
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 14 March 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.807934
This article is part of the Research TopicThe Immunological Role of Platelet Activation in the Pathophysiology of COVID-19View all 11 articles
 Stefan D. Jevtic1
Stefan D. Jevtic1 Ishac Nazy1,2*
Ishac Nazy1,2*Coronavirus disease 2019 (COVID-19) is a highly prothrombotic viral infection that primarily manifests as an acute respiratory syndrome. However, critically ill COVID-19 patients will often develop venous thromboembolism with associated increases in morbidity and mortality. The cause for this prothrombotic state is unclear but is likely related to platelet hyperactivation. In this review, we summarize the current evidence surrounding COVID-19 thrombosis and platelet hyperactivation. We highlight the fact that several studies have identified a soluble factor in COVID-19 patient plasma that is capable of altering platelet phenotype in vitro. Furthermore, this soluble factor appears to be an immune complex, which may be composed of COVID-19 Spike protein and related antibodies. We suggest that these Spike-specific immune complexes contribute to COVID-19 platelet activation and thrombosis in a manner similar to heparin-induced thrombocytopenia. Understanding this underlying pathobiology will be critical for advancement of future research and therapeutic options.
Coronavirus disease 2019 (COVID-19) is a respiratory infection caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1, 2). It has resulted in a global pandemic and is characterized by a highly inflammatory and prothrombotic state. Pulmonary involvement is the primary clinical manifestation but subsequent multi-organ failure and death can occur in severe cases (3–5). The prevalence of COVID-19 thromboembolism is quite variable across studies but appears to be highest in the critical care population, where estimates range from 20-65% (5–9). These can be both arterial and venous thromboses, making COVID-19 a unique prothrombotic state. Although the exact mechanisms underlying thrombosis are likely multifactorial, mounting evidence suggests that platelets play a crucial role.
Platelets have long been known to function as mediators of thrombosis and hemostasis but have only recently gained recognition in their role as immune mediators (10, 11). These anucleate cells mediate various immune related roles throughout the body, from antigen presentation to immune complex signaling. Of particular interest is their role in viral infection, where platelets are able to internalize and degrade pathogen as well as release soluble immune mediators (12). This likely contributes to their important role in COVID-19 and explains, at least in part, how platelet hyperactivation increases thrombotic risk.
In this review, we summarize the general thrombotic nature of COVID-19 and the importance of understanding this process in regards to therapeutic options. Subsequently, we review platelet physiology and their current recognized functions as immune cells. We also review platelet activation in heparin-induced thrombocytopenia (HIT) and how this parallels platelet activity in COVID-19. Finally, we discuss the novel entity of vaccine-induced thrombotic thrombocytopenia (VITT) and the role of platelets in this unique COVID-19 associated disorder.
COVID-19 is a severe viral infection that was identified in Wuhan, China in late 2019. It initially presents as a respiratory tract infection, including fever, dyspnea, and myalgias, but can rapidly progress to a more severe form (1). These critically ill COVID-19 patients are characterized by excess inflammation and a prothrombotic state. COVID-19 thrombosis features both arterial and venous thromboembolic events, often with concomitant thrombocytopenia (7, 13). In one retrospective study of 1476 hospitalized COVID-19 patients, 20.7% were found to have thrombocytopenia (where 125 x 109/L was the lower limit of normal) (14). The degree of thrombocytopenia has also been shown to independently associate with mortality outcomes, suggesting that platelet activation plays an important role in disease severity (14).
Thrombosis is particularly prevalent amongst critically ill COVID-19 patients, with some studies identifying deep vein thrombosis in up to 79% of patients through ultrasound screening (9). Unusual thrombi are also more prevalent amongst COVID-19 patients, including ischemic stroke, limb ischemia, and aortic thrombi (15). Up to 27.6% of thromboembolic events in critically ill patients occur even in the setting of prophylactic anticoagulation, emphasizing the extreme nature of this thrombotic state (16). Therapeutic dosing of anticoagulation may thus be required in COVID-19 hospitalized patients.
This observation has resulted in several randomized clinical trials demonstrating a benefit of therapeutic-dose low molecular weight heparin in hospitalized COVID-19 patients. In the ATTACC, ACTIV-4a, and REMAP-CAP multi-platform trial, therapeutic doses of heparin increased the probability of organ support-free days in non-critically ill patients (odds ratio 1.27, 95% credible interval 1.03-1.58) (17). However, there was no significant benefit for survival until hospital discharge, although there was a trend towards benefit (adjusted odds ratio 1.21, 95% credible interval 0.87-1.68). Interestingly, the RAPID trial did demonstrate a significant mortality benefit from therapeutic heparin in hospitalized, non-critically ill patients (odds ratio 0.22, 95% confidence interval 0.07-0.65) (18). Therefore, therapeutic anticoagulation is likely to benefit COVID-19 patients who are hospitalized without critical illness. It should be noted, however, that therapeutic anticoagulation showed no benefit to mortality or reduced organ support in critically ill COVID-19 patients (19). It may be that anticoagulation in these patients was introduced at an overly advanced stage of disease. Nonetheless, it implies that critically ill patients differ significantly in their underlying physiology and require unique therapies.
Prior to delving into platelet activation in COVID-19, it is important to gain a basic understanding of how platelet synthesis and function are intimately related to immunity. Platelets are produced in the bone marrow from progenitor cells, termed megakaryocytes, through a complex process of hematopoietic stem cell differentiation (20). Their production is primarily driven by the cytokine mediator, thrombopoietin (TPO), which is synthesized by both the liver and kidneys. TPO is known to be upregulated by inflammatory cytokines, such as IL-6, and contributes to the rapid platelet production seen with inflammation (21). This is secondary to a subgroup of “pre-differentiated” stem cells that are biased towards the megakaryocyte lineage and rapidly differentiate on TPO exposure (22–24). The hematopoietic system is thus efficiently designed to produce platelets in the context of infection, suggesting an important role in immunity.
Once released into circulation, platelets are equipped with various intracellular materials (over 300) to mediate their effector functions (25). These include inflammatory cytokines (e.g. IL-1β), procoagulant factors (tissue factor, serotonin), and angiostatic molecules (platelet-factor 4/PF4) (26–28). These molecules are released upon platelet activation, which is mediated through various cell surface receptors. Many of these cell surface receptors also contribute to immune cell interaction and function. For example, the GPIb receptor is normally involved in platelet adhesion at sites of vascular injury through von Willebrand factor binding. However, GPIb is also capable of binding to von Willebrand factor exposed on immune cells infected with bacterial pathogen, such as hepatic Kupffer cells (29). It has been shown in a mouse model that this interaction is crucial for platelet aggregation around infected cells and host survival. P-selectin is another platelet surface receptor that is known to be upregulated with platelet activation. It is capable of binding to leukocytes through the P-selectin glycoprotein ligand-1 to mediate intracellular leukocyte signaling and neutrophil rolling (30–32). This process is crucial for leukocyte mobilization and concentration at sites of infection. Indeed, the P-selectin dependent interaction between neutrophils and platelets has been shown to contribute to acute lung injury in mouse models (33, 34). Platelet depletion or P-selectin inhibition both reduced subsequent neutrophil recruitment and lung injury. Platelets are thus equipped, through both intracellular and cell surface proteins, to mediate various immune functions. These interactions may contribute to the lung pathology seen in COVID-19 through immune cell recruitment.
Immune complexes are important initial defenses against pathogen infection and are formed from antibody binding to soluble antigen. They often consist of immunoglobulins (Ig) of the IgG or IgM type but can also be IgA (35, 36). Immune complexes primarily mediate function through binding to cell surface receptors found on various cell types, including platelets. Most binding occurs through the Fcγ receptors II (FcγRII) and III, which are either activating (a) or inhibiting (b), respectively (35). Platelets contain only one Fc receptor (FcγRIIa) on their surface and thus are able to bind IgG-specific immune complexes (37). Immune complex binding to the platelet receptor leads to subsequent activation and release of intracellular molecules such as serotonin. This promotes a prothrombotic state and has been implicated in various autoimmune conditions. The most well-characterized platelet-mediated immune complex disorder is heparin-induced thrombocytopenia (HIT) (38, 39).
HIT is a prothrombotic autoimmune disorder characterized by the presence of thrombocytopenia (low platelets) and thrombosis that shares many features with COVID-19. It most commonly presents in hospitalized patients who are receiving unfractionated heparin anticoagulation and is characterized by antibodies targeting platelet factor 4 (PF4)-heparin complexes (39). PF4 is a positively charged protein released from platelets that is capable of binding negatively charged molecules, such as heparin (40). Certain individuals develop anti-PF4/heparin IgG antibodies that form immune complexes. These immune complexes activate the FcγRIIa on platelets resulting in thrombocytopenia that is often accompanied by thrombosis, which is secondary to the release of serotonin and other procoagulant platelet microparticles. Circulating anti-PF4/heparin antibodies can be found in up to 50% of patients exposed to heparin (41, 42). However, only a minority of these will be functional and lead to disease presentation. This is secondary to the unique epitope specificity required for immune complex formation and platelet activation (43). Therefore, it is important to use functional platelet activation assays to diagnose HIT.
One of the international reference assays for diagnosing HIT, developed at our institution, is the serotonin release assay (SRA). Briefly, platelets from healthy donors are incubated with radioactive 14C-serotonin allowing uptake into platelets. These modified platelets are subsequently exposed to patient plasma, in the presence and absence of heparin, which allows formation of anti-PF4/heparin immune complexes (44). These immune complexes subsequently activate platelets through the FcγRIIa leading to release of 14C-serotonin, which is then measured by beta radioactivity. The addition of exogenous heparin is vital for this activation in HIT to facilitate formation of appropriate antigen complexes. However, certain samples tested in the SRA for HIT will demonstrate heparin-independent platelet activation (45). This is inconsistent with a diagnosis of classical HIT, meaning that the assay may detect additional mechanisms of platelet activation. Therefore, the SRA can be modified to study platelet hyperactivation in novel diseases, such as COVID-19 (see Figure 1).
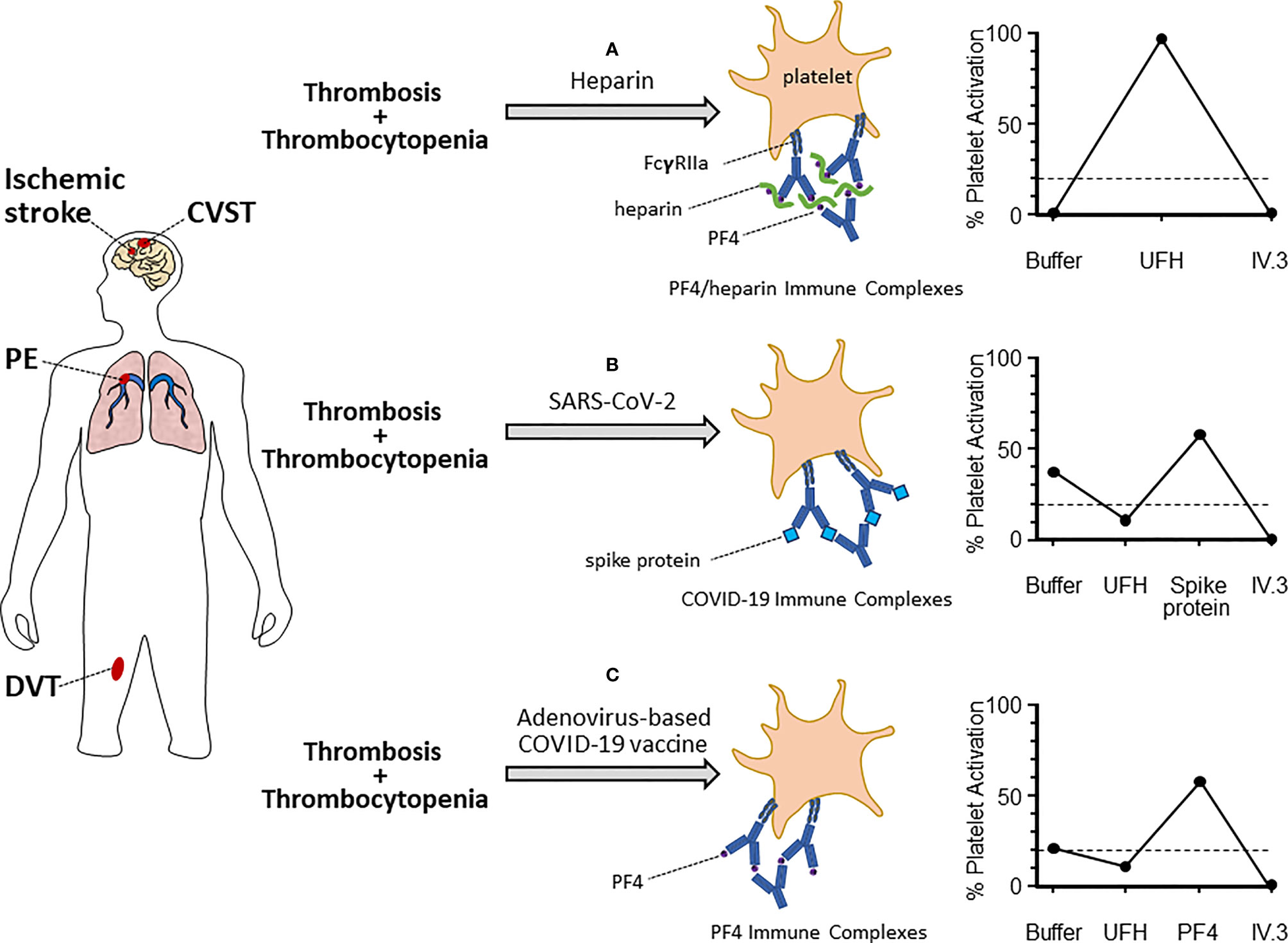
Figure 1 Platelet activation mechanisms and assay patterns in various thrombotic-thrombocytopenic syndromes. Patients who present with evidence of thrombosis (arterial or venous) and thrombocytopenia should be considered for hematology referral and specialized testing for platelet activation disorders. These can include heparin-induced thrombocytopenia (HIT, A), COVID-19-related platelet activation (B), and vaccine-induced thrombotic thrombocytopenia (VITT, C). The suspected platelet activation disorder will depend on the clinical context and known exposure to antigen (e.g. heparin, SARS-CoV-2, or adenovirus-based COVID-19 vaccine). Each of these disorders is characterized by platelet activation through the FcγRIIa via unique immune complexes. These include: anti-PF4/heparin (HIT), unidentified immune complexes (COVID-19), or anti-PF4 (VITT). Serum testing from patients will also feature unique platelet activation schemas in functional activation assays, such as the serotonin release assay (examples shown on the right). In the classic HIT-SRA (top right), addition of exogenous UFH significantly increases immune complex formation and platelet activation, which is inhibited by IV.3. Contrastingly, UFH inhibits immune complex formation in COVID-19 and VITT thrombotic patients. Instead, alternate antigens (Spike protein and PF4, respectively) are required for significant platelet activation. Dashed lined represents 20% platelet activation, which is the positive cut-off for the SRA.
To this point, many studies have confirmed that platelets in COVID-19 patients display a hyperactivated phenotype with altered gene expression. In a cohort of 115 COVID-19 patients, featuring both non-severe and severe disease, platelets secreted increased IL-1beta and soluble CD40 ligand compared to healthy controls (46). Furthermore, circulating levels of serotonin and PF4 were increased in patient serum, suggesting platelet degranulation. Common cell markers of platelet activation, including P-selectin and CD63, are also increased in critically ill COVID-19 patients, but not those with mild disease (47). Platelets from critically ill COVID-19 patients also demonstrated increased markers of apoptosis, such as phosphatidylserine externalization and cleaved-caspase 9, which correlate with thromboembolic events (48). In addition to platelet activation, there is evidence of unique transcriptome changes that occur in platelets from COVID-19 patients. Using RNA-seq analysis on platelets from 10 COVID-19 patients, Manne et al. demonstrated significant upregulation of genes involved in antigen presentation (49). Platelets are thus significantly altered to a more active phenotype in COVID-19, particularly in critically ill patients, and may contribute to clinical presentation.
One mechanism by which platelets may contribute to COVID-19 presentation is through neutrophil recruitment and aggregation. As previously noted, platelet P-selectin is able to bind neutrophil ligands to induce rolling and aggregation at sites of activation (50). This interaction can lead to prothrombotic platelet-neutrophil aggregates as well as the formation of neutrophil extracellular traps. For example, plasma from hospitalized COVID-19 patients demonstrates increased circulating platelet-neutrophil aggregates on flow cytometry compared to healthy controls (51). Furthermore, autopsies in COVID-19 patients confirm the presence of microvascular thrombi consisting of neutrophil extracellular traps and platelets (52, 53). These platelet-neutrophil interactions are more prominent in critically ill COVID-19 patients, where there is evidence of a hyperactivated platelet phenotype (52, 54). Therefore, hyperactivated platelets in COVID-19 also contribute to neutrophil activation, which fuels the thrombo-inflammatory milieu.
It is still unclear as to what triggers such drastic platelet changes in critically ill patients. Some have hypothesized that SARS-CoV-2 directly interacts with platelets to mediate these observed effects. Evidence for this is supported by the presence of viral RNA in platelets of infected individuals, although this is only seen in up to 24% of patients (46, 49, 55). However, aside from a single study (55), multiple studies have failed to demonstrate ACE2 expression on the platelet surface or evidence of ACE2 RNA in platelets (46, 49). The cause of this discrepancy is unclear and may be related to different techniques for platelet isolation (56). Regardless, SARS-CoV-2 RNA has been consistently found within platelets and thus suggests that ACE2-independent mechanisms of entry exist. Interestingly, when critically ill COVID-19 patient plasma is incubated with platelets from healthy volunteers, there is a similar increase in platelet activation markers (P-selectin, CD63) (47). While circulating virus may account for this change as well, other soluble mediators should be considered.
Immune complexes are one potential circulating factor that could contribute to platelet activation in COVID-19. As previously mentioned, immune complexes activate platelets through the FcγRIIa and may be formed from antibodies against self or exogenous antigens. Viral illnesses are well documented to produce antibodies against self-antigens, such as antiphospholipid antibodies, through a process called molecular mimicry. Early reports in COVID-19 patients highlighted the presence of these antibodies in association with thrombosis, including anti-beta-2 glycoprotein and non-specific inhibitor (57–59). Injection of the serum IgG fraction from these patients into mice resulted in significantly increased thrombus formation compared to controls (59). However, this thrombus formation was also seen with COVID-19 patient serum that had low levels of antiphospholipid antibodies. This suggests that antiphospholipid antibodies are not the sole antibodies associated with this prothrombotic state.
Another potential hypothesis is that HIT antibodies are contributing to the IgG-mediated platelet activation seen in COVID-19 patients. This is supported by the observations that COVID-19 and HIT share many clinical similarities; COVID-19 patients are often exposed to heparin in the context of hospitalization; and a high proportion of COVID-19 patients test positive for anti-PF4/heparin antibodies on further testing (60, 61). However, in a cohort of ten critically ill COVID-19 patients with high suspicion of HIT, we found no evidence of platelet-activating HIT antibodies, which has been replicated by others (61, 62). Interestingly, six of these samples were able to activate platelets in the serotonin release assay in the absence of heparin. This activation was inhibited by IV.3, an FcγRIIa inhibitor, thus confirming immune complex mediated platelet activation. Furthermore, all patients with platelet activation also contained anti-Spike IgG antibodies targeting SARS-CoV-2. It is plausible that Spike-specific IgG antibodies bind circulating Spike protein in viremic, critically ill patients to form platelet-activating immune complexes. This mechanism has previously been shown to occur with H1N1 influenza virus whereby influenza antibodies bind to virus to form immune complexes (63). These immune complexes activate platelets through the FcγRIIa and likely contribute to the pulmonary thrombosis seen with H1N1 infection (64). Most recently, one in vitro study confirmed that recombinant anti-Spike IgG is able to activate platelets through the FcγRIIa (65). This was determined through in vitro thrombus measurement using microfluidic flow chips and confocal microscopy. Thrombus formation only occurred in the presence of Spike protein and an “inflammatory signal” (von Willebrand factor in this study). Interestingly, anti-Spike IgG and Spike protein alone did not lead to significant thrombus formation. How exactly von Willebrand factor interacts to promote platelet activation is unclear but may be through facilitating platelet aggregation. Glycosylation status of anti-Spike IgG was also found to be a significant factor in the ability of these complexes to activate platelets. Therefore, certain anti-Spike IgG activate platelets in the context of COVID-19 infection, but this remains to be validated in the clinical context.
It would be remiss to avoid a discussion of vaccine-induced thrombotic thrombocytopenia (VITT) in the context of platelet activation and COVID-19. Although this platelet activation is not directly related to SARS-CoV-2 viral infection, it has important clinical and public health implications. VITT is a novel, “drug” related disorder attributed to vaccination by adenoviral vector-based SARS-CoV-2 vaccines. This primarily includes the ChAd-Ox1 (produced by AstraZeneca) and Ad26.COV2.S (Johnson and Johnson) vaccines. VITT was first described in eleven patients, predominantly female, who presented with unusual thromboses (cerebral venous sinus thrombosis, splanchnic-vein thrombosis) and thrombocytopenia (66). Patients often present a median of 14 days from vaccination and can be critically ill – estimated mortality is 22% in one cohort of 220 VITT cases (67). This mortality is significantly reduced from initial reports (55%), likely due to a combination of increased recognition and better treatment implementation (66). Given the parallels to HIT, it was hypothesized that similar platelet activating antibodies may be the underlying cause. This proved to be the case, with all patients featuring high titers of anti-PF4/heparin antibodies that were able to activate platelets in functional assays (66). Interestingly, these antibodies did not require the presence of heparin to form immune complexes and thus are able to target PF4 independently (i.e. anti-PF4 antibodies). The binding site for these antibodies is located in the heparin-binding site on PF4, as shown by alanine-scanning mutagenesis, thus allowing them to form tetrameric immune complexes and activate platelets through FcγRIIa (68). This competitive binding to the heparin site likely explains why heparin inhibits VITT platelet activation in vitro (unlike in HIT, where heparin facilitates activation; see Figure 1). As previously mentioned, anti-PF4/heparin antibodies do not appear to be responsible for the thrombosis and platelet activation seen in COVID-19. In a cohort of 222 COVID-19 patients with thrombosis, only nineteen (8.6%) tested positive for anti-PF4/heparin antibodies (69). None of these were able to activate platelets in the functional platelet assay. Therefore, anti-PF4 antibodies are likely not responsible for the thrombosis seen in COVID-19 patients and do not demonstrate cross reactivity with the Spike protein. This understanding is important for future vaccine development and management of these rare cases.
Although the field of COVID-19 thrombosis is in its infancy, there is sufficient evidence to support a major role for platelets in disease pathogenesis. Platelets have been shown to be hyperactivated in critically ill patients and secrete excessive procoagulant molecules. Furthermore, they are able to interact with other immune cells to mediate the immune response. However, this excess inflammation may also contribute to tissue damage and ultimately, mortality. Targeting these pathways in order to dampen the excess immune response may thus present attractive therapeutic targets.
Immune complexes also appear to contribute significantly to these platelet changes in a manner similar to HIT and other immune-complex mediated disorders. The antigen specificity and additional characteristics of these immune complexes remain to be determined but will be crucial to the development of therapeutic targets. Identifying the specific antibodies involved may also allow clinicians to risk stratify patients who are at high risk of severe disease or thrombosis, thus offering the potential for prophylactic anticoagulation.
Both SJ and IN contributed equally to this work, including initial manuscript preparation, editing, and final drafting. All authors contributed to the article and approved the submitted version.
Funding was received from the Canadian Institutes of Health Research (CIHR 452655) awarded to IN.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Jo-Ann Shepherd for her contributions as an illustrator.
1. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A Novel Coronavirus From Patients With Pneumonia in China, 2019. N. Engl J Med (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
2. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
3. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383(2):120–128. doi: 10.1056/NEJMoa2015432
4. Connors JM, Levy JH. COVID-19 and its Implications for Thrombosis and Anticoagulation. Blood (2020) 135:2033–40. doi: 10.1182/blood.2020006000
5. Malas MB, Naazie IN, Elsayed N, Mathlouthi A, Marmor R, Clary B. Thromboembolism Risk of COVID-19 is High and Associated With a Higher Risk of Mortality: A Systematic Review and Meta-Analysis. EClinicalMedicine (2020) 29:100639. doi: 10.1016/j.eclinm.2020.100639
6. Boonyawat K, Chantrathammachart P, Numthavaj P, Nanthatanti N, Phusanti S, Phuphuakrat A, et al. Incidence of Thromboembolism in Patients With COVID-19: A Systematic Review and Meta-Analysis. Thromb J (2020) 18:34. doi: 10.1186/s12959-020-00248-5
7. Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of Thrombotic Complications in Critically Ill ICU Patients With COVID-19. Thromb Res (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
8. Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Confirmation of the High Cumulative Incidence of Thrombotic Complications in Critically Ill ICU Patients With COVID-19: An Updated Analysis. Thromb Res (2020) 191:148–50. doi: 10.1016/j.thromres.2020.04.041
9. Nahum J, Morichau-Beauchant T, Daviaud F, Echegut P, Fichet J, Maillet J-M, et al. Venous Thrombosis Among Critically Ill Patients With Coronavirus Disease 2019 (COVID-19). JAMA Netw Open (2020) 3:e2010478. doi: 10.1001/jamanetworkopen.2020.10478
10. Hottz ED, Bozza FA, Bozza PT. Platelets in Immune Response to Virus and Immunopathology of Viral Infections. Front Med (2018) 5:121. doi: 10.3389/fmed.2018.00121
11. Mezger M, Nording H, Sauter R, Graf T, Heim C, von Bubnoff N, et al. Platelets and Immune Responses During Thromboinflammation. Front Immunol (2019) 10:1731. doi: 10.3389/fimmu.2019.01731
12. Worth RG, Chien CD, Chien P, Reilly MP, McKenzie SE, Schreiber AD. Platelet Fcγriia Binds and Internalizes IgG-Containing Complexes. Exp Hematol (2006) 34:1490–5. doi: 10.1016/j.exphem.2006.06.015
13. Levi M, Thachil J, Iba T, Levy JH. Coagulation Abnormalities and Thrombosis in Patients With COVID-19. Lancet Haematol (2020) 7:e438–40. doi: 10.1016/S2352-3026(20)30145-9
14. Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, et al. Thrombocytopenia and its Association With Mortality in Patients With COVID-19. J Thromb Haemost JTH (2020) 18:1469–72. doi: 10.1111/jth.14848
15. Kashi M, Jacquin A, Dakhil B, Zaimi R, Mahé E, Tella E, et al. Severe Arterial Thrombosis Associated With Covid-19 Infection. Thromb Res (2020) 192:75–7. doi: 10.1016/j.thromres.2020.05.025
16. Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, et al. Venous and Arterial Thromboembolic Complications in COVID-19 Patients Admitted to an Academic Hospital in Milan, Italy. Thromb Res (2020) 191:9–14. doi: 10.1016/j.thromres.2020.04.024
17. Lawler PR, Goligher EC, Berger JS, Neal MD, McVerry BJ, Nicolau JC. Therapeutic Anticoagulation With Heparin in Noncritically Ill Patients With Covid-19. N Engl J Med (2021) 385:790–802. doi: 10.1056/NEJMoa2105911
18. Sholzberg M, Tang GH, Rahhal H, AlHamzah M, Kreuziger LB, Áinle FN, et al. Effectiveness of Therapeutic Heparin Versus Prophylactic Heparin on Death, Mechanical Ventilation, or Intensive Care Unit Admission in Moderately Ill Patients With Covid-19 Admitted to Hospital: RAPID Randomised Clinical Trial. BMJ (2021) 375:n2400. doi: 10.1136/bmj.n2400
19. Goligher EC, Bradbury CA, McVerry BJ, Berger JS, Gong MN. Therapeutic Anticoagulation With Heparin in Critically Ill Patients With Covid-19. N Engl J Med (2021) 385:777–89. doi: 10.1056/NEJMoa2103417
20. van der Meijden PEJ, Heemskerk JWM. Platelet Biology and Functions: New Concepts and Clinical Perspectives. Nat Rev Cardiol (2019) 16:166–79. doi: 10.1038/s41569-018-0110-0
21. Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, et al. Interleukin-6 Stimulates Thrombopoiesis Through Thrombopoietin: Role in Inflammatory Thrombocytosis. Blood (2001) 98:2720–5. doi: 10.1182/blood.v98.9.2720
22. Shin JY, Hu W, Naramura M, Park CY. High C-Kit Expression Identifies Hematopoietic Stem Cells With Impaired Self-Renewal and Megakaryocytic Bias. J Exp Med (2014) 211:217–31. doi: 10.1084/jem.20131128
23. Grinenko T, Arndt K, Portz M, Mende N, Günther M, Cosgun KN, et al. Clonal Expansion Capacity Defines Two Consecutive Developmental Stages of Long-Term Hematopoietic Stem Cells. J Exp Med (2014) 211:209–15. doi: 10.1084/jem.20131115
24. Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, et al. Platelet-Biased Stem Cells Reside at the Apex of the Haematopoietic Stem-Cell Hierarchy. Nature (2013) 502:232–6. doi: 10.1038/nature12495
25. Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the Proteins Released From Activated Platelets Leads to Localization of Novel Platelet Proteins in Human Atherosclerotic Lesions. Blood (2004) 103:2096–104. doi: 10.1182/blood-2003-08-2804
26. Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Möpps B, et al. SDF-1α/CXCR4 Axis Is Instrumental in Neointimal Hyperplasia and Recruitment of Smooth Muscle Progenitor Cells. Circ Res (2005) 96:784–91. doi: 10.1161/01.RES.0000162100.52009.38
27. Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, et al. Platelets Release Pathogenic Serotonin and Return to Circulation After Immune Complex-Mediated Sequestration. Proc Natl Acad Sci (2018) 115:E1550–9. doi: 10.1073/pnas.1720553115
28. Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, et al. Escaping the Nuclear Confines: Signal-Dependent Pre-mRNA Splicing in Anucleate Platelets. Cell (2005) 122:379–91. doi: 10.1016/j.cell.2005.06.015
29. Wong CHY, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of Platelets With Blood-Borne Pathogens on Kupffer Cells Precedes Other Innate Immunity and Contributes to Bacterial Clearance. Nat Immunol (2013) 14:785–92. doi: 10.1038/ni.2631
30. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, et al. Platelet/Polymorphonuclear Leukocyte Interaction: P-Selectin Triggers Protein-Tyrosine Phosphorylation–Dependent CD11b/CD18 Adhesion: Role of PSGL-1 as a Signaling Molecule. Blood (1999) 93:876–85. doi: 10.1182/blood.V93.3.876
31. Frenette PS, Denis CV, Weiss L, Jurk K, Subbarao S, Kehrel B, et al. P-Selectin Glycoprotein Ligand 1 (Psgl-1) Is Expressed on Platelets and Can Mediate Platelet–Endothelial Interactions in Vivo. J Exp Med (2000) 191:1413–22. doi: 10.1084/jem.191.8.1413
32. Buttrum S, Hatton R, Nash G. Selectin-Mediated Rolling of Neutrophils on Immobilized Platelets. Blood (1993) 82:1165–74. doi: 10.1182/blood.V82.4.1165.1165
33. Asaduzzaman M, Lavasani S, Rahman M, Zhang S, Braun OO, Jeppsson B, et al. Platelets Support Pulmonary Recruitment of Neutrophils in Abdominal Sepsis. Crit Care Med (2009) 37:1389–96. doi: 10.1097/CCM.0b013e31819ceb71
34. Zarbock A, Singbartl K, Ley K. Complete Reversal of Acid-Induced Acute Lung Injury by Blocking of Platelet-Neutrophil Aggregation. J Clin Invest (2006) 116:3211–9. doi: 10.1172/JCI29499
35. Krishna M, Nadler SG. Immunogenicity to Biotherapeutics – The Role of Anti-Drug Immune Complexes. Front Immunol (2016) 7:21. doi: 10.3389/fimmu.2016.00021
36. Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, et al. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front Immunol (2016) 7:117. doi: 10.3389/fimmu.2016.00117
37. McKenzie SE, Taylor SM, Malladi P, Yuhan H, Cassel DL, Chien P, et al. The Role of the Human Fc Receptor Fcγriia in the Immune Clearance of Platelets: A Transgenic Mouse Model. J Immunol (1999) 162:4311–8.
38. Warkentin TE. How I Diagnose and Manage HIT. Hematol Am Soc Hematol Educ Program (2011) 2011:143–9. doi: 10.1182/asheducation-2011.1.143
39. Warkentin TE, Levine MN, Hirsh J, Horsewood P, Roberts RS, Gent M, et al. Heparin-Induced Thrombocytopenia in Patients Treated With Low-Molecular-Weight Heparin or Unfractionated Heparin. N Engl J Med (1995) 332:1330–6. doi: 10.1056/NEJM199505183322003
40. Zucker MB, Katz IR. Platelet Factor 4: Production, Structure, and Physiologic and Immunologic Action. Proc Soc Exp Biol Med Soc Exp Biol Med N Y N (1991) 198:693–702. doi: 10.3181/00379727-198-43309
41. Selleng S, Selleng K, Friesecke S, Gründling M, Kuhn S-O, Raschke R, et al. Prevalence and Clinical Implications of Anti-PF4/heparin Antibodies in Intensive Care Patients: A Prospective Observational Study. J Thromb Thromb (2015) 39:60–7. doi: 10.1007/s11239-014-1105-2
42. Huynh A, Arnold DM, Kelton JG, Clare R, Ivanova M, Nazy I. Pathogenic Antibodies in Heparin-Induced Thrombocytopenia Specifically Target an Immunodominant Region on Platelet Factor 4. Blood (2019) 134:219. doi: 10.1182/blood-2019-131462
43. Huynh A, Arnold DM, Kelton JG, Smith JW, Horsewood P, Clare R, et al. Characterization of Platelet Factor 4 Amino Acids That Bind Pathogenic Antibodies in Heparin-Induced Thrombocytopenia. J Thromb Haemost JTH (2019) 17:389–99. doi: 10.1111/jth.14369
44. Warkentin TE, Arnold DM, Nazi I, Kelton JG. The Platelet Serotonin-Release Assay. Am J Hematol (2015) 90:564–72. doi: 10.1002/ajh.24006
45. Warkentin TE. Laboratory Diagnosis of Heparin-Induced Thrombocytopenia. Int J Lab Hematol (2019) 41:15–25. doi: 10.1111/ijlh.12993
46. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets Can Associate With SARS-CoV-2 RNA and Are Hyperactivated in COVID-19. Circ Res (2020) 127:1404–18. doi: 10.1161/CIRCRESAHA.120.317703
47. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, et al. Platelet Activation and Platelet-Monocyte Aggregate Formation Trigger Tissue Factor Expression in Patients With Severe COVID-19. Blood (2020) 136:1330–41. doi: 10.1182/blood.2020007252
48. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, et al. Antibody-Induced Procoagulant Platelets in Severe COVID-19 Infection. Blood (2021) 137:1061–71. doi: 10.1182/blood.2020008762
49. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet Gene Expression and Function in Patients With COVID-19. Blood (2020) 136:1317–29. doi: 10.1182/blood.2020007214
50. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils Scan for Activated Platelets to Initiate Inflammation. Science (2014) 346:1234–8. doi: 10.1126/science.1256478
51. Taus F, Salvagno G, Canè S, Fava C, Mazzaferri F, Carrara E, et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arterioscler Thromb Vasc Biol (2020) 40:2975–89. doi: 10.1161/ATVBAHA.120.315175
52. Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation (2020) 142:1176–89. doi: 10.1161/CIRCULATIONAHA.120.048488
53. Middleton EA, He X-Y, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136:1169–79. doi: 10.1182/blood.2020007008
54. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5:e138999. doi: 10.1172/jci.insight.138999
55. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J Hematol OncolJ Hematol Oncol (2020) 13:120. doi: 10.1186/s13045-020-00954-7
56. Campbell RA, Boilard E, Rondina MT. Is There a Role for the ACE2 Receptor in SARS-CoV-2 Interactions With Platelets? J Thromb Haemost (2021) 19:46–50. doi: 10.1111/jth.15156
57. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and Antiphospholipid Antibodies in Patients With Covid-19. N Engl J Med (2020) 382:e38. doi: 10.1056/NEJMc2007575
58. Bowles L, Platton S, Yartey N, Dave M, Lee K, Hart DP, et al. Lupus Anticoagulant and Abnormal Coagulation Tests in Patients With Covid-19. N Engl J Med (2020) 383:288–90. doi: 10.1056/NEJMc2013656
59. Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H, et al. Prothrombotic Autoantibodies in Serum From Patients Hospitalized With COVID-19. Sci Transl Med (2020) 12:eabd3876. doi: 10.1126/scitranslmed.abd3876
60. Warkentin TE, Kaatz S. COVID-19 Versus HIT Hypercoagulability. Thromb Res (2020) 196:38–51. doi: 10.1016/j.thromres.2020.08.017
61. Brodard J, Kremer Hovinga JA, Fontana P, Studt J-D, Gruel Y, Greinacher A. COVID-19 Patients Often Show High-Titer Non-Platelet-Activating Anti-PF4/heparin IgG Antibodies. J Thromb Haemost JTH (2021) 19:1294–8. doi: 10.1111/jth.15262
62. Nazy I, Jevtic SD, Moore JC, Huynh A, Smith JW, Kelton JG, et al. Platelet-Activating Immune Complexes Identified in Critically Ill COVID-19 Patients Suspected of Heparin-Induced Thrombocytopenia. J Thromb Haemost JTH (2021) 19:1342–7. doi: 10.1111/jth.15283
63. Boilard E, Paré G, Rousseau M, Cloutier N, Dubuc I, Lévesque T, et al. Influenza Virus H1N1 Activates Platelets Through Fcγriia Signaling and Thrombin Generation. Blood (2014) 123:2854–63. doi: 10.1182/blood-2013-07-515536
64. Harms PW, Schmidt LA, Smith LB, Newton DW, Pletneva MA, Walters LL, et al. Autopsy Findings in Eight Patients With Fatal H1N1 Influenza. Am J Clin Pathol (2010) 134:27–35. doi: 10.1309/AJCP35KOZSAVNQZW
65. Bye AP, Hoepel W, Mitchell JL, Jégouic S, Loureiro S, Sage T, et al. Aberrant Glycosylation of Anti-SARS-CoV-2 Spike IgG is a Prothrombotic Stimulus for Platelets. Blood (2021) 138:1481–9. doi: 10.1182/blood.2021011871
66. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic Thrombocytopenia After ChAdOx1 Ncov-19 Vaccination. N Engl J Med (2021) 384(22):2092–101. doi: 10.1056/NEJMoa2104840
67. Pavord S, Scully M, Hunt BJ, Lester W, Bagot C, Craven B, et al. Clinical Features of Vaccine-Induced Immune Thrombocytopenia and Thrombosis. N Engl J Med (2021) 385:1680–9. doi: 10.1056/NEJMoa2109908. 0:null.
68. Huynh A, Kelton JG, Arnold DM, Daka M, Nazy I. Antibody Epitopes in Vaccine-Induced Immune Thrombotic Thrombocytopaenia. Nature (2021) 596:565–9. doi: 10.1038/s41586-021-03744-4
Keywords: COVID-19, platelet, antigen-antibody complex, immune complex, thrombosis, thrombocytopenia, heparin, VITT
Citation: Jevtic SD and Nazy I (2022) The COVID Complex: A Review of Platelet Activation and Immune Complexes in COVID-19. Front. Immunol. 13:807934. doi: 10.3389/fimmu.2022.807934
Received: 02 November 2021; Accepted: 31 January 2022;
Published: 14 March 2022.
Edited by:
Geoffrey Wool, University of Chicago, United StatesReviewed by:
Carsten Deppermann, Johannes Gutenberg University Mainz, GermanyCopyright © 2022 Jevtic and Nazy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ishac Nazy, bmF6eWlAbWNtYXN0ZXIuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.