance revascularization or angiogenesis after ischemic stroke (57). IL-6 increases CNS neuronal survival and decreases excitotoxic neuronal damage against NMDA-mediated injury and protects neurons against apoptosis (54). Continuously injection of recombinant for 7 days IL-6 into the lateral ventricle of gerbils subjected to transient cerebral ischemia, IL-6 injection was found to prevent learning disabilities and delayed neuronal loss (58). In conclusion, IL-6 has a dual effect in ischemic stroke, acting as an inflammatory factor in the acute stage and a neurotrophic mediator in the subacute and prolonged phase.
6 IL-8 in Ischemic Stroke
IL-8 is a chemotactic cytokine that promotes the chemotaxis of inflammatory cells and induces cell proliferation. After ischemic stroke, IL-8 levels are increased (59, 60), mobilizes and activates neutrophils, causing neutrophils to infiltrate into the ischemic area, aggravating the local inflammatory response, leading to the expansion of ischemic lesions, and leading to severe morbidity and disability (2, 61–63). Endothelin-1 may be a stimulator of IL-8 (62). IL-8 may be also involved in recruiting blood polymorphonuclear leukocytes to the sites of cerebral ischemia (59). The expression of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, TNF-α) in the cortex of ischemic stroke mice was detected after the occurrence of cerebral ischemia (64). One study showed that the levels of these pro-inflammatory cytokines in patients with acute cerebral ischemia were observably higher than those in the control normal group, and the degree of disability in early phase of acute stroke was positively correlated with the level of serum IL-8 (65). IL-8 gene knockout has been shown to promote neuroglial cells activation while inhibit neuroinflammation through the PI3K/Akt/NF-κB-signaling pathway in mice with ischemic stroke (66). The high serum IL-8 levels are associated with prognosis. IL-8 exaggerates the ischemic stroke injury through inducing neutrophil-mediated-inflammation (61). The development of new neuroprotective treatments aimed to prevent neutrophil-mediated-inflammation induced by IL-8 is critical in the treatment of stroke, and prevention of clinical worsening. IL-8 can be used as important indicator to judge the severity of the early condition of acute ischemic stroke patients (67). However, IL-8 stimulates VEGF production in human bone marrow mesenchymal stem cells partially via the PI3K/Akt and MAPK/ERK signal transduction pathways and that administration of IL-8-treated human bone marrow mesenchymal stem cells increases angiogenesis after stroke (68).
7 IL-10
IL-10 is a significant anti-inflammatory cytokine that has inhibitory effects on a variety of immune cells. IL-10 was first identified in mouse Th2 cells, and was subsequently found to be secreted in astrocytes, neurons, B cells, monocytes/macrophages, keratinocytes, and human Th1 cells. IL-10 has strong anti-inflammatory and immunosuppressive activity. It can inhibit the production and release of IL-2, IFN-γ and pro-inflammatory factors, reduce the expression of immune receptors, inhibit human Th2 cells, lead to cell proliferation, cytokine production reduction (69). IL-10 binds to IL-10 receptors (IL-10R) to decrease inflammation and limit apoptosis (70). These effects make it a very important role in the protection of cerebral ischemia.
7.1 IL-10 in Ischemic Stroke
The neuroprotection of IL-10 on ischemic stroke has always been a research hotspot. A meta-analysis exploring the relationship between IL-10 gene polymorphism and ischemic stroke risk revealed no overall significant association of IL-10 with ischemic stroke risk, but an association was found with macrovascular disease and microvascular disease (71), demonstrating that certain subtypes of ischemic stroke are correlated to IL-10 gene polymorphisms.
In experimental stroke, the levels of IL-10 mRNA and protein and IL-10R mRNA were elevated, with IL-10 observed in microglia and IL-10R on astrocytes in the ischemic penumbra (72). In transgenic mice that overexpressed IL-10, infarct size was reduced, and apoptosis was limited 4 days post ischemic stroke (73). Additionally, Overexpression of IL-10 enhances the neuroprotective effect of mesenchymal stem cell transplantation through anti-inflammatory regulation, thus supporting the survival of neurons during acute ischemia (74). Both systemic intravenous (IV) and central intracerebroventricular (ICV) exogenous administration of IL-10 reduced infarct size after permanent MCAO (pMCAO) (75). Moreover, low IL-10 levels were related to poor stroke outcomes and a delayed, exacerbated inflammatory response that was alleviated by IL-10 administration after pMCAO (76). Lower levels of IL-10 and IL-33 may also be used to predict post stroke depression (77, 78). The IL-10 expression in the brain tissue increases with pathological changes of the central nervous system, promotes the survival of gliocyte and neurons, and inhibits inflammatory responses through multiple signaling pathways. Previous research showed that the significant decrease of IL-10 was significantly associated with the degree of neurological impairment, and the concentration of IL-10 had a high predictive value on the early neurobehavioral performance post-acute stroke (79). However, stroke patients are susceptible to infection as a result of stroke-induced immunosuppression, and elevated serum IL-10 levels have been identified as an independent predictor of post-stroke infection (80, 81). IL-10 overreaction can lead to immunosuppression and worsening neurological prognosis after stroke, indicating that IL-10 therapy should be used with caution (82). Elevated IL-10 levels may be associated with higher incidence of post-stroke urinary tract infection, leading to poorer recovery after ischemic stroke in women (83, 84). In addition, IL-10 can mediate the function of Th2 cells, exert a protective effect, and lead to the reduction of ischemic infarction lesions (85). Future studies should be aimed at differentiating between central and peripheral IL-10 effects post-stroke.
7.1.1 The Mechanism of IL-10 in Inhibiting Inflammatory Responses After Stroke
Immune cells, including T and B cells, are important in ameliorating neuroinflammation via the modulation of varieties of cytokines and chemokines, with IL-10 playing a central immunomodulatory role (86, 87). The protective effect of IL-10 on stroke is mainly achieved by inhibiting inflammatory reactions. Firstly, IL-10 decreased the expression and activity of pro-inflammatory cytokines such as IFN-γ, IL-1β and TNF-α through PI3K and STAT3 activation (Figure 3) (88). Secondly, IL-10 inhibits the synthesis and activity of Th1 lymphocytes (89). In addition, IL-10 treatment can effectively down-regulate the up-regulated pro-inflammatory signals in acute ischemic lesions after stroke, and can provide neuroprotection for ischemic stroke (90). IL-10 gene transduction before cerebral artery ischemia can alleviate brain damage induced by ischemia/reperfusion in rats through increasing the expression of heme oxygenase (91). IL-10 also exert its anti-inflammatory effects partially through inhibition of NF-κB (92). Hydrogen sulfide donor administration during reperfusion protects the integrity of BBB after ischemia/reperfusion and is accompanied by increased IL-10 expression, reduced NF-κB nuclear translocation, and MMP-9 and NOX4 activity (93). In MCAO mice, by reducing the release of neuroinflammatory factors (IL-6, IL-1β, TNF-α) and astrocyte activation, IL-32α overexpressing transgenic mice showed reduced cell death of ischemic neurons and enhanced anti-neuroinflammatory factor (IL-10), indicating a crosstalk between IL-32α and IL-6, IL-1β, IL-10 (94). IL-10-secreting CD4+ T cells induced by nasal MOG reduce injury following stroke. IL-10 secreted from CD4+ T cells may be the reason of the neuroprotection of oligodendrocyte glycoprotein administration in MCAO mice (95, 96). Increased IL-10 levels also decreased the number of CD11b+ cells that may contribute to secondary infarct expansion via nitric oxide pathways (96). Expansion of the CNS Treg cell population by administration of the CD28 superagonist monoclonal antibody at the start of reperfusion decreased the infarct volume 7 days after MCAO, and its effect was attributed to the increased IL-10 (97). Transfer of IL-10-producing B cells into B cell-deficient mice 24 h after MCAO attenuated cerebral ischemia-reperfusion injury, reduced the amount of T cells and monocytes in cerebral parenchyma, and improved the peripheral proinflammatory homeostasis (87). Interesting, IL-10-producing B cells also upregulated the number of Tregs (87), suggesting that there may be a positive feedback loop between B cells and Tregs, both of which play a neuroprotective role through IL-10 production. These facts indicate a complicated network between IL-10 and immune cells in ischemic stroke. These methods of targeting IL-10 to prevent recurrence of stroke may be realized in the interventional treatment of stroke.
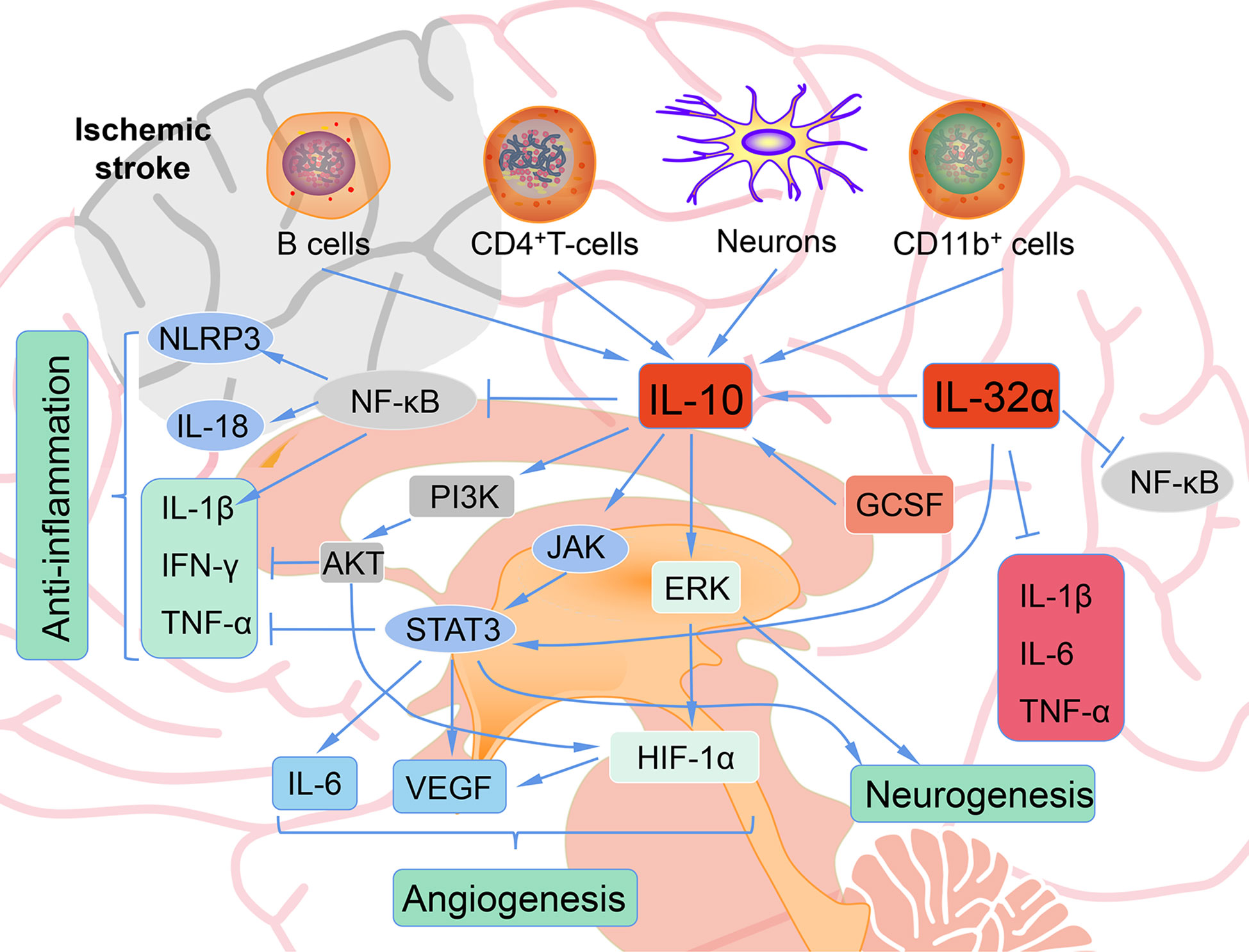
Figure 3 The role of IL-10 after ischemic stroke. B cells, CD4+ T cells, neurons, CD11b+ cells produce IL-10 after ischemic. IL-10 shows anti-inflammatory, angiogenesis, and neurogenesis role after ischemic stroke.
7.1.2 The Role of IL-10 in Neurogenesis After Ischemic Stroke
Injection of activated Tregs into the lateral ventricle of C57BL/6 mice 60 min after of transient ischemia promotes the proliferation of neural stem cell in the subventricular region in ischemic brain tissues. Moreover, this effect was abolished by blocking IL-10 with a neutralizing antibody, suggesting that activated Tregs act through IL-10 to facilitate the proliferation of neural stem cells (98). Hematopoietic cytokines such as GCSF and stem cell factor have been confirmed to promote neurogenesis (99), and also may be required to provide the initial signals for IL-10 production in ischemic stroke (100). Administration of these cytokines early (1–10 days) and later (11–20 days) post MCAO significantly elevated the mRNA expression of IL-10, reduced the activation of microglia/macrophages, and did not change proinflammatory cytokine levels in C57BL/6J mice (100). A study where bone marrow-derived mesenchymal stem cells were transplanted into the lateral ventricle of Sprague-Dawley rats before pMCAO yielded similar results, where IL-10 mRNA and protein levels were elevated 4 days post-stroke, TNFα was reduced, infarct size was smaller, and neurologic function was improved (101). IL-10 targets Nestin+ progenitors and activates neurogenesis by modulating ERK and STAT3 activity in adult subventricular zone (102). Either administration of stem cells themselves or hematopoietic cytokines may ameliorate ischemic stroke injury partially through the increase of IL-10. Additionally, neuroprotection after dysbiosis depends on IL-10 and IL-17. IL-10 is required for Treg mediated IL-17+ γδ T suppression (103). Recently, it has been reported that IL-10 acts differentially on αβ and γδ T cells. IL-17A producing CD4+ αβ T cells are directly controlled via their IL-10-receptor (IL-10R), whereas IL-10 by itself has no direct effect on the IL-17A production in γδ T cells. The control of the IL-17A production in γδ T cells depended on an intact IL-10R signaling in Tregs (104).
8 IL-12 and ischemic stroke
During antigen presentation to naive T cells, IL-12, IL-23, and IL-27 are produced by activated antigen-presenting cells, while IL-35 is a product of B cells and Tregs (105). The primary target cells of IL-12 are NK and T cells, which are stimulated to produce cytokines, proliferative and cytotoxic activities (106). IL-12 is produced early in infection and plays a pro-inflammatory role in the immune response, and serve as a cofactor in the polarization of T cell response to cell-mediated immunity (107).
Previous studies have confirmed that IL-12 plays an essential role in the pathological process of acute ischemic stroke (108). Cytokines are usually released in response to tissue injury, so increase levels of IL-12 in serum in patients with acute cerebral infarction are consistent with a rapid increase IL-12 levels in the serum of patients with acute myocardial infarction and severe brain injury (109). The increase of IL-12 in serum of stroke patients appears to be a local or systemic immune response to ischemic brain injury, and the increase of IL-12 in serum may be caused by the release of cytokines from the infarcted brain region or cerebrospinal fluid to the periphery. On the other hand, stroke-activated cerebral endothelial cells secrete cytokines that may activate peripheral blood monocytes, leading to systemic expression of cytokines (110). An increase in the number of IL-12 secreting monocytes and monocytes isolated from peripheral blood of patients with cerebral ischemia has been demonstrated (111, 112). In this case, the increase in serum IL-12 levels in patients with stroke may be caused by cytokines.
IL-12 may promote the deterioration of ischemic brain injury via cytokines by activating the ability of immunoreactive cells and modulating their abilities in response to inflammation. IL-12 increases the production and action of several pro-inflammatory cytokines and chemokines and promotes endothelial cells to release adhesion molecules, which are potent chemical attractors for different subsets of white blood cells, including monocytes and neutrophils (113, 114). Additionally, in a mouse cancer model, IL-12 gene therapy is associated with increased tissue infiltration and apoptosis of NK and T cells, which are important mechanisms of neuronal death induced by aggressive inflammatory cells in the ischemic brain (115). At present, we can know exactly that there is close correlation between increased levels of IL-12 in the ischemic stroke patients and reaction intensity in acute phase, the size of the early brain injury, neurological stroke severity and functional disability, suggesting IL-12 may play a critical role in pathophysiology of cerebral ischemia.
9 IL-13 and Ischemic Stroke
IL-13 is a protein secreted by activated T cells and is a powerful regulator of human monocyte and B cell function in vitro (116). IL-13 shares a common biological activity with IL-4 (117). IL-13 can induce the differentiation of mononuclear cell, inhibit LPS-induced mononuclear factor secretion, control inflammatory response, induce the proliferation of B cell and synthesize IgE antibody, cooperate with IL-2 to stimulate NK cells to produce IFN, thereby promoting mononuclear macrophage activation.
Previous studies have confirmed the indispensable role of IL-13 in ischemic stroke. IL-13 exerts an effect on microglia and infiltrating macrophages in the brain after stroke, and it can regulate the spontaneous polarization transition from anti-inflammatory to pro-inflammatory phenotype of microglia and macrophages (118). As a well-known modulator of immune response in vitro, IL-13 has been shown to have neuro-protective abilities in several experimental models of neurodegenerative diseases by significantly reducing the secretion of pro-inflammatory factors, reducing inflammatory cell infiltration, and inhibiting axonal loss as well as inducing anti-inflammatory microglial/macrophage responses (119, 120). Interestingly, interleukin-13 can also improve ischemic liver gluconeogenesis and hyperglycemia in stroke model rats (121), exerting a salutary action. That is, it has been demonstrated that mesenchymal stem cells that continuously secrete IL 13 can differentiate microglia and macrophages into neuroprotective M2 phenotypes in the pro-inflammatory state of ischemic stroke (122).
10 IL-15 and Ischemic Stroke
IL-15 can be produced by activated mononuclear macrophages, epidermal cells and fibroblasts. Its molecular structure has many similarities with that of IL-2, and it plays a similar biological activity to IL-2 (123). IL-15 also has the ability to chemotaxis and promote survival, and it can be involved in neuroinflammation. IL-15 also acts as an effective chemotactic agent for T cells, promoting the migration of T cells to inflammatory tissues (124). In addition, IL-15 maintains homeostasis and cytotoxic activity in lymphocytes (NK and CD8+ T cells) carrying its receptor (125).
Although recent studies have shown that astrocytes are a major source of IL-15 in the inflammatory central nervous system (126, 127), the potential role of IL-15 in astrocytes in cerebral ischemic injury is not completely clear. However, significant increase in IL-15 expression in astrocytes post ischemia reperfusion has been observed. Subsequent studies have shown that IL-15 is a key factor for astrocytes to control the degree of central nervous system inflammation and brain injury following ischemic stroke (126). Astrocytes produce inflammatory cytokines such as IL-15, which promote the cell-mediated immune reaction to ischemic stroke, increase the number of CD8+ T cells and NK cells, participating in ischemic nerve injury. In addition, blockage of IL-15 decreased the effector capacity of NK, CD8+ T and CD4+ T cells in WT mice after CIRI, and the elimination of IL-15 response after CIRI improved brain damage in adult mice (127). Moreover, IL-15 as a mediator of the crosstalk between astrocytes and microglia that exacerbates brain injury after intracerebral hemorrhage (128). Recently, IL-15 has been reported to modulates the response of cortical neurons to ischemia through alleviating endoplasmic reticulum stress and increasing cell survival (129). Therefore, therapy targeted IL-15 is a potential strategy for cerebral ischemia.
11 IL-16 in Ischemic Stroke
IL-16 is a pro-inflammatory cytokine produced by activated CD8+ T cells and activates CD4+ T cells, monocytes, macrophages, and dendritic cells by binding to the CD4 molecule (130). In addition, IL-16 promotes the production of inflammatory cytokines such as TNF-α, IL-1β, and IL-6, which has key effects in immune responses after ischemic stroke (131). Although the mechanism by which IL-16 acts as a mediator of inflammation is not fully understood, previous study has shown that IL-16 is involved in inflammatory disease through the activation of T lymphocytes and the expression of inflammatory cytokines (132). In the early stage of cerebral ischemia, T lymphocytes are activated to release reactive oxygen species, which eventually lead to brain damage. In later stages, T lymphocytes regulate brain recovery and regeneration. The depletion of T cells in the acute phase of ischemia reduces the infarction size and has a sustained protective action against ischemic stroke in the later stages of infarction development (133). IL-16 accumulates during injury-related response areas and perivascular areas through the infiltrated immune cells (e.g., neutrophils, CD8+ lymphocytes, and activated CD68+ microglia/macrophages) (134). The recruitment and activation of immune cells lead to microvascular aggregation and disorder of BBB, leading to secondary injury.
12 IL-18 in Ischemic Stroke
IL-18 is known as a pro-inflammatory cytokine. IL-18 expression is mainly observed in neuronal cells at early phase and in microglia at a later stage. IL-18 is associated with stroke-induced inflammation and that initial serum IL-18 levels may be predictive of stroke outcome. IL-18 KO mice exhibit the resistance to spatial restraint stress and CIRI (135). Caspase-1 activated by NLRP3 inflammasome, cleavage pro-IL-1β and pro-IL-18 to mature forms (IL-1β and IL-18), and mediate the inflammatory response or initiate the process of inflammatory cell death and pyrolysis. In addition, increased IL-18 in the brain causes depression-like behaviors by promoting the IL-18 receptor/NKCC1 (a sodium-potassium chloride co-transporter) signaling pathway. Hence, agents that inhibits NLRP3 inflammasome exert a neuroprotective effect on ischemic stroke and post-stroke depression via suppressing the expression of IL-18.
13 IL-19 and Ischemic Stroke
IL-19 is a member of the IL-10 family, which includes IL-10, IL-19, IL-20, IL-22, IL-24, and IL-26 (136). IL-19 was first discovered in primary human monocytes stimulated by LPS and GM-CSF (137). Subsequent reports on IL-19 mainly focused on its role as a product of immune cells. In immune cells, IL-19 is mainly secreted by monocytes, and a small part is expressed by B cells. It has been reported that IL-19 treatment can mature human T cell polarize them from pro-inflammatory Th1 phenotype to anti-inflammatory Th2 phenotype (138, 139). In addition, the anti-inflammatory effect of IL-19 on vascular diseases has also been clearly demonstrated (140).
As an anti-inflammatory factor, IL-19 also exerts a critical action in the immune reaction after the onset of ischemic stroke. Studies have shown that IL-19 can reduce infarct size and reduce neurological impairment after ischemic stroke through its anti-inflammatory ability (141). Moreover, IL-19 treatment can significantly reduce the up-regulation of TNF-α and IL-6 mRNA expression after ischemic stroke, inhibit the increase of microglia, macrophages, CD4+ T cells, CD8+ T cells, and B cells, and suppress the activation of macrophages and neutrophils. The administration of IL-19 also contributes to preserve the reduced number of immune cells, including macrophages, CD4+ T cells, CD8+ T cells, and B cells in peripheral blood compared to controls. In conclusion, IL-19 reduces cerebral infarction and neurologic deficits after cerebral ischemia in mice, possibly by inhibiting the infiltration and activation of immune cells and the increased expression of pro-inflammatory cytokine genes. Therefore, IL-19 may be identified as a new therapeutic agent to suppress the development of neuroinflammation after ischemic stroke.
14 IL-20 in Ischemic Stroke
IL-20 is also a member of the IL-10 family, and is produced by monocytes, epithelial cells, and endothelial cells. IL-20 has been related to a variety of inflammatory diseases, such as psoriasis, rheumatoid arthritis, atherosclerosis, and renal failure (142). IL-20 also induces the production of IL-6 (143), which is also a major pro-inflammatory cytokine. In addition, the levels of IL-6 in serum are correlated with cerebral infarction volume and stroke severity. IL-20 may be associated with the increased IL-6 levels in serum after ischemic stroke.
14.1 IL-20 Promotes Inflammation After Ischemic Stroke
The pathogenicity of IL-20 in ischemic brain injury has been demonstrated in transient MCAO animal models. After cerebral ischemia reperfusion, the levels of IL-20 in serum and ischemic penumbra were significantly elevated than sham groups, and glial cells were the main source of IL-20. After cerebral ischemia, hypoxia also induces the production of IL-20 in endothelial cells (144). The upregulation of IL-20 on glial pro-inflammatory cytokines and chemokines (may cooperate with IL-1β to promote inflammatory activity) is associated with inflammatory response and brain damage after ischemic stroke (145). Inflammatory cytokines and chemokines such as IL-1β, IL-8 and monocyte chemotactic protein 1 (MCP-1) are involved in the inflammatory response of infarcts (146). In conclusion, IL-20 is a novel hypoxia response factor that is upregulated in gliocyte after experimental ischemic stroke and mediates cell proliferation, signal transduction, and cytokine production. These suggest that IL-20 is related to the pathogenesis of cerebral ischemia, and IL-20 antagonists may have clinical therapeutic effects on ischemic stroke.
15 IL-23/IL-17
The IL-23/IL-17 axis has essential effect on the development of chronic inflammation and host defense against bacterial infection (147). In chronic inflammation, antigen-stimulated dendritic cells and macrophages produce IL-23, promoting the development of Th17 cells (148). Th17 cells produce IL-17, which promotes T cell activation by inducing the expression of various of inflammatory cytokines and triggers a powerful inflammatory response. IL-23 also acts on dendritic cells and macrophages in an autocrine/paracrine manner to promote the production of inflammatory cytokines, such as IL-1, IL-6, and TNF-α (149).
15.1 IL-23/IL-17 in Ischemic Stroke
Studies have shown that routine CD172a+/IRF4+ 2 type dendritic cells (CDC2s) are the main source of IL-23 in the brain following ischemic stroke, and are essential for IL-17 expression in γδT cells (148). Dendritic cells infiltrate the peri-infarcted area near the blood vessels after stroke. These cells induce γδT cells to produce IL-17, promoting neutrophil recruitment to the ischemic hemisphere (150). However, IL-23R gene knockout has no significant effect on the mortality in mice, suggesting that DC cells exert their adverse effects not only through IL-23, but also through other mechanisms (148). Additionally, IL-23 and IL-17 have been reported to increase after stroke, but there is insufficient clinical discriminatory power to predict the outcome of stroke (151). Vγ4 T cell-derived IL-17A, and IL-1β/IL-23 in infract hemisphere coordinately to exaggerate the inflammatory cascades and exacerbate ischemic tissue injury (152).
16 IL-33 and ischemic stroke
As a member of the IL-1 family, IL-33 can bind to membrane receptors on target cells to mediate downstream signaling pathways, or be transported to the nucleus of target cells to function as a DNA-binding factor. After IL-33 binds to its receptor complex, activated signals transmits into cells, and activates NF-κB and mitogen-activated protein kinase (MAPK) through a series of downstream signaling molecules such as IL-1-associated protein kinase, myeloid differentiation factor 88, and TNF receptor-associated factor 6 (153).
IL-33 has been reported to have neuroprotective effects through inhibiting inflammation via ST2 (a member of the IL-1 receptor family)/IL-33 signaling (154, 155). After ischemic brain injury, IL-33 expression in mature oligodendrocytes and astrocytes is increased. Interleukin-33 also protects against ischemic brain injury by regulating microglia and regulatory T cell activity (156). Serum IL-33 has been proved to be a novel predictive biomarker of hemorrhage transformation and outcome in acute ischemic stroke (157). The expression of ST2 on microglia/macrophages increases after MCAO. ST2 deficiency can exacerbate neurobehavioral disorders and brain damage via shifting microglial polarization toward M1. Some traditional oriental medicine, such as celastrol, ameliorate ischemic stroke injury through promoting IL-33/ST2 axis-mediated microglia/macrophage M2 polarization (158). IL-33 also protects ischemic stroke injury by regulating specific microglial activities (159). IL-10 is an essential protective factor for the neuroprotection of IL-33/ST2 signaling. In the ischemic brain, intracerebroventricular IL-33 can activate the downstream Foxp3 via ST2 receptor to increase Treg proportions, which produce amphiregulin to activate epidermal growth factor receptor located in neurons, leading to better outcomes (160). In addition, systemic administration of Th2 cells to promote cytokines IL-33 and IL-4 can reduce acute brain injury after CIRI (154). Astrocyte lipogenesis increases IL-33 production in the peri-infarct region, which promotes BBB repair in the subacute phase of cerebral ischemic injury and improves long-term functional recovery (161). The long-term protective role of IL-33 in ischemic stroke may be partly associated to its regulation of splenic T-cell immune responses via inhibiting Th1 response and promoting Treg response (162). Although IL-17 has these neuroprotective effects, mice treated with IL-33 showed an exacerbation of post-stroke pulmonary bacterial infection associated with greater functional impairment and mortality after 24 hours, suggesting exacerbation of systemic immunosuppression after ischemic stroke (163).
17 Other ILs in Ischemic Stroke
IL-5 and IL-9 are decreased in severe stroke patients acute ischemic stroke patients with poor outcome than mild stroke (164). It had been indicated that IL-5 and IL-7 may be predictors of edema and infarct volume (165). In experimental stroke, expressions of IL-9 and its upstream stimulating factors has been confirmed to be increased (166). Anti-IL-9-neutralizing antibody can ameliorate ischemic stroke injury partially by alleviating the destruction of the BBB via down-regulation of astrocyte-derived VEGF-A (167). In OGD, IL-9 has a destructive effect on the BBB, partly by decreasing eNOS production (168). IL-21 polymorphism is related to the increased susceptibility to ischemic stroke possibly by upregulating gene expression (169). IL-21R-deficient mice have reduced collateral vascular connections and increased brain infarct volume, suggesting that IL-21R regulates collateral vascular anatomy and innate neuroprotection. The neuroprotective effects of IL-21R are mediated through the JAK/STAT signaling pathway and upregulation of caspase 3 (170). IL-22 exerts a protective action through regulating the JAK2-STAT3 pathway to improve oxidative stress, inflammation, and neuronal apoptosis following CIRI (171). IL-32 silence protects PC12 cells against OGD/R-induced injury via activation of Nrf2/NF-κB pathway (172). Increased serum IL-34 levels may be a novel diagnostic and prognostic biomarker in patients with acute ischemic stroke (173). Increased serum IL-37 in ischemic stroke patients is correlated with stroke recurrence (174) and 3-month functional prognosis (175). However, another study has illustrated that IL-37 exert protective effects by modulating post-stroke inflammation in the brain and periphery (176). Large randomized controlled trials are needed to further verify the role of IL-37 in ischemic stroke. The lower changes in IL-38 serum level lead to a poorer prognosis, indicating that IL-38 serum changes might be a novel early predictor factor for ischemic stroke prognosis (177) (141).
18 Conclusions and Perspectives
In summary, we have briefly discussed the functional role of interleukins and their relationships with ischemic stroke. Based on the classification of the effect of interleukin on the immune response after stroke, interleukins can be roughly divided into anti-inflammatory and pro-inflammatory categories. IL-1β, IL-6, IL-8, IL-12, IL-15, IL-16, IL-20, IL-18, IL-23/IL-17 and so on play a pro-inflammatory role after ischemic stroke (Figure 4). ILs that have anti-inflammatory effects on ischemic stroke include IL-2, IL-4, IL-10, IL-13, IL-19, IL-33 and so on (Figure 5). However, the IL family contains so many ILs, the complicated roles of ILs in ischemic stroke cannot be discussed in detail in this brief overview. After ischemic stroke, ischemia leads to vascular endothelial damage and induces immune responses. The action of these ILs on local or systemic immune cells, or the interaction of these ILs, determines the progress of immune response in the ischemic brain. From a macroscopic perspective, it is their interactions that determine the degree of neurological impairment and clinical prognosis of ischemic stroke patients. Therefore, interleukins play an important role in the immune mechanism of ischemic stroke. This also urges us to make continuous progress and search in this field, in order to find a breakthrough method for clinical treatment of ischemic stroke, which is a worldwide problem, and bring hope to stroke patients.
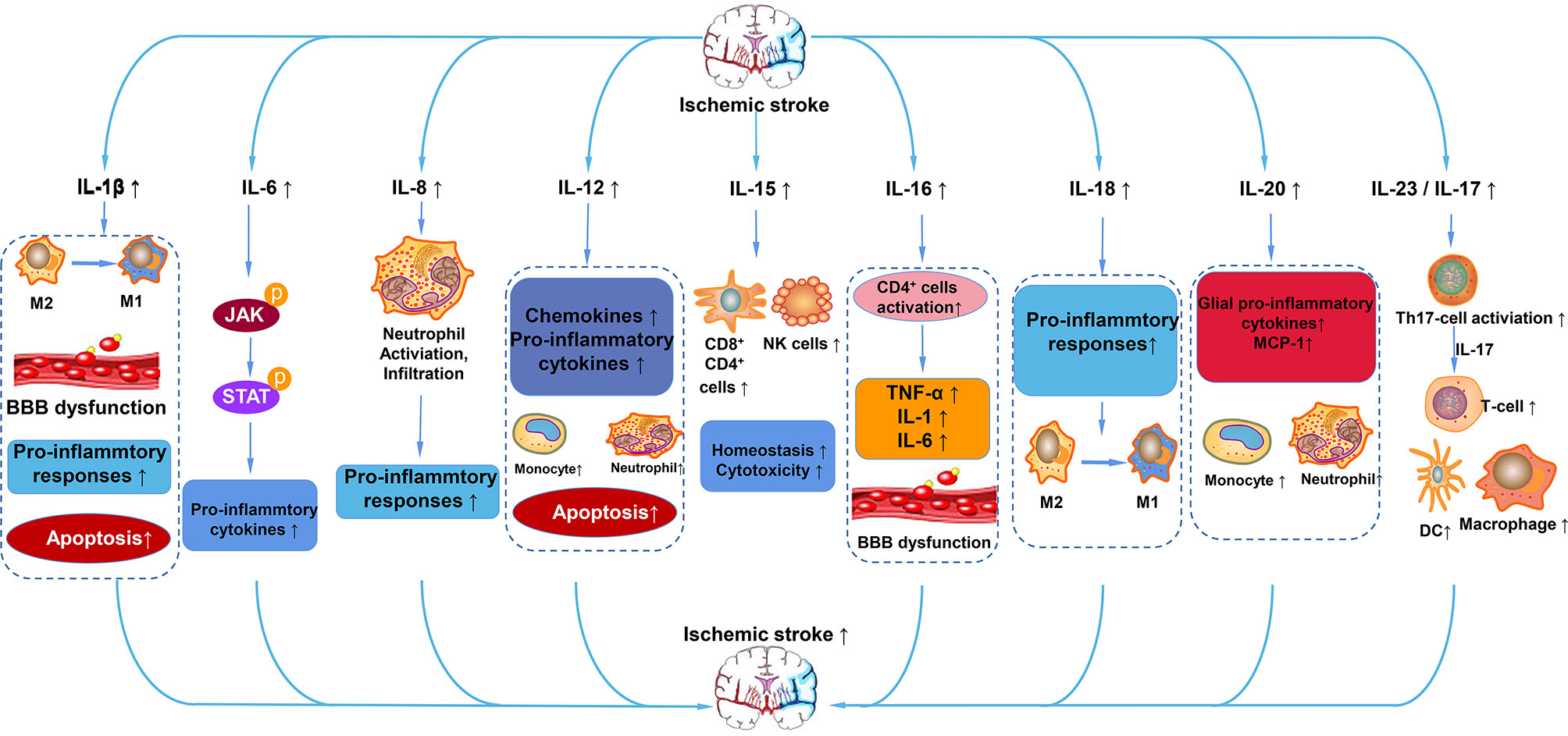
Figure 4 Pro-inflammatory ILs in ischemic stroke. After ischemic stroke, IL-1β aggravates cerebral infarction injury by polarization of microglia/macrophages to M1 phenotype, BBB dysfunction, and apoptosis. IL-6 activates the JAK/STAT pathway to promote the expression of pro-cytokines. IL-8 promotes the activation and infiltration of neutrophils into cerebral infarction. IL-12 promotes the expression of chemokines and pro-inflammatory mediators, promotes apoptosis, exerting a pro-inflammatory effect. IL-15 increases the number of CD4+, CD8+, and NK cells. IL-16 activates CD4+ cells and increases the levels of TNF-α, IL-1, and IL-6. IL-18 promotes the polarization of microglia/macrophages to the M1 phenotype and enhances the pro-inflammatory response. IL-20 promotes the expression of pro-inflammatory cytokines of MCP-1 and glia pro-inflammatory cytokines. IL-23/17 activates Th17 cells, which secrete IL-17 to increase the number of T cells, macrophages, and DC cells.
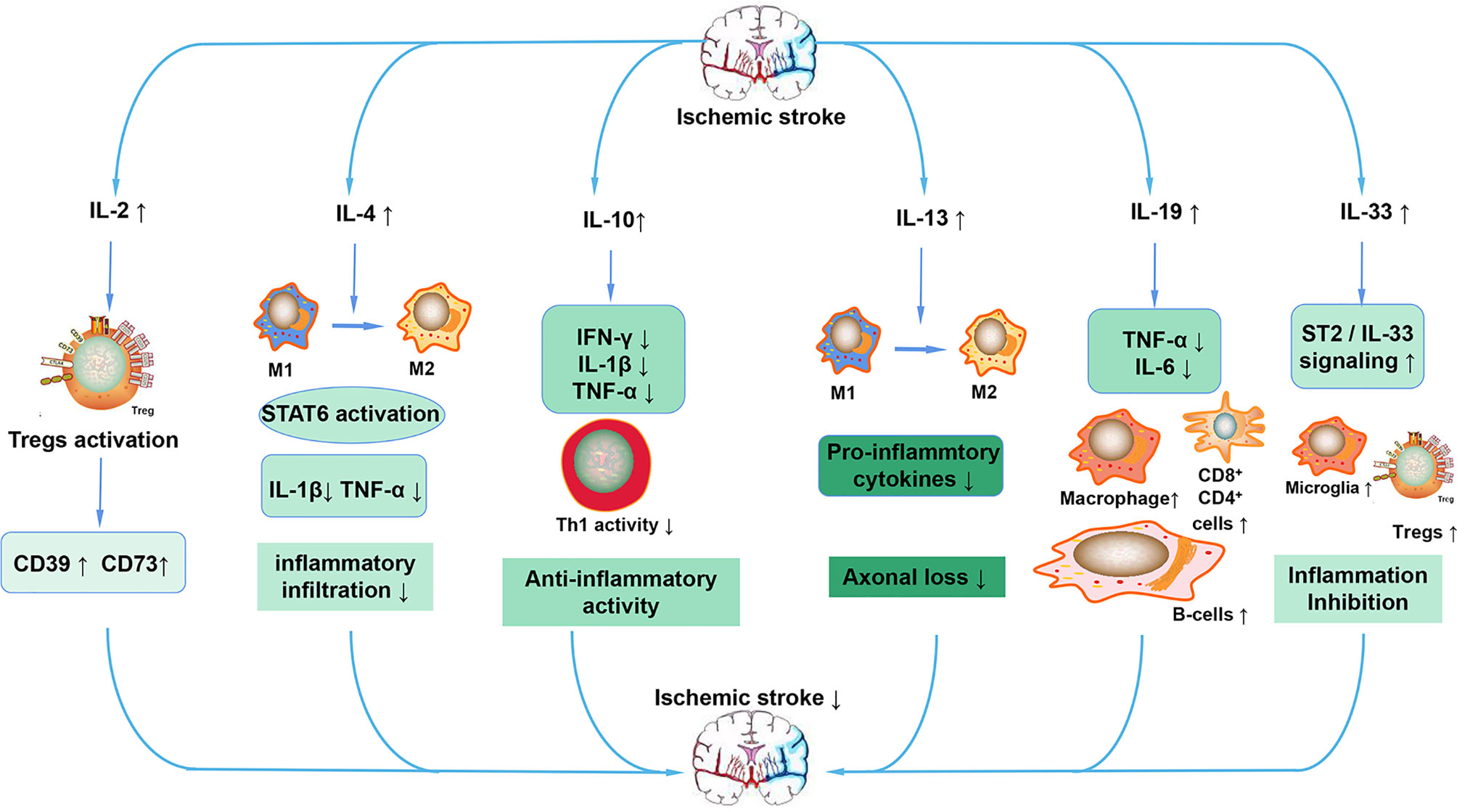
Figure 5 Anti-inflammatory ILs in ischemic stroke. After stroke, increased IL-2 activates Tregs, leading to the increased expression of CD39 and CD73. The increased IL-4 promotes the polarization of macrophages to the M2 phenotype and decreases the expression of pro-inflammatory cytokines such as IL-1β and TNF-α by activating STAT6. IL-10 also reduces the expression of pro-inflammatory cytokines, maintains the activity of Th1, ultimately exerting an anti-inflammatory action. IL-13 also increases the polarization of macrophages to the M2 phenotype, decreases the expression of pro-inflammatory cytokines and decreases the loss of neurons. IL-19 mainly preserves the reduced the number of CD4+, CD8+, and B cells and reduces the expression of pro-inflammatory cytokines. IL-33 inhibits inflammation after stroke mainly through the ST2/IL-33 signaling pathway.
Author Contributions
HZ wrote the manuscript. JC, SQ, and SH designed this work. The figures were prepared by HZ. YL, YS, XH, and XX revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (81870939 to XX).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Al-Bahrani A, Taha S, Shaath H, Bakhiet M. TNF-Alpha and IL-8 in Acute Stroke and the Modulation of These Cytokines by Antiplatelet Agents. Curr Neurovasc Res (2007) 4:31–7. doi: 10.2174/156720207779940716
3. Vignali DA, Kuchroo VK. IL-12 Family Cytokines: Immunological Playmakers. Nat Immunol (2012) 13:722–8. doi: 10.1038/ni.2366
4. Smith DE, Renshaw BR, Ketchem RR, Kubin M, Garka KE, Sims JE. Four New Members Expand the Interleukin-1 Superfamily. J Biol Chem (2000) 275:1169–75. doi: 10.1074/jbc.275.2.1169
5. Patterson D, Jones C, Hart I, Bleskan J, Berger R, Geyer D, et al. The Human Interleukin-1 Receptor Antagonist (IL1RN) Gene is Located in the Chromosome 2q14 Region. Genomics (1993) 15:173–6. doi: 10.1006/geno.1993.1025
6. Tarlow JK, Blakemore AI, Lennard A, Solari R, Hughes HN, Steinkasserer A, et al. Polymorphism in Human IL-1 Receptor Antagonist Gene Intron 2 is Caused by Variable Numbers of an 86-Bp Tandem Repeat. Hum Genet (1993) 91:403–4. doi: 10.1007/bf00217368
7. Murray KN, Parry-Jones AR, Allan SM. Interleukin-1 and Acute Brain Injury. Front Cell Neurosci (2015) 9:18. doi: 10.3389/fncel.2015.00018
8. Brough D, Tyrrell PJ, Allan SM. Regulation of Interleukin-1 in Acute Brain Injury. Trends Pharmacol Sci (2011) 32:617–22. doi: 10.1016/j.tips.2011.06.002
9. Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and Neuronal Injury. Nat Rev Immunol (2005) 5:629–40. doi: 10.1038/nri1664
10. John GR, Lee SC, Song X, Rivieccio M, Brosnan CF. IL-1-Regulated Responses in Astrocytes: Relevance to Injury and Recovery. Glia (2005) 49:161–76. doi: 10.1002/glia.20109
11. Konsman JP, Drukarch B, Van Dam AM. (Peri)vascular Production and Action of Pro-Inflammatory Cytokines in Brain Pathology. Clin Sci (Lond) (2007) 112:1–25. doi: 10.1042/cs20060043
12. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta Enhances NMDA Receptor-Mediated Intracellular Calcium Increase Through Activation of the Src Family of Kinases. J Neurosci (2003) 23:8692–700. doi: 10.1523/jneurosci.23-25-08692.2003
13. Sobowale OA, Parry-Jones AR, Smith CJ, Tyrrell PJ, Rothwell NJ, Allan SM. Interleukin-1 in Stroke: From Bench to Bedside. Stroke (2016) 47:2160–7. doi: 10.1161/strokeaha.115.010001
14. Simi A, Tsakiri N, Wang P, Rothwell NJ. Interleukin-1 and Inflammatory Neurodegeneration. Biochem Soc Trans (2007) 35:1122–6. doi: 10.1042/bst0351122
15. Strijbos PJ, Rothwell NJ. Interleukin-1 Beta Attenuates Excitatory Amino Acid-Induced Neurodegeneration In Vitro: Involvement of Nerve Growth Factor. J Neurosci (1995) 15:3468–74. doi: 10.1523/jneurosci.15-05-03468.1995
16. Barnes J, Mondelli V, Pariante CM. Genetic Contributions of Inflammation to Depression. Neuropsychopharmacology (2017) 42:81–98. doi: 10.1038/npp.2016.169
17. Yang J, Ma K, Zhang C, Liu Y, Liang F, Hu W, et al. Burns Impair Blood-Brain Barrier and Mesenchymal Stem Cells Can Reverse the Process in Mice. Front Immunol (2020) 11:578879. doi: 10.3389/fimmu.2020.578879
18. Dinarello CA. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
19. Liu G, Tsuruta Y, Gao Z, Park YJ, Abraham E. Variant IL-1 Receptor-Associated Kinase-1 Mediates Increased NF-Kappa B Activity. J Immunol (2007) 179:4125–34. doi: 10.4049/jimmunol.179.6.4125
20. Cahill CM, Rogers JT. Interleukin (IL) 1beta Induction of IL-6 is Mediated by a Novel Phosphatidylinositol 3-Kinase-Dependent AKT/IkappaB Kinase Alpha Pathway Targeting Activator Protein-1. J Biol Chem (2008) 283:25900–12. doi: 10.1074/jbc.M707692200
21. Kong X, Gong Z, Zhang L, Sun X, Ou Z, Xu B, et al. JAK2/STAT3 Signaling Mediates IL-6-Inhibited Neurogenesis of Neural Stem Cells Through DNA Demethylation/Methylation. Brain Behav Immun (2019) 79:159–73. doi: 10.1016/j.bbi.2019.01.027
22. Zhu H, Jian Z, Zhong Y, Ye Y, Zhang Y, Hu X, et al. Janus Kinase Inhibition Ameliorates Ischemic Stroke Injury and Neuroinflammation Through Reducing NLRP3 Inflammasome Activation via JAK2/STAT3 Pathway Inhibition. Front Immunol (2021) 12:714943. doi: 10.3389/fimmu.2021.714943
23. Chaparro-Huerta V, Rivera-Cervantes MC, Flores-Soto ME, Gómez-Pinedo U, Beas-Zárate C. Proinflammatory Cytokines and Apoptosis Following Glutamate-Induced Excitotoxicity Mediated by P38 MAPK in the Hippocampus of Neonatal Rats. J Neuroimmunol (2005) 165:53–62. doi: 10.1016/j.jneuroim.2005.04.025
24. Li W, Zheng S, Tang C, Zhu Y, Wang X. JNK-AP-1 Pathway Involved in Interleukin-1beta-Induced Calcitonin Gene-Related Peptide Secretion in Human Type II Alveolar Epithelial Cells. Peptides (2007) 28:1252–9. doi: 10.1016/j.peptides.2007.03.021
25. Roth S, Cao J, Singh V, Tiedt S, Hundeshagen G, Li T, et al. Post-Injury Immunosuppression and Secondary Infections are Caused by an AIM2 Inflammasome-Driven Signaling Cascade. Immunity (2021) 54:648–59.e8. doi: 10.1016/j.immuni.2021.02.004
26. Nayak AR, Kashyap RS, Purohit HJ, Kabra D, Taori GM, Daginawala HF. Evaluation of the Inflammatory Response in Sera From Acute Ischemic Stroke Patients by Measurement of IL-2 and IL-10. Inflammation Res (2009) 58:687–91. doi: 10.1007/s00011-009-0036-4
27. Zhang H, Xia Y, Ye Q, Yu F, Zhu W, Li P, et al. In Vivo Expansion of Regulatory T Cells With IL-2/IL-2 Antibody Complex Protects Against Transient Ischemic Stroke. J Neurosci (2018) 38:10168–79. doi: 10.1523/jneurosci.3411-17.2018
28. Zhou YX, Wang X, Tang D, Li Y, Jiao YF, Gan Y, et al. IL-2mab Reduces Demyelination After Focal Cerebral Ischemia by Suppressing CD8(+) T Cells. CNS Neurosci Ther (2019) 25:532–43. doi: 10.1111/cns.13084
29. Shi L, Sun Z, Su W, Xu F, Xie D, Zhang Q, et al. Treg Cell-Derived Osteopontin Promotes Microglia-Mediated White Matter Repair After Ischemic Stroke. Immunity (2021) 54:1527–42.e8. doi: 10.1016/j.immuni.2021.04.022
30. Zhao H, Li F, Huang Y, Zhang S, Li L, Yang Z, et al. Prognostic Significance of Plasma IL-2 and sIL-2rα in Patients With First-Ever Ischaemic Stroke. J Neuroinflamm (2020) 17:237. doi: 10.1186/s12974-020-01920-3
31. Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the Brain: A Cytokine to Remember. J Immunol (2012) 189:4213–9. doi: 10.4049/jimmunol.1202246
32. Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, et al. Regulation of Learning and Memory by Meningeal Immunity: A Key Role for IL-4. J Exp Med (2010) 207:1067–80. doi: 10.1084/jem.20091419
33. Zhang Q, Zhu W, Xu F, Dai X, Shi L, Cai W, et al. The Interleukin-4/Pparγ Signaling Axis Promotes Oligodendrocyte Differentiation and Remyelination After Brain Injury. PloS Biol (2019) 17:e3000330. doi: 10.1371/journal.pbio.3000330
34. Chen X, Zhang J, Song Y, Yang P, Yang Y, Huang Z, et al. Deficiency of Anti-Inflammatory Cytokine IL-4 Leads to Neural Hyperexcitability and Aggravates Cerebral Ischemia-Reperfusion Injury. Acta Pharm Sin B (2020) 10:1634–45. doi: 10.1016/j.apsb.2020.05.002
35. García-Berrocoso T, Giralt D, Bustamante A, Llombart V, Rubiera M, Penalba A, et al. Role of Beta-Defensin 2 and Interleukin-4 Receptor as Stroke Outcome Biomarkers. J Neurochem (2014) 129:463–72. doi: 10.1111/jnc.12649
36. Kim HM, Shin HY, Jeong HJ, An HJ, Kim NS, Chae HJ, et al. Reduced IL-2 But Elevated IL-4, IL-6, and IgE Serum Levels in Patients With Cerebral Infarction During the Acute Stage. J Mol Neurosci (2000) 14:191–6. doi: 10.1385/jmn:14:3:191
37. Xiong X, Xu L, Wei L, White RE, Ouyang YB, Giffard RG. IL-4 Is Required for Sex Differences in Vulnerability to Focal Ischemia in Mice. Stroke (2015) 46:2271–6. doi: 10.1161/strokeaha.115.008897
38. Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, et al. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke (2016) 47:498–504. doi: 10.1161/strokeaha.115.012079
39. Tian Y, Zhu P, Liu S, Jin Z, Li D, Zhao H, et al. IL-4-Polarized BV2 Microglia Cells Promote Angiogenesis by Secreting Exosomes. Adv Clin Exp Med (2019) 28:421–30. doi: 10.17219/acem/91826
40. Shi Q, Cai X, Shi G, Lv X, Yu J, Wang F. Interleukin-4 Protects From Chemotherapy-Induced Peripheral Neuropathy in Mice Modal via the Stimulation of IL-4/STAT6 Signaling. Acta Cir Bras (2018) 33:491–8. doi: 10.1590/s0102-865020180060000003
41. Akira S, Taga T, Kishimoto T. Interleukin-6 in Biology and Medicine. Adv Immunol (1993) 54:1–78. doi: 10.1016/s0065-2776(08)60532-5
42. Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric Structure and Assembly of the Interleukin-6/IL-6 Alpha-Receptor/Gp130 Complex. Science (2003) 300:2101–4. doi: 10.1126/science.1083901
43. Uyama N, Tsutsui H, Wu S, Yasuda K, Hatano E, Qin XY, et al. Anti-Interleukin-6 Receptor Antibody Treatment Ameliorates Postoperative Adhesion Formation. Sci Rep (2019) 9:17558. doi: 10.1038/s41598-019-54175-1
44. Zimmermann M, Aguilera FB, Castellucci M, Rossato M, Costa S, Lunardi C, et al. Chromatin Remodelling and Autocrine Tnfα are Required for Optimal Interleukin-6 Expression in Activated Human Neutrophils. Nat Commun (2015) 6:6061. doi: 10.1038/ncomms7061
45. Erta M, Quintana A, Hidalgo J. Interleukin-6, a Major Cytokine in the Central Nervous System. Int J Biol Sci (2012) 8:1254–66. doi: 10.7150/ijbs.4679
46. Su JH, Luo MY, Liang N, Gong SX, Chen W, Huang WQ, et al. Interleukin-6: A Novel Target for Cardio-Cerebrovascular Diseases. Front Pharmacol (2021) 12:745061. doi: 10.3389/fphar.2021.745061
47. Jenny NS, Callas PW, Judd SE, McClure LA, Kissela B, Zakai NA, et al. Inflammatory Cytokines and Ischemic Stroke Risk: The REGARDS Cohort. Neurology (2019) 92:e2375–84. doi: 10.1212/wnl.0000000000007416
48. Kowalska K, Klimiec E, Weglarczyk K, Pera J, Slowik A, Siedlar M, et al. Reduced Ex Vivo Release of Pro-Inflammatory Cytokines and Elevated Plasma Interleukin-6 are Inflammatory Signatures of Post-Stroke Delirium. J Neuroinflamm (2018) 15:111. doi: 10.1186/s12974-018-1156-y
49. Mechtouff L, Bochaton T, Paccalet A, Da Silva CC, Buisson M, Amaz C, et al. Association of Interleukin-6 Levels and Futile Reperfusion After Mechanical Thrombectomy. Neurology (2021) 96:e752–7. doi: 10.1212/wnl.0000000000011268
50. Mechtouff L, Bochaton T, Paccalet A, Crola Da Silva C, Buisson M, Amaz C, et al. A Lower Admission Level of Interleukin-6 Is Associated With First-Pass Effect in Ischemic Stroke Patients. J Neurointerv Surg (2021). doi: 10.1136/neurintsurg-2021-017334
51. Yao H, Zhang Y, Shu H, Xie B, Tao Y, Yuan Y, et al. Hyperforin Promotes Post-Stroke Neuroangiogenesis via Astrocytic IL-6-Mediated Negative Immune Regulation in the Ischemic Brain. Front Cell Neurosci (2019) 13:201. doi: 10.3389/fncel.2019.00201
52. Armstead WM, Hekierski H, Pastor P, Yarovoi S, Higazi AA, Cines DB. Release of IL-6 After Stroke Contributes to Impaired Cerebral Autoregulation and Hippocampal Neuronal Necrosis Through NMDA Receptor Activation and Upregulation of ET-1 and JNK. Transl Stroke Res (2019) 10:104–11. doi: 10.1007/s12975-018-0617-z
53. Tarkowski E, Rosengren L, Blomstrand C, Wikkelsö C, Jensen C, Ekholm S, et al. Early Intrathecal Production of Interleukin-6 Predicts the Size of Brain Lesion in Stroke. Stroke (1995) 26:1393–8. doi: 10.1161/01.str.26.8.1393
54. Suzuki S, Tanaka K, Suzuki N. Ambivalent Aspects of Interleukin-6 in Cerebral Ischemia: Inflammatory Versus Neurotrophic Aspects. J Cereb Blood Flow Metab (2009) 29:464–79. doi: 10.1038/jcbfm.2008.141
55. Rose-John S. Interleukin-6 Family Cytokines. Cold Spring Harb Perspect Biol (2018) 10:a028415. doi: 10.1101/cshperspect.a028415
56. Zhu H, Zhang Y, Zhong Y, Ye Y, Hu X, Gu L, et al. Inflammation-Mediated Angiogenesis in Ischemic Stroke. Front Cell Neurosci (2021) 15:652647. doi: 10.3389/fncel.2021.652647
57. Swartz KR, Liu F, Sewell D, Schochet T, Campbell I, Sandor M, et al. Interleukin-6 Promotes Post-Traumatic Healing in the Central Nervous System. Brain Res (2001) 896:86–95. doi: 10.1016/s0006-8993(01)02013-3
58. Matsuda S, Wen TC, Morita F, Otsuka H, Igase K, Yoshimura H, et al. Interleukin-6 Prevents Ischemia-Induced Learning Disability and Neuronal and Synaptic Loss in Gerbils. Neurosci Lett (1996) 204:109–12. doi: 10.1016/0304-3940(96)12340-5
59. Kostulas N, Kivisäkk P, Huang Y, Matusevicius D, Kostulas V, Link H. Ischemic Stroke is Associated With a Systemic Increase of Blood Mononuclear Cells Expressing Interleukin-8 mRNA. Stroke (1998) 29:462–6. doi: 10.1161/01.str.29.2.462
60. Kostulas N, Pelidou SH, Kivisäkk P, Kostulas V, Link H. Increased IL-1beta, IL-8, and IL-17 mRNA Expression in Blood Mononuclear Cells Observed in a Prospective Ischemic Stroke Study. Stroke (1999) 30:2174–9. doi: 10.1161/01.str.30.10.2174
61. Domac FM, Misirli H. The Role of Neutrophils and Interleukin-8 in Acute Ischemic Stroke. Neurosci (Riyadh) (2008) 13:136–41.
62. Zidovetzki R, Chen P, Chen M, Hofman FM. Endothelin-1-Induced Interleukin-8 Production in Human Brain-Derived Endothelial Cells Is Mediated by the Protein Kinase C and Protein Tyrosine Kinase Pathways. Blood (1999) 94:1291–9. doi: 10.1182/blood.V94.4.1291
63. Yamasaki Y, Matsuo Y, Matsuura N, Onodera H, Itoyama Y, Kogure K. Transient Increase of Cytokine-Induced Neutrophil Chemoattractant, a Member of the Interleukin-8 Family, in Ischemic Brain Areas After Focal Ischemia in Rats. Stroke (1995) 26:318–22; discussion 322-3. doi: 10.1161/01.str.26.2.318
64. Wang HJ, Wei JY, Liu DX, Zhuang SF, Li Y, Liu H, et al. Endothelial Atg7 Deficiency Ameliorates Acute Cerebral Injury Induced by Ischemia/Reperfusion. Front Neurol (2018) 9:998. doi: 10.3389/fneur.2018.00998
65. Shaheen HA, Daker LI, Abbass MM, Abd El Fattah AA. The Relationship Between the Severity of Disability and Serum IL-8 in Acute Ischemic Stroke Patients. Egypt J Neurol Psychiatr Neurosurg (2018) 54:26. doi: 10.1186/s41983-018-0025-z
66. Lv H, Li J, Che YQ. CXCL8 Gene Silencing Promotes Neuroglial Cells Activation While Inhibiting Neuroinflammation Through the PI3K/Akt/NF-κb-Signaling Pathway in Mice With Ischemic Stroke. J Cell Physiol (2019) 234:7341–55. doi: 10.1002/jcp.27493
67. Zhang L, Xu D, Zhang T, Hou W, Yixi L. Correlation Between Interleukin-6, Interleukin-8, and Modified Early Warning Score of Patients With Acute Ischemic Stroke and Their Condition and Prognosis. Ann Palliat Med (2021) 10:148–55. doi: 10.21037/apm-20-2200
68. Hou Y, Ryu CH, Jun JA, Kim SM, Jeong CH, Jeun SS. IL-8 Enhances the Angiogenic Potential of Human Bone Marrow Mesenchymal Stem Cells by Increasing Vascular Endothelial Growth Factor. Cell Biol Int (2014) 38:1050–9. doi: 10.1002/cbin.10294
69. van Scott MR, Justice JP, Bradfield JF, Enright E, Sigounas A, Sur S. IL-10 Reduces Th2 Cytokine Production and Eosinophilia But Augments Airway Reactivity in Allergic Mice. Am J Physiol Lung Cell Mol Physiol (2000) 278:L667–74. doi: 10.1152/ajplung.2000.278.4.L667
70. Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of Interleukin-10. Cytokine Growth Factor Rev (2010) 21:331–44. doi: 10.1016/j.cytogfr.2010.09.002
71. Kumar P, Yadav AK, Misra S, Kumar A, Chakravarty K, Prasad K. Role of Interleukin-10 (-1082a/G) Gene Polymorphism With the Risk of Ischemic Stroke: A Meta-Analysis. Neurol Res (2016) 38:823–30. doi: 10.1080/01616412.2016.1202395
72. Nguyen TV, Frye JB, Zbesko JC, Stepanovic K, Hayes M, Urzua A, et al. Multiplex Immunoassay Characterization and Species Comparison of Inflammation in Acute and Non-Acute Ischemic Infarcts in Human and Mouse Brain Tissue. Acta Neuropathol Commun (2016) 4:100. doi: 10.1186/s40478-016-0371-y
73. de Bilbao F, Arsenijevic D, Moll T, Garcia-Gabay I, Vallet P, Langhans W, et al. In Vivo Over-Expression of Interleukin-10 Increases Resistance to Focal Brain Ischemia in Mice. J Neurochem (2009) 110:12–22. doi: 10.1111/j.1471-4159.2009.06098.x
74. Nakajima M, Nito C, Sowa K, Suda S, Nishiyama Y, Nakamura-Takahashi A, et al. Mesenchymal Stem Cells Overexpressing Interleukin-10 Promote Neuroprotection in Experimental Acute Ischemic Stroke. Mol Ther Methods Clin Dev (2017) 6:102–11. doi: 10.1016/j.omtm.2017.06.005
75. Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 Reduces Rat Brain Injury Following Focal Stroke. Neurosci Lett (1998) 251:189–92. doi: 10.1016/s0304-3940(98)00537-0
76. Pérez-de Puig I, Miró F, Salas-Perdomo A, Bonfill-Teixidor E, Ferrer-Ferrer M, Márquez-Kisinousky L, et al. IL-10 Deficiency Exacerbates the Brain Inflammatory Response to Permanent Ischemia Without Preventing Resolution of the Lesion. J Cereb Blood Flow Metab (2013) 33:1955–66. doi: 10.1038/jcbfm.2013.155
77. Chi CH, Huang YY, Ye SZ, Shao MM, Jiang MX, Yang MY, et al. Interleukin-10 Level is Associated With Post-Stroke Depression in Acute Ischaemic Stroke Patients. J Affect Disord (2021) 293:254–60. doi: 10.1016/j.jad.2021.06.037
78. Chen Z, Zhang R, Wu Y, Fu Q, Qin X. Serum Interleukin-33 is a Predictor of Depression in Patients With Acute Ischemic Stroke. Curr Neurovasc Res (2020) 17:719–24. doi: 10.2174/1567202617999210101223635
79. Chang LT, Yuen CM, Liou CW, Lu CH, Chang WN, Youssef AA, et al. Link Between Interleukin-10 Level and Outcome After Ischemic Stroke. Neuroimmunomodulation (2010) 17:223–8. doi: 10.1159/000290038
80. Worthmann H, Tryc AB, Dirks M, Schuppner R, Brand K, Klawonn F, et al. Lipopolysaccharide Binding Protein, Interleukin-10, Interleukin-6 and C-Reactive Protein Blood Levels in Acute Ischemic Stroke Patients With Post-Stroke Infection. J Neuroinflamm (2015) 12:13. doi: 10.1186/s12974-014-0231-2
81. Chamorro A, Amaro S, Vargas M, Obach V, Cervera A, Torres F, et al. Interleukin 10, Monocytes and Increased Risk of Early Infection in Ischaemic Stroke. J Neurol Neurosurg Psychiatry (2006) 77:1279–81. doi: 10.1136/jnnp.2006.100800
82. Conway SE, Roy-O'Reilly M, Friedler B, Staff I, Fortunato G, McCullough LD. Sex Differences and the Role of IL-10 in Ischemic Stroke Recovery. Biol Sex Differ (2015) 6:17. doi: 10.1186/s13293-015-0035-9
83. Lambertsen KL, Finsen B, Clausen BH. Post-Stroke Inflammation-Target or Tool for Therapy? Acta Neuropathol (2019) 137:693–714. doi: 10.1007/s00401-018-1930-z
84. Bushnell C, McCullough LD, Awad IA, Chireau MV, Fedder WN, Furie KL, et al. Guidelines for the Prevention of Stroke in Women: A Statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke (2014) 45:1545–88. doi: 10.1161/01.str.0000442009.06663.48
85. Pelidou SH, Kostulas N, Matusevicius D, Kivisäkk P, Kostulas V, Link H. High Levels of IL-10 Secreting Cells are Present in Blood in Cerebrovascular Diseases. Eur J Neurol (1999) 6:437–42. doi: 10.1046/j.1468-1331.1999.640437.x
86. Liesz A, Zhou W, Na SY, Hämmerling GJ, Garbi N, Karcher S, et al. Boosting Regulatory T Cells Limits Neuroinflammation in Permanent Cortical Stroke. J Neurosci (2013) 33:17350–62. doi: 10.1523/jneurosci.4901-12.2013
87. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL-10-Producing B-Cells Limit CNS Inflammation and Infarct Volume in Experimental Stroke. Metab Brain Dis (2013) 28:375–86. doi: 10.1007/s11011-013-9413-3
88. Sharma S, Yang B, Xi X, Grotta JC, Aronowski J, Savitz SI. IL-10 Directly Protects Cortical Neurons by Activating PI-3 Kinase and STAT-3 Pathways. Brain Res (2011) 1373:189–94. doi: 10.1016/j.brainres.2010.11.096
89. Oleszycka E, McCluskey S, Sharp FA, Muñoz-Wolf N, Hams E, Gorman AL, et al. The Vaccine Adjuvant Alum Promotes IL-10 Production That Suppresses Th1 Responses. Eur J Immunol (2018) 48:705–15. doi: 10.1002/eji.201747150
90. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. Treatment of Experimental Stroke With IL-10-Producing B-Cells Reduces Infarct Size and Peripheral and CNS Inflammation in Wild-Type B-Cell-Sufficient Mice. Metab Brain Dis (2014) 29:59–73. doi: 10.1007/s11011-013-9474-3
91. Liang QJ, Jiang M, Wang XH, Le LL, Xiang M, Sun N, et al. Pre-Existing Interleukin 10 in Cerebral Arteries Attenuates Subsequent Brain Injury Caused by Ischemia/Reperfusion. IUBMB Life (2015) 67:710–9. doi: 10.1002/iub.1429
92. Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular Mechanisms of Interleukin-10-Mediated Inhibition of NF-kappaB Activity: A Role for P50. Clin Exp Immunol (2004) 135:64–73. doi: 10.1111/j.1365-2249.2004.02342.x
93. Wang Y, Jia J, Ao G, Hu L, Liu H, Xiao Y, et al. Hydrogen Sulfide Protects Blood-Brain Barrier Integrity Following Cerebral Ischemia. J Neurochem (2014) 129:827–38. doi: 10.1111/jnc.12695
94. Hwang CJ, Yun HM, Jung YY, Lee DH, Yoon NY, Seo HO, et al. Reducing Effect of IL-32α in the Development of Stroke Through Blocking of NF-κb, But Enhancement of STAT3 Pathways. Mol Neurobiol (2015) 51:648–60. doi: 10.1007/s12035-014-8739-0
95. Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by IL-10-Producing MOG CD4+ T Cells Following Ischemic Stroke. J Neurol Sci (2005) 233:125–32. doi: 10.1016/j.jns.2005.03.022
96. Frenkel D, Huang Z, Maron R, Koldzic DN, Hancock WW, Moskowitz MA, et al. Nasal Vaccination With Myelin Oligodendrocyte Glycoprotein Reduces Stroke Size by Inducing IL-10-Producing CD4+ T Cells. J Immunol (2003) 171:6549–55. doi: 10.4049/jimmunol.171.12.6549
97. Na SY, Mracsko E, Liesz A, Hünig T, Veltkamp R. Amplification of Regulatory T Cells Using a CD28 Superagonist Reduces Brain Damage After Ischemic Stroke in Mice. Stroke (2015) 46:212–20. doi: 10.1161/strokeaha.114.007756
98. Wang J, Xie L, Yang C, Ren C, Zhou K, Wang B, et al. Activated Regulatory T Cell Regulates Neural Stem Cell Proliferation in the Subventricular Zone of Normal and Ischemic Mouse Brain Through Interleukin 10. Front Cell Neurosci (2015) 9:361. doi: 10.3389/fncel.2015.00361
99. Kawada H, Takizawa S, Takanashi T, Morita Y, Fujita J, Fukuda K, et al. Administration of Hematopoietic Cytokines in the Subacute Phase After Cerebral Infarction is Effective for Functional Recovery Facilitating Proliferation of Intrinsic Neural Stem/Progenitor Cells and Transition of Bone Marrow-Derived Neuronal Cells. Circulation (2006) 113:701–10. doi: 10.1161/circulationaha.105.563668
100. Morita Y, Takizawa S, Kamiguchi H, Uesugi T, Kawada H, Takagi S. Administration of Hematopoietic Cytokines Increases the Expression of Anti-Inflammatory Cytokine (IL-10) mRNA in the Subacute Phase After Stroke. Neurosci Res (2007) 58:356–60. doi: 10.1016/j.neures.2007.04.006
101. Liu N, Chen R, Du H, Wang J, Zhang Y, Wen J. Expression of IL-10 and TNF-Alpha in Rats With Cerebral Infarction After Transplantation With Mesenchymal Stem Cells. Cell Mol Immunol (2009) 6:207–13. doi: 10.1038/cmi.2009.28
102. Pereira L, Font-Nieves M, Van den Haute C, Baekelandt V, Planas AM, Pozas E. IL-10 Regulates Adult Neurogenesis by Modulating ERK and STAT3 Activity. Front Cell Neurosci (2015) 9:57. doi: 10.3389/fncel.2015.00057
103. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal Microbiota Affects Ischemic Stroke Outcome by Regulating Intestinal γδ T Cells. Nat Med (2016) 22:516–23. doi: 10.1038/nm.4068
104. Piepke M, Clausen BH, Ludewig P, Vienhues JH, Bedke T, Javidi E, et al. Interleukin-10 Improves Stroke Outcome by Controlling the Detrimental Interleukin-17A Response. J Neuroinflamm (2021) 18:265. doi: 10.1186/s12974-021-02316-7
105. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The Inhibitory Cytokine IL-35 Contributes to Regulatory T-Cell Function. Nature (2007) 450:566–9. doi: 10.1038/nature06306
106. Reading PC, Whitney PG, Barr DP, Wojtasiak M, Mintern JD, Waithman J, et al. IL-18, But Not IL-12, Regulates NK Cell Activity Following Intranasal Herpes Simplex Virus Type 1 Infection. J Immunol (2007) 179:3214–21. doi: 10.4049/jimmunol.179.5.3214
107. Noble A, Thomas MJ, Kemeny DM. Early Th1/Th2 Cell Polarization in the Absence of IL-4 and IL-12: T Cell Receptor Signaling Regulates the Response to Cytokines in CD4 and CD8 T Cells. Eur J Immunol (2001) 31:2227–35. doi: 10.1002/1521-4141(200107)31:7<2227::aid-immu2227>3.0.co;2-c
108. Zaremba J, Losy J. Interleukin-12 in Acute Ischemic Stroke Patients. Folia Neuropathol (2006) 44:59–66.
109. Zykov MV, Barbarash OL, Kashtalap VV, Kutikhin AG, Barbarash LS. Interleukin-12 Serum Level has Prognostic Value in Patients With ST-Segment Elevation Myocardial Infarction. Heart Lung (2016) 45:336–40. doi: 10.1016/j.hrtlng.2016.03.007
110. Han D, Liu H, Gao Y. The Role of Peripheral Monocytes and Macrophages in Ischemic Stroke. Neurol Sci (2020) 41:3589–607. doi: 10.1007/s10072-020-04777-9
111. Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism Against Stroke. Int J Mol Sci (2017) 18:2135. doi: 10.3390/ijms18102135
112. Kouwenhoven M, Carlström C, Ozenci V, Link H. Matrix Metalloproteinase and Cytokine Profiles in Monocytes Over the Course of Stroke. J Clin Immunol (2001) 21:365–75. doi: 10.1023/a:1012244820709
113. Ogawa H, Nishimura N, Nishioka Y, Azuma M, Yanagawa H, Sone S. Adenoviral Interleukin-12 Gene Transduction Into Human Bronchial Epithelial Cells: Up-Regulation of Pro-Inflammatory Cytokines and its Prevention by Corticosteroids. Clin Exp Allergy (2003) 33:921–9. doi: 10.1046/j.1365-2222.2003.01702.x
114. Yago T, Tsukuda M, Fukushima H, Yamaoka H, Kurata-Miura K, Nishi T, et al. IL-12 Promotes the Adhesion of NK Cells to Endothelial Selectins Under Flow Conditions. J Immunol (1998) 161:1140–5.
115. Zhu S, Lee DA, Li S. IL-12 and IL-27 Sequential Gene Therapy via Intramuscular Electroporation Delivery for Eliminating Distal Aggressive Tumors. J Immunol (2010) 184:2348–54. doi: 10.4049/jimmunol.0902371
116. Hart PH, Bonder CS, Balogh J, Dickensheets HL, Vazquez N, Davies KV, et al. Diminished Responses to IL-13 by Human Monocytes Differentiated In Vitro: Role of the IL-13Ralpha1 Chain and STAT6. Eur J Immunol (1999) 29:2087–97. doi: 10.1002/(sici)1521-4141(199907)29:07<2087::Aid-immu2087>3.0.Co;2-j
117. Zurawski G, de Vries JE. Interleukin 13, an Interleukin 4-Like Cytokine That Acts on Monocytes and B Cells, But Not on T Cells. Immunol Today (1994) 15:19–26. doi: 10.1016/0167-5699(94)90021-3
118. Chung HS, Lee BS, Ma JY. Ethanol Extract of Mylabris Phalerata Inhibits M2 Polarization Induced by Recombinant IL-4 and IL-13 in Murine Macrophages. Evid Based Complement Alternat Med (2017) 2017:4218468. doi: 10.1155/2017/4218468
119. Hamzei Taj S, Le Blon D, Hoornaert C, Daans J, Quarta A, Praet J, et al. Targeted Intracerebral Delivery of the Anti-Inflammatory Cytokine IL13 Promotes Alternative Activation of Both Microglia and Macrophages After Stroke. J Neuroinflamm (2018) 15:174. doi: 10.1186/s12974-018-1212-7
120. Kolosowska N, Keuters MH, Wojciechowski S, Keksa-Goldsteine V, Laine M, Malm T, et al. Peripheral Administration of IL-13 Induces Anti-Inflammatory Microglial/Macrophage Responses and Provides Neuroprotection in Ischemic Stroke. Neurotherapeutics (2019) 16:1304–19. doi: 10.1007/s13311-019-00761-0
121. Liao KY, Chen CJ, Hsieh SK, Pan PH, Chen WY. Interleukin-13 Ameliorates Postischemic Hepatic Gluconeogenesis and Hyperglycemia in Rat Model of Stroke. Metab Brain Dis (2020) 35:1201–10. doi: 10.1007/s11011-020-00596-1
122. Hawkins KE, Corcelli M, Dowding K, Ranzoni AM, Vlahova F, Hau KL, et al. Embryonic Stem Cell-Derived Mesenchymal Stem Cells (MSCs) Have a Superior Neuroprotective Capacity Over Fetal MSCs in the Hypoxic-Ischemic Mouse Brain. Stem Cells Transl Med (2018) 7:439–49. doi: 10.1002/sctm.17-0260
123. Sim GC, Radvanyi L. The IL-2 Cytokine Family in Cancer Immunotherapy. Cytokine Growth Factor Rev (2014) 25:377–90. doi: 10.1016/j.cytogfr.2014.07.018
124. Perera LP, Goldman CK, Waldmann TA. IL-15 Induces the Expression of Chemokines and Their Receptors in T Lymphocytes. J Immunol (1999) 162:2606–12.
125. Zhou X, Yu J, Cheng X, Zhao B, Manyam GC, Zhang L, et al. The Deubiquitinase Otub1 Controls the Activation of CD8(+) T Cells and NK Cells by Regulating IL-15-Mediated Priming. Nat Immunol (2019) 20:879–89. doi: 10.1038/s41590-019-0405-2
126. Li M, Li Z, Yao Y, Jin WN, Wood K, Liu Q, et al. Astrocyte-Derived Interleukin-15 Exacerbates Ischemic Brain Injury via Propagation of Cellular Immunity. Proc Natl Acad Sci USA (2017) 114:E396–e405. doi: 10.1073/pnas.1612930114
127. Lee GA, Lin TN, Chen CY, Mau SY, Huang WZ, Kao YC, et al. Interleukin 15 Blockade Protects the Brain From Cerebral Ischemia-Reperfusion Injury. Brain Behav Immun (2018) 73:562–70. doi: 10.1016/j.bbi.2018.06.021
128. Shi SX, Li YJ, Shi K, Wood K, Ducruet AF, Liu Q. IL (Interleukin)-15 Bridges Astrocyte-Microglia Crosstalk and Exacerbates Brain Injury Following Intracerebral Hemorrhage. Stroke (2020) 51:967–74. doi: 10.1161/strokeaha.119.028638
129. Nguyen V, Ameri K, Huynh K, Fredkin M, Grona R, Larpthaveesarp A, et al. Interleukin-15 Modulates the Response of Cortical Neurons to Ischemia. Mol Cell Neurosci (2021) 115:103658. doi: 10.1016/j.mcn.2021.103658
130. Liu XL, Du JZ, Zhou YM, Shu QF, Li YG. Interleukin-16 Polymorphism is Associated With an Increased Risk of Ischemic Stroke. Mediators Inflammation (2013) 2013:564750. doi: 10.1155/2013/564750
131. Mathy NL, Scheuer W, Lanzendörfer M, Honold K, Ambrosius D, Norley S, et al. Interleukin-16 Stimulates the Expression and Production of Pro-Inflammatory Cytokines by Human Monocytes. Immunology (2000) 100:63–9. doi: 10.1046/j.1365-2567.2000.00997.x
132. Skundric DS, Cruikshank WW, Drulovic J. Role of IL-16 in CD4+ T Cell-Mediated Regulation of Relapsing Multiple Sclerosis. J Neuroinflamm (2015) 12:78. doi: 10.1186/s12974-015-0292-x
133. Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Göbel K, Schuhmann MK, et al. Regulatory T Cells are Strong Promoters of Acute Ischemic Stroke in Mice by Inducing Dysfunction of the Cerebral Microvasculature. Blood (2013) 121:679–91. doi: 10.1182/blood-2012-04-426734
134. Schwab JM, Nguyen TD, Meyermann R, Schluesener HJ. Human Focal Cerebral Infarctions Induce Differential Lesional Interleukin-16 (IL-16) Expression Confined to Infiltrating Granulocytes, CD8+ T-Lymphocytes and Activated Microglia/Macrophages. J Neuroimmunol (2001) 114:232–41. doi: 10.1016/s0165-5728(00)00433-1
135. Wu D, Zhang G, Zhao C, Yang Y, Miao Z, Xu X. Interleukin-18 From Neurons and Microglia Mediates Depressive Behaviors in Mice With Post-Stroke Depression. Brain Behav Immun (2020) 88:411–20. doi: 10.1016/j.bbi.2020.04.004
136. Wolk K, Kunz S, Asadullah K, Sabat R. Cutting Edge: Immune Cells as Sources and Targets of the IL-10 Family Members? J Immunol (2002) 168:5397–402. doi: 10.4049/jimmunol.168.11.5397
137. Gallagher G. Interleukin-19: Multiple Roles in Immune Regulation and Disease. Cytokine Growth Factor Rev (2010) 21:345–52. doi: 10.1016/j.cytogfr.2010.08.005
138. Gallagher G, Eskdale J, Jordan W, Peat J, Campbell J, Boniotto M, et al. Human Interleukin-19 and its Receptor: A Potential Role in the Induction of Th2 Responses. Int Immunopharmacol (2004) 4:615–26. doi: 10.1016/j.intimp.2004.01.005
139. Oral HB, Kotenko SV, Yilmaz M, Mani O, Zumkehr J, Blaser K, et al. Regulation of T Cells and Cytokines by the Interleukin-10 (IL-10)-Family Cytokines IL-19, IL-20, IL-22, IL-24 and IL-26. Eur J Immunol (2006) 36:380–8. doi: 10.1002/eji.200425523
140. England RN, Autieri MV. Anti-Inflammatory Effects of Interleukin-19 in Vascular Disease. Int J Inflam (2012) 2012:253583. doi: 10.1155/2012/253583
141. Xie W, Fang L, Gan S, Xuan H. Interleukin-19 Alleviates Brain Injury by Anti-Inflammatory Effects in a Mice Model of Focal Cerebral Ischemia. Brain Res (2016) 1650:172–7. doi: 10.1016/j.brainres.2016.09.006
142. Autieri MV. IL-19 and Other IL-20 Family Member Cytokines in Vascular Inflammatory Diseases. Front Immunol (2018) 9:700. doi: 10.3389/fimmu.2018.00700
143. Hsu YH, Yang YY, Huwang MH, Weng YH, Jou IM, Wu PT, et al. Anti-IL-20 Monoclonal Antibody Inhibited Inflammation and Protected Against Cartilage Destruction in Murine Models of Osteoarthritis. PloS One (2017) 12:e0175802. doi: 10.1371/journal.pone.0175802
144. Chen WY, Chang MS. IL-20 is Regulated by Hypoxia-Inducible Factor and Up-Regulated After Experimental Ischemic Stroke. J Immunol (2009) 182:5003–12. doi: 10.4049/jimmunol.0803653
145. Hsu YH, Lin RM, Chiu YS, Liu WL, Huang KY. Effects of IL-1β, IL-20, and BMP-2 on Intervertebral Disc Inflammation Under Hypoxia. J Clin Med (2020) 9:140. doi: 10.3390/jcm9010140
146. Lee JS, Hsu YH, Chiu YS, Jou IM, Chang MS. Anti-IL-20 Antibody Improved Motor Function and Reduced Glial Scar Formation After Traumatic Spinal Cord Injury in Rats. J Neuroinflamm (2020) 17:156. doi: 10.1186/s12974-020-01814-4
147. Iwakura Y, Ishigame H. The IL-23/IL-17 Axis in Inflammation. J Clin Invest (2006) 116:1218–22. doi: 10.1172/jci28508
148. Gelderblom M, Gallizioli M, Ludewig P, Thom V, Arunachalam P, Rissiek B, et al. IL-23 (Interleukin-23)-Producing Conventional Dendritic Cells Control the Detrimental IL-17 (Interleukin-17) Response in Stroke. Stroke (2018) 49:155–64. doi: 10.1161/strokeaha.117.019101
149. Gooderham MJ, Papp KA, Lynde CW. Shifting the Focus - the Primary Role of IL-23 in Psoriasis and Other Inflammatory Disorders. J Eur Acad Dermatol Venereol (2018) 32:1111–9. doi: 10.1111/jdv.14868
150. Chien YH, Meyer C, Bonneville M. γδ T Cells: First Line of Defense and Beyond. Annu Rev Immunol (2014) 32:121–55. doi: 10.1146/annurev-immunol-032713-120216
151. Backes FN, de Souza A, Bianchin MM. IL-23 and IL-17 in Acute Ischemic Stroke: Correlation With Stroke Scales and Prognostic Value. Clin Biochem (2021) 98:29–34. doi: 10.1016/j.clinbiochem.2021.09.003
152. Lu L, Wang Y, Zhou L, Li Y, Zhang X, Hu X, et al. Vγ4 T Cell-Derived IL-17A is Essential for Amplification of Inflammatory Cascades in Ischemic Brain Tissue After Stroke. Int Immunopharmacol (2021) 96:107678. doi: 10.1016/j.intimp.2021.107678
153. Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU. IL-1 Receptor Accessory Protein is Essential for IL-33-Induced Activation of T Lymphocytes and Mast Cells. Proc Natl Acad Sci USA (2007) 104:18660–5. doi: 10.1073/pnas.0705939104
154. Drieu A, Martinez de Lizarrondo S, Rubio M. Stopping Inflammation in Stroke: Role of ST2/IL-33 Signaling. J Neurosci (2017) 37:9614–6. doi: 10.1523/jneurosci.1863-17.2017
155. Sun Y, Wen Y, Wang L, Wen L, You W, Wei S, et al. Therapeutic Opportunities of Interleukin-33 in the Central Nervous System. Front Immunol (2021) 12:654626. doi: 10.3389/fimmu.2021.654626
156. Liu X, Hu R, Pei L, Si P, Wang C, Tian X, et al. Regulatory T Cell is Critical for Interleukin-33-Mediated Neuroprotection Against Stroke. Exp Neurol (2020) 328:113233. doi: 10.1016/j.expneurol.2020.113233
157. Chen Z, Hu Q, Huo Y, Zhang R, Fu Q, Qin X. Serum Interleukin-33 is a Novel Predictive Biomarker of Hemorrhage Transformation and Outcome in Acute Ischemic Stroke. J Stroke Cerebrovasc Dis (2021) 30:105506. doi: 10.1016/j.jstrokecerebrovasdis.2020.105506
158. Jiang M, Liu X, Zhang D, Wang Y, Hu X, Xu F, et al. Celastrol Treatment Protects Against Acute Ischemic Stroke-Induced Brain Injury by Promoting an IL-33/ST2 Axis-Mediated Microglia/Macrophage M2 Polarization. J Neuroinflamm (2018) 15:78. doi: 10.1186/s12974-018-1124-6
159. Luo Q, Fan Y, Lin L, Wei J, Li Z, Li Y, et al. Interleukin-33 Protects Ischemic Brain Injury by Regulating Specific Microglial Activities. Neuroscience (2018) 385:75–89. doi: 10.1016/j.neuroscience.2018.05.047
160. Guo S, Luo Y. Brain Foxp3(+) Regulatory T Cells Can Be Expanded by Interleukin-33 in Mouse Ischemic Stroke. Int Immunopharmacol (2020) 81:106027. doi: 10.1016/j.intimp.2019.106027
161. Wei H, Zhen L, Wang S, Zhang Y, Wang K, Jia P, et al. De Novo Lipogenesis in Astrocytes Promotes the Repair of Blood-Brain Barrier After Transient Cerebral Ischemia Through Interleukin-33. Neuroscience (2021) 481:85–98. doi: 10.1016/j.neuroscience.2021.11.026
162. Xiao W, Guo S, Chen L, Luo Y. The Role of Interleukin-33 in the Modulation of Splenic T-Cell Immune Responses After Experimental Ischemic Stroke. J Neuroimmunol (2019) 333:576970. doi: 10.1016/j.jneuroim.2019.576970
163. Zhang SR, Piepke M, Chu HX, Broughton BR, Shim R, Wong CH, et al. IL-33 Modulates Inflammatory Brain Injury But Exacerbates Systemic Immunosuppression Following Ischemic Stroke. JCI Insight (2018) 3:e121560. doi: 10.1172/jci.insight.121560
164. Li X, Lin S, Chen X, Huang W, Li Q, Zhang H, et al. The Prognostic Value of Serum Cytokines in Patients With Acute Ischemic Stroke. Aging Dis (2019) 10:544–56. doi: 10.14336/ad.2018.0820
165. Martha SR, Cheng Q, Fraser JF, Gong L, Collier LA, Davis SM, et al. Expression of Cytokines and Chemokines as Predictors of Stroke Outcomes in Acute Ischemic Stroke. Front Neurol (2019) 10:1391:1391. doi: 10.3389/fneur.2019.01391
166. Lin Y, Zhang L, Dai Y, Li H, Wang Y, Zhang B, et al. Expression of Interleukin-9 and its Upstream Stimulating Factors in Rats With Ischemic Stroke. Neurol Sci (2015) 36:913–20. doi: 10.1007/s10072-015-2096-2
167. Tan S, Shan Y, Lin Y, Liao S, Zhang B, Zeng Q, et al. Neutralization of Interleukin-9 Ameliorates Experimental Stroke by Repairing the Blood-Brain Barrier via Down-Regulation of Astrocyte-Derived Vascular Endothelial Growth Factor-a. FASEB J (2019) 33:4376–87. doi: 10.1096/fj.201801595RR
168. Tan S, Shan Y, Wang Y, Lin Y, Liao S, Deng Z, et al. Exacerbation of Oxygen-Glucose Deprivation-Induced Blood-Brain Barrier Disruption: Potential Pathogenic Role of Interleukin-9 in Ischemic Stroke. Clin Sci (Lond) (2017) 131:1499–513. doi: 10.1042/cs20170984
169. Li G, Xu R, Cao Y, Xie X, Zheng Z. Interleukin-21 Polymorphism Affects Gene Expression and is Associated With Risk of Ischemic Stroke. Inflammation (2014) 37:2030–9. doi: 10.1007/s10753-014-9935-9
170. Lee HK, Keum S, Sheng H, Warner DS, Lo DC, Marchuk DA. Natural Allelic Variation of the IL-21 Receptor Modulates Ischemic Stroke Infarct Volume. J Clin Invest (2016) 126:2827–38. doi: 10.1172/jci84491
171. Dong Y, Hu C, Huang C, Gao J, Niu W, Wang D, et al. Interleukin-22 Plays a Protective Role by Regulating the JAK2-STAT3 Pathway to Improve Inflammation, Oxidative Stress, and Neuronal Apoptosis Following Cerebral Ischemia-Reperfusion Injury. Mediators Inflammation (2021) 2021:6621296. doi: 10.1155/2021/6621296
172. Yin H, Wu M, Jia Y. Knockdown of IL-32 Protects PC12 Cells Against Oxygen-Glucose Deprivation/Reoxygenation-Induced Injury via Activation of Nrf2/NF-κb Pathway. Metab Brain Dis (2020) 35:363–71. doi: 10.1007/s11011-019-00530-0
173. Huang X, Li F, Yang T, Li H, Liu T, Wang Y, et al. Increased Serum Interleukin-34 Levels as a Novel Diagnostic and Prognostic Biomarker in Patients With Acute Ischemic Stroke. J Neuroimmunol (2021) 358:577652. doi: 10.1016/j.jneuroim.2021.577652
174. Zhang Y, Xu C, Wang H, Nan S. Serum Interleukin-37 Increases in Patients After Ischemic Stroke and Is Associated With Stroke Recurrence. Oxid Med Cell Longev (2021) 2021:5546991. doi: 10.1155/2021/5546991
175. Zhang F, Zhu T, Li H, He Y, Zhang Y, Huang N, et al. Plasma Interleukin-37 is Elevated in Acute Ischemic Stroke Patients and Probably Associated With 3-Month Functional Prognosis. Clin Interv Aging (2020) 15:1285–94. doi: 10.2147/cia.S230186
176. Zhang SR, Nold MF, Tang SC, Bui CB, Nold CA, Arumugam TV, et al. IL-37 Increases in Patients After Ischemic Stroke and Protects From Inflammatory Brain Injury, Motor Impairment and Lung Infection in Mice. Sci Rep (2019) 9:6922. doi: 10.1038/s41598-019-43364-7
Keywords: interleukins, ischemic stroke, inflammation, cytokines, neuro-immune
Citation: Zhu H, Hu S, Li Y, Sun Y, Xiong X, Hu X, Chen J and Qiu S (2022) Interleukins and Ischemic Stroke. Front. Immunol. 13:828447. doi: 10.3389/fimmu.2022.828447
Received: 03 December 2021; Accepted: 12 January 2022;
Published: 31 January 2022.
Edited by:
Yan Wang, Peking University Third Hospital, ChinaReviewed by:
Xinchun Jin, Capital Medical University, ChinaZongjian Liu, Capital Medical University, China
Copyright © 2022 Zhu, Hu, Li, Sun, Xiong, Hu, Chen and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junjing Chen, Y2hlbmp1bmppbmcxOTgzQDE2My5jb20=; Sheng Qiu, cWl1czIwMDFAMTI2LmNvbQ==
†These authors have contributed equally to this work