 Sinduya Krishnarajah
Sinduya Krishnarajah Burkhard Becher
Burkhard Becher
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 07 March 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.822919
This article is part of the Research Topic Neuroimmunology in Africa View all 16 articles
The invasion of immune cells into the central nervous system (CNS) is a hallmark of the process we call neuroinflammation. Diseases such as encephalitides or multiple sclerosis (MS) are characterised by the dramatic influx of T lymphocytes and monocytes. The communication between inflammatory infiltrates and CNS resident cells is primarily mediated through cytokines. Over the years, numerous cytokine networks have been assessed to better understand the development of immunopathology in neuroinflammation. In MS for instance, many studies have shown that CD4+ T cells infiltrate the CNS and subsequently lead to immunopathology. Inflammatory CD4+ T cells, such as TH1, TH17, GM-CSF-producing helper T cells are big players in chronic neuroinflammation. Conversely, encephalitogenic or meningeal regulatory T cells (TREGs) and TH2 cells have been shown to drive a decrease in inflammatory functions in microglial cells and thus promote a neuroprotective microenvironment. Recent studies report overlapping as well as differential roles of these cells in tissue inflammation. Taken together, this suggests a more complex relationship between effector T cell subsets in neuroinflammation than has hitherto been established. In this overview, we review the interplay between helper T cell subsets infiltrating the CNS and how they actively contribute to neuroinflammation and degeneration. Importantly, in this context, we will especially focus on the current knowledge regarding the contribution of various helper cell subsets to neuroinflammation by referring to their helper T cell profile in the context of their target cell.
T cell mediated immunity is reliant on the differentiation of naïve T cells into their effector T cell counterparts. Upon activation, these cells bifurcate into their two major lineages – CD8-expressing cytotoxic T lymphocytes (CTL), and CD4-expressing helper T cells (TH) (1). CD4+ cells are important in the regulation of the adaptive immune response against a plethora of pathogens. Through differentiation and the secretion of cytokines, these cells help activate antigen-specific B cells to produce antibodies, and hence drive humoral immunity.
About 4 decades ago, it was postulated that CD4 T cells can differentiate into subsets with characteristic effector functions (2). Effector T cells are classified and differentiated based on i) the type of pathogen that elicited the activation and ii) the subsequent group of cytokines secreted by these cells. The main effector subsets of CD4 T cells were historically described to only bifurcate into two distinct populations, driven by their inflammatory milieu (3). Briefly, type 1 versus type 2 immunity was grossly classified as immune responses towards intracellular pathogens versus extracellular parasites and helminths. However, this historical classification has now been revised to include many further helper T cell subsets extending beyond the scope of the original TH1 and TH2 cells.
Further Helper T cell subsets include T follicular helper (TFH) and Regulatory T (TREG) cells. TFH cells work alongside TH1, TH2, or TH17 cells to help B cells generate class-switched immunoglobulins of different isotypes, which are recognised by different innate immune effector cells through cell characteristic expression of cell surface Fc receptors. TREG cells, characterised by their expression of the IL-2 receptor alpha chain CD25 (4) alongside with the transcription factor (TF) FoxP3 (5), have immunoregulatory functions and promote tolerance towards the antigens they recognise, usually self-antigens.
The above-mentioned descriptions of helper T cell subsets fit the historical classification. However, with increasing advances in the field of immunophenotyping, it has become clear that helper T cell nomenclature in the context of a single lead effector cytokine fails to capture the functional diversity of these cells. Thus, we and others propose that T cells should be rather categorised into the kind of help that these cells provide at a site of injury – based on whether their downstream functions affect i) phagocytes (henceforth referred to as type 1 immunity), ii) polymorph-nucleated cells (type 2), or iii) non-immune cells (type 3) (6). This model of naming and classifying T cells is summarised in the form of a schematic as seen in Figure 1. Taking this into account, in this review, we describe the role of helper T cells in the context of their target and effector cells in neuroinflammation.
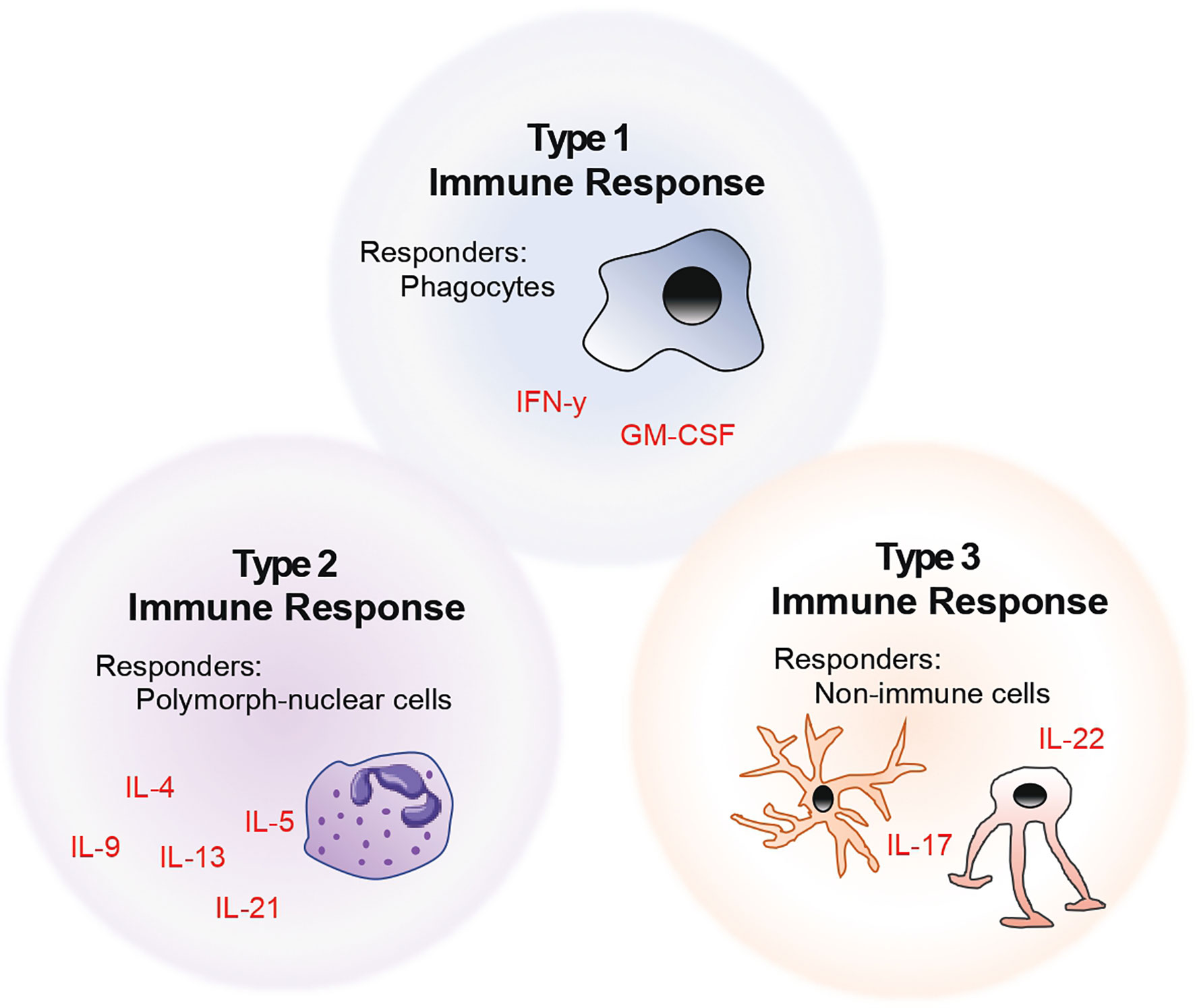
Figure 1 Model of helper T cell classification by considering the role of helper T cells in the context of their target and effector cells.
TH1 cells are the most prominent members of the type 1 TH cell family. TH1 cells were first characterised by their ability to produce interferon gamma (IFN-γ), a potent cytokine with important immunomodulatory functions. TH1 cells help orchestrate the adaptive immune response against intracellular pathogens (e.g. viruses) through direct activation of phagocytic cells or CTLs. These cells in turn directly kill the pathogen or virus infected or transformed host cell in question and can further promote antibody-dependent cellular cytotoxicity (ADCC) and opsonisation.
In addition to IFN-γ, TH1 cells can also be recognised by their cell surface expression of the IL-12 receptor (R) β chains (1 and 2) and the chemokine receptor type 3 (CXCR3). Further work from the late 20th century revealed that there are also key TFs which play important roles in TH1/TH2 polarisation – and thus T-bet was associated with TH1, and GATA-3 with TH2 cells (7–9). The TH1 signal is self-regulating through a positive feedback loop, as IL-12 and IFN-γ both induce T-bet, which in turn induces IFN-γ and T-bet, too (10).
Early studies in an animal model of multiple sclerosis (MS), termed experimental autoimmune encephalomyelitis (EAE), showed that IFN-γ positive cells were the biggest immune cell population in the diseased brain (11, 12), suggesting that TH1 cells were potentially very important in the neuro-pathogenesis of the disease. Furthermore, the adoptive transfer of TH1 cells into naïve animals was shown to drive neuroinflammation, further supporting this notion (13).
The exact role of these brain-infiltrating CD4+ T cells in the context of neuroinflammatory disease is still under investigation. However, a potential downstream target of TH1 mediated effector functions in the central nervous system (CNS) are the resident macrophages of the brain called microglia. Like most other resident macrophages of the body, several studies have suggested that TH1 cells secreting their signature cytokine cocktail leads to the activation of microglia into an inflammatory phenotype (14). In the parenchyma of the brain, microglia are the only resident leukocytes, which makes them a solid contender to interact with T cells invading the CNS in neuroinflammatory conditions (15).
The capacity of these cells to present antigens has been shown in several in vitro studies (16–19). Subsequently, several follow-up studies suggested that microglial activation is directly linked to immune infiltration of the CNS and the maintenance of encephalitogenicity during the effector phase of EAE (20–22). However, in the non-inflamed brain, most cell types including microglia do not express MHC class II or costimulatory molecules. This makes them unlikely to be responsible for the initial reactivation of encephalitogenic T cells.
Key studies were carried out to investigate the bona-fide antigen presentation capabilities of CNS-resident cells, using mouse models where MHC class II expression could be restricted to certain antigen presenting cell (APC) subsets. These experiments revealed that in vivo, neither microglia, nor any other parenchymal elements are required to mediate interactions between APCs and helpers T cells (23). Building on these findings, systematic interrogation of each potential APC within the brain revealed that among the conventional dendritic cell (cDC) subsets, cDC2s in particular are powerful APCs in bridging CNS-T cell interactions (24). Whilst microglia may not be the main players in initiating neuroinflammatory pathology, it is however feasible that during the chronic phases of the disease, microglia play a role in chronification and disease perpetuation.
The most likely immune cell target for type 1 cytokines such as IFN-γ is in fact not resident to the CNS, but instead may invade the CNS from the circulation, namely monocytes. In mice and humans, monocytes come in two flavours. One that is patrolling in the blood (in mice, Ly6Clow) and another capable of reacting to inflammatory stimuli and invading tissues (Ly6Chigh). IFN-γ has been shown to be important for the monocyte to macrophage transition in inflamed sites (25). Nevertheless, the functional consequences of this IFN-γ induced maturation of monocytes remain unclear.
Further studies in animals revealed the extent of the role of TH1 cells in neuroinflammation. IFN-γ is heavily present in the brain lesions present in EAE mice, and the same holds true for MS patients. Clinical trial data revealed the IFN-γ administration to patients suffering with MS made their symptoms worse, and led to increased relapses (26). In contrast though, mice lacking the IL12R β2 chain (27, 28), or the p35 subunit (29), are susceptible to EAE. The same holds true for animals deficient in IFN-γ (30). Moreover, IL-12 administration to mice suffering from early stages of EAE suppressed the disease – the authors of this study also showed that this was an IFN-γ dependent phenomenon (28). Whilst the majority of historical evidence points towards an overall pathogenic role for IFN-γ producing TH1 cells (31), many contradictory studies reveal a potential protective role of these same cells in neuroinflammation (32, 33). To date, the mechanisms by which IL-12 and IFN-γ regulate or suppress neuroinflammation remain completely unknown.
In the context of autoimmunity, studies revealed that IL-23, a cytokine with a shared p40 subunit with IL-12 (34), is important in driving inflammation in models of multiple sclerosis and psoriasiform inflammation. Additionally, the IL-23R comprises the IL12R β1 chain (35) – and these observations helped to clarify the contradictory data described in the previous section. It was then established that IL-23 is a driver of neuroinflammation by the induction of a subset of helper T cells which secrete IL-17 and therefore also activate a type 1 response (36, 37).
Hence, the way was paved for the coining of TH17 cells (36). TH17 cells produce the cytokines IL-17A, IL-17F, IL-21 and IL-22 as lead cytokines (38). The cells are further characterised by the expression of CCR4, CCR6, CD161 as well as IL23R and IL-1R. In addition, these cells express retinoic acid receptor-elated orphan nuclear receptor γτ (RORγτ) intracellularly.
The main reason we call these cells type 3 immune cells is because their primary targets are non-immune cells. Receptors for IL-17 and IL-22 are expressed in various densities throughout the immune as well as stromal compartments. Dysregulation of IL-17 for instance, leads to inflammation of tissues of the body lining, rich in epithelial cells (39). While these mice developed severe skin inflammation, most solid tissues including the CNS were unaffected. In line with this, dysregulation of any members from this group of cytokines, such as IL-17A/F, IL-21 or IL-22, generally leads to pathologies restricted to barrier tissues, like the skin, lung or gut (40–42).
IL-21 was initially described to play an important role in encephalitogenicity (43) – however, this claim was rebuked by many follow-up studies (44, 45).
Whilst these responses are important to curb off an imminent infection, the flipside of a sustained TH17 response is tissue inflammation and damage. In neuroinflammation specifically, these cells have been described to be involved in the pathogenesis of EAE and MS. There have been claims that helper T cells which secrete IL-17 are abundant in both the peripheral blood as well as the cerebrospinal fluid (CSF) of MS patients (46). However, overall, there is no evidence of overt dysregulation of IL-17 signalling itself in MS. Even though a clinical trial neutralising IL-17 in MS has shown some early signs of efficacy, it has not been pursued further and approval was never sought for (47).
Even though disease progression and active disease have also been linked with the increased presence of TH17 cells in patients, the most likely contribution from IL-17 in neuroinflammation may be its effects on the blood brain barrier (BBB). Evidence links IL-17 with barrier function in other organs such as the lung and gut (48, 49), with further experimental data pointing towards IL-17 playing a role in altering of the neurovascular junction being convincing (50, 51). In addition, TH17 cells from patients in relapsing MS are associated with inflammatory lesions and have increased migratory capacities (52).
Astrocytes are a potential neurological cell type which has been investigated in recent years as an effector cell of TH17 responses. They are a subtype of glial cells which reside between the BBB and resident brain cells, are characteristically histologically star-shaped (53), and perform a vast range of functions including tissue maintenance, repair, and regulating cerebral flow. Their main function is directly linked to their location within the brain, where they can monitor and regulate the exchange between the CNS and the systemic circulation (54). Increased expression of a functional IL-17 receptor was demonstrated in vitro (55), as well as under EAE conditions (56, 57). Disruption of IL-17 signalling in these cells was shown to improve EAE in mice (58). However, the signalling pathway targeted in these studies is by no means IL-17 specific, and thus the contribution of IL-17 via astrocytes towards neuroinflammation remains a subject of debate.
Finally, IL-17 also has an effect on a final CNS resident cell type, known as oligodendrocytes. These cells assemble myelin, which is a multi-layered sheath of lipidous membrane around axonal segments. Studies have shown that TH17 cells interfere and inhibit the maturation cycle as well as the survival rate of oligodendrocytes (59, 60).
As discussed previously, recent mounting evidence has led to the belief that helper T cell subsets may not be rigid and cemented in their functional and expression profiles, but that they may adapt according to environmental cues. This is at least true for TH17 cells. There is a strong propensity for these to differentiate into cells that secrete IFN-γ or play the opposing role by producing non-inflammatory IL-10 (61).
A study by Capone and colleagues demonstrated this principle. In relapsing MS patients, TH17 cells upregulate the expression of IL-1R and produce higher levels of IL-21,IL-2, and TNF-β (62). Similarly, within the TH17 compartment of MS patients with active symptoms, another study found elevated expression of IFN-γ and CXCR3 together with reduced expression of IL-10 (63). Conversely, TREGs have been shown to be highly stable (64).
Recent studies have gone a step further and suggested the notion that these subsets may be overlapping in such a manner that their current naming is largely redundant. Cells that secrete both IFN-γ as well as IL-17, hence sitting on the fence between a TH1 and TH17 phenotype (65, 66), have been reported on several occasions. These cells express the receptor for IL-23R. In addition, they co-express CXCR3 and T-bet together with CCR6 and RORγτ. Interestingly, they have been described to produce lower amounts of IL-17A compared to classical TH17 cells but high levels of IFN-γ [reviewed in (67)]. Specifically, in the context of neuroinflammation, cells characterised by the expression of TNF, IFN-γ, IL-2, the CXC chemokine receptor type 4 (CXCR4) and very late antigen 4 (VLA4) were convergent in the blood of patients with MS. These cells were also enriched within the CNS, and were drastically reduced upon therapeutic intervention (68). During acute EAE, cells with a similar mixed helper T cell phenotype can cross the BBB and accumulate in the CNS. Finally, cells with a similar phenotypic profile were also found in brain tissues from MS patients and upregulated in patients during relapse (69, 70).
The observed plasticity across TH cells is clearly beneficial to immunity in the fight against infections. An overly rigid, hard-wired program makes little sense given that the primary role of TH cells is providing ‘help’. This is why we believe that, in the future, a categorisation based on single cytokines or even multiple cytokines will fade in favour of a more nimble and logical description across their specific helper function (6).
In line with a categorisation of TH cells towards their helper function, another prominent cytokine produced by type 1 TH cells is the granulocyte macrophage colony-stimulating factor (GM-CSF). GM-CSF was originally classified as a growth factor contributing to haematopoiesis upon its discovery, as it was shown to lead to the differentiation of bone marrow progenitors into granulocytes and macrophages in vitro (71–73). What makes GM-CSF unique among other CSFs is that lack of either the cytokine, or its receptor, does not lead to any disturbance to myeloid cell development or maintenance in mice (74–76), despite its receptor being almost exclusively expressed within the myeloid compartment.
In vitro, there are compelling data to suggest that GM-CSF promotes DC differentiation from both human and mouse progenitor cells (73, 77). However, the same could not be readily replicated in vivo (78). What was clear is the role of GM-CSF in tissue inflammation, due to evidence pointing to its role in activation and survival of many myeloid cell subtypes such as neutrophils, monocytes and macrophages (79, 80).
GM-CSF expression originates from a plethora of cell types, including haematopoietic cells as well as epithelial or endothelial cells, fibroblasts and stromal cells. Under steady state, healthy physiological conditions, GM-CSF is rarely detected in physiological conditions in vivo – rather, its secretion has also been associated with sites of inflammatory injury (81–83). TH cells secreting GM-CSF were shown to be induced by IL-23 (84), and El-Behi et al. showed that GM-CSF producing cells promote a positive-feedback loop to keep stimulating IL-23 secretion (85). The evidence that GM-CSF is a mandatory cytokine produced by encephalitogenic T cells is overwhelming. IL-1β can further elicit GM-CSF secretion in TH17 cells in vitro, while IL-27, IFN-γ and IL-12 counteracts GM-CSF production (21, 84).
Using a fate-mapping and reporter system for GM-CSF expressing cells, it was shown that secretion of GM-CSF was both IL-23 and IL-1β dependent (86). The specific role of each of these individual cytokines on the expression of GM-CSF is yet to be elucidated. In the same study, cells that formerly secreted GM-CSF were shown to be more likely to express GM-CSF once again in a recall setting as opposed to their GM-CSF naïve counterparts (86). Another study revealed that antigen-independent GM-CSF release by TH cells, and this cytokine alone, was enough to induce neuroinflammation. Interestingly, whilst GM-CSF lead to severe neurological symptoms, other organs were not affected (87). In this study, the authors showed that GM-CSF-induced infiltration of inflammatory phagocytes was confined to the CNS, liver, and lung. Conversely, the skin, colon, and pancreas were spared. This suggests that the specific tissue microenvironments harbour different cues for the invasion of myeloid cells. In addition, it seems that the microenvironment of the target tissue itself influences the effector function of these cells, since the inflammatory phagocytes found in the CNS had a unique genetic signature when compared to the phagocytes within the other tissues. Microarray analysis of in vitro-differentiated cytokine-secreting TH cells identified a large portfolio of genes that were exclusively expressed in GM-CSF-secreting TH cells (88). Altogether, these findings support the notion of a distinct TH subset related to GM-CSF driving neuroinflammation.
There is no doubt that encephalitogenic TH cells play an important role of in propagating neuroinflammation. Even though there is a heavy debate as to whether MS is primarily driven by type 1 or type 3 cytokines, if one considers the cellular composition within neuroinflammatory lesions, it should be termed a type 1-driven immune response. However, the ability of type 3 cytokines (e.g. IL-17) to interact with epithelial and endothelial cells, suggests a role of type 3 immunity in BBB dysfunction. The interplay of other factors and the rest of the cytokine network in neuroinflammation remains to be established. Currently ongoing research is targeted towards elucidating these unanswered questions. Among the most pressing questions is the relative role of CNS resident versus invading cells in immunopathology, and how this intertwines with the instruction delivered by CNS invading TH cells. Equally, among the biggest challenges will be to identify unique molecular patterns of encephalitogenic TH cells which allows for their targeting and neutralisation without collateral broad immunosuppression.
SK conceptualised the manuscript and wrote the first draft. Both SK and BB critically reviewed and revised the manuscript.
Funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme grant agreement No 882424, the Swiss National Science Foundation (733 310030_170320, 310030_188450, and CRSII5_183478 to BB), the CRPP Immunocure and the LOOP Zurich.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Meuer SC, Schlossman SF, Reinherz EL. Clonal Analysis of Human Cytotoxic T Lymphocytes: T4+ and T8+ Effector T Cells Recognize Products of Different Major Histocompatibility Complex Regions. Proc Natl Acad Sci U S A (1982) 79:4395–9. doi: 10.1073/pnas.79.14.4395
2. Parish CR, Liew FY. Immune Response to Chemically Modified Flagellin: III. Enhanced Cell-Mediated Immunity During High and Low Zone Antibody Tolerance to Flagellin. J Exp Med (1972) 135:298–311. doi: 10.1084/jem.135.2.298
3. Cherwinski HM, Schumacher JH, Brown KD, Mosmann TR. Two Types of Mouse Helper T Cell Clone: III. Further Differences in Lymphokine Synthesis Between Th1 and Th2 Clones Revealed by RNA Hybridization, Functionally Monospecific Bioassays, and Monoclonal Antibodies. J Exp Med (1987) 166:1229–44. doi: 10.1084/jem.166.5.1229
4. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155:1151–64.
5. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. J Immunol (2017) 198:986–92. doi: 10.1038/ni904
6. Tuzlak S, Dejean AS, Iannacone M, Quintana FJ, Waisman A, Ginhoux F, et al. Repositioning TH Cell Polarization From Single Cytokines to Complex Help. Nat Immunol (2021) 22:1210–7. doi: 10.1038/s41590-021-01009-w
7. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
8. Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription Factor GATA-3 is Differentially Expressed in Murine Th1 and Th2 Cells and Controls Th2-Specific Expression of the Interleukin-5 Gene. J Biol Chem (1997) 272:21597–603. doi: 10.1074/jbc.272.34.21597
9. Zheng WP, Flavell RA. The Transcription Factor GATA-3 Is Necessary and Sufficient for Th2 Cytokine Gene Expression in CD4 T Cells. Cell (1997) 89:587–96. doi: 10.1016/S0092-8674(00)80240-8
10. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T Cells Through IL-12 Produced by Listeria-Induced Macrophages. Sci (80- ) (1993) 260:547–9. doi: 10.1126/science.8097338
11. Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T Cells in the B10.PL Model of Experimental Allergic Encephalomyelitis (EAE) are of the Th-1 Lymphokine Subtype. Cell Immunol (1989) 124:132–43. doi: 10.1016/0008-8749(89)90117-2
12. Voskuhl RR, Martin R, Bergman C, Dalal M, Ruddle NH, Mcfarland HF. T Helper 1 (TH1) Functional Phenotype of Human Myelin Basic Protein-Specific T Lymphocytes. Autoimmunity (1993) 15:137–43. doi: 10.3109/08916939309043888
13. Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA. Surface Expression of α4 Integrin by CD4 T Cells is Required for Their Entry Into Brain Parenehyma. J Exp Med (1993) 177:57–68. doi: 10.1084/jem.177.1.57
14. Murphy ÁC, Lalor SJ, Lynch MA, Mills KHG. Infiltration of Th1 and Th17 Cells and Activation of Microglia in the CNS During the Course of Experimental Autoimmune Encephalomyelitis. Brain Behav Immun (2010) 24:641–51. doi: 10.1016/j.bbi.2010.01.014
15. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate Mapping Analysis Reveals That Adult Microglia Derive From Primitive Macrophages. Sci (80- ) (2010) 330:841–5. doi: 10.1126/science.1194637
16. Becher B, Antel JP. Comparison of Phenotypic and Functional Properties of Immediately Ex Vivo and Cultured Human Adult Microglia. Glia (1996) 18:1–10. doi: 10.1002/(SICI)1098-1136(199609)18:1<1::AID-GLIA1>3.0.CO;2-6
17. Juedes AE, Ruddle NH. Resident and Infiltrating Central Nervous System APCs Regulate the Emergence and Resolution of Experimental Autoimmune Encephalomyelitis. J Immunol (2001) 166:5168–75. doi: 10.4049/jimmunol.166.8.5168
18. Matyszak MK, Denis-Donini S, Citterio S, Longhi R, Granucci F, Ricciardi-Castagnoli P. Microglia Induce Myelin Basic Protein-Specific T Cell Anergy or T Cell Activation, According to Their State of Activation. Eur J Immunol (1999) 29:3063–76. doi: 10.1002/(SICI)1521-4141(199910)29:10<3063::AID-IMMU3063>3.0.CO;2-G
19. Ulvestad E, Williams K, Bjerkvig R, Tiekotter K, Antel J, Matre R. Human Microglial Cells Have Phenotypic and Functional Characteristics in Common With Both Macrophages and Dendritic Antigen-Presenting Cells. J Leukoc Biol (1994) 56:732–40. doi: 10.1002/jlb.56.6.732
20. Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ. The Clinical Course of Experimental Autoimmune Encephalomyelitis and Inflammation is Controlled by the Expression of CD40 Within the Central Nervous System. J Exp Med (2001) 193:967–74. doi: 10.1084/jem.193.8.967
21. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 Rather Than Interleukin-12 is the Critical Cytokine for Autoimmune Inflammation of the Brain. Nature (2003) 421:744–8. doi: 10.1038/nature01355
22. Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, et al. Experimental Autoimmune Encephalomyelitis Repressed by Microglial Paralysis. Nat Med (2005) 11:146–52. doi: 10.1038/nm1177
23. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, et al. Dendritic Cells Permit Immune Invasion of the CNS in an Animal Model of Multiple Sclerosis. Nat Med (2005) 11:328–34. doi: 10.1038/nm1197
24. Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, Becher B. Conventional DCs Sample and Present Myelin Antigens in the Healthy CNS and Allow Parenchymal T Cell Entry to Initiate Neuroinflammation. Sci Immunol (2019) 4:8380. doi: 10.1126/sciimmunol.aau8380
25. Amorim A, De Feo D, Friebel E, Ingelfinger F, Anderfuhren CD, Krishnarajah S, et al. IFNγ and GM-CSF Control Complementary Differentiation Programs in the Monocyte-to-Phagocyte Transition During Neuroinflammation. Nat Immunol (2022) 23(2):217–28. doi: 10.1038/s41590-021-01117-7
26. Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of Multiple Sclerosis With Gamma Interferon: Exacerbations Associated With Activation of the Immune System. Neurology (1987) 37:1097–102. doi: 10.1212/WNL.37.7.1097
27. Becher B, Durell BG, Noelle RJ. Experimental Autoimmune Encephalitis and Inflammation in the Absence of Interleukin-12. J Clin Invest (2002) 110:493–7. doi: 10.1172/JCI0215751
28. Gran B, Chu N, Zhang GX, Yu S, Li Y, Chen XH, et al. Early Administration of IL-12 Suppresses EAE Through Induction of Interferon-γ. J Neuroimmunol (2004) 156:123–31. doi: 10.1016/j.jneuroim.2004.07.019
29. Zhang G-X, Gran B, Yu S, Li J, Siglienti I, Chen X, et al. Induction of Experimental Autoimmune Encephalomyelitis in IL-12 Receptor-β2-Deficient Mice: IL-12 Responsiveness Is Not Required in the Pathogenesis of Inflammatory Demyelination in the Central Nervous System. J Immunol (2003) 170:2153–60. doi: 10.4049/jimmunol.170.4.2153
30. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice With a Disrupted IFN-Gamma Gene are Susceptible to the Induction of Experimental Autoimmune Encephalomyelitis (EAE). J Immunol (1996) 156:5–7.
31. Pettinelli CB, McFarlin DE. Adoptive Transfer of Experimental Allergic Encephalomyelitis in SJL/J Mice After In Vitro Activation of Lymph Node Cells by Myelin Basic Protein: Requirement for Lyt 1+ 2- T Lymphocytes. J Immunol (1981) 127:1420–3.
32. Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, et al. Enhancement of Experimental Allergic Encephalomyelitis in Mice by Antibodies Against IFN-Gamma. J Immunol (1988) 140:1506–10.
33. Sabatino JJ, Shires J, Altman JD, Ford ML, Evavold BD. Loss of IFN-γ Enables the Expansion of Autoreactive CD4 + T Cells to Induce Experimental Autoimmune Encephalomyelitis by a Nonencephalitogenic Myelin Variant Antigen. J Immunol (2008) 180:4451–7. doi: 10.4049/jimmunol.180.7.4451
34. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel P19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, With Biological Activities Similar as Well as Distinct From IL-12. Immunity (2000) 13:715–25. doi: 10.1016/S1074-7613(00)00070-4
35. Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A Receptor for the Heterodimeric Cytokine IL-23 Is Composed of IL-12rβ1 and a Novel Cytokine Receptor Subunit, IL-23r. J Immunol (2002) 168:5699–708. doi: 10.4049/jimmunol.168.11.5699
36. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 Drives a Pathogenic T Cell Population That Induces Autoimmune Inflammation. J Exp Med (2005) 201:233–40. doi: 10.1084/jem.20041257
37. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol (2005) 6:1133–41. doi: 10.1038/ni1261
38. Stockinger B, Veldhoen M. Differentiation and Function of Th17 T Cells. Curr Opin Immunol (2007) 19:281–6. doi: 10.1016/j.coi.2007.04.005
39. Croxford AL, Karbach S, Kurschus FC, Wörtge S, Nikolaev A, Yogev N, et al. IL-6 Regulates Neutrophil Microabscess Formation in IL-17A-Driven Psoriasiform Lesions. J Invest Dermatol (2014) 134:728–35. doi: 10.1038/jid.2013.404
40. Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 Cells and IL-17 Receptor Signaling are Essential for Mucosal Host Defense Against Oral Candidiasis. J Exp Med (2009) 206:299–311. doi: 10.1084/jem.20081463
41. O’Connor W, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, et al. A Protective Function for Interleukin 17A in T Cell-Mediated Intestinal Inflammation. Nat Immunol (2009) 10:603–9. doi: 10.1038/ni.1736
42. Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological Versus Protective Functions of IL-22 in Airway Inflammation are Regulated by IL-17a. J Exp Med (2010) 207:1293–305. doi: 10.1084/jem.20092054
43. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential Autocrine Regulation by IL-21 in the Generation of Inflammatory T Cells. Nature (2007) 448:480–3. doi: 10.1038/nature05969
44. Coquet JM, Chakravarti S, Smyth MJ, Godfrey DI. Cutting Edge: IL-21 Is Not Essential for Th17 Differentiation or Experimental Autoimmune Encephalomyelitis. J Immunol (2008) 180:7097–101. doi: 10.4049/jimmunol.180.11.7097
45. Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are Not Required for Development of Th17 Cells and Autoimmunity In Vivo. Eur J Immunol (2008) 38:1833–8. doi: 10.1002/eji.200838511
46. Matusevicius D, Kivisäkk P, He B, Kostulas N, Özenci V, Fredrikson S, et al. Interleukin-17 mRNA Expression in Blood and CSF Mononuclear Cells is Augmented in Multiple Sclerosis. Mult Scler (1999) 5:101–4. doi: 10.1177/135245859900500206
47. Havrdová E, Belova A, Goloborodko A, Tisserant A, Wright A, Wallstroem E, et al. Activity of Secukinumab, an Anti-IL-17A Antibody, on Brain Lesions in RRMS: Results From a Randomized, Proof-of-Concept Study. J Neurol (2016) 263:1287–95. doi: 10.1007/s00415-016-8128-x
48. Aden K, Rehman A, Falk-Paulsen M, Secher T, Kuiper J, Tran F, et al. Epithelial IL-23r Signaling Licenses Protective IL-22 Responses in Intestinal Inflammation. Cell Rep (2016) 16:2208–18. doi: 10.1016/j.celrep.2016.07.054
49. Chen K, Eddens T, Trevejo-Nunez G, Way EE, Elsegeiny W, Ricks DM, et al. IL-17 Receptor Signaling in the Lung Epithelium Is Required for Mucosal Chemokine Gradients and Pulmonary Host Defense Against K. Pneumoniae. Cell Host Microbe (2016) 20:596–605. doi: 10.1016/j.chom.2016.10.003
50. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular Mechanisms of IL-17-Induced Blood-Brain Barrier Disruption. FASEB J (2010) 24:1023–34. doi: 10.1096/fj.09-141978
51. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 Lymphocytes Promote Blood-Brain Barrier Disruption and Central Nervous System Inflammation. Nat Med (2007) 13:1173–5. doi: 10.1038/nm1651
52. Colamatteo A, Maggioli E, Azevedo Loiola R, Hamid Sheikh M, Calì G, Bruzzese D, et al. Reduced Annexin A1 Expression Associates With Disease Severity and Inflammation in Multiple Sclerosis Patients. J Immunol (2019) 203:1753–65. doi: 10.4049/jimmunol.1801683
53. Somjen GG. Nervenkitt: Notes on the History of the Concept of Neuroglia. Glia (1988) 1:2–9. doi: 10.1002/glia.440010103
54. Wang DD, Bordey A. The Astrocyte Odyssey. Prog Neurobiol (2008) 86:342–67. doi: 10.1016/j.pneurobio.2008.09.015
55. Das Sarma J, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, et al. Functional Interleukin-17 Receptor A is Expressed in Central Nervous System Glia and Upregulated in Experimental Autoimmune Encephalomyelitis. J Neuroinflamm (2009) 6:1–12. doi: 10.1186/1742-2094-6-14
56. Xiao Y, Jin J, Chang M, Nakaya M, Hu H, Zou Q, et al. TPL2 Mediates Autoimmune Inflammation Through Activation of the TAK1 Axis of IL-17 Signaling. J Exp Med (2014) 211:1689–702. doi: 10.1084/jem.20132640
57. Yi H, Bai Y, Zhu X, Lin L, Zhao L, Wu X, et al. IL-17a Induces MIP-1α Expression in Primary Astrocytes via Src/MAPK/PI3K/NF-kB Pathways: Implications for Multiple Sclerosis. J Neuroimmune Pharmacol (2014) 9:629–41. doi: 10.1007/s11481-014-9553-1
58. Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, et al. Astrocyte-Restricted Ablation of Interleukin-17-Induced Act1-Mediated Signaling Ameliorates Autoimmune Encephalomyelitis. Immunity (2010) 32:414–25. doi: 10.1016/j.immuni.2010.03.004
59. Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, et al. Act1 Mediates IL-17-Induced EAE Pathogenesis Selectively in NG2 + Glial Cells. Nat Neurosci (2013) 16:1401–8. doi: 10.1038/nn.3505
60. Paintlia MK, Paintlia AS, Singh AK, Singh I. Synergistic Activity of Interleukin-17 and Tumor Necrosis Factor-α Enhances Oxidative Stress-Mediated Oligodendrocyte Apoptosis. J Neurochem (2011) 116:508–21. doi: 10.1111/j.1471-4159.2010.07136.x
61. Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of Human CD4+ Cells With CD3 and CD46 Induces a T-Regulatory Cell 1 Phenotype. Nature (2003) 421:388–92. doi: 10.1038/nature01315
62. Capone A, Bianco M, Ruocco G, De Bardi M, Battistini L, Ruggieri S, et al. Distinct Expression of Inflammatory Features in T Helper 17 Cells From Multiple Sclerosis Patients. Cells (2019) 8:533. doi: 10.3390/cells8060533
63. Hu D, Notarbartolo S, Croonenborghs T, Patel B, Cialic R, Yang TH, et al. Transcriptional Signature of Human Pro-Inflammatory TH17 Cells Identifies Reduced IL10 Gene Expression in Multiple Sclerosis. Nat Commun (2017) 8:1–14. doi: 10.1038/s41467-017-01571-8
64. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the Regulatory T Cell Lineage In Vivo. Sci (80- ) (2010) 329:1667–71. doi: 10.1126/science.1191996
65. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface Phenotype and Antigenic Specificity of Human Interleukin 17-Producing T Helper Memory Cells. Nat Immunol (2007) 8:639–46. doi: 10.1038/ni1467
66. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming Growth Factor-β Induces Development of the T H17 Lineage. Nature (2006) 441:231–4. doi: 10.1038/nature04754
67. Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Defining the Human T Helper 17 Cell Phenotype. Trends Immunol (2012) 33:505–12. doi: 10.1016/j.it.2012.05.004
68. Galli E, Hartmann FJ, Schreiner B, Ingelfinger F, Arvaniti E, Diebold M, et al. GM-CSF and CXCR4 Define a T Helper Cell Signature in Multiple Sclerosis. Nat Med (2019) 25:1290–300. doi: 10.1038/s41591-019-0521-4
69. Edwards LJ, Robins RA, Constantinescu CS. Th17/Th1 Phenotype in Demyelinating Disease. Cytokine (2010) 50:19–23. doi: 10.1016/j.cyto.2009.12.003
70. Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, et al. Preferential Recruitment of Interferon-γ-Expressing TH17 Cells in Multiple Sclerosis. Ann Neurol (2009) 66:390–402. doi: 10.1002/ana.21748
71. Burgess AW, Metcalf D. The Nature and Action of Granulocyte - Macrophage Colony Stimulating Factors. Blood (1980) 56:947–58. doi: 10.1182/blood.V56.6.947.947
72. Guthridge MA, Stomski FC, Thomas D, Woodcock JM, Bagley CJ, Berndt MC, et al. Mechanism of Activation of the GM-CSF, IL-3, and IL-5 Family of Receptors. Stem Cells (1998) 16:301–13. doi: 10.1002/stem.160301
73. Inba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of Large Numbers of Dendritic Cells From Mouse Bone Marrow Cultures Supplemented With Granulocyte/Macrophage Colony-Stimulating Factor. J Exp Med (1992) 176:1693–702. doi: 10.1084/jem.176.6.1693
74. Becher B, Tugues S, Greter M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity (2016) 45:963–73. doi: 10.1016/j.immuni.2016.10.026
75. Louis C, Cook AD, Lacey D, Fleetwood AJ, Vlahos R, Anderson GP, et al. Specific Contributions of CSF-1 and GM-CSF to the Dynamics of the Mononuclear Phagocyte System. J Immunol (2015) 195:134–44. doi: 10.4049/jimmunol.1500369
76. Manz MG, Boettcher S. Emergency Granulopoiesis. Nat Rev Immunol (2014) 14:302–14. doi: 10.1038/nri3660
77. Sallusto F, Lanzavecchi A. Efficient Presentation of Soluble Antigen by Cultured Human Dendritic Cells is Maintained by Granulocyte/Macrophage Colony-Stimulating Factor Plus Iuterleukin 4 and Downregulated by Tumor Necrosis Factor α. J Exp Med (1994) 179:1109–18. doi: 10.1084/jem.179.4.1109
78. Vremec D, Lieschke GJ, Dunn AR, Robb L, Metcalf D, Shortman K. The Influence of Granulocyte/Macrophage Colony-Stimulating Factor on Dendritic Cell Levels in Mouse Lymphoid Organs. Eur J Immunol (1997) 27:40–4. doi: 10.1002/eji.1830270107
79. Hamilton JA, Stanley ER, Burgess AW, Shadduck RK. Stimulation of Macrophage Plasminogen Activator Activity by Colony-Stimulating Factors. J Cell Physiol (1980) 103:435–45. doi: 10.1002/jcp.1041030309
80. Handman E, Burgess AW. Stimulation by Granulocyte-Macrophage Colony-Stimulating Factor of Leishmania Tropica Killing by Macrophages. J Immunol (1979) 122:1134–117.
81. Ingelfinger F, Krishnarajah S, Kramer M, Utz SG, Galli E, Lutz M, et al. Single-Cell Profiling of Myasthenia Gravis Identifies a Pathogenic T Cell Signature. Acta Neuropathol (2021) 141:901–15. doi: 10.1007/s00401-021-02299-y
82. Tugues S, Amorim A, Spath S, Martin-Blondel G, Schreiner B, De Feo D, et al. Graft-Versus-Host Disease, But Not Graft-Versus-Leukemia Immunity, Is Mediated by GM-CSF-Licensed Myeloid Cells. Sci Transl Med (2018) 10:8410. doi: 10.1126/scitranslmed.aat8410
83. Williamson DJ, Begley CG, Vadas MA, Metcalf D. The Detection and Initial Characterization of Colony-Stimulating Factors in Synovial Fluid. Clin Exp Immunol (1988) 72:67–73.
84. Codarri L, Gyülvészii G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. Rorγ3t Drives Production of the Cytokine GM-CSF in Helper T Cells, Which is Essential for the Effector Phase of Autoimmune Neuroinflammation. Nat Immunol (2011) 12:560–7. doi: 10.1038/ni.2027
85. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The Encephalitogenicity of TH 17 Cells is Dependent on IL-1- and IL-23-Induced Production of the Cytokine GM-CSF. Nat Immunol (2011) 12:568–75. doi: 10.1038/ni.2031
86. Komuczki J, Tuzlak S, Friebel E, Hartwig T, Spath S, Rosenstiel P, et al. Fate-Mapping of GM-CSF Expression Identifies a Discrete Subset of Inflammation-Driving T Helper Cells Regulated by Cytokines IL-23 and IL-1β. Immunity (2019) 50:1289–304.e6. doi: 10.1016/j.immuni.2019.04.006
87. Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, et al. Dysregulation of the Cytokine GM-CSF Induces Spontaneous Phagocyte Invasion and Immunopathology in the Central Nervous System. Immunity (2017) 46:245–60. doi: 10.1016/j.immuni.2017.01.007
Keywords: helper T (TH) cells, neuroinflammation, cytokines, multiple sclerosis, EAE (experimental autoimmune encephalitis), GMCSF, granulocyte macrophage colony-stimulating factor
Citation: Krishnarajah S and Becher B (2022) TH Cells and Cytokines in Encephalitogenic Disorders. Front. Immunol. 13:822919. doi: 10.3389/fimmu.2022.822919
Received: 26 November 2021; Accepted: 15 February 2022;
Published: 07 March 2022.
Edited by:
Richard Idro, Makerere University, UgandaCopyright © 2022 Krishnarajah and Becher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sinduya Krishnarajah, a3Jpc2huYXJhamFoQGltbXVub2xvZ3kudXpoLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.