 Katherine E. Herman1
Katherine E. Herman1 Takeshi Yoshida2
Takeshi Yoshida2 Angela Hughson1Alex Grier3
Angela Hughson1Alex Grier3 Steven R. Gill3Lisa A. Beck2
Steven R. Gill3Lisa A. Beck2 Deborah J. Fowell1,4*
Deborah J. Fowell1,4*- 1David H. Smith Center for Vaccine Biology and Immunology, Aab Institute of Biomedical Sciences, University of Rochester Medical Center, Rochester, NY, United States
- 2Department of Dermatology, University of Rochester Medical Center, Rochester, NY, United States
- 3Department of Microbiology and Immunology, University of Rochester Medical Center, Rochester, NY, United States
- 4Department of Microbiology and Immunology, Cornell University, Ithaca, NY, United States
Wiskott–Aldrich Syndrome (WAS) is characterized by recurrent infections, thrombocytopenia, and eczema. Here, we show that WASp-deficient mice on a BALB/c background have dysregulated cutaneous immune homeostasis with increased leukocyte accumulation in the skin, 1 week after birth. Increased cutaneous inflammation was associated with epithelial abnormalities, namely, altered keratinization, abnormal epidermal tight junctional morphology and increased trans-epidermal water loss; consistent with epidermal barrier dysfunction. Immune and physical barrier disruption was accompanied by progressive skin dysbiosis, highlighting the functional significance of the disrupted cutaneous homeostasis. Interestingly, the dysregulated immunity in the skin preceded the systemic elevation in IgE and lymphocytic infiltration of the colonic lamina propria associated with WASp deficiency. Mechanistically, the enhanced immune cell accumulation in the skin was lymphocyte dependent. Elevated levels of both Type 2 (IL-4, IL-5) and Type 17 (IL-17, IL-22, IL-23) cytokines were present in the skin, as well as the ‘itch’ factor IL-31. Unexpectedly, the canonical WAS-associated cytokine IL-4 did not play a role in the immune dysfunction. Instead, IL-17 was critical for skin immune infiltration and elevation of both Type 2 and Type 17 cytokines. Our findings reveal a previously unrecognized IL-17-dependent breakdown in immune homeostasis and cutaneous barrier integrity in the absence of WASp, targeting of which may provide new therapeutic possibilities for the treatment of skin pathologies in WAS patients.
Introduction
Among patients with primary immunodeficiency disorders (PIDD), 40–70% have cutaneous pathology (1–4). Several PIDDs, including Wiskott–Aldrich Syndrome (WAS), present clinically with eczematous lesions (5). Over 70% of WAS patients develop severe, difficult-to-treat eczema which leads to significant morbidity, namely, disseminated cutaneous viral infections and fulminant sepsis (6, 7). The etiology of WAS eczema is unclear but the microbial complications are likely secondary to impaired anti-microbial immunity (8–10). In contrast to a defective IFN-γ response, patients with WAS deficiency develop robust type 2 responses, namely, elevated serum IgE, atopic-like eczema, and food allergy (11, 12).
Human WAS is caused by mutations in the WAS gene, a key regulator of actin cytoskeletal dynamics and gene transcription (13, 14). WASp expression, thought to be restricted to cells of the hematopoietic lineage, regulates many actin-dependent events such as leukocyte migration, adhesion, cellular polarization, and receptor signaling and also actin-independent transcriptional functions (15–20). In the absence of WASp, both innate and adaptive immunity is compromised with defects in phagocytosis, impaired CD8 T cell and NK cytotoxicity, reduced cytokine secretion, and altered migration (21, 22). Defects in regulatory function (Tregs, Bregs and anti-inflammatory, IL-10-producing, macrophages) also contribute to WAS pathologies resulting in unrestrained immune responses (11, 23–28).
Humans and mice lacking WASp display increased sensitization to food antigens (11) and develop poorly-restrained type 2 responses. Up to 10% of WAS patients and 100% of WASKO mice on the 129SvEv background develop inflammatory bowel disease (29). In WASp-deficient mice, the development of spontaneous colitis is dependent on both lymphocyte and innate cell subsets (22) and partially dependent on IL-4 (29). Defects in anti-inflammatory macrophages in WASp-deficient mice also lead to dysregulated intestinal homeostasis (28). Dysregulated immune cell trafficking may compromise immune homeostasis in the skin (30), with the failure of WAS−/− dendritic cells to migrate out of the skin following immune challenge leading to aberrant local immune activation and inflammation (31, 32). However, a spontaneous skin pathology in the WASp-deficient mouse similar to that of human WAS patients has not been characterized.
Here we show that WASp is necessary for the maintenance of skin barrier homeostasis. In BALB/c mice, the absence of WASp resulted in epidermal barrier disruption with tight junction aberrancies, increased transepidermal water loss (TEWL) and upregulation of the atopic dermatitis (AD) associated factor IL-31. This pathology was associated with innate and adaptive immune cell accumulation in the skin and a type 2- and type 17-skewed inflammatory milieu. Cutaneous immune dysfunction was abrogated in Rag−/− WAS−/− mice demonstrating a lymphocyte-dependent dermatitis. Unexpectedly, IL-17, and not IL-4, was required for the immune dysfunction. Reduced cutaneous inflammation and reduced expression of both type 2 and type 17 cytokines in WAS−/− IL-17−/− mice suggests a critical role for type 17 inflammation in WAS-associated skin changes. These data show that WASp regulates the maintenance of epidermal barrier integrity and immune cell homeostasis in the skin through regulating homeostatic IL-17 responses.
Methods
Mice
Wild-type (WT) BALB/c were obtained from the NCI. WAS−/− C57BL/6 mice were provided by Dr. Janis Burkhardt (U. Penn). WAS−/− BALB/c were generated by backcross of WAS−/− C57BL/6 for at least 10 generations. IL17−/− BALB/c mice were provided by Dr. Anna Valujskikh (Cleveland Clinic). IL-4−/− BALB/c animals were purchased from Jackson Animal Laboratories. Rag2−/− BALB/c mice were obtained from Dr. Terry Wright (U. Rochester). All animals were housed in the specific pathogen-free facility at the University of Rochester. All animal experimentation was reviewed and approved by the University of Rochester’s University Committee on Animal Resources and the Institutional Animal Care and Use Committee.
Trans-Epidermal Water Loss
Fur on mouse flank skin was trimmed manually, then TEWL was measured using TM300 device from Courage-Khazaka Electronics (Cologne, Germany).
Whole Mount Immunofluorescent Staining
For whole mount IF of epidermal sheets, hair was first removed from ears with depilatory cream (VEET). Ear dorsal and ventral sheets were separated and incubated in 0.5 M ammonium thiocyanate at 30°C for 20 min. Epidermis and dermis were separated and epidermal sheets fixed in 4% paraformaldehyde, washed and permeabilized with cold 100% methanol at −20°C. Epidermal sheets were blocked (1% BSA) and stained with polyclonal rabbit anti-claudin 1 Ab (Thermo Fisher; PAD : JAY.8) and species-specific AlexaFluor-tagged secondary Ab (Invitrogen) or AlexaFluor-tagged anti-mouse I-A/I-E (Biolegend: M5/114.15.2) and imaged using laser scanning confocal microscopy (Olympus FV1000). Three to four fields of view per epidermal sheet were captured and analyzed using Imaris v8.3.
Flow Cytometry
Ears and flank skin were digested for 30 min at 37°C with collagenase/dispase. Colonic LP cells were isolated as previously described (33). Single cell suspensions were stained with a combination of the following antibodies: CD3 (BD; 145-2C11), CD4 (RM4-4, RM4-5), CD5 (53-7.3), CD8α (53-6.7), CD8β (53-5.8), CD11b (M1/70), CD11c (N418), CD45R/B220 (RA3-6B2), CD45 (30-F11), CD49b (Dx5), CD103 (2E7), CD127 (A7R34), CD205 (NLDC-145), CD207 (4C7), FcϵR1 (MAR-1), Siglec-F (E50-2440), Gr-1 (RB6-8C5), Ly6G (1A8), I-A/I-E (M5/114.15.2), T1/ST2 (DJ8), ICOS (C398.4A), NKp46 (29A1.4), KLRG1 (2F1), TER-119 (TER-119), γδ TCR(eBioGL3), Foxp3 (FJK-16s), GATA3 (L50-823), RORγt (Q31-378) and Live/Dead (Invitrogen). LSR-II flow cytometer acquisition and analysis using FlowJo software (Tree Star). Cell counts were determined by flow cytometry, from whole ear homogenates. The total homogenate from the ear pinna of each mouse was collected by flow cytometry and the total number of live, CD45+ cells calculated using 20,000 AccuCheck (Invitrogen) counting beads/sample for calibration, according to the manufacturers protocol.
Cytokine Measurement
Ear tissue was digested in collagenase/dispase and homogenized in 500 μl PBS. Supernatants were assayed using the Milliplex Mouse Cytokine/Chemokine kits, per manufacturer’s instructions. Plates were read using Bio-Plex 200 instrument (Bio-Rad). For LN intracellular cytokine staining, 1 × 106 LN cells in RPMI/10%FCS were stimulated at 37°C with PMA (Sigma; 50 ng/ml) and ionomycin (Calbiochem; 500 ng/ml) for 4 h. Brefeldin A (BD; 1 ug/ml) was added after 1 h of culture, for the remaining 3 h of culture. Cells were washed and surface stained of CD45 and CD4 before fixation and permeabilization (BD Cytofix/cytoperm). Cells were stained for intracellular cytokines IFNγ (eBioscience; XMG1.2), IL-4 (eBioscience; 11B11) and IL-17A (Biolegend; TC11-18H10.1) and analyzed using the LSR-II flow cytometer and FlowJo software.
Serum IgE Measurement
Serum IgE was measured by ELISA.
Skin Microbiome Analysis
Catch-All Sample Collection swabs (Epicentre) of mouse ears were stored in 2 ml Eppendorf Safe-Lock Biopur tubes containing 100 μl of Yeast cell lysis buffer (Epicentre) at −80°C. DNA was extracted using MasterPure Yeast DNA Purification Kit (Epicentre) and PureLink Genomic DNA Mini Kit (Thermo Fisher) and eluted with 40 μl MoBio PCR water (certified DNA-free). 16S rRNA V1-V3 amplicon libraries were sequenced using the Illumina HiSeq2500 Sequencer (San Diego, CA). Illumina reads were assessed for quality and then analyzed using phylogenetic and operational taxonomic unit methods in the Quantitative Insights into Microbial Ecology (QIIME) open source software v1.9.1.
Statistical Analysis
Prism software (GraphPad) was used for all statistical tests except microbiome analyses. All data presented are mean ± SEM unless otherwise indicated. Statistical analysis of microbiome data was performed in R software. Alpha diversity: Shannon index and PD whole tree. Beta diversity: Bray–Curtis dissimilarity index and Unweighted UniFrac. Relative abundance of taxa were binned for WT vs. WAS−/−; p-values by Student’s t-test and 999 Monte Carlo permutations, adjusted for multiple comparison using the p.adjust function in R (method = ‘fdr’). *p <.05, **p <.01, ***p <.001, ****p <.0001, not significant (n.s.) p >.05.
Results
Spontaneous Skin Pathology and Epidermal Barrier Dysfunction in the Absence of WASp
We assessed homeostatic immunity in the ear skin of both adult (8-week old) C57BL/6 and BALB/c WAS−/− mice. No overt changes in immune cell numbers in the skin of WAS−/− C57BL/6 mice were found, however, WAS−/− BALB/c mice had significantly elevated numbers of CD45+ hematopoietic cells in the steady state (Figure 1A). We noted some variability between experiments in the overall magnitude of inflammation, but in all cases the difference between WT and WAS−/− mice was significant within each individual experiment (Figure 1B). Increases in CD45+ cells were also observed in the dorsal flank skin of WAS−/− BALB/c animals (Figure 1C, left panel). In contrast, no increase in CD45+ cells in the cervical skin-draining lymph node (LN) was observed, suggesting immune dysregulation was cutaneously, and not centrally, driven (Figure 1C, right panel). Moreover, cutaneous inflammation was only observed in the full absence of WASp, WAS+/− mice showing no overt skin inflammation (Figure 1D). Accompanying the increase in leukocyte infiltration was the upregulation of the atopic dermatitis-associated pruritogenic ‘itch’ factor IL-31 (Figure 1E).
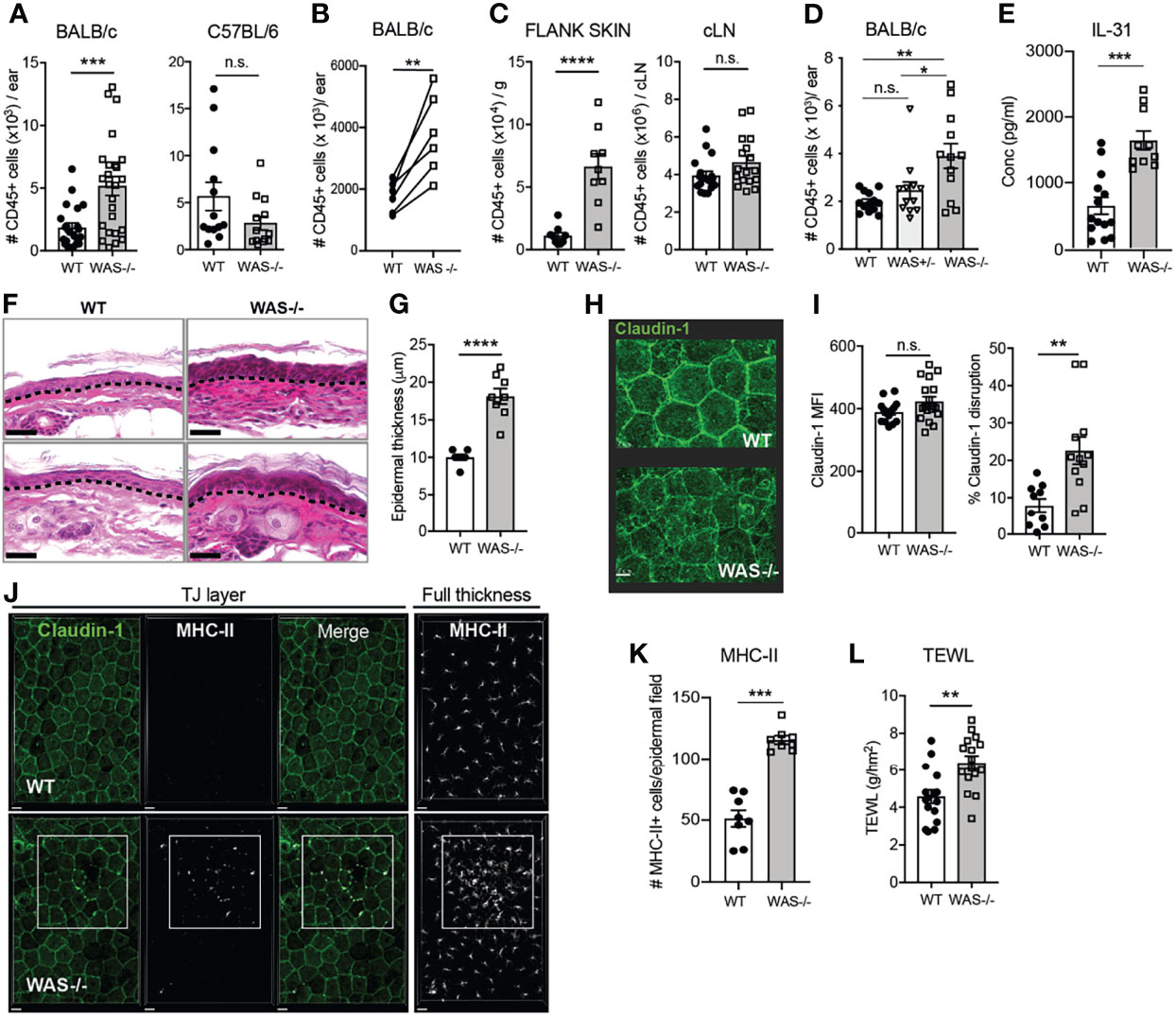
Figure 1 Spontaneous skin pathology and barrier dysfunction in WASp-deficient mice. (A) Number of CD45+ cells per ear pinna in 8-week BALB/c (left) and C57BL/6 (right) mice, measured by flow cytometry, combined data from 4 independent experiments. (B) Paired 8 week BALB/c WT and WAS−/− mice in individual experiments, each set of paired symbols represents the mean CD45+ cell count per ear pinna in a given experiment. (C) Number of CD45+ cells/g of dorsal flank skin (left) or cervical skin-draining LN of BALB/c mice. (D) Number of CD45+ cells per ear pinna WT, WAS+/− and WAS−/− mice. (E) IL-31 measured by luminex assay from WT and WAS−/− BALB/c ear skin supernatants at eight weeks of age. (A–E), n = 3–6/group, 2–6 independent experiments. (F) Hematoxylin and eosin-stained ear skin from WT BALB/c and WAS−/− mice. Dotted line, dermal–epidermal junction. Scale bar = 20 μm. (G) Epidermal thickness quantified from (F), measured at thickest point. (H) Representative images of Claudin-1 expression in WT and WAS−/− BALB/c epidermal sheets, by confocal microscopy. Scale bar = 15 μm. (I) Quantification of total claudin-1 expression intensity from panel (F). Right, quantification of claudin-1 disruption along length of epithelial cell border (see Supplementary Figure 1). (J) Representative images of MHC-II and claudin-1 staining in the TJ layer of epidermal sheets from WT and WAS−/− mice. Right panel, MHC-II staining in the full thickness epidermal sheet. (K) Quantitation of the number of MHC-II+ cells in each imaging field from panel (J). Scale bar = 20 μm. (J, K) n = 3–4/group, 2 independent experiments. (L) TEWL measured from flank skin of WT and WAS−/− animals, n = 3–4/group, 4 independent experiments. All animals 8 weeks of age. *p <.05, **p <.01, ***p < .001, ****p <.0001, n.s., not significant p > .05, Student’s t-test.
To determine if the elevated immune cell accumulation in the skin had physiological effects on the skin barrier, we first examined histological sections of mouse ear skin. At eight weeks of age, we observed dysmorphology of WAS−/− mouse skin architecture, with parakeratosis and significant thickening of the epidermis compared to WT (Figures 1F, G). Parakeratosis may indicate altered keratinocyte differentiation and changes to the epidermal tight junction (TJ) network (34–38). We used confocal microscopy to visualize expression patterns of claudin-1 deposition, a major skin TJ protein, in epidermal sheets prepared from WT and WAS−/− mouse skin (Figure 1H). The WT epidermis showed clear claudin-1 stain localized at interepithelial junctions in a typical honeycomb pattern (39), while the epidermis from WASp-deficient mice showed regions of claudin-1 staining with poorly defined intercellular borders. There were no differences in total claudin-1 epidermal expression between WT and WAS−/− skin (MFI) (Figure 1I, left panel), but quantification of the contiguous intensity of claudin-1 staining along the epithelial cell borders revealed WAS−/− claudin-1 expression was significantly more irregular than WT (Figure 1I, right panel; Supplementary Figure 1). In response to TJ disruption, Langerhan cells (LCs) have been reported to extend dendrites out into the TJ area to ‘plug’ holes in a disrupted epidermis. Consistent with a TJ breach, we observed an increase in MHC Class II+ projections (predominantly from CD11b+CD11c+Langerin+ cells) between epithelial cells in WAS−/− epidermal sheets (Figure 1J, TJ layer panels). Moreover, there was a marked accumulation of MHC-II+ cells in the epidermal sheets (Figure 1J, right panel; Figure 1K), as previously noted (31, 32).
These data suggest a disruption in the interface between the physical and immune barriers of the skin in the absence of WASp. Disrupted tight junctions in the epidermis in human atopic dermatitis corresponds to alterations in skin permeability to water (37, 39). WAS−/− mice had significantly elevated trans-epidermal water loss (TEWL) values compared to WT mice, indicating increased skin permeability and epidermal barrier dysfunction (Figure 1L). Thus, we find loss of WASp leads to both cutaneous immune dysregulation and skin barrier disruption.
Enhanced Immune Cell Accumulation in the Skin of Mice Lacking WASp
Immune composition by flow cytometry in the skin at eight weeks of age revealed a general increase in many immune cell subsets in WAS−/− mice (subsets defined in Supplementary Table 1). Most innate cell subsets tested were enhanced including neutrophils, basophils, CD11c+ dendritic cells (DCs), LCs, NK cells, and CD11b+ monocytes/macrophages (Figure 2A) and increases in group 2 and 3 innate lymphoid cells (ILC2 and ILC3, Figure 2A). Eosinophils were, notably, not elevated in the absence of WASp (Figure 2A). CD4+ T cells, CD8+ T cells, and γδ TCR+ T cells were also significantly increased in WAS−/− skin (Figure 2B). Foxp3+ CD4+ T regulatory cell (Treg) numbers were only modestly enhanced in WAS−/− skin (Figure 2C, left panel) resulting in a significant reduction in the proportion of Tregs among CD4+ T cells (Figure 2C, right panel). Aside from Tregs, we found no significant alterations in the relative frequencies of the elevated immune cell subsets we evaluated (Figure 2D and Supplementary Figure 2). Thus, we observed significant increases in multiple skin immune cell populations, indicating broad dysregulation of skin immunity in WASp-deficient mice.
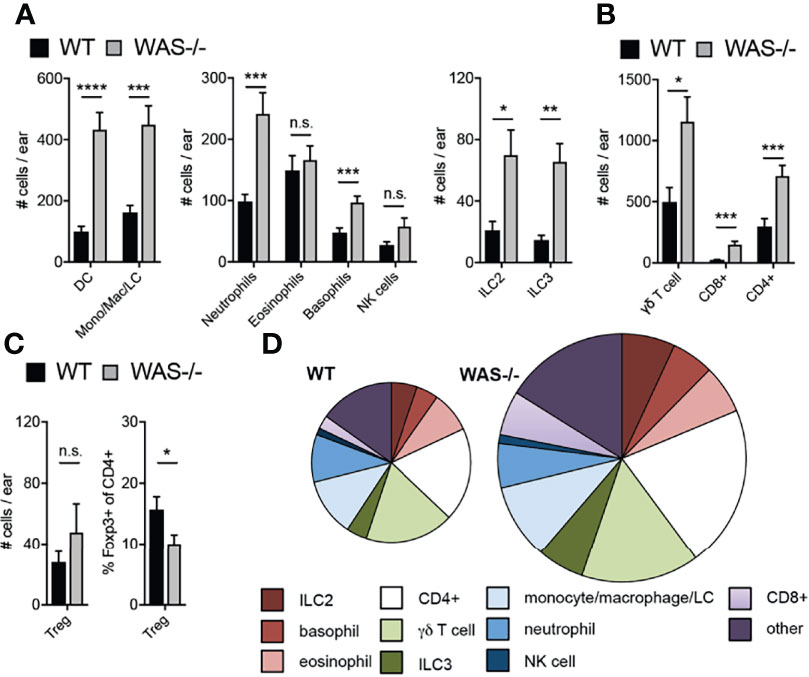
Figure 2 Accumulation of immune cells in skin of mice lacking WASp. (A–C) Number of immune cell subsets in the skin of the ear pinna, assessed by multiparameter flow cytometry. (D) Pie graphs represent relative frequencies of immune subsets (% of CD45+) in WT (left) and WAS−/− skin of the ear pinna (right). Size of pie represents relative number of CD45+ cells. All mice 8 weeks of age. n = 3–6/group, 2–5 independent experiments. *p <.05, **p <.01, ***p <.001, ****p <.0001, n.s., not significant p > .05 Student’s t-test.
Early Accumulation of Immune Cells in Skin of Mice Lacking WASp
Immune cell cytokines could induce the observed epidermal barrier defects (40–43), or early barrier disruption could enhance immune cell accumulation. To gain mechanistic insight into the skin pathology, we assessed the concordance of immunologic changes and epidermal barrier dysfunction in the skin of WASp-deficient mice kinetically starting at 1 week of age. We detected increased CD45+ cell infiltration in WAS−/− skin as early as one week of age (Figure 3A). At this time, a modest, but not significant, increase in TEWL was observed in WAS−/− mice (Figure 3B) but no change was seen in the young (1 wk) skin architecture between WT and WAS−/− mice (Figure 3C). In contrast, physical barrier dysfunctions (TEWL, epidermal thickness) were significantly elevated by 4 weeks of age (Figures 3B, D) at the height of immune accumulation (Figure 3A). Thus, altered immune cell accumulation appears to precede the epidermal skin barrier dysfunction.
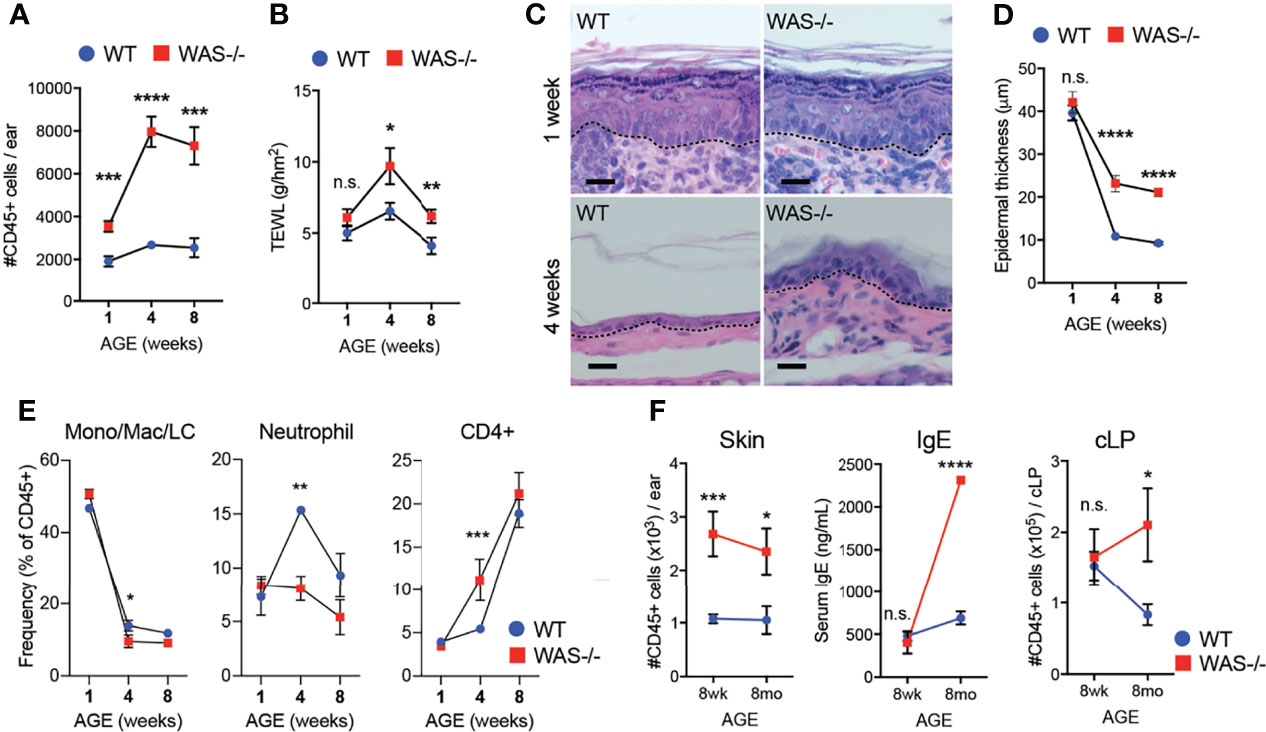
Figure 3 Immune cell accumulation precedes development of barrier dysfunction in mice lacking WASp. (A) Number of CD45+ cells per ear in mice aged 1, 4, and 8 weeks. (B) TEWL measured from flank skin of WT and WAS−/− animals from panel (A). (C) Hematoxylin and eosin-stained ear skin from 1 and 4 week-old WT BALB/c and WAS−/− mice. Dotted line denotes dermal–epidermal junction. Scale bar = 20 μm. (D) Epidermal thickness quantified from ear skin sections in panel (C), measured at thickest point. (A–D) n = 3–5/group, 2 experiments. (E) Frequencies of immune subsets at 1, 4, and 8 weeks of age in WT and WAS−/− mice. (F) CD45+ cellular infiltrate in the skin (left), serum IgE (middle) and CD45+ cellular infiltrate in the colonic lamina propria (cLP) (right) of mice aged 8–10 weeks and 8 months. n = 3–5/genotype/age group, 4 independent experiments. *p <.05, **p <.01, ***p <.001, ****p <.0001, n.s., not significant p > .05 Student’s t-test.
With respect to immune cells, neonatal WT and WAS−/− had a similar balance of immune cells, heavily skewed to monocyte/macrophage populations (Figure 3E). However, at four weeks of age, WAS−/− mice showed an enhanced frequency of CD4+ cells in the skin, with a concomitant decrease in the proportion of neutrophils and monocytes/macrophages present in the skin (Figure 3E). Interestingly, these early defects in skin immunity came weeks before the WASp-associated elevation in serum IgE and before lymphocytic infiltration of the colonic lamina propria (Figure 3F).
Skin Dysbiosis in Mice Lacking WASp
Significant alterations in the skin microbiome have been reported in humans with primary immunodeficiencies and in AD (44–46). To investigate whether the immune and barrier dysregulation in the WAS−/− skin impacted the skin microbiome, we performed 16S ribosomal RNA (rRNA) sequencing on skin swabs from ear skin. Using the Illumina MiSeq platform, we obtained an average of 143,474 ± 40,444 V1–V3 reads/sample for 10 WT and 10 WAS−/− mice at eight weeks of age. Rarefaction curves indicated good coverage for dominant species present in the skin (Supplementary Figure 3). We detected no significant differences in alpha diversity between WT and WAS−/− (WT mean: 4.7 ± 1.8, WAS−/− mean: 4.5 ± 0.9 p = .773) using the Shannon diversity index (which assesses microbial community richness and evenness within a single sample) (Figure 4A). However, we observed significant clustering of WAS−/− samples separate from WT using the unweighted Unifrac method and principle coordinate analysis for assessing beta diversity (Figure 4B) (p = 1.41E−18). Changes in relative abundance of a number of bacterial communities was observed with WASp-deficiency (Figure 4C) with enrichment of the Streptococcus and Helicobacter genera, and also the Deferribacteres phylum (Figure 4D). Furthermore, there was colonization of a particular Gammaproteobacteria species, Aggregatibacter pneumotropica, on WAS−/− mouse skin that was not detected on WT skin (Figure 4D).
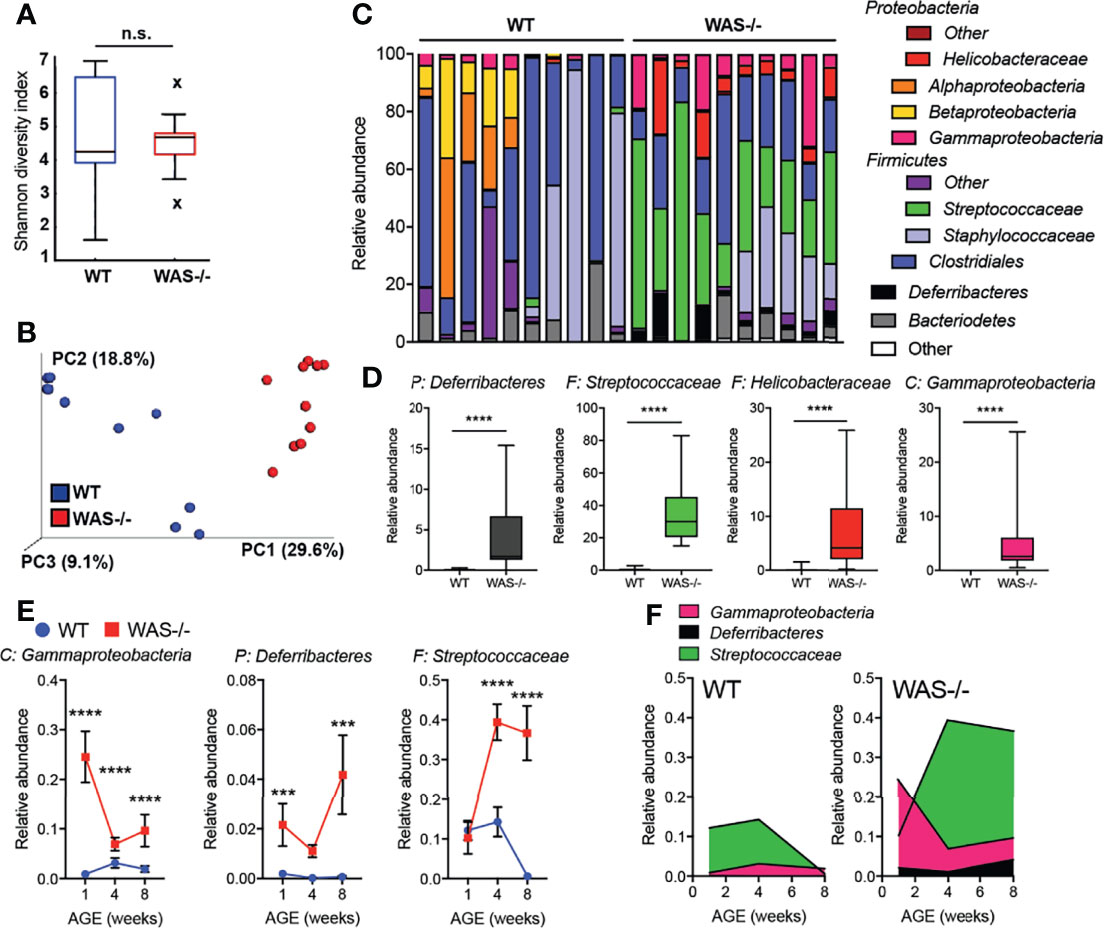
Figure 4 Dysbiosis in WAS−/− mouse skin. (A) Shannon diversity index of WT and WAS−/− skin microbiome samples collected at 8 weeks of age. (B) Principle coordinate analysis generated using unweighted UniFrac matrix. (C) Relative abundances of 12 major phyla/class/family taxonomies in 10 WT and 10 WAS−/− microbiome samples. (D) Taxa significantly enhanced in WAS−/− skin over WT by multivariate analysis (p <.05, beta coefficient >.1). P, phylum, C, class, F, family. (E) Kinetics of changes in WAS−/− signature microbial taxa in WT and WAS−/− animals at 1, 4, and 8 weeks of age. (F) Relative abundance of selected taxa in panel (E). One-way ANOVA with Tukey’s post-test, ***p <.001, ****p <.0001, n.s., not significant p > .05.
To assess the concordance of dysbiosis with the onset of skin pathology, we analyzed the skin microbiome at 1, 4, and 8 weeks of age (Figures 4E, F and Supplemental Figure 4). At one week of age, a striking change in the representation of microbial taxa in WAS−/− mice was already evident (Figures 4E, F). Elevations in Gammaproteobacteria and members of the Deferribacteres phyla at 1 week, remained elevated above WT at all timepoints (Figures 4E, F). The relative abundance of Streptococcus spp. in the WAS−/− mice was not seen as a neonate but increased in representation with age (Figures 4E, F), possibly as a secondary consequence of cutaneous inflammation. Therefore, the dysbiosis in WAS−/− mice appears coincident with the early immune dysfunction in the skin of WASp-deficient mice. Preliminary co-housing experiments suggest that the altered microbiome of WAS−/− skin is not sufficient to induce skin inflammation in WAS-sufficient (WT) mice (Supplementary Figure 5). Four week old WT and WAS−/− mice were co-housed for two weeks and then assessed for ear skin inflammation. After two weeks of co-housing, WT and WAS−/− skin microbiomes had equilibrated, containing a similar microbial profile (Supplementary Figure 5). This microbial mixing was mainly attributed to the acquisition of elevated WASp-associated Gammaproteobacteria, Deferribacteres, and Streptococcaceae by WT mice, with no significant impact of the WT microbiome on the co-housed WAS−/− mice. However, we observed no induction of skin inflammation associated with these changes in the skin microbiome in WT cohoused mice (Supplementary Figure 5), suggesting that the microbiome of WAS−/− mice is not sufficient to drive cutaneous inflammation in immunocompetent WT hosts.
Type 2- and Type 17-Biased Inflammatory Milieu in Skin of Mice Lacking WASp
We next measured inflammatory mediators in the mouse ear skin. We found marked differences in cytokines and chemokines in the skin of WAS−/− mice using a multiplex Luminex assay (Figure 5A). Out of 55 analytes tested (Supplementary Table 2), 18 analytes were significantly upregulated in WAS−/− versus WT skin (Figure 5A, highlighted in magenta) and no factors were downregulated. IL-4 and IL-17 were both significantly enhanced in the WAS−/− skin, but there was no increase in IFN-γ, suggesting a bias toward type 2 and/or 17 responses (Figure 5B). Indeed, type 2 cytokines (IL-4, IL-5) and type 17 cytokines (IL-17, TNF, IL-22, IL-23) were among the most upregulated analytes (Figure 5C). CCL17, whose cognate receptor CCR4 is expressed by Th2/Th17 CD4+ cells, was particularly enriched in WAS−/− skin (Figure 5C). This chemoattractant cue may promote the preferential accumulation of type 2/17 effectors in the skin (47) along with the AD-associated pruritogenic cytokine IL-31 (Figure 1C) (48). To determine whether the type 2/17 inflammatory bias present in the skin was associated with changes in LN Th differentiation, we assessed cytokine production by CD4+ T cells from the skin-draining LN. Upon in vitro re-stimulation, IL-4 was upregulated 14.62 fold, IL-17 by 4.887 fold and IFN-γ by 2.853 fold in WAS−/− CD4+ T cells compared to WT (Figure 5D). Thus, the type 2 cytokine enrichment (and to a lesser extent type 17) we observed in the skin may be associated with enhanced priming or potential for IL-4/IL-17 production in the skin draining LN. In the skin, γδ T cells were also sources of elevated IL-17 (data not shown). Kinetically, the elevated inflammatory cytokines in the skin at 8 weeks (Figure 5C), were not enhanced at 1 week of age. However, IL-5 and IL-17 cytokines were upregulated in the WAS−/− skin by 4 weeks of age (Figure 5E).
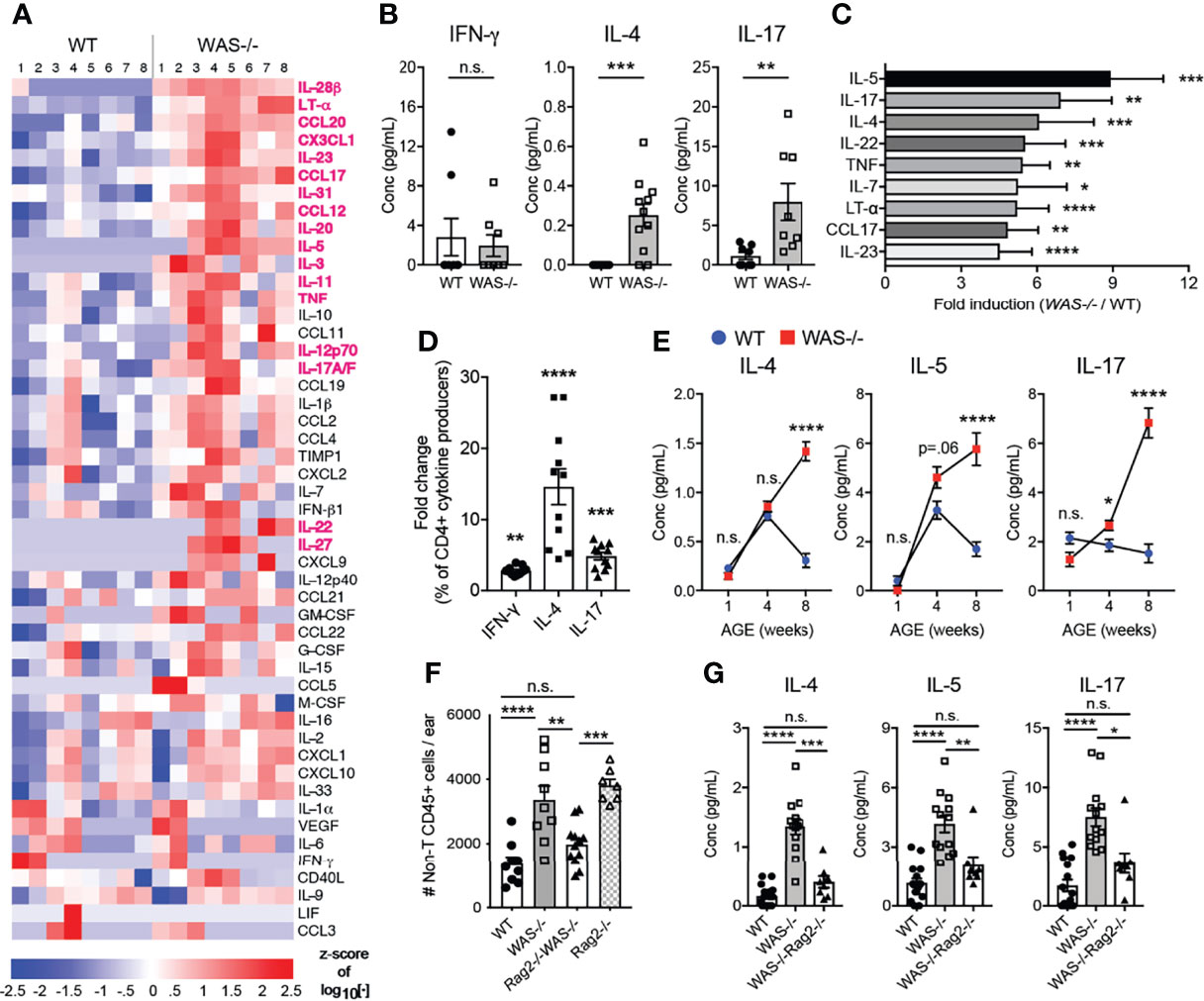
Figure 5 Biased type 2 and 17 inflammation in WAS−/− skin at eight weeks of age requires adaptive immune cells for development. (A) Heat map of Z-score of analytes measured by luminex assay from WT and WAS-/- BALB/c ear skin supernatants. Factors in magenta p <0.01. (B) Absolute values of cytokines measured by luminex as in panel (A). (C) All factors with fold change greater than four (WAS−/−:WT). (A–C) n = 8–11/group, 2 independent experiments. (D) Cytokine frequencies among CD4+ cells from the skin-draining LN following PMA/ionomycin stimulation and intracellular cytokine staining, fold change (WAS−/−:WT) n = 3–5/group, 3 independent experiments. (E) Cytokines in ear skin homogenates at 1, 4, and 8 weeks of age. (A–D) All mice 8 weeks of age. Statistics: Mann–Whitney U test. Statistics for (E): One-way ANOVA with Tukey’s post test. *p <.05, **p <.01, ***p <.001, ****p <.0001. (F) Total CD45+CD4−CD8β−γδ− cells in ears of WT, WAS−/−, WAS−/−Rag2−/− and Rag2−/− mice. (G) Absolute values of cytokines measured by Luminex. All mice 8 weeks of age, n = 2–5/group, 4 independent experiments. Statistics (F, G) One-way ANOVA with Tukey’s post test. *p <.05, **p <.01, ***p <.001, ****p <.0001, n.s., not significant p > .05.
To assess the role of the adaptive immune compartment in the cutaneous immune dysregulation, we crossed WAS−/− mice to Rag2−/− mice on the BALB/c background (WAS−/−Rag2−/−). Loss of T and B cells abolished WAS-associated skin inflammation, as determined by normalizing for the non-T cell CD45+ cell (CD45+CD4−CD8β−γδ−) compartment between WAS−/− and WAS−/−Rag2−/− (Figure 5F). The results suggest the adaptive immune compartment actively drives the enhanced innate cell recruitment in WAS−/− skin. However, interpretation of these experiments is complicated by the presence of an unexpected skin inflammation in the Rag2−/− mice themselves. Loss of both T cells and WASp, WAS−/−Rag2−/−, led to the amelioration of inflammation. At the cytokine level, elevated cytokine production in the WAS−/− skin was reduced to WT levels in the WAS−/−Rag2−/− mice (Figure 5G and Supplementary Table 3). Thus, lymphocytes appear to be essential for the initiation and/or amplification of cutaneous pathology in WASp-deficient mice.
IL-17, But Not IL-4, Contributes to the Immune Pathology in Mice Lacking WASp
In the gut, elevated IL-4 has been previously associated with enhanced serum IgE and colitis in WASp-deficient mice (29). Recent studies in an adoptive transfer system of Th1/Th17 colitis have shown elevated IFNγ and IL-17 in the gut when macrophages lack WASp, but the functional significance of IL-17 was not explored (28, 29). Therefore, we assessed the role of IL-4 and IL-17 in the development of skin inflammation using WAS−/−IL-4-/- and WAS−/−IL-17−/− mice (Figures 6A, B). IL-4 deficiency failed to alleviate the dysregulated accumulation of immune cells in the WAS−/− skin (Figure 6A). Furthermore, we found little change in composition of immune cell subsets or the cytokine/chemokine milieu in skin of WAS−/−IL-4−/− compared to WAS−/− mice (Supplementary Figure 6). In contrast, we found that total CD45+ cells were significantly decreased in WASp-deficient skin in the absence of IL-17 (Figure 6B). Lack of IL-17A in WASp-deficient animals led to significant reductions in the accumulation of innate cell types such as monocytes/macrophages/LCs, basophils and CD11c+ dendritic cells (Figure 6C), suggesting that IL-17A may play a role in the tissue recruitment or expansion of these populations in WAS−/− animals. CD4+ T cells counts trended down in WAS−/− groups in the absence of IL-17, but the WAS−/−IL-17−/− phenotype was intermediate between WT and WAS−/− groups, with no significant differences when compared to either group.
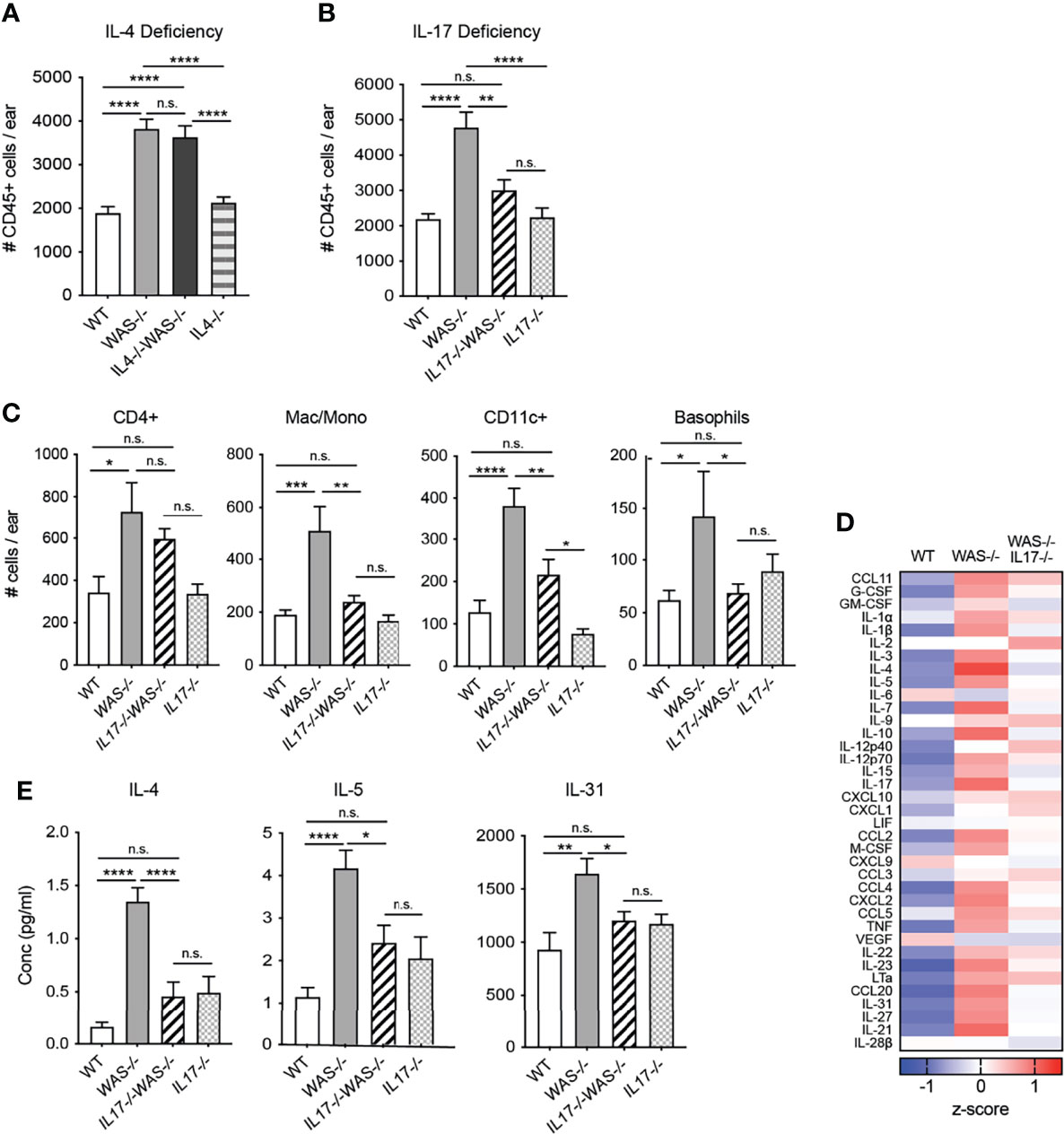
Figure 6 IL-17, but not IL-4, necessary for the development and/or maintenance of skin inflammation in mice lacking WASp. (A) Total CD45+ cells in ears of WT, WAS−/− and WAS−/− IL-4−/− and IL-4−/− BALB/c mice. (B) Total CD45+ cells in ears of WT, WAS−/− and WAS−/− IL-17−/− and IL-17−/− BALB/c mice. (A, B) Total CD45+ cells in ear measured by flow cytometry. (C) Number of immune cell subsets in ear skin. (D) Heat map of Z-scores of analytes measured by luminex assay from ear skin supernatants. (E) Absolute values of cytokines measured by Luminex assay as in panel (E). n = 3–5/genotype, 3 independent experiments. *p <.05, **p <.01, ***p <.001, ****p <.0001, n.s., not significant p > .05, One-way ANOVA with Tukey’s post test. All mice at 8 weeks of age.
Most notably, loss of IL-17 was accompanied by significant reductions in many of the inflammatory mediators found elevated in the WAS−/− skin (Figure 6D), to levels similar to that of immunocompetent WT and IL-17−/− mice. In particular, we found a striking reduction in the type 2 cytokines IL-4 and IL-5 in the WAS−/−IL-17−/− skin compared to WAS−/− controls (Figure 6E). The pruritogenic factor IL-31 was also significantly reduced to WT levels in the skin of WAS−/−IL-17−/− mice (Figure 6E). Our observations point to IL-17A, but not IL-4, contributing to the development and/or maintenance of the skin inflammation described in WAS−/− animals. Interestingly, as noted in other murine models of asthma and eczema (49–51), IL-17 appears to support or amplify type 2 cytokines in WASp-deficient animals. Our results uncover a previously unappreciated spontaneous breakdown in cutaneous immune homeostasis in the absence of WASp driven by elevated IL-17. Immune dysregulation occurred soon after birth and was associated with altered skin TJ morphology and barrier function resulting in dysbiosis.
Discussion
Despite the prominence of eczema in human WAS patients, little is known about the pathogenesis of skin inflammation. This may be due in part to a lack of a cutaneous pathological phenotype in WASp-deficient mice. While accumulation of Langerhans cells in the steady-state epidermis has been previously observed, to our knowledge no overt changes in total immune cell number or changes in skin architecture have been reported (32). In this study, we showed that integrating the WASp deficiency into the BALB/c background led to the development of a spontaneous dermatitis associated with a type 2 and type 17 inflammatory milieu, significant changes in epidermal morphology and barrier dysfunction (see model, Supplementary Figure 7). This immune and physical barrier dysfunction was concomitant with microbial dysbiosis. IL-17, and not IL-4, was a key driver of the immune inflammation in the skin.
From a very young age, mice lacking WASp displayed a marked enrichment of immune cells in the skin. While we do not yet know the inciting factor, a number of studies in WAS−/− mice have demonstrated impaired LC and DC trafficking out of the skin in response to cutaneous sensitization, suggesting the early leukocyte accumulation may arise due to initial defects in immune cell egress from the skin (31, 32, 52). Aberrant immune cell accumulation in the skin was associated with marked changes in physical epidermal organization and barrier function. Inflammatory cytokines are known to reduce skin barrier function by modulating keratinocyte expression of tight junctional proteins and altering keratinocyte maturation/differentiation (40–42, 53). These physical changes may alter the topography of niche environments available to skin microbiota and/or enhance commensal translocation and access to the immune system (54). Our findings suggest that in the absence of WASp, Langerhan cells are distinctly positioned to respond to environmental antigens. Enhanced contact with microbial stimuli may amplify the homeostatic disruption leading to the overexpression of the type 2 and type 17 cytokines/chemokines. Reduced immunosuppression (26) with WASp-deficiency likely conspires to propagate the inflammatory response in the skin.
The resident skin immune system tunes responses to commensals to maintain barrier immunity and protection without overt inflammation (55). In turn, commensal organisms have been associated both with initiation of, and protection from, mucosal pathology (56–58). Similar to our results in the skin, Helicobacter spp. was strongly associated with the development of inflammatory colitis in 129SvEv WAS−/− mice (59). The presence of Streptococcus in the WAS−/− mice is also seen in AD-prone skin (60). In contrast, Gammaproteobacteria have been linked to protection from allergy through IL-10 induction (61). Our studies showed dominant Gammaproteobacteria early in WAS−/− skin was rapidly replaced by an abundance of Streptococcus. Whether dysbiosis elicits or is a consequence of barrier dysfunction is a long-outstanding question in AD. Preliminary studies co-housing WT mice in the same microbial environment as the WAS−/− BALB/c mice was not sufficient to induce skin inflammation (Supplementary Figure 5). Moreover, pilot experiments with transfer of the BALB/c WAS−/− skin microbiome to WT germ-free mice also failed, by itself, to promote skin inflammation. Our results suggest that WAS-mediated cutaneous immune dysregulation may initially occur independently of microbial cues.
The combination of WAS-deficiency and the BALB/c genetic background was necessary to reveal dysregulated cutaneous immunity. There are significant differences between C57BL/6 and BALB/c strains including major histocompatibility complex (MHC) haplotype and type 1 and type 2 cytokine biases (62, 63). Given the predisposition of BALB/c animals to develop type 2 responses, it is possible that genetic background strain polymorphisms (at non-WAS loci) associated with heightened type 2 immunity synergize with WASp-deficiency to precipitate overt type 2 and type17 cutaneous inflammation. Our data suggest that in human WAS, additional genetic factors may contribute to differences in disease heterogeneity or severity.
Perhaps our most provocative observation is that cutaneous dysregulation preceded both LP inflammation and serum IgE elevation. Disrupted skin barrier in AD is thought to contribute to the development of food allergy and asthma through epicutaneous sensitization to environmental antigens (64–66). Indeed, in WASp-deficient mice many of the circulating IgE antibodies appear to be specific for chow components (11). Cutaneous exposure to food and environmental antigens via disrupted eczematous skin is thought to reduce tolerance to these antigens and lead to the development of specific IgE (67). Cutaneous sensitization is the major hypothesis behind the “atopic march” in patients with AD (68–72): the progression of eczema in infancy to food allergy and asthma/allergic rhinitis later in life (73). The spontaneous early skin immune dysregulation, in addition to food allergy and serum IgE elevation, in the WAS−/− mouse model may be a useful tool for understanding the evolution of atopy and determining ways to halt its progression.
Notably, the spontaneous dermatitis in WASp-deficient mice was dependent on IL-17 and not IL-4. Loss of IL-17 also abrogated the dysregulated IL-4 response. At this stage, we do not know if the elevated IL-17 directly drives the elevated Type 2 cytokines or whether it contributes indirectly by disrupting skin homeostasis and facilitating aberrant Type 2 responses. IL-17A can induce Th2 responses in murine models of atopic dermatitis and Th2 cells making both IL-4 and IL-17 have been seen in human allergic asthma and AD and may correlate with disease activity (74–76). Interestingly, recent work suggests that IL-17/IL-22 responses are particularly elevated in pediatric atopic dermatitis (77, 78). While a direct comparison of our mouse studies to human WAS is not possible without further investigation of the cutaneous pathology in humans, assessment of IL-17 in the difficult-to-treat eczema experienced in WAS patients could provide a new pathway for targeted treatment to alleviate the cutaneous disease.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI, PRJNA790974.
Ethics Statement
All mice were maintained in a pathogen-free facility at the University of Rochester Medical Center. All mouse procedures were performed with approval of the University of Rochester’s Institutional Animal Care and Use Committee.
Author Contributions
KH designed and conducted experiments, analyzed the data and wrote the manuscript. TY conducted experiments and analyzed the data. AH conducted experiments and analyzed the data. LB helped design the experiments, provided expertise/reagents and edited the manuscript. AG analyzed the microbiome data. SG helped design the experiments, provided expertise/reagents and edited the manuscript. DF designed the experiments, analyzed the data and wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by grants from the NIH NIAID: AI072690 and AI102851 awarded to DF and the T32 AI118689 to KH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aknowledgments
We thank the members of the Fowell lab for helpful discussion of the data and review of the manuscript. We gratefully acknowledge support from the National Institutes of Health, National Institute of Allergy and Infectious Diseases: Grants P01 AI02851 and R01 AI070826 to DF and T32 AI118689 to KH.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.817427/full#supplementary-material
References
1. Berron-Ruiz A, Berron-Perez R, Ruiz-Maldonado R. Cutaneous Markers of Primary Immunodeficiency Diseases in Children. Pediatr Dermatol (2000) 17(2):91–6. doi: 10.1046/j.1525-1470.2000.01721.x
2. Al-Herz W, Nanda A. Skin Manifestations in Primary Immunodeficient Children. Pediatr Dermatol (2011) 28(5):494–501. doi: 10.1111/j.1525-1470.2011.01409.x
3. Moin A, Farhoudi A, Moin M, Pourpak Z, Bazargan N. Cutaneous Manifestations of Primary Immunodeficiency Diseases in Children. Iran J Allergy Asthma Immunol (2006) 5(3):121–6.
4. Lehman H. Skin Manifestations of Primary Immune Deficiency. Clin Rev Allergy Immunol (2014) 46(2):112–9. doi: 10.1007/s12016-013-8377-8
5. Pichard DC, Freeman AF, Cowen EW. Primary Immunodeficiency Update: Part I. Syndromes Associated With Eczematous Dermatitis. J Am Acad Dermatol (2015) 73(3):355–364; quiz 365-356. doi: 10.1016/j.jaad.2015.01.054
6. Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, et al. Autoimmunity in Wiskott-Aldrich Syndrome: Risk Factors, Clinical Features, and Outcome in a Single-Center Cohort of 55 Patients. Pediatrics (2003) 111(5 Pt 1):e622–7. doi: 10.1542/peds.111.5.e622
7. Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich Syndrome: A Comprehensive Review. Ann NY Acad Sci (2013) 1285:26–43. doi: 10.1111/nyas.12049
8. Andreansky S, Liu H, Turner S, McCullers JA, Lang R, Rutschman R, et al. WASP- Mice Exhibit Defective Immune Responses to Influenza A Virus, Streptococcus Pneumoniae, and Mycobacterium Bovis BCG. Exp Hematol (2005) 33(4):443–51. doi: 10.1016/j.exphem.2004.12.006
9. Trifari S, Sitia G, Aiuti A, Scaramuzza S, Marangoni F, Guidotti LG, et al. Defective Th1 Cytokine Gene Transcription in CD4+ and CD8+ T Cells From Wiskott-Aldrich Syndrome Patients. J Immunol (2006) 177(10):7451–61. doi: 10.4049/jimmunol.177.10.7451
10. Lang PA, Shaabani N, Borkens S, Honke N, Scheu S, Booth S, et al. Reduced Type I Interferon Production by Dendritic Cells and Weakened Antiviral Immunity in Patients With Wiskott-Aldrich Syndrome Protein Deficiency. J Allergy Clin Immunol (2013) 131(3):815–24. doi: 10.1016/j.jaci.2012.08.050
11. Lexmond WS, Goettel JA, Lyons JJ, Jacobse J, Deken MM, Lawrence MG, et al. FOXP3+ Tregs Require WASP to Restrain Th2-Mediated Food Allergy. J Clin Invest (2016) 126(10):4030–44. doi: 10.1172/JCI85129
12. Ozcan E, Notarangelo LD, Geha RS. Primary Immune Deficiencies With Aberrant IgE Production. J Allergy Clin Immunol (2008) 122(6):1054–1062; quiz 1063-1054. doi: 10.1016/j.jaci.2008.10.023
13. Ochs HD. Mutations of the Wiskott-Aldrich Syndrome Protein Affect Protein Expression and Dictate the Clinical Phenotypes. Immunol Res (2009) 44(1-3):84–8. doi: 10.1007/s12026-008-8084-3
14. Sadhukhan S, Sarkar K, Taylor M, Candotti F, Vyas YM. Nuclear Role of WASp in Gene Transcription Is Uncoupled From Its ARP2/3-Dependent Cytoplasmic Role in Actin Polymerization. J Immunol (2014) 193(1):150–60. doi: 10.4049/jimmunol.1302923
15. Blundell MP, Worth A, Bouma G, Thrasher AJ. The Wiskott-Aldrich Syndrome: The Actin Cytoskeleton and Immune Cell Function. Dis Markers (2010) 29(3-4):157–75. doi: 10.1155/2010/781523
16. Bouma G, Burns SO, Thrasher AJ. Wiskott-Aldrich Syndrome: Immunodeficiency Resulting From Defective Cell Migration and Impaired Immunostimulatory Activation. Immunobiology (2009) 214(9-10):778–90. doi: 10.1016/j.imbio.2009.06.009
17. Derry JM, Ochs HD, Francke U. Isolation of a Novel Gene Mutated in Wiskott-Aldrich Syndrome. Cell (1994) 79(5):635–4. doi: 10.1016/0092-8674(94)90528-2
18. Taylor MD, Sadhukhan S, Kottangada P, Ramgopal A, Sarkar K, D'Silva S, et al. Nuclear Role of WASp in the Pathogenesis of Dysregulated TH1 Immunity in Human Wiskott-Aldrich Syndrome. Sci Transl Med (2010) 2(37):37ra44. doi: 10.1126/scitranslmed.3000813
19. Sarkar K, Han SS, Wen KK, Ochs HD, Dupre L, Seidman MM, et al. R-Loops Cause Genomic Instability in T Helper Lymphocytes From Patients With Wiskott-Aldrich Syndrome. J Allergy Clin Immunol (2018) 142(1):219–34. doi: 10.1016/j.jaci.2017.11.023
20. Babich A, Burkhardt JK. Coordinate Control of Cytoskeletal Remodeling and Calcium Mobilization During T-Cell Activation. Immunol Rev (2013) 256(1):80–94. doi: 10.1111/imr.12123
21. Thrasher AJ, Burns SO. WASP: A Key Immunological Multitasker. Nat Rev Immunol (2010) 10(3):182–92. doi: 10.1038/nri2724
22. Nguyen DD, Wurbel MA, Goettel JA, Eston MA, Ahmed OS, Marin R, et al. Wiskott-Aldrich Syndrome Protein Deficiency in Innate Immune Cells Leads to Mucosal Immune Dysregulation and Colitis in Mice. Gastroenterology (2012) 143(3):719–29.e711-712. doi: 10.1053/j.gastro.2012.06.008
23. Maillard MH, Cotta-de-Almeida V, Takeshima F, Nguyen DD, Michetti P, Nagler C, et al. The Wiskott-Aldrich Syndrome Protein Is Required for the Function of CD4(+)CD25(+)Foxp3(+) Regulatory T Cells. J Exp Med (2007) 204(2):381–91. doi: 10.1084/jem.20061338
24. Adriani M, Aoki J, Horai R, Thornton AM, Konno A, Kirby M, et al. Impaired In Vitro Regulatory T Cell Function Associated With Wiskott-Aldrich Syndrome. Clin Immunol (2007) 124(1):41–8. doi: 10.1016/j.clim.2007.02.001
25. Humblet-Baron S, Sather B, Anover S, Becker-Herman S, Kasprowicz DJ, Khim S, et al. Wiskott-Aldrich Syndrome Protein Is Required for Regulatory T Cell Homeostasis. J Clin Invest (2007) 117(2):407–18. doi: 10.1172/JCI29539
26. Marangoni F, Trifari S, Scaramuzza S, Panaroni C, Martino S, Notarangelo LD, et al. WASP Regulates Suppressor Activity of Human and Murine CD4(+)CD25(+)FOXP3(+) Natural Regulatory T Cells. J Exp Med (2007) 204(2):369–80. doi: 10.1084/jem.20061334
27. Yokoyama T, Yoshizaki A, Simon KL, Kirby MR, Anderson SM, Candotti F. Age-Dependent Defects of Regulatory B Cells in Wiskott-Aldrich Syndrome Gene Knockout Mice. PLoS One (2015) 10(10):e0139729. doi: 10.1371/journal.pone.0139729
28. Biswas A, Shouval DS, Griffith A, Goettel JA, Field M, Kang YH, et al. WASP-Mediated Regulation of Anti-Inflammatory Macrophages Is IL-10 Dependent and Is Critical for Intestinal Homeostasis. Nat Commun (2018) 9(1):1779. doi: 10.1038/s41467-018-03670-6
29. Nguyen DD, Maillard MH, Cotta-de-Almeida V, Mizoguchi E, Klein C, Fuss I, et al. Lymphocyte-Dependent and Th2 Cytokine-Associated Colitis in Mice Deficient in Wiskott-Aldrich Syndrome Protein. Gastroenterology (2007) 133(4):1188–97. doi: 10.1053/j.gastro.2007.07.010
30. Thrasher AJ, Jones GE, Kinnon C, Brickell PM, Katz DR. Is Wiskott–Aldrich Syndrome a Cell Trafficking Disorder? Immunol Today (1998) 19(12):537–9. doi: 10.1016/S0167-5699(98)01350-4
31. de Noronha S, Hardy S, Sinclair J, Blundell MP, Strid J, Schulz O, et al. Impaired Dendritic-Cell Homing In Vivo in the Absence of Wiskott-Aldrich Syndrome Protein. Blood (2005) 105(4):1590–7. doi: 10.1182/blood-2004-06-2332
32. Baptista MA, Keszei M, Oliveira M, Sunahara KK, Andersson J, Dahlberg CI, et al. Deletion of Wiskott-Aldrich Syndrome Protein Triggers Rac2 Activity and Increased Cross-Presentation by Dendritic Cells. Nat Commun (2016) 7:12175. doi: 10.1038/ncomms12175
33. Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and Subsequent Analysis of Murine Lamina Propria Mononuclear Cells From Colonic Tissue. Nat Protoc (2007) 2(10):2307–11. doi: 10.1038/nprot.2007.315
34. Kirschner N, Poetzl C, von den Driesch P, Wladykowski E, Moll I, Behne MJ, et al. Alteration of Tight Junction Proteins Is an Early Event in Psoriasis: Putative Involvement of Proinflammatory Cytokines. Am J Pathol (2009) 175(3):1095–106. doi: 10.2353/ajpath.2009.080973
35. Brandner JM, Zorn-Kruppa M, Yoshida T, Moll I, Beck LA, De Benedetto A. Epidermal Tight Junctions in Health and Disease. Tissue Barriers (2015) 3(1-2):e974451. doi: 10.4161/21688370.2014.974451
36. Kirschner N, Rosenthal R, Furuse M, Moll I, Fromm M, Brandner JM. Contribution of Tight Junction Proteins to Ion, Macromolecule, and Water Barrier in Keratinocytes. J Invest Dermatol (2013) 133(5):1161–9. doi: 10.1038/jid.2012.507
37. De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight Junction Defects in Patients With Atopic Dermatitis. J Allergy Clin Immunol (2011) 127(3):773–86.e771-777. doi: 10.1016/j.jaci.2010.10.018
38. Ohnemus U, Kohrmeyer K, Houdek P, Rohde H, Wladykowski E, Vidal S, et al. Regulation of Epidermal Tight-Junctions (TJ) During Infection With Exfoliative Toxin-Negative Staphylococcus Strains. J Invest Dermatol (2008) 128(4):906–16. doi: 10.1038/sj.jid.5701070
39. Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, et al. Claudin-Based Tight Junctions Are Crucial for the Mammalian Epidermal Barrier: A Lesson From Claudin-1-Deficient Mice. J Cell Biol (2002) 156(6):1099–111. doi: 10.1083/jcb.200110122
40. Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, et al. Cytokine Modulation of Atopic Dermatitis Filaggrin Skin Expression. J Allergy Clin Immunol (2007) 120(1):150–5. doi: 10.1016/j.jaci.2007.04.031
41. Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, DeBenedetto A, et al. Cytokine Modulation of Atopic Dermatitis Filaggrin Skin Expression. J Allergy Clin Immunol (2009) 124(3 Suppl 2):R7–R12. doi: 10.1016/j.jaci.2009.07.012
42. Donetti E, Cornaghi L, Gualerzi A, Baruffaldi Preis FW, Prignano F. An Innovative Three-Dimensional Model of Normal Human Skin to Study the Proinflammatory Psoriatic Effects of Tumor Necrosis Factor-Alpha and Interleukin-17. Cytokine (2014) 68(1):1–8. doi: 10.1016/j.cyto.2014.03.003
43. Prignano F, Arnaboldi F, Cornaghi L, Landoni F, Tripo L, Preis FW, et al. Tumor Necrosis Factor-Alpha and Interleukin-17 Differently Affects Langerhans Cell Distribution and Activation in an Innovative Three-Dimensional Model of Normal Human Skin. Eur J Cell Biol (2015) 94(2):71–7. doi: 10.1016/j.ejcb.2014.12.003
44. Oh J, Freeman AF, Program NCS, Park M, Sokolic R, Candotti F, et al. The Altered Landscape of the Human Skin Microbiome in Patients With Primary Immunodeficiencies. Genome Res (2013) 23(12):2103–14. doi: 10.1101/gr.159467.113
45. Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and Staphylococcus Aureus Colonization Drives Inflammation in Atopic Dermatitis. Immunity (2015) 42(4):756–66. doi: 10.1016/j.immuni.2015.03.014
46. Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal Shifts in the Skin Microbiome Associated With Disease Flares and Treatment in Children With Atopic Dermatitis. Genome Res (2012) 22(5):850–9. doi: 10.1101/gr.131029.111
47. Yoshie O, Matsushima K. CCR4 and Its Ligands: From Bench to Bedside. Int Immunol (2015) 27(1):11–20. doi: 10.1093/intimm/dxu079
48. Nakashima C, Otsuka A, Kabashima K. Interleukin-31 and Interleukin-31 Receptor: New Therapeutic Targets for Atopic Dermatitis. Exp Dermatol (2018) 27(4):327–31. doi: 10.1111/exd.13533
49. Nakajima S, Kitoh A, Egawa G, Natsuaki Y, Nakamizo S, Moniaga CS, et al. IL-17A as an Inducer for Th2 Immune Responses in Murine Atopic Dermatitis Models. J Invest Dermatol (2014) 134(8):2122–30. doi: 10.1038/jid.2014.51
50. Zhao J, Lloyd CM, Noble A. Th17 Responses in Chronic Allergic Airway Inflammation Abrogate Regulatory T-Cell-Mediated Tolerance and Contribute to Airway Remodeling. Mucosal Immunol (2013) 6(2):335–46. doi: 10.1038/mi.2012.76
51. Eyerich K, Pennino D, Scarponi C, Foerster S, Nasorri F, Behrendt H, et al. IL-17 in Atopic Eczema: Linking Allergen-Specific Adaptive and Microbial-Triggered Innate Immune Response. J Allergy Clin Immunol (2009) 123(1):59–66.e54. doi: 10.1016/j.jaci.2008.10.031
52. Bouma G, Burns S, Thrasher AJ. Impaired T-Cell Priming In Vivo Resulting From Dysfunction of WASp-Deficient Dendritic Cells. Blood (2007) 110(13):4278–84. doi: 10.1182/blood-2007-06-096875
53. Yuki T, Tobiishi M, Kusaka-Kikushima A, Ota Y, Tokura Y. Impaired Tight Junctions in Atopic Dermatitis Skin and in a Skin-Equivalent Model Treated With Interleukin-17. PLoS One (2016) 11(9):e0161759. doi: 10.1371/journal.pone.0161759
54. Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial Community Variation in Human Body Habitats Across Space and Time. Science (2009) 326(5960):1694–7. doi: 10.1126/science.1177486
55. Chen YE, Fischbach MA, Belkaid Y. Skin Microbiota-Host Interactions. Nature (2018) 553(7689):427–36. doi: 10.1038/nature25177
56. Sanford JA, Gallo RL. Functions of the Skin Microbiota in Health and Disease. Semin Immunol (2013) 25(5):370–7. doi: 10.1016/j.smim.2013.09.005
57. Eyerich S, Eyerich K, Traidl-Hoffmann C, Biedermann T. Cutaneous Barriers and Skin Immunity: Differentiating A Connected Network. Trends Immunol (2018) 39(4):315–27. doi: 10.1016/j.it.2018.02.004
58. Belkaid Y, Segre JA. Dialogue Between Skin Microbiota and Immunity. Science (2014) 346(6212):954–9. doi: 10.1126/science.1260144
59. Nguyen DD, Muthupalani S, Goettel JA, Eston MA, Mobley M, Taylor NS, et al. Colitis and Colon Cancer in WASP-Deficient Mice Require Helicobacter Species. Inflamm Bowel Dis (2013) 19(10):2041–50. doi: 10.1097/MIB.0b013e318295fd8f
60. Chng KR, Tay AS, Li C, Ng AH, Wang J, Suri BK, et al. Whole Metagenome Profiling Reveals Skin Microbiome-Dependent Susceptibility to Atopic Dermatitis Flare. Nat Microbiol (2016) 1(9):16106. doi: 10.1038/nmicrobiol.2016.106
61. Hanski I, von Hertzen L, Fyhrquist N, Koskinen K, Torppa K, Laatikainen T, et al. Environmental Biodiversity, Human Microbiota, and Allergy Are Interrelated. Proc Natl Acad Sci USA (2012) 109(21):8334–9. doi: 10.1073/pnas.1205624109
62. Sellers RS, Clifford CB, Treuting PM, Brayton C. Immunological Variation Between Inbred Laboratory Mouse Strains: Points to Consider in Phenotyping Genetically Immunomodified Mice. Vet Pathol (2012) 49(1):32–43. doi: 10.1177/0300985811429314
63. Bix M, Wang ZE, Thiel B, Schork NJ, Locksley RM. Genetic Regulation of Commitment to Interleukin 4 Production by a CD4(+) T Cell-Intrinsic Mechanism. J Exp Med (1998) 188(12):2289–99. doi: 10.1084/jem.188.12.2289
64. Noti M, Kim BS, Siracusa MC, Rak GD, Kubo M, Moghaddam AE, et al. Exposure to Food Allergens Through Inflamed Skin Promotes Intestinal Food Allergy Through the Thymic Stromal Lymphopoietin-Basophil Axis. J Allergy Clin Immunol (2014) 133(5):1390–1399, 1399.e1391-1396. doi: 10.1016/j.jaci.2014.01.021
65. Bartnikas LM, Gurish MF, Burton OT, Leisten S, Janssen E, Oettgen HC, et al. Epicutaneous Sensitization Results in IgE-Dependent Intestinal Mast Cell Expansion and Food-Induced Anaphylaxis. J Allergy Clin Immunol (2013) 131(2):451–60.e451-456. doi: 10.1016/j.jaci.2012.11.032
66. Sampson HA, O'Mahony L, Burks AW, Plaut M, Lack G, Akdis CA. Mechanisms of Food Allergy. J Allergy Clin Immunol (2018) 141(1):11–9. doi: 10.1016/j.jaci.2017.11.005
67. Walker MT, Green JE, Ferrie RP, Queener AM, Kaplan MH, Cook-Mills JM. Mechanism for Initiation of Food Allergy: Dependence on Skin Barrier Mutations and Environmental Allergen Costimulation. J Allergy Clin Immunol (2018) 141(5):1711–25.e1719. doi: 10.1016/j.jaci.2018.02.003
68. Akei HS, Brandt EB, Mishra A, Strait RT, Finkelman FD, Warrier MR, et al. Epicutaneous Aeroallergen Exposure Induces Systemic TH2 Immunity That Predisposes to Allergic Nasal Responses. J Allergy Clin Immunol (2006) 118(1):62–9. doi: 10.1016/j.jaci.2006.04.046
69. Lin JY, Chen JS, Hsu CJ, Miaw SC, Liu CY, Lee SJ, et al. Epicutaneous Sensitization With Protein Antigen Induces Th9 Cells. J Invest Dermatol (2012) 132(3 Pt 1):739–41. doi: 10.1038/jid.2011.382
70. Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous Sensitization With Protein Antigen Induces Localized Allergic Dermatitis and Hyperresponsiveness to Methacholine After Single Exposure to Aerosolized Antigen in Mice. J Clin Invest (1998) 101(8):1614–22. doi: 10.1172/JCI1647
71. Matsumoto K, Saito H. Epicutaneous Immunity and Onset of Allergic Diseases - Per-“Eczema”Tous Sensitization Drives the Allergy March. Allergol Int (2013) 62(3):291–6. doi: 10.2332/allergolint.13-RAI-0603
72. Lack G. Epidemiologic Risks for Food Allergy. J Allergy Clin Immunol (2008) 121(6):1331–6. doi: 10.1016/j.jaci.2008.04.032
73. Bantz SK, Zhu Z, Zheng T. The Atopic March: Progression From Atopic Dermatitis to Allergic Rhinitis and Asthma. J Clin Cell Immunol (2014) 5(2):202. doi: 10.4172/2155-9899.1000202
74. Heo WI, Lee KE, Hong JY, Kim MN, Oh MS, Kim YS, et al. The Role of Interleukin-17 in Mouse Models of Atopic Dermatitis and Contact Dermatitis. Clin Exp Dermatol (2015) 40(6):665–71. doi: 10.1111/ced.12567
75. Dhingra N, Guttman-Yassky E. A Possible Role for IL-17A in Establishing Th2 Inflammation in Murine Models of Atopic Dermatitis. J Invest Dermatol (2014) 134(8):2071–4. doi: 10.1038/jid.2014.141
76. Hasegawa T, Uga H, Mori A, Kurata H. Increased Serum IL-17A and Th2 Cytokine Levels in Patients With Severe Uncontrolled Asthma. Eur Cytokine Netw (2017) 28(1):8–18. doi: 10.1684/ecn.2017.0390
77. Esaki H, Brunner PM, Renert-Yuval Y, Czarnowicki T, Huynh T, Tran G, et al. Early-Onset Pediatric Atopic Dermatitis Is TH2 But Also TH17 Polarized in Skin. J Allergy Clin Immunol (2016) 138(6):1639–51. doi: 10.1016/j.jaci.2016.07.013
Keywords: Wiskott–Aldrich syndrome, skin, immune homeostasis, IL-17, inflammation
Citation: Herman KE, Yoshida T, Hughson A, Grier A, Gill SR, Beck LA and Fowell DJ (2022) IL-17-Dependent Dysregulated Cutaneous Immune Homeostasis in the Absence of the Wiskott–Aldrich Syndrome Protein. Front. Immunol. 13:817427. doi: 10.3389/fimmu.2022.817427
Received: 18 November 2021; Accepted: 17 January 2022;
Published: 21 February 2022.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Tomohiro Morio, Tokyo Medical and Dental University, JapanRaif Geha, Boston Children's Hospital and Harvard Medical School, United States
Copyright © 2022 Herman, Yoshida, Hughson, Grier, Gill, Beck and Fowell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah J. Fowell, ZGpmMjczQGNvcm5lbGwuZWR1