 Shivangi Rastogi
Shivangi Rastogi Volker Briken
Volker Briken- Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, MD, United States
The inflammasome complex is important for host defense against intracellular bacterial infections. Mycobacterium tuberculosis (Mtb) is a facultative intracellular bacterium which is able to survive in infected macrophages. Here we discuss how the host cell inflammasomes sense Mtb and other related mycobacterial species. Furthermore, we describe the molecular mechanisms of NLRP3 inflammasome sensing of Mtb which involve the type VII secretion system ESX-1, cell surface lipids (TDM/TDB), secreted effector proteins (LpqH, PPE13, EST12, EsxA) and double-stranded RNA acting on the priming and/or activation steps of inflammasome activation. In contrast, Mtb also mediates inhibition of the NLRP3 inflammasome by limiting exposure of cell surface ligands via its hydrolase, Hip1, by inhibiting the host cell cathepsin G protease via the secreted Mtb effector Rv3364c and finally, by limiting intracellular triggers (K+ and Cl- efflux and cytosolic reactive oxygen species production) via its serine/threonine kinase PknF. In addition, Mtb inhibits the AIM2 inflammasome activation via an unknown mechanism. Overall, there is good evidence for a tug-of-war between Mtb trying to limit inflammasome activation and the host cell trying to sense Mtb and activate the inflammasome. The detailed molecular mechanisms and the importance of inflammasome activation for virulence of Mtb or host susceptibility have not been fully investigated.
Introduction
Tuberculosis (TB) is a major cause of morbidity and mortality with approximately 10 million new cases and 1-2 million deaths, annually (1). The disease is caused by the human pathogen Mycobacterium tuberculosis (Mtb) which is transmitted via aerosol from the lung of an infected individual to the naïve bystander. Current chemotherapy leads to positive outcomes in about 85% of the patients but takes 6-9 months to complete and the success rate drops dramatically if drug resistant strains of Mtb are the cause of the infection (2). Consequently, the search for better antibiotics and more efficient treatment regiments is of great interest. In addition, host-directed therapy (HDT) is a complementary approach to improving clinical outcomes by targeting host signaling pathways that, for example, support bacterial replication or cause immune pathologies (3). For the latter approach to be successful, a more detailed understanding of host responses to infection with Mtb and their importance for host protection or susceptibility is required. The cytokine Interleukin (IL)-1β is of crucial importance for host resistance to Mtb. In this review we will provide some background information on mechanisms of inflammasome activation which leads to the generation of IL-1β, summarize the findings of the importance of IL-1β for host resistance to Mtb infections and their potential for host-targeted therapeutic approaches and finally we will focus on how the inflammasome detects various mycobacterial species and how Mtb is able to inhibit inflammasome activation (summarized in Table 1 and Figure 2).
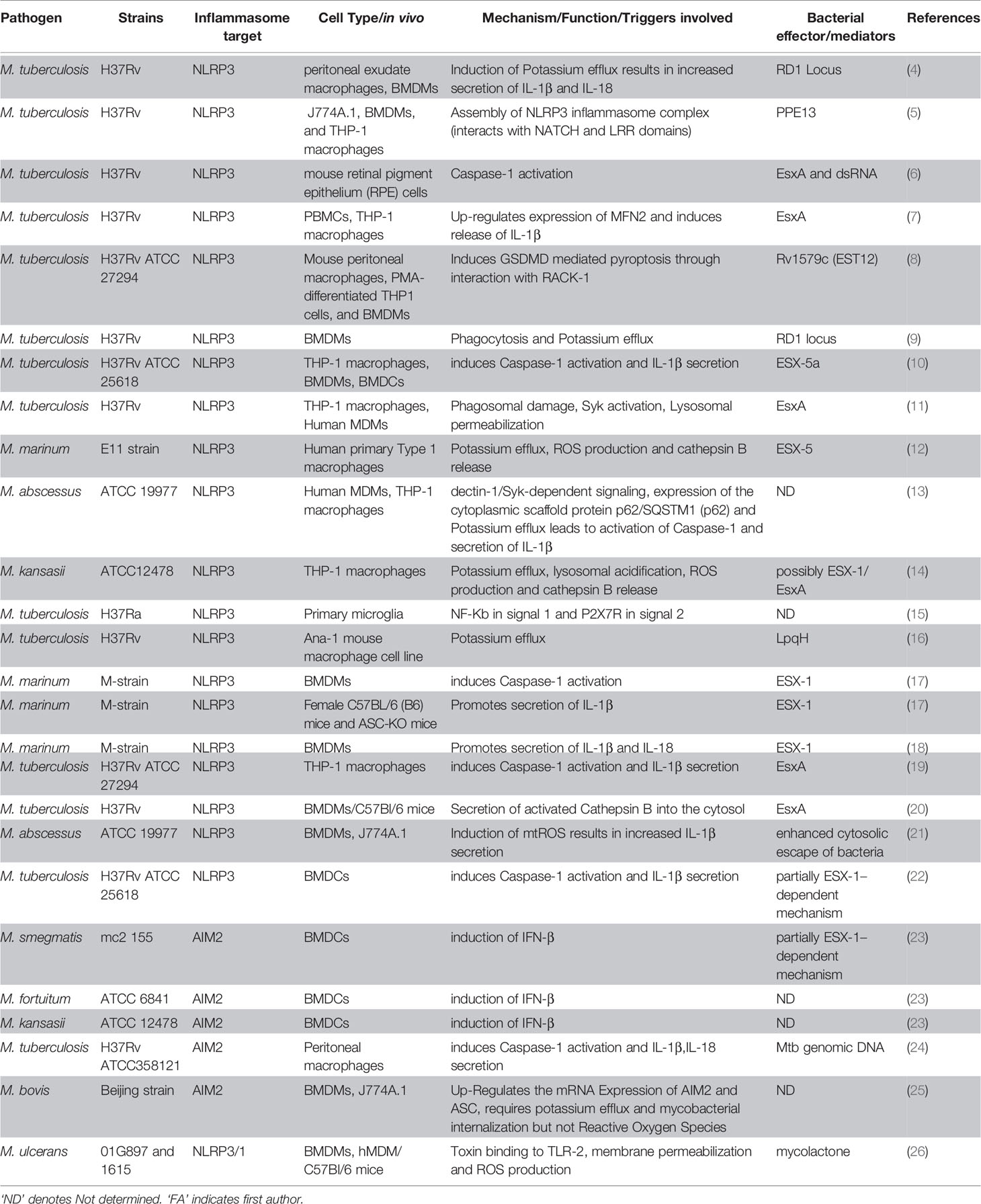
Table 1 Summary overview of different mycobacterial species and effectors involved in activation of either NLRP3 or AIM2 inflammasome.
Overview of Mechanisms of Inflammasome Activation and Its Consequences
In this section we will provide a concise overview of the components and signaling pathways involving mainly two (NLRP3, AIM2) kinds of inflammasomes since they are most relevant to our subsequent discussion on interaction of mycobacteria with inflammasomes (see also Figure 1). We would encourage the interested reader to follow-up on this brief overview with any of the excellent specialized reviews on the topic for an in-depth discussion (27–36).
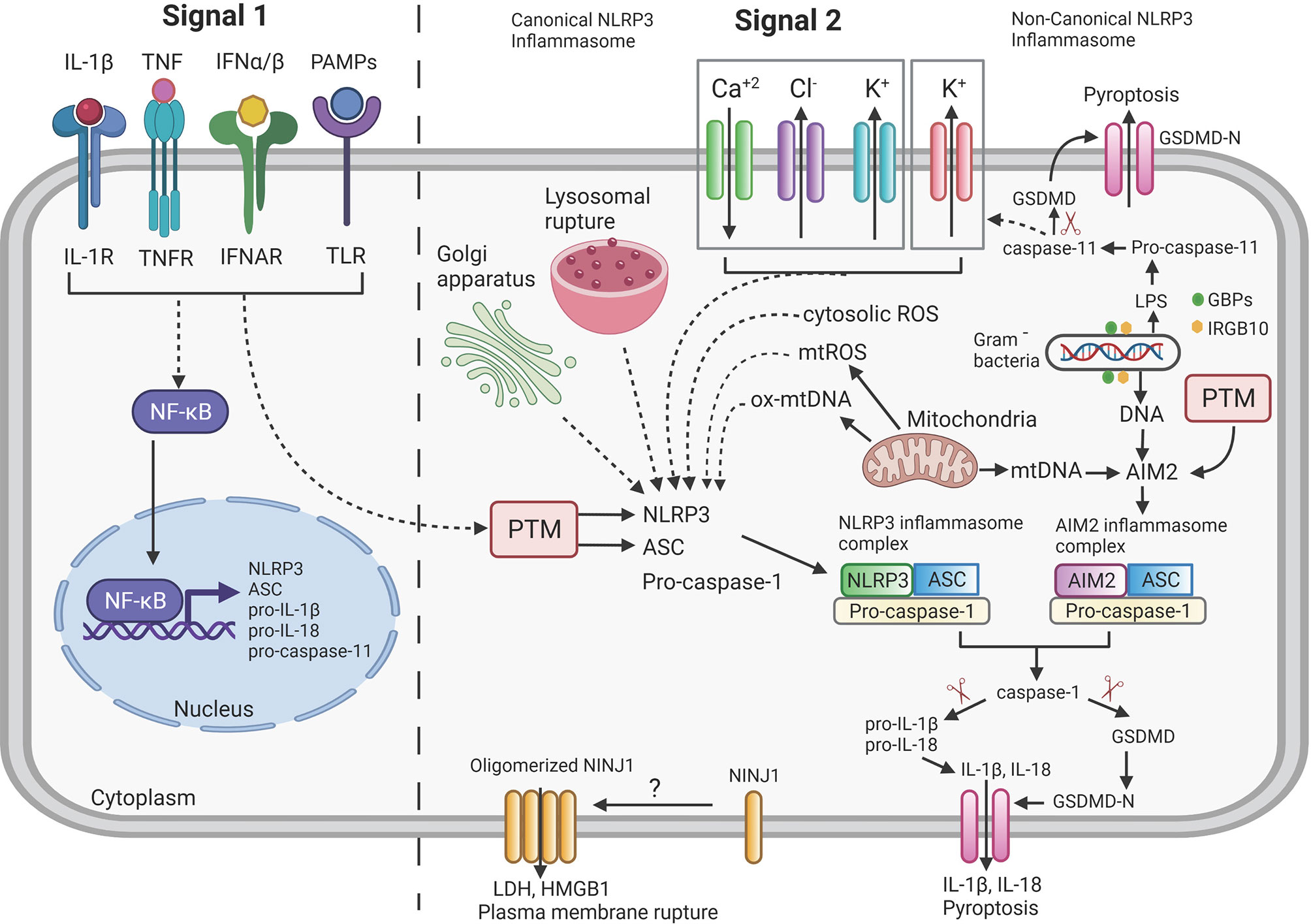
Figure 1 Overview of mechanism of Inflammasome signaling pathway. NLRP3 and AIM2 inflammasome activation requires two distinct signals: Signal 1 (priming signal, left) is induced by the detection of pathogen-associated molecular patterns (PAMPs) or endogenous cytokines by the Toll-like receptor (TLR) or cytokine receptors (IL-1R, TNFR, IFNAR) and thus leading to increased transcription of NLRP3, ASC, pro-IL-1β, pro-IL-18 and pro-caspase-11 through activation of NF-κB. Signal 2 (activation signal, right) for the NLRP3 inflammasome is triggered by various stimuli such as potassium (K+) efflux, chloride (Cl-) efflux, calcium (Ca+2) influx, oxidized mitochondrial DNA (ox-mtDNA), lysosomal rupture and intracellular reactive oxygen species (ROS) production. All these triggers lead to oligomerization and assembly of NLRP3 inflammasome complex. AIM2 directly recognizes either DNA released from Gram negative bacteria or mtDNA released from mitochondria and lead to assembly of AIM2 inflammasome complex. Activated inflammasome complexes (NLRP3 or AIM2) recruit and cleave pro-caspase-1 to active caspase-1 that further results in the proteolytic cleavage of pro-IL-1β and pro-IL-18 to the mature forms IL-1β and IL-18. Caspase-1 also cleaves gasdermin D (GSDMD) to its pore-forming N-terminal fragment GSDMD-N which results in pyroptosis. Ninjurin-1 (NINJ1) by unknown mechanism as indicated by “?” oligomerizes and forms a pore to facilitate release of LDH, HMGB1 and the accumulation of these pores ultimately led to plasma membrane rupture which is not achieved by the GSDMD-N pores. Post-translational modifications (PTM) of cytosolic sensors (NLRP3/AIM2) and adaptor protein (ASC) regulate the activity of inflammasome. Gram negative bacteria are lysed to releases LPS and DNA via a mechanism requiring various interferon-inducible Guanylate-binding proteins (GBPs) and IRGB10. The LPS binds to pro-caspase-11 to initiate autocleavage into active caspase-11 which further cleaves GSDMD to GSDMD-N and results in pyroptosis (indicated as Non-Canonical NLRP3 inflammasome) and increased efflux of K+, thus further activating the canonical NLRP3 inflammasome pathway (dashed lines = indirect interaction; solid lines = direct interaction; arrowhead = activation). Created with Biorender.com.
As opposed to the membrane-bound pathogen recognition receptors (PRRs) such as Toll-like receptors (TLRs) that survey extracellular pathogen components, the nucleotide binding and oligomerization domain-like receptors (NLRs, e.g. NLRP3) and Absent in Melanoma 2 (AIM2)-like receptors (ALRs, e.g. AIM2) survey the host cell cytosol for the presence of pathogens (27, 37–41). Upon binding of NLRs/ALRs to pathogen or danger associated molecular patterns (P/DAMP), they initiate the formation the inflammasome (Signal 2: Figure 1). IL-1β is a potent immunomodulator and its overproduction may cause rheumatoid arthritis and other pathologies (42, 43). Consequently, IL-1β production is highly regulated and in addition to the inflammasome activation pathway another signaling pathway (Signal 1: Figure 1) needs to be engaged to achieve production of mature IL-1β. This pathway involves other PRRs, for example, TLRs or cytokine receptors such as receptors for IL-1β and Tumor Necrosis Factor (TNF) which, after ligand binding, activate NFκB, resulting in the transcriptional activation of, for example, the IL1B gene to increase protein levels of pro-IL-1β (44). There is also a crosstalk of Signal 1 with Signal 2 via the activation of proteins that perform posttranslational modifications (PTM) of inflammasome components (44–46) (Figure 1).
Once Signal 1 and Signal 2 are activated, in case of AIM2 or NLRP3, they associate with the adapter molecule Apoptosis-associated speck-like protein containing a CARD (ASC) which recruits pro-caspase-1 into a complex that continues to oligomerize to form in some cases structures called “specks”, measuring 1-2μm in diameter (47–49). The formed inflammasome complex supports the self-cleavage of pro-caspase-1 into active caspase-1 which cleaves pro-IL-1β and pro-IL-18 and gasdermin D (GSDMD) to release the N-terminal fragment that is capable of oligomerization and membrane pore formation (50–52). The GSDMD pores in the cell membrane will lead to pyroptosis and cytokine release but they do not allow for plasma membrane rupture and secretion of higher molecular weight proteins and protein complexes (e.g., HMGB1) (53) (Figure 1). More recently, a complementary role in pore generation after inflammasome activation has been determined for gasdermin E (54).
Signal 2: Activation of the NLRP3 and AIM2 Inflammasomes
The activation of the NLRP3 inflammasome is complex and one of the reasons is that no ligand that physically binds to NLPR3 has yet been identified. Instead, the prevailing model is that NLRP3 is a stress sensor of the cell which reacts to the increase of various cellular stress signals (32) (Figure 1). A major trigger shared across many activating stimuli is the efflux of potassium ions (K+) (55). Additional triggers are the efflux of chloride ions (Cl-) (56–58), mobilization of intracellular calcium ions (Ca2+) (59–61), increase in intracellular reactive oxygen species (ROS) (62) or the release of oxidized mitochondrial DNA (63) [for review (41, 45, 46, 64)] (Figure 1). The non-canonical NLRP3 inflammasome pathway (for review (65)) targets caspase-11 in mice and caspases-4/5 in humans (66) and is dependent on the TRIF-mediated induction of interferon (IFN) production and subsequent IFN-receptor mediated signaling (67, 68). Ultimately, the activation of non-canonical pathway is triggered by the presence of intracellular lipopolysaccharide (LPS) of gram-negative bacteria (69, 70) which directly binds to and activates caspases-4/5/11 (71) (Figure 1). IFN-stimulated genes, such as the family of guanylate-binding proteins (GBPs), are critical for non-canonical NLRP3 inflammasome activation (72, 73) via mechanisms that involve: attacking the outer membrane of cytosolic bacteria such as Shigella or Fransicella (74–76), releasing intracellular phagosomal bacteria (e.g. Salmonella) into the cytosol (73) or assembly to provide a platform for caspase-4 recruitment and activation on the surface of the bacteria (77, 78). Another IFN-stimulated gene product, IRGB10, is also involved in release LPS from intracellular bacteria (75). After activation, caspase-11, like caspase-1, cleaves GSDMD which will lead to pore formation and pyroptosis but not cytokine maturation (51, 52). The activation of the non-canonical pathway will ultimately also lead to efflux of K+ which will activate the canonical NLRP3 inflammasome pathway (79) (Figure 1).
The signal transduction of the activation of the AIM2 inflammasome is fairly simple because it is mediated by the binding of the HIN-200 domain of AIM2 to DNA (80–82) (Figure 1). The pathogen DNA can be accumulating in the cytosol due to infection of the cell with viruses or they can be generated by degrading bacteria in the cytosol via action of GBPs and IRGB10 (73–76). Cell stress leading to the release of non-oxidized mitochondrial DNA can also activate the AIM2 inflammasome (83) (Figure 1).
Two other important aspects to inflammasome regulation, namely post translational modifications (PTMs) and intracellular location of inflammasome components, will not be discussed in detail here because very little is known about the effect of mycobacterial infection on these two parameters. PTMs in inflammasome activation involves phosphorylation, ubiquitination, sumoylation, S-nitrosylation and ADP-ribosylation of inflammasome components which may lead to either activation or inhibition of the inflammasome formation (44–46, 64, 84). Consequently, the proteins involved in mediating the PTMs are themselves important components of the inflammasome regulatory network. In addition, the subcellular localization of inflammasome components and their association with specific organelles impact activation of the inflammasome (35, 46) (Figure 1).
Inflammasome-Independent Production of IL-1β
It is important to mention that in certain settings mature IL-1β and IL-18 can be generated mostly without the activation of the inflammasome [for review (85, 86)]. The inflammasome-independent IL-1β and IL-18 production is most relevant in an in vivo setting where neutrophils dominate (85, 86). The proteases produced in neutrophils involved in cleavage of pro-IL-1β are: proteinase 3 (87, 88), zinc-dependent metalloproteinase meprin A and its monomer meprin α (89), matrix metalloproteinases-2, -3 and -9 (90) and granzyme A (91).
Another pathway for inflammasome independent generation of mature IL-1β and IL-18 is via activation of the caspase-8 and its subsequent cleavage of pro-IL-1β and pro-IL-18 (31, 92). One possible pathway for caspase-8 activation is through ligand binding to TLR3 or TLR4 which leads to recruitment of TRIF (Toll/IL-1R domain-containing adapter-inducing IFN-β) and subsequent recruitment of receptor interacting protein 1/receptor interacting protein serine/threonine kinase 1 (RIP1/RIPK1), FAS-associated death domain (FADD) and caspase-8 (93–95). The TNF receptor family member Fas can also activate the FADD/caspase-8 pathway to induce mature IL-1β and IL-18 (96, 97). Other studies also implicated the RIPK3 in the caspase-8 activation pathway (98, 99).
Another inflammasome-independent pathway involving caspase-8 is important for IL-1β and IL-18 production in response to fungal pathogens (100). Dectin-1 signals via the tyrosine kinase SYK to induce the formation of a CARD9, Bcl-10, MALT1 and caspase-8 complex which recruits the ASC protein to finally mediate cleavage of pro-IL-1β and -IL-18 (100, 101). Active caspase-8 cleaves pro-IL-1β at the same site that caspase-1 does (93).
The Impact of IL-1β on Host Response During Mtb Infection
Protective Role of IL-1β During Mtb Infections
The role of IL-1β throughout infection of the host with Mtb is complex with some evidence in support of a host protective role and other data supporting a role in increasing host susceptibility (3). First, we will discuss the data demonstrating that IL-1β is of importance for host resistance against infections with Mtb and the possible mechanisms (3, 102–106). Mouse studies demonstrate the hyper susceptibility of mice deficient in the expression of either IL-1α/-β or the IL-1β receptor (107–112). The mechanism of protection conferring host resistance by IL-1β has been proposed to involve cell intrinsic mechanisms via the increase in host cell apoptosis (113) or autophagy signaling (114). The inflammasome activation has been linked to increasing maturation of Mtb-containing phagosomes and thus limiting bacterial growth (115, 116). Nevertheless, another study demonstrates that cell intrinsic mechanisms are not how IL-1β confers host resistance but instead it is mediated via trans-protection of infected cells (117). Although the precise mechanism is unclear, in vivo, a major function of IL-1β seems to be to suppress necrosis of lung cells (110, 118). This seems counterintuitive since inflammasome-mediated IL-1β production is associated with cell death (pyroptosis) but it was shown that, at least in the mouse model, IL-1β production is independent of the inflammasome during Mtb infections (111).
It is well-established that, at least partially, the protective effect of IL-1β in vivo is linked to its capacity to suppress IFN-β expression since increased IFN-β production increases host susceptibility (for review (3, 102, 104, 106, 119, 120). Importantly, IL-1β can suppress IFN-β expression and vice versa (121). This interdependence can be exploited for HDT approaches to boost IL-1β production, reduce IFN-β expression and reduce the associated morbidities and mortalities (118). Interestingly, IFN-β-mediated signaling is associated with increased necrotic cell death during ex vivo Mtb infections which could provide a mechanism for the in vivo observed immunopathologies associated with high IFN-β expression (122). Mtb clinical isolates associated with severe TB evade NLRP3 inflammasome activation suggesting a host protective role of IL-1β (123). Macrophages isolated from patients with inflammatory disease carrying gain-of-function genetic variants in inflammasome genes (NLRP3 and/or CARD8) subsequently infected with Mtb displayed increased growth restriction of Mtb in human macrophages (116). In the zebrafish/Mmar infection model, treatment with the drug clemastine modulates the host innate immunity via potentiation of P2RX7 that enhances calcium transients within infected macrophages in vivo. P2RX7 potentiation augments inflammasome activation, resulting in constraint of mycobacterial growth in zebrafish larvae (124).
Detrimental Role of IL-1β During Mtb Infections
In contrast, other data from mouse and human studies point towards a role of IL-1β in increasing host susceptibility (3). Some of the strongest evidence for a positive correlation between increased IL-1β and severity of disease in humans comes from several studies analyzing genetic variability and clinical outcomes. The analysis of single nucleotide polymorphisms (SNP) in the human IL1B gene identified 3 SNPs in the genes promoter region that results in increased IL-1β expression and was associated with more severe tuberculosis possibly due to the increased infiltration of neutrophils (125). A polymorphism in the IL-1 receptor agonist (IL1RA) gene resulted in population with decreased IL1RA and increased IL1B gene expression that was more commonly found in patients with tuberculoid pleurisy (126). Furthermore, several studies using the mouse model suggest a detrimental role of IL-1β to host defense. For example, it was demonstrated that the primary protective mechanism of nitric oxide (NO) during Mtb infection is not antibacterial activity but instead the suppression of inflammasome activation (127, 128). MCC950 inhibits the NLRP3 inflammasome activation in Mtb-infected BMDMs and results in decreased survival of Mtb in addition to reduced processing of IL-1β (129).
In conclusion, it is very likely that, similar to the situation with TNF and IFN-β (130, 131), also for the production of the IL-1β the Goldilocks principle applies with just the right amount of cytokine being produced in the right context at the right time during infection in order to produce a host protective outcome.
Inflammasome Recognition of Mycobacteria
Recognition of Mtb by the NLRP3 Inflammasome
Different mycobacterial species express different proteins and lipids which may affect their recognition by NLR/ALR proteins (132) (Table 1). Secretion of proinflammatory mature IL-1β or IL-18 during Mtb infection requires activation of NLRP3 inflammasome (9–11, 17, 19, 22, 133). The activation of the NLRP3 inflammasome after Mtb infection is conserved across various cell types: mouse bone marrow derived macrophages (BMDMs) (4, 5, 8–10, 20), peritoneal exudate macrophages (4, 8), THP-1 human macrophages (5, 7, 8, 10, 11, 19), mouse retinal pigment epithelium (RPE) cells (6), primary microglia (15), Ana-1 mouse macrophage (16), mouse bone marrow derived dendritic cells (BMDCs) (10, 22), J774A.1 mouse macrophages (5), PBMCs (7) and human monocyte derived macrophages (hMDMs) (11) (see also Table 1). The adaptor ASC, NLRP3 and caspase-1/11 are required for the secretion of IL-1β in Mtb-infected BMDCs (22). Mtb infection induces increased K+ and Cl- efflux but does not affect Ca2+ flux (134). The phagosomal and mitochondrial ROS are not involved in the activation of the NLRP3 inflammasome upon Mtb infection but instead it is cytosolic ROS, generated by the xanthine oxidase (XO) (134). Interestingly, plasma membrane damage mediated by ESX-1 system triggers increase in K+ efflux, consequently activating the NLRP3 inflammasome and pyroptosis, which permits the spreading of Mtb to neighboring cells (135).
Recognition of NTM by the NLRP3 Inflammasome
The activation of the NLRP3 inflammasome has also been demonstrated for non-tuberculous mycobacteria (NTM) species including Mycobacterium marinum (Mmar) (12, 17), Mycobacterium abscessus (Mab) (13, 21) and Mycobacterium kansasii (Mkan) (14) and Mycobacterium ulcerans (26) (Table 1). More specifically, Mab leads to caspase-1 activation and release of IL-1β in human macrophages. Dectin-1/Syk-dependent signaling, increased expression of the cytoplasmic scaffold protein p62/SQSTM1 and potassium efflux are implicated in Mab mediated NLRP3 inflammasome activation (13). Consistently, it has been demonstrated that Mab results in increased production of IL-1β in murine macrophages (21). Mab induces mitochondrial ROS and thereby leads to enhanced NLRP3 inflammasome activation (21). A recent study has demonstrated that infection of microglia with the attenuated Mtb H37Ra strain triggers NLRP3 mediated secretion of IL-1β and IL-18 (15). Moreover, they also found that by inhibiting NF-κB (signal 1) and P2X7R (signal 2) they can alter the secretion of IL-1β and IL-18 and thereby regulate the NLRP3 inflammasome pathway in microglia during Mtb infection (15) (Table 1). Interestingly, irrespective of the overexpression of NLRP3 and inflammatory caspases-4/5 detected in the lepromatous pole, low expression of caspase-1, IL-1β, and IL-18 were observed in leprosy and therefore these results indicate that NLRP3 inflammasome does not actively contribute to the innate immune response in leprosy, suggesting immune evasion of M. leprae (136).
Recognition of Mycobacteria by the AIM2 Inflammasome
In addition to NLRP3 inflammasome, different reports have implicated the critical role for the AIM2 inflammasome following mycobacterial infection. Activation of AIM2 inflammasome has been reported in several cell types during infection with various mycobacterial species, including Mycobacterium smegmatis (Msme) (23), Mycobacterium fortuitum (Mfor) and Mkan in BMDCs (23) (Table 1). In BMDCs about 40–50% of the production of IL-1β following Msme, Mfor, and Mkan infection was dependent on the AIM2 inflammasome and moreover there was an inverse correlation between virulence of the mycobacterial species and the amount of IL-1β release, with the least virulent species inducing the highest levels of IL-1β (23). Mycobacterium bovis (Mbov) infection activated the AIM2 inflammasome in BMDMs and J774A.1 mouse macrophage (25) (Table 1). Mbov infection augments the mRNA expression of AIM2 and ASC in both BMDMs and J774A.1 mouse macrophage (25). Potassium efflux and mycobacterial escape into the cytosol are the two essential triggers in the activation of AIM2 inflammasome during Mbov infection. Additionally, infection of J774A.1 mouse macrophage with Mbov results in activation of caspase-1 as early as 6h post-infection (25). The transfection of Mtb genomic DNA into LPS-primed peritoneal macrophages results in AIM2-dependent caspase-1 activation and subsequent secretion of mature IL-1β and IL-18 (24). This is not surprising since AIM2 recognizes any type of dsDNA but interesting because Mtb does not activate the AIM2 inflammasome upon infection of BMDCs or BMDMs (23).
Activation of the NLRP3 Inflammasome by Mycobacterial Proteins and Lipids
Mtb contains several secretion systems to export proteins into the cell well and beyond (137, 138). In order for these proteins to potentially reach the host cell cytosol, the ESX-1-secreted effector EsxA is involved in permeabilizing the phagosomal membrane (Figure 2) (139, 140). A small number of mycobacterial secreted protein effectors have been identified that are involved in activation of NLRP3 inflammasome (5–8, 10, 11, 16, 19, 20) (Figure 2 and Table 1).
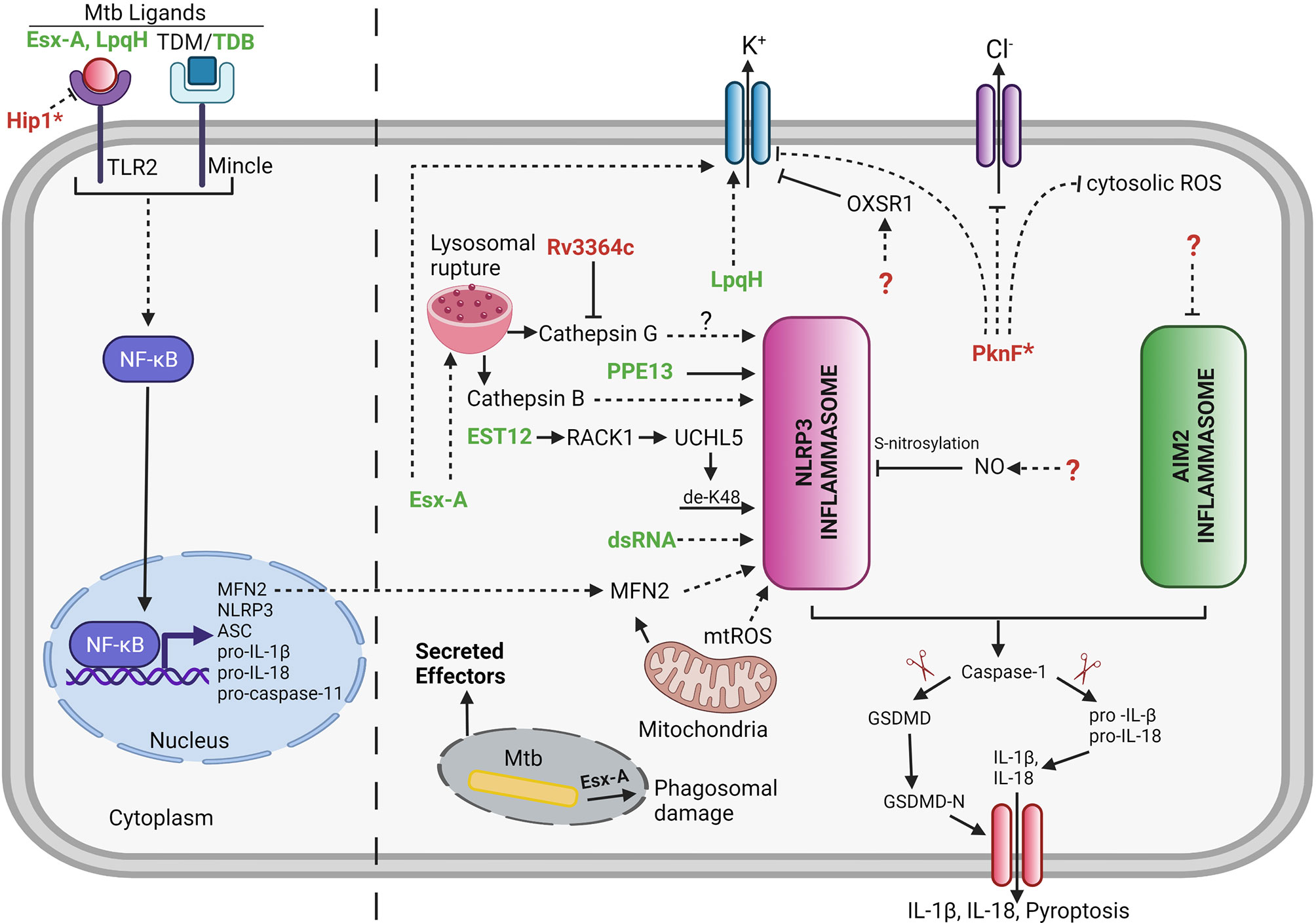
Figure 2 Mycobacterial effectors involved in regulation of host cell inflammasome. Several Mtb effectors either secreted or non-secreted (as indicated by *) are known to be implicated in manipulation of the host cell inflammasome pathway. Bold green color denotes the Mtb effectors involved either directly or indirectly in activation of inflammasome. Bold red color denotes the Mtb effectors involved in inhibition of inflammasome. Unknown Mtb effectors are represented by ? (dashed lines = indirect interaction; solid lines = direct interaction; arrowhead = activation; blunt end = inhibition). LpqH,19 kDa Lipoprotein antigen precursor; PPE13, PPE family protein 13; EST12, Estimated 12kDa (Rv1579c); EsxA, 6 kDa Early secretory antigenic target; dsRNA, double stranded Ribonucleic Acid; TDB, Trehalose-6,6-dibehenate; TDM, Trehalose dimycolate; PknF, Protein kinase F; Hip1, Hydrolase important for pathogenesis 1; NO, Nitric Oxide; RACK1, Receptor for Activated C Kinase 1; UCHL5, Ubiquitin C-Terminal Hydrolase L5; MFN2, Mitofusin 2; OXSR1, Oxidative Stress Responsive Kinase 1. Created with Biorender.com.
PPE13
The Proline-Proline-Glutamate (PPE) family protein, PPE13, participates in the assembly of NLRP3 inflammasome complex via its C-terminal repetitive major polymorphic tandem repeat (MPTR) domain by directly interacting with the NACHT and Leucine-rich repeat (LRR) domains of NLRP3 (5) (Figure 2 and Table 1). A recombinant Msme strain expressing the Mbov PPE13 induces increased cell death and increased secretion of IL-1β in J774A.1, BMDMs, and THP-1 macrophages but to different levels depending on the cell type (5). The release of IL-1β was dependent on activation of NLRP3 inflammasome as confirmed by caspase-1 and NLRP3 inflammasome inhibitor studies (5). The role of PPE13 in inflammasome signaling needs further validation by creating a gene specific knockout in Mtb.
EST12
Recent studies show that Rv1579c, located within the Mtb H37Rv region of difference 3 (RD3), encodes for a protein (EST12) which acts as a pyroptosis-inducing protein (8) (Figure 2 and Table 1). Indeed, EST12 interacts with the host protein receptor for activated C kinase 1 (RACK1) and forms a EST12-RACK1 complex in macrophages. The EST12-RACK1 dimer recruits the deubiquitinase UCHL5 to stimulate the K48-linked deubiquitination of NLRP3 and consequently triggers the NLRP3 inflammasome mediated pyroptosis and IL-1β secretion (8) (Figure 2). Mice infected with an Mtb strain lacking EST12 showed significant increase in the bacterial growth in the lungs and lower levels of serum IL-1β compared to wild-type Mtb-infected mice. Consistently, mice infected with BCG or Msme strains overexpressing EST12 showed lower bacterial burden in lungs or spleen and increased levels of IL-1β compared to mice infected with control bacteria. Consequently, Mtb EST12 increases mycobacterial clearance in mice and is responsible to activate the host’s immunity (8). It will be interesting to see in future studies which Mtb secretion system is responsive for the secretion of EST12.
LpqH
The Mtb lipoprotein, LpqH, activates the NLRP3 inflammasome via a mechanism that involves activation of the TLR-2 receptor (16) (Figure 2 and Table 1). The treatment of LPS-primed Ana-1 mouse macrophages with purified LpqH protein results in increased expression of NLRP3, ASC and caspase-1 proteins in a dose-dependent manner (16). Moreover, potassium efflux acts as an important trigger for LpqH-mediated activation of the NLRP3 inflammasome (16). However, the underlying mechanism of how LpqH influences potassium efflux has not been explored in this study. The work was performed in mouse macrophages and thus further validation in human macrophages would be valuable. It will also be crucial to test a lpqH deletion mutant of Mtb for changes in NLRP3 inflammasome activation to confirm that the observed activity of purified proteins is conserved within the context of a whole bacterium.
ESX-1 and ESX-5
The Mtb RD1 locus which encodes for the ESX-1 secretion system is required for activation of NLRP3 inflammasome and subsequent release of IL-1β in human PBMCs, THP-1 cells and mouse BMDMs and retinal pigment epithelium (RPE) cells (4, 6, 7, 9, 11, 14, 19, 20) (Figure 2 and Table 1). The ESX-5a is a duplicated region of 4 genes out of the ESX-5 secretion system which is important for the secretion of subset of ESX-5-secreted proteins and its deletion results in reduced inflammasome activation (10) (Table 1).
EsxA
EsxA is one of the main substrates secreted by the ESX-1 system and consequently many studies have been performed by adding purified EsxA to cells during ex vivo experimentations. EsxA interacts with TLR-2 and TLR-4 receptors to induce host cell signaling (141–144). One report shows that treatment of mouse RPE cells with different doses of EsxA results in caspase-1 activation in a dose dependent manner and that this activation is dependent on TLR/MyD88 signaling and the NLRP3 inflammasome (6). Other studies reveal that stimulation of either PBMCs (7) or THP-1 macrophages (7, 19) with EsxA results in increased release of proinflammatory cytokine IL-1β. Furthermore, stimulation of PBMCs and THP-1 derived macrophages with either Mtb protein EsxA or heat inactivated Mtb lysates induces increase in expression of MFN2 and results in increased release of IL-1β. Therefore, these findings suggest that MFN2 is required for the assembly and activation of NLRP3 inflammasome during Mtb infection (7). Transcriptional profiling in human PBMCs from active tuberculosis patients and healthy controls determined that a mitochondrial outer membrane protein, MFN2 expression was significantly upregulated in TB patients compared to healthy controls (7). Intriguingly, another study reported the importance of EsxA and RD1 locus in secretion of IL-1β, since BMDMs when infected with Mtb strains lacking esxA or RD1 showed a significant reduction in the secretion of IL-1β compared to the BMDMs infected with Mtb (20). EsxA was partially responsible for the release of mature cathepsin B by the lysosomes during Mtb infection and further leads to NLRP3 inflammasome activation in BMDMs (20). In addition to lysosomal permeabilization, EsxA also triggers phagosome damage and Syk activation in human macrophages that result in NLRP3 mediated necrotic cell death (11). Additionally, EsxA facilitates the translocation of other immunostimulatory Mtb components such as Ag85 into the macrophage cytosol, resulting in increased activation of caspase-1 and subsequent secretion of IL-1β (19).
Critical role for the ESX-1/EsxA in NLRP3 activation has also been reported during the infection with non-tuberculous mycobacteria species including Mmar (12, 17, 18), and Mkan (14) (Table 1). In addition to ESX-1, ESX-5 secreted substrates also play a role in activation of NLRP3 inflammasome in response to infection with Mmar (12) (Table 1). Additionally, transfection of mouse RPE cells with mycobacterial dsRNA induces NLRP3 inflammasome-dependent caspase-1 activation via an uncharacterized mechanism (6).
TDB/TDM
The role of mycobacterial cell wall lipids in NLRP3 inflammasome activation has also been studied in addition to mycobacterial secreted effector proteins (145). Trehalose-6,6’-dibehenate (TDB), a synthetic analogue of Trehalose-6,6’-dimycolate (TDM) also known as mycobacterial cord factor has been developed as an effective adjuvant for tuberculosis subunit vaccine and both act as a potent proinflammatory pathogen-associated molecular pattern (PAMP) which is recognized by macrophage inducible C-type lectin (Mincle) receptor on innate immune cells and triggers host innate immune response. TDB induces the NLRP3/ASC/Caspase-1 mediated increase in production of IL-1β in BMDCs. Activation of NLRP3 inflammasome by TDB involves numerous triggers such as lysosomal permeabilization, increased ROS generation and increased potassium efflux (145).
It is important to remember that the NLRP3 inflammasome is acting most likely as the cells stress or danger sensor and thus any bacterial induction of the NRLP3 inflammasome might not be mediated by a direct interaction with the inflammasome complex but through interaction with other cellular components/pathways that then trigger the stress/danger signal.
AIM2 Inflammasome Inhibition by Mtb
Mycobacterial extracellular DNA enters the host cell cytosol in an ESX-1 secretion system dependent manner (146). Intriguingly, AIM2 recognizes and binds to cytosolic DNA of intracellular pathogens such as Francisella and Listeria (82, 147) and even Mtb (24). However, AIM2 is not activated during the course of ex vivo Mtb H37Rv infection of BMDM and BMDCs (23). Nonvirulent mycobacterial species such as Msme induce the activation of AIM2 inflammasome in contrast to virulent Mtb that inhibits the AIM2 inflammasome activation induced by either Msme or AIM2 agonists (23). Moreover, Mtb-mediated AIM2 inflammasome inhibition is dependent on a functional ESX-1 secretion system since infection with the Mtb strain deficient in esxA fails to inhibit IL-1β secretion induced by Msme (23). Intriguingly, Mtb inhibits the secretion of IFN-β in infected cells, which may provide one of the mechanisms to suppresses the activation of AIM2 inflammasome (23). Consequently, co-secretion of a putative AIM2 inhibitor and/or IFN-β inhibitor through ESX-1 secretion system into the host cell cytosol together with cytosolic Mtb DNA may play an important role in Mtb-mediated evasion of AIM2 inflammasome activation. The inhibition of the AIM2 inflammasome activation might be of importance for virulence of Mtb because aim2-/- mice are highly susceptible to Mtb infections and showing impaired production of pro-inflammatory cytokines IL-1β and IL-18 (24). An area of interest will be the discovery of the Mtb genes involved in the AIM2 inflammasome inhibition in order to assess the importance of AIM2-inflammasoem evasion for the virulence of Mtb.
NLRP3 Inflammasome Inhibition by Mtb
Mtb PknF
As discussed before many studies have shown that Mtb causes NLRP3 inflammasome activation and even that the deletion of certain Mtb genes led to an increased activation of the NLRP3 inflammasome (Figure 2 and Table 1). Nevertheless, not until recently was it shown that Mtb infection can inhibit activation of the NLRP3 inflammasome via either LPS/Nigericin or LPS/ATP stimuli (134). Mtb inhibits the NLRP3 inflammasome activation via a mechanism that is independent of the ESX-1 secretion system which is opposed to the capacity of Mtb to inhibit the AIM2 inflammasome in an ESX-1 dependent mechanism (23, 134). Mtb infection inhibits the LPS/ATP-induced K+ efflux and increase in xanthine oxidase (XO) activity leading to decreased cytosolic ROS levels (134). The Mtb serine threonine kinase, PknF, mediates inhibition of NLRP3 inflammasome dependent production of IL-1β and pyroptosis in both mouse and human derived primary macrophages (134). Moreover, K+ efflux, Cl2+ efflux and ROS generation are implicated in the PknF-mediated NLRP3 inflammasome inhibition (134) (Figure 2). Additionally, the Mtb pknF mutant induces an increase in the XO activity compared to Mtb-infected cells and thus increasing XO-mediated ROS production (134). Altogether it seems that PknF is inhibiting the exact NLRP3 inflammasome activation pathway that is being only slightly activated upon Mtb infection.
Mtb Zmp1
The BCG gene zmp1 encodes a zinc metalloprotease that has been shown to inhibit the NLRP3 inflammasome dependent processing of IL-1β (115). However, these findings could not be independently confirmed since generation of the zmp1 Mtb deletion mutant strain did not show any effect on pyroptosis, on the caspase-1 activation nor the release of IL-1β (11). The inconsistency between these two publications might be due to a difference in the background of the zmp1 mutant strain because most of the studies performed by Master et al. were done using the BCG strain that, unlike Mtb, lacks a functional ESX-1 secretion system. Another possibility may be the difference in cell types used to study the strains lacking zmp1.
Mtb Hip1 and Rv3364c
The Mtb serine hydrolase, Hip1, inhibits the NLRP3 inflammasome activation by dampening the TLR2-dependent cell signaling in BMDMs (148) (Figure 2). Hip1 not only affects the secretion of inflammasome dependent cytokines but also result in decrease of other proinflammatory cytokines thus suggesting Hip1 modulates the proinflammatory responses in macrophages by preventing the activation of TLR2 dependent cell signaling (148). The mechanism involves Hip1-mediated proteolytical cleavage of GroEL2 from multimer to monomer and the cleaved monomeric GroEL2 subsequently contributes to dampening of proinflammatory responses in macrophages mediated by TLR2 signaling (149). Therefore, it is not possible to attribute the in vivo attenuation of the hip1 Mtb deletion mutant to its increase in inflammasome activation since also other important proinflammatory cytokines such as TNF are upregulated (148).The Mtb protein, Rv3364c, binds to and inhibits the membrane associated host serine protease cathepsin G which leads to suppression of caspase-1 activity and pyroptosis in macrophages (150) (Figure 2).
Nitric Oxide (NO)
Host cell derived NO acts as a negative regulator of NLRP3 inflammasome activation and inhibits processing of IL-1β (127, 151) (Figure 2). Stimulation of Mtb-infected macrophages with IFN-γ showed iNOS-dependent thiol nitrosylation of NLRP3 which leads to inhibition of NLRP3 inflammasome dependent maturation of IL-1β and minimize inflammatory tissue damage during chronic Mtb infection (127). Indeed, the NO-mediated nitrosylation inhibits the assembly of NLRP3 inflammasome complex (127, 151).
OXSR1
During mycobacterial infection, a host serine/threonine protein kinase, the oxidative stress responsive kinase 1 (OXSR1), inhibits K+ channels responsible for K+ efflux (152). Indeed, the mycobacterial infection leads to the upregulation of host OXSR1 and this host cell manipulation is dependent on the mycobacterial ESX-1 secretion system. The immunomodulatory role of OXSR1 is conserved in both zebrafish and humans (152). Furthermore, inhibition or depletion of OXSR1 results in diminished levels of intracellular potassium and hence limits the growth of mycobacteria. Therefore, targeting of OXSR1 might be a valuable approach for host-directed therapy. Micheliolide (MCL), a sesquiterpene lactone, act as an anti-inflammatory molecule by inhibiting PI3K/Akt/NF-κB and NLRP3 inflammasome signaling during Mtb infection (153). However, previous report showed that RAW264.7 cells do not release mature IL-1β since they do not express ASC as determined by immunoblot analysis with an ASC-specific antibody (154). The study by Zhang et al. did not address this problem since all experiments were performed in RAW264.7 macrophages and hence there remains some questions on the validity of their results.
Overall, there has been notable progress but still the molecular mechanisms by which Mtb evades NLRP3 inflammasome activation and the importance of this manipulation for virulence of Mtb remain poorly understood.
Conclusion
Since we last reviewed the literature on the subject of Mtb-host cell inflammasome interactions in 2013 (155) a tremendous amount of progress has been made in our understanding of the molecular mechanisms of inflammasome activation and subsequent pathways of pyroptosis induction (Figure 1). Also, our knowledge of the role of IL-1β during Mtb infections has been greatly expended. Nevertheless, important questions remain; for example, what is the in vivo mechanisms of host resistance that is mediated by IL-1β? We know that protective effects are mediated by bystander cells but what is IL-1β/IL-1β-signaling doing onto those cells that conveys the protective effect? We now know that Mtb is able to inhibit the AIM2- and NLRP3-inflammasome during ex vivo infections (Figure 2) but is there a role for the inflammasome for increasing host resistance or susceptibility during in vivo Mtb infections? The herein described progress made in identifying various Mtb effectors activating or inhibiting the host cell inflammasome (Figure 2 and Table 1) and the subsequent availability of specific Mtb mutants perturbing the inflammasome activation will provide the tools necessary to start answering that question.
Author Contributions
SR and VB wrote and edited the manuscript. SR created the figures and table. All authors contributed to the article and approved the submitted version.
Funding
SR and VB are funded by NIH/NIAID grants AI139492 and AI147630.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. WHO. Global Tuberculosis Report 2020 (2020). Available at: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2020.
2. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Primers (2016) 2:16076. doi: 10.1038/nrdp.2016.76
3. Mayer-Barber KD, Sassetti CM. Type I Interferon and Interleukin-1 Driven Inflammatory Pathways as Targets for HDT in Tuberculosis. In: Karakousis PC, Hafner R, Gennaro ML, editors. Advances in Host-Directed Therapies Against Tuberculosis. Springer International Publishing (2021). p. 219–32.
4. Kurenuma T, Kawamura I, Hara H, Uchiyama R, Daim S, Dewamitta SR, et al. The RD1 Locus in the Mycobacterium Tuberculosis Genome Contributes to Activation of Caspase-1 via Induction of Potassium Ion Efflux in Infected Macrophages ▿. Infect Immun (2009) 77:3992–4001. doi: 10.1128/iai.00015-09
5. Yang Y, Xu P, He P, Shi F, Tang Y, Guan C, et al. Mycobacterial PPE13 Activates Inflammasome by Interacting With the NATCH and LRR Domains of NLRP3. FASEB J (2020) 34:12820–33. doi: 10.1096/fj.202000200rr
6. Basu S, Fowler BJ, Kerur N, Arnvig KB, Rao NA. NLRP3 Inflammasome Activation by Mycobacterial ESAT-6 and Dsrna in Intraocular Tuberculosis. Microb Pathogen (2018) 114:219–24. doi: 10.1016/j.micpath.2017.11.044
7. Xu F, Qi H, Li J, Sun L, Gong J, Chen Y, et al. Mycobacterium Tuberculosis Infection Up-Regulates MFN2 Expression to Promote NLRP3 Inflammasome Formation. J Biol Chem (2020) 295:17684–97. doi: 10.1074/jbc.ra120.014077
8. Qu Z, Zhou J, Zhou Y, Xie Y, Jiang Y, Wu J, et al. Mycobacterial EST12 Activates a RACK1–NLRP3–Gasdermin D Pyroptosis–IL-1β Immune Pathway. Sci Adv (2020) 6:eaba4733. doi: 10.1126/sciadv.aba4733
9. Dorhoi A, Nouailles G, Jörg S, Hagens K, Heinemann E, Pradl L, et al. Activation of the NLRP3 Inflammasome by Mycobacterium Tuberculosis is Uncoupled From Susceptibility to Active Tuberculosis. Eur J Immunol (2012) 42:374–84. doi: 10.1002/eji.201141548
10. Shah S, Cannon JR, Fenselau C, Briken V. A Duplicated ESAT-6 Region of ESX-5 is Involved in Protein Export and Virulence of Mycobacteria. Infect Immun (2015) 83:4349–61. doi: 10.1128/iai.00827-15
11. Wong K, Jr WRJ. Critical Role for NLRP3 in Necrotic Death Triggered by Mycobacterium Tuberculosis. Cell Microbiol (2011) 13:1371–84. doi: 10.1111/j.1462-5822.2011.01625.x
12. Abdallah AM, Bestebroer J, Savage NDL, de Punder KZ, van M, Wilson L, et al. Mycobacterial Secretion Systems ESX-1 and ESX-5 Play Distinct Roles in Host Cell Death and Inflammasome Activation. J Immunol (2011) 187:4744–53. doi: 10.4049/jimmunol.1101457
13. Lee H, Yuk J, Kim K, Jang J, Kang G, Park JB, et al. Mycobacterium Abscessus Activates the Nlrp3 Inflammasome via Dectin-1–Syk and P62/Sqstm1. Immunol Cell Biol (2012) 90:601–10. doi: 10.1038/icb.2011.72
14. Chen C-C, Tsai S-H, Lu C-C, Hu S-T, Wu T-S, Huang T-T, et al. Activation of an NLRP3 Inflammasome Restricts Mycobacterium Kansasii Infection. PloS One (2012) 7:e36292. doi: 10.1371/journal.pone.0036292
15. Xie Z, Hui H, Yao Q, Duan Y, Li W, Cheng Y, et al. By Regulating the NLRP3 Inflammasome can Reduce the Release of Inflammatory Factors in the Co-Culture Model of Tuberculosis H37Ra Strain and Rat Microglia. Front Cell Infect Mi (2021) 11:637769:637769. doi: 10.3389/fcimb.2021.637769
16. Liu L, Zhai K, Chen Y, Chen X, Wang G, Wu L. Effect and Mechanism of Mycobacterium Tuberculosis Lipoprotein Lpqh in NLRP3 Inflammasome Activation in Mouse Ana-1 Macrophage. BioMed Res Int (2021) 2021:1–8. doi: 10.1155/2021/8239135
17. Carlsson F, Kim J, Dumitru C, Barck KH, Carano RAD, Sun M, et al. Host-Detrimental Role of Esx-1-Mediated Inflammasome Activation in Mycobacterial Infection. PloS Pathog (2010) 6:e1000895. doi: 10.1371/journal.ppat.1000895
18. Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ. ESX-1-Dependent Cytolysis in Lysosome Secretion and Inflammasome Activation During Mycobacterial Infection. Cell Microbiol (2008) 10:1866–78. doi: 10.1111/j.1462-5822.2008.01177.x
19. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium Tuberculosis Protein ESAT-6 is a Potent Activator of the NLRP3/ASC Inflammasome. Cell Microbiol (2010) 12:1046–63. doi: 10.1111/j.1462-5822.2010.01450.x
20. Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, et al. Lysosomal Cathepsin Release is Required for NLRP3-Inflammasome Activation by Mycobacterium Tuberculosis in Infected Macrophages. Front Immunol (2018) 9:1427. doi: 10.3389/fimmu.2018.01427
21. Kim B-R, Kim B-J, Kook Y-H, Kim B-J. Mycobacterium Abscessus Infection Leads to Enhanced Production of Type 1 Interferon and NLRP3 Inflammasome Activation in Murine Macrophages via Mitochondrial Oxidative Stress. PloS Pathog (2020) 16:e1008294. doi: 10.1371/journal.ppat.1008294
22. Abdalla H, Srinivasan L, Shah S, Mayer-Barber KD, Sher A, Sutterwala FS, et al. Mycobacterium Tuberculosis Infection of Dendritic Cells Leads to Partially Caspase-1/11-Independent IL-1β and IL-18 Secretion But Not to Pyroptosis. PloS One (2012) 7:e40722. doi: 10.1371/journal.pone.0040722
23. Shah S, Bohsali A, Ahlbrand SE, Srinivasan L, Rathinam VAK, Vogel SN, et al. Cutting Edge: Mycobacterium Tuberculosis But Not Nonvirulent Mycobacteria Inhibits IFN-B and AIM2 Inflammasome–Dependent IL-1β Production via its ESX-1 Secretion System. J Immunol (2013) 191:3514–8. doi: 10.4049/jimmunol.1301331
24. Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, et al. Critical Role of AIM2 in Mycobacterium Tuberculosis Infection. Int Immunol (2012) 24:637–44. doi: 10.1093/intimm/dxs062
25. Yang Y, Zhou X, Kouadir M, Shi F, Ding T, Liu C, et al. The AIM2 Inflammasome is Involved in Macrophage Activation During Infection With Virulent Mycobacterium Bovis Strain. J Infect Dis (2013) 208:1849–58. doi: 10.1093/infdis/jit347
26. Foulon M, Robbe-Saule M, Manry J, Esnault L, Boucaud Y, Alcaïs A, et al. Mycolactone Toxin Induces an Inflammatory Response by Targeting the IL-1β Pathway: Mechanistic Insight Into Buruli Ulcer Pathophysiology. PloS Pathog (2020) 16:e1009107. doi: 10.1371/journal.ppat.1009107
27. Rathinam VAK, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
28. Sharma D, Kanneganti T-D. The Cell Biology of Inflammasomes: Mechanisms of Inflammasome Activation and Regulation. J Cell Biol (2016) 213:617–29. doi: 10.1083/jcb.201602089
29. Man SM, Karki R, Kanneganti T-D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol Rev (2017) 277:61–75. doi: 10.1111/imr.12534
30. Broz P. Recognition of Intracellular Bacteria by Inflammasomes. Microbiol Spectr (2019) 7. doi: 10.1128/microbiolspec.bai-0003-2019
31. Chan AH, Schroder K. Inflammasome Signaling and Regulation of Interleukin-1 Family Cytokines. J Exp Med (2019) 217:e20190314. doi: 10.1084/jem.20190314
32. Evavold CL, Kagan JC. Inflammasomes: Threat-Assessment Organelles of the Innate Immune System. Immunity (2019) 51:609–24. doi: 10.1016/j.immuni.2019.08.005
33. Broz P, Pelegrín P, Shao F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat Rev Immunol (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
34. Linder A, Hornung V. Irgm2 and Gate-16 Put a Break on non-Canonical Inflammasome Activation. EMBO Rep (2020) 11:561948–3. doi: 10.15252/embr.202051787
35. Seoane PI, Lee B, Hoyle C, Yu S, Lopez-Castejon G, Lowe M, et al. The NLRP3–Inflammasome as a Sensor of Organelle Dysfunction. J Cell Biol (2020) 219:e202006194. doi: 10.1083/jcb.202006194
36. Deets KA, Vance RE. Inflammasomes and Adaptive Immune Responses. Nat Immunol (2021) 22:412–22. doi: 10.1038/s41590-021-00869-6
37. Lamkanfi M, Dixit VM. Inflammasomes: Guardians of Cytosolic Sanctity. Immunol Rev (2009) 227:95–105. doi: 10.1111/j.1600-065x.2008.00730.x
38. Broz P, Monack DM. Molecular Mechanisms of Inflammasome Activation During Microbial Infections. Immunol Rev (2011) 243:174–90. doi: 10.1111/j.1600-065x.2011.01041.x
39. Bauernfeind F, Hornung V. Of Inflammasomes and Pathogens–Sensing of Microbes by the Inflammasome. EMBO Mol Med (2013) 5:814–26. doi: 10.1002/emmm.201201771
40. Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. Recognition of Bacteria by Inflammasomes. Annu Rev Immunol (2013) 31:73–106. doi: 10.1146/annurev-immunol-032712-095944
41. Hayward JA, Mathur A, Ngo C, Man SM. Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol Mol Biol R (2018) 82:e00015–18. doi: 10.1128/mmbr.00015-18
42. Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity (2013) 39:1003–18. doi: 10.1016/j.immuni.2013.11.010
43. Jesus AA, Goldbach-Mansky R. IL-1 Blockade in Autoinflammatory Syndromes1. Annu Rev Med (2014) 65:223–44. doi: 10.1146/annurev-med-061512-150641
44. McKee CM, Coll RC. NLRP3 Inflammasome Priming: A Riddle Wrapped in a Mystery Inside an Enigma. J Leukocyte Biol (2020) 108:937–52. doi: 10.1002/jlb.3mr0720-513r
45. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and its Inhibitors. Cell Death Dis (2019) 10:128. doi: 10.1038/s41419-019-1413-8
46. Weber ANR, Bittner ZA, Shankar S, Liu X, Chang T-H, Jin T, et al. Recent Insights Into the Regulatory Networks of NLRP3 Inflammasome Activation. J Cell Sci (2020) 133:jcs248344. doi: 10.1242/jcs.248344
47. Fernandes-Alnemri T, Wu J, Yu J-W, Datta P, Miller B, Jankowski W, et al. The Pyroptosome: A Supramolecular Assembly of ASC Dimers Mediating Inflammatory Cell Death via Caspase-1 Activation. Cell Death Differ (2007) 14:1590–604. doi: 10.1038/sj.cdd.4402194
48. Cai X, Chen J, Xu H, Liu S, Jiang Q-X, Halfmann R, et al. Prion-Like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation. Cell (2014) 156:1207–22. doi: 10.1016/j.cell.2014.01.063
49. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
50. He W, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
51. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for non-Canonical Inflammasome Signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
52. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
53. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 Mediates Plasma Membrane Rupture During Lytic Cell Death. Nature (2021), 591(7848):131–6. doi: 10.1038/s41586-021-03218-7
54. Zhou B, Abbott DW. Gasdermin E Permits Interleukin-1 Beta Release in Distinct Sublytic and Pyroptotic Phases. Cell Rep (2021) 35:108998. doi: 10.1016/j.celrep.2021.108998
55. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ Efflux is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
56. Verhoef PA, Kertesy SB, Lundberg K, Kahlenberg JM, Dubyak GR. Inhibitory Effects of Chloride on the Activation of Caspase-1, IL-1β Secretion, and Cytolysis by the P2X7 Receptor. J Immunol (2005) 175:7623–34. doi: 10.4049/jimmunol.175.11.7623
57. Compan V, Baroja-Mazo A, López-Castejón G, Gomez AI, Martínez CM, Angosto D, et al. Cell Volume Regulation Modulates Nlrp3 Inflammasome Activation. Immunity (2012) 37:487–500. doi: 10.1016/j.immuni.2012.06.013
58. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. Clics-Dependent Chloride Efflux Is an Essential and Proximal Upstream Event for NLRP3 Inflammasome Activation. Nat Commun (2017) 8:202. doi: 10.1038/s41467-017-00227-x
59. Lee G-S, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The Calcium-Sensing Receptor Regulates the Nlrp3 Inflammasome Through Ca2+ and Camp. Nature (2012) 492:123–7. doi: 10.1038/nature11588
60. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc Natl Acad Sci (2012) 109:11282–7. doi: 10.1073/pnas.1117765109
61. Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, et al. Extracellular Ca2+ is a Danger Signal Activating the NLRP3 Inflammasome Through G Protein-Coupled Calcium Sensing Receptors. Nat Commun (2012) 3:1329. doi: 10.1038/ncomms2339
62. Zhou R, Yazdi AS, Menu P, Tschopp J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
63. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome During Apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
64. Groslambert M, Py BF. Spotlight on the NLRP3 Inflammasome Pathway. J Inflammation Res (2018) 11:359–74. doi: 10.2147/jir.s141220
65. Yang J, Zhao Y, Shao F. Non-Canonical Activation of Inflammatory Caspases by Cytosolic LPS in Innate Immunity. Curr Opin Immunol (2015) 32:78–83. doi: 10.1016/j.coi.2015.01.007
66. Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
67. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 Increases Susceptibility to Salmonella Infection in the Absence of Caspase-1. Nature (2012) 490:288–91. doi: 10.1038/nature11419
68. Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell (2012) 150:606–19. doi: 10.1016/j.cell.2012.07.007
69. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science (2013) 341:1250–3. doi: 10.1126/science.1240988
70. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science (2013) 341:1246–9. doi: 10.1126/science.1240248
71. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory Caspases are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
72. Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP5 Promotes NLRP3 Inflammasome Assembly and Immunity in Mammals. Science (2012) 336:481–5. doi: 10.1126/science.1217141
73. Meunier E, Dick MS, Dreier RF, Schürmann N, Broz DK, Warming S, et al. Caspase-11 Activation Requires Lysis of Pathogen-Containing Vacuoles by IFN-Induced Gtpases. Nature (2014) 509:366–70. doi: 10.1038/nature13157
74. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science (2013) 339:975–8. doi: 10.1126/science.1230751
75. Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell (2016) 167:382–396.e17. doi: 10.1016/j.cell.2016.09.012
76. Kutsch M, Sistemich L, Lesser CF, Goldberg MB, Herrmann C, Coers J. Direct Binding of Polymeric GBP1 to LPS Disrupts Bacterial Cell Envelope Functions. EMBO J (2020) 39:e104926. doi: 10.15252/embj.2020104926
77. Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, et al. Human GBP1 Binds LPS to Initiate Assembly of a Caspase-4 Activating Platform on Cytosolic Bacteria. Nat Commun (2020) 11:3276. doi: 10.1038/s41467-020-16889-z
78. Wandel MP, Kim B-H, Park E-S, Boyle KB, Nayak K, Lagrange B, et al. Guanylate-Binding Proteins Convert Cytosolic Bacteria Into Caspase-4 Signaling Platforms. Nat Immunol (2020) 21:880–91. doi: 10.1038/s41590-020-0697-2
79. Yi Y. Functional Crosstalk Between non-Canonical Caspase-11 and Canonical NLRP3 Inflammasomes During Infection-Mediated Inflammation. Immunology (2020) 159:142–55. doi: 10.1111/imm.13134
80. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An Orthogonal Proteomic-Genomic Screen Identifies AIM2 as a Cytoplasmic DNA Sensor for the Inflammasome. Nat Immunol (2009) 10:266–72. doi: 10.1038/ni.1702
81. Fernandes-Alnemri T, Yu J-W, Wu J, Datta P, Alnemri ES. AIM2 Activates the Inflammasome and Cell Death in Response to Cytoplasmic DNA. Nature (2009) 458:509–13. doi: 10.1038/nature07710
82. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 Recognizes Cytosolic Dsdna and Forms a Caspase-1 Activating Inflammasome With ASC. Nature (2009) 458:514–8. doi: 10.1038/nature07725
83. Wang L-Q, Liu T, Yang S, Sun L, Zhao Z-Y, Li L-Y, et al. Perfluoroalkyl Substance Pollutants Activate the Innate Immune System Through the AIM2 Inflammasome. Nat Commun (2021) 12:2915. doi: 10.1038/s41467-021-23201-0
84. Moretti J, Blander JM. Increasing Complexity of NLRP3 Inflammasome Regulation. J Leukocyte Biol (2021) 109:561–71. doi: 10.1002/jlb.3mr0520-104rr
85. Netea MG, Simon A, van de Veerdonk F, Kullberg B-J, der Meer JWMV, Joosten LAB. IL-1β Processing in Host Defense: Beyond the Inflammasomes. PloS Pathog (2010) 6:e1000661. doi: 10.1371/journal.ppat.1000661
86. Netea MG, van de Veerdonk FL, van der Meer JWM, Dinarello CA, Joosten LAB. Inflammasome-Independent Regulation of IL-1-Family Cytokines. Annu Rev Immunol (2014) 33:1–29. doi: 10.1146/annurev-immunol-032414-112306
87. Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, et al. Converting Enzyme-Independent Release of Tumor Necrosis Factor A and IL-1β From a Stimulated Human Monocytic Cell Line in the Presence of Activated Neutrophils or Purified Proteinase 3. Proc Natl Acad Sci (1999) 96:6261–6. doi: 10.1073/pnas.96.11.6261
88. Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A, et al. Neutrophil Proteinase 3-Mediated Induction of Bioactive IL-18 Secretion by Human Oral Epithelial Cells. J Immunol (2001) 167:6568–75. doi: 10.4049/jimmunol.167.11.6568
89. Herzog C, Haun RS, Kaushal V, Mayeux PR, Shah SV, Kaushal GP. Meprin a and Meprin A Generate Biologically Functional IL-1β From Pro-IL-1β. Biochem Bioph Res Co (2009) 379:904–8. doi: 10.1016/j.bbrc.2008.12.161
90. Schönbeck U, Mach F, Libby P. Generation of Biologically Active IL-1 Beta by Matrix Metalloproteinases: A Novel Caspase-1-Independent Pathway of IL-1 Beta Processing. J Immunol Baltim Md 1950 (1998) 161:3340–6.
91. Hildebrand D, Bode KA, Rieß D, Cerny D, Waldhuber A, Römmler F, et al. Granzyme a Produces Bioactive IL-1β Through a Nonapoptotic Inflammasome-Independent Pathway. Cell Rep (2014) 9:910–7. doi: 10.1016/j.celrep.2014.10.003
92. Orning P, Lien E. Multiple Roles of Caspase-8 in Cell Death, Inflammation, and Innate Immunity. J Leukocyte Biol (2021) 109:121–41. doi: 10.1002/jlb.3mr0420-305r
93. Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, et al. Stimulation of Toll-Like Receptor 3 and 4 Induces Interleukin-1β Maturation by Caspase-8. J Exp Med (2008) 205:1967–73. doi: 10.1084/jem.20071632
94. Antonopoulos C, Sanadi CE, Kaiser WJ, Mocarski ES, Dubyak GR. Proapoptotic Chemotherapeutic Drugs Induce Noncanonical Processing and Release of IL-1β via Caspase-8 in Dendritic Cells. J Immunol (2013) 191:4789–803. doi: 10.4049/jimmunol.1300645
95. Shenderov K, Riteau N, Yip R, Mayer-Barber KD, Oland S, Hieny S, et al. Cutting Edge: Endoplasmic Reticulum Stress Licenses Macrophages to Produce Mature IL-1β in Response to TLR4 Stimulation Through a Caspase-8– and TRIF-Dependent Pathway. J Immunol (2014) 192:2029–33. doi: 10.4049/jimmunol.1302549
96. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas Triggers an Alternative, Caspase-8–Independent Cell Death Pathway Using the Kinase RIP as Effector Molecule. Nat Immunol (2000) 1:489–95. doi: 10.1038/82732
97. Bossaller L, Chiang P-I, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VAK, et al. Cutting Edge: FAS (CD95) Mediates Noncanonical IL-1β and IL-18 Maturation via Caspase-8 in an RIP3-Independent Manner. J Immunol (2012) 189:5508–12. doi: 10.4049/jimmunol.1202121
98. Vince JE, Wong WW-L, Gentle I, Lawlor KE, Allam R, O’Reilly L, et al. Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity (2012) 36:215–27. doi: 10.1016/j.immuni.2012.01.012
99. Moriwaki K, Bertin J, Gough PJ, Chan FK-M. A RIPK3–Caspase 8 Complex Mediates Atypical Pro–IL-1β Processing. J Immunol (2015) 194:1938–44. doi: 10.4049/jimmunol.1402167
100. Briard B, Malireddi RKS, Kanneganti T-D. Role of Inflammasomes/Pyroptosis and Panoptosis During Fungal Infection. PloS Pathog (2021) 17:e1009358. doi: 10.1371/journal.ppat.1009358
101. Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, et al. Dectin-1 is an Extracellular Pathogen Sensor for the Induction and Processing of IL-1β via a Noncanonical Caspase-8 Inflammasome. Nat Immunol (2012) 13:246–54. doi: 10.1038/ni.2222
102. Mayer-Barber KD, Sher A. Cytokine and Lipid Mediator Networks in Tuberculosis. Immunol Rev (2015) 264:264–75. doi: 10.1111/imr.12249
103. Orme IM, Robinson RT, Cooper AM. The Balance Between Protective and Pathogenic Immune Responses in the TB-Infected Lung. Nat Immunol (2015) 16:57–63. doi: 10.1038/ni.3048
104. Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and Chemokines in Mycobacterium Tuberculosis Infection. Microbiol Spectr (2016) 4(5):10.1128/microbiolspec.TBTB2-0018-2016. doi: 10.1128/microbiolspec.tbtb2-0018-2016
105. Simmons JD, Stein CM, Seshadri C, Campo M, Alter G, Fortune S, et al. Immunological Mechanisms of Human Resistance to Persistent Mycobacterium Tuberculosis Infection. Nat Rev Immunol (2018) 18:575–89. doi: 10.1038/s41577-018-0025-3
106. Sia JK, Rengarajan J. Immunology of Mycobacterium Tuberculosis Infections. Microbiol Spectr (2019) 7(4):10.1128/microbiolspec.GPP3-0022-2018. doi: 10.1128/microbiolspec.gpp3-0022-2018
107. Juffermans NP, Florquin S, Camoglio L, Verbon A, Kolk AH, Speelman P, et al. Interleukin-1 Signaling is Essential for Host Defense During Murine Pulmonary Tuberculosis. J Infect Dis (2000) 182:902–8. doi: 10.1086/315771
108. Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I. Protective Role of Interleukin-1 in Mycobacterial Infection in IL-1 Alpha/Beta Double-Knockout Mice. Lab Invest (2000) 80:759–67. doi: 10.1038/labinvest.3780079
109. Sugawara I, Yamada H, Hua S, Mizuno S. Role of Interleukin (IL)-1 Type 1 Receptor in Mycobacterial Infection. Microbiol Immunol (2001) 45:743–50. doi: 10.1111/j.1348-0421.2001.tb01310.x
110. Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, et al. IL-1 Receptor-Mediated Signal is an Essential Component of Myd88-Dependent Innate Response to Mycobacterium Tuberculosis Infection. J Immunol (2007) 179:1178–89. doi: 10.4049/jimmunol.179.2.1178
111. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, et al. Caspase-1 Independent IL-1beta Production is Critical for Host Resistance to Mycobacterium Tuberculosis and Does Not Require TLR Signaling in Vivo. J Immunol (Baltimore Md: 1950) (2010) 184:3326–30. doi: 10.4049/jimmunol.0904189
112. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and Adaptive Interferons Suppress IL-1α and IL-1β Production by Distinct Pulmonary Myeloid Subsets During Mycobacterium Tuberculosis Infection. Immunity (2011) 35:1023–34. doi: 10.1016/j.immuni.2011.12.002
113. Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, et al. IL-1β Promotes Antimicrobial Immunity in Macrophages by Regulating TNFR Signaling and Caspase-3 Activation. J Immunol (Baltimore Md. : 1950) (2013) 190:4196–204. doi: 10.4049/jimmunol.1202688
114. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity (2012) 37:223–34. doi: 10.1016/j.immuni.2012.04.015
115. Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, et al. Mycobacterium Tuberculosis Prevents Inflammasome Activation. Cell Host Microbe (2008) 3:224–32. doi: 10.1016/j.chom.2008.03.003
116. Eklund D, Welin A, Andersson H, Verma D, Söderkvist P, Stendahl O, et al. Human Gene Variants Linked to Enhanced NLRP3 Activity Limit Intramacrophage Growth of Mycobacterium Tuberculosis. J Infect Dis (2014) 209:749–53. doi: 10.1093/infdis/jit572
117. Bohrer AC, Tocheny C, Assmann M, Ganusov VV, Mayer-Barber KD. Cutting Edge: IL-1R1 Mediates Host Resistance to Mycobacterium Tuberculosis by Trans-Protection of Infected Cells. J Immunol (Baltimore Md. : 1950) (2018) 201:1645–50. doi: 10.4049/jimmunol.1800438
118. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-Directed Therapy of Tuberculosis Based on Interleukin-1 and Type I Interferon Crosstalk. Nature (2014) 511:99–103. doi: 10.1038/nature13489
119. Dorhoi A, Kaufmann SHE. Pathology and Immune Reactivity: Understanding Multidimensionality in Pulmonary Tuberculosis. Semin Immunopathol (2016) 38:153–66. doi: 10.1007/s00281-015-0531-3
120. Moreira-Teixeira L, Mayer-Barber K, Sher A, O’Garra A. Type I Interferons in Tuberculosis: Foe and Occasionally Friend. J Exp Med (2018) 215:1273–85. doi: 10.1084/jem.20180325
121. Mayer-Barber KD, Yan B. Clash of the Cytokine Titans: Counter-Regulation of Interleukin-1 and Type I Interferon-Mediated Inflammatory Responses. Cell Mol Immunol (2017) 14:22–35. doi: 10.1038/cmi.2016.25
122. Zhang L, Jiang X, Pfau D, Ling Y, Nathan CF. Type I Interferon Signaling Mediates Mycobacterium Tuberculosis–Induced Macrophage Death. J Exp Med (2020) 218:556–16. doi: 10.1084/jem.20200887
123. Sousa J, Cá B, Maceiras AR, Simões-Costa L, Fonseca KL, Fernandes AI, et al. Mycobacterium Tuberculosis Associated With Severe Tuberculosis Evades Cytosolic Surveillance Systems and Modulates IL-1β Production. Nat Commun (2020) 11:1949. doi: 10.1038/s41467-020-15832-6
124. Matty MA, Knudsen DR, Walton EM, Beerman RW, Cronan MR, Pyle CJ, et al. Potentiation of P2RX7 as a Host-Directed Strategy for Control of Mycobacterial Infection. Elife (2019) 8:e39123. doi: 10.7554/elife.39123
125. Zhang G, Zhou B, Li S, Yue J, Yang H, Wen Y, et al. Allele-Specific Induction of IL-1β Expression by C/Ebpβ and PU.1 Contributes to Increased Tuberculosis Susceptibility. PloS Pathog (2014) 10:e1004426. doi: 10.1371/journal.ppat.1004426
126. Wilkinson RJ, Patel P, Llewelyn M, Hirsch CS, Pasvol G, Snounou G, et al. Influence of Polymorphism in the Genes for the Interleukin (IL)-1 Receptor Antagonist and IL-1β on Tuberculosis. J Exp Med (1999) 189:1863–74. doi: 10.1084/jem.189.12.1863
127. Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric Oxide Controls the Immunopathology of Tuberculosis by Inhibiting NLRP3 Inflammasome–Dependent Processing of IL-1β. Nat Immunol (2012) 14:52–60. doi: 10.1038/ni.2474
128. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric Oxide Prevents a Pathogen-Permissive Granulocytic Inflammation During Tuberculosis. Nat Microbiol (2017) 2:17072. doi: 10.1038/nmicrobiol.2017.72
129. Subbarao S, Sanchez-Garrido J, Krishnan N, Shenoy AR, Robertson BD. Genetic and Pharmacological Inhibition of Inflammasomes Reduces the Survival of Mycobacterium Tuberculosis Strains in Macrophages. Sci Rep-uk (2020) 10:3709. doi: 10.1038/s41598-020-60560-y
130. Roca FJ, Ramakrishnan L. TNF Dually Mediates Resistance and Susceptibility to Mycobacteria via Mitochondrial Reactive Oxygen Species. Cell (2013) 153:521–34. doi: 10.1016/j.cell.2013.03.022
131. Taft J, Bogunovic D. The Goldilocks Zone of Type I Ifns: Lessons From Human Genetics. J Immunol (Baltimore Md: 1950) (2018) 201:3479–85. doi: 10.4049/jimmunol.1800764
132. Zheng D, Liwinski T, Elinav E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov (2020) 6:36. doi: 10.1038/s41421-020-0167-x
133. TeKippe EM, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, et al. Granuloma Formation and Host Defense in Chronic Mycobacterium Tuberculosis Infection Requires PYCARD/ASC But Not NLRP3 or Caspase-1. PloS One (2010) 5:e12320. doi: 10.1371/journal.pone.0012320
134. Rastogi S, Ellinwood S, Augenstreich J, Mayer-Barber KD, Briken V. Mycobacterium Tuberculosis Inhibits the NLRP3 Inflammasome Activation via its Phosphokinase Pknf. PloS Pathog (2021) 17:e1009712. doi: 10.1371/journal.ppat.1009712
135. Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, et al. Plasma Membrane Damage Causes NLRP3 Activation and Pyroptosis During Mycobacterium Tuberculosis Infection. Nat Commun (2020) 11:2270. doi: 10.1038/s41467-020-16143-6
136. Mendes ALG, Joaquim HDM, Zamae MIS, Assis RM, Peixoto JR, de M, et al. Expression of NLRP3 Inflammasome in Leprosy Indicates Immune Evasion of Mycobacterium Leprae. Memórias Inst Oswaldo Cruz (2020) 115:e190324. doi: 10.1590/0074-02760190324
137. Ligon LS, Hayden JD, Braunstein M. The Ins and Outs of Mycobacterium Tuberculosis Protein Export. Tuberc (Edinburgh Scotland) (2012) 92:121–32. doi: 10.1016/j.tube.2011.11.005
138. Majlessi L, Rosales RP, Casadevall A, Brosch R. Release of Mycobacterial Antigens. Immunol Rev (2015) 264:25–45. doi: 10.1111/imr.12251
139. Vaziri F, Brosch R. ESX/Type VII Secretion Systems—An Important Way Out for Mycobacterial Proteins. Microbiol Spectr (2019) 7:351–62. doi: 10.1128/microbiolspec.psib-0029-2019
140. Augenstreich J, Briken V. Host Cell Targets of Released Lipid and Secreted Protein Effectors of Mycobacterium Tuberculosis. Front Cell Infect Microbiol (2020) 10:595029. doi: 10.3389/fcimb.2020.595029
141. Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, et al. Direct Extracellular Interaction Between the Early Secreted Antigen ESAT-6 of Mycobacterium Tuberculosis and TLR2 Inhibits TLR Signaling in Macrophages. Nat Immunol (2007) 8:610–8. doi: 10.1038/ni1468
142. Chatterjee S, Dwivedi VP, Singh Y, Siddiqui I, Sharma P, Kaer LV, et al. Early Secreted Antigen ESAT-6 of Mycobacterium Tuberculosis Promotes Protective T Helper 17 Cell Responses in a Toll-Like Receptor-2-Dependent Manner. PloS Pathog (2011) 7:e1002378. doi: 10.1371/journal.ppat.1002378
143. Kumar P, Agarwal R, Siddiqui I, Vora H, Das G, Sharma P. ESAT6 Differentially Inhibits IFN-Γ-Inducible Class II Transactivator Isoforms in Both a TLR2-Dependent and -Independent Manner. Immunol Cell Biol (2012) 90:411–20. doi: 10.1038/icb.2011.54
144. Jang A-R, Choi J-H, Shin SJ, Park J-H. Mycobacterium Tuberculosis ESAT6 Induces IFN-B Gene Expression in Macrophages via Tlrs-Mediated Signaling. Cytokine (2017) 104:104–9. doi: 10.1016/j.cyto.2017.10.006
145. Schweneker K, Gorka O, Schweneker M, Poeck H, Tschopp J, Peschel C, et al. The Mycobacterial Cord Factor Adjuvant Analogue Trehalose-6,6′-Dibehenate (TDB) Activates the Nlrp3 Inflammasome. Immunobiology (2013) 218:664–73. doi: 10.1016/j.imbio.2012.07.029
146. Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium Tuberculosis Activates the DNA-Dependent Cytosolic Surveillance Pathway Within Macrophages. Cell Host Microbe (2012) 11:469–80. doi: 10.1016/j.chom.2012.03.007
147. Sauer J-D, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria Monocytogenes Triggers AIM2-Mediated Pyroptosis Upon Infrequent Bacteriolysis in the Macrophage Cytosol. Cell Host Microbe (2010) 7:412–9. doi: 10.1016/j.chom.2010.04.004
148. Madan-Lala R, Peixoto KV, Re F, Rengarajan J. Mycobacterium Tuberculosis Hip1 Dampens Macrophage Proinflammatory Responses by Limiting Toll-Like Receptor 2 Activation. Infect Immun (2011) 79:4828–38. doi: 10.1128/iai.05574-11
149. Naffin-Olivos JL, Georgieva M, Goldfarb N, Madan-Lala R, Dong L, Bizzell E, et al. Mycobacterium Tuberculosis Hip1 Modulates Macrophage Responses Through Proteolysis of Groel2. PloS Pathog (2014) 10:e1004132. doi: 10.1371/journal.ppat.1004132
150. Danelishvili L, Everman JL, McNamara MJ, Bermudez LE. Inhibition of the Plasma-Membrane-Associated Serine Protease Cathepsin G by Mycobacterium Tuberculosis Rv3364c Suppresses Caspase-1 and Pyroptosis in Macrophages. Front Microbiol (2012) 2:281. doi: 10.3389/fmicb.2011.00281
151. Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, et al. Cutting Edge: Nitric Oxide Inhibits the NLRP3 Inflammasome. J Immunol (2012) 189:5113–7. doi: 10.4049/jimmunol.1202479
152. Hortle E, Tran LV, Fontaine AR, Pinello N, Wong JJ-L, Britton WJ, et al. OXSR1 Inhibits Inflammasome Activation by Limiting Potassium Efflux During Mycobacterial Infection. Biorxiv (2021) 2021.04.21.440692. doi: 10.1101/2021.04.21.440692
153. Zhang Q, Jiang X, He W, Wei K, Sun J, Qin X, et al. MCL Plays an Anti-Inflammatory Role in Mycobacterium Tuberculosis-Induced Immune Response by Inhibiting NF-Kb and NLRP3 Inflammasome Activation. Mediat Inflammation (2017) 2017:1–12. doi: 10.1155/2017/2432904
154. Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 Receptor Differentially Couples to Distinct Release Pathways for IL-1β in Mouse Macrophage. J Immunol (2008) 180:7147–57. doi: 10.4049/jimmunol.180.11.7147
Keywords: Mycobacterium tuberculosis, inflammasome, NLRP3, AIM2, ESX-1, IL-1b, NTM = nontuberculous mycobacteria
Citation: Rastogi S and Briken V (2022) Interaction of Mycobacteria With Host Cell Inflammasomes. Front. Immunol. 13:791136. doi: 10.3389/fimmu.2022.791136
Received: 08 October 2021; Accepted: 13 January 2022;
Published: 14 February 2022.
Edited by:
Suzie Hingley-Wilson, University of Surrey, United KingdomReviewed by:
Gang Pei, Friedrich-Loeffler-Institute, GermanyRachel Simmonds, University of Surrey, United Kingdom
Copyright © 2022 Rastogi and Briken. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Volker Briken, dmJyaWtlbkB1bWQuZWR1