
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 02 March 2022
Sec. Vaccines and Molecular Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.826045
This article is part of the Research Topic Crystal Clear: Visualizing the Immune Recognition for the Mechanism and Intervention View all 16 articles
 Dan Lu1,2†
Dan Lu1,2† Zepeng Xu2,3†Ding Zhang2,4†Min Jiang1,2Kefang Liu2Juanhua He2,5Dongli Ma6Xiaopeng Ma6
Zepeng Xu2,3†Ding Zhang2,4†Min Jiang1,2Kefang Liu2Juanhua He2,5Dongli Ma6Xiaopeng Ma6 Shuguang Tan2
Shuguang Tan2 George F. Gao1,2*Yan Chai2*
George F. Gao1,2*Yan Chai2*Immune checkpoint therapy (ICT) with a monoclonal antibody (MAb) against programmed cell death protein 1 (PD-1) is a powerful clinical treatment for tumors. Cemiplimab is a human IgG4 antibody approved in 2018 and is the first MAb proven to be effective for locally advanced basal cell carcinoma. Here, we report the crystal structure of cemiplimab bound to PD-1 and the effects of PD-1 N-glycosylation on the interactions with cemiplimab. The structure of the cemiplimab/PD-1 complex shows that cemiplimab mainly binds to PD-1 with its heavy chain, whereas the light chain serves as the predominant region to compete with the binding of PD-L1 to PD-1. The interaction network of cemiplimab to PD-1 resembles that of camrelizumab (another PD-1-binding MAb), and the N58 glycan on the BC loop of PD-1 may be involved in the interaction with cemiplimab. The binding affinity of cemiplimab with PD-1 was substantially decreased with N58-glycan-deficient PD-1, whereas the PD-1/PD-L1 blocking efficiency of cemiplimab was attenuated upon binding to the N58-glycosylation-deficient PD-1. These results indicate that both the binding and blocking efficacy of cemiplimab require the N58 glycosylation of PD-1. Taken together, these findings expand our understanding of the significance of PD-1 glycosylation in the interaction with cemiplimab.
Immune checkpoint therapy (ICT), also called immune checkpoint blockade (ICB), has been widely used in tumor immunotherapy since the approval of the CTLA-4-specific ipilimumab in 2011 (1–3). Encouragingly, the blocking of the programmed cell death protein-1 (PD-1)/PD-1 ligand 1 (PD-L1) pathway with monoclonal antibodies (MAbs) has dramatically improved the treatment prospects for multiple tumors (2, 4). PD-1 belongs to the immunoglobulin gene superfamily, which was identified in T cells upon programmed cell death (5). PD-1 is mainly expressed in immune cells, including activated dendritic cells, natural killer cells, T cells, and B cells (6). The ligand for PD-1, PD-L1, is upregulated in a broad range of tumor cells and mediates tumor immune escape through interaction with PD-1 (7). Inhibition of the PD-1/PD-L1 interaction with MAbs restores T-cell function to retain preexisting antitumor activity (4, 8). Currently, there are 10 clinically approved anti-PD-1 MAbs: nivolumab (Bristol-Myers Squibb, 2014), pembrolizumab (Merck Sharp & Dohme, 2014), cemiplimab (Sanofi and Regeneron, 2018), toripalimab (Junshi, 2018), sintilimab (Innovent, 2018), camrelizumab (HengRui, 2019), tislelizumab (BeiGene, 2019), dostarlimab (Tesaro, 2021), penpulimab (Chia Tai-Tianqing, 2021), and zimberelimab (Gloria Biosciences, Arcus Biosciences, and Taiho, 2021).
Glycosylation is a common protein post-translational modification, and it plays critical roles in multiple biological processes (9). For instance, N-glycosylation is important in the maintenance of the surface expression of PD-1 protein and the regulation of the interaction with PD-L1 (10). Our previous work demonstrates that the extracellular domain of PD-1 (PD-1-ECD) is extensively glycosylated, and N-glycosylation is found in all the four potential N-glycosylation sites (N49, N58, N74, and N116) (11, 12).
Structural evidence suggests the impact of N-glycans on PD-1 interaction with MAbs. Among the four N-glycosylation sites, structural evidence suggests that N58 in the BC loop of PD-1 is located near the interface between PD-1 and PD-L1, whereas glycosylation at N58 is not involved in the binding of PD-1 to PD-L1 (11, 13). The reported complex structures of PD-1 with nivolumab, pembrolizumab, tislelizumab, or toripalimab show that PD-1 glycosylation does not engage with these MAbs (11, 14–16). In contrast, PD-1 N-glycans are involved in the binding to some anti-PD1 antibodies (11, 17). We previously reported that PD-1 glycosylation at N58 promotes the interaction with camrelizumab, and the blocking efficacy of camrelizumab is dampened upon binding to N58 glycosylation-deficient PD-1 (17). Furthermore, the binding of other PD-1-specific MAbs (e.g., MW11-h317, mAb059c, and STM418) also involves the glycans at the N58 site (12, 18, 19). Structural evidence shows that these MAbs mainly engage with the conserved core region of the glycan chains at N58.
Cemiplimab (REGN2810, Libtayo®), co-developed by Sanofi and Regeneron, is a fully human IgG4 MAb specific to the PD-1 receptor (18). It was approved by the US Food and Drug Administration (FDA) for clinical treatment of metastatic cutaneous squamous cell carcinoma (CSCC) in 2018 (19, 20). Cemiplimab is the first MAb proven to be effective for locally advanced basal cell carcinoma, a tumor with no standard treatment regimen after first-line hedgehog inhibitor therapy (21). Here, we report the molecular basis of cemiplimab binding to PD-1 through the determination of the cemiplimab/PD-1 complex structure, and we investigated the roles of PD-1 N-glycosylation in the cemiplimab interaction. We found that both the binding and inhibition efficacies of cemiplimab to PD-1 were promoted by PD-1 N58 glycosylation. The findings observed in the present study expand our knowledge of the interaction mechanisms of glycosylation for antibodies to PD-1 in the context of tumor ICT.
To investigate the binding mechanisms of cemiplimab to PD-1, the PD-1-ECD (PD-1-E. coli) protein and single-chain variable fragment (scFv) of cemiplimab were expressed in Escherichia coli cells as inclusion bodies and renatured by in vitro refolding (11, 17). The cemiplimab-scFv/PD-1 complex was prepared after in vitro co-refolding and used for crystal screening (Supplementary Figure 1). Diffractable crystals were obtained, and the structure of the cemiplimab-scFv/PD-1 complex was solved at a resolution of 3.4 Å, with Rwork and Rfree values of 0.245 and 0.285, respectively (Table 1). The overall structure reveals that PD-1-ECD and cemiplimab-scFv form a 1:1 complex, and the interaction of cemiplimab with PD-1 buries a total surface area of 1,614 Å. Cemiplimab binds to PD-1 with all three complementarity-determining regions (CDRs) from its heavy chain and the LCDR3 from the light chain variable domain (VL) (Figure 1A and Table 2). Specifically, there are multiple hydrogen bond interactions between residues from LCDR3 (S92) of cemiplimab and the FG loop of PD-1 (K131 and A132) and between residues from HCDR2 (S52, R56, D57, and Y59) of cemiplimab and the BC loop (E61 and S62) and FG loop (A129) of PD-1 (Figure 1B and Table 2; Supplementary Figure 2).

Table 1 Crystallographic data collection and refinement statistics.
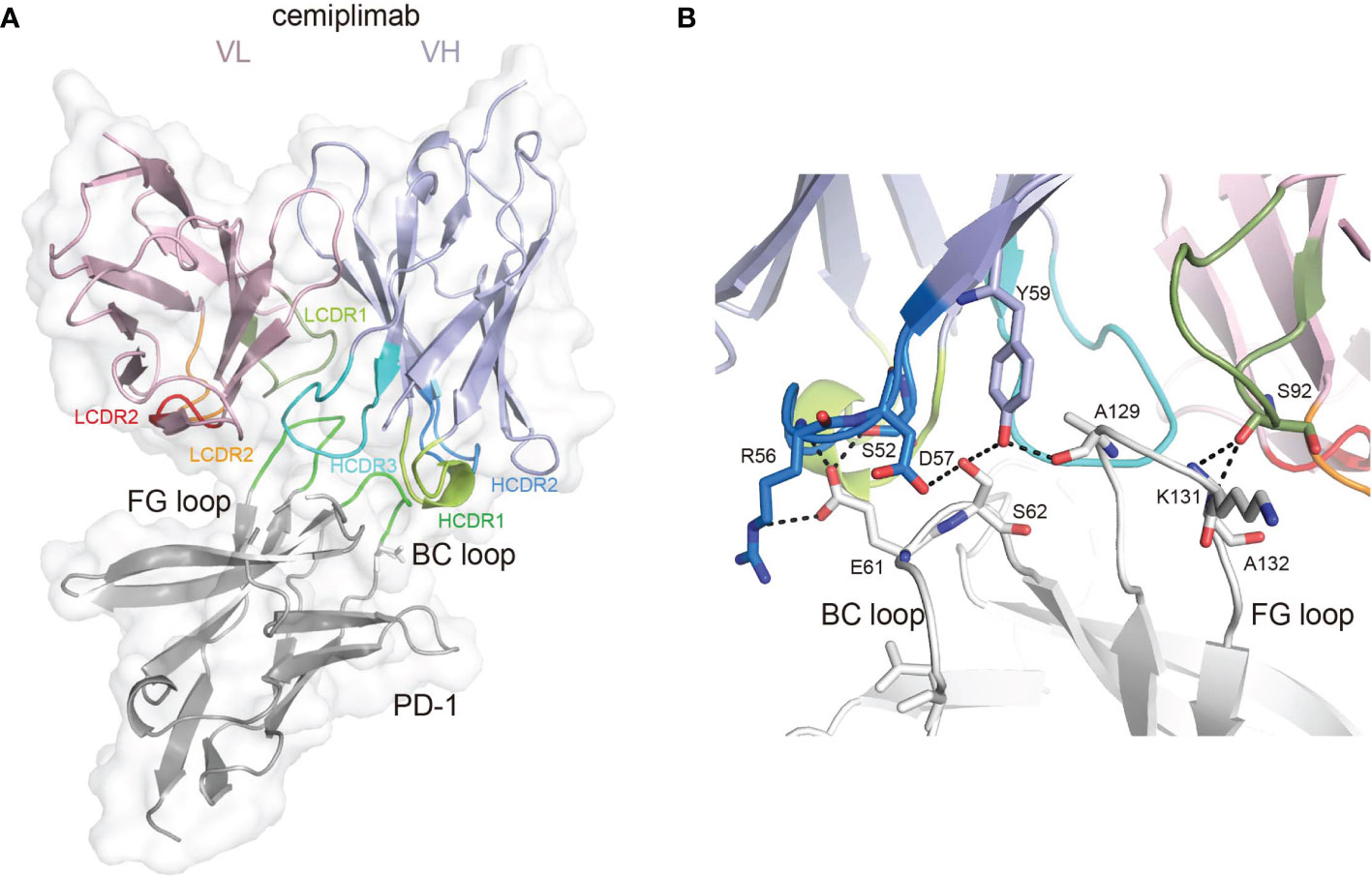
Figure 1 The binding mechanism of cemiplimab to PD-1. (A) Overall structure of cemiplimab bound to PD-1. PD-1 colored in gray is shown as surface representation, while the heavy chain (VH) and light chain (VL) of cemiplimab-scFv are shown as cartoon colored in light blue and light pink. The CDR1, CDR2, and CDR3 loops of the heavy chain are colored in green (HCDR1), blue (HCDR2), and cyan (HCDR3), respectively. The CDR1, CDR2, and CDR3 loops of the light chain are colored in limon (LCDR1), orange (LCDR2), and red (LCDR3), respectively. The BC and FG loops are colored in green. (B) The detailed binding of cemiplimab to the FG and BC loops of PD-1. The residues taking part in forming hydrogen bonds are shown as sticks. The hydrogen bonds between residues are shown as a dashed line in black.
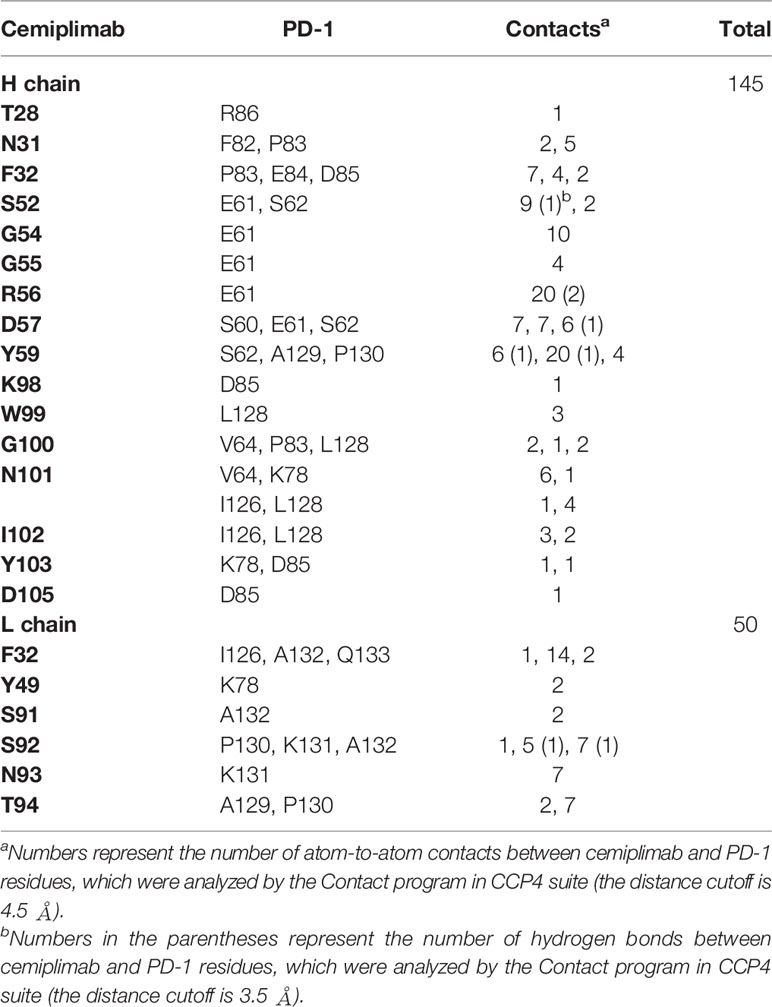
Table 2 Residues contributed interaction between cemiplimab and PD-1.
Cemiplimab mainly binds to the BC and FG loops of PD-1 through its HCDR2, HCDR3, and LCDR3 loops (Figure 2A). Of note, the HCDR2 (G53 and G54) of cemiplimab is close to N58 of PD-1. The structure of PD-1 molecules with PD-L1, nivolumab, pembrolizumab, toripalimab, camrelizumab, and MW11-h317 were then superimposed with that from the cemiplimab/PD-1 complex to investigate the conformational changes upon binding to different MAbs (Figure 2B). Pembrolizumab predominantly binds to the C’D loop of PD-1, toripalimab mainly binds to the FG loop, and the binding of nivolumab is mainly located on the N-terminal loop of PD-1. In contrast, camrelizumab, MW11-h317, and cemiplimab mainly bind to the BC and FG loops, while camrelizumab and MW11-h317 contact the N58 glycan chains at the BC loop of PD-1. The FG loop exhibits substantial conformational variation when bound to different MAbs, while the BC loop shows limited conformational changes (Figure 2B). Of note, the FG loop of PD-1 in the cemiplimab/PD-1 complex exhibits a similar conformation to that in the PD-1/PD-L1 complex, while varied conformational changes were induced upon binding to the other MAbs (Figure 2C).
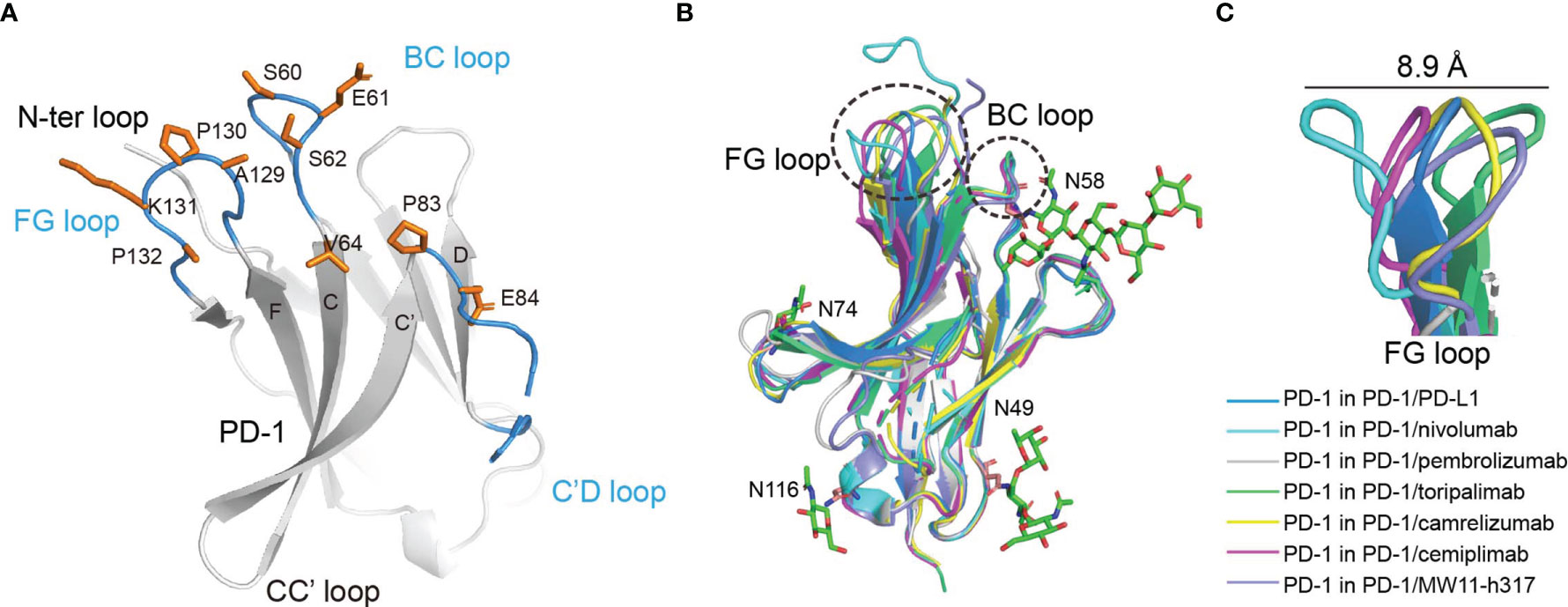
Figure 2 The structure characterization of PD-1 upon binding to MAbs. (A) The key region of PD-1 binding to cemiplimab. The β-strands of PD-1 are represented as the capital characters C, C′, D, F, and G, respectively. The CC′, C′D, and FG loops of PD-1 are highlighted in the blue cartoon, and the key epitopes of PD-1 binding to cemiplimab are shown as orange sticks, respectively. (B) Superposition of PD-1 upon binding to the PD-L1 ligand or different MAbs, including the PD-1 extracted from the complex structures of PD-1/PD-L1 (blue) (PDB code: 4ZQK), PD-1/nivolumab (cyan) (PDB code: 5WT9), PD-1/pembrolizumab (gray) (PDB code: 5JXE), PD-1/toripalimab (green) (PDB code: 6JBT), PD-1/camrelizumab (yellow) (PDB code: 7CU5), PD-1/cemiplimab (magenta) and PD-1/MW11-h317 (light blue) (PDB code: 6JJP). FG loop and BC loop of PD-1 which contributed vital binding to the cemiplimab are shown as a dashed line in black. (C) Flexible conformations of the FG loop of PD-1 upon binding to PD-L1 or different MAbs.
The structure of the cemiplimab/PD-1 complex was next superimposed with that of the PD-1/PD-L1 complex (PDB: 4ZQK) to analyze the PD-1/PD-L1 inhibition mechanism of cemiplimab. The analysis revealed that the major domain of cemiplimab responsible for inducing stereospecific hindrance to the binding of PD-L1 is the VL domain (Figure 3A). Additionally, the binding area of cemiplimab on PD-1 substantially overlaps with that of PD-L1 (Figure 3B). Together with the superior binding affinity of cemiplimab over PD-L1 to PD-1, the binding of cemiplimab would abrogate PD-L1 binding to PD-1 and inhibit PD-L1-mediated signaling. Among the clinically approved MAbs, the binding area of cemiplimab is similar to that of toripalimab, camrelizumab, nivolumab, and MAbs engaging mainly with the FG loop of PD-1 (e.g., MW11-h317, mAb059c, and GY5), whereas it is distinct from that of pembrolizumab and tislelizumab (Figure 3C and Supplementary Figure S2). Taken together, our structural analyses indicate that cemiplimab blocks the PD-1/PD-L1 interaction mainly through its VL domain.
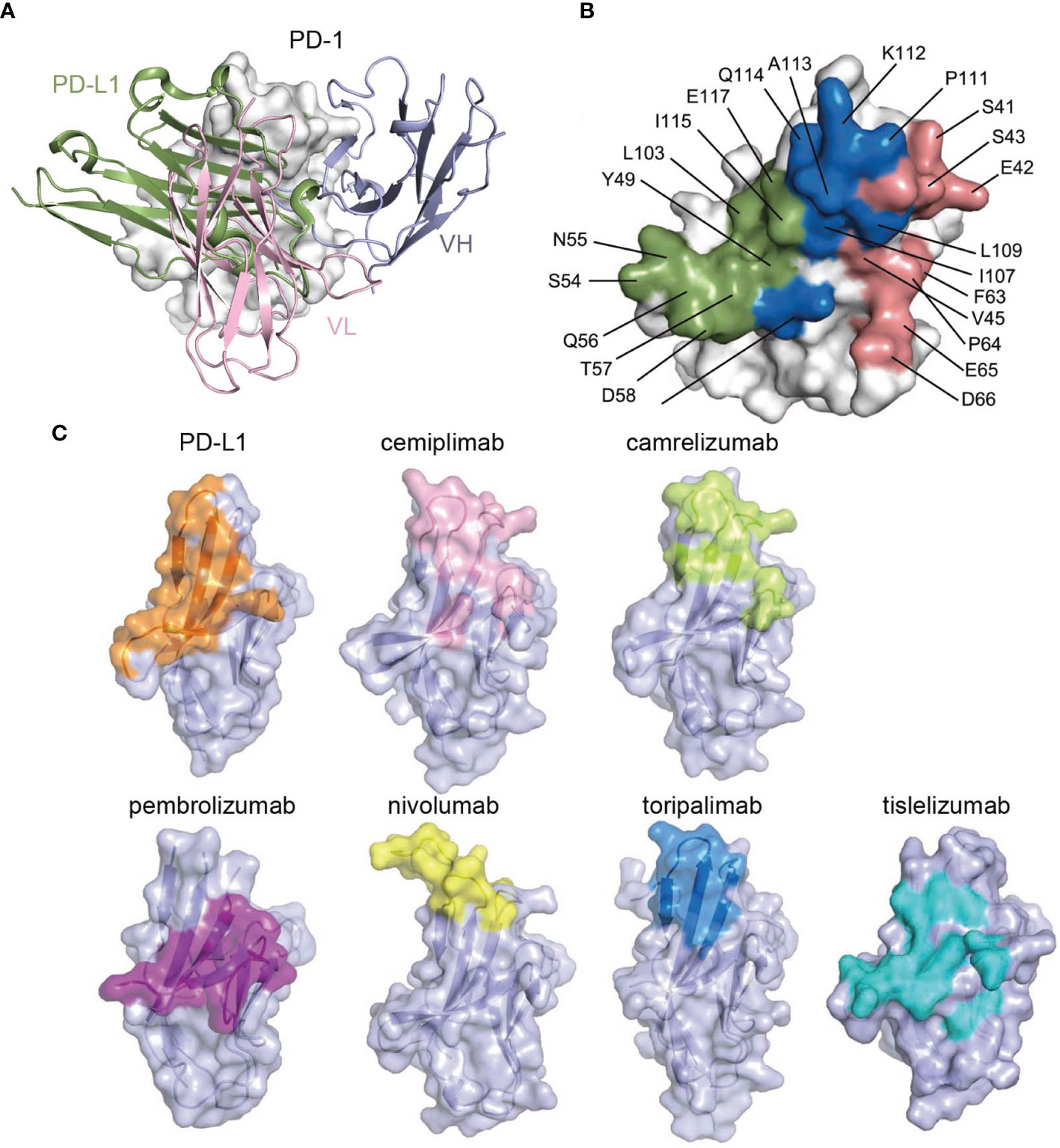
Figure 3 Structural basis of the blockade binding of cemiplimab with PD-L1. (A) Comparison of cemiplimab/PD-1 with PD-L1 extracted from PD-1/PD-L1 complex structure (PDB code: 4ZQK). PD-L1 is shown as smudge cartoon, while PD-1 is shown as surface diagram in white. VH and VL of cemiplimab-scFv are shown as cartoons in light blue and light pink, respectively. (B) The competitive binding surfaces of cemiplimab with PD-L1 on PD-1. The residues bound to cemiplimab alone are colored in deep salmon, while the residues contact with PD-L1 alone are colored in smudge, and the residues contacted by both cemiplimab and PD-L1 are colored in blue. The epitope residues in PD-1 are pointed out in black characters. (C) The binding surface of PD-L1 and structurally known clinically approved MAbs on PD-1 is shown in different colors. The binding surface of PD-L1 and other MAbs, e.g., cemiplimab, camrelizumab, pembrolizumab, nivolumab, toripalimab, and tislelizumab are colored in orange, light pink, limon, purple, yellow, blue, and teal, respectively.
To compare the binding mode of PD-1-targeting MAbs, the structure of the cemiplimab/PD-1 complex was then superimposed with the nivolumab/PD-1 and pembrolizumab/PD-1 complexes, with the structure of PD-1 fixed. These comparative analyses show that nivolumab and pembrolizumab adopt distinct binding orientations compared to cemiplimab, although the binding surface with nivolumab highly overlaps (Figure 4A). Comparative analysis with other N58 glycan-engaged MAbs revealed that the orientation of cemiplimab upon binding to PD-1 resembles that of camrelizumab and MW11-h317, while the binding of mAb059c is substantially biased toward the FG-loop (Figure 4B). Alignment of these MAbs’ sequences reveals that the heavy chains of the glycosylation-engaged MAbs camrelizumab, MW11-h317, and cemiplimab share similar HCDR2s compared to those from other glycosylation-independent MAbs, except for mAb059c that also contacts the N-glycan at N58 (Figure 4C). In the MW11-h317/PD-1 and camrelizumab/PD-1 complex structures, the N-acetylglucosamine (NAG) and mannose (MAN) form multiple hydrogen bond interactions with residues from HCDR1 (S31) and HCDR2 (G53 and G54) (Figures 4D, E). Although the E. coli-expressed PD-1-ECD proteins used in this study for crystal growing did not contain any glycan modification, the conserved conformation of the HCDR1 and HCDR2 of cemiplimab with that of camrelizumab indicates that the glycan chains of PD-1 N58 would form a similar interaction network through amino acids from HCDR1 and HCDR2 of cemiplimab (Figure 4F).
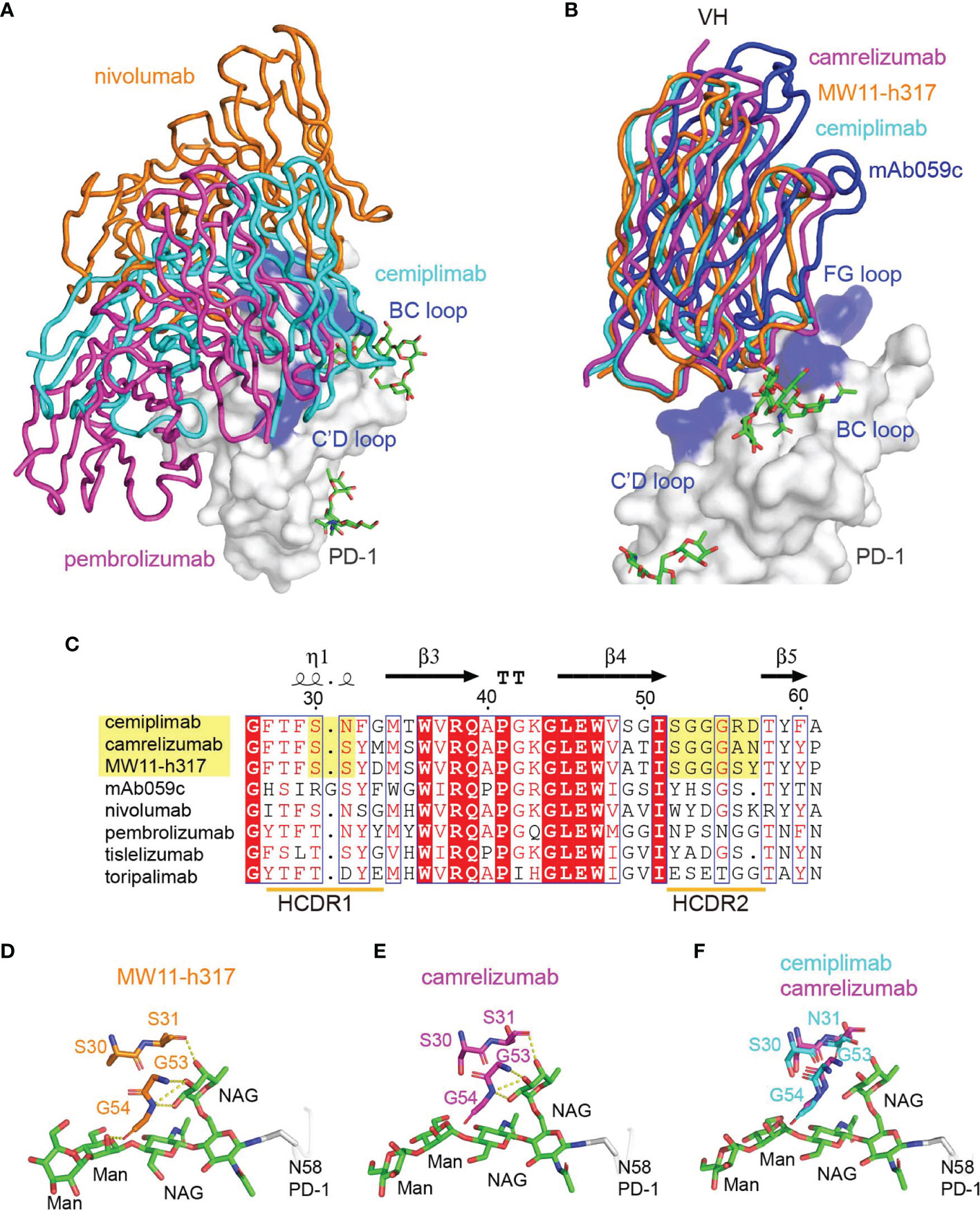
Figure 4 Interaction between MAbs and N58 glycosylation on BC loop. (A) The comparison of the overall binding of cemiplimab, nivolumab, and pembrolizumab to PD-1. Superimposition of cemiplimab/PD-1 complex with that of pembrolizumab/PD-1 (PDB: 5JXE) and nivolumab/PD-1 (PDB: 5WT9). The cemiplimab, pembrolizumab, and nivolumab are shown as ribbon and colored in cyan, magenta, and orange, respectively. PD-1 extracted from camrelizumab/PD-1 (PDB code: 7CU5) complex is shown as surface representation colored in white. (B) Superimposition of cemiplimab/PD-1 complex with that of camrelizumab (PDB: 7CU5), MW11-h317 (PDB: 6JJP) and mAb059c (PDB: 6K0Y). The VH domains of the MAbs are shown as ribbons while the VL domains are not shown. PD-1, cemiplimab, camrelizumab, MW11-h317, and mAb059c are colored in gray, cyan, magenta, orange and blue, respectively. The CC′, C′D, and FG loops of PD-1, which participate in binding to Mabs are highlighted in blue. (C) Structure-based sequence alignment of cemiplimab and other anti-PD-1 MAbs. Coils above the sequences indicate α-helices, and the lines with arrowhead represent the β sheets. Residues highlighted in yellow are highly conserved. The sequence alignment was generated with ClustalX and ESPript. (D, E) The interaction of N-glycosylation N58 with MW11-h317 (D) or camrelizumab (E). The amino acid residues involved in hydrogen bond interaction and N58 glycans are shown as sticks, with amino acids in MW11-h317 colored in orange, camrelizumab colored in magenta, and the glycans in PD-1 colored in green. Hydrogen bonds are labeled by yellow dashed lines. (F) Comparison of cemiplimab/PD-1 complex with that of camrelizumab (PDB: 7CU5), and the amino acids in cemiplimab are colored in cyan and residues in camrelizumab are colored in magenta.
Based on this structural information, we speculated that the N58 glycan of PD-1 potentially plays a role in binding to cemiplimab, although the N58 glycan chains were not observed with the PD-1-E. coli proteins used in the structural study. Therefore, we further evaluated the binding profiles of cemiplimab with wild type (WT) PD-1 protein (PD-1-WT) expressed in 293F cells, which enabled full glycosylation on proteins similarly to host cells using surface plasmon resonance (SPR). Additionally, N58A-mutated PD-1 protein (PD-1-N58A) expressed in 293F cells, which is specifically deficient in N58 glycosylation, was also investigated. The binding affinity (evaluated as KD) of cemiplimab to N58-mutated PD-1 protein substantially decreased to 106 nM, a 60-fold reduction compared to PD-1-WT (KD = 1.68 nM) (Figure 5A). Moreover, we analyzed the binding profiles of cemiplimab to PD-1-ECD proteins obtained from E. coli cells, which carried no post-translational modifications. The SPR analysis revealed a similar reduction in binding affinity to PD-1 protein from E. coli (KD = 691.38 nM) compared to that from 293F cells (KD = 1.68 nM) (Figure 5A and Supplementary Table S1). The binding of camrelizumab, which is promoted by glycosylation of PD-1 N58, was tested in parallel as a control (Figure 5B). The decreased binding affinities of camrelizumab with N58A-mutated PD-1 proteins from 293F cells (KD = 492.85 nM) was similar to that of cemiplimab, and the binding affinity of camrelizumab with non-glycosylated PD-1 proteins from E. coli (KD = 2.63 μM) is substantially decreased compared with glycosylated PD-1 (KD = 4.8 nM) like cemiplimab (Figure 5B). Based on these findings, we concluded that N-glycosylation at N58 promotes the binding of cemiplimab to PD-1.
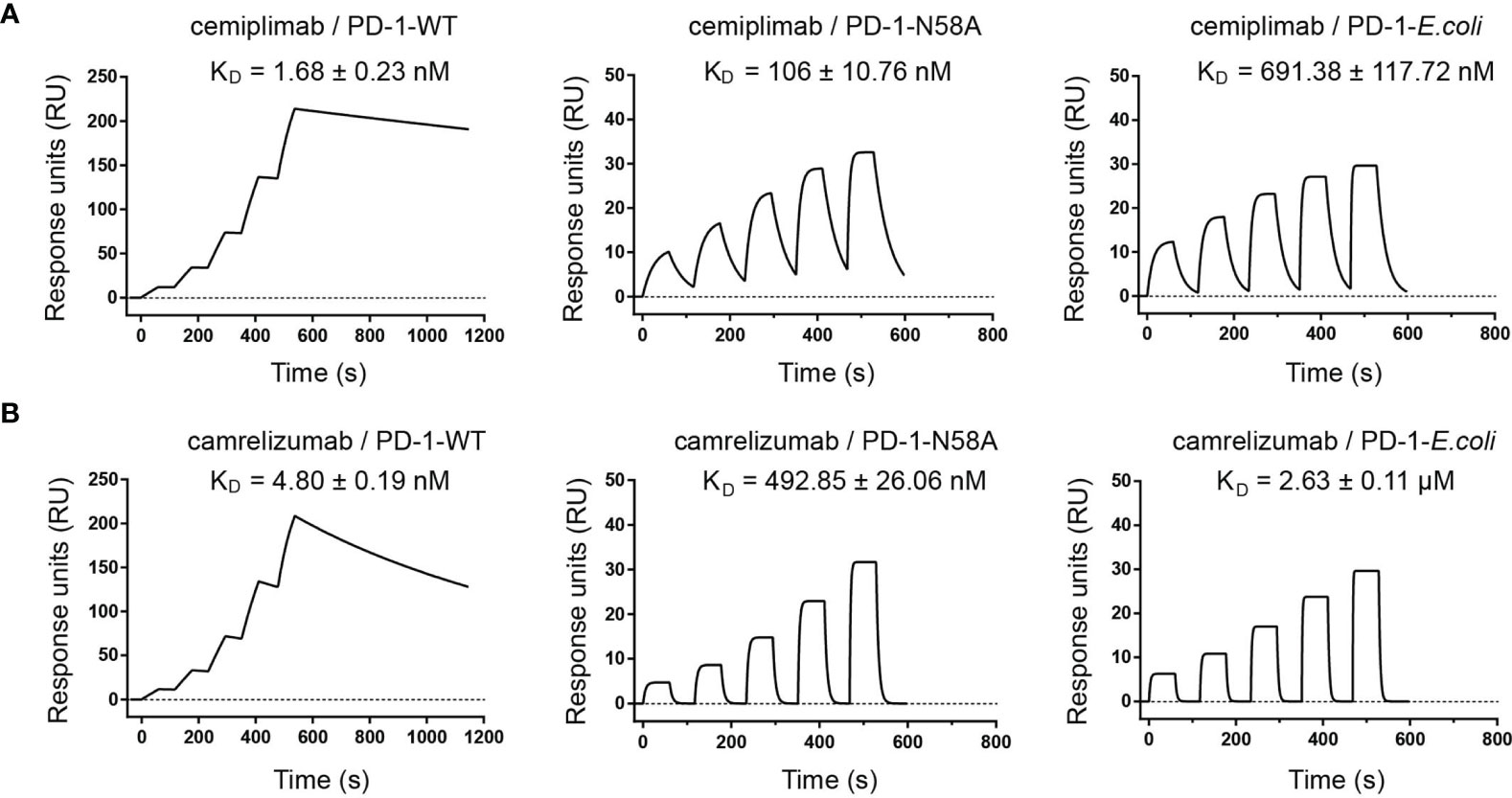
Figure 5 N-glycosylation of N58 remotes the binding to cemiplimab. (A) SPR assay characterization of the binding profiles of cemiplimab with PD-1-WT (left), PD-1-N58A (middle) proteins expressed in 293F cells, and PD-1-E. coli (right) expressed in E. coli cells. (B) SPR assay characterization of the binding of camrelizumab with PD-1-WT (left), PD-1-N58A (middle) expressed in 293F cells, and PD-1-E. coli (right) expressed in E. coli cells. The mean value of the KD was recorded after repeating each experiment three times.
To verify the roles of N58 glycosylation in PD-1/PD-L1 blocking by cemiplimab, a mechanism believed to be the key aspect for the restoration of antitumor efficacy for MAb-based ICT, we further tested the blocking efficiency of the full-length cemiplimab to N58 glycosylation-deficient PD-1. His-tagged PD-1-WT and PD-1-N58A recombinant proteins were prepared from 293F cells and were used to stain 293T cells transiently expressing PD-L1. The blocking of the PD-1/PD-L1 interaction was analyzed by staining the PD-L1-expressing 293T cells with a mixture of serial dilutions of the full-length cemiplimab or camrelizumab proteins pre-incubated with the same concentrations of WT or N58A-mutated PD-1-His proteins (2 μg/ml). As controls, mock-transfected 293T cells stained with PD-1-WT and PD-L1-transfected 293T cells stained with isotype antibody were enrolled as controls (Figure 6A). We found that the frequency of protein-staining-positive cells with PD-1-WT-His protein substantially decreased from 65.0% to 5.0% in the presence of 20 μg/ml cemiplimab, indicating the complete blockade of the PD-1/PD-L1 interaction (Figures 6B, C). However, the blocking efficacy of cemiplimab to PD-1-N58A mutant protein with PD-L1 was decreased compared to that of PD-1-WT. No substantial blocking efficacy was observed for cemiplimab with N58A mutant protein, even at the high concentration of 80 μg/ml (Figure 6B). The decreased blocking scenario of camrelizumab to N58A mutant PD-1 is similar to that of cemiplimab (Figures 6B, D). These results indicate that N58 glycosylation promotes both the binding and blocking of cemiplimab.
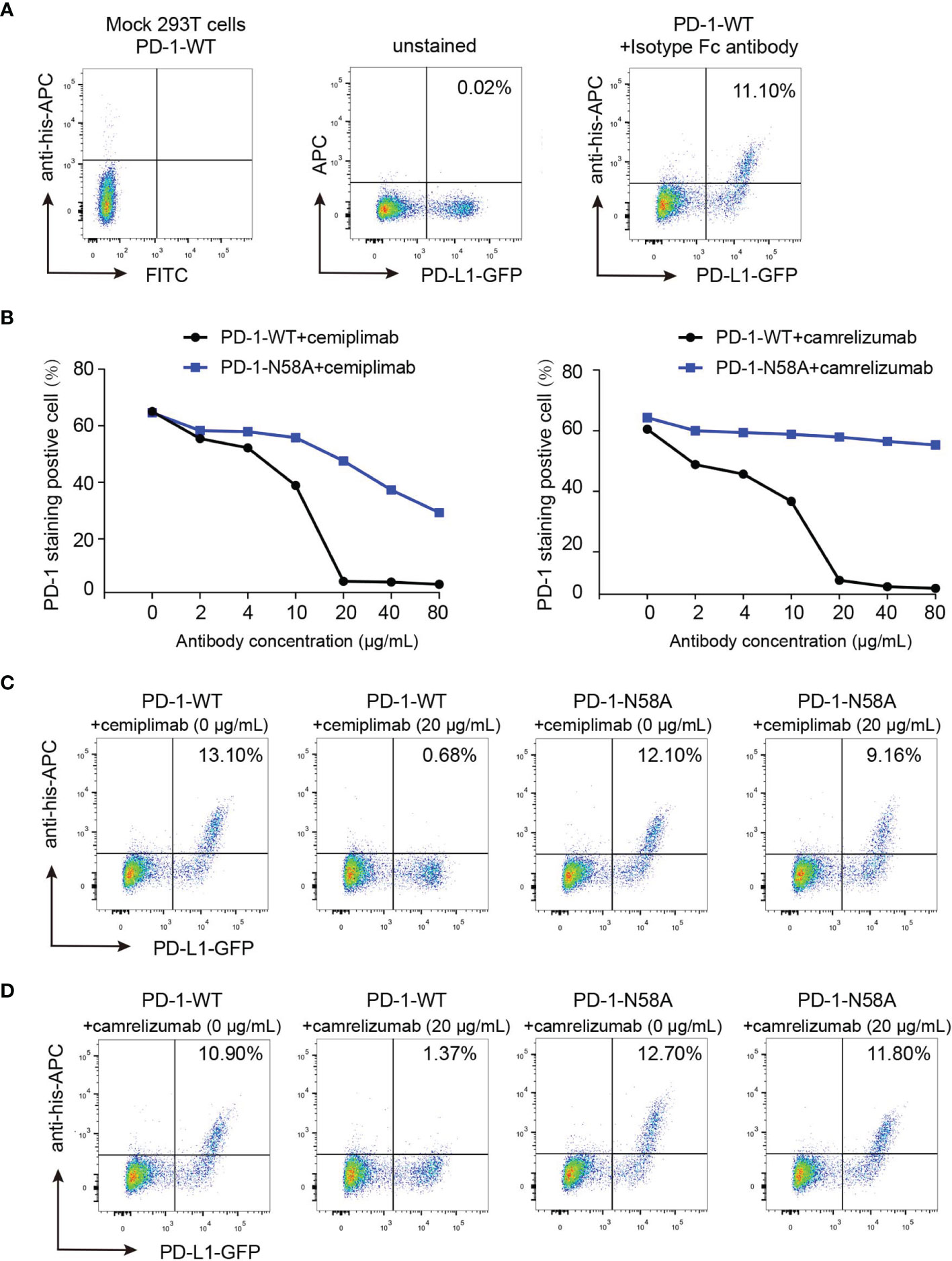
Figure 6 Reduced blocking efficiency of cemiplimab to N58 glycosylation-deficient PD-1. (A) Untransfected 293T cells and transfected 293T cells incubated with isotype antibody as negative control. (B) The blocking of the binding of His-tagged PD-1-WT (blank) or PD-1-N58A (blue) proteins to PD-L1 expressing 293T cells is analyzed with varying concentrations (0, 2, 4, 10, 20, 40, and 80 μg/ml) of full-length cemiplimab (left) or camrelizumab (right). The PD-L1 expressing 293T cells staining with His-tagged PD-1-WT or PD-1-N58A are prepared as a positive control. (C, D) The frequencies of the His-tagged PD-1-WT or PD-1-N58A protein staining positive subpopulations in the absence (0 μg/ml) or presence (20 μg/ml) of cemiplimab (C) or camrelizumab (D). At the same concentration (20 μg/ml) of cemiplimab or camrelizumab, the frequency of His-tagged PD-1-WT or PD-1-N58A staining positive cells was calculated based on PD-L1-GFP-positive cells. The experiment was repeated twice in the results averaged.
In this study, we report the interaction mechanisms between cemiplimab and PD-1. Overall, the binding of cemiplimab to PD-1 resembles that of camrelizumab, as we reported earlier (17). Structural analysis indicates that cemiplimab competes with the binding of PD-L1 to PD-1 with overlapping binding surface areas of PD-L1 resulting in steric hindrance. Cemiplimab mainly binds to the BC and FG loops of PD-1, whereas the FG loop of PD-1 contributes to multiple interactions with PD-L1 and therefore mediates the competitive interaction with cemiplimab or PD-L1 (22). Comparative structural analyses with other solved MAb/PD-1 complex structures suggest that the binding of cemiplimab resembles that of camrelizumab and may be promoted by the glycan chains at N58 on the BC loop. Subsequent analyses indicated that the deficiency of N58 glycosylation substantially decreases both the PD-1 binding affinity and blocking of cemiplimab, which is similar to that of camrelizumab.
The previously reported complex structures of clinically approved MAbs (e.g., nivolumab, pembrolizumab, camrelizumab, toripalimab, and tislelizumab) reveal that these MAbs predominantly bind to the surface or terminal loops of PD-1, whereas PD-L1 mainly binds to the surface on PD-1 constituted by β sheets. Comparative analysis revealed that the flexible surface or terminal loops of PD-1 exhibit distinct conformations upon binding to varied MAbs. The FG loop of PD-1 engages with PD-L1 (22, 23) and is a hot spot loop for PD-1 specific therapeutic MAbs, e.g., toripalimab, camrelizumab, GY-5, and GY-14 (15, 17, 24). Structural analysis revealed that the binding region of cemiplimab on PD-1 is similar to that of camrelizumab, which mainly binds to the BC loop, FG loop, and C′D loop.
Glycosylation is involved in fundamental biological processes and plays pivotal roles in tumor development and progression, immune modulation, and metastasis (25). PD-1 protein is not only upregulated in T cells to mediate immune suppression but is also expressed across a broad range of tumor cells to promote tumor suppression (26). Dysregulated protein glycosylation occurs in tumor cells and tumor-associated dysregulated glycosylation includes fucosylation, sialylation, N− and O−linked glycan branching, and O−glycan truncation (25, 27). Furthermore, abnormal glycosylation also occurs in the tumor microenvironment due to hypoxia, inflammatory events, and metabolism, and it plays a crucial functional role in tumor progression and metastasis. Therefore, the glycosylation of PD-1 may not only affect the immune regulatory roles of the MAbs targeting PD-1 in T cells but may also interfere with the tumor regulatory roles of the MAbs when binding to PD-1 in tumor cells. The PD-1-specific blocking MAbs camrelizumab, MW11-h317, mAb059c, and STM418 contact the N58 glycan when binding to PD-1 (10, 17, 28, 29). Although clinical evidence supports improved overall survival rates across multiple cancer types with camrelizumab, unexpected binding of camrelizumab to VEGFR2 has been reported and may correlate with the side effects of capillary hemangiomas usually observed in clinical studies with camrelizumab (30). Structural analysis reveals that camrelizumab binds to the core region of the N-glycan of PD-1, which is conserved in the N-glycosylation of some proteins. Therefore, the binding of these MAbs to the conserved N-glycan on PD-1 may reduce the binding specificity, although further systemic investigations should be performed to evaluate the binding specificities of the MAbs that engage with the N58 glycan. Both the binding affinities and PD-1/PD-L1 blocking efficiencies of cemiplimab to N58 glycan-deficient PD-1 were similar to that of camrelizumab, as revealed in the present study. Structural analysis and sequence alignment also indicate that cemiplimab binds to N58 glycan chains with conserved HCDR2 regions similar to camrelizumab and MW11-h317. However, clinical studies for cemiplimab do not report a high prevalence of capillary hemangiomas as observed for camrelizumab (58.6%) (31). This indicates that although the conserved N58 glycan promotes the binding of these two MAbs to PD-1 in a similar mode, the binding specificities of the MAbs may vary due to the variable regions responsible for the binding to residues in PD-1.
Taken together, we report the molecular basis of cemiplimab binding to PD-1. Cemiplimab mainly utilizes its heavy chain to bind to the binding “hotspot” for therapeutic MAbs targeting PD-1, i.e., the FG loop of PD-1. Cemiplimab binds to PD-1 in a similar mode to camrelizumab, and the N58 glycan on the BC loop of PD-1 was verified to promote both the binding and blocking of cemiplimab. All of these findings facilitate our understanding of the interaction between cemiplimab and PD-1 and will benefit the future design of agents targeting glycosylated PD-1.
For E. coli cell expression, the extracellular domain of PD-1 (UniProt: Q15116, residues L25-R147) and cemiplimab-scFv [designed as a format of VL-GGGGS (4)-VH] were cloned into Novagen’s prokaryotic expression vector pET-21a(+). The two plasmids above were transformed into E. coli strain BL21 (DE3) pLysS cells and overexpressed as inclusion bodies under IPTG (1 mM) induction, which were verified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The inclusion bodies were then dissolved by dissolution buffer [6 M Gua-HCl, 10% v/v glycerol, 50 mM Tris-HCl, 100 mM NaCl, 10 mM ethylenediaminetetraacetic acid (EDTA), pH 8.0] and co-refolded as previously described (32–34). Briefly, a solution of PD-1 and cemiplimab-scFv was mixed in 1:1 molar ratio, and then, 5 ml of the mixture (30 mg/ml) added drop by drop to 2.5 L refolding buffer (100 mM Tris–HCl, 400 mM L-Arg-HCl, 2 mM EDTA, 5 mM glutathione (GSH), and 0.5 mM oxidized glutathione (GSSG), pH 8.0]. After gently stirring for 8 h, the solution was concentrated and exchanged to protein buffer (20 mM Tris–HCl, 150 mM NaCl, pH 8.0). Subsequentially, the cemiplimab-scFv/PD-1 complex protein was purified via size exclusion using an AKTA Pure system with Superdex™ 200 Increase 10/300 GL column.
For mammalian cell expression, the extracellular domains of PD-1 (residues L25-R147) and PD-1 N58A residue substitution mutant gene (obtained by site-directed mutagenesis) were cloned into an expression vector pCAGGS with signal peptide at the N-terminal and six histidines at the C-terminal, named as PD-1-WT and PD-1-N58A, respectively. The full-length heavy- and light-chain genes of cemiplimab and camrelizumab were cloned into the pCAGGS vector individually with EcoRI and XhoI sites, named as cemiplimab-Fc and camrelizumab-Fc. Plasmids were transiently transfected into 293F cells and incubated at 37°C for 72 h. The culture was centrifuged, and supernatant was then collected and filtered with a 0.22-μm filter. The PD-1-WT or PD-1-N58A proteins were purified first by His-Trap HP column (GE Healthcare) followed by Superdex™ 200 10/300 GL (GE Healthcare). The proteins of full-length cemiplimab and camrelizumab were purified with protein A column (GE Healthcare) before loading on a Superdex™ 200 10/300 GL (GE Healthcare, Chicago, United States). The purified protein was stored in the protein buffer (20 mM Tris–HCl, 150 mM NaCl, pH8.0). The protein purity was assessed by SDS-PAGE, and proteins were stained with Coomassie brilliant blue (Supplementary Figure S4). The human PD-L1 gene (full length) was cloned into (Clontech’s, Beijing, China) pEGFP-N1 vector, which was named pEGFP-PD-L1.
For crystal screening, 100 μl of crystallization solution is added to the reservoir of the crystallization chamber. One microliter of cemiplimab-scFv/PD-1 complex protein at a concentration of 5 mg/ml and 1 μl of the crystallization solution are pipetted onto the sitting drop post that is located at the center of beside chamber. Crystallization plates were sealed and placed at 4 or 18°C to perform a sitting drop vapor diffusion experiment. Crystals of cemiplimab-scFv/PD-1 complex were grown in 0.1 M sodium acetate, pH 5.0, 5% w/v γ-PGA (Na+ form, LM), and 20% w/v PEG 2000 MME. The diffraction data were collected at 100 K on the beamlines BL19U1 of the Shanghai Synchrotron Radiation Facility (SSRF). The collected intensities were processed and scaled using the HKL2000 software package (HKL Research). The structures were determined using molecular replacement with the program Phaser MR in CCP4 (35). The search model used in this complex was from Protein Data Bank (PDB) codes 5GGU and 6KTR with the most similar sequences. Model building was performed using COOT by hand (36). Structure refinement was done by using Phenix (37). Structure-related figures in this article were generated using PyMOL (http://www.pymol.org/). The buried surface between MAbs and PD-1 was calculated on the web server (https://www.molnac.unisa.it/BioTools/cocomaps/index.psp).
The SPR measurements between different forms of PD-1 and MAbs were performed on the BIAcore8000 system (GE Healthcare) with Sensor Chip CM5 (GE Healthcare) at room temperature. To measure the binding characteristics between PD-1 antibodies (cemiplimab or camrelizumab) and different forms of PD-1 proteins (PD-1-WT, PD-1-N58A, and PD-1-E. coli), cemiplimab-scFv and camrelizumab-scFv were individually immobilized on the CM5 chip to 695 and 569 response units, respectively. Then, serially diluted PD-1-WT samples and blank control, prepared as 0 , 6.26 , 12.5, 25, 50, and 100 nM, were flowed over Sensor Chip CM5. After regeneration, PD-1-N58A protein, expressed by 293F cells, was flowed over the CM5 sensor chip with various concentrations (50–800 nM, five gradients, twofold dilution). Similarly, different concentrations of PD-1-E. coli (0, 0.5, 1, 2, 4, and 8 μM) were flowed over the CM5 chip. The binding kinetics were all analyzed with the Biacore™ insight evaluation software (GE Healthcare) using a 1:1 Langmuir binding model.
The pEGFP-PD-L1 plasmid was transfected into human embryonic kidney 293 cells (293T) with polyethyleneimine transfection reagent. After 24 h, cell density was adjusted to 1 × 107 cells/ml with phosphate-buffered saline (PBS). PD-1-WT or PD-1-N58A protein (2 ug/ml) was respectively preincubated with different concentrations (0, 2, 4, 10, 20, 40, and 80 μg/ml) of full-length cemiplimab or camrelizumab at room temperature for 30 min. Subsequently, the 293T cells expressing PD-L1 fused enhanced green fluorescent protein (EGFP) were incubated with these mixed samples for a further 30 min at room temperature. The 293T cells were washed three times with PBS and stained with secondary APC mouse anti-His antibody (Cat: 130-119-782; clone: GG11-8F3.5.1; Miltenyi Biotec, Beijing, China) for 30 min, then washed twice with PBS, and resuspended with 300 μl of PBS for flow cytometry (BD FACS Canto Flow Cytometer, Franklin Lakes, USA). The FACS files were analyzed by FlowJo 7.6.
Atomic coordinates have been deposited in the Protein Data Bank (PDB, http://www.rcsb.org) under the accession code 7WVM.
GFG, ST, YC, DL, ZX, and DZ contributed to the conception and design of the study. DL, ZX, DZ, MJ, and JH performed the experiment. DL, KL, ST, and YC performed the statistical analysis. DM, XM, and GFG helped perform the analysis with constructive discussions. ST, DL, and YC wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (NSFC, 31830097 and 32100752).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the staff of BL02U and BL19U beamline at the Shanghai Synchrotron Radiation Facility for assistance with data collection. We thank Zheng Fan and Wei Zhang from the Institute of Microbiology, CAS, for their technical support in the SPR assay. We also thank Ting Li from the Institute of Genetics and Developmental Biology, CAS, for her technical support in the flow cytometry analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.826045/full#supplementary-material
1. Herzberg B, Campo MJ, Gainor JF. Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer. Oncologist (2017) 22(1):81–8. doi: 10.1634/theoncologist.2016-0189
2. Chae YK, Arya A, Iams W, Cruz MR, Chandra S, Choi J, et al. Current Landscape and Future of Dual Anti-CTLA4 and PD-1/PD-L1 Blockade Immunotherapy in Cancer; Lessons Learned From Clinical Trials With Melanoma and Non-Small Cell Lung Cancer (NSCLC). J Immunother Cancer (2018) 6(1):39. doi: 10.1186/s40425-018-0349-3
3. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
4. Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, et al. Liver Metastasis and Treatment Outcome With Anti-PD-1 Monoclonal Antibody in Patients With Melanoma and NSCLC. Cancer Immunol Res (2017) 5(5):417–24. doi: 10.1158/2326-6066.CIR-16-0325
5. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, Upon Programmed Cell Death. EMBO J (1992) 11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
6. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
7. Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
8. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and Activity of Anti-PD-L1 Antibody in Patients With Advanced Cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
9. Helenius A, Markus A. Intracellular Functions of N-Linked Glycans. Science (2001) 291(5512):2364–9. doi: 10.1126/science.291.5512.2364
10. Sun L, Li C-W, Chung EM, Yang R, Kim Y-S, Park AH, et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res (2020) 80(11):2298–310. doi: 10.1158/0008-5472.CAN-19-3133
11. Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, et al. An Unexpected N-Terminal Loop in PD-1 Dominates Binding by Nivolumab. Nat Commun (2017) 8:14369. doi: 10.1038/ncomms14369
12. Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, et al. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-Tumor Immune Responses of T Cells. Cell Rep (2017) 20(5):1017–28. doi: 10.1016/j.celrep.2017.07.027
13. Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, et al. Structure of the Complex of Human Programmed Death 1, PD-1, and its Ligand PD-L1. Structure (2015) 23(12):2341–8. doi: 10.1016/j.str.2015.09.010
14. Na Z, Yeo SP, Bharath SR, Bowler MW, Balikci E, Wang CI, et al. Structural Basis for Blocking PD-1-Mediated Immune Suppression by Therapeutic Antibody Pembrolizumab. Cell Res (2017) 27(1):147–50. doi: 10.1038/cr.2016.77
15. Liu H, Guo L, Zhang J, Zhou Y, Zhou J, Yao J, et al. Glycosylation-Independent Binding of Monoclonal Antibody Toripalimab to FG Loop of PD-1 for Tumor Immune Checkpoint Therapy. MAbs (2019) 11(4):681–90. doi: 10.1080/19420862.2019.1596513
16. Hong Y, Feng Y, Sun H, Zhang B, Wu H, Zhu Q, et al. Tislelizumab Uniquely Binds to the CC' Loop of PD-1 With Slow-Dissociated Rate and Complete PD-L1 Blockage. FEBS Open Bio (2021) 11(3):782–92. doi: 10.1002/2211-5463.13102
17. Liu K, Tan S, Jin W, Guan J, Wang Q, Sun H, et al. N-Glycosylation of PD-1 Promotes Binding of Camrelizumab. EMBO Rep (2020) 21(12):e51444. doi: 10.15252/embr.202051444
18. Burova E, Hermann A, Waite J, Potocky T, Lai V, Hong S, et al. Characterization of the Anti-PD-1 Antibody REGN2810 and its Antitumor Activity in Human PD-1 Knock-In Mice. Mol Cancer Ther (2017) 16(5):861–70. doi: 10.1158/1535-7163.MCT-16-0665
19. Migden MR, Khushalani NI, Chang ALS, Lewis KD, Schmults CD, Hernandez-Aya L, et al. Cemiplimab in Locally Advanced Cutaneous Squamous Cell Carcinoma: Results From an Open-Label, Phase 2, Single-Arm Trial. Lancet Oncol (2020) 21(2):294–305. doi: 10.1016/s1470-2045(19)30728-4
20. Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 Blockade With Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N Engl J Med (2018) 379(4):341–51. doi: 10.1056/NEJMoa1805131
21. Ahmed SR, Petersen E, Patel R, Migden MR. Cemiplimab-Rwlc as First and Only Treatment for Advanced Cutaneous Squamous Cell Carcinoma. Expert Rev Clin Pharmacol (2019) 12(10):947–51. doi: 10.1080/17512433.2019.1665026
22. Lin DY-W, Tanaka Y, Iwasaki M, Gittis AG, Su H-P, Mikami B, et al. The PD-1/PD-L1 Complex Resembles the Antigen-Binding Fv Domains of Antibodies and T Cell Receptors. Proc Natl Acad Sci U S A (2008) 105(8):3011–6. doi: 10.1073/pnas.0712278105
23. Lázár-Molnár E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal Structure of the Complex Between Programmed Death-1 (PD-1) and its Ligand PD-L2. Proc Natl Acad Sci U S A (2008) 105(30):10483. doi: 10.1073/pnas.0804453105
24. Chen D, Tan S, Zhang H, Wang H, He W, Shi R, et al. The FG Loop of PD-1 Serves as a “Hotspot” for Therapeutic Monoclonal Antibodies in Tumor Immune Checkpoint Therapy. iScience (2019) 14:113–24. doi: 10.1016/j.isci.2019.03.017
25. Pinho SS, Reis CA. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat Rev Cancer (2015) 15(9):540–55. doi: 10.1038/nrc3982
26. Wang X, Yang XA-O, Zhang C, Wang Y, Cheng T, Duan L, et al. Tumor Cell-Intrinsic PD-1 Receptor is a Tumor Suppressor and Mediates Resistance to PD-1 Blockade Therapy. Proc Natl Acad Sci U S A (2020) 117(12):6640–50. doi: 10.1073/pnas.1921445117
27. Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Cell Surface Protein Glycosylation in Cancer. Proteomics (2014) 14(4-5):525–46. doi: 10.1002/pmic.201300387
28. Wang M, Wang J, Wang R, Jiao S, Wang S, Zhang J, et al. Identification of a Monoclonal Antibody That Targets PD-1 in a Manner Requiring PD-1 Asn58 Glycosylation. Commun Biol (2019) 2:392. doi: 10.1038/s42003-019-0642-9
29. Liu J, Wang G, Liu L, Wu R, Wu Y, Fang C, et al. Study of the Interactions of a Novel Monoclonal Antibody, Mab059c, With the hPD-1 Receptor. Sci Rep (2019) 9(1):17830. doi: 10.1038/s41598-019-54231-w
30. Finlay WJJ, Coleman JE, Edwards JS, Johnson KS. Anti-PD1 'SHR-1210' Aberrantly Targets Pro-Angiogenic Receptors and This Polyspecificity can be Ablated by Paratope Refinement. MAbs (2019) 11(1):26–44. doi: 10.1080/19420862.2018.1550321
31. Li L, Li G, Rao B, Dong AH, Liang W, Zhu JX, et al. Landscape of Immune Checkpoint Inhibitor-Related Adverse Events in Chinese Population. Sci Rep (2020) 10(1):15567. doi: 10.1038/s41598-020-72649-5
32. Liu WJ, Lan J, Liu K, Deng Y, Yao Y, Wu S, et al. Protective T Cell Responses Featured by Concordant Recognition of Middle East Respiratory Syndrome Coronavirus-Derived CD8+ T Cell Epitopes and Host MHC. J Immunol (2017) 198(2):873–82. doi: 10.4049/jimmunol.1601542
33. Zhang D, Liu K, Lu D, Wang P, Zhang Q, Liu P, et al. Stability and Structure of Bat Major Histocompatibility Complex Class I With Heterologous β2-Microglobulin. JoVE (2021) 169):e61462. doi: 10.3791/61462
34. Lu D, Liu K, Zhang D, Yue C, Lu Q, Cheng H, et al. Peptide Presentation by Bat MHC Class I Provides New Insight Into the Antiviral Immunity of Bats. PloS Biol (2019) 17(9):e3000436. doi: 10.1371/journal.pbio.3000436
35. Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr D Biol Crystallogr (1998) 54(Pt 5):905–21. doi: 10.1107/s0907444998003254
36. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Crystallogr D Biol Crystallogr (2010) 66(Pt 4):486–501. doi: 10.1107/S0907444910007493
Keywords: PD-1, antibody, N58 glycosylation, cemiplimab, immune checkpoint therapy (ICT)
Citation: Lu D, Xu Z, Zhang D, Jiang M, Liu K, He J, Ma D, Ma X, Tan S, Gao GF and Chai Y (2022) PD-1 N58-Glycosylation-Dependent Binding of Monoclonal Antibody Cemiplimab for Immune Checkpoint Therapy. Front. Immunol. 13:826045. doi: 10.3389/fimmu.2022.826045
Received: 30 November 2021; Accepted: 04 February 2022;
Published: 02 March 2022.
Edited by:
Peter Sun, National Institute of Allergy and Infectious Diseases (NIH), United StatesReviewed by:
Gongyi Zhang, National Jewish Health (United States), United StatesCopyright © 2022 Lu, Xu, Zhang, Jiang, Liu, He, Ma, Ma, Tan, Gao and Chai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George F. Gao, Z2FvZkBpbS5hYy5jbg==; Yan Chai, Y2hhaXlhbkBpbS5hYy5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.