 Carlos Wagner S. Wanderley1,2
Carlos Wagner S. Wanderley1,2 Tatiana Strava Correa3
Tatiana Strava Correa3 Mariana Scaranti4
Mariana Scaranti4 Fernando Queiroz Cunha1,2
Fernando Queiroz Cunha1,2 Romualdo Barroso-Sousa3*
Romualdo Barroso-Sousa3*- 1Center for Research in Inflammatory Diseases (CRID), Ribeirao Preto Medical School, Ribeirao Preto, Brazil
- 2Department of Pharmacology, Ribeirao Preto Medical School, University of São Paulo, Ribeirao Preto, Brazil
- 3Oncology Center, Hospital Sírio-Libanês, Brasília, Brazil
- 4Oncologia DASA, Hospital Nove de Julho, São Paulo, Brazil
Reinvigorating the antitumor immune response using immune checkpoint inhibitors (ICIs) has revolutionized the treatment of several malignancies. However, extended use of ICIs has resulted in a cancer-specific response. In tumors considered to be less immunogenic, the response rates were low or null. To overcome resistance and improve the beneficial effects of ICIs, novel strategies focused on ICI-combined therapies have been tested. In particular, poly ADP-ribose polymerase inhibitors (PARPi) are a class of agents with potential for ICI combined therapy. PARPi impairs single-strand break DNA repair; this mechanism involves synthetic lethality in tumor cells with deficient homologous recombination. More recently, novel evidence indicated that PAPRi has the potential to modulate the antitumor immune response by activating antigen-presenting cells, infiltrating effector lymphocytes, and upregulating programmed death ligand-1 in tumors. This review covers the current advances in the immune effects of PARPi, explores the potential rationale for combined therapy with ICIs, and discusses ongoing clinical trials.
Introduction
Cancer immunotherapy has revolutionized the field of oncology by demonstrating that the use of immune checkpoint inhibitors (ICIs) alone or in combination with other therapies prolongs the survival of patients with advanced disease, including melanoma, genitourinary, lung, gastric, and more recently breast cancer (1–4). However, the efficacy of ICIs varies depending on the type of cancer and within the same tumor tissue cohort (5). Ultimately, the benefits of ICI therapy in the overall population could be considered low, especially in some common tumor types, such as prostate and breast cancers (6, 7).
In this context, strategies to enhance the benefit of ICIs have focused on patient selection based on biomarkers such as programmed death ligand-1 (PD-L1) or the use of ICIs combined with other agents, including chemotherapy or targeted therapy (4, 5).
Poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) are a class of drugs that inhibit single-strand DNA repair, leading to DNA damage and apoptosis (8). Notably, this process of DNA damage can modulate the antitumor immune response by activating antigen-presenting cells (APCs), infiltrating effector lymphocytes, and upregulating PD-L1 in tumors. In this review, we summarize the current knowledge on the immune-mediated effects of PARPi and the rationale for clinical trials that combine these agents with ICIs.
DNA Damage Repair Pathways and Cancer
The genome of every cell is constantly exposed to endogenous and/or exogenous sources of DNA damage. Usually, a chemical addition or disruption to a base of DNA or a break in one or both chains of DNA strands is characterized as DNA damage (9). DNA damage mechanisms for detecting and repairing DNA, collectively termed DNA damage response (DDR), are activated to ensure cell survival (10). Thus, dysregulation and mutations in these DDR factors and their modulators have implications for human health and disease, including increased susceptibility to DNA mutations that can lead to neoplastic transformation (9, 10). High levels of replication stress often induce DNA damage in cancer cells and their survival relies on certain DNA repair pathways (11). Understanding the broader role of DDR pathways in cancers has led to the development of pharmacological interventions for cancer therapy, such as drugs targeting poly (ADP-ribose) polymerases (PARP) (12).
DNA Repair and PARP
PARP belongs to a family of 17 enzymes involved in several cellular processes, including DDR (13). Poly (ADP-ribose) polymerase 1 (PARP1), the most well-known enzyme in this family, is involved in the detection and repair of DNA single-strand breaks (13, 14). Functionally, PARP1 can rapidly detect DNA damage. The binding of PARP1 to DNA alters its catalytic domains, causing PARP1 to catalyze the post-translational polymerization of ADP-ribose units (15). PARP1 enables the auto-PARylation and PARylation of histones and other chromatin-associated proteins. Finally, PARP1 recruits additional DNA repair molecules, such as X-ray repair cross complementing 1 (XRCC1), to the site of damage, promoting the effective repair of DNA (8, 16, 17). However, when PARP fails or is pharmacologically inhibited, single-stranded breaks accumulate and become double-stranded breaks (18). Cells with an increasing number of double-strand breaks become more dependent on other repair pathways, mainly homologous recombination (HR) and non-homologous end joining (NHEJ) (19). The two main pathways involved in DNA double-strand break repair are described below:
Homologous Recombination (HR)
HR is an efficient and high-fidelity DNA repair mechanism based on a homologous template (8, 20). The HR pathway mainly occurs during the S/G2 phase of the cell cycle (21). HR is initiated by the MRN-complex, composed of meiotic recombination 11 (MRE11), RAD50 homolog (RAD50), and Nijmegen breakage syndrome 1 (NBS1), which is recruited to the sites of double-strand breaks (22). The MRN complex produces a 3 overhang of single-stranded DNA that is coated by replication protein A (RPA) to avoid DNA secondary structure formation (8, 20). Breast cancer susceptibility genes 1 and 2 (BRCA1 and BRCA2) enable DNA repair protein RAD51 homolog 1 (RAD51) recombinase to displace RPA and stabilize RAD51-single-stranded DNA filaments (20, 23). These filaments invade a sister chromatid to execute the homology search, and repair-associated DNA synthesis is terminated by the generation of a double-Holliday junction, which leads to the effective repair of the DNA double-strand break (21). Therefore, tumor cells with defective HR, such as those with a BRCA1/2 mutation, are susceptible to impairment of PARP, facilitating cell death, or can be alternatively repaired by the error-prone NHEJ pathway, resulting in genomic instability before cell death (24, 25).
Non-Homologous End Joining (NHEJ)
NHEJ repairs double-strand breaks in DNA without a template strand. Consequently, NHEJ is an error-prone double-strand break repair mechanism. It does not require a template strand and can be activated in all phases of the cell cycle (8, 26). The initial step in NHEJ is recognition and binding of the Ku heterodimer protein (Ku70/80) to double-strand breaks. The Ku-DNA complex acts as a scaffold for DNA-dependent protein kinase (DNA-PKcs) and enzymes such as X-ray cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF), and DNA ligase IV, which ligate DNA and mediate the ligation of the double-strand break (8, 26). In this context, PARPi in HR-deficient cells promotes NHEJ DNA repair and induces genomic instability or cell death.
PARP Inhibitors (PARPi)
Currently, four agents (olaparib, rucaparib, niraparib, and talazoparib) are approved for the treatment of different tumors, including ovarian, breast, prostate, and pancreatic cancers. The success of PARPi in cancer treatment is believed to originate from their ability to induce synthetic lethality (27, 28). Synthetic lethality arises when the co-occurrence of two gene conditions causes cell death, whereas a deficiency in only one of the genes does not determine cell lethality (29). Mechanistically, PARPi anticancer agents compete with nicotinamide (NAD+) for the PARP catalytic site, inhibiting single-strand break repair (8, 30, 31). This effect promotes the accumulation of single-strand breaks that result in the collapse of the replication fork and replication associated with the double-strand break. Subsequently, tumor cells become more dependent on the HR or NHEJ repair pathways (18, 19). In tumors with HR defects, such as those with BRCA1/2 mutations, PARPi induces synthetic lethality. BRCA2-deficient cells compared to BRCA2-proficient cells are 90 times more sensitive to PARP inhibition (18, 32). Although there is a consensus that PARPi mechanisms of action rely on inducing synthetic lethality in tumors with defective HR, more recent findings suggest that PARPi also modulates the antitumor immune response.
Immune Effects of PARP Inhibitors
Previous reports have noted an association between DDR defects or failure with the activation of anticancer immunity through the response-dependent type I interferon (IFN) pathway or via the accumulation of mutations and neoantigens (33–35). In this context, novel findings have demonstrated that the pharmacological inhibition of PARP can mimic this condition, dramatically affecting the balance of the immune response in the tumor microenvironment.
The DNA Damage Induced by PARPi Induces Antitumor Immune Response
DNA sensing through the cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS)/stimulator of interferon genes (STING) pathway participates in host defense by detecting aberrant entry of DNA into the cytosol (36). This pathway is classically involved in defense against viruses; however, new evidence indicates that the cGAS-STING pathway is also activated by fragments of endogenous DNA generated by cancer treatment, driving an effective antitumor immune response (36–38).
In preclinical studies, PARPi effectiveness in BRCA1-deficient tumors was found to be dependent on CD8 T-cell recruitment via intratumoral cGAS/STING pathway activation. The use of PARPi in DRR-defective tumors produces single-and double-strand breaks in DNA that bind to cGAS, leading to the production of a second messenger molecule that stimulates the adapter protein STING. STING, via kinases TANK-binding kinase 1 (TBK1) and IkappaB kinase (IKK), activates transcription factor interferon regulatory transcription factor 3 (IRF3) and factor nuclear kappa B (NF-κB), which translocate into the nucleus to trigger type I IFN signaling (39).
It is well known that IFNs play a central role in antitumor immunity (40). The seminal demonstration that interferon-α/β receptor (IFNAR) or signal transducer and activator of transcription 1 (STAT1) knockout mice fail to reject immunogenic tumors (41, 42). Numerous studies have shown that the expression levels of IFNs are positively correlated with CD8 + T cell lymphocyte infiltration in the tumor microenvironment (39, 43, 44). Thus, the introduction of DNA damage by PARPi can trigger the transformation of tumors from cold to hot (39). Moreover, CD8 T lymphocytes kill malignant cells upon recognition by the T-cell receptor (TCR) of specific antigenic peptides present on the surface of the target cells (45). In this context, effective antitumor immunity relies on cross-presentation of tumor antigens by APCs to CD8 T lymphocytes. APC activation requires type I IFN signaling, which can be initiated by cGAS-STING activation (40, 42, 46, 47). Therefore, cGAS-STING signaling can act as a bridge between DNA damage and the activation of anticancer immune responses.
However, in parallel, type I IFNs activate pathways that control the exacerbated inflammatory immune responses. For example, IFN-β has been shown to induce the expression of PD-L1 in tumor cells, which contributes to the immune escape by cancer cells (48). In line with our premise, PARPi induces upregulation of PD-L1 in tumor cells (49).
The Genomic Instability Induced by PARPi Triggers Antitumor Immune Response
In tumor cells, DDR failure can result in the accumulation of mutations in drive genes that produce survival advantages and accelerate tumor development (50). However, this genomic instability can encode tumor-specific neoantigens, which may make tumors more attractive to the immune response (51, 52). There is a correlation between tumor mutational burden and the likelihood of response to ICIs. Preclinical studies have shown that cancer cells with microsatellite instability (MSI) or defective mismatch repair (dMMR) grow in immune-deficient mice but are unable to grow in immune-competent mice (53). In clinics, MSI or dMMR are biomarkers for predicting responses to ICIs approved by the FDA (54). In this context, it has been discussed whether drugs that modulate DDR pathways, such as PARPis, can promote genetic instability and neoantigen formation.
It has been experimentally demonstrated that in BRCA-deficient cells, PARPi induces chromosomal instability typified by the accumulation of chromosomal breaks and eventual lethality via NHEJ (24). In another study, genomic instability and cell death induced in BRCA1-deficient cells by PARPi were found to be dependent on the NHEJ factor p53-binding protein 1 (53BP1) (55). Although this mechanism still needs to be further explored clinically, these primary findings suggest that the pharmacological blockade of PARP has the potential to increase genomic instability and lead to dynamic mutational profiles, resulting in the persistent renewal of neoantigens and engagements of an immune response.
The Rationale for the Combination of ICIs and PARPi
Immune checkpoints represent a set of modulatory pathways essential for exacerbating inflammatory responses and maintaining self-tolerance (56). The receptors cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and PD-1, expressed mainly in lymphocytes, and PD-L1, expressed in APCs, are part of the immunological checkpoint system (57). The interaction between CTLA-4 and CD80, CD86, or PD-L1/PD-1 reduces T cell activity, leading to suppression of the inflammatory response and preservation of tissues (57, 58). However, this mechanism favors cancer progression, enabling the escape of the anti-tumor immune response. Therefore, the use of monoclonal antibodies (mAbs) to block CTLA-4, PD-1, or its ligand PD-L1, ICIs, reactivates and drives the immune response to detect and destroy tumors by overcoming the negative feedback mechanism of the immune response (59).
Although there is no doubt that ICI therapy positively impacts cancer treatment in several neoplasms, ICIs may not be sufficient for optimal antitumor activity in some patients, particularly those with a defect in cancer antigen-specific T-cell activation or impairment of T-cell infiltration into tumors (60). Thus, efforts to enhance these responses are needed. The interaction between tumor DNA damage and the immune system plays a role in driving the response to ICI. DNA-damaging agents include chemotherapy (CT), ionizing radiation (RDT), and targeted DNA repair therapies. CT activates the immune system by inducing immunogenic cell death pathways. RT causes several types of DNA damage. DNA repair targeted agents include PARPi. In particular, combination strategies with PARPi can potentially maximize the benefit from ICIs, and its plausible synergistic effect resides in the immune properties of PARPi at different points in the cancer immune response. PARPi may facilitate a more profound antitumor immune response and synergize with ICIs by inducing DNA damage, producing a T helper 1 (Th1) immune-mediated response via IFN signaling, activation of APC cells, increased recruitment of effector lymphocytes, and promoting upregulation of PD-L1 in tumor cells (39, 49, 61).
In summary, DNA damage induced by a PARPi can promote antitumor immunity via the cGAS-STING/type I IFN/CD8 army (positive effect). In contrast, type I IFN induces PD-L1 expression and promotes tumor immune escape (a negative effect). In this context, the combination of PARPi and ICIs has particular translational appeal owing to its potent immune-stimulatory anticancer effects (Figure 1).
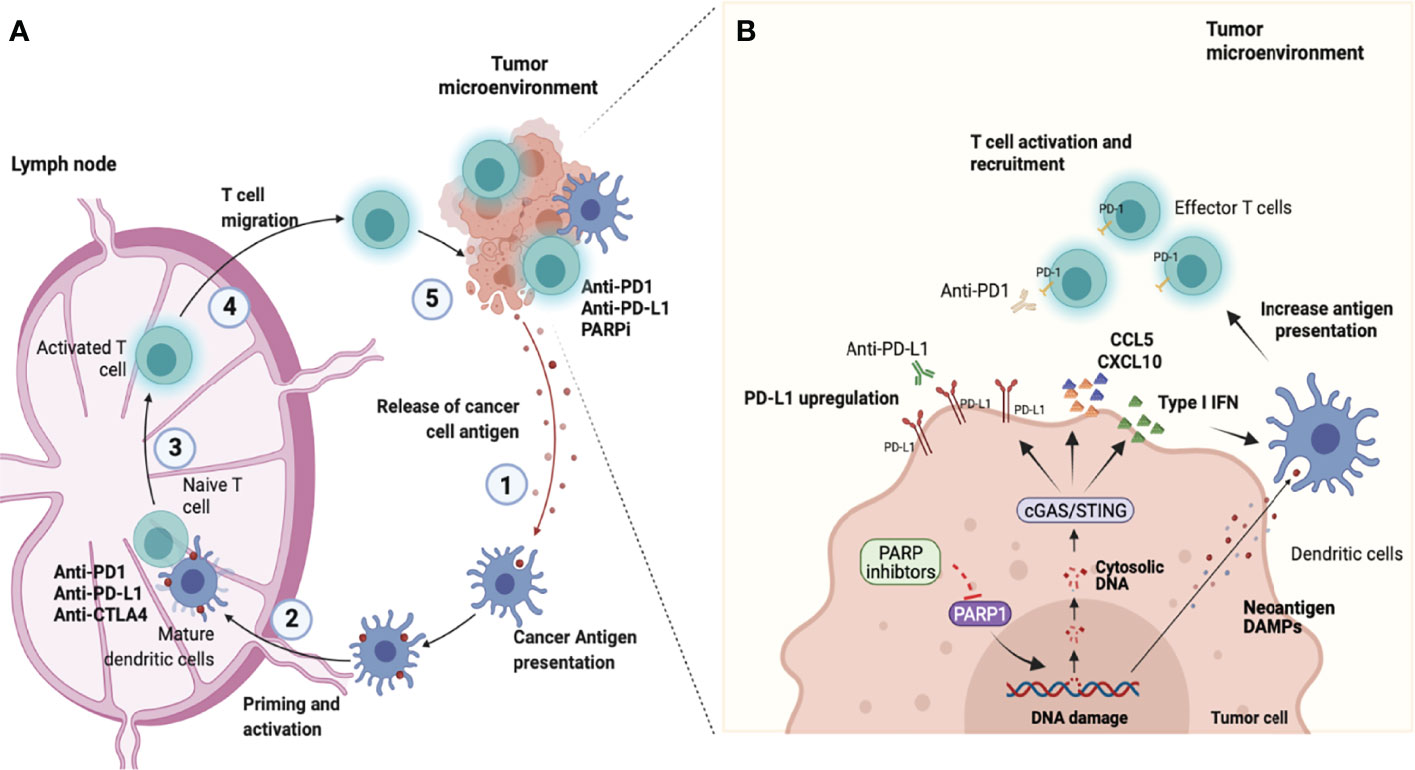
Figure 1 Combining PARP inhibition and immune checkpoint blockade. (A) Antitumor immunity depends on a series of stepwise events. Primarily this process includes the capture and processing of Tumor-associated antigens (TAAs) by Antigen-presenting cells (APCs), such as dendritic cells or macrophages in the tumor microenvironment (step 1). Next, APCs cells presented antigen to CD8+ T cells at the lymph nodes (step 2). This process promotes the prime and activation of effector CD8 T cells (step 3). Finally, the activated effector T cells migrate from lymphocytes (step 4) and infiltrate into the tumor microenvironment to recognize and eliminate tumor cells (step 5), completing the cancer-immune cycle. However, the continued immune attack may enable cancer cells to evolve mechanisms for the escape of immune attacks. Molecules that negatively regulate T lymphocyte activation, called immune checkpoints are central players involved in tumor immune escape. In the cancer-immune cell cycle the ICIs (anti-CTLA-4, anti-PD-1, or anti-PD-L1) reactivate and drive the immune response to detect and destroy tumors by overcoming the negative feedback mechanism of the immune response acting in steps 3 and 5. (B) Poly-ADP-ribose polymerase inhibitors (PARPi) have effects in the early steps of the cancer-immune cell cycle. PARPi induce DNA breaks in BRCA1/2-deficient cells which can result in cell death or genomic instability and neoantigen formation. Furthermore, the DNA damage induces the release of DNA fragments into the cytosol which causes the cGAS/STING pathway activation in tumor cells and the production of type I IFN and chemokines (CCL5 and CXCL10). This effect culminates with paracrine activation of APCs such as dendritic cells (step 1 and 2) and with the recruitment of CD8 cells for the tumor microenvironment (step 4). Another important immune effect of PARPi is associated with the increased expression of Programmed death ligand-1 (PD-L1) in tumor cells (step 5). Therefore, the combined use of PARPi with Immune checkpoint inhibitors (ICIs) has the potential to amplify the entire cancer immune cycle (image created at Biorender).
PARPi and ICIs Combination in Preclinical Studies
In preclinical models, PARPi has demonstrated synergy with ICIs in a variety of tumor models regardless of BRCA1/2-defect. It was demonstrated that PARPi-based therapy synergizes with anti-PD-1 against both MSI and microsatellite stable (MSS) colon cancer models, with a potential sensitizing effect of anti-PD-1 therapy against MSS tumors (61). In another study, PARPi led to the accumulation of cytosolic double-stranded DNA, thereby activating type I IFN-related immune response. Shen et al. (2019) (62) demonstrated the combined use of PARPi and ICIs against colon and ovarian experimental tumors, regardless of the BRCA1/2 mutation status of the cell lines assessed both in vitro and in vivo. Furthermore, PARPi treatment upregulated PD-L1 expression in vitro and in vivo in breast cancer cell lines, xenograft tumors, and syngeneic tumors. Although PARPi attenuated anticancer immunity via upregulation of PD-L1, the combination of PARPi and anti-PD-L1 therapy compared with each agent alone significantly increased therapeutic efficacy (49). Investigating the effects of the PARP1/2 inhibitor niraparib in combination with ICI therapy in BRCA-deficient and BRCA-proficient breast cancer tumor models, it was observed that the combined regimen demonstrated synergistic antitumor activity in both BRCA-proficient and BRCA-deficient tumors. Interestingly, mice with tumors cured by single-agent niraparib completely rejected tumor growth upon rechallenge with the same tumor cell line, suggesting the potential establishment of immune memory (63).
Together, these data reinforce that PARPi in combination with ICIs may be beneficial in tumors, regardless of DNA repair status, which has important clinical implications.
PARPi and ICIs Combination in Clinical Studies
Combination of a PARPi With Anti-PD1/PD-L1 ICIs: What Do We Already Know?
Phase I study analysis of this combination showed toxicities manageable with supportive care, and no new adverse events were noted compared with the PARPi or ICI toxicities in monotherapy (64, 65) A phase I study of solid tumors tested a combination of durvalumab, an anti-PD-L1 agent, and olaparib. Durvalumab was administered at 10 mg/kg every 2 weeks or 1,500 mg every 4 weeks, and olaparib tablets were administered twice daily. No dose-limiting toxicity was observed for durvalumab plus olaparib. Two partial responses (≥15 months and ≥ 11 months) and eight stable diseases ≥ 4 months (median, 8 months [4–14.5 months]) were seen in patients who received this combination, generating an 83% disease control rate (65).
Here, we explored clinical trials evaluating the efficacy of PARPi and anti-PD1/PD-L1 ICIs in ovarian and breast cancers. Studies with breast cancer and ovarian cancer patients, as summarized in Table 1, demonstrate interesting response rates with acceptable toxicity.
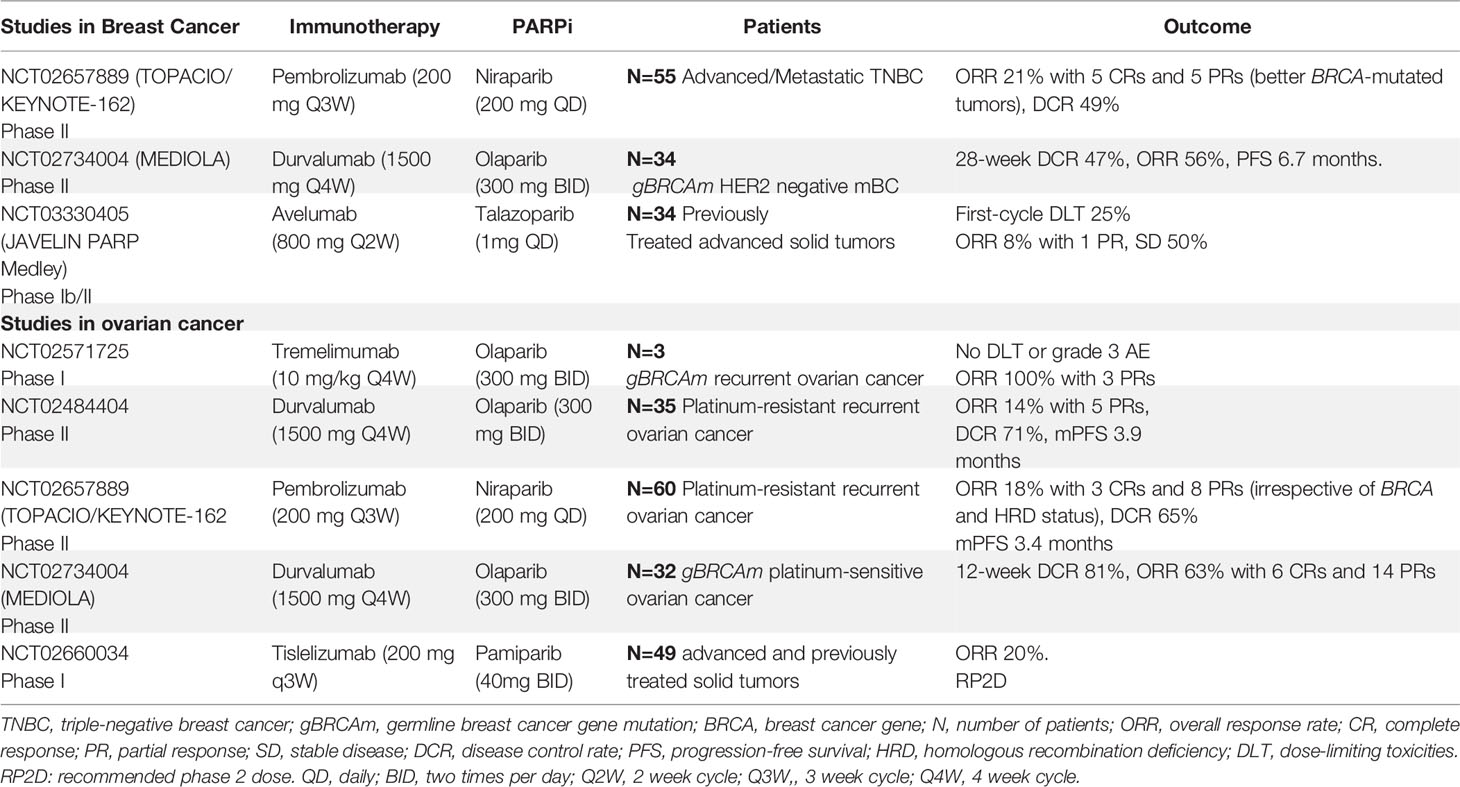
Table 1 Clinical trials evaluating the combination of PARP inhibitors and immune checkpoint inhibitors in breast cancer ovarian cancer.
MEDIOLA is a phase II basket study assessing the efficacy and safety of a chemo-free combination of olaparib and durvalumab in patients with solid tumors (NCT02734004) and germline BRCA1/2 (gBRCA1/2) mutations. Patients received olaparib for 4 weeks, followed by a combination of olaparib and durvalumab until disease progression. The primary endpoints were the disease control rate (DCR) at 12 weeks, safety, and tolerability. Patients with platinum-sensitive recurrent ovarian cancer (n=34) received at least one prior line of platinum therapy. The 28-week DCR was 65.6%, while the overall response rate (ORR) was 71.9%, with a total of seven complete responses (CRs). The median progression-free survival (PFS) was 11.1 months (95% CI: 8.2, 15.9), with a median duration of response (DOR) of 10.2 months. The median overall survival (OS) for all patients is not yet reached, with 87.0% of patients alive at 24 months (66). Thirty-four patients were enrolled in the human epidermal growth factor 2 receptor (HER2)-negative metastatic breast cancer group. The 12- and 28-week DCRs were 81% and 47%, respectively. The ORR for the overall cohort was 56%, with one patient with CR and six (19%) patients with progressive disease (PD). The median PFS was 6.7 months (95% CI: 4.6, 11.7 months). The most common grade 3 or 4 adverse events reported were anemia (11.8%), neutropenia (8.8%), and pancreatitis (5.9%) (67). Therefore, we concluded that the combination of olaparib and durvalumab was well tolerated and showed promising median PFS and DOR for ovarian cancer, breast cancer, and gBRCA1/2 mutations.
The TOPACIO/KEYNOTE-162 phase I/II study evaluated the efficacy and safety of nivolumab plus pembrolizumab in platinum-resistant recurrent ovarian cancer and metastatic triple-negative breast cancer (TNBC). This trial included patients with or without gBRCA mutations. The primary outcome was ORR. In the ovarian cancer group (n = 60), ORR 18% with 3 CRs and 8 PRs (irrespective of BRCA and HRD status), and DCR 65%. The median PFS was 3.4 months, with acceptable toxicity. Responses in patients without tumor BRCA mutations were higher than expected with either agent as monotherapy (68). Of 46 breast cancer evaluable patients, 20 (49%) achieved durable clinical benefit (any complete response/partial response or stable disease ≥16 weeks), with stronger activity in BRCA-mutated tumors (69).
The PARPi talazoparib was also evaluated in the phase Ib/II study. Patients with advanced solid tumors who had received ≥1 prior standard of care chemotherapy regimen were treated with Avelumab in combination with Talazoparib. In phase 2 cohorts, eligible patients had metastatic TNBC (cohort 2A) or hormone receptor-positive (HR+), HER2 negative, DNA damage repair defect-positive breast cancer (cohort 2B). Patients in cohort 2A/B had received 0 to 2 prior therapies (no progression on prior platinum-based chemotherapy). The primary endpoint was the objective response. A total of 22 patients had been treated in both cohorts. In cohort 2A, 12 patients were evaluable for disease assessment: PR in 1, SD in 6, and PD in 5. All 3 patients in cohort 2B were non-evaluable for response at data cutoff. Treatment-related Adverse events (AEs) of any grade occurred in 94.7% of patients, the most common AEs were anemia, nausea, fatigue, and thrombocytopenia; 9 patients (47.4%) had grade ≥3 AEs. Therefore, Avelumab administered in combination with Talazoparib in patients with advanced solid tumors showed preliminary antitumor activity and a manageable safety profile. The study is ongoing (70).
Michael Friedlander and colleagues reported the findings of a phase 1a/b trial of the combination of a PARPi (Pamiparib) and ICI (Tislelizumab) in 49 patients with previously treated, advanced solid tumors. The results from the dose-escalation stage, phase 1a/b trial, show that the combination was generally well tolerated and associated with antitumor responses (20%) in patients with advanced solid tumors supporting further investigation of the combination (64).
Combination of PARPi With ICIs: Ongoing Studies
There are numerous ongoing trials (phases I-III) exploring the combination of PARPi and anti-PD1/anti-PD-L1 agents, and some trials with new immunotherapy agents such as TSR-022. TSR-022 is a monoclonal antibody against T-cell immunoglobulin and mucin domain molecule 3 (TIM-3) (also called HAVCR2), an immune checkpoint receptor. Table 2 summarizes the ongoing phase III studies with a combination of immunotherapy and PARPi.
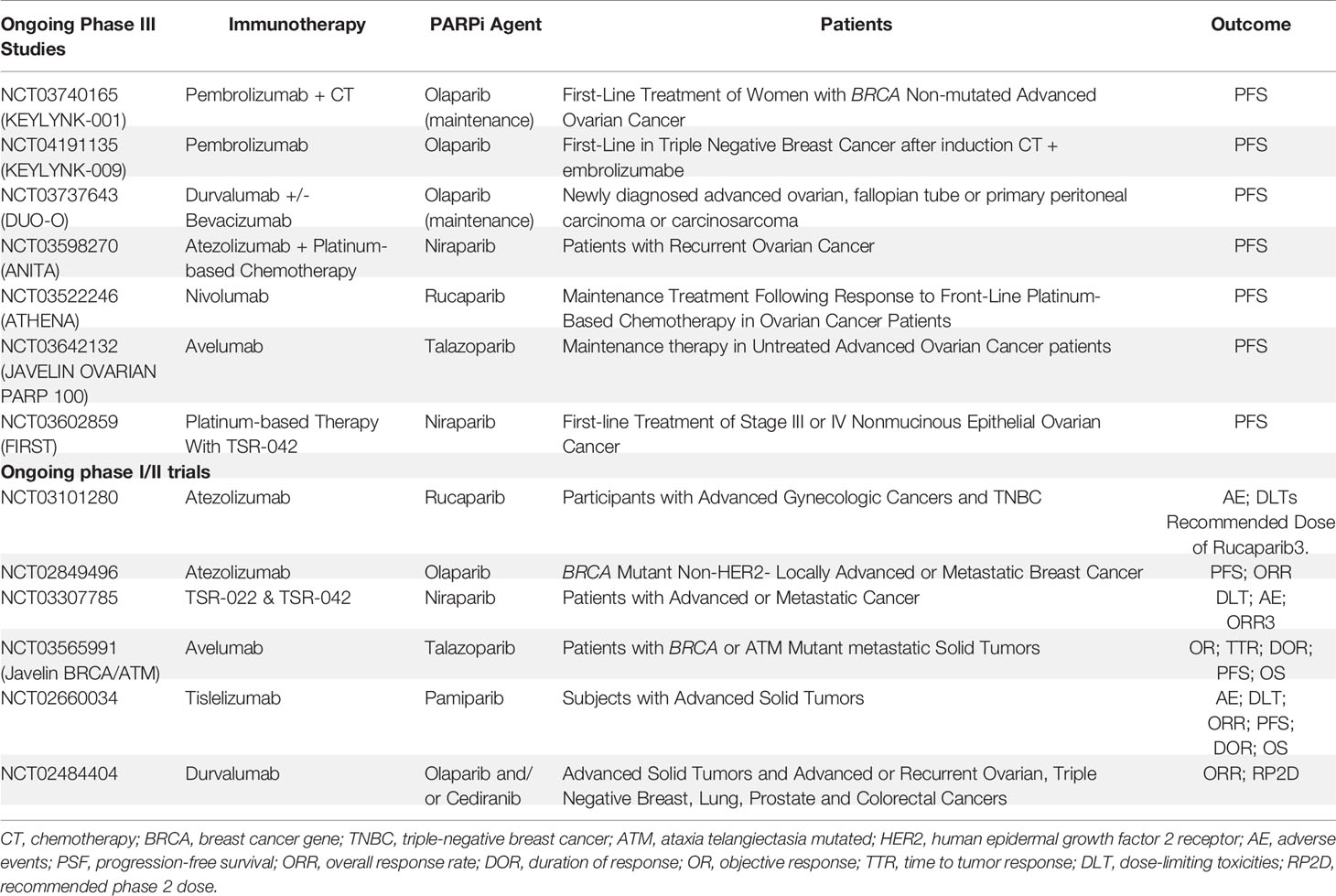
Table 2 Ongoing studies with a combination of immunotherapy and PARP inhibitors.
The association between PARPi and anti-CTLA-4 has been less studied. The combination of PARPi and CTLA-4 blockade is tolerable in heavily pretreated women with recurrent BRCA-associated ovarian cancer (62). Preliminary results of a phase I study combining olaparib and tremelimumab demonstrated evidence of therapeutic effects, supporting the ongoing evaluation of this regimen in phase II trials: NCT02571725 (71).
Targeting DNA Damage Signaling Proteins: Beyond PARP Inhibitors
The mechanisms that have inspired numerous PARPi-based combination therapies, including immunotherapy, also capitalize on the potential synergistic effects of different inhibitors of the DDR pathway, such as ataxia telangiectasia and Rad3-related (ATR), ataxia telangiectasia mutated (ATM), and Checkpoint kinase 1 (CHK1) inhibitors.
Preclinical data have demonstrated a synthetic lethal interaction between ATR and the ATM-p53 pathway in cells that respond to DNA damage. In a large proportion of cancer cells, where ATM-p53 signaling is defective, initiation of DNA replication continues and DNA damage accumulates, leading to cell death (72). It was demonstrated that combined treatment with ATR and CHK1 inhibitors leads to replication fork arrest, single-stranded DNA accumulation, replication collapse, and synergistic cell death in cancer cells in vitro and in vivo (73).
Strikingly, in addition to direct cytotoxic effects, ATM, ATR, and CHK1/2 inhibitors potentiate antitumor immunity. Inhibition of ATM/Chk2 led to replication stress and accumulation of cytosolic DNA, which subsequently activated the STING-mediated immune response (74). Vendetii et al. (2018) and Sheng et al. (2018), the ATR kinase inhibitor AZD6738 combined with radiation therapy boosted infiltration, increased cell proliferation, enhanced IFNγ production by CD8 T cells, and caused a decrease in the number of Tregs and exhausted T cells in the tumor in mouse models. Mechanistically, this study revealed that the antitumor effect of AZD6738 relied on the activation of the cGAS/STING pathway. These findings indicate that inhibitors of key DRR mechanisms, beyond PARP, promote the antitumor immune response through activation of the STING pathway (75, 76).
The proposed rational approach to enhance the efficacy of ICIs to utilize DRR inhibitors, to increase tumor DNA damage and thereby ‘prime’ tumors for response to immune ICIs have been explored in mouse models. The genic deletion of ATM induced IFN response and enhanced lymphocyte infiltration into the tumor microenvironment via cGAS/STING activation. This effect potentiated ICI therapy in mouse melanoma (B16) and breast cancer (4T1) tumors (77). In another study, tumor immunogenicity was evaluated after the pharmacological inhibition of ATM following PD-L1/PD-1 checkpoint inhibition. ATM inhibition increased the tumoral expression of type-I IFN in a TBK1- and SRC-dependent manner. Furthermore, ATM silencing increased PD-L1 expression, tumoral CD8 cells, and the sensitivity of pancreatic tumors to ICIs, suggesting that the efficacy of ICIs in pancreatic cancer can be enhanced by ATM inhibition (78). Similarly, ATM inhibition in tumors with a mutation in AT-rich interactive domain-containing protein 1A (ARID1A), a component of the chromatin-remodeling complex switch/sucrose-nonfermentable (SWI/SNF), selectively potentiates replication stress and accumulation of cytosolic DNA, which subsequently activates the DNA sensor STING-mediated innate immune response in ARID1A-deficient tumors. In patients, tumors with mutations or low expression of both ARID1A and ATM/CHK2 exhibit increased tumor-infiltrating lymphocytes and are associated with longer patient survival (74).
The combination of SRA737, an oral CHK1 inhibitor, with or without anti–PD-L1/anti-PD-1 leads to an antitumor response in multiple cancer models, including Small Cells Lung Cancer (SCLC). The combination of low-dose non-cytotoxic gemcitabine with SRA737 plus anti–PD-L1 increased the expression of type I IFN genes and chemokines (CCL5 and CXCL10), and the number of CD8, dendritic cells, and M1 macrophages in the tumor microenvironment. Using the PARPi (olaparib) or the CHK1 inhibitor (prexasertib) in combination with anti-PD-L1, a significant increase in cytotoxic T-cell infiltration inducing tumor regression was observed in the SCLC mouse model (79). Mechanistically, it was demonstrated that the treatment with DDR inhibitors activated the STING/TBK1/IRF3 pathway, leading to increased levels of chemokines (CXCL10 and CCL5), which recruited and activated CD8 T lymphocytes into the tumors (80).
In the clinics the ATR inhibitor ceralasertib has been tested in phase I in combination with chemotherapy, olaparib, or an anti-PD-L1 antibody. The durvalumab plus ceralasertib combination arm enrolled 25 patients with advanced head and neck squamous cell carcinoma or non-small cell lung cancer (NSCLC). The primary objective was to recommend a phase 2 dose of ceralasertib. Of the 21 patients evaluated, one complete response and three partial responses were observed, independent of tumor PD-L1 expression. They concluded that this combination is tolerated in dose escalation, with preliminary signals of antitumor activity in patients with advanced solid tumors (81). Berzosertib, another ATR inhibitor, has been tested in a phase IB/II study of combination chemotherapy and pembrolizumab in patients with advanced NSCLC with squamous cell histology; the estimated enrollment was 18 participants (NCT04216316).
Conclusion and Perspectives
The combination of PARPi and ICIs is promising and has been explored in various clinical trials. While most studies with this combination have focused on patients with ovarian or breast cancer harboring germline pathogenic variants in BRCA1/2 genes, other tumor histologies, including prostate cancer and pancreatic cancer, have been studied (82). Biomarkers trying to identify patients whose tumors have HR defects without germline BRCA mutations that could benefit from this combinatorial approach have also been explored. The results of ongoing phase III studies are awaited and can change the landscape of treatment for these patients.
Authors Contributions
RB-S conceived the work. CW, TC, FC, MS, and RB-S wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Instituto de Pesquisa e Ensino (IEP) - Hospital Sírio Libanês and Fundação de Amparo à Pesquisa do Estado de São Paulo - FAPESP (2017/25308-9).
Author Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation with the authors CW and FC at the time of review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
53BP1, p53-binding protein 1; AEs, Adverse events; APCs, Antigen present cells; ARID1A, AT-rich interactive domain-containing protein 1A; ATM, Ataxia telangiectasia mutated; ATR, Ataxia Telangiectasia Rad3-related; BC, Breast cancer; BRCA, Breast cancer gene; BRCA1, Breast cancer gene 1; BRCA2, Breast cancer gene 2; cGAS, Cyclic guanosine monophosphate–adenosine monophosphate synthase; CHK1, Checkpoint kinase1; CHK2, Checkpoint kinase2; CRs, Complete responses; CT, Chemotherapy; CTLA-4, Cytotoxic T-lymphocyte-associated protein 4; DCR, Disease control Rate; DDR, DNA damage response; DLT, Dose-limiting toxicities; dMMR, Defective mismatch repair; DNA-PKcs, DNA-dependent protein kinase; DOR, Duration of response; HER2, Human epidermal growth factor 2 receptor; HR, Homologous recombination; ICIs, Immune checkpoint inhibitors; IFNAR, Interferon-α/β receptor; IFNs, type I interferons; IKK, IkappaB kinase; IRF3, Interferon regulatory transcription factor 3; Ku70/80, Ku heterodimer protein; MRE11, Meiotic recombination 11; MSI, Microsatellite instability; MSS, Microsatellite stable; NAD+, Nicotinamide; NBS1, Nijmegen breakage syndrome 1; NF-κB, Factor nuclear kappa B; NHEJ, Non-homologous end-joining; OC, Ovarian cancer; OR, Objective Response; ORR, Overall response rate; OS, Overall survival; PARP, Poly (ADP-ribose) polymerases; PARP1, Poly (ADP-ribose) polymerase 1; PARPi, Poly-ADP-ribose polymerase inhibitors; PD, Progressive disease; PD-1, Receptor of programed death 1; PD-L1, Programmed death ligand-1; PFS, Progression-free survival; PR, Partial response; RAD50, RAD50 homolog; RPA, Replication protein A; SCLC, Small Cells Lung Cancer; SD, Stable disease; SOC, Serous ovarian cancers; STAT1, Signal transducer and activator of transcription 1; STING, Stimulator of interferon genes; SWI/SNF, Switch/Sucrose-Nonfermentable; TAAs, Tumor-associated antigens; TBK1, TANK-binding kinase 1; TCR, T-cell receptor; TFAM, Mitochondrial transcription factor A; Th1, T helper 1; TILs, Tumor-infiltrating lymphocytes; TTR, Time to tumor response; XLF, XRCC4-like factor; XRCC1, X-Ray Repair Cross Complementing 1; XRCC4, X-ray cross-complementing protein 4.
References
1. Robert C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat Commun (2020) 11(1):3801. doi: 10.1038/s41467-020-17670-y
2. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, et al. Safety and Activity of Anti-PD-L1 Antibody in Patients With Advanced Cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
3. Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med (2018) 378(21):1976–86. doi: 10.1056/NEJMoa1716078
4. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med (2018) 379(22):2108–21. doi: 10.1056/NEJMoa1809615
5. Wang S, Xie K, Liu T. Cancer Immunotherapies: From Efficacy to Resistance Mechanisms - Not Only Checkpoint Matters. Front Immunol (2021) 12:690112. doi: 10.3389/fimmu.2021.690112
6. Fay EK, Graff JN. Immunotherapy in Prostate Cancer. Cancers (Basel) (2020) 12(7):1752. doi: 10.3390/cancers12071752
7. Emens LA. Breast Cancer Immunotherapy: Facts and Hopes. Clin Cancer Res (2018) 24(3):511–20. doi: 10.1158/1078-0432.CCR-16-3001
8. Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol (2020) 8:564601. doi: 10.3389/fcell.2020.564601
9. Bernstein C, Prasad AR, Nfonsam V, Harris Bernstein H. "DNA Damage, DNA Repair and Cancer". IntechOpen (2013). doi: 10.5772/53919
10. Jackson SP, Bartek J. The DNA-Damage Response in Human Biology and Disease. Nature (2009) 461(7267):1071–8. doi: 10.1038/nature08467
11. Curtin NJ. DNA Repair Dysregulation From Cancer Driver to Therapeutic Target. Nat Rev Cancer (2012) 12(12):801–17. doi: 10.1038/nrc3399
12. Pilger D, Seymour LW, Jackson SP. Interfaces Between Cellular Responses to DNA Damage and Cancer Immunotherapy. Genes Dev (2021) 35(9-10):602–18. doi: 10.1101/gad.348314.121
13. Langelier MF, Eisemann T, Riccio AA, Pascal JM. PARP Family Enzymes: Regulation and Catalysis of the Poly(ADP-Ribose) Posttranslational Modification. Curr Opin Struct Biol (2018) 53:187–98. doi: 10.1016/j.sbi.2018.11.002
14. Pascal JM. The Comings and Goings of PARP-1 in Response to DNA Damage. DNA Repair (Amst) (2018) 71:177–82. doi: 10.1016/j.dnarep.2018.08.022
15. Eustermann S, Wu WF, Langelier MF, Yang JC, Easton LE, Riccio AA, et al. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol Cell (2015) 60(5):742–54. doi: 10.1016/j.molcel.2015.10.032
16. Ray-Chaudhuri A, Nussenzweig A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat Rev Mol Cell Biol (2017) 18(10):610–21. doi: 10.1038/nrm.2017.53
17. Liu L, Kong M, Gassman NR, Freudenthal BD, Prasad R, Zhen S, et al. PARP1 Changes From Three-Dimensional DNA Damage Searching to One-Dimensional Diffusion After Auto-PARylation or in the Presence of APE1. Nucleic Acids Res (2017) 45(22):12834–47. doi: 10.1093/nar/gkx1047
18. Chen A. PARP Inhibitors: Its Role in Treatment of Cancer. Chin J Cancer (2011) 30(7):463–71. doi: 10.5732/cjc.011.10111
19. Lieber MR. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu Rev Biochem (2010) 79:181–211. doi: 10.1146/annurev.biochem.052308.093131
20. Brandsma I, Gent DC. Pathway Choice in DNA Double Strand Break Repair: Observations of a Balancing Act. Genome Integr (2012) 3(1):9. doi: 10.1186/2041-9414-3-9
21. Choi EH, Yoon S, Koh YE, Seo YJ, Kim KP. Maintenance of Genome Integrity and Active Homologous Recombination in Embryonic Stem Cells. Exp Mol Med (2020) 52(8):1220–9. doi: 10.1038/s12276-020-0481-2
22. Lamarche BJ, Orazio NI, Weitzman MD. The MRN Complex in Double-Strand Break Repair and Telomere Maintenance. FEBS Lett (2010) 584(17):3682–95. doi: 10.1016/j.febslet.2010.07.029
23. Zhao W, Steinfeld JB, Liang F, Chen X, Maranon DG, Jian Ma C, et al. BRCA1-BARD1 Promotes RAD51-Mediated Homologous DNA Pairing. Nature (2017) 550(7676):360–5. doi: 10.1038/nature24060
24. Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous End Joining Drives Poly(ADP-Ribose) Polymerase (PARP) Inhibitor Lethality in Homologous Recombination-Deficient Cells. Proc Natl Acad Sci USA (2011) 108(8):3406–11. doi: 10.1073/pnas.1013715108
25. Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 Mutation-Associated and BRCA-Like Malignancies. Ann Oncol (2014) 25(1):32–40. doi: 10.1093/annonc/mdt384
26. Davis AJ, Chen DJ. DNA Double Strand Break Repair via Non-Homologous End-Joining. Transl Cancer Res (2013) 2(3):130–43. doi: 10.3978/j.issn.2218-676X.2013.04.02
27. Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, et al. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann Oncol (2019) 30(9):1437–47. doi: 10.1093/annonc/mdz192
28. Lord CJ, Ashworth A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science (2017) 355(6330):1152–8. doi: 10.1126/science.aam7344
29. Nijman SM. Synthetic Lethality: General Principles, Utility and Detection Using Genetic Screens in Human Cells. FEBS Lett (2011) 585(1):1–6. doi: 10.1016/j.febslet.2010.11.024
30. Purnell MR, Whish WJ. Novel Inhibitors of Poly(ADP-Ribose) Synthetase. Biochem J (1980) 185(3):775–7. doi: 10.1042/bj1850775
31. Canan Koch SS, Thoresen LH, Tikhe JG, Maegley KA, Almassy RJ, Li J, et al. Novel Tricyclic Poly(ADP-Ribose) Polymerase-1 Inhibitors With Potent Anticancer Chemopotentiating Activity: Design, Synthesis, and X-Ray Cocrystal Structure. J Med Chem (2002) 45(23):4961–74. doi: 10.1021/jm020259n
32. Evers B, Drost R, Schut E, de Bruin M, Derksen PW, et al. Selective Inhibition of BRCA2-Deficient Mammary Tumor Cell Growth by AZD2281 and Cisplatin. Clin Cancer Res (2008) 14(12):3916–25. doi: 10.1158/1078-0432.CCR-07-4953
33. Nakad R, Schumacher B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front Genet (2016) 7:147. doi: 10.3389/fgene.2016.00147
34. Mboko WP, Mounce BC, Wood BM, Kulinski JM, Corbett JA, Tarakanova VL. Coordinate Regulation of DNA Damage and Type I Interferon Responses Imposes an Antiviral State That Attenuates Mouse Gammaherpesvirus Type 68 Replication in Primary Macrophages. J Virol (2012) 86(12):6899–912. doi: 10.1128/JVI.07119-11
35. Chabanon RM, Rouanne M, Lord CJ, Soria JC, Pasero P, Postel-Vinay S. Targeting the DNA Damage Response in Immuno-Oncology: Developments and Opportunities. Nat Rev Cancer (2021) 21(11):701–17. doi: 10.1038/s41568-021-00386-6
36. Gao M, He Y, Tang H, Chen X, Liu S, Tao Y.. cGAS/STING: Novel Perspectives of the Classic Pathway. Mol BioMed (2020) 1:7. doi: 10.1186/s43556-020-00006-z
37. Hoong BYD, Gan YH, Liu H, Chen ES. cGAS-STING Pathway in Oncogenesis and Cancer Therapeutics. Oncotarget (2020) 11(30):2930–55. doi: 10.18632/oncotarget.27673
38. Pu F, Chen F, Liu J, Zhang Z, Shao Z. Immune Regulation of the cGAS-STING Signaling Pathway in the Tumor Microenvironment and Its Clinical Application. Onco Targets Ther (2021) 14:1501–16. doi: 10.2147/OTT.S298958
39. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP Inhibitor Efficacy Depends on CD8+ T-Cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov (2019) 9(6):722–37. doi: 10.1158/2159-8290.CD-18-1218
40. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G.. Type I Interferons in Anticancer Immunity. Nat Rev Immunol (2015) 15:405–14. doi: 10.1038/nri3845
41. Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an Interferon Gamma-Dependent Tumor Surveillance System in Immunocompetent Mice. Proc Natl Acad Sci USA (1998) 95(13):7556–61. doi: 10.1073/pnas.95.13.7556
42. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host Type I IFN Signals are Required for Antitumor CD8+ T Cell Responses Through CD8{alpha}+ Dendritic Cells. J Exp Med (2011) 208(10):2005–16. doi: 10.1084/jem.20101159
43. Lim JY, Gerber SA, Murphy SP, Lord EM. Type I Interferons Induced by Radiation Therapy Mediate Recruitment and Effector Function of CD8(+) T Cells. Cancer Immunol Immunother (2014) 63(3):259–71. doi: 10.1007/s00262-013-1506-7
44. Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 Blockade Enhances T-Cell Migration to Tumors by Elevating IFN-γ Inducible Chemokines. Cancer Res (2012) 72(20):5209–18. doi: 10.1158/0008-5472.CAN-12-1187
45. Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front Immunol (2018) 9:14. doi: 10.3389/fimmu.2018.00014
46. Karimi K, Karimi Y, Chan J, Boudreau JE, Basset J, Chew MV, et al. Type I IFN Signaling on Dendritic Cells Is Required for NK Cell-Mediated Anti-Tumor Immunity. Innate Immun (2015) 21(6):626–34. doi: 10.1177/1753425915575078
47. Müller E, Speth M, Christopoulos PF, Lunde A, Avdagic A, Øynebråten I, et al. Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined With TLR Stimulation. Front Immunol (2018) 9:2520. doi: 10.3389/fimmu.2018.02520
48. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep (2017) 19(6):1189–201. doi: 10.1016/j.celrep.2017.04.031
49. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res (2017) 23(14):3711–20. doi: 10.1158/1078-0432.CCR-16-3215
50. Torgovnick A, Schumacher B. DNA Repair Mechanisms in Cancer Development and Therapy. Front Genet (2015) 6:157. doi: 10.3389/fgene.2015.00157
51. Aguadé-Gorgorió G, Solé R. Genetic Instability as a Driver for Immune Surveillance. J Immunother Cancer (2019) 7(1):345. doi: 10.1186/s40425-019-0795-6
52. Yarchoan M, Lutz ER, Laheru DA, Jaffee EM. Targeting Neoantigens to Augment Antitumour Immunity. Nat Rev Cancer (2017) 17(4):209–22. doi: 10.1038/nrc.2016.154
53. Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, Russo M, et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature (2017) 552(7683):116–20. doi: 10.1038/nature24673
54. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin Cancer Res (2019) 25(13):3753–8. doi: 10.1158/1078-0432.CCR-18-4070
55. Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell (2010) 141(2):243–54. doi: 10.1016/j.cell.2010.03.012
56. Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5
57. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
58. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front Immunol (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
59. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
60. Vukadin S, Khaznadar F, Kizivat T, Vcev A, Smolic M. Molecular Mechanisms of Resistance to Immune Checkpoint Inhibitors in Melanoma Treatment: An Update. Biomedicines (2021) 9(7):835. doi: 10.3390/biomedicines9070835
61. Ghonim MA, Ibba SV, Tarhuni AF, Errami Y, Luu HH, Dean MJ, et al. Targeting PARP-1 With Metronomic Therapy Modulates MDSC Suppressive Function and Enhances Anti-PD-1 Immunotherapy in Colon Cancer. J Immunother Cancer (2021) 9(1):e001643. doi: 10.1136/jitc-2020-001643
62. Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res (2019) 79(2):311–9. doi: 10.1158/0008-5472.CAN-18-1003
63. Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, et al. Niraparib Activates Interferon Signaling and Potentiates Anti-PD-1 Antibody Efficacy in Tumor Models. Sci Rep (2019) 9(1):1853. doi: 10.1038/s41598-019-38534-6
64. Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in Combination With Tislelizumab in Patients With Advanced Solid Tumours: Results From the Dose-Escalation Stage of a Multicentre, Open-Label, Phase 1a/B Trial. Lancet Oncol (2019) 20(9):1306–15. doi: 10.1016/S1470-2045(19)30396-1
65. Lee JM, Cimino-Mathews A, Peer CJ, Zimmer A, Lipkowitz S, Annunziata CM, et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination With Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1-3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. J Clin Oncol (2017) 35(19):2193–202. doi: 10.1200/JCO.2016.72.1340
66. Drew Y, de Jonge M, Hong SH, Park YH, Wolfer A, Brown J, et al. An Open-Label, Phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): Results in Germline BRCA-Mutated (gBRCAm) Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Gynecol Oncol (2018) 149:246–7. doi: 10.1016/j.ygyno.2018.04.555
67. Domchek S, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Phase II Study of Olaparib (O) and Durvalumab (D) (MEDIOLA): Updated Results in Patients (Pts) With Germline BRCA-Mutated (gBRCAm) Metastatic Breast Cancer (MBC). Ann Oncol (2019) 30:v477. doi: 10.1093/annonc/mdz253.017
68. Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol (2019) 5:1141. doi: 10.1001/jamaoncol.2019.1048
69. Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-Label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMAOncol (2019) 5:1132. doi: 10.1001/jamaoncol.2019.1029
70. Yap TA, Konstantinopoulos P, Telli ML, Saraykar S, Beck JT, Galsky MD, et al. Abstract P1-19-03: JAVELIN PARP Medley, a Phase 1b/2 Study of Avelumab Plus Talazoparib: Results From Advanced Breast Cancer Cohorts. Cancer Res (2020) 80:P1‐P1‐19–03. doi: 10.1158/1538-7445.SABCS19-P1-19-03
71. Adams SF, Rixe O, Lee JH, McCance DJ, Westgate S, Eberhardt SC, et al. Phase I Study Combining Olaparib and Tremelimumab for the Treatment of Women With BRCA-Deficient Recurrent Ovarian Cancer. JCO (2017) 35:e17052. doi: 10.1200/JCO.2017.35.15_suppl.e17052
72. Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective Killing of ATM- or P53-Deficient Cancer Cells Through Inhibition of ATR. Nat Chem Biol (2011) 7(7):428–30. doi: 10.1038/nchembio.573
73. Sanjiv K, Hagenkort A, Calderón-Montaño JM, Koolmeister T, Reaper P, Mortusewicz O, et al. Cancer-Specific Synthetic Lethality Between ATR and CHK1 Kinase Activities. Cell Rep (2016) 14(2):298–309. doi: 10.1016/j.celrep.2015.12.032
74. Wang L, Yang L, Wang C, Zhao W, Ju Z, Zhang W, et al. Inhibition of the ATM/Chk2 Axis Promotes cGAS/STING Signaling in ARID1A-Deficient Tumors. J Clin Invest (2020) 130(11):5951–66. doi: 10.1172/JCI130445
75. Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, et al. ATR Kinase Inhibitor AZD6738 Potentiates CD8+ T Cell-Dependent Antitumor Activity Following Radiation. J Clin Invest (2018) 128(9):3926–40. doi: 10.1172/JCI96519
76. Sheng H, Huang Y, Xiao Y, Zhu Z, Shen M, Zhou P, et al. ATR Inhibitor AZD6738 Enhances the Antitumor Activity of Radiotherapy and Immune Checkpoint Inhibitors by Potentiating the Tumor Immune Microenvironment in Hepatocellular Carcinoma. J immunotherapy Cancer (2020) 8(1):e000340. doi: 10.1136/jitc-2019-000340
77. Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, et al. ATM Inhibition Enhances Cancer Immunotherapy by Promoting mtDNA Leakage and cGAS/STING Activation. J Clin Invest (2021) 131(3):e139333. doi: 10.1172/JCI139333
78. Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res (2019) 79(15):3940–51. doi: 10.1158/0008-5472.CAN-19-0761
79. Sen T, Della Corte CM, Milutinovic S, Cardnell RJ, Diao L, Ramkumar K, et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J Thorac Oncology: Off Publ Int Assoc Study Lung Cancer (2019) 14(12):2152–63. doi: 10.1016/j.jtho.2019.08.009
80. Sen T, Rodriguez BL, Chen L, Corte MD, et al. Targeting DNA Damage Response Promotes Antitumor Immunity Through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov (2019) 9(5):646–61. doi: 10.1158/2159-8290.CD-18-1020
81. Krebs MG, Lopez J, El-Khoueiry A, Bang Y-J, postel-Vinay S, Abida W, et al. Phase I Study of AZD6738, an Inhibitor of Ataxia Telangiectasia Rad3-Related (ATR), in Combination With Olaparib or Durvalumab in Patients (Pts) With Advanced Solid Cancers. Cancer Res (2018) 78(13 Suppl):CT026. In : Proceedings of the American Association for Cancer Research Annual Meeting 2018; 2018 Apr 14–18; Chicago, IL. Philadelphia (PA): AACR. doi: 10.1158/1538-7445.AM2018-CT026
Keywords: cancer, immunotherapy, DNA damage, immune response, PARP (poly(ADP-ribose), polymerase
Citation: Wanderley CWS, Correa TS, Scaranti M, Cunha FQ and Barroso-Sousa R (2022) Targeting PARP1 to Enhance Anticancer Checkpoint Immunotherapy Response: Rationale and Clinical Implications. Front. Immunol. 13:816642. doi: 10.3389/fimmu.2022.816642
Received: 16 November 2021; Accepted: 25 March 2022;
Published: 27 April 2022.
Edited by:
Niels Olsen Saraiva Camara, University of São Paulo, BrazilReviewed by:
Alice Chen, Independent Researcher, Bethesda, MD, United StatesNahum Puebla-Osorio, University of Texas MD Anderson Cancer Center, United States
Copyright © 2022 Wanderley, Correa, Scaranti, Cunha and Barroso-Sousa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Romualdo Barroso-Sousa, cm9tdWFsZG8uYmFycm9zb0Boc2wub3JnLmJy