 Matthew Ayokunle Olude1*
Matthew Ayokunle Olude1* Abdeslam Mouihate2
Abdeslam Mouihate2 Oluwaseun Ahmed Mustapha1
Oluwaseun Ahmed Mustapha1 Cinthia Farina3
Cinthia Farina3 Francisco Javier Quintana4
Francisco Javier Quintana4 James Olukayode Olopade5
James Olukayode Olopade5- 1Vertebrate Morphology, Environmental Toxicology and Neuroscience Unit, College of Veterinary Medicine, Federal University of Agriculture, Abeokuta, Nigeria
- 2Department of Physiology, Faculty of Medicine, Health Sciences Centre, Kuwait University, Kuwait City, Kuwait
- 3Istituto di Ricovero e Cura a Carattere Scientifico (IRCSS) San Raffaele Scientific Institute, Institute of Experimental Neurology (INSPE) and Division of Neuroscience, Milan, Italy
- 4Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States
- 5Neuroscience Unit, Department of Veterinary Anatomy, Faculty of Veterinary Medicine, University of Ibadan, Ibadan, Nigeria
Background: Africa is laden with a youthful population, vast mineral resources and rich fauna. However, decades of unfortunate historical, sociocultural and leadership challenges make the continent a hotspot for poverty, indoor and outdoor pollutants with attendant stress factors such as violence, malnutrition, infectious outbreaks and psychological perturbations. The burden of these stressors initiate neuroinflammatory responses but the pattern and mechanisms of glial activation in these scenarios are yet to be properly elucidated. Africa is therefore most vulnerable to neurological stressors when placed against a backdrop of demographics that favor explosive childbearing, a vast population of unemployed youths making up a projected 42% of global youth population by 2030, repressive sociocultural policies towards women, poor access to healthcare, malnutrition, rapid urbanization, climate change and pollution. Early life stress, whether physical or psychological, induces neuroinflammatory response in developing nervous system and consequently leads to the emergence of mental health problems during adulthood. Brain inflammatory response is driven largely by inflammatory mediators released by glial cells; namely astrocytes and microglia. These inflammatory mediators alter the developmental trajectory of fetal and neonatal brain and results in long-lasting maladaptive behaviors and cognitive deficits. This review seeks to highlight the patterns and mechanisms of stressors such as poverty, developmental stress, environmental pollutions as well as malnutrition stress on astrocytes and microglia in neuroinflammation within the African context.
Introduction
Astrocytes: From Physiology to Neuroinflammation
Astrocytes are glial cells of the central nervous system (CNS) of neuroectodermal origin. In fact, neurons, oligodendrocytes and astrocytes derive from a common multipotent self-renewable neural stem cell in a process that occurs with precise timing (1). While neurogenesis takes place early during embryonic development and is accomplished at about birth, gliogenesis follows neurogenesis and is finalized in postnatal life (1), with synaptogenesis and neuronal function depending on astrocyte morphology, maturation and regional specification (1).
Astrocytes play key physiological functions in the CNS that, if altered, may lead to or amplify tissue damage and neuroinflammation and hamper relevant brain functions, such as cognition and memory.
First, astrocytes contribute to the complexity of CNS structure. Evolution has led to the relative expansion of astrocytes, especially in the human brain where the number of astrocytes exceeds that of neurons and astrocytes present complex arborisation architectures (2). CNS damage triggers a complex response in astrocytes, which become reactive and undergo transcriptional remodelling, early upregulation of the intermediate filament glial fibrillary acidic protein (GFAP), morphological changes and proliferation (3). This reaction alters physiological tissue topology and may lead to scar formation as observed in traumatic, neuroinflammatory and neurodegenerative CNS disorders (3, 4). By contrast, decreased astrocyte numbers and GFAP signals have been evidenced in mental disorders (5), underlying a distinct pattern of astrocyte pathology. Astrocytes are involved in the control of neuroinflammation, as conventional or inducible GFAP deficient mice display exacerbated expression of Toxoplasma encephalitis, Staphylococcus aureus-induced brain abscess, spinal cord and brain injury (reviewed in 6). In the experimental model of multiple sclerosis, the experimental autoimmune encephalomyelitis (EAE), astrocyte depletion may worsen or attenuate disease depending on whether depletion occurs immediately after EAE induction or during the chronic phase respectively (7, 8), indicating that GFAP positive cells may display protective or detrimental functions at distinct stages of disease.
Second, astrocytes regulate neuronal survival via release of and/or response to crucial mediators like neurotrophins, well known growth factors for neurons to which astrocytes may become sensitive under pathological conditions and react with the synthesis of neurotoxic nitric oxide (9, 10). Furthermore, astrocytes sense neuronal activity as their fine extensions take contact with pre- and post-synaptic neurons (forming the so-called tripartite synapse) and bear neurotransmitter receptors (11). They may modulate the concentration of glutamate, the main excitatory neurotransmitter in the brain, present in the synaptic cleft via specific transporters called glutamate/aspartate transporter (GLAST) and Astrocytic Glutamate Transporter 1 (GLT1) (11). In vitro and in vivo evidences indicate that neuroinflammation is characterized by alterations in expression of glutamate transporters and glutamate buffering (12, 13). The accumulation of glutamate in the extracellular space causes neuronal damage by excitotoxicity, a phenomenon observed in neurological and psychiatric disorders (reviewed in 14). Furthermore, astrocytes can provide metabolic precursors of glutamate back to the neurons through monocarboxylate transporters (MCT), thus satisfying neuronal metabolic needs and limiting novel neurotransmitter synthesis (15, 16) Though scarce information is available about astrocyte metabolism in neuroinflammatory mouse models, it is known that respiration-deficient astrocytes may survive as glycolytic cells in vivo in the absence of tissue inflammation and damage and that inflammatory cytokines increase glycolytic rates of astrocytes in vitro (17, 18), suggesting sustained glycolytic proficiency of the astrocyte in neuroinflammation. Interestingly, disruption of MCT transporters in astrocytes in vivo causes amnesia, underlying a key role for astrocyte-neuron metabolic coupling in long term memory formation (19).
Third, neuronal activity strongly depends on continuous supply of oxygen and glucose through the cerebral blood flow (15). Astrocytes cover most of the cerebral vasculature and create neurovascular units which link synaptic activity to vessel tone, thus regulating microcirculation (15). Further, astrocytes are key constituents of the blood-brain barrier (BBB) and their interaction with endothelial cells regulates BBB development and function (20). On the one hand, astrocyte factors control formation of tight junctions, blood flow, microvascular permeability, cell matrix and angiogenesis; on the other hand, endothelial signals regulate astrocyte maturation and expression of receptor proteins and ion channels on the glial membrane (20). Thus, for example Aquaporin-4 (AQP4) expression at astrocytic endfeet in contact with the vasculature together with the inward rectifying K+ channel Kir 4.1 provides local control of water and ion homeostasis (20). Autoantibodies directed to AQP4 are at the basis of the pathogenesis of neuromyelitis optica (NMO), an inflammatory CNS disorder characterized by astrocyte loss, axonal damage and demyelination (21–23), while the occurrence of anti-Kir 4.1 antibodies in MS is controversial (24). Further, Kir 4.1 has been reported as downregulated in ALS and epilepsy (25, 26), while upregulated in animal models of depression (27). Notably, mice lacking astrocyte Kir 4.1 display ataxia and seizures and die prematurely (28) while animals overexpressing astrocytic Kir4.1 develop a depression-like phenotype (27).
Fourth, astrocytes can exert and control immune reactions in the CNS (29–31). Similarly, to microglia, they bear a repertoire of pattern recognition receptors (PRR), which allow recognition of genome, proteins, and glycolipids of microbial origin (aka pathogen-associated molecular patterns, PAMP) (29). PRR include toll-like receptors, scavenger receptors and complement factors and have been initially identified as tools of the innate immune system to fight infections (29). However, PRR also recognize danger signals, that are endogenous molecules released or activated during stress or damage under sterile conditions (and collectively called DAMP, damage-associated molecular patterns) (32). DAMP include molecules from the extracellular matrix (e.g. biglycan and fibrinogen), cytosol (e.g. S100 proteins and heat shock proteins), and nucleus (e.g. histones) (reviewed in 32) Activation of innate immune pathways has been demonstrated in infectious, autoimmune, neurodegenerative disorders (29, for review). PRR engagement on glia cells activates pro-inflammatory responses required to eliminate or, at least, to contain infectious agents or damage. Critical is the activation of the transcription factor NF-kB, which controls gene expression of inflammatory cytokines, chemokines, nitric oxide synthase, apoptosis regulators (6, 29). In fact, in vivo inhibition of NFkB signalling in astrocytes protects from spinal cord and brain injury, EAE and toxic demyelination (33–36).
While rare in physiology, under pathological settings reactive astrocytes may modulate adaptive immunity in the CNS in several ways. Reactive astrocytes may release chemokines, such as CXCL10 and CCL2, which attract T cells from the circulation into the CNS parenchyma (37, 38) and in vivo deletion of astrocyte CCL2 and CXCL10 protects from EAE (39, 40). Next, T cell-derived factors such as IFN γ stimulate the expression of MHC class II molecules on astrocyte membrane, so that these glia cells become efficient in presentation of CNS antigens (e.g. myelin proteins) to T cells (41, 42), thus potentially sustaining local autoreactive adaptive immune responses. On the other hand, IFN γ can be released also by regulatory T cells and IFN γ signalling has been shown to be protective in vivo in EAE (43–45). Astrocytes are also great producers of TGFbeta, a known immunosuppressive mediator, and blockade of TGFbeta synthesis in astrocytes enhances tissue pathology in stroke and infectious CNS models (46). Further, astrocytes have been shown to express CTLA4, CD39 and CD73 (47, 48), which limit T cell activation, and FasL and TRAIL which trigger T cell deletion (31, 49). Regarding the interaction with B lymphocytes, astrocytes may release CXCL12, which promotes B cell recruitment to the CNS (50) and BAFF, a mediator important for B cell development, survival and function (51). Overall, these observations indicate a key role for astrocytes in regulating adaptive immune reactions. Activated astrocytes support B cell survival and activation, in turn, activated B cells induce a better T cell proliferation (52).
Microglia in Health and Disease
Microglia, a set of small glial cells within the CNS, were first described by del Río Hortega (53). Several decades passed before the importance of microglial functions in the CNS were appreciated. In 1920s, del Río Hortega had provided histological evidence that these cells derive from the mesoderm and not the ectoderm; the source of all other neural cells (oligodendrocytes, neurons and astrocytes) (53–55). It is now accepted that these glial cells originate in the yolk sac during fetal development and emerge at an earlier stage than tissue macrophages (56–58). Under basal condition, microglia display a multitude of physiological effects in such cellular processes as neurogenesis, cerebral angiogenesis, synaptic pruning, and oligodendrogenesis during brain development in both rodents and primates (59–61). Indeed, microglia contribute to neurogenesis and olidendrogenesis during prenatal and neonatal period (59, 62, 63). They emerge concomitantly with newly-born neurons and heavily invade neurogenic niches such as the ventricular and subventricular zones. This spatiotemporal co-existence between microglia and newly-born neural cells (neurons and oligodendrocytes) indicate the potential role of microglia in the regulation of neurogenesis and oligodendrogenesis (62, 63). For example, a subgroup of microglia expressing CD11c play a major role in the initial phase of myelination in developing brain (61). Through their phagocytic activity, microglia contribute to the removal of cell debris of dying neural cells and create optimal environment for neuronal connectivity.
Microglia affect cell survival/death programs of neural cells and remodel synaptic connection between developing neurons by secreting a variety of pro- and anti-inflammatory cytokines (Tumor Necrosis Factor (TNFα), Interleukin 1β (IL-1β), IL-6 and IL-10) and growth factors such as insulin growth factor 1 (IGF1) and brain-derived neurotrophic factor (BDNF) (61, 64–67). Thus, any alteration to these developmental effects of microglial can have long-lasting impact on brain structure and function (65, 67–69).
In addition to their homeostatic effects, microglia play a major role in the immune response to a variety of insults including pathogens (viral, bacterial or parasitic), trauma, stroke, and neurodegenerative diseases (70). For their immune-related function, microglia are referred to as the immune-competent cells of the CNS. Under basal condition, microglia form a network of cells characterized with small perikarya and long thin processes. These processes dynamically “sniff” their environment for signs of tissue damage such as high extracellular concentrations of calcium ions or adenosine triphosphate (ATP) (71). A damage to neural tissues triggers a cascade of cellular events within microglia. These cells send long processes towards the site of damage and adopt morphological changes whereby their cell bodies become enlarged and adopt an amoeboid shape. Their processes become short and thick. The genetic program of activated microglia is shifted towards cell division and phagocytosis and is characterized by the synthesis and release of a myriad of inflammatory cytokines and trophic factors (71). These cellular and molecular responses appear to be beneficial during the acute phase of insult. However, a prolonged activation of microglia can become deleterious (72, 73). While microglial cellular response appears to be non-specific, many line of evidence suggest that these cells exert a strong pro-inflammatory response during the initial phase of insult followed later by a regulatory response that consist mainly in the production of anti-inflammatory cytokines (IL-4 and IL10) and trophic factors (73, 74). The secondary wave of anti-inflammatory cytokines and trophic factors contributes to the recovery process of CNS tissue from the injury.
Astrocytes and Microglia Cross-Talk
Cell-cell interactions control CNS physiology and pathology (6, 75–84). Astrocyte-microglia interactions, for example, play important roles in CNS development, health and disease (31, 85, 86). In 2012 Ben Barres and colleagues reported that LPS induces a neurotoxic phenotype in astrocytes (87). Follow up mechanistic studies stablished that LPS induces the production of IL-1α, TNF-α, and complement component 1q (C1q) by microglia, which act on astrocytes to induce neurotoxic activity mediated by a lipid and additional as-yet unidentified neurotoxic factor (79, 88). In addition, these microglia-induced neurotoxic astrocytes display decreased phagocytic activity, and the reduced expression of neurotrophic factors (79). Finally, the analysis of patient samples suggest that these neurotoxic astrocytes contribute to the pathology of multiple neurologic diseases, including Huntington’s disease, Alzheimer’s disease, and multiple sclerosis (MS), among others (79). Collectively, these findings opened new avenues about the microglial regulation of astrocyte responses, and its contribution to CNS pathology. For instance, microglia can likely produce both positive and negative regulators of astrocyte pathogenic responses (see 80). Several molecules have been found to be involved in astrocyte-microglia communication, and the control of these cell-cell interactions by the commensal flora in specific diseases such as MS (89–96). For instance, VEGF-B was identified as a microglial product that boosts disease-promoting astrocyte responses. The transcription factor aryl hydrocarbon receptor (AHR) in microglia boosts TGFα while repressing the production of VEGF-B. Furthermore, AHR can also be activated in the CNS by metabolites produced by the commensal gut flora (which as a result of their chemical structure cross the BBB) and induced by environmental chemicals (83, 97–99). These thus contribute to regulation of astrocyte-microglia communication and CNS pathology (see 100).
Astrocytes can control microglia responses (100). Although multiple mechanisms likely mediate the control of microglial responses by astrocytes, some candidate pathways have already been identified. For example, fate-mapping and other studies established that astrocytes produce GM-CSF (100), 101), a known regulator of microglial activation (84, 102, 103). Astrocytes have been shown to modulate microglial responses via the production of GM-CSF (8, 104). Similar observations have been made for IL-6 (105–109). The above-mentioned findings exemplify the important role astrocyte-microglia interactions in CNS physiology. The recently developed RABID-seq (Rabies Barcoding In Droplets) which uses a library of genetically barcoded rabies virus in combination with single-cell RNA-seq to study CNS cell-cell interactions in vivo (110), identifying interacting cells, the mechanisms involved, and the biologic consequences of those interactions, has helped to highlight an important role for microglial-astrocyte interactions mediated by EphrinB3 and EphB3 in the promotion of CNS pathology (110) by inducing proinflammatory gene expression in the CNS (111), potentially via the activation of MAPK and the NLRP3 inflammasome (112, 113). In addition, EphB3 signaling in astrocytes induces the production of D-serine (114), which promotes synaptic damage via NMDA receptors (115). We also found that EphB3 in astrocytes is activated by its membrane-bound ligand EphrinB3 expressed by microglia. Interestingly, EphrinB3 harbors an intracellular domain that can trigger specific signaling pathways. Indeed, reverse signaling via EphrinB3 boosts the expression of NF-kB-driven transcriptional programs in microglia that promote inflammation and neurodegeneration (see 104, 110).
Early-Life Immune Challenge
In Africa, vast populations are exposed to stressors across all age groups with early life exposures carrying the greatest neurological burdens. These early life challenges alter the developmental trajectory of the CNS and consequently result in neurodevelopmental disorders (116). Epidemiological studies have shown a correlation between early life immune challenge and brain related diseases such as schizophrenia (117, 118), autism spectrum disorder (119, 120) and attention deficit hyperactivity disorder (121). It was suggested that the emergence of these brain related diseases are linked to altered early life function of microglia, as these cells play a pivotal role in synaptic pruning, neuronal connectivity and removal of dying neurons during brain development (122). Furthermore, depletion of microglia during early life induces persistent changes in social behavior such as reduced anxiety-like behavior and impaired working memory (123, 124). These effects were absent when microglial activity was inhibited during adulthood (125).
Experimental studies have shown that early life exposure to pathogens such as bacteria or viruses alters brain development trajectory and consequently leads to persistent cognitive deficits and behavioral dysfunctions. Indeed prenatal or neonatal exposures to either viral mimetics (polyinosinic:polycytidylic acid: PolyI:C) or bacterial active ingredient (Lipopolysaccharide: LPS) reprograms the hypothalamic-pituitary adrenal axis and affects brain development and plasticity that lasts into adulthood (126–129). These long-lasting effects are not due to the pathogens per se, but are triggered by maternal immune response to these pathogens (130, 131). We and others have shown that maternal immune activation alters adult brain plasticity and cognitive functions via maternally borne mediators such as interleukin-6 (IL-6) (132–134) and transforming growth factor-β (TGF-β) (135).
In addition to pathogens, non-infectious agents such as stress (136–138) or exposure to air pollution (diesel exhaust particles) can also activate maternal immune system and consequently alters fetal brain development (139). Indeed, exposure to these non-infectious agents induces microgliosis in the fetal brain and leads to an enhanced reactivity of microglia later in life, which is accompanied with cognitive dysfunction such as learning and memory deficits (140).
Long-Lasting Impact of Maternal Infection in Africa
The major cause of deaths in sub-Saharan Africa is infectious diseases (69%). A significant percentage of these deaths is associated with infection during pregnancy because pregnancy is characterized by an immune tolerant state to prevent rejection of the fetus (141–143). A relatively large epidemiological study has shown that the frequency of maternal infection and its resulting complication was higher in African low-income countries (15 African countries), when compared to high-income non-African countries. Indeed, obstetric infection led to maternal mortality of about 10.7% in low-income countries when compared to that seen in high income countries (about 4.7%). These infections include urinary tract infections, chorioamnionitis and abortion related infections (144, 145). While pregnancy-associated maternal death had received much attention, data concerning the impact of maternal infection on the brain development of children born to surviving mothers is scarce. As discussed above, experimental evidence strongly suggest that maternal infection can alter the developmental trajectory of fetal brain mainly by microglia. Few epidemiological studies have focused on pediatric patients in Africa (Gambia, Nigeria). In a cohort of 128 children in Gambia, a sizable fraction of these pediatric patients showed brain related delay such as learning difficulties (55%) and speech disorder (42%) (146). A similar set of studies in Nigeria show that children showed signs of epilepsy (60%), intellectual disability (7.2%) (147) and cerebral palsy (16.2%) likely due to early life events such as birth asphyxia and infection (148). These correlative studies strongly suggest that the prevalence of adult behavioral dysfunction and cognitive deficits in this African population is due, at least in part, to early life exposure to infectious pathogens.
Despite the prevalence of maternal infection, and its potential role in the emergence of such diseases as schizophrenia and autism spectrum disorder, few epidemiological and clinical trials have addressed these developmental diseases in Africa (149, 150). A recent study has shown that schizophrenic patients from South Africa (Xhosa ethnic group) carry damaging mutations in genes involved in synaptic function, such as receptors for glutamate and γ-amino-butyric acid (GABA) as well as postsynaptic proteins, scaffold proteins, and cell adhesion molecules (151). These mutations are comparable in nature to those observed in schizophrenic patients in Sweden (152). The mutation of these genes has been associated with intellectual disability, schizophrenia and autism spectrum disorders (153). It appears that the prevalence of schizophrenia is related to early life challenges such as childhood trauma (154). Similarly, maternal malaria has been associated with altered placental-fetal barrier by macrophage inflammatory mediators and complement factors (C5a), which can lead to altered fetal brain development (155).
The long term consequences of maternal infection on fetal brain development and function in sub-Saharan Africa has received little attention despite the overwhelming prevalence of infection during perinatal period. There is a need for studies that focus on the mechanistic link between perinatal infections and adult brain plasticity and function in Africa. These studies should take into consideration that subsaharan African mothers frequently experience multiple infections (parasitic, viral, bacterial) throughout pregnancy, which could be compounded with such factors as stress and malnutrition (156).
Malnutrition and Neuroinflammation
Besides the lack or shortage of food, several sociocultural factors e.g. poverty, poor social infrastructure, food security, uncontrolled population explosion, land and crop degradation, and lack of access to health services, contribute to the rising levels of malnutrition in Africa (157, 158). Others factors include famine, limited knowledge about safe hygiene practices, pediatric environmental enteropathy (PEE), natural disasters as well as internal population displacements as a result of civil (religious or ethnic) unrest leading to children staying in unhygienic camps amongst others (159, 160). In particular, a strong link has been established between nutrition, inflammation and neurodevelopment from foetal life to adolescence on the continent (161). Malnutrition can be generally defined as the intake of insufficient, excess or disproportionate amount of energy and/or nutrients (162, 163). In all its manifestations, malnutrition presents as either (i) undernutrition, (ii) micronutrient imbalance, (iii) overnutrition and, (iv) diet-related non-communicable diseases (e.g. cardiovascular disease, stroke, diabetes etc.) (157, 163).
Scope of the Problem
The statistics of numbers and people group affected by malnutrition creates a wide scope of problems in Africa. As a major global public health burden, the greatest concern is among infants, children, adolescents and women (particularly pregnant women) representing the most vulnerable category at greater risk of malnutrition (164–166). Globally in 2014, about 462 million adults worldwide were underweight, while 1.9 billion were either overweight or obese. An epidemiological study performed in 2016 showed that approximately about 155 million children under the age of 5 years were suffering from stunting, whereas 41 million were overweight or obese. In 2020, 40% of 149 million (59.6 million) stunted children under 5, 27% of 45 million (12.2 million) estimated to be wasted, and 24% out of 38.9 million estimated to be overweight or obese were from Africa (167; 163). The complicating fact however is that while there is a global decline in malnutrition, Africa has continued to record an increase in all forms of malnutrition, and for the most part, cases of undernutrition (168). This trend remains a serious concern as one of the leading cause of early child morbidity and mortality (157, 164).
The most common form of malnutrition recorded in developing countries most especially in Africa is undernutrition. Key indicators of undernutrition are wasting (low weight-for-height), stunting (low height-for-age) and underweight (low weight-for-age) (169, 170). Children under the age of five are the most severely affected of these vulnerable groups, with an estimated 45% of deaths attributed to undernutrition in this age group, mostly in low- and middle-income countries (170–172). Malnutrition is also responsible for significant abnormalities in physical and mental development with undernourished children usually having cognitive performance deficits and serious learning challenges (167, 173).
The continuous exposure of children and vulnerable groups to infectious agents under poor sanitary and unhygienic environment is of particular interest and has been shown to permanently weaken the immune systems and also cause a chronic inflammation of the intestine referred to as pediatric environmental enteropathy (PEE) in children (174–179). This gut disorder is as a result of both structural and functional changes in the intestinal mucosa characterized by intestinal villi atrophy, malabsorption, disruption of the intestinal gut barrier and an increased permeability (180, 181). This then makes it easier for microbes to translocate through the altered intestinal barrier. Over 75% of children in developing countries have been reported to affected by PEE (182).
Maternal Malnutrition
Gressens et al. (183) noted a reduction on cortical astrogenesis in mice pups fed with low protein diet during the first fourteen days of gestation. Although the effect of malnutrition on the permeability of the blood-barrier (BBB) is not yet fully understood, alterations in astrocyte development might affect BBB formation (184, 185). Malnutrition during pregnancy causes a reduction on GFAP expression on rat hypothalamus (186) and hippocampus (187), and mice cerebellum (188). While several studies have reported a reduction on synaptic contacts after a period of malnutrition (189), recent data suggests that microglia dysfunction in their ability to respond to environmental stimuli during gestation and lactation affects synaptic plasticity via epigenetic regulatory mechanisms (67). In the adult brain, synaptic plasticity and basal neurotransmission has been found to be affected by certain soluble factors (e.g. BNDF) released by microglia (185).
In general, early‐life malnutrition in the form of overnutrition or undernutrition can have a lasting impact on astrocytes. Abbink et al. (190) posited that both overnutrition and undernutrition present with a very similar phenotype, specifically increased GFAP expression and glucose transporters. It is worthy of note that in the case of overnutrition, although energy levels remain high, a lack in nutrients might still occur. This could suggest that the observed changes are associated with alterations or shortages in circulating nutrients, changes in the metabolic profile, or just general energy imbalance, rather than it being a specific effect of either a lack or excess of energy. In a recent study conducted by Kogel et al. (191) on the effect of long-term semi-starvation on primary cortical rat astrocytes using an undernutrition model, authors provided morphological and genetic evidence for pro-inflammatory astrocyte subtype-induction suggesting that inflammatory processes are a relevant factor in undernutrition. This response is characterized by elevated pro-inflammatory cytokines and genes associated with starvation. Furthermore, a shift toward the pro-inflammatory A1-like phenotype and an altered morphology suggest an increased astrocytic reactivity.
Crosstalk Between Malnutrition, Maternal Immune Activation and Neuroinflammation
Maternal immune activation (MIA) occurs when the measured levels of inflammatory markers in the dam exceeds normal range (192). It is usually a result of triggering of the maternal immune system by either infectious or non-infectious (malnutrition in this context) stimuli (193). This often leads to the release of inflammatory cytokines and immunologic alterations, and their transmission via innate placental immune activation to the developing foetus leading to adverse phenotypes particularly in the central nervous system (193–195).
There are strong emerging data from both animal and human studies that malnutrition-induced MIA results in foetal brain programming and modifications of their immune and metabolic genes through inflammatory and epigenetic mechanisms during critical periods of CNS astrocytes, microglial and immune system development (190, 194–197). Indeed, malnutrition during in utero and early life, notably due to undernutrition in the mothers, can affect the children’s growth, metabolism, immune function, brain, and cognitive development (198–200). Interestingly, neuroinflammation has recently been revealed as one of the key underlying mechanism responsible for deleterious consequences of diet-induced MIA on offspring neurodevelopment.
Microglial priming has been proposed as a major consequence of MIA, representing a vital connexion in a causal chain that leads to the wide spectrum of neuronal dysfunctions and behavioural phenotypes observed in the juvenile, adult or aged offspring. (201). In a study conducted by Ozaki et al. (202), authors observed maternal immune activation in mid-pregnancy led to an increase in IL-6 expression in embryonic microglia, but did not cause any marked changes in their morphologies either at E18 or after birth. However, they observed a sustained alteration in the microglial process motility pattern and deficits in behaviour when MIA was induced earlier (at E12).
These observations further strengthen the notion of the existence of a connecting link between maternal immune activation during pregnancy, and neuroinflammation and neurodevelopment disorders in the offspring. A significantly programmed imbalance in the expression of inflammatory mediators such as interleukin 6 (IL-6), IL-1 α, IL-10, tumor necrosis factor-α (TNFα), C-reactive protein or the complement system has been insinuated to play a role (118, 131, 134, 203–205).
Together, malnutrition-induced MIA induces the release of damage-associated molecular patterns (DAMPs), which then activates Toll-like receptors on maternal innate immune system and placental cells to produce pro-inflammatory cytokines (206–208). Following this, placental innate immune activation occurs and by means of passive transport as well as active placental production, cytokines across the placenta barrier with resultant interaction and activation of transplacental metabolic, hypothalamic–pituitary–adrenal (stress) and neuroendocrine signaling pathways (209). This consequently leads to foetal microglial priming, activation and neuroinflammation in the developing brain and also, the induction of immunological memory on the foetal microglia and the peripheral immune cells (194, 197). The resultant outcome is the occurrence of a dynamic crosstalk between the CNS immune cells (microglia) and peripheral immune cells (monocytes) (210). Second “hits” or wave of stress after birth (for instance by malnutrition) usually results in exaggerated responses and chronic inflammation in both the brain and periphery, manifesting as lifelong neurobehavioural deficits and may perpetuate a continuous cycle (194, 201, 211; Figure 1).
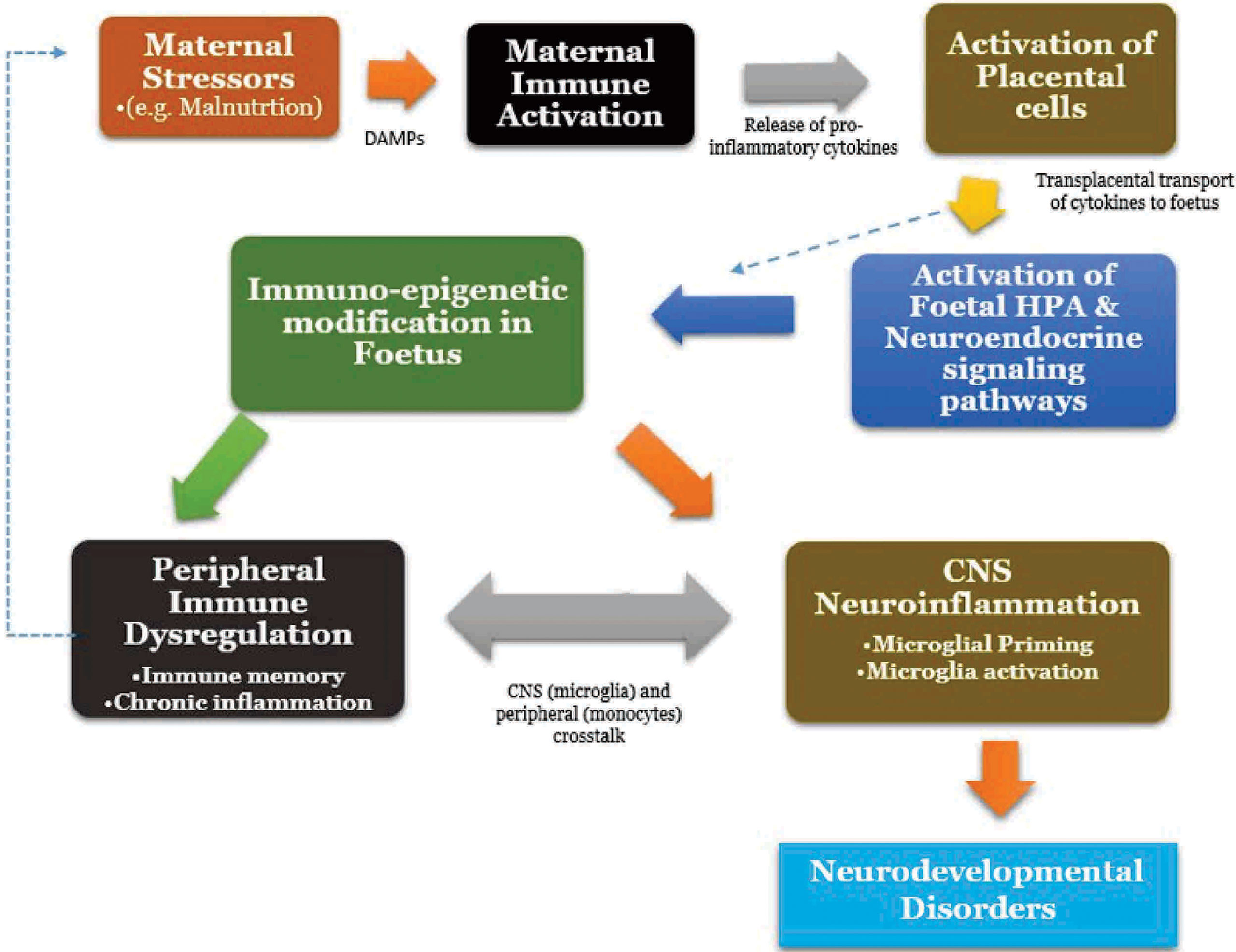
Figure 1 Malnutrition and Maternal immune activation, Neuroinflammation Crosstalk. HPA, Hypothalamic-Pituitary-Adrenal Axis.
The Vicious Tripartite Cycle of Malnutrition, Poverty and Neuroinflammation
The relationships between nutrition, inflammation and neurodevelopment has been noted to be reciprocal; this further supports the concept of the vicious cycle posed by malnutrition (161, 163, 196). Poverty amplifies the risk of, and risks from, malnutrition. People who are poor are more likely to be affected by different forms of malnutrition. Also, malnutrition increases health care costs, reduces productivity, and slows economic growth, which can perpetuate a cycle of poverty and ill-health (161, 212; Figure 2). This portends significant risk for the African population viz neuroinflammation.
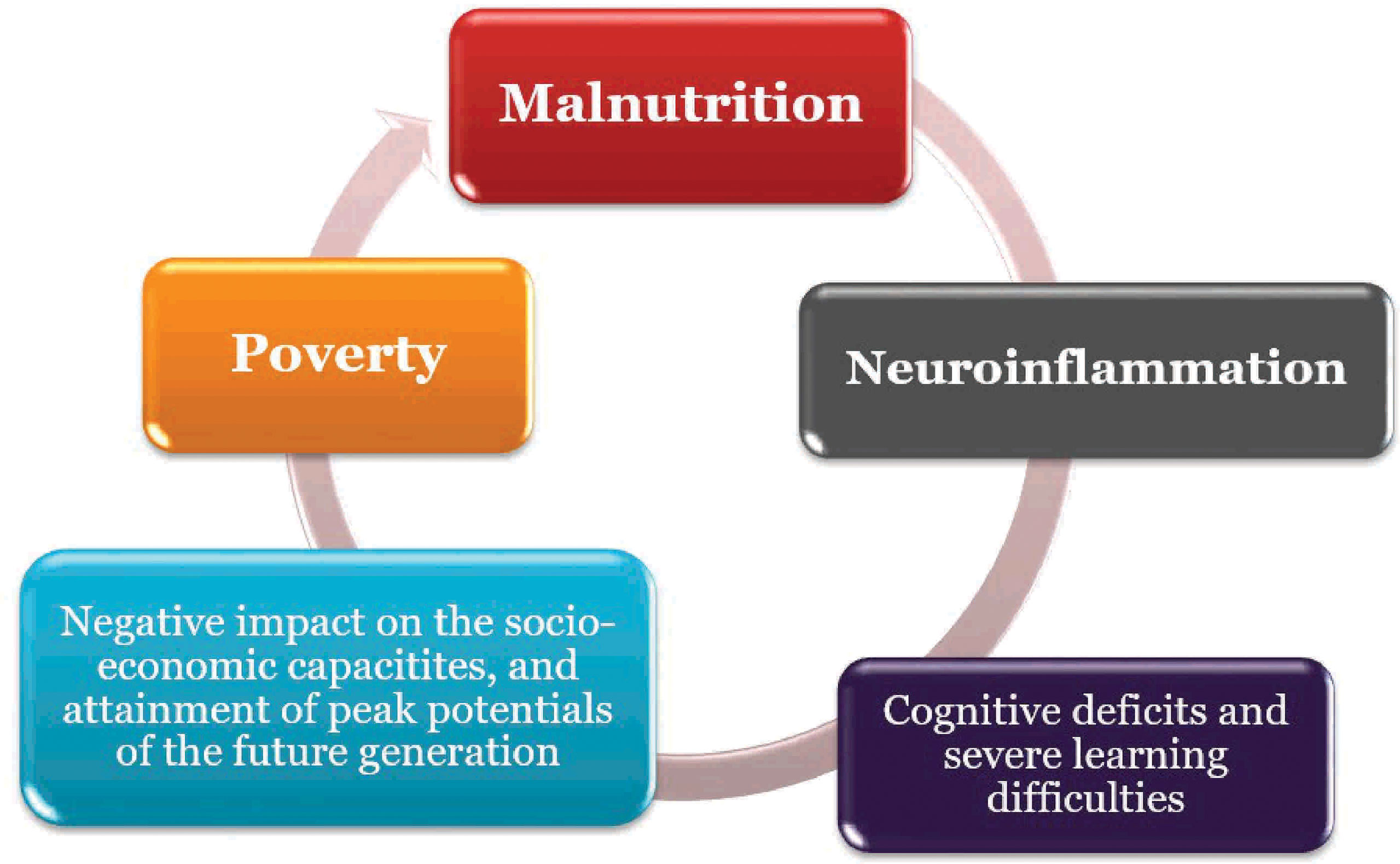
Figure 2 Vicious cycle of Malnutrition, Poverty and Neuroinflammation.
Pollution-Induced Neuroinflammation in Africa
Africa is home to major stressors of the CNS that are known to alter the microglia-astrocyte physiology. This section reviews existing knowledge on glia interaction in the face of pollutants (metals, pesticides and contaminated air. Other than infectious diseases which are not fully addressed in the review, these pollutants pose as predisposing factors to CNS disorders in Africa, owing to the immense exponential rise in use and impact of chemicals in health, economic growth and sustainability especially in sectors of agriculture, mining, education and several other industrial processes. This has come with grave complications on communities of both users and non-users when exposed to these pollutants with bio-accumulation in the soil, water and in the air (213, 214). In a twist of tales, Africa is neither a major producer nor a consumer of chemicals in global terms, but has the highest levels of pollution because of non-existent or poorly implemented government environmental laws and waste disposal policies, poorly regulated mining sector, fossil fuel burning and wrong agriculture practices in the pharmaceutical, beverage, and food industries (215–217 and 214). From the lead (Pb) and cadmium (Cd) rich electronic dumpsite of Agbobloshie in Ghana, vanadium (V) rich crude oil and gas flares in Nigeria’s Niger-Delta, illegal mining in Congo and several other African countries, communities release multiple neurotoxic factors daily with the young and women being most vulnerable (218). Since early life stress produces altered neurobehavioural deficits in adult life (128). Many of these stress factors directly or indirectly easily cross the placenta, and the blood–brain barrier (which is not fully developed in in humans until about 6 months post-partum) and may lead to congenital malformations and risk of neonatal neurotoxicity (219–221). These pollutants exist in the form of several sulfides, sulfates, hydroxides, phosphates, silicates oxides, and organic compounds (222, 223) and may also cause acute or chronic effects on the CNS in the general population (216, 224, 225).
Neuroinflammatory Mechanisms of Pollutants
Increasing evidence shows that astrocytes-microglia interplay may determine the phenotypic outcomes of the innate immune cells in disease conditions of the CNS. Glial activation can either aggravate tissue injury or promotes brain repair, most likely due to the nature of stress factors like the pollutant, dose and time course of exposure, and precise interplay of signals from the environment (226). Chemical pollutants include metals such as selenium, cadmium, arsenic, nickel, mercury, chromium, lead, zinc, and cobalt which are of paramount attention due to their potential role in toxicity when in trace amounts as well as other toxic pollutants such vanadium, tin oxide, copper etc. (227, 228). Other chemical pollutants include pesticides and air pollutants.
In metal pollution, microglia and astrocytes are known to express endogenous pattern recognition receptors (PRRs) in response to signals released by necrotic neurons or other pathologic products produced during disease including oxidized proteins and lipids (229), messenger ribonucleic acid (mRNA), fibronectin, hyaluronic acid, heat shock proteins, amyloid-beta, neuromelanin, and alpha-synuclein (230, 231). These are capable of responding to a variety of damage-associated molecular patterns (DAMPs) and in turn activate inflammation and neurodegeneration promoting molecular signaling events (232). The production of inflammatory mediators is further increased by activated glia, leading to a feed-forward cycle of inflammation and further release of neurotoxic mediators of tissue injury. The activated glia release diverse inflammatory factors including cytokines, chemokines, reactive oxygen species (ROS), and nitric oxide (NO) that are toxic to neurons (233). Cytokines such as tumor necrosis factor-alpha (TNFα) and interleukin-6 (IL-6) are often upregulated very quickly in activated glial cells and can directly amplify inflammation through recruitment of both innate and adaptive immune cells, leading to neuronal apoptosis (231).
When exposed to pollutants, microglia and astrocytes typically increase the production and release of inflammatory cytokines which enhance (ROS) generation, impede antioxidant activity, and result in neuronal injury or neuronal loss in the brain or other parts of the CNS (234). While the precise mechanisms are not yet fully understood, microglia have been shown as the first line of action and they respond in a dose-dependent fashion, while astrocytes are known to accumulate more toxic elements and express cytokines much later. Exposure to metals such Lead (Pb), Methyl mercury (MeHg), Vanadium (V), Tin Oxide (TO) results in gliosis by activating Toll-like receptor 4 (TLR4) -myeloid differentiation primary response 88 (MyD88) -nuclear factor (NF)-κB signaling cascade, increasing receptor phosphorylation and the activation of Mitogen-activated protein kinase (MAPK) cascades with subsequent initiation of signal transduction some of which are responsible for the production of pro-inflammatory cytokines (218, 234–236). This exposure is associated with upregulated activation of nuclear factor erythroid 2–related factor 2 (Nrf2) which acts against electrophiles and oxidants in the detoxification of ROS to maintain homeostasis (237). When exposed to ROS, Nrf2 acts by separating from the cytoplasmic repressor protein - Kelch-like ECH-associated protein 1 (Keap1), transferring to the nucleus, and activating the expression of antioxidant response elements (ARE)-dependent genes, including the phase II detoxifying/antioxidant enzyme HO-1 and NQO1 (238). The activation of the apoptotic caspase-3 pathway, which results in neuronal damage neurons is also suggested (239). This mechanism induces primary microglial toxicity and may be the pathological basis of metal pollution induced neurological dysfunction.
Mercury
MeHg pollution inhibits the astrocytic uptake of cysteine; an essential precursor for glutathione (GSH) synthesis. Implying that MeHg pollution induces neuronal oxidative damage (240). Part of the inhibitory mechanisms of MeHg include astrocytic glutamate uptake inhibition and glutamate efflux (241). This results in excessive glutamate in the synaptic cleft and, consequently leads to neuronal excitotoxicity. In vivo, mercury has been shown to induce microglial production and secretion of lysosomal proteases, leading to neuronal toxicity while astrocytes, when co-cultured with neurons, increase neuronal resistance to the damaging effect of MeHg (242, 243). Astrocytes and microglia therefore mediate protective effects against MeHg-induced neuronal toxicity. Microglia increase interleukin-6 (IL-6) production and release (244).
In organotins however, In-vitro studies have shown increased expression of IL-1β, tumor necrosis factor (TNF-α), IL-6, and nitric oxide synthase (iNOS) in the cultured astrocytes and microglia (245, 246).
Manganese (Mn)
Molecular mechanisms involved in Mn-induced neurotoxicity involve direct damage to the substantia nigra, globus pallidus, basal ganglia, striatum, and various other cellular components of the nervous system. Mn accumulates in the mitochondria of various cellular components in the brain, causing F0/F1 synthase and succinate dehydrogenase abnormality, leading to reduced ATP production (247). Diminishing ATP levels increase intracellular calcium levels and induce severe oxidative stress, forming ROS. Manganese was also shown to oxidize dopamine into reactive quinone species and disruption of antioxidant enzymes via binding to their thiol and hydroxyl groups (248). Glia activation has however been shown to occur in astrocytes and microglia in manganism (249) as it potentiates the effects of LPS and cytokines on activation of both microglia and astrocytes leading to increased production of TNFα, IL-1β, ROS, and NOS2 expression that can cause neuronal injury (250).
Manganese activates NF-κB and mitogen-activated protein kinase (MAPK) in microglia resulting in inflammatory gene expression and production of inflammatory mediators (251). The inflammatory effects are tightly regulated both at the level of IKK activation as well as by nuclear proteins that modulate transcriptional activity of inflammatory genes- NR4A1 (Nurr1) (252). Microglia then release neuroinflammatory mediators and pro-inflammatory cytokines, as well as reactive oxygen and nitrogen species (ROS and RNS), all of which can act on astrocytes to amplify inflammatory responses in the CNS (250).
In astrocytes, higher levels of accumulation of Mn occur than in neurons. This makes them target cells for transport of Mn into the brain as well as for initiating inflammatory signaling during neuronal stress and injury. Since astrocytes are a heterogeneous population of cells with different morphological and physiological characteristics depending on their location with the brain (253), they invariably serve as the major homeostatic regulator and storage site for Mn in the brain and a prominent contributor to Mn-stimulated nitric oxide (NO) production through NOS2 (254, 255). The regulation of astrocyte activation is under the control of many factors including cytokines IL-6, IFNγ, tumor necrosis factor-alpha (TNFα), toll-like receptor activators, neurotransmitters, ATP, reactive oxygen species, hypoxia, glucose deprivation, ammonia, and protein aggregates (256). Frequently, these activators are by-products of already injured neurons or factors released by activated microglia which indicate that astrocyte activation is often later in disease progression (257).
Cell culture models of glia cross talk in managanism indicate that removal of microglia or use of antioxidants has shown to reduce neuronal loss indicating microglial activation may serve as a critical step in mediating neuronal injury during Mn exposure and that microglia also likely directly promote activation of astrocytes that then amplify neuronal damage (258). However, astrogliosis is often more persistent than microgliosis and is believed to be important in amplifying inflammatory processes and thereby inducing greater damage (259).
Pesticides
Over 45% of neurotoxic chemicals are pesticides. Exposure to toxic doses of these chemicals activates the CNS immune system by reducing Nrf2 activation, activating the NF-kB pathway, or the opening of voltage gated calcium channels in neurons. These lead to increased oxidative stress, neuroinflammation, neuronal apoptosis, activation of p38MAPks, nucleotide-binding domain, leucine-rich repeat (NLR) family pyrin domain containing 3 (NLRP3) inflammasome, and reduced serotonin. Examples include the organophosphates which primarily cause accumulation of acetylcholine at cholinergic synapses, resulting in muscarinic and nicotinic receptor over-stimulation leading to oxidative stress, lipid peroxidation (260). Organophosphates can also alter the cyclic-AMP-protein kinase A signaling pathway of which affects the expression and function of several nuclear transcription factors such as c-fos, p53, AP-1, Sp1 and CREB (Ca2+/cAMP response element binding protein) involved in the switch from proliferation to differentiation of neural cells (260).
Dieldrin is an organochlorine extensively used as pesticides for corn, cotton, and citrus crops has been reported to induce severe alteration in the function of dopaminergic neurons and GABAA receptor (261) with evidence of significant oxidative stress, mitochondrial dysfunctions, and generation of pro-apoptotic proteins such as caspase-3 and Bcl-2 in the dopaminergic neurons (262). Endosulfan is an off-patent organochlorine insecticide and acaricide. It has been used globally as a pesticide since the 1950s to control a variety of insects including whiteflies, aphids, leafhoppers, Colorado potato beetles, and cabbage worms applied extensively to coffee, tea, and cotton crops, among others (263). It induces severe oxidative stress, induces the expression of pro-apoptotic proteins and inflammatory cytokines, and activation of glial cells (264). Pyrethroids are synthetic insecticides, which are used for the controlling insect pests in agriculture, public health, and animal health. They mediate prolongation of the kinetics of voltage-gated sodium channels, which are responsible for generation of the inward sodium current that produces the action potential in excitable cells leading to a hyperexcitable state, damage BBB and cause induction of severe endoplasmic reticulum stress, neuronal apoptosis, microglial activation, and neuroinflammation (265, 266).
Rotenone and pyridaben are two mitochondrial complex I inhibitors and are highly lipophilic. They easily cross BBB and produce ROS, Ca2+-mediated hyperexcitation, nuclear translocation of NF-kB, activation of p38 MAPKS, the formation of NLRP3 inflammasome, and mitochondrial dysfunctions (267–270).
Traffic Related Air Pollutants
TRAP exposure induces oxidative stress products, such as malondialdehydes (MDA), thiobarbituric acid reactive substances (TBARs) as well as ROS such as H2O2. (Nrf2), superoxide dismutase (SOD), glutathione (GSH), heme oxygenase 1 (HO-1), and catalase (CAT) are commonly elevated in the central nervous system, indicating need for detoxification. This induction activates glia response with astrocytic activation usually occurring either concomitantly with, or immediately after microglia stimulation, thus contributing to the release of oxidant species and pro-inflammatory cytokines (222, 271, 272).
Diesel Exhaust (DE): has been shown to induce oxidative stress, to activate microglia and to enhance levels of several pro-inflammatory cytokines (IL-1a, IL-1b, IL-3, IL-6, TNF-α) in the olfactory bulb and the hippocampus and microglia activation resulting in decreased adult neurogenesis in the hippocampal subgranular zone (SGZ) and the subventricular zone (SVZ) [Reviewed in (222, 273)].
Conclusion
In summary, multiple factors not limited to those discussed in this review may modulate neuroinflammation within the African context. These stressors assault the CNS through several cellular and molecular pathways to modulate neuroinflammatory responses that can be traced back to early development, with possible persistence into adult life and risk of mortality (274, 275). The most usually implicated pathways have oxidative stress, cerebral vascular damage, neurodegeneration and infiltrating systemic inflammation or nanoparticles as major route to damage. Microglia and astroglia respond to these stressors via multiple mechanisms that are still a subject of intense investigations. With the continent being home to over 1.3 billion people with myriads of stressors, the increasing burden of neurological disorders may be a ticking time bomb for neurological disorders (276). Therefore, further research in collaboration with Africans on epidemiological and mechanistic studies into the association of stressors and neuroinflammation will go a long way in understanding pathways that may be beneficial in treating or managing cases. Such studies will help to determine the neurological disease burden and to what extent these stressors contribute to neurological disease progression, co-morbidities with other neurodegenerative diseases and mortalities, by looking at the genetic and molecular adaptations and or vulnerabilities that exist in the African space compare to their Western cohorts.
Author Contributions
MO, AM, CF and FQ contributed to concept note development. MO wrote the abstract and conclusion, AM wrote section on microglia, CF wrote on astrocytes, FQ contributed the astrocyte and microglia cross talk section, OM wrote on malnutrition, MO and JO wrote on environmental pollutants, JO supervised all contributions of MO. OM worked on references with the assistance of MO. All authors contributed equally to manuscript proof reading and editing.
Funding
CF’s research is supported by Italian Ministry for Health (Ricerca Corrente and RF-2018-12367731), FISM (Fondazione Italiana Sclerosi Multipla, grant number 2016/R/14) and cofinanced with the 5 per mille public funding. AM was supported by research grants from Kuwait University, Research Sector (YM04/21 and YM11/17).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Freeman MR. Specification and Morphogenesis of Astrocytes. Science (2010) 330:774–8. doi: 10.1126/science.1190928
2. Allaman I, Fiumelli H, Magistretti PJ, Martin JL. Fluoxetine Regulates the Expression of Neurotrophic/Growth Factors and Glucose Metabolism in Astrocytes. Psychopharmacol (Berl) (2011) 216(1):75–84. doi: BF02740665/s00213-011-2190-y
3. Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat Neurosci (2021) 24:312–25. doi: 10.1038/s41593-020-00783-4
4. D’Ambrosi N, Apolloni S. Fibrotic Scar in Neurodegenerative Diseases. Front Immunol (2020) 11:1394. doi: 10.3389/fimmu.2020.01394
5. Koyama Y. Functional Alterations of Astrocytes in Mental Disorders: Pharmacological Significance as a Drug Target. Front Cell Neurosci (2015) 9:261. doi: 10.3389/fncel.2015.00261
6. Colombo E, Farina C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol (2016) 37:608–20. doi: 10.1016/j.it.2016.06.006
7. Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, et al. Reactive Astrocytes Form Scar-Like Perivascular Barriers to Leukocytes During Adaptive Immune Inflammation of the CNS. J Neurosci (2009) 29(37):11511–22. doi: 10.1523/JNEUROSCI.1514-09.2009
8. Mayo L, Trauger SA, Blain M, Nadeau M, Patel B, Alvarez JI, et al. Regulation of Astrocyte Activation by Glycolipids Drives Chronic CNS Inflammation. Nat Med (2014) 20:1147–56. doi: 10.1038/nm.3681
9. Colombo E, Cordiglieri C, Melli G, Newcombe J, Krumbholz M, Parada LF, et al. Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J Exp Med (2012) 209(3):521–35. doi: 10.1084/jem.20110698
10. Farina C. The role of neurotrophins in neuron-glia interaction. Book Chapter in Neuron-Glia Interaction in Neuroinflammation. In: Suzumura A, Ikenaka K, editors. Series Advances in Neurobiology, vol. 7. New York, NY: Springer: (2013). doi: BF02740665/978-1-4614-8313-7_7
11. Sancho L, Contreras M, Allen NJ. Glia as Sculptors of Synaptic Plasticity. Neurosci Res (2021) 167:17–29. doi: 10.1016/j.neures.2020.11.005
12. Ohgoh M, Hanada T, Smith T, Hashimoto T, Ueno M, Yamanishi Y, et al. Altered Expression of Glutamate Transporters in Experimental Autoimmune Encephalomyelitis. J Neuroimmunol (2002) 125(1-2):170–8. doi: 10.1016/s0165-5728(02)00029-2
13. Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL, et al. Altered Glutamate Reuptake in Relapsing-Remitting and Secondary Progressive Multiple Sclerosis Cortex: Correlation With Microglia Infiltration, Demyelination, and Neuronal and Synaptic Damage. J Neuropathol Exp Neurol (2007) 66(8):732–9. doi: 10.1097/nen.0b013e31812571b0
14. Olloquequi J, Cornejo-Córdova E, Verdaguer E, Soriano FX, Binvignat O, Auladell C, et al. Excitotoxicity in the Pathogenesis of Neurological and Psychiatric Disorders: Therapeutic Implications. J Psychopharmacol (2018) 32(3):265–75. doi: 10.1177/0269881118754680
15. Bélanger M, Allaman I, Magistretti PJ. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab (2011) 14(6):724–38. doi: 10.1016/j.cmet.2011.08.016
16. Magistretti PJ, Allaman IA. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron (2015) 86(4):883–901. doi: 10.1016/j.neuron.2015.03.035
17. Gavillet M, Allaman I, Magistretti PJ. Modulation of Astrocytic Metabolic Phenotype by Proinflammatory Cytokines. Glia (2008) 56(9):975–89. doi: 10.1002/glia.20671
18. Supplie LM, Düking T, Campbell G, Diaz F, Moraes CT, Götz M, et al. Respiration-Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J Neurosci (2017) 37(16):4231–42. doi: 10.1523/JNEUROSCI.0756-16.2017
19. Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, et al. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell (2011) 144(5):810–23. doi: 10.1016/j.cell.2011.02.018
20. Abbott N, Rönnbäck L, Hansson E. Astrocyte–Endothelial Interactions at the Blood–Brain Barrier. Nat Rev Neurosci (2006) 7:41–53. doi: 10.1038/nrn1824
21. Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, et al. Loss of Aquaporin 4 in Lesions of Neuromyelitis Optica: Distinction From Multiple Sclerosis. Brain (2007) 130(Pt 5):1224–34. doi: 10.1093/brain/awm047
22. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis Optica: Pathogenicity of Patient Immunoglobulin In Vivo. Ann Neurol (2009) 66(5):630–43. doi: 10.1002/ana.21837
23. Herwerth M, Kalluri SR, Srivastava R, Kleele T, Kenet S, Illes Z, et al. In Vivo Imaging Reveals Rapid Astrocyte Depletion and Axon Damage in a Model of Neuromyelitis Optica-Related Pathology. Ann Neurol (2016) 79(5):794–805. doi: 10.1002/ana.24630
24. Imamura M, Higuchi O, Maeda Y, Mukaino A, Ueda M, Matsuo H, et al. Anti-Kir4.1 Antibodies in Multiple Sclerosis: Specificity and Pathogenicity. Int J Mol Sci (2020) 21(24):9632. doi: 10.3390/ijms21249632
25. Zurolo E, de Groot M, Iyer A, Anink J, van Vliet EA, Heimans JJ, et al. Regulation of Kir4.1 Expression in Astrocytes and Astrocytic Tumors: A Role for Interleukin-1 β. J Neuroinflamm (2012) 9:280. doi: 10.1186/1742-2094-9-280
26. Kelley KW, Ben HL, Schirmer L, Tyzack GE, Tolman M, Miller JG, et al. Kir4.1-Dependent Astrocyte-Fast Motor Neuron Interactions Are Required for Peak Strength. Neuron (2018) 98(2):306–19.e7. doi: 10.1016/j.neuron.2018.03.010
27. Cui Y, Yang Y, Ni Z, Dong Y, Cai G, Foncelle A, et al. Astroglial Kir4.1 in the Lateral Habenula Drives Neuronal Bursts in Depression. Nature (2018) 554(7692):323–27. doi: 10.1038/nature25752
28. Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional Knock-Out of Kir4.1 Leads to Glial Membrane Depolarization, Inhibition of Potassium and Glutamate Uptake, and Enhanced Short-Term Synaptic Potentiation. J Neurosci (2007) 27(42):11354–65. doi: 10.1523/JNEUROSCI.0723-07.2007
29. Farina C, Aloisi F, Meinl E. Astrocytes are Active Players in Cerebral Innate Immunity. Trends Immunol (2007) 28(3):138–45. doi: 10.1016/j.it.2007.01.005
30. Cordiglieri C, Farina C. Astrocytes Exert and Control Immune Responses in the Brain. Curr Immunol Rev (Discontinued) (2010) 6(3):150–9. doi: 10.2174/157339510791823655
31. Sanmarco LM, Polonio CM, Wheeler MA, Quintana FJ. Functional Immune Cell-Astrocyte Interactions. J Exp Med (2021) 218(9):e20202715. doi: 10.1084/jem.20202715
32. Roh JS, Sohn DH. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw (2018) 18(4):e27. doi: 10.4110/in.2018
33. Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, et al. Inhibition of Astroglial Nuclear Factor kappaB Reduces Inflammation and Improves Functional Recovery After Spinal Cord Injury. J Exp Med (2005) 202(1):145–56. doi: 10.1084/jem.20041918
34. Brambilla R, Hurtado A, Persaud T, Esham K, Pearse DD, Oudega M, et al. Transgenic Inhibition of Astroglial NF-Kappa B Leads to Increased Axonal Sparing and Sprouting Following Spinal Cord Injury. J Neurochem (2009) 110:765–78. doi: 10.1111/j.1471-4159.2009.06190.x
35. Fuchtbauer L, Groth-Rasmussen M, Holm TH, Lobner M, Toft-Hansen H, Khorooshi R, et al. Angiotensin II Type 1 Receptor (AT1) Signaling in Astrocytes Regulates Synaptic Degeneration-Induced Leukocyte Entry to the Central Nervous System. Brain Behav Immun (2011) 25(5):897–904. doi: 10.1016/j.bbi.2010.09.015
36. Raasch J, Zeller N, van Loo G, Merkler D, Mildner A, Erny D, et al. IkappaB Kinase 2 Determines Oligodendrocyte Loss by Non-Cell-Autonomous Activation of NF-kappaB in the Central Nervous System. Brain (2011) 134(Pt 4):1184–98. doi: 10.1093/brain/awq359
37. Phares TW, Stohlman SA, Hinton DR, Bergmann CC. Astrocyte-Derived CXCL10 Drives Accumulation of Antibody-Secreting Cells in the Central Nervous System During Viral Encephalomyelitis. J Virol (2013) 87(6):3382–92. doi: 10.1128/JVI.03307-12
38. Cédile O, Wlodarczyk A, Owens T. CCL2 Recruits T Cells Into the Brain in a CCR2-Independent Manner. APMIS (2017) 125(11):945–56. doi: 10.1111/apm.12740
39. Paul D, Ge S, Lemire Y, Jellison ER, Serwanski DR, Ruddle NH, et al. Cell-Selective Knockout and 3D Confocal Image Analysis Reveals Separate Roles for Astrocyte- and Endothelial-Derived CCL2 in Neuroinflammation. J Neuroinflamm (2014) 11:10. doi: 10.1186/1742-2094-11-10
40. Ko EM, Ma JH, Guo F, Miers L, Lee E, Bannerman P, et al. Deletion of Astroglial CXCL10 Delays Clinical Onset But Does Not Affect Progressive Axon Loss in a Murine Autoimmune Multiple Sclerosis Model. J Neuroinflamm (2014) 11:105. doi: 10.1186/1742-2094-11-105
41. Soos JM, Morrow J, Ashley TA, Szente BE, Bikoff EK, Zamvil SS. Astrocytes Express Elements of the Class II Endocytic Pathway and Process Central Nervous System Autoantigen for Presentation to Encephalitogenic T Cells. J Immunol (1998) 161(11):5959–66. doi: 10.1016/S0165-5728(98)91399-6
42. Dong Y, Rohn WM, Benveniste EN. IFN-Gamma Regulation of the Type IV Class II Transactivator Promoter in Astrocytes. J Immunol (1999) 162(8):4731–9.
43. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic Control of Type 1 Regulatory T Cell Differentiation by AHR and HIF1-α. Nat Med (2015) 21(6):638–46. doi: 10.1038/nm.3868
44. Hindinger C, Bergmann CC, Hinton DR, Phares TW, Parra GI, Hussain S, et al. IFN-γ Signaling to Astrocytes Protects From Autoimmune Mediated Neurological Disability. PloS One (2012) 7(7):e42088. doi: 10.1371/journal.pone.0042088
45. Smith BC, Sinyuk M, Jenkins JE, Psenicka MW, Williams JL.. The impact of regional astrocyte interferon-γ signaling during chronic autoimmunity: a novel role for the immunoproteasome. J Neuroinflamm (2020) 17:184. doi: 10.1186/s12974-020-01861-x
46. Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic Transforming Growth Factor-Beta Signaling Reduces Subacute Neuroinflammation After Stroke in Mice. Glia (2014) 62(8):1227–40. doi: 10.1002/glia.22675
47. Filipello F, Pozzi D, Proietti M, Romagnani A, Mazzitelli S, Matteoli M, et al. Ectonucleotidase Activity and Immunosuppression in Astrocyte-CD4 T Cell Bidirectional Signaling. Oncotarget (2016) 7(5):5143–56. doi: 10.18632/oncotarget.6914
48. Gimsa U, ØRen A, Pandiyan P, Teichmann D, Bechmann I, Nitsch R, et al. Astrocytes Protect the CNS: Antigen Specific T Helper Cell Responses Are Inhibited by Astrocyte-Induced Upregulation of CTLA-4 (Cd152). J Mol Med (Berl) (2004) 82:364–72. doi: BF02740665/s00109-004-0531-6
49. Wang X, Fahad H, Saoussen K, Martina D, Dirk S. Astrocytic Fas Ligand Expression is Required to Induce T-Cell Apoptosis and Recovery From Experimental Autoimmune Encephalomyelitis. Eur J Immunol. (2013) 43:115–24. doi: 10.1002/eji.201242679
50. Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisäkk P, Ransohoff RM, et al. Chemokines in Multiple Sclerosis: CXCL12 and CXCL13 Up-Regulation Is Differentially Linked to CNS Immune Cell Recruitment. Brain (2006) 129(Pt 1):200–11. doi: 10.1093/brain/awh680
51. Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, et al. BAFF is Produced by Astrocytes and Upregulated in Multiple Sclerosis Lesions and Primary Central Nervous System Lymphoma. J Exp Med (2005) 201:195–200. doi: 10.1084/jem.20041674
52. Touil H, Kobert A, Lebeurrier N, Rieger A, Saikali P, Lambert C, et al. Human Central Nervous System Astrocytes Support Survival and Activation of B Cells: Implications for MS Pathogenesis. J Neuroinflamm (2018) 15(1):1–11. doi: 10.1186/s12974-018-1136-2
53. Del Río Hortega P. Estudios Sobre La Neuroglía. La Microglía Y Su Transformación En Células En Bastoncito Y Cuerpos Granuloadiposos. Trab Lab Invest Biol (1920) 18:37–82.
54. Del Río Hortega P. Lo Que Debe Entenderse Por "Tercer Elemento" De Los Centros Nerviosos. Bol Soc Esp Biol (1924) 11:33–5.
55. Del Río Hortega P. Tercera Aportación Al Conocimiento Morfológico E Interpretación Funcional De La Oligodendroglia. Mem Real Soc Esp Hist Nat (1928) 14:5–122.
56. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
57. Monier A, Evrard P, Gressens P, Verney C. Distribution and differentiation of microglia in the human encephalon during the first two trimesters of gestation. J Comp Neurol (2006) 499(4):565–82. doi: 10.1002/cne.21123
58. Verney C, Monier A, Fallet-Bianco C, Gressens P. Early Microglial Colonization of the Human Forebrain and Possible Involvement in Periventricular White-Matter Injury of Preterm Infants. J Anat (2010) 217(4):436–48. doi: 10.1111/j.1469-7580.2010.01245.x
59. Shigemoto-Mogami Y, Hoshikawa K, Goldman JE, Sekino Y, Sato K. Microglia Enhance Neurogenesis and Oligodendrogenesis in the Early Postnatal Subventricular Zone. J Neurosci (2014) 34:2231–43. doi: 10.1523/JNEUROSCI.1619-13.2014
60. Tay TL, Savage JC, Hui CW, Bisht K, Tremblay MÃ. Microglia Across the Lifespan: From Origin to Function in Brain Development, Plasticity and Cognition. J Physiol (2017) 595:1929–45. doi: 10.1113/JP272134
61. Wlodarczyk A, Holtman IR, Krueger M, Yogev N, Bruttger J, Khorooshi R, et al. A Novel Microglial Subset Plays a Key Role in Myelinogenesis in Developing Brain. EMBO J (2017) 36(22):3292–308. doi: 10.15252/embj.201696056
62. Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia Regulate the Number of Neural Precursor Cells in the Developing Cerebral Cortex. J Neurosci (2013) 33:4216–33. doi: 10.1523/JNEUROSCI.3441-12.2013
63. Tong CK, Vidyadaran S. Role of Microglia in Embryonic Neurogenesis. Exp Biol Med (Maywood) (2016) 241(15):1669–75. doi: 10.1177/1535370216664430
64. Hagemeyer N, Hanft KM, Akriditou MA, Unger N, Park ES, Stanley ER, et al. Microglia Contribute to Normal Myelinogenesis and to Oligodendrocyte Progenitor Maintenance During Adulthood. Acta Neuropathol (2017) 134(3):441–58. doi: BF02740665/s00401-017-1747-1
65. Lenz KM, Nugent BM, Haliyur R, McCarthy MM. Microglia Are Essential to Masculinization of Brain and Behavior. J Neurosci (2013) 33(7):2761–72. doi: 10.1523/JNEUROSCI.1268-12.2013
66. Lim SH, Park EE, You B, Jung Y, Park AR, Park SG, et al. Neuronal Synapse Formation Induced by Microglia and Interleukin 10. PLoS One (2013) 8(11):e81218. doi: 10.1371/journal.pone.0081218
67. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. Microglia Promote Learning-Dependent Synapse Formation Through Brain-Derived Neurotrophic Factor. Cell (2013) 155(7):1596–609. doi: 10.1016/j.cell.2013.11.030
68. Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E. Deficiency of the Microglial Receptor CX3CR1 Impairs Postnatal Functional Development of Thalamocortical Synapses in the Barrel Cortex. J Neurosci (2012) 32(43):15106–11. doi: 10.1523/JNEUROSCI.1167-12.2012
69. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia Contact Induces Synapse Formation in Developing Somatosensory Cortex. Nat Commun (2016) 7:12540. doi: 10.1038/ncomms12540
70. Bachiller S, Jimenez-Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, et al. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front Cell Neurosci (2018) 12:488. doi: 10.3389/fncel.2018.00488
71. Prinz M, Priller J. Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease. Nat Rev Neurosci (2014) 15:300–12. doi: 10.1038/nrn3722
72. Lloyd AF, Miron VE. The Pro-Remyelination Properties of Microglia in the Central Nervous System. Nat Rev Neurol (2019) 15:447–58. doi: 10.1038/s41582-019-0184-2
73. Loane DJ, Kumar A. Microglia in the TBI Brain: The Good, the Bad, and the Dysregulated. Exp Neurol (2016) 275 Pt3(03):316–27. doi: 10.1016/j.expneurol.2015.08.018
74. Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 Microglia: The Good, the Bad, and the Inflamed. J Neuroinflamm (2014) 11:98. doi: 10.1186/1742-2094-11-98
75. Sofroniew MV. Molecular Dissection of Reactive Astrogliosis and Glial Scar Formation. Trends Neurosci (2009) 32:638–47. doi: 10.1016/j.tins.2009.08.002
76. Glass CK, Saijo K. Nuclear Receptor Transrepression Pathways That Regulate Inflammation in Macrophages and T Cells. Nat Rev Immunol (2010) 10:365–76. doi: 10.1038/nri2748
77. Goldmann T, Prinz M. Role of Microglia in CNS Autoimmunity. Clin Dev Immunol (2013) 2013:208093. doi: 10.1155/2013/208093
78. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique Microglia Type Associated With Restricting Development of Alzheimer’s Disease. Cell (2017) 169:1276–90.e1217. doi: 10.1016/j.cell.2017.05.018
79. Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity (2017) 46:957–67. doi: 10.1016/j.immuni.2017.06.006
80. Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao CC, Ardura-Fabregat A, et al. Microglial Control of Astrocytes in Response to Microbial Metabolites. Nature (2018) 557(7707):724–8. doi: 10.1038/s41586-018-0119-x
81. Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-Derived Interleukin-33 Promotes Microglial Synapse Engulfment and Neural Circuit Development. Science (2018) 359(6831):1269–73. doi: 10.1126/science.aal3589
82. Prinz M, Jung S, Priller J. Microglia Biology: One Century of Evolving Concepts. Cell (2019) 179:292–311. doi: 10.1016/j.cell.2019.08.053
83. Rothhammer V, Quintana FJ. The Aryl Hydrocarbon Receptor: An Environmental Sensor Integrating Immune Responses in Health and Disease. Nat Rev Immunol (2019) 19(3):184–97. doi: 10.1038/s41577-019-0125-8
84. Wheeler MA, Clark IC, Tjon EC, Li Z, Zandee SEJ, Couturier CP, et al. MAFG-Driven Astrocytes Promote CNS Inflammation. Nature (2020) 578(7796):593–99. doi: 10.1038/s41586-020-1999-0
85. Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte Crosstalk in CNS Inflammation. Neuron (2020) 108:608–22. doi: 10.1016/j.neuron.2020.08.012
86. Prinz M, Masuda T, Wheeler MA, Quintana FJ. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu Rev Immunol (2021) 39:251–77. doi: 10.1146/annurev-immunol-093019-110159
87. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic Analysis of Reactive Astrogliosis. J Neurosci (2012) 32:6391–410. doi: 10.1523/jneurosci.6221-11.2012
88. Guttenplan KA, Weigel MK, Prakash P, Wijewardhane PR, Hasel P, Rufen-Blanchette U, et al. Neurotoxic Reactive Astrocytes Induce Cell Death via Saturated Lipids. Nature (2021) 599:102–7. doi: 10.1038/s41586-021-03960-y
89. Cirac A, Tsaktanis T, Beyer T, Linnerbauer M, Andlauer T, Grummel V, et al. The Aryl Hydrocarbon Receptor-Dependent TGF-α/VEGF-B Ratio Correlates With Disease Subtype and Prognosis in Multiple Sclerosis. Neurol Neuroimmunol Neuroinflamm (2021) 8(5):e1043. doi: 10.1212/NXI.0000000000001043
90. Tsaktanis T, Beyer T, Nirschl L, Linnerbauer M, Grummel V, Bussas M, et al. Aryl Hydrocarbon Receptor Plasma Agonist Activity Correlates With Disease Activity in Progressive Ms. Neurol Neuroimmunol Neuroinflamm (2020) 8(2):e933.. doi: 10.1212/NXI.0000000000000933
91. Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-Mediated Codelivery of Myelin Antigen and a Tolerogenic Small Molecule Suppresses Experimental Autoimmune Encephalomyelitis. Proc Natl Acad Sci USA (2012) 109:11270–275. doi: 10.1073/pnas.1120611109
92. Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, et al. AHR Activation Is Protective Against Colitis Driven by T Cells in Humanized Mice. Cell Rep (2016) 17:1318–29. doi: 10.1016/j.celrep.2016.09.082
93. Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, et al. Tolerogenic Nanoparticles Inhibit T Cell-Mediated Autoimmunity Through SOCS2. Sci Signal (2016) 9:ra61. doi: 10.1126/scisignal.aad0612
94. Kenison JE, Jhaveri A, Li Z, Khadse N, Tjon E, Tezza S, et al. Tolerogenic Nanoparticles Suppress Central Nervous System Inflammation. Proc Natl Acad Sci USA (2020) 117:32017–28. doi: 10.1073/pnas.2016451117
95. Barroso A, Mahler JV, Fonseca-Castro PH, Quintana FJ. The Aryl Hydrocarbon Receptor and the Gut-Brain Axis. Cell Mol Immunol (2021a) 18(2):259–68. doi: 10.1038/s41423-020-00585-5
96. Rothhammer V, Kenison JE, Li Z, Tjon E, Takenaka MC, Chao CC, et al. Aryl Hydrocarbon Receptor Activation in Astrocytes by Laquinimod Ameliorates Autoimmune Inflammation in the CNS. Neurol Neuroimmunol Neuroinflamm (2021) 8(2):e946. doi: 10.1212/nxi.0000000000000946
97. Quintana FJ, Sherr DH. Aryl Hydrocarbon Receptor Control of Adaptive Immunity. Pharmacol Rev (2013) 65:1148–61. doi: 10.1124/pr.113.007823
98. Gutiérrez-Vázquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity (2018) 48:19–33. doi: 10.1016/j.immuni.2017.12.012
99. Barroso A, Mahler JV, Fonseca-Castro PH, Quintana FJ. Therapeutic Induction of Tolerogenic Dendritic Cells via Aryl Hydrocarbon Receptor Signaling. Curr Opin Immunol (2021b) 70:33–9. doi: 10.1016/j.coi.2021.02.003
100. Wheeler MA, Jaronen M, Covacu R, Zandee SEJ, Scalisi G, Rothhammer V, et al. Environmental Control of Astrocyte Pathogenic Activities in CNS Inflammation. Cell (2019) 176(3):581–96.e518. doi: 10.1016/j.cell.2018.12.012
101. Komuczki J, Tuzlak S, Friebel E, Hartwig T, Spath S, Rosenstiel P, et al. Fate-Mapping of GM-CSF Expression Identifies a Discrete Subset of Inflammation-Driving T Helper Cells Regulated by Cytokines IL-23 and IL-1β. Immunity (2019) 50:1289–304.e1286. doi: 10.1016/j.immuni.2019.04.006
102. Hamilton JA. Colony-Stimulating Factors in Inflammation and Autoimmunity. Nat Rev Immunol (2008) 8:533–44. doi: 10.1038/nri2356
103. McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte Macrophage Colony-Stimulating Factor: A New Putative Therapeutic Target in Multiple Sclerosis. J Exp Med (2001) 194(7):873–82. doi: 10.1084/jem.194.7.873
104. Chao CC, Gutiérrez-Vázquez C, Rothhammer V, Mayo L, Wheeler MA, Tjon EC, et al. Metabolic Control of Astrocyte Pathogenic Activity via Cpla2-MAVS. Cell (2019) 179:1483–98. e1422. doi: 10.1016/j.cell.2019.11.016
105. Gruol DL, Nelson TE. Physiological and Pathological Roles of Interleukin-6 in the Central Nervous System. Mol Neurobiol (1997) 15:307–39. doi: 10.1007/BF02740665
106. Samoilova EB, Horton JL, Hilliard B, Liu TS, Chen Y. IL-6-Deficient Mice are Resistant to Experimental Autoimmune Encephalomyelitis: Roles of IL-6 in the Activation and Differentiation of Autoreactive T Cells. J Immunol (1998) 161:6480–86.
107. Atreya R, Mudter J, Finotto S, Müllberg J, Jostock T, Wirtz S, et al. Blockade of Interleukin 6 Trans Signaling Suppresses T-Cell Resistance Against Apoptosis in Chronic Intestinal Inflammation: Evidence in Crohn Disease and Experimental Colitis In Vivo. Nat Med (2000) 6:583–8. doi: 10.1038/75068
108. Savarin C, Hinton DR, Valentin-Torres A, Chen Z, Trapp BD, Bergmann CC, et al. Astrocyte Response to IFN-γ Limits IL-6-Mediated Microglia Activation and Progressive Autoimmune Encephalomyelitis. J Neuroinflamm (2015) 12:79. doi: 10.1186/s12974-015-0293-9
109. Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, et al. Trans-Presentation of IL-6 by Dendritic Cells Is Required for the Priming of Pathogenic T(H)17 Cells. Nat Immunol (2017) 18:74–85. doi: 10.1038/ni.3632
110. Clark IC, Gutiérrez-Vázquez C, Wheeler MA, Li Z, Rothhammer V, Linnerbauer M, et al. Barcoded Viral Tracing of Single-Cell Interactions in Central Nervous System Inflammation. Science (2021) 372:eabf1230. doi: 10.1126/science.abf1230
111. Mendiola AS, Ryu JK, Bardehle S, Meyer-Franke A, Ang KK, Wilson C, et al. Transcriptional Profiling and Therapeutic Targeting of Oxidative Stress in Neuroinflammation. Nat Immunol (2020) 21(5):513–24. doi: 10.1038/s41590-020-0654-0
112. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial Reactive Oxygen Species Promote Production of Proinflammatory Cytokines and Are Elevated in TNFR1-Associated Periodic Syndrome (TRAPS). J Exp Med (2011) 208(3):519–33. doi: 10.1084/jem.20102049
113. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-Inflammatory Effect of IL-10 Mediated by Metabolic Reprogramming of Macrophages. Science (2017) 356:513–9. doi: 10.1126/science.aal3535
114. Zhuang Z, Yang B, Theus MH, Sick JT, Bethea JR, Sick TJ, et al. EphrinBs Regulate D-Serine Synthesis and Release in Astrocytes. J Neurosci (2010) 30(47):16015–24. doi: 10.1523/JNEUROSCI.0481-10.2010
115. Perez EJ, Tapanes SA, Loris ZB, Balu DT, Sick TJ, Coyle JT, et al. Enhanced Astrocytic D-Serine Underlies Synaptic Damage After Traumatic Brain Injury. J Clin Invest (2017) 127(8):3114–25. doi: 10.1172/jci92300
116. Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, et al. Early Life Programming and Neurodevelopmental Disorders. Biol Psychiatry (2010) 68:314–9. doi: 10.1016/j.biopsych.2010.05.028
117. Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, et al. Serologic Evidence of Prenatal Influenza in the Etiology of Schizophrenia, Arch. Gen Psychiatry (2004) 61:774–80. doi: 10.1001/archpsyc.61.8.774
118. Canetta S, Sourander A, Surcel HM, Hinkka-Yli-Salomaki S, Leiviska J, Kellendonk C, et al. Elevated Maternal C-Reactive Protein and Increased Risk of Schizophrenia in a National Birth Cohort. Am J Psychiatry (2014) 171:960–68. doi: 10.1176/appi.ajp.2014.13121579
119. Brown AS, Sourander A, Hinkka-Yli-Salomäki S, McKeague IW, Sundvall J, Surcel HM. Elevated Maternal C-Reactive Protein and Autism in a National Birth Cohort. Mol Psychiatry (2014) 19(2):259–64. doi: 10.1038/mp.2012.197
120. Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX, et al. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav Immun (2016) 58:165–72. doi: 10.1016/j.bbi.2016.06.005
121. Instanes JT, Halmøy A, Engeland A, Haavik J, Furu K, Klungsøyr K. Attention-Deficit/Hyperactivity Disorder in Offspring of Mothers With Inflammatory and Immune System Diseases. Biol Psychiatry (2017) 81(5):452–9. doi: 10.1016/j.biopsych.2015.11.024
122. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science (2011) 333(6048):1456–8. doi: 10.1126/science.1202529
123. Nelson LH, Lenz KM. Microglia Depletion in Early Life Programs Persistent Changes in Social, Mood-Related, and Locomotor Behavior in Male and Female Rats. Behav Brain Res (2017) 316:279–93. doi: 10.1016/j.bbr.2016.09.006
124. VanRyzin JW, Yu SJ, Perez-Pouchoulen M, McCarthy MM. Temporary Depletion of Microglia During the Early Postnatal Period Induces Lasting Sex-Dependent and Sex-Independent Effects on Behavior in Rats. eNeuro (2016) 3(6):ENEURO.0297-16.2016. doi: 10.1523/ENEURO.0297-16.2016
125. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-Stimulating Factor 1 Receptor Signaling Is Necessary for Microglia Viability, Unmasking a Microglia Progenitor Cell in the Adult Brain. Neuron (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
126. Ellis S, Mouihate A, Pittman QJ. Neonatal Programming of the Rat Neuroimmune Response: Stimulus Specific Changes Elicited by Bacterial and Viral Mimetics. J Physiol (2006) 15:695–701. doi: 10.1113/jphysiol.2005.102939
127. Meyer U. Prenatal Poly(I:C) Exposure and Other Developmental Immune Activation Models in Rodent Systems. Biol Psychiatry (2014) 75(4):307–15. doi: 10.1016/j.biopsych.2013.07.011
128. Mouihate A, Galic MA, Ellis SL, Spencer SJ, Tsutsui S, Pittman QJ. Early Life Activation of Toll-Like Receptor 4 Reprograms Neural Anti-Inflammatory Pathways. J Neurosci (2010) 30(23):7975–83. doi: 10.1523/JNEUROSCI.6078-09.2010
129. Patterson PH. Maternal Infection: Window on Neuroimmune Interactions in Fetal Brain Development and Mental Illness. Curr Opin Neurobiol (2002) 12(1):115–8. doi: 10.1016/s0959-4388(02)00299-4
130. Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN. The Role of Cytokines in Mediating Effects of Prenatal Infection on the Fetus: Implications for Schizophrenia. Mol Psychiatry (2006) 11:47–55. doi: 10.1038/sj.mp.4001748
131. Boksa P. Effects of Prenatal Infection on Brain Development and Behavior: A Review of Findings From Animal Models. Brain Behav Immun (2010) 24(6):881–97. doi: 10.1016/j.bbi.2010.03.005
132. Mouihate A. Prenatal Activation of Toll-Like Receptor-4 Dampens Adult Hippocampal Neurogenesis in An IL-6 Dependent Manner. Front Cell Neurosci (2016) 10:173. doi: 10.3389/fncel.2016.00173
133. Mouihate A, Mehdawi H. Toll-Like Receptor 4-Mediated Immune Stress in Pregnant Rats Activates STAT3 in the Fetal Brain: Role of Interleukin-6. Pediatr Res (2016) 79(5):781–87. doi: 10.1038/pr.2015.86
134. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal Immune Activation Alters Fetal Brain Development Through Interleukin-6. J Neurosci (2007) 27(40):10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
135. Graciarena M, Roca V, Mathieu P, Depino AM, Pitossi FJ. Differential Vulnerability of Adult Neurogenesis by Adult and Prenatal Inflammation: Role of TGF-Beta1. Brain Behav Immun (2013) 34:17–28. doi: 10.1016/j.bbi.2013.05.007
136. Diz-Chaves Y, Pernia O, Carrero P, Garcia-Segura LM. Prenatal Stress Causes Alterations in the Morphology of Microglia and the Inflammatory Response of the Hippocampus of Adult Female Mice. J Neuroinflamm (2012) 9:71. doi: 10.1186/1742-2094-9-71
137. Diz-Chaves Y, Astiz M, Bellini MJ, Garcia-Segura LM. Prenatal Stress Increases the Expression of Proinflammatory Cytokines and Exacerbates the Inflammatory Response to LPS in the Hippocampal Formation of Adult Male Mice. Brain Behav Immun (2013) 28:196–206. doi: 10.1016/j.bbi.2012.11.013
138. Gómez-González B, Escobar A. Prenatal Stress Alters Microglial Development and Distribution in Postnatal Rat Brain. Acta Neuropathol (2010) 119:303–15. doi: 10.1007/s00401-009-0590-4
139. Bolton JL, Marinero S, Hassanzadeh T, Natesan D, Le D, Belliveau C, et al. Gestational Exposure to Air Pollution Alters Cortical Volume, Microglial Morphology, and Microglia-Neuron Interactions in a Sex-Specific Manner. Front Synaptic Neurosci (2017) 9:10. doi: 10.3389/fnsyn.2017.00010
140. Lenz KM, Nelson LH. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front Immunol (2018) 9:698. doi: 10.3389/fimmu.2018.00698
141. Kieffer TEC, Laskewitz A, Scherjon SA, Faas MM, Prins JR. Memory T Cells in Pregnancy. Front Immunol (2019) 10:625. doi: 10.3389/fimmu.2019.00625
142. Singer M. Development, Coinfection, and the Syndemics of Pregnancy in Sub-Saharan Africa. Infect Dis Poverty (2013) 2(1):26. doi: 10.1186/2049-9957-2-26
143. Tsuda S, Nakashima A, Shima T, Saito S. New Paradigm in the Role of Regulatory T Cells During Pregnancy. Front Immunol (2019) 10:573. doi: 10.3389/fimmu.2019.00573
144. Say L, Chou D, Gemmill A, Tunçalp Ö, Moller AB, Daniels J, et al. Global Causes of Maternal Death: A WHO Systematic Analysis. Lancet Glob Health (2014) 2(6):e323–33. doi: 10.1016/S2214-109X(14)70227-X
145. WHO Global Maternal Sepsis Study (GLOSS) Research Group. Frequency and Management of Maternal Infection in Health Facilities in 52 Countries (GLOSS): A 1-Week Inception Cohort Study. Lancet Glob Health (2020) 8:e661–71. doi: 10.1016/S2214-109X(20)30109-1
146. Burton KJ, Allen S. A Review of Neurological Disorders Presenting at a Paediatric Neurology Clinic and Response to Anticonvulsant Therapy in Gambian Children. Ann Trop Paediatr (2003) 23:139–43. doi: 10.1179/027249303235002215
147. Izuora GI, Iloeje SO. A Review of Neurological Disorders Seen at the Paediatric Neurology Clinic of the University of Nigeria Teaching Hospital, Enugu. Ann Trop Paediatr (1989) 9(4):185–90. doi: 10.1080/02724936.1989.11748629
148. Nottidge VA, Okogbo ME. Cerebral Palsy in Ibadan, Nigeria. Dev Med Child Neurol (1991) 33(3):241–45. doi: 10.1111/j.1469-8749.1991.tb05113.x
149. Franz L, Chambers N, von Isenburg M, de Vries PJ. Autism Spectrum Disorder in Sub-Saharan Africa: A Comprehensive Scoping Review. Autism Res (2017) 10:723–49. doi: 10.1002/aur.1766
150. Purgato M, Adams C, Barbui C. Schizophrenia Trials Conducted in African Countries: A Drop of Evidence in the Ocean of Morbidity? Int J Ment Health Syst (2012) 6(1):9. doi: 10.1186/1752-4458-6-9
151. Gulsuner S, Stein DJ, Susser ES, Sibeko G, Pretorius A, Walsh T, et al. Genetics of Schizophrenia in the South African Xhosa. Science (2020) 367:569–73. doi: 10.1126/science.aay8833
152. Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landén M, et al. Increased Burden of Ultra-Rare Protein-Altering Variants Among 4,877 Individuals With Schizophrenia. Nat Neurosci (2016) 19:1433–41. doi: 10.1038/nn.4402
153. Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, et al. SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron (2019) 103:217–34. doi: 10.1016/j.neuron.2019.05.002
154. Mall S, Platt JM, Temmingh H, Musenge E, Campbell M, Susser E, et al. The Relationship Between Childhood Trauma and Schizophrenia in the Genomics of Schizophrenia in the Xhosa People (SAX) Study in South Africa. Psychol Med (2020) 50:1570–77. doi: 10.1017/S0033291719001703
155. McDonald CR, Elphinstone RE, Kain KC. The Impact of Placental Malaria on Neurodevelopment of Exposed Infants: A Role for the Complement System? Trends Parasitol (2013) 29(5):213–9. doi: 10.1016/j.pt.2013.03.005
156. Nachega JB, Sam-Agudu NA, Budhram S, Taha TE, Vannevel V, Somapillay P, et al. Effect of SARS-CoV-2 Infection in Pregnancy on Maternal and Neonatal Outcomes in Africa: An AFREhealth Call for Evidence Through Multi-Country Research Collaboration. Am J Trop Med Hyg (2020) 104(2):461–5. doi: 10.4269/ajtmh.20-1553
157. Müller O, Krawinkel M. Malnutrition and Health in Developing Countries. Can Med Assoc J (2005) 173(3):279–86. doi: 10.1503/cmaj.050342
158. Tette EM, Sifah EK, Nartey ET. Factors Affecting Malnutrition in Children and the Uptake of Interventions to Prevent the Condition. BMC Pediatr (2015) 15:189. doi: 10.1186/s12887-015-0496-3
159. Thomas D, Frankenberg E. Health, Nutrition and Prosperity: A Microeconomic Perspective. Bull World Health Organ (2002) 80(2):106–13.
160. Sachs JD, McArthur JW. The Millennium Project: A Plan for Meeting the Millennium Development Goals. Lancet (2005) 365(9456):347–53. doi: 10.1016/S0140-6736(05)17791-5
161. Suchdev PS, Boivin MJ, Forsyth BW, Georgieff MK, Guerrant RL, Nelson CA 3rd. Assessment of Neurodevelopment, Nutrition, and Inflammation From Fetal Life to Adolescence in Low-Resource Settings. Pediatrics (2017) 139(Suppl 1):S23–37. doi: 10.1542/peds.2016-2828E
162. McKenna CG, Bartels SA, Pablo LA, Walker M. Women’s Decision-Making Power and Undernutrition in Their Children Under Age Five in the Democratic Republic of the Congo: A Cross-Sectional Study. PloS One (2019) 14:e0226041. doi: 10.1371/journal.pone.0226041
163. World Health Organization. Malnutrition (2021). Available at: https://www.who.int/news-room/fact-sheets/detail/malnutrition (Accessed September 25, 2021).
164. Galgamuwa LS, Iddawela D, Dharmaratne SD, Galgamuwa GLS. Nutritional Status and Correlated Socio-Economic Factors Among Preschool and School Children in Plantation Communities, Sri Lanka. BMC Public Health (2017) 17:377. doi: 10.1186/s12889-017-4311-y
165. Christian P, Smith ER. Adolescent Undernutrition: Global Burden, Physiology, and Nutritional Risks. Ann Nutr Metab (2018) 72:316–28. doi: 10.1159/000488865
166. Dukhi N. “Global Prevalence of Malnutrition: Evidence From Literature. In: Imran M, Imran A, editors. Malnutrition. London, UK: IntechOpen (2020). Available at: https://www.intechopen.com/chapters/71665. doi: 10.5772/intechopen.92006
167. Galler JR, Koethe JR, Yolken RH. Neurodevelopment: The Impact of Nutrition and Inflammation During Adolescence in Low-Resource Settings. Pediatrics (2017) 139:S72. doi: 10.1542/peds.2016-2828I
168. United Nations International Children’s Emergency Fund, UNICEF. Children, Food and Nutrition: Growing Well in a Changing World. The State of the World’s Children (2019). Available at: https://www.unicef.org/reports/state-of-worlds-children-2019 (Accessed September 25, 2021).
169. Akombi BJ, Agho KE, Hall JJ, Merom D, Astell-Burt T, Renzaho AM. Stunting and Severe Stunting Among Children Under-5 Years in Nigeria: A Multilevel Analysis. BMC Pediatr (2017) 17:15. doi: 10.1186/s12887-016-0770-z
170. Obasohan PE, Walters SJ, Jacques R, Khatab K. Risk Factors Associated With Malnutrition Among Children Under-Five Years in Sub-Saharan African Countries: A Scoping Review. Int J Environ Res Public Health (2020) 17:8782. doi: 10.3390/ijerph17238782
171. Simonyan H, Sargsyan A, Balalian AA, Davtyan K, Gupte HA. Short-Term Nutrition and Growth Indicators in 6-Month to 6-Year-Old Children are Improved Following Implementation of a Multidisciplinary Community-Based Programme in a Chronic Conflict Setting. Public Health Nutr (2020) 23:134–45. doi: 10.1017/S1368980019002969
172. World Health Organisation. Fact Sheets—Malnutrition (2020). Available at: https://www.who.int/news-room/fact-sheets/detail/malnutrition (Accessed May 31, 2020).
173. Institut Pasteur. Childhood Malnutrition (2020). Available at: https://www.pasteur.fr/en/institut-pasteur/institut-pasteur-throughout-world/international-research-programs/childhood-malnutrition (Accessed September 25, 2021).
174. Sullivan PB, Lunn PG, Northrop-Clewes CA, Crowe PT, Marsh MN, Neale G. Persistent Diarrhea and Malnutrition—The Impact of Treatment on Small Bowel Structure and Permeability. J Pediatr Gastroenterol Nutr (1992) 14:208–15. doi: 10.1097/00005176-199202000-00016
175. Walker SP, Wachs TD, Meeks Gardner J, Lozoff B, Wasserman GA, Pollitt E, et al. Child Development: Risk Factors for Adverse Outcomes in Developing Countries. Lancet (2007) 369:145–57. doi: 10.1016/S0140-6736(07)60076-2
176. Mondal D, Minak J, Alam M, Liu Y, Dai J, Korpe P, et al. Contribution of Enteric Infection, Altered Intestinal Barrier Function, and Maternal Malnutrition to Infant Malnutrition in Bangladesh. Clin Infect Dis (2012) 54(2):185–92. doi: 10.1093/cid/cir807
177. Korpe PS, Petri J, William A. Environmental Enteropathy: Critical Implication of a Poorly Understood Condition. Trends Mol Med (2012) 18:328–36. doi: 10.1016/j.molmed.2012.04.007
178. Prendergast A, Kelly P. Enteropathies in the Developing World: Neglected Effects on Global Health. Am J Trop Med Hyg (2012) 86:756–63. doi: 10.4269/ajtmh.2012.11-0743
179. Keusch GT, Denno DM, Black RE, Duggan C, Guerrant RL, Lavery JV, et al. Environmental Enteric Dysfunction: Pathogenesis, Diagnosis, and Clinical Consequences. Clin Infect Dis (2014) 59:S207–12. doi: 10.1093/cid/ciu485
180. Vonaesch P, Randremanana R, Gody JC, Collard JM, Giles-Vernick T, Doria M, et al. Identifying the Etiology and Pathophysiology Underlying Stunting and Environmental Enteropathy: Study Protocol of the AFRIBIOTA Project. BMC Pediatr (2018) 18(1):236. doi: 10.1186/s12887-018-1189-5
181. Gilmartin AA, Petri WA. Exploring the Role of Environmental Enteropathy in Malnutrition, Infant Development and Oral Vaccine Response. Phil Trans R Soc Lond B Biol Sci (2015) 370(1671):20140143. doi: 10.1098/rstb.2014.0143
182. George CM, Burrowes V, Perin J, Oldja L, Biswas S, Sack D, et al. Enteric Infections in Young Children Are Associated With Environmental Enteropathy and Impaired Growth. Trop Med Int Health (2018) 23(1):26–33. doi: 10.1111/tmi.13002
183. Gressens P, Muaku SM, Besse L, Nsegbe E, Gallego J, Delpech B, et al. Maternal Protein Restriction Early in Rat Pregnancy Alters Brain Development in the Progeny. Dev Brain Res (1997) 103(1):21–35. doi: 10.1016/s0165-3806(97)00109-0
184. Yusuf HK, Haque Z, Mozaffar Z. Effect of Malnutrition and Subsequent Rehabilitation on the Development of Mouse Brain Myelin. J Neurochem (1981) 36:924–30. doi: 10.1111/j.1471-4159.1981.tb01683.x
185. Chertoff M. Protein Malnutrition and Brain Development. Brain Disord Ther (2014) 4:168. doi: 10.4172/2168-975X.1000171
186. Plagemann A, Harder T, Rake A, Melchior K, Rohde W, Dörner G. Hypothalamic Nuclei Are Malformed in Weanling Offspring of Low Protein Malnourished Rat Dams. J Nutr (2000) 130:2582–89. doi: 10.1093/jn/130.10.2582
187. Cordero ME, Zvaighaft A, Muzzo S, Brunser O. Histological Maturation of Astroglial Cells in the Archicortex of Early Malnourished Rats. Pediatr Res (1982) 16(3):187–91. doi: 10.1203/00006450-198203000-00005
188. Ranade SC, Sarfaraz Nawaz M, Kumar Rambtla P, Rose AJ, Gressens P, Mani S. Early Protein Malnutrition Disrupts Cerebellar Development and Impairs Motor Coordination. Br J Nutr (2012) 107(8):1167–75. doi: 10.1017/S0007114511004119
189. Díaz-Cintra S, Cintra L, Ortega A, Kemper T, Morgane PJ. Effects of Protein Deprivation on Pyramidal Cells of the Visual Cortex in Rats of Three Age Groups. J Comp Neurol (1990) 292:117–26. doi: 10.1002/cne.902920108
190. Abbink MR, van Deijk AF, Heine VM, Verheijen MH, Korosi A. The Involvement of Astrocytes in Early-Life Adversity Induced Programming of the Brain. Glia (2019) 67(9):1637–53. doi: 10.1002/glia.23625
191. Kogel V, Trinh S, Gasterich N, Beyer C, Seitz J. Long-Term Glucose Starvation Induces Inflammatory Responses and Phenotype Switch in Primary Cortical Rat Astrocytes. J Mol Neurosci (2021) 71:2368–82. doi: 10.1007/s12031-021-01800-2
192. Boulanger-Bertolus J, Pancaro C, Mashour GA. Increasing Role of Maternal Immune Activation in Neurodevelopmental Disorders. Front Behav Neurosci (2018) 12:230. doi: 10.3389/fnbeh.2018.00230
193. Minakova E, Warner BB. Maternal Immune Activation, Central Nervous System Development and Behavioral Phenotypes. Birth Defects Res (2018) 110(20):1539–50. doi: 10.1002/bdr2.1416
194. Estes ML, McAllister AK. Maternal Immune Activation: Implications for Neuropsychiatric Disorders. Sci (New York N.Y.) (2016) 353(6301):772–7. doi: 10.1126/science.aag3194
195. Han VX, Patel S, Jones HF, Dale RC. Maternal Immune Activation and Neuroinflammation in Human Neurodevelopmental Disorders. Nat Rev Neurol (2021) 17:564–79. doi: 10.1038/s41582-021-00530-8
196. Bourke CD, Berkley JA, Prendergast AJ. Immune Dysfunction as a Cause and Consequence of Malnutrition. Trends Immunol (2016) 37(6):386–98. doi: 10.1016/j.it.2016.04.003
197. Cheray M, Joseph B. Epigenetics Control Microglia Plasticity. Front Cell Neurosci (2018) 12:243. doi: 10.3389/fncel.2018.00243
198. Ahmed T, Hossain M, Sanin KI. Global Burden of Maternal and Child Undernutrition and Micronutrient Deficiencies. Ann Nutr Metab (2012) 61:8–17. doi: 10.1159/000345165
199. Godfrey KM, Cutfield W, Chan S-Y, Baker PN, Chong Y-S. Nutritional Intervention Preconception and During Pregnancy to Maintain Healthy Glucose Metabolism and Offspring Health (“NiPPeR”): Study Protocol for a Randomised Controlled Trial. Trials (2017) 18:131. doi: 10.1186/s13063-017-1875-x
200. Chen C, Xu X, Yan Y. Estimated Global Overweight and Obesity Burden in Pregnant Women Based on Panel Data Model. PloS One (2018) 13:e0202183. doi: 10.1371/journal.pone.0202183
201. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, et al. Maternal Immune Activation and Abnormal Brain Development Across CNS Disorders. Nat Rev Neurol (2014) 10(11):643–60. doi: 10.1038/nrneurol.2014.187
202. Ozaki K, Kato D, Ikegami A, Hashimoto A, Sugio S, Guo Z, et al. Maternal Immune Activation Induces Sustained Changes in Fetal Microglia Motility. Sci Rep (2020) 10(1):21378. doi: 10.1038/s41598-020-78294-2
203. Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult Behavioral and Pharmacological Dysfunctions Following Disruption of the Fetal Brain Balance Between Pro-Inflammatory and IL-10-Mediated Anti-Inflammatory Signaling. Mol Psychiatry (2008) 13(2):208–21. doi: 10.1038/sj.mp.4002042
204. Girard S, Tremblay L, Lepage M, Sébire G. IL-1 Receptor Antagonist Protects Against Placental and Neurodevelopmental Defects Induced by Maternal Inflammation. J Immunol (2010) 184:3997–4005. doi: 10.4049/jimmunol.0903349
205. Coulthard LG, Hawksworth OA, Woodruff TM. Complement: The Emerging Architect of the Developing Brain. Trends Neurosci (2018) 41:373–84. doi: 10.1016/j.tins.2018.03.009
206. Akira S, Takeda K. Toll- Like Receptor Signalling. Nat Rev Immunol (2004) 4:499–511. doi: 10.1038/nri1391
207. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: Signal 0s That Spur Autophagy and Immunity. Immunol Rev (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
208. Afkham A, Eghbal-Fard S, Heydarlou H, Azizi R, Aghebati-Maleki L, Yousefi M. Toll- Like Receptors Signaling Network in Pre- Eclampsia: An Updated Review. J Cel Physiol (2019) 234:2229–40. doi: 10.1002/jcp.27189
209. Howerton CL, Bale TL. Prenatal Programing: At the Intersection of Maternal Stress and Immune Activation. Horm Behav (2012) 62:237–42. doi: 10.1016/j.yhbeh.2012.03.007
210. Wohleb ES, Delpech JC. Dynamic Cross- Talk Between Microglia and Peripheral Monocytes Underlies Stress-Induced Neuroinflammation and Behavioral Consequences. Prog Neuropsychopharmacol Biol Psychiatry (2017) 79:40–8. doi: 10.1016/j.pnpbp.2016.04.013
211. Martino D, Johnson I, Leckman JF. What Does Immunology Have to do With Normal Brain Development and the Pathophysiology Underlying Tourette Syndrome and Related Neuropsychiatric Disorders? Front Neurol (2020) 11:567407. doi: 10.3389/fneur.2020.567407
212. Vorster H. The Link Between Poverty and Malnutrition: A South African Perspective. Health SA Gesondheid (2010) 15(1):6. doi: 10.4102/hsag.v15i1.435
213. Yabe J, Ishizuka M, Umemura T. Current Levels of Heavy Metal Pollution in Africa. Vet Med Sci (2010) 72(10):1257–63. doi: 10.1292/jvms.10-0058
214. Bhargava V, Goldstein CD, Russell L, Xu L, Ahmed M, Li W, et al. GCNA Preserves Genome Integrity and Fertility Across Species. Dev Cell (2020) 52(1):38–52. doi: 10.1016/j.devcel.2019.11.007
215. World Health Organization. WHO Guidelines for Indoor Air Quality: Household Fuel Combustion (2014). Available at: http://apps.who.int/iris/bitstream10665/141496/1/9789241548885_eng.pdf (Accessed August 15, 2021).
216. Vargas R, Ponce-Canchihuamán J. Emerging Various Environmental Threats to Brain and Overview of Surveillance System With Zebrafish Model. Toxicol Rep (2017) 4:467–73. doi: 10.1016/j.toxrep.2017.08.002
217. Putra M, Gage M, Sharma S, Gardner C, Gasser G, Anantharam V, et al. Diapocynin, an NADPH Oxidase Inhibitor, Counteracts Diisopropylfluorophosphate-Induced Long-Term Neurotoxicity in the Rat Model. Ann N Y Acad Sci (2020) 1479(1):75–93. doi: 10.1111/nyas.14314
218. Fatola OI, Olaolorun FA, Olopade FE, Olopade JO. Trends in Vanadium Neurotoxicity. Brain Res Bull (2019) 145:75–80. doi: 10.1016/j.brainresbull.2018.03.010
219. Petronilho F, Goldman JL, Barichello T. Evans Blue-Albumin as a Marker to Evaluate Blood-Brain Barrier Integrity in Neonatal and Adult Rodents. In: Barichello T, editor. Blood-Brain Barrier. Neuromethods, vol. 142 . New York, NY: Humana Press (2019). p. 197–203. doi: 10.1007/978-1-4939-8946-1_12
220. Mortuza TB, Edwards GL, White CA, Patel V, Cummings BS, Bruckner JV. Age Dependency of Blood-Brain Barrier Penetration by Cis-and Trans-Permethrin in the Rat. Drug Metab Dispos (2019) 47(3):234–37. doi: 10.1124/dmd.118.084822
221. Aaseth J, Wallace D, Vejrup K, Alexander J. Methylmercury and Developmental Neurotoxicity: A Global Concern. Curr Opin Toxicol (2020) 19:80–7. doi: 10.1016/j.cotox.2020.01.005
222. Costa LG, Cole TB, Coburn J, Chang Y-C, Dao K, Roqué PJ. Neurotoxicity of Traffic-Related Air Pollution. Neurotoxicology (2017) 59:133–39. doi: 10.1016/j.neuro.2015.11.008
223. Mishra S, Bharagava RN, More N, Yadav A, Zainith S, Mani S, et al. “Heavy Metal Contamination: An Alarming Threat to Environment and Human Health,”. In: Sobti RC, Arora NK, Kothari R, editors. Environmental Biotechnology: For Sustainable Future. Singapore: Springer (2019). p. 103–25. doi: 10.1007/978-981-10-7284-0_5
224. Iqubal A, Ahmed M, Ahmad S, Sahoo CR, Iqubal MK, Haque SE. Environmental Neurotoxic Pollutants: Review. Environmental Science and Pollution Research (2020) 27:41175–98. doi: 10.1007/s11356-020-10539-z
225. Bertotto LB, Catron TR, Tal TJN. Exploring Interactions Between Xenobiotics, Microbiota, and Neurotoxicity in Zebrafish. NeuroToxicology (2020) 76:235–44. doi: 10.1016/j.neuro.2019.11.008
226. Matejuk A, Ransohoff RM. Crosstalk Between Astrocytes and Microglia: An Overview. Front Immunol (2020) 11:1416. doi: 10.3389/fimmu.2020.01416
227. Belyaeva EA, Sokolova TV, Emelyanova LV, Zakharova IO. Mitochondrial Electron Transport Chain in Heavy Metal-Induced Neurotoxicity: Effects of Cadmium, Mercury, and Copper. Sci World J (2012) 136063:14. doi: 10.1100/2012/136063
228. Ferraz da Silva I, Freitas-Lima LC, Graceli JB, Rodrigues LCM. Organotins in Neuronal Damage, Brain Function, and Behavior: A Short Review. Front Endocrinol (2018) 8:366. doi: 10.3389/fendo.2017.00366
229. Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger Receptors in Neurobiology and Neuropathology: Their Role on Microglia and Other Cells of the Nervous System. Glia (2002) 40:195–205. doi: 10.1002/glia.10148
230. Block ML, Hong JS. Microglia and Inflammation-Mediated Neurodegeneration: Multiple Triggers With a Common Mechanism. Prog Neurobiol (2005) 76:77–98. doi: 10.1016/j.pneurobio.2005.06.004
231. Gensel JC, Kigerl KA, Mandrekar-Colucci SS, Gaudet AD, Popovich PG. Achieving CNS Axon Regeneration by Manipulating Convergent Neuro-Immune Signaling. Cell Tissue Res (2012) 349:201–13. doi: 10.1007/s00441-012-1425-5
232. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms Underlying Inflammation in Neurodegeneration. Cell (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
233. Kim YS, Kim SS, Cho JJ, Choi DH, Hwang O, Shin DH, et al. Matrix Metalloproteinase-3: A Novel Signaling Proteinase From Apoptotic Neuronal Cells That Activates Microglia. J Neurosci (2005) 25:3701–11. doi: 10.1523/JNeurosci.4346-04.2005
234. Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz M, et al. Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell (2015) 160(1-2):177–90. doi: 10.1016/j.cell.2014.12.019
235. Chibowska K, Baranowska-Bosiacka I, Falkowska A, Gutowska I, Goschorska M, Chlubek D. Effect of Lead (Pb) on Inflammatory Processes in the Brain. Int J Mol Sci (2016) 17(12):2140. doi: 10.3390/ijms17122140
236. Peng Q, Deng Z, Pan H, Gu L, Liu O, Tang Z. Mitogen-Activated Protein Kinase Signaling Pathway in Oral Cancer. Oncol Lett (2018) 15(2):1379–88. doi: 10.3892/ol.2017.7491
237. Shokeir AA, Hussein AM, Barakat N, Abdelaziz A, Elgarba M, Awadalla A. Activation of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) and Nrf-2-Dependent Genes by Ischaemic Pre-Conditioning and Post-Conditioning: New Adaptive Endogenous Protective Responses Against Renal Ischaemia/Reperfusion Injury. Acta Physiol (Oxf) (2014) 210(2):342–53. doi: 10.1111/apha.12164
238. Baird L, Dinkova-Kostova AT. The Cytoprotective Role of the Keap1-Nrf2 Pathway. Arch Toxicol (2011) 85(4):241–72. doi: 10.1007/s00204-011-0674-5
239. Kumawat KL, Kaushik DK, Goswami P, Basu A. Acute Exposure to Lead Acetate Activates Microglia and Induces Subsequent Bystander Neuronal Death via Caspase-3 Activation. Neurotoxicology (2014) 41:143–53. doi: 10.1016/j.neuro.2014.02.002
240. Allen JW, Shanker G, Tan KH, Aschner M. The Consequences of Methylmercury Exposure on Interactive Functions Between Astrocytes and Neurons. Neurotoxicology (2002) 23:755–59. doi: 10.1016/S0161-813X(01)00076-6
241. Aschner M, Du YL, Gannon M, Kimelberg HK. Methylmercury-Induced Alterations in Excitatory Amino Acid Transport in Rat Primary Astrocyte Cultures. Brain Res (1993) 602(2):181–6. doi: 10.1016/0006-8993(93)90680-l
242. Morken TS, Sonnewald U, Aschner M, Tore Syversen T. Effects of Methylmercury on Primary Brain Cells in Mono- and Co-Culture. Toxicol Sci (2005) 87:169–75. doi: 10.1093/toxsci/kfi227
243. Sakamoto M, Miyamoto K, Wu Z, Nakanishi H. Possible Involvement of Cathepsin B Released by Microglia in Methylmercury-Induced Cerebellar Pathological Changes in the Adult Rat. Neurosci Lett. (2008) 442:292–96. doi: 10.1016/j.neulet.2008.07.019
244. Eskes C, Honegger P, Juillerat-Jeanneret L, Monnet-Tschudi F. Microglial Reaction Induced by Noncytotoxic Methylmercury Treatment Leads to Neuroprotection via Interactions With Astrocytes and IL-6 Release. Glia (2002) 37(1):43–52 doi: 10.1002/glia.10019
245. Ceccariglia S, Alvino A, Del Fà A, Parolini O, Michetti F, Gangitano C. Autophagy Is Activated In Vivo During Trimethyltin-Induced Apoptotic Neurodegeneration: A Study in the Rat Hippocampus. Int J Mol Sci (2020) 21(1):175. doi: 10.3390/ijms21010175
246. Vickers NJ. Animal Communication: When I’m Calling You, Will You Answer Too? Curr Biol (2017) 27(14):R713–5. doi: 10.1016/j.cub.2017.05.064
247. Gunter TE, Gerstner B, Lester T, Wojtovich AP, Malecki J, Swarts SG, et al. An Analysis of the Effects of Mn2+ on Oxidative Phosphorylation in Liver, Brain, and Heart Mitochondria Using State 3 Oxidation Rate Assays. Toxicol Appl Pharmacol (2010) 249(1):65–75. doi: 10.1016/j.taap.2010.08.018
248. Iqubal A, Iqubal MK, Sharma S, Ansari MA, Najmi AK, Ali SM, et al. Molecular Mechanism Involved in Cyclophosphamide-Induced Cardiotoxicity: Old Drug With a New Vision. Life Sci (2019) 218:112–31. doi: 10.1016/j.lfs.2018.12.018
249. Filipov NM, Seegal RF, Lawrence DA. Manganese Potentiates In Vitro Production of Proinflammatory Cytokines and Nitric Oxide by Microglia Through a Nuclear Factor Kappa B-Dependent Mechanism. Toxicol Sci (2005) 84:139–48. doi: 10.1093/toxsci/kfi055
250. Tjalkens RB, Popichak KA, Kirkley KA. Inflammatory Activation of Microglia and Astrocytes in Manganese Neurotoxicity. Adv Neurobiol (2017) 18:159–81. doi: 10.1007/978-3-319-60189-2_8
251. Filipov NM, Dodd CA. Role of Glial Cells in Manganese Neurotoxicity. J Appl Toxicol (2012) 32:310–7. doi: 10.1002/jat.1762
252. De Miranda BR, Popichak KA, Hammond SL, Jorgensen BA, Phillips AT, Safe S, et al. The Nurr1 Activator 1,1-Bis(3′-Indolyl)-1-(P-Chlorophenyl)methane Blocks Inflammatory Gene Expression in BV-2 Microglial Cells by Inhibiting Nuclear Factor Kappab. Mol Pharmacol (2015) 87:1021–34. doi: 10.1124/mol.114.095398
253. Olude MA, Mustapha O, Aderounmu T, Olopade JO, Ihunwo AO. Astrocyte Morphology, Heterogeneity and Density in the Developing African Giant Rat (Cricetomys Gambianus). Front Neuroanat (2015) 9:67. doi: 10.3389/fnana.2015.00067
254. Zhang P, Lokuta KM, Turner DE, Liu B. Synergistic Dopaminergic Neurotoxicity of Manganese and Lipopolysaccharide: Differential Involvement of Microglia and Astroglia. J Neurochem (2010) 112:434–43. doi: 10.1111/j.1471-4159.2009.06477.x
255. Aschner M, Aschner JL. Manganese Neurotoxicity: Cellular Effects and Blood-Brain Barrier Transport. Neurosci Biobehav Rev (1991) 15:333–40. doi: 10.1016/S0149-7634(05)80026-0
256. Sofroniew MV, Vinters HV. Astrocytes: Biology and Pathology. Acta Neuropathol (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
257. Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s Disease: A Target for Neuroprotection? Lancet Neurol (2009) 8:382–97. doi: 10.1016/S1474-4422(09)70062-6
258. Zhao F, Cai T, Liu M, Zheng G, Luo W, Chen J. Manganese Induces Dopaminergic Neurodegeneration via Microglial Activation in a Rat Model of Manganism. Toxicol Sci (2009) 107(1):156–64. doi: 10.1093/toxsci/kfn213
259. Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST Pathway in Microglia and Astrocytes Protects Dopaminergic Neurons From Inflammation-Induced Death. Cell (2009) 137(1):47–59
260. Costa LG. Current Issues in Organophosphate Toxicology. Clin Chim Acta (2006) 366:1–13. doi: 10.1016/j.cca.2005.10.008
261. Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin-Induced Oxidative Stress and Neurochemical Changes Contribute to Apoptotic Cell Death in Dopaminergic Cells. Free Radic Biol Med (2001) 31(11):1473–85. doi: 10.1016/S0891-5849(01)00726-2
262. Park E, Chun HS. Protective Effects of Quercetin on Dieldrin-Induced Endoplasmic Reticulum Stress and Apoptosis in Dopaminergic Neuronal Cells. Neuroreport (2016) 27:1140–46. doi: 10.1097/WNR.0000000000000667
263. Berntssen MHG, Maage A, Lundebye AK. Chemical Contamination of Finfish with Organic Pollutants and Metals. Chem Contam Residues Food (2017) 2;517–51. doi: 10.1016/B978-0-08-100674-0.00020-5
264. Chan MP, Morisawa S, Nakayama A, Kawamoto Y, Yoneda M. Development of an In Vitro Blood–Brain Barrier Model to Study the Effects of Endosulfan on the Permeability of Tight Junctions and a Comparative Study of the Cytotoxic Effects of Endosulfan on Rat and Human Glial and Neuronal Cell Cultures. Environ Toxicol (2006) 21:223–35. doi: 10.1002/tox.20175
265. Hossain MM, DiCicco-Bloom E, Richardson JR. Hippocampal ER Stress and Learning Deficits Following Repeated Pyrethroid Exposure. Toxicol Sci (2015) 143:220–8. doi: 10.1093/toxsci/kfu226
266. Soderlund DM. Molecular Mechanisms of Pyrethroid Insecticide Neurotoxicity: Recent Advances. Arch Toxicol (2012) 86(2):165–81. doi: 10.1007/s00204-011-0726-x
267. Charli A, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. Alterations in Mitochondrial Dynamics Induced by Tebufenpyrad and Pyridaben in a Dopaminergic Neuronal Cell Culture Model. Neurotoxicology (2016) 53:302–13. doi: 10.1016/j.neuro.2015.06.007
268. Mursaleen L, Somavarapu S, Zariwala MG. Deferoxamine and Curcumin Loaded Nanocarriers Protect Against Rotenone-Induced Neurotoxicity. J Parkinsons Dis (2020) 10(1):99–111. doi: 10.3233/JPD-191754
269. Zeng R, Zhou Q, Zhang W, Fu X, Wu Q, Lu Y, et al. Icariin-Mediated Activation of Autophagy Confers Protective Effect on Rotenone Induced Neurotoxicity In Vivo and In Vitro. Toxicol Rep (2019) 6:637–44. doi: 10.1016/j.toxrep.2019.06.014
270. Garabadu D, Agrawal N. Naringin Exhibits Neuroprotection Against Rotenone-Induced Neurotoxicity in Experimental Rodents. Neuro Mol Med (2020) 22(2):314–30. doi: 10.1007/s12017-019-08590-2
271. Patten KT, González EA, Valenzuela A, Berg E, Wallis C, Garbow JR, et al. Effects of Early Life Exposure to Traffic-Related Air Pollution on Brain Development in Juvenile Sprague-Dawley Rats. Transl Psychiatry (2020) 10(1);166. doi: 10.1038/s41398-020-0845-3
272. Block ML, Calderon-Garciduenas L. Air Pollution: Mechanisms of Neuroinflammation and CNS Disease. Trends Neurosci (2009) 32;506–16.
273. Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation Is Detrimental for Neurogenesis in Adult Brain. Proc Natl Acad Sci USA (2003) 100(23):13632–7. doi: 10.1073/pnas.2234031100
274. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic Inflammation in the Etiology of Disease Across the Life Span. Nat Med (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
275. Slavich GM. Understanding Inflammation, its Regulation, and Relevance for Health: A Top Scientific and Public Priority. Brain Behav Immun (2015) 45:13–4. doi: 10.1016/j.bbi.2014.10.012
276. Silberberg D, Katabira E. Neurological Disorders. In: Jamison DT, Feachem RG, Makgoba MW, Bos ER, Baingana FK, Hofman KJ, Rogo KO, editors. Disease and Mortality in Sub-Saharan Africa, 2nd ed. Washington (DC: The International Bank for Reconstruction and Development / The World Bank (2006). Chapter 23.
Keywords: astrocytes, microglia, reactive oxygen species, malnutrition, environmental pollution
Citation: Olude MA, Mouihate A, Mustapha OA, Farina C, Quintana FJ and Olopade JO (2022) Astrocytes and Microglia in Stress-Induced Neuroinflammation: The African Perspective. Front. Immunol. 13:795089. doi: 10.3389/fimmu.2022.795089
Received: 14 October 2021; Accepted: 13 April 2022;
Published: 30 May 2022.
Edited by:
Richard Idro, Makerere University, UgandaReviewed by:
Hanane Touil, Columbia University Irving Medical Center, United StatesHuanmin Yang, Heilongjiang Bayi Agricultural University, China
Copyright © 2022 Olude, Mouihate, Mustapha, Farina, Quintana and Olopade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew Ayokunle Olude, b2x1ZGVtYUBmdW5hYWIuZWR1Lm5n