 Luca Hensen1
Luca Hensen1 Patricia T. Illing2
Patricia T. Illing2 Louise C. Rowntree1
Louise C. Rowntree1 Jane Davies3Adrian Miller4
Jane Davies3Adrian Miller4 Steven Y. C. Tong3,5,6Jennifer R. Habel1
Steven Y. C. Tong3,5,6Jennifer R. Habel1 Carolien E. van de Sandt1,7
Carolien E. van de Sandt1,7 Katie L. Flanagan8,9,10,11
Katie L. Flanagan8,9,10,11 Anthony W. Purcell2
Anthony W. Purcell2 Katherine Kedzierska1*†
Katherine Kedzierska1*† E. Bridie Clemens1†
E. Bridie Clemens1†- 1Department of Microbiology and Immunology, University of Melbourne, at the Peter Doherty Institute for Infection and Immunity, Parkville, VIC, Australia
- 2Department of Biochemistry and Molecular Biology & Infection and Immunity Program, Biomedicine Discovery Institute, Monash University, Clayton, VIC, Australia
- 3Menzies School of Health Research, Darwin, NT, Australia
- 4Indigenous Engagement, CQUniversity, Townsville, QLD, Australia
- 5Victorian Infectious Diseases Service, The Royal Melbourne Hospital at the Peter Doherty Institute for Infection and Immunity, Melbourne, VIC, Australia
- 6Department of Infectious Diseases, The University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Melbourne, VIC, Australia
- 7Department of Hematopoiesis, Sanquin Research and Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 8Department of Infectious Diseases and Tasmanian Vaccine Trial Centre, Launceston General Hospital, Launceston, TAS, Australia
- 9School of Health Sciences and School of Medicine, University of Tasmania, Launceston, TAS, Australia
- 10Department of Immunology and Pathology, Monash University, Melbourne, VIC, Australia
- 11School of Health and Biomedical Science, RMIT University, Melbourne, VIC, Australia
CD8+ T cells are a pivotal part of the immune response to viruses, playing a key role in disease outcome and providing long-lasting immunity to conserved pathogen epitopes. Understanding CD8+ T cell immunity in humans is complex due to CD8+ T cell restriction by highly polymorphic Human Leukocyte Antigen (HLA) proteins, requiring T cell epitopes to be defined for different HLA allotypes across different ethnicities. Here we evaluate strategies that have been developed to facilitate epitope identification and study immunogenic T cell responses. We describe an immunopeptidomics approach to sequence HLA-bound peptides presented on virus-infected cells by liquid chromatography with tandem mass spectrometry (LC-MS/MS). Using antigen presenting cell lines that stably express the HLA alleles characteristic of Indigenous Australians, this approach has been successfully used to comprehensively identify influenza-specific CD8+ T cell epitopes restricted by HLA allotypes predominant in Indigenous Australians, including HLA-A*24:02 and HLA-A*11:01. This is an essential step in ensuring high vaccine coverage and efficacy in Indigenous populations globally, known to be at high risk from influenza disease and other respiratory infections.
Introduction
Novel respiratory viruses pose a major global pandemic threat associated with high disease burden and mortality. To date, the 2019 SARS-CoV-2 virus has caused more than 470 million confirmed cases and more than 6.1 million deaths reported to WHO across the world (1). Likewise, influenza viruses are responsible for annual seasonal epidemics that cause 243,000-645,000 deaths (2) and sporadic pandemics, such as the 1918-1919 H1N1 pandemic, which lead to an estimated 50 million deaths worldwide (3, 4). Outbreaks of the avian-derived H7N9 and H5N1 viruses have been associated with high rates of hospitalisation (both >99%) and death (39% H7N9 and 56% H5N1 case fatality ratio (CFR) (5)), highlighting the pandemic potential of these emerging novel influenza viruses. Indigenous populations worldwide are disproportionately affected by greater infection rates, disease severity and mortality when novel pandemic virus strains enter human circulation. During the 1918-1919 H1N1 influenza pandemic, higher rates of mortality were reported amongst the Māori people of New Zealand (7-fold higher than the European population (6)), First Nations people of Canada (8-fold higher than non-First Nations (7)), Alaskan Natives (100% of adults in some populations (8)), Indigenous Australians (10-20% compared to <1% of other Australians (9)) and Western Samoans (19-22% of total population (10)). Although less severe, the 2009 H1N1 pandemic virus also caused higher hospitalisation and mortality rates for Indigenous Australians (6.6-fold higher hospitalisation and 5.2-fold higher mortality than Non-Indigenous Australians (11)), New Zealand Māori (5-fold higher hospitalization (12)), Pacific Islanders (7-fold higher hospitalization (12, 13)), American Indians and Alaskan Natives (4-fold higher mortality (14)) and First Nations people of Canada (3-fold higher hospitalisation (15)). Emerging evidence from the UK and USA suggests a possible association between ethnicity and mortality due to SARS-CoV-2 infection (16–19), with mortality rates 3.3 times higher for Indigenous Americans compared to White Americans (20), though more global data are needed that specifically consider Indigenous populations. In the Brazilian Amazon region, the combination of SARS-CoV-2, an immunologically vulnerable Indigenous population and limited healthcare facilities has been associated with disproportionate case numbers and lethality rates among Indigenous people (21, 22). It is important to acknowledge the roles of socioeconomic disparity, high rates of comorbidities, reduced access to health services, occupation and household/community/environmental characteristics in contributing to higher rates of exposure and disease severity in ethnic minorities and Indigenous populations (16–19, 23, 24). However, underlying hereditary host and immunological factors may also contribute, including HLA genotype and polymorphisms in angiotensin-converting enzyme 2 (ACE2), transmembrane protease serine-type 2 (TMPRSS2) and interferon-induced transmembrane protein 3 (IFITM3) genes (reviewed in (25–27)). With the continuing threat of emerging respiratory viruses, targeted measures to boost immunity through vaccines, particularly those that activate long-lasting, cross-strain protective CD8+ T cells offer a means of protecting high risk groups, including the ~370-500 million Indigenous Peoples worldwide (28).
T cells play a key role in the resolution and modulation of disease severity in acute respiratory virus infections such as influenza and SARS-CoV-2. Mild to moderate influenza and SARS-CoV-2 infections are associated with prototypical antiviral immune responses involving transient, co-ordinated activation and contraction of virus-specific CD4+ and CD8+ T cells, B cells and antibodies (29–32). Meanwhile, severe SARS-CoV-2 infection is associated with dysregulation of cytokines and ligands (IL-18 and sIL-6R), hyperactivation of innate and adaptive immune cell compartments (marked by CD38 and HLA-DR expression), high SARS-CoV-2-specific antibody titres as well delayed SARS-CoV-2-specific CD4+ and CD8+ T cell responses (31, 32). Similarly, severe influenza disease is characterised by increased inflammatory cytokines and diminished influenza-specific responses from CD8+ T cells, CD4+ T cells, NK cells, MAIT cells, γδ T cells and antibody secreting cells (ASC) (33). While antibody-based vaccines targeting surface glycoproteins (e.g. Spike protein (S) of SARS-CoV-2 and hemagglutinin (HA) and neuraminidase (NA) of influenza) are an effective way to combat infection, potentially providing sterilizing immunity, they can fail to provide protection when antigenically different strains of virus emerge. These new strains can arise through gradual changes in the antigenicity of viral proteins, driven by rapid virus mutation and immune selection of viral mutants that can evade antibody binding (antigenic drift), or via recombination of viral genomes, resulting in a substantially new virus that escapes pre-existing antibody responses (antigenic shift) (34). This is seen during influenza pandemics (antigenic shift) and epidemics (antigenic drift) and now with emerging SARS-CoV-2 variants that are less susceptible to natural and vaccine-induced antibody responses (35). The unpredictable and evolving nature of these acute respiratory viruses undermines the efficacy of existing antibody-based vaccines and necessitates the annual reformulation of influenza vaccines to protect against unpredictable emerging strains, with varying efficacy (36). In contrast to antibody immunity, T cell immunity is mediated by CD8+ T cells that lyse virus-infected cells expressing Class I Major Histocompatibility Complex (MHC-I) molecules presenting virus peptides and CD4+ T cells that recognise Class II MHC (MHC-II) molecules presenting virus peptides and can kill infected cells or play a role in co-ordinating the immune response, including B cell responses and memory responses. Unlike neutralizing antibodies, the antigenic targets of T cells can be peptide+MHC epitopes derived from internal virus proteins that are often functionally significant and more highly conserved across strains, providing the basis for heterologous or “universal” immunity across unrelated strains (30, 37, 38). In situations where new viruses emerge that evade existing antibody responses, the recall of cross-reactive memory T cells that provide broadly heterotypic protection can reduce the severity of infection. This was demonstrated during the 2013 H7N9 influenza outbreak, where a shorter recovery time from severe H7N9 disease was associated with early and robust CD8+ T cell responses, and prolonged hospital stays with late recruitment of CD4+ and CD8+ T cells (39). Thus, vaccines that activate cross-strain protective cellular immunity from T cells represent an effective means of protecting against the threat of unpredictable and evolving acute respiratory viruses.
Human MHC, known as Human Leukocyte Antigen (HLA) molecules, display a high degree of polymorphism in the residues that line the peptide-binding pocket, which influences the array of peptides that can be bound in the pocket based on favourable peptide-HLA interactions. Different HLA alleles will bind different peptides which can often be defined by characteristic motifs compatible for binding. Indigenous populations express distinct HLA profiles that differ from the profiles of other ethnic groups. For instance, HLA-A*34:01 (30%), A*24:02 (24%) and B*13:01 (24%) are among the most common HLA Class I (HLA-I) alleles expressed by Indigenous Australians (40, 41) and, while shared to some extent with other Indigenous populations (often in geographic proximity), these are found at lower frequencies at a global population level (A*34:01 0.3%, A*24:02 10% and B*13:01 0.6%) (Allele Frequency Net Database (42)). Other alleles such as A*24:06, A*24:13 and HLA-B*56:56 are uniquely described in Indigenous Australians, though at low frequency (<1%) (40). Thus, the CD8+ T cell responses of Indigenous Australians are likely to target very distinct peptide epitopes compared to other ethnicities, a key consideration for the design and evaluation of T cell vaccines that can provide coverage and efficacy across ethnically diverse global populations. This is highlighted by the finding that some current influenza vaccine candidates lack key components for immunogenicity in HLA-A*24:02+ individuals (41). HLA-A*24:02 is associated with risk of severe influenza disease (43) and notably is expressed by a significant proportion of Indigenous Australians (24%) (40, 41) and Alaskans (58%) (42). Whilst CD8+ T cell responses to IAV typically focus on epitopes derived from the virus Nucleoprotein (NP), Polymerase basic subunit B1 (PB1) and Matrix protein 1 (M1) (30, 37, 44, 45), immunogenic HLA-A*24:02-restricted IAV epitopes are derived from PB1 and Polymerase basic subunit B2 (PB2) proteins (41), representing a different focus in the viral proteins targeted. HLA-A*68:01, which is highly expressed in Indigenous populations from Alaska (15% among Alaskan Yup’ik) (46) and Southern and Central America (range 2.5%-61.54%, median 12% among Amerindians) (42), was also associated with severe influenza disease during the 2009 H1N1 influenza pandemic (43, 47). This may be linked to difficulties in the recruitment of CD8+ T cells specific for the HLA-A*68:01-NP145-156 epitope during IAV infection and could potentially be overcome with repeated boosting (48).
Associations between HLA genotype and disease severity are seen for several viruses including human immunodeficiency virus 1 (HIV-1) (49, 50), dengue (51, 52), human papilloma virus (HPV) (53), hepatitis C virus (HCV) (54, 55) and SARS-CoV-1 (56) (reviewed in (57)). Likewise, certain alleles are associated with risk of tuberculosis infection (58), susceptibility to type 1 Diabetes (59, 60) or increased risk of developing autoimmune responses directed, for instance, to myelin (61) or dietary antigens (62). For influenza, five HLA-I alleles (A*02:01, A*03:01, B*57:01, B*18:01 and B*08:01) are linked with robust, cross-protective CD8+ T cell responses against all human influenza A viruses (63). Conversely, the aforementioned A*24:02 and A*68:01 alleles are associated with increased mortality to the 2009 H1N1 virus (43) and diminished (41) or poorly recruited (48) memory CD8+ T cell populations in uninfected donors, respectively. In a cohort of healthy individuals who received a seasonal influenza A vaccine, HLA-DRB1*11:04, DRB1*16:01, DQB1*05:02 and DPA*02:02 were marginally associated with higher antibody titres, while DRB1*13:03 was associated with lower antibody titre post vaccination (64). Emerging data indicate that expression of certain HLA alleles may also influence the outcome of SARS-CoV-2 infection (65–67), though this association is not seen in other studies (68, 69) (reviewed in (70)). Using HLA frequency data and in silico HLA-binding predictions applied across the SARS-CoV-2 proteome, HLA-B*15:03 (most highly expressed in Sub-Saharan African populations (42)) showed the greatest capacity to present SARS-CoV-2 peptides that are highly conserved among other pathogenic human coronaviruses (66). Although not validated experimentally in human donors, the findings suggest that expression of HLA-B*15:03 is associated with broadly protective CD8+ T cell immunity to SARS-CoV-2. Meanwhile, HLA-B*46:01 (most highly expressed in South-East Asian populations (42)) was predicted to bind the fewest SARS-CoV-2 peptides (66), suggesting diminished capacity for CD8+ T cell responses and increased susceptibility to severe disease, as observed in SARS-CoV-1 (56). Given that HLA profiles are heritable and influenced by ethnicity, such HLA-related effects on adaptive immunity may have greater impact on populations with distinct or highly restricted HLA profiles, such as Indigenous populations. For instance, the five HLA-I alleles associated with “universal” immunity to IAVs are reasonably common in Caucasian populations (57%), but less so in Indigenous Alaskans and Australians (both 16%) (63) which are populations with a history of disproportionate influenza disease.
Thus, to avoid potential immunological gaps and optimize protective immunity across different Indigenous groups, the development and evaluation of T cell-activating vaccines may benefit from consideration of the particular HLA expressed and the specific array of peptides they present during infection. Whilst vaccine approaches are logically aimed at achieving broad coverage of global populations, either through inclusion of large epitope rich portions of the target pathogen (71, 72) or polyepitopes (41), this may result in exclusion of the best epitope candidates for some HLA profiles, potentially impacting vaccine efficacy in some populations. Tailoring vaccines based on population HLA profiles could theoretically help achieve optimal immunity and protection in these situations, yet implementing this across the HLA diverse human population on a global scale currently poses significant challenges. Broadening our understanding of immunologically relevant epitopes for Indigenous populations can potentially guide the search for optimal vaccine targets, ensure wide population coverage of vaccines and form the basis for evaluating vaccine responses which can then inform the next generation of vaccines, vaccine regimens (adjuvanting/dose frequency/high dose), development of public health measures and use of immunotherapies.
The identification and study of CD8+ T cell epitopes has traditionally focused on common and widely-expressed HLA alleles, such as HLA-A*02:01, which is highly prevalent in Caucasian populations (26%) (42). However, these well-studied HLA alleles are not always representative of ethnically diverse populations and are often found at lower frequencies in Indigenous populations. In addition, these studies may focus on one or a few immunodominant epitopes rather than comprehensively mapping the hierarchy of immunogenic epitope specificities for a given HLA. Overall, the HLA alleles most prevalent in Indigenous Australians are under-represented in epitope discovery, which typically reflects a focus on globally frequent HLA alleles (Figure 1 and Table 1). There are currently no epitopes from any pathogen reported in the Immune Epitope Database (IEDB) (73) for HLA-A*34:01, the most prominent HLA allele expressed by ~30% of Indigenous Australians (up to 68% in some populations (74)) (Figure 1 and Table 1). Of the eight most common (>10%) HLA I alleles expressed by Indigenous Australians, only the four most globally frequent (HLA-A*02:01, A*24:02, A*11:01 and B*40:01) currently have T cell epitopes reported for influenza A virus in the IEBD, while five (HLA-A*02:01, A*24:02, B*13:01, A*11:01 and B*40:01) have epitopes from SARS-CoV-2 reported (Table 1) (41). Comprehensively defining CD8+ T cell epitope specificities for a set of HLAs representative of Indigenous populations could assist the rational design and evaluation of vaccines that include suitable antigenic targets for priming relevant CD8+ T cells. Furthermore, understanding the breadth (number) and hierarchy of epitope-specific responses for these HLA allotypes is important to avoid vaccine-concentrated immune pressure on one or two epitopes leading to viral escape (75, 76). Epitope change in the presence of CD8+ T cell immune pressure has been observed for influenza virus in a mouse model, where mutations at anchor sites and TCR contacts for CD8+ T cell epitopes were identified and observed to revert in the absence of epitope-specific immune pressure (76). Furthermore, anchor mutations have been identified in the HLA-B*27:05- and B*08:01-restricted NP383–391/380–388 epitope that abrogate presentation and enable escape from T cell recognition (77, 78), indicating that T cell-mediated antigenic drift (44, 79) could subvert the efficacy of a T cell-based vaccine that concentrates on only a few epitopes in populations with limited HLA diversity. Comprehensively defining CD8+ T cell epitopes also enables consideration of how epitope-specific CD8+ T cell response hierarchies and immunodominance for a given HLA are influenced by HLA co-expression, cross-reactivity and infection history, which may be relevant to vaccine antigen selection and uncover associations between HLA expression and disease protection versus susceptibility.
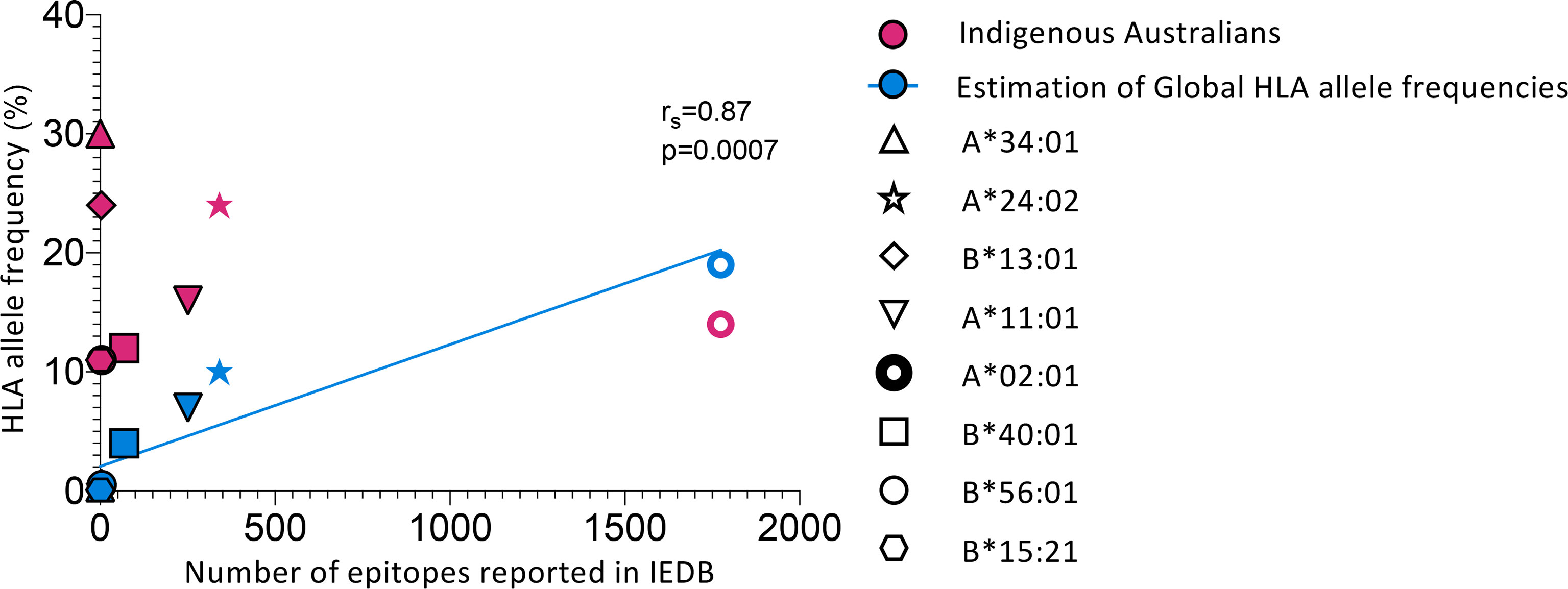
Figure 1 Indigenous Australian HLA alleles are underrepresented in epitope discovery. Comparing HLA allele frequencies from global population estimates (42) and Indigenous Australians (LIFT cohort) (40) with the number of epitopes reported in the Immune Epitope Database (IEDB) (73). Search filters for reported epitopes in the IEDB were: Epitope : Any, Host : Human, Assay:T cell, MHC restriction:Specific alleles indicated in the figure (HLA-A*34:01, A*24:02, B*13:01, A*11:01, A*02:01, B*40:01, B*56:01 and B*15:21), Disease : Any. Only epitopes from organisms other than homo sapiens were counted. See Table 1 for full details of organisms and number of epitopes reported for each allele considered (except HLA-A*02:01). Blue line shows correlation (Spearman’s test) of estimates of global HLA allele frequencies and reported epitopes. HLA-A*34:01, A*24:02, B*13:01, A*11:01, A*02:01, B*40:01, B*56:01 and B*15:21 represent the eight most common (>10%) HLA-I alleles expressed by Indigenous Australians (40).

Table 1 Indigenous Australian HLA alleles are underrepresented in T cell epitope discovery.
While several experimental methodologies and in silico prediction tools for epitope discovery have been developed, comprehensively defining novel T cell epitopes presented by the distinct and unique HLA expressed by Indigenous populations presents specific challenges. Here we review these current methodologies in the context of distinct Indigenous HLA profiles and describe in detail an immunopeptidomics approach to sequence HLA-bound peptides presented on HLA-defined virus-infected cell lines by liquid chromatography with tandem mass spectrometry (LC-MS/MS). This approach is being successfully used to comprehensively identify novel immunogenic CD8+ T cell epitopes for influenza A and influenza B viruses restricted by HLAs predominant in Indigenous Australians (41, 80). Given the shift in vaccine design towards targeted strategies that harness antibody and cellular immunity, understanding the T cell epitopes for HLAs representative of vulnerable Indigenous populations is critical to ensure equality of vaccine coverage and efficacy across the full diversity of global populations.
Approaches for Epitope Identification
Extensive HLA polymorphism poses a considerable challenge for epitope identification. Various experimental and in silico approaches (Figure 2) have been developed that provide a framework for dealing with the large diversity of HLA alleles (24,009 Class I HLA and 8,888 Class II HLA alleles identified as of February 2022 (IPD-IMGT/HLA Database (81)) and the array of peptides they can present. These approaches have advantages and disadvantages when considering the distinct sets of HLA alleles prevalent within Indigenous populations that are not necessarily shared at high frequency across other ethnicities (Table 2).
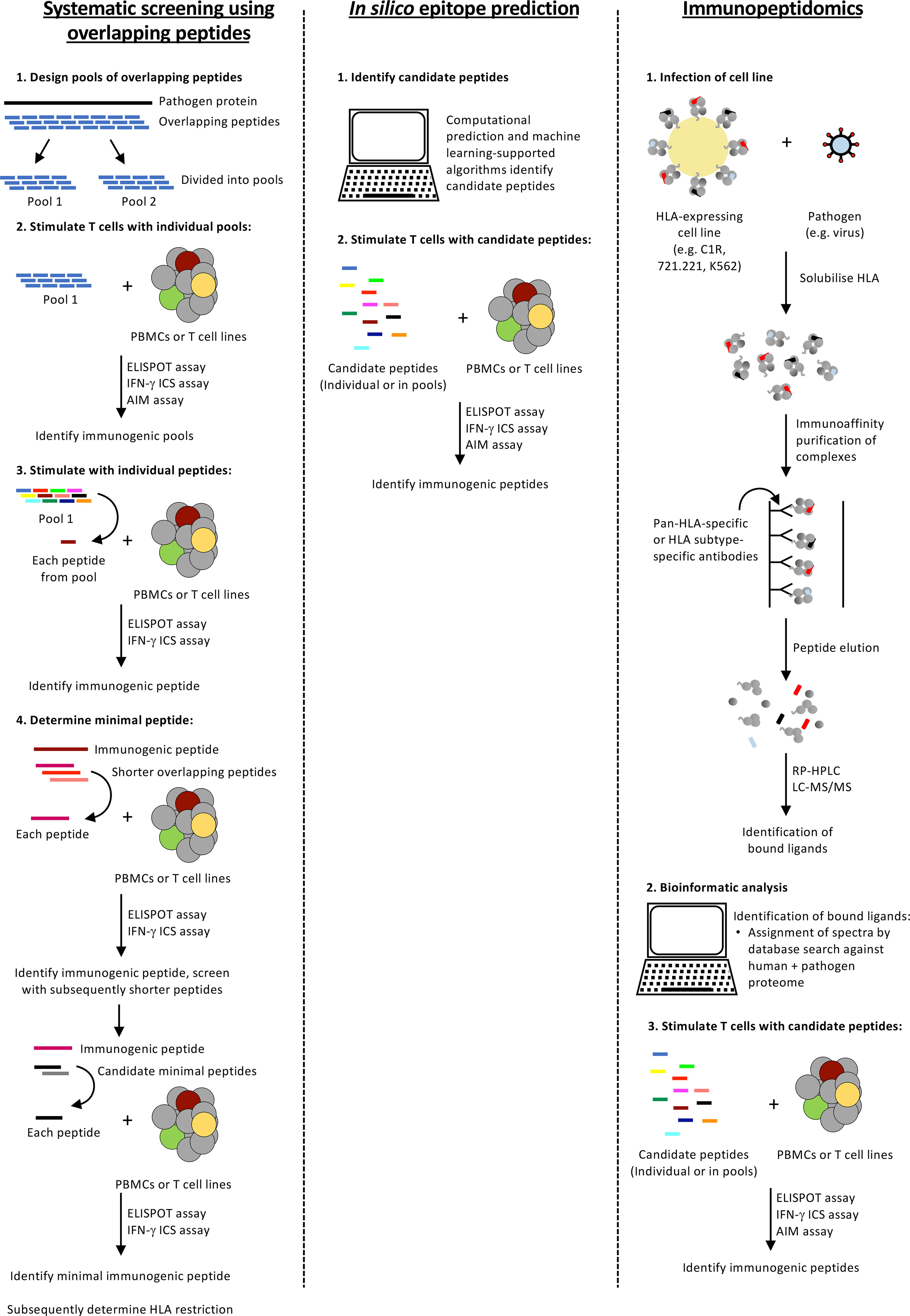
Figure 2 Approaches for identifying T cell epitopes for HLA alleles expressed by Indigenous populations. A summary of three epitope identification approaches.
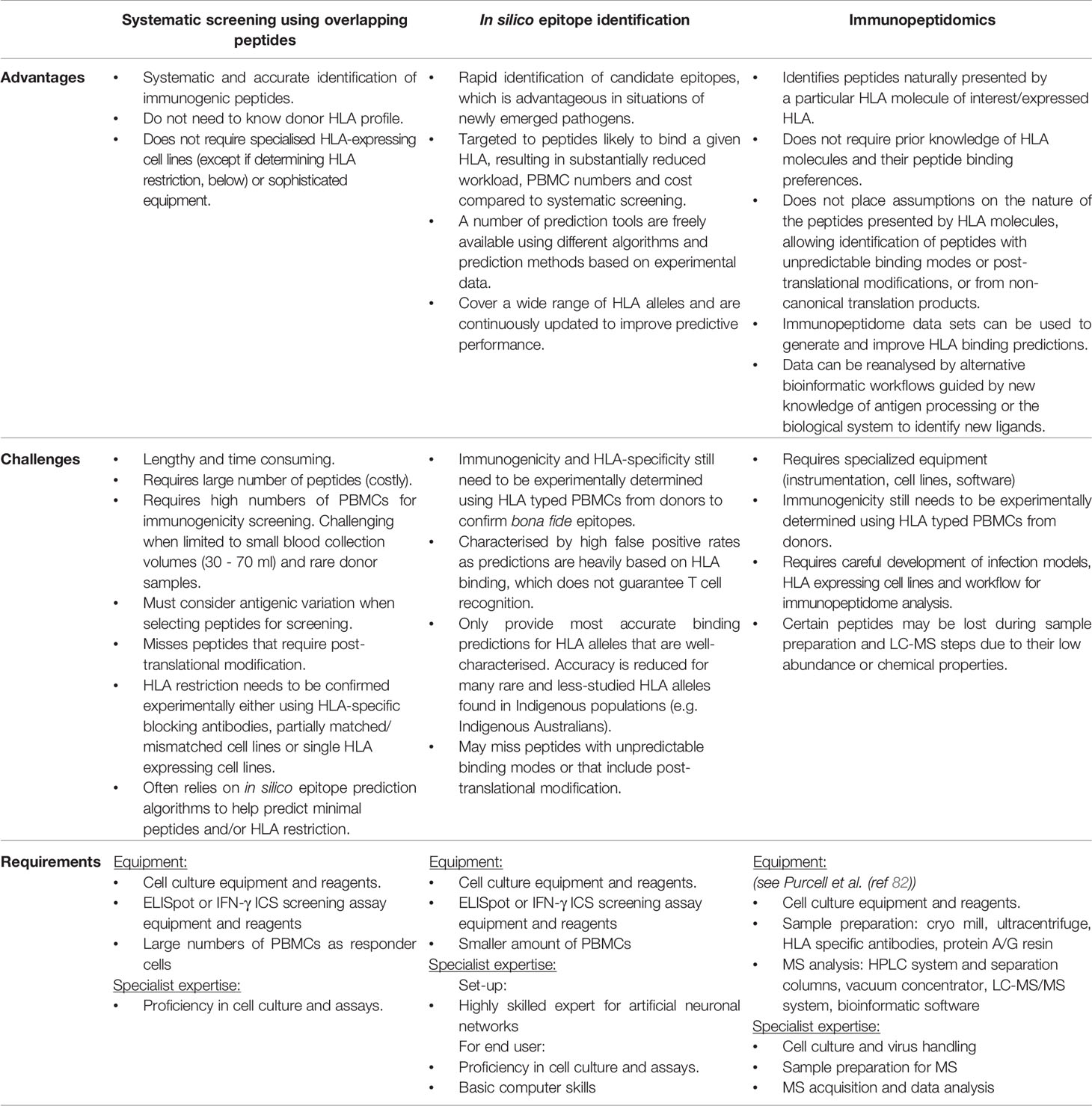
Table 2 Comparing epitope identification approaches, their advantages and challenges when applied to distinct sets of HLA alleles expressed by Indigenous populations.
Systematic Screening Using Overlapping Peptides
A robust and accurate approach for epitope identification is systematic screening for epitope-specific T cell responses by stimulating peripheral blood mononuclear cells (PBMCs) with pools of overlapping peptides (Figure 2). Whilst 15- or 18-mer peptides (overlapping by 10 or 12 amino acids (aa), with 5 or 6 aa shifts, respectively) are often used, Class HLA I molecules prefer shorter peptides (8-10 aa) due to the closed ends of the peptide binding groove and consequently, screening with overlong peptides can miss responses (83). Thus, depending on the budget and responder cell availability larger pools of shorter peptides (10 aa) are often used. T cell responses to peptide pools can be characterised directly following in vitro peptide stimulation by detection of cytokine (ELISpot assay or intracellular cytokine staining (ICS)), degranulation (CD107a) or upregulation of specific activation-induced markers (activation induced marker (AIM) assay) (Figure 3 and Table 3). Notably, the readout of cytokine assays (e.g. IFN-γ, TNF, IL-2 IL-4, IL-5, IL-9 and/or IL-17 production) needs to be tailored to the pathophysiologic state under study (84, 85). If only small numbers of PBMCs are available or the epitope-specific T cells are rare, responses can be characterised after initial in vitro expansion by co-culturing the PBMCs for 10-15 days with matched PBMCs pulsed with the overlapping peptide pools or infected with the pathogen of interest (86). Notably, when choosing to expand epitope-specific T cells using pathogen infection, it is important to consider the possible implications of the infection model and immunodominance hierarchies on the responses measured (e.g. the timing of protein/peptide expression, efficiency of epitope presentation in the cell line infected, epitope abundance and co-expressed HLA alleles). Expanded T cell lines are then screened for responses to the pools of overlapping peptides as above and immunogenic pools identified. The same T cell lines or PBMCs are then stimulated with individual peptides within the immunogenic pools to identify immunogenic peptides, most often using an IFN-γ ICS assay to define the peptide-specificity and CD4+ or CD8+ phenotype of the T cell response. Once an immunogenic peptide is identified, shorter overlapping peptides spanning the immunogenic peptide are synthesised (e.g. 13-mer peptides overlapping by 11 aa with 2 aa shifts) and tested for responses to determine the minimal immunogenic peptide sequence. In some cases, it may be possible to use algorithms to help predict the minimal peptide sequence, however, this relies on knowing the HLA profile of the donor and the availability of reliable data to predict the binding preferences of these HLA, something that is often lacking for the distinct HLA alleles expressed in Indigenous populations.
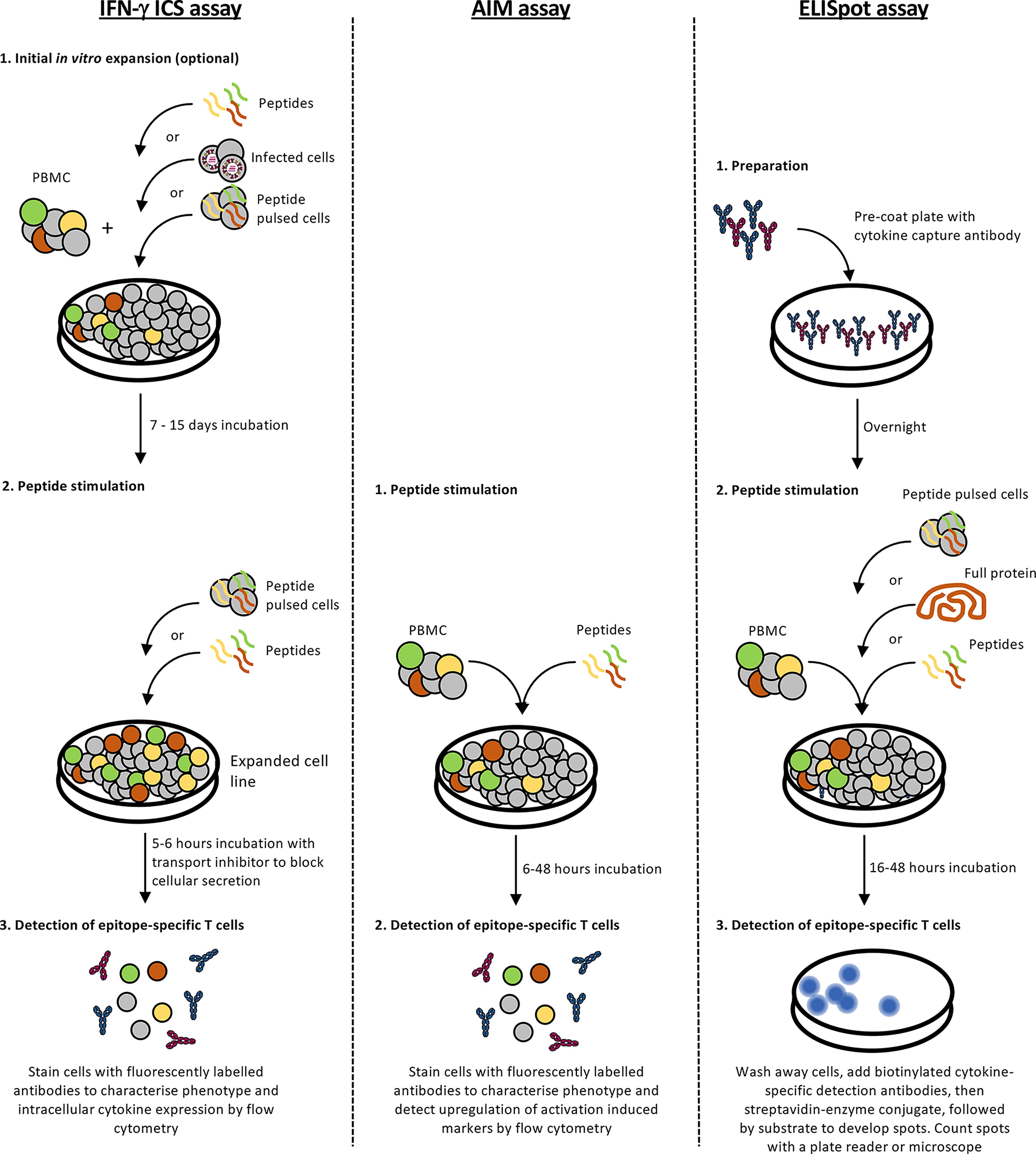
Figure 3 Screening for immunogenic epitopes. A summary of enzyme-linked immunospot (ELISpot), intracellular cytokine staining (ICS) and activation-induced marker (AIM) assay protocols for validating immunogenic epitopes.
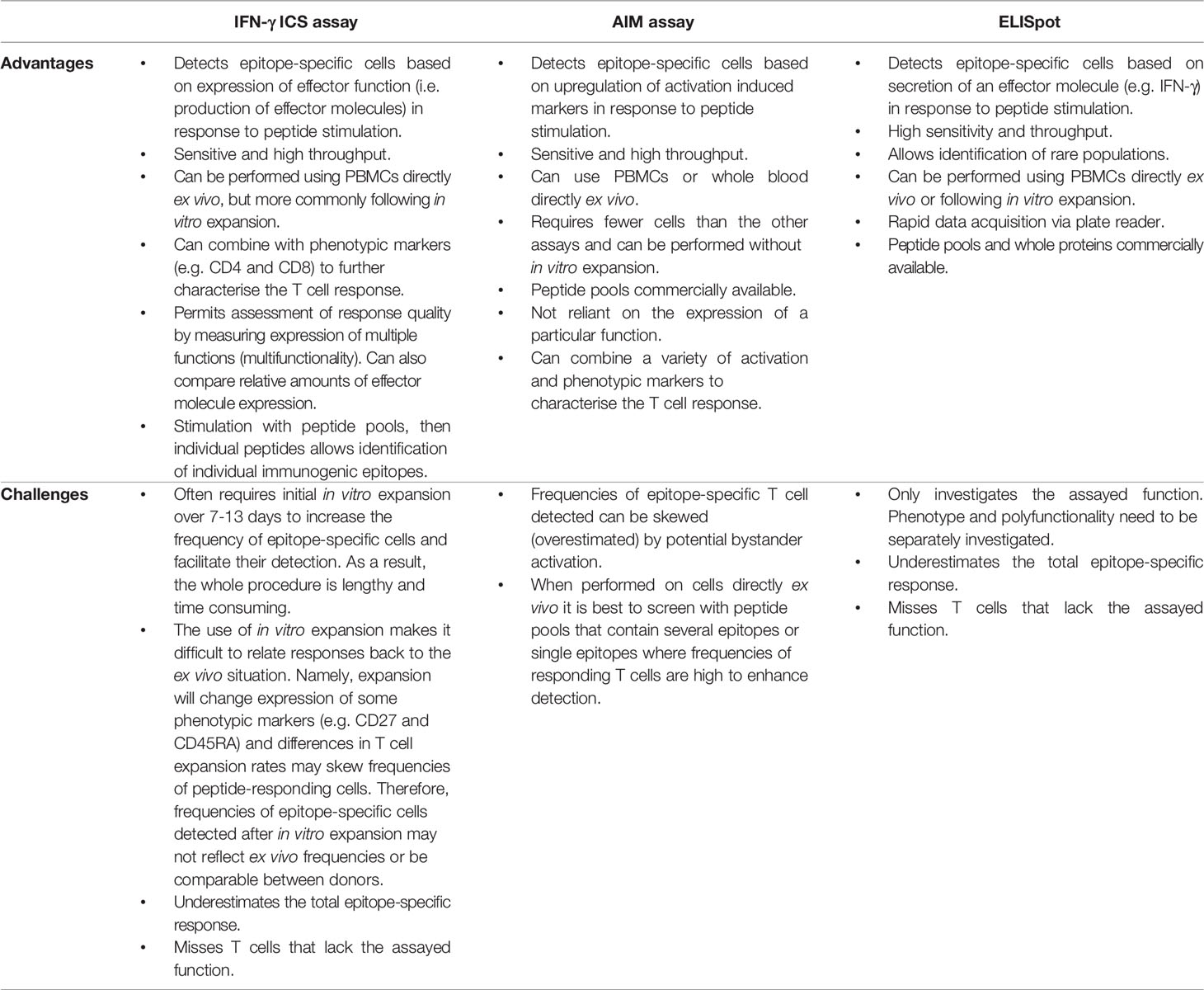
Table 3 Comparing assays used to measure T cell responses to candidate epitopes.
Determination of HLA restriction is an important step in characterising novel immunogenic peptides. As mentioned above, in some cases this can be predicted using algorithms trained to identify characteristic HLA-binding motifs or determined experimentally using antibodies that bind to specific HLA, blocking peptide presentation and T cell recognition. Alternatively, partially HLA matched/mismatched cell lines (or PBMCs) or a panel of single HLA transfected cell lines can be used as antigen presenting cells in a peptide stimulation IFN-γ ICS assay with the peptide-specific T cells to determine whether a peptide can be presented or not, allowing HLA restriction to be identified (86). These approaches are not always useful in the context of distinct or rare HLA alleles from Indigenous populations due to a lack of training data to boost the predictive power of algorithms, and lack of specific HLA blocking antibodies and suitable cell lines expressing these particular HLA.
While the systematic screening approach is lengthy, time consuming and involves large numbers of peptides which can be costly (~$50-100 per commercially synthesised peptide), it can allow for the accurate identification of immunogenic peptides independently of knowing the donor HLA profile and the peptide binding characteristics of the restricting HLA. However, reasonably high numbers of PBMCs are needed to systematically screen the full length of multiple viral proteins. Buffy coats obtained from routine blood donations (~470 ml whole blood) provide large numbers of PBMCs (>1-2x108 cells), enabling epitope screening across multiple peptide pools containing relatively small numbers of peptides (10-30 peptides) (86). Meanwhile, the study of Indigenous HLA alleles requires PBMCs isolated from Indigenous individuals, which in our experience come from smaller volumes of blood (30-70 ml) kindly donated by consenting study participants. Thus, sample cell numbers (<2x107 cells) become the major limiting factor for screening multiple peptide pools in Indigenous donors. Furthermore, our study of Indigenous Australians identified two new HLA alleles (HLA-A*02:741 and HLA-A*56:56) that, to our knowledge, have not been found expressed in any other ethnic group worldwide (40). These, along with other previously described unique Indigenous Australian alleles (74) occur at very low frequency (<1% Indigenous Australians), meaning that few donors with these HLA alleles are available to study, necessitating a more targeted approach. Another challenge that must be considered across all epitope identification approaches, but especially the systematic screening approach, is antigenic variation across different virus strains and lineages that may influence the detection of immunogenic epitopes depending on patterns of virus circulation and exposure history. As antigenic variation can occur particularly in epitope regions under immune pressure, leading to evasion of existing immune responses, it is necessary to consider the particular peptide sequences selected for epitope screening and potentially include pools of variant peptides to ensure responses to variable epitopes are not overlooked. For instance with the immunodominant HLA-B*07/B*35:01-restricted NP418-426 epitope, sequential viral variation has generated over 20 different peptide variants at TCR contact sites, some of which prime distinct CD8+ T cells with variable patterns of cross-reactivity to other variants (87).
Overall, systematic screening using overlapping peptides across multiple virus proteins offers the advantages of being systematic and accurate without the need for donor HLA typing or specialised equipment (laboratory equipment, engineered cell lines or software) (Table 2). However, is not feasible for defining T cell epitopes restricted by many of the distinct and rare HLAs found in Indigenous populations, mainly due to the high numbers of PBMCs and peptides required for thorough screening.
In Silico Epitope Prediction
The threat of unpredictable novel respiratory viruses, highlighted by SARS-CoV-2, underscores the need for more accelerated methods of epitope discovery that facilitate rapid vaccine design and evaluation. The development of in silico epitope prediction tools enables identification of candidate peptides using computational prediction and machine learning-supported algorithms (Figure 2). Candidate peptides can then be screened for immunogenicity by stimulating PBMCs from suitable donors with the peptide and measuring epitope-specific T cell responses either by ELISpot, ICS or AIM assay (Figure 3). As HLA binding is a prerequisite for a peptide to be recognised as an epitope, in silico epitope prediction tools select peptide candidates based mainly on predicted HLA binding affinity which is determined through evaluation of the amino acid sequence of the peptide to forecast its compatibility for binding HLA. Such forecasting is possible because the structural and chemical properties of the HLA peptide-binding groove dictate the selective binding of peptides with compatible amino acid features. Various tools are available that differ in the experimental data or ‘training data sets’ used to develop algorithms and their methods of calculation. Training data sets generally consist of experimental data obtained from MHC binding assays measuring binding affinities of peptides to specific MHC molecules, or MHC elution data from sets of naturally processed MHC ligands eluted from cell surface MHC molecules and identified by mass spectrometry. More recent prediction tools such as NetMHCpan-4.1 and NetMHCIIpan-4.1 combine both data sets to improve prediction performance (88). MHC binding can be predicted using one of two approaches. Systems using linear regression such as PickPocket, SMM and TEPITOPE apply a fitted weight matrix to calculate a binding affinity value to a selected peptide sequence (89). The current state-of the-art methods NetMHCpan-4.0, NetMHCIIpan-4.0, MHCflurry and NNAlign use artificial neural networks to simulate MHC-binding with a variety of extra features in addition to the input peptide sequence and also allow for calculation of binding-affinity for untrained HLA alleles by comparison with closest neighbouring MHC (89). Comparison of these algorithms revealed the superiority of artificial neural network-based approaches in predicting MHC binding, but lower correlations of predicted versus measured binding affinities for strong binding peptides (89). Some algorithms such as NetChop and NetCTL-1.2 can improve epitope predictions by not only predicting the binding affinity of peptides trained on endogenously presented peptide data sets and binding affinities, but also the likelihood of cleavage by the proteasome and transport by TAP (transporter associated with antigen processing) into the ER to predict immunogenic CD8+ T cell targets (90). Computational tools can also be used to further refine predicted epitopes based on their potential immunogenicity, robustness, allergenicity, toxicity and autoimmunity. One of these algorithms, NetCTLpan showed up to 40% reduced experimental workload to identify 90% of new epitopes compared to prediction tools that only assess MHC binding affinity (90).
In silico epitope prediction is of high interest for quickly identifying T cell epitopes when new pathogens emerge, speeding up identification of novel T cell targets that allow for the assessment of immune responses or the development of T cell activating vaccines. These tools were utilized for example to efficiently predict epitopes for various HLAs during the COVID-19 pandemic, highlighting their emerging importance in research (see below). Another situation where epitope prediction tools are superior to classical screening is in personalized cancer treatment. Cancer cells can be sequenced and neo-epitopes predicted for use in a vaccine to induce cancer-specific T cells. This allows a level of personalized treatment that was unachievable previously and is already efficiently used in clinical trials. BioNTech was able to show that vaccination with personalised melanoma neo-epitopes could induce T cell responses in all participants and resulted in killing of neo-epitope-expressing melanoma cells (91). Likewise, Ott et al. (92) demonstrated induction of highly specific, polyfunctional CD4+ and CD8+ T cell responses that targeted a broader range of neo-epitopes than induced by existing immunotherapeutics and prevented disease recurrence in 4/6 patients up to 25 months post-vaccination. Thus, aided by epitope prediction tools, this treatment potentially overcomes key challenges to effective cancer therapy including individual tumour heterogeneity, selective targeting of tumour versus healthy tissue and tumour escape through loss of antigen. An intriguing idea is that computation tools could potentially be adapted to use self-peptidome data to establish rules that predict peptides to be avoided for use for infectious disease or cancer indications, perhaps as they have motifs or properties that are too similar to self and potentially could cause autoimmunity. Lastly, the usage of epitope prediction tools can substantially reduce workload and cost by limiting the amount of peptides required to screen, an attractive feature in the context of rare and limited donor samples.
In silico epitope prediction methods are characterised by high false positive rates, in that many of the predicted epitopes are found to be non-immunogenic. For example, Zheng and colleagues used in silico analysis to predict novel hepatitis B virus polymerase epitopes for the most common human MHC-I, HLA-A*02:01 (93). Of the pre-selected epitopes, 25% (2 out of 8) demonstrated good ability to induce immune responses and suppress hepatitis B virus replication in vitro. In a different study, only 13 out of 225 (6%) predicted tumour-associated antigens showed immunogenicity in an in vivo experiment using transgenic mice (94). The efficiency of in silico epitope prediction can depend on factors such as the protein sequence analysed, the MHC molecule of interest and the data set used for training. However, a key limitation of the algorithms is that they predict HLA binding as a surrogate for epitope presentation, which does not guarantee T cell recognition and activation, i.e. immunogenicity. Future algorithms could potentially be enhanced by incorporating information on potential T cell receptor (TCR) interactions as well as comparison to other endogenously presented peptides on which the immune system is trained. Inclusion of these parameters could increase the frequency of correctly predicted immunogenic T cell epitopes.
A further limitation is that most in silico epitope prediction studies and experimental validations are biased towards certain HLA alleles. All methods of in silico prediction rely on previously published data and, as already outlined, the identification and study of T cell epitopes often focuses on common and widely-expressed HLA alleles. As such, these algorithms are constrained by the training data and can therefore only provide most accurate guidance for MHC alleles that are well-characterised or similar to well-characterised MHCs (95, 96). Their accuracy is reduced for rare or less-studied HLA alleles that are poorly represented in databases, which is the case for certain HLA alleles found in Indigenous Australians. Inclusion of additional HLA-specific binding and elution data sets is needed to further improve the capacity of these algorithms to accurately predict T cell epitopes for Indigenous populations.
While initial programming of artificial neural networks and generation of training data is both expensive as well as requiring highly trained personnel, if well programmed the resulting epitope prediction tools are extremely easy to use for the end user (Table 2). They significantly reduce the budget required to purchase and screen candidate peptides, facilitating research on smaller populations like Indigenous Australians or less common pathogens that might not be of high commercial interest for bigger pharma companies or attract sufficient funding for high-scale experiments.
In Silico Epitope Prediction and COVID-19
In silico epitope prediction is highly advantageous in situations of newly emerged pathogens, utilizing previously determined peptide data sets and the novel pathogen’s genetic sequence. Shortly after the genetic sequence of SARS-CoV-2 was published in early 2020 (97), in silico methods were used to predict T cell epitopes as potential targets for SARS-CoV-2 vaccines. Two rationales for in silico prediction have been used to predict SARS-CoV-2 epitopes, i) utilizing the genetic similarities between SARS-CoV-1 and SARS-CoV-2 and ii) peptide-HLA binding prediction methods. Epitopes derived from SARS-CoV-1 structural proteins induce long lasting T cell immunity (98, 99) and these proteins share high genetic similarity with SARS-CoV-2, ~76% for spike and >90% for nucleoprotein, membrane, and envelope proteins (100). The high genetic similarity prompted in silico studies to use T cell epitopes from SARS-CoV-1 immunological studies to predict likely SARS-CoV-2 T cell targets (98, 100–103). As sequencing data from new SARS-CoV-2 variants continued to emerge, a web-based platform, COVIDep, was developed to enable the identification of experimentally-derived SARS-CoV-1 epitopes that have a close genetic match with the latest available sequencing data (104). Nonetheless, the large majority of in silico SARS-CoV-2 studies have predicted T cell epitopes using existing peptide-HLA binding prediction methods, which benefit from years of peptide-HLA research. In silico predicted epitopes have been derived from all 12 SARS-CoV-2 proteins, with the spike protein being the most analysed protein, though unsurprisingly, there is considerable overlap among the sets of epitopes for SARS-CoV-2 predicted using different methods. As of September 2020, 2,239 HLA class I and 2,580 HLA class II epitopes had been predicted using in silico methods for SARS-CoV-2 (105). However, a low proportion of these have been experimentally validated, reflecting a disconnect between in silico studies and validation studies. Of interest, a study focusing on South American populations used updated HLA frequencies and in silico epitope prediction tools to establish a tailored list of 27 HLA-I and 34 HLA-II candidate epitopes that would provide high regional coverage if incorporated in a vaccine against SARS-CoV-2 (106). These candidates achieved better regional coverage than other approaches that aimed to identify epitopes that cover the global population. This highlights the strength of epitope prediction tools to rapidly identify candidate epitopes for a defined HLA profile. However, it also shows how approaches based on broad population coverage can result in reduced regional coverage due to the associated HLA profile.
While in silico epitope prediction is incredibly effective at generating large databases of potential epitopes, immunogenicity and HLA-specificity need to be experimentally determined to confirm bona fide epitopes. PBMCs from individuals recovered from COVID-19 have been used to experimentally characterise in silico predicted peptides. T cell activation can be measured following in vitro peptide stimulation (ELISpot, ICS or AIM assay, see below) either directly (102, 107, 108) or after initial in vitro expansion by co-culturing (109–111). Additionally, HLA restrictions of the reported epitopes can be experimentally determined by using multimer qualitative binding (112–114). Following experimental validation, over 700 unique SARS-CoV-2 T cell epitopes with known HLA allele restriction have been reported, including 20 immunoprevalent epitopes (>50% of tested recovered individuals responded) registered across multiple cohorts (115). This is highlighted by the experimental-verification of peptide-HLA binding predicted epitopes restricted to HLA-A*02:01 and HLA-A*24:02. From epitopes predicted by NetCTLpan and/or NetMHCpan, three HLA-A*02:01 (S269–277, S976–984 and Orf1ab3183–3191) and three HLA-A*24:02 (S1208–1218, S448–456 and S193–201) epitopes were found to be immunogenic following in vitro expansion of PBMCs from COVID-19 donors for 10-12 days (110, 111). Importantly, while these HLA alleles are common and widely-expressed globally, they are also highly expressed in Indigenous Australians. Following identification of novel SARS-CoV-2 epitopes, T cells tested directly ex vivo from COVID-19 patients and pre-pandemic healthy individuals can be assessed for their phenotype, function and TCR. Different SARS-CoV-2 epitopes have been found to have different immunodominances, with A1/ORF1a1637 and B7/N107 currently identified at higher precursor frequencies compared to the known HLA-A*02:01 and A*24:02 epitopes (112, 114, 115). T cells specific to SARS-CoV-2 epitopes restricted to both HLA-A*02:01 and A*24:02 have a predominantly naïve phenotype in pre-pandemic individuals, whereas the phenotypic profile in COVID-19 donors is more varied with skewing towards memory phenotypes (111, 112). The clonal composition and diversity of the TCR repertoire can impact immunodominance, functionality and protection. Recent studies have suggested that TCRαβ diversity might be linked with the prominence of SARS-CoV-2 CD8+ T-cell responses during the primary infection. The subdominant A2/S269+ and A24/S448+ CD8+ TCRαβ repertoires were driven by restricted motifs, whereas the A1/ORF1a1637+CD8+, B7/N105+CD8+ and A24/S1208+CD8+ TCRαβ repertoires were more diverse across COVID-19 patients (111, 112, 116).
Characterization of SARS-CoV-2 T cell epitopes is important in the fight against COVID-19 and for protecting Indigenous populations. In silico epitope prediction and experimental verification of SARS-CoV-2 T cell responses has assembled a data set of key T cell epitopes presented by a range of HLA alleles. Analysis of epitopes for mutations in SARS-CoV-2 variants of concern can help with monitoring potential viral escape from T cell responses, with the identification of epitopes capable of eliciting broadly cross-reactive immunity across emerging variants of upmost importance. Furthermore, novel antiviral vaccines developed for COVID-19 can elicit both antibody and T cells responses (117, 118). Therefore, the discovery of SARS-CoV-2 epitopes allows epitope-specific T cells to be tracked and correlates of SARS-CoV-2 protection defined following infection and vaccination. Finally, the identification of immunogenic T cell epitopes could aid the rational design and evaluation of the next-generation universal COVID-19 vaccines, especially for high-risk groups such as Indigenous people.
Immunopeptidomics for Identification of Candidate T Cell Epitopes
Mass spectrometry-based characterisation of the peptides presented by HLA/MHC molecules, or immunopeptidomics, has come to the forefront in the last 10 years as a means to detect naturally processed and presented peptide ligands. The great benefit of these analyses is that they do not place assumptions on the nature of the peptides that can be liberated through enzymatic processing, nor do they rely on sufficient knowledge of the interrogated HLA/MHC molecules for accurate binding predictions. Not only does this allow for identification of ligands that adopt unpredictable binding modes (e.g. overhanging/extended HLA-I ligands (119–121)), but it also allows detection of post-translationally modified peptides of biological relevance (e.g. glycosylation, deamidation, phosphorylation) as well as those from non-canonical sources (alternative reading frames, UTRs) that would not necessarily be considered when designing overlapping peptide libraries or performing binding predictions.
HLA-I molecules constitutively present peptides derived from the enzymatic processing of proteins produced within the cell. Characterisation of these peptides via mass spectrometry, especially from cell lines engineered to express single HLA-I alleles, has been used to help define peptide binding preferences of numerous HLA variants (121–126). However, in the context of viral infection, immunopeptidomics also represents a method for the identification of naturally presented candidate epitopes for subsequent immunogenicity screening (for example (41, 127–130)). In-depth descriptions of the methodologies for immunopeptidome analysis can be found elsewhere (82, 131). The basic process (Figure 2) generally involves:
1. Solubilisation of the MHC/HLA from cellular material via non-denaturing lysis to maintain complex conformation. Alternatively, cells can be engineered to express soluble forms of MHC/HLA which can be harvested from cell culture supernatant (125, 126, 128, 129, 132), while soluble HLA can also be isolated from blood serum (133, 134).
2. Immunoaffinity purification of complexes using either pan-specific (e.g. W6/32 for all HLA-I) or sub-type specific (e.g. BB7.2 for HLA-A2) HLA/MHC antibodies.
3. Peptide elution via denaturation (usually acid elution).
4. Removal of HLA/MHC heavy and light chains. For example, via reversed-phase high performance liquid chromatography (RP-HPLC) (including fractionation the peptide ligands) or using molecular weight cut-off filters.
5. Liquid chromatography-tandem mass spectrometry (LC-MS/MS).
6. Bioinformatic analysis.
Workflow modifications can be incorporated to increase robustness of quantitative comparisons (e.g. targeted analysis using Multiple Reaction Monitoring) (135), to enrich for specific modifications within the sample such as phosphorylation (136), or to interrogate samples for specific post-translational modifications (e.g. spliced peptides, glycosylation) (137, 138). Currently (February, 2022) more than 970,000 MHC ligands have been collated within the IEDB (73) that have been identified using mass spectrometry (Search filters: Include positive Assays, MHC Assays : Ligand presentation|mass spectrometry, ligand presentation|cellular MHC/mass spectrometry, ligand presentation|secreted MHC/mass spectrometry). Amongst these are more than 3000 (non-redundant by sequence and modification) derived from viruses including vaccinia (>900), SARS-CoV-2 (>800), and influenza viruses (>700).
As mentioned above, screening of large numbers of peptide pools is often prohibitive due to low sample volumes available for immunogenicity screening. An immunopeptidomics approach to sequence HLA-bound peptides by LC-MS/MS generates a more highly curated and biologically relevant list of peptides to screen via ELISpot, ICS or AIM assays compared to overlapping peptide libraries or binding predictions alone. Applying this approach to specific HLAs relevant to Indigenous populations enables comprehensive analyses of peptide presentation regardless of how well-studied the HLA allele is or the availability of relevant PBMCs. Nevertheless, whilst an in-depth mass-spectrometry-based immunopeptidomics approach is very successful at defining large numbers of peptides presented by HLA/MHC molecules during infection (41, 80, 119, 127, 130), many peptides eluted from HLA molecules are not immunogenic. For instance, in our immunopeptidomic study of HLA-A*24:02, dominant responses were detected against 3-4 (depending on ethnicity) out of 54 IAV peptides tested, and 6 out of 41 IBV peptides tested (41) (data from peptide elution studies are available through IEDB (http://www.iedb.org/refId/1039181) and the ProteomeXchange Consortium via the PRIDE (139) partner repository (accession PXD020292)). However, another study of ~170 vaccinia virus pMHCI presented on infected mouse cells found nearly 40% were immunogenic in more than half of the C57BL/6 mice screened (140). This again reflects the fact that HLA binding is necessary but not sufficient for immunogenicity and screening peptides identified using an immunopeptidomics approach with relevant PBMC samples will always be necessary to identify immunologically relevant T cell epitopes.
While LC-MS/MS-based analysis of the immunopeptidome can achieve identification of many thousands of peptides in a single experiment (including a subset of viral peptides in infection experiments), certain limitations and challenges should be acknowledged. Firstly, relatively large cell numbers are required for broad coverage of the immunopeptidome due to the overall low abundance of many bound species. Secondly, certain peptides may be lost during sample preparation due to lack of binding to chromatographic stationary phases (e.g. highly hydrophilic peptides may display low interaction with the stationary phase used during RP-HPLC separation, which can be further exacerbated by oxidation at methionine residues), while highly hydrophobic peptides (e.g. the immunodominant HLA-A*02:01-restricted IAV epitope M158-66 GILGFVFTL) may display low solubility in aqueous buffers. Thirdly, MHC-bound peptides which lack positively charged residues may exist as singly-charged ions and are often excluded from fragmentation in traditional proteome analysis based LC-MS methods which are deliberately biased to charge states of 2 or greater (typical of tryptic peptides which terminate in positively charged residues) to avoid fragmentation of singly charged contaminants common in mass spectrometry analysis. Inclusion of singly charged species has been shown to increase the number of MHC ligands identified for certain HLA alleles, although not incorporated by all studies (131). Indeed, differences in preparative, analytical and bioinformatic workflows have been shown to impact the properties of ligands identified by immunopeptidomics, with different pipelines inducing biases for/against specific amino acids (e.g., hydrophobic/basic/proline) that should be considered during workflow development (141, 142). Moreover, these workflows are susceptible to contamination by non-HLA bound protein fragments. Possible contaminants can be highlighted bioinformatically using strategies including GibbsCluster 2.0 (to identify outliers) (143), assessment of peptide ladders generated by proteolytic degradation (144) or comparison with similar data sets for common contaminants. Ultimately however, when searching for novel epitopes, application of immunopeptidome analysis for epitope discovery can be considered hypothesis generation, with immunogenicity testing as the final validation of biological relevance.
Finally, compared to using overlapping peptide libraries or performing binding predictions, immunopeptidome analysis requires access to specific materials such as appropriate cell lines, HLA-specific antibodies and immunoaffinity purification reagents, as well as instrumentation including a HPLC system, vacuum concentrator and high resolution LC-MS/MS. Furthermore, specialist expertise in sample preparation for MS analysis to avoid introduction of problematic contaminants, LC-MS/MS acquisition and bioinformatic analysis, as well as initial infection protocols are also required (Table 2).
Although we have stated that one of the great strengths of immunopeptidome analysis is detection of naturally processed and presented peptides that might be missed by binding predictions, immunopeptidome data sets can also be used to generate and improve MHC/HLA binding predictions. Indeed, as mentioned above, prediction tools such as NetMHCpan and HLAthena preferentially or even exclusively incorporate elution data to train their prediction algorithms (88, 123), while others seek to enable input of user defined data sets to generate bespoke MHC/HLA binding predictions (145). In the context of understudied HLA/MHC with limited or no published ligands/affinity data/immunopeptidome data, such as several of those expressed by Indigenous Australians, new immunopeptidome data sets represent a critical avenue to generate and improve HLA-binding predictions. Thus, in-depth immunopeptidomic investigation of a single infection model in the context of an understudied HLA, while directly detecting potential epitopes for downstream analyses, can also be repurposed, utilising the large data sets of host and pathogen derived peptide ligands to train binding models for application to alternate pathogens which are not as amenable to analysis (e.g. high biosafety level, poor infection efficiency).
Considerations for Single HLA Analysis via an Immunopeptidomics Approach
When available, the use of HLA-specific antibodies for isolation of peptide-bound HLA-I molecules can enhance the accuracy of assigning HLA binding (for example we utilised the anti-HLA-A2 antibody BB7.2 to specifically isolate HLA-A*02:01 from the C1R in our study of IBV peptide presentation (127)), which is of greater importance against more complex HLA backgrounds. However specific antibodies are not available for most HLA alleles. Normal human cell lines can express up to 6 distinct classical HLA-I at the cell surface due to HLA co-expression and heterozygosity. This generates a mix of HLA binding specificities when performing isolations with pan HLA-I antibodies. Several bioinformatic approaches have been generated to deconvolute multiple HLA-binding motifs from multi-allele data sets and represent one avenue to distinguish individual binding specificities (e.g (143, 146, 147)). Alternatively, single HLA-expressing cell lines with no or low endogenous HLA simplify this problem and facilitate the assignment of peptide ligands to a specific HLA allomorph with less bioinformatic processing. Single HLA-expressing cell lines are often generated via electroporation with a plasmid encoding the HLA of interest and cultured under conditions of antibiotic selection to maintain stable transfectants (148), or more recently by adapting retroviral transduction protocols (149) to stably transduce HLA genes (150) (L. Hensen, E. B. Clemens and K. Kedzierska, unpublished protocol. See reagent availability note.). Classical HLA-I-reduced (e.g. C1R) or null (721.221, K562) cell lines are valuable tools for both characterisation of peptides presented by a transfected HLA of interest and functional dissection of T cell epitope restriction (122–124). C1R cells are derived from EBV-transformed B cells passaged after three rounds of γ-irradiation and immunoselection for reduced HLA-I expression (151). C1R cells do not express any HLA-A molecules, have a markedly reduced expression of HLA-B*35:03 and stably express HLA-C*04:01, and our frequent use of this line in immunopeptidome characterisation has given us in-depth knowledge of the peptides presented by these endogenous HLA molecules (41, 127, 152). Permissivity to infection is an important feature of the cell line to be used and notably, while influenza virus strains differ in their ability to infect B cells (153–155), C1R cells can be readily infected with laboratory strains of IAV and IBV in vitro (41). Through insertion of single HLA genes into C1R cell lines, cell surface presentation of peptides by HLAs of interest can be assessed during infection without the need for HLA-I-subtype specific antibodies to enable identification of CD8+ T cell epitopes presented under physiological conditions of cellular virus infection. Alternatively, transfection of cells with DNA constructs to express soluble HLA, which is then harvested from the supernatant, is an alternative approach for interrogating peptides from single HLA variants (125, 126, 128, 129).
It should be acknowledged that presentation by single HLA-expressing cell lines such as C1R may not be a perfect mimic of presentation in the in vivo context, and may either miss epitopes generated in in vivo infected cell types (e.g. lung epithelia), or over-represent certain peptides. During infection in vivo, presentation by professional antigen presenting cells such as dendritic cells is important for T cell priming, whilst presentation by different infected cell types (e.g. epithelial cells of the airways during influenza (156) and SARS-CoV-2 (157) infection) is critical for their clearance. Altered quantitative hierarchies of epitope presentation via cross presentation on dendritic cells versus direct presentation on infected cells have been shown for influenza epitopes in mice, suggesting presentation pathway and presenting cell types may be factors that influence the immune response (158). Furthermore, processing enzyme expression also impacts peptide presentation with C1R cells, for instance, expressing standard and immunoproteasomal subunits (159) and specific ERAP1 and ERAP2 variants for which polymorphisms alter cleavage efficiencies (160).
During in vivo infection antigen presentation at different anatomical sites (e.g. the airways) is likely changeable dependent on the cellular response to infection and the inflammatory environment at different stages of infection. Inflammatory cytokines have been shown to cause marked changes in the immunopeptidome through modulation of components of antigen processing and presentation, including expression levels from different HLA loci and of the standard proteasome and immunoproteasome subunits, which can alter the levels of specific epitopes at the cell surface (161–165). Timing post-infection should also be considered, as in our own studies involving IAV and IBV infection we observed broadest peptide identification 8-12 hrs post infection (41), while down regulation of HLA molecules, a common strategy for viral evasion of cellular immune responses (166) (reviewed in (167, 168)), is observed at late stages of IAV and IBV infection in vitro (169), potentially impacting the breadth of peptide presentation. This may change for different viruses, strains and infection models. Thus, the development of models that more closely mimic the in vivo setting is of great interest (170) and a consideration for the success of this strategy. For viruses with low infection efficiency of cell lines, or for which high physical containment is required, analysis of presentation of specific viral proteins can also be performed by transfection with expression constructs for viral antigens (171). Moreover, the self-immunopeptidome of these lines can be used to build peptide binding predictions for these understudied HLA molecules, as mentioned above. These binding predictions provide a secondary lens through which to assess identified candidates to increase confidence of assignment as binders of a given HLA and differentiate contaminants. For understudied HLA allomorphs, where limited training data is available for binding predictions, the co-isolated self-derived immunopeptidome is of increased importance for understanding binding preferences and triaging candidate epitopes.
Validation and Characterisation of T Cell Responses to Candidate Epitopes
Specific T cell responses to candidate epitopes can be measured using a variety of different assays (Figure 3 and Table 3). ELISpot and IFN-γ ICS assays are sensitive and high throughput methods that measure epitope specificity based on cytokine production by T cells in response to peptide stimulation. These assays are particularly useful for screening responses initially to peptide pools, then smaller pools or individual peptides to identify minimal immunogenic epitopes. The assays can be performed using PBMCs directly ex vivo or following in vitro expansion using autologous PBMCs pulsed with peptide pools to expand epitope-specific T cells, particularly if the epitope-specific T cells of interest are present at low frequencies. Other methodologies for expansion such as virus infection or whole antigens would also be suitable. T cell lines expanded in vitro can then be stimulated with the corresponding peptide pool or individual peptides for detection of immunogenic peptides. The ICS assay has the advantage of discriminating CD4 and CD8 phenotype and detecting expression of multiple functional molecules (e.g. IFN-γ, TNF, MIP-1α, IL-2 and CD107a), allowing measurement of polyfunctionality and phenotypic subsetting. However, ELISpot is more sensitive at measuring low-level responses (172).
Using an ICS approach, we have recently identified human CD8+ T cell epitopes restricted by HLA-A*11:01 for influenza A and influenza B viruses in Indigenous and non-Indigenous people (80). HLA-A*11:01 is highly prevalent in Asian and Indigenous Australian populations, making it an ideal candidate for targeting CD8+ T cell immunity in a large proportion of the global population. Multiple influenza A virus CD8+ T cell epitopes restricted by HLA-A*11:01, or the HLA-A*03 supertype, have been reported (45, 173–178). HLA supertypes, defined by similarity in anchor pockets responsible for binding the primary anchor residues of peptide ligands, can be useful starting points for less studied alleles (179–181). Due to shared peptide binding features, HLA alleles within the same HLA supertype are likely bind similar peptides, potentially simplifying the need to perform epitope analyses for each of the >30,000 HLA allelic variants that exist (81). However, polymorphisms across the antigen binding cleft can impact both the binding and conformation of bound peptides within supertypes (182, 183), resulting in very different outcomes for T cell activation (184, 185). Thus, the idea that HLA molecules from the same supertype will bind the same peptide and activate an immune response does not always hold true (181). We endeavoured to confirm the immunogenicity of previously reported HLA-A*11:01-restricted IAV peptides and identify novel peptides using immunopeptidomics. Similar to the work on HLA-A*24:02 (41), IAV peptides were screened by pulsing HLA-A*11:01+ PBMCs with peptide pools containing 10 IAV peptides each, and co-culturing with unstimulated, autologous PBMCs. After culturing for 10-12 days in vitro, cells were restimulated with pooled or individual peptides and ICS performed to determine CD8+ T cell activation. Two peptide variants, PB2320-331 and PB2323-331, were found to be immunogenic in 2 of 7 individuals tested. Overall, the immunogenicity of IAV peptides restricted by HLA-A*11:01 was relatively low, suggesting that robust CD8+ T cell mediated immunity toward IAV may not be best directed by HLA-A*11:01. Unlike the known HLA-A*11:01-restricted IAV epitopes, influenza B epitopes have only recently been identified for HLA-A*11:01 using our immunopeptidomics approach. The immunogenicity of identified HLA-A*11:01-restricted IBV peptides was determined using an IBV infection method for T cell expansion. This involved infection of HLA-A*11:01-expressing C1R cells with B/Malaysia/2506/2004 to stimulate and expand antigen-specific CD8+ T cells from HLA-A*11:01-expressing individuals in vitro (41, 80). CD8+ T cells were restimulated with pooled or individual IBV peptides and their activation was determined via ICS. To this end, three IBV peptides were found to be immunogenic, the strongest of which was M141-49, while subdominant responses were observed with NS1186-195 and NP511-510.
ICS approaches are highly useful and readily adaptable for screening and analysing T cell responses to candidate peptides. However, it is important to note that ELISpot and ICS assays can underestimate the magnitude of epitope-specific responses compared to assays that measure TCR binding to pMHC (118), presumably because some cells may lack the particular function assayed (186, 187). Furthermore, any screening strategy using PBMCs from human donors must consider variability in pathogen strains commonly circulating in the geographic regions of the donor cohort and the presence of naturally occurring peptide variants. Tools such as the Influenza Research Database “Identify short peptides in proteins” analysis tool (188) are useful to search for naturally occurring peptide variants to include in epitope mapping and assess the cross-reactivity of CD8+ T cell responses to epitope variants. In addition, when screening a large number of peptides, it is advantageous to arrange the peptides in pools relative to their predicted binding affinity for the HLA of interest (using a prediction tool such as NetMHCpan4.0) to potentially avoid high affinity peptides outcompeting low affinity peptides for HLA binding which might result in reduced sensitivity of the assay.
Aside from cytokine expression, epitope-specific T cells can also be detected following peptide stimulation by cell surface expression of CD107a, a marker of degranulation, or activation induced markers. Since in vitro culture and peptide-driven expansion can alter expression of activation, phenotypic and memory markers, an AIM assay is often performed ex vivo with the use of peptide pools to enhance detection. PBMCs or whole blood can be used, and cells are stimulated with peptides for 24 to 72 hrs. A variety of different markers can be assessed to determine T cell activation. The most common markers for CD4+ T cells include OX40 (CD134), CD25 or CD137 (189–191). Other markers that have been tested include CD69 and PD-L1 (192). These markers overlap with activation markers for CD8+ T cells where CD69 and CD137 have been shown to deliver robust results (190, 191). The AIM assay allows for rapid detection of epitope-specific cells and is not reliant on the expression of a particular function or proliferation in contrast to in vitro expansion of epitope-specific T cells, while also requiring fewer cells. However, due to the low frequency of epitope-specific T cells ex vivo, it is only suitable for screening for responses to bigger peptide pools that contain several epitopes or single epitopes where frequencies of epitope-specific T cells are high. It is often unsuitable for the identification of individual epitope-specific populations of T cells in donors. It is, however, very useful to compare total T cell responses towards peptide pools derived from whole proteins when epitopes or the donor HLAs are unknown, for example when comparing vaccine responses or responses induced by infection derived from novel pathogens (189, 190). In particular, assessing the total magnitude of T cell responses to vaccination may be more informative for gauging the overall robustness of vaccine responses than measuring responses to individual epitopes. Screening T cell responses across different populations could be used to determine vaccine responses in various ethnicities or identify vulnerable populations that might require modified vaccines, such as adjuvanted, high dose or inclusion of additional immune cell targets.
Snyder et al. (193) have combined the AIM assay with a prior stimulation of memory T cells using an anti-CD3 antibody for 8-13 days. This can increase the total number of available cells for screening, facilitating detection of low frequency epitope-specific populations or overcoming constraints due to limited donor blood volumes. The suitability of this protocol modification however depends on the research question. One of the advantages with the AIM assay is that the results are very representative of ex vivo responses. However, stimulation for at least 8 days with a stimulatory antibody will substantially alter the phenotype of the cells. Furthermore, it is unclear if different T cells expand at different rates, skewing the frequency of peptide responding cells measured, especially if they are derived from donors that had recent contact with the antigen. Unfortunately the authors do not compare the frequency of their epitope-specific populations prior to and post expansion (193). However, in the context of novel antigens, where the amount of relevant donors and samples are limited, this adaptation of the AIM assay could be extremely useful for initial screens followed by more in-depth analysis.
HLA tetramer/multimer assays detect epitope-specific T cells based on TCR binding of a given peptide-HLA complex. This approach necessitates the specific synthesis of peptide-HLA complexes of interest, hence it is not used for large scale screening of many candidate epitopes and is instead reserved for subsequent in-depth characterisation of selected epitope-specific T cells. HLA tetramer/multimer enrichment techniques can be used to enumerate and isolate low frequency epitope-specific T cells, including naïve epitope-specific T cells. The identified populations can be used for multiparameter phenotypic analysis, functional assays, TCR sequencing and transcriptomic profiling. These can provide powerful insights into the specificity, function and quality of epitope-specific CD8+ T cells and how these can best be harnessed for stronger, durable and cross-protective immunity.
Approaches for Epitope Identification – Pros and Cons
In summary, systematic screening using overlapping peptides, in silico epitope predictions and immunopeptidome analysis each have their advantages and challenges (Table 2). Whilst overlapping libraries encompass the full sequence of the tested antigen, they are reagent greedy and fail to encompass post-translational modifications. Similarly, in silico methods rely on sufficient training data for high accuracy predictions and may miss peptides that adopt unanticipated binding modes or incorporate post-translation modifications. In contrast, generation of lists of candidate epitopes from immunopeptidome analysis focusses analyses on peptides that are naturally processed for presentation by the HLA and can encompass post-translational modifications. However this workflow may miss less abundant species, or peptide species that are lost during sample preparation due to biophysical properties that reduce interaction with chromatographic columns. Overall, whilst each technique has blind spots, they each have the capacity to reveal important epitopes to inform vaccine development, whether in the form of polyepitopes or large epitope rich portions of the target pathogen.
Concluding Remarks
The importance of T cell epitope identification has never been more clear as it is now in the face of an ongoing COVID-19 pandemic. T cell epitope identification is highly relevant to situations where new viruses capable of infecting humans emerge or re-emerge. It is also of great importance in the context of globally diverse populations and ethnicities, including immunologically vulnerable Indigenous populations. A number of successful strategies for epitope identification are now available, but HLA alleles prominent in Indigenous populations require special consideration, because of the often distinct, rare and understudied nature of these molecules. Studies to understand the peptide repertoires and immunogenic epitopes presented by HLA alleles prevalent in Indigenous people provide vital insights that inform the rational design and evaluation of T cell-based vaccines to ensure they provide Indigenous populations worldwide with effective protection from infectious diseases.
Reagent Availability
C1R cell lines expressing key Indigenous Australian HLA (HLA-A*11:01 (80), 24:06, 24:13, 34:01, B*13:01, 15:21 (150), 15:25 (150), 40:01, 40:02, 56:01, 56:02, 56:56) are available upon requestion from KK. Requires a materials transfer agreement (MTA) from The University of Melbourne.
Author Contributions
EC, KK, LH, PI and AP planned the manuscript. EC, KK, LH, PI, LR and JH wrote sections of the manuscript and generated figures. JD, AM, ST, CS, KF and AP contributed specialist expertise, perspectives and data for the manuscript and contributed to modification of the original and revised versions of the manuscript. EC, KK, LH, PI and AP wrote and modified the original manuscript and the revised versions.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
LH received funding from a Melbourne International Research Scholarship (MIRS) and Melbourne International Fee Remission Scholarship (MIFRS) from The University of Melbourne. ST is a NHMRC Career Development Fellow (#1145033). JH is supported by a Melbourne Research Scholarship from the University of Melbourne. CS has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement (#792532). KF received funding from a Clifford Craig Foundation Grant (#145). AP is supported by an NHMRC Principal Research Fellowship (#1137739) and NHMRC Project Grant (#1085018). EC is a NHMRC Peter Doherty Fellow (#1091516). KK is supported by a NHMRC Senior Research Fellowship (#1102792), NHMRC Program Grant (#1071916), NHMRC Investigator Grant (#1173871) and NHMRC Project Grant (#1122524) to KK, ST, AM, and AP.
References
1. WHO. WHO Coronavirus (COVID-19) Dashboard (2021). Available at: https://covid19.who.int/.
2. Iuliano AD, Roguski KM, Chang HH, Muscatello DJ, Palekar R, Tempia S, et al. Estimates of Global Seasonal Influenza-Associated Respiratory Mortality: A Modelling Study. Lancet (2018) 391:1285–300. doi: 10.1016/S0140-6736(17)33293-2
3. Jester B, Uyeki TM, Jernigan DB, Tumpey TM. Historical and Clinical Aspects of the 1918 H1N1 Pandemic in the United States. Virology (2019) 527:32–7. doi: 10.1016/j.virol.2018.10.019
4. Johnson NP, Mueller J. Updating the Accounts: Global Mortality of the 1918-1920 "Spanish" Influenza Pandemic. Bull Hist Med (2002) 76:105–15. doi: 10.1353/bhm.2002.0022
5. WHO. Avian Influenza Weekly Update Number 805. (2021). Available at: https://www.who.int/
6. Summers JA, Baker M, Wilson N. New Zealand’s Experience of the 1918-19 Influenza Pandemic: A Systematic Review After 100 Years. N Z Med J (2018) 131(1487):54–69.
7. Kelm M. British Columbia First Nations and the Influenza Pandemic of 1918–1919. Br Columbian Q (1999) 122(Summer).
8. Ahmed R, Oldstone MB, Palese P. Protective Immunity and Susceptibility to Infectious Diseases: Lessons From the 1918 Influenza Pandemic. Nat Immunol (2007) 8:1188–93. doi: 10.1038/ni1530
9. Briscoe G. Disease Health & Healing – Aspects of Indigenous Health in WA & Queensland, 1900-1940. PhD Thesis. Aust Natl Univ (1996).
10. Herda PS. Disease and the Colonial Narrative: The 1918 Influenza Pandemic in Western Polynesia. New Z J Hist (2000) 34:133–44.
11. Kelly H, Mercer G, Cheng A. Quantifying the Risk of Pandemic Influenza in Pregnancy and Indigenous People in Australia in 2009. Euro Surveill (2009) 14(50):19441. doi: 10.2807/ese.14.50.19441-en
12. Verrall A, Norton K, Rooker S, Dee S, Olsen L, Tan CE, et al. Hospitalizations for Pandemic (H1N1) 2009 Among Maori and Pacific Islanders, New Zealand. Emerg Infect Dis (2010) 16:100–2. doi: 10.3201/eid1601.090994
13. Mousseau M. H1N1 in Retrospect: A Review of Risk Factors and Policy Recommendations. Int Indigenous Policy J (2013) 4. doi: 10.18584/iipj.2013.4.2.4
14. CDC. Deaths Related to 2009 Pandemic Influenza A (H1N1) Among American Indian/Alaska Natives - 12 States, 2009. Morbidity Mortality Weekly Rep (MMWR) (2009) 58:1341–4.
15. Boggild AK, Yuan L, Low DE, McGeer AJ. The Impact of Influenza on the Canadian First Nations. Can J Public Health (2011) 102:345–8. doi: 10.1007/BF03404174
16. Pan D, Sze S, Minhas JS, Bangash MN, Pareek N, Divall P, et al. The Impact of Ethnicity on Clinical Outcomes in COVID-19: A Systematic Review. EClinicalMedicine (2020) 23:100404. doi: 10.1016/j.eclinm.2020.100404
17. Mathur R, Rentsch CT, Morton CE, Hulme WJ, Schultze A, MacKenna B, et al. Ethnic Differences in SARS-CoV-2 Infection and COVID-19-Related Hospitalisation, Intensive Care Unit Admission, and Death in 17 Million Adults in England: An Observational Cohort Study Using the OpenSAFELY Platform. Lancet (2021) 397:1711–24. doi: 10.1016/S0140-6736(21)00634-6
18. Pan D, Martin CA, Nazareth J, Nevill CR, Minhas JS, Divall P, et al. Ethnic Disparities in COVID-19: Increased Risk of Infection or Severe Disease? Lancet (2021) 398:389–90. doi: 10.1016/S0140-6736(21)01428-8
19. Mathur R, Rentsch CT, Morton CE, Eggo RM, Bhaskaran K, Tomlinsonn L, et al. Ethnic Disparities in COVID-19: Increased Risk of Infection or Severe Disease? - Authors’ Reply. Lancet (2021) 398:390. doi: 10.1016/S0140-6736(21)01424-0
20. AMP Research Lab. The Colour of Coronavirus: COVID-19 Deaths by Race and Ethnicity in the US (2021). Available at: https://www.apmresearchlab.org/covid/deaths-by-race.
21. Ramírez JD, Sordillo EM, Gotuzzo E, Zavaleta C, Caplivski D, Navarro JC, et al. SARS-CoV-2 in the Amazon Region: A Harbinger of Doom for Amerindians. PloS Negl Trop Dis (2020) 14:e0008686. doi: 10.1371/journal.pntd.0008686
22. Santos VS, Souza Araújo AA, de Oliveira JR, Quintans-Júnior LJ, Martins-Filho PR. COVID-19 Mortality Among Indigenous People in Brazil: A Nationwide Register-Based Study. J Public Health (Oxf) (2021) 43:e250–1. doi: 10.1093/pubmed/fdaa176
23. Randall DA, Lujic S, Havard A, Eades SJ, Jorm L. Multimorbidity Among Aboriginal People in New South Wales Contributes Significantly to Their Higher Mortality. Med J Aust (2018) 209:19–23. doi: 10.5694/mja17.00878
24. Mathur R, Bear L, Khunti K, Eggo RM. Urgent Actions and Policies Needed to Address COVID-19 Among UK Ethnic Minorities. Lancet (2020) 396:1866–8. doi: 10.1016/S0140-6736(20)32465-X
25. Ovsyannikova IG, Haralambieva IH, Crooke SN, Poland GA, Kennedy RB. The Role of Host Genetics in the Immune Response to SARS-CoV-2 and COVID-19 Susceptibility and Severity. Immunol Rev (2020) 296:205–19. doi: 10.1111/imr.12897
26. Jafarpour R, Pashangzadeh S, Dowran R. Host Factors: Implications in Immunopathogenesis of COVID-19. Pathol Res Pract (2021) 228:153647. doi: 10.1016/j.prp.2021.153647
27. Suh S, Lee S, Gym H, Yoon S, Park S, Cha J, et al. A Systematic Review on Papers That Study on Single Nucleotide Polymorphism That Affects Coronavirus 2019 Severity. BMC Infect Dis (2022) 22:47. doi: 10.1186/s12879-022-07034-w
28. United Nations, Department of Economic and Social Affairs. State of the Worlds Indigenous Peoples (2009). Available at: https://www.un.org/esa/socdev/unpfii/documents/SOWIP/en/SOWIP_web.pdf.
29. Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, et al. Cellular Immune Correlates of Protection Against Symptomatic Pandemic Influenza. Nat Med (2013) 19:1305–12. doi: 10.1038/nm.3350
30. Hayward AC, Wang L, Goonetilleke N, Fragaszy EB, Bermingham A, Copas A, et al. Natural T Cell-Mediated Protection Against Seasonal and Pandemic Influenza. Results of the Flu Watch Cohort Study. Am J Respir Crit Care Med (2015) 191:1422–31. doi: 10.1164/rccm.201411-1988OC
31. Koutsakos M, Rowntree LC, Hensen L, Chua BY, van de Sandt CE, Habel JR, et al. Integrated Immune Dynamics Define Correlates of COVID-19 Severity and Antibody Responses. Cell Rep Med (2021) 2:100208. doi: 10.1016/j.xcrm.2021.100208
32. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations With Age and Disease Severity. Cell (2020) 183:996–1012.e1019. doi: 10.1016/j.cell.2020.09.038
33. Nguyen THO, Koutsakos M, van de Sandt CE, Crawford JC, Loh L, Sant S, et al. Immune Cellular Networks Underlying Recovery From Influenza Virus Infection in Acute Hospitalized Patients. Nat Commun (2021) 12:2691. doi: 10.1038/s41467-021-23018-x
34. Yewdell JW. Antigenic Drift: Understanding COVID-19. Immunity (2021) 54:2681–7. doi: 10.1016/j.immuni.2021.11.016
35. Dejnirattisai W, Zhou D, Supasa P, Liu C, Mentzer AJ, Ginn HM, et al. Antibody Evasion by the P.1 Strain of SARS-CoV-2. Cell (2021) 184:2939–54.e2939. doi: 10.1016/j.cell.2021.03.055
36. Nuwarda RF, Alharbi AA, Kayser V. An Overview of Influenza Viruses and Vaccines. Vaccines (Basel) (2021) 9(9):1032. doi: 10.3390/vaccines9091032
37. Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, et al. Preexisting Influenza-Specific CD4+ T Cells Correlate With Disease Protection Against Influenza Challenge in Humans. Nat Med (2012) 18:274–80. doi: 10.1038/nm.2612
38. Kundu R, Narean JS, Wang L, Fenn J, Pillay T, Fernandez ND, et al. Cross-Reactive Memory T Cells Associate With Protection Against SARS-CoV-2 Infection in COVID-19 Contacts. Nat Commun (2022) 13:80. doi: 10.1038/s41467-021-27674-x
39. Wang Z, Wan Y, Qiu C, Quiñones-Parra S, Zhu Z, Loh L, et al. Recovery From Severe H7N9 Disease Is Associated With Diverse Response Mechanisms Dominated by CD8⁺ T Cells. Nat Commun (2015) 6:6833. doi: 10.1038/ncomms7833
40. Clemens EB, Grant EJ, Wang Z, Gras S, Tipping P, Rossjohn J, et al. Towards Identification of Immune and Genetic Correlates of Severe Influenza Disease in Indigenous Australians. Immunol Cell Biol (2016) 94:367–77. doi: 10.1038/icb.2015.93
41. Hensen L, Illing PT, Bridie Clemens E, Nguyen THO, Koutsakos M, van de Sandt CE, et al. CD8(+) T Cell Landscape in Indigenous and non-Indigenous People Restricted by Influenza Mortality-Associated HLA-A*24:02 Allomorph. Nat Commun (2021) 12:2931. doi: 10.1038/s41467-021-23212-x
42. Gonzalez-Galarza FF, McCabe A, Santos E, Jones J, Takeshita L, Ortega-Rivera ND, et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acids Res (2020) 48:D783–d788. doi: 10.1093/nar/gkz1029
43. Hertz T, Oshansky CM, Roddam PL, DeVincenzo JP, Caniza MA, Jojic N, et al. HLA Targeting Efficiency Correlates With Human T-Cell Response Magnitude and With Mortality From Influenza A Infection. Proc Natl Acad Sci USA (2013) 110:13492–7. doi: 10.1073/pnas.1221555110
44. Woolthuis RG, van Dorp CH, Keşmir C, de Boer RJ, van Boven M. Long-Term Adaptation of the Influenza A Virus by Escaping Cytotoxic T-Cell Recognition. Sci Rep (2016) 6:33334. doi: 10.1038/srep33334
45. Assarsson E, Bui HH, Sidney J, Zhang Q, Glenn J, Oseroff C, et al. Immunomic Analysis of the Repertoire of T-Cell Specificities for Influenza A Virus in Humans. J Virol (2008) 82:12241–51. doi: 10.1128/JVI.01563-08
46. Leffell MS, Fallin MD, Erlich HA, Fernandez-Vĩna M, Hildebrand WH, Mack SJ, et al. HLA Antigens, Alleles and Haplotypes Among the Yup’ik Alaska Natives: Report of the ASHI Minority Workshops, Part II. Hum Immunol (2002) 63:614–25. doi: 10.1016/S0198-8859(02)00415-9
47. Falfán-Valencia R, Narayanankutty A, Reséndiz-Hernández JM, Pérez-Rubio G, Ramírez-Venegas A, Nava-Quiroz KJ, et al. An Increased Frequency in HLA Class I Alleles and Haplotypes Suggests Genetic Susceptibility to Influenza A (H1N1) 2009 Pandemic: A Case-Control Study. J Immunol Res (2018) 2018:3174868. doi: 10.1155/2018/3174868
48. van de Sandt CE, Clemens EB, Grant EJ, Rowntree LC, Sant S, Halim H, et al. Challenging Immunodominance of Influenza-Specific CD8(+) T Cell Responses Restricted by the Risk-Associated HLA-A*68:01 Allomorph. Nat Commun (2019) 10:5579. doi: 10.1038/s41467-019-13346-4
49. MacDonald KS, Fowke KR, Kimani J, Dunand VA, Nagelkerke NJ, Ball TB, et al. Influence of HLA Supertypes on Susceptibility and Resistance to Human Immunodeficiency Virus Type 1 Infection. J Infect Dis (2000) 181:1581–9. doi: 10.1086/315472
50. Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, et al. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science (2010) 330:1551–7. doi: 10.1126/science.1195271
51. Stephens HA, Klaythong R, Sirikong M, Vaughn DW, Green S, Kalayanarooj S, et al. HLA-A and -B Allele Associations With Secondary Dengue Virus Infections Correlate With Disease Severity and the Infecting Viral Serotype in Ethnic Thais. Tissue Antigens (2002) 60:309–18. doi: 10.1034/j.1399-0039.2002.600405.x
52. Khor CC, Chau TN, Pang J, Davila S, Long HT, Ong RT, et al. Genome-Wide Association Study Identifies Susceptibility Loci for Dengue Shock Syndrome at MICB and PLCE1. Nat Genet (2011) 43:1139–41. doi: 10.1038/ng.960
53. Chen D, Gaborieau V, Zhao Y, Chabrier A, Wang H, Waterboer T, et al. A Systematic Investigation of the Contribution of Genetic Variation Within the MHC Region to HPV Seropositivity. Hum Mol Genet (2015) 24:2681–8. doi: 10.1093/hmg/ddv015
54. Duggal P, Thio CL, Wojcik GL, Goedert JJ, Mangia A, Latanich R, et al. Genome-Wide Association Study of Spontaneous Resolution of Hepatitis C Virus Infection: Data From Multiple Cohorts. Ann Intern Med (2013) 158:235–45. doi: 10.7326/0003-4819-158-4-201302190-00003
55. Miki D, Ochi H, Takahashi A, Hayes CN, Urabe Y, Abe H, et al. HLA-DQB1*03 Confers Susceptibility to Chronic Hepatitis C in Japanese: A Genome-Wide Association Study. PloS One (2013) 8:e84226. doi: 10.1371/journal.pone.0084226
56. Lin M, Tseng HK, Trejaut JA, Lee HL, Loo JH, Chu CC, et al. Association of HLA Class I With Severe Acute Respiratory Syndrome Coronavirus Infection. BMC Med Genet (2003) 4:9. doi: 10.1186/1471-2350-4-9
57. Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. The MHC Locus and Genetic Susceptibility to Autoimmune and Infectious Diseases. Genome Biol (2017) 18:76. doi: 10.1186/s13059-017-1207-1
58. Sveinbjornsson G, Gudbjartsson DF, Halldorsson BV, Kristinsson KG, Gottfredsson M, Barrett JC, et al. HLA Class II Sequence Variants Influence Tuberculosis Risk in Populations of European Ancestry. Nat Genet (2016) 48:318–22. doi: 10.1038/ng.3498
59. Hu X, Deutsch AJ, Lenz TL, Onengut-Gumuscu S, Han B, Chen WM, et al. Additive and Interaction Effects at Three Amino Acid Positions in HLA-DQ and HLA-DR Molecules Drive Type 1 Diabetes Risk. Nat Genet (2015) 47:898–905. doi: 10.1038/ng.3353
60. Howson JM, Walker NM, Clayton D, Todd JA. Confirmation of HLA Class II Independent Type 1 Diabetes Associations in the Major Histocompatibility Complex Including HLA-B and HLA-A. Diabetes Obes Metab (2009) 11 Suppl 1:31–45. doi: 10.1111/j.1463-1326.2008.01001.x
61. Kaushansky N, Ben-Nun A. DQB1*06:02-Associated Pathogenic Anti-Myelin Autoimmunity in Multiple Sclerosis-Like Disease: Potential Function of DQB1*06:02 as a Disease-Predisposing Allele. Front Oncol (2014) 4:280. doi: 10.3389/fonc.2014.00280
62. Gutierrez-Achury J, Zhernakova A, Pulit SL, Trynka G, Hunt KA, Romanos J, et al. Fine Mapping in the MHC Region Accounts for 18% Additional Genetic Risk for Celiac Disease. Nat Genet (2015) 47:577–8. doi: 10.1038/ng.3268
63. Quiñones-Parra S, Grant E, Loh L, Nguyen TH, Campbell KA, Tong SY, et al. Preexisting CD8+ T-Cell Immunity to the H7N9 Influenza A Virus Varies Across Ethnicities. Proc Natl Acad Sci USA (2014) 111:1049–54. doi: 10.1073/pnas.1322229111
64. Poland GA, Ovsyannikova IG, Jacobson RM. Immunogenetics of Seasonal Influenza Vaccine Response. Vaccine (2008) 26(Suppl 4):D35–40. doi: 10.1016/j.vaccine.2008.07.065
65. Iturrieta-Zuazo I, Rita CG, García-Soidán A, de Malet Pintos-Fonseca A, Alonso-Alarcón N, Pariente-Rodríguez R, et al. Possible Role of HLA Class-I Genotype in SARS-CoV-2 Infection and Progression: A Pilot Study in a Cohort of Covid-19 Spanish Patients. Clin Immunol (2020) 219:108572. doi: 10.1016/j.clim.2020.108572
66. Nguyen A, David JK, Maden SK, Wood MA, Weeder BR, Nellore A, et al. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J Virol (2020) 94(13):e00510–20. doi: 10.1128/JVI.00510-20
67. Castelli EC, de Castro MV, Naslavsky MS, Scliar MO, Silva NSB, Andrade HS, et al. MHC Variants Associated With Symptomatic Versus Asymptomatic SARS-CoV-2 Infection in Highly Exposed Individuals. Front Immunol (2021) 12:742881. doi: 10.3389/fimmu.2021.742881
68. Gutiérrez-Bautista JF, Rodriguez-Nicolas A, Rosales-Castillo A, López-Ruz M, Martín-Casares AM, Fernández-Rubiales A, et al. Study of HLA-A, -B, -C, -DRB1 and -DQB1 Polymorphisms in COVID-19 Patients. J Microbiol Immunol Infect (2021). doi: 10.1016/j.jmii.2021.08.009
69. Ben Shachar S, Barda N, Manor S, Israeli S, Dagan N, Carmi S, et al. MHC Haplotyping of SARS-CoV-2 Patients: HLA Subtypes Are Not Associated With the Presence and Severity of COVID-19 in the Israeli Population. J Clin Immunol (2021) 41:1154–61. doi: 10.1007/s10875-021-01071-x
70. Douillard V, Castelli EC, Mack SJ, Hollenbach JA, Gourraud PA, Vince N, et al. Current HLA Investigations on SARS-CoV-2 and Perspectives. Front Genet (2021) 12:774922. doi: 10.3389/fgene.2021.774922
71. Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med (2020) 383:2603–15. doi: 10.1056/NEJMoa2034577
72. Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. Safety and Efficacy of the ChAdOx1 Ncov-19 Vaccine (AZD1222) Against SARS-CoV-2: An Interim Analysis of Four Randomised Controlled Trials in Brazil, South Africa, and the UK. Lancet (2021) 397:99–111. doi: 10.1016/S0140-6736(20)32661-1
73. Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res (2019) 47:D339–d343. doi: 10.1093/nar/gky1006
74. Gao X, Lester S, Veale A, Boettcher B, Currie B, McCluskey J, et al. HLA Class I Alleles in Australian Aborigines and Their Peptide Binding Profiles. In: Kasahara M, editor. Major Histocompatibility Complex: Evolution, Struction, and Function, 1 ed. Tokyo: Springer (2000). p. 446–62. doi: 10.1007/978-4-431-65868-9_33
75. Dolton G, Rius C, Hasan MS, Wall A, Szomolay B, Behiry E, et al. Emergence of Immune Escape at Dominant SARS-CoV-2 Killer T-Cell Epitope. medRxiv (2021) 2021.2006.2021.21259010. doi: 10.1101/2021.06.21.21259010
76. Valkenburg SA, Quiñones-Parra S, Gras S, Komadina N, McVernon J, Wang Z, et al. Acute Emergence and Reversion of Influenza A Virus Quasispecies Within CD8+ T Cell Antigenic Peptides. Nat Commun (2013) 4:2663. doi: 10.1038/ncomms3663
77. Voeten JT, Bestebroer TM, Nieuwkoop NJ, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. Antigenic Drift in the Influenza A Virus (H3N2) Nucleoprotein and Escape From Recognition by Cytotoxic T Lymphocytes. J Virol (2000) 74:6800–7. doi: 10.1128/JVI.74.15.6800-6807.2000
78. Berkhoff EG, Boon AC, Nieuwkoop NJ, Fouchier RA, Sintnicolaas K, Osterhaus AD, et al. A Mutation in the HLA-B*2705-Restricted NP383-391 Epitope Affects the Human Influenza A Virus-Specific Cytotoxic T-Lymphocyte Response In Vitro. J Virol (2004) 78:5216–22. doi: 10.1128/JVI.78.10.5216-5222.2004
79. Machkovech HM, Bedford T, Suchard MA, Bloom JD. Positive Selection in CD8+ T-Cell Epitopes of Influenza Virus Nucleoprotein Revealed by a Comparative Analysis of Human and Swine Viral Lineages. J Virol (2015) 89:11275–83. doi: 10.1128/JVI.01571-15
80. Habel JR, Nguyen AT, Rowntree LC, Szeto C, Mifsud NA, Clemens EB, et al. HLA-A*11:01-Restricted CD8+ T Cell Immunity Against Influenza A and Influenza B Viruses in Indigenous and non-Indigenous People. PloS Pathog (2022) 18(3):e1010337. doi: 10.1371/journal.ppat.1010337
81. Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE. IPD-IMGT/HLA Database. Nucleic Acids Res (2020) 48:D948–d955. doi: 10.1093/nar/gkz950
82. Purcell AW, Ramarathinam SH, Ternette N. Mass Spectrometry-Based Identification of MHC-Bound Peptides for Immunopeptidomics. Nat Protoc (2019) 14:1687–707. doi: 10.1038/s41596-019-0133-y
83. Singh SK, Meyering M, Ramwadhdoebe TH, Stynenbosch LF, Redeker A, Kuppen PJ, et al. The Simultaneous Ex Vivo Detection of Low-Frequency Antigen-Specific CD4+ and CD8+ T-Cell Responses Using Overlapping Peptide Pools. Cancer Immunol Immunother (2012) 61:1953–63. doi: 10.1007/s00262-012-1251-3
84. Bancroft T, Dillon MB, da Silva Antunes R, Paul S, Peters B, Crotty S, et al. Th1 Versus Th2 T Cell Polarization by Whole-Cell and Acellular Childhood Pertussis Vaccines Persists Upon Re-Immunization in Adolescence and Adulthood. Cell Immunol (2016) 304-305:35–43. doi: 10.1016/j.cellimm.2016.05.002
85. da Silva Antunes R, Babor M, Carpenter C, Khalil N, Cortese M, Mentzer AJ, et al. Th1/Th17 Polarization Persists Following Whole-Cell Pertussis Vaccination Despite Repeated Acellular Boosters. J Clin Invest (2018) 128:3853–65. doi: 10.1172/JCI121309
86. Grant E, Wu C, Chan KF, Eckle S, Bharadwaj M, Zou QM, et al. Nucleoprotein of Influenza A Virus Is a Major Target of Immunodominant CD8+ T-Cell Responses. Immunol Cell Biol (2013) 91:184–94. doi: 10.1038/icb.2012.78
87. Gras S, Kedzierski L, Valkenburg SA, Laurie K, Liu YC, Denholm JT, et al. Cross-Reactive CD8+ T-Cell Immunity Between the Pandemic H1N1-2009 and H1N1-1918 Influenza A Viruses. Proc Natl Acad Sci USA (2010) 107:12599–604. doi: 10.1073/pnas.1007270107
88. Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Res (2020) 48:W449–w454. doi: 10.1093/nar/gkaa379
89. Zhao W, Sher X. Systematically Benchmarking Peptide-MHC Binding Predictors: From Synthetic to Naturally Processed Epitopes. PloS Comput Biol (2018) 14:e1006457. doi: 10.1371/journal.pcbi.1006457
90. Stranzl T, Larsen MV, Lundegaard C, Nielsen M. NetCTLpan: Pan-Specific MHC Class I Pathway Epitope Predictions. Immunogenetics (2010) 62:357–68. doi: 10.1007/s00251-010-0441-4
91. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity Against Cancer. Nature (2017) 547:222–6. doi: 10.1038/nature23003
92. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An Immunogenic Personal Neoantigen Vaccine for Patients With Melanoma. Nature (2017) 547:217–21. 10.1038/nature22991
93. Zheng J, Xia Z, Xu Y, Ou Z, Lin X, Jin S, et al. Identification of Novel Hepatitis B Virus Therapeutic Vaccine Candidates Derived From Polymerase Protein. Aging (Albany NY) (2021) 13:14372–84. doi: 10.18632/aging.203053
94. Fridman A, Finnefrock AC, Peruzzi D, Pak I, La Monica N, Bagchi A, et al. An Efficient T-Cell Epitope Discovery Strategy Using in Silico Prediction and the Itopia Assay Platform. Oncoimmunology (2012) 1:1258–70. doi: 10.4161/onci.21355
95. Gfeller D, Bassani-Sternberg M. Predicting Antigen Presentation-What Could We Learn From a Million Peptides? Front Immunol (2018) 9:1716. doi: 10.3389/fimmu.2018.01716
96. Yu K, Petrovsky N, Schönbach C, Koh JY, Brusic V. Methods for Prediction of Peptide Binding to MHC Molecules: A Comparative Study. Mol Med (2002) 8:137–48. doi: 10.1007/BF03402006
97. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet (2020) 395:565–74. doi: 10.1016/S0140-6736(20)30251-8
98. Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, et al. SARS-CoV-2-Specific T Cell Immunity in Cases of COVID-19 and SARS, and Uninfected Controls. Nature (2020) 584:457–62. doi: 10.1038/s41586-020-2550-z
99. Ng OW, Chia A, Tan AT, Jadi RS, Leong HN, Bertoletti A, et al. Memory T Cell Responses Targeting the SARS Coronavirus Persist Up to 11 Years Post-Infection. Vaccine (2016) 34:2008–14. doi: 10.1016/j.vaccine.2016.02.063
100. Ahmed SF, Quadeer AA, McKay MR. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses (2020) 12(3):254. doi: 10.1101/2020.02.03.933226
101. Lee CH, Koohy H. In Silico Identification of Vaccine Targets for 2019-Ncov. F1000Res (2020) 9:145. doi: 10.12688/f1000research.22507.2
102. Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe (2020) 27:671–80.e672. doi: 10.1016/j.chom.2020.03.002
103. Ranga V, Niemelä E, Tamirat MZ, Eriksson JE, Airenne TT, Johnson MS. Immunogenic SARS-CoV-2 Epitopes: In Silico Study Towards Better Understanding of COVID-19 Disease-Paving the Way for Vaccine Development. Vaccines (Basel) (2020) 8(3):408. doi: 10.3390/vaccines8030408
104. Ahmed SF, Quadeer AA, McKay MR. COVIDep: A Web-Based Platform for Real-Time Reporting of Vaccine Target Recommendations for SARS-CoV-2. Nat Protoc (2020) 15:2141–2. doi: 10.1038/s41596-020-0358-9
105. Sohail MS, Ahmed SF, Quadeer AA, McKay MR. In Silico T Cell Epitope Identification for SARS-CoV-2: Progress and Perspectives. Adv Drug Delivery Rev (2021) 171:29–47. doi: 10.1016/j.addr.2021.01.007
106. Requena D, Médico A, Chacón RD, Ramírez M, Marín-Sánchez O. Identification of Novel Candidate Epitopes on SARS-CoV-2 Proteins for South America: A Review of HLA Frequencies by Country. Front Immunol (2020) 11:2008. doi: 10.3389/fimmu.2020.02008
107. Nelde A, Bilich T, Heitmann JS, Maringer Y, Salih HR, Roerden M, et al. SARS-CoV-2-Derived Peptides Define Heterologous and COVID-19-Induced T Cell Recognition. Nat Immunol (2021) 22:74–85. doi: 10.1038/s41590-020-00808-x
108. Tarke A, Sidney J, Kidd CK, Dan JM, Ramirez SI, Yu ED, et al. Comprehensive Analysis of T Cell Immunodominance and Immunoprevalence of SARS-CoV-2 Epitopes in COVID-19 Cases. Cell Rep Med (2021) 2:100204. doi: 10.1016/j.xcrm.2021.100204
109. Hu C, Shen M, Han X, Chen Q, Li L, Chen S, et al. Identification of Cross-Reactive CD8(+) T Cell Receptors With High Functional Avidity to a SARS-CoV-2 Immunodominant Epitope and Its Natural Mutant Variants. Genes Dis (2021) 9(1):216–29. doi: 10.1016/j.gendis.2021.05.006
110. Habel JR, Nguyen THO, van de Sandt CE, Juno JA, Chaurasia P, Wragg K, et al. Suboptimal SARS-CoV-2-Specific CD8(+) T Cell Response Associated With the Prominent HLA-A*02:01 Phenotype. Proc Natl Acad Sci USA (2020) 117:24384–91. doi: 10.1073/pnas.2015486117
111. Rowntree LC, Petersen J, Juno JA, Chaurasia P, Wragg K, Koutsakos M, et al. SARS-CoV-2-Specific CD8(+) T-Cell Responses and TCR Signatures in the Context of a Prominent HLA-A*24:02 Allomorph. Immunol Cell Biol (2021) 99(9):990–1000. doi: 10.1111/imcb.12482
112. Nguyen THO, Rowntree LC, Petersen J, Chua BY, Hensen L, Kedzierski L, et al. CD8(+) T Cells Specific for an Immunodominant SARS-CoV-2 Nucleocapsid Epitope Display High Naive Precursor Frequency and TCR Promiscuity. Immunity (2021) 54:1066–82.e1065. doi: 10.1016/j.immuni.2021.04.009
113. Saini SK, Hersby DS, Tamhane T, Povlsen HR, Amaya Hernandez SP, Nielsen M, et al. SARS-CoV-2 Genome-Wide T Cell Epitope Mapping Reveals Immunodominance and Substantial CD8(+) T Cell Activation in COVID-19 Patients. Sci Immunol (2021) 6(58):eabf7550. doi: 10.1126/sciimmunol.abf7550
114. Schulien I, Kemming J, Oberhardt V, Wild K, Seidel LM, Killmer S, et al. Characterization of Pre-Existing and Induced SARS-CoV-2-Specific CD8(+) T Cells. Nat Med (2021) 27:78–85. doi: 10.1038/s41591-020-01143-2
115. Quadeer AA, Ahmed SF, McKay MR. Landscape of Epitopes Targeted by T Cells in 852 Individuals Recovered From COVID-19: Meta-Analysis, Immunoprevalence, and Web Platform. Cell Rep Med (2021) 2:100312. doi: 10.1016/j.xcrm.2021.100312
116. Gangaev A, Ketelaars SLC, Isaeva OI, Patiwael S, Dopler A, Hoefakker K, et al. Identification and Characterization of a SARS-CoV-2 Specific CD8(+) T Cell Response With Immunodominant Features. Nat Commun (2021) 12:2593. doi: 10.1038/s41467-021-22811-y
117. Ewer KJ, Barrett JR, Belij-Rammerstorfer S, Sharpe H, Makinson R, Morter R, et al. T Cell and Antibody Responses Induced by a Single Dose of ChAdOx1 Ncov-19 (AZD1222) Vaccine in a Phase 1/2 Clinical Trial. Nat Med (2021) 27:270–8. doi: 10.1038/s41591-020-01194-5
118. Sahin U, Muik A, Vogler I, Derhovanessian E, Kranz LM, Vormehr M, et al. BNT162b2 Vaccine Induces Neutralizing Antibodies and Poly-Specific T Cells in Humans. Nature (2021) 595:572–7. doi: 10.1038/s41586-021-03653-6
119. McMurtrey C, Trolle T, Sansom T, Remesh SG, Kaever T, Bardet W, et al. Toxoplasma Gondii Peptide Ligands Open the Gate of the HLA Class I Binding Groove. Elife (2016) 5:e12556. doi: 10.7554/eLife.12556
120. Pymm P, Illing PT, Ramarathinam SH, O’Connor GM, Hughes VA, Hitchen C, et al. MHC-I Peptides Get Out of the Groove and Enable a Novel Mechanism of HIV-1 Escape. Nat Struct Mol Biol (2017) 24:387–94. doi: 10.1038/nsmb.3381
121. Escobar H, Crockett DK, Reyes-Vargas E, Baena A, Rockwood AL, Jensen PE, et al. Large Scale Mass Spectrometric Profiling of Peptides Eluted From HLA Molecules Reveals N-Terminal-Extended Peptide Motifs. J Immunol (2008) 181:4874–82. doi: 10.4049/jimmunol.181.7.4874
122. Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-Allelic Cells Enables More Accurate Epitope Prediction. Immunity (2017) 46:315–26. doi: 10.1016/j.immuni.2017.02.007
123. Sarkizova S, Klaeger S, Le PM, Li LW, Oliveira G, Keshishian H, et al. A Large Peptidome Dataset Improves HLA Class I Epitope Prediction Across Most of the Human Population. Nat Biotechnol (2020) 38:199–209. doi: 10.1038/s41587-019-0322-9
124. Schittenhelm RB, Sivaneswaran S, Lim Kam Sian TC, Croft NP, Purcell AW. Human Leukocyte Antigen (HLA) B27 Allotype-Specific Binding and Candidate Arthritogenic Peptides Revealed Through Heuristic Clustering of Data-Independent Acquisition Mass Spectrometry (DIA-MS) Data. Mol Cell Proteomics (2016) 15:1867–76. doi: 10.1074/mcp.M115.056358
125. Hickman HD, Luis AD, Buchli R, Few SR, Sathiamurthy M, VanGundy RS, et al. Toward a Definition of Self: Proteomic Evaluation of the Class I Peptide Repertoire. J Immunol (2004) 172:2944–52. doi: 10.4049/jimmunol.172.5.2944
126. Prilliman K, Lindsey M, Zuo Y, Jackson KW, Zhang Y, Hildebrand W. Large-Scale Production of Class I Bound Peptides: Assigning a Signature to HLA-B*1501. Immunogenetics (1997) 45:379–85. doi: 10.1007/s002510050219
127. Koutsakos M, Illing PT, Nguyen THO, Mifsud NA, Crawford JC, Rizzetto S, et al. Human CD8(+) T Cell Cross-Reactivity Across Influenza A, B and C Viruses. Nat Immunol (2019) 20:613–25. doi: 10.1038/s41590-019-0320-6
128. Wahl A, Schafer F, Bardet W, Buchli R, Air GM, Hildebrand WH. HLA Class I Molecules Consistently Present Internal Influenza Epitopes. Proc Natl Acad Sci USA (2009) 106:540–5. doi: 10.1073/pnas.0811271106
129. Yaciuk JC, Skaley M, Bardet W, Schafer F, Mojsilovic D, Cate S, et al. Direct Interrogation of Viral Peptides Presented by the Class I HLA of HIV-Infected T Cells. J Virol (2014) 88:12992–3004. doi: 10.1128/JVI.01914-14
130. Lübke M, Spalt S, Kowalewski DJ, Zimmermann C, Bauersfeld L, Nelde A, et al. Identification of HCMV-Derived T Cell Epitopes in Seropositive Individuals Through Viral Deletion Models. J Exp Med (2020) 217(3):jem.20191164. doi: 10.1084/jem.20191164
131. Pandey K, Mifsud NA, Lim Kam Sian TCC, Ayala R, Ternette N, Ramarathinam SH, et al. In-Depth Mining of the Immunopeptidome of an Acute Myeloid Leukemia Cell Line Using Complementary Ligand Enrichment and Data Acquisition Strategies. Mol Immunol (2020) 123:7–17. doi: 10.1016/j.molimm.2020.04.008
132. Scull KE, Dudek NL, Corbett AJ, Ramarathinam SH, Gorasia DG, Williamson NA, et al. Secreted HLA Recapitulates the Immunopeptidome and Allows in-Depth Coverage of HLA A*02:01 Ligands. Mol Immunol (2012) 51:136–42. doi: 10.1016/j.molimm.2012.02.117
133. Ritz D, Gloger A, Weide B, Garbe C, Neri D, Fugmann T. High-Sensitivity HLA Class I Peptidome Analysis Enables a Precise Definition of Peptide Motifs and the Identification of Peptides From Cell Lines and Patients’ Sera. Proteomics (2016) 16:1570–80. doi: 10.1002/pmic.201500445
134. Bassani-Sternberg M, Barnea E, Beer I, Avivi I, Katz T, Admon A. Soluble Plasma HLA Peptidome as a Potential Source for Cancer Biomarkers. Proc Natl Acad Sci USA (2010) 107:18769–76. doi: 10.1073/pnas.1008501107
135. Croft NP, Purcell AW, Tscharke DC. Quantifying Epitope Presentation Using Mass Spectrometry. Mol Immunol (2015) 68:77–80. doi: 10.1016/j.molimm.2015.06.010
136. Zarling AL, Ficarro SB, White FM, Shabanowitz J, Hunt DF, Engelhard VH. Phosphorylated Peptides are Naturally Processed and Presented by Major Histocompatibility Complex Class I Molecules In Vivo. J Exp Med (2000) 192:1755–62. doi: 10.1084/jem.192.12.1755
137. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A Large Fraction of HLA Class I Ligands are Proteasome-Generated Spliced Peptides. Science (2016) 354:354–8. doi: 10.1126/science.aaf4384
138. Malaker SA, Ferracane MJ, Depontieu FR, Zarling AL, Shabanowitz J, Bai DL, et al. Identification and Characterization of Complex Glycosylated Peptides Presented by the MHC Class II Processing Pathway in Melanoma. J Proteome Res (2017) 16:228–37. doi: 10.1021/acs.jproteome.6b00496
139. Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res (2019) 47:D442–d450. doi: 10.1093/nar/gky1106
140. Croft NP, Smith SA, Pickering J, Sidney J, Peters B, Faridi P, et al. Most Viral Peptides Displayed by Class I MHC on Infected Cells are Immunogenic. Proc Natl Acad Sci USA (2019) 116:3112–7. doi: 10.1073/pnas.1815239116
141. Parker R, Tailor A, Peng X, Nicastri A, Zerweck J, Reimer U, et al. The Choice of Search Engine Affects Sequencing Depth and HLA Class I Allele-Specific Peptide Repertoires. Mol Cell Proteomics (2021) 20:100124. doi: 10.1016/j.mcpro.2021.100124
142. Nicastri A, Liao H, Muller J, Purcell AW, Ternette N. The Choice of HLA-Associated Peptide Enrichment and Purification Strategy Affects Peptide Yields and Creates a Bias in Detected Sequence Repertoire. Proteomics (2020) 20:e1900401. doi: 10.1002/pmic.201900401
143. Andreatta M, Alvarez B, Nielsen M. GibbsCluster: Unsupervised Clustering and Alignment of Peptide Sequences. Nucleic Acids Res (2017) 45:W458–w463. doi: 10.1093/nar/gkx248
144. Fritsche J, Kowalewski DJ, Backert L, Gwinner F, Dorner S, Priemer M, et al. Pitfalls in HLA Ligandomics-How to Catch a Li(e)Gand. Mol Cell Proteomics (2021) 20:100110. doi: 10.1016/j.mcpro.2021.100110
145. Mei S, Li F, Xiang D, Ayala R, Faridi P, Webb GI, et al. Anthem: A User Customised Tool for Fast and Accurate Prediction of Binding Between Peptides and HLA Class I Molecules. Brief Bioinform (2021) 22(5):bbaa415. doi: 10.1093/bib/bbaa415
146. Alvarez B, Reynisson B, Barra C, Buus S, Ternette N, Connelley T, et al. NNAlign_MA; MHC Peptidome Deconvolution for Accurate MHC Binding Motif Characterization and Improved T-Cell Epitope Predictions. Mol Cell Proteomics (2019) 18:2459–77. doi: 10.1074/mcp.TIR119.001658
147. Bassani-Sternberg M, Gfeller D. Unsupervised HLA Peptidome Deconvolution Improves Ligand Prediction Accuracy and Predicts Cooperative Effects in Peptide-HLA Interactions. J Immunol (2016) 197:2492–9. doi: 10.4049/jimmunol.1600808
148. Storkus WJ, Alexander J, Payne JA, Dawson JR, Cresswell P. Reversal of Natural Killing Susceptibility in Target Cells Expressing Transfected Class I HLA Genes. Proc Natl Acad Sci USA (1989) 86:2361–4. doi: 10.1073/pnas.86.7.2361
149. Holst J, Vignali KM, Burton AR, Vignali DA. Rapid Analysis of T-Cell Selection In Vivo Using T Cell-Receptor Retrogenic Mice. Nat Methods (2006) 3:191–7. doi: 10.1038/nmeth858
150. Mifsud NA, Illing PT, Lai JW, Fettke H, Hensen L, Huang Z, et al. Carbamazepine Induces Focused T Cell Responses in Resolved Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis Cases But Does Not Perturb the Immunopeptidome for T Cell Recognition. Front Immunol (2021) 12:653710. doi: 10.3389/fimmu.2021.653710
151. Zemmour J, Little AM, Schendel DJ, Parham P. The HLA-A,B "Negative" Mutant Cell Line C1R Expresses a Novel HLA-B35 Allele, Which Also has a Point Mutation in the Translation Initiation Codon. J Immunol (1992) 148(6):1941–8.
152. Schittenhelm RB, Dudek NL, Croft NP, Ramarathinam SH, Purcell AW. A Comprehensive Analysis of Constitutive Naturally Processed and Presented HLA-C*04:01 (Cw4)-Specific Peptides. Tissue Antigens (2014) 83:174–9. doi: 10.1111/tan.12282
153. Dougan SK, Ashour J, Karssemeijer RA, Popp MW, Avalos AM, Barisa M, et al. Antigen-Specific B-Cell Receptor Sensitizes B Cells to Infection by Influenza Virus. Nature (2013) 503:406–9. doi: 10.1038/nature12637
154. Lee ACY, To KKW, Zhu H, Chu H, Li C, Mak WWN, et al. Avian Influenza Virus A H7N9 Infects Multiple Mononuclear Cell Types in Peripheral Blood and Induces Dysregulated Cytokine Responses and Apoptosis in Infected Monocytes. J Gen Virol (2017) 98:922–34. doi: 10.1099/jgv.0.000751
155. Lersritwimanmaen P, Na-Ek P, Thanunchai M, Thewsoongnoen J, Sa-Ard-Iam N, Wiboon-ut S, et al. The Presence of Monocytes Enhances the Susceptibility of B Cells to Highly Pathogenic Avian Influenza (HPAI) H5N1 Virus Possibly Through the Increased Expression of α2,3 SA Receptor. Biochem Biophys Res Commun (2015) 464:888–93. doi: 10.1016/j.bbrc.2015.07.061
156. Hui KPY, Ching RHH, Chan SKH, Nicholls JM, Sachs N, Clevers H, et al. Tropism, Replication Competence, and Innate Immune Responses of Influenza Virus: An Analysis of Human Airway Organoids and Ex-Vivo Bronchus Cultures. Lancet Respir Med (2018) 6:846–54. doi: 10.1016/S2213-2600(18)30236-4
157. Liu J, Li Y, Liu Q, Yao Q, Wang X, Zhang H, et al. SARS-CoV-2 Cell Tropism and Multiorgan Infection. Cell Discov (2021) 7:17. doi: 10.1038/s41421-021-00249-2
158. Wu T, Guan J, Handel A, Tscharke DC, Sidney J, Sette A, et al. Quantification of Epitope Abundance Reveals the Effect of Direct and Cross-Presentation on Influenza CTL Responses. Nat Commun (2019) 10:2846. doi: 10.1038/s41467-019-10661-8
159. Sesma L, Alvarez I, Marcilla M, Paradela A, López de Castro JA. Species-Specific Differences in Proteasomal Processing and Tapasin-Mediated Loading Influence Peptide Presentation by HLA-B27 in Murine Cells. J Biol Chem (2003) 278:46461–72. doi: 10.1074/jbc.M308816200
160. García-Medel N, Sanz-Bravo A, Van Nguyen D, Galocha B, Gómez-Molina P, Martín-Esteban A, et al. Functional Interaction of the Ankylosing Spondylitis-Associated Endoplasmic Reticulum Aminopeptidase 1 Polymorphism and HLA-B27 In Vivo. Mol Cell Proteomics (2012) 11:1416–29. doi: 10.1074/mcp.M112.019588
161. Javitt A, Barnea E, Kramer MP, Wolf-Levy H, Levin Y, Admon A, et al. Pro-Inflammatory Cytokines Alter the Immunopeptidome Landscape by Modulation of HLA-B Expression. Front Immunol (2019) 10:141. doi: 10.3389/fimmu.2019.00141
162. Goncalves G, Mullan KA, Duscharla D, Ayala R, Croft NP, Faridi P, et al. Ifnγ Modulates the Immunopeptidome of Triple Negative Breast Cancer Cells by Enhancing and Diversifying Antigen Processing and Presentation. Front Immunol (2021) 12:645770. doi: 10.3389/fimmu.2021.645770
163. Komov L, Kadosh DM, Barnea E, Milner E, Hendler A, Admon A. Cell Surface MHC Class I Expression Is Limited by the Availability of Peptide-Receptive "Empty" Molecules Rather Than by the Supply of Peptide Ligands. Proteomics (2018) 18:e1700248. doi: 10.1002/pmic.201700248
164. Woods K, Knights AJ, Anaka M, Schittenhelm RB, Purcell AW, Behren A, et al. Mismatch in Epitope Specificities Between Ifnγ Inflamed and Uninflamed Conditions Leads to Escape From T Lymphocyte Killing in Melanoma. J Immunother Cancer (2016) 4:10. doi: 10.1186/s40425-016-0111-7
165. Chapiro J, Claverol S, Piette F, Ma W, Stroobant V, Guillaume B, et al. Destructive Cleavage of Antigenic Peptides Either by the Immunoproteasome or by the Standard Proteasome Results in Differential Antigen Presentation. J Immunol (2006) 176:1053–61. doi: 10.4049/jimmunol.176.2.1053
166. Burgert HG, Kvist S. An Adenovirus Type 2 Glycoprotein Blocks Cell Surface Expression of Human Histocompatibility Class I Antigens. Cell (1985) 41:987–97. doi: 10.1016/S0092-8674(85)80079-9
167. Schuren AB, Costa AI, Wiertz EJ. Recent Advances in Viral Evasion of the MHC Class I Processing Pathway. Curr Opin Immunol (2016) 40:43–50. doi: 10.1016/j.coi.2016.02.007
168. van de Weijer ML, Luteijn RD, Wiertz EJ. Viral Immune Evasion: Lessons in MHC Class I Antigen Presentation. Semin Immunol (2015) 27:125–37. doi: 10.1016/j.smim.2015.03.010
169. Koutsakos M, McWilliam HEG, Aktepe TE, Fritzlar S, Illing PT, Mifsud NA, et al. Downregulation of MHC Class I Expression by Influenza A and B Viruses. Front Immunol (2019) 10:1158. doi: 10.3389/fimmu.2019.01158
170. Nicholas B, Bailey A, Staples KJ, Wilkinson T, Elliott T, Skipp P. Immunopeptidomic Analysis of Influenza A Virus Infected Human Tissues Identifies Internal Proteins as a Rich Source of HLA Ligands. PloS Pathog (2022) 18:e1009894. doi: 10.1371/journal.ppat.1009894
171. Nagler A, Kalaora S, Barbolin C, Gangaev A, Ketelaars SLC, Alon M, et al. Identification of Presented SARS-CoV-2 HLA Class I and HLA Class II Peptides Using HLA Peptidomics. Cell Rep (2021) 35:109305. doi: 10.1016/j.celrep.2021.109305
172. Karlsson AC, Martin JN, Younger SR, Bredt BM, Epling L, Ronquillo R, et al. Comparison of the ELISPOT and Cytokine Flow Cytometry Assays for the Enumeration of Antigen-Specific T Cells. J Immunol Methods (2003) 283:141–53. doi: 10.1016/j.jim.2003.09.001
173. Liu J, Zhang S, Tan S, Yi Y, Wu B, Cao B, et al. Cross-Allele Cytotoxic T Lymphocyte Responses Against 2009 Pandemic H1N1 Influenza A Virus Among HLA-A24 and HLA-A3 Supertype-Positive Individuals. J Virol (2012) 86:13281–94. doi: 10.1128/JVI.01841-12
174. Liu WJ, Tan S, Zhao M, Quan C, Bi Y, Wu Y, et al. Cross-Immunity Against Avian Influenza A(H7N9) Virus in the Healthy Population Is Affected by Antigenicity-Dependent Substitutions. J Infect Dis (2016) 214:1937–46. doi: 10.1093/infdis/jiw471
175. Alexander J, Bilsel P, del Guercio MF, Marinkovic-Petrovic A, Southwood S, Stewart S, et al. Identification of Broad Binding Class I HLA Supertype Epitopes to Provide Universal Coverage of Influenza A Virus. Hum Immunol (2010) 71:468–74. doi: 10.1016/j.humimm.2010.02.014
176. Alexander J, Oseroff C, Sidney J, Wentworth P, Keogh E, Hermanson G, et al. Derivation of HLA-A11/Kb Transgenic Mice: Functional CTL Repertoire and Recognition of Human A11-Restricted CTL Epitopes. J Immunol (1997) 159(10):4753–61.
177. Chang CX, Tan AT, Or MY, Toh KY, Lim PY, Chia AS, et al. Conditional Ligands for Asian HLA Variants Facilitate the Definition of CD8+ T-Cell Responses in Acute and Chronic Viral Diseases. Eur J Immunol (2013) 43:1109–20. doi: 10.1002/eji.201243088
178. Gianfrani C, Oseroff C, Sidney J, Chesnut RW, Sette A. Human Memory CTL Response Specific for Influenza A Virus is Broad and Multispecific. Hum Immunol (2000) 61:438–52. doi: 10.1016/S0198-8859(00)00105-1
179. Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA Class I Supertypes: A Revised and Updated Classification. BMC Immunol (2008) 9:1. doi: 10.1186/1471-2172-9-1
180. Wang M, Claesson MH. Classification of Human Leukocyte Antigen (HLA) Supertypes. Methods Mol Biol (2014) 1184:309–17. doi: 10.1007/978-1-4939-1115-8_17
181. Nguyen AT, Szeto C, Gras S. The Pockets Guide to HLA Class I Molecules. Biochem Soc Trans (2021) 49:2319–31. doi: 10.1042/BST20210410
182. Illing PT, Pymm P, Croft NP, Hilton HG, Jojic V, Han AS, et al. HLA-B57 Micropolymorphism Defines the Sequence and Conformational Breadth of the Immunopeptidome. Nat Commun (2018) 9:4693. doi: 10.1038/s41467-018-07109-w
183. Macdonald WA, Purcell AW, Mifsud NA, Ely LK, Williams DS, Chang L, et al. A Naturally Selected Dimorphism Within the HLA-B44 Supertype Alters Class I Structure, Peptide Repertoire, and T Cell Recognition. J Exp Med (2003) 198:679–91. doi: 10.1084/jem.20030066
184. Kløverpris HN, Cole DK, Fuller A, Carlson J, Beck K, Schauenburg AJ, et al. A Molecular Switch in Immunodominant HIV-1-Specific CD8 T-Cell Epitopes Shapes Differential HLA-Restricted Escape. Retrovirology (2015) 12:20. doi: 10.1186/s12977-015-0149-5
185. Geldmacher C, Metzler IS, Tovanabutra S, Asher TE, Gostick E, Ambrozak DR, et al. Minor Viral and Host Genetic Polymorphisms can Dramatically Impact the Biologic Outcome of an Epitope-Specific CD8 T-Cell Response. Blood (2009) 114:1553–62. doi: 10.1182/blood-2009-02-206193
186. Yuen TJ, Flesch IE, Hollett NA, Dobson BM, Russell TA, Fahrer AM, et al. Analysis of A47, an Immunoprevalent Protein of Vaccinia Virus, Leads to a Reevaluation of the Total Antiviral CD8+ T Cell Response. J Virol (2010) 84:10220–9. doi: 10.1128/JVI.01281-10
187. Nakiboneka R, Mugaba S, Auma BO, Kintu C, Lindan C, Nanteza MB, et al. Interferon Gamma (IFN-γ) Negative CD4+ and CD8+ T-Cells can Produce Immune Mediators in Response to Viral Antigens. Vaccine (2019) 37:113–22. doi: 10.1016/j.vaccine.2018.11.024
188. Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, et al. The Influenza Virus Resource at the National Center for Biotechnology Information. J Virol (2008) 82:596–601. doi: 10.1128/JVI.02005-07
189. Juno JA, Tan HX, Lee WS, Reynaldi A, Kelly HG, Wragg K, et al. Humoral and Circulating Follicular Helper T Cell Responses in Recovered Patients With COVID-19. Nat Med (2020) 26:1428–34. doi: 10.1038/s41591-020-0995-0
190. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans With COVID-19 Disease and Unexposed Individuals. Cell (2020) 181:1489–501.e1415. doi: 10.1016/j.cell.2020.05.015
191. Zaunders JJ, Munier ML, Seddiki N, Pett S, Ip S, Bailey M, et al. High Levels of Human Antigen-Specific CD4+ T Cells in Peripheral Blood Revealed by Stimulated Coexpression of CD25 and CD134 (Ox40). J Immunol (2009) 183:2827–36. doi: 10.4049/jimmunol.0803548
192. Reiss S, Baxter AE, Cirelli KM, Dan JM, Morou A, Daigneault A, et al. Comparative Analysis of Activation Induced Marker (AIM) Assays for Sensitive Identification of Antigen-Specific CD4 T Cells. PloS One (2017) 12:e0186998. doi: 10.1371/journal.pone.0186998
Keywords: CD8+ T cell epitopes, Human Leucocyte Antigen (HLA), influenza, SARS-CoV-2, Indigenous populations, epitope discovery, antigen presentation, immunopeptidome
Citation: Hensen L, Illing PT, Rowntree LC, Davies J, Miller A, Tong SYC, Habel JR, van de Sandt CE, Flanagan K L, Purcell AW, Kedzierska K and Clemens EB (2022) T Cell Epitope Discovery in the Context of Distinct and Unique Indigenous HLA Profiles. Front. Immunol. 13:812393. doi: 10.3389/fimmu.2022.812393
Received: 10 November 2021; Accepted: 28 March 2022;
Published: 06 May 2022.
Edited by:
Susan Prockop, Boston Children’s Hospital, United StatesReviewed by:
David Koelle, University of Washington, United StatesArie Admon, Technion Israel Institute of Technology, Israel
Copyright © 2022 Hensen, Illing, Rowntree, Davies, Miller, Tong, Habel, van de Sandt, Flanagan, Purcell, Kedzierska and Clemens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine Kedzierska, a2tlZHpAdW5pbWVsYi5lZHUuYXU=
†These authors have contributed equally to this work and share senior authorship