 Mengyu Dai1,2,3
Mengyu Dai1,2,3 Xiaoying Yang
Xiaoying Yang Yinghua Yu
Yinghua Yu Wei Pan
Wei Pan- 1Jiangsu Key Laboratory of Immunity and Metabolism, Department of Pathogen Biology and Immunology, Xuzhou Medical University, Xuzhou, China
- 2The Second Clinical Medicine, Xuzhou Medical University, Xuzhou, China
- 3National Demonstration Center for Experimental Basic Medical Science Education (Xuzhou Medical University), Xuzhou, China
Obesity and its associated Metabolic Syndromes (Mets) represent a global epidemic health problem. Metabolic inflammation, lipid accumulation and insulin resistance contribute to the progression of these diseases, thereby becoming targets for drug development. Epidemiological data have showed that the rate of helminth infection negatively correlates with the incidence of obesity and Mets. Correspondingly, numerous animal experiments and a few of clinic trials in human demonstrate that helminth infection or its derived molecules can mitigate obesity and Mets via induction of macrophage M2 polarization, inhibition of adipogenesis, promotion of fat browning, and improvement of glucose tolerance, insulin resistance and metabolic inflammation. Interestingly, sporadic studies also uncover that several helminth infections can reshape gut microbiota of hosts, which is intimately implicated in the pathogenesis of obesity and Mets. Overall, these findings indicate that the crosstalk between helminth and hosts may be a novel direction for obesity and Mets therapy. The present article reviews the molecular mechanism of how helminth masters immunity and metabolism in obesity.
Introduction
Obesity, an epidemic and systemic metabolic disease, is characterized by excessive fat accumulation and low-grade chronic inflammation. The prevalence of obesity is increasing at an alarming rate in many parts of the world since 1975, arising public awareness (1). Notably, obesity is well recognized to increase the risk of Metabolic Syndromes (Mets) including type 2 diabetes (T2DM), cardiovascular diseases (CVD), non-alcoholic fatty liver diseases (NAFLD) and other metabolic disorders (2, 3). Particularly, accumulating evidence links obesity as a crucial factor with neurodegenerative diseases such as Alzheimer’s disease (AD) (4), which still lacks effective therapy, although numerous money has been invested in this area (5). Considering the grave consequences, it is therefore imperative to seek novel strategies against obesity and its associated Mets.
Inappropriate diets have been identified as the key factor for obesity and Mets. The findings from animal experiments and population epidemiological investigations firmly support that the long-term intake of western diet (WD) is closely associated with the incidence of obesity and Mets (6–8). As an extremely active endocrine organ in the body, adipose tissue can release a large variety of adipokines that regulate diverse biological processes such as glucolipid metabolism, energy expenditure, appetite control, insulin sensitivity, and inflammation (9). In obese state, excessive fat accumulation produces multiple metabolites including palmitic acid (PA) and adipokines, which activate pro-inflammatory pathways and release inflammatory factors (10, 11). They can jointly promote the progression of obesity and Mets. Interestingly, recent studies have uncovered that gut microbiota is also implicated in the pathogenesis of obesity, diabetes and neurological disorders (12, 13). In view of the essential role of these factors mentioned above in obesity and Mets, reprogramming these key events therefore benefits for development of intervention strategies.
Parasites are categorized into protozoa (Trypanosoma, Toxoplasma, etc.) and helminths (cestodes, nematodes, trematodes). As one of the relatively successful pathogens, helminth infects approximately a quarter of the world’s population, seriously endangering public health and causing social and economic problems. However, the mortality due to helminth infection is rare, suggesting a long evolutionary co-adaptation between parasites and human. Helminth is recognized to be the strongest natural stimuli of type-2 immune responses, which can down-regulate the anti-infective immunity, thereby allowing the long-term survival of the parasites in hosts (14–16). In recent years, epidemiological evidence shows that there is an inverse correlation between the exposure to helminth and the prevalence of obesity and Mets (17–21). The Hygiene Hypothesis proposes that the fewer infections (especially helminth infection) in early childhood lead to the greater possibilities of developing allergic, inflammatory and metabolic diseases in the future, which implies the ability of helminths to master immunopathology and the potential therapeutic effects on diseases (22, 23). According to published studies, the infections of several helminths have been reported to alleviate obesity and Mets via inhibition of adipogenesis, improvement of glucose tolerance and insulin resistance (IR) (24–27). Furthermore, accumulating studies have showed that helminth derived molecules or excretory-secretory products (ESPs) can act as key modulators to exert metabolic and immune modifying functions (28). For example, ES-62, a protein secreted by filarial nematode Acanthocheilonema viteae, is reported to prevent metabolic dysfunction via promotion of the recruitment of eosinophils and M2 type macrophages in retroperitoneal adipose tissues of infected mice (29–31). Notably, not all parasitic infection is beneficial for improving metabolic disorders. Infection stage and parasite species are the key factors that determine the protective or harmful effect in the condition of obesity. It is recently reported that Trypanosoma cruzi (T. cruzi) infection induces adipogenic signaling and promotes the accumulation of lipid in the hearts during the early chronic stage in infected mice (32). Moreover, the parasite can exacerbate inflammation and aggravate obesity related metabolic disorders (e.g. atherosclerosis and NAFLD) during the acute phase of infection in obese mice, due to a high affinity for host cholesterol (33, 34). Since the relationship between protozoa and adipose tissue has been reviewed (25, 35), we herein did not have too much discussion about it. In the present article, we mainly focused on the underlying mechanism of how helminth infection or their derived molecules reprogram metabolic inflammation, fat browning, adipokine production, IR and gut microbiota in WD-induced obesity, which could provide a basis for helminth therapy against obesity and Mets.
Helminth-Induced M2 Macrophage Polarization via Metabolic Reprogramming Creates an Anti-Inflammatory Environment
Macrophages, one of dominant immune cells in adipose tissues, have the characteristics of pluripotency and plasticity that can differentiate into different phenotypes after exposure to endogenous or exogenous stimuli. In obese mice, PA, lipopolysaccharide (LPS), and tumor necrosis factor-α (TNF-α) can polarize macrophages towards “classically activated” phenotype (M1 type) that releases pro-inflammatory cytokines (such as IL-6, TNF-α, IL-1β) (10, 36, 37). In lean adipose tissues, resident macrophages are alternatively activated, exhibiting an anti-inflammatory and M2-like phenotype that involves in homeostasis maintenance of adipose tissues (38). Macrophage polarization is featured by the change of cell surface marker expression. CD80 and CD86 are universally acknowledged markers for M1 macrophages. In contrast, the level of arginase-1 (Arg-1), mannose receptor (CD206), and chemokine (C-C) motif ligand 17 (CCL17) and CCL22 are significantly increased in M2 macrophages (39). M1 macrophages sense intracellular pathogens mainly through the expression of toll-like receptors (TLRs) (40), whereas M2 macrophages sense extracellular pathogens through expression of scavenger receptors (41). In infected tissues, pro-inflammatory M1 phenotype are first polarized to protect the host against pathogens, followed by M2 polarization to form an anti-inflammatory response and promote tissue repair. M2 macrophages are reported to participate in eliminating dead adipocytes for the melioration of inflammatory milieu, and recruiting adipocyte progenitors for the regulation of their proliferation and differentiation, finally controlling fat hypertrophy as well as effectively improving obesity (42). Therefore, targeting adipose tissue inflammation and inducing M2 macrophages have emerged as potential therapeutic strategies for obesity-related metabolic disorders (43).
The helminth infection is accompanied by the setting of complex metabolic reprogramming events and the induction of macrophages M2 polarization in adipose tissues (44, 45), which provides a basis for obesity intervention. Prior observations have reported that Heligmosomoides polygyrus (H. polygyrus) infection induces polarization of M2 macrophages with upregulation of anti-inflammatory cytokines (Arg1, IL-10), to resist metabolic inflammation mediated by M1 macrophages in WD fed mice (46, 47). Moreover, WD mice had less body weight gain after administration of H. polygyrus-induced M2 macrophages (46). Similarly, Cortes-Selva et al. found that Schistosoma mansoni (S. mansoni) infection induces M2 polarization of macrophages to improve hyperlipidemia and atherosclerosis (48). Interestingly, acute T. cruzi infection potentiates adipose tissue inflammation accompanied by M1 macrophage infiltration (49), whereas chronic T. cruzi infection can cause a shift in the M2/M1 ratio towards an anti-inflammatory phenotype (35, 50). Besides live parasite infection, their ESPs or derived molecules can regulate macrophage polarization. Omega-1, one of major immunomodulatory glycoproteins in the eggs of S. mansoni, induces type 2 immune response, and improves metabolic homeostasis through independent inhibition of food intake in WD fed mice (51). Moreover, Hussaarts et al. reported that chronic S. mansoni infection and its soluble egg antigens (SEA) promote the infiltration of eosinophils and the accumulation of M2 macrophages in adipose tissues, thereby ameliorating WD-induced obesity (24).
As the component of ESPs released by helminth, extracellular vesicles (EVs), a group of heterogeneous lipid-enclosed particles derived from different cells ranging from nano to micrometer in size, have emerged as a new mediator for intercellular communication (52). Notably, EVs derived from helminths have spurred a new paradigm in studying host-helminth interaction (53). Several studies have showed that parasite derived EVs can regulate pro- or anti-inflammatory responses and induce macrophage polarization (54). For example, Echinococcus multilocularis EVs is reported to trigger production of anti-inflammatory cytokines by activation of M2 macrophages (55). Trichinella spiralis EVs can improve colitis via induction of M2 macrophages infiltration (56). Importantly, a growing body of findings highlight that adipose tissue-secreted EVs can maintain metabolic homeostasis through polarizing M2 macrophages, inhibiting adipocyte hypertrophy and promoting fat browning, which ameliorates obesity and Mets (57, 58). Thus, it is most likely that helminth EVs may be a novel direction to discover intervention strategies for obesity and Mets.
In recent years, the emerging immunometabolism has attracted considerable research interest (59, 60). The discipline shows that the changes in intracellular metabolic pathways can determine the differentiation and effector function of immune cells including macrophages (61). M1 macrophages express high levels of glycolysis and pentose phosphate pathways, whereas M2 macrophages depend on mitochondrial respiration and oxidative phosphorylation (OXPHOS) for energy supply (62). During the chronic or late stage of helminth infection, the host immune response is characterized by type 2 immune response, in which M2 macrophage is dominant. Helminth infection differentiates macrophages into M2 phenotype through increasing expression of IL-4 and maintaining the IL-4 signaling pathways (e.g. fatty acid oxidation, FAO and OXPHOS) (63–65). It is well established that LPS-induced M1 activated cells are characterized by elevated glycolysis rate, enhanced pentose phosphate pathway, and attenuated OXPHOS level (66). Interestingly, Trypanosoma. brucei metabolite indolepyruvate can inhibit this effect mentioned above, and decrease the pro-inflammatory cytokine IL-1β production in macrophages, thereby contributing to immune evasion (67). Concurrently, we previously found that mice infected with the larval Echinococcus granulosus (E. granulosus) show enhanced lipolysis in adipose tissue, which is accompanied by increased arginine metabolism (26). It is generally known that arginine metabolism has an intimate association with M2 polarization. A recent study also discovers that blockage of tricarboxylic acid cycle (TCA cycle) can reprogram metabolic flux, resulting in the accumulation of metabolites such as succinate and fumarate, which in turn act as metabolic signals to modulate macrophage function (68). Succinate has been demonstrated to hyperpolarize M2 macrophages via interacting with its receptor succinate receptor 1 (SUCNR1) (69–71). It has been reported that succinate level is elevated prior to host cells invasion by T. cruzi (72), but the association between succinate in T. cruzi infection and M2 macrophages polarization remains to be elucidated. Succinate dehydrogenase (SDH) is recently demonstrated to be a major energetic metabolic node and a crucial regulator of activation of M1 macrophages. SDH inhibition is found to cut off pro-inflammatory signal in mitochondria, thereby driving anti-inflammatory phenotype (73). For instance, Lampropoulou et al. found that itaconate, one of the most easily induced metabolites in activated macrophages, exerts anti-inflammatory effects by suppressing SDH-catalyzed oxidation of succinate (74, 75). Of note, SDH enzyme activities were significantly decreased after S. mansoni infection (76), causing succinate accumulation followed by preventing the induction of a range of pro-inflammatory factors (IL-1β) and enhancing a range of anti-inflammatory factors (IL-1RA and IL-10) (73). Thus, helminth infection may promote M2 polarization of macrophages by reprogramming metabolism (Figure 1). Exploitation of M2 macrophages induced by parasitic infection to inhibit metabolic inflammation and adipogenesis may provide a niche for the intervention of obesity and Mets.
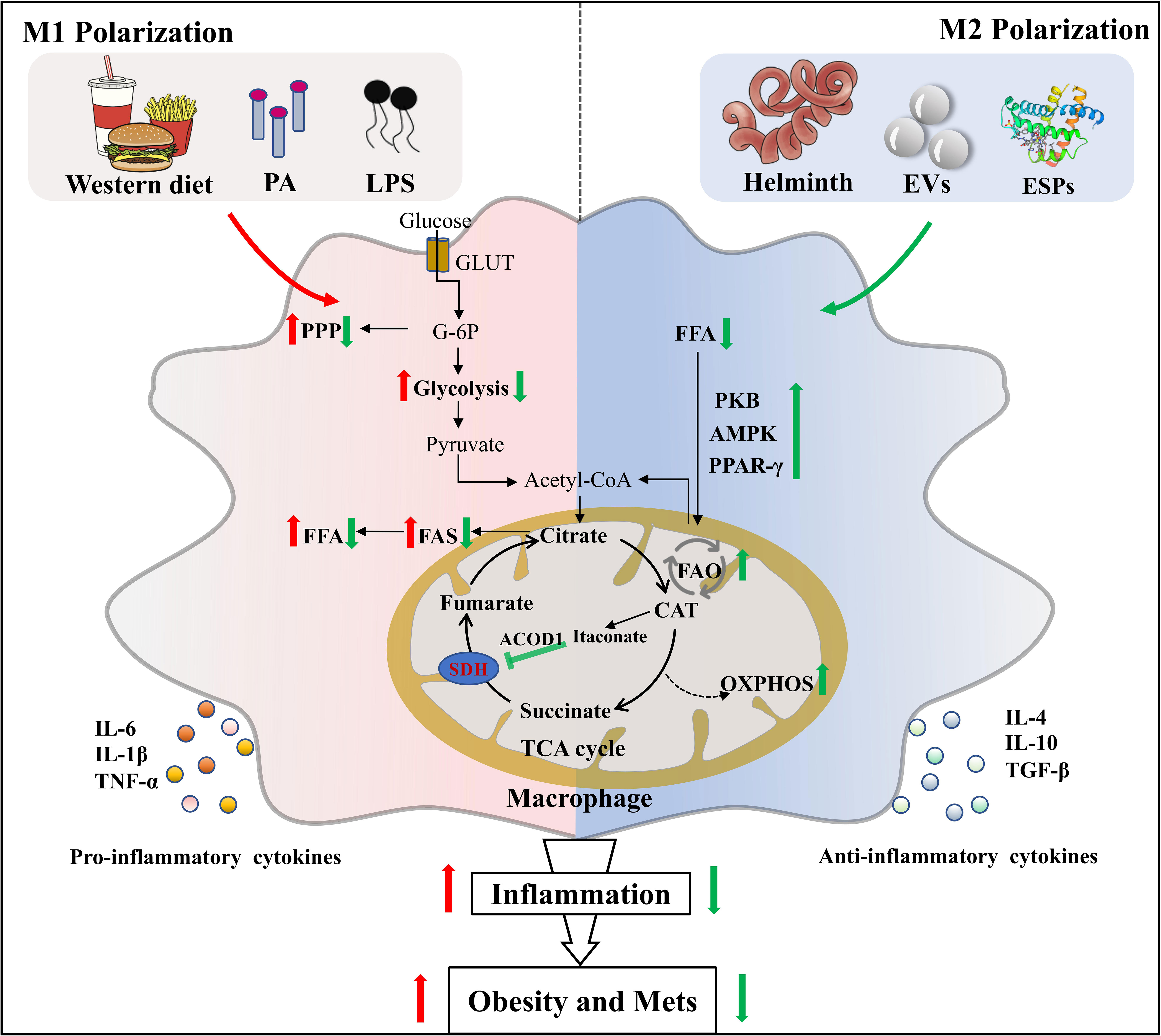
Figure 1 Helminth or its derived molecules induce M2 polarization of macrophages via metabolic reprogramming in obesity. Helminth infection or their derived molecules can induce M2 macrophages polarization along with the setting of complex metabolic reprogramming events in adipose tissue of mice fed by western diet, which provides therapeutic potential for obesity and Mets. PA, palmitic acid; LPS, lipopolysaccharide; GLUT, glucose transporter; G-6P, glucose-6-phosphate; PPP, pentose phosphate pathway; FFA, free fatty acid; FAS, fatty acid synthesis; SDH, succinate dehydrogenase; ACOD1, aconitate decarboxylase 1; CAT, cis-aconitate; EVs, extracellular vesicles; ESPs, excretory-secretory products; PKB, protein kinase B; AMPK, adenosine monophosphate (AMP)-activated protein kinase; PPAR-γ, peroxisome proliferator–activated receptor-γ; FAO, fatty acid oxidation; OXPHOS, oxidative phosphorylation.
Helminth Infection May Recruit Other Immune Cells to Regulate Homeostasis of Metabolism and Immunity
In adipose tissues, T cells, B cells and eosinophils are also important mediators for homeostasis of metabolism and immunity. Generally, T helper1 (Th1) cells, Th17 cells, CD8+ T cells and B2 cells are responsible for the obesity-induced inflammation, while regulatory T cells (Tregs), regulatory B cells (Bregs), Th2 cells, B1 cells, and eosinophils contribute to the anti-inflammatory response (77, 78). However, the balance is often broken after long term intake of WD, which is accompanied by IR (79, 80). Several studies have showed that helminth infection or ESPs can suppress the differentiation of Th1 and Th17, and promote the induction of Tregs and Bregs to resist the anti-infectious immunity (16, 81, 82). It is reported that S.mansoni infection alleviates allergy airway inflammation via induction of Treg population (83, 84). Moreover, induction of Bregs post Schistosoma japonicum (S. japonicum) regulates the systematic inflammation induced by WD (85). In addition, S. japonicum-activated Bregs can control the levels of proinflammatory chemokines and cytokines by IL-10 secretion to protect against the parasite induced liver inflammation and fibrosis (86). Finally, chronic S. mansoni infection and SEA injection promote eosinophil activation via IL-4 secretion to maintain M2 macrophages polarization in adipose tissue (24) (Figure 2). Thus, it is rational that these anti-inflammatory cell populations also contribute to the beneficial effect of helminth infection on obesity and Mets, although the underlying mechanism has not been investigated.
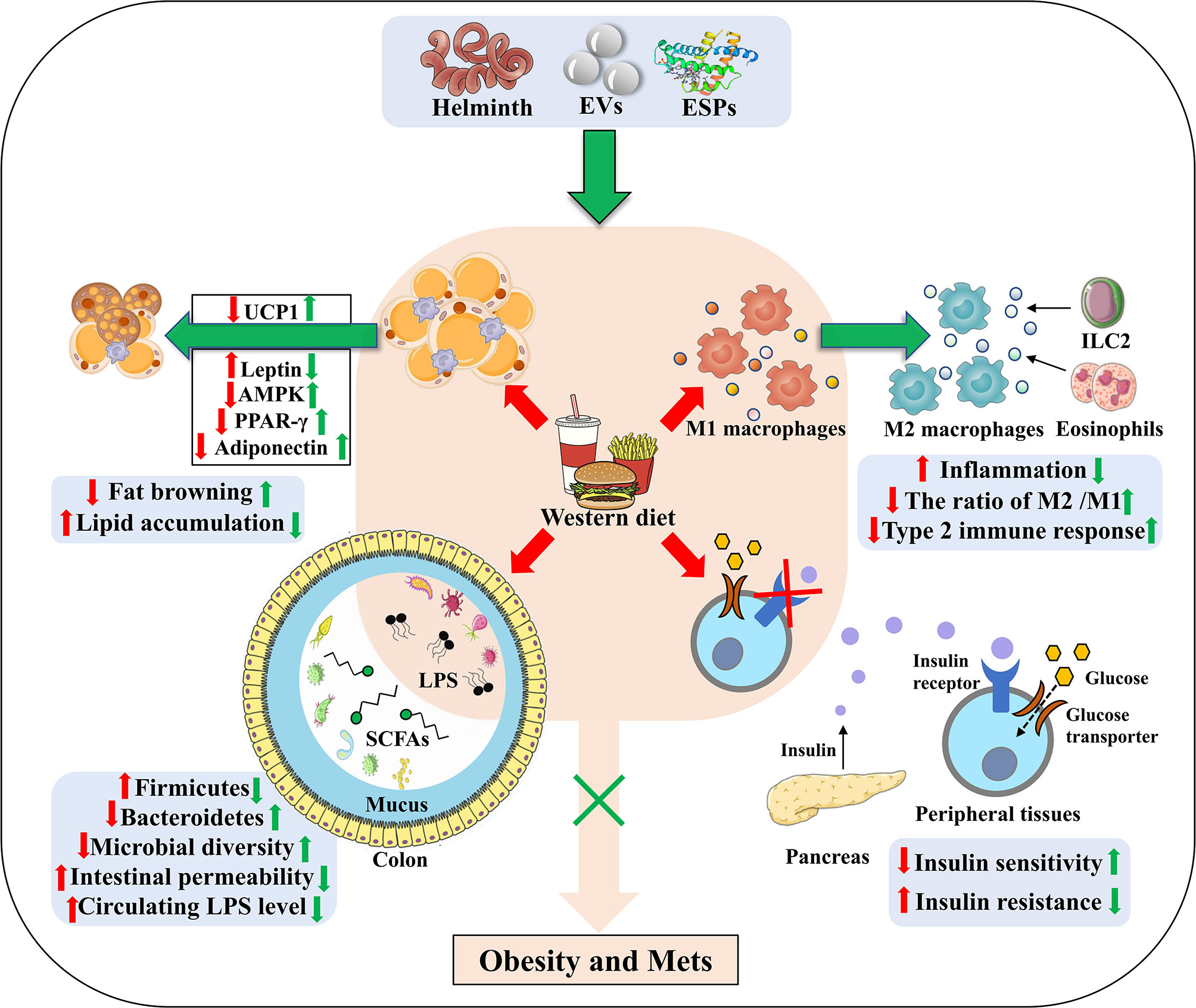
Figure 2 The underlying strategies of how helminth or its derived molecules modulate the tissue-specific homeostasis in obesity. Helminth infection or their derived molecules can ameliorate western diet-induced obesity and its associated Mets through inducing M2 macrophage polarization, down-regulating metabolic inflammation, promoting fat browning, attenuating lipid accumulation, ameliorating insulin resistance (through improving the impaired ability of glucose uptake and promoting insulin binding to its receptors) and relieving the dysbiosis of gut microbiota. EVs, extracellular vesicles; ESPs, excretory-secretory products; UCP1, uncoupling protein 1; AMPK, adenosine monophosphate (AMP)-activated protein kinase; PPAR-γ, peroxisome proliferator–activated receptor-γ; LPS, lipopolysaccharide; SCFAs, short-chain fatty acids; ILC2, group 2 innate lymphoid cells.
Helminth Infection Promotes Fat Browning via Elevation of UCP1 Expression
Adipose tissue, which can maintain a dynamic balance between energy storage in the form of lipids and energy utilization (87), has been traditionally subclassified into white adipose tissue (WAT) and brown adipose tissue (BAT) based on morphological and functional difference. WAT, the main site for the body to store lipids, stores excess energy in the form of triglycerides; whereas, BAT plays an important role in regulating energy balance and protecting against obesity by virtue of the capability for energy expenditure through thermogenesis mediated by a BAT-specific mitochondrial protein, uncoupling protein1 (UCP1) (88, 89). Recent data suggest that there are two distinct types of BAT: classical BAT derived from a myogenic factor 5 (myf-5) cellular lineage, and “brown-like” cells that reside in WAT from a non-myf-5 lineage, also called beige or brite cells (90). When mice are chronically exposed to cold environment, β3-adrenergic receptor agonists or peroxisome proliferator–activated receptor (PPAR)-γ agonists (90), the pre-existing beige adipocytes will go through phenotypic “transdifferentiation”, and “fat browning” (a switch from energy-storing white adipocytes to thermogenic brown fat-like cells) will occur (91–93). In contrast to extremely low basal expression of UCP1 within WAT, the UCP1 expression in beige adipocytes after exposure to the same stimuli is upregulated to the levels that can resemble classic brown adipocytes, which is consistent with increased BAT and a greater capacity for energy dissipation through thermogenesis. The potential contribution of BAT thermogenesis and fat browning to whole body energy expenditure, consequently, can be considered as a therapeutic target to combat obesity and its related comorbidities (94, 95).
It is reported that H. polygyrus infection elevates UCP1 expression, promoting the browning of WAT in mice, by which can increase energy expenditure and attenuate obesity in mice (46). Moreover, S. mansoni egg-derived ω1, a predominant type 2-inducing molecule, induces systemic and localized release of the type 2 initiator cytokine IL-33 that is involved in maintaining glucose homeostasis and promoting browning of WAT (96). In addition, group 2 innate lymphoid cells (ILC2s) activated by IL-33 are demonstrated to produce methionine-enkephalin peptides and catecholamines, which can directly upregulate UCP1 expression in adipocytes, thereby enhancing fat browning and improving metabolic parameters in obese mice (97–99). Helminth infection, therefore, holds the promise for increasing fat browning to ameliorate obesity (Figure 2).
Helminth Infection Alters the Levels of Adipokines and the Expression of Enzymes in Lipid Metabolism
Far from hormonally inert, adipose tissue has been, in recent years, recognized as a major endocrine organ, as it produces a wide spectrum of adipokines such as adiponectin, leptin and resistin that play a role in glucolipid homeostasis and immune regulation (100–103). Notably, leptin and resistin have pro-inflammatory effects via increasing the production of IL-6 and other pro-inflammatory factors, whilst adiponectin exerts anti-inflammatory properties due to the inhibition of TNF-α (104). Leptin, a hormone that is capable of effectively reducing food intake and body weight, was initially considered for obesity treatment. Indeed, obese mice have since been found to exhibit higher leptin mRNA levels, directly associated with the increased adiposity (101). Due to defects in the blood-brain barrier transduction pathway, obese mice often develop hyperleptinemia and central leptin resistance (105, 106). The inability of leptin to exert its anorexigenic effects in obese individuals, and therefore, the lack of clinical utility of leptin in obesity, is defined as leptin resistance (107). It has been reported that chronic T.cruzi infection can improve leptin resistance in obese mice (50, 106). Additionally, leptin and other adipocytokines can jointly induce the recruitment and activation of immune cells during WAT expansion in obese mice, create a pro-inflammatory environment and promote the release of free fatty acids, so as to exacerbate obesity-associated metabolic inflammation (50) (Figure 2). When obese mice were infected with H. polygyrus, the gene expression of leptin was markedly decreased (46), thereby reducing the production of fatty acids and fighting against obesity.
The physiological function of adipose tissue depends on fat synthesis and lipolysis, and both the progresses are strictly manipulated by local adipokines (such as PPAR-γ, adiponectin, TNF-α) (108, 109). When mice were infected with T. cruzi at an acute phase, adipose tissue displayed a significant decrease of lipid accumulation, adipocyte size and fat mass, which was correlated with increased expression of lipolytic enzymes (49). As a dominant transcription factor of adipogenesis and a master modulator of adipocyte differentiation, PPAR-γ promotes lipid storage in adipose tissue through stimulating the expression of lipogenic enzymes and inhibits the secretion of inflammatory mediators when activated by the PPAR-γ agonist (110). González FB et al. showed that experimental acute T. cruzi infection downregulated PPAR-γ expression (50, 111), coming to a state compatible with the adipose tissue atrophy and M1 macrophage polarization (112). Moreover, H. polygyrus infection may ameliorate diet-induced obesity via modulating gene expression of key transcription factors in adipogenesis, such as PPAR-γ and CCAAT enhancer-binding proteins α (C/EBPα) (46). Nevertheless, Schistosomal-derived lysophosphatidylcholine can induce M2 macrophage polarization secondary to increased PPAR-γ expression (113). It is reported that adenosine monophosphate (AMP)- activated protein kinase (AMPK), a potent cellular energy sensor for maintenance of metabolism homeostasis, can favor FAO and limit fatty acid synthesis to regulate lipid accumulation (114). Xu et al. reported that S. japonicum infection can exert a strong metabolic effect via activating the AMPK and protein kinase B (PKB, also known as AKT) signaling molecules in S. japonicum SEA-stimulated macrophages, which further promote FAO and suppress fatty acid synthesis (115). Moreover, S. japonicum infection induces upregulated expression of the FAO-related genes while downregulating the expression of the genes associated with fatty acid synthesis and lipid uptake, which is consistent with SEA-induced anti-inflammatory M2 phenotype (116, 117). It is reported that M2 macrophages shifts into FAO and OXPHOS states, directed by signaling via IL-4 (118). Therefore, parasite infection or its derived molecules may reduce fat mass and improve obesity by regulating lipid metabolism in infected hosts (Figure 2).
Helminth Infection Ameliorates Insulin Sensitivity and Insulin Resistance
In obesity, abnormal adipokine secretion and excessive lipid accumulation can cause a decrease in the expression or activity of glucose transporter 4 (GLUT4) via phosphoinositide 3-kinase (PI3K)/AKT signaling pathway. Alterations in GLUT4 translocation impair glucose uptake and insulin sensitivity, finally contributing to IR (119–122) (Figure 2). IR is characterized by glucose dysregulation with elevated serum insulin level, which increases the risk for metabolic syndromes such as T2DM, CVD and polycystic ovary syndrome (PCOS) (122–125). Recent data have showed that in obese animals, proinflammatory mediators (namely TNF-α, IL-6, and IL-1β) can damage the pancreatic β cells insulin secretion function in autocrine and paracrine manners, and down-regulate insulin sensitivity in liver and skeletal muscle, jointly inducing the occurrence of IR that is closely related to the development of T2DM (126–128).
Emerging evidence has demonstrated that parasitic infection can improve IR and glucose tolerance in obese mice (129). Eosinophils are reported to improve glucose homeostasis by inducing macrophage M2 polarization in adipose tissue of obese mice (130). In line with this, Nippostrongylus brasiliensis (N. brasiliensis) infection can increase percentages of ILC2s and accumulation of eosinophils in visceral adipose tissues to enhance insulin sensitivity (131). Simultaneously, infection of obese mice with N. brasiliensis can attenuate body weight gain, decrease adipose tissue mass, and ameliorate glucose metabolism and insulin sensitivity, accompanied by a dramatic decline of insulin levels (132). In addition, Filarial nematode Litomosoides sigmodontis (L. sigmodontis) infection and L.sigmodontis antigen (LsAg) administration is reported to increased numbers of eosinophils and M2 macrophages within adipose tissues and improve glucose tolerance in obese mice by the eosinophil-dependent mechanism (133). Notably, LsAg administration can also increase the level of adiponectin that related to insulin sensitization and inhibit the expression of proinflammatory factor interferon-γ (IFN-γ) and IL-17 to improve IR (134). Obese mice treated with LsAg injections show significant upregulation of gene expression that are linked to insulin sensitivity, such as GLUT4 and hexokinase 2 (HK2), which may further support insulin signaling and improve IR (133). Furthermore, administration of lacto-N-fucopentaose III (LNFPIII), an immunomodulatory glycan derived from S. mansoni SEA, is shown to improve insulin sensitivity and glucose tolerance in obese mice, which is mediated partly via IL-10 production in macrophages and dendritic cells (51, 135). It is well recognized that high level of IL-10 reduces the risk for Mets, particularly T2DM (136). Correspondingly, a cross-sectional study performed by Hays et al. showed a negative correlation between the infection rate of Strongyloides stercoralis and the occurrence of T2DM (137). As a consequence, helminth infection can improve IR and prevent obesity-related Mets (Figure 2).
Helminth Infection Reshapes the Composition of Gut Microbiota
The gut microbiota act as an important factor in the progression of obesity via maintenance of energy homeostasis and host immunity (138, 139). It has been reported that the decrease in the richness and diversity of gut microbiota in obese mice induced by WD is accompanied by a reduction in expression of intestinal tight junction proteins, which is linked to increased intestinal permeability, thereby resulting in a malfunctioning gut barrier (140) (Figure 2). Moreover, increased circulating LPS levels due to a “leaky gut”, can induce an inflammatory state and metabolic hyperendotoxinemia, eventually driving the development of obesity-associated IR and cognitive impairment (141–143). The association between the composition of the gut microbiota and metabolic dysfunction is becoming clear and has been extensively reported. Therefore, modulation of the gut microbiota may be a potential therapeutic way for treating obesity and Mets (144).
H. polygyrus infection can induce significant alterations in gut microbiome composition as evidenced by a marked increase in Bacteroidetes and a decrease in Firmicutes (145). Similarly, Walk ST et al. found a significant shift in the abundance and relative distribution of bacterial species in the ileum of mice post H. polygyrus infection (146). H. polygyrus-modulated microbiota exhibit levels of short-chain fatty acids (SCFAs) and upregulate expression of G protein coupled receptors (GPRs) (145). SCFAs (mainly acetate, butyrate and propionate), the key bacterial metabolites, can participate in host energy homeostasis and immune function, playing a beneficial role in preventing obesity via interacting with GPRs. It is reported that acetate can improve appetite control through the interaction with the central nervous system (147), and that butyrate and propionate can not only induce the production of gut hormones associated with the reduction of food intake, but also enhance gut epithelial barrier integrity as well as promote an anti-inflammatory milieu (148–150). A recent study also shows that SCFAs-induced protection against HFD-induced obesity is mediated by down-regulation of PPARγ, promoting a switch from lipid synthesis to lipid oxidation (151). Moreover, H. polygyrus affects the composition of the intestinal microbiota to increase norepinephrine and then enhance UCP1 expression in adipose tissues, which is responsible for limiting weight gain (152). Infection with Strongyloides venezuelensis results in modifications of the gut microbiota, most notably by increasing Lactobacillus spp. These modifications in the microbiota may alter host metabolism by switching macrophages from M1 to M2 in the adipose tissue, increasing the levels of anti-inflammatory cytokines, upregulating the expression of tight junction proteins (thereby reducing the permeability) and decreasing LPS in the sera. Furthermore, these changes correlate with improved insulin signaling and sensitivity, suggesting that modulation of the microbiota by helminth infection has a positive effect on the glucose homeostasis of hosts (153). In line with others reports, our latest work showed that the ESPs derived from the larval E. granulosus improves cognitive decline, mitigates the gut microbiota dysbiosis, and reverses gut barrier dysfunction in WD fed mice (154). Notably, ablation of gut microbiota abolishes the effect of ESPs on brain and gut. This is the first time to utilize parasite model to treat obesity induced cognitive decline via microbiota-gut-brain axis (154). It is therefore proposed that helminth or its derived molecules-induced alterations of microbiota composition, and microbiota-produced metabolites may play a vital but neglected role in the protective effects of helminth infection on obesity (Figure 2).
Summary and Prospect
As one of relatively successful pathogens, parasites have coevolved with human over millennia, developing elegant and intricate immune escape mechanisms to manipulate the equilibrium of immune and metabolism in hosts. Helminth infection or their derived molecules (mainly ESPs) can lead to a broad range of outcomes that ameliorate WD-induced obesity and metabolic disorders, including M2 polarization of macrophages (Figure 1), down-regulation of metabolic inflammation by triggering the release of type 2 cytokines (such as IL-4, IL-10, and IL-13), the increase of thermogenesis and energy consumption by promoting fat browning, modification of adipokines and lipid metabolism, improvement of insulin sensitivity and glucose tolerance, and modulation of gut microbiota (Figure 2).
Fully understanding and harnessing characteristics of helminth-driven immunomodulation may become an important therapeutic insight for human diseases including obesity and Mets (155). In 2013, the safety and tolerability of Trichuris suis ova (TSO) have been confirmed in randomised clinical trials (156), and the United States Food and Drug Administration has approved TSO to treat patients with inflammatory bowel diseases (IBD). A recent randomized, double-blind, placebo-controlled Phase 1b clinical trial conducted by Pierce et al. (157), showed that it is safe and well-tolerated to inject a certain dose of larvae III stage of Necator americanus into patients with central obesity and metabolic syndrome, identifying hookworm infection as a potentially alternative therapy for obesity. Overall, these clinical trials lay a foundation for further exploiting helminth-inspired therapies against obesity and Mets.
However, helminth therapy also presents a challenge to the drug development community. This results from the fact that live parasitic administration has the possibility to increase the infective risk and bring biosafety issues. Alternatively, animal models have demonstrated that the ESPs released by parasites can evoke type 2 immune responses, alleviate adipose tissue inflammation and enhance glucose homeostasis, thereby reducing the body weight of obese animals (155). The use of ESPs instead of active helminth infection potentially addresses some of the drawbacks and obstacles currently faced by experimental helminth therapy. However, type 2 immunity induced by parasite derived molecules may also have adverse effects. For example, a higher occurrence rate of asthma is observed in obese individuals (158, 159), while the tropomyosin of Ascaris lumbricoides is reported to have strong allergenic activity (160). Administration of such molecules is speculated to increase the incidence rate of asthma in obesity. What’s more, it is well known that parasite infection is closely associated with accumulation of eosinophils (24, 131), and eosinophil recruitment represents one of the pathogenesis for asthma (161). Thus, to frame the possibility that helminth derived molecules could be developed as drugs, more animals and clinical trials should be tested.
Furthermore, a more elaborated description of the definitive immunomodulatory components of helminth could facilitate a more precise therapeutic approach against obesity and its associated Mets. The molecular diversity of helminth products with therapeutic potential is noticeable. As such, identifying specific molecules, targeted receptors, and downstream signaling pathways that work in therapy or prevention of obesity and its related Mets, constitutes important future directions. Even if the molecules involved in their immunometabolic effects are identified, immune suppression to human should be avoided, in order to prevent other complications. Of note, recombinant expression platform is overwhelmingly vital for the production of developing helminth biologics. Moreover, if helminthic therapy comes true as innovative therapeutic avenues for obesity and its associated disorders, the drug dose, the frequency and route of administration and duration of treatment will be likely to be different in a therapeutic setting (155). More experiments especially clinical trials are needed to determine these parameters. Currently, on account of limited available information of helminth therapy, many problems are not clear such as whether other immune cells or cell subsets are involved in immunomodulation, as well as how helminth-induced type 2 immunity affects metabolic organs other than adipose tissue, including liver, skeletal muscle, gut and pancreas (162). Thus, there is a long way to clarify the mechanism and find more effective strategies against obesity and its associated diseases.
In addition, the emerging immunometabolism, provides a novel insight on addressing these scientific puzzles mentioned above. Accumulating evidence has indicated that helminth or its derived molecules can reprogram the metabolic events in macrophages of host, thereby modulating the anti-infective immunity and providing a moderate living environment for the growth and development of helminth (163). Nevertheless, only sporadic mechanistic investigations in the view of immunometabolism have been reported (164, 165). Several metabolites may be implicated in the inner workings of helminth infections and immunity. For example, SUCNR1 has recently been reported to increase the expression of the anti-inflammatory markers related to M2 macrophages in WD induced obesity (70). The enzyme aconitate decarboxylase 1 (ACOD1, originally named by immune responsive gene 1, IRG-1), is responsible for itaconate production through the decarboxylation of cis-aconitate in the TCA cycle. The latest studies have uncovered that ACOD1/itaconate axis links metabolism to immunity in macrophages (166), and has gained lots of interests in immunometabolism field and inflammatory disease models (167, 168). Of note, our lncRNA microarray analysis showed that larval E. granulosus infection can upregulate the expression of SUCNR1 and ACOD1 in adipose tissues of mice (26), which is paralleled with the M2 macrophage polarization post infection. Interestingly, a recent study reported that injection of soluble egg derived from S. japonicum exhibited inhibitory effects on the expression of lipogenesis-related genes in mice fed with WD, thereby contributing to the treatment for obesity-related fatty liver disease (169). These studies emphasize the intimate crosstalk between immune and metabolism of host and helminth or derived molecules. It is believed that immunometabolism can improve our understanding of the inherent mechanisms of host-helminth/derived molecules interplay, promising to contribute to a stream of innovative therapeutic avenues for obesity and its related Mets.
Author Contributions
WP conceived the manuscript. MD, XY, YY, and WP wrote the manuscript. All authors have seen and approved the submitted version of this manuscript.
Funding
Project support was provided in part by the National Natural Science Foundation of China (Nos. 81871670, 81800718, 82071184), Natural Science Foundation of Jiangsu Province (No. BK20201459), the Training Programs of Innovation and Entrepreneurship for College Students in Jiangsu Province (Nos. 202010313035Z, 202010313009), Jiangsu Planned Projects for Postdoctoral Research Funds (No. 2019K063) and Jiangsu Qinglan Project. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Collaboration N R F. Trends in Adult Body-Mass Index in 200 Countries From 1975 to 2014: A Pooled Analysis of 1698 Population-Based Measurement Studies With 19·2 Million Participants. Lancet (Lond Engl) (2016) 387(10026):1377–96. doi: 10.1016/s0140-6736(16)30054-x
2. Bray GA, Kim KK, Wilding JPH. Obesity: A Chronic Relapsing Progressive Disease Process. A Position Statement of the World Obesity Federation. Obes Rev (2017) 18(7):715–23. doi: 10.1111/obr.12551
3. Lumeng CN, Saltiel AR. Inflammatory Links Between Obesity and Metabolic Disease. J Clin Invest (2011) 121(6):2111–7. doi: 10.1172/jci57132
4. Miller AA, Spencer SJ. Obesity and Neuroinflammation: A Pathway to Cognitive Impairment. Brain Behav Immun (2014) 42:10–21. doi: 10.1016/j.bbi.2014.04.001
5. Cummings JL, Morstorf T, Zhong K. Alzheimer's Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimers Res Ther (2014) 6(4):37. doi: 10.1186/alzrt269
6. Grooms KN, Ommerborn MJ, Pham DQ, Djoussé L, Clark CR. Dietary Fiber Intake and Cardiometabolic Risks Among US Adults, NHANES 1999-2010. Am J Med (2013) 126(12):1059–67.e1-4. doi: 10.1016/j.amjmed.2013.07.023
7. Sekgala MD, Mchiza ZJ, Parker WA, Monyeki KD. Dietary Fiber Intake and Metabolic Syndrome Risk Factors Among Young South African Adults. Nutrients (2018) 10(4):504. doi: 10.3390/nu10040504
8. Bortolin RC, Vargas AR, Gasparotto J. A New Animal Diet Based on Human Western Diet Is a Robust Diet-Induced Obesity Model: Comparison to High-Fat and Cafeteria Diets in Term of Metabolic and Gut Microbiota Disruption. Int J Obes (Lond) (2018) 42(3):525–34. doi: 10.1038/ijo.2017.225
9. Scheja L, Heeren J. The Endocrine Function of Adipose Tissues in Health and Cardiometabolic Disease. Nat Rev Endocrinol (2019) 15(9):507–24. doi: 10.1038/s41574-019-0230-6
10. Engin AB, Engin A, Gonul Ii. The Effect of Adipocyte-Macrophage Crosstalk in Obesity-Related Breast Cancer. J Mol Endocrinol (2019) 62(3):R201–22. doi: 10.1530/jme-18-0252
11. Zinöcker MK, Lindseth IA. The Western Diet-Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients (2018) 10(3):365. doi: 10.3390/nu10030365
12. Díez-Sainz E, Milagro FI, Riezu-Boj JI, Lorente-Cebrián S. Effects of Gut Microbiota-Derived Extracellular Vesicles on Obesity and Diabetes and Their Potential Modulation Through Diet. J Physiol Biochem (2021), 1–15. doi: 10.1007/s13105-021-00837-6
13. Shi H, Yu Y, Lin D, Zheng P, Zhang P, Hu M, et al. β-Glucan Attenuates Cognitive Impairment via the Gut-Brain Axis in Diet-Induced Obese Mice. Microbiome (2020) 8(1):143. doi: 10.1186/s40168-020-00920-y
14. Shea-Donohue T, Sun R, Bohl JA, Mclean LP, Zhao A. Enteric Nematodes and the Path to Up-Regulation of Type 2 Cytokines IL-4 and IL-13. Cytokine (2015) 75(1):62–7. doi: 10.1016/j.cyto.2015.06.007
15. Wammes LJ, Mpairwe H, Elliott AM, Yazdanbakhsh M. Helminth Therapy or Elimination: Epidemiological, Immunological, and Clinical Considerations. Lancet Infect Dis (2014) 14(11):1150–62. doi: 10.1016/s1473-3099(14)70771-6
16. Maizels RM, Mcsorley HJ. Regulation of the Host Immune System by Helminth Parasites. J Allergy Clin Immunol (2016) 138(3):666–75. doi: 10.1016/j.jaci.2016.07.007
17. Chen Y, Lu J, Huang Y, Wang T, Xu Y, Xu M, et al. Association of Previous Schistosome Infection With Diabetes and Metabolic Syndrome: A Cross-Sectional Study in Rural China. J Clin Endocrinol Metab (2013) 98(2):E283–7. doi: 10.1210/jc.2012-2517
18. Wolde M, Berhe N, Medhin G, Chala F, Van Die I, Tsegaye A. Inverse Associations of Schistosoma Mansoni Infection and Metabolic Syndromes in Humans: A Cross-Sectional Study in Northeast Ethiopia. Microbiol Insights (2019) 12:1178636119849934. doi: 10.1177/1178636119849934
19. Wiria AE, Wammes LJ, Hamid F, Dekkers OM, Prasetyani MA, May L, et al. Relationship Between Carotid Intima Media Thickness and Helminth Infections on Flores Island, Indonesia. PloS One (2013) 8(1):e54855. doi: 10.1371/journal.pone.0054855
20. Shen SW, Lu Y, Li F, Shen ZH, Xu M, Yao WF, et al. The Potential Long-Term Effect of Previous Schistosome Infection Reduces the Risk of Metabolic Syndrome Among Chinese Men. Parasit Immunol (2015) 37(7):333–9. doi: 10.1111/pim.12187
21. Tracey EF, Mcdermott RA, Mcdonald MI. Do Worms Protect Against the Metabolic Syndrome? A Systematic Review and Meta-Analysis. Diabetes Res Clin Pract (2016) 120:209–20. doi: 10.1016/j.diabres.2016.08.014
22. Maizels RM, Mcsorley HJ, Smyth DJ. Helminths in the Hygiene Hypothesis: Sooner or Later? Clin Exp Immunol (2014) 177(1):38–46. doi: 10.1111/cei.12353
23. Yazdanbakhsh M, Kremsner PG, Van Ree R. Allergy, Parasites, and the Hygiene Hypothesis. Science (2002) 296(5567):490–4. doi: 10.1126/science.296.5567.490
24. Hussaarts L, García-Tardón N, Van Beek L, Heemskerk MM, Haeberlein S, van der Zon GC, et al. Chronic Helminth Infection and Helminth-Derived Egg Antigens Promote Adipose Tissue M2 Macrophages and Improve Insulin Sensitivity in Obese Mice. FASEB J (2015) 29(7):3027–39. doi: 10.1096/fj.14-266239
25. Attar N. Parasite Physiology: Trypanosomes Chew the Fat. Nat Rev Microbiol (2016) 14(7):402–3. doi: 10.1038/nrmicro.2016.86
26. Lu Y, Liu H, Yang XY, Liu JX, Dai MY, Wu JC, et al. Microarray Analysis of lncRNA and mRNA Reveals Enhanced Lipolysis Along With Metabolic Remodeling in Mice Infected With Larval Echinococcus Granulosus. Front Physiol (2020) 11:1078. doi: 10.3389/fphys.2020.01078
27. Yang X, Ding W, Qian X, Jiang P, Chen Q, Zhang X, et al. Schistosoma Japonicum Infection Leads to the Reprogramming of Glucose and Lipid Metabolism in the Colon of Mice. Front Vet Sci (2021) 8:645807. doi: 10.3389/fvets.2021.645807
28. Harnett W. Secretory Products of Helminth Parasites as Immunomodulators. Mol Biochem Parasitol (2014) 195(2):130–6. doi: 10.1016/j.molbiopara.2014.03.007
29. Crowe J, Lumb FE. The Parasitic Worm Product ES-62 Promotes Health- and Life-Span in a High Calorie Diet-Accelerated Mouse Model of Ageing. PloS Pathog (2020) 16(3):e1008391. doi: 10.1371/journal.ppat.1008391
30. Al-Riyami L, Harnett W. Immunomodulatory Properties of ES-62, a Phosphorylcholine-Containing Glycoprotein Secreted by Acanthocheilonema Viteae. Endocr Metab Immune Disord Drug Targets (2012) 12(1):45–52. doi: 10.2174/187153012799278893
31. Pineda MA, Lumb F, Harnett MM, Harnett W. ES-62, a Therapeutic Anti-Inflammatory Agent Evolved by the Filarial Nematode Acanthocheilonema Viteae. Mol Biochem Parasitol (2014) 194(1-2):1–8. doi: 10.1016/j.molbiopara.2014.03.003
32. Lizardo K, Ayyappan JP, Oswal N, Weiss LM. Fat Tissue Regulates the Pathogenesis and Severity of Cardiomyopathy in Murine Chagas Disease. PloS Negl Trop Dis (2021) 15(4):e0008964. doi: 10.1371/journal.pntd.0008964
33. Lucchetti BFC, Boaretto N, Lopes FNC, Malvezi AD, Lovo-Martins MI, Tatakihara VLH, et al. Metabolic Syndrome Agravates Cardiovascular, Oxidative and Inflammatory Dysfunction During the Acute Phase of Trypanosoma Cruzi Infection in Mice. Sci Rep (2019) 9(1):18885. doi: 10.1038/s41598-019-55363-9
34. Onofrio LI, Arocena AR, Paroli AF, Cabalén ME, Andrada MC, Cano RC, et al. Trypanosoma Cruzi Infection Is a Potent Risk Factor for Non-Alcoholic Steatohepatitis Enhancing Local and Systemic Inflammation Associated With Strong Oxidative Stress and Metabolic Disorders. PloS Negl Trop Dis (2015) 9(2):e0003464. doi: 10.1371/journal.pntd.0003464
35. Tanowitz HB, Scherer PE, Mota MM, Figueiredo LM. Adipose Tissue: A Safe Haven for Parasites? Trends Parasitol (2017) 33(4):276–84. doi: 10.1016/j.pt.2016.11.008
36. Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, et al. Local Proliferation of Macrophages Contributes to Obesity-Associated Adipose Tissue Inflammation. Cell Metab (2014) 19(1):162–71. doi: 10.1016/j.cmet.2013.11.017
37. Hersoug LG, Møller P, Loft S. Role of Microbiota-Derived Lipopolysaccharide in Adipose Tissue Inflammation, Adipocyte Size and Pyroptosis During Obesity. Nutr Res Rev (2018) 31(2):153–63. doi: 10.1017/s0954422417000269
38. Hill AA, Reid Bolus W, Hasty AH. A Decade of Progress in Adipose Tissue Macrophage Biology. Immunol Rev (2014) 262(1):134–52. doi: 10.1111/imr.12216
39. Van Dyken SJ, Locksley RM. Interleukin-4- and Interleukin-13-Mediated Alternatively Activated Macrophages: Roles in Homeostasis and Disease. Annu Rev Immunol (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
40. Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
41. Rőszer T. Understanding the Mysterious M2 Macrophage Through Activation Markers and Effector Mechanisms. Mediators Inflammation (2015) 2015:816460. doi: 10.1155/2015/816460
42. Nawaz A, Tobe K. M2-Like Macrophages Serve as a Niche for Adipocyte Progenitors in Adipose Tissue. J Diabetes Invest (2019) 10(6):1394–400. doi: 10.1111/jdi.13114
43. Peterson KR, Cottam MA, Kennedy AJ, Hasty AH. Macrophage-Targeted Therapeutics for Metabolic Disease. Trends Pharmacol Sci (2018) 39(6):536–46. doi: 10.1016/j.tips.2018.03.001
44. Coakley G, Harris NL. Interactions Between Macrophages and Helminths. Parasit Immunol (2020) 42(7):e12717. doi: 10.1111/pim.12717
45. Nair MG, Herbert DR. Immune Polarization by Hookworms: Taking Cues From T Helper Type 2, Type 2 Innate Lymphoid Cells and Alternatively Activated Macrophages. Immunology (2016) 148(2):115–24. doi: 10.1111/imm.12601
46. Su CW, Chen CY, Li Y, Long SR, Massey W, Kumar DV, et al. Helminth Infection Protects Against High Fat Diet-Induced Obesity via Induction of Alternatively Activated Macrophages. Sci Rep (2018) 8(1):4607. doi: 10.1038/s41598-018-22920-7
47. Reynolds LA, Filbey KJ, Maizels RM. Immunity to the Model Intestinal Helminth Parasite Heligmosomoides Polygyrus. Semin Immunopathol (2012) 34(6):829–46. doi: 10.1007/s00281-012-0347-3
48. Cortes-Selva D, Elvington AF, Ready A, Rajwa B, Pearce EJ, Randolph GJ, et al. Schistosoma Mansoni Infection-Induced Transcriptional Changes in Hepatic Macrophage Metabolism Correlate With an Athero-Protective Phenotype. Front Immunol (2018) 9:2580. doi: 10.3389/fimmu.2018.02580
49. Nagajyothi F, Desruisseaux MS, Machado FS, Upadhya R, Zhao D, Schwartz GJ, et al. Response of Adipose Tissue to Early Infection With Trypanosoma Cruzi (Brazil Strain). J Infect Dis (2012) 205(5):830–40. doi: 10.1093/infdis/jir840
50. Cabalén ME, Cabral MF, Sanmarco LM, Andrada MC, Onofrio LI, Ponce NE, et al. Chronic Trypanosoma Cruzi Infection Potentiates Adipose Tissue Macrophage Polarization Toward an Anti-Inflammatory M2 Phenotype and Contributes to Diabetes Progression in a Dietinduced Obesity Model. Oncotarget (2016) 7(12):13400–15. doi: 10.18632/oncotarget.7630
51. Van Der Zande HJP, Gonzalez MA, De Ruiter K, Wilbers RHP, García-Tardón N, Van Huizen M, et al. The Helminth Glycoprotein Omega-1 Improves Metabolic Homeostasis in Obese Mice Through Type 2 Immunity-Independent Inhibition of Food Intake. FASEB J (2021) 35(2):e21331. doi: 10.1096/fj.202001973R
52. Szatanek R, Baj-Krzyworzeka M, Zimoch J, Lekka M, Siedlar M, Baran J. The Methods of Choice for Extracellular Vesicles (EVs) Characterization. Int J Mol Sci (2017) 18(6):1153. doi: 10.3390/ijms18061153
53. Eichenberger RM, Sotillo J, Loukas A. Immunobiology of Parasitic Worm Extracellular Vesicles. Immunol Cell Biol (2018). doi: 10.1111/imcb.12171
54. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage Polarization and Function With Emphasis on the Evolving Roles of Coordinated Regulation of Cellular Signaling Pathways. Cell Signal (2014) 26(2):192–7. doi: 10.1016/j.cellsig.2013.11.004
55. Zheng Y, Guo X, Su M, Guo A, Ding J, Yang J, et al. Regulatory Effects of Echinococcus Multilocularis Extracellular Vesicles on RAW264.7 Macrophages. Vet Parasitol (2017) 235:29–36. doi: 10.1016/j.vetpar.2017.01.012
56. Gao X, Yang Y, Liu X, Wang Y, Yang Y, Boireau P, et al. Extracellular Vesicles Derived From Trichinella Spiralis Prevent Colitis by Inhibiting M1 Macrophage Polarization. Acta Trop (2021) 213:105761. doi: 10.1016/j.actatropica.2020.105761
57. Zhao H, Shang Q, Pan Z, Bai Y, Li Z, Zhang H, et al. Exosomes From Adipose-Derived Stem Cells Attenuate Adipose Inflammation and Obesity Through Polarizing M2 Macrophages and Beiging in White Adipose Tissue. Diabetes (2018) 67(2):235–47. doi: 10.2337/db17-0356
58. Li CJ, Fang QH, Liu ML, Lin JN. Current Understanding of the Role of Adipose-Derived Extracellular Vesicles in Metabolic Homeostasis and Diseases: Communication From the Distance Between Cells/Tissues. Theranostics (2020) 10(16):7422–35. doi: 10.7150/thno.42167
59. Van Den Bossche J, O'neill LA, Menon D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol (2017) 38(6):395–406. doi: 10.1016/j.it.2017.03.001
60. Tabas I, Bornfeldt KE. Intracellular and Intercellular Aspects of Macrophage Immunometabolism in Atherosclerosis. Circ Res (2020) 126(9):1209–27. doi: 10.1161/circresaha.119.315939
61. Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W, et al. Metabolic Reprogramming in Macrophage Responses. Biomark Res (2021) 9(1):1. doi: 10.1186/s40364-020-00251-y
62. O'neill LA, Kishton RJ, Rathmell J. A Guide to Immunometabolism for Immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
63. Odegaard JI, Chawla A. Alternative Macrophage Activation and Metabolism. Annu Rev Pathol (2011) 6:275–97. doi: 10.1146/annurev-pathol-011110-130138
64. O'neill LA, Pearce EJ. Immunometabolism Governs Dendritic Cell and Macrophage Function. J Exp Med (2016) 213(1):15–23. doi: 10.1084/jem.20151570
65. Pearce EL, Pearce EJ. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity (2013) 38(4):633–43. doi: 10.1016/j.immuni.2013.04.005
66. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, Mcgettrick AF, Goel G, et al. Succinate Is an Inflammatory Signal That Induces IL-1β Through HIF-1α. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
67. Mcgettrick AF, Corcoran SE, Barry PJ, Mcfarland J, Crès C, Curtis AM, et al. Trypanosoma Brucei Metabolite Indolepyruvate Decreases HIF-1α and Glycolysis in Macrophages as a Mechanism of Innate Immune Evasion. Proc Natl Acad Sci USA (2016) 113(48):E7778–e87. doi: 10.1073/pnas.1608221113
68. Ryan DG, O'neill L. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu Rev Immunol (2020) 38:289–313. doi: 10.1146/annurev-immunol-081619-104850
69. Trauelsen M, Hiron TK, Lin D, Petersen JE, Breton B, Husted AS, et al. Extracellular Succinate Hyperpolarizes M2 Macrophages Through SUCNR1/GPR91-Mediated Gq Signaling. Cell Rep (2021) 35(11):109246. doi: 10.1016/j.celrep.2021.109246
70. Keiran N, Ceperuelo-Mallafré V, Calvo E, Hernández-Alvarez MI, Ejarque M, Núñez-Roa C, et al. SUCNR1 Controls an Anti-Inflammatory Program in Macrophages to Regulate the Metabolic Response to Obesity. Nat Immunol (2019) 20(5):581–92. doi: 10.1038/s41590-019-0372-7
71. Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa ASH, Kwok CK, Leonardi T, et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell (2018) 22(3):355–68.e13. doi: 10.1016/j.stem.2018.01.020
72. Mattos EC, Canuto G. Reprogramming of Trypanosoma Cruzi Metabolism Triggered by Parasite Interaction With the Host Cell Extracellular Matrix. PloS Negl Trop Dis (2019) 13(2):e0007103. doi: 10.1371/journal.pntd.0007103
73. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell (2016) 167(2):457–70.e13. doi: 10.1016/j.cell.2016.08.064
74. Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, et al. Itaconic Acid Is a Mammalian Metabolite Induced During Macrophage Activation. J Am Chem Soc (2011) 133(41):16386–9. doi: 10.1021/ja2070889
75. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase With Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24(1):158–66. doi: 10.1016/j.cmet.2016.06.004
76. Hamed MA, Ali SA, Aly HF, El-Rigal NS, Rizk MZ. Biomphalaria Alexandrina Snails as Immunogens Against Schistosoma Mansoni Infection in Mice. Mem Inst Oswaldo Cruz (2010) 105(7):879–88. doi: 10.1590/s0074-02762010000700007
77. Lee BC, Lee J. Cellular and Molecular Players in Adipose Tissue Inflammation in the Development of Obesity-Induced Insulin Resistance. Biochim Biophys Acta (2014) 1842(3):446–62. doi: 10.1016/j.bbadis.2013.05.017
78. Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, et al. Adipose Tissue B2 Cells Promote Insulin Resistance Through Leukotriene LTB4/LTB4R1 Signaling. J Clin Invest (2017) 127(3):1019–30. doi: 10.1172/jci90350
79. Liu L, Hu J, Wang Y, Lei H, Xu D. The Role and Research Progress of the Balance and Interaction Between Regulatory T Cells and Other Immune Cells in Obesity With Insulin Resistance. Adipocyte (2021) 10(1):66–79. doi: 10.1080/21623945.2021.1876375
80. García-Hernández MH, Rodríguez-Varela E, García-Jacobo RE, Hernández-De La Torre M, Uresti-Rivera EE, González-Amaro R, et al. Frequency of Regulatory B Cells in Adipose Tissue and Peripheral Blood From Individuals With Overweight, Obesity and Normal-Weight. Obes Res Clin Pract (2018) 12(6):513–9. doi: 10.1016/j.orcp.2018.07.001
81. Pan W, Zhou HJ, Shen YJ, Wang Y, Xu YX, Hu Y, et al. Surveillance on the Status of Immune Cells After Echinnococcus Granulosus Protoscoleces Infection in Balb/c Mice. PloS One (2013) 8(3):e59746. doi: 10.1371/journal.pone.0059746
82. Pan W, Hao WT, Shen YJ, Li XY, Wang YJ, Sun FF, et al. The Excretory-Secretory Products of Echinococcus Granulosus Protoscoleces Directly Regulate the Differentiation of B10, B17 and Th17 Cells. Parasit Vectors (2017) 10(1):348. doi: 10.1186/s13071-017-2263-9
83. Layland LE, Straubinger K, Ritter M, Loffredo-Verde E, Garn H, Sparwasser T, et al. Schistosoma Mansoni-Mediated Suppression of Allergic Airway Inflammation Requires Patency and Foxp3+ Treg Cells. PloS Negl Trop Dis (2013) 7(8):e2379. doi: 10.1371/journal.pntd.0002379
84. White MPJ, Mcmanus CM, Maizels RM. Regulatory T-Cells in Helminth Infection: Induction, Function and Therapeutic Potential. Immunology (2020) 160(3):248–60. doi: 10.1111/imm.13190
85. Li M, Wang H, Ni Y, Li C, Xu X, Chang H, et al. Helminth-Induced CD9(+) B-Cell Subset Alleviates Obesity-Associated Inflammation via IL-10 Production. Int J Parasitol (2021) 52(2–3):111–23. doi: 10.1016/j.ijpara.2021.08.009
86. Yong L, Tang Y, Ren C, Liu M, Shen J, Hou X. B1 Cells Protect Against Schistosoma Japonicum-Induced Liver Inflammation and Fibrosis by Controlling Monocyte Infiltration. PloS Negl Trop Dis (2019) 13(6):e0007474. doi: 10.1371/journal.pntd.0007474
87. Rosen ED, Spiegelman BM. Adipocytes as Regulators of Energy Balance and Glucose Homeostasis. Nature (2006) 444(7121):847–53. doi: 10.1038/nature05483
88. Mcqueen AE, Koliwad SK, Wang JC. Fighting Obesity by Targeting Factors Regulating Beige Adipocytes. Curr Opin Clin Nutr Metab Care (2018) 21(6):437–43. doi: 10.1097/mco.0000000000000509
89. Inagaki T, Sakai J, Kajimura S. Transcriptional and Epigenetic Control of Brown and Beige Adipose Cell Fate and Function. Nat Rev Mol Cell Biol (2016) 17(8):480–95. doi: 10.1038/nrm.2016.62
90. Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell (2012) 150(2):366–76. doi: 10.1016/j.cell.2012.05.016
91. Jung SM, Sanchez-Gurmaches J, Guertin DA. Brown Adipose Tissue Development and Metabolism. Handb Exp Pharmacol (2019) 251:3–36. doi: 10.1007/164_2018_168
92. Carobbio S, Guénantin AC, Samuelson I, Bahri M, Vidal-Puig A. Brown and Beige Fat: From Molecules to Physiology and Pathophysiology. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(1):37–50. doi: 10.1016/j.bbalip.2018.05.013
93. Herz CT, Kiefer FW. Adipose Tissue Browning in Mice and Humans. J Endocrinol (2019) 241(3):R97–R109. doi: 10.1530/joe-18-0598
94. Cui Z, Liu Y, Wan W, Xu Y, Hu Y, Ding M, et al. Ethacrynic Acid Targets GSTM1 to Ameliorate Obesity by Promoting Browning of White Adipocytes. Protein Cell (2021) 12(6):493–501. doi: 10.1007/s13238-020-00717-7
95. White JD, Dewal RS, Stanford KI. The Beneficial Effects of Brown Adipose Tissue Transplantation. Mol Aspects Med (2019) 68:74–81. doi: 10.1016/j.mam.2019.06.004
96. Hams E, Bermingham R, Wurlod FA, Hogan AE, O'shea D, Preston RJ, et al. The Helminth T2 RNase ω1 Promotes Metabolic Homeostasis in an IL-33- and Group 2 Innate Lymphoid Cell-Dependent Mechanism. FASEB J (2016) 30(2):824–35. doi: 10.1096/fj.15-277822
97. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 Innate Lymphoid Cells Promote Beiging of White Adipose Tissue and Limit Obesity. Nature (2015) 519(7542):242–6. doi: 10.1038/nature14115
98. Obi S, Shimokawa C. IL-33 Is Essential to Prevent High-Fat Diet-Induced Obesity in Mice Infected With an Intestinal Helminth. Parasit Immunol (2020) 42(9):e12700. doi: 10.1111/pim.12700
99. Igarashi Y, Nawaz A, Kado T, Bilal M, Kuwano T, Yamamoto S, et al. Partial Depletion of CD206-Positive M2-Like Macrophages Induces Proliferation of Beige Progenitors and Enhances Browning After Cold Stimulation. Sci Rep (2018) 8(1):14567. doi: 10.1038/s41598-018-32803-6
100. Tanowitz HB, Jelicks LA, Machado FS, Esper L, Qi X, Desruisseaux MS, et al. Adipose Tissue, Diabetes and Chagas Disease. Adv Parasitol (2011) 76:235–50. doi: 10.1016/b978-0-12-385895-5.00010-4
101. Smitka K, Marešová D. Adipose Tissue as an Endocrine Organ: An Update on Pro-Inflammatory and Anti-Inflammatory Microenvironment. Prague Med Rep (2015) 116(2):87–111. doi: 10.14712/23362936.2015.49
102. Booth A, Magnuson A, Fouts J, Foster MT. Adipose Tissue: An Endocrine Organ Playing a Role in Metabolic Regulation. Horm Mol Biol Clin Investig (2016) 26(1):25–42. doi: 10.1515/hmbci-2015-0073
103. Song Z, Xiaoli AM, Yang F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients (2018) 10(10):1383. doi: 10.3390/nu10101383
104. Wong Y, Nakamizo S, Tan KJ, Kabashima K. An Update on the Role of Adipose Tissues in Psoriasis. Front Immunol (2019) 10:1507. doi: 10.3389/fimmu.2019.01507
105. Izquierdo AG, Crujeiras AB. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients (2019) 11(11):2704. doi: 10.3390/nu11112704
106. Monteiro L, Pereira J, Palhinha L, Moraes-Vieira PMM. Leptin in the Regulation of the Immunometabolism of Adipose Tissue-Macrophages. J Leukoc Biol (2019) 106(3):703–16. doi: 10.1002/jlb.mr1218-478r
107. De Git KCG, Peterse C, Beerens S, Luijendijk MCM, van der Plasse G, La Fleur SE. Is Leptin Resistance the Cause or the Consequence of Diet-Induced Obesity? Int J Obes (Lond) (2018) 42(8):1445–57. doi: 10.1038/s41366-018-0111-4
108. Piao L, Dorotea D, Jiang S, Koh EH, Oh GT, Ha H. Impaired Peroxisomal Fitness in Obese Mice, a Vicious Cycle Exacerbating Adipocyte Dysfunction via Oxidative Stress. Antioxid Redox Signal (2019) 31(18):1339–51. doi: 10.1089/ars.2018.7614
109. Kuri-Harcuch W, Velez-Delvalle C. A Cellular Perspective of Adipogenesis Transcriptional Regulation. J Cell Physiol (2019) 234(2):1111–29. doi: 10.1002/jcp.27060
110. Derosa G, Maffioli P. Peroxisome Proliferator-Activated Receptor-γ (PPAR-γ) Agonists on Glycemic Control, Lipid Profile and Cardiovascular Risk. Curr Mol Pharmacol (2012) 5(2):272–81. doi: 10.2174/1874467211205020272
111. Nagajyothi F, Desruisseaux MS, Thiruvur N, Weiss LM, Braunstein VL, Albanese C, et al. Trypanosoma Cruzi Infection of Cultured Adipocytes Results in an Inflammatory Phenotype. Obes (Silver Spring Md) (2008) 16(9):1992–7. doi: 10.1038/oby.2008.331
112. González FB, Villar SR, Toneatto J, Pacini MF, Márquez J, D'attilio L, et al. Immune Response Triggered by Trypanosoma Cruzi Infection Strikes Adipose Tissue Homeostasis Altering Lipid Storage, Enzyme Profile and Adipokine Expression. Med Microbiol Immunol (2019) 208(5):651–66. doi: 10.1007/s00430-018-0572-z
113. Assunção LS, Magalhães KG, Carneiro AB, Molinaro R, Almeida PE, Atella GC, et al. Schistosomal-Derived Lysophosphatidylcholine Triggers M2 Polarization of Macrophages Through Pparγ Dependent Mechanisms. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862(2):246–54. doi: 10.1016/j.bbalip.2016.11.006
114. Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-Activated Protein Kinase (AMPK) and Cancer: Many Faces of a Metabolic Regulator. Cancer Lett (2015) 356(2 Pt A):165–70. doi: 10.1016/j.canlet.2014.01.018
115. Xu ZP, Chang H, Ni YY, Li C, Chen L, Hou M, et al. Schistosoma Japonicum Infection Causes a Reprogramming of Glycolipid Metabolism in the Liver. Parasit Vectors (2019) 12(1):388. doi: 10.1186/s13071-019-3621-6
116. Zhu J, Xu Z, Chen X, Zhou S, Zhang W, Chi Y, et al. Parasitic Antigens Alter Macrophage Polarization During Schistosoma Japonicum Infection in Mice. Parasit Vectors (2014) 7:122. doi: 10.1186/1756-3305-7-122
117. Qian XY, Ding WM, Chen QQ, Zhang X, Jiang WQ, Sun FF, et al. The Metabolic Reprogramming Profiles in the Liver Fibrosis of Mice Infected With Schistosoma Japonicum. Inflammation (2020) 43(2):731–43. doi: 10.1007/s10753-019-01160-5
118. Nguyen-Lefebvre AT, Horuzsko A. Kupffer Cell Metabolism and Function. J Enzymol Metab (2015) 1(1):101.
119. Zhu Y, Pereira RO, O'neill BT, Riehle C, Ilkun O, Wende AR, et al. Cardiac PI3K-Akt Impairs Insulin-Stimulated Glucose Uptake Independent of Mtorc1 and GLUT4 Translocation. Mol Endocrinol (2013) 27(1):172–84. doi: 10.1210/me.2012-1210
120. Haeusler RA, Mcgraw TE, Accili D. Biochemical and Cellular Properties of Insulin Receptor Signalling. Nat Rev Mol Cell Biol (2018) 19(1):31–44. doi: 10.1038/nrm.2017.89
121. Yazıcı D, Sezer H. Insulin Resistance, Obesity and Lipotoxicity. Adv Exp Med Biol (2017) 960:277–304. doi: 10.1007/978-3-319-48382-5_12
122. Makki K, Froguel P, Wolowczuk I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflammation (2013) 2013:139239. doi: 10.1155/2013/139239
123. Patel TP, Rawal K, Bagchi AK, Akolkar G, Bernardes N, Dias DDS, et al. Insulin Resistance: An Additional Risk Factor in the Pathogenesis of Cardiovascular Disease in Type 2 Diabetes. Heart Fail Rev (2016) 21(1):11–23. doi: 10.1007/s10741-015-9515-6
124. Hong SH, Sung YA, Hong YS, Jeong K, Chung H, Lee H. Polycystic Ovary Morphology Is Associated With Insulin Resistance in Women With Polycystic Ovary Syndrome. Clin Endocrinol (Oxf) (2017) 87(4):375–80. doi: 10.1111/cen.13380
125. Polak AM, Adamska A. Body Composition, Serum Concentrations of Androgens and Insulin Resistance in Different Polycystic Ovary Syndrome Phenotypes. J Clin Med (2020) 9(3):732. doi: 10.3390/jcm9030732
126. Kuryłowicz A, Koźniewski K. Anti-Inflammatory Strategies Targeting Metaflammation in Type 2 Diabetes. Mol (Basel Switzerland) (2020) 25(9):2224. doi: 10.3390/molecules25092224
127. Fujisaka S. The Role of Adipose Tissue M1/M2 Macrophages in Type 2 Diabetes Mellitus. Diabetol Int (2021) 12(1):74–9. doi: 10.1007/s13340-020-00482-2
128. Mcgillicuddy FC, Harford KA, Reynolds CM, Oliver E, Claessens M, Mills KH, et al. Lack of Interleukin-1 Receptor I (IL-1RI) Protects Mice From High-Fat Diet-Induced Adipose Tissue Inflammation Coincident With Improved Glucose Homeostasis. Diabetes (2011) 60(6):1688–98. doi: 10.2337/db10-1278
129. Geach T. Diabetes: Helminths Improve Insulin Sensitivity and Enhance M2 Macrophage Numbers in WAT of Obese Mice. Nat Rev Endocrinol (2015) 11(6):316. doi: 10.1038/nrendo.2015.68
130. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils Sustain Adipose Alternatively Activated Macrophages Associated With Glucose Homeostasis. Science (2011) 332(6026):243–7. doi: 10.1126/science.1201475
131. Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate Lymphoid Type 2 Cells Sustain Visceral Adipose Tissue Eosinophils and Alternatively Activated Macrophages. J Exp Med (2013) 210(3):535–49. doi: 10.1084/jem.20121964
132. Yang Z, Grinchuk V, Smith A, Qin B, Bohl JA, Sun R, et al. Parasitic Nematode-Induced Modulation of Body Weight and Associated Metabolic Dysfunction in Mouse Models of Obesity. Infect Immun (2013) 81(6):1905–14. doi: 10.1128/iai.00053-13
133. Berbudi A, Surendar J, Ajendra J, Gondorf F, Schmidt D, Neumann AL, et al. Filarial Infection or Antigen Administration Improves Glucose Tolerance in Diet-Induced Obese Mice. J Innate Immun (2016) 8(6):601–16. doi: 10.1159/000448401
134. Surendar J, Frohberger SJ, Karunakaran I, Schmitt V, Stamminger W, Neumann AL, et al. Adiponectin Limits IFN-γ and IL-17 Producing CD4 T Cells in Obesity by Restraining Cell Intrinsic Glycolysis. Front Immunol (2019) 10:2555. doi: 10.3389/fimmu.2019.02555
135. Bhargava P, Li C, Stanya KJ, Jacobi D, Dai L, Liu S, et al. Immunomodulatory Glycan LNFPIII Alleviates Hepatosteatosis and Insulin Resistance Through Direct and Indirect Control of Metabolic Pathways. Nat Med (2012) 18(11):1665–72. doi: 10.1038/nm.2962
136. Barry JC, Shakibakho S, Durrer C, Simtchouk S, Jawanda KK, Cheung ST, et al. Hyporesponsiveness to the Anti-Inflammatory Action of Interleukin-10 in Type 2 Diabetes. Sci Rep (2016) 6:21244. doi: 10.1038/srep21244
137. Hays R, Esterman A, Giacomin P, Loukas A, Mcdermott R. Does Strongyloides Stercoralis Infection Protect Against Type 2 Diabetes in Humans? Evidence From Australian Aboriginal Adults. Diabetes Res Clin Pract (2015) 107(3):355–61. doi: 10.1016/j.diabres.2015.01.012
138. Manco M, Putignani L, Bottazzo GF. Gut Microbiota, Lipopolysaccharides, and Innate Immunity in the Pathogenesis of Obesity and Cardiovascular Risk. Endocr Rev (2010) 31(6):817–44. doi: 10.1210/er.2009-0030
139. Mcphee JB, Schertzer JD. Immunometabolism of Obesity and Diabetes: Microbiota Link Compartmentalized Immunity in the Gut to Metabolic Tissue Inflammation. Clin Sci (Lond) (2015) 129(12):1083–96. doi: 10.1042/cs20150431
140. Wei YX, Zheng KY, Wang YG. Gut Microbiota-Derived Metabolites as Key Mucosal Barrier Modulators in Obesity. World J Gastroenterol (2021) 27(33):5555–65. doi: 10.3748/wjg.v27.i33.5555
141. Zhang P, Yu Y, Qin Y, Zhou Y, Tang R, Wang Q, et al. Alterations to the Microbiota-Colon-Brain Axis in High-Fat-Diet-Induced Obese Mice Compared to Diet-Resistant Mice. J Nutr Biochem (2019) 65:54–65. doi: 10.1016/j.jnutbio.2018.08.016
142. Shi H, Wang Q, Zheng M, Hao S, Lum JS, Chen X, et al. Supplement of Microbiota-Accessible Carbohydrates Prevents Neuroinflammation and Cognitive Decline by Improving the Gut Microbiota-Brain Axis in Diet-Induced Obese Mice. J Neuroinflamm (2020) 17(1):77. doi: 10.1186/s12974-020-01760-1
143. Shi H, Ge X, Ma X, Zheng M, Cui X, Pan W, et al. A Fiber-Deprived Diet Causes Cognitive Impairment and Hippocampal Microglia-Mediated Synaptic Loss Through the Gut Microbiota and Metabolites. Microbiome (2021) 9(1):223. doi: 10.1186/s40168-021-01172-0
144. Torres-Fuentes C, Schellekens H, Dinan TG, Cryan JF. The Microbiota-Gut-Brain Axis in Obesity. Lancet Gastroenterol Hepatol (2017) 2(10):747–56. doi: 10.1016/s2468-1253(17)30147-4
145. Su CW, Chen CY, Jiao L, Long SR, Mao T, Ji Q, et al. Helminth-Induced and Th2-Dependent Alterations of the Gut Microbiota Attenuate Obesity Caused by High-Fat Diet. Cell Mol Gastroenterol Hepatol (2020) 10(4):763–78. doi: 10.1016/j.jcmgh.2020.06.010
146. Walk ST, Blum AM, Ewing SA, Weinstock JV, Young VB. Alteration of the Murine Gut Microbiota During Infection With the Parasitic Helminth Heligmosomoides Polygyrus. Inflammation Bowel Dis (2010) 16(11):1841–9. doi: 10.1002/ibd.21299
147. Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, et al. The Short-Chain Fatty Acid Acetate Reduces Appetite via a Central Homeostatic Mechanism. Nat Commun (2014) 5:3611. doi: 10.1038/ncomms4611
148. Yan H, Ajuwon KM. Butyrate Modifies Intestinal Barrier Function in IPEC-J2 Cells Through a Selective Upregulation of Tight Junction Proteins and Activation of the Akt Signaling Pathway. PloS One (2017) 12(6):e0179586. doi: 10.1371/journal.pone.0179586
149. Lin HV, Frassetto A, Kowalik EJ Jr, Nawrocki AR, Lu MM, Kosinski JR, et al. Butyrate and Propionate Protect Against Diet-Induced Obesity and Regulate Gut Hormones via Free Fatty Acid Receptor 3-Independent Mechanisms. PloS One (2012) 7(4):e35240. doi: 10.1371/journal.pone.0035240
150. Xia Z, Han Y, Wang K, Guo S, Wu D, Huang X, et al. Oral Administration of Propionic Acid During Lactation Enhances the Colonic Barrier Function. Lipids Health Dis (2017) 16(1):62. doi: 10.1186/s12944-017-0452-3
151. Den Besten G, Bleeker A, Gerding A, Van Eunen K, Havinga R, Van Dijk TH, et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a Pparγ-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes (2015) 64(7):2398–408. doi: 10.2337/db14-1213
152. Shimokawa C, Obi S, Shibata M, Olia A, Imai T, Suzue K, et al. Suppression of Obesity by an Intestinal Helminth Through Interactions With Intestinal Microbiota. Infect Immun (2019) 87(6):e00042–19. doi: 10.1128/iai.00042-19
153. Pace F, Carvalho BM, Zanotto TM, Santos A, Guadagnini D, Silva KLC, et al. Helminth Infection in Mice Improves Insulin Sensitivity via Modulation of Gut Microbiota and Fatty Acid Metabolism. Pharmacol Res (2018) 132:33–46. doi: 10.1016/j.phrs.2018.04.008
154. Wu J, Zhu Y, Zhou L, Lu Y, Feng T, Dai M, et al. Parasite-Derived Excretory-Secretory Products Alleviate Gut Microbiota Dysbiosis and Improve Cognitive Impairment Induced by a High-Fat Diet. Front Immunol (2021) 12:710513. doi: 10.3389/fimmu.2021.710513
155. Ryan SM, Eichenberger RM. Harnessing Helminth-Driven Immunoregulation in the Search for Novel Therapeutic Modalities. PloS Pathog (2020) 16(5):e1008508. doi: 10.1371/journal.ppat.1008508
156. Sandborn WJ, Elliott DE, Weinstock J, Summers RW, Landry-Wheeler A, Silver N, et al. Randomised Clinical Trial: The Safety and Tolerability of Trichuris Suis Ova in Patients With Crohn's Disease. Aliment Pharmacol Ther (2013) 38(3):255–63. doi: 10.1111/apt.12366
157. Pierce D, Merone L, Lewis C, Rahman T, Croese J, Loukas A, et al. Safety and Tolerability of Experimental Hookworm Infection in Humans With Metabolic Disease: Study Protocol for a Phase 1b Randomised Controlled Clinical Trial. BMC Endocr Disord (2019) 19(1):136. doi: 10.1186/s12902-019-0461-5
158. Ali Z, Ulrik CS. Obesity and Asthma: A Coincidence or a Causal Relationship? A Systematic Review. Respir Med (2013) 107(9):1287–300. doi: 10.1016/j.rmed.2013.03.019
159. Shore SA, Johnston RA. Obesity and Asthma. Pharmacol Ther (2006) 110(1):83–102. doi: 10.1016/j.pharmthera.2005.10.002
160. Caraballo L, Coronado S. Parasite Allergens. Mol Immunol (2018) 100:113–9. doi: 10.1016/j.molimm.2018.03.014
161. Rzepecka J, Harnett W. Impact of Worms and Their Products on Eosinophils and Neutrophils in Experimental Asthma. Curr Mol Med (2013) 13(7):1192–202. doi: 10.2174/15665240113139990057
162. Van Der Zande HJP, Zawistowska-Deniziak A, Guigas B. Immune Regulation of Metabolic Homeostasis by Helminths and Their Molecules. Trends Parasitol (2019) 35(10):795–808. doi: 10.1016/j.pt.2019.07.014
163. Chen JY, Zhou JK, Pan W. Immunometabolism: Towards a Better Understanding the Mechanism of Parasitic Infection and Immunity. Front Immunol (2021) 12:661241. doi: 10.3389/fimmu.2021.661241
164. Saunders EC, Mcconville MJ. Immunometabolism of Leishmania Granulomas. Immunol Cell Biol (2020) 98(10):832–44. doi: 10.1111/imcb.12394
165. Cortes-Selva D, Fairfax K. Schistosome and Intestinal Helminth Modulation of Macrophage Immunometabolism. Immunology (2021) 162(2):123–34. doi: 10.1111/imm.13231
166. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-Responsive Gene 1 Protein Links Metabolism to Immunity by Catalyzing Itaconic Acid Production. Proc Natl Acad Sci USA (2013) 110(19):7820–5. doi: 10.1073/pnas.1218599110
167. O'neill L, Artyomov MN. Itaconate: The Poster Child of Metabolic Reprogramming in Macrophage Function. Nat Rev Immunol (2019) 19(5):273–81. doi: 10.1038/s41577-019-0128-5
168. Domínguez-Andrés J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, Arts RJW, et al. The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab (2019) 29(1):211–20.e5. doi: 10.1016/j.cmet.2018.09.003
Keywords: parasite, obesity, metabolic inflammation, macrophages, fat browning, adipokine, insulin resistance, microbiota
Citation: Dai M, Yang X, Yu Y and Pan W (2022) Helminth and Host Crosstalk: New Insight Into Treatment of Obesity and Its Associated Metabolic Syndromes. Front. Immunol. 13:827486. doi: 10.3389/fimmu.2022.827486
Received: 02 December 2021; Accepted: 02 February 2022;
Published: 25 February 2022.
Edited by:
Christoph Hölscher, Research Center Borstel (LG), GermanyReviewed by:
Isabelle Wolowczuk, U1019 Centre d’Infection et Immunité de Lille (CIIL)(INSERM), FranceBarbara Nikolajczyk, University of Kentucky, United States
Copyright © 2022 Dai, Yang, Yu and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Pan, cGFud2VpNTI1QDEyNi5jb20=; Yinghua Yu, eWluZ2h1YUB1b3cuZWR1LmF1