 Tianhao Duan
Tianhao Duan Yang Du
Yang Du Changsheng Xing1
Changsheng Xing1 Helen Y. Wang
Helen Y. Wang Rong-Fu Wang
Rong-Fu Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 March 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.812774
This article is part of the Research TopicStructural Immunology of Molecular Innate ImmunityView all 14 articles
Innate immunity is the first defense system against invading pathogens. Toll-like receptors (TLRs) are well-defined pattern recognition receptors responsible for pathogen recognition and induction of innate immune responses. Since their discovery, TLRs have revolutionized the field of immunology by filling the gap between the initial recognition of pathogens by innate immune cells and the activation of the adaptive immune response. TLRs critically link innate immunity to adaptive immunity by regulating the activation of antigen-presenting cells and key cytokines. Furthermore, recent studies also have shown that TLR signaling can directly regulate the T cell activation, growth, differentiation, development, and function under diverse physiological conditions. This review provides an overview of TLR signaling pathways and their regulators and discusses how TLR signaling, directly and indirectly, regulates cell-mediated immunity. In addition, we also discuss how TLR signaling is critically important in the host’s defense against infectious diseases, autoimmune diseases, and cancer.
The innate immune system is the first line of defense against infectious pathogens and cancer by sensing and responding to the structure-conserved molecules of the pathogens (pathogen-associated molecular patterns, or PAMPs) as well as the endogenous ligands released from damaged cells (damage-associated molecular patterns, or DAMPs). The pattern recognition receptors (PRRs) are a key element of the immune system, including Toll-like receptors (TLRs), RIG-I-like receptors, Nod-like receptors (NLRs), AIM2-like receptors, C-type lectin receptors, and intracellular DNA and RNA sensors (1–3). Upon the recognition of their specific ligands from the invasive pathogens or damaged cells, PRRs initiate a variety of downstream signaling cascades, including nuclear factor kappa B (NF-κB), type I interferon (IFN) and inflammasome signaling pathways, leading to the production of corresponding proinflammatory or antiviral cytokines and chemokines (2, 4). The activation of TLR signaling is also crucial to the induction of antigen-specific adaptive immune responses by promoting the maturation of dendritic cells (DCs) and activating the adaptive immune cells for the clearance of invading pathogens (4–6).
TLRs belong to the family of Type I integral membrane glycoproteins characterized by the extracellular domains containing variable numbers of leucine-rich-repeat (LRR) motifs and a cytoplasmic Toll/interleukin 1 (IL-1) receptor (TIR) homology domain (7). Toll was identified initially as a gene controlling dorsoventral axis formation of the Drosophila embryo in the 1980s (8), and its crucial anti-fungal function in Drosophila was demonstrated in 1996 (9). The mammalian homolog of the Toll receptor (now termed TLR4) was first discovered in 1997 to play a critical role the in innate immunity by inducing the expression of inflammatory responses-related genes (10). These findings revolutionized our understanding of the immune system and triggered an explosion of research in PRRs. To date, 10 TLRs have been identified in humans (TLR1–TLR10) and 12 in mice (TLR1–TLR9 and TLR11–TLR13). TLR1–TLR10 are conserved between mice and humans, although mouse TLR10 is not functional, while TLR11–TLR13 are expressed only in mice but not in humans. These receptors are localized on the cell surface (TLR1, TLR2, TLR4, and TLR5) or in intracellular compartments, such as the endoplasmic reticulum, endosome, lysosome, or endolysosome (TLR3, TLR7, TLR8, and TLR9) (6).
Cell surface TLRs mainly recognize membrane components of the microorganisms such as lipids, lipoproteins, and proteins (2). For example, TLR4 recognizes lipopolysaccharide (LPS). TLR2 forms a heterodimer with either TLR1 or TLR6 and recognizes different PAMPs of pathogens (including lipoproteins, peptidoglycans, lipoteichoic acids, zymosan, mannan, and glycosylphosphatidylinositol-anchored mucin-like glycoproteins from Trypanosoma cruzi trypomastigotes) (11). TLR5 recognizes the flagellin of bacteria (2). Human TLR10 can homodimerize or heterodimerize with TLR1, TLR2, and TLR6 (12), and sense HIV proteins (13). Intracellular TLRs mainly recognize nucleic acids derived from pathogens or self-nucleic acids in a disease condition. TLR3 recognizes double-stranded viral RNA and self RNAs derived from damaged cells; TLR7, TLR8, and TLR13 recognize fragments of single-stranded RNA with distinct sequence preferences, and TLR7 is predominantly expressed in plasmacytoid dendritic cells (pDCs). In addition, TLR9 recognizes single-stranded DNA containing unmethylated cytidine-phosphateguanosine (CpG) motifs from bacteria or viruses (6, 14). TLR10 was recently identified to sense HIV-1 gp41 protein but its biological functions in humans haven't been fully elucidated (12, 13).
Each TLR contains a similar cytoplasmic portion known as the TIR domain, which is highly similar to that of the IL-1 receptor family. The extracellular portion of TLRs is the ectodomain, with LRRs displayed as a horseshoe-like structure. The characteristic feature of these LRRs is the consensus sequence motif—L(X2)LXL(X2)NXL(X2)L(X7)L(X2)—in which X can be any amino acid (15). The ectodomain of TLRs forms a homo- or hetero-dimer along with a co-receptor or accessory molecule to interact with their respective PAMPs or DAMPs (16). TLRs are expressed on all the innate immune cells and a large majority of non-hematopoietic cells, such as macrophages, neutrophils, DCs, natural killer cells, mast cells, basophils, eosinophil, and epithelial cells (4, 17). Importantly, TLRs can also be detected on adaptive immune cells, including T and B cells (18, 19).
Adaptive immunity consists of humoral immunity and cell-mediated immunity, which are mainly mediated by B lymphocytes and T lymphocytes, respectively. Cell-mediated immunity (also called cellular immunity) is responsible for generating a cluster of differentiation 8 (CD8)+ cytotoxic T-lymphocytes (CTLs) and an antigen-specific cluster of differentiation 4 (CD4)+ T helper (Th) cells, which help B cells produce antibodies. CTLs recognize and produce molecules that directly kill infected host cells. In contrast, Th cells release various cytokines that influence the function of other cells involved in both adaptive and innate immune responses (20). To induce efficient activation and clonal expansion of antigen-specific T cells, antigen presentation and co-stimulatory signaling are essential, which must be simultaneously provided by the antigen-presenting cells (APCs) to T cells. Importantly, the production of cytokines, expression of costimulatory molecules, and antigen-presenting activity in APCs are induced or enhanced by microbe-derived adjuvants, which are recognized by TLRs expressed on APCs and boost the APC signaling to promote activation of immune responses in T cells (21). Therefore, TLRs play a critical role in linking the innate immunity and cell-mediated immunity. This review article mainly summarizes the recent progress on TLR signaling pathways and their crucial role in cell-mediated immunity.
Innate immunity was formerly thought to be a nonspecific immune response. However, the discovery of TLRs led to the realization of the considerable specificity of innate immunity and its capability to discriminate between self and nonself (22–24). Cell surface TLRs (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11) mainly recognize microbial membrane components to induce an inflammatory response (11). By contrast, intracellular TLRs (TLR3, TLR7, TLR8, and TLR9) mainly recognize microbial nucleic acids derived from bacteria or viruses and induce Type I IFN responses and inflammatory responses. However, the misrecognition of self-nucleic acids may cause autoimmune diseases (25).
Upon binding by specific ligands, ligand-mediated dimerization of TLR ectodomains results in the coordinate dimerization of the cytosolic TIR domains of each TLR (26). Dimerized receptor TIR domains are detected by two receptor-proximal membrane adaptor proteins: the TIR domain-containing adapter protein (TIRAP; also known as MAL) (27, 28) and the TIRAP-inducing IFN-β (TRIF)-related adaptor molecule (TRAM) (29, 30). These peripheral membrane proteins survey the inner leaflets of the plasma and endosomal membranes through the actions of an N-terminal phosphoinositide binding domain of TIRAP or a bipartite localization domain of TRAM consisting of an N-terminal myristoylation motif and a phosphoinositide-binding motif (31–33).
TIRAP and TRAM can further recruit myeloid differentiation primary response protein 88 (MyD88) and TRIF, respectively (34), and stimulate the assembly of a large oligomeric scaffold called Myddosome or Triffosome (35). These supramolecular complexes consist of downstream signaling components and kinase enzymes. Increased local concentrations of signaling molecules promote the intrinsically weak allosteric interactions and initiate cytosolic signaling transduction (36). Depending on the distinct supramolecular complexes formed, TLR signaling pathways can be mainly classified as either MyD88-dependent pathways, which drive the induction of inflammatory cytokines, or TRIF-dependent pathways, which are responsible for the induction of Type I IFN as well as inflammatory cytokines (2) (Figure 1).
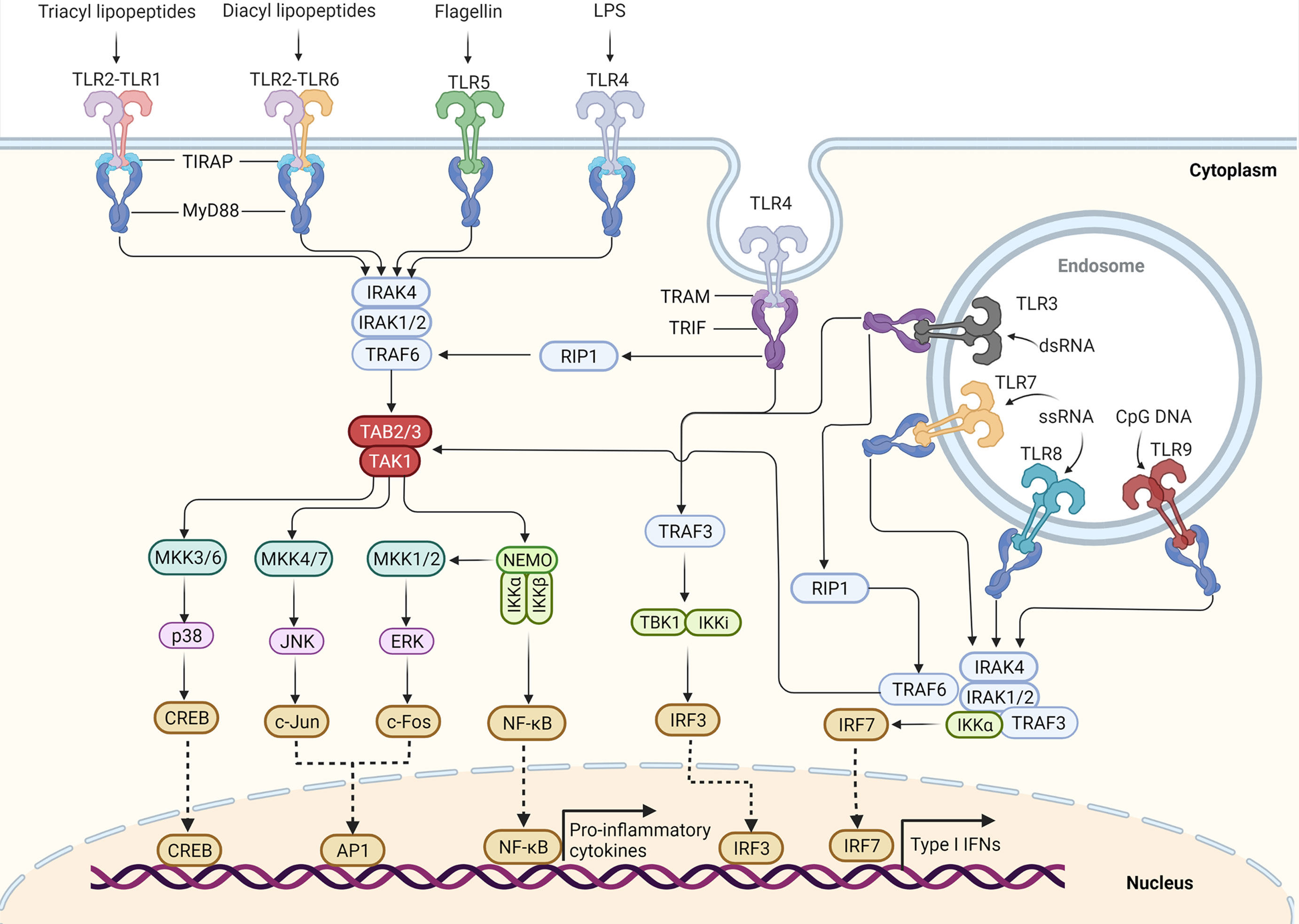
Figure 1 TLR signaling pathway in innate immune cells. TLR5, TLR4, and the heterodimers of TLR2–TLR1 or TLR2–TLR6 prefer to recognize the membrane components of pathogens at the cell surface, whereas TLR3, TLR7–TLR8, and TLR9 localize to the endosomes, where they recognize the nucleic acids from both the host and foreign microorganisms. TLR4 localizes at the plasma membrane, but it is endocytosed into endosomes upon activation. Upon binding to their respective ligands, TLR signaling is initiated by dimerization of receptors, leading to the engagement of TIR domains of TLRs with TIRAP and MyD88 (or directly interact with MyD88) or with TRAM and TRIF (or directly interact with TRIF). The TLR4 signaling switches from MyD88 to TRIF once TLR4 moves to the endosomes. Engagement of MyD88 recruits the downstream signaling molecules to form Myddosome, which is based on MyD88 and contains IRAK4 and IRAK1/2. IRAK1 further activates the E3 ubiquitin ligase-TRAF6 to synthesize the K63-linked polyubiquitin chains, leading to the recruitment and activation of the TAK1 complex. The activated TAK1 further phosphorylates and activates the canonical IKK complex, ultimately leading to the activation factor NF-κB. The activation of TAK1 also leads to the activation of MAPKs, including MKK4/7 and MKK3/6, which further activate JNK and p38, respectively. The activation of IKKβ also leads to the activation of MKK1 and MKK2, which further activate ERK1/2. The activation of these MAPKs leads to some important transcription factor activations, such as CREB, AP1. These transcription factors cooperate with NF-κB to promote the induction of pro-inflammatory cytokines. Engagement of TRIF recruits the TRAF6 and TRAF3. Activated TRAF6 can recruit the kinase RIP1 and activate the TAK1 complex and IKK complex, leading to the activation of NF-κB and MAPKs. TRIF also promotes the TRAF3-dependent activation of the TBK1 and IKKϵ (originally IKKi), which further phosphorylates and activates IRF3. Among TLR7, TLR8, and TLR9 signaling in pDCs, IRF7 can bind to the Myddosome and is directly activated by IRAK1 and IKKϵ. Activation of IRF3 and IRF7 leads to the induction of Type I IFN.
MyD88 is the first identified member of the TIR family; it is commonly used by all the TLRs except TLR3, and it activates the NF-κB signaling pathway (11). Upon activation by specific ligands, MyD88 recruits IL-1 receptor-associated kinases (IRAK)—IRAK4, IRAK1, IRAK2, and IRAK-M—which form a complex with IRAK kinase family members, referred to as the Myddosome (37–39). During Myddosome formation, IRAK4 is activated initially by MyD88 through its N-terminal death domain, which is also contained in IRAK4. Similar to MyD88, IRAK4 is also essential for the activation of NF-κB and mitogen-activated protein kinases (MAPKs) in the MyD88-dependent pathway (40, 41). The activated IRAK4 can sequentially activate IRAK1 and IRAK2, which are then autophosphorylated at several sites (42). Although activation of both kinases is required for robust activation of TLR-induced NF-κB and MAPK signaling, the relative importance of IRAK1 and IRAK2 may differ in humans and mice (43).
Activated IRAK1 can interact with tumor necrosis factor (TNF) receptor-associated factors 6 (TRAF6), an E3 ligase that catalyzes the synthesis of Lys63 (K63)-linked polyubiquitin, resulting in activation of TRAF6. TRAF6, along with E2 ubiquitin-conjugating enzymes Ubc13 and Uev1A, generates the K63-linked polyubiquitin chains and promotes K63-linked polyubiquitination of both TRAF6 itself and IRAK1. Early studies suggested that K63-linked polyubiquitination of TRAF6 and IRAK1 might serve as a platform for activation of downstream TGFβ-activated protein kinase 1 (TAK1) or IκB kinase (IKK) (44–48). However, the direct biochemical evidence is missing and the conflicting results have been reported that ubiquitination of TRAF6 may be dispensable for the downstream protein kinase activation (49). Therefore, whether the K63-linked polyubiquitination of TRAF6 and IRAK1 can directly activate downstream protein kinases or it just serves as a marker of signaling pathway activation still requires further investigations. Recent biochemical studies revealed that the free K63 polyubiquitin chains synthesized by TRAF6 and Ubc13/Uev1A, which are not conjugated to any cellular protein, could directly activate TAK1 in vitro by binding to the novel zinc finger-type ubiquitin-binding domain of TAB2 and TAB3 (50), leading to close proximity-dependent transphosphorylation of TAK1 at Thr-187 (50, 51). However, whether and how these free polyubiquitin chains activate downstream protein kinases in vivo remains to be determined. Phosphorylated TAK1 then activates the IKK complex-NF-κB pathway and -MAPK pathway (6).
The IKK complex is comprised of the catalytic subunits IKKα and IKKβ and the regulatory subunit nuclear factor-κB essential modulator (NEMO) (also called IKKγ) (44). K63 polyubiquitin chains might bridge TAK1 to form a complex with IKK, thus allowing TAK1 to phosphorylate IKKβ through its close proximity to the IKK complex, which leads to activation of the IKK complex (52–54). Recently, Met1-linked ubiquitin dimers (also known as linear ubiquitin dimers) were shown to bind with 100-fold higher affinity to NEMO compared with K63-linked ubiquitin (55, 56), indicating that linear ubiquitination, catalyzed by the linear ubiquitin chain assembly complex (LUBAC), also contributes to the activation of IKK (57–63). The activated IKK complex can further phosphorylate the NF-κB inhibitory protein IκBα, which undergoes proteasome degradation, allowing NF-κB to translocate into the nucleus to induce proinflammatory gene expression (6).
In the MAPK pathway, the activated TAK1 simultaneously activates the MAPK family members Jun N-terminal kinases (JNKs) and p38 by inducing the phosphorylation of MAPK kinases 4/7 (MKK4/7) and MKK3/6. The IKKβ also catalyzes the phosphorylation of p105 to cause its degradation by the Skp1-Cul1-F-box ubiquitin ligase (SCFβTrCP) complex, producing p50 and releasing tumor progression locus 2 (TPL2) to activate MKK1/2, which further phosphorylates and activates extracellular signal−regulated protein kinase 1 (ERK1) and ERK2. These MAPKs then phosphorylate cyclic AMP-responsive element-binding protein (CREB) and activator protein 1 (AP-1) transcription factors consisted of a heterodimer of c-Fos and c-Jun subunits to regulate inflammatory responses (44). TAK1 is a central component of MyD88-dependent NF-κB and MAPK signaling pathways. An earlier study suggested that TAK1 is required for the activation of the NF-κB and MAPK signaling pathway in both mouse embryonic fibroblast cells, B cells and T cells (64–68). However, we found that TAK1 serves as a negative regulator in mouse neutrophils (69, 70). By contrast, TAK1 might serve as a positive regulator in human neutrophils (71), suggesting a cell type-specific role for TAK1 in TLR-induced signaling (72). Interestingly, we recently found that Tak1 deficiency in mice alters the intestinal microbiome, which can drive protective immunity against colitis and colorectal cancer (73).
Among TLR7, TLR8, and TLR9 signaling in pDCs, MyD88 also activates NF-κB signaling and interacts with interferon regulatory factor (IRF)-5 and IRF-7 for the induction of proinflammatory cytokines or Type I IFN (IFN-α and IFN-β) responses (74–76). IRF7 is highly expressed by pDCs, which can bind to the Myddosome containing IRAK4, TRAF6, TRAF3, IRAK1, and IKKα (77). IRAK1 and IKKα further phosphorylate the IRF7 protein, leading to its dissociation from the Myddosome and dimerization. The IRF7 homodimer translocates into the nucleus and drives IFNα expression (11). By contract, IRF5 is phosphorylated by IKKβ on Ser462 and contributes to proinflammatory cytokine transcription but not IFNα production (76, 78–80).
In macrophages and conventional DCs (cDCs), TLR3- or TLR4-induced IFN expression is not dependent on MyD88 but instead is driven by TRIF as well as the proteins TRAM and TRAF3 (29, 30, 81, 82). Upon detection of dimerized TLR4 in endosomes, TRAM is thought to interact with TRIF to induce the formation of the putative Triffosome (35), in which TRIF interacts with TRAF6 and TRAF3. Activated TRAF6 can recruit the kinase receptor-interacting protein 1 (RIP1), which in turn recruits and activates the TAK1 complex and IKK complex, leading to activation of NF-κB and MAPKs and the induction of inflammatory cytokines (6). An earlier study suggested that TRAF6 might mediate RIP1 ubiquitination (83). However, TRAF6 has also been reported to be dispensable for TRIF-dependent TLR signaling (84), suggesting that additional E3(s) might be responsible for RIP1 ubiquitination. Recently, an E3 ubiquitin ligase Peli1 was found to facilitate TRIF-dependent TLR signaling and proinflammatory cytokine production by inducing the ubiquitination of RIP1 (85), indicating that Peli1 might share a redundant role with TRAF6.
TRIF also promotes the TRAF3-dependent activation of the IKK-related kinase TANK-binding kinase 1 (TBK1). TRAF3 activates the TBK1 and inhibitor of NF-κB kinase (IKKi) along with NEMO for phosphorylation and dimerization of the IFN-inducing transcription factor IFN regulatory factor 3 (IRF3). Subsequently, the IRF3 homodimer translocates into the nucleus from the cytoplasm, where it drives the expression of Type I IFN genes and IFN-stimulated genes (ISGs) (86–88). Recently, a 39-amino-acid pLxIS motif was identified within TRIF (but not MyD88), which can be phosphorylated by TBK1. The phosphorylated motif can recruit IRF3, leading to the phosphorylation and activation of IRF3 by TBK1 (89, 90). Therefore, the TLR4 uses TRIF but not MyD88 to promote IRF3-induced IFN expression in the endosome. Unlike TLR4, TRAM cannot interact with TLR3 or regulate TLR3 signaling (29), indicating that TLR3 might directly interact with TRIF or use another sorting adaptor to link TRIF to TLR3.
TLR-mediated activation and maturation of DCs and macrophages are critical links between innate and adaptive immunity (21). DCs are professional APCs and play a central role in inducing the activation and differentiation of naïve T cells into Th type 1 (Th1) cells, Th2 cells, and CTL effectors (91). Once DCs take up the antigen, the activated DCs can migrate to local lymphoid tissues to present the antigenic peptides on the relevant major histocompatibility complex (MHC) molecules (4). This process is regulated by recognizing pathogens via the variety of PRRs expressed by DCs. Among these PRRs, TLR family members play a critical role in generating effector T cell responses (4, 92, 93). The production of “innate” cytokines (type I IFN, IL-1, IL-6, IL-12, TNF-α), up-regulation of costimulatory molecules (CD40, CD80, and CD86), and altered expression of chemokine receptors (CCR2, CCR5, and CCR7) are the characteristics of DC maturation (21), which can be induced by ligands of TLRs, including LPS, lipoproteins, and CpG DNA (4, 75, 81, 94–96) (Figure 2).
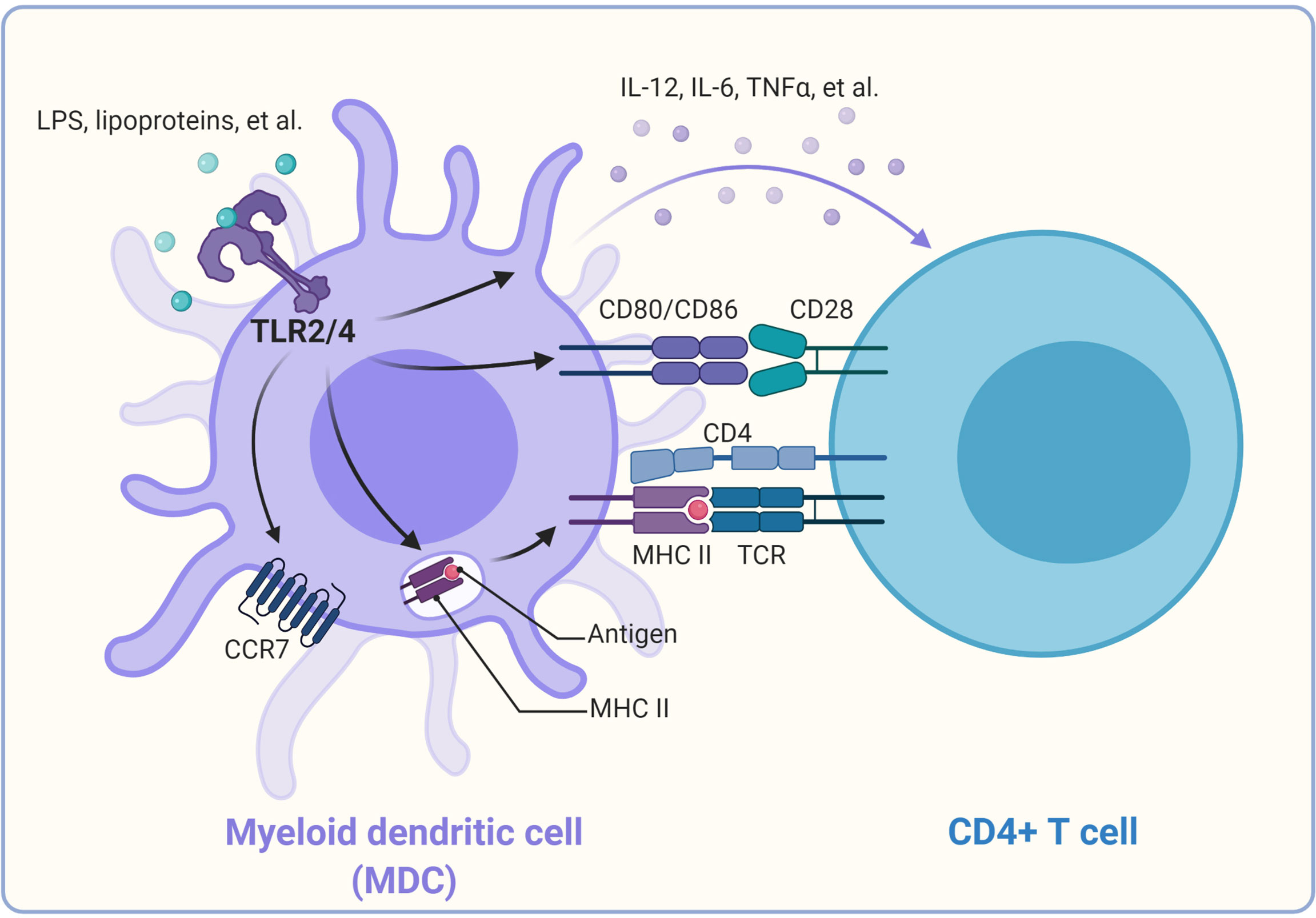
Figure 2 Promotion of CD4+ T cell activation by TLRs on dendritic cells. Once TLR2/4 recognize their individual ligands, they can alter the expression of chemokine receptors (CCR2, CCR5, and CCR7), leading to DC migration from the infected tissue to the draining lymph node, where naïve T cells are stimulated. TLR2/4 signaling can promote the antigen process and bind to the major histocompatibility complex II and be presented to the CD4+ T cells, thus providing the first signal for activation of the CD4+ T cells. In addition, TLR signaling triggers the up-regulation of costimulatory molecules on the cell surface of DCs, which provide the second signal to activate the antigen-specific CD4+ T cells. TLR signaling can also induce the production of cytokines such as IL-12, TNF-α in DCs. These cytokines function as “instructive” cytokines and drive the activation and differentiation of CD4+ T cells.
Moreover, TLR signals can also facilitate peptide loading onto MHC molecules or the cross-presentation of exogenous antigens for the stimulation of CD8+ T cell responses by promoting the acidification of endosomes or the fusion of MHC Class I-containing endosomes with phagosomes in DCs (97–99). In addition, LPS-induced TLR signaling can promote the redistribution of MHC Class I and II molecules to the surface of DCs (100). TLR4 activation on DCs promotes cytosolic routing of dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN)-targeted antigens for presentation on MHC Class I and increased CD8+ T cell activation (101). Recently, TLR3, TLR4, and TLR9 ligands were reported to induce autocrine C3a receptor and C5a receptor (C3ar1/C5ar1) signaling in DCs, which causes the expansion of effector T cells and instability of regulatory T cells and contributes to T cell-dependent transplant rejection (102). IFN-γ combined with TLR ligation TLR2, TLR4, or TLR9 agonists can enhance DC activation and function to increase antigen-specific T cell responses (103).
Interestingly, TLR2 seems more critical than TLR4 in mouse DC-derived IL-10 responses to schistosome antigens (104). TLR2 signaling activation on DCs can promote higher frequency effector and memory CD4+ T cell responses than TLR4 signaling activation. The novel TLR2 agonist SUP3 also showed a heightened ability to enhance DC-mediated antigen presentation and T cell activation (105). By contrast, the TLR5 ligand flagellin was most effective at activating neonatal lung APCs by inducing significantly higher expression of maturation markers on CD103+ (cDC1) and CD11b+ (cDC2) subsets (106). Monocyte-derived DCs stimulated with TLR4 and TLR7/8 ligands induce naive allogeneic CD4+ T cells to secrete IL-10 and IFN-γ sequentially and eventually IL-17A (107). The activation of TLR9 and IL-12 pathways in CD8α+ DCs can drive CD4+ T cells to act as Th cells or induce rapid polyclonal conversion to immunosuppressive Treg during Listeria infection (108). Interestingly, although all TLRs on DCs are able to induce CD8+ T cell activation in vitro, the abilities of surface and endosomal TLRs to activate CD8+ T cells might be different in vivo. The nucleic acid recognizing endosomal TLRs potently induce CD8+ T cell activation, whereas the bacterial ligands recognizing surface TLRs were incapable of inducing CD8+ T cell priming. Moreover, surface TLRs might have a dominant effect of inhibiting CD8+ T cell expansion induced by activation of endosomal TLRs (109).
Based on the particular cell surface markers, DCs can be divided into different subsets, including myeloid DCs, pDCs, CD8α+ DCs, and CD11b+ DCs (4). Human pDCs express TLR7 and TLR9, whereas CD11c+ human myeloid DCs express TLR1, TLR2, TLR3, TLR5, TLR6, and TLR8 (110–112). Human blood monocytes express TLR1, TLR2, TLR4, and TLR5, but progressively lose these receptors and acquire the expression of TLR3 as they differentiate into mature DCs in the presence of granulocyte-macrophage colony-stimulating factor and IL-4 (113). Mice splenic DC subsets express TLR1, TLR2, TLR4, TLR6, TLR8, and TLR9, but not TLR3 (114). Interestingly, freshly isolated mouse splenic DC subsets or macrophages only express low amounts of TLR4 and do not respond to LPS stimulation. By contrast, bone marrow-derived DCs or macrophages have high expression of TLR4 and respond robustly to LPS (115).
Since different DC subsets express subset-specific PRRs, DCs are functionally heterogeneous (110, 114, 116). Different DC subsets respond to different stimuli and activate distinct signaling pathways, leading to the release of specific cytokines, which in turn determine the specific Th cell subsets that are generated and activated (117). pDCs express TLR7 and TLR9, which recognize ssRNA and CpG DNA, respectively, but pDCs do not express other TLRs that detect bacterial cell wall components. Therefore, pDCs are thought to specifically detect viral infections to induce Type I IFNs and control antiviral immunity (35, 116).
TLR signaling in innate immune cells indirectly regulates T cell differentiation and proliferation by promoting DC maturation and regulatory cytokine production (118). Since T cells also express different TLRs, recent studies have revealed that TLR-mediated signaling can directly regulate effector T cells and Treg cells (119).
CD4+ Th cells play a critical role in initiating and maintaining adaptive immune responses against cancer (120). CD4+ Th cells are required for the expansion and maintenance of memory CD8+ T cells (121). Naïve murine or human CD4+ T cells can express TLR2 after their stimulation (122, 123). TLR2 signaling can promote the proliferation and production of IFNγ in Th1 cells (124). Costimulation of TCR and TLR2 in naïve murine CD4+ T cells increases their differentiation to proinflammatory Th1 cells and secretion of cytokines and chemokines (122, 125). Costimulation of neonatal CD4+ T cells with TLR2 ligand and anti-CD3 also show an increased proinflammatory Th1 immune response (IFN-γ and TNF-α production) and IL-2 production (126). Moreover, TLR2 signaling on CD4+ T cells exerts a protective action by increasing the population of Mycobacterium tuberculosis Ag-specific T cells during mycobacterial tuberculosis (127).
Similarly, TLR2 signaling can also regulate the immune response of the CD8+ T cell. TLR2 agonists can enhance cell survival, proliferation, IFN-γ production, and memory cell formation of CD8+ T cells in response to a suboptimal TCR signal by reducing the threshold for costimulatory signals from APCs (128–130). The TLR2/MyD88-dependent signaling pathway in CD8+ T cells also can increase their survival, clonal expansion, and differentiation into long-lived memory T cells by activating the phosphatidylinositol 3-kinase (PI3K)-Akt pathway during vaccinia virus infection (130). Interestingly, MyD88-dependent signaling is also essential for CD4+ T cell-promoted IFN-γ production and hematopoietic progenitor cell expansion during intracellular bacterial infection (131). In addition, MyD88-dependent signaling in the host can protect against acute allogenic graft versus host disease after bone marrow transplantation (132). However, activating the MyD88 signaling pathway in donor CD4+ T cells promotes the survival and differentiation of T cells toward Th1, Tc1, and Th17. It increased the severity of graft versus host disease in a mouse model of allogeneic hematopoietic stem cell transplantation (133). MyD88-dependent signaling is also reported to promote differentiation and proliferation of CD4+ T cells toward Th17 cells by linking IL-1 and IL-23 signaling and sustaining mTOR signaling (134).
Besides TLR2, TLR4 is also expressed on CD4+ T cells, and the TLR4 ligation could enhance both the in vitro cell proliferation and survival of CD4+ T cells (135). However, the activation of TLR4 signaling could affect the phenotype and ability of CD4+ T cells to provoke the intestinal inflammation, through the induction of MAPK phosphatase 3 (MKP-3) to inhibit TCR stimulation-induced activation of ERK1/2 (136). Moreover, LPS can induce the adhesion of human T cells to fibronectin and the up-regulated expression of suppressor of cytokine signaling 3 (SOCS3), which further led to the inhibition of T cell chemotaxis toward the chemokine stromal cell-derived factor 1α (CXCL12) (137, 138). By contrast, CD4+ T cells are pathologic and contribute to an exaggerated immune activation in the mice that is absence of functional Tregs, resulting in the mortality to a nonlethal dose of LPS or Escherichia coli challenge (139). Recently, it was reported that the TLR4 expression on T cells goes down during TCR and mitogenic activation (140). However, the VIPER peptide (VP), an established inhibitor of TLR4 signaling, restores TLR4 expression and regulates the activation of naive T cell, indicating that TLR4 responses might be associated with the acute-stage T cell responses (140).
The agonist of TLR9 (CpG-ODNs) was found to promote the release of IL-8 in purified CD8+ T cells (110). Additionally, the expression of TLR7 is increased in the mesenteric lymph node CD4+ and CD8+ T cells after Schistosoma japonicum infection. The TLR7 agonist can enhance the production of IFN-γ in CD8+ T cells from mesenteric lymph node T cells in infected mice (141). Moreover, TLR7/MyD88-dependent signaling activation in CD8+ T cells can promote cellular glycolysis and enhance T cell effector functions (142). TLR3 is constitutively expressed on CD8+ T effector cells. Furthermore, the TLR3 agonist polyinosinic-polycytidylic acid [Poly (I:C)] increases IFN-γ production in Ag-specific CD8+ T cells (143). Poly (I:C) treatment significantly increases the IL-2 and IFN-γ production of chimeric antigen receptor-modified T (CAR T) cells along with improving their lytic action against tumor or cancer cells (144). CAR T cells also show an increased anti-tumor action against refractory or relapsed B cell acute lymphoblastic leukemia upon co-stimulation with TLR2 signaling by introducing the TIR domain of TLR2 into the CAR construct (145). The third-generation anti-CD19 CAR T cells incorporated with the intracellular signaling domains of CD28 and TLR2 are under clinical trial for relapsed or refractory B-cell non-Hodgkin’s lymphoma (146).
Treg cells are critical for maintaining peripheral tolerance, preventing autoimmune diseases, and limiting chronic inflammatory diseases by suppressing host immune responses and inducing self-tolerance (147). CD4+ Treg cells are a small subset (5–6%) of the overall CD4+ T cell population (121). Foxp3 is a specific marker of CD4+ Treg cells in both mice and humans (148–152). In previous studies, the elevated proportion of CD4+ CD25+ Treg cells in the total CD4+ T cell population was observed in several different human cancers, including lung, breast, and ovarian tumors (153–155). We also demonstrated the presence of antigen-specific CD4+ Treg cells at tumor sites (152, 156). We showed that Treg cells could suppress the proliferation of naive CD4+ T cells and inhibit IL-2 secretion of CD4+ effector cells upon activation by tumor-specific antigens (157). In addition, we identified CD8+ Treg and γδ-TCR Treg cells in prostate and breast cancer (158, 159). Notably, the CD8+ Treg cells expressed Foxp3 molecules, while the γδ-TCR Treg cells did not. Like CD4+ Treg cells, both of these CD8+ and γδ-TCR Treg cell subtypes have immune suppression ability and inhibit anti-tumor immunity.
To abrogate Treg cell-mediated immune suppression, we sought to identify the TLR ligands that could reverse Treg cell suppressive activity. We found that Poly-G10 oligonucleotides can directly change their suppressive function in the absence of DCs. The TLR8-MyD88 signaling pathway is required to reverse Treg cell function by Poly-G oligonucleotides (158, 160). Moreover, we found that the natural ligands for human TLR8—ssRNA40 and ssRNA33, which are derived from HIV viral sequences (161)—could completely reverse the suppressive function of Treg cells, indicating that activation of the TLR8-dependent signaling pathway is critical for the reversal of Treg-suppressive functions. Besides different subsets of CD4+ Treg cells, we found that the CD8+ Treg cells and γδ-TCR Treg cells in prostate and breast cancer also express a low level of human TLR8 molecules (158, 159). Interestingly, we demonstrated that Poly-G oligonucleotide treatment could also reverse the suppressive function of CD8+ Treg cells and γδ-TCR Treg cells, suggesting that these cells might share the same TLR8/MyD88 signaling pathway-mediated mechanism with previously characterized CD4+ Treg cell subsets (158, 159).
Recent studies show that TLR8 stimulation in humans reverses Tregs’ immunosuppressive function and enhances their anti-tumor function by inhibiting glycolysis and glucose uptake (162). CD4+ T cells stimulation with TLR8 ligand ssRNA 40 in a co-culture system with ovarian cancer cells (SKOV3) inhibited the glycolysis metabolism and downregulated the percentage of Treg cells (163). Therefore, the TLR8 signaling pathway may regulate Treg by reprogramming the glycolysis metabolism. These findings raise an intriguing possibility that the activation of the TLR8 signaling pathway could block the suppressive function of different subsets of Treg cells to improve the efficacy of cancer immunotherapy.
Since TLR8 is non-functional in mice (164), Poly-G oligonucleotides cannot reverse the suppressive activity of murine Treg cells. However, recent studies showed that other TLR signaling in mice could also mediate the regulation of Treg cells. TLR2-deficient mice showed a reduced number of CD4+ CD25+ Treg cells (165). Additionally, stimulation of mouse Treg cells with TLR2 ligand Pam3Cys increased its proliferation and temporarily reversed its suppressive function (166, 167). The activation of TLR9 signaling has been reported to inhibit the immunosuppressive function of Treg through direct MyD88-dependent costimulation of effector CD4+ T cells (168). However, another study showed that human CD4+ CD25+ Treg or effector Th1 and Th2 cells did not highly express TLR9 naturally, but 25-dihydroxyvitamin D3 (1α25VitD3)—the active form of Vitamin D—could induce it (169). Stimulation of 1α25VitD3-induced IL-10–secreting Treg with TLR9 agonists showed a decreased IL-10 and IFN-γ production, indicating the reduction of their immunoregulatory function (169). In contrast, stimulation of human Treg cells with the TLR5 ligand flagellin increased rather than reversed their suppressive function (170). The TLR4 ligand LPS was also reported to induce proliferation and enhance the suppressive function of Treg cells (171).
Uncontrolled TLR signaling activation can be harmful or even fatal (172). Therefore, the stringent and precise regulation of TLR signaling pathways is essential to maintaining immune balance in the host. In the last few years, many positive and negative regulators have been identified to control TLR-induced NF-κB signaling pathways at multiple levels through different mechanisms (173). These regulators include co-receptors, such as CD14 (174, 175); soluble receptors, such as sTLR (176, 177); transmembrane proteins, such as ST2L (178), SIGIRR (179, 180), and TRAILR (181); and intracellular regulators, such as SOCS-1 (182, 183), MyD88s (184, 185), TOLLIP (186), IRAK-M (187), A20 (188, 189), CYLD (the familial cylindromatosis tumor suppressor gene) (190–194), Nrdp1 (195), regulatory Nod proteins (196–206), TRIAD3A (207), and tripartite motif-containing proteins (TRIMs) (208). These molecules maintain the balance between activation and inhibition of TLR signaling in response to diverse PAMPs (172).
We also participated in the identification of some critical regulators of TLRs and the NF-κB signaling pathway. NLRs were originally believed to function as pathogen sensors and cellular danger signals. However, alongside other groups, we recently found that several NLRs, known as regulatory NLRs, negatively regulate TLR and RIG-I-like receptor signaling. NLR family member X1 (NLRX1) is the first NLR that was identified to negatively modulate RIG-I-mediated antiviral responses by binding to mitochondrial antiviral-signaling protein (MAVS) and disrupting RIG-I-MAVS (209). Then, we found that it could also negatively regulate TLR-induced NF-κB signaling by targeting the TRAF6 and IKKα/β-NEMO complex (199, 201). Besides NLRX1, NLR family CARD domain containing 5 (NLRC5) is another member of the NLR protein family that is recognized as a novel regulator of both adaptive and innate immune responses (210). We identified NLRC5 as a negative regulator of both NF-κB and Type I IFN signaling (196, 200, 202, 203, 206). NLRC5 inhibits IKK phosphorylation and NF-κB signaling by interacting with IKKα/β but not NEMO. NLRC5 inhibits Type I IFN signaling by targeting RIG-I/MDA5 after viral infection and blocking the RIG-I–MAVS interaction. We recently identified NLR family pyrin domain-containing 11 (NLRP11) as a regulatory NLR to attenuate TLR signaling by targeting TRAF6 for degradation via the ubiquitin ligase RNF19A (205).
Besides the NLR family, we also discovered some regulators from the LRR-containing (LRRC) family, ubiquitin-specific protease (USP) family, and tripartite motif family (TRIM) family. We found that LRRC25 negatively regulates the TLR-induced NF-κB signaling pathway by promoting p65/RelA for autophagic degradation (211). Interestingly, LRRC25 also inhibits Type I IFN signaling by targeting IFN-stimulated gene 15 (ISG15)-associated RIG-I for autophagic degradation (212). We found that USP38 could also negatively regulate TLR and RIG-I signaling through different mechanisms (213, 214). In contrast, TRIM14 functions as a positive regulator in the noncanonical NF-κB signaling pathway and cGAS- and RIG-I-mediated Type I IFN signaling pathway (215–218).
Deidier observed that patients infected with syphilis had remission of malignant tumors, revealing the correlation between immune system activation triggered by infection and cancer remission (219). Studies on TLRs involved in cancer have shown that TLR signaling has not only anti-tumor effects but also pro-tumor functions on carcinogenesis, which is dependent on the individual TLR and cancer type (220, 221). TLR stimulation enhances the anti-tumor immune response either through immune cells or directly targeting tumor cells to induce apoptosis. In murine models of hepatocellular carcinoma, TLR2-deficient mice showed a decrease in the expression of IFN-γ, TNF-α, (IL)-1α/β, IL-6, and Cxcl-2, which attenuate p21- and p16/pRb-dependent senescence, leading to the increased proliferation of tumor cells (222). We found that TLR8 ligand treatment suppresses prostate and breast cancer by reversing the function of CD8+ Treg cells and γδ-TCR Treg cells (160). Shanshan Qi et al. (223) generated hTLR8 mice by replacing exon 3 of mouse Tlr8 with human TLR8 to analyze the role of TLR8 in tumor progression. They found that the MC38 tumor grew slower in hTLR8 mice compared with naïve mice. hTLR8 mice also exhibit increased IFN-γ and TNF-α positive CD4+ T cells and effector T cells (223).
In addition, a synthetic bacterial lipoprotein (a TLR1/TLR2 agonist) was reported to reduce the suppressive function of Foxp3+ Treg cells and enhance the cytotoxicity of tumor-specific CTL (224). Combination treatment with the TLR1/2 ligand Pam3CSK4 and anti-CTLA4 mAb improved the anti-tumor immunity compared with anti-CTLA4 mAb alone. This study showed that TLR1/2 increased FcgR IV expression in macrophages, which led to Treg cell depletion and augmentation of T cell/Treg ratios within the tumor (225). Besides their anti-tumor effects, TLRs have also shown pro-tumor functions. Stimulation of TLR4 by LPS promoted immunosuppressive cytokine production, resulting in tumor immune evasion in lung cancer cells (226). In breast cancer, a stimulation expressed-TLR4 tumor with LPS promoted cancer cell proliferation via upregulation of IL-8 and IL-6 production (227, 228). Interestingly, TLR6 signaling was recently reported to prevent the inflammation by impacting the composition of microbiota during inflammation-induced colorectal cancer (229). Besides TLRs, MyD88 is also involved in cancer development. MyD88-dependent signaling is reported to control the expression of several key modifier genes of intestinal tumorigenesis and play a crucial role in both spontaneous and carcinogen-induced tumor development (230). Besides, diethylnitrosamine (DEN) administration induced higher serum interleukin-6 (IL-6) production in males than it did in females in DEN-induced hepatocellular carcinoma model. Further study showed that DEN exposure promoted the production of IL-6 in Kupffer cells (KCs) in a MyD88-dependent manner and depletion of MyD88 protected male mice from DEN-induced hepatocarcinogenesis (231). In the activated B-cell-like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL), MyD88 L265P is reported to contribute to the constitutive NF-κB and JAK kinase signaling, which promotes malignant cell survival in these lymphomas (232). MyD88 L265P somatic mutation is identified as a commonly recurring mutation in patients with Waldenström’s macroglobulinemia (233). 69% of patients with cutaneous diffuse large B cell lymphoma (CBCL) carry MyD88 L265P mutation, which is significantly associated with shorter disease-specific survival (234). In addition, the MyD88/IL1 receptor (IL1R) axis upregulates programmed cell death (PD)-1 expression on tumor-associated macrophages (TAMs) via promoting recruitment of NF-κB to the Pdcd1 promoter, which sustains their immunosuppressive function in melanoma (235). Based on the critical role of TLRs and TLRs-mediated signaling pathways in cancer development, researchers have taken advantage of agonists and antagonists of TLRs to treat some types of cancer (Table 1). Various agonists of TLRs are currently under investigation in clinical trials for cancer treatments (Table 2). Due to the double-edged role of TLRs in tumor biology, it is essential to understand how TLRs manipulate the immune system and tumor cell characteristics, which may provide us with new therapeutic strategies against cancer.
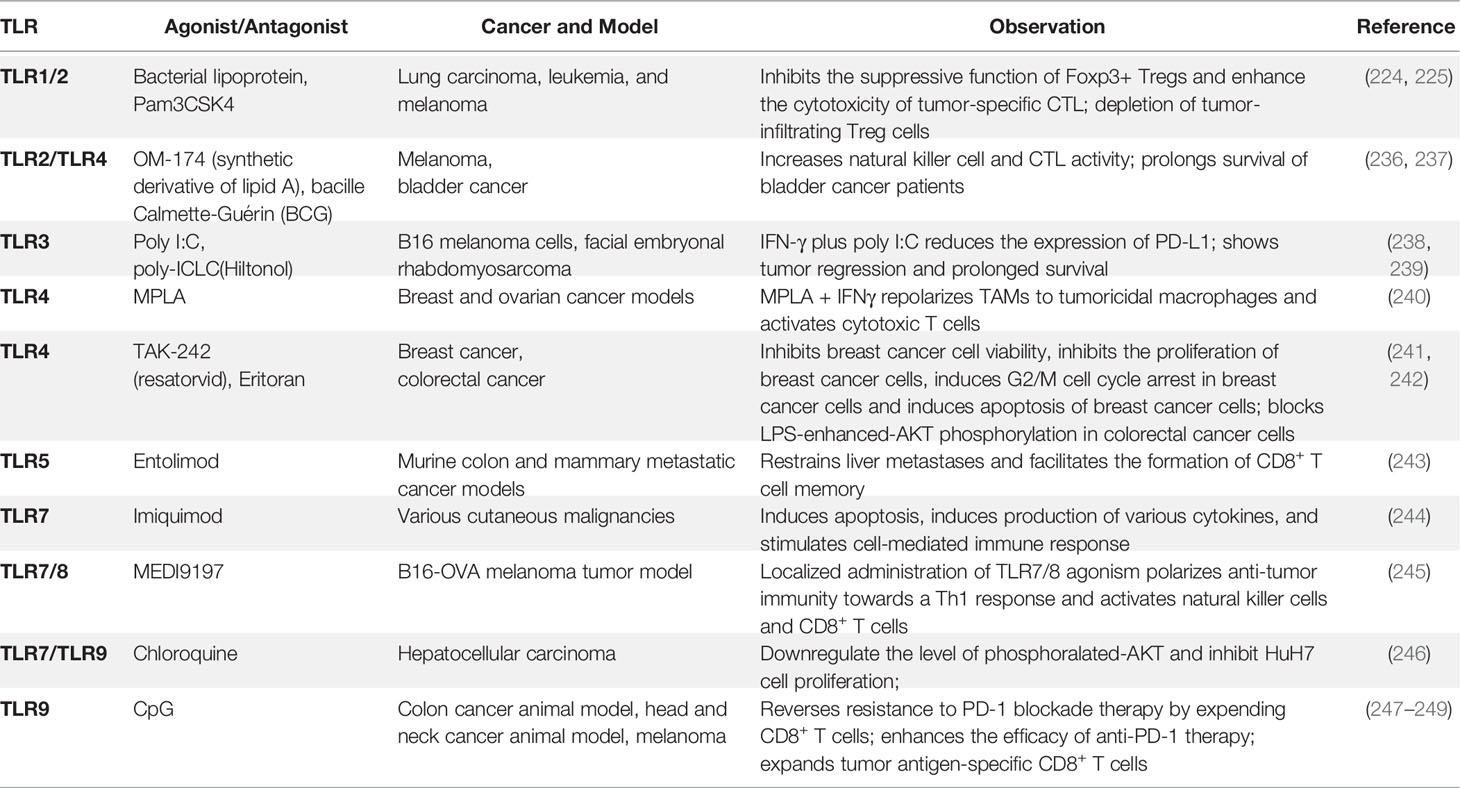
Table 1 TLR agonists, antagonists and cancer.
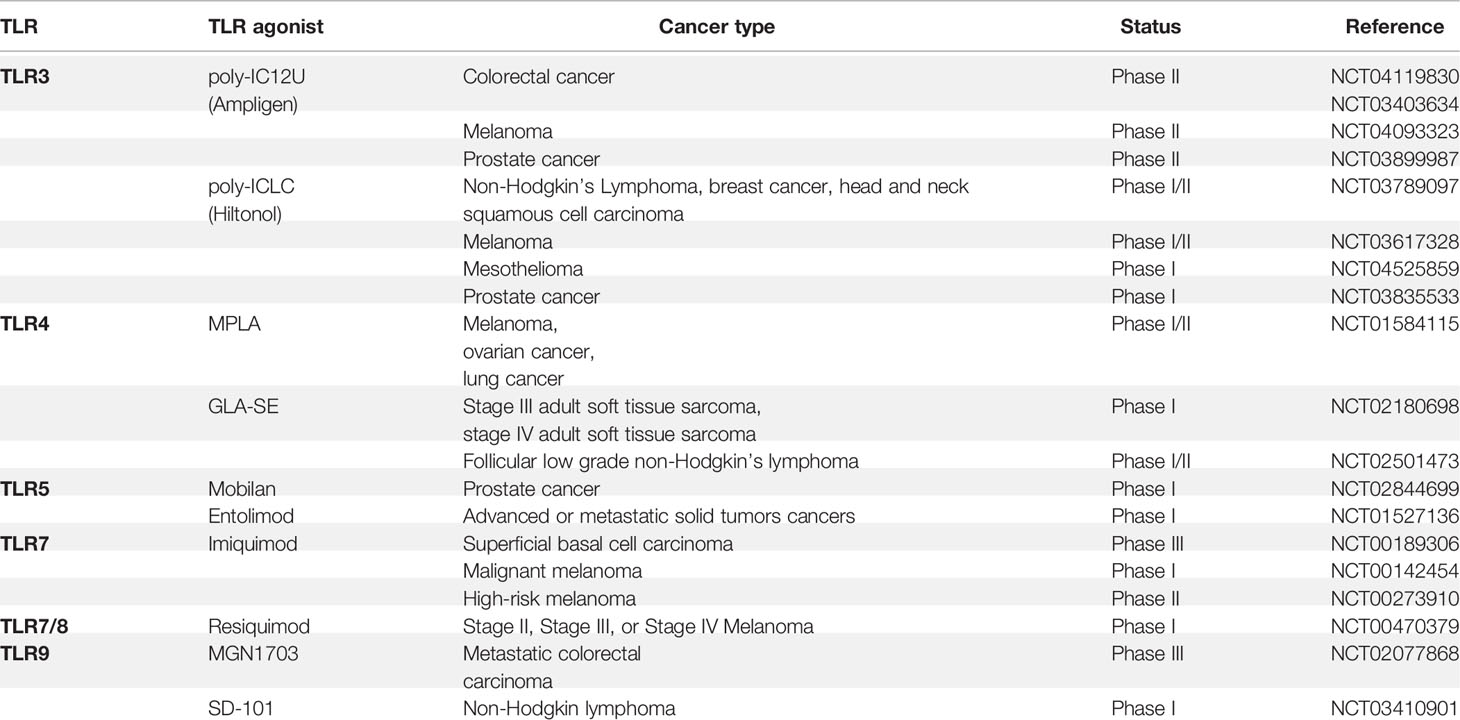
Table 2 Clinical trials of TLR agonists TLR in cancer.
TLRs are supposed to sense pathogenic components and initiate the immune response that contributes to host homeostasis. However, in the specific scenario, TLRs are improperly activated by self-antigens, leading to chronic systemic inflammatory disorders and the occurrence of autoimmunity. Numerous studies have demonstrated that TLRs are involved in the pathogenesis of various autoimmune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), multiple sclerosis, and Crohn’s disease (250) (Table 3). RA is an autoimmune disorder that affects the synovial joints, causing chronic and persistent inflammation and the destruction of articular tissues. MyD88 also has been demonstrated to be crucial for the production of MMPs (the major enzymes involved in joint tissue destruction) in RA synovial membrane cultures (275). TLR2 and TLR4 expression were reported to be associated with the levels of IL-12 and IL-8 in the synovial tissue of RA patients (251). The surface expression of TLR4 on CD8+ T cells directly correlates with the disease severity of RA. And the TLR4-expressing CD8+ T cells can respond to LPS and express robust amounts of cytolytic and inflammatory molecules including TNFα and IFNγ (256). Besides RA, TLR2 and TLR4 are also involved in heat shock proteins-associated atherosclerosis (257, 258). Moreover, emerging evidence indicates that TLR2 is strongly associated with diabetes (259–261).
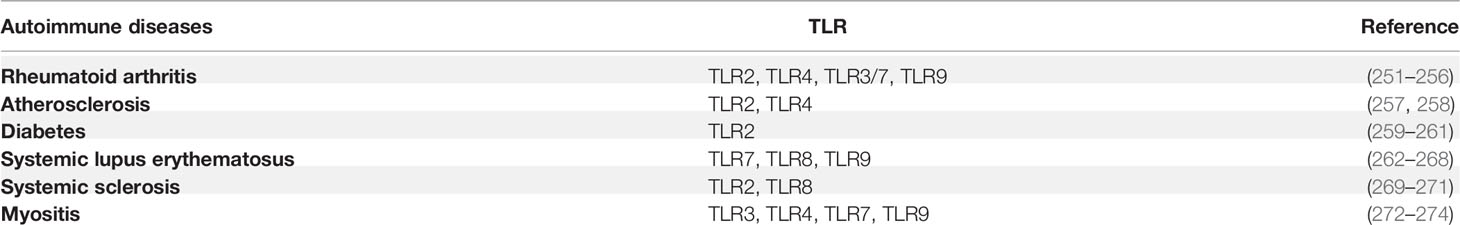
Table 3 TLRs implicated in autoimmune diseases.
SLE is characterized by the presence of autoantibodies triggered by CpG DNA and ssRNA-associated self-antigens. In endosomes, the self-antigens are sensed by TLR7 and TLR9. TLR7 is essential for generating the germinal center and drives the extrafollicular pathway, which is associated with pathogenic antibody secretion. Notably, TLR9 has been demonstrated to have a protective function in SLE by limiting the stimulatory activity of TLR7 (262). Genetic studies have shown that copy number variation of TLR7 is associated with SLE development (263, 264). Additionally, TLR7 localizes on the X chromosome escapes X inactivation in B cells and myeloid cells in females, resulting in the gender difference in TLR7 expression (265), leading to a higher incidence in women than men. Moreover, SLE patients with increased expression of TLR7 showed significant expansion of CD19+ IgD+CD38++ transitional B cells and increased IgG auto-Ab production (266). Recent data show that the expression of TLR7 in mild and severe lupus-prone models is dependent on the activity of IRAK4 (the TLR7-downstream signaling molecule) and the pathogenic environment. Impairments of IRAK4 signaling refrain from all pathological characteristics associated with murine lupus. These data suggest a feedback loop of TLR7 expression and pathological changes in SLE patients (267).
A study in experimental autoimmune encephalomyelitis (EAE) mice models showed that deficiency of MyD88 conferred complete resistance to EAE in mice, indicating that a TLR-mediated immune response is required to induce EAE (276). Consistently, depletion of TLR4 solely in CD4+ T cells impairs Th17 and markedly abolishes the disease symptoms (135). In addition, accumulating evidence suggests that TLR8 contributes to autoimmune diseases as well. It is reported that human monocytes that lack CD14 (CD14dim) and express CD16 do not produce cytokines in response to bacterial cues that are sensed by cell-surface TLRs. Instead, they trigger the production of TNF-α, IL-1β, and C-C motif chemokine ligand 3 in response to viruses through the TLR7-TLR8-MyD88-MEK pathway. Further study showed that these CD14dim monocytes recognize self-nucleic acids and drive the production of inflammatory cytokines in patients with lupus (268).
Cristiana et al. (277) used human TLR8-transgenic mice to show that high copy number chimeras developed the multiorgan inflammatory syndrome through DC-intrinsic huTLR8 activation and subsequent T cell activation. The severity of the inflammation was associated with the expression level of huTLR8. They observed spontaneous arthritis in high-expressing human TLR8 mice. Furthermore, they demonstrated that TLR8 mRNA expression was much higher in blood from both SoJIA and Still’s disease donors than healthy donors. Finally, the mRNA level of TLR8 was associated with the transcription level of inflammatory cytokines in these patients.
In addition, the ectopic expression of TLR8 on pDCs in systemic sclerosis patients induces the production of CXCL4, which in turn enhances TLR8- and TLR9-induced IFN production by pDCs. Both CXCL4 and IFNs are the featured cytokines in systemic sclerosis (269). These data suggest that TLR8 is the key RNA-sensing TLR in the pathogenesis of autoimmune disease, demonstrating the potential of TLR8 for clinical development. Emerging evidence suggests numerous autoimmune diseases are triggered by the dysregulation of TLR. Some TLR antagonists have already been applied to autoimmune disease treatment in mice models. Due to redundancy between different TLRs in different disease-affected tissues, it is crucial to dissect the detailed molecular mechanism and cell-mediated immune regulation in the specific disease context to facilitate drug development for clinic treatments.
TLRs play an essential role in host immune responses to various invading pathogens, including bacteria, fungi, viruses, and parasites (Table 4). TLR1 is crucial for the induction of mucosal Th17 immunity and IgA responses during Yersinia enterocolitica infection (316, 317). The I602S mutant of TLR1 results in the deficiency of TLR1 trafficking from the cytosol to the cell surface, potentially impairing blood monocytes’ immune functions against pathogenic Mycobacterium tuberculosis (318). Recently, it was reported that mice deficient in both TLR2 and TLR4 were highly susceptible to intracellular Salmonella typhimurium infection (319). However, Tlr2/4-deficient mice lacking additional TLR9 involved in S. typhimurium recognition were less susceptible to infection (319). Notably, TLR2 was also reported to recognize the envelope (E) protein of SARS-CoV-2 to induce a hyperinflammatory response in mice bone-marrow-derived macrophages (283). Besides TLR2, TLR4 was also reported to recognize the spike (S) protein of SARS-CoV-2 and activate the NF-κB signaling to produce IL-1β (320).
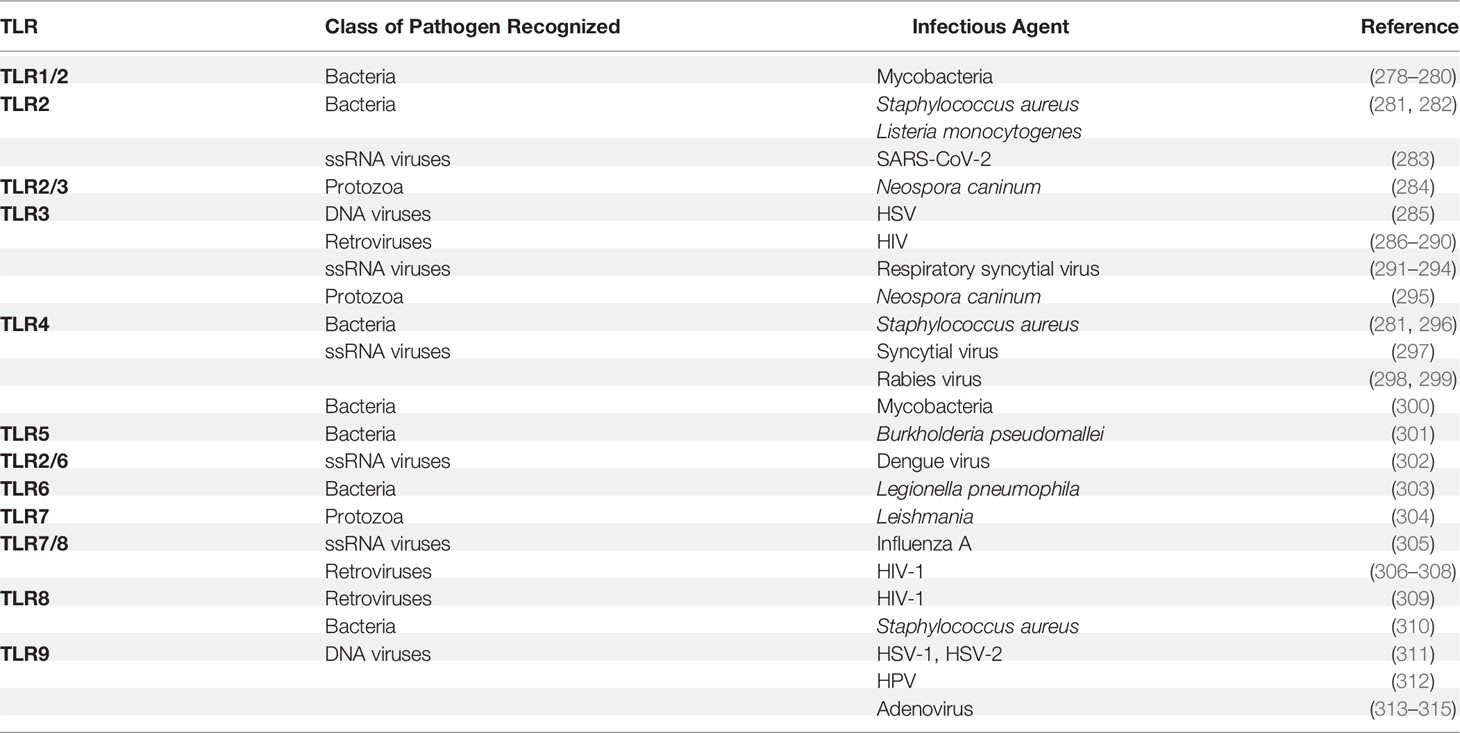
Table 4 TLRs and infectious diseases.
Numerous studies have shown that TLR4 is also involved in various infectious diseases. Infants carrying D299G and T399I polymorphisms are more vulnerable to respiratory syncytial virus infection (321). The single nucleotide polymorphism rs11536889 of TLR4 is involved in organ failure in sepsis patients (322). However, the effects of TLR4 remain controversial during M. tuberculosis infection. It was reported that Tlr4-deficient mice exhibited the same sensitivity compared to congenic control mice (323). By contrast, another study found that TLR4 mutant mice showed reduced macrophage recruitment and failure to develop a protective immune response against chronic M. tuberculosis infection (324). Melioidosis is a high-mortality infectious disease caused by Burkholderia pseudomallei, a flagellated, Gram-negative bacterium. TLR5 c.1174C>T (a TLR5 variant carrying a nonsense mutation) is associated with lower IL-10 and TNF-α production and prolonged survival in human melioidosis.
Influenza A virus is a contagious agent that causes respiratory disease. TLR7 is responsible for influenza A virus sensing in the endosome, while RIG-I senses influenza A virus in the cytosol (161, 325–327). The sensing mechanisms for TLR7 and RIG-I are different. TLR7 directly recognizes virus ssRNA in a virus-replication-independent manner. By contrast, RIG-I recognizes the viral replication intermediates in certain cell types (328). Both signaling pathways move toward the activation of IRF3/7 and NF-κB to trigger the production of Type I IFN and proinflammatory cytokines and downstream IFN-stimulated genes (1). Furthermore, it has been shown that intranasal administration of the TLR7 agonist (imiquimod) can significantly reduce airway and pulmonary inflammation in mice during influenza A virus infection (329).
Different single nucleotide polymorphisms of TLR8 and TLR9 confer varying degrees of risk in the development of tuberculosis, suggesting that TLR8 and TLR9 are involved in tuberculosis (330). The most characteristic role of TLRs is sensing the PAMPs from pathogens and initiating immune responses against infectious agents. Notably, in certain scenarios, TLRs may be subverted by the pathogens to alter the host cytokine pattern for their own benefit (319, 331). Thus, further studies on the interplay between pathogen evasion and TLR subversion will have implications for human health.
This review provides an updated overview of TLR signaling and its critical role in cell-mediated immunity. The fundamental mechanisms of TLR signaling transduction have been identified by cell biological and biochemical approaches, as well as loss-of-function genetic analysis. Significant progress has also been made in the structural elucidation of TLRs and their downstream signaling supramolecular complex (332, 333). The essential role of TLR signaling in activating innate immune cells to initiate adaptive immunity was illustrated. Importantly, the direct regulatory roles of TLR signaling in effector T cells and Treg cells have been identified. In addition, the individual TLRs signaling involved in infectious disease, autoimmune disease and cancer have been extensively studied (Figure 3).
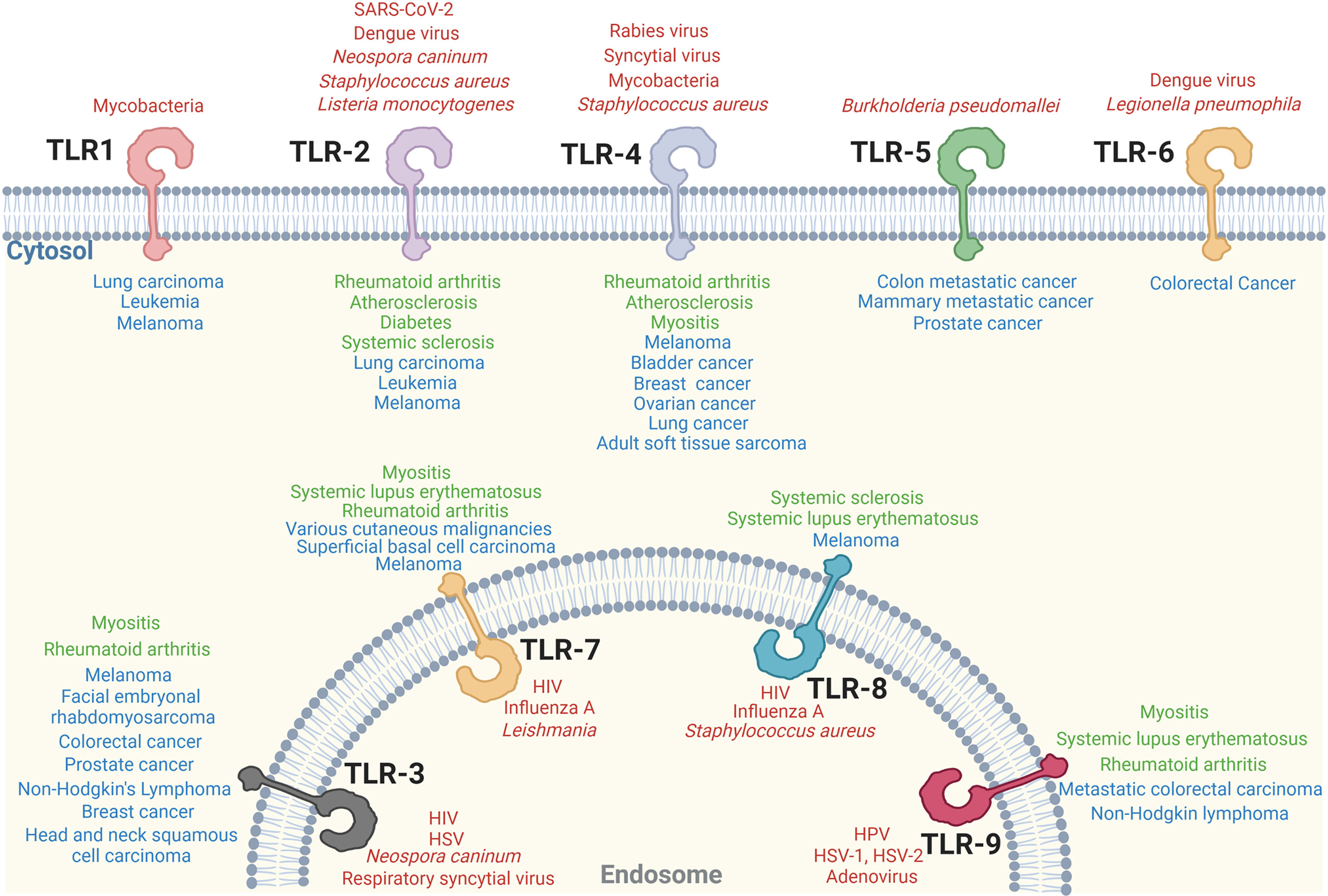
Figure 3 Individual TLR singaling involves in various diseases. Individual TLR-associated infectious diseases (red), autoimmune diseases (green) and cancer (blue) are shown.
Despite the rapid advancement of our knowledge, there are still large gaps in our understanding of TLR signaling. For example, although free unanchored K63-Ub chains have been demonstrated as a kind of indispensable “second messenger” to activate downstream protein kinases during TLR-induced NF-κB signaling activation by biochemical experiments, how and whether these free unanchored K63-Ub chains activate downstream protein kinases in cells is still unknown. Especially, how to control the activation specificity if these chains are just free in the cytoplasm still remains elusive. Therefore, the detailed mechanisms of how these polyubiquitin chains activate the kinase complex during TLR signaling pathway activation warrant further investigation.
Due to the vital role of TLR signaling in T cell activation, growth, differentiation, and function, it would be necessary to dissect the T cell-specific TLR signaling pathway. In the past few years, T cell-based cancer immunotherapy has made significant progress (334). The Food Drug Administration (FDA) has approved four CD19-CAR-engineered T cell products for blood cancer. Cancer vaccines along with TLR signaling activation could become a more effective therapeutic approach to inhibiting or even eliminating cancer cells. Cancer vaccines along with TLR signaling activation could become a more effective therapeutic approach to inhibiting or even eliminating cancer cells (335).
Besides cancer immunity, TLRs are also involved in many infectious diseases by recognizing the PAMPs of pathogens, initiating inflammatory responses, and eliminating invasive microorganisms at the early stage. However, prolonged or excessive inflammatory responses are harmful or even fatal for the host at the late stage. In the current COVID-19 pandemic, fatal hyperinflammation, but not SARS-CoV-2 directly, is the primary cause of mortality in severe COVID-19 patients. Therefore, drugs targeting viral replication might be ineffective for severe COVID-19 patients since most hospitalized patients are at the late stage of disease. In this case, drugs targeting TLR-dependent inflammatory signaling pathways might be more effective in reducing the mortality of severe COVID-19 patients.
As the field has developed, multidisciplinary approaches have been used in the study of TLR signaling pathways. Integrated methods, combined with transcriptomics, genetic/chemical perturbations, and phosphoproteomics, have been used to systematically discover TLR signaling regulatory components. The m6A RNA sequence technique led to the discovery that mRNA stability is an essential mechanism for regulating TLR-dependent innate immune responses. The single-cell sequencing (scRNA-seq) approach is used to dissect the characteristics of TLR-mediated immune responses at the single-cell level. The development of super-resolution single-molecule localization microscopy empowers the ability to directly observe the supramolecular signaling complex during TLR signaling activation at the single-molecule level. Finally, the advances in cryo-electron microscopy have facilitated our understanding how TLRs recognize their ligands and initiate immune signaling at the atom level. These recent advancements markedly increase our ability to understand TLR signaling pathways and develop new therapeutic strategies against various infectious, autoimmune diseases, and cancers.
R-FW, TD, and YD designed and wrote the manuscript. TD, YD, CX, HW, and R-FW discussed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was in part supported by grants from the NCI, NIH (R01CA101795, R01CA246547 and U54CA210181), Department of Defense (DoD) CDMRP BCRP (BC151081) and LCRP (LC200368) to R-FW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
2. Akira S, Uematsu S, Takeuchi O. Pathogen Recognition and Innate Immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
3. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol Cell (2014) 54(2):289–96. doi: 10.1016/j.molcel.2014.03.040
4. Iwasaki A, Medzhitov R. Toll-Like Receptor Control of the Adaptive Immune Responses. Nat Immunol (2004) 5(10):987–95. doi: 10.1038/ni1112
5. Hayden MS, Ghosh S. NF-kappaB in Immunobiology. Cell Res (2011) 21(2):223–44. doi: 10.1038/cr.2011.13
6. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
7. Wang HY, Wang R-F. Innate Immune Signaling and Negative Regulators in Cancer. In: Wang R, editor. Innate Immune Regulation and Cancer Immunotherapy. New York, NY: Springer New York (2012). doi: 10.1007/978-1-4419-9914-6_6
8. Anderson KV, Jürgens G, Nüsslein-Volhard C. Establishment of Dorsal-Ventral Polarity in the Drosophila Embryo: Genetic Studies on the Role of the Toll Gene Product. Cell (1985) 42(3):779–89. doi: 10.1016/0092-8674(85)90274-0
9. Lemaitre B, Nicolas E, Michaut L, Reichhart J-M, Hoffmann JA. The Dorsoventral Regulatory Gene Cassette Spätzle/Toll/cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell (1996) 86(6):973–83. doi: 10.1016/S0092-8674(00)80172-5
10. Medzhitov R, Preston-Hurlburt P, Janeway CA. A Human Homologue of the Drosophila Toll Protein Signals Activation of Adaptive Immunity. Nature (1997) 388(6640):394–7. doi: 10.1038/41131
11. Kawai T, Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
12. Fore F, Indriputri C, Mamutse J, Nugraha J. TLR10 and Its Unique Anti-Inflammatory Properties and Potential Use as a Target in Therapeutics. Immune Netw (2020) 20(3):e21–1. doi: 10.4110/in.2020.20.e21
13. Henrick BM, Yao X-D, Zahoor MA, l. Abimiku A, Osawe S, Rosenthal KL. TLR10 Senses HIV-1 Proteins and Significantly Enhances HIV-1 Infection. Front Immunol (2019) 10:482. doi: 10.3389/fimmu.2019.00482
14. Lind NA, Rael VE, Pestal K, Liu B, Barton GM. Regulation of the Nucleic Acid-Sensing Toll-Like Receptors. Nat Rev Immunol (2021) 16:1–12. doi: 10.1038/s41577-021-00577-0
15. Medzhitov R. Toll-Like Receptors and Innate Immunity. Nat Rev Immunol (2001) 1(2):135–45. doi: 10.1038/35100529
16. Botos I, Segal DM, Davies DR. The Structural Biology of Toll-Like Receptors. Structure (2011) 19(4):447–59. doi: 10.1016/j.str.2011.02.004
17. McClure R, Massari P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front Immunol (2014) 5:386. doi: 10.3389/fimmu.2014.00386
18. Sutmuller RPM, Morgan ME, Netea MG, Grauer O, Adema GJ. Toll-Like Receptors on Regulatory T Cells: Expanding Immune Regulation. Trends Immunol (2006) 27(8):387–93. doi: 10.1016/j.it.2006.06.005
19. Hua Z, Hou B. TLR Signaling in B-Cell Development and Activation. Cell Mol Immunol (2013) 10(2):103–6. doi: 10.1038/cmi.2012.61
20. Bhagavan NV, Ha C-E. Chapter 33 - Immunology. In: Bhagavan NV, Ha C-E, editors. Essentials of Medical Biochemistry. San Diego: Academic Press (2011). doi: 10.1016/B978-0-12-095461-2.00033-3
21. Akira S, Takeda K, Kaisho T. Toll-Like Receptors: Critical Proteins Linking Innate and Acquired Immunity. Nat Immunol (2001) 2(8):675–80. doi: 10.1038/90609
22. Medzhitov R, Janeway CA. Innate Immunity: The Virtues of a Nonclonal System of Recognition. Cell (1997) 91(3):295–8. doi: 10.1016/S0092-8674(00)80412-2
23. Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RAB. Phylogenetic Perspectives in Innate Immunity. Science (1999) 284(5418):1313–8. doi: 10.1126/science.284.5418.1313
24. Aderem A, Ulevitch RJ. Toll-Like Receptors in the Induction of the Innate Immune Response. Nature (2000) 406(6797):782–7. doi: 10.1038/35021228
25. Blasius AL, Beutler B. Intracellular Toll-Like Receptors. Immunity (2010) 32(3):305–15. doi: 10.1016/j.immuni.2010.03.012
26. Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DCG, et al. Ligand-Induced Conformational Changes Allosterically Activate Toll-Like Receptor 9. Nat Immunol (2007) 8(7):772–9. doi: 10.1038/ni1479
27. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-Adapter-Like) Is Required for Toll-Like Receptor-4 Signal Transduction. Nature (2001) 413(6851):78–83. doi: 10.1038/35092578
28. Horng T, Barton GM, Medzhitov R. TIRAP: An Adapter Molecule in the Toll Signaling Pathway. Nat Immunol (2001) 2(9):835–41. doi: 10.1038/ni0901-835
29. Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, et al. TRAM is Specifically Involved in the Toll-Like Receptor 4-Mediated MyD88-Independent Signaling Pathway. Nat Immunol (2003) 4(11):1144–50. doi: 10.1038/ni986
30. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 Signaling to IRF-3/7 and NF-Kappa B Involves the Toll Adapters TRAM and TRIF. J Exp Med (2003) 198(7):1043–55. doi: 10.1084/jem.20031023
31. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM Couples Endocytosis of Toll-Like Receptor 4 to the Induction of Interferon-Beta. Nat Immunol (2008) 9(4):361–8. doi: 10.1038/ni1569
32. Rowe DC, McGettrick AF, Latz E, Monks BG, Gay NJ, Yamamoto M, et al. The Myristoylation of TRIF-Related Adaptor Molecule Is Essential for Toll-Like Receptor 4 Signal Transduction. Proc Natl Acad Sci USA (2006) 103(16):6299–304. doi: 10.1073/pnas.0510041103
33. Kagan JC, Medzhitov R. Phosphoinositide-Mediated Adaptor Recruitment Controls Toll-Like Receptor Signaling. Cell (2006) 125(5):943–55. doi: 10.1016/j.cell.2006.03.047
34. O’Neill LA, Bowie AG. The Family of Five: TIR-Domain-Containing Adaptors in Toll-Like Receptor Signalling. Nat Rev Immunol (2007) 7(5):353–64. doi: 10.1038/nri2079
35. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
36. Kagan JC, Magupalli VG, Wu H. SMOCs: Supramolecular Organizing Centres That Control Innate Immunity. Nat Rev Immunol (2014) 14(12):821–6. doi: 10.1038/nri3757
37. Lin SC, Lo YC, Wu H. Helical Assembly in the MyD88-IRAK4-IRAK2 Complex in TLR/IL-1R Signalling. Nature (2010) 465(7300):885–U2. doi: 10.1038/nature09121
38. Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, et al. An Oligomeric Signaling Platform Formed by the Toll-Like Receptor Signal Transducers MyD88 and IRAK-4. J Biol Chem (2009) 284(37):25404–11. doi: 10.1074/jbc.M109.022392
39. Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, et al. A Promiscuous Lipid-Binding Protein Diversifies the Subcellular Sites of Toll-Like Receptor Signal Transduction. Cell (2014) 156(4):705–16. doi: 10.1016/j.cell.2014.01.019
40. Cao ZD, Henzel WJ, Gao XO. IRAK: A Kinase Associated With the Interleukin-1 Receptor. Science (1996) 271(5252):1128–31. doi: 10.1126/science.271.5252.1128
41. Li SY, Strelow A, Fontana EJ, Wesche H. IRAK-4: A Novel Member of the IRAK Family With the Properties of an IRAK-Kinase. Proc Natl Acad Sci USA (2002) 99(8):5567–72. doi: 10.1073/pnas.082100399
42. Kollewe C, Mackensen AC, Neumann D, Knop J, Cao P, Li SU, et al. Sequential Autophosphorylation Steps in the Interleukin-1 Receptor-Associated Kinase-1 Regulate its Availability as an Adapter in Interleukin-1 Signaling. J Biol Chem (2004) 279(7):5227–36. doi: 10.1074/jbc.M309251200
43. Sun J, Li N, Oh KS, Dutta B, Vayttaden SJ, Lin B, et al. Comprehensive RNAi-Based Screening of Human and Mouse TLR Pathways Identifies Species-Specific Preferences in Signaling Protein Use. Sci Signal (2016) 9(409):ra3. doi: 10.1126/scisignal.aab2191
44. Clark K, Nanda S, Cohen P. Molecular Control of the NEMO Family of Ubiquitin-Binding Proteins. Nat Rev Mol Cell Biol (2013) 14(10):673–85. doi: 10.1038/nrm3644
45. Windheim M, Stafford M, Peggie M, Cohen P. Interleukin-1 (IL-1) Induces the Lys63-Linked Polyubiquitination of IL-1 Receptor-Associated Kinase 1 to Facilitate NEMO Binding and the Activation of IkappaBalpha Kinase. Mol Cell Biol (2008) 28(5):1783–91. doi: 10.1128/MCB.02380-06
46. Gottipati S, Rao NL, Fung-Leung WP. IRAK1: A Critical Signaling Mediator of Innate Immunity. Cell Signal (2008) 20(2):269–76. doi: 10.1016/j.cellsig.2007.08.009
47. Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD. Lys63-Linked Polyubiquitination of IRAK-1 is Required for Interleukin-1 Receptor- and Toll-Like Receptor-Mediated NF-kappaB Activation. Mol Cell Biol (2008) 28(10):3538–47. doi: 10.1128/MCB.02098-07
48. Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-Specific Lys-63-Linked Tumor Necrosis Factor Receptor-Associated Factor 6 Auto-Ubiquitination is a Critical Determinant of I Kappa B Kinase Activation. J Biol Chem (2007) 282(6):4102–12. doi: 10.1074/jbc.M609503200
49. Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y. TRAF6 Autoubiquitination-Independent Activation of the Nfκb and MAPK Pathways in Response to IL-1 and RANKL. PloS One (2008) 3(12):e4064. doi: 10.1371/journal.pone.0004064
50. Hu L, Xu J, Xie X, Zhou Y, Tao P, Li H, et al. Oligomerization-Primed Coiled-Coil Domain Interaction With Ubc13 Confers Processivity to TRAF6 Ubiquitin Ligase Activity. Nat Commun (2017) 8(1):814. doi: 10.1038/s41467-017-01290-0
51. Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, et al. Direct Activation of Protein Kinases by Unanchored Polyubiquitin Chains. Nature (2009) 461(7260):114–9. doi: 10.1038/nature08247
52. Ea C-K, Deng L, Xia Z-P, Pineda G, Chen ZJ. Activation of IKK by TNFα Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Mol Cell (2006) 22(2):245–57. doi: 10.1016/j.molcel.2006.03.026
53. Wu C-J, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-Linked Polyubiquitination by NEMO is a Key Event in NF-κB Activation. Nat Cell Biol (2006) 8(4):398–406. doi: 10.1038/ncb1384
54. Bhoj VG, Chen ZJ. Ubiquitylation in Innate and Adaptive Immunity. Nature (2009) 458(7237):430–7. doi: 10.1038/nature07959
55. Lo Y-C, Lin S-C, Rospigliosi CC, Conze DB, Wu C-J, Ashwell JD, et al. Structural Basis for Recognition of Diubiquitins by NEMO. Mol Cell (2009) 33(5):602–15. doi: 10.1016/j.molcel.2009.01.012
56. Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific Recognition of Linear Ubiquitin Chains by NEMO is Important for NF-kappaB Activation. Cell (2009) 136(6):1098–109. doi: 10.1016/j.cell.2009.03.007
57. Tokunaga F, Sakata SI, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of Linear Polyubiquitylation of NEMO in NF-κb Activation. Nat Cell Biol (2009) 11(2):123–32. doi: 10.1038/ncb1821
58. Fujita H, Rahighi S, Akita M, Kato R, Sasaki Y, Wakatsuki S, et al. Mechanism Underlying IkappaB Kinase Activation Mediated by the Linear Ubiquitin Chain Assembly Complex. Mol Cell Biol (2014) 34(7):1322–35. doi: 10.1128/MCB.01538-13
59. Tokunaga F. Linear Ubiquitination-Mediated NF-κb Regulation and its Related Disorders. J Biochem (2013) 154(4):313–23. doi: 10.1093/jb/mvt079
60. Noad J, von der Malsburg A, Pathe C, Michel MA, D. Komander and F. Randow: LUBAC-Synthesized Linear Ubiquitin Chains Restrict Cytosol-Invading Bacteria by Activating Autophagy and NF-KappaB. Nat Microbiol (2017) 2:17063. doi: 10.1038/nmicrobiol.2017.63
61. Shimizu Y, Taraborrelli L, Walczak H. Linear Ubiquitination in Immunity. Immunol Rev (2015) 266(1):190–207. doi: 10.1111/imr.12309
62. Sasaki K, Iwai K. Roles of Linear Ubiquitinylation, a Crucial Regulator of NF-κb and Cell Death, in the Immune System. Immunol Rev (2015) 266(1):175–89. doi: 10.1111/imr.12308
63. Iwai K, Fujita H, Sasaki Y. Linear Ubiquitin Chains: NF-κb Signalling, Cell Death and Beyond. Nat Rev Mol Cell Biol (2014) 15(8):503–8. doi: 10.1038/nrm3836
64. Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. Essential Function for the Kinase TAK1 in Innate and Adaptive Immune Responses. Nat Immunol (2005) 6(11):1087–95. doi: 10.1038/ni1255
65. Schuman J, Chen Y, Podd A, Yu M, Liu H-H, Wen R, et al. A Critical Role of TAK1 in B-Cell Receptor–Mediated Nuclear Factor κb Activation. Blood (2009) 113(19):4566–74. doi: 10.1182/blood-2008-08-176057
66. Liu H-H, Xie M, Schneider MD, Chen ZJ. Essential Role of TAK1 in Thymocyte Development and Activation. Proc Natl Acad Sci (2006) 103(31):11677. doi: 10.1073/pnas.0603089103
67. Sato S, Sanjo H, Tsujimura T, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. TAK1 is Indispensable for Development of T Cells and Prevention of Colitis by the Generation of Regulatory T Cells. Int Immunol (2006) 18(10):1405–11. doi: 10.1093/intimm/dxl082
68. Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. The Kinase TAK1 Integrates Antigen and Cytokine Receptor Signaling for T Cell Development, Survival and Function. Nat Immunol (2006) 7(8):851–8. doi: 10.1038/ni1355
69. Ajibade AA, Wang QF, Cui J, Zou J, Xia XJ, Wang MJ, et al. TAK1 Negatively Regulates NF-Kappa B and P38 MAP Kinase Activation in Gr-1(+)CD11b(+) Neutrophils. Immunity (2012) 36(1):43–54. doi: 10.1016/j.immuni.2011.12.010
70. Lamothe B, Lai YJ, Hur L, Orozco NM, Wang J, Campos AD, et al. Deletion of TAK1 in the Myeloid Lineage Results in the Spontaneous Development of Myelomonocytic Leukemia in Mice. PloS One (2012) 7(12):e51228. doi: 10.1371/journal.pone.0051228
71. Ear T, Fortin CF, Simard FA, McDonald PP. Constitutive Association of TGF-β–Activated Kinase 1 With the IκB Kinase Complex in the Nucleus and Cytoplasm of Human Neutrophils and Its Impact on Downstream Processes. J Immunol (2010) 184(7):3897–906. doi: 10.4049/jimmunol.0902958
72. Ajibade AA, Wang HY, Wang RF. Cell Type-Specific Function of TAK1 in Innate Immune Signaling. Trends Immunol (2013) 34(7):307–16. doi: 10.1016/j.it.2013.03.007
73. Xing C, Wang M, Ajibade AA, Tan P, Fu C, Chen L, et al. Microbiota Regulate Innate Immune Signaling and Protective Immunity Against Cancer. Cell Host Microbe (2021) 29(6):959–974.e7. doi: 10.1016/j.chom.2021.03.016
74. Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal Regulation of MyD88–IRF-7 Signalling for Robust Type-I Interferon Induction. Nature (2005) 434(7036):1035–40. doi: 10.1038/nature03547
75. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the Master Regulator of Type-I Interferon-Dependent Immune Responses. Nature (2005) 434(7034):772–7. doi: 10.1038/nature03464
76. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral Role of IRF-5 in the Gene Induction Programme Activated by Toll-Like Receptors. Nature (2005) 434(7030):243–9. doi: 10.1038/nature03308
77. Kawai T, Akira S. Toll-Like Receptor and RIG-1-Like Receptor Signaling. Ann N Y Acad Sci (2008) 1143(1):1–20. doi: 10.1196/annals.1443.020
78. Ren JY, Chen X, Chen ZJJ. IKK Beta is an IRF5 Kinase That Instigates Inflammation. Proc Natl Acad Sci USA (2014) 111(49):17438–43. doi: 10.1073/pnas.1418516111
79. Lopez-Pelaez M, Lamont DJ, Peggie M, Shpiro N, Gray NS, Cohen P. Protein Kinase IKK Beta-Catalyzed Phosphorylation of IRF5 at Ser462 Induces its Dimerization and Nuclear Translocation in Myeloid Cells. Proc Natl Acad Sci USA (2014) 111(49):17432–7. doi: 10.1073/pnas.1418399111
80. Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, et al. The Interferon Regulatory Factor, IRF5, Is a Central Mediator of Toll-Like Receptor 7 Signaling. J Biol Chem (2005) 280(17):17005–12. doi: 10.1074/jbc.M412584200
81. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science (2003) 301(5633):640–3. doi: 10.1126/science.1087262
82. Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han JH, et al. Upregulation of Costimulatory Molecules Induced by Lipopolysaccharide and Double-Stranded RNA Occurs by Trif-Dependent and Trif-Independent Pathways. Nat Immunol (2003) 4(12):1223–9. doi: 10.1038/ni1010
83. Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA. Rip1 Mediates the Trif-Dependent Toll-Like Receptor 3- and 4-Induced NF-κB Activation But Does Not Contribute to Interferon Regulatory Factor 3 Activation*. J Biol Chem (2005) 280(44):36560–6. doi: 10.1074/jbc.M506831200
84. Gohda J, Matsumura T, Inoue J-I. Cutting Edge: TNFR-Associated Factor (TRAF) 6 Is Essential for MyD88-Dependent Pathway But Not Toll/IL-1 Receptor Domain-Containing Adaptor-Inducing IFN-β (TRIF)-Dependent Pathway in TLR Signaling. J Immunol (2004) 173(5):2913. doi: 10.4049/jimmunol.173.5.2913
85. Chang M, Jin W, Sun S-C. Peli1 Facilitates TRIF-Dependent Toll-Like Receptor Signaling and Proinflammatory Cytokine Production. Nat Immunol (2009) 10(10):1089–95. doi: 10.1038/ni.1777
86. McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-Regulatory Factor 3-Dependent Gene Expression is Defective in Tbk1-Deficient Mouse Embryonic Fibroblasts. Proc Natl Acad Sci USA (2004) 101(1):233–8. doi: 10.1073/pnas.2237236100
87. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin RT, Hiscott J. Triggering the Interferon Antiviral Response Through an IKK-Related Pathway. Science (2003) 300(5622):1148–51. doi: 10.1126/science.1081315
88. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKK Epsilon and TBK1 are Essential Components of the IRF3 Signaling Pathway. Nat Immunol (2003) 4(5):491–6. doi: 10.1038/ni921
89. Liu SQ, Cai X, Wu JX, Cong Q, Chen X, Li T, et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science (2015) 347(6227):1217–U17. doi: 10.1126/science.aaa2630
90. Zhao B, Shu C, Gao X, Sankaran B, Du F, Shelton CL, et al. Structural Basis for Concerted Recruitment and Activation of IRF-3 by Innate Immune Adaptor Proteins. Proc Natl Acad Sci (2016) 113(24):E3403. doi: 10.1073/pnas.1603269113
91. Banchereau J, Steinman RM. Dendritic Cells and the Control of Immunity. Nature (1998) 392(6673):245–52. doi: 10.1038/32588
92. Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-Like Receptors Control Activation of Adaptive Immune Responses. Nat Immunol (2001) 2(10):947–50. doi: 10.1038/ni712
93. Palliser D, Ploegh H, Boes M. Myeloid Differentiation Factor 88 Is Required for Cross-Priming In Vivo. J Immunol (2004) 172(6):3415. doi: 10.4049/jimmunol.172.6.3415
94. Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, et al. IκB Kinase-α is Critical for Interferon-α Production Induced by Toll-Like Receptors 7 and 9. Nature (2006) 440(7086):949–53. doi: 10.1038/nature04641
95. Ito T, Amakawa R, Kaisho T, Hemmi H, Tajima K, Uehira K, et al. Interferon-α and Interleukin-12 Are Induced Differentially by Toll-Like Receptor 7 Ligands in Human Blood Dendritic Cell Subsets. J Exp Med (2002) 195(11):1507–12. doi: 10.1084/jem.20020207
96. Reis e Sousa C, Sher A, Kaye P. The Role of Dendritic Cells in the Induction and Regulation of Immunity to Microbial Infection. Curr Opin Immunol (1999) 11(4):392–9. doi: 10.1016/S0952-7915(99)80066-1
97. Nair-Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, Huang YJ, et al. TLR Signals Induce Phagosomal MHC-I Delivery From the Endosomal Recycling Compartment to Allow Cross-Presentation. Cell (2014) 158(3):506–21. doi: 10.1016/j.cell.2014.04.054
98. Blander JM, Medzhitov R. Toll-Dependent Selection of Microbial Antigens for Presentation by Dendritic Cells. Nature (2006) 440(7085):808–12. doi: 10.1038/nature04596
99. Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of Lysosomal Function During Dendritic Cell Maturation. Science (2003) 299(5611):1400–3. doi: 10.1126/science.1080106
100. Turley SJ, Inaba K, Garrett WS, Ebersold M, Unternaehrer J, Steinman RM, et al. Transport of Peptide-MHC Class II Complexes in Developing Dendritic Cells. Science (2000) 288(5465):522–7. doi: 10.1126/science.288.5465.522
101. Horrevorts SK, Duinkerken S, Bloem K, Secades P, Kalay H, Musters RJ, et al. Toll-Like Receptor 4 Triggering Promotes Cytosolic Routing of DC-SIGN-Targeted Antigens for Presentation on MHC Class I. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01231
102. Sheen J-H, Strainic MG, Liu J, Zhang W, Yi Z, Medof ME, et al. TLR-Induced Murine Dendritic Cell (DC) Activation Requires DC-Intrinsic Complement. J Immunol (2017) 199(1):278–91. doi: 10.4049/jimmunol.1700339
103. Sheng K-C, Day S, Wright MD, Stojanovska L, Apostolopoulos V. Enhanced Dendritic Cell-Mediated Antigen-Specific CD4+ T Cell Responses: IFN-Gamma Aids TLR Stimulation. J Drug Deliv (2013) 2013:516749. doi: 10.1155/2013/516749
104. Gao Y, Zhang M, Chen L, Hou M, Ji M, Wu G. Deficiency in TLR2 But Not in TLR4 Impairs Dendritic Cells Derived IL-10 Responses to Schistosome Antigens. Cell Immunol (2012) 272(2):242–50. doi: 10.1016/j.cellimm.2011.10.007
105. Guo X, Wu N, Shang Y, Liu X, Wu T, Zhou Y, et al. The Novel Toll-Like Receptor 2 Agonist SUP3 Enhances Antigen Presentation and T Cell Activation by Dendritic Cells. Front Immunol (2017) 8:158. doi: 10.3389/fimmu.2017.00158
106. Sharma P, Levy O, Dowling DJ. The TLR5 Agonist Flagellin Shapes Phenotypical and Functional Activation of Lung Mucosal Antigen Presenting Cells in Neonatal Mice. Front Immunol (2020) 11:171. doi: 10.3389/fimmu.2020.00171
107. Lombardi V, Van Overtvelt L, Horiot S, Moingeon P. Human Dendritic Cells Stimulated via TLR7 and/or TLR8 Induce the Sequential Production of Il-10, IFN-γ, and IL-17A by Naive CD4+ T Cells. J Immunol (2009) 182(6):3372–9. doi: 10.4049/jimmunol.0801969
108. Dolina JS, Lee J, Griswold RQ, Labarta-Bajo L, Kannan S, Greenbaum JA, et al. TLR9 Sensing of Self-DNA Controls Cell-Mediated Immunity to Listeria Infection via Rapid Conversion of Conventional CD4+ T Cells to Treg. Cell Rep (2020) 31(1):107249. doi: 10.1016/j.celrep.2020.01.040
109. Mandraju R, Murray S, Forman J, Pasare C. Differential Ability of Surface and Endosomal TLRs to Induce CD8 T Cell Responses In Vivo. J Immunol (Baltimore Md 1950) (2014) 192(9):4303–15. doi: 10.4049/jimmunol.1302244
110. Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdörfer B, Giese T, et al. Quantitative Expression of Toll-Like Receptor 1–10 mRNA in Cellular Subsets of Human Peripheral Blood Mononuclear Cells and Sensitivity to CpG Oligodeoxynucleotides. J Immunol (2002) 168(9):4531. doi: 10.4049/jimmunol.168.9.4531
111. Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and Complementarity in Microbial Molecule Recognition by Human Myeloid and Plasmacytoid Dendritic Cells. Eur J Immunol (2001) 31(11):3388–93. doi: 10.1002/1521-4141(200111)31:11<3388::AID-IMMU3388>3.0.CO;2-Q
112. Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, et al. Subsets of Human Dendritic Cell Precursors Express Different Toll-Like Receptors and Respond to Different Microbial Antigens. J Exp Med (2001) 194(6):863–9. doi: 10.1084/jem.194.6.863
113. Visintin A, Mazzoni A, Spitzer JH, Wyllie DH, Dower SK, Segal DM. Regulation of Toll-Like Receptors in Human Monocytes and Dendritic Cells. J Immunol (2001) 166(1):249. doi: 10.4049/jimmunol.166.1.249
114. Edwards AD, Diebold SS, Slack EMC, Tomizawa H, Hemmi H, Kaisho T, et al. Toll-Like Receptor Expression in Murine DC Subsets: Lack of TLR7 Expression by CD8 Alpha(+) DC Correlates With Unresponsiveness to Imidazoquinolines. Eur J Immunol (2003) 33(4):827–33. doi: 10.1002/eji.200323797
115. Boonstra A, Asselin-Paturel C, Gilliet M, Crain C, Trinchieri G, Liu Y-J, et al. Flexibility of Mouse Classical and Plasmacytoid-Derived Dendritic Cells in Directing T Helper Type 1 and 2 Cell Development: Dependency on Antigen Dose and Differential Toll-Like Receptor Ligation. J Exp Med (2003) 197(1):101–9. doi: 10.1084/jem.20021908
116. Merad M, Sathe P, Helft J, Miller J, Mortha A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu Rev Immunol (2013) 31. doi: 10.1146/annurev-immunol-020711-074950
117. Anderson DA, Murphy KM. Models of Dendritic Cell Development Correlate Ontogeny With Function. Adv Immunol (2019) 143. doi: 10.1016/bs.ai.2019.09.001
118. Mills KH. TLR-Dependent T Cell Activation in Autoimmunity. Nat Rev Immunol (2011) 11(12):807–22. doi: 10.1038/nri3095
119. Kumar V. Toll-Like Receptors in Adaptive Immunity. Handb Exp Pharmacol (2021) 225(3):510–9. doi: 10.1007/164_2021_543
120. Wang RF. The Role of MHC Class II-Restricted Tumor Antigens and CD4(+) T Cells in Antitumor Immunity. Trends Immunol (2001) 22(5):269–76. doi: 10.1016/s1471-4906(01)01896-8
121. Wang RF, Miyahara Y, Wang HY. Toll-Like Receptors and Immune Regulation: Implications for Cancer Therapy. Oncogene (2008) 27(2):181–9. doi: 10.1038/sj.onc.1210906
122. Karim AF, Reba SM, Li Q, Boom WH, Rojas RE. Toll Like Receptor 2 Engagement on CD4(+) T Cells Promotes TH9 Differentiation and Function. Eur J Immunol (2017) 47(9):1513–24. doi: 10.1002/eji.201646846
123. Komai-Koma M, Jones L, Ogg GS, Xu DM, Liew FY. TLR2 is Expressed on Activated T Cells as a Costimulatory Receptor. Proc Natl Acad Sci USA (2004) 101(9):3029–34. doi: 10.1073/pnas.0400171101
124. Imanishi T, Hara H, Suzuki S, Suzuki N, Akira S, Saito T. Cutting Edge: TLR2 Directly Triggers Th1 Effector Functions. J Immunol (2007) 178(11):6715–9. doi: 10.4049/jimmunol.178.11.6715
125. Biswas A, Banerjee P, Biswas T. Porin of Shigella Dysenteriae Directly Promotes Toll-Like Receptor 2-Mediated CD4(+) T Cell Survival and Effector Function. Mol Immunol (2009) 46(15):3076–85. doi: 10.1016/j.molimm.2009.06.006
126. Sinnott BD, Park B, Boer MC, Lewinsohn DA, Lancioni CL. Direct TLR-2 Costimulation Unmasks the Proinflammatory Potential of Neonatal CD4(+) T Cells. J Immunol (2016) 197(1):68–77. doi: 10.4049/jimmunol.1501297
127. Reba SM, Li Q, Onwuzulike S, Ding XD, Karim AF, Hernandez Y, et al. TLR2 Engagement on CD4+T Cells Enhances Effector Functions and Protective Responses to Mycobacterium Tuberculosis. Eur J Immunol (2014) 44(5):1410–21. doi: 10.1002/eji.201344100
128. Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S, et al. TLR2 Engagement on CD8 T Cells Lowers the Threshold for Optimal Antigen-Induced T Cell Activation. Eur J Immunol (2006) 36(7):1684–93. doi: 10.1002/eji.200636181
129. Lee S-M, Joo Y-D, Seo S-K. Expression and Function of TLR2 on CD4 Versus CD8 T Cells. Immune Netw (2009) 9(4):127–32. doi: 10.4110/in.2009.9.4.127
130. Mercier BC, Cottalorda A, Coupet CA, Marvel J, Bonnefoy-Berard N. TLR2 Engagement on CD8 T Cells Enables Generation of Functional Memory Cells in Response to a Suboptimal TCR Signal. J Immunol (2009) 182(4):1860–7. doi: 10.4049/jimmunol.0801167
131. Zhang Y, Jones M, McCabe A, Winslow GM, Avram D, MacNamara KC. MyD88 Signaling in CD4 T Cells Promotes IFN-γ Production and Hematopoietic Progenitor Cell Expansion in Response to Intracellular Bacterial Infection. J Immunol (Baltimore Md 1950) (2013) 190(9):4725–35. doi: 10.4049/jimmunol.1203024
132. Xing S, Zhang X, Liu JH, Huang X, Zhou P. Host MyD88 Signaling Protects Against Acute Graft-Versus-Host Disease After Allogeneic Bone Marrow Transplantation. Clin Exp Immunol (2019) 195(1):121–31. doi: 10.1111/cei.13215
133. Satomi M, Daigo H, Masanori K, Hiroyuki O, Eiko H, Emi Y, et al. Myeloid Differentiation Factor 88 Signaling in Donor T Cells Accelerates Graft-Versus-Host Disease. Haematologica (2020) 105(1):226–34. doi: 10.3324/haematol.2018.203380
134. Chang J, Burkett PR, Borges CM, Kuchroo VK, Turka LA, Chang C-H. MyD88 is Essential to Sustain mTOR Activation Necessary to Promote T Helper 17 Cell Proliferation by Linking IL-1 and IL-23 Signaling. Proc Natl Acad Sci USA (2013) 110(6):2270–5. doi: 10.1073/pnas.1206048110
135. Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-Like Receptor 4 Signaling in T Cells Promotes Autoimmune Inflammation. Proc Natl Acad Sci USA (2012) 109(32):13064–9. doi: 10.1073/pnas.1120585109
136. González-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, et al. TLR4 Signaling in Effector CD4+ T Cells Regulates TCR Activation and Experimental Colitis in Mice. J Clin Invest (2010) 120(2):570–81. doi: 10.1172/JCI40055
137. Zanin-Zhorov A, Tal-Lapidot G, Cahalon L, Cohen-Sfady M, Pevsner-Fischer M, Lider O, et al. Cutting Edge: T Cells Respond to Lipopolysaccharide Innately via TLR4 Signaling. J Immunol (2007) 179(1):41. doi: 10.4049/jimmunol.179.1.41
138. Cohen I, Zanin-Zhorov A. Signaling via TLR2 and TLR4 Directly Down-Regulates T Cell Effector Functions: The Regulatory Face of Danger Signals. Front Immunol (2013) 4:211. doi: 10.3389/fimmu.2013.00211
139. Okeke EB, Okwor I, Uzonna JE. Regulatory T Cells Restrain CD4<sup<+</sup< T Cells From Causing Unregulated Immune Activation and Hypersensitivity to Lipopolysaccharide Challenge. J Immunol (2014) 193(2):655. doi: 10.4049/jimmunol.1303064
140. Sahoo SS, Pratheek BM, Meena VS, Nayak TK, Kumar PS, Bandyopadhyay S, et al. VIPER Regulates Naive T Cell Activation and Effector Responses: Implication in TLR4 Associated Acute Stage T Cell Responses. Sci Rep (2018) 8(1):7118. doi: 10.1038/s41598-018-25549-8
141. Qu J, Yu X, Jin C, Feng Y, Xie S, Xie H, et al. TLR7 Modulated T Cell Response in the Mesenteric Lymph Node of Schistosoma Japonicum-Infected C57BL/6 Mice. J Immunol Res (2019) 2019:2691808. doi: 10.1155/2019/2691808
142. Li Q, Yan Y, Liu J, Huang X, Zhang X, Kirschning C, et al. Toll-Like Receptor 7 Activation Enhances CD8+ T Cell Effector Functions by Promoting Cellular Glycolysis. Front Immunol (2019) 10:2191(2191). doi: 10.3389/fimmu.2019.02191
143. Tabiasco J, Devêvre E, Rufer N, Salaun B, Cerottini J-C, Speiser D, et al. Human Effector CD8+ T Lymphocytes Express TLR3 as a Functional Coreceptor. J Immunol (2006) 177(12):8708–13. doi: 10.4049/jimmunol.177.12.8708
144. Di S, Zhou M, Pan Z, Sun R, Chen M, Jiang H, et al. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells. Front Oncol (2019) 9:241. doi: 10.3389/fonc.2019.00241
145. Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, et al. Toll-Like Receptor 2 Costimulation Potentiates the Antitumor Efficacy of CAR T Cells. Leukemia (2018) 32(3):801–8. doi: 10.1038/leu.2017.249
146. George P, Dasyam N, Giunti G, Mester B, Bauer E, Andrews B, et al. Third-Generation Anti-CD19 Chimeric Antigen Receptor T-Cells Incorporating a TLR2 Domain for Relapsed or Refractory B-Cell Lymphoma: A Phase I Clinical Trial Protocol (ENABLE). BMJ Open (2020) 10(2):e034629. doi: 10.1136/bmjopen-2019-034629
147. Vignali DAA, Collison LW, Workman CJ. How Regulatory T Cells Work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
148. Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, et al. Induction of FoxP3 and Acquisition of T Regulatory Activity by Stimulated Human CD4(+)CD25(-) T Cells. J Clin Invest (2003) 112(9):1437–43. doi: 10.1172/jci200319441
149. Khattri R, Cox T, Yasayko SA, Ramsdell F. An Essential Role for Scurfin in CD4(+)CD25(+) T Regulatory Cells. Nat Immunol (2003) 4(4):337–42. doi: 10.1038/ni909
150. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
151. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4(+)CD25(+) Regulatory T Cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
152. Wang HY, Lee DA, Peng GY, Guo Z, Li YC, Kiniwa Y, et al. Tumor-Specific Human CD4(+) Regulatory T Cells and Their Ligands: Implications for Immunotherapy. Immunity (2004) 20(1):107–18. doi: 10.1016/s1074-7613(03)00359-5
153. Curiel TJ, Coukos G, Zou LH, Alvarez X, Cheng P, Mottram P, et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat Med (2004) 10(9):942–9. doi: 10.1038/nm1093
154. Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of Regulatory T Cells is Increased in Peripheral Blood and Tumor Microenvironment of Patients With Pancreas or Breast Adenocarcinoma. J Immunol (2002) 169(5):2756–61. doi: 10.4049/jimmunol.169.5.2756
155. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T Cells in Tumors From Patients With Early-Stage non-Small Cell Lung Cancer and Late-Stage Ovarian Cancer. Cancer Res (2001) 61(12):4766–72.
156. Wang HY, Peng GY, Guo Z, Shevach EM, Wang RF. Recognition of a New ARTC1 Peptide Ligand Uniquely Expressed in Tumor Cells by Antigen-Specific CD4(+) Regulatory T Cells. J Immunol (2005) 174(5):2661–70. doi: 10.4049/jimmunol.174.5.2661
157. Wang HY, Wang RF. Regulatory T Cells and Cancer. Curr Opin Immunol (2007) 19(2):217–23. doi: 10.1016/j.coi.2007.02.004
158. Peng G, Wang HY, Peng W, Kiniwa Y, Seo KH, Wang RF. Tumor-Infiltrating Gamma Delta T Cells Suppress T and Dendritic Cell Function via Mechanisms Controlled by a Unique Toll-Like Receptor Signaling Pathway. Immunity (2007) 27(2):334–48. doi: 10.1016/j.immuni.2007.05.020
159. Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, et al. CD8+ Foxp3+ Regulatory T Cells Mediate Immunosuppression in Prostate Cancer. Clin Cancer Res (2007) 13(23):6947–58. doi: 10.1158/1078-0432.ccr-07-0842
160. Peng GY, Guo Z, Kiniwa Y, Voo KS, Peng WY, Fu TH, et al. Toll-Like, Receptor 8-Mediated Reversal of CD4(+) Regulatory T Cell Function. Science (2005) 309(5739):1380–4. doi: 10.1126/science.1113401
161. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-Specific Recognition of Single-Stranded RNA via Toll-Like Receptor 7 and 8. Science (2004) 303(5663):1526–9. doi: 10.1126/science.1093620
162. Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab (2019) 29(1):103–123.e5. doi: 10.1016/j.cmet.2018.09.020
163. Shang W, Xu R, Xu T, Wu M, Xu J, Wang F. Ovarian Cancer Cells Promote Glycolysis Metabolism and TLR8-Mediated Metabolic Control of Human CD4+ T Cells. Front Oncol (2020) 10:570899(1956). doi: 10.3389/fonc.2020.570899
164. Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 Independently Confer Responsiveness to the Antiviral Compound R-848. Nat Immunol (2002) 3(6):499–9. doi: 10.1038/ni0602-499
165. Netea MG, Sutmuller R, Hermann C, van der Graaf CAA, van der Meer JWM, van Krieken JH, et al. Toll-Like Receptor 2 Suppresses Immunity Against Candida Albicans Through Induction of IL-10 and Regulatory T Cells. J Immunol (2004) 172(6):3712–8. doi: 10.4049/jimmunol.172.6.3712
166. Sutmuller RPM, den Brok M, Kramer M, Bennink EJ, Toonen LWJ, Kullberg BJ, et al. Toll-Like Receptor 2 Controls Expansion and Function of Regulatory T Cells. J Clin Invest (2006) 116(2):485–94. doi: 10.1172/jci25439
167. Liu H, Komai-Koma M, Xu D, Liew FY. Toll-Like Receptor 2 Signaling Modulates the Functions of CD4+ CD25+ Regulatory T Cells. Proc Natl Acad Sci USA (2006) 103(18):7048–53. doi: 10.1073/pnas.0601554103
168. LaRosa DF, Gelman AE, Rahman AH, Zhang J, Turka LA, Walsh PT. CpG DNA Inhibits CD4+CD25+ Treg Suppression Through Direct MyD88-Dependent Costimulation of Effector CD4+ T Cells. Immunol Lett (2007) 108(2):183–8. doi: 10.1016/j.imlet.2006.12.007
169. Urry Z, Xystrakis E, Richards DF, McDonald J, Sattar Z, Cousins DJ, et al. Ligation of TLR9 Induced on Human IL-10-Secreting Tregs by 1alpha,25-Dihydroxyvitamin D3 Abrogates Regulatory Function. J Clin Invest (2009) 119(2):387–98. doi: 10.1172/JCI32354
170. Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4(+) T Cells Express TLR5 and its Ligand Flagellin Enhances the Suppressive Capacity and Expression of FOXP3 in CD4(+)CD25(+) T Regulatory Cells. J Immunol (2005) 175(12):8051–9. doi: 10.4049/jimmunol.175.12.8051
171. Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T Cells Selectively Express Toll-Like Receptors and are Activated by Lipopolysaccharide. J Exp Med (2003) 197(4):403–11. doi: 10.1084/jem.20021633
172. Liew FY, Xu DM, Brint EK, O’Neill LAJ. Negative Regulation of Toll-Like Receptor-Mediated Immune Responses. Nat Rev Immunol (2005) 5(6):446–58. doi: 10.1038/nri1630
173. Cui J, Chen YJ, Wang HY, Wang RF. Mechanisms and Pathways of Innate Immune Activation and Regulation in Health and Cancer. Hum Vaccines Immunother (2014) 10(11):3270–85. doi: 10.4161/21645515.2014.979640
174. Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, et al. CD14 Controls the LPS-Induced Endocytosis of Toll-Like Receptor 4. Cell (2011) 147(4):868–80. doi: 10.1016/j.cell.2011.09.051
175. Baumann CL, Aspalter IM, Sharif O, Pichlmair A, Bluml S, Grebien F, et al. CD14 is a Coreceptor of Toll-Like Receptors 7 and 9. J Exp Med (2010) 207(12):2689–701. doi: 10.1084/jem.20101111
176. LeBouder E, Rey-Nores JE, Rushmere NK, Grigorov M, Lawn SD, Affolter M, et al. Soluble Forms of Toll-Like Receptor (TLR)2 Capable of Modulating TLR2 Signaling are Present in Human Plasma and Breast Milk. J Immunol (2003) 171(12):6680–9. doi: 10.4049/jimmunol.171.12.6680
177. Iwami K, Matsuguchi T, Masuda A, Kikuchi T, Musikacharoen T, Yoshikai Y. Cutting Edge: Naturally Occurring Soluble Form of Mouse Toll-Like Receptor 4 Inhibits Lipopolysaccharide Signaling. J Immunol (2000) 165(12):6682–6. doi: 10.4049/jimmunol.165.12.6682
178. Brint EK, Xu DM, Liu HY, Dunne A, McKenzie ANJ, O’Neill LAJ, et al. ST2 is an Inhibitor of Interleukin 1 Receptor and Toll-Like Receptor 4 Signaling and Maintains Endotoxin Tolerance. Nat Immunol (2004) 5(4):373–9. doi: 10.1038/ni1050
179. Garlanda C, Riva F, Polentarutti N, Buracchi C, Sironi M, De Bortoli M, et al. Intestinal Inflammation in Mice Deficient in Tir8, an Inhibitory Member of the IL-1 Receptor Family. Proc Natl Acad Sci USA (2004) 101(10):3522–6. doi: 10.1073/pnas.0308680101
180. Wald D, Qin JZ, Zhao ZD, Qian YC, Naramura M, Tian LP, et al. SIGIRR, a Negative Regulator of Toll-Like Receptor-Interleukin 1 Receptor Signaling. Nat Immunol (2003) 4(9):920–7. doi: 10.1038/ni968
181. Diehl GE, Yue HH, Hsieh K, Kuang AA, Ho M, Morici LA, et al. TRAIL-R as a Negative Regulator of Innate Immune Cell Responses. Immunity (2004) 21(6):877–89. doi: 10.1016/j.immuni.2004.11.008
182. Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, et al. SOCS-1 Participates in Negative Regulation of LPS Responses. Immunity (2002) 17(5):677–87. doi: 10.1016/s1074-7613(02)00449-1
183. Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, et al. SOCS1/JAB is a Negative Regulator of LPS-Induced Macrophage Activation. Immunity (2002) 17(5):583–91. doi: 10.1016/s1074-7613(02)00446-6
184. Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of Interleukin 1 Receptor/Toll-Like Receptor Signaling Through the Alternatively Spliced, Short Form of MyD88 is Due to its Failure to Recruit IRAK-4. J Exp Med (2003) 197(2):263–8. doi: 10.1084/jem.20021790
185. Janssens S, Burns K, Tschopp J, Beyaert R. Regulation of Interleukin-1-and Lipopolysaccharide-Induced NF-Kappa B Activation by Alternative Splicing of Myd88. Curr Biol (2002) 12(6):467–71. doi: 10.1016/s0960-9822(02)00712-1
186. Zhang GL, Ghosh S. Negative Regulation of Toll-Like Receptor-Mediated Signaling by Tollip. J Biol Chem (2002) 277(9):7059–65. doi: 10.1074/jbc.M109537200
187. Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Medzhitov R, Flavell RA. IRAK-M is a Negative Regulator of Toll-Like Receptor Signaling. Cell (2002) 110(2):191–202. doi: 10.1016/s0092-8674(02)00827-9
188. Shembade N, Ma A, Harhaj EW. Inhibition of NF-Kappa B Signaling by A20 Through Disruption of Ubiquitin Enzyme Complexes. Science (2010) 327(5969):1135–9. doi: 10.1126/science.1182364
189. Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The Ubiquitin-Modifying Enzyme A20 is Required for Termination of Toll-Like Receptor Responses. Nat Immunol (2004) 5(10):1052–60. doi: 10.1038/ni1110
190. Lee AJ, Zhou XF, Chang M, Hunzeker J, Bonneau RH, Zhou DP, et al. Regulation of Natural Killer T-Cell Development by Deubiquitinase CYLD. EMBO J (2010) 29(9):1600–12. doi: 10.1038/emboj.2010.31
191. Reiley WW, Jin W, Lee AJ, Wright A, Wu XF, Tewalt EF, et al. Deubiquitinating Enzyme CYLD Negatively Regulates the Ubiquitin-Dependent Kinase Tak1 and Prevents Abnormal T Cell Responses. J Exp Med (2007) 204(6):1475–85. doi: 10.1084/jem.20062694
192. Reiley WW, Zhang MY, Jin W, Losiewicz M, Donohue KB, Norbury CC, et al. Regulation of T Cell Development by the Deubiquitinating Enzyme CYLD. Nat Immunol (2006) 7(4):411–7. doi: 10.1038/ni1315
193. Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD Is a Deubiquitinating Enzyme That Negatively Regulates NF-Kappa B Activation by TNFR Family Members. Nature (2003) 424(6950):793–6. doi: 10.1038/nature01803
194. Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The Tumour Suppressor CYLD Negatively Regulates NF-Kappa B Signalling by Deubiquitination. Nature (2003) 424(6950):801–5. doi: 10.1038/nature01802
195. Wang C, Chen TY, Zhang J, Yang MJ, Li N, Xu XF, et al. The E3 Ubiquitin Ligase Nrdp1 ’Preferentially’ Promotes TLR-Mediated Production of Type I Interferon. Nat Immunol (2009) 10(7):744–U100. doi: 10.1038/ni.1742
196. Tong YZ, Cui J, Li QT, Zou J, Wang HY, Wang RF. Enhanced TLR-Induced NF-Kappa B Signaling and Type I Interferon Responses in NLRC5 Deficient Mice. Cell Res (2012) 22(5):822–35. doi: 10.1038/cr.2012.53
197. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen HT, et al. The Innate Immune Sensor NLRC3 Attenuates Toll-Like Receptor Signaling via Modification of the Signaling Adaptor TRAF6 and Transcription Factor NF-Kappa B. Nat Immunol (2012) 13(9):823–31. doi: 10.1038/ni.2378
198. Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 Negatively Regulates Innate Immunity and Host Defence Against Bacterial Pathogens. Nature (2012) 488(7411):389–+. doi: 10.1038/nature11250
199. Xia XJ, Cui J, Wang HLY, Zhu L, Matsueda S, Wang QF, et al. NLRX1 Negatively Regulates TLR-Induced NF-Kappa B Signaling by Targeting TRAF6 and IKK. Immunity (2011) 34(6):843–53. doi: 10.1016/j.immuni.2011.02.022
200. Kumar H, Pandey S, Zou JA, Kumagai Y, Takahashi K, Akira S, et al. NLRC5 Deficiency Does Not Influence Cytokine Induction by Virus and Bacteria Infections. J Immunol (2011) 186(2):994–1000. doi: 10.4049/jimmunol.1002094
201. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 Protein Attenuates Inflammatory Responses to Infection by Interfering With the RIG-I-MAVS and TRAF6-NF-Kappa B Signaling Pathways. Immunity (2011) 34(6):854–65. doi: 10.1016/j.immuni.2011.03.026
202. Cui J, Zhu L, Xia XJ, Wang HY, Legras X, Hong J, et al. NLRC5 Negatively Regulates the NF-Kappa B and Type I Interferon Signaling Pathways. Cell (2010) 141(3):483–96. doi: 10.1016/j.cell.2010.03.040
203. Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 Limits the Activation of Inflammatory Pathways. J Immunol (2010) 185(3):1681–91. doi: 10.4049/jimmunol.0903900
204. Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a Mitochondrial NOD-Like Receptor That Amplifies NF-Kappa B and JNK Pathways by Inducing Reactive Oxygen Species Production. EMBO Rep (2008) 9(3):293–300. doi: 10.1038/sj.embor.7401161
205. Wu C, Su Z, Lin M, Ou J, Zhao W, Cui J, et al. NLRP11 Attenuates Toll-Like Receptor Signalling by Targeting TRAF6 for Degradation via the Ubiquitin Ligase RNF19A. Nat Commun (2017) 8(1):1977–7. doi: 10.1038/s41467-017-02073-3
206. Meng Q, Cai C, Sun T, Wang Q, Xie W, Wang R, et al. Reversible Ubiquitination Shapes NLRC5 Function and Modulates NF-κb Activation Switch. J Cell Biol (2015) 211(5):1025–40. doi: 10.1083/jcb.201505091
207. Chuang TH, Ulevitch RJ. Triad3A, an E3 Ubiquitin-Protein Ligase Regulating Toll-Like Receptors. Nat Immunol (2004) 5(5):495–502. doi: 10.1038/ni1066
208. Shi M, Deng WW, Bi EG, Mao KR, Ji YY, Lin GM, et al. TRIM30 Alpha Negatively Regulates TLR-Mediated NF-Kappa B Activation by Targeting TAB2 and TAB3 for Degradation. Nat Immunol (2008) 9(4):369–77. doi: 10.1038/ni1577
209. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a Regulator of Mitochondrial Antiviral Immunity. Nature (2008) 451(7178):573–U8. doi: 10.1038/nature06501
210. Kobayashi KS, van den Elsen PJ. NLRC5: A Key Regulator of MHC Class I-Dependent Immune Responses. Nat Rev Immunol (2012) 12(12):813–20. doi: 10.1038/nri3339
211. Feng Y, Duan T, Du Y, Jin S, Wang M, Cui J, et al. LRRC25 Functions as an Inhibitor of NF-κb Signaling Pathway by Promoting P65/RelA for Autophagic Degradation. Sci Rep (2017) 7(1):13448–8. doi: 10.1038/s41598-017-12573-3
212. Du Y, Duan T, Feng Y, Liu Q, Lin M, Cui J, et al. LRRC25 Inhibits Type I IFN Signaling by Targeting ISG15-Associated RIG-I for Autophagic Degradation. EMBO J (2018) 37(3):351–66. doi: 10.15252/embj.201796781
213. Zhao Z, Su Z, Liang P, Liu D, Yang S, Wu Y, et al. USP38 Couples Histone Ubiquitination and Methylation via KDM5B to Resolve Inflammation. Adv Sci (Weinheim Baden-Wurttemberg Germany) (2020) 7(22):2002680–2002680. doi: 10.1002/advs.202002680
214. Lin M, Zhao Z, Yang Z, Meng Q, Tan P, Xie W, et al. USP38 Inhibits Type I Interferon Signaling by Editing TBK1 Ubiquitination Through NLRP4 Signalosome. Mol Cell (2016) 64(2):267–81. doi: 10.1016/j.molcel.2016.08.029
215. Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor P62 to Promote Innate Immune Responses. Mol Cell (2016) 64(1):105–19. doi: 10.1016/j.molcel.2016.08.025
216. Chen M, Zhao Z, Meng Q, Liang P, Su Z, Wu Y, et al. TRIM14 Promotes Noncanonical NF-κb Activation by Modulating P100/P52 Stability via Selective Autophagy. Adv Sci (Weinheim Baden-Wurttemberg Germany) (2019) 7(1):1901261–1901261. doi: 10.1002/advs.201901261
217. Tan P, He L, Cui J, Qian C, Cao X, Lin M, et al. Assembly of the WHIP-TRIM14-PPP6C Mitochondrial Complex Promotes RIG-I-Mediated Antiviral Signaling. Mol Cell (2017) 68(2):293–307.e5. doi: 10.1016/j.molcel.2017.09.035
218. Zhou Z, Jia X, Xue Q, Dou Z, Ma Y, Zhao Z, et al. TRIM14 is a Mitochondrial Adaptor That Facilitates Retinoic Acid-Inducible Gene-I–like Receptor-Mediated Innate Immune Response. Proc Natl Acad Sci (2014) 111(2):E245–54. doi: 10.1073/pnas.1316941111
219. Deidier A. Deux Dissertations Medecinales et Chirurgicales, L’une Sur La Maladie Venerienne, L’autre Sur La Nature & La Curation Des Tumeurs. Chez Charles Maurice d’Houry (1725).
220. Dajon M, Iribarren K, Cremer I. Toll-Like Receptor Stimulation in Cancer: A Pro- and Anti-Tumor Double-Edged Sword. Immunobiology (2017) 222(1):89–100. doi: 10.1016/j.imbio.2016.06.009
221. Urban-Wojciuk Z, Khan MM, Oyler BL, Fåhraeus R, Marek-Trzonkowska N, Nita-Lazar A, et al. The Role of TLRs in Anti-Cancer Immunity and Tumor Rejection. Front Immunol (2019) 10:2388(2388). doi: 10.3389/fimmu.2019.02388
222. Lin H, Yan J, Wang Z, Hua F, Yu J, Sun W, et al. Loss of Immunity-Supported Senescence Enhances Susceptibility to Hepatocellular Carcinogenesis and Progression in Toll-Like Receptor 2-Deficient Mice. Hepatology (2013) 57(1):171–82. doi: 10.1002/hep.25991
223. Qi S, Zhang H, Sun R, An A, Li H, Ouyang D. 245 Human TLR8 Knock-in Mice Potentiate Immunotherapy Responses of MC38 Syngeneic Tumors. J Immunother Cancer (2020) 8(Suppl 3):A146. doi: 10.1136/jitc-2020-SITC2020.0245
224. Zhang Y, Luo F, Cai Y, Liu N, Wang L, Xu D, et al. TLR1/TLR2 Agonist Induces Tumor Regression by Reciprocal Modulation of Effector and Regulatory T Cells. J Immunol (2011) 186(4):1963–9. doi: 10.4049/jimmunol.1002320
225. Sharma N, Vacher J, Allison JP. TLR1/2 Ligand Enhances Antitumor Efficacy of CTLA-4 Blockade by Increasing Intratumoral Treg Depletion. Proc Natl Acad Sci USA (2019) 116(21):10453–62. doi: 10.1073/pnas.1819004116
226. He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 Signaling Promotes Immune Escape of Human Lung Cancer Cells by Inducing Immunosuppressive Cytokines and Apoptosis Resistance. Mol Immunol (2007) 44(11):2850–9. doi: 10.1016/j.molimm.2007.01.022
227. Yang H, Zhou H, Feng P, Zhou X, Wen H, Xie X, et al. Reduced Expression of Toll-Like Receptor 4 Inhibits Human Breast Cancer Cells Proliferation and Inflammatory Cytokines Secretion. J Exp Clin Cancer Res (2010) 29:92. doi: 10.1186/1756-9966-29-92
228. Yang H, Wang B, Wang T, Xu L, He C, Wen H, et al. Toll-Like Receptor 4 Prompts Human Breast Cancer Cells Invasiveness via Lipopolysaccharide Stimulation and is Overexpressed in Patients With Lymph Node Metastasis. PloS One (2014) 9(10):e109980. doi: 10.1371/journal.pone.0109980
229. Kim J-H, Kordahi MC, Chac D, DePaolo RW. Toll-Like Receptor-6 Signaling Prevents Inflammation and Impacts Composition of the Microbiota During Inflammation-Induced Colorectal Cancer. Cancer Prev Res (2020) 13(1):25. doi: 10.1158/1940-6207.CAPR-19-0286
230. Rakoff-Nahoum S, Medzhitov R. Regulation of Spontaneous Intestinal Tumorigenesis Through the Adaptor Protein Myd88. Science (2007) 317: (5834):124–7. doi: 10.1126/science.1140488
231. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science (2007) 317: (5834):121–4. doi: 10.1126/science.1140485
232. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically Active MYD88 Mutations in Human Lymphoma. Nature (2011) 470(7332):115–9. doi: 10.1038/nature09671
233. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N Engl J Med (2012) 367: (9):826–33. doi: 10.1056/NEJMoa1200710
234. Pham-Ledard A, Beylot-Barry M, Barbe C, Leduc M, Petrella T, Vergier B, et al. High Frequency and Clinical Prognostic Value of MYD88 L265P Mutation in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg-Type. JAMA Dermatol (2014) 150(11):1173–9. doi: 10.1001/jamadermatol.2014.821
235. Tartey S, Neale G, Vogel P, Malireddi RKS, Kanneganti T-D. A MyD88/IL1R Axis Regulates PD-1 Expression on Tumor-Associated Macrophages and Sustains Their Immunosuppressive Function in Melanoma. J Cancer Res (2021) 81(9):2358–72. doi: 10.1158/0008-5472.CAN-20-3510
236. D’Agostini C, Pica F, Febbraro G, Grelli S, Chiavaroli C, Garaci E. Antitumour Effect of OM-174 and Cyclophosphamide on Murine B16 Melanoma in Different Experimental Conditions. Int Immunopharmacol (2005) 5(7-8):1205–12. doi: 10.1016/j.intimp.2005.02.013
237. Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette-Guerin in the Treatment of Superficial Bladder Tumors. J Urol (1976) 116(2):180–3. doi: 10.1016/s0022-5347(17)58737-6
238. Guinn ZP, Petro TM. IFN-Gamma Synergism With Poly I:C Reduces Growth of Murine and Human Cancer Cells With Simultaneous Changes in Cell Cycle and Immune Checkpoint Proteins. Cancer Lett (2018) 438:1–9. doi: 10.1016/j.canlet.2018.09.003
239. Salazar AM, Erlich RB, Mark A, Bhardwaj N, Herberman RB. Therapeutic in Situ Autovaccination Against Solid Cancers With Intratumoral Poly-ICLC: Case Report, Hypothesis, and Clinical Trial. Cancer Immunol Res (2014) 2(8):720–4. doi: 10.1158/2326-6066.CIR-14-0024
240. Sun L, Kees T, Almeida AS, Liu B, He XY, Ng D, et al. Activating a Collaborative Innate-Adaptive Immune Response to Control Metastasis. Cancer Cell (2021) 39(10):1361–74.e9. doi: 10.1016/j.ccell.2021.08.005
241. Zandi Z, Kashani B, Bashash D, Poursani EM, Mousavi SA, Chahardoli B, et al. The Anticancer Effect of the TLR4 Inhibition Using TAK-242 (Resatorvid) Either as a Single Agent or in Combination With Chemotherapy: A Novel Therapeutic Potential for Breast Cancer. J Cell Biochem (2020) 121: (2):1623–34. doi: 10.1002/jcb.29397
242. Hsu RYC, Chan CHF, Spicer JD, Rousseau MC, Giannias B, Rousseau S, et al. LPS-Induced TLR4 Signaling in Human Colorectal Cancer Cells Increases β1 Integrin-Mediated Cell Adhesion and Liver Metastasis. J Cancer Res (2011) 71(5):1989–98. doi: 10.1158/0008-5472.CAN-10-2833
243. Brackett CM, Kojouharov B, Veith J, Greene KF, Burdelya LG, Gollnick SO, et al. Toll-Like Receptor-5 Agonist, Entolimod, Suppresses Metastasis and Induces Immunity by Stimulating an NK-Dendritic-CD8+ T-Cell Axis. Proc Natl Acad Sci USA (2016) 113(7):E874–83. doi: 10.1073/pnas.1521359113
244. Bubna AK. Imiquimod - Its Role in the Treatment of Cutaneous Malignancies. Indian J Pharmacol (2015) 47(4):354–9. doi: 10.4103/0253-7613.161249
245. Mullins SR, Vasilakos JP, Deschler K, Grigsby I, Gillis P, John J, et al. Intratumoral Immunotherapy With TLR7/8 Agonist MEDI9197 Modulates the Tumor Microenvironment Leading to Enhanced Activity When Combined With Other Immunotherapies. J Immunother Cancer (2019) 7(1):244. doi: 10.1186/s40425-019-0724-8
246. Mohamed FE, Al-Jehani RM, Minogue SS, Andreola F, Winstanley A, Olde Damink SWM, et al. Effect of Toll-Like Receptor 7 and 9 Targeted Therapy to Prevent the Development of Hepatocellular Carcinoma. Liver Int (2015) 35: (3):1063–76. doi: 10.1111/liv.12626
247. Wang S, Campos J, Gallotta M, Gong M, Crain C, Naik E, et al. Intratumoral Injection of a CpG Oligonucleotide Reverts Resistance to PD-1 Blockade by Expanding Multifunctional CD8+ T Cells. Proc Natl Acad Sci USA (2016) 113(46):E7240–9. doi: 10.1073/pnas.1608555113
248. Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, et al. Combination Immunotherapy With TLR Agonists and Checkpoint Inhibitors Suppresses Head and Neck Cancer. JCI Insight (2017) 2(18):e93397. doi: 10.1172/jci.insight.93397
249. Fourcade J, Kudela P, Andrade Filho PA, Janjic B, Land SR, Sander C, et al. Immunization With Analog Peptide in Combination With CpG and Montanide Expands Tumor Antigen-Specific CD8+ T Cells in Melanoma Patients. J Immunother (2008) 31(8):781–91. doi: 10.1097/CJI.0b013e318183af0b
250. Farrugia M, Baron B. The Role of Toll-Like Receptors in Autoimmune Diseases Through Failure of the Self-Recognition Mechanism. Int J Inflam (2017) 2017:8391230. doi: 10.1155/2017/8391230
251. Radstake TR, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, van Riel PL, Barrera P, et al. Expression of Toll-Like Receptors 2 and 4 in Rheumatoid Synovial Tissue and Regulation by Proinflammatory Cytokines Interleukin-12 and Interleukin-18 via Interferon-Gamma. Arthritis Rheum (2004) 50(12):3856–65. doi: 10.1002/art.20678
252. Roelofs MF, Wenink MH, Brentano F, Abdollahi-Roodsaz S, Oppers-Walgreen B, Barrera P, et al. Type I Interferons Might Form the Link Between Toll-Like Receptor (TLR) 3/7 and TLR4-Mediated Synovial Inflammation in Rheumatoid Arthritis (RA). Ann Rheum Dis (2009) 68(9):1486–93. doi: 10.1136/ard.2007.086421
253. Li X, Xu T, Wang Y, Huang C, Li J. Toll-Like Receptor (TLR)-3: A Potent Driving Force Behind Rheumatoid Arthritis. Clin Rheumatol (2014) 33(2):291–2. doi: 10.1007/s10067-013-2418-9
254. Kim W, Kim TH, Oh SJ, Kim HJ, Kim JH, Kim HA, et al. Association of TLR 9 Gene Polymorphisms With Remission in Patients With Rheumatoid Arthritis Receiving TNF-Alpha Inhibitors and Development of Machine Learning Models. Sci Rep (2021) 11(1):20169. doi: 10.1038/s41598-021-99625-x
255. Davis MLR, LeVan TD, Yu F, Sayles H, Sokolove J, Robinson W, et al. Associations of Toll-Like Receptor (TLR)-4 Single Nucleotide Polymorphisms and Rheumatoid Arthritis Disease Progression: An Observational Cohort Study. Int Immunopharmacol (2015) 24(2):346–52. doi: 10.1016/j.intimp.2014.12.030
256. Tripathy A, Khanna S, Padhan P, Smita S, Raghav S, Gupta B. Direct Recognition of LPS Drive TLR4 Expressing CD8+ T Cell Activation in Patients With Rheumatoid Arthritis. Sci Rep (2017) 7(1):933. doi: 10.1038/s41598-017-01033-7
257. Xu Q, Metzler B, Jahangiri M, Mandal K. Molecular Chaperones and Heat Shock Proteins in Atherosclerosis. Am J Physiol Heart Circ Physiol (2012) 302(3):H506–14. doi: 10.1152/ajpheart.00646.2011
258. Wick G, Jakic B, Buszko M, Wick MC, Grundtman C. The Role of Heat Shock Proteins in Atherosclerosis. Nat Rev Cardiol (2014) 11(9):516–29. doi: 10.1038/nrcardio.2014.91
259. Park Y, Park S, Yoo E, Kim D, Shin H. Association of the Polymorphism for Toll-Like Receptor 2 With Type 1 Diabetes Susceptibility. Ann N Y Acad Sci (2004) 1037:170–4. doi: 10.1196/annals.1337.028
260. Sepehri Z, Kiani Z, Nasiri AA, Kohan F. Toll-Like Receptor 2 and Type 2 Diabetes. Cell Mol Biol Lett (2016) 21:2. doi: 10.1186/s11658-016-0002-4
261. Guo Z, Zhang Y, Liu C, Youn JY, Cai H. Toll-Like Receptor 2 (TLR2) Knockout Abrogates Diabetic and Obese Phenotypes While Restoring Endothelial Function via Inhibition of NOX1. Diabetes (2021) 70(9):2107–19. doi: 10.2337/db20-0591
262. Fillatreau S, Manfroi B, Dorner T. Toll-Like Receptor Signalling in B Cells During Systemic Lupus Erythematosus. Nat Rev Rheumatol (2021) 17(2):98–108. doi: 10.1038/s41584-020-00544-4
263. Garcia-Ortiz H, Velazquez-Cruz R, Espinosa-Rosales F, Jimenez-Morales S, Baca V, Orozco L. Association of TLR7 Copy Number Variation With Susceptibility to Childhood-Onset Systemic Lupus Erythematosus in Mexican Population. Ann Rheum Dis (2010) 69(10):1861–5. doi: 10.1136/ard.2009.124313
264. Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and Functional Impact of Copy Number Variation in the Human Genome. Nature (2010) 464(7289):704–12. doi: 10.1038/nature08516
265. Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, et al. TLR7 Escapes X Chromosome Inactivation in Immune Cells. Sci Immunol (2018) 3(19):eaap8855. doi: 10.1126/sciimmunol.aap8855
266. Wang T, Marken J, Chen J, Tran VB, Li QZ, Li M, et al. High TLR7 Expression Drives the Expansion of CD19(+)CD24(hi)CD38(hi) Transitional B Cells and Autoantibody Production in SLE Patients. Front Immunol (2019) 10:1243:1243. doi: 10.3389/fimmu.2019.01243
267. Celhar T, Lu HK, Benso L, Rakhilina L, Lee HY, Tripathi S, et al. TLR7 Protein Expression in Mild and Severe Lupus-Prone Models Is Regulated in a Leukocyte, Genetic, and IRAK4 Dependent Manner. Front Immunol (2019) 10:1546:1546. doi: 10.3389/fimmu.2019.01546
268. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14dim Monocytes Patrol and Sense Nucleic Acids and Viruses via TLR7 and TLR8 Receptors. Immunity (2010) 33(3):375–86. doi: 10.1016/j.immuni.2010.08.012
269. Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid Dendritic Cells Promote Systemic Sclerosis With a Key Role for TLR8. Sci Transl Med (2018) 10(423):eaam8458. doi: 10.1126/scitranslmed.aam8458
270. Ciechomska M, O’Reilly S, Przyborski S, Oakley F, Bogunia-Kubik K, van Laar JM. Histone Demethylation and Toll-Like Receptor 8-Dependent Cross-Talk in Monocytes Promotes Transdifferentiation of Fibroblasts in Systemic Sclerosis Via Fra-2. Arthritis Rheumatol (2016) 68(6):1493–504. doi: 10.1002/art.39602
271. O’Reilly S, Cant R, Ciechomska M, Finnigan J, Oakley F, Hambleton S, et al. Serum Amyloid A Induces Interleukin-6 in Dermal Fibroblasts via Toll-Like Receptor 2, Interleukin-1 Receptor-Associated Kinase 4 and Nuclear Factor-Kappab. Immunology (2014) 143(3):331–40. doi: 10.1111/imm.12260
272. Zong M, Bruton JD, Grundtman C, Yang H, Li JH, Alexanderson H, et al. TLR4 as Receptor for HMGB1 Induced Muscle Dysfunction in Myositis. Ann Rheum Dis (2013) 72(8):1390–9. doi: 10.1136/annrheumdis-2012-202207
273. Tournadre A, Lenief V, Eljaafari A, Miossec P. Immature Muscle Precursors are a Source of Interferon-Beta in Myositis: Role of Toll-Like Receptor 3 Activation and Contribution to HLA Class I Up-Regulation. Arthritis Rheum (2012) 64(2):533–41. doi: 10.1002/art.33350
274. Cappelletti C, Baggi F, Zolezzi F, Biancolini D, Beretta O, Severa M, et al. Type I Interferon and Toll-Like Receptor Expression Characterizes Inflammatory Myopathies. Neurology (2011) 76(24):2079–88. doi: 10.1212/WNL.0b013e31821f440a
275. Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, et al. The Toll-Like Receptor Adaptor Proteins MyD88 and Mal/TIRAP Contribute to the Inflammatory and Destructive Processes in a Human Model of Rheumatoid Arthritis. Am J Pathol (2007) 170(2):518–25. doi: 10.2353/ajpath.2007.060657
276. Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, et al. Innate Immunity Mediated by TLR9 Modulates Pathogenicity in an Animal Model of Multiple Sclerosis. J Clin Invest (2006) 116(2):456–64. doi: 10.1172/JCI26078
277. Guiducci C, Gong M, Cepika AM, Xu Z, Tripodo C, Bennett L, et al. RNA Recognition by Human TLR8 can Lead to Autoimmune Inflammation. J Exp Med (2013) 210(13):2903–19. doi: 10.1084/jem.20131044
278. Tapping RI, Tobias PS. Mycobacterial Lipoarabinomannan Mediates Physical Interactions Between TLR1 and TLR2 to Induce Signaling. J Endotoxin Res (2003) 9(4):264–8. doi: 10.1179/096805103225001477
279. Misch EA, Macdonald M, Ranjit C, Sapkota BR, Wells RD, Siddiqui MR, et al. Human TLR1 Deficiency Is Associated With Impaired Mycobacterial Signaling and Protection From Leprosy Reversal Reaction. PloS Negl Trop Dis (2008) 2(5):e231. doi: 10.1371/journal.pntd.0000231
280. Elass E, Aubry L, Masson M, Denys A, Guerardel Y, Maes E, et al. Mycobacterial Lipomannan Induces Matrix Metalloproteinase-9 Expression in Human Macrophagic Cells Through a Toll-Like Receptor 1 (TLR1)/TLR2- and CD14-Dependent Mechanism. Infect Immun (2005) 73(10):7064–8. doi: 10.1128/IAI.73.10.7064-7068.2005
281. Stenzel W, Soltek S, Sanchez-Ruiz M, Akira S, Miletic H, Schluter D, et al. Both TLR2 and TLR4 are Required for the Effective Immune Response in Staphylococcus Aureus-Induced Experimental Murine Brain Abscess. Am J Pathol (2008) 172(1):132–45. doi: 10.2353/ajpath.2008.070567
282. Nguyen BN, Chavez-Arroyo A, Cheng MI, Krasilnikov M, Louie A, Portnoy DA. TLR2 and Endosomal TLR-Mediated Secretion of IL-10 and Immune Suppression in Response to Phagosome-Confined Listeria Monocytogenes. PloS Pathog (2020) 16(7):e1008622. doi: 10.1371/journal.ppat.1008622
283. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 Senses the SARS-CoV-2 Envelope Protein to Produce Inflammatory Cytokines. Nat Immunol (2021) 22(7):829–38. doi: 10.1038/s41590-021-00937-x
284. Zhang X, Li X, Gong P, Wang X, Zhang N, Chen M, et al. Host Defense Against Neospora Caninum Infection via IL-12p40 Production Through TLR2/TLR3-AKT-ERK Signaling Pathway in C57BL/6 Mice. Mol Immunol (2021) 139:140–52. doi: 10.1016/j.molimm.2021.08.019
285. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 Deficiency in Patients With Herpes Simplex Encephalitis. Science (2007) 317(5844):1522–7. doi: 10.1126/science.1139522
286. Fujimoto C, Nakagawa Y, Ohara K, Takahashi H. Polyriboinosinic Polyribocytidylic Acid [Poly(I:C)]/TLR3 Signaling Allows Class I Processing of Exogenous Protein and Induction of HIV-Specific CD8+ Cytotoxic T Lymphocytes. Int Immunol (2004) 16(1):55–63. doi: 10.1093/intimm/dxh025
287. Sironi M, Biasin M, Cagliani R, Forni D, De Luca M, Saulle I, et al. A Common Polymorphism in TLR3 Confers Natural Resistance to HIV-1 Infection. J Immunol (2012) 188(2):818–23. doi: 10.4049/jimmunol.1102179
288. Huik K, Avi R, Pauskar M, Kallas E, Jogeda EL, Karki T, et al. Association Between TLR3 Rs3775291 and Resistance to HIV Among Highly Exposed Caucasian Intravenous Drug Users. Infect Genet Evol (2013) 20:78–82. doi: 10.1016/j.meegid.2013.08.008
289. Cheng L, Wang Q, Li G, Banga R, Ma J, Yu H, et al. TLR3 Agonist and CD40-Targeting Vaccination Induces Immune Responses and Reduces HIV-1 Reservoirs. J Clin Invest (2018) 128(10):4387–96. doi: 10.1172/JCI99005
290. Liu H, Zhou RH, Liu Y, Guo L, Wang X, Hu WH, et al. HIV Infection Suppresses TLR3 Activation-Mediated Antiviral Immunity in Microglia and Macrophages. Immunology (2020) 160(3):269–79. doi: 10.1111/imm.13181
291. Rudd BD, Smit JJ, Flavell RA, Alexopoulou L, Schaller MA, Gruber A, et al. Deletion of TLR3 Alters the Pulmonary Immune Environment and Mucus Production During Respiratory Syncytial Virus Infection. J Immunol (2006) 176(3):1937–42. doi: 10.4049/jimmunol.176.3.1937
292. Huang S, Wei W, Yun Y. Upregulation of TLR7 and TLR3 Gene Expression in the Lung of Respiratory Syncytial Virus Infected Mice. Wei Sheng Wu Xue Bao (2009) 49(2):239–45.
293. Wu J, Wang Z, Guo YN, Dong LQ. Activation of TLR3 Pathway in the Pathogenesis of Nephrotic Syndrome Induced by Respiratory Syncytial Virus in Rat Model. Sichuan Da Xue Xue Bao Yi Xue Ban (2010) 41(4):600–3, 625.
294. Satkunanathan S, Kumar N, Bajorek M, Purbhoo MA, Culley FJ. Respiratory Syncytial Virus Infection, TLR3 Ligands, and Proinflammatory Cytokines Induce CD161 Ligand LLT1 Expression on the Respiratory Epithelium. J Virol (2014) 88(5):2366–73. doi: 10.1128/JVI.02789-13
295. Miranda VDS, Franca FBF, da Costa MS, Silva VRS, Mota CM, Barros P, et al. Toll-Like Receptor 3-TRIF Pathway Activation by Neospora Caninum RNA Enhances Infection Control in Mice. Infect Immun (2019) 87(4):e00739-18. doi: 10.1128/IAI.00739-18
296. Chantratita N, Tandhavanant S, Seal S, Wikraiphat C, Wongsuvan G, Ariyaprasert P, et al. TLR4 Genetic Variation is Associated With Inflammatory Responses in Gram-Positive Sepsis. Clin Microbiol Infect (2017) 23(1):47 e1–47 e10. doi: 10.1016/j.cmi.2016.08.028
297. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern Recognition Receptors TLR4 and CD14 Mediate Response to Respiratory Syncytial Virus. Nat Immunol (2000) 1(5):398–401. doi: 10.1038/80833
298. Tang HB, Lu ZL, Wei XK, Zhong TZ, Zhong YZ, Ouyang LX, et al. Viperin Inhibits Rabies Virus Replication via Reduced Cholesterol and Sphingomyelin and is Regulated Upstream by TLR4. Sci Rep (2016) 6:30529. doi: 10.1038/srep30529
299. Chen C, Zhang C, Li H, Wang Z, Yuan Y, Zhou M, et al. TLR4 Regulates Rabies Virus-Induced Humoral Immunity Through Recruitment of cDC2 to Lymph Organs. J Virol (2021) 95(24):JVI0082921. doi: 10.1128/JVI.00829-21
300. Zhang ZM, Zhang AR, Xu M, Lou J, Qiu WQ. TLR-4/miRNA-32-5p/FSTL1 Signaling Regulates Mycobacterial Survival and Inflammatory Responses in Mycobacterium Tuberculosis-Infected Macrophages. Exp Cell Res (2017) 352(2):313–21. doi: 10.1016/j.yexcr.2017.02.025
301. Chaichana P, Chantratita N, Brod F, Koosakulnirand S, Jenjaroen K, Chumseng S, et al. A Nonsense Mutation in TLR5 is Associated With Survival and Reduced IL-10 and TNF-Alpha Levels in Human Melioidosis. PloS Negl Trop Dis (2017) 11(5):e0005587. doi: 10.1371/journal.pntd.0005587
302. Chen J, Ng MM, Chu JJ. Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection. PloS Pathog (2015) 11(7):e1005053. doi: 10.1371/journal.ppat.1005053
303. Misch EA, Verbon A, Prins JM, Skerrett SJ, Hawn TR. A TLR6 Polymorphism is Associated With Increased Risk of Legionnaires’ Disease. Genes Immun (2013) 14(7):420–6. doi: 10.1038/gene.2013.34
304. Regli IB, Passelli K, Martinez-Salazar B, Amore J, Hurrell BP, Muller AJ, et al. TLR7 Sensing by Neutrophils Is Critical for the Control of Cutaneous Leishmaniasis. Cell Rep (2020) 31(10):107746. doi: 10.1016/j.celrep.2020.107746
305. Pang IK, Pillai PS, Iwasaki A. Efficient Influenza A Virus Replication in the Respiratory Tract Requires Signals From TLR7 and RIG-I. Proc Natl Acad Sci USA (2013) 110(34):13910–5. doi: 10.1073/pnas.1303275110
306. Valencia Pacheco GJ, Pinzon Herrera F, Cruz Lopez JJ, Vera Gamboa Ldel C, Pavia Ruiz N, Santos Rivero A, et al. Expression and Activation of Intracellular Receptors TLR7, TLR8 and TLR9 in Peripheral Blood Monocytes From HIV-Infected Patients. Colomb Med (Cali) (2013) 44(2):92–9. doi: 10.25100/cm.v44i2.1183
307. Cardoso EC, Pereira NZ, Mitsunari GE, Oliveira LM, Ruocco RM, Francisco RP, et al. TLR7/TLR8 Activation Restores Defective Cytokine Secretion by Myeloid Dendritic Cells But Not by Plasmacytoid Dendritic Cells in HIV-Infected Pregnant Women and Newborns. PloS One (2013) 8(6):e67036. doi: 10.1371/journal.pone.0067036
308. Pietrobon AJ, Yoshikawa FSY, Oliveira LM, Pereira NZ, Matozo T, de Alencar BC, et al. Antiviral Response Induced by TLR7/TLR8 Activation Inhibits HIV-1 Infection in Cord Blood Macrophages. J Infect Dis (2021) 225(3):510–9. doi: 10.1093/infdis/jiab389
309. Gringhuis SI, van der Vlist M, van den Berg LM, den Dunnen J, Litjens M, Geijtenbeek TB. HIV-1 Exploits Innate Signaling by TLR8 and DC-SIGN for Productive Infection of Dendritic Cells. Nat Immunol (2010) 11(5):419–26. doi: 10.1038/ni.1858
310. Greulich W, Wagner M, Gaidt MM, Stafford C, Cheng Y, Linder A, et al. TLR8 Is a Sensor of RNase T2 Degradation Products. Cell (2019) 179(6):1264–1275 e13. doi: 10.1016/j.cell.2019.11.001
311. Ma Y, He B. Recognition of Herpes Simplex Viruses: Toll-Like Receptors and Beyond. J Mol Biol (2014) 426(6):1133–47. doi: 10.1016/j.jmb.2013.11.012
312. Lai ZZ, Ni Z, Pan XL, Song L. Toll-Like Receptor 9 (TLR9) Gene Polymorphisms Associated With Increased Susceptibility of Human Papillomavirus-16 Infection in Patients With Cervical Cancer. J Int Med Res (2013) 41(4):1027–36. doi: 10.1177/0300060513483398
313. Basner-Tschakarjan E, Gaffal E, O’Keeffe M, Tormo D, Limmer A, Wagner H, et al. Adenovirus Efficiently Transduces Plasmacytoid Dendritic Cells Resulting in TLR9-Dependent Maturation and IFN-Alpha Production. J Gene Med (2006) 8(11):1300–6. doi: 10.1002/jgm.964
314. Pahl JH, Verhoeven DH, Kwappenberg KM, Vellinga J, Lankester AC, van Tol MJ, et al. Adenovirus Type 35, But Not Type 5, Stimulates NK Cell Activation via Plasmacytoid Dendritic Cells and TLR9 Signaling. Mol Immunol (2012) 51(1):91–100. doi: 10.1016/j.molimm.2012.02.119
315. Zang J, Zheng MH, Cao XL, Zhang YZ, Zhang YF, Gao XY, et al. Adenovirus Infection Promotes the Formation of Glioma Stem Cells From Glioblastoma Cells Through the TLR9/NEAT1/STAT3 Pathway. Cell Commun Signal (2020) 18(1):135. doi: 10.1186/s12964-020-00598-7
316. DePaolo RW, Kamdar K, Khakpour S, Sugiura Y, Wang W, Jabri B. A Specific Role for TLR1 in Protective T(H)17 Immunity During Mucosal Infection. J Exp Med (2012) 209(8):1437–44. doi: 10.1084/jem.20112339
317. Sugiura Y, Kamdar K, Khakpour S, Young G, Karpus WJ, DePaolo RW. TLR1-Induced Chemokine Production is Critical for Mucosal Immunity Against Yersinia Enterocolitica. Mucosal Immunol (2013) 6(6):1101–9. doi: 10.1038/mi.2013.5
318. Johnson CM, Lyle EA, Omueti KO, Stepensky VA, Yegin O, Alpsoy E, et al. Cutting Edge: A Common Polymorphism Impairs Cell Surface Trafficking and Functional Responses of TLR1 But Protects Against Leprosy. J Immunol (2007) 178(12):7520–4. doi: 10.4049/jimmunol.178.12.7520
319. Arpaia N, Godec J, Lau L, Sivick KE, McLaughlin LM, Jones MB, et al. TLR Signaling is Required for Salmonella Typhimurium Virulence. Cell (2011) 144(5):675–88. doi: 10.1016/j.cell.2011.01.031
320. Zhao Y, Kuang M, Li J, Zhu L, Jia Z, Guo X, et al. SARS-CoV-2 Spike Protein Interacts With and Activates TLR41. Cell Res (2021) 31(7):818–20. doi: 10.1038/s41422-021-00495-9
321. Awomoyi AA, Rallabhandi P, Pollin TI, Lorenz E, Sztein MB, Boukhvalova MS, et al. Association of TLR4 Polymorphisms With Symptomatic Respiratory Syncytial Virus Infection in High-Risk Infants and Young Children. J Immunol (2007) 179(5):3171–7. doi: 10.4049/jimmunol.179.5.3171
322. Mansur A, von Gruben L, Popov AF, Steinau M, Bergmann I, Ross D, et al. The Regulatory Toll-Like Receptor 4 Genetic Polymorphism Rs11536889 is Associated With Renal, Coagulation and Hepatic Organ Failure in Sepsis Patients. J Transl Med (2014) 12:177. doi: 10.1186/1479-5876-12-177
323. Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, et al. Cutting Edge: Toll-Like Receptor (TLR)2- and TLR4-Mediated Pathogen Recognition in Resistance to Airborne Infection With Mycobacterium Tuberculosis. J Immunol (2002) 169(7):3480–4. doi: 10.4049/jimmunol.169.7.3480
324. Abel B, Thieblemont N, Quesniaux VJ, Brown N, Mpagi J, Miyake K, et al. Toll-Like Receptor 4 Expression is Required to Control Chronic Mycobacterium Tuberculosis Infection in Mice. J Immunol (2002) 169(6):3155–62. doi: 10.4049/jimmunol.169.6.3155
325. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of Single-Stranded RNA Viruses by Toll-Like Receptor 7. Proc Natl Acad Sci USA (2004) 101(15):5598–603. doi: 10.1073/pnas.0400937101
326. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science (2004) 303(5663):1529–31. doi: 10.1126/science.1093616
327. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, et al. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5’-Phosphates. Science (2006) 314(5801):997–1001. doi: 10.1126/science.1132998
328. Iwasaki A, Medzhitov R. Regulation of Adaptive Immunity by the Innate Immune System. Science (2010) 327(5963):291–5. doi: 10.1126/science.1183021
329. To EE, Erlich J, Liong F, Luong R, Liong S, Bozinovski S, et al. Intranasal and Epicutaneous Administration of Toll-Like Receptor 7 (TLR7) Agonists Provides Protection Against Influenza A Virus-Induced Morbidity in Mice. Sci Rep (2019) 9(1):2366. doi: 10.1038/s41598-019-38864-5
330. Wang MG, Zhang MM, Wang Y, Wu SQ, Zhang M, He JQ. Association of TLR8 and TLR9 Polymorphisms With Tuberculosis in a Chinese Han Population: A Case-Control Study. BMC Infect Dis (2018) 18(1):561. doi: 10.1186/s12879-018-3485-y
331. Jude BA, Pobezinskaya Y, Bishop J, Parke S, Medzhitov RM, Chervonsky AV, et al. Subversion of the Innate Immune System by a Retrovirus. Nat Immunol (2003) 4(6):573–8. doi: 10.1038/ni926
332. Asami J, Shimizu T. Structural and Functional Understanding of the Toll-Like Receptors. Protein Sci (2021) 30(4):761–72. doi: 10.1002/pro.4043
333. Behzadi P, García-Perdomo HA, Karpiński TM. Toll-Like Receptors: General Molecular and Structural Biology. J Immunol Res (2021) 2021:9914854. doi: 10.1155/2021/9914854
334. Wang R-F, Wang HY. Immune Targets and Neoantigens for Cancer Immunotherapy and Precision Medicine. Cell Res (2017) 27(1):11–37. doi: 10.1038/cr.2016.155
Keywords: toll-like receptors, cell-mediated immunity, T cells, signaling pathway, infectious diseases, autoimmune diseases, cancer
Citation: Duan T, Du Y, Xing C, Wang HY and Wang R-F (2022) Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 13:812774. doi: 10.3389/fimmu.2022.812774
Received: 10 November 2021; Accepted: 08 February 2022;
Published: 03 March 2022.
Edited by:
Subhasis Chattopadhyay, National Institute of Science Education and Research (NISER), IndiaReviewed by:
Subhransu Sekhar Sahoo, Purdue University, United StatesCopyright © 2022 Duan, Du, Xing, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong-Fu Wang, cm9uZ2Z1d2FAdXNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.