 Tae Seok Oh
Tae Seok Oh Damian C. Hutchins
Damian C. Hutchins Rabina Mainali
Rabina Mainali Kevin H. Goslen
Kevin H. Goslen Matthew A. Quinn
Matthew A. Quinn- 1Department of Pathology, Section on Comparative Medicine, Winston-Salem, NC, United States
- 2Department of Internal Medicine, Section on Molecular Medicine Wake Forest School of Medicine, Winston-Salem, NC, United States
A Krebs cycle intermediate metabolite, itaconate, has gained attention as a potential antimicrobial and autoimmune disease treatment due to its anti-inflammatory effects. While itaconate and its derivatives pose an attractive therapeutic option for the treatment of inflammatory diseases, the effects outside the immune system still remain limited, particularly in the muscle. Therefore, we endeavored to determine if itaconate signaling impacts muscle differentiation. Utilizing the well-established C2C12 model of in vitro myogenesis, we evaluated the effects of itaconate and its derivatives on transcriptional and protein markers of muscle differentiation as well as mitochondrial function. We found itaconate and the derivatives dimethyl itaconate and 4-octyl itaconate disrupt differentiation media-induced myogenesis. A primary biological effect of itaconate is a succinate dehydrogenase (SDH) inhibitor. We find the SDH inhibitors dimethyl malonate and harzianopyridone phenocopie the anti-myogenic effects of itaconate. Furthermore, we find treatment with exogenous succinate results in blunted myogenesis. Together our data indicate itaconate and its derivatives interfere with in vitro myogenesis, potentially through inhibition of SDH and subsequent succinate accumulation. We also show 4-octyl itaconate suppresses injury-induced MYOG expression in vivo. More importantly, our findings suggest the therapeutic potential of itaconate, and its derivatives could be limited due to deleterious effects on myogenesis.
Introduction
It is well appreciated that dramatic metabolic reprogramming occurs in response to inflammatory challenges. One of the most robust metabolic adaptations made in myeloid cells in response to inflammatory cues is the diversion of cis-aconitate away from α-ketoglutarate towards the synthesis of itaconate (1, 2). One primary function itaconate serves is to reduce inflammation via the inhibition of succinate dehydrogenase (SDH) thereby reducing accumulation of reactive oxygen species (ROS) (1). Due to its potent anti-inflammatory properties, itaconate and its derivatives such as dimethyl itaconate (DMI) and 4-octyl itaconate (4-OI) have gained attention as potential treatments for autoimmune conditions such as psoriasis (3), multiple sclerosis (4), rheumatoid arthritis (5), and systemic lupus erythematosus (6).
While the signaling attributes of itaconate with regards to immune function are well delineated, not much is known on the actions of itaconate in non-immune organs. What is known is that itaconate possesses anti-inflammatory, anti-fibrotic and pro-survival attributes in organs such as the liver (7) and kidney (8). Furthermore, amassing evidence has shown that itaconate derivatives have therapeutic effects on idiopathic pulmonary fibrosis (9), ischemia-reperfusion injury (10, 11), abdominal aortic aneurysm (12), and sepsis (13). However, the effects of itaconate regulation of muscle function are yet unknown.
Recent studies have shown side effects of commonly used drugs on muscle atrophy (14) and toxicity (15, 16). Nevertheless, knowledge concerning potential side effects of itaconate and its derivatives on muscle is still limited. Given that chronic inflammation is hypothesized to incur sustained itaconate production, coupled to its potential therapeutic use for the treatment of a variety of diseases, it is of the upmost importance to understand potential off target tissues and effects in the face of elevated itaconate levels. To address this critical gap in knowledge we utilized the well-established C2C12 murine myoblast model system for in vitro myogenesis as well as barium chloride-induced muscle regeneration and studied the effects of itaconate and its derivatives on altering myoblast differentiation.
Results
Itaconate Derivatives Inhibits Myogenesis in C2C12 Cells
Past research has shown that Krebs cycle metabolites serve as signal transducers in the immune response within macrophages (17). We endeavored to evaluate how the presence of itaconate affects myogenic differentiation within C2C12 cells. Given its recent focus as a potential treatment for autoimmune conditions, the function of dimethyl itaconate (DMI) was first investigated. Gene expression markers for myogenesis (i.e. Myh1, Myh2, Myh7, Tnnt1, and Tnnt3) were measured in the presence of DMI versus vehicle over a 4-day differentiation protocol. We looked at day 1 and 2 to confirm initiation and induction of myogenesis and investigated day 4 to analyze myotube formation. As expected, vehicle group cells had a time-dependent increase in the transcription of myogenic genes (Figure 1A). However, treatment with DMI significantly inhibited the induction of these myogenic markers during our differentiation protocol (Figure 1A). Another analog of itaconate was also utilized since DMI cannot be endogenously converted to intracellular itaconate (18). 4-octyl itaconate (4-OI) was subsequently used because it has been suggested as a possible treatment for autoimmune conditions, but unlike DMI, it can be endogenously converted to itaconate (18, 19). We analyzed C2C12 cells treated with 4-OI over 4 days in the presence of differentiation media (DM). 4-OI significantly inhibited the induction of myogenic gene transcription as well (Figure 1B).
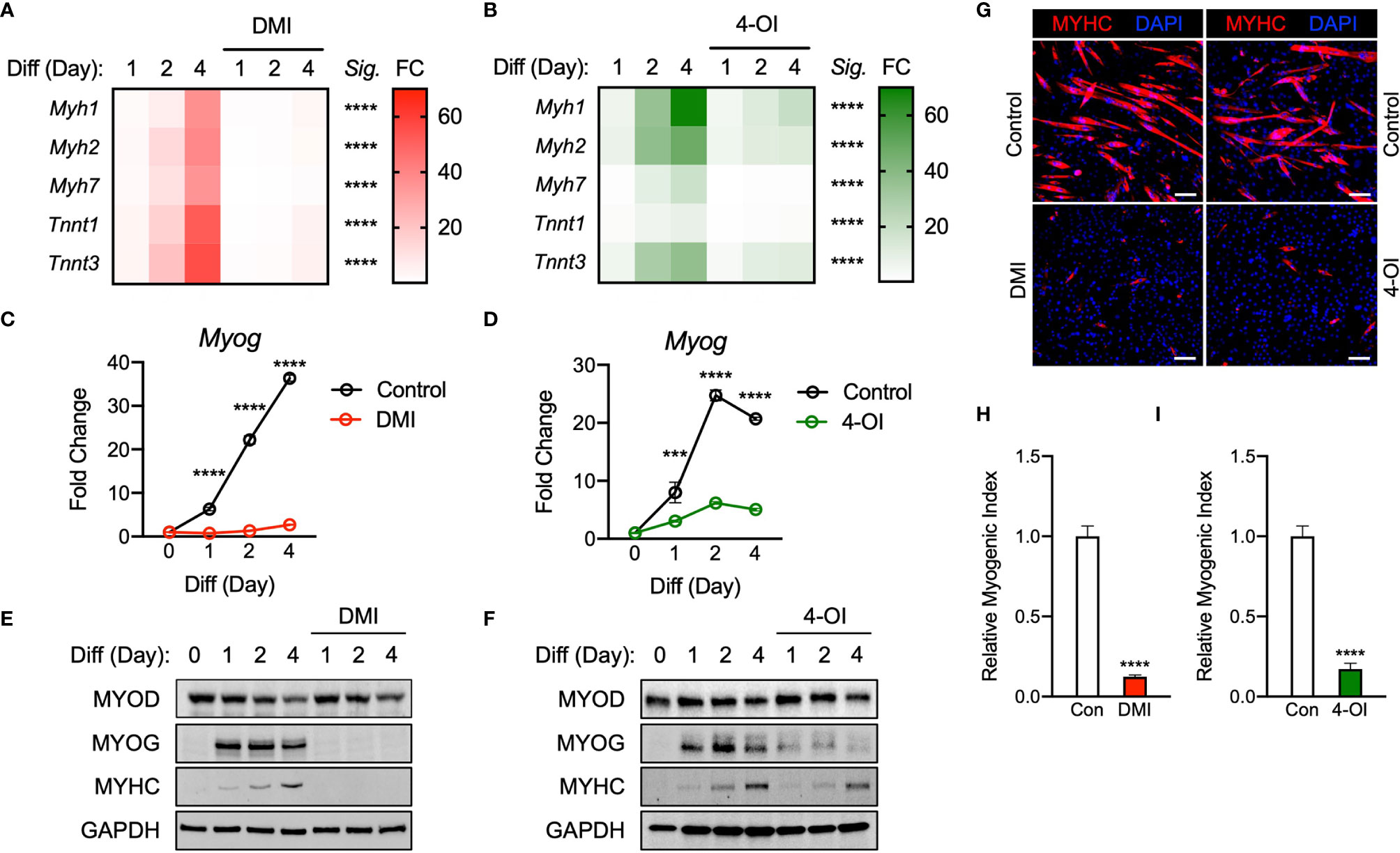
Figure 1 Itaconate derivatives repress C2C12 myogenesis by modulating myogenic transcription mechanisms. Heatmap depiction of average fold change (Sig. denotes statistical differences at day 4, n = 3) in myogenic gene transcription levels detected via RT-qPCR when exposed to differential media with or without (A) dimethyl itaconate (DMI: 125 µM) and (B) 4-octyl itaconate (4-OI: 125 µM) relative to the untreated group over a 4-day time course. Fold change of Myog mRNA expression at various time points comparing differentiation media (DM) control groups to (C) DMI and (D) 4-OI groups relative to expression without either treatment (n = 3). Western blotting data showing differences in myogenic transcription factor expression between DM treated groups and groups treated with DM plus (E) DMI and (F) 4-OI over a 4 day time course as well as groups which were not exposed to either treatment for any period. (G) MYHC staining at day 4 of differentiation. Bar: 100 μm. Relative myogenic index (percentage of total nuclei associated with myotubes) of (H) DMI and (I) 4-OI. *** denotes p < 0.001, ****p < 0.0001 compared to control based on t-test.
To further determine how DMI and 4-OI impairs myogenesis, we next assessed the expression of the essential myogenic transcription factors Myog and Myod. The expression of Myog mRNA was significantly impaired by DMI throughout day 1 to day 4 of differentiation compared to the control (Figure 1C). Transcription of Myog was also blunted in response to 4-OI exposure (Figure 1D). Protein levels of MYOD were not different in the DMI groups compared to the control in response to DM. However, MYOG expression was almost completely abolished by DMI (Figure 1E). Consequently, DMI-treated C2C12 myoblasts failed to differentiate into myotubes. DMI-treated groups showed no trace of MYHC in response to DM cues, while vehicle-treated cells displayed a time-dependent increase in MYHC protein (Figure 1E). 4-OI significantly blocked MYOG and MYHC expression levels although it did not change MYOD levels (Figure 1F). MYHC was visualized by immunofluorescence at day 4 of differentiation (Figure 1G) and myogenic index was significantly impaired by DMI (Figure 1H) and 4-OI (Figure 1I).
Physiological Itaconate Inhibits Myogenesis
To investigate a role of physiological itaconate, C2C12 myoblast cells were then treated with DM only or DM plus itaconate and then examined over 4 days. It was reported that exogenous itaconate readily enters cells (17). Itaconate was used to portray physiological mechanisms more accurately. Transcription levels of Myh2 and Myh7 were not significantly impacted by itaconate exposure. However, the blunted induction of Myh1, Tnnt1, and Tnnt3 was significant on the 4th day of exposure (Figure 2A). Significant inhibition of Myog transcription on days 2 and 4 was also observed in itaconate groups (Figure 2B). MYOG and MYHC expression on day 2 were also inhibited in itaconate groups (Figure 2C). Itaconate treatment reduced MYHC staining (Figure 2D) and myogenic index (Figure 2E).
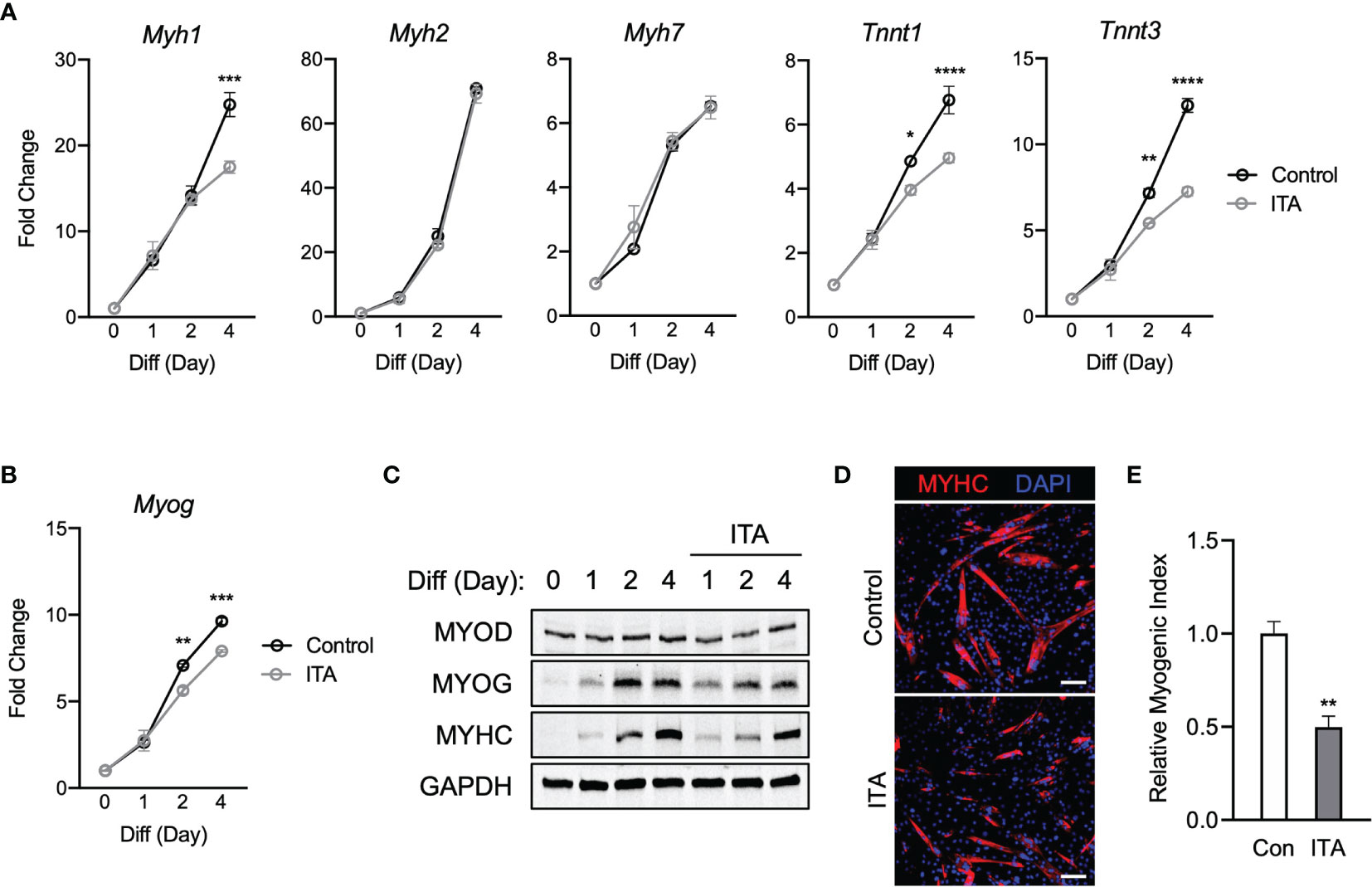
Figure 2 Itaconate represses C2C12 myogenesis by modulating myogenic transcription mechanisms. (A) 4-day time course showcasing fold change differences of myogenic gene transcription levels detected via RT-qPCR when exposed to differential media with or without itaconate (7.5 mM) relative to the untreated group (n = 3). (B) Fold change of Myog mRNA expression at various time points comparing DM control groups to DM and itaconate groups relative to expression without either treatment (n = 3). (C) Western blotting data showing differences in myogenic transcription factor expression between DM treated groups and groups treated with both DM and itaconate over a 4 day time course as well as groups which were not exposed to either treatment for any period. (D) MYHC staining at day 4 of differentiation. Bar: 100 μm. (E) Relative myogenic index (percentage of total nuclei associated with myotubes). * denotes p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control based on t-test.
Overall DMI shows the strongest myogenic inhibitory response and itaconate has the weakest amount of myogenic suppression among itaconate and derivatives tested. Taken together, these results suggest that itaconate and its derivatives are sufficient to inhibit myogenesis in C2C12 cells, modulating myogenic regulating factors at the transcription and protein levels.
Itaconate Derivatives Lead to Inhibition of Succinate Dehydrogenase and Accumulation of Intracellular Succinate During Myogenesis
Considering itaconate can lead to accumulation of succinate via SDH inhibition (20–22), we tested if this happens in our model with itaconate derivatives. As expected, SDH activity was reduced significantly at day 1 of differentiation when treated with DMI and 4-OI compared to control (Figure 3A). However, this inhibition was not maintained at day 2 of differentiation (Figure 3B). In line with decreased SDH activity in response to itaconate derivatives during myogenesis, intracellular succinate levels were higher when treated with itaconate derivatives compared to control at day 1 and 2 of differentiation (Figures 3C, D). Taken together, inhibition of SDH by itaconate derivatives during myogenesis on day 1 was sufficient to keep succinate accumulated in C2C12 cells.
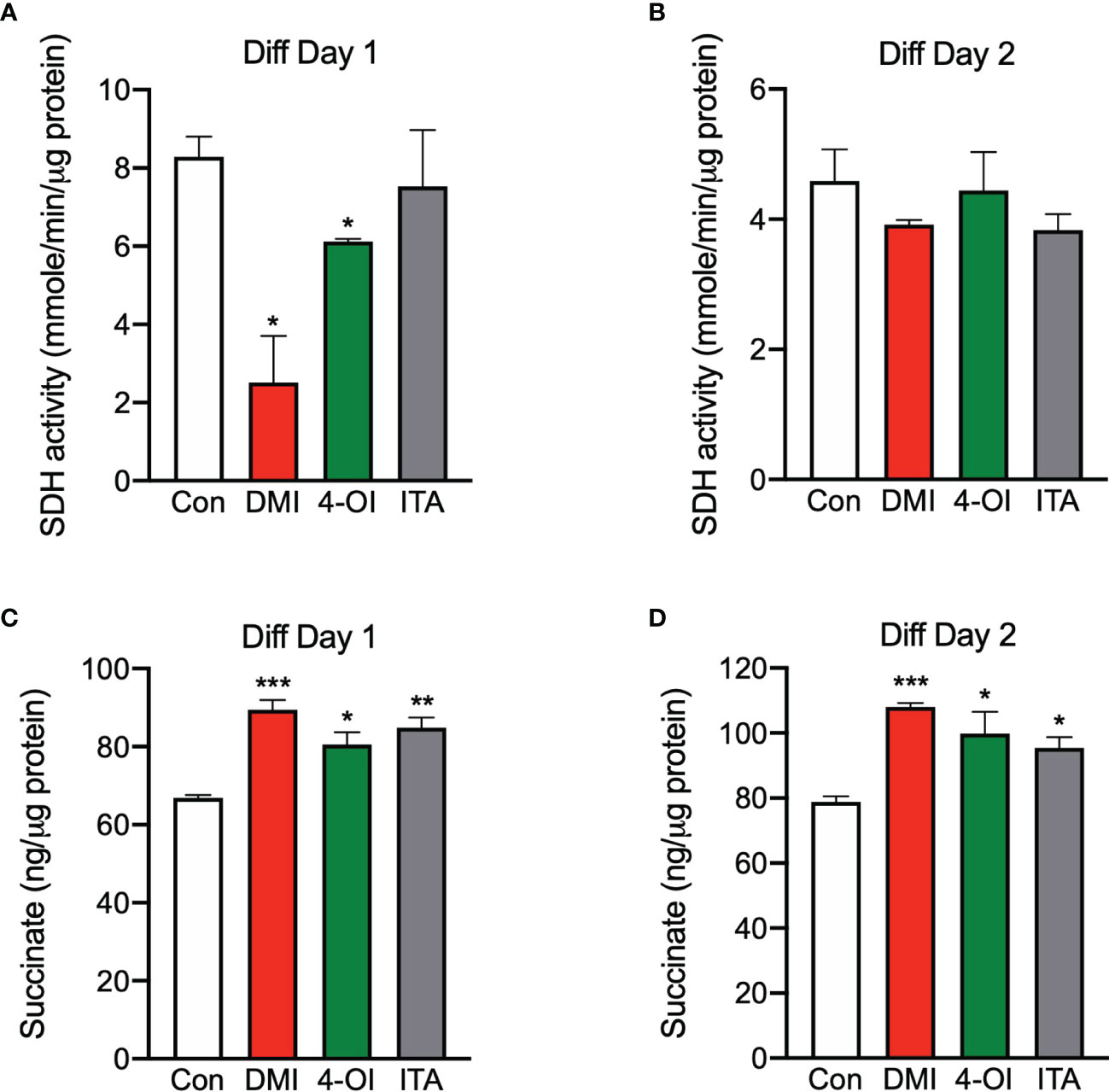
Figure 3 Itaconate derivatives inhibit succinate dehydrogenase and accumulate intracellular succinate during C2C12 myogenesis. Succinate dehydrogenase (SDH) activities in the presence of itaconate derivatives at day 1 (A) and day 2 (B) of differentiation (n = 6). Succinate levels in the presence of itaconate derivatives at day 1 (C) and day 2 (D) of differentiation (n = 6). * denotes p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control based on t-test.
Malonate and Pharmacological Succinate Dehydrogenase Inhibitor Obstruct Myogenesis
Given that both itaconate and malonate are Krebs cycle metabolites that are shown to inhibit SDH (1, 23), our next step was to determine whether malonate exposure could also inhibit C2C12 myogenesis. Dimethyl malonate (DMM) was administrated instead of itaconate under similar parameters to those discussed in Figures 1 through 2. Malonate inhibited the induction of myogenic markers during myogenesis (Figure 4A). This inhibition was more severe than itaconate but not as robust as the inhibition caused by itaconate derivatives. In parallel, a potent and specific inhibitor of SDH, Harzianopyridone (Harz) (24), was tested to determine whether this specific shared inhibition was a mechanism underlying impaired myogenesis. We examined C2C12 cells exposed to Harz using the same methods to elucidate whether SDH inhibition independent of the Krebs cycle might also elicit a dramatic obstruction of myogenesis. Harz treatment resulted in significant suppression of myogenic markers (Figure 4B). Malonate lowered Myog levels compared to the control at all the time points (Figure 4C). Consistent suppression of Myog expression was also observed following Harz treatment throughout the time course (Figure 4D). MYOD levels were similar in malonate-treated cells during differentiation, but malonate led to a reduction in MYOG levels and a significant decrease in MYHC levels (Figure 4E). Harz showed significantly reduced MYOG and MYHC levels as well (Figure 4F). Malonate showed similar extent of reduction in MYHC staining (Figure 4G) and myogenic index (Figure 4H) as itaconate. Harz displayed the most robust inhibition of myogenesis shown by MYHC staining (Figure 4G) and myogenic index (Figure 4I). The degree of suppression caused by Harz was most comparable to the amount of inhibition by DMI. In tandem, these findings support that inhibition of SDH affects myogenesis in C2C12 cells.
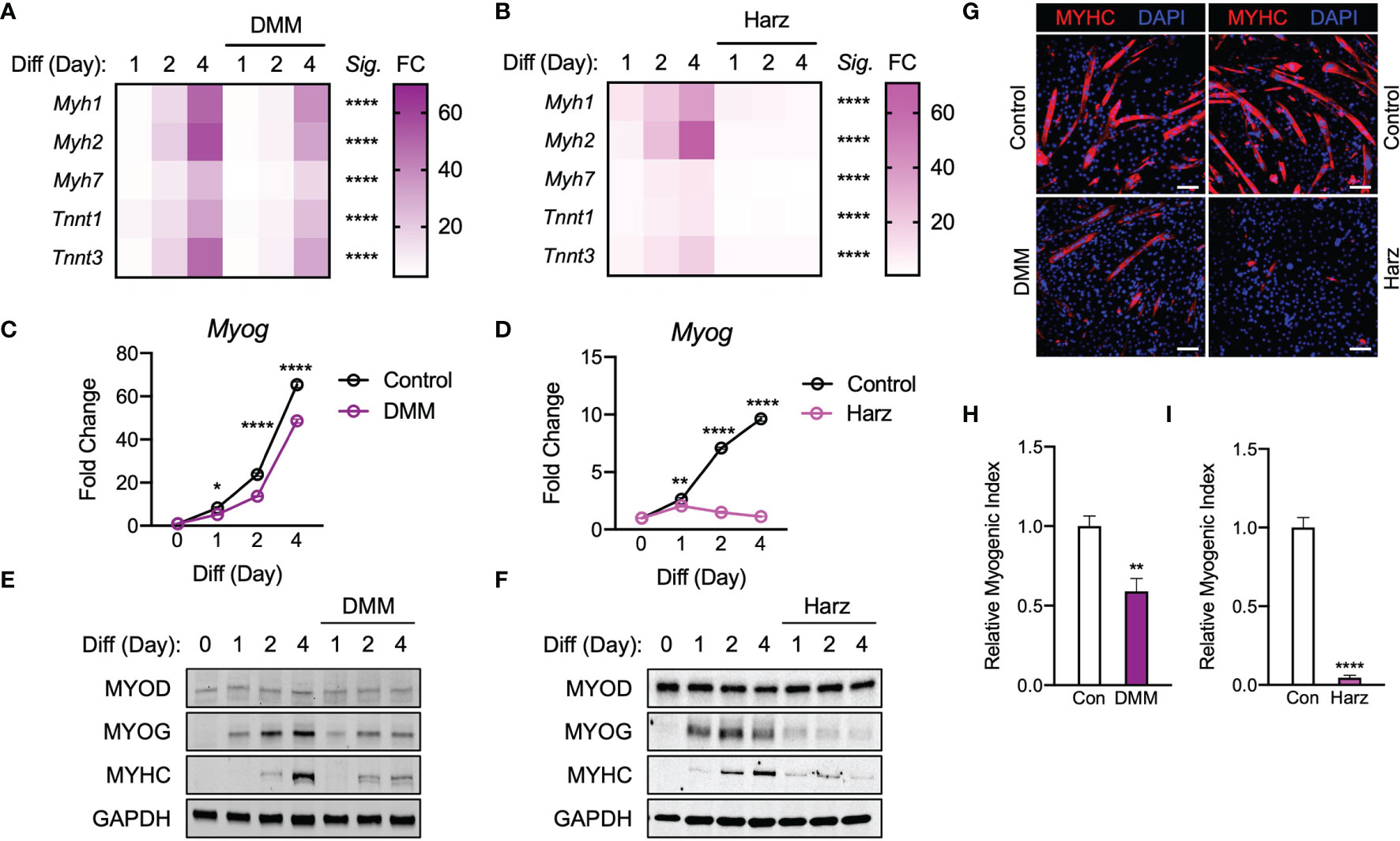
Figure 4 Malonate and pharmacological inhibitor of succinate dehydrogenase repress C2C12 myogenesis via similar mechanism of action to that of itaconate and its derivatives. Heatmap depiction of average fold change (Sig. denotes statistical differences at day 4, n = 3) in myogenic gene transcription levels detected via RT-qPCR when exposed to differential media with or without (A) dimethyl malonate (DMM: 5 mM) and (B) harzianopyridone (Harz: 4 µM) relative to the untreated group over a 4-day time course. Fold change of Myog mRNA expression at various time points comparing differentiation media (DM) control groups to (C) DMI and (D) 4-OI groups relative to expression without either treatment (n = 3). Western blotting data showing differences in myogenic transcription factor expression between DM treated groups and groups treated with DM plus (E) DMM and (F) Harz over a 4 day time course as well as groups which were not exposed to either treatment for any period. (G) MYHC staining at day 4 of differentiation. Bar: 100 μm. Relative myogenic index (percentage of total nuclei associated with myotubes) of (H) DMM and (I) Harz. * denotes p < 0.05, **p < 0.01, ****p < 0.0001 compared to control based on t-test.
Myogenesis Is Inhibited by Exogenous Succinate
Inhibition of SDH activity can lead to an increase in intracellular succinate levels (20). Given that SDH inhibition alone is able to significantly inhibit myogenesis, we next asked if succinate accumulation could be the connecting factor leading to myogenic inhibition. Thus, we tested if exogenous succinate could cause myogenic failure in C2C12 cells treated with DM. All the data with diethyl succinate (DES) represented a very similar trend to previous findings. Myogenic gene expression (Figure 5A), Myog (Figure 5B), and myogenic proteins (Figure 5C) were all inhibited by the exogenous succinate treatment. Reduced MYHC expression (Figure 5D) and myogenic index (Figure 5E) by succinate indicate its inhibitory potency of myogenesis. The degree of suppression was most similar to those results observed in groups treated with itaconate or malonate. These results support a possibility that inhibition of myogenesis by itaconate or malonate is at least partly due to accumulated succinate caused by SDH inhibition.
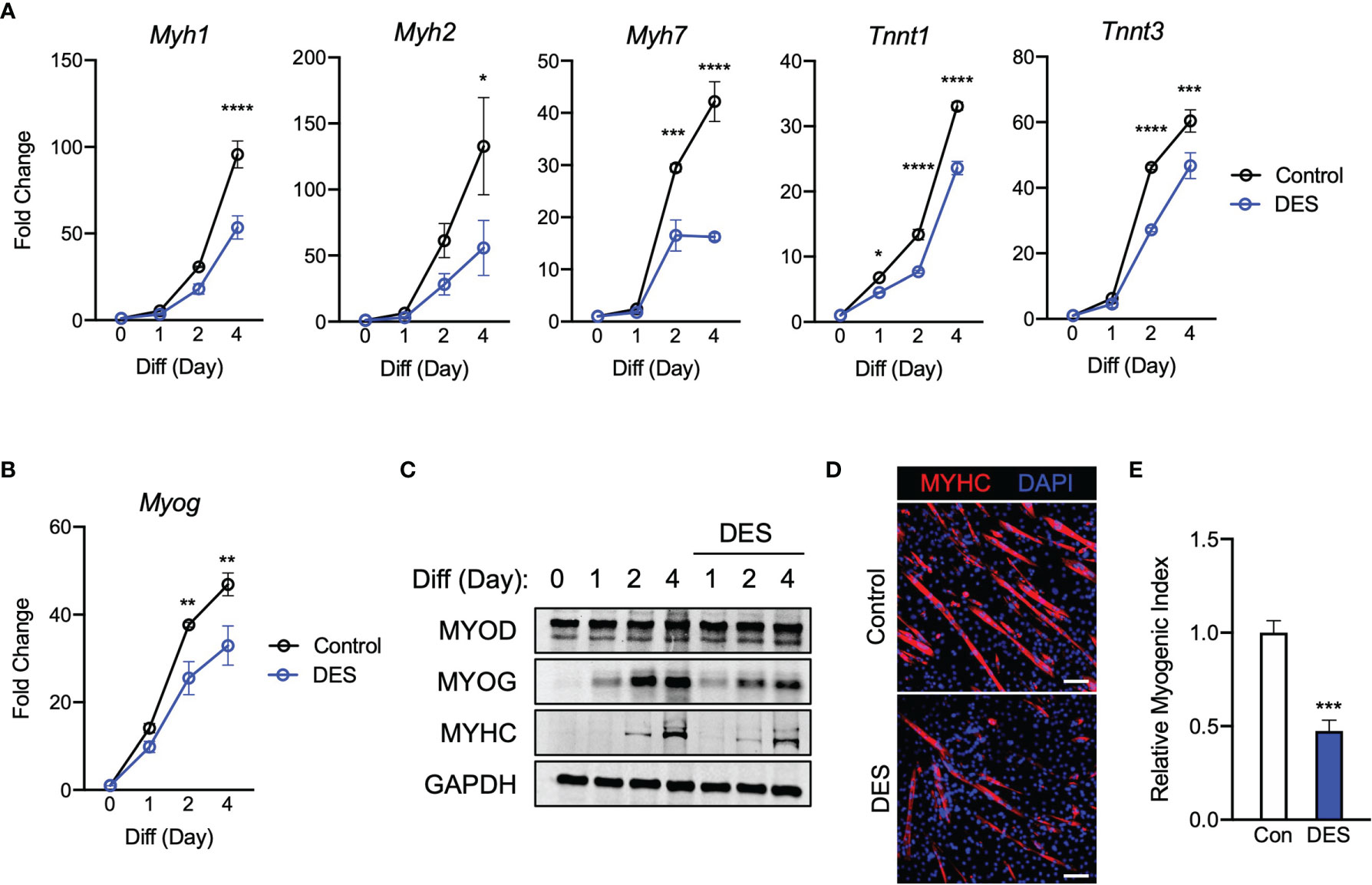
Figure 5 Elevated exposure to succinate elicits inhibition of C2C12 myogenesis via mechanism similar to those observed when exposing cells to itaconate and malonate. (A) 4-day time course showcasing fold change differences of myogenic gene transcription levels detected via RT-qPCR when exposed to differential media with or without diethyl succinate (DES: 5 mM) relative to the untreated group (n = 3). (B) Fold change of Myog expression at various time points comparing DM control groups to DM and DES groups relative to expression without either treatment (n = 3). (C) Western blotting data showing differences in myogenic transcription factor expression between DM treated groups and groups treated with both DM and DES over a 4 day time course as well as groups which were not exposed to either treatment for any period. (D) MYHC staining at day 4 of differentiation. Bar: 100 μm. (E) Relative myogenic index (percentage of total nuclei associated with myotubes). * denotes p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control based on t-test.
4-OI Inhibits Myogenic Induction During Post-Injury Muscle Regeneration
To gain insight on the effect of itaconate on myogenesis in vivo, we utilized a post-injury muscle regeneration model by injecting barium chloride (BaCl2) in TA muscle of mice and administrated 4-OI or control (Figure 6A). Myog gene expression was significantly increased on day four in injured TA muscle compared to vehicle (Figure 6B). Importantly, injury-induced Myog induction was blunted in the presence of 4-OI (Figure 6B). Muscle injury strongly increased protein levels of MYOD and 4-OI did not result in alteration of MYOD levels in BaCl2 treated muscle in accordance with in vitro findings (Figures 6C, D). In response to increases in MYOD, its downstream target MYOG was also elevated in injured TA muscle. Interestingly, protein levels of MYOG in 4-OI administrated mice were indeed significantly reduced compared to control in the BaCl2 group (Figures 6C, D). Collectively, these results indicate that 4-OI blunts myogenic induction during post-injury muscle regeneration in vivo by regulating one of the master myogenic transcription factors.
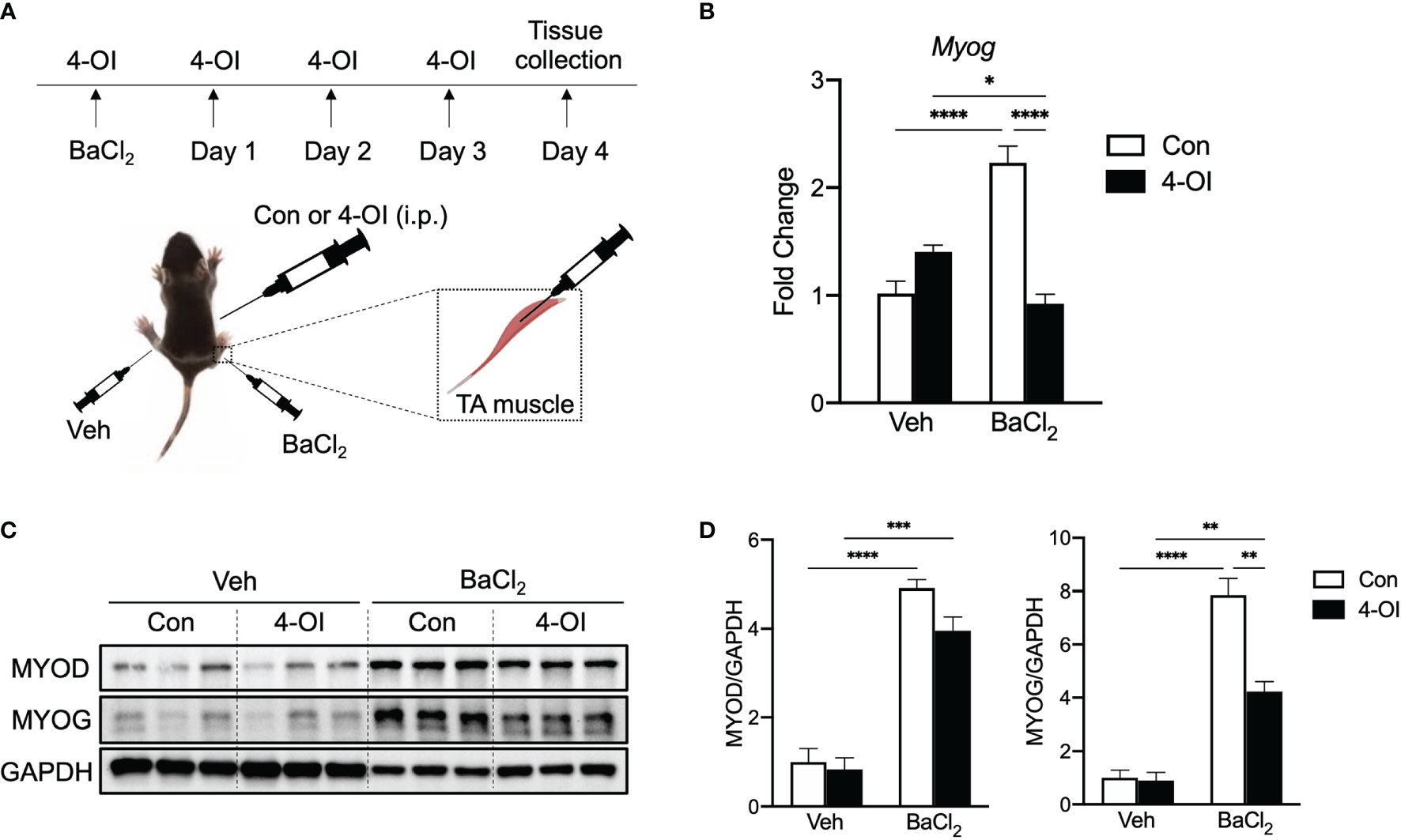
Figure 6 4-OI suppresses gene and protein expression of MYOG in muscle after injury. (A) Experimental scheme of muscle injury in mice. (B) Fold change of Myog mRNA expression at day 4 of muscle injury by barium chloride (BaCl2) with or without 4-OI (n = 4). (C) Western blotting data showing differences in myogenic transcription factor expression between vehicle (Veh) treated groups and BaCl2 treated groups with or without 4-OI. (D) Quantification of MYOD and MYOG. * denotes p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 based on one-way ANOVA.
Discussion
Itaconate has become the epitome of metabolites that also have immunomodulation properties. It is a decarboxylated cis-aconitate which is primarily known for its SDH inhibiting activity in the TCA cycle. While initially used as a polymer for industrial purposes, it has been researched to have both antimicrobial and anti-inflammatory properties. Some bacteria (e.g. Yersinia pestis) have itaconate degrading enzymes and itaconate itself has been observed to restrict the growth of bacteria by inhibiting isocitrate lyase activity (25). It also regulates host mechanisms. Its anti-inflammatory role has gained the most attention recently. Itaconate is observed to significantly increase in response to macrophage activation (23). It acts to reduce inflammation by inhibiting the release of proinflammatory cytokines through a plethora of mechanisms, inhibiting ROS production through suppression of SDH activity (1), activating the regulator of antioxidant expression NRF2 (26), and activating the anti-inflammatory signaling transcription factor ATF3 (3). These effects in tandem have attracted scientists to get approval for itaconate derivatives as potential treatments to limit microbial pathogenicity and to attenuate autoimmune disease symptoms.
DMI and 4-OI have been researched to have promising effects on psoriasis, rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus due to their ability to suppress IL-17 signaling and reduce proinflammatory cytokine production (3, 6). While seemingly propitious as a potential treatment, the fact is that knowledge is very limited on the effects of itaconate and its derivates outside of those relating to the immunomodulatory axis. A consequence of itaconate derivative use may be one or more harmful side effects which could lessen enthusiasm for their utilization as treatments. Blocking itaconate signaling, which has yet to be proposed, might also be used to attenuate symptoms of other conditions. The focus of our research is to showcase the additional effects of itaconate on muscle regulation. Chronic inflammation has been linked to muscle wasting. Prolonged itaconate exposure resulting from chronic inflammation may cause or exacerbate muscle wasting.
Our hypothesis is that itaconate obstructs myogenesis via inhibition of SDH. This hypothesis arose from research done on the effects of succinate abundance on muscle wasting. Succinate is a substrate for SDH, so its intracellular concentration is directly linked to SDH activity. Succinate abundance has been proposed as a causative agent of muscle wasting. Treatments to elevate intracellular succinate concentration has been observed to decrease total muscle protein expression by ~25%, inhibit in vitro C2C12 myoblast cell myogenesis, decrease myofiber diameter in murine models, inhibit myogenesis in vivo following barium chloride injury, reduce respiration capacity by ~35% in myoblasts, and also disrupt a number of other metabolic processes within muscle cells (27). SDH has also been shown to have significantly reduced activity in sarcopenic muscle (28). Given that anti-inflammatory activity of itaconate is incurred via SDH inhibition, we endeavored to determine if itaconate treatment led to similar effects seen in succinate abundant cells.
We first tested the proposed treatment forms of itaconate, DMI, and 4-OI. We then examined itaconate alone followed by malonate, a pharmacological inhibitor of SDH, and then lastly diethyl succinate. These molecules did not show significant alterations in cell viability or proliferation, indicating inhibition of myogenesis by these molecules is independent of cytotoxicity (Figure S1). Treatment incurred transcriptional modulation of Myh1, Myh2, Myh7, Tnnt1, and Tnnt3 along with alteration of myogenic transcription factor (mainly MYOG) and myogenesis marker MYHC protein expression were examined. Our results showcased substantial inhibition of myogenesis in C2C12 cells by DMI. 4-OI treatment also exhibited significant inhibition, but not as aggressive as DMI. Itaconate treatment also resulted in significant myogenesis inhibition, but not to the degree seen in either DMI or 4-OI treated groups. It is possible that inability of DMI conversion to itaconate leads to a strong inhibition of myogenesis exhibiting sustained activity because 4-OI can be converted to itaconate, but DMI cannot (18). It is established that itaconate can be converted to itaconyl-CoA by succinate-CoA ligase (21). A possible mechanism to explain this tiered order of inhibition severity may involve itaconate conversion to itaconyl-CoA. Regardless of potency, all itaconate treatments inhibited in vitro myogenesis.
We show that SDH suppression and subsequent succinate accumulation significantly inhibit myogenesis via myogenic transcription suppression. Given that MYOG expression is dependent upon MYOD activity, it is hypothesized that MYOD activity was also suppressed in the treatment groups, but via post translational modification that leads to changes in MYOD activity such as phosphorylation and acetylation (29, 30) rather than transcriptional inhibition. Inactive MYOD in response to SDH inhibition and succinate causes blunted induction of Myog thereby suppressing the transcription of myogenic genes. Together, our results support that itaconate may contribute to impairment of myogenesis via its effects on SDH which modulates myogenic transcription mechanisms. Therefore, future studies should be focused on elucidating further molecular mechanisms by which itaconate and its derivatives inhibit transcription of myogenic genes.
Sarcopenia patients have been observed to have inhibited mitochondrial functioning via transcriptional downregulation of key genes involved in oxidative phosphorylation and mitochondria proteostasis (28). 4-OI has been shown to increase aerobic respiration by reducing glycolytic function through inhibition of GAPDH (19). This data may implicate that in addition to subsequent muscle wasting induced by increased succinate concentration, prolonged itaconate activity may exacerbate issues relating to mitochondrial function. What demands further study is the determination of whether itaconate and its derivatives induce mitochondrial activity suppression which may cause subsequent inhibition of the oxidative phosphorylation pathway and thus reduce energy demand accommodation capability.
In this study, we find that itaconate and derivatives contribute to suppression of in vitro and in vivo myogenesis. Administrating 4-OI during post-injury muscle regeneration clearly contributes to suppression of MYOG in muscle of mice. It is speculated that skeletal muscle regeneration might be delayed in the presence of itaconate derivatives followed by lowered MYOG. Future studies about prolonged effects of itaconate during muscle regeneration will provide further insight about actual influence on muscle regeneration in the presence of itaconate. What remains to be researched next is how itaconate affects muscle regulation outside of succinate accumulation. For example, investigating the effects of succinylation on myogenic promotor binding proteins or transcription factors is warranted. Our results showcase functions of itaconate outside of immunomodulation and implicate that derivative use to treat autoimmune diseases or microbial infections should be cautioned due to a potential for deleterious effects on myogenesis. Further elucidation of these functions may lead to the development of treatments to attenuate muscle wasting via possible itaconate signaling inhibition.
Materials and Methods
Antibodies and Chemical Reagents
Primary antibodies used in this study are as follows: mouse monoclonal anti-GAPDH (Santa Cruz Biotechnology, sc-32233), mouse monoclonal anti-MYHC (Sigma, 05-716), mouse monoclonal anti-MYOD (Santa Cruz Biotechnology, sc-377460), and mouse monoclonal anti-MYOG (Santa Cruz Biotechnology, sc-12732). Reagents added to the media are as follows: Diethyl succinate (Sigma, 112402), Dimethyl itaconate (Sigma, 592498), Dimethyl malonate (Sigma 136441), Harzianopyridone (Santa Cruz Biotechnology, sc-280769), Itaconic acid (Sigma, I29204), and 4-octyl itaconate (Tocris Bioscience, 6662).
Cell Culture and Differentiation
The mouse myogenic C2C12 myoblasts were maintained on plastic cell culture plates in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in a humidified incubator kept at 37°C and 5% CO2. Cells were used up until passage 9. For differentiation, cells at 90% confluency were serum restricted with differentiation medium (DMEM, 2% horse serum, 1% penicillin-streptomycin) for up to 4 days. Growth or differentiation medium was replenished every day. For treatment in differentiation medium, following concentration was used and freshly added every day until the end of experiments: DMI (125 µM), 4-OI (125 µM), itaconate (7.5 mM), DMM (5 mM), Harz (4 µM), and DES (5 mM).
RNA Isolation and RT-qPCR
RNA isolation was performed using the commercially available Aurum RNA miniprep kit (Bio-rad, 732-6820). Gene expression analysis was conducted with 50 ng RNA using iTaq Universal One-Step RT-qPCR Kit (Bio-rad, 1725140). The reaction was carried out according to the manufacturer’s instructions using CFX Connect Real-Time PCR Detection System (Bio-rad, 1855200). Probes used for TaqMan® Gene Expression Assays (ThermoFisher) were as follows: Myog (Mm00446194_m1), Myh1 (Mm01332489_m1), Myh2 (Mm01332564_m1), Myh7 (Mm00600555_m1), Ppib (Mm00478295_m1), Tnnt1 (Mm00449089_m1), and Tnnt3 (Mm01137842_g1).
Western Blot
Cells were lysed in RIPA buffer (ThermoFisher, 89901) in the presence of Halt™ Protease and Phosphatase Inhibitor Cocktail (ThermoFisher, 78440), and protein concentration was measured using Pierce™ BCA Protein Assay Kit (ThermoFisher, 23227). Lysates were boiled for 5 min with 4x Laemmli Sample Buffer (Bio-Rad, 1610747) and 2-mercaptoethanol (Bio-Rad, 1610710). Lysates were resolved on SDS polyacrylamide gels and blotted onto PVDF membranes using Trans-Blot Turbo RTA Midi 0.45 µm LF PVDF Transfer Kit (Bio-Rad, 1704275). Transfer was run in Trans-Blot Turbo Transfer System (Bio-Rad, 1704150) using Mixed MW (1.3A-25V-7M) protocol. The membranes were blocked with Intercept™ (TBS) Blocking Buffer (LI-COR, 927-60001) for 1 h at room temperature and incubated with appropriate primary antibodies diluted 1:1000 in blocking buffer at 4°C overnight. After 3 washes with 0.05% tween20 (Bio-Rad) in TBS buffer (Bio-Rad), the membrane was incubated with IRDye® 800CW Goat anti-Mouse IgG (LI-COR, 926-32210) and IRDye® 680RD Goat anti-Rabbit IgG (LI-COR, 926-68071) secondary antibodies diluted 1:10000 in blocking buffer for 1 h at room temperature. Band images were visualized using ChemiDoc™ MP Imaging System (Bio-Rad, 12003154).
Immunofluorescence and Calculation of Myogenic Index
Cells were seeded in a 24-well cell culture plate and followed by above-mentioned differentiation protocol. At day 4 of differentiation, cells were rinsed with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. Then, cells were permeablized with 0.5% Triton™ X-100 (Fisher, BP151-100) in PBS for 10 min at room temperature. Next, cells were submerged with Intercept® (PBS) Blocking Buffer (LI-COR, 927-70001) for 1 h at room temperature. MYHC primary antibody was diluted to 1:100 in blocking buffer and incubated overnight at 4°C with the fixed cells. Next day, after 3 washes with 0.05% tween20 in PBS, Alexa Fluor 594 secondary antibody (Fisher, A11005) was diluted to 1:500 in blocking buffer and incubated with the cells for 1 h at room temperature. Followed by further washes, cells were mounted with VECTASHIELD® Antifade Mounting Medium with DAPI (Vectalabs, H-1200) and covered with circular cover glass. Images were taken using ZOE™ Fluorescent Cell Imager (Bio-rad, 1450031). Myogenic index was calculated as the percentage of nuclei in fused myotubes (MYHC positive) out of the total nuclei in given images. Distribution of nuclei in myoblasts and myotubes was measured by counting over 100 nuclei at 4 distinct locations. The number of nuclei was counted using imageJ.
Succinate Dehydrogenase Activity and Succinate Assay
SDH activity was measured using Succinate Dehydrogenase Assay Kit (Sigma, MAK197) according to manufacturer’s protocol. Briefly, at day 1 or 2 of differentiation in the presence of itaconate derivatives, cells were rinsed with PBS and lysed with SDH assay buffer. The whole cell lysate was freshly used for the measurement. SDH activity was calculated by measuring product absorbance at 600 nm every 5 min for 30 min at 25°C. SDH activity was normalized by protein contents in each sample measured by BCA assay. Succinate levels were determined using Succinate Colorimetric Assay Kit (Sigma, MAK184). After differentiation in the presence of itaconate derivatives, cells were rinsed with PBS and lysed with succinate assay buffer. The cell lysate was diluted 1:40 in assay buffer for analysis. After incubating samples with the reaction mix for 30 min at 37°C, the absorbance was measured at 450 nm. Concentration of succinate was normalized by protein contents in each sample measured by BCA assay.
Muscle Injury in Mice
Male C57BL/6J mice aged 8 weeks were purchased from The Jackson Laboratory (Bar Harbor, ME). All animals were subjected to a 12:12 hr dark/light cycle with ad libitum access to standard rodent chow and water. To induce muscle injury in vivo, mice were anesthetized with 2-3% of isoflurane, and then 50 µl of 1.2% BaCl2 (Sigma, 202738) was slowly injected unilaterally into the TA muscle alongside tibia. Mice were kept warm during recovery and then returned to their cage. 4-OI (25mg/kg; Tocris, 6662) dissolved in saline plus (2-Hydroxypropyl)-β-cyclodextrin (Sigma, H107) was pretreated intraperitoneally 2 h beforehand and given daily for 3 consecutive days. At day 4, tissues were collected for further analysis. All protocols and experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Wake Forest School of Medicine (Winston-Salem, NC).
Statistical Analysis
Unpaired t-test and one-way ANOVA using analysis software in Graphpad Prism 9 were performed to determine significant relationships between groups in the experiments. Data are represented as mean ± SEM.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
TO planned, executed, analyzed data, and drafted manuscript. DH planned, executed, analyzed data, and drafted manuscript. RM executed, analyzed data, and drafted manuscript. KG executed and analyzed data. MQ planned, executed, analyzed data and drafted manuscript, and supervised the project. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.748375/full#supplementary-material
References
1. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase With Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
2. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate Is an Anti-Inflammatory Metabolite That Activates Nrf2 via Alkylation of KEAP1. Nature (2018) 556:113–7. doi: 10.1038/nature25986
3. Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic Properties of Itaconate and Derivatives Regulate the IkappaBzeta-ATF3 Inflammatory Axis. Nature (2018) 556:501–4. doi: 10.1038/s41586-018-0052-z
4. Kuo PC, Weng WT, Scofield BA, Paraiso HC, Brown DA, Wang PY, et al. Dimethyl Itaconate, an Itaconate Derivative, Exhibits Immunomodulatory Effects on Neuroinflammation in Experimental Autoimmune Encephalomyelitis. J Neuroinflamm (2020) 17:138. doi: 10.1186/s12974-020-01768-7
5. Daly R, Blackburn G, Best C, Goodyear CS, Mudaliar M, Burgess K, et al. Changes in Plasma Itaconate Elevation in Early Rheumatoid Arthritis Patients Elucidates Disease Activity Associated Macrophage Activation. Metabolites (2020) 10:241. doi: 10.3390/metabo10060241
6. Tang C, Wang X, Xie Y, Cai X, Yu N, Hu Y, et al. 4-Octyl Itaconate Activates Nrf2 Signaling to Inhibit Pro-Inflammatory Cytokine Production in Peripheral Blood Mononuclear Cells of Systemic Lupus Erythematosus Patients. Cell Physiol Biochem (2018) 51:979–90. doi: 10.1159/000495400
7. Yi Z, Deng M, Scott MJ, Fu G, Loughran PA, Lei Z, et al. IRG1/Itaconate Activates Nrf2 in Hepatocytes to Protect Against Liver Ischemia-Reperfusion Injury. Hepatology (2020) 72:1394–411. doi: 10.1002/hep.31147
8. Tian F, Wang Z, He J, Zhang Z, Tan N. 4-Octyl Itaconate Protects Against Renal Fibrosis via Inhibiting TGF-Beta/Smad Pathway, Autophagy and Reducing Generation of Reactive Oxygen Species. Eur J Pharmacol (2020) 873:172989. doi: 10.1016/j.ejphar.2020.172989
9. Ogger PP, Albers GJ, Hewitt RJ, O'Sullivan BJ, Powell JE, Calamita E, et al. Itaconate Controls the Severity of Pulmonary Fibrosis. Sci Immunol (2020) 5:eabc1884. doi: 10.1126/sciimmunol.abc1884
10. Cordes T, Lucas A, Divakaruni AS, Murphy AN, Cabrales P, Metallo CM. Itaconate Modulates Tricarboxylic Acid and Redox Metabolism to Mitigate Reperfusion Injury. Mol Metab (2020) 32:122–35. doi: 10.1016/j.molmet.2019.11.019
11. Zhang D, Lu Z, Zhang Z, Man J, Guo R, Liu C, et al. A Likely Protective Effect of Dimethyl Itaconate on Cerebral Ischemia/Reperfusion Injury. Int Immunopharmacol (2019) 77:105924. doi: 10.1016/j.intimp.2019.105924
12. Song H, Xu T, Feng X, Lai Y, Yang Y, Zheng H, et al. Itaconate Prevents Abdominal Aortic Aneurysm Formation Through Inhibiting Inflammation via Activation of Nrf2. EBioMedicine (2020) 57:102832. doi: 10.1016/j.ebiom.2020.102832
13. Zhang S, Jiao Y, Li C, Liang X, Jia H, Nie Z, et al. Dimethyl Itaconate Alleviates the Inflammatory Responses of Macrophages in Sepsis. Inflammation (2021) 44:549–57. doi: 10.1007/s10753-020-01352-4
14. Campins L, Camps M, Riera A, Pleguezuelos E, Yebenes JC, Serra-Prat M. Oral Drugs Related With Muscle Wasting and Sarcopenia. A Review. Pharmacology (2017) 99:1–8. doi: 10.1159/000448247
15. Jones JD, Kirsch HL, Wortmann RL, Pillinger MH. The Causes of Drug-Induced Muscle Toxicity. Curr Opin Rheumatol (2014) 26:697–703. doi: 10.1097/BOR.0000000000000108
16. Janssen L, Allard NAE, Saris CGJ, Keijer J, Hopman MTE, Timmers S. Muscle Toxicity of Drugs: When Drugs Turn Physiology Into Pathophysiology. Physiol Rev (2020) 100:633–72. doi: 10.1152/physrev.00002.2019
17. Swain A, Bambouskova M, Kim H, Andhey PS, Duncan D, Auclair K, et al. Comparative Evaluation of Itaconate and Its Derivatives Reveals Divergent Inflammasome and Type I Interferon Regulation in Macrophages. Nat Metab (2020) 2:594–602. doi: 10.1038/s42255-020-0210-0
18. ElAzzouny M, Tom CT, Evans CR, Olson LL, Tanga MJ, Gallagher KA, et al. Dimethyl Itaconate Is Not Metabolized Into Itaconate Intracellularly. J Biol Chem (2017) 292:4766–9. doi: 10.1074/jbc.C117.775270
19. Liao ST, Han C, Xu DQ, Fu XW, Wang JS, Kong LY. 4-Octyl Itaconate Inhibits Aerobic Glycolysis by Targeting GAPDH to Exert Anti-Inflammatory Effects. Nat Commun (2019) 10:5091. doi: 10.1038/s41467-019-13078-5
20. Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem (2016) 291:14274–84. doi: 10.1074/jbc.M115.685792
21. Nemeth B, Doczi J, Csete D, Kacso G, Ravasz D, Adams D, et al. Abolition of Mitochondrial Substrate-Level Phosphorylation by Itaconic Acid Produced by LPS-Induced Irg1 Expression in Cells of Murine Macrophage Lineage. FASEB J (2016) 30:286–300. doi: 10.1096/fj.15-279398
22. Cordes T, Metallo CM. Itaconate Alters Succinate and Coenzyme A Metabolism via Inhibition of Mitochondrial Complex II and Methylmalonyl-CoA Mutase. Metabolites (2021) 11:117. doi: 10.3390/metabo11020117
23. O'Neill LAJ, Artyomov MN. Itaconate: The Poster Child of Metabolic Reprogramming in Macrophage Function. Nat Rev Immunol (2019) 19:273–81. doi: 10.1038/s41577-019-0128-5
24. Dickinson JM, Hanson JR, Hitchcock PB, Claydon N. Structure and Biosynthesis of Harzianopyridone, an Antifungal Metabolite of Trichoderma Harzianum. J Chem Society Perkin Trans (1989) 1:1885–7. doi: 10.1039/p19890001885
25. Sasikaran J, Ziemski M, Zadora PK, Fleig A, Berg IA. Bacterial Itaconate Degradation Promotes Pathogenicity. Nat Chem Biol (2014) 10:371–7. doi: 10.1038/nchembio.1482
26. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell (2016) 167:457–70.e413. doi: 10.1016/j.cell.2016.08.064
27. Arneson PC, Hogan KA, Shin AM, Samani A, Jatoi A, Doles JD. The Wasting-Associated Metabolite Succinate Disrupts Myogenesis and Impairs Skeletal Muscle Regeneration. JCSM Rapid Commun (2020) 3:56–69. doi: 10.1002/rco2.14
28. Migliavacca E, Tay SKH, Patel HP, Sonntag T, Civiletto G, McFarlane C, et al. Mitochondrial Oxidative Capacity and NAD(+) Biosynthesis Are Reduced in Human Sarcopenia Across Ethnicities. Nat Commun (2019) 10:5808. doi: 10.1038/s41467-019-13694-1
29. Di Padova M, Caretti G, Zhao P, Hoffman EP, Sartorelli V. MyoD Acetylation Influences Temporal Patterns of Skeletal Muscle Gene Expression. J Biol Chem (2007) 282:37650–9. doi: 10.1074/jbc.M707309200
Keywords: C2C12, myogenesis, itaconate, succinate, succinate dehydrogenase
Citation: Oh TS, Hutchins DC, Mainali R, Goslen KH and Quinn MA (2022) Itaconate and Its Derivatives Repress Early Myogenesis In Vitro and In Vivo. Front. Immunol. 13:748375. doi: 10.3389/fimmu.2022.748375
Received: 27 July 2021; Accepted: 01 February 2022;
Published: 21 February 2022.
Edited by:
Naeem K. Patil, Vanderbilt University Medical Center, United StatesReviewed by:
Bhalchandra Mirlekar, University of North Carolina at Chapel Hill, United StatesUzma Saqib, Indian Institute of Technology Indore, India
Allison M. Owen, Vanderbilt University Medical Center, United States
Copyright © 2022 Oh, Hutchins, Mainali, Goslen and Quinn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew A. Quinn, bXF1aW5uQHdha2VoZWFsdGguZWR1
†These authors have contributed equally to this work