 Constance Renault1
Constance Renault1 Nicolas Veyrenche1,2
Nicolas Veyrenche1,2 Franck Mennechet1
Franck Mennechet1 Anne-Sophie Bedin1
Anne-Sophie Bedin1 Jean-Pierre Routy3
Jean-Pierre Routy3 Philippe Van de Perre1,2Jacques Reynes2,4,5
Philippe Van de Perre1,2Jacques Reynes2,4,5 Edouard Tuaillon1,2*
Edouard Tuaillon1,2*- 1Pathogenesis and Control of Chronic and Emerging Infections, INSERM U1058, University of Montpellier, Etablissement Français du Sang, Antilles University, Montpellier, France
- 2Virology Laboratory, CHU de Montpellier, Montpellier, France
- 3Chronic Viral Illness Service and Research Institute and Division of Hematology, McGill University Health Centre, Montreal, QC, Canada
- 4IRD UMI 233, INSERM U1175, University of Montpellier, Montpellier, France
- 5Infectious Diseases Department, CHU de Montpellier, Montpellier, France
Among CD4+ T-cells, T helper 17 (Th17) cells play a sentinel role in the defense against bacterial/fungal pathogens at mucosal barriers. However, Th17 cells are also highly susceptible to HIV-1 infection and are rapidly depleted from gut mucosal sites, causing an imbalance of the Th17/Treg ratio and impairing cytokines production. Consequently, damage to the gut mucosal barrier leads to an enhanced microbial translocation and systemic inflammation, a hallmark of HIV-1 disease progression. Th17 cells’ expression of mucosal homing receptors (CCR6 and α4β7), as well as HIV receptors and co-receptors (CD4, α4β7, CCR5, and CXCR4), contributes to susceptibility to HIV infection. The up-regulation of numerous intracellular factors facilitating HIV production, alongside the downregulation of factors inhibiting HIV, helps to explain the frequency of HIV DNA within Th17 cells. Th17 cells harbor long-lived viral reservoirs in people living with HIV (PLWH) receiving antiretroviral therapy (ART). Moreover, cell longevity and the proliferation of a fraction of Th17 CD4 T cells allow HIV reservoirs to be maintained in ART patients.
Introduction
Toward the Progressive Definition of Th17-Oriented CD4+ T Cells
Over the past decade, functional and phenotype characterizations of T helper (Th) cells have allowed the subsets of lymphocytes to be defined. Th cells’ subpopulations were initially identified among effector cells through their cytokine secretion profiles, eliciting functional characteristics for each type. Hence, subpopulations of effector CD4+ T lymphocytes are referred to as Th1, Th2, Th9, Th17, Th22, T follicular helper cells (Tfh), and Treg regulatory profiles.
The Th17 profile was first described in 2005 by Harrington et al. (1) from a subpopulation previously included in the Th1 profile. Th17 effector cells are characterized by interleukin 17 secretion, expression of the transcription factor RORC2 (retinoic acid receptor-related orphan receptor 2), and cell surface expression of CCR4, CCR6, CD161, and IL-23R (2–4). RORC2 is an isoform of RORγ (nuclear receptor ROR-gamma) encoded by the RORC gene, known as RORγt, in mice (5).
Different homologous genes encode the cytokine members of the IL-17 family (6). This family is composed of six members: IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F (7). IL-17A is the most studied and is directly called IL-17 in many studies (6). Th17 cells also can produce other cytokines, such as IL-21 and IL-22, whereas cells producing IL-17 and IFNγ are referred to as Th1/Th17 cells (3). Th22 cells producing IL-22, IL-13, IL-26, and TNFα but not IL-17 and IFNγ were separated from the Th17 cell subset in 2009 (8).
Th17 cells are located primarily at barrier surfaces in the gastrointestinal tract, lung, and skin, participating in mucosal immunity (9). In the GALT (Gut-Associated Lymphoid Tissue), mainly composed of Peyer’s patches, other lymphoid follicles, and lamina propria, Th17 cells represent 80 to 90% of the total CD4+ T cells (10). Th17 cells also represent a significant quantity of the T cells located in female genital mucosal tissues (11).
Extensive evidence has shown that Th17 CD4+ T cells are highly susceptible to HIV-1 (human immunodeficiency virus) infection and are rapidly depleted from gut mucosal sites (12, 13). Numerous mechanisms recently have been proposed to explain the susceptibility of Th17 cells to HIV infection. This review examines the current state of knowledge about: i) the definition and characteristics of conventional and unconventional Th17 CD4+ T cells; ii) data proving that Th17 CD4+ T cells are a preferential target for HIV reservoirs; and iii) cellular and molecular mechanisms contributing to HIV infection and HIV replication in Th17 CD4+ T cells.
Conventional and Unconventional IL-17–Producing Cells
Conventional and unconventional CD4 T cells can produce IL-17. Conventional CD4 T lymphocytes are characterized by αβ TCR chains (1), whereas unconventional T cells harbor γδ TCR chains (14), semi invariant TCR (Vα7.2) on MAIT cells (Mucosal-Associated Invariant T cells), and invariant TCR (Va24-Ja18 paired with Vb11) on iNKT (invariant Natural Killer cells). Some studies have described γδ T cells as the most significant source of IL-17 secretion during infection (14, 15). MAIT cells, which are mainly CD8 T cells located in the GALT and peripheral organs, produce IL-17 once activated (16, 17). NKT cells (18) (Natural Killer T cell), and particularly iNKT cells (19, 20), also produce IL-17 upon appropriate stimulation.
Th17 Profile’s Development
In an appropriate cytokine environment and under co-stimulation, the recognition of a peptide antigen-class II major histocompatibility complex (MHC), which takes place through interaction with the TCR (T cells receptor), activates naïve T cells and can induce Th17 polarization (Figure 1) (21). Expression of the CD161 receptor by naive T cells may represent an initial condition for the fate of Th17 (4, 22). Among the factors involved in Th17 differentiation, TGF-β (23, 24) (transforming growth factor beta) and IL-6 (25) are essential. IL-6 induces the phosphorylation and dimerization of the transcription factor STAT3 (signal transducer and activator of transcription 3) (26). In turn, STAT3 induces the expression of the transcription factor RORγt and RORα, which are crucial for the orientation of Th17 by binding the IL-17A, IL-21, and IL-22 genes to the promoter regions (26). Activation by IL-6 requires support from TGF-β to induce the expression of RORγt (24), although the role of TGF-β has been questioned (25, 27). IL-1β may also be involved in the upregulation of RORγt (25).
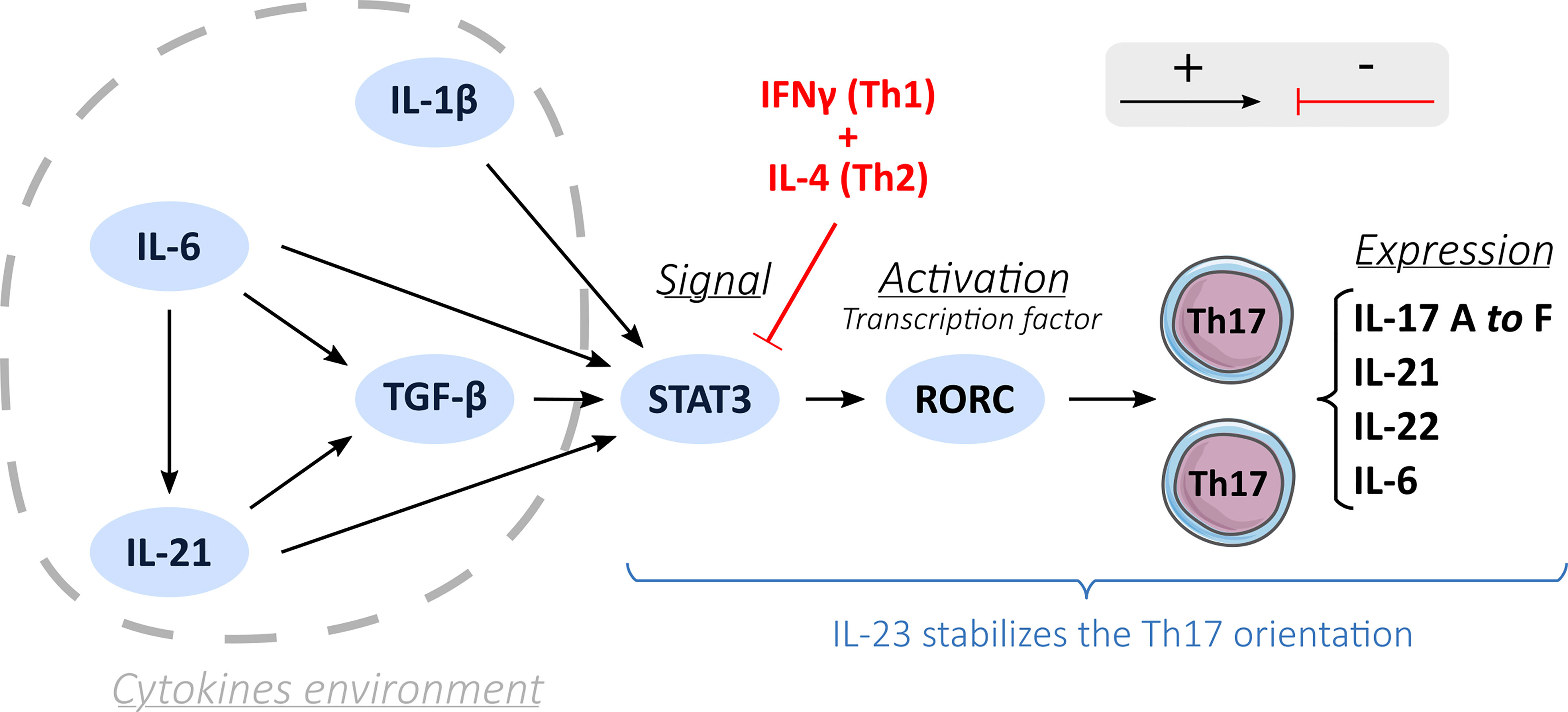
Figure 1 Th17 differentiation process. A description of the Th17 orientation process through different activation factors (black arrows): cytokines environment, induction by STAT3 and activation by a transcription factor RORC and the inhibition of the Th17 orientation process by the Th1 and Th2 cells (red arrows). IL-23 stabilizes the Th17 orientation.
It is thought that Th17 differentiation involves three pathways: i) the classical pathway involving IL-6 alongside TGF-β to induce the transcription of RORγt; ii) the alternative pathway, where IL-21 replaces IL-6; iii) and the third pathway involving IL-23 (24). TGF-β is also part of this third pathway by allowing the expression of the IL-23 receptor (IL-23R) on naïve CD4+ T lymphocytes (24). IL-23 then could engage and amplify the differentiation and growth of Th17 cells (28) and acts on their immune and proinflammatory functions by helping the recruitment of Th17 cells at the inflammation sites and increasing the production of IL-22 cytokines (29).
There is a crucial balance between proinflammatory Th17 and regulatory T cells (Treg) from a developmental perspective (13, 30). Th17 and Treg oriented CD4 T cells have interconnected development and antagonist functions (10, 31). The Foxp3 transcription factor allows the development of Treg cells and inhibits RORγt functions driving Th17 polarization (32). However, under certain immune circumstances, even a Treg cell may become Th17-like, producing IL-17 cytokines and expressing CCR6 at high levels, but a strong activation in the presence of IL-1β and IL-6 could stop this capacity (10).
Immune Functions of Th17 Cells
Th17 cells are abundant in mucosal-associated lymphoid tissues and play a vital role in the immune system’s defense against intestinal bacterial and fungi infections (33, 34). IL-23 produced by dendritic cells in contact with pathogens triggers the activation of mucosal resident memory Th17 cells (34) (Figure 2). The Th17 activated cells up-regulate IL-17 and IL-22 production (24, 30, 34), inducing mucosal epithelial cells to secrete antimicrobial peptides, such as members of the S100 family, HβD-2 (human β-defensin 2), and LCN2 (lipocalin 2) (34). The production of IL-17 also induces the expression of chemotactic factors, such as IL-6, CXCL2, and CXCL8, to recruit mainly neutrophils and macrophages to the infection site. IL-17 also activates the production of G-CSF (granulocyte colony-stimulating factor) and GM-CSF (granulocyte-monocyte colony-stimulating factor) by epithelial cells, fibroblasts, and keratinocytes (33, 34). These two growth factors allow the differentiation of mucosal resident monocytes into macrophages and increase the production and egress of macrophages and neutrophils from the bone marrow. The accumulation of neutrophils and macrophages in the mucosa with the activation of epithelial cells, endothelial cells, and fibroblasts promotes the secretion of proinflammatory cytokines (IL-1, IL-6, IL-23). These cytokines fuel the production of antimicrobial peptides and the local inflammatory response to attract other immune cells to the site of inflammation (34).
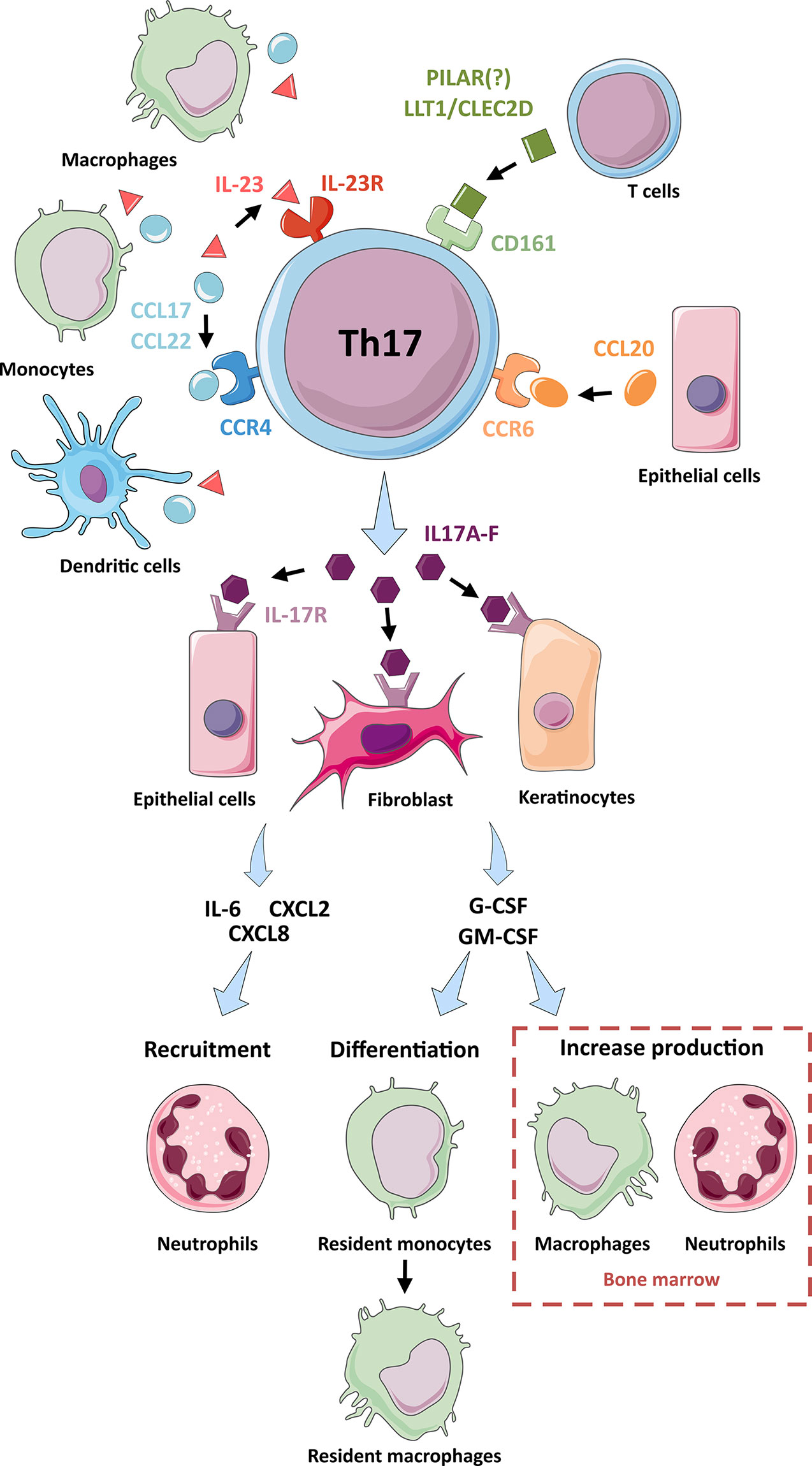
Figure 2 Interactions and functions of Th17 cells. A representation of the interactions of Th17 cells with other cell types: Through their production ligands for receptors on the surface of Th17 cells (IL-23R, CD161, CCR4 and CCR6), macrophages, monocytes and dendritic cells (CCL17, CCL22 and IL-23 production), T cells (LLT1, CLEC2D and PILAR) and epithelial cells (CCL20) participate in the activation of Th17 cells and their production of IL-17 cytokines. The IL-17 cytokines produced by Th17 cells activate epithelial cells, fibroblasts, and keratinocytes via their IL-17R surface receptor. Following this activation, these cells produce chemotactic factors (IL-6, CXCL2 and CXCL8) which participate in the recruitment of neutrophils. These cells also produce G-CSF and GM-CSF, which are involved in the differentiation of resident monocytes into resident macrophages and the increased production of macrophages and neutrophils in the bone marrow.
Laboratory Methods for Identification of Th17-Oriented CD4 T Cells
In addition to identifying Th17 cells by their cytokine phenotype, cell surface phenotypic expression is used to define and study this cell subset (Table 1 and Figure 2). The CCR6 chemokine receptor is the main surface marker of Th17 lymphocytes (2). However, CCR6 expression is not limited to Th17 cells since it also is present on B cells and dendritic cells located in lymphoid tissues of the intestinal mucosa. CCR6 is a receptor of the β-chemokine family (66). Unlike other chemokine receptors, CCR6 has CCL20 as the sole chemokine ligand. CCL20 is expressed at a low basal level, but it increases in the presence of proinflammatory signals. CCR6 also has a low affinity for two microbial peptides: ligands β-defensin 1 and β-defensin 2 (66). CCL20 signaling causes the chemotaxis of Th17 cells towards the gut and other barrier surfaces. In this pathway, this homing receptor is essential for the recruitment of Th17 lymphocytes (66). A study in 2016 reported four Th17 sub-populations: classical Th17 (CCR6+ CXCR3- CCR4+), Th1/Th17 (CCR6+ CXCR3+ CCR4-), double-negative (CCR6+ CXCR3- CCR4-) and double-positive (CCR6+ CXCR3+ CCR4+.) (60) These four sub-populations produce IL-17A but differ in their capacity to produce other cytokines such as IL-17F, IL-10, IL-13, IL-22, IFNγ, GM-CSF, and the CCR6+ double-negative population is described as a significant producer of IL-17A and IL-17F (60).

Table 1 Phenotypes used to characterize and sort Th17 cells.
The CD161 surface receptor, formerly called NKR-P1 (Natural Killer Receptor), is the second hallmark of Th17 cells. CD161 can be expressed on αβ and γδ T cells, on the majority of NK cells, and NKT and MAIT cells (22). The physiological ligand of CD161 is LLT1 (the lectin-like transcript-1), belonging to the CLEC2 family (C-type lectin domain 2 family). This ligand could promote the transendothelial migration of Th17 effectors into tissues after recruitment via the CCL20-CCR6 interaction (22).
The first description of the CD161 receptor as a marker for Th17 cells was published in 2008 (67). In the CD161+ cell population, we can identify the Th17, Th1, and Th1/Th17 profiles, whereas in the CD161- population, we can only find the Th1 and Th1/Th17 profiles. The association of CCR6 and CD161 surface markers can allow, therefore, the identification of the vast majority of Th17-oriented cells.
IL23-R and CD26 can also be used to complete the cell surface phenotype of Th17 cells. IL-23R is a receptor expressed on the surface of Th17 cells (2, 3, 55). IL-23 is a heterodimer with a p40 subunit common to the cytokine IL-12 and a different p19 subunit, produced by monocytes, macrophages, and activated dendritic cells (68). On Th17 cells, the binding of IL-23 to its receptor IL-23R induces their activation (69). Hence, the cytokine IL-23 plays a role in the proinflammatory process mediated by Th17 cells. CD26 (dipeptidyl-peptidase IV) expression also can be used to identify Th17 oriented cells (57). CD26 is an active enzyme involved in the activation of T cells and found in tissue inflammatory responses. Th17 oriented cells have a high expression of CD26, which also is expressed to a lesser extent by Th1 and Th2 oriented cells.
HIV Infection and Th17 Cells
Susceptibility of Th17 Cells to HIV Infection
Mael-A-Nantawat’s team first demonstrated the susceptibility of Th17 cells to HIV infection in 2007 (35). Compared to Th1 and Th2 oriented CD4 T cells, Th17 cells show a superior permissivity and the highest level of HIV DNA integration in vitro and in vivo (12, 39). In contrast, during SIV (simian immunodeficiency virus) infection of rhesus macaques, Th17 cells are not preferentially infected compared to CD4+ memory T cells (70). These data suggest a limitation of using the SIV model to study differences in infectivity between Th17 cells and other CD4+ T cells. Th17 cells are enriched for different HIV dependency factors (HDFs) essential for virus replication (58, 71). The mechanisms allowing this enhanced permissibility can be separate into the entry and post-entry phases of HIV infection.
Entry level
HIV entry requires an attachment with the CD4 receptor, an interaction with either the CCR5 or CXCR4 co-receptors, and is facilitated by the presence of the α4β7 receptor (Figure 3). CD4+ helper T cells expressing these receptors are the primary target of HIV (11, 13, 44, 72). Whereas CD4 T cells express mainly CXCR4 and infrequently CXCR4 plus CCR5 co-receptors, CD4 Th17-oriented cells frequently express both CCR5 (3, 21, 54) and CXCR4 (3, 44). Consequently, Th17 cells are permissive in vitro to infection by viruses that have R5 (CCR5) or X4 (CXCR4) tropism (39, 56).
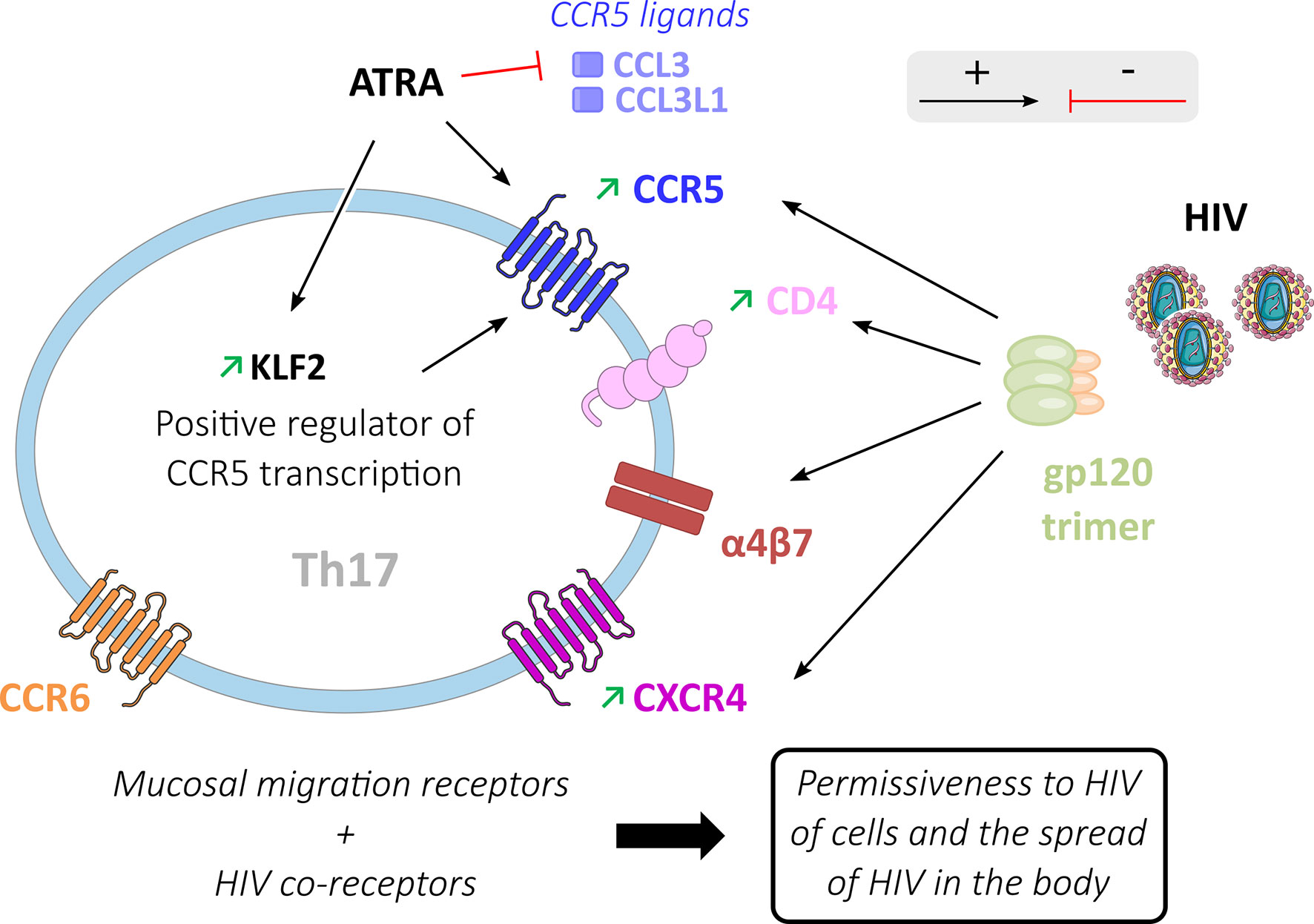
Figure 3 HIV entry process in Th17 cells. Activating (black arrows) or inhibiting (red arrows) factors are involved in HIV entry into Th17 cells. Mucosal migration receptors (α4β7 and CCR6) and HIV gp120 binding to co-receptors (CD4, CCR5, CXCR4 and α4β7) increase HIV permissiveness in these Th17 cells where these receptors are preferentially expressed. ATRA inhibits the production of classical ligands of the CCR5 receptor (CCL3, CCL3L1) thereby promoting the interaction of CCR5 with gp120. ATRA also increases the expression of KLF2 in Th17 cells, a positive regulator of CCR5 transcription.
The CD4 and CXCR4 expression in Th17 oriented cells may be modulated by cell activation. Following in vitro polyclonal stimulation by anti-CD3/CD28, IL1-β, and IL-23, the subsets of CCR6+ CD4 T cells show higher expression of CD4 and CXCR4 than CCR6- cells, which may contribute to the greater permissiveness of Th17 cells to HIV infection (44). CCR5+ CD4+ memory T cells in healthy subjects are composed of 10 to 20% of Treg cells, 20 to 50% of Th1 cells, and 20 to 40% of Th17 cells (65). Different teams have shown that Th17 cells present a significantly higher expression of the CCR5 co-receptor compared to CCR6- cell subtypes (12, 56, 61, 63, 73). ATRA (All-Trans Retinoic Acid), a vitamin A metabolite produced by dendritic cells in the GALT (74), increases the expression of CCR5 in Th17 cells (21). ATRA induces a higher expression of CCR5 on memory CCR6 CD4 T cells co-stimulated by CD3/CD28, whereas its effect is modest on CCR6 negative cells (21). ATRA up-regulates transcription of KLF2 (Krüppel-like Factor 2) (63), a positive regulator of CCR5 transcription (75), and down-regulates the CCR5 ligands CCL3 and CCL3L1 (63), which have been shown to decrease the risk of HIV infection in vivo (76). Furthermore, ATRA plays a key role in the homing capacity of inflammatory Th17 cells in the intestine through a positive effect on CCR9 and α4β7 expression (77), which are required for cell migration to the gut. Hence, ATRA could be one of the factors involved in the permissiveness of Th17 cells to HIV infection by CCR5 expression and homing to the GALT.
The homing receptor α4β7 is strongly expressed on gut Th17 cells. The α4β7 receptor is an accessory binding target of the gp120 receptor (44, 63, 77, 78). Compared with the CD4 receptor, the α4β7 heterodimer is prominent because of its larger size. This characteristic makes α4β7 an efficient receptor for the capture of HIV particles (79). HIV attachment to the cell facilitates ensuing interaction with the CD4 entry receptor and the dissemination of the virus between cells (78). α4β7+ cells have been reported to be a preferential target of HIV during acute HIV-1 infection (80). Th17 cells are more permissive to HIV than CCR6- cells with low α4β7 expression (44). Th17 cell expression of mucosal migration receptors (CCR6 and α4β7), as well as HIV co-receptors (CD4, α4β7, CCR5, and CXCR4), participates in the remarkable capacity of these cells to disseminate HIV following initial infection (21).
Post-Entry Level and Replication
After cell entry, the virus uses its enzymes and host machinery to reverse transcribe HIV RNA, integrate into the human genome, produce viral proteins, and form new viruses. Signaling pathways of Th17 cells such as mTORc1 (mammalian Target Of Rapamycin Complex 1) are involved in the post-entry steps of HIV replication (63) (Figure 4).
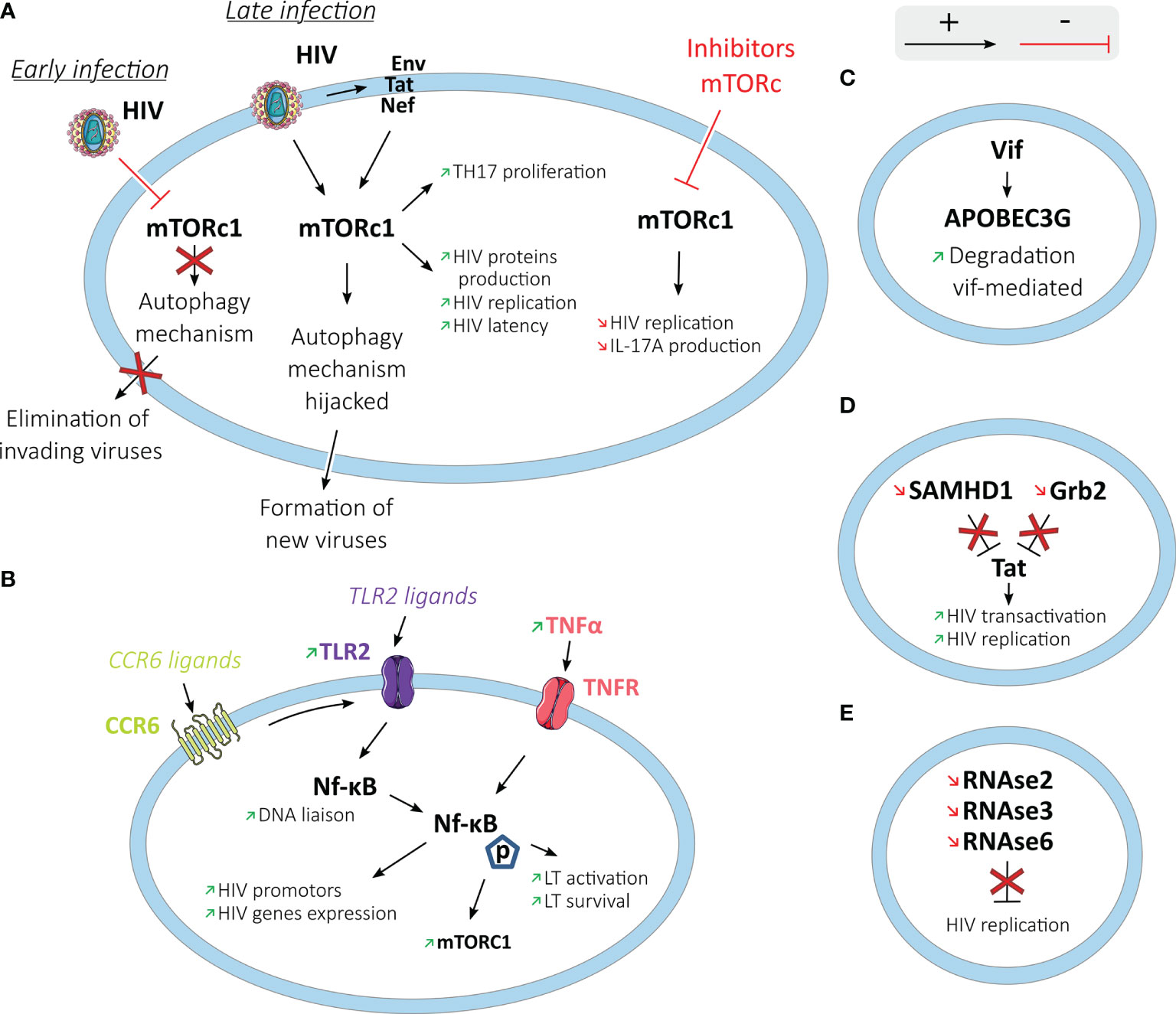
Figure 4 HIV post-entry process and replication in Th17 cells. Th17 cells and HIV have different interactions (activating in black arrows and inhibiting in red arrows) to promote HIV replication in these cells. (A) In the mTOR pathway: at an early stage of the infection, HIV stops the autophagy mechanism directed by mTORc1 to block the elimination of invading viruses. At a later stage, HIV interacts with mTORc1 to hijack the autophagy mechanism to help form new viruses. By interacting with HIV or its proteins (Env, Tat, or Nef), mTORc1 also increases Th17 proliferation, HIV protein production, HIV replication, and latency. (B) by interaction with NF-κB: increased production of TNFα and TLR2 ligation increase in Th17 cells NF-κB translocation and activity, leading to increased HIV replication. Increased NF-κB activity also increases the activity of the mTORc1 pathway and the activation and survival of T cells. (C) Th17 cells exhibit Vif-mediated degradation of the HIV restriction factor, APOBEC3G. (D) SAMHD1 and Grb2 are downregulated in Th17 cells, allowing increased HIV transactivation and replication. (E) RNase2, RNAse3, and RNAse6 genes known to inhibit viral replication are downregulated in Th17 cells.
Activation of mTORc1 enhances Th17 differentiation, whereas mTORc1 dysfunction impairs it (81). In contrast, mTORc1 and mTORc2 inhibit Treg differentiation (82, 83). mTORc1 controls autophagy, which is an intracellular protective mechanism recycling cellular elements and eliminating newly formed viruses (84).
HIV-1 interacts with mTORc1, inhibiting or activating the mTORc1 complex (85). During the acute infection stage, different HIV-1 proteins inhibit mTORc1 activity, limiting autophagy. At a later stage of the infection in T cells, macrophages, dendritic and neuronal cells, HIV-1 hijacks the autophagy mechanism using the autophagosomal membrane to assemble viruses (86, 87). It has been shown that ATRA exposure increases mTOR expression and phosphorylation and HIV replication in Th17 cells (63). In contrast, metformin, which has an indirect inhibiting activity on mTOR, is shown to significantly decrease its activation through its phosphorylation in colonic CCR6+ cells (88). mTORc1 inhibitors such as Rapamycin and INK128 block HIV transcription through Tat-dependent and independent mechanisms (89). Tat is an HIV protein that acts as a viral transactivator.
Several HIV dependency factors (HDFs) were overexpressed in Th17 oriented CD4 T cells compared to Th1, Th2, and Th1/Th17 cells (58). Hence, higher expression of PAK2 (p21 Activated Kinase 2), PI3K (Phosphoinositide 3-Kinase), ZAP-70, and Lck (lymphocyte-specific protein tyrosine kinase) were observed using genome-wide transcriptional profiling (58). Binding of PAK2 by the HIV protein Nef impairs cell functions and facilitates virus replication by disrupting the dynamic rearrangements of the actin cytoskeleton in response to stimulation (90). PI3K participates in the negative control of MHC1 (Major Histocompatibility Complex 1) molecules mediated by the HIV protein Nef (91). ZAP-70 and Lck increase TCR signal and facilitate HIV replication by inducing cell activation upon weak TCR signals (92–94).
Some transcripts associated with a restriction of HIV replication are down-regulated in Th17 cells compared to Th1 cells, like Grb2 (Growth factor Receptor-Bound protein 2), known to inhibit HIV-1 LTR transactivation mediated by Tat during HIV replication (58, 95). Th17 cells present an enhanced Vif-mediated degradation of APOBEC3G (Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G), an HIV restriction factor (73, 96). SAMHD1 (SAM domain and HD domain-containing protein 1) is also found to be down-regulated in Th17 cells (51). The reverse transcriptase activity requires the intracellular pool of dNTPs to replicate HIV; however, SAMHD1 can block this replication in dendritic cells, macrophages, monocytes, and CD4+ T cells by depleting the intracellular pool of dNTPs (97, 98). SAMHD1low circulating memory CD4+ T cells are enriched in Th17 cells (median at 41%) and demonstrate high levels of HIV-1 DNA (52). Higher levels of HIV-1 DNA were observed in the CD45RO+ SAMHD1low memory population (4.5 HIV-1 DNA log copies/106cells) compared to CD45RO+ SAMHD1+ memory cells (3.8 HIV-1 DNA log copies/106cells, p=0.009) and CD45RO- SAMHD1+ naive cells (3.1 HIV-1 DNA log copies/106cells, p<0.0001). Likewise, a higher population of p24 producing cells was detected within the SAMHD1low memory population in viremic and treated individuals (52). SAMHD1low memory cells harbored a higher percentage of Ki67 expression (15.2%) compared to SAMHD1+ memory cells (3.37%) and SAMHD1+ naïve cells (0.23%). A positive correlation was observed between HIV-1 DNA levels and Ki67 expression.
NF-κB is another transcription factor involved in T cells activation and cell survival (99). NF-κB also can initiate HIV genome transcription by liaison with the HIV LTR enhancer region promotor, which contains two NF-κB binding sites (100). Th17 cells have been found to have an increased nuclear translocation activity of NF-κB (nuclear factor-kappa B) and DNA liaison compared to Th1 cells (58). The increased activity of NF-κB in Th17 cells compared to Th1 cells is associated with higher production of TNF-α (63). TNF-α (Tumor Necrosis Factor-α) can increase HIV replication by activating NF-κB (101). Once activated, TLR2 (Toll-like receptor 2) leads to NF-κB translocation (102). TLR2 is a member of the TLR family, which can recognize different PAMPs (Pathogen-associated molecular patterns), and thus different types of pathogens (103). TLR2 activity could enhance the susceptibility of CD4+ T cells to HIV-1 productive infection, notably by significantly increasing p65 phosphorylation (NF-κB activity marker) in Th17 cells (61, 104). Th17 cells, quiescent or activated, present higher levels of TLR2 expression in FACS analysis (61). Following in vitro activation, TLR2 ligation induces a higher level of HIV-1 replication in Th17 cells compared to CCR6- cells (61).
Increased Lck and ZAP-70 expressions in Th17 cells compared to Th1 cells can enhance the capacity of Th17 cells to be activated in response to weak TCR signals (58). This enhanced capacity to increase cell metabolism creates an environment suitable for HIV replication. RORγt, the key transcription factor for Th17 cell orientation, can promote HIV-1 replication (105). RORγt is thought to regulate viral gene expression through its binding to the HIV-1 LTR. Th17 cells can also increase intracellular HIV replication by a lower expression of some members of the RNase A (Ribonuclease A) superfamily previously described as factors inhibiting viral replication (60, 73). RNase2, RNase3, and RNase6 genes, all known to have viral replication inhibition activity, are expressed at lower levels in Th17 cells compared to CCR6- cells on RNA microarray analysis (73).
Overall, the up-regulation of numerous factors facilitating HIV replication, such as PAK2, ZAP-70, Lck, NF-κB with TNF-α or TLR2, alongside the down-regulation of factors inhibiting HIV, such as Grb2 and SAMHD1, in Th17 oriented CD4 helps explain why these cells represent a favored target for HIV.
Quantitative Depletion to Th17 Cells and Effects of Antiretroviral Therapy
Depletion of Th17 Cell Pool
The severe depletion of Th17 polarized CD4 T cells is observed early during HIV infection (73). Many studies have shown that the depletion of Th17 cells occurs in the blood compartment (13, 36, 40, 54), the intestinal mucosa (13, 36, 40, 64), and the genital mucosa (42, 45). Furthermore, the number of Th17 cells circulating is inversely correlated with HIV viral load (59).
Impact of ART in Th17 Cell Recovery
ART reduces plasma HIV to undetectable levels and induces a progressive replenishment of CD4+ T cells (38). However, after four years of effective therapy, ART appears to be only partially effective in restoring Th17 CD4 T cells (40). After six years on ART, the Th17 cell frequency can be normalized in blood, but recovery of Th17 function is highly variable between individuals (48). Early versus delayed initiation of ART also affects Th17 cell recovery. The number of Th17 cells in the intestinal mucosa could return to a normal level when treatment is initiated in the acute infection phase but not when initiated during the chronic phase of HIV infection (13, 40, 44).
Th17 CD4 T cell recovery also appears variable in the different anatomical compartments and between the different Th17 cell subsets (40). The pool of mucosal Th17 cells is less rapidly restored than the blood counterpart (40, 106). A study of Th17 CD4 T cells in female genital mucosa has observed partial recovery of Th17 cells under ART with a low capacity of Th17 related cytokine production (48). The difficulty encountered by Th17 cells in restoring their cell pool may be partially explained by the decrease in CD4+ CD161+ cells, considered Th17 cell precursors with mucosal homing capacity (22, 67). In untreated subjects with a chronic HIV infection, the proportion of circulating CD4+ CD161+ cells is lower than in healthy controls (39).
The CCR6+ CXCR3- CCR4- (double negative) cell subtype may be preserved during HIV infection in blood and lymph nodes, unlike the Th17 (CCR6+ CXCR3- CCR4+), Th1/Th17 (CCR6+ CXCR3+ CCR4-), and double-positive (CCR6+ CXCR3+ CCR4+) subtypes, which are more severely depleted and only partially restored under ART (60). CCR6+ double negative cells may be oriented to the three other subtypes based on the differentiation signals emitted (60).
CD4+ T cells have been explored by transcriptional analysis in HIV-infected individuals maintaining an undetectable HIV-1 viral load over time in the absence of ART (elite controllers) (107). Genes associated with Th1, Th17, and Th22 orientation were highly expressed in HIV-specific CD4+ T cells in elite controllers compared to untreated patients with a progressive HIV infection, in particular the genes enabling the production of the transcription factor RORC driving Th17 differentiation and the cytokines IL-17 and IL-22 (107). A high production of IL-17A, IL-17F, and IL-22 is therefore found in elite controllers. In contrast, in untreated subjects with a progressive disease, the production of these cytokines is low or undetectable. After initiation of ART, the difference persists between the elites controllers and subjects with a chronic infection, with a low expression in untreated subjects of genes associated with Th17 cells amongst HIV-specific CD4+ T cells (107).
Functional Impairments Related to Th17 Cell Depletion
The impaired functionality of the Th17 cell population on ART may contribute to functional problems observed in people living with HIV.
Intestinal Barrier Function
Th17 cells play a crucial role in regulating gut mucosal immune defense against microbial pathogens (108). Depletion of CD4+ T cells in gut-associated lymphoid tissues occurs early during HIV infection and persists durably on ART, disrupting the intestinal barrier’s integrity.
The early decrease of mucosal Th17 cells from the acute HIV infection, and incomplete replenishment of Th17 cells on ART, lead to a frequent and prolonged impairment of the antimicrobial immune defense (54). Plasma levels of LPS (lipopolysaccharide) and soluble CD14 levels, markers of microbial translocation, are increased in untreated and treated HIV-infected subjects (40). Plasma levels of BDG ((1➔3)-β-D-Glucan), a compound of the fungi membrane and a marker of fungal translocation, are also elevated in HIV-infected individuals (109). These bacterial and fungal translocations are associated with systemic immune activation and tissue inflammation (109). Cells in inflamed gut tissue are described as having a 27-fold higher frequency of HIV DNA than cells in non-inflamed gut tissue (110). This immune activation and inflammation is thought to lead to an increase in the frequency of other conditions such as metabolic and cardiovascular diseases (111).
In turn, bacterial translocation and chronic immune activation impair the cytokine expression of Th17 cells. Th17-oriented cells naturally produce IL-10 in minimal quantities and proinflammatory cytokines (IL-17, IL-22, TNFα, and IFNγ) in large quantities (13). In uninfected controls, a median of 69.7% of sigmoid Th17 cells produce TNFα, and 0.3% produce IL-10 (13). In the early stages of an HIV infection, IL-10 production increases in sigmoid Th17 cells (3.5%), whereas TNFα production decreases (43.4%), allowing an IL-10/TNFα imbalance (13). This ratio in IL-10/TNFα production normalizes in the chronic stage of HIV infection and under long-term ART, with increased production of TNFα in sigmoid colon Th17 cells (88.0%) in ART subjects (versus 69.7% in uninfected controls at p=0.002).
The Dynamic of Th17/Treg Balance During HIV Infection and on ART
The relative proportion of Th17 versus Treg cells is essential to maintain immune homeostasis under physiological conditions. In healthy controls, the Th17/Treg cells ratio is usually between 1.0 and 1.2. HIV infection disrupts the Th17/Treg ratio axis (10) with a balance shifted in favor of Treg cells. A rapid decrease of Th17 cells is observed in ART naïve subjects, while the decline in the number of Treg cells is gradual (39). Both the number and frequency of Th17 cells decrease during chronic HIV infection, whereas Treg cell numbers decrease slowly and the Treg proportion increases. The Th17/Treg ratio decay becomes severe at a late stage of HIV infection, with values ranging from 0.75 to 0.2. In contrast, data suggest that Th17/Treg balance may be preserved in long-term non-progressors and maintained at an average level in elite controllers (47, 49). Tryptophan catabolism into immunosuppressive kynurenine (Kyn) by indoleamine 2,3-dioxygenase (IDO) could contribute to this difference in the Th17/Treg ratio (112). The enzyme IDO, which catabolizes Trp to Kyn, is also described as a marker of inflammation for the study of HIV-1 progression (112). IDO is thought to impact the Th17/Treg balance by stimulating Treg cells and blocking their reprogramming into Th17 (112). Early administration of ART could block this Treg cell stimulation and restore the Kyn/Trp ratio (113).
Monitoring Treg cells in HIV-infected patients beginning ART shows a rise in the frequency of Treg cells circulating in the first weeks and a subsequent decrease to reach values similar to healthy controls (114). While Treg recovery is rapid during the first months of ART, the gain of Th17 oriented cells is delayed, contributing to the imbalance of the Th17/Treg ratio (43). Th17/Treg balance can be restored when treatment is initiated in the early phase of the infection and after prolonged ART associated with a gradual rise of Th17 cell numbers (43).
HIV Reservoirs in Th17 Cells
One of the main characteristics of an HIV reservoir is its persistence under viral suppressive treatment (115). Different cell subsets located in different anatomical compartments comprise the pool of HIV-infected cells. Mucosal tissues such as the intestinal mucosa are enriched sites for HIV infection, replication, and reservoir (48, 64, 115, 116). HIV infection impairs intestinal mucosal immunity from the early phase of infection via a profound depletion of mucosal CD4 + T cells (117). The persistence of HIV reservoirs in the GALT in patients undergoing ART is a major obstacle to the eradication of HIV (118). GALT lymphocytes comprise a high proportion of effector memory and activated CD4+ lymphocytes (115). Hence, it is of prime importance to study Th17 cells for their ability to serve as an HIV reservoir (72).
Quantity and Heterogeneity of HIV Reservoirs in Th17-Oriented Cells
HIV persists in circulating and gut located CD4+ CCR6+ T cells in patients under treatment (62). In patients on ART, the CCR6+ population has significantly higher levels of HIV DNA in the blood (1.5-fold) and colon (2.8-fold) than CCR6- cells. CCR6+ cells also have a higher expression of the activation marker HLA-DR than CCR6- cells in the colon (62). The size of the HIV reservoir has been compared in CD161+ and CD161- cells from patients on ART (56). The quantity of HIV-1 DNA measured by qPCR was found to be 6.7-fold higher in CD161+ cells than in CD161- cells (56). The level of intact HIV reservoirs measured by Intact Proviral DNA Assay (IPDA) was 13.0-fold higher in the CD161+ cells than in the CD161 negative counterpart (56). Likewise, the number of replication-competent latent HIV measured by quantitative viral outgrowth assay (QVOA) was 2.1-fold higher in CD161+ cells than CD161- cells (56).
The different subsets of Th17 cells, classical Th17 (CCR6+ CXCR3- CCR4+), Th1/Th17 (CCR6+ CXCR3+ CCR4-), double negative (CCR6+ CXCR3- CCR4-) and double positive (CCR6+ CXCR3+ CCR4+), harbor HIV reservoirs. These different subsets of IL-17 producing cells contain integrated HIV DNA and are replication-competent HIV-infected cells (60).
Th17 cells can produce viral particles after four days of CD3/CD28 stimulation (60). Using a modified viral outgrowth assay, the authors have shown expression of HIV-p24 associated alongside IL-17A (60). In vitro infection of CD4 T cells shows that Th17 oriented cells account for 18% of the total population of infected cells and constitute a fifth of p24+ cells, whereas they represent only 6.2% of CD4 T cells (73). This observation has been confirmed in vivo using cytometry and single-cell analysis; among HIV-infected cells, Th17 cells (CCR6+ CXCR3- CCR4+) accounted for a quarter of p24-producing cells (53). Th17 cells presented higher p24 secretion in cell supernatants and a higher level of HIV RNA in sorted cells (73).
PCR quantification of HIV-1 DNA from patients under prolonged ART confirmed that Th17 and Th1/Th17 cells significantly contribute to the HIV reservoir relative to their frequency among T CD4 memory cells (119). In comparison, the Th1-oriented cells appeared to have a proportionate contribution to the HIV reservoir relative to their frequency, while Th2 cells appeared to have a minor contribution to the HIV reservoir. In this study, the follow-up of patients showed that the contribution of Th17 and Th1/Th17 cells to the HIV reservoir increased from 29% on short-term ART to 53% under long-term therapy, whereas it slightly lowered in Th1 cells (119).
Longevity, Proliferation Capacity, and Clonal Expansion
A long lifespan is an essential characteristic of the cells participating in the latent reservoir. Furthermore, the proliferation of latently infected cells generates clones of CD4 T cells that carry integrated proviruses (120).
In 2011, a team described Th17 cells sharing the characteristics of longevity and plasticity of stem cells (121). The Transcription Factor 7 protein (Tcf7) plays a crucial role in regulating self-renewal capacity versus differentiation of stem cells through its association with beta-catenin that operates in the absence of autocrine Wnt signaling (122, 123). In vitro Th17 cells have a higher expression of Tcf7 associated with a significant accumulation of the protein β-catenin compared to Th1 cells and naive cells (121). By testing apoptosis resistance and survival capacity in vitro, Th17 cells showed less activation-induced cell death (AICD) and a remarkable ability to persist the pool of memory cells compared to Th1 cells (121).
The subset of CCR6+ double negative (CXCR3- CCR4-) cells regarded as CD4 T cells engaged toward the early development of Th17 orientation presents a transcriptional signature of self-renewal (60). Hence, some pathways and markers of human stem cells, such as NANOG, LEF1, and MYC, were found up-regulated in CCR6+ double negative cells, as well as TORC, an anti-senescence marker (60).
The link between long-term survival, proliferation capacities, and clonal expansion of the latent HIV-1 reservoir also was studied in CD161+ Th17-oriented cells and compared to CD161- cells (56). CD161+ cells presented a significantly higher expression of c-kit and Bcl-2, two critical molecules for cell survival (56, 124, 125). CD161+ cells also showed a higher expression OX40 (56), a protein associated with long-term survival and clonal proliferation of CD4+ cells (126). In patients on ART, clonal HIV-1 sequences were more frequently detected in OX40 positive CD4 T cells (127).
Conclusion
In many respects, Th17 cells are a critical cell compartment of the HIV reservoir. Through their prominent location in the gastrointestinal tract and their participation in the mucosal tissues’ immunity, Th17 cells are actively implicated in a preferential anatomical site and cellular subset for HIV infection. Th17 cells participate in the dissemination of HIV following infection through their expression of mucosal migration receptors (CCR6, α4β7) and HIV co-receptors (CD4, α4β7, CCR5, and CXCR4) through different biological pathways. Th17 cells facilitate HIV replication at the entry-level as well as the post-entry and productive levels. The long-term cell survival and proliferation capacities of Th17 cells are characteristics that contribute to the persistence and clonal expansion of HIV in ART-treated persons. Hence, Th17 cells account for a large proportion of HIV reservoirs compared to their relative frequency among CD4 memory T cells. Innovative therapeutic approaches aiming to achieve an HIV cure should consider and take advantage of the characteristics and particularities of Th17 cells (128).
Author Contributions
Writing, original draft preparation, figure creation, CR. Writing, review and editing, NV, FM, A-SB, JPR, PP, JR, and ET. All authors contributed to the article and approved the submitted version.
Funding
CR was supported by the University of Montpellier, the Centre Hospitalier Universitaire (CHU) de Montpellier, Fédération Hospitalo-Universitaire Infections Chroniques (InCH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-Producing CD4+ Effector T Cells Develop via a Lineage Distinct From the T Helper Type 1 and 2 Lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
2. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface Phenotype and Antigenic Specificity of Human Interleukin 17-Producing T Helper Memory Cells. Nat Immunol (2007) 8(6):639–46. doi: 10.1038/ni1467
3. Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and Functional Features of Human Th17 Cells. J Exp Med (2007) 204(8):1849–61. doi: 10.1084/jem.20070663
4. Cosmi L, Cimaz R, Maggi L, Santarlasci V, Capone M, Borriello F, et al. Evidence of the Transient Nature of the Th17 Phenotype of CD4+CD161+ T Cells in the Synovial Fluid of Patients With Juvenile Idiopathic Arthritis. Arthritis Rheumatol (2011) 63(8):2504–15. doi: 10.1002/art.30332
5. Jetten AM. Retinoid-Related Orphan Receptors (RORs): Critical Roles in Development, Immunity, Circadian Rhythm, and Cellular Metabolism. Nucl Recept Signal (2009) 7:e003. doi: 10.1621/nrs.07003
6. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 Family of Cytokines in Health and Disease. Immunity (2019) 50(4):892–906. doi: 10.1016/j.immuni.2019.03.021
7. Wacleche VS, Landay A, Routy JP, Ancuta P. The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis. Viruses (2017) 9(10):303. doi: 10.3390/v9100303
8. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of Interleukin 22 But Not Interleukin 17 by a Subset of Human Skin-Homing Memory T Cells. Nat Immunol (2009) 10(8):857–63. doi: 10.1038/ni.1767
9. Klatt NR, Brenchley JM. Th17 Cell Dynamics in HIV Infection. Curr Opin HIV AIDS (2010) 5(2):135–40. doi: 10.1097/COH.0b013e3283364846
10. Valverde-Villegas JM, Matte MCC, de Medeiros RM, Chies JAB. New Insights About Treg and Th17 Cells in HIV Infection and Disease Progression. J Immunol Res (2015) 2015:647916. doi: 10.1155/2015/647916
11. Rodriguez-Garcia M, Barr FD, Crist SG, Fahey JV, Wira CR. Phenotype and Susceptibility to HIV Infection of CD4+ Th17 Cells in the Human Female Reproductive Tract. Mucosal Immunol (2014) 7(6):1375–85. doi: 10.1038/mi.2014.26
12. Gosselin A, Monteiro P, Chomont N, Diaz-Griffero F, Said EA, Fonseca S, et al. Peripheral Blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T Cells Are Highly Permissive to HIV-1 Infection. J Immunol (2010) 184(3):1604–16. doi: 10.4049/jimmunol.0903058
13. Kim CJ, McKinnon LR, Kovacs C, Kandel G, Huibner S, Chege D, et al. Mucosal Th17 Cell Function Is Altered During HIV Infection and is an Independent Predictor of Systemic Immune Activation. J Immunol (2013) 191(5):2164–73. doi: 10.4049/jimmunol.1300829
14. Chien Yh, Zeng X, Prinz I. The Natural and the Inducible: Interleukin (IL)-17-Producing γδ T Cells. Trends Immunol (2013) 34(4):151–4. doi: 10.1016/j.it.2012.11.004
15. Price AE, Reinhardt RL, Liang HE, Locksley RM. Marking and Quantifying IL-17A-Producing Cells In Vivo. PloS One (2012) 7(6):e39750. doi: 10.1371/journal.pone.0039750
16. Dusseaux M, Martin E, Serriari N, Péguillet I, Premel V, Louis D, et al. Human MAIT Cells are Xenobiotic-Resistant, Tissue-Targeted, CD161hi IL-17-Secreting T Cells. Blood (2011) 117(4):1250–9. doi: 10.1182/blood-2010-08-303339
17. Gibbs A, Leeansyah E, Introini A, Paquin-Proulx D, Hasselrot K, Andersson E, et al. MAIT Cells Reside in the Female Genital Mucosa and are Biased Towards IL-17 and IL-22 Production in Response to Bacterial Stimulation. Mucosal Immunol (2017) 10(1):35–45. doi: 10.1038/mi.2016.30
18. Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, et al. Cutting Edge: NKT Cells Constitutively Express IL-23 Receptor and RORgammat and Rapidly Produce IL-17 Upon Receptor Ligation in an IL-6-Independent Fashion. J Immunol (2008) 180(8):5167–71. doi: 10.4049/jimmunol.180.8.5167
19. Michel M-L, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-Producing NK1.1(neg) iNKT Cell Population Involved in Airway Neutrophilia. J Exp Med (2007) 204(5):995–1001. doi: 10.1084/jem.20061551
20. Moreira-Teixeira L, Resende M, Coffre M, Devergne O, Herbeuval J-P, Hermine O, et al. Proinflammatory Environment Dictates the IL-17-Producing Capacity of Human Invariant NKT Cells. J Immunol (2011) 186(10):5758–65. doi: 10.4049/jimmunol.1003043
21. Monteiro P, Gosselin A, Wacleche VS, El-Far M, Said EA, Kared H, et al. Memory CCR6+CD4+ T Cells are Preferential Targets for Productive HIV Type 1 Infection Regardless of Their Expression of Integrin β7. J Immunol (2011) 186(8):4618–30. doi: 10.4049/jimmunol.1004151
22. Fergusson JR, Fleming VM, Klenerman P. CD161-Expressing Human T Cells. Front Immunol (2011) 2:36. doi: 10.3389/fimmu.2011.00036
23. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
24. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
25. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 But Not Transforming Growth Factor-Beta are Essential for the Differentiation of Interleukin 17-Producing Human T Helper Cells. Nat Immunol (2007) 8(9):942–9. doi: 10.1038/ni1496
26. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity (2008) 29(1):44–56. doi: 10.1016/j.immuni.2008.05.007
27. Evans HG, Suddason T, Jackson I, Taams LS, Lord GM. Optimal Induction of T Helper 17 Cells in Humans Requires T Cell Receptor Ligation in the Context of Toll-Like Receptor-Activated Monocytes. Proc Natl Acad Sci USA (2007) 104(43):17034–9. doi: 10.1073/pnas.0708426104
28. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J Biol Chem (2003) 278(3):1910–4. doi: 10.1074/jbc.M207577200
29. Stockinger B, Veldhoen M. Differentiation and Function of Th17 T Cells. Curr Opin Immunol (2007) 19(3):281–6. doi: 10.1016/j.coi.2007.04.005
30. Cambier L, Defaweux V, Baldo A, Mathy A, Tabart J, Bagut lena T, et al. Rôle Des Cellules Th17 Dans Les Maladies Infectieuses Et Auto-Immunes. Ann Méd Vét (2010) 154(154):104–12.
31. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic Conversion of Foxp3+ T Cells Into TH17 Cells in Autoimmune Arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
32. Zhou L, Lopes JE, Chong MMW, Ivanov II, Min R, Victora GD, et al. TGF-β-Induced Foxp3 Inhibits T H 17 Cell Differentiation by Antagonizing Rorγt Function. Nature (2008) 453(7192):236–40. doi: 10.1038/nature06878
33. Khader SA, Gaffen SL, Kolls JK. Th17 Cells at the Crossroads of Innate and Adaptive Immunity Against Infectious Diseases at the Mucosa. Mucosal Immunol (2009) 2(5):403–11. doi: 10.1038/mi.2009.100
34. Peck A, Mellins ED. Precarious Balance: Th17 Cells in Host Defense. Infect Immun (2010) 78(1):32–8. doi: 10.1128/IAI.00929-09
35. Maek-A-Nantawat W, Buranapraditkun S, Klaewsongkram J, Ruxrungtham K, Ruxrungthum K. Increased Interleukin-17 Production Both in Helper T Cell Subset Th17 and CD4-Negative T Cells in Human Immunodeficiency Virus Infection. Viral Immunol (2007) 20(1):66–75. doi: 10.1089/vim.2006.0063
36. Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, et al. Differential Th17 CD4 T-Cell Depletion in Pathogenic and Nonpathogenic Lentiviral Infections. Blood (2008) 112(7):2826–35. doi: 10.1182/blood-2008-05-159301
37. Macal M, Sankaran S, Chun T-W, Reay E, Flamm J, Prindiville TJ, et al. Effective CD4+ T-Cell Restoration in Gut-Associated Lymphoid Tissue of HIV-Infected Patients Is Associated With Enhanced Th17 Cells and Polyfunctional HIV-Specific T-Cell Responses. Mucosal Immunol (2008) 1(6):475–88. doi: 10.1038/mi.2008.35
38. Ndhlovu LC, Chapman JM, Jha AR, Snyder-Cappione JE, Pagán M, Leal FE, et al. Suppression of HIV-1 Plasma Viral Load Below Detection Preserves IL-17 Producing T Cells in HIV-1 Infection. AIDS (2008) 22(8):990–2. doi: 10.1097/QAD.0b013e3282ff884e
39. Prendergast A, Prado JG, Kang Y-H, Chen F, Riddell LA, Luzzi G, et al. HIV-1 Infection is Characterized by Profound Depletion of CD161+ Th17 Cells and Gradual Decline in Regulatory T Cells. AIDS (2010) 24(4):491–502. doi: 10.1097/QAD.0b013e3283344895
40. Chege D, Sheth PM, Kain T, Kim CJ, Kovacs C, Loutfy M, et al. Sigmoid Th17 Populations, the HIV Latent Reservoir, and Microbial Translocation in Men on Long-Term Antiretroviral Therapy. AIDS (2011) 25(6):741–9. doi: 10.1097/QAD.0b013e328344cefb
41. Brandt L, Benfield T, Mens H, Clausen LN, Katzenstein TL, Fomsgaard A, et al. Low Level of Regulatory T Cells and Maintenance of Balance Between Regulatory T Cells and TH17 Cells in HIV-1-Infected Elite Controllers. J Acquir Immune Defic Syndr (2011) 57(2):101–8. doi: 10.1097/QAI.0b013e318215a991
42. McKinnon LR, Nyanga B, Chege D, Izulla P, Kimani M, Huibner S, et al. Characterization of a Human Cervical CD4+ T Cell Subset Coexpressing Multiple Markers of HIV Susceptibility. J Immunol (2011) 187(11):6032–42. doi: 10.4049/jimmunol.1101836
43. He Y, Li J, Zheng Y, Luo Y, Zhou H, Yao Y, et al. A Randomized Case-Control Study of Dynamic Changes in Peripheral Blood Th17/Treg Cell Balance and Interleukin-17 Levels in Highly Active Antiretroviral-Treated HIV Type 1/AIDS Patients. AIDS Res Hum Retroviruses (2012) 28(4):339–45. doi: 10.1089/AID.2011.0140
44. Alvarez Y, Tuen M, Shen G, Nawaz F, Arthos J, Wolff MJ, et al. Preferential HIV Infection of CCR6+ Th17 Cells is Associated With Higher Levels of Virus Receptor Expression and Lack of CCR5 Ligands. J Virol (2013) 87(19):10843–54. doi: 10.1128/JVI.01838-13
45. McKinnon LR, Nyanga B, Kim CJ, Izulla P, Kwatampora J, Kimani M, et al. Early HIV-1 Infection is Associated With Reduced Frequencies of Cervical Th17 Cells. J Acquir Immune Defic Syndr (2015) 68(1):6–12. doi: 10.1097/QAI.0000000000000389
46. Li Y, Sun W. Effects of Th17/Treg Cll Imbalance on HIV Replication in Patients With AIDS Complicated With Tuberculosis. Exp Ther Med (2018) 15(3):2879–93. doi: 10.3892/etm.2018.5768
47. Saxena V, Patil A, Tayde R, Bichare S, Chinchkar V, Bagul R, et al. HIV-Specific CD4+Th17 Cells From HIV Infected Long-Term Non-Progressors Exhibit Lower CTLA-4 Expression and Reduced Apoptosis. Immunobiology (2018) 223(11):658–62. doi: 10.1016/j.imbio.2018.07.011
48. Caruso MP, Falivene J, Holgado MP, Zurita DH, Laufer N, Castro C, et al. Impact of HIV-ART on the Restoration of Th17 and Treg Cells in Blood and Female Genital Mucosa. Sci Rep (2019) 9(1):1978. doi: 10.1038/s41598-019-38547-1
49. Caetano DG, de Paula HHS, Bello G, Hoagland B, Villela LM, Grinsztejn B, et al. HIV-1 Elite Controllers Present a High Frequency of Activated Regulatory T and Th17 Cells. PloS One (2020) 15(2):e0228745. doi: 10.1371/journal.pone.0228745
50. Becattini S, Latorre D, Mele F, Foglierini M, De Gregorio C, Cassotta A, et al. T Cell Immunity. Functional Heterogeneity of Human Memory CD4+ T Cell Clones Primed by Pathogens or Vaccines. Science (2015) 347(6220):400–6. doi: 10.1126/science.1260668
51. Ruffin N, Brezar V, Ayinde D, Lefebvre C, Schulze Zur Wiesch J, van Lunzen J, et al. Low SAMHD1 Expression Following T-Cell Activation and Proliferation Renders CD4+ T Cells Susceptible to HIV-1. AIDS (2015) 29(5):519–30. doi: 10.1097/QAD.0000000000000594
52. Hani L, Chaillon A, Nere ML, Ruffin N, Alameddine J, Salmona M, et al. Proliferative Memory SAMHD1low CD4+ T Cells Harbour High Levels of HIV-1 With Compartmentalized Viral Populations. PloS Pathog (2019) 15(6):e1007868. doi: 10.1371/journal.ppat.1007868
53. Pardons M, Baxter AE, Massanella M, Pagliuzza A, Fromentin R, Dufour C, et al. Single-Cell Characterization and Quantification of Translation-Competent Viral Reservoirs in Treated and Untreated HIV Infection. PloS Pathog (2019) 15(2):e1007619. doi: 10.1371/journal.ppat.1007619
54. El Hed A, Khaitan A, Kozhaya L, Manel N, Daskalakis D, Borkowsky W, et al. Susceptibility of Human Th17 Cells to Human Immunodeficiency Virus and Their Perturbation During Infection. J Infect Dis (2010) 201(6):843–54. doi: 10.1086/651021
55. Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, et al. CD161 is a Marker of All Human IL-17-Producing T-Cell Subsets and Is Induced by RORC. Eur J Immunol (2010) 40(8):2174–81. doi: 10.1002/eji.200940257
56. Li X, Liu Z, Li Q, Hu R, Zhao L, Yang Y, et al. CD161+ CD4+ T Cells Harbor Clonally Expanded Replication-Competent HIV-1 in Antiretroviral Therapy-Suppressed Individuals. mBio (2019) 10(5):e02121–19. doi: 10.1128/mBio.02121-19
57. Bengsch B, Seigel B, Flecken T, Wolanski J, Blum HE, Thimme R. Human Th17 Cells Express High Levels of Enzymatically Active Dipeptidylpeptidase IV (Cd26). J Immunol (2012) 188(11):5438–47. doi: 10.4049/jimmunol.1103801
58. Cleret-Buhot A, Zhang Y, Planas D, Goulet J-P, Monteiro P, Gosselin A, et al. Identification of Novel HIV-1 Dependency Factors in Primary CCR4(+)CCR6(+)Th17 Cells via a Genome-Wide Transcriptional Approach. Retrovirology (2015) 12:102. doi: 10.1186/s12977-015-0226-9
59. Dunay GA, Tóth I, Eberhard JM, Degen O, Tolosa E, van Lunzen J, et al. Parallel Assessment of Th17 Cell Frequencies by Surface Marker Co-Expression Versus Ex Vivo IL-17 Production in HIV-1 Infection. Cytom B Clin Cytom (2016) 90(6):486–92. doi: 10.1002/cyto.b.21352
60. Wacleche VS, Goulet J-P, Gosselin A, Monteiro P, Soudeyns H, Fromentin R, et al. New Insights Into the Heterogeneity of Th17 Subsets Contributing to HIV-1 Persistence During Antiretroviral Therapy. Retrovirology (2016) 13(1):59. doi: 10.1186/s12977-016-0293-6
61. Bolduc JF, Ouellet M, Hany L, Tremblay MJ. Toll-Like Receptor 2 Ligation Enhances HIV-1 Replication in Activated CCR6+ CD4+ T Cells by Increasing Virus Entry and Establishing a More Permissive Environment to Infection. J Virol (2017) 91(4):e01402–16. doi: 10.1128/JVI.01402-16
62. Gosselin A, Wiche Salinas TR, Planas D, Wacleche VS, Zhang Y, Fromentin R, et al. HIV Persists in CCR6+CD4+ T Cells From Colon and Blood During Antiretroviral Therapy. AIDS (2017) 31(1):35–48. doi: 10.1097/QAD.0000000000001309
63. Planas D, Zhang Y, Monteiro P, Goulet J-P, Gosselin A, Grandvaux N, et al. HIV-1 Selectively Targets Gut-Homing CCR6+CD4+ T Cells via mTOR-Dependent Mechanisms. JCI Insight (2017) 2(15):e93230. doi: 10.1172/jci.insight.93230
64. Côté SC, Stilla A, Burke Schinkel SC, Berthoud TK, Angel JB. IL-7-Induced Proliferation of Peripheral Th17 Cells is Impaired in HAART-Controlled HIV Infection. AIDS (2019) 33(6):985–91. doi: 10.1097/QAD.0000000000002164
65. Zaunders J, Munier CML, McGuire HM, Law H, Howe A, Xu Y, et al. Mapping the Extent of Heterogeneity of Human CCR5+ CD4+ T Cells in Peripheral Blood and Lymph Nodes. AIDS (2020) 34(6):833–48. doi: 10.1097/QAD.0000000000002503
66. Ito T, Carson WF, Cavassani KA, Connett JM, Kunkel SL. CCR6 as a Mediator of Immunity in the Lung and Gut. Exp Cell Res (2011) 317(5):613–9. doi: 10.1016/j.yexcr.2010.12.018
67. Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, et al. Human Interleukin 17-Producing Cells Originate From a CD161+CD4+ T Cell Precursor. J Exp Med (2008) 205(8):1903–16. doi: 10.1084/jem.20080397
68. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel P19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, With Biological Activities Similar as Well as Distinct From IL-12. Immunity (2000) 13(5):715–25. doi: 10.1016/S1074-7613(00)00070-4
69. Malamut G. [IL-17/IL-23: A New Therapeutic Target in Inflammatory Bowel Diseases]. Gastroenterol Clin Biol (2008) 32(3):354–6. doi: 10.1016/j.gcb.2008.02.017
70. Lai SH, Starke CE, Flynn JK, Vinton CL, Ortiz AM, Mudd JC, et al. Simian Immunodeficiency Virus Infects Functionally Polarized Memory CD4 T Cells Equivalently In Vivo. J Virol (2019) 93(9):e02163-18. doi: 10.1128/JVI.02163-18
71. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, et al. Pro-Inflammatory Human Th17 Cells Selectively Express P-Glycoprotein and are Refractory to Glucocorticoids. J Exp Med (2014) 211(1):89–104. doi: 10.1084/jem.20130301
72. Ivanov II, Frutos R de L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific Microbiota Direct the Differentiation of IL-17-Producing T-Helper Cells in the Mucosa of the Small Intestine. Cell Host Microbe (2008) 4(4):337–49. doi: 10.1016/j.chom.2008.09.009
73. Christensen-Quick A, Lafferty M, Sun L, Marchionni L, DeVico A, Garzino-Demo A. Human Th17 Cells Lack HIV-Inhibitory RNases and Are Highly Permissive to Productive HIV Infection. J Virol (2016) 90(17):7833–47. doi: 10.1128/JVI.02869-15
74. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The Role of Retinoic Acid in Tolerance and Immunity. Immunity (2011) 35(1):13–22. doi: 10.1016/j.immuni.2011.07.002
75. Richardson MW, Jadlowsky J, Didigu CA, Doms RW, Riley JL. Kruppel-Like Factor 2 Modulates CCR5 Expression and Susceptibility to HIV-1 Infection. J Immunol (2012) 189(8):3815–21. doi: 10.4049/jimmunol.1201431
76. Casazza JP, Brenchley JM, Hill BJ, Ayana R, Ambrozak D, Roederer M, et al. Autocrine Production of Beta-Chemokines Protects CMV-Specific CD4 T Cells From HIV Infection. PloS Pathog (2009) 5(10):e1000646. doi: 10.1371/journal.ppat.1000646
77. Wang C, Kang SG, HogenEsch H, Love PE, Kim CH. Retinoic Acid Determines the Precise Tissue Tropism of Inflammatory Th17 Cells in the Intestine. J Immunol (2010) 184(10):5519–26. doi: 10.4049/jimmunol.0903942
78. Arthos J, Cicala C, Martinelli E, Macleod K, Van Ryk D, Wei D, et al. HIV-1 Envelope Protein Binds to and Signals Through Integrin Alpha4beta7, the Gut Mucosal Homing Receptor for Peripheral T Cells. Nat Immunol (2008) 9(3):301–9. doi: 10.1038/ni1566
79. Cicala C, Arthos J, Fauci AS. HIV-1 Envelope, Integrins and Co-Receptor Use in Mucosal Transmission of HIV. J Transl Med (2011) 9 Suppl 1:S2. doi: 10.1186/1479-5876-9-S1-S2
80. Tokarev A, McKinnon LR, Pagliuzza A, Sivro A, Omole TE, Kroon E, et al. Preferential Infection of α4β7+ Memory CD4+ T Cells During Early Acute Human Immunodeficiency Virus Type 1 Infection. Clin Infect Dis (2020) 71(11):e735–43. doi: 10.1093/cid/ciaa497
81. Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-Mtorc1-S6K1/2 Axis Controls Th17 Differentiation by Regulating Gfi1 Expression and Nuclear Translocation of Rorγ. Cell Rep (2012) 1(4):360–73. doi: 10.1016/j.celrep.2012.02.007
82. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T Cell Receptor Signaling Controls Foxp3 Expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A (2008) 105(22):7797–802. doi: 10.1073/pnas.0800928105
83. Nagai S, Kurebayashi Y, Koyasu S. Role of PI3K/Akt and mTOR Complexes in Th17 Cell Differentiation. Ann N Y Acad Sci (2013) 1280:30–4. doi: 10.1111/nyas.12059
84. Hay N, Sonenberg N. Upstream and Downstream of mTOR. Genes Dev (2004) 18(16):1926–45. doi: 10.1101/gad.1212704
85. Akbay B, Shmakova A, Vassetzky Y, Dokudovskaya S. Modulation of Mtorc1 Signaling Pathway by HIV-1. Cells (2020) 9(5):1090. doi: 10.3390/cells9051090
86. Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, et al. Autophagy Pathway Intersects With HIV-1 Biosynthesis and Regulates Viral Yields in Macrophages. J Cell Biol (2009) 186(2):255–68. doi: 10.1083/jcb.200903070
87. Nardacci R, Ciccosanti F, Marsella C, Ippolito G, Piacentini M, Fimia GM. Role of Autophagy in HIV Infection and Pathogenesis. J Intern Med (2017) 281(5):422–32. doi: 10.1111/joim.12596
88. Planas D, Pagliuzza A, Ponte R, Fert A, Marchand LR, Massanella M, et al. LILAC Pilot Study: Effects of Metformin on mTOR Activation and HIV Reservoir Persistence During Antiretroviral Therapy. EBioMedicine (2021) 65:103270. doi: 10.1016/j.ebiom.2021.103270
89. Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe (2016) 20(6):785–97. doi: 10.1016/j.chom.2016.11.001
90. Van den Broeke C, Radu M, Chernoff J, Favoreel HW. An Emerging Role for P21-Activated Kinases (Paks) in Viral Infections. Trends Cell Biol (2010) 20(3):160–9. doi: 10.1016/j.tcb.2009.12.005
91. Blagoveshchenskaya AD, Thomas L, Feliciangeli SF, Hung CH, Thomas G. HIV-1 Nef Downregulates MHC-I by a PACS-1- and PI3K-Regulated ARF6 Endocytic Pathway. Cell (2002) 111(6):853–66. doi: 10.1016/s0092-8674(02)01162-5
92. Sol-Foulon N, Sourisseau M, Porrot F, Thoulouze M-I, Trouillet C, Nobile C, et al. ZAP-70 Kinase Regulates HIV Cell-to-Cell Spread and Virological Synapse Formation. EMBO J (2007) 26(2):516–26. doi: 10.1038/sj.emboj.7601509
93. Weil R, Veillette A. Signal Transduction by the Lymphocyte-Specific Tyrosine Protein Kinase P56lck. Curr Top Microbiol Immunol (1996) 205:63–87. doi: 10.1007/978-3-642-79798-9_4
94. Bushman FD, Malani N, Fernandes J, D’Orso I, Cagney G, Diamond TL, et al. Host Cell Factors in HIV Replication: Meta-Analysis of Genome-Wide Studies. PloS Pathog (2009) 5(5):e1000437. doi: 10.1371/journal.ppat.1000437
95. Rom S, Pacifici M, Passiatore G, Aprea S, Waligorska A, Del Valle L, et al. HIV-1 Tat Binds to SH3 Domains: Cellular and Viral Outcome of Tat/Grb2 Interaction. Biochim Biophys Acta (2011) 1813(10):1836–44. doi: 10.1016/j.bbamcr.2011.06.012
96. Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Mol Cell (2003) 12(3):591–601. doi: 10.1016/s1097-2765(03)00353-8
97. Pan X, Baldauf HM, Keppler OT, Fackler OT. Restrictions to HIV-1 Replication in Resting CD4 + T Lymphocytes. Cell Res (2013) 23(7):876–85. doi: 10.1038/cr.2013.74
98. Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, et al. SAMHD1 Restricts HIV-1 by Reducing the Intracellular Pool of Deoxynucleotide Triphosphates. Nat Immunol (2012) 13(3):223–8. doi: 10.1038/ni.2236
99. Ruland J, Mak TW. From Antigen to Activation: Specific Signal Transduction Pathways Linking Antigen Receptors to NF-kappaB. Semin Immunol (2003) 15(3):177–83. doi: 10.1016/s1044-5323(03)00034-4
100. Hiscott J, Kwon H, Génin P. Hostile Takeovers: Viral Appropriation of the NF-kappaB Pathway. J Clin Invest (2001) 107(2):143–51. doi: 10.1172/JCI11918
101. Schütze S, Wiegmann K, Machleidt T, Krönke M. TNF-Induced Activation of NF-κb. Immunobiology (1995) 193(2):193–203. doi: 10.1016/S0171-2985(11)80543-7
102. Takeda K, Akira S. TLR Signaling Pathways. Semin Immunol (2004) 16(1):3–9. doi: 10.1016/j.smim.2003.10.003
103. Huet O, Choukroun G, Mira J. Récepteurs De Type Toll, Réponse Inflammatoire Et Sepsis. Réanimation (2004) 13(3):167–75. doi: 10.1016/j.reaurg.2004.02.002
104. Thibault S, Tardif MR, Barat C, Tremblay MJ. TLR2 Signaling Renders Quiescent Naive and Memory CD4+ T Cells More Susceptible to Productive Infection With X4 and R5 HIV-Type 1. J Immunol (2007) 179(7):4357–66. doi: 10.4049/jimmunol.179.7.4357
105. Wiche Salinas TR, Zhang Y, Sarnello D, Zhyvoloup A, Marchand LR, Fert A, et al. Th17 Cell Master Transcription Factor RORC2 Regulates HIV-1 Gene Expression and Viral Outgrowth. bioRxiv (2021) 118(48):e2105927118. doi: 10.1101/2021.03.27.435072
106. Mehandru S, Poles MA, Tenner-Racz K, Jean-Pierre P, Manuelli V, Lopez P, et al. Lack of Mucosal Immune Reconstitution During Prolonged Treatment of Acute and Early HIV-1 Infection. PloS Med (2006) 3(12):e484. doi: 10.1371/journal.pmed.0030484
107. Morou A, Brunet-Ratnasingham E, Dubé M, Charlebois R, Mercier E, Darko S, et al. Altered Differentiation is Central to HIV-Specific CD4+ T Cell Dysfunction in Progressive Disease. Nat Immunol (2019) 20(8):1059–70. doi: 10.1038/s41590-019-0418-x
108. Aujla SJ, Dubin PJ, Kolls JK. Th17 Cells and Mucosal Host Defense. Semin Immunol (2007) 19(6):377–82. doi: 10.1016/j.smim.2007.10.009
109. Isnard S, Lin J, Bu S, Fombuena B, Royston L, Routy JP. Gut Leakage of Fungal-Related Products: Turning Up the Heat for HIV Infection. Front Immunol (2021) 12:656414. doi: 10.3389/fimmu.2021.656414
110. Peng X, Isnard S, Lin J, Fombuena B, Bessissow T, Chomont N, et al. Differences in HIV Burden in the Inflamed and Non-Inflamed Colon From a Person Living With HIV and Ulcerative Colitis. J Virus Erad (2021) 7(1):100033. doi: 10.1016/j.jve.2021.100033
111. El-Far M, Durand M, Turcotte I, Larouche-Anctil E, Sylla M, Zaidan S, et al. Upregulated IL-32 Expression And Reduced Gut Short Chain Fatty Acid Caproic Acid in People Living With HIV With Subclinical Atherosclerosis. Front Immunol (2021) 12:664371. doi: 10.3389/fimmu.2021.664371
112. Jenabian M-A, Patel M, Kema I, Kanagaratham C, Radzioch D, Thébault P, et al. Distinct Tryptophan Catabolism and Th17/Treg Balance in HIV Progressors and Elite Controllers. PloS One (2013) 8(10):e78146. doi: 10.1371/journal.pone.0078146
113. Jenabian M-A, El-Far M, Vyboh K, Kema I, Costiniuk CT, Thomas R, et al. Immunosuppressive Tryptophan Catabolism and Gut Mucosal Dysfunction Following Early HIV Infection. J Infect Dis (2015) 212(3):355–66. doi: 10.1093/infdis/jiv037
114. Presicce P, Orsborn K, King E, Pratt J, Fichtenbaum CJ, Chougnet CA. Frequency of Circulating Regulatory T Cells Increases During Chronic HIV Infection and is Largely Controlled by Highly Active Antiretroviral Therapy. PloS One (2011) 6(12):e28118. doi: 10.1371/journal.pone.0028118
115. Chun T-W, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, et al. Persistence of HIV in Gut-Associated Lymphoid Tissue Despite Long-Term Antiretroviral Therapy. J Infect Dis (2008) 197(5):714–20. doi: 10.1086/527324
116. DuPont HL, Marshall GD. HIV-Associated Diarrhoea and Wasting. Lancet (1995) 346(8971):352–6. doi: 10.1016/s0140-6736(95)92229-6
117. Haase AT. Early Events in Sexual Transmission of HIV and SIV and Opportunities for Interventions. Annu Rev Med (2011) 62:127–39. doi: 10.1146/annurev-med-080709-124959
118. Brenchley JM, Price DA, Douek DC. HIV Disease: Fallout From a Mucosal Catastrophe? Nat Immunol (2006) 7(3):235–9. doi: 10.1038/ni1316
119. Sun H, Kim D, Li X, Kiselinova M, Ouyang Z, Vandekerckhove L, et al. Th1/17 Polarization of CD4 T Cells Supports HIV-1 Persistence During Antiretroviral Therapy. J Virol (2015) 89(22):11284–93. doi: 10.1128/JVI.01595-15
120. Halvas EK, Joseph KW, Brandt LD, Guo S, Sobolewski MD, Jacobs JL, et al. HIV-1 Viremia Not Suppressible by Antiretroviral Therapy can Originate From Large T Cell Clones Producing Infectious Virus. J Clin Invest (2020) 130(11):5847–57. doi: 10.1172/JCI138099
121. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 Cells are Long Lived and Retain a Stem Cell-Like Molecular Signature. Immunity (2011) 35(6):972–85. doi: 10.1016/j.immuni.2011.09.019
122. Gattinoni L, Ji Y, Restifo NP. Wnt/beta-Catenin Signaling in T-Cell Immunity and Cancer Immunotherapy. Clin Cancer Res (2010) 16(19):4695–701. doi: 10.1158/1078-0432.CCR-10-0356
123. Staal FJT, Sen JM. The Canonical Wnt Signaling Pathway Plays an Important Role in Lymphopoiesis and Hematopoiesis. Eur J Immunol (2008) 38(7):1788–94. doi: 10.1002/eji.200738118
124. Lennartsson J, Rönnstrand L. Stem Cell Factor Receptor/c-Kit: From Basic Science to Clinical Implications. Physiol Rev (2012) 92(4):1619–49. doi: 10.1152/physrev.00046.2011
125. Mekori YA, Gilfillan AM, Akin C, Hartmann K, Metcalfe DD. Human Mast Cell Apoptosis Is Regulated Through Bcl-2 and Bcl-XL. J Clin Immunol (2001) 21(3):171–4. doi: 10.1023/A:1011083031272
126. Song J, So T, Cheng M, Tang X, Croft M. Sustained Survivin Expression From OX40 Costimulatory Signals Drives T Cell Clonal Expansion. Immunity (2005) 22(5):621–31. doi: 10.1016/j.immuni.2005.03.012
127. Kuo H-H, Ahmad R, Lee GQ, Gao C, Chen H-R, Ouyang Z, et al. Anti-Apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4+ T Cells. Immunity (2018) 48(6):1183–1194.e5. doi: 10.1016/j.immuni.2018.04.004
Keywords: T-helper 17 cells, HIV infections, HIV reservoir, mucosal immunology, CD4-positive T cells, lymphocytes
Citation: Renault C, Veyrenche N, Mennechet F, Bedin A-S, Routy J-P, Van de Perre P, Reynes J and Tuaillon E (2022) Th17 CD4+ T-Cell as a Preferential Target for HIV Reservoirs. Front. Immunol. 13:822576. doi: 10.3389/fimmu.2022.822576
Received: 26 November 2021; Accepted: 14 January 2022;
Published: 07 February 2022.
Edited by:
Joanne E Konkel, The University of Manchester, United KingdomReviewed by:
Elizabeth Connick, University of Arizona, United StatesAndrea Introini, University of Milan, Italy
Copyright © 2022 Renault, Veyrenche, Mennechet, Bedin, Routy, Van de Perre, Reynes and Tuaillon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edouard Tuaillon, ZS10dWFpbGxvbkBjaHUtbW9udHBlbGxpZXIuZnI=