 Xiaoxia Zhang1,2
Xiaoxia Zhang1,2 Yumin Li
Yumin Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 February 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.793610
This article is part of the Research Topic Post-Translational Modifications of Proteins in Cancer Immunity and Immunotherapy View all 7 articles
Ubiquitin conjugating enzyme E2 is an important component of the post-translational protein ubiquitination pathway, which mediates the transfer of activated ubiquitin to substrate proteins. UBE2L3, also called UBcH7, is one of many E2 ubiquitin conjugating enzymes that participate in the ubiquitination of many substrate proteins and regulate many signaling pathways, such as the NF-κB, GSK3β/p65, and DSB repair pathways. Studies on UBE2L3 have found that it has an abnormal expression in many diseases, mainly immune diseases, tumors and Parkinson’s disease. It can also promote the occurrence and development of these diseases. Resultantly, UBE2L3 may become an important target for some diseases. Herein, we review the structure of UBE2L3, and its mechanism in diseases, as well as diseases related to UBE2L3 and discuss the related challenges.
Ubiquitination is an important posttranslational modification that regulates many cellular processes, including protein turnover and the stress response, the cell cycle, organelle synthesis, and the intracellular homeostasis maintenance (1). Thus, abnormities in ubiquitination can lead to the development of many serious diseases-related processes, such as tumors, neurodegeneration, immune diseases, and susceptibility to infections (2–4).
Small molecules of ubiquitin arrive at the substrate proteins to complete ubiquitination through a complex three-step enzyme cascade (5). Using ATP, the E1 ubiquitin-activating enzymes activate ubiquitin and form the ubiquitin-E1 complex. Ubiquitin, which is activated by adenylate, is transferred to a E2 ubiquitin-conjugating enzyme via a thioester bond to form the E2-ubiquitin complex (6). The E3 ubiquitin ligase enzyme then promotes the ubiquitin transfer from E2 to the substrate lysine to complete the ubiquitination process (6) (Figure 1). This process forms the functional site of ubiquitination- the isopeptide bond formed between the C- terminal glycine (Gly) of ubiquitin and the lysine on the substrate protein (6–8). Deubiquitin enzymes (DUBs) process ubiquitin precursors, edit the chain topologies, or cut ubiquitin from the substrates to terminate the signal transduction (9, 10).
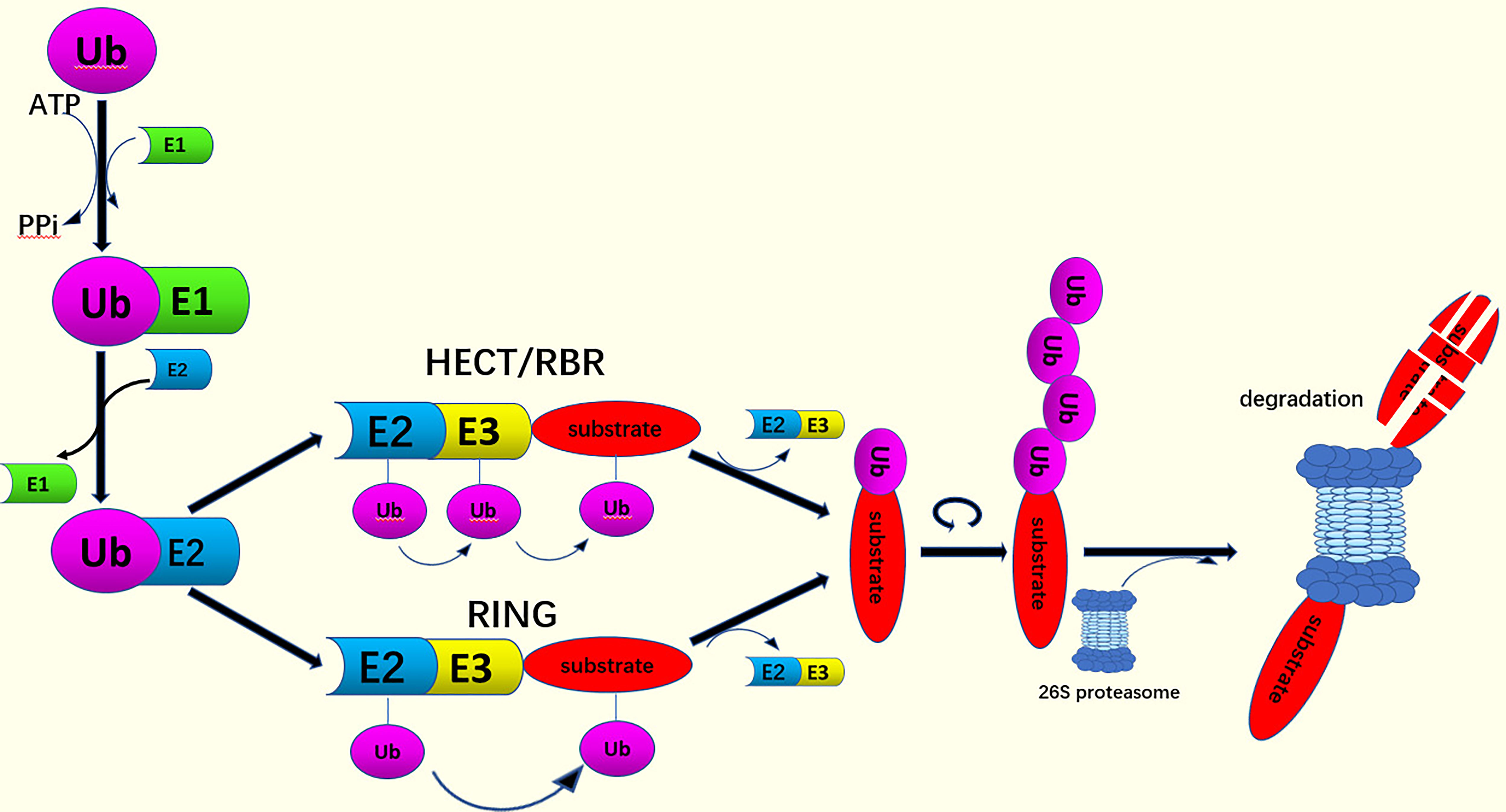
Figure 1 The process of ubiquitination. Ub is activated by E1 through an ATP-dependent step. The activated Ub attaches to the E2. The binding of Ub to target proteins can be catalyzed by HECT and RBR-type E3 ligases, or RING domain E3 ligases. This acts as a bridge between the activated E2 and the substrate indicating that the ubiquitin signals form polyubiquitin chains. Finally, the substrate is degraded by the 26S proteasome.
Ubiquitin is an 8.9 kDa protein with 76 amino acids that is, attached covalently to the lysine residues on the substrates during the ubiquitination process (11). Moreover, in the absence of lysine residues, the substrates can be ubiquitinated in a cysteine-dependent manner (12). Therefore ubiquitin has many potential post-translational modification sites (13). Ubiquitin has a compact structure and comprises a short 310-helix, a 3.5 turn of α-helix, and a β-sheet containing five strands and seven reverse circles (14). The β-sheet of the ubiquitin is a hydrophobic patch layer that includes Leu8, Ile44 and Val70 (15). The other structural centers of ubiquitin are located in Ile36, homologous to the C-terminal of E6-AP E3(HETC) and are recognized by the presence of the ubiquitin binding domain(UBD) and as well as the DUBs (16–18). Ubiquitin has eight ubiquitination sites in which seven lysine residues(K6、K11、K27、K29、K33、K48、K63) are used as receptor sites for the polyubiquitin chain formation enabling the attachment to different substrates to complete the ubiquitination process (19–21). When ubiquitin is connected to the N-terminus of the second ubiquitin, an eighth chain type, named Met1 or the “linear” chains, is created (21). Due to the complexity of ubiquitination, different Ub lysine linkages can form homotypic (linked by a single residue), heterotypic, or branched chains (22, 23). Proteomic studies have found that all possible ubiquitin chain types may coexist in the cells (19, 24–27). The results of the different ubiquitination processes in the substrate are diverse. Monubiquitination promotes the recognition of protein, the formation of complex, or allosteric regulation (21, 28, 29). While, Lys48-linked polyubiquitin chains, which are the predominant linkage type in the cells (19, 24), can target proteins for proteasome degradation (7). Conversely, Lys63-linked polyubiquitin chains, the second-most abundant chain type, perform non-biodegradable functions such as cell signaling, intracellular transport, protein kinase activation and DNA damage responses (29, 30). Furthermore, chains linked by other residues, such as K6, K27, K33, and linear ubiquitin chains, often perform various non-degradative functions, such as selective autophagy, DNA repair, and innate immunity (31). Different links among the ubiquitin molecules can form different categories of ubiquitin chains and generate different signals in the cells for the performance of various biological functions (32–35).
The E2 enzyme, which is different from E1 and E3 enzymes, plays a key transferase role in ubiquitination. UBE2L3, also called UBcH7, is one of the many E2 ubiquitin conjugating enzymes that participate in the ubiquitination of many substrate proteins. In the current study, we review the structure of UBE2L3 and its mechanism in diseases, as well as diseases associated with UBE2L3, and discuss the related challenges.
Deep proteomic studies on the protein copy number in vivo have shown that UBE2L3, with 153 amino acid residues encoded, is one of the richest E2s in mammalian cell lines (36). The current three-dimensional data of the protein have indicated that UBE2L3 is highly conservative (37). The UBE2L3 enzyme retains some of the structural characteristics of universal UBC folding and is composed of a UBC domain, with up to 35% being conserved between the different family members, to provide a platform for the combination of the E1s, E3s, and activated Ub/UBL (38). The catalytic crack in this UBE2L3 structure contains a histidine (His119) similar to the acid residue in the α3-α4 ring (D117 in UBE2D1), which is directed into the lysine of the substrate protein (24, 28, 29).
Normally, while most E2-Ub complexes transfer Ub to the RING E3 ubiquitin ligases, UBE2L3-Ub transfers Ub to HECTs because it lacks any inherent E3-independent lysine reactivity (39). Early biochemical and structural studies have also demonstrated that UBE2L3 plays a biological role in conjunction mainly with HECT E3 (40–42). A landmark study compared the E2-Ub reactivity curve independent of E3 and found that UBE2L3-Ub was sensitive only to the thiol (cysteine) receptors (43). Furthermore, the inherent capability of UBE2L3 was not affected by the RING E3s in the ubiquitin chain. Analysis of the UBE2L3 surface revealed the presence of hot-spot residues on UBE2L3, including Lys9 in the α1, and Glu93, Lys96, and Lys100 in the 310-α2 circle, as well as Phe63 in the β2-β3 circle, which contributes to the reaction of UBE2L3 with HECT E3s and the subsequent ubiquitination (44, 45). Since UBE2L3 is inherently catalytic, which means it can only interact with certain parts of E3, such as HECT’s E3 ligase and a special class of RBR E3 (43, 44). In Figure 2A, the structure of the E6AP Hect domain and its complex with UBE2L3 provides a preliminary view of the UBE2L3 and E2-E3 complex (46). Another study reported on the crystal structure of the RBR E3 (HHARI) and UBE2L3-Ub complexes, revealing the molecular basis of the specificity of the homologous E2/RBR E3 pairs (47), as shown in Figure 2B. UBE2L3 can also be associated with the disease-related E3 ligase LUBAC to form high-yield E2-E3 pairs in vitro (48). The LUBAC (Linear ubiquitin chain Assembly Complex), which belongs to the RBR E3 enzyme, binds to UBE2L3 to form a specific linear ubiquitin chain linked by MET1 in vitro. LUBAC is composed of HOIL-1L-interacting protein (HOIP), Sharpin (SHANK-associated RH domain interaction protein in postsynaptic density) and heme-oxidized IRP2 ubiquitin ligase-1 (HOIL-1L), of which HOIP is identified as the key catalytic site of LUBAC (49). In recent years, emerging evidence has confirmed that LUBAC is a targeted part of the typical NF-κB signaling pathway that is critical for inflammation and immune development (50, 51). When stimulated by pro-inflammatory signals, the key substrates of LUBAC, IKK (NEMO), RIPK1, RIPK2, IRAKs, MyD88 and ASC (50, 52, 53) attach to the linear Ub chains through LUBAC (54). The loss of LUBAC results in impaired biological functions, including the attenuation of NF-KB and mitogen-activated protein kinase (MAPK)-mediated signaling pathways and increased cell death (54). In summary, UBE2L3 reacts mainly with HECT or RBR (RING-in-between-RING) E3 ligase on the ubiquitin chain. The E3 ubiquitin ligases linked to UBE2L3 and their corresponding functions are summarized in Table 1 (40, 46, 48, 55–72). In the current study, we review the structure of UBE2L3, and its mechanism in diseases, as well as diseases related to UBE2L3 and discuss the related challenges.
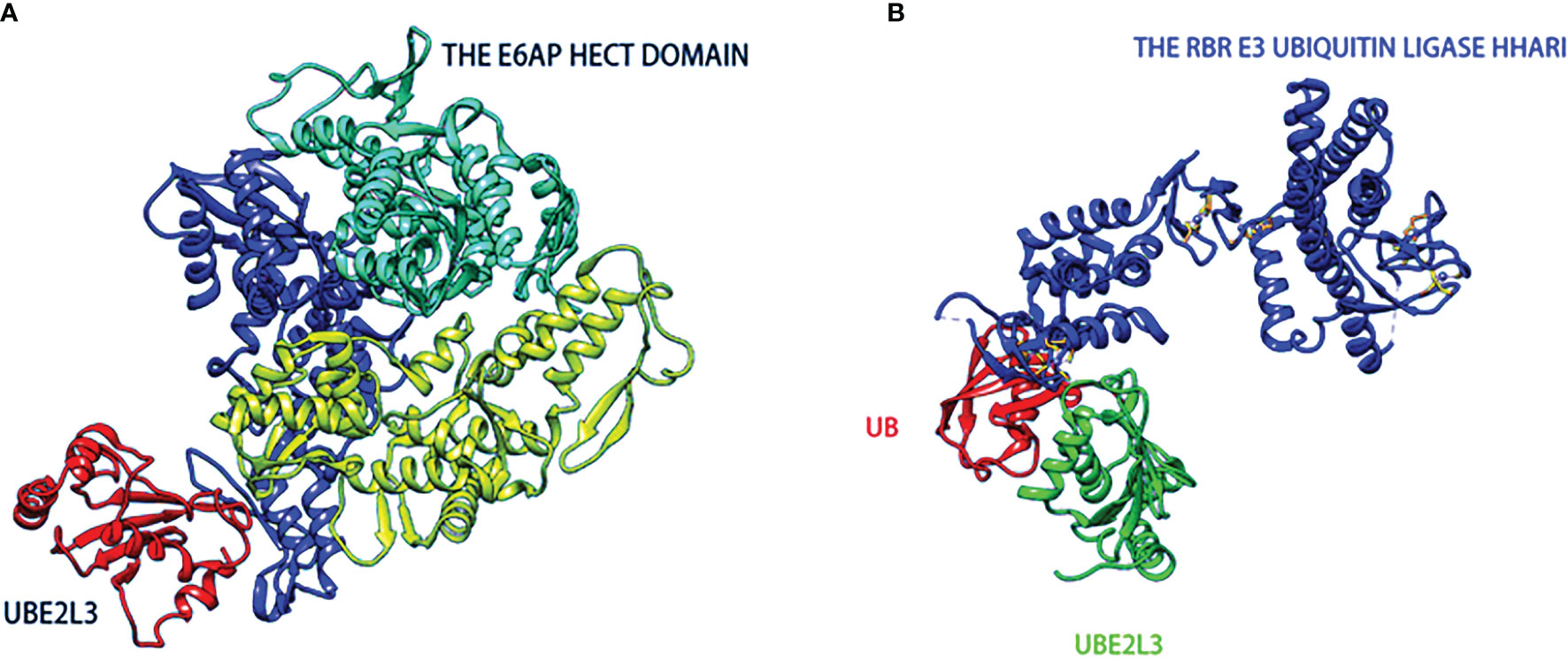
Figure 2 The structures of HECT-type (A) and RBR family (B) E3 ubiquitin-protein ligases in complexes with UBE2L3. (A) The catalytic HECT structure of E6AP is double-lobed (yellow, blue), and there is a large catalytic crack at the junction of the two lobed structures. The crack contains conserved residues(green) formed by mutated interference with the ubiquitin-thioester bond. (B) Ring-in-between-ring (RBR) ubiquitin E3 ligase(blue) binds to Ub(red)-UBE2L3(green) through RING/HECT hybridization mechanism.

Table 1 The E3 Ubiquitin ligases linked to UBE2L3 and their related functions.
As a ubiquitin conjugating E2 enzyme, UBE2L3 can work with E1s and E3s to catalyze multiple substrate proteins to complete the ubiquitination process. A large number of reports have indicated that UBE2L3 is involved in the occurrence and development of tumors, immune diseases, and Parkinson’s disease, through mainly the following signaling pathways: The nuclear transcription factor-kappa B (NF-κB) signaling pathway, GSK3β/p65 signaling pathway, p53 signaling pathway, autophagy mediated by p62, and DSB repair pathway.
NF-κB includes a number of transcription factors involved in regulating biological responses and it regulates a variety of cellular functions in the immune system, including cell survival, differentiation, and proliferation (73). Ubiquitination has an essential effect on the regulation of NF-κB signaling pathways. Under normal conditions, NF-κB, combined with an inhibitory protein of the IκB family, is sequestrated in the cytoplasm. When cells are stimulated by different types of agents, including inflammatory cytokines such as interleukin-1 (IL-1), tumor necrosis factor α (TNFα), and microbial products such as lipopolysaccharide (LPS), the IκB kinase (IKK) complex is activated. The IKK complex consists of IKKα, IKKβ, and NEMO (also called IKKγ or IKKAP). The activated IKK phosphorylates IκB and promotes the degradation of these inhibitors through the ubiquitin proteasome system, that allows the NF-κB to translocate to the nucleus and to initiate the transcription of a large number of target genes (74, 75). Some of the key components that are generated after NF-κB activation in turn regulate NF-κB activity negatively. However, ubiquitination is a highly dynamic and reversible process in which the ubiquitin chains can be removed from the substrates by DUBs (9). Several DUBs, such as CYLD (76, 77), A20 (78), and OTULIN (79), act as negative regulators to inhibit the activity of NF-κB (77).
In the field of ubiquitin regulation of NF-κB activation, the E2 ubiquitin conjugating enzymes UBE2N and UBE2D1 have been the most highly studied. UBE2N regulates NF-kB activation through the IL-1 pathway. In the IL-1 pathway, IL-1 binds with the corresponding receptors to recruit many different cellular signaling molecules, such as MyD88 and TRAF6. TRAF6 is a RING E3 ubiquitin ligase, which functions together with UBE2N to catalyze the synthesis of the K63 - polyubiquitin chain (80). This K63 polyubiquitin chain activates the TGF-β activated kinase 1 (TAK1) kinase complex, which in turn phosphorylates IKKβ leading to the activation of IKK (81), while UBE2D1 regulates the NF-kB activation through the TNFα pathway. In the TNFα pathway, TNFα binds to the corresponding receptors to recruit TRAF2, TRAF5, cIAP1, cIAP2 and RIP, and cIAP1 together with the UBE2D1 synthesized polyubiquitin chains with different linking modes, including the K63, K11 and linear ubiquitin chains. The heterogeneous polyubiquitin chains promote ubiquitin RIP, which activates the TAK1 kinase complex, leading to the activation of IKK and the phosphorylation of IKKβ (82).
UBE2L3, an E2 ubiquitin conjugating enzyme, is also involved in the regulation of the NF-κB signaling pathway. Current studies have indicated that there are many, although controversial mechanisms through which UBE2L3 regulates NF-κB activation. Tax, a transactivator encoded by human T-cell leukemia virus Type 1 (HTLV-1), is critical in the life cycle of the virus (83). UBE2L3 combines with Tax, a novel E3 ubiquitin ligase, for the assembly of the mixed-linked polyubiquitin chain in a variety of linkages, although not for the K63-polyubiquitin chain, which activates IKK directly (72). UBE2L3, together with Tax, can synthesize the mixed-linked polyubiquitin chain that activate IKK directly. However, it cannot activate the TKA1 complex, as this can be activated only by the K63-polyubiquitin chain (84). However, UBE2L3 linked to different E3 ubiquitin ligases play different roles in NF-κB regulation. OspG kinase, one of the host invasion and cell-disrupting shigella effectors, inhibits the NF-κB pathway activation by blocking phospho-IκBα degradation (85). OspG can only interact with several E2-conjugating enzymes that contain Ub (85). A structural and biochemical study showed that OspG binds preferentially to the UBE2L3-Ub conjugate (46). First, the stable UBE2L3-Ub complex is formed by the disulfide bond between the catalytic cysteine (C86) in UBE2L3 and the C-terminus of Ub (G76C) (86). The ubiquitin is located in the “open” conformation of UBE2L3 and interacts with the C-terminus of OspG via its I44 patch. UBE2L3 then binds to OspG via the two conserved rings that are necessary for the E3 ligase recruitment. Although UBE2L3-Ub binds to OspG at sites far from the active kinase site, it increases its kinase activity and inhibits NF-κB activation (46). UBE2L3 is also the main E2 conjugating enzyme linked to LUBAC to regulate NF-κB activity. When TNFα binds to its receptor to recruit the LUBAC, UBE2L3 binds to HOIL-1L in LUBAC to promote the linear ubiquitination of NEMO, which in turn recruits the TNF signaling complex. NEMO contains two ubiquitin binding sites that not only cross-link and stabilize other ubiquitin components associated with the activated TNFR1 complex, but also acts as an adaptor of IKKα/IKKβ kinase. IKKs are recruited into the TNFR1 complex and activated, then the activated IKKs phosphorylates IκBα to promote the activity of NF-κB (20). UBE2L3 interacts with the three E3 ligases to regulate NF-κB activity, as shown in Figure 3. According to additional studies on the regulation of UBE2L3 in NF-κB, a UBE2L3 inhibitor has been found. Dimethyl fumarate (DMF), a derivative of fumaric acid, has neuroprotective and immunomodulatory effects (87–89). TLR7 stimulation leads to intracellular accumulation of the linear ubiquitin chains, similar to TNFα. Co-overexpression of UBE2L3 and LUBAC enhanced NF-KB activation by TLR7-driven, and DMF as an antagonist of UBE2L3 inhibited the response to TLR7 activation. Unfortunately, these effects of DMF on UBE2L3 were only published in the conference abstract FRI0271 without relevant research articles, so the specific mechanism of DMF on UBE2L3 is not very clear.
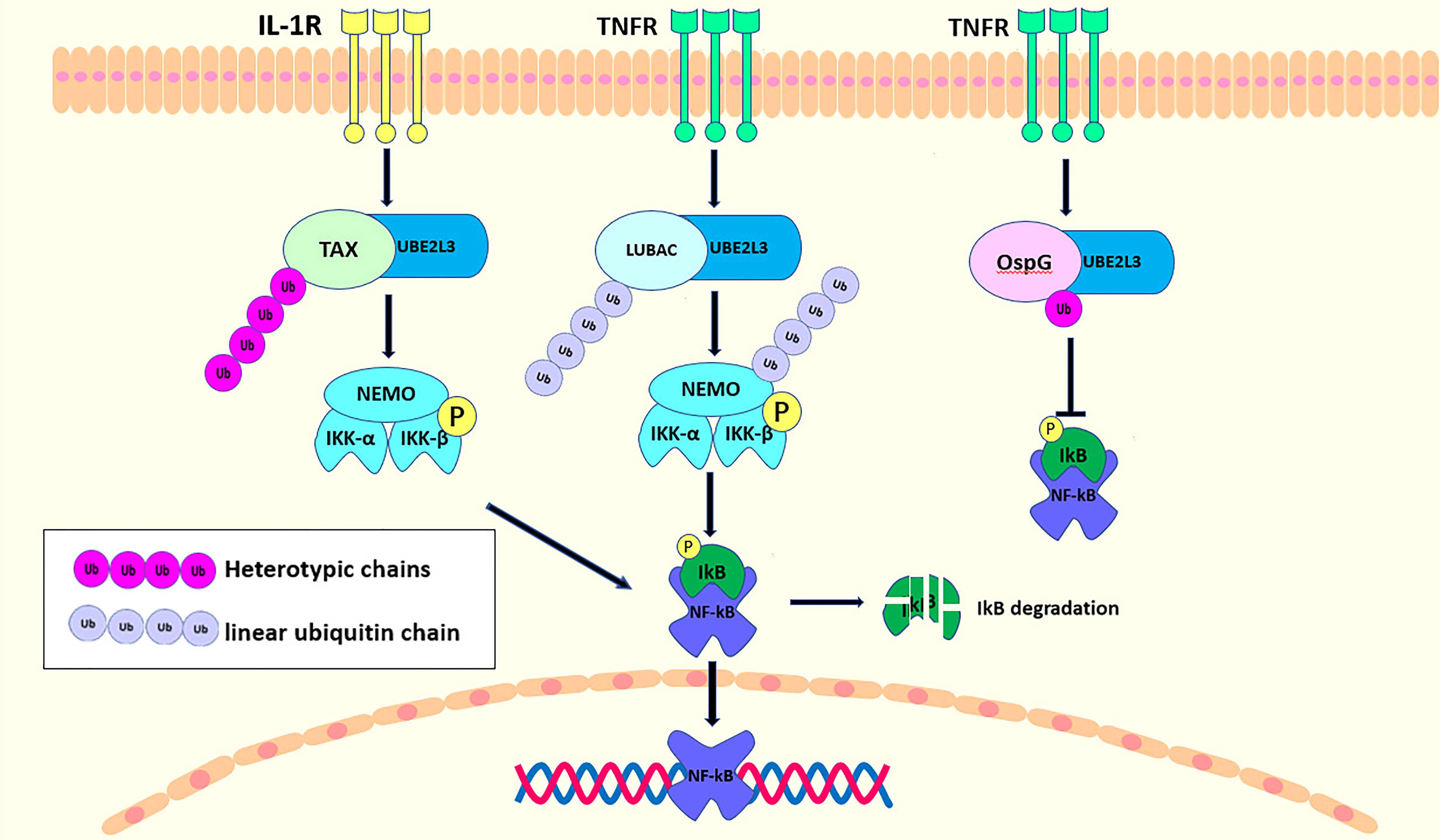
Figure 3 UBE2L3 binding with different E3 ubiquitin ligases acts on NF-κB signaling pathway. Upon the stimulation of IL-1R, TAX interacts with UBE2L3 to synthesize the heterotypic chains, resulting in IκB phosphorylation, and the degradation and activation of NF-κB. In the TNFR pathway, UBE2L3 interacts with LUBAC to synthesize first, a linear ubiquitin chain, then there is a linear ubiquitination of NEMO and a phosphorylation of IKKβ, ultimately leading to the activation of NF-κB. While OspG can interact with UBE2L3-Ub conjugates to improve the kinase activity of OspG, which inhibit the activation of NF-κB.
Additional studies have shown that UBE2L3 regulates NF-κB by binding with different E3 ubiquitin ligases to form different types of ubiquitin chains, leading to positive or negative regulatory reactions. Therefore, future research should pay more attention to the structure and biochemical mechanisms of UBE2L3 that link the different E3 ligases. This will provide a theoretical basis for the discovery of additional antagonists.
Glycogen synthase kinase 3β (GSK3β) is a serine-threonine kinase that phosphorylates and inactivates glycogen synthase (90). GSK3β participates in a variety of signaling pathways to regulate a plethora of cellular activities, such as metabolic activities, transcriptional regulation, neuronal functions, vesicle transport, cell cycles, as well as tumorigenesis and tumor development (91). GSK3β upregulates the activity of NF-κB, and the enhanced activity of NF-κB further stimulates and participates in the pathways of cell proliferation, the production of the tumor promoting cytokines and the promotion of anti-apoptosis (92–94). The inhibition of GSK3β enhances the activity of transcription factors that promote epithelial to mesenchymal transformation (94, 95).
Studies have shown that the knockdown of UBE2L3 and the overexpression of GSK3β can increase NF-κB activity. Moreover, further research has demonstrated that there is a negative feedback regulation between UBE2L3 and GSK3β showing an interaction between UBE2L3 and GSK3β. p65 is the most important subunit and active part of NF-κB. The knockdown of GSK3β can reduce the expression of p65, inhibit the phosphorylation of p65, and further inhibit NF-κB activity. Therefore, the knockdown of UBE2L3 promotes the expression of GSK3β, which activates p65 and leads to NF-κB activation (96) (Figure 4). In summary, UBE2L3 regulates NF-κB activity through the GSK3β/p65 signaling pathway. In this process, in order to clarify whether the degradation of GSK3β by UBE2L3 depends on the ubiquitin-proteasome system, the protein expression level of GSK3β significantly decreases when UBE2L3 is overexpressed, while the protein level of GSK3β significantly recovers when MG132, a proteasome inhibitor, is added. This suggests that the degradation of GSK3β by UBE2L3 depends on the ubiquitin proteasome pathway (96); however, the specific mechanism of GSK3β degradation by UBE2L3 was not clarified in this study. Therefore, further studies should evaluate which of the E3 ubiquitin ligases UBE2L3 interacts with to complete the ubiquitination of GSK3β and which ubiquitin chains are formed during the ubiquitination of GSK3β.
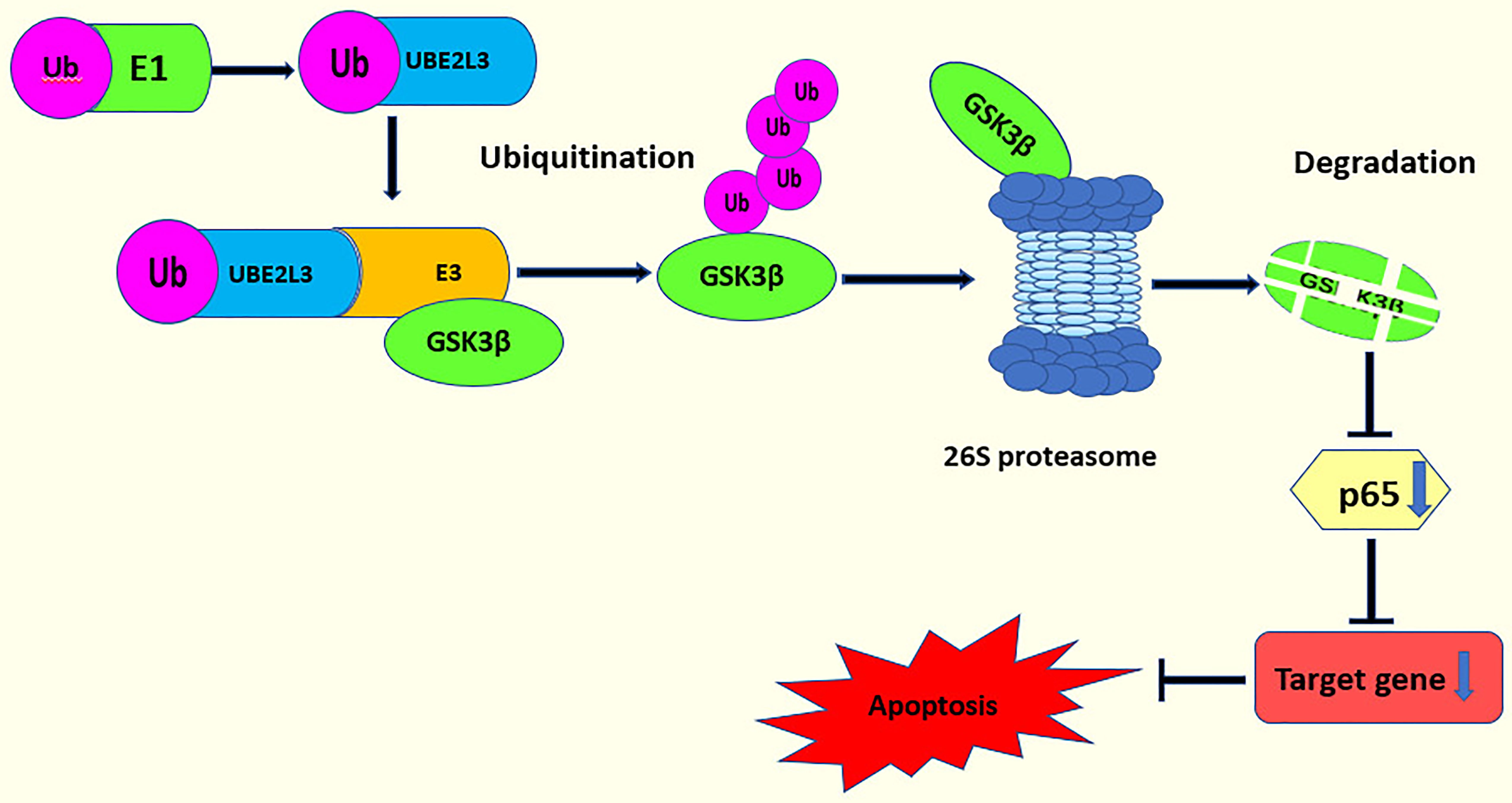
Figure 4 UBE2L3 acts on the GSK3β/p65 signaling pathway. UBE2L3 participates in the ubiquitination of GSK3β and promotes the degradation of GSK3β, leading to a decrease on the expression of p65, thereby inhibiting apoptosis.
Organisms depend on genetic code to perform various cellular functions, so DNA damage poses a threat to organisms. In the face of DNA damage, specialized DNA repair mechanisms can maintain genome integrity (97). In eukaryotic cells, the most destructive type of DNA damage is DSB damage, which may be repaired by two primary, mechanically different pathways: homologous recombination (HR) and non-homologous terminal joining (NHEJ) (97). HR replaces the missing or damaged DNA sequences in a high-fidelity manner that depends mainly on the intact sister chromatid as a template. Therefore, HR only functions in the S and G2 phases of the cell cycle in which a sister chromatid is present (97, 98). By comparison, NHEJ remains active during the whole cell cycle, repairing DSB by connecting directly to the ends of chromosomes without using sister chromatids as templates. However, because of the way NHEJ works, it is prone to errors in the repair process (97, 99). The failure of DSB repair pathways not only affects the stability of the genome but also leads to the development of cancer and resistance to anticancer therapy (100).
UBE2L3 is involved in regulating DSB repair through different mechanisms. BRCA1, a tumor suppressor protein of breast and ovarian cancer, binds to a RING finger protein BARD1 to form a ring heterodimer (69) and it functions as an E3 ubiquitin ligase for ubiquitination (101). BRAC1 is involved in several processes, including DSB repair, cell cycle progression and transcription. It has been reported that BRCA1 undergoes autoubiquitination and interacts with a phosphorylated histone 2AX (pH2AX), in an early cellular response induced by DSB (102). Several E2 ubiquitin conjugating enzymes in BRCA1 that depend on ligase reactions were screened out, including UBE2H, UBE2D2, UBE2D3, UBE2R1, and UBE2L3 (103); however, only UBE2D3 and UBE2L3 combine with BRCA1 (69). In contrast, the complex formed by UBE2D3 binding with BRCA1 catalyzes the ubiquitination of H2A/H2AX (104, 105), while UBE2L3 was inactive in the Ub ligase activity test (69). Therefore, the specific mechanism of UBE2L3 binding to BRAC1 without catalytic ubiquitination needs to be studied further.
Another important regulator in DSB repair is the tumor suppressor protein p53 binding protein 1 (53BP1) (100). 53BP1, combined with Rif1, co-inhibit BRCA1-dependent HR, thereby facilitating NHEJ in the G1 phase (106, 107). Conversely, 53BP1/Rif1, antagonized by BRCA1, favors HR in the S and G2 phases (108, 109). UBE2L3 regulates the protein stability of 53BP1 through ubiquitination to determine DSB repair mode. The deletion of UBE2L3 stabilizes the protein level of 53BP1, causing the cells to choose NHEJ over HR in the repair of DSB, which can threaten the DNA-damaged cells. These DNA-damaged cells create obstacles in the process of creating the DNA replication fork, leading to the breakdown of the DNA replication fork and the generation of a one-ended DSB (100). This one-ended DSB requires repair by HR rather than by NHEJ (100, 109). A one-ended DSB repaired by NHEJ results in radial chromosomes and cell death (100, 110–112). Therefore, the overexpression of UBE2L3 downregulate the protein levels of 53BP1. Then, the cells choose HR to repair the one-ended DSB (100) (Figure 5). However, whether UBE2L3 regulates 53BP1 during DSB repair through the ubiquitin proteasome system has not been clearly stated, so further studies should determine this point.
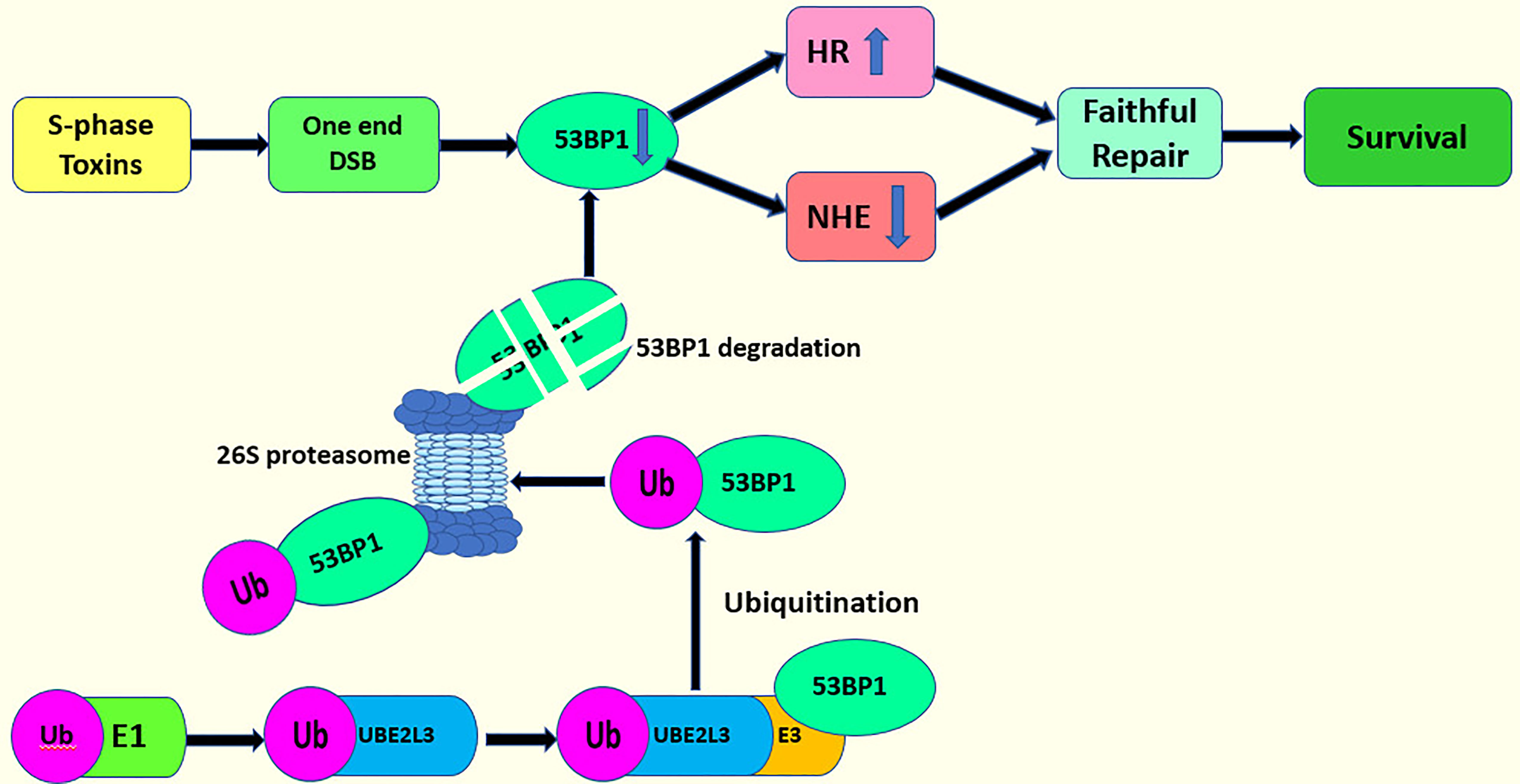
Figure 5 UBE2L3 acts on DSB repair and cell survival.
The ubiquitin–proteasome system (UPS) and autophagy are two different interlinked pathways, which play a key role in cellular proteostasis under normal and stressed conditions. In eukaryotes, two interconnected pathways remove misfolded proteins (113). In UPS, the misfolded proteins are linked to the polyubiquitin chains via lysine residues to form a ubiquitin-protein complex. Ubiquitinated proteins are recognized by the proteasome, deubiquitinated by DUB, and ultimately degraded (114). However, when the UPS is damaged or the misfolded proteins exceed its repair capacity, these proteins can be stored to create larger structures that then serve as substrates for autophagy (113, 115). This procedure is also referred to as aggregation. During aggregation, the double-membraned organelles known as autophagosomes insulate the protein substrates (116). After the fusion of the autophagosome and lysosome, the substrate proteins degrade (116).
p62 is an autophagy receptor that was discovered in mammals (117). Shin named it Sequestosome 1 (SQSTM 1) because it can create aggregates (118). p62 links the UPS and autophagy (113, 119). Under conditions of stress such as heat shock, the overexpression of ubiquitin and the inhibition of proteasome, the amount of ubiquitin increases abnormally, causing UB+ stress and further inducing p62 ubiquitination. A large number of ubiquitinated proteins exceed the capacity of the UPS, and the body finally unlocks the autophagy pathway to degrade the ubiquitinated proteins (6, 96, 120) (Figure 6). In a previous report, UBE2L3 interacted with Parkin (60). Parkin consists of two domains with different functions: a C-terminal ring-box domain and an N-terminal Ubl domain (60). UBE2L3 can bind to the C-terminal ring-box of Parkin to specifically discharge ubiquitin into the active sites of Parkin (43). In another study, however, a set of UBE2 enzyme may regulate Parkin-mediated autophagy by p62 signaling pathway. The autophagy clearance of depolarized mitochondria can be reduced significantly by inhibiting the E2 enzymes, UBE2L3, UBE2N, or UBE2D2 and UBE2D3 (UBE2D2/3). However, after single UBE2 knockdowns, polyubiquitin and p62 were still found in mitochondria indicating that UBE2N, UBE2L3, and UBE2D2/3 participate cooperatively in the Parkin-mediated autophagy via the p62 signaling pathway (121). Further, another study confirmed that UBE2L3, UBE2D and UBE2N were positive regulators of Parkin mitochondrial translocation. However, this study confirmed that the knockout of UBE2L3, UBE2D, and UBE2N alone could delay Parkin translocation to and p62 recruitment by damaged mitochondria. The double knockout experiment showed that the combined silencing UBE2L3 and UBE2D had an additive effect, which confirmed the redundancy of UBE2D and UBE2L3. However, the combined knockout of UBE2N with UBE2D or UBE2L3 showed no strong additive effect, indicating that UBE2N, UBE2D, and UBE2L3 were not redundant, but acted in different stages. Moreover, although these three E2s catalyzed the formation of the ubiquitin modified Parkin in vitro, only UBE2L3 promoted the obvious self-ubiquitination of Parkin (122).
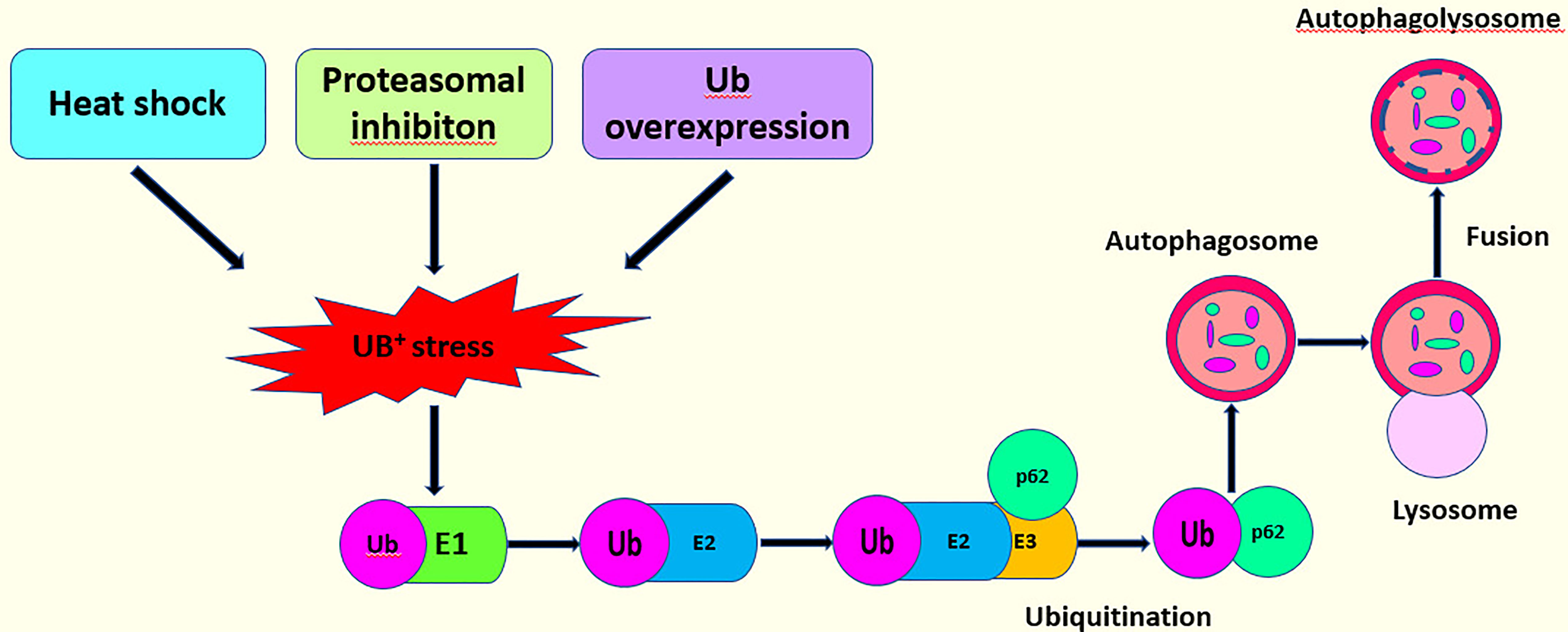
Figure 6 This model describes the interaction of the p62 signaling pathway with the UPS to activate autophagy.
p53 is a protein that inhibits tumor growth and plays an important role in tumor growth by DNA replication, controlling cell cycle and uncontrolled cell division (123). p53 is formed from 393 amino acids, and has several primary functional domains: DNA binding, transcription, regulatory domain and tetramerization (124). The structure of this protein comprises a loop helical structure (L, S, H) and five conserved regions (I, II, III, IV, V). The five conserved domains overlap with the loop helix domains, and together, they form part of the protein’s three-dimensional structure. Furthermore, abnormalities in the three-dimensional domain of p53 may lead to mutations (124).
Although p53 has many biological functions, its expression levels are low in unstressed cells due to the strict control of the ubiquitin-proteasome degradation system (125). Several ubiquitin ligases, such as E6AP (40), ARF-BP1 (126), MDM2 (127), COP1 (126), and PIRH2 (126, 128), can bind to p53 and lead to its degradation, in which EA6P plays a leading role. E6 oncoprotein, derived from HPV, promotes the development of cervical cancer by degrading p53 (129). Furthermore, several ubiquitin-conjugating enzymes, including UBE2D1, UBE2E1, UBE2L3, UBE2D2, and UBE2D3 are involved in the ubiquitination of p53. However, only UBE2D1 and UBE2L3 can interact with E6AP in the ubiquitination of p53, indicating that UBE2D1 and UBE2L3 are E6AP ubiquitin ligase E2s (130, 131). The aromatic hydrocarbon receptor, a ligand-activated receptor, acts as a regulator of p53 and promotes the ubiquitination and degradation of p53 by controlling the expression of the UBE2L3 protein (132). However, further studies are required to determine whether there are other E3s and regulators involved in the interactions between UBE2L3 and p53 in the ubiquitin proteasome system.
The localization of UBE2L3, in Chromosome (Chr) 22q11.2-13.1 (133), is unusual for E2 ubiquitin conjugating enzymes, because it has no direct homolog in yeast, which limits the exploration of its biological functions. UBE2L3 has important mammalian functions since the decrease of this enzyme in mice leads to growth retardation and placental vascular defects causing prenatal or perinatal death (134). The expression levels and activation statuses of UBE2L3 alter during cell differentiation and in the cell cycle. The downregulation expression of UBE2L3 prolongs the proportion of cells in the S phase while the upregulation expression of UBE2L3 increases the proportion of cells in the G1 phase relative to the S phase (135, 136). UBE2L3 also acts in several inflammatory disease (137–139) and the formation and progression of tumors (96, 100, 140). Therefore, this review summarizes the role of UBE2L3 in different types of diseases.
The immune system is important for self-protection. It enables the body to remove antigens for the purpose of self-protection (141). Immunodeficiencies can lead to a variety of viral and bacterial infections, while over immunity always results in autoimmune diseases (142). The NF-κB signaling pathway regulates the expression of immune-related genes extensively, and the role of UBE2L3 in immune-related diseases relies mainly on the NF-κB pathway (143). It has been reported that in addition to the NF-κB signaling pathway, UBE2L3 regulates immunity through several other pathways.
In genome-wide association studies (GWAS) and other genetic studies, UBE2L3 has been to be implicated in a variety of autoimmune diseases (Table 2) (139, 144–158). A few of the single nucleotide polymorphisms (SNPs) depict risk alleles related to several autoimmune diseases, including systemic lupus erythematosus(SLE) (145, 146, 148), Crohn’s disease (155), rheumatoid arthritis(RA) (150), inflammatory bowel disease(IBD) (153), celiac disease(CD) (158), psoriasis (156), diffuse systemic skin sclerosis (dcSSc) (159), and juvenile idiopathic arthritis(JIA) (152). Moreover, the risk allele rs5754217, located on UBE2L3, is related significantly to SLE and RA, indicating that UBE2L3 may be a common susceptibility site for these two autoimmune rheumatic diseases (137). A haplotype analysis has shown that the structure of UBE2L3 loci is relatively simple, and two haplotypes may extend to the whole gene length and cover most of the genetic variations. A recent study also showed that a single haplotype block, which is related to multiple other autoimmune diseases, is relevant to SLE (147). UBE2L3 was associated recently with the GWAS of chronic hepatitis B in the Han community (157), showing that UBE2L3 is required for overcoming hepatitis B virus infections. In the original GWAS and in a combined analysis of two independent Dutch replication cohorts, two novel potential risk genes for Crohn’s disease were identified: UBE2L3 and BCL3. The high expression of the UBE2L3 gene was related closely to the risk allele(rs2298428), located on the UBE2L3 gene. The overexpression of UBE2L3 results in a faster degradation of the NF-κB precursor and reduced NF-κB production, further inhibiting the innate immune response (139). TNFAIP3 and TNIP1, which are ubiquitin-related genes, have been shown to be correlated with SLE, RA, and systemic sclerosis (SSc). Moreover, diffuse cutaneous systemic sclerosis(dcSSc) has been reported to have a strong association with these two genes (159–161). This is supported by a case-control study on the genetic background of dcSSc in Japanese that showed that UBE2L3 may also be involved in the genetic background of SSc through ubiquitination, and that this mechanism may act on the pathogenesis of dcSSc (159). Due to limited understanding of the overall genetic structure of juvenile idiopathic arthritis, relatively few loci associated with the disease have been identified (162). In addition, an immunomicroarray sequence analysis, the association of JIA with six loci, which were C5orf56IRF1(rs4705862), RUNX1 (rs9979383), RUNX3(rs4648881), ERAP2LNPEP(rs27290), PRR5L(rs4755450), and UBE2L3(rs2266959), was confirmed (163). Using an analysis of expression quantitative trait loci, a large number SNPs of UBE2L3 was associated with disease risks relevant to the overexpression of UBE2L3. Therefore, a higher expression and higher activity of UBE2L3 may be the cause of autoimmune diseases caused by altered immune response pathways. Furthermore, the specific mechanism of UBE2L3 in autoimmune diseases needs to be studied further.
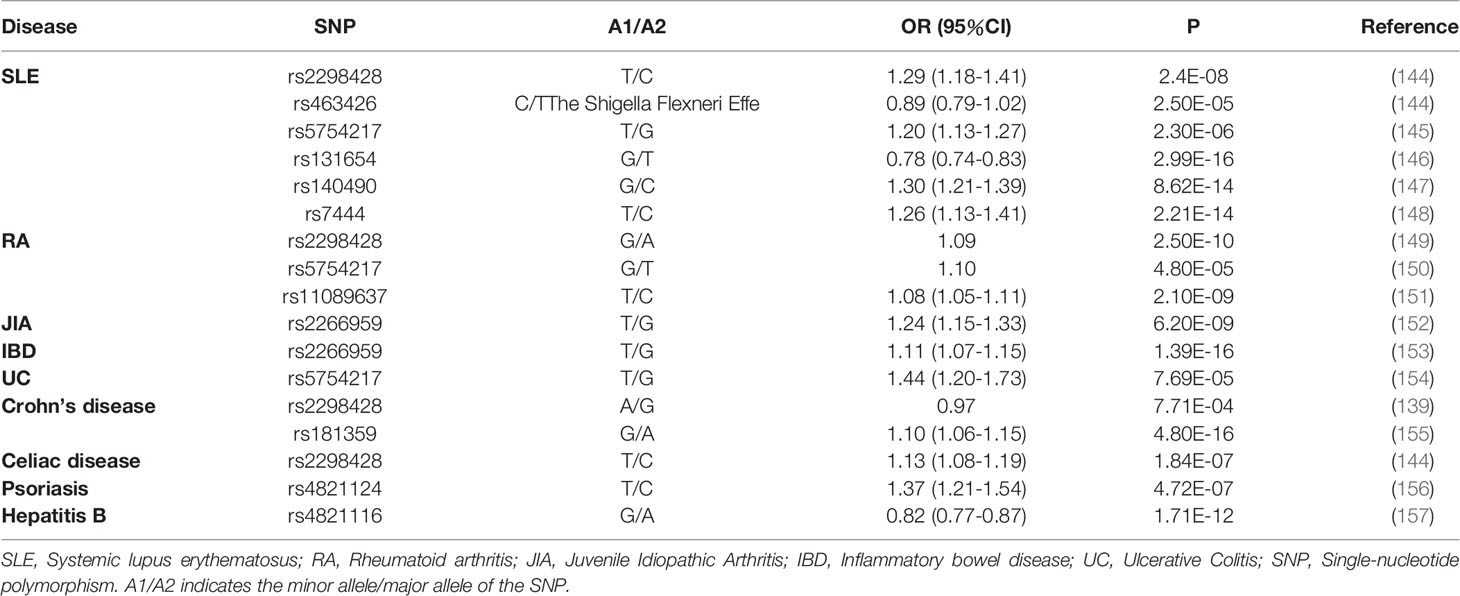
Table 2 Genetic studies showing an association between UBE2L3 and autoimmune and infectious diseases.
In recent years, an increasing number of studies have shown the correlation between UBE2L3 and tumors. Studies have found an abnormal expression of UBE2L3 in the cell lines and tumor tissues from many patients with a tumor indicating that UBE2L3 is involved in a number of cancer-related signaling pathways.
GSK3β reportedly stimulates the development of certain tumors, including those in pancreatic cancer, colorectal cancer and myeloma cancer, while inhibiting the development of other tumors, including those in lung, esophageal, and breast cancer (164–166). A high level of UBE2L3 in hepatocellular carcinoma inhibits p65 by down-regulating GSK3β through the ubiquitin-mediated proteasome degradation, rather than by the classical NF-κB signaling pathway, suggesting that an overexpression of UBE2L3 in hepatocellular carcinoma promotes apoptosis evasion through the inhibition of the GSK3β/p65 pathway (96).
The p53 pathway plays an important role in a many of the biological functions of the body, including cell cycle regulation, metabolism, aging and development, reproduction, and the inhibition of tumor expression (167–169), and has been considered as a crucial anticancer target (170). The E6 oncoprotein derived from HPV promotes the growth of cervical cancer by degrading p53 and blocking the p53-mediated growth arrest and apoptosis (129, 171). This relied mainly on UBE2L3 and E6AP (129, 172–174). Other studies have shown that UBE2L3 may reduce HeLa cell apoptosis by stimulating ubiquitination and the degradation of p53 (132, 175).
Abnormalities in the DSB repair pathway result in the growth of tumors and resistance to antineoplastic treatment (100). The formation of the tumor suppressor signal 53BP1 foci plays a role in selecting the DSB repair pathway. Controlling the level of 53BP1 showed an impact almost as powerful as lesion formation in DSB repair and in establishing cell sensitivity to anticancer therapies (176). A study that analyzed protein expression levels of UBE2L3 and 53BP1 in several breast cell lines showed a negative correlation between the two proteins, particularly in triple-negative breast cancer cells. All of these data reveal that the UBE2L3/53BP1 axis may act on the occurrence and development of tumors. Through further research, the regulation of UBE2L3 can stabilize the levels of 53BP1 protein, resulting in reduced HRs for DSB repair (100).
UBE2L3 controls the protein stability of several signaling molecules, which acts on cancer regulation. A study reported that UBE2L3 promotes the ubiquitination and degradation of p27kip1 in non-small-cell lung cancer (NSCLC), and further study suggested that high expression of UBE2L3 led to the down-regulation of p27KIP1 protein levels, which are relevant to a poor prognosis for NSCLC (177). Another study identified the oncogenic UBE2L3-KRAS fusion in DU145 prostate cancer cells using an integrative genomics approach, and further investigation demonstrated that the fusion of UBE2L3 with KRAS may lead to the progression of metastases in rare subsets of prostate cancer (140).
Parkinson’s disease (PD) is a common neurodegenerative disease with complex clinical features. Genotypic and linkage analyses have shown that the most common cause of sporadic early and late PD cases was mutations in the PARK2 gene encoding Parkin which act as an RBR E3 ligase (178, 179). Parkin may have a neuroprotective function by regulating mitochondrial autophagy. The N-terminal UBL domain of Parkin can inhibit its activity automatically; thus, the interaction between the E2-loaded ubiquitin and Parkin is affected (180–183). When Parkin’s autoinhibitory state is removed, the dual PINK1 kinase-mediated phosphorylation activates Parkin (184–188). The activated Parkin can then interact with the various E2-loaded ubiquitin, including UBE2L3, to induce a process of mitochondrial autophagy. Studies have suggested that UBE2L3 may act on Parkin-dependent autophagy through the p62 signaling pathway (121, 122). However, there remains controversy with respect to the interactions between Parkin and UBE2L3, UBE2N and UBE2D for the induction of mitochondrial autophagy, which was described in the section on the p62 signaling pathway. This detailed mechanism should be studied further.
Ubiquitination is an important post-translational modification that regulates the various biological functions of cells by modifying proteins. In recent years, an increasing number of researchers have begun paying attention to ubiquitination due to an increase in studies on tumor progression and treatment, and in order to seek effective antineoplastic treatment (189). UBE2L3 is an E2 conjugating enzyme that exists widely in eukaryotes. It plays a critical role in the UPS. UBE2L3 is expressed abnormally in several human cancers, including hepatocellular carcinoma, NSCLC and cervical cancer. UBE2L3 can stimulate the occurrence, development, and metastasis of human tumors by regulating different signaling pathways relevant to human tumors and a variety of proteins that do not participate in the above signaling pathways. p53 can promote tumor cell apoptosis and is an important tumor suppressor. Inhibiting UBE2L3 can activate p53, which may provide a direction for the development of new anticancer drugs. However, the regulation of UBE2L3 on GSK3β/p65, DSB repair and other signaling pathways also requires further study, and the specific mechanisms of UBE2L3 in other tumors and their potential as novel antineoplastic treatment should be investigated. Although the findings of previous studies, have shown that UBE2L3 is a promising target for therapies for immune diseases and Parkinson’s disease, the specific molecular mechanism of the involvement of UBE2L3 in regulating immune diseases and Parkinson’s disease should be studied further. We recommend that future studies focus on the development and evaluation of UBE2L3 inhibitors and demonstrate their efficacy and safety in the treatment of these diseases.
XZ: Writing—original draft preparation, investigation, and figure preparation. CH: Investigation and figure preparation. YLiu: Investigation. RS: Investigation. YZ: Investigation. YLi: Conceptualization, methodology, and supervision. All authors contributed to the article and approved the submitted version.
This work was supported by Fundamental Research Funds from the Central Universities (lzujbky-2021-ct18), the Development and Reform Commission Project of Gansu Province (2020-2022), the Education Department of Gansu Province (2021jyjbgs) and the Major Science and Technology Special Project of Gansu Province (20ZD7FA003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Glickman MH, Ciechanover A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol Rev (2002) 82(2):373–428. doi: 10.1152/physrev.00027.2001
2. Popovic D, Vucic D, Dikic I. Ubiquitination in Disease Pathogenesis and Treatment. Nat Med (2014) 20(11):1242–53. doi: 10.1038/nm.3739
3. Heaton SM, Borg NA, Dixit VM. Ubiquitin in the Activation and Attenuation of Innate Antiviral Immunity. J Exp Med (2015) 213(1):1–13. doi: 10.1084/jem.20151531
4. Gilberto S, Peter M. Dynamic Ubiquitin Signaling in Cell Cycle Regulation. J Cell Biol (2017) 216(8):2259–71. doi: 10.1083/jcb.201703170
5. Pickart CM. Mechanisms Underlying Ubiquitination. Annu Rev Biochem (2001) 70(1):503–33. doi: 10.1146/annurev.biochem.70.1.503
6. Peng H, Yang J, Li G, You Q, Han W, Li T, et al. Ubiquitylation of P62/Sequestosome1 Activates Its Autophagy Receptor Function and Controls Selective Autophagy Upon Ubiquitin Stress. Cell Res (2017) 27(5):657–74. doi: 10.1038/cr.2017.40
7. Hershko A, Ciechanover A. THE UBIQUITIN SYSTEM. Annu Rev Biochem (1998) 67(1):425–79. doi: 10.1146/annurev.biochem.67.1.425
8. Ciechanover A. Intracellular Protein Degradation: From a Vague Idea Through the Lysosome and the Ubiquitin–Proteasome System and Onto Human Diseases and Drug Targeting. Bioorganic Medicinal Chem (2013) 21(12):3400–10. doi: 10.1016/j.bmc.2013.01.056
9. Komander D, Clague MJ, Urbé S. Breaking the Chains: Structure and Function of the Deubiquitinases. Nat Rev Mol Cell Biol (2009) 10(8):550–63. doi: 10.1038/nrm2731
10. Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbé S. Deubiquitylases From Genes to Organism. Physiol Rev (2013) 93(3):1289–315. doi: 10.1152/physrev.00002.2013
11. Hershko A, Ciechanover A, Rose IA. Resolution of the ATP-Dependent Proteolytic System From Reticulocytes: A Component That Interacts With ATP. Proc Natl Acad Sci USA (1979) 76(7):3107–10. doi: 10.1073/pnas.76.7.3107
12. Cadwell K, Coscoy L. Ubiquitination on Nonlysine Residues by a Viral E3 Ubiquitin Ligase. Science (2005) 309(5731):127–30. doi: 10.1126/science.1110340
13. Callis J, Carpenter T, Sun CW, Vierstra RD. Structure and Evolution of Genes Encoding Polyubiquitin and Ubiquitin-Like Proteins in Arabidopsis Thaliana Ecotype Columbia. Genetics (1995) 139(2):921–39. doi: 10.1093/genetics/139.2.921
14. Vijay-Kumar S, Bugg CE, Cook WJ. Structure of Ubiquitin Refined at 1.8Åresolution. J Mol Biol (1987) 194(3):531–44. doi: 10.1016/0022-2836(87)90679-6
15. Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-Binding Domains — From Structures to Functions. Nat Rev Mol Cell Biol (2009) 10(10):659–71. doi: 10.1038/nrm2767
16. Kamadurai HB, Souphron J, Scott DC, Duda DM, Miller DJ, Stringer D, et al. Insights Into Ubiquitin Transfer Cascades From a Structure of a UbcH5B∼Ubiquitin-HECTNEDD4L Complex. Mol Cell (2009) 36(6):1095–102. doi: 10.1016/j.molcel.2009.11.010
17. Hu M, Li P, Li M, Li W, Yao T, Wu JW, et al. Crystal Structure of a UBP-Family Deubiquitinating Enzyme in Isolation and in Complex With Ubiquitin Aldehyde. Cell (2002) 111(7):1041–54. doi: 10.1016/S0092-8674(02)01199-6
18. Reyes-Turcu FE, Horton JR, Mullally JE, Heroux A, Cheng X, Wilkinson KD. The Ubiquitin Binding Domain ZnF UBP Recognizes the C-Terminal Diglycine Motif of Unanchored Ubiquitin. Cell (2006) 124(6):1197–208. doi: 10.1016/j.cell.2006.02.038
19. Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A. Systematic and Quantitative Assessment of the Ubiquitin-Modified Proteome. Mol Cell (2011) 44(2):325–40. doi: 10.1016/j.molcel.2011.08.025
20. Fu B, Li S, Wang L, Berman MA, Dorf ME. The Ubiquitin Conjugating Enzyme UBE2L3 Regulates Tnfα-Induced Linear Ubiquitination. Cell Res (2014) 24(3):376–9. doi: 10.1038/cr.2013.133
21. Swatek KN, Komander D. Ubiquitin Modifications. Cell Res (2016) 26(4):399–422. doi: 10.1038/cr.2016.39
22. Yau RG, Doerner K, Castellanos ER, Haakonsen DL, Werner A, Wang N, et al. Assembly and Function of Heterotypic Ubiquitin Chains in Cell-Cycle and Protein Quality Control. Cell (2017) 171(4):918–933.e20. doi: 10.1016/j.cell.2017.09.040
23. Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, et al. Activation of the Canonical IKK Complex by K63/M1-Linked Hybrid Ubiquitin Chains. Proc Natl Acad Sci USA (2013) 110(38):15247–52. doi: 10.1073/pnas.1314715110
24. Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, et al. A Proteome-Wide, Quantitative Survey of In Vivo Ubiquitylation Sites Reveals Widespread Regulatory Roles. Mol Cell Proteomics (2011) 10(10):M111.013284. doi: 10.1074/mcp.M111.013284
25. Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, et al. Quantitative Proteomics Reveals the Function of Unconventional Ubiquitin Chains in Proteasomal Degradation. Cell (2009) 137(1):133–45. doi: 10.1016/j.cell.2009.01.041
26. Dammer EB, Na CH, Xu P, Seyfried NT, Duong DM, Cheng D, et al. Polyubiquitin Linkage Profiles in Three Models of Proteolytic Stress Suggest the Etiology of Alzheimer Disease. J Biol Chem (2011) 286(12):10457–65. doi: 10.1074/jbc.M110.149633
27. Ziv I, Matiuhin Y, Kirkpatrick DS, Erpapazoglou Z, Leon S, Pantazopoulou M, et al. A Perturbed Ubiquitin Landscape Distinguishes Between Ubiquitin in Trafficking and in Proteolysis. Mol Cell Proteomics (2011) 10(5):M111.009753. doi: 10.1074/mcp.M111.009753
28. Komander D, Rape M. The Ubiquitin Code. Annu Rev Biochem (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
29. Yau R, Rape M. The Increasing Complexity of the Ubiquitin Code. Nat Cell Biol (2016) 18(6):579–86. doi: 10.1038/ncb3358
30. Chen ZJJ, Sun LJJ. Nonproteolytic Functions of Ubiquitin in Cell Signaling. Mol Cell (2009) 33(3):275–86. doi: 10.1016/j.molcel.2009.01.014
31. Behrends C, Harper JW. Constructing and Decoding Unconventional Ubiquitin Chains. Nat Struct Mol Biol (2011) 18(5):520–8. doi: 10.1038/nsmb.2066
32. Trempe J-F. Reading the Ubiquitin Postal Code. Curr Opin Struct Biol (2011) 21(6):792–801. doi: 10.1016/j.sbi.2011.09.009
33. Husnjak K, Dikic I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu Rev Biochem (2012) 81(1):291–322. doi: 10.1146/annurev-biochem-051810-094654
34. Baumann K. Lys33-Linked Ubiquitin in Post-Golgi Transport. Nat Rev Mol Cell Biol (2014) 15(6):365–5. doi: 10.1038/nrm3812
35. Kristariyanto YA, Choi SY, Rehman SA, Ritorto MS, Campbell DG, Morrice NA, et al. Assembly and Structure of Lys33-Linked Polyubiquitin Reveals Distinct Conformations. Biochem J (2015) 467(2):345–52. doi: 10.1042/BJ20141502
36. Clague MJ, Heride C, Urbé S. The Demographics of the Ubiquitin System. Trends Cell Biol (2015) 25(7):417–26. doi: 10.1016/j.tcb.2015.03.002
37. Michelle C, Vourc'h P, Mignon L, Andres CR. What was the Set of Ubiquitin and Ubiquitin-Like Conjugating Enzymes in the Eukaryote Common Ancestor? J Mol Evol (2009) 68(6):616–28. doi: 10.1007/s00239-009-9225-6
38. Burroughs AM, Jaffee M, Iyer LM, Aravind L. Anatomy of the E2 Ligase Fold: Implications for Enzymology and Evolution of Ubiquitin/Ub-Like Protein Conjugation. J Struct Biol (2008) 162(2):205–18. doi: 10.1016/j.jsb.2007.12.006
39. Rankin CA, Galeva NA, Bae K, Ahmad MN, Witte TM, Richter ML. Isolated RING2 Domain of Parkin Is Sufficient for E2-Dependent E3 Ligase Activity. Biochemistry (2014) 53(1):225–34. doi: 10.1021/bi401378p
40. Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, et al. Structure of an E6AP-UbcH7 Complex: Insights Into Ubiquitination by the E2-E3 Enzyme Cascade. Sci (New York NY) (1999) 286(5443):1321–6. doi: 10.1126/science.286.5443.1321
41. Kumar S, Kao WH, Howley PM. Physical Interaction Between Specific E2 and Hect E3 Enzymes Determines Functional Cooperativity. J Biol Chem (1997) 272(21):13548–54. doi: 10.1074/jbc.272.21.13548
42. Nuber U, Scheffner M. Identification of Determinants in E2 Ubiquitin-Conjugating Enzymes Required for Hect E3 Ubiquitin-Protein Ligase Interaction. J Biol Chem (1999) 274(11):7576–82. doi: 10.1074/jbc.274.11.7576
43. Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 Reactivity Profile Reveals Parkin and HHARI to be RING/HECT Hybrids. Nature (2011) 474(7349):105–8. doi: 10.1038/nature09966
44. Sheng Y, Hong JH, Doherty R, Srikumar T, Shloush J, Avvakumov GV, et al. A Human Ubiquitin Conjugating Enzyme (E2)-HECT E3 Ligase Structure-Function Screen. Mol Cell Proteomics: MCP (2012) 11(8):329–41. doi: 10.1074/mcp.O111.013706
45. Eletr ZM, Kuhlman B. Sequence Determinants of E2-E6AP Binding Affinity and Specificity. J Mol Biol (2007) 369(2):419–28. doi: 10.1016/j.jmb.2007.03.026
46. Grishin AM, Condos TE, Barber KR, Campbell-Valois FX, Parsot C, Shaw GS, et al. Structural Basis for the Inhibition of Host Protein Ubiquitination by Shigella Effector Kinase OspG. Structure (2014) 22(6):878–88. doi: 10.1016/j.str.2014.04.010
47. Yuan L, Lv Z, Atkison JH, Olsen SK. Structural Insights Into the Mechanism and E2 Specificity of the RBR E3 Ubiquitin Ligase HHARI. Nat Commun (2017) 8(1):211. doi: 10.1038/s41467-017-00272-6
48. Stieglitz B, Morris-Davies AC, Koliopoulos MG, Christodoulou E, Rittinger K. LUBAC Synthesizes Linear Ubiquitin Chains via a Thioester Intermediate. EMBO Rep (2012) 13(9):840–6. doi: 10.1038/embor.2012.105
49. Alpi AF, Chaugule V, Walden H. Mechanism and Disease Association of E2-Conjugating Enzymes: Lessons From UBE2T and UBE2L3. Biochem J (2016) 473(20):3401–19. doi: 10.1042/BCJ20160028
50. Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of Linear Polyubiquitylation of NEMO in NF-kappaB Activation. Nat Cell Biol (2009) 11(2):123–32. doi: 10.1038/ncb1821
51. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear Ubiquitination Prevents Inflammation and Regulates Immune Signalling. Nature (2011) 471(7340):591–6. doi: 10.1038/nature09816
52. Fiil BK, Gyrd-Hansen M. Met1-Linked Ubiquitination in Immune Signalling. FEBS J (2014) 281(19):4337–50. doi: 10.1111/febs.12944
53. Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific Recognition of Linear Ubiquitin Chains by NEMO Is Important for NF-kappaB Activation. Cell (2009) 136(6):1098–109. doi: 10.1016/j.cell.2009.03.007
54. Aksentijevich I, Zhou Q. NF-kappaB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases. Front Immunol (2017) 8:399. doi: 10.3389/fimmu.2017.00399
55. Zhang Y, Higashide WM, McCormick BA, Chen J, Zhou D. The Inflammation-Associated Salmonella SopA Is a HECT-Like E3 Ubiquitin Ligase. Mol Microbiol (2006) 62(3):786–93. doi: 10.1111/j.1365-2958.2006.05407.x
56. Diao J, Zhang Y, Huibregtse JM, Zhou D, Chen J. Crystal Structure of SopA, a Salmonella Effector Protein Mimicking a Eukaryotic Ubiquitin Ligase. Nat Struct Mol Biol (2008) 15(1):65–70. doi: 10.1038/nsmb1346
57. Lin DYW, Diao JB, Chen J. Crystal Structures of Two Bacterial HECT-Like E3 Ligases in Complex With a Human E2 Reveal Atomic Details of Pathogen-Host Interactions. Proc Natl Acad Sci USA (2012) 109(6):1925–30. doi: 10.1073/pnas.1115025109
58. Kathania M, Zeng M, Yadav VN, Moghaddam SJ, Yang B, Venuprasad K. Ndfip1 Regulates Itch Ligase Activity and Airway Inflammation via Ubch7. J Immunol (2015) 194(5):2160–7. doi: 10.4049/jimmunol.1402742
59. Sauve V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, et al. A Ubl/Ubiquitin Switch in the Activation of Parkin. EMBO J (2015) 34(20):2492–505. doi: 10.15252/embj.201592237
60. Shimura H, Hattori N, Si K, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson Disease Gene Product, Parkin, Is a Ubiquitin-Protein Ligase. Nat Genet (2000) 25(3):302–5. doi: 10.1038/77060
61. Ardley HC, Tan NG, Rose SA, Markham AF, Robinson PA. Features of the Parkin/ariadne-Like Ubiquitin Ligase, HHARI, That Regulate Its Interaction With the Ubiquitin-Conjugating Enzyme, Ubch7. J Biol Chem (2001) 276(22):19640–7. doi: 10.1074/jbc.M011028200
62. Marteijn JA, van der Meer LT, Smit JJ, Noordermeer SM, Wissink W, Jansen P, et al. The Ubiquitin Ligase Triad1 Inhibits Myelopoiesis Through UbcH7 and Ubc13 Interacting Domains. Leukemia (2009) 23(8):1480–9. doi: 10.1038/leu.2009.57
63. Niwa J, Ishigaki S, Doyu M, Suzuki T, Tanaka K, Sobue G. A Novel Centrosomal RING-Finger Protein, Dorfin, Mediates Ubiquitin Ligase Activity. Biochem Biophys Res Commun (2001) 281(3):706–13. doi: 10.1006/bbrc.2001.4414
64. Fortier JM, Kornbluth J. NK Lytic-Associated Molecule, Involved in NK Cytotoxic Function, Is an E3 Ligase. J Immunol (2006) 176(11):6454–63. doi: 10.4049/jimmunol.176.11.6454
65. Huang J, Xu LG, Liu T, Zhai Z, Shu HB. The P53-Inducible E3 Ubiquitin Ligase P53rfp Induces P53-Dependent Apoptosis. FEBS Lett (2006) 580(3):940–7. doi: 10.1016/j.febslet.2005.09.105
66. Toby GG, Gherraby W, Coleman TR, Golemis EA. A Novel RING Finger Protein, Human Enhancer of Invasion 10, Alters Mitotic Progression Through Regulation of Cyclin B Levels. Mol Cell Biol (2003) 23(6):2109–22. doi: 10.1128/MCB.23.6.2109-2122.2003
67. Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a C-Cbl-UbcH7 Complex: RING Domain Function in Ubiquitin-Protein Ligases. Cell (2000) 102(4):533–9. doi: 10.1016/S0092-8674(00)00057-X
68. Geetha T, Jiang JX, Wooten MW. Lysine 63 Polyubiquitination of the Nerve Growth Factor Receptor TrkA Directs Internalization and Signaling. Mol Cell (2005) 20(2):301–12. doi: 10.1016/j.molcel.2005.09.014
69. Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fukuda M, Ohta T, et al. Binding and Recognition in the Assembly of an Active BRCA1/BARD1 Ubiquitin-Ligase Complex. Proc Natl Acad Sci USA (2003) 100(10):5646–51. doi: 10.1073/pnas.0836054100
70. Ahel J, Lehner A, Vogel A, Schleiffer A, Meinhart A, Haselbach D, et al. Moyamoya Disease Factor RNF213 Is a Giant E3 Ligase With a Dynein-Like Core and a Distinct Ubiquitin-Transfer Mechanism. Elife (2020) 9:e56185 doi: 10.7554/eLife.56185
71. Peris-Moreno D, Malige M, Claustre A, Armani A, Coudy-Gandilhon C, Deval C, et al. UBE2L3, a Partner of MuRF1/TRIM63, Is Involved in the Degradation of Myofibrillar Actin and Myosin. Cells (2021) 10(8):1974. doi: 10.3390/cells10081974
72. Wang C, Long W, Peng C, Hu L, Zhang Q, Wu A, et al. HTLV-1 Tax Functions as a Ubiquitin E3 Ligase for Direct IKK Activation via Synthesis of Mixed-Linkage Polyubiquitin Chains. PloS Pathog (2016) 12(4):e1005584. doi: 10.1371/journal.ppat.1005584
73. Hayden MS, Ghosh S. NF-κb, the First Quarter-Century: Remarkable Progress and Outstanding Questions. Genes Dev (2012) 26(3):203–34. doi: 10.1101/gad.183434.111
74. Scheidereit C. I Kappa B Kinase Complexes: Gateways to NF-Kappa B Activation and Transcription. Oncogene (2006) 25(51):6685–705. doi: 10.1038/sj.onc.1209934
75. Hayden MS, Ghosh S. Shared Principles in NF-kappaB Signaling. Cell (2008) 132(3):344–62. doi: 10.1016/j.cell.2008.01.020
76. Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD Is a Deubiquitinating Enzyme That Negatively Regulates NF-kappaB Activation by TNFR Family Members. Nature (2003) 424(6950):793–6. doi: 10.1038/nature01803
77. Harhaj EW, Dixit VM. Regulation of NF-κb by Deubiquitinases. Immunol Rev (2012) 246(1):107–24. doi: 10.1111/j.1600-065X.2012.01100.x
78. Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB Signaling by A20 Through Disruption of Ubiquitin Enzyme Complexes. Science (2010) 327(5969):1135–9. doi: 10.1126/science.1182364
79. Lork M, Verhelst K, Beyaert R. CYLD, A20 and OTULIN Deubiquitinases in NF-κb Signaling and Cell Death: So Similar, Yet So Different. Cell Death Differ (2017) 24(7):1172–83. doi: 10.1038/cdd.2017.46
80. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IkappaB Kinase Complex by TRAF6 Requires a Dimeric Ubiquitin-Conjugating Enzyme Complex and a Unique Polyubiquitin Chain. Cell (2000) 103(2):351–61. doi: 10.1016/S0092-8674(00)00126-4
81. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 Is a Ubiquitin-Dependent Kinase of MKK and IKK. Nature (2001) 412(6844):346–51. doi: 10.1038/35085597
82. Xu M, Skaug B, Zeng W, Chen ZJ. A Ubiquitin Replacement Strategy in Human Cells Reveals Distinct Mechanisms of IKK Activation by TNFalpha and IL-1beta. Mol Cell (2009) 36(2):302–14. doi: 10.1016/j.molcel.2009.10.002
83. Giam CZ, Jeang KT. HTLV-1 Tax and Adult T-Cell Leukemia. Front Biosci (2007) 12:1496–507. doi: 10.2741/2163
84. Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, et al. Direct Activation of Protein Kinases by Unanchored Polyubiquitin Chains. Nature (2009) 461(7260):114–9. doi: 10.1038/nature08247
85. Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C. The Shigella Flexneri Effector OspG Interferes With Innate Immune Responses by Targeting Ubiquitin-Conjugating Enzymes. Proc Natl Acad Sci USA (2005) 102(39):14046–51. doi: 10.1073/pnas.0504466102
86. Serniwka SA, Shaw GS. 1H, 13C and 15N Resonance Assignments for the Human E2 Conjugating Enzyme, Ubch7. Biomol NMR Assign (2008) 2(1):21–3. doi: 10.1007/s12104-007-9074-4
87. Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, et al. Fumarates Promote Cytoprotection of Central Nervous System Cells Against Oxidative Stress via the Nuclear Factor (Erythroid-Derived 2)-Like 2 Pathway. J Pharmacol Exp Ther (2012) 341(1):274–84. doi: 10.1124/jpet.111.190132
88. Albrecht P, Bouchachia I, Goebels N, Henke N, Hofstetter HH, Issberner A, et al. Effects of Dimethyl Fumarate on Neuroprotection and Immunomodulation. J Neuroinflamm (2012) 9:163. doi: 10.1186/1742-2094-9-163
89. Phillips JT, Fox RJ. BG-12 in Multiple Sclerosis. Semin Neurol (2013) 33(1):56–65. doi: 10.1055/s-0033-1343796
90. Lei CQ, Zhong B, Zhang Y, Zhang J, Wang S, Shu HB. Glycogen Synthase Kinase 3β Regulates IRF3 Transcription Factor-Mediated Antiviral Response via Activation of the Kinase TBK1. Immunity (2010) 33(6):878–89. doi: 10.1016/j.immuni.2010.11.021
91. Jope RS, Johnson GV. The Glamour and Gloom of Glycogen Synthase Kinase-3. Trends Biochem Sci (2004) 29(2):95–102. doi: 10.1016/j.tibs.2003.12.004
92. Mamaghani S, Patel S, Hedley DW. Glycogen Synthase Kinase-3 Inhibition Disrupts Nuclear factor-kappaB Activity in Pancreatic Cancer, But Fails to Sensitize to Gemcitabine Chemotherapy. BMC Cancer (2009) 9:132. doi: 10.1186/1471-2407-9-132
93. Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen Synthase Kinase-3beta Participates in Nuclear Factor kappaB-Mediated Gene Transcription and Cell Survival in Pancreatic Cancer Cells. Cancer Res (2005) 65(6):2076–81. doi: 10.1158/0008-5472.CAN-04-3642
94. Edderkaoui M, Chheda C, Soufi B, Zayou F, Hu RW, Ramanujan VK, et al. An Inhibitor of GSK3B and HDACs Kills Pancreatic Cancer Cells and Slows Pancreatic Tumor Growth and Metastasis in Mice. Gastroenterology (2018) 155(6):1985–98.e5. doi: 10.1053/j.gastro.2018.08.028
95. Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX, Shetuni B, et al. ERalpha Signaling Through Slug Regulates E-Cadherin and EMT. Oncogene (2010) 29(10):1451–62. doi: 10.1038/onc.2009.433
96. Tao NN, Zhang ZZ, Ren JH, Zhang J, Zhou YJ, Wai Wong VK, et al. Overexpression of Ubiquitin-Conjugating Enzyme E2 L3 in Hepatocellular Carcinoma Potentiates Apoptosis Evasion by Inhibiting the GSK3β/P65 Pathway. Cancer Lett (2020) 481:1–14. doi: 10.1016/j.canlet.2020.03.028
97. Ishida N, Nakagawa T, Iemura SI, Yasui A, Shima H, Katoh Y, et al. Ubiquitylation of Ku80 by RNF126 Promotes Completion of Nonhomologous End Joining-Mediated DNA Repair. Mol Cell Biol (2017) 37(4):e00347–16. doi: 10.1128/MCB.00347-16
98. Heyer W-D, Ehmsen KT, Liu J. Regulation of Homologous Recombination in Eukaryotes. Annu Rev Genet (2010) 44:113–39. doi: 10.1146/annurev-genet-051710-150955
99. Lieber MR. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu Rev Biochem (2010) 79:181–211. doi: 10.1146/annurev.biochem.052308.093131
100. Han X, Zhang L, Chung J, Mayca Pozo F, Tran A, Seachrist DD, et al. UbcH7 Regulates 53BP1 Stability and DSB Repair. Proc Natl Acad Sci USA (2014) 111(49):17456–61. doi: 10.1073/pnas.1408538111
101. Kentsis A, Gordon RE, Borden KLB. Control of Biochemical Reactions Through Supramolecular RING Domain Self-Assembly. Proc Natl Acad Sci USA (2002) 99(24):15404–9. doi: 10.1073/pnas.202608799
102. Mah LJ, El-Osta A, Karagiannis TC. Gammah2ax: A Sensitive Molecular Marker of DNA Damage and Repair. Leukemia (2010) 24(4):679–86. doi: 10.1038/leu.2010.6
103. Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM. RING Fingers Mediate Ubiquitin-Conjugating Enzyme (E2)-Dependent Ubiquitination. Proc Natl Acad Sci USA (1999) 96(20):11364–9. doi: 10.1073/pnas.96.20.11364
104. Chen A, Kleiman FE, Manley JL, Ouchi T, Pan ZQ. Autoubiquitination of the BRCA1*BARD1 RING Ubiquitin Ligase. J Biol Chem (2002) 277(24):22085–92. doi: 10.1074/jbc.M201252200
105. Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, Klevit RE. A UbcH5/ubiquitin Noncovalent Complex Is Required for Processive BRCA1-Directed Ubiquitination. Mol Cell (2006) 21(6):873–80. doi: 10.1016/j.molcel.2006.02.008
106. Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, et al. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol Cell (2013) 49(5):858–71. doi: 10.1016/j.molcel.2013.01.002
107. Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol Cell (2013) 49(5):872–83. doi: 10.1016/j.molcel.2013.01.001
108. Panier S, Boulton SJ. Double-Strand Break Repair: 53BP1 Comes Into Focus. Nat Rev Mol Cell Biol (2014) 15(1):7–18. doi: 10.1038/nrm3719
109. Goodarzi AA, Jeggo PA. The Repair and Signaling Responses to DNA Double-Strand Breaks. Adv Genet (2013) 82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9
110. Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated With Triple-Negative and BRCA-Mutated Breast Cancers. Nat Struct Mol Biol (2010) 17(6):688–95. doi: 10.1038/nsmb.1831
111. Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell (2010) 141(2):243–54. doi: 10.1016/j.cell.2010.03.012
112. Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, et al. A Selective Requirement for 53BP1 in the Biological Response to Genomic Instability Induced by Brca1 Deficiency. Mol Cell (2009) 35(4):534–41. doi: 10.1016/j.molcel.2009.06.037
113. Dikic I. Proteasomal and Autophagic Degradation Systems. Annu Rev Biochem (2017) 86:193–224. doi: 10.1146/annurev-biochem-061516-044908
114. Finley D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu Rev Biochem (2009) 78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607
115. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular Definitions of Autophagy and Related Processes. EMBO J (2017) 36(13):1811–36. doi: 10.15252/embj.201796697
116. Zaffagnini G, Martens S. Mechanisms of Selective Autophagy. J Mol Biol (2016) 428(9 Pt A):1714–24. doi: 10.1016/j.jmb.2016.02.004
117. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic Levels of P62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell (2007) 131(6):1149–63. doi: 10.1016/j.cell.2007.10.035
118. Shin J. P62 and the Sequestosome, a Novel Mechanism for Protein Metabolism. Arch Pharmacal Res (1998) 21(6):629–33. doi: 10.1007/BF02976748
119. Demishtein A, Fraiberg M, Berko D, Tirosh B, Elazar Z, Navon A. SQSTM1/p62-Mediated Autophagy Compensates for Loss of Proteasome Polyubiquitin Recruiting Capacity. Autophagy (2017) 13(10):1697–708. doi: 10.1080/15548627.2017.1356549
120. Yang J, Peng H, Xu Y, Xie X, Hu R. SQSTM1/p62 (Sequestosome 1) Senses Cellular Ubiquitin Stress Through E2-Mediated Ubiquitination. Autophagy (2018) 14(6):1072–3. doi: 10.1080/15548627.2017.1332566
121. Geisler S, Vollmer S, Golombek S, Kahle PJ. The Ubiquitin-Conjugating Enzymes UBE2N, UBE2L3 and UBE2D2/3 Are Essential for Parkin-Dependent Mitophagy. J Cell Sci (2014) 127(Pt 15):3280–93. doi: 10.1242/jcs.146035
122. Fiesel FC, Moussaud-Lamodière EL, Ando M, Springer W. A Specific Subset of E2 Ubiquitin-Conjugating Enzymes Regulate Parkin Activation and Mitophagy Differently. J Cell Sci (2014) 127(Pt 16):3488–504. doi: 10.1242/jcs.147520
123. Luo Q, Beaver JM, Liu Y, Zhang Z. Dynamics of P53: A Master Decider of Cell Fate. Genes (2017) 8(2):66. doi: 10.3390/genes8020066
124. May P, May E. Twenty Years of P53 Research: Structural and Functional Aspects of the P53 Protein. Oncogene (1999) 18(53):7621–36. doi: 10.1038/sj.onc.1203285
125. Kwon SK, Saindane M, Baek KH. P53 Stability Is Regulated by Diverse Deubiquitinating Enzymes. Biochim Biophys Acta Rev Cancer (2017) 1868(2):404–11. doi: 10.1016/j.bbcan.2017.08.001
126. Wang L, He G, Zhang P, Wang X, Jiang M, Yu L. Interplay Between MDM2, MDMX, Pirh2 and COP1: The Negative Regulators of P53. Mol Biol Rep (2011) 38(1):229–36. doi: 10.1007/s11033-010-0099-x
128. Wang Z, Yang B, Dong L, Peng B, He X, Liu W. A Novel Oncoprotein Pirh2: Rising From the Shadow of MDM2. Cancer Sci (2011) 102(5):909–17. doi: 10.1111/j.1349-7006.2011.01899.x
129. Yi SA, Lee DH, Kim GW, Ryu HW, Park JW, Lee J, et al. HPV-Mediated Nuclear Export of HP1γ Drives Cervical Tumorigenesis by Downregulation of P53. Cell Death Differentiation (2020) 27(9):2537–51. doi: 10.1038/s41418-020-0520-5
130. Ciechanover A, Shkedy D, Oren M, Bercovich B. Degradation of the Tumor Suppressor Protein P53 by the Ubiquitin-Mediated Proteolytic System Requires a Novel Species of Ubiquitin-Carrier Protein, E2. J Biol Chem (1994) 269(13):9582–9. doi: 10.1016/S0021-9258(17)36921-1
131. Nuber U, Schwarz S, Kaiser P, Schneider R, Scheffner M. Cloning of Human Ubiquitin-Conjugating Enzymes UbcH6 and UbcH7 (E2-F1) and Characterization of Their Interaction With E6-AP and RSP5. J Biol Chem (1996) 271(5):2795–800. doi: 10.1074/jbc.271.5.2795
132. Reyes-Hernández OD, Mejía-García A, Sánchez-Ocampo EM, Cabañas-Cortés MA, Ramírez P, Chávez-González L, et al. Ube2l3 Gene Expression Is Modulated by Activation of the Aryl Hydrocarbon Receptor: Implications for P53 Ubiquitination. Biochem Pharmacol (2010) 80(6):932–40. doi: 10.1016/j.bcp.2010.05.007
133. Moynihan TP, Ardley HC, Leek JP, Thompson J, Brindle NS, Markham AF, et al. Characterization of a Human Ubiquitin-Conjugating Enzyme Gene UBE2L3. Mamm Genome (1996) 7(7):520–5. doi: 10.1007/s003359900155
134. Harbers K, Müller U, Grams A, Li E, Jaenisch R, Franz T. Provirus Integration Into a Gene Encoding a Ubiquitin-Conjugating Enzyme Results in a Placental Defect and Embryonic Lethality. Proc Natl Acad Sci USA (1996) 93(22):12412–7. doi: 10.1073/pnas.93.22.12412
135. Whitcomb EA, Dudek EJ, Liu Q, Taylor A. Novel Control of S Phase of the Cell Cycle by Ubiquitin-Conjugating Enzyme H7. Mol Biol Cell (2009) 20(1):1–9. doi: 10.1091/mbc.e08-01-0036
136. Guo W, Shang F, Liu Q, Urim L, West-Mays J, Taylor A. Differential Regulation of Components of the Ubiquitin-Proteasome Pathway During Lens Cell Differentiation. Invest Ophthalmol Vis Sci (2004) 45(4):1194–201. doi: 10.1167/iovs.03-0830
137. Orozco G, Eyre S, Hinks A, Bowes J, Morgan AW, Wilson AG, et al. Study of the Common Genetic Background for Rheumatoid Arthritis and Systemic Lupus Erythematosus. Ann Rheum Dis (2011) 70(3):463–8. doi: 10.1136/ard.2010.137174
138. Wang Y, Zhu YF, Wang Q, Xu J, Yan N, Xu J, et al. The Haplotype of UBE2L3 Gene Is Associated With Hashimoto’s Thyroiditis in a Chinese Han Population. BMC Endocr Disord (2016) 16:18. doi: 10.1186/s12902-016-0098-6
139. Fransen K, Visschedijk MC, van Sommeren S, Fu JY, Franke L, Festen EA, et al. Analysis of SNPs With an Effect on Gene Expression Identifies UBE2L3 and BCL3 as Potential New Risk Genes for Crohn’s Disease. Hum Mol Genet (2010) 19(17):3482–8. doi: 10.1093/hmg/ddq264
140. Wang X-S, Shankar S, Dhanasekaran SM, Ateeq B, Sasaki AT, Jing X, et al. Characterization of KRAS Rearrangements in Metastatic Prostate Cancer. Cancer Discovery (2011) 1(1):35–43. doi: 10.1158/2159-8274.CD-10-0022
141. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing Innate Immunity in Cancer Therapy. Nature (2019) 574(7776):45–56. doi: 10.1038/s41586-019-1593-5
142. Vlachiotis S, Andreakos E. Lambda Interferons in Immunity and Autoimmunity. J Autoimmun (2019) 104:102319. doi: 10.1016/j.jaut.2019.102319
143. Taniguchi K, Karin M. NF-κb, Inflammation, Immunity and Cancer: Coming of Age. Nat Rev Immunol (2018) 18(5):309–24. doi: 10.1038/nri.2017.142
144. Zhang Y, Wang YF, Yang J, Zhang J, Sun L, Hirankarn N, et al. Meta-Analysis of Two Chinese Populations Identifies an Autoimmune Disease Risk Allele in 22q11.21 as Associated With Systemic Lupus Erythematosus. Arthritis Res Ther (2015) 17:67. doi: 10.1186/s13075-015-0577-6
145. Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A Large-Scale Replication Study Identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as Risk Loci for Systemic Lupus Erythematosus. Nat Genet (2009) 41(11):1228–33. doi: 10.1038/ng.468
146. Han J-W, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-Wide Association Study in a Chinese Han Population Identifies Nine New Susceptibility Loci for Systemic Lupus Erythematosus. Nat Genet (2009) 41(11):1234–7. doi: 10.1038/ng.472
147. Lewis MJ, Vyse S, Shields AM, Boeltz S, Gordon PA, Spector TD, et al. UBE2L3 Polymorphism Amplifies NF-κb Activation and Promotes Plasma Cell Development, Linking Linear Ubiquitination to Multiple Autoimmune Diseases. Am J Hum Genet (2015) 96(2):221–34. doi: 10.1016/j.ajhg.2014.12.024
148. Wang S, Adrianto I, Wiley GB, Lessard CJ, Kelly JA, Adler AJ, et al. A Functional Haplotype of UBE2L3 Confers Risk for Systemic Lupus Erythematosus. Genes Immun (2012) 13(5):380–7. doi: 10.1038/gene.2012.6
149. Zhernakova A, Stahl EA, Trynka G, Raychaudhuri S, Festen EA, Franke L, et al. Meta-Analysis of Genome-Wide Association Studies in Celiac Disease and Rheumatoid Arthritis Identifies Fourteen Non-HLA Shared Loci. PloS Genet (2011) 7(2):e1002004. doi: 10.1371/journal.pgen.1002004
150. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-Wide Association Study Meta-Analysis Identifies Seven New Rheumatoid Arthritis Risk Loci. Nat Genet (2010) 42(6):508–14. doi: 10.1038/ng.582
151. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of Rheumatoid Arthritis Contributes to Biology and Drug Discovery. Nature (2014) 506(7488):376–81. doi: 10.1038/nature12873
152. Hinks A, Cobb J, Marion MC, Prahalad S, Sudman M, Bowes J, et al. Dense Genotyping of Immune-Related Disease Regions Identifies 14 New Susceptibility Loci for Juvenile Idiopathic Arthritis. Nat Genet (2013) 45(6):664–9. doi: 10.1038/ng.2614
153. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
154. Yang S-K, Hong M, Zhao W, Jung Y, Tayebi N, Ye BD, et al. Genome-Wide Association Study of Ulcerative Colitis in Koreans Suggests Extensive Overlapping of Genetic Susceptibility With Caucasians. Inflamm Bowel Dis (2013) 19(5):954–66. doi: 10.1097/MIB.0b013e3182802ab6
155. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-Wide Meta-Analysis Increases to 71 the Number of Confirmed Crohn’s Disease Susceptibility Loci. Nat Genet (2010) 42(12):1118–25. doi: 10.1038/ng.717
156. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 New Psoriasis Susceptibility Loci Highlights the Role of Innate Immunity. Nat Genet (2012) 44(12):1341–8. doi: 10.1038/ng.2467
157. Hu Z, Liu Y, Zhai X, Dai J, Jin G, Wang L, et al. New Loci Associated With Chronic Hepatitis B Virus Infection in Han Chinese. Nat Genet (2013) 45(12):1499–503. doi: 10.1038/ng.2809
158. Dubois PCA, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple Common Variants for Celiac Disease Influencing Immune Gene Expression. Nat Genet (2010) 42(4):295–302. doi: 10.1038/ng.543
159. Hasebe N, Kawasaki A, Ito I, Kawamoto M, Hasegawa M, Fujimoto M, et al. Association of UBE2L3 Polymorphisms With Diffuse Cutaneous Systemic Sclerosis in a Japanese Population. Ann Rheum Dis (2012) 71(7):1259–60. doi: 10.1136/annrheumdis-2011-201091
160. Dieudé P, Guedj M, Wipff J, Ruiz B, Riemekasten G, Matucci-Cerinic M, et al. Association of the TNFAIP3 Rs5029939 Variant With Systemic Sclerosis in the European Caucasian Population. Ann Rheum Dis (2010) 69(11):1958–64. doi: 10.1136/ard.2009.127928
161. Allanore Y, Saad M, Dieudé P, Avouac J, Distler JH, Amouyel P, et al. Genome-Wide Scan Identifies TNIP1, PSORS1C1, and RHOB as Novel Risk Loci for Systemic Sclerosis. PloS Genet (2011) 7(7):e1002091. doi: 10.1371/journal.pgen.1002091
162. Chistiakov DA, Savost’anov KV, Baranov AA. Genetic Background of Juvenile Idiopathic Arthritis. Autoimmunity (2014) 47(6):351–60. doi: 10.3109/08916934.2014.889119
163. Chiaroni-Clarke RC, Munro JE, Chavez RA, Pezic A, Allen RC, Akikusa JD, et al. Independent Confirmation of Juvenile Idiopathic Arthritis Genetic Risk Loci Previously Identified by Immunochip Array Analysis. Pediatr Rheumatol Online J (2014) 12:53. doi: 10.1186/1546-0096-12-53
164. Garcea G, Manson MM, Neal CP, Pattenden CJ, Sutton CD, Dennison AR, et al. Glycogen Synthase Kinase-3 Beta; a New Target in Pancreatic Cancer? Curr Cancer Drug Targets (2007) 7(3):209–15. doi: 10.2174/156800907780618266
165. Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, et al. GSK-3 Beta Targets Cdc25A for Ubiquitin-Mediated Proteolysis, and GSK-3 Beta Inactivation Correlates With Cdc25A Overproduction in Human Cancers. Cancer Cell (2008) 13(1):36–47. doi: 10.1016/j.ccr.2007.12.002
166. Zhou Y, Uddin S, Zimmerman T, Kang JA, Ulaszek J, Wickrema A. Growth Control of Multiple Myeloma Cells Through Inhibition of Glycogen Synthase Kinase-3. Leukemia Lymphoma (2008) 49(10):1945–53. doi: 10.1080/10428190802304966
167. Hu W, Feng Z, Teresky AK, Levine AJ. P53 Regulates Maternal Reproduction Through LIF. Nature (2007) 450(7170):721–4. doi: 10.1038/nature05993
168. Kruiswijk F, Labuschagne CF, Vousden KH. P53 in Survival, Death and Metabolic Health: A Lifeguard With a Licence to Kill. Nat Rev Mol Cell Biol (2015) 16(7):393–405. doi: 10.1038/nrm4007
169. Tanikawa C, Zhang YZ, Yamamoto R, Tsuda Y, Tanaka M, Funauchi Y, et al. The Transcriptional Landscape of P53 Signalling Pathway. EBioMedicine (2017) 20:109–19. doi: 10.1016/j.ebiom.2017.05.017
170. Khan H, Reale M, Ullah H, Sureda A, Tejada S, Wang Y, et al. Anti-Cancer Effects of Polyphenols via Targeting P53 Signaling Pathway: Updates and Future Directions. Biotechnol Adv (2020) 38:107385. doi: 10.1016/j.biotechadv.2019.04.007
171. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 Oncoprotein Encoded by Human Papillomavirus Types 16 and 18 Promotes the Degradation of P53. Cell (1990) 63(6):1129–36. doi: 10.1016/0092-8674(90)90409-8
172. Werness BA, Levine AJ, Howley PM. Association of Human Papillomavirus Types 16 and 18 E6 Proteins With P53. Sci (New York NY) (1990) 248(4951):76–9. doi: 10.1126/science.2157286
173. Martinez-Zapien D, Ruiz FX, Poirson J, Mitschler A, Ramirez J, Forster A, et al. Structure of the E6/E6AP/p53 Complex Required for HPV-Mediated Degradation of P53. Nature (2016) 529(7587):541–5. doi: 10.1038/nature16481
174. Vogt M, Butz K, Dymalla S, Semzow J, Hoppe-Seyler F. Inhibition of Bax Activity Is Crucial for the Antiapoptotic Function of the Human Papillomavirus E6 Oncoprotein. Oncogene (2006) 25(29):4009–15. doi: 10.1038/sj.onc.1209429
175. Flores-Pérez A, Elizondo G. Apoptosis Induction and Inhibition of HeLa Cell Proliferation by Alpha-Naphthoflavone and Resveratrol Are Aryl Hydrocarbon Receptor-Independent. Chemico Biol Interact (2018) 281:98–105. doi: 10.1016/j.cbi.2017.12.029
176. Mayca Pozo F, Tang J, Bonk KW, Keri RA, Yao X, Zhang Y. Regulatory Cross-Talk Determines the Cellular Levels of 53BP1 Protein, a Critical Factor in DNA Repair. J Biol Chem (2017) 292(14):5992–6003. doi: 10.1074/jbc.M116.760645
177. Ma X, Zhao J, Yang F, Liu H, Qi W. Ubiquitin Conjugating Enzyme E2 L3 Promoted Tumor Growth of NSCLC Through Accelerating P27kip1 Ubiquitination and Degradation. Oncotarget (2017) 8(48):84193–203. doi: 10.18632/oncotarget.20449
178. Dawson TM, Dawson VL. The Role of Parkin in Familial and Sporadic Parkinson’s Disease. Movement Disorders: Off J Movement Disord Soc (2010) 25 Suppl 1:S32–9. doi: 10.1002/mds.22798
179. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature (1998) 392(6676):605–8. doi: 10.1038/33416
180. Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, et al. Autoregulation of Parkin Activity Through Its Ubiquitin-Like Domain. EMBO J (2011) 30(14):2853–67. doi: 10.1038/emboj.2011.204
181. Spratt DE, Martinez-Torres RJ, Noh YJ, Mercier P, Manczyk N, Barber KR, et al. A Molecular Explanation for the Recessive Nature of Parkin-Linked Parkinson’s Disease. Nat Commun (2013) 4:1983. doi: 10.1038/ncomms2983
182. Trempe J-F, Sauvé V, Grenier K, Seirafi M, Tang MY, Ménade M, et al. Structure of Parkin Reveals Mechanisms for Ubiquitin Ligase Activation. Sci (New York NY) (2013) 340(6139):1451–5. doi: 10.1126/science.1237908
183. Wauer T, Komander D. Structure of the Human Parkin Ligase Domain in an Autoinhibited State. EMBO J (2013) 32(15):2099–112. doi: 10.1038/emboj.2013.125
184. Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 Is Activated by Mitochondrial Membrane Potential Depolarization and Stimulates Parkin E3 Ligase Activity by Phosphorylating Serine 65. Open Biol (2012) 2(5):120080. doi: 10.1098/rsob.120080
185. Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, et al. Parkin-Catalyzed Ubiquitin-Ester Transfer Is Triggered by PINK1-Dependent Phosphorylation. J Biol Chem (2013) 288(30):22019–32. doi: 10.1074/jbc.M113.467530
186. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity. J Cell Biol (2014) 205(2):143–53. doi: 10.1083/jcb.201402104
187. Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, et al. Parkin Is Activated by PINK1-Dependent Phosphorylation of Ubiquitin at Ser65. Biochem J (2014) 460(1):127–39. doi: 10.1042/BJ20140334
188. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin Is Phosphorylated by PINK1 to Activate Parkin. Nature (2014) 510(7503):162–6. doi: 10.1038/nature13392
Keywords: Ubiquitination, UBE2L3, NF-κB, p53, p62, cancer, immune diseases, Parkinson’s disease
Citation: Zhang X, Huo C, Liu Y, Su R, Zhao Y and Li Y (2022) Mechanism and Disease Association With a Ubiquitin Conjugating E2 Enzyme: UBE2L3. Front. Immunol. 13:793610. doi: 10.3389/fimmu.2022.793610
Received: 12 October 2021; Accepted: 24 January 2022;
Published: 21 February 2022.
Edited by:
Brian North, Creighton University, United StatesReviewed by:
Hasem Habelhah, The University of Iowa, United StatesCopyright © 2022 Zhang, Huo, Liu, Su, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yumin Li, bGl5bUBsenUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.