 Ravi Vattepu
Ravi Vattepu Sunny Lyn Sneed
Sunny Lyn Sneed Robert M. Anthony
Robert M. Anthony- Center for Immunology and Inflammatory Diseases, Division of Rheumatology, Allergy and Immunology, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA, United States
Antibodies play a critical role in linking the adaptive immune response to the innate immune system. In humans, antibodies are categorized into five classes, IgG, IgM, IgA, IgE, and IgD, based on constant region sequence, structure, and tropism. In serum, IgG is the most abundant antibody, comprising 75% of antibodies in circulation, followed by IgA at 15%, IgM at 10%, and IgD and IgE are the least abundant. All human antibody classes are post-translationally modified by sugars. The resulting glycans take on many divergent structures and can be attached in an N-linked or O-linked manner, and are distinct by antibody class, and by position on each antibody. Many of these glycan structures on antibodies are capped by sialic acid. It is well established that the composition of the N-linked glycans on IgG exert a profound influence on its effector functions. However, recent studies have described the influence of glycans, particularly sialic acid for other antibody classes. Here, we discuss the role of glycosylation, with a focus on terminal sialylation, in the biology and function across all antibody classes. Sialylation has been shown to influence not only IgG, but IgE, IgM, and IgA biology, making it an important and unappreciated regulator of antibody function.
Introduction
Antibodies are critical mediators of to host defense and homeostasis. Further, the therapeutic potential of antibodies has been fundamental in advancing basic immunology and biotechnology. Antibodies have two unique functions that are responsible for their biological function. They can recognize specific structures with high affinity and specificity, and simultaneously engage with cells of the innate immune system. Antibodies can be passively transferred across individuals or species, which gives them therapeutic utility. Indeed, polyclonal IgG from tens of thousands of healthy individuals, intravenous immunoglobulin (IVIG), is administered as IgG replacement therapy to immunocompromised individuals, and as an anti-inflammatory agent to some patients suffering from inflammatory and autoimmune diseases (1–3). Hybridoma technologies enable generation of monoclonal antibody preparations. The molecular biology revolution following the invention of PCR enabled cloning and recombinant production of monoclonal antibodies. Currently, there are 100 monoclonal antibodies (mAbs) approved by the FDA and monoclonal antibodies are the fastest growing class of therapeutics (4).
The monomeric structures of antibodies are composed of two identical heavy chains and two identical light chains connected by disulfide bonds (5, 6). In humans, antibodies are categorized into five classes: IgG, IgM, IgA, IgE, and IgD, based on constant region structure, properties, and oligomerization (Figure 1A) (7). In serum, IgG is the most abundant antibody comprising 75% of all antibodies in circulation, followed by IgA at 15%, IgM at 10%, and IgD and IgE are the least abundant (6, 8). These antibodies are structurally different and elicit different effector functions through interactions between their fragment crystallizable (Fc) portion and different Fc receptors (6).
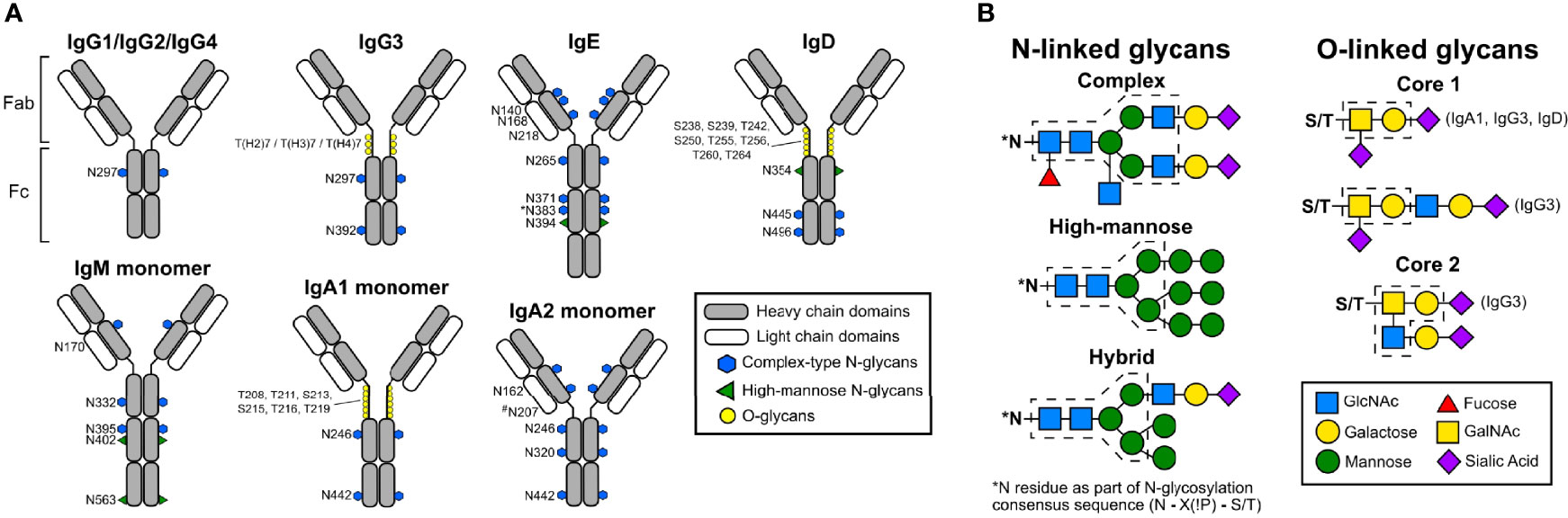
Figure 1 Schematic representations of human antibody structures and attached glycans. (A) Human immunoglobulin structures with corresponding glycosylation sites (human numbering) are shown. Each antibody isotype is comprised identical heavy chains (gray) and light chains (white), forming the antigen-binding fragment (Fab) and fragment crystallizable region (Fc). While monomeric structures are shown, IgM typically multimerizes into a pentamers or hexamers, and IgA1 and IgA2 can form dimers. Blue hexagons denote sites occupied by complex-type N-glycans. Green triangles indicate sites occupied by oligomannose N-glycans. Yellow circles mark sites occupied by O-linked glycans. The N383 site on human IgE is unoccupied, and N207 on human IgA2 is present only in specific allotypes. (B) Structures of N- and O-linked glycans found on human antibodies are represented. N-linked glycans are attached to asparagine residues containing the consensus sequence N-X-S/T. N-linked glycans can form complex, high-mannose, or hybrid types. Blue squares; GlcNAc. Yellow circles; galactose. Green circles; mannose. Red triangles; fucose. Yellow squares; GalNAc. Purple diamonds; sialic acid. Dashed lines indicate core structures, and sugar residues outside the core are variable additions.
Therapeutic antibodies currently used in the clinic belong only to the IgG class because of their functional characteristics such as long half-life, strong antigen binding, and effector functions such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). There is interest in developing therapeutic IgM, IgE, and IgA antibodies to explore novel targets and harness the effector functions of different immune cells. In contrast to IgG antibodies, IgM antibodies do not bind to Fc gamma receptors (FcγRs), but do bind the Fc mu receptor (FcμR) and can trigger potent CDC activity by binding to C1q (9). Currently, there are some IgM antibodies in clinical development (9). Further, IgE-based therapeutic antibodies are being evaluated in different studies (10). Advantages of using IgE include higher affinity interactions with the Fc epsilon receptor I (FcϵRI), longer tissue residence time, and ability to infiltrate the tumor microenvironment (10). Similarly, the development of IgA therapeutics poses advantages such as the ability to recruit neutrophils to kill tumors cells, targeting IgA interactions with the Fc alpha receptor FcαRI, and mucosal applications (11). However, there are numerous challenges in the expression, purification, and characterization of IgE, IgM, and IgA, including heterogeneous products and low yields following production, due in part to complex structures and multiple glycosylation sites (9, 11–13).
Studies over the last several years have revealed that glycosylation, both N-linked and O-linked, can play profound roles in modifying the effector functions for each antibody class. This ranges from maintaining the structure, tuning effector functions, engaging co-receptors and co-factors, improving their stability, and modifying antigenicity (14, 15). In this review, we will focus on the impact of glycosylation across the antibody classes, with a particular focus on the impact of sialic acid.
Antibody N-Linked and O-Linked Glycosylation
Glycosylation of antibodies is initiated in the endoplasmic reticulum (ER) of antibody-producing cells and further trimming and remodeling steps are processed by glycosidases and glycosyltransferases in the Golgi apparatus (16). N-linked glycosylation occurs on the amino acid asparagine within the consensus sequence of Asn-X-Ser/Thr, where X is any amino acid except proline. The glycosylation process starts with the transfer of the preassembled, lipid-linked glycan precursor, triglucosylated high-mannose-type tetradecasaccharide, to the asparagine residue by oligosaccharyl transferase in the ER (17). The glycan precursor undergoes further processing by glycosidases, which remove the glucose residues, one mannose residue, and three N-acetylglucosamine (GlcNAc) residues. Once the antibody is translocated into the cis-Golgi, the glycan undergoes further trimming by glycosidases and further remodeling is performed by the glycosyltransferases in the median Golgi.
Based on their composition, glycans are classified into three types: oligomannose, hybrid, and complex (Figure 1B) (16–18). Oligomannose glycans contain 5 to 9 branching mannose residues which are not further cleaved during processing. Complex glycans possess a core structure composed of two inner GlcNAc residues, three mannose residues, and two outer GlcNAc residues each β-1,2-linked to the α-3 and the α-6 mannose residues, forming two antennae (19). The innermost GlcNAc can be further modified by addition of an α-1,6 linked fucose, which is catalyzed by α-1,6 fucosyltransferase (FUT8) (16, 20). One galactose residue may be added to each of the β-1,2 GlcNAc by β -1,4 galactosyl transferase-1, and this addition can be further extended by the addition of sialic acid by α-2,6 sialyltransferase (16, 21). Hybrid type glycans are a combination of the high-mannose and complex types, with one antenna possessing only mannose residues and the other arm including one GlcNAc residue with further addition of galactose and then sialic acid. Variations in antibody glycosylation depend on substrate availability, steric hindrance, and expression level of glycosyltransferases (22). This leads to heterogeneity in glycosylation, which the immune system uses to fine-tune immune responses, and certain glycosylation patterns can be used as biomarkers for certain diseases.
O-linked glycosylation is present in the hinge region of IgA1, IgG3, and in the Fc portions of human IgD (23–25). O-linked glycosylation occurs on serine and threonine residues, but the consensus sequence for this modification has not yet been identified, the regulation of O-linked glycosylation is less well understood compared to the N-linked pathways, and it occurs exclusively in the Golgi. In the first step, GalNAc is attached to serine or threonine by UDP-N-acetylgalactosaminyl transferase 2 (GalNAc-T2) in the cis-Golgi. Next, the addition of galactose is catalyzed by β1,3-galactosyltransferase (C1GalT1), an enzyme which also requires the molecular chaperone COSMC for stability and function. O-linked glycans are further extended by the addition of sialic acid to the GalNAc and/or galactose residues by α2,6-sialyltransferase (ST6GalNAc-I)or α2,3-sialyltransferase (ST3Gal-1), respectively (26–29) in medial- and trans-Golgi.
Antibody Structure, Glycosylation Across Classes and Subclasses
IgG Antibodies
The monomeric structures of all antibodies, including IgG, contain two functional domains. The antigen-binding region (Fab) is involved in the recognition of antigens and the fragment crystallizable region (Fc) interacts with FcγRs and other effector proteins, including complement component C1q to mediate effector functions. The Fab region is composed of one variable and one constant domain (CH1) of both the heavy and light chains, while the Fc domain is a homodimer consisting of the heavy chain constant domains (CH2 and CH3). A flexible hinge region connects the Fab and Fc domains.
The four subclasses of IgG antibodies are numbered based on their prevalence in the serum: IgG1, IgG2, IgG3, and IgG4 (30–32). These four IgG subclasses share more than 90% sequence similarity, but each subclass has a distinct functional profile in mediating effector function, complement activation, half-life, and placental transport (33, 34). The Fc domain of each subclass carries a conserved N-linked glycosylation site at asparagine 297 (N297) in the CH2 domain. The glycan occupying this site has a complex biantennary structure. Several studies have shown roles for Fc glycosylation in mediating immune effector functions, including anti-inflammatory responses, ADCC, CDC, and antibody-dependent cell-mediated phagocytosis (ADCP) (34–38). Structural studies showed Fc glycans play a crucial role in maintaining an open conformation of the Fc domain and mediating interaction with FcγRs and C1q (39, 40). Removal of this glycosylation site abrogates effector functions and abolishes binding to FcγRs and C1q (41–43). Intriguingly, IgG Fc glycans are highly heterogeneous with more than 30 distinct glycoforms detected on IgG in healthy individuals (44, 45). The composition of the IgG Fc glycan has been demonstrated to significantly impact effector functions, pharmacokinetics, immunogenicity, stability, and aggregation (46–48).
IgG Sialylation
In a healthy individuals, approximately 10-15% of serum IgG are sialylated, with the majority of those possessing monosialylated glycoforms (49). Indeed, this percentage is reduced during inflammation and autoimmune disease flares (49, 50). IVIG is a therapeutic preparation of monomeric IgG pooled from the plasma of thousands of donors. It is administered as an IgG-replacement to immunocompromised individuals at 400-600mg/kg. Paradoxically, it is used to treat autoimmune and inflammatory diseases such as Myasthenia gravis (MG), immune thrombocytopenia (ITP), multiple sclerosis (MS), systemic lupus erythematosus (SLE), chronic inflammatory demyelinating neuropathy, and Kawasaki disease at a high dose of 1-2g/kg (51–53). Multiple studies support α-2,6 sialylated IgG as the biologically active component of immunomodulatory high dose IVIG (Figure 2) (36, 37, 54, 55). A series of mechanistic studies revealed that the immunomodulatory and anti-inflammatory activity of IVIG requires the inhibitory receptor FcγRIIB, and that FcγRIIB surface expression is increased on macrophages, dendritic cells, and B cells following IVIG infusion (56–58).
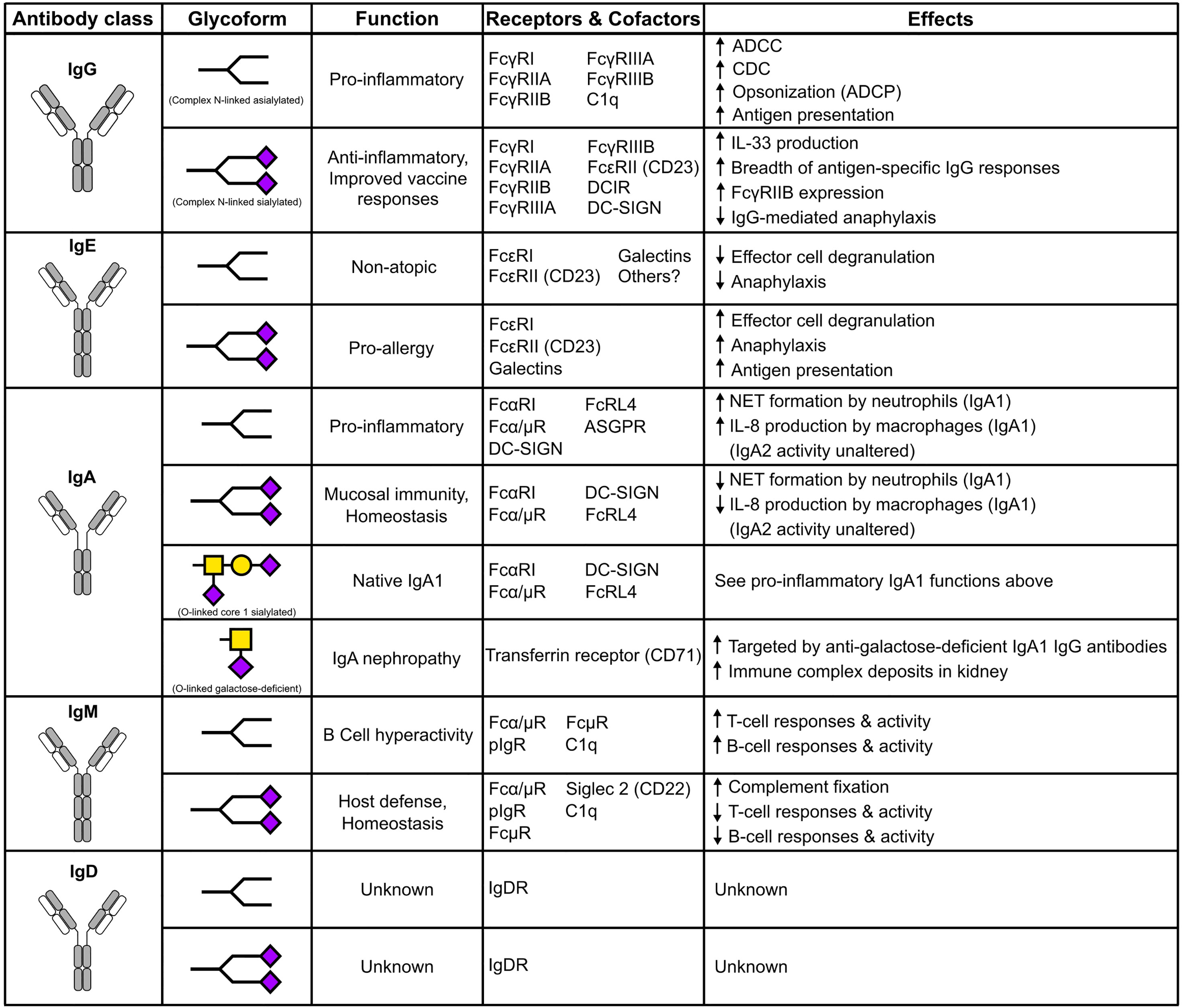
Figure 2 The impact of N- and O-linked sialylation across antibody classes. Antibody schematics are shown, along with skeleton sialylated (terminating in purple diamonds) or asialylated N-linked glycans. O-glycans and IgA1 are comprised of GalNAc (Yellow squares), galactose (yellow circles), and sialic acid (purple diamonds).
Further studies revealed terminal α-2,6 sialic acid on the IgG Fc glycans were required for anti-inflammatory activity, and C type lectins Specific ICAM-3 Grabbing Non-Integrin-Related 1 (SIGN-R1) and Dendritic Cell-Specific ICAM-3 Grabbing Non-Integrin (DC-SIGN) in mice and humans, respectively, were also required (36, 37, 39, 54). Administration of sialylated IgG Fc induces IL-33 production, which in turn triggers the expansion of IL-4-producing basophils and T regulatory cells, culminating in upregulation of FcγRIIB expression on effector cells (59, 60). While functional models support a role for DC-SIGN in the anti-inflammatory activity of sialylated IgG, whether there is a direct interaction between these entities is under debate (61, 62). Insertion of additional N-linked glycosylation sites in the hinge region of IgG1 Fc via mutants D221N/N297A/N563A and D221N/N563A increases sialyation 75% and 44% respectively, resulting in increased binding to Siglec-1 (Sialoadhesion), and these mutants also inhibited demyelination by anti-MOG antibodies in an ex vivo model when multimerized (63–65). Another study demonstrated that sialylated IVIG mediates anti-inflammatory activity by induction of T regulatory cells through engagement of the dendritic cell immunoreceptor (DCIR) (66).
IgG sialylation is emerging as a therapeutic option to treat autoimmune diseases. A phase 1/2 clinical trial (NCT03866577) reported sialylated IVIG offered protection to patients with ITP (67). Confirmation of previous murine studies came following IVIG infusion in chronic inflammatory demyelinating polyneuropathy (CIDP) patients, which led to increased surface expression of FcγRIIB (68, 69). Further, IL-33 has been detected in the serum following IVIG infusion (70). Finally, sialylated IgG attenuated IgG-mediated anaphylaxis in murine models, but additional experiments are required to determine whether sialylated IgG is a viable therapeutic for allergic disease (71, 72).
While sialylation conveys anti-inflammatory activity to monomeric IgG, it imparts unique functions to antigen-specific IgG. Indeed, increased antigen-specific IgG was detected following vaccination to Streptococcus pneumonia and SARS-CoV-2, and administration of sialylated IgG immune complexes led to an enhanced breadth of IgG responses in a FcϵRII (CD23)-dependent manner (73–75). IgG sialylation appears to have little impact on ADCC, or FcγRI and FcγRIIIA binding, and a only small enhancement of binding to FcγRIIA (76). Some functional studies have reported that sialylation impairs IgG-mediated ADCC and CDC (77–80). Consistently, asialylated IgG1 displayed enhanced C1q binding and CDC activity compared to sialylated IgG1 (80). In fact, a randomized clinical trial of patients with CIDP receiving high dose IVIG showed increased IgG sialyation and significantly lower levels of complement activation associated with disease remission (80). One contrasting study reported IgG1 Fc sialylation resulted in increased C1q binding (81). Further characterization of the role of IgG Fc sialylation and other glycoforms in FcγR binding, C1q binding, and functional assays is needed to resolve some of these discrepancies.
IgG Fucosylation, Galactosylation, and Bisecting GlcNAc
It is well-established that non-sialic acid sugars also exert important and profound influence over IgG effector functions, as has been reviewed extensively elsewhere (82–85). Briefly, more than 90% of IgG glycans in human serum contain a fucose residue attached to the innermost GlcNAc core structure (86). Antibody fucosylation is catalyzed by FUT8 in the medial/trans-Golgi. Several studies have demonstrated that removal of the core fucose residue increases IgG binding affinity to the human FcγRIIIA receptor expressed on macrophages, dendritic cells, and natural killer cells, resulting in enhanced ADCC (87, 88). This generated tremendous interest in the roles of IgG glycosylation in Fc receptor binding and antibody function. Afucosylated therapeutic antibodies, including anti-HER2 and anti-CD20 IgG1, show 100-fold higher ADCC compared to fucosylated anti-HER2 antibodies (89). Thus, there is a huge potential in immunotherapy for afucosylated antibodies with increased ADCC activity (90–92). Further, the severity of viral diseases caused by Dengue virus and SARS-CoV-2 are associated with the percentage and titers of viral-specific afucosylated IgG (93, 94).
The IgG N-glycan may be agalactosylated (G0F), mono-galactosylated (G1F), or di-galactosylated (G2F), and the effector functions of each glycoform can vary significantly. In the serum of a healthy individual, G0F, G1F, and G2F each account for approximately 35%, 35%, and 15% of IgG glycoforms, respectively (49, 95). A decrease in IgG galactosylation in human serum is associated with chronic inflammatory and autoimmune diseases, such as SLE, MS, rheumatoid arthritis (RA), auto-immune vasculitis, active spondyloarthropathy, Crohn’s disease, inflammatory bowel disease, and psoriatic arthritis (49, 96–100). IgG galactosylation level also decreases with age and inflammation, but the rate of change is faster during the development of inflammatory conditions compared to typical aging (49). Galactosylated IgG1 immune complexes showed anti-inflammatory activity in mice through interactions with FcγRIIB and the receptor Dectin-1 (101). Galactosylated IgG have also been shown to bind complement effectively (102).
In healthy individuals, only a small fraction (10-15%) of antibodies contain bisecting GlcNAc (biGlcNAc) (49). In the Golgi, β-1,4-N-acetylglucosaminyltransferase III (GnT-III) catalyzes the addition of biGlcNAc to the internal mannose residue. Recombinant IgG Fcs with biGlcNAc possess an increase in ADCC activity and FcγRIIIA binding (103). However, the addition of biGlcNAc inhibits the addition of a core fucose residue due to steric hinderance, and increased ADCC is also attributed to the inhibition of fucose attachment (83).
IgE Antibodies
In comparison with other antibodies, IgE is the least abundant antibody in serum with a concentration of 50–100 ng/ml, and a half-life of only two to three days; binding to the high affinity receptor FcϵRI expressed on mast cells extends tissue half-life to three weeks (104). IgE is involved in triggering allergic reactions to food and pollen, and is involved in atopic diseases such as asthma and atopic dermatitis. The incidence of allergic disease is significantly increasing worldwide, with an estimated of 30-40% of the population having one or more allergic diseases. Thus, there has been significant interest in developing IgE-targeted therapeutic antibodies to treat IgE-mediated allergies. For example, Xolair (Omalizumab) is an FDA approved monoclonal antibody to treat IgE-mediated allergic disease. On the other hand, IgE antibodies also provide a defense against parasite infections and insect venoms (105–107).
IgE is composed of two identical heavy chains and two light chains. Each heavy chain consists of a single variable domain and four constant domains, and the light chain is composed of one variable and one constant domain. IgE is a heavily glycosylated monomeric antibody, with seven N-linked glycosylation sites spread across the four constant domains of the heavy chain. N394 is a conserved glycosylation site with an oligomannose glycan, N383 is unoccupied, and the other five sites (N140, N168, N218, N265, N371) are occupied by complex type biantennary glycans (108–111). The major structural differences of IgE compared to IgG are a shorter hinge region, the presence of an extra constant region, and an increased number of glycosylation sites.
Similar to IgG, the constant region of IgE mediates various effector functions through binding to the high-affinity and low-affinity receptors expressed on innate immune cells. The IgE high affinity receptor FcϵRI is expressed on mast cells and basophils shows a significantly high binding affinity, approximately 1011 M−1 (112). The binding of an allergen to IgE followed by crosslinking the FcϵRI receptors triggers cellular degranulation and symptoms of allergic inflammation (113). The low-affinity IgE receptor, FcεRII (CD23), has a binding affinity coefficient of 106 M-1. Interactions with FcεRI or FcεRII depend on IgE’s Fc conformation. In the open conformation, IgE binds to FcϵRI at the CH3 domain, while in the closed conformation IgE binds to FcεRII at both the CH3 and CH4 domains (114, 115). Binding to the high affinity and low affinity receptors is mutually exclusive; conformation changes upon binding to one receptor hinder the binding to another receptor (116).
IgE Sialylation
With the emergence of glycosylation’s role in regulating IgG structure and function, there is growing interest in the role of glycosylation across other antibody classes. Initial work indicated that IgE glycosylation is critical for FcϵRI binding and effector functions (117–119). Subsequently, other studies showed that aglycosylated IgE produced in E. coli binds to FcϵRI and can trigger effector functions, suggesting a null effect of glycosylation (120–122). However, studies coupling in vitro, cell-based, and mice model experiments have shown an important role for IgE glycosylation. The point mutation N384Q in murine IgE (mIgE), equivalent to human N383Q, abolished binding to FcϵRI in ear mast cells and cell-based assays, and similar results were also observed with the enzymatic treatment of mIgE using PNGaseF to remove all N-glycans (123). These results suggest an absolute requirement of mIgE N384 glycosylation for the initiation of anaphylaxis. Further, circular dichroism results showed enzymatic removal of N-linked glycans altered the secondary structure of IgE and abrogated binding to FcϵRI (123). These results highlight the role of glycosylation in binding to the high-affinity receptor similar to the role of IgG glycosylation in FcγR binding. This also suggests that IgE produced in E. coli might adopt a different conformation when refolding than a glycosylated IgE and thus can still result in FcϵRI binding.
Disease-specific IgE glycosylation patterns have been characterized in allergic and atopic cohorts (111). Between the two cohorts, mannose, fucose, and biGlcNAc content in complex bi-antennary type glycans were largely similar. However, terminal galactose was enriched on atopic total IgE while increased sialylation was observed in allergic IgE. Functional experiments revealed increased mast cell degranulation was observed after sensitization with peanut-allergic IgE compared to the atopic IgE. The role of IgE sialylation was further confirmed by the passive cutaneous anaphylaxis model. Indeed, sialylated IgE increased anaphylaxis compared to asialylated IgE (111). The emerging patterns of the effects of antibody sialylation are intriguing; IgG sialylation mediates anti-inflammatory activity, but IgE sialyation promotes allergy (Figure 2).
IgM Antibodies
There are generally thought to be two types of IgM produced, called natural IgM and immune IgM. Natural IgM antibodies are produced spontaneously and act as the first line of defense during microbial infections, prior to the adaptive immune response; its repertoire is largely unaffected by external antigens (124, 125). In humans, natural IgM antibodies are produced by B1 cells without exposure to exogenous antigens, are polyreactive, and constitute the majority of circulating IgM in serum (126, 127). Natural IgM antibodies can recognize specific neoepitopes on apoptotic cells to remove them selectively and to maintain tissue homeostasis. Antigens recognized by natural IgM include specific carbohydrates, phospholipids, and double-stranded DNA (128–130). The other IgM class, immune IgM, is produced after exposure to external antigens (131). While the source of the two IgM classes is different, the differences in their structural and functional properties are minimal (132).
IgM antibodies are found in a pentamer or hexamer format, in which each monomer is composed of a heavy chain with one variable region and four constant regions (CH1, CH2, CH3, CH4), and a light chain (133). In contrast to IgG, the molecular structure of IgM possesses a short hinge region, similar to IgE. A short peptide sequence of 18 amino acids, called a tailpiece, is essential for IgM multimerization. The tailpiece of each heavy chain forms disulfide bonds with each heavy chain monomer to form IgM and another disulfide bond between C414 residues in the CH3 region of each heavy chain is involved in the formation of the multimer (134, 135). Tailpiece multimerization has also been successfully used to form IgG hexamers (136, 137). IgM also contains a J chain in the pentameric structure which is involved in the transportation of IgM through binding to specific receptors (9, 138, 139).
Apart from multimerization, glycosylation sites add further complexity to the IgM structure. IgM is heavily glycosylated in comparison with IgG; it has five N-linked glycosylation sites in the heavy chain (N171 (CH1), N332 (CH2), N395, N402 (both in CH3)), one in the tailpiece (N563), and one on the J chain (140, 141). The N-linked glycosylation pattern of IgM includes complex type and high-mannose type glycans; as shown in Figure 1A, N171, N332, and N395 are occupied by complex type glycans and N402 and N563 contain high-mannose type glycans (141, 142). Similar to IgG, IgM complex type glycans also may contain terminal sialic acid.
IgM Sialylation
Natural anti-T lymphocyte IgM antibody levels are increased in patients with inflammatory conditions, HIV infections, and SLE compared to healthy individuals (143, 144). These antibodies mediate the inhibitory effects on anti-CD3–induced T cell activation but for a time the mechanism was not completely understood (145, 146). A recent study by Colucci et al. shows that IgM’s inhibitory effects depend on antibody glycosylation (147). Five different IgM antibodies purified from healthy donors (n=2) or myeloma patients (n=3) were incubated with human PBMCs. Results showed that the IgM antibody from healthy donors (IgMh) bound to the T cells and were then internalized and inhibited T cell proliferation. Meanwhile, IgM from myeloma patients (IgMm) remained on the surface and did not inhibit T cell proliferation. Glycosylation analysis of these antibodies showed only the IgMh, which mediated the inhibitory effects, contained sialic acid but the IgMm antibodies did not. Furthermore, removal of sialic acid using neuraminidase enzyme completely abrogated internalization of IgM and this asialylated IgM did not inhibit T-cell proliferation. The proposed mechanism is that both sialylated and asialylated IgM bind to FcμR, but only the sialylated antibody is internalized and triggers the inhibitory pathways (147, 148).
IgA Antibodies
In the serum, IgA is the second most prevalent circulating antibody after IgG and it is the predominant antibody found in external secretions such as those that bathe mucosal surfaces (8). Interestingly, the molecular form of IgA varies between the serum and mucosal surfaces. In the serum IgA exists in the monomeric form, but on mucosal surfaces IgA exists as a dimer called secretory IgA (SIgA). SIgA contains two IgA molecules joined by a J chain, similarly to IgM, to form a dimer, and this complex is also bound to a secretory component (149). Serum IgA contains two heavy chains composed of one variable region and three constant regions, and two light chains.
Based on its structure, IgA is divided into subclasses IgA1 and IgA2. IgA1 contains a hinge region with six O-linked glycosylation sites and two N-Linked glycosylation sites on each heavy chain as shown in Figure 1A. In contrast, IgA2 lacks O-linked glycosylation due to a shortened hinge region and has two N-linked glycosylation sites per heavy chain. The glycosylation pattern of the O-linked glycans may vary, but the majority of the O-linked glycans contain GalNAc and galactose with one or two sialic acids (11, 26).
IgA blocks the entry of pathogens through antigen-binding sites and the IgA-specific receptor FcαRI mediates the clearance mechanism (150). The SIgA molecule, with its multiple antigen-binding sites, provides high avidity to cross-link antigens in a highly efficient manner, which can block pathogen activity. Both serum IgA and secretory SIgA bind to the FcαRI receptor with similar binding affinity but cause divergent downstream effects. To evade the immune system, pathogenic bacteria such as Staphylococcus aureus produce IgA binding proteins to inhibit IgA and FcαRI (151). In another strategy, pathogenic bacteria produce specific bacterial proteases against IgA, which specifically cleaves inside the hinge region of IgA leading to the inefficient clearance of pathogens (11, 150).
Several studies have shown that variations in IgA1 O-linked glycosylation play a crucial role in the pathogenesis of IgA Nephropathy (IgAN) and kidney failure (152). Analysis of IgAN patient samples showed a galactose deficiency in O-linked glycans, resulting in immune complex formation stimulated by anti-IgA1 IgG antibodies specific for this O-linked glycan. The large immune complexes escape clearance mechanisms and deposit in the renal mesangium, leading to glomerular injury (26, 152–154). As mentioned previously, glycosylation patterns are driven by the availability of substrate and enzyme expression during biosynthesis. In the case of a IgAN, there is deficiency in the galactosyltransferase enzyme C1GALT1, resulting in IgA1 with galactose-deficient O-linked glycosylation in the hinge region (26).
IgA Sialylation
Recent studies have shown IgA subclasses isolated from serum display different effector functions (Figure 2). IgA2 elicits pro-inflammatory effects on macrophages and neutrophils, but this is not observed with IgA1. These differences are attributed to variations in N-linked glycosylation, where IgA2 has 25% less sialylation compared to IgA1. These results were further confirmed by the enzymatic removal of total sialic acid using neuraminidase and the complete removal of N-linked glycans using PNGaseF, both of which resulted in increased pro-inflammatory activity (155). Another study showed IgA mediates anti-viral activity through sialic acid from the complex N-linked glycan at N459, where sialic acid interfered with cell surface attachment of influenza A (156).
Glycoengineering
Monoclonal antibody (mAb) based therapeutics are the fastest-growing group of therapeutics for several clinical indications including oncology, autoimmune diseases, inflammatory disease, and bacterial and viral infections. This field is dominated by IgG-based therapeutics. However, IgE has been developed as an immunotherpeautic agent for cancer, and IgA-based therapeutics have been proposed for treatment of bacterial and viral infections, and as anti-inflammatory and anti-tumor agents (10, 11, 150, 157). More testing is needed to determine the best suited glycoforms for these antibody classes. Much of the glycoengineering focus thus far has revolved around generation of afucosylated IgG, which is reviewed extensively elsewhere (82, 92, 158, 159).
Mammalian host expression systems are used for the production of antibodies to maintain mammalian antibody glycosylation patterns and minimize immunogenicity. However, there are differences in glycosylation patterns between endogenous antibodies produced in humans and mammalian cell lines. Antibody glycosylation is influenced by the host cell, glycosyltransferase expression, and cell culture conditions, such as low dissolved oxygen concentration and a reduced culture reduction potential (160, 161). Glycoforms of therapeutic mAbs produced in CHO, HEK293, mouse myeloma NS0, and Sp2/0 cell lines are heterogeneous with a predominance of the G0F glycoform produced, along with limited galactosylated and sialylated glycoforms. Therapeutic mAbs produced in CHO cells are less galactosylated compared to mAbs produced in mouse myeloma cell lines (162). The rat hybridoma cell line YB2/0 has low α-1,6 fucosyltransferase activity; therefore, this cell line is used for the production of mAbs with lower fucosylation levels. Specific glycosylation patterns, such as the absence of fucose, bisecting GlcNAc, and the presence of high mannose glycans increase ADCC, while galactose increases CDCC and sialic acid increases anti-inflammatory activity (163–167). Since different antibody glycoforms have distinct effects on biological activity, the production of homogenous glycoforms is necessary to improve therapeutic outcomes and maintain treatment quality. Enrichment of specific glycoforms and separation of said glycoforms using chromatography is challenging. Alternatively, two strategies have been employed to generate homogenous glycoforms with tailor-made sugar residues to modify antibody function. Host cell glycosylation pathway manipulation, called mammalian cell line glycoengineering, and in vitro glycoengineering are widely used to produce homogeneously glycosylated antibodies (91, 92, 158).
Mammalian Cell Line Engineering
Biosynthetic pathways in mammalian cell lines can be manipulated to produce afucosylated antibodies (Figure 3A). This strategy has focused on non-sialylated glycoforms by overexpression of different enzymes, generating gene knockout lines, and using monosaccharides as inhibitors (82, 92, 158). Recombinant glycoproteins and antibodies produced in CHO cells only produce α-2,3 sialylated glycans due to lack of ST6GAL1 gene expression. To increase sialylation, different strategies have been developed, including overexpression of the CMP-sialic acid transporter, overexpression of sialyltransferases, and knock out of genes (168–171). Production cell lines engineered to overexpress trans-Golgi enzymes B4GALT1 and ST6GAL1 have been described to generate IgG with enriched ~70% biantennary sialylated glycoforms (169). Co-expression of B4GALT1 and ST6GAL1 with IgG increased the amount of sialylation to 32% (170). Using gene editing technology, a CHO-Mgat2/-Stgal4/-Stgal6/+B4GALT1/+ST6GAL1 clone was generated by knocking out the ST3GAL4, ST3GAL6, and Mgat2 genes to inhibit α-2,3 sialylation and knocked in the B4GALT1 and ST6GAL1 genes to increase α-2,6 sialylation (171).
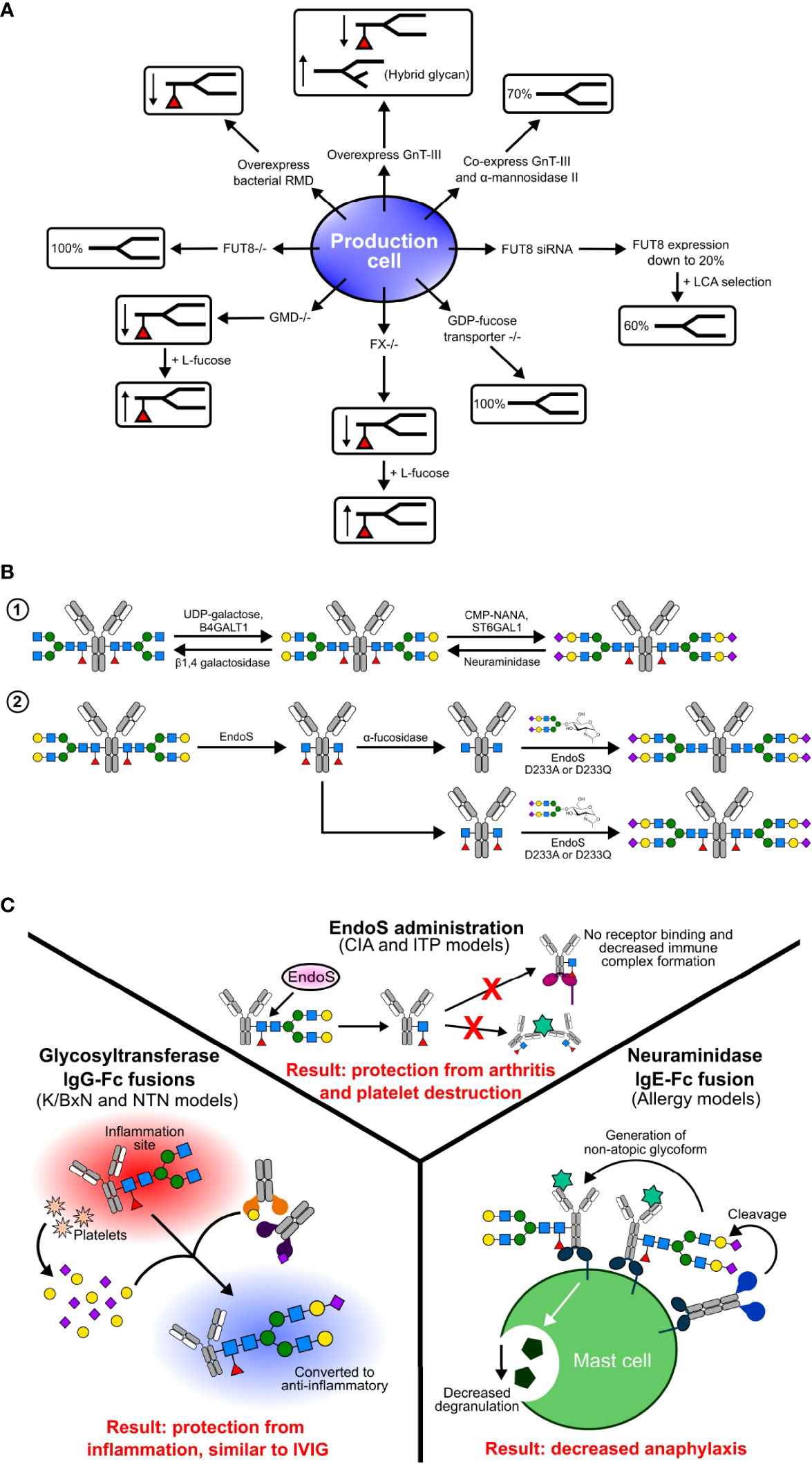
Figure 3 Glycoengineering methods in production cell lines (A), in vitro (B), and in vivo (C). (A) Various genetic manipulations have been performed in production cells to produce differentially fucosylated antibodies. In some cell lines, such as GMD and FX knockouts, fucosylation can be rescued by the addition of substrates such as L-fucose. (B) In vitro methods of glycoengineering and sialylation include step-wise enzymatic building of glycans and chemoenzymatic engineering. (C) In vivo glycoengineering involves administration of enzymes, and have had success in some pre-clinical animal models and on human cells.
Antibody fucosylation is decreased by overexpression of GnT-III. GnT-III is expressed in the Golgi and catalyzes the addition of biGlcNAc on to a β-linked mannose residue of an N-linked glycan. The addition of biGlcNAc increases steric hindrance, inhibits the addition of fucose, and produces hybrid type N-glycans. Although CHO cells do not express GnT-III, overexpression of it in CHO cells resulted in reduced antibody Fc fucosylation due to the addition of biGlcNAc (103, 172). This method is further improved by co-expression of GnT-III and Golgi α-mannosidase II, which results in a 70% decrease in core fucosylation (103, 173).
Gene knock out cell lines were developed by targeting specific genes involved in core fucosylation. The FUT8 gene encoding α-1,6 fucosyltransferase catalyzes the addition of α-1,6 fucose to the innermost GlcNAc. In the CHO-DG44 cell line, small interfering RNAs (siRNAs) targeting FUT8 reduced mRNA expression levels to 20%. Coupling this method with Lens culinaris agglutinin (LCA) selection produced 60% afucosylated antibodies with a resulting 100-fold increase in ADCC (174).
Afucosylated antibodies can also be generated by manipulation of GDP-fucose biosynthetic pathways (175). GDP-mannose 4,6-dehydratase (GMD) and GDP-4-keto-6-deoxymannose-3,5-epimerase-4-reductase (FX) catalyze the conversion of GDP-mannose into GDP-fucose. In knockout lines for each of these enzymes, core fucosylation is absent due to a lack of GDP-fucose but can be rescued by addition of L-fucose to the media (176, 177). Alternatively, overexpression of the bacterial GDP-6-deoxy-D-lyxo-4-hexulose reductase (RMD) enzyme reduces core fucosylation by depleting the intermediate GDP-4-keto-6-deoxy-D-mannose used in the synthesis GDP-fucose (175, 178, 179). An additional strategy to generate afucosylated IgG involves the addition of fucose analogs 2-fluorofucose and 5-alkynylfucose to inhibit the biosynthetic pathway of fucosylation (180).
In Vitro Glycoengineering
There is a tremendous improvement in the generation of glycoengineered antibodies through biosynthetic pathways and genetic manipulations in mammalian and non-mammalian cells. However, these methods require complex protocols and special cell line development for each type of glycan addition or deletion. These problems can be overcome by using in vitro glycoengineering, where trimming or extending of antibody glycans is performed using substrates, glycosyltransferases, and glycosidases (Figure 3B). These approaches have been successfully applied to antibodies generated in both mammalian and non-mammalian cells. There are two types of in vitro glycoengineering methods: enzymatic and chemoenzymatic.
In the enzymatic method, the antibody of interest is treated with uridine diphosphate galactose (UDP-Gal) and β-1,4 galactosyltransferase-1 (B4GalT1) to generate the G2F glycoform, which is further modified by treating the product with cytidine 5′ mono-phospho-N-acetyl neuraminic acid (CMP-NANA) and α 2,6 sialyltransferase (ST6Gal1) to produce the G2S2F glycoform (55). This method can also be applied to remove sialic acid and galactose by treatment with neuraminidase and β-1,4 galactosidase. In early studies, lectin enrichment was used to prepare sialylated Fc, but later studies used enzymatic glycoengineering methods to add galactose followed by sialic acid (36, 37). Several studies have shown sialylated IgG1 Fc and sialylated IVIG protect mice from induced arthritis at lower doses compared to IVIG (37, 55). This data is further supported by the use of sialylated IgG Fc clinical trials for the successful treatment of ITP (67). This method is limited by the stepwise addition of sugars.
Chemoenzymatic glycoengineering of IgG antibodies is a two-step process, consisting first of deglycosylation of N-linked glycans using native endoglycosidases followed by the addition of homogenous N-linked glycans using mutant endoglycosidases. This strategy was improved by the discovery of the novel endoglycosidase endo-β-N-acetylglucosaminidase (EndoS) mutants D233A and EndoS D233Q, which allowed the addition of N-linked glycans to GlcNAc or core-fucosylated GlcNAc (181). This is a very robust method that allows for the addition of the entire glycan complex at one time. Using the chemoenzymatic method, Rituximab has been glycoengineered to prepare homogenous G2, S2G2, G2F, and S2G2F glycoforms. Results from in vivo and in vitro studies have shown that only afucosylated glycoforms increased FcγRIIIA binding and elevated ADCC compared to the G2F and G2S2F glycoforms (77). Enzymatic glycoengineering and biosynthetic pathway manipulation are limited to the addition of complex biantennary type glycans, but the chemoenzymatic method allows selective addition of branched glycans. The D165A and D165Q mutants of the bacterial endoglycosidase Endo-F3 allow selective addition of bi- and triantennary N-glycans to the fucose core (182). The Endo-F3 D165A and Endo-S D233A allow site-specific glycoengineering of the Fab and Fc domains, respectively (183).
Currently, antibody glycoengineering efforts are exclusively focused on the Fc-core due to its role in mediating effector functions (63, 77, 82), but it should be noted that the antibody Fab domain also undergoes glycosylation for antigen recognition and other functions (184–186). The therapeutic antibody Cetuximab undergoes glycosylation in the Fab domain at N88 and in the Fc at N297. Glycoengineered Cetuximab without fucose at the Fc core and with sialic acid at Fab region showed increased binding affinity to FcγRIIIA and increased ADCC (183). Other applications of chemoenzymatic methods include bioconjugation, site specific labelling, and the synthesis of antibody-drug conjugates.
In Vivo Glycoengineering
Biosynthetic pathway manipulation of cell lines requires more time to establish knock out cell lines but is faster once the cell line is established, whereas in vitro glycoengineering methods require more time and effort in the purification steps. Another emerging strategy is glycoengineering circulating IgG antibodies in vivo using glycosidases and glycosyltransferases (Figure 3C). The first time deglycosylation of circulating IgG antibodies was shown was by in vivo administration of EndoS from the human pathogen Streptococcus pyogenes (187). EndoS selectively catalyzes the hydrolysis of the glycosidic bond between the first two N-acetylglucosamine residues of the of the N-linked glycan on the Fc of each IgG subclass (IgG1-4). In the collagen-induced arthritis mouse model, pretreatment of antibodies with EndoS enzyme inhibited the advancement of arthritis, abolished IgG binding to Fc receptors, and disturbed immune complex formation (188). Furthermore, the administration of EndoS has shown a protective effect in a mouse model of IgG-driven ITP (189). However, repeated administration of EndoS triggers an immune response, and using bacterial enzymes in a therapeutic setting raises a safety concern. Alternatively, the bacterial protease IdeS was successful in a trial targeting IgG mediated autoimmune conditions (190–192)
In another strategy of in vivo sialylation, human glycosyltransferases B4GalT1 and ST6Gal1 have been fused to IgG Fc to glycoengineer pathogenic antibodies (193). This approach recapitulates sialylated IVIG or IgG anti-inflammatory activity by converting pathogenic antibodies into sialylated, non-pathogenic antibodies. Interestingly, Fc-fused enzymes specifically sialylated pathogenic antibodies only at sites of inflammation, but not the antibodies or other glycoproteins in circulation (193). This specificity of in vivo glycoengineering is due to platelets releasing sugar donors, sialic acid and galactose, at sites of inflammation. This strategy has been successfully demonstrated in murine autoimmune disease models, in which Fc-fused enzymes attenuated auto-antibody inflammation by converting autoantibodies into anti-inflammatory mediators (193). By fusing glycoenzymes to IgG Fc, the enzymes’ stability and half-life is increased. In vivo sialyation has the potential to be applied to those diseases currently treated using IVIG and offers a potential therapeutic strategy for autoimmune and inflammatory conditions.
Removing sialic acid from IgE is an appealing therapeutic strategy for allergic disease. With that in mind, a bacterial neuraminidase enzyme was fused to the IgE Fc Cϵ2–4 domains (NEUFcϵ), thereby targeting neuraminidase to IgE-bearing cells (111). Administration of this enzyme fusion during murine models of allergic inflammation resulted in attenuation of anaphylaxis (111). However, further testing of modulating antibody sialic acid content in vivo is needed.
Conclusions
Endogenous antibody glycosylation is heterogeneous and varies with age and pathophysiological conditions. Harnessing antibody glycosylation is in its infancy, but applications that are engaged in this endeavor include the development of biologic therapeutics and using certain glycosylation patterns as biomarkers. Therapeutic antibodies are a rapidly growing class for the treatment of cancer, autoimmune diseases, infectious diseases, and other conditions. Glycosylation of therapeutic antibodies is very sensitive to cell culture and other production conditions and variations in glycosylation impacts the quality of the product. Therapeutic antibody glycosylation is a critical quality attribute because of its impact on effector functions and half-life. In depth understanding of IgG glycosylation has revealed the role of each glycan in modulating IgG function and resulted in the development of therapeutic afucosylated and sialylated antibodies.
Glycosylation across antibody classes regulates the structure and function of antibodies. Several studies have shown that sialic acid has divergent functions in antibody classes. For instance, sialylated IgG1 mediates anti-inflammatory activity, sialylated IgE is associated with allergic pathogenicity, sialylated IgA shows anti-viral activity, and sialylated IgM shows inhibitory effects on T-cell proliferation. For most antibody classes, sialylation exerts profound influence over the effector functions. There are multiple, non-mutually exclusive reasons for this, including influencing receptor binding and introducing addition receptors and co-factors. Further studies are required to characterize specific mechanisms of antibody activity and the involvement of additional receptors. Also, the role of sialylation across all antibody subclasses is in need of more examination. In vitro sialylation of IgG markedly enriches sialylated glycoform for desire effector functions. This method has been successfully applied to generate sialylated antibodies to mediate anti-inflammatory activity. Indeed, glycoengineering has applicability across antibody classes is a major advantage. More understanding of the role and regulation of antibody sialylation and glycosylation will likely uncover additional applications for glycoengineering.
Author Contributions
RV, SS, and RA wrote the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants (R01 AI155662, R01 AI153441, and R01 AI139669) to RA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Imbach P. Treatment of Immune Thrombocytopenia With Intravenous Immunoglobulin and Insights for Other Diseases. A Historical Review. Swiss Med Wkly (2012) 142:w13593. doi: 10.4414/smw.2012.13593
2. Bayry J, Negi VS, Kaveri SV. Intravenous Immunoglobulin Therapy in Rheumatic Diseases. Nat Rev Rheumatol (2011) 7(6):349–59. doi: 10.1038/nrrheum.2011.61
3. Schwab I, Nimmerjahn F. Intravenous Immunoglobulin Therapy: How Does IgG Modulate the Immune System? Nat Rev Immunol (2013) 13(3):176–89. doi: 10.1038/nri3401
4. Mullard A. FDA Approves 100th Monoclonal Antibody Product. Nat Rev Drug Discovery (2021) 20(7):491–5. doi: 10.1038/d41573-021-00079-7
5. Harris LJ, Larson SB, Hasel KW, Day J, Greenwood A, McPherson A. The Three-Dimensional Structure of an Intact Monoclonal Antibody for Canine Lymphoma. Nature (1992) 360(6402):369–72. doi: 10.1038/360369a0
6. Schroeder HW Jr, Cavacini L. Structure and Function of Immunoglobulins. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S41–52. doi: 10.1016/j.jaci.2009.09.046
7. Amzel LM, Poljak RJ. Three-Dimensional Structure of Immunoglobulins. Annu Rev Biochem (1979) 48:961–97. doi: 10.1146/annurev.bi.48.070179.004525
8. Kerr MA. The Structure and Function of Human IgA. Biochem J (1990) 271(2):285–96. doi: 10.1042/bj2710285
9. Keyt BA, Baliga R, Sinclair AM, Carroll SF, Peterson MS. Structure, Function, and Therapeutic Use of IgM Antibodies. Antib (Basel) (2020) 9(4):53. doi: 10.3390/antib9040053
10. Sutton BJ, Davies AM, Bax HJ, Karagiannis SN. IgE Antibodies: From Structure to Function and Clinical Translation. Antib (Basel) (2019) 8(1):19. doi: 10.3390/antib8010019
11. de Sousa-Pereira P, Woof JM. IgA: Structure, Function, and Developability. Antib (Basel) (2019) 8(4):57. doi: 10.3390/antib8040057
12. Rouwendal GJ, van der Lee MM, Meyer S, Reiding KR, Schouten J, de Roo G, et al. A Comparison of Anti-HER2 IgA and IgG1 In Vivo Efficacy is Facilitated by High N-Glycan Sialylation of the IgA. MAbs (2016) 8(1):74–86. doi: 10.1080/19420862.2015.1102812
13. Tchoudakova A, Hensel F, Murillo A, Eng B, Foley M, Smith L, et al. High Level Expression of Functional Human IgMs in Human PER. C6 Cells MAbs (2009) 1(2):163–71. doi: 10.4161/mabs.1.2.7945
14. Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural Analysis of Human IgG-Fc Glycoforms Reveals a Correlation Between Glycosylation and Structural Integrity. J Mol Biol (2003) 325(5):979–89. doi: 10.1016/S0022-2836(02)01250-0
15. Yamaguchi Y, Nishimura M, Nagano M, Yagi H, Sasakawa H, Uchida K, et al. Glycoform-Dependent Conformational Alteration of the Fc Region of Human Immunoglobulin G1 as Revealed by NMR Spectroscopy. Biochim Biophys Acta (2006) 1760(4):693–700. doi: 10.1016/j.bbagen.2005.10.002
16. Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, et al. eds. Essentials of Glycobiology. Cold Spring Harbor (NY (2015).
17. Stanley P, Schachter H, Taniguchi N, Varki A, Cummings RD, Esko JD, et al. N-Glycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, et al, editors. Essentials of Glycobiology. Cold Spring Harbor (NY (2009).
18. Schwarz F, Aebi M. Mechanisms and Principles of N-Linked Protein Glycosylation. Curr Opin Struct Biol (2011) 21(5):576–82. doi: 10.1016/j.sbi.2011.08.005
19. Kornfeld R, Kornfeld S. Assembly of Asparagine-Linked Oligosaccharides. Annu Rev Biochem (1985) 54:631–64. doi: 10.1146/annurev.bi.54.070185.003215
20. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozi CR, et al. Essentials of Glycobiology. Cold Spring Harbor (NY (2009).
21. Butters TD. Control in the N-Linked Glycoprotein Biosynthesis Pathway. Chem Biol (2002) 9(12):1266–8. doi: 10.1016/S1074-5521(02)00290-9
22. Nishima W, Miyashita N, Yamaguchi Y, Sugita Y, Re S. Effect of Bisecting GlcNAc and Core Fucosylation on Conformational Properties of Biantennary Complex-Type N-Glycans in Solution. J Phys Chem B (2012) 116(29):8504–12. doi: 10.1021/jp212550z
23. Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, et al. The Glycosylation and Structure of Human Serum IgA1, Fab, and Fc Regions and the Role of N-Glycosylation on Fcalpha Receptor Interactions. J Biol Chem (1998) 273(4):2260–72. doi: 10.1074/jbc.273.4.2260
24. Takayasu T, Suzuki S, Kametani F, Takahashi N, Shinoda T, Okuyama T, et al. Amino Acid Sequence of Galactosamine-Containing Glycopeptides in the Hinge Region of a Human Immunoglobulin D. Biochem Biophys Res Commun (1982) 105(3):1066–71. doi: 10.1016/0006-291X(82)91078-6
25. Plomp R, Dekkers G, Rombouts Y, Visser R, Koeleman CA, Kammeijer GS, et al. Hinge-Region O-Glycosylation of Human Immunoglobulin G3 (Igg3). Mol Cell Proteomics (2015) 14(5):1373–84. doi: 10.1074/mcp.M114.047381
26. Novak J, Julian BA, Tomana M, Mestecky J. IgA Glycosylation and IgA Immune Complexes in the Pathogenesis of IgA Nephropathy. Semin Nephrol (2008) 28(1):78–87. doi: 10.1016/j.semnephrol.2007.10.009
27. Iwasaki H, Zhang Y, Tachibana K, Gotoh M, Kikuchi N, Kwon YD, et al. Initiation of O-Glycan Synthesis in IgA1 Hinge Region is Determined by a Single Enzyme, UDP-N-Acetyl-Alpha-D-Galactosamine:Polypeptide N-Acetylgalactosaminyltransferase 2. J Biol Chem (2003) 278(8):5613–21. doi: 10.1074/jbc.M211097200
28. Stewart TJ, Takahashi K, Xu N, Prakash A, Brown R, Raska M, et al. Quantitative Assessment of Successive Carbohydrate Additions to the Clustered O-Glycosylation Sites of IgA1 by Glycosyltransferases. Glycobiology (2021) 31(5):540–56. doi: 10.1093/glycob/cwaa111
29. Ohyama Y, Renfrow MB, Novak J, Takahashi K. Aberrantly Glycosylated IgA1 in IgA Nephropathy: What We Know and What We Don't Know. J Clin Med (2021) 10(16):3467. doi: 10.3390/jcm10163467
30. Nimmerjahn F, Ravetch JV. Divergent Immunoglobulin G Subclass Activity Through Selective Fc Receptor Binding. Science (2005) 310(5753):1510–2. doi: 10.1126/science.1118948
31. Franklin EC. Structure and Function of Immunoglobulins. Acta Endocrinol Suppl (Copenh) (1975) 194:77–95. doi: 10.1530/acta.0.080S077
32. Spiegelberg HL. Biological Role of Different Antibody Classes. Int Arch Allergy Appl Immunol (1989) 90(Suppl 1):22–7. doi: 10.1159/000235071
33. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and Affinity of Human Fcgamma Receptors and Their Polymorphic Variants for Human IgG Subclasses. Blood (2009) 113(16):3716–25. doi: 10.1182/blood-2008-09-179754
34. Vidarsson G, Dekkers G, Rispens T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front Immunol (2014) 5:520. doi: 10.3389/fimmu.2014.00520
35. Bournazos S, Wang TT, Dahan R, Maamary J, Ravetch JV. Signaling by Antibodies: Recent Progress. Annu Rev Immunol (2017) 35:285–311. doi: 10.1146/annurev-immunol-051116-052433
36. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-Inflammatory Activity of Immunoglobulin G Resulting From Fc Sialylation. Science (2006) 313(5787):670–3. doi: 10.1126/science.1129594
37. Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIG Anti-Inflammatory Activity With a Recombinant IgG Fc. Science (2008) 320(5874):373–6. doi: 10.1126/science.1154315
38. Wang X, Mathieu M, Brezski RJ. IgG Fc Engineering to Modulate Antibody Effector Functions. Protein Cell (2018) 9(1):63–73. doi: 10.1007/s13238-017-0473-8
39. Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General Mechanism for Modulating Immunoglobulin Effector Function. Proc Natl Acad Sci USA (2013) 110(24):9868–72. doi: 10.1073/pnas.1307864110
40. Subedi GP, Barb AW. The Structural Role of Antibody N-Glycosylation in Receptor Interactions. Structure (2015) 23(9):1573–83. doi: 10.1016/j.str.2015.06.015
41. Nose M, Wigzell H. Biological Significance of Carbohydrate Chains on Monoclonal Antibodies. Proc Natl Acad Sci USA (1983) 80(21):6632–6. doi: 10.1073/pnas.80.21.6632
42. Pound JD, Lund J, Jefferis R. Aglycosylated Chimaeric Human IgG3 can Trigger the Human Phagocyte Respiratory Burst. Mol Immunol (1993) 30(3):233–41. doi: 10.1016/0161-5890(93)90052-D
43. Tao MH, Morrison SL. Studies of Aglycosylated Chimeric Mouse-Human IgG. Role of Carbohydrate in the Structure and Effector Functions Mediated by the Human IgG Constant Region. J Immunol (1989) 143(8):2595–601.
44. Pucic M, Knezevic A, Vidic J, Adamczyk B, Novokmet M, Polasek O, et al. High Throughput Isolation and Glycosylation Analysis of IgG-Variability and Heritability of the IgG Glycome in Three Isolated Human Populations. Mol Cell Proteomics (2011) 10(10):M111 010090. doi: 10.1074/mcp.M111.010090
45. Jefferis R. Glycosylation of Recombinant Antibody Therapeutics. Biotechnol Prog (2005) 21(1):11–6. doi: 10.1021/bp040016j
46. Liu L. Pharmacokinetics of Monoclonal Antibodies and Fc-Fusion Proteins. Protein Cell (2018) 9(1):15–32. doi: 10.1007/s13238-017-0408-4
47. Liu H, Nowak C, Andrien B, Shao M, Ponniah G, Neill A. Impact of IgG Fc-Oligosaccharides on Recombinant Monoclonal Antibody Structure, Stability, Safety, and Efficacy. Biotechnol Prog (2017) 33(5):1173–81. doi: 10.1002/btpr.2498
48. Higel F, Seidl A, Sorgel F, Friess W. N-Glycosylation Heterogeneity and the Influence on Structure, Function and Pharmacokinetics of Monoclonal Antibodies and Fc Fusion Proteins. Eur J Pharm Biopharm (2016) 100:94–100. doi: 10.1016/j.ejpb.2016.01.005
49. Gudelj I, Lauc G, Pezer M. Immunoglobulin G Glycosylation in Aging and Diseases. Cell Immunol (2018) 333:65–79. doi: 10.1016/j.cellimm.2018.07.009
50. Li D, Lou Y, Zhang Y, Liu S, Li J, Tao J. Sialylated Immunoglobulin G: A Promising Diagnostic and Therapeutic Strategy for Autoimmune Diseases. Theranostics (2021) 11(11):5430–46. doi: 10.7150/thno.53961
51. Imbach P, Wagner HP, Berchtold W, Gaedicke G, Hirt A, Joller P, et al. Intravenous Immunoglobulin Versus Oral Corticosteroids in Acute Immune Thrombocytopenic Purpura in Childhood. Lancet (1985) 2(8453):464–8. doi: 10.1016/s0140-6736(85)90400-3
52. Levy Y, Sherer Y, Ahmed A, Langevitz P, George J, Fabbrizzi F, et al. A Study of 20 SLE Patients With Intravenous Immunoglobulin–Clinical and Serologic Response. Lupus (1999) 8(9):705–12. doi: 10.1191/096120399678841007
53. Hughes RA, Donofrio P, Bril V, Dalakas MC, Deng C, Hanna K, et al. Intravenous Immune Globulin (10% Caprylate-Chromatography Purified) for the Treatment of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (ICE Study): A Randomised Placebo-Controlled Trial. Lancet Neurol (2008) 7(2):136–44. doi: 10.1016/S1474-4422(07)70329-0
54. Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a Receptor Required for the Anti-Inflammatory Activity of IVIG. Proc Natl Acad Sci USA (2008) 105(50):19571–8. doi: 10.1073/pnas.0810163105
55. Washburn N, Schwab I, Ortiz D, Bhatnagar N, Lansing JC, Medeiros A, et al. Controlled Tetra-Fc Sialylation of IVIg Results in a Drug Candidate With Consistent Enhanced Anti-Inflammatory Activity. Proc Natl Acad Sci USA (2015) 112(11):E1297–306. doi: 10.1073/pnas.1422481112
56. Samuelsson A, Towers TL, Ravetch JV. Anti-Inflammatory Activity of IVIG Mediated Through the Inhibitory Fc Receptor. Science (2001) 291(5503):484–6. doi: 10.1126/science.291.5503.484
57. Nimmerjahn F, Ravetch JV. The Antiinflammatory Activity of IgG: The Intravenous IgG Paradox. J Exp Med (2007) 204(1):11–5. doi: 10.1084/jem.20061788
58. Crow AR, Song S, Freedman J, Helgason CD, Humphries RK, Siminovitch KA, et al. IVIg-Mediated Amelioration of Murine ITP via FcgammaRIIB is Independent of SHIP1, SHP-1, and Btk Activity. Blood (2003) 102(2):558–60. doi: 10.1182/blood-2003-01-0023
59. Fiebiger BM, Maamary J, Pincetic A, Ravetch JV. Protection in Antibody- and T Cell-Mediated Autoimmune Diseases by Antiinflammatory IgG Fcs Requires Type II FcRs. Proc Natl Acad Sci USA (2015) 112(18):E2385–94. doi: 10.1073/pnas.1505292112
60. Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous Gammaglobulin Suppresses Inflammation Through a Novel T(H)2 Pathway. Nature (2011) 475(7354):110–3. doi: 10.1038/nature10134
61. Temming AR, Dekkers G, van de Bovenkamp FS, Plomp HR, Bentlage AEH, Szittner Z, et al. Human DC-SIGN and CD23 do Not Interact With Human IgG. Sci Rep (2019) 9(1):9995. doi: 10.1038/s41598-019-46484-2
62. Yu X, Vasiljevic S, Mitchell DA, Crispin M, Scanlan CN. Dissecting the Molecular Mechanism of IVIg Therapy: The Interaction Between Serum IgG and DC-SIGN is Independent of Antibody Glycoform or Fc Domain. J Mol Biol (2013) 425(8):1253–8. doi: 10.1016/j.jmb.2013.02.006
63. Blundell PA, Le NPL, Allen J, Watanabe Y, Pleass RJ. Engineering the Fragment Crystallizable (Fc) Region of Human IgG1 Multimers and Monomers to Fine-Tune Interactions With Sialic Acid-Dependent Receptors. J Biol Chem (2017) 292(31):12994–3007. doi: 10.1074/jbc.M117.795047
64. Pleass RJ. The Therapeutic Potential of Sialylated Fc Domains of Human IgG. MAbs (2021) 13(1):1953220. doi: 10.1080/19420862.2021.1953220
65. Baksmeier C, Blundell P, Steckel J, Schultz V, Gu Q, Da Silva Filipe A, et al. Modified Recombinant Human IgG1-Fc is Superior to Natural Intravenous Immunoglobulin at Inhibiting Immune-Mediated Demyelination. Immunology (2021) 164(1):90–105. doi: 10.1111/imm.13341
66. Massoud AH, Yona M, Xue D, Chouiali F, Alturaihi H, Ablona A, et al. Dendritic Cell Immunoreceptor: A Novel Receptor for Intravenous Immunoglobulin Mediates Induction of Regulatory T Cells. J Allergy Clin Immunol (2014) 133(3):853–63.e5. doi: 10.1016/j.jaci.2013.09.029
67. Arroyo S, Tiessen RG, Denney WS, Jin J, van Iersel MP, Zeitz H, et al. Hyper-Sialylated IgG M254, an Innovative Therapeutic Candidate, Evaluated in Healthy Volunteers and in Patients With Immune Thrombocytopenia Purpura: Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics. Blood (2019) 134(Supplement_1):1090–. doi: 10.1182/blood-2019-125993
68. Nimmerjahn F, Lunemann JD. Expression and Function of the Inhibitory Fcgamma-Receptor in CIDP. J Peripher Nerv Syst (2011) 16(Suppl 1):41–4. doi: 10.1111/j.1529-8027.2011.00305.x
69. Tackenberg B, Jelcic I, Baerenwaldt A, Oertel WH, Sommer N, Nimmerjahn F, et al. Impaired Inhibitory Fcgamma Receptor IIB Expression on B Cells in Chronic Inflammatory Demyelinating Polyneuropathy. Proc Natl Acad Sci USA (2009) 106(12):4788–92. doi: 10.1073/pnas.0807319106
70. Tjon AS, van Gent R, Jaadar H, Martin van Hagen P, Mancham S, van der Laan LJ, et al. Intravenous Immunoglobulin Treatment in Humans Suppresses Dendritic Cell Function via Stimulation of IL-4 and IL-13 Production. J Immunol (2014) 192(12):5625–34. doi: 10.4049/jimmunol.1301260
71. Bartsch YC, Eschweiler S, Leliavski A, Lunding HB, Wagt S, Petry J, et al. IgG Fc Sialylation is Regulated During the Germinal Center Reaction Following Immunization With Different Adjuvants. J Allergy Clin Immunol (2020) 146(3):652–66.e11. doi: 10.1016/j.jaci.2020.04.059
72. Petry J, Rahmoller J, Duhring L, Lilienthal GM, Lehrian S, Buhre JS, et al. Enriched Blood IgG Sialylation Attenuates IgG-Mediated and IgG-Controlled-IgE-Mediated Allergic Reactions. J Allergy Clin Immunol (2021) 147(2):763–7. doi: 10.1016/j.jaci.2020.05.056
73. Wang TT, Maamary J, Tan GS, Bournazos S, Davis CW, Krammer F, et al. Anti-HA Glycoforms Drive B Cell Affinity Selection and Determine Influenza Vaccine Efficacy. Cell (2015) 162(1):160–9. doi: 10.1016/j.cell.2015.06.026
74. Chakraborty S, Gonzalez JC, Sievers BL, Mallajosyula V, Chakraborty S, Dubey M, et al. Early Non-Neutralizing, Afucosylated Antibody Responses Are Associated With COVID-19 Severity. Sci Transl Med (2022) 14(635):eabm7853. doi: 10.1126/scitranslmed.abm7853
75. Wang TT, Ravetch JV. Functional Diversification of IgGs Through Fc Glycosylation. J Clin Invest (2019) 129(9):3492–8. doi: 10.1172/JCI130029
76. Thomann M, Schlothauer T, Dashivets T, Malik S, Avenal C, Bulau P, et al. In Vitro Glycoengineering of IgG1 and its Effect on Fc Receptor Binding and ADCC Activity. PloS One (2015) 10(8):e0134949. doi: 10.1371/journal.pone.0134949
77. Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Modulating IgG Effector Function by Fc Glycan Engineering. Proc Natl Acad Sci USA (2017) 114(13):3485–90. doi: 10.1073/pnas.1702173114
78. Scallon BJ, Tam SH, McCarthy SG, Cai AN, Raju TS. Higher Levels of Sialylated Fc Glycans in Immunoglobulin G Molecules can Adversely Impact Functionality. Mol Immunol (2007) 44(7):1524–34. doi: 10.1016/j.molimm.2006.09.005
79. Wada R, Matsui M, Kawasaki N. Influence of N-Glycosylation on Effector Functions and Thermal Stability of Glycoengineered IgG1 Monoclonal Antibody With Homogeneous Glycoforms. MAbs (2019) 11(2):350–72. doi: 10.1080/19420862.2018.1551044
80. Quast I, Keller CW, Maurer MA, Giddens JP, Tackenberg B, Wang LX, et al. Sialylation of IgG Fc Domain Impairs Complement-Dependent Cytotoxicity. J Clin Invest (2015) 125(11):4160–70. doi: 10.1172/JCI82695
81. Dekkers G, Treffers L, Plomp R, Bentlage AEH, de Boer M, Koeleman CAM, et al. Decoding the Human Immunoglobulin G-Glycan Repertoire Reveals a Spectrum of Fc-Receptor- and Complement-Mediated-Effector Activities. Front Immunol (2017) 8:877. doi: 10.3389/fimmu.2017.00877
82. Pereira NA, Chan KF, Lin PC, Song Z. The "Less-is-More" in Therapeutic Antibodies: Afucosylated Anti-Cancer Antibodies With Enhanced Antibody-Dependent Cellular Cytotoxicity. MAbs (2018) 10(5):693–711. doi: 10.1080/19420862.2018.1466767
83. Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The Absence of Fucose But Not the Presence of Galactose or Bisecting N-Acetylglucosamine of Human IgG1 Complex-Type Oligosaccharides Shows the Critical Role of Enhancing Antibody-Dependent Cellular Cytotoxicity. J Biol Chem (2003) 278(5):3466–73. doi: 10.1074/jbc.M210665200
84. Mimura Y, Katoh T, Saldova R, O'Flaherty R, Izumi T, Mimura-Kimura Y, et al. Glycosylation Engineering of Therapeutic IgG Antibodies: Challenges for the Safety, Functionality and Efficacy. Protein Cell (2018) 9(1):47–62. doi: 10.1007/s13238-017-0433-3
85. Satoh M, Iida S, Shitara K. Non-Fucosylated Therapeutic Antibodies as Next-Generation Therapeutic Antibodies. Expert Opin Biol Ther (2006) 6(11):1161–73. doi: 10.1517/14712598.6.11.1161
86. Kobata A. The N-Linked Sugar Chains of Human Immunoglobulin G: Their Unique Pattern, and Their Functional Roles. Biochim Biophys Acta (2008) 1780(3):472–8. doi: 10.1016/j.bbagen.2007.06.012
87. Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A Crystal Structure of the Human IgG1 Fc Fragment-Fc gammaRIII Complex. Nature (2000) 406(6793):267–73. doi: 10.1038/35018508
88. Ferrara C, Stuart F, Sondermann P, Brunker P, Umana P. The Carbohydrate at FcgammaRIIIa Asn-162. An Element Required for High Affinity Binding to Non-Fucosylated IgG Glycoforms. J Biol Chem (2006) 281(8):5032–6. doi: 10.1074/jbc.M510171200
89. Shields RL, Lai J, Keck R, O'Connell LY, Hong K, Meng YG, et al. Lack of Fucose on Human IgG1 N-Linked Oligosaccharide Improves Binding to Human Fcgamma RIII and Antibody-Dependent Cellular Toxicity. J Biol Chem (2002) 277(30):26733–40. doi: 10.1074/jbc.M202069200
90. Mastrangeli R, Palinsky W, Bierau H. Glycoengineered Antibodies: Towards the Next-Generation of Immunotherapeutics. Glycobiology (2019) 29(3):199–210. doi: 10.1093/glycob/cwy092
91. Li W, Zhu Z, Chen W, Feng Y, Dimitrov DS. Crystallizable Fragment Glycoengineering for Therapeutic Antibodies Development. Front Immunol (2017) 8:1554. doi: 10.3389/fimmu.2017.01554
92. Wang LX, Tong X, Li C, Giddens JP, Li T. Glycoengineering of Antibodies for Modulating Functions. Annu Rev Biochem (2019) 88:433–59. doi: 10.1146/annurev-biochem-062917-012911
93. Chakraborty S, Gonzalez J, Edwards K, Mallajosyula V, Buzzanco AS, Sherwood R, et al. Proinflammatory IgG Fc Structures in Patients With Severe COVID-19. Nat Immunol (2021) 22(1):67–73. doi: 10.1038/s41590-020-00828-7
94. Larsen MD, de Graaf EL, Sonneveld ME, Plomp HR, Nouta J, Hoepel W, et al. Afucosylated IgG Characterizes Enveloped Viral Responses and Correlates With COVID-19 Severity. Science (2021) 371(6532):eabc8378. doi: 10.1126/science.abc8378
95. Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu Rev Immunol (2007) 25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702
96. Parekh RB, Dwek RA, Sutton BJ, Fernandes DL, Leung A, Stanworth D, et al. Association of Rheumatoid Arthritis and Primary Osteoarthritis With Changes in the Glycosylation Pattern of Total Serum IgG. Nature (1985) 316(6027):452–7. doi: 10.1038/316452a0
97. Tomana M, Schrohenloher RE, Koopman WJ, Alarcon GS, Paul WA. Abnormal Glycosylation of Serum IgG From Patients With Chronic Inflammatory Diseases. Arthritis Rheumatol (1988) 31(3):333–8. doi: 10.1002/art.1780310304
98. Leirisalo-Repo M, Hernandez-Munoz HE, Rook GA. Agalactosyl IgG is Elevated in Patients With Active Spondyloarthropathy. Rheumatol Int (1999) 18(5-6):171–6. doi: 10.1007/s002960050080
99. Dube R, Rook GA, Steele J, Brealey R, Dwek R, Rademacher T, et al. Agalactosyl IgG in Inflammatory Bowel Disease: Correlation With C-Reactive Protein. Gut (1990) 31(4):431–4. doi: 10.1136/gut.31.4.431
100. Ercan A, Cui J, Chatterton DE, Deane KD, Hazen MM, Brintnell W, et al. Aberrant IgG Galactosylation Precedes Disease Onset, Correlates With Disease Activity, and is Prevalent in Autoantibodies in Rheumatoid Arthritis. Arthritis Rheumatol (2010) 62(8):2239–48. doi: 10.1002/art.27533
101. Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, et al. Anti-Inflammatory Activity of IgG1 Mediated by Fc Galactosylation and Association of FcgammaRIIB and Dectin-1. Nat Med (2012) 18(9):1401–6. doi: 10.1038/nm.2862
102. van Osch TLJ, Nouta J, Derksen NIL, van Mierlo G, van der Schoot CE, Wuhrer M, et al. Fc Galactosylation Promotes Hexamerization of Human IgG1, Leading to Enhanced Classical Complement Activation. J Immunol (2021) 207(6):1545–54. doi: 10.4049/jimmunol.2100399
103. Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered Glycoforms of an Antineuroblastoma IgG1 With Optimized Antibody-Dependent Cellular Cytotoxic Activity. Nat Biotechnol (1999) 17(2):176–80. doi: 10.1038/6179
104. Vieira P, Rajewsky K. The Half-Lives of Serum Immunoglobulins in Adult Mice. Eur J Immunol (1988) 18(2):313–6. doi: 10.1002/eji.1830180221
105. Dombrowicz D, Brini AT, Flamand V, Hicks E, Snouwaert JN, Kinet JP, et al. Anaphylaxis Mediated Through a Humanized High Affinity IgE Receptor. J Immunol (1996) 157(4):1645–51.
106. Gould HJ, Sutton BJ, Beavil AJ, Beavil RL, McCloskey N, Coker HA, et al. The Biology of IGE and the Basis of Allergic Disease. Annu Rev Immunol (2003) 21:579–628. doi: 10.1146/annurev.immunol.21.120601.141103
107. Matsumoto M, Sasaki Y, Yasuda K, Takai T, Muramatsu M, Yoshimoto T, et al. IgG and IgE Collaboratively Accelerate Expulsion of Strongyloides Venezuelensis in a Primary Infection. Infect Immun (2013) 81(7):2518–27. doi: 10.1128/IAI.00285-13
108. Arnold JN, Radcliffe CM, Wormald MR, Royle L, Harvey DJ, Crispin M, et al. The Glycosylation of Human Serum IgD and IgE and the Accessibility of Identified Oligomannose Structures for Interaction With Mannan-Binding Lectin. J Immunol (2004) 173(11):6831–40. doi: 10.4049/jimmunol.173.11.6831
109. Plomp R, Hensbergen PJ, Rombouts Y, Zauner G, Dragan I, Koeleman CA, et al. Site-Specific N-Glycosylation Analysis of Human Immunoglobulin E. J Proteome Res (2014) 13(2):536–46. doi: 10.1021/pr400714w
110. Wu G, Hitchen PG, Panico M, North SJ, Barbouche MR, Binet D, et al. Glycoproteomic Studies of IgE From a Novel Hyper IgE Syndrome Linked to PGM3 Mutation. Glycoconj J (2016) 33(3):447–56. doi: 10.1007/s10719-015-9638-y
111. Shade KC, Conroy ME, Washburn N, Kitaoka M, Huynh DJ, Laprise E, et al. Sialylation of Immunoglobulin E is a Determinant of Allergic Pathogenicity. Nature (2020) 582(7811):265–70. doi: 10.1038/s41586-020-2311-z
112. Chang TW. The Pharmacological Basis of Anti-IgE Therapy. Nat Biotechnol (2000) 18(2):157–62. doi: 10.1038/72601
113. Anvari S, Miller J, Yeh CY, Davis CM. IgE-Mediated Food Allergy. Clin Rev Allergy Immunol (2019) 57(2):244–60. doi: 10.1007/s12016-018-8710-3
114. Garman SC, Wurzburg BA, Tarchevskaya SS, Kinet JP, Jardetzky TS. Structure of the Fc Fragment of Human IgE Bound to its High-Affinity Receptor Fc epsilonRI Alpha. Nature (2000) 406(6793):259–66. doi: 10.1038/35018500
115. Holdom MD, Davies AM, Nettleship JE, Bagby SC, Dhaliwal B, Girardi E, et al. Conformational Changes in IgE Contribute to its Uniquely Slow Dissociation Rate From Receptor FcvarepsilonRI. Nat Struct Mol Biol (2011) 18(5):571–6. doi: 10.1038/nsmb.2044
116. Borthakur S, Hibbert RG, Pang MO, Yahya N, Bax HJ, Kao MW, et al. Mapping of the CD23 Binding Site on Immunoglobulin E (IgE) and Allosteric Control of the IgE-Fc epsilonRI Interaction. J Biol Chem (2012) 287(37):31457–61. doi: 10.1074/jbc.C112.397059
117. Nettleton MY, Kochan JP. Role of Glycosylation Sites in the IgE Fc Molecule. Int Arch Allergy Immunol (1995) 107(1-3):328–9. doi: 10.1159/000237017
118. Sayers I, Cain SA, Swan JR, Pickett MA, Watt PJ, Holgate ST, et al. Amino Acid Residues That Influence Fc Epsilon RI-Mediated Effector Functions of Human Immunoglobulin E. Biochemistry (1998) 37(46):16152–64. doi: 10.1021/bi981456k
119. Hunt J, Beavil RL, Calvert RA, Gould HJ, Sutton BJ, Beavil AJ. Disulfide Linkage Controls the Affinity and Stoichiometry of IgE Fcepsilon3-4 Binding to FcepsilonRI. J Biol Chem (2005) 280(17):16808–14. doi: 10.1074/jbc.M500965200
120. Helm B, Marsh P, Vercelli D, Padlan E, Gould H, Geha R. The Mast Cell Binding Site on Human Immunoglobulin E. Nature (1988) 331(6152):180–3. doi: 10.1038/331180a0
121. Helm BA, Sayers I, Padlan EA, McKendrick JE, Spivey AC. Structure/function Studies on IgE as a Basis for the Development of Rational IgE Antagonists. Allergy (1998) 53(45 Suppl):77–82. doi: 10.1111/j.1398-9995.1998.tb04945.x
122. Henry AJ, McDonnell JM, Ghirlando R, Sutton BJ, Gould HJ. Conformation of the Isolated Cepsilon3 Domain of IgE and its Complex With the High-Affinity Receptor, FcepsilonRI. Biochemistry (2000) 39(25):7406–13. doi: 10.1021/bi9928391
123. Shade KT, Platzer B, Washburn N, Mani V, Bartsch YC, Conroy M, et al. A Single Glycan on IgE is Indispensable for Initiation of Anaphylaxis. J Exp Med (2015) 212(4):457–67. doi: 10.1084/jem.20142182
124. Haury M, Sundblad A, Grandien A, Barreau C, Coutinho A, Nobrega A. The Repertoire of Serum IgM in Normal Mice is Largely Independent of External Antigenic Contact. Eur J Immunol (1997) 27(6):1557–63. doi: 10.1002/eji.1830270635
125. Jayasekera JP, Moseman EA, Carroll MC. Natural Antibody and Complement Mediate Neutralization of Influenza Virus in the Absence of Prior Immunity. J Virol (2007) 81(7):3487–94. doi: 10.1128/JVI.02128-06
126. Griffin DO, Holodick NE, Rothstein TL. Human B1 Cells in Umbilical Cord and Adult Peripheral Blood Express the Novel Phenotype CD20+ CD27+ CD43+ Cd70. J Exp Med (2011) 208(1):67–80. doi: 10.1084/jem.20101499
127. Casali P, Schettino EW. Structure and Function of Natural Antibodies. Curr Top Microbiol Immunol (1996) 210:167–79. doi: 10.1007/978-3-642-85226-8_17
128. Gronwall C, Vas J, Silverman GJ. Protective Roles of Natural IgM Antibodies. Front Immunol (2012) 3:66. doi: 10.3389/fimmu.2012.00066
129. Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation-Specific Epitopes Are Dominant Targets of Innate Natural Antibodies in Mice and Humans. J Clin Invest (2009) 119(5):1335–49. doi: 10.1172/JCI36800
130. Baumgarth N. The Double Life of a B-1 Cell: Self-Reactivity Selects for Protective Effector Functions. Nat Rev Immunol (2011) 11(1):34–46. doi: 10.1038/nri2901
131. Ehrenstein MR, Notley CA. The Importance of Natural IgM: Scavenger, Protector and Regulator. Nat Rev Immunol (2010) 10(11):778–86. doi: 10.1038/nri2849
132. Racine R, Winslow GM. IgM in Microbial Infections: Taken for Granted? Immunol Lett (2009) 125(2):79–85. doi: 10.1016/j.imlet.2009.06.003
133. Davis AC, Roux KH, Shulman MJ. On the Structure of Polymeric IgM. Eur J Immunol (1988) 18(7):1001–8. doi: 10.1002/eji.1830180705
134. Sorensen V, Sundvold V, Michaelsen TE, Sandlie I. Polymerization of IgA and IgM: Roles of Cys309/Cys414 and the Secretory Tailpiece. J Immunol (1999) 162(6):3448–55.
135. Pasalic D, Weber B, Giannone C, Anelli T, Muller R, Fagioli C, et al. A Peptide Extension Dictates IgM Assembly. Proc Natl Acad Sci USA (2017) 114(41):E8575–E84. doi: 10.1073/pnas.1701797114
136. Sopp JM, Peters SJ, Rowley TF, Oldham RJ, James S, Mockridge I, et al. On-Target IgG Hexamerisation Driven by a C-Terminal IgM Tail-Piece Fusion Variant Confers Augmented Complement Activation. Commun Biol (2021) 4(1):1031. doi: 10.1038/s42003-021-02513-3
137. Smith RI, Coloma MJ, Morrison SL. Addition of a Mu-Tailpiece to IgG Results in Polymeric Antibodies With Enhanced Effector Functions Including Complement-Mediated Cytolysis by Igg4. J Immunol (1995) 154(5):2226–36.
138. Frutiger S, Hughes GJ, Paquet N, Luthy R, Jaton JC. Disulfide Bond Assignment in Human J Chain and its Covalent Pairing With Immunoglobulin M. Biochemistry (1992) 31(50):12643–7. doi: 10.1021/bi00165a014
139. Sorensen V, Rasmussen IB, Sundvold V, Michaelsen TE, Sandlie I. Structural Requirements for Incorporation of J Chain Into Human IgM and IgA. Int Immunol (2000) 12(1):19–27. doi: 10.1093/intimm/12.1.19
140. Moh ES, Lin CH, Thaysen-Andersen M, Packer NH. Site-Specific N-Glycosylation of Recombinant Pentameric and Hexameric Human IgM. J Am Soc Mass Spectrom (2016) 27(7):1143–55. doi: 10.1007/s13361-016-1378-0
141. Muraoka S, Shulman MJ. Structural Requirements for IgM Assembly and Cytolytic Activity. Effects of Mutations in the Oligosaccharide Acceptor Site at Asn402. J Immunol (1989) 142(2):695–701.
142. Arnold JN, Wormald MR, Suter DM, Radcliffe CM, Harvey DJ, Dwek RA, et al. Human Serum IgM Glycosylation: Identification of Glycoforms That can Bind to Mannan-Binding Lectin. J Biol Chem (2005) 280(32):29080–7. doi: 10.1074/jbc.M504528200
143. Dorsett B, Cronin W, Chuma V, Ioachim HL. Anti-Lymphocyte Antibodies in Patients With the Acquired Immune Deficiency Syndrome. Am J Med (1985) 78(4):621–6. doi: 10.1016/0002-9343(85)90405-X
144. Winfield JB, Fernsten PD, Czyzyk JK. Anti-Lymphocyte Autoantibodies in Systemic Lupus Erythematosus. Trans Am Clin Climatol Assoc (1997) 108:127–35.
145. Lobo PI, Schlegel KH, Spencer CE, Okusa MD, Chisholm C, McHedlishvili N, et al. Naturally Occurring IgM Anti-Leukocyte Autoantibodies (IgM-ALA) Inhibit T Cell Activation and Chemotaxis. J Immunol (2008) 180(3):1780–91. doi: 10.4049/jimmunol.180.3.1780
146. Lobo PI, Brayman KL, Okusa MD. Natural IgM Anti-Leucocyte Autoantibodies (IgM-ALA) Regulate Inflammation Induced by Innate and Adaptive Immune Mechanisms. J Clin Immunol (2014) 34(Suppl 1):S22–9. doi: 10.1007/s10875-014-0027-2
147. Colucci M, Stockmann H, Butera A, Masotti A, Baldassarre A, Giorda E, et al. Sialylation of N-Linked Glycans Influences the Immunomodulatory Effects of IgM on T Cells. J Immunol (2015) 194(1):151–7. doi: 10.4049/jimmunol.1402025
148. Lloyd KA, Wang J, Urban BC, Czajkowsky DM, Pleass RJ. Glycan-Independent Binding and Internalization of Human IgM to FCMR, its Cognate Cellular Receptor. Sci Rep (2017) 7:42989. doi: 10.1038/srep42989
149. Kumar Bharathkar S, Parker BW, Malyutin AG, Haloi N, Huey-Tubman KE, Tajkhorshid E, et al. The Structures of Secretory and Dimeric Immunoglobulin A. Elife (2020) 9:e56098. doi: 10.7554/eLife.56098
150. Breedveld A, van Egmond M. IgA and FcalphaRI: Pathological Roles and Therapeutic Opportunities. Front Immunol (2019) 10:553. doi: 10.3389/fimmu.2019.00553
151. Ramsland PA, Willoughby N, Trist HM, Farrugia W, Hogarth PM, Fraser JD, et al. Structural Basis for Evasion of IgA Immunity by Staphylococcus Aureus Revealed in the Complex of SSL7 With Fc of Human Iga1. Proc Natl Acad Sci USA (2007) 104(38):15051–6. doi: 10.1073/pnas.0706028104
152. Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J. Galactose-Deficient IgA1 in Sera of IgA Nephropathy Patients is Present in Complexes With IgG. Kidney Int (1997) 52(2):509–16. doi: 10.1038/ki.1997.361
153. Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating Immune Complexes in IgA Nephropathy Consist of IgA1 With Galactose-Deficient Hinge Region and Antiglycan Antibodies. J Clin Invest (1999) 104(1):73–81. doi: 10.1172/JCI5535
154. Novak J, Vu HL, Novak L, Julian BA, Mestecky J, Tomana M. Interactions of Human Mesangial Cells With IgA and IgA-Containing Immune Complexes. Kidney Int (2002) 62(2):465–75. doi: 10.1046/j.1523-1755.2002.00477.x
155. Steffen U, Koeleman CA, Sokolova MV, Bang H, Kleyer A, Rech J, et al. IgA Subclasses Have Different Effector Functions Associated With Distinct Glycosylation Profiles. Nat Commun (2020) 11(1):120. doi: 10.1038/s41467-019-13992-8
156. Maurer MA, Meyer L, Bianchi M, Turner HL, Le NPL, Steck M, et al. Glycosylation of Human IgA Directly Inhibits Influenza A and Other Sialic-Acid-Binding Viruses. Cell Rep (2018) 23(1):90–9. doi: 10.1016/j.celrep.2018.03.027
157. Sterlin D, Gorochov G. When Therapeutic IgA Antibodies Might Come of Age. Pharmacology (2021) 106(1-2):9–19. doi: 10.1159/000510251
158. Wang Q, Chung CY, Chough S, Betenbaugh MJ. Antibody Glycoengineering Strategies in Mammalian Cells. Biotechnol Bioeng (2018) 115(6):1378–93. doi: 10.1002/bit.26567
159. Li S, McCraw AJ, Gardner RA, Spencer DIR, Karagiannis SN, Wagner GK. Glycoengineering of Therapeutic Antibodies With Small Molecule Inhibitors. Antib (Basel) (2021) 10(4):44. doi: 10.3390/antib10040044
160. Dionne B, Mishra N, Butler M. A Low Redox Potential Affects Monoclonal Antibody Assembly and Glycosylation in Cell Culture. J Biotechnol (2017) 246:71–80. doi: 10.1016/j.jbiotec.2017.01.016
161. Blundell PA, Lu D, Dell A, Haslam S, Pleass RJ. Choice of Host Cell Line Is Essential for the Functional Glycosylation of the Fc Region of Human IgG1 Inhibitors of Influenza B Viruses. J Immunol (2020) 204(4):1022–34. doi: 10.4049/jimmunol.1901145
162. Raju TS, Jordan RE. Galactosylation Variations in Marketed Therapeutic Antibodies. MAbs (2012) 4(3):385–91. doi: 10.4161/mabs.19868
163. Yu M, Brown D, Reed C, Chung S, Lutman J, Stefanich E, et al. Production, Characterization, and Pharmacokinetic Properties of Antibodies With N-Linked Mannose-5 Glycans. MAbs (2012) 4(4):475–87. doi: 10.4161/mabs.20737
164. Jefferis R. Glycosylation as a Strategy to Improve Antibody-Based Therapeutics. Nat Rev Drug Discovery (2009) 8(3):226–34. doi: 10.1038/nrd2804
165. Zhong X, Cooley C, Seth N, Juo ZS, Presman E, Resendes N, et al. Engineering Novel Lec1 Glycosylation Mutants in CHO-DUKX Cells: Molecular Insights and Effector Modulation of N-Acetylglucosaminyltransferase I. Biotechnol Bioeng (2012) 109(7):1723–34. doi: 10.1002/bit.24448
166. Shi HH, Goudar CT. Recent Advances in the Understanding of Biological Implications and Modulation Methodologies of Monoclonal Antibody N-Linked High Mannose Glycans. Biotechnol Bioeng (2014) 111(10):1907–19. doi: 10.1002/bit.25318
167. Liu L. Antibody Glycosylation and its Impact on the Pharmacokinetics and Pharmacodynamics of Monoclonal Antibodies and Fc-Fusion Proteins. J Pharm Sci (2015) 104(6):1866–84. doi: 10.1002/jps.24444
168. Wong NS, Yap MG, Wang DI. Enhancing Recombinant Glycoprotein Sialylation Through CMP-Sialic Acid Transporter Over Expression in Chinese Hamster Ovary Cells. Biotechnol Bioeng (2006) 93(5):1005–16. doi: 10.1002/bit.20815
169. Nguyen NTB, Lin J, Tay SJ, Mariati, Yeo J, Nguyen-Khuong T, et al. Multiplexed Engineering Glycosyltransferase Genes in CHO Cells via Targeted Integration for Producing Antibodies With Diverse Complex-Type N-Glycans. Sci Rep (2021) 11(1):12969. doi: 10.1038/s41598-021-92320-x
170. Raymond C, Robotham A, Spearman M, Butler M, Kelly J, Durocher Y. Production of Alpha2,6-Sialylated IgG1 in CHO Cells. MAbs (2015) 7(3):571–83. doi: 10.1080/19420862.2015.1029215
171. Schulz MA, Tian W, Mao Y, Van Coillie J, Sun L, Larsen JS, et al. Glycoengineering Design Options for IgG1 in CHO Cells Using Precise Gene Editing. Glycobiology (2018) 28(7):542–9. doi: 10.1093/glycob/cwy022
172. Davies J, Jiang L, Pan LZ, LaBarre MJ, Anderson D, Reff M. Expression of GnTIII in a Recombinant Anti-CD20 CHO Production Cell Line: Expression of Antibodies With Altered Glycoforms Leads to an Increase in ADCC Through Higher Affinity for FC Gamma RIII. Biotechnol Bioeng (2001) 74(4):288–94. doi: 10.1002/bit.1119
173. Ferrara C, Brünker P, Suter T, Moser S, Püntener U, Umaña P. Modulation of Therapeutic Antibody Effector Functions by Glycosylation Engineering: Influence of Golgi Enzyme Localization Domain and Co-Expression of Heterologous Beta1, 4-N-Acetylglucosaminyltransferase III and Golgi Alpha-Mannosidase II. Biotechnol Bioeng (2006) 93(5):851–61. doi: 10.1002/bit.20777
174. Mori K, Kuni-Kamochi R, Yamane-Ohnuki N, Wakitani M, Yamano K, Imai H, et al. Engineering Chinese Hamster Ovary Cells to Maximize Effector Function of Produced Antibodies Using FUT8 siRNA. Biotechnol Bioeng (2004) 88(7):901–8. doi: 10.1002/bit.20326
175. Becker DJ, Lowe JB. Fucose: Biosynthesis and Biological Function in Mammals. Glycobiology (2003) 13(7):41R–53R. doi: 10.1093/glycob/cwg054
176. Louie S, Haley B, Marshall B, Heidersbach A, Yim M, Brozynski M, et al. FX Knockout CHO Hosts can Express Desired Ratios of Fucosylated or Afucosylated Antibodies With High Titers and Comparable Product Quality. Biotechnol Bioeng (2017) 114(3):632–44. doi: 10.1002/bit.26188
177. Kanda Y, Imai-Nishiya H, Kuni-Kamochi R, Mori K, Inoue M, Kitajima-Miyama K, et al. Establishment of a GDP-Mannose 4,6-Dehydratase (GMD) Knockout Host Cell Line: A New Strategy for Generating Completely Non-Fucosylated Recombinant Therapeutics. J Biotechnol (2007) 130(3):300–10. doi: 10.1016/j.jbiotec.2007.04.025
178. von Horsten HH, Ogorek C, Blanchard V, Demmler C, Giese C, Winkler K, et al. Production of Non-Fucosylated Antibodies by Co-Expression of Heterologous GDP-6-Deoxy-D-Lyxo-4-Hexulose Reductase. Glycobiology (2010) 20(12):1607–18. doi: 10.1093/glycob/cwq109
179. Chan KF, Shahreel W, Wan C, Teo G, Hayati N, Tay SJ, et al. Inactivation of GDP-Fucose Transporter Gene (Slc35c1) in CHO Cells by ZFNs, TALENs and CRISPR-Cas9 for Production of Fucose-Free Antibodies. Biotechnol J (2016) 11(3):399–414. doi: 10.1002/biot.201500331
180. Okeley NM, Alley SC, Anderson ME, Boursalian TE, Burke PJ, Emmerton KM, et al. Development of Orally Active Inhibitors of Protein and Cellular Fucosylation. Proc Natl Acad Sci USA (2013) 110(14):5404–9. doi: 10.1073/pnas.1222263110
181. Huang W, Giddens J, Fan SQ, Toonstra C, Wang LX. Chemoenzymatic Glycoengineering of Intact IgG Antibodies for Gain of Functions. J Am Chem Soc (2012) 134(29):12308–18. doi: 10.1021/ja3051266
182. Giddens JP, Lomino JV, Amin MN, Wang LX. Endo-F3 Glycosynthase Mutants Enable Chemoenzymatic Synthesis of Core-Fucosylated Triantennary Complex Type Glycopeptides and Glycoproteins. J Biol Chem (2016) 291(17):9356–70. doi: 10.1074/jbc.M116.721597
183. Giddens JP, Lomino JV, DiLillo DJ, Ravetch JV, Wang LX. Site-Selective Chemoenzymatic Glycoengineering of Fab and Fc Glycans of a Therapeutic Antibody. Proc Natl Acad Sci USA (2018) 115(47):12023–7. doi: 10.1073/pnas.1812833115
184. van de Bovenkamp FS, Hafkenscheid L, Rispens T, Rombouts Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J Immunol (2016) 196(4):1435–41. doi: 10.4049/jimmunol.1502136
185. van de Bovenkamp FS, Derksen NIL, Ooijevaar-de Heer P, van Schie KA, Kruithof S, Berkowska MA, et al. Adaptive Antibody Diversification Through N-Linked Glycosylation of the Immunoglobulin Variable Region. Proc Natl Acad Sci USA (2018) 115(8):1901–6. doi: 10.1073/pnas.1711720115
186. van de Bovenkamp FS, Derksen NIL, van Breemen MJ, de Taeye SW, Ooijevaar-de Heer P, Sanders RW, et al. Variable Domain N-Linked Glycans Acquired During Antigen-Specific Immune Responses Can Contribute to Immunoglobulin G Antibody Stability. Front Immunol (2018) 9:740. doi: 10.3389/fimmu.2018.00740
187. Collin M, Svensson MD, Sjoholm AG, Jensenius JC, Sjobring U, Olsen A. EndoS and SpeB From Streptococcus Pyogenes Inhibit Immunoglobulin-Mediated Opsonophagocytosis. Infect Immun (2002) 70(12):6646–51. doi: 10.1128/IAI.70.12.6646-6651.2002
188. Nandakumar KS, Collin M, Olsen A, Nimmerjahn F, Blom AM, Ravetch JV, et al. Endoglycosidase Treatment Abrogates IgG Arthritogenicity: Importance of IgG Glycosylation in Arthritis. Eur J Immunol (2007) 37(10):2973–82. doi: 10.1002/eji.200737581
189. Collin M, Shannon O, Bjorck L. IgG Glycan Hydrolysis by a Bacterial Enzyme as a Therapy Against Autoimmune Conditions. Proc Natl Acad Sci USA (2008) 105(11):4265–70. doi: 10.1073/pnas.0711271105
190. Stubbs MJ, Thomas M, Vendramin C, Sonesson E, Kjellman C, Jarnum S, et al. Administration of Immunoglobulin G-Degrading Enzyme of Streptococcus Pyogenes (IdeS) for Persistent Anti-ADAMTS13 Antibodies in Patients With Thrombotic Thrombocytopenic Purpura in Clinical Remission. Br J Haematol (2019) 186(1):137–40. doi: 10.1111/bjh.15706
191. Jordan SC, Lorant T, Choi J, Kjellman C, Winstedt L, Bengtsson M, et al. IgG Endopeptidase in Highly Sensitized Patients Undergoing Transplantation. N Engl J Med (2017) 377(5):442–53. doi: 10.1056/NEJMoa1612567
192. Jordan SC, Legendre C, Desai NM, Lorant T, Bengtsson M, Lonze BE, et al. Imlifidase Desensitization in Crossmatch-Positive, Highly Sensitized Kidney Transplant Recipients: Results of an International Phase 2 Trial (Highdes). Transplantation (2021) 105(8):1808–17. doi: 10.1097/TP.0000000000003496
Keywords: antibody, glycosylation, sialylation, in vitro glycoengineering, in vivo glyco engineering, sialic acid, Immunoglobulins
Citation: Vattepu R, Sneed SL and Anthony RM (2022) Sialylation as an Important Regulator of Antibody Function. Front. Immunol. 13:818736. doi: 10.3389/fimmu.2022.818736
Received: 19 November 2021; Accepted: 17 March 2022;
Published: 07 April 2022.
Edited by:
David Falck, Leiden University Medical Center, NetherlandsReviewed by:
Richard John Pleass, Liverpool School of Tropical Medicine, United KingdomSteven William De Taeye, Academic Medical Center, Netherlands
Copyright © 2022 Vattepu, Sneed and Anthony. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert M. Anthony, cm9iZXJ0LmFudGhvbnlAbWdoLmhhcnZhcmQuZWR1