 Byunghyuk Lee
Byunghyuk Lee Seung-Hyo Lee
Seung-Hyo Lee Kihyuk Shin
Kihyuk Shin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 09 January 2023
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1103823
This article is part of the Research TopicThe Immunomodulatory Role of Fibroblast in Tissue ImmunopathologyView all 6 articles
Fibroblasts are primarily considered as cells that support organ structures and are currently receiving attention for their roles in regulating immune responses in health and disease. Fibroblasts are assigned distinct phenotypes and functions in different organs owing to their diverse origins and functions. Their roles in the immune system are multifaceted, ranging from supporting homeostasis to inducing or suppressing inflammatory responses of immune cells. As a major component of immune cells, T cells are responsible for adaptive immune responses and are involved in the exacerbation or alleviation of various inflammatory diseases. In this review, we discuss the mechanisms by which fibroblasts regulate immune responses by interacting with T cells in host health and diseases, as well as their potential as advanced therapeutic targets.
As the principal components of connective tissues, fibroblasts are known to contribute to the formation and maintenance of extracellular matrix components and their remodeling, consequently playing pivotal roles in tissue development, differentiation, and repair. Therefore, their roles in immune responses have long been overlooked. Previous studies have identified the heterogeneity of fibroblasts based on their origins, surface markers, and functions using genetic lineage-tracing and single-cell RNA sequencing, and suggested the mechanism underlying the interaction of local immune cells in cancers and autoimmune disorders (1). At the site of inflammation, fibroblasts communicate with immune cells via secretion of cytokines and chemokines and direct cell-cell contact (2, 3). These interactions induce the recruitment of immune cells and produce inflammatory mediators; infiltrated immune cells play important roles in the phenotypes and functions of fibroblasts (1, 3). This suggests that fibroblasts have additional roles as members of immune networks while being a key structural compartment in different organs.
Fibroblasts play different roles in pathogenesis at different anatomical positions in diseases. They have diverse phenotypes with flexible responses to signals from microenvironmental niches. Disease-specific fibroblasts, such as cancer-associated fibroblasts (CAFs) and fibroblast-like synoviocytes in rheumatoid arthritis (RA), can directly interact with immune cells. T cells are important representatives of immune cells and are involved in the pathogenesis of cancers and autoimmune disorders. In addition, many studies have demonstrated that T cells participate in the deterioration or alleviation of diseases by interacting with tissue-specific fibroblasts.
Here, we discuss the complex crosstalk between fibroblasts and T cells in host health and disease. Interestingly, targeting fibroblast-T cell crosstalk could be a noteworthy therapeutic strategy for the treatment of diseases.
Lymph nodes (LNs) are specialized organs wherein adaptive immune response is initiated and organize into distinctive compartments by highly specialized fibroblastic reticular cells (FRCs) (1, 4). FRCs contribute to the maintenance of immune homeostasis by supporting LN structures that offer distinct microenvironments and interact with immune cells by recruiting them and presenting antigens (5–7). Recent studies, such as single-cell analysis, high-resolution imaging, and various reporter mice studies, have identified the heterogeneity of FRCs. Depending on their position, phenotypes, and functions, FRC subtypes consist of marginal reticular cells, interfollicular FRCs, T-B border reticular cells, T-zone reticular cells (TRC), deep cortex periphery reticular cells, and medullary reticular cells. To participate in immune responses, FRCs produce cytokines, chemokines, and growth factors, thereby supporting the survival, activation, proliferation, and differentiation of immune cells. TRCs control T cell positioning, survival, and differentiation. TRCs directly regulate T effector functions via nitric oxide or constitutive cyclooxygenase enzymes (8–11). Co-culture of human FRCs with CD8+ or CD4+ T cells shows that FRCs can induce T cell anergy by producing indoleamine-2,3-dioxygenase, adenosine 2A receptor, prostaglandin E2, and transforming growth factor β (TGF-β) receptor, which limits T cell proliferation (12). In a co-culture of mouse FRCs with CD8+ T cells, FRCs induce an increased production of IL-2 and tumor necrosis factor (TNF) in CD8+ T cells (13). T cell survival, proliferation, and migration are affected by TRC-derived molecules such as IL-7, IL-15, IL-33, delta-like 4, CXCL12, CCL19, CCL21, and CD40 (5). T cells, which are regulated by FRCs, can influence the FRC phenotype and function. Activated CD8+ T cells can elicit TRCs to produce more immunostimulatory molecules, including the ICOS ligand, CD40, and IL-6 in a co-culture system (5, 13).
LN-FRCs have received much attention as key participants in tumor progression. In the colon adenocarcinoma model, LN-FRCs suppress an anti-tumor immune response, consequently contributing to the premetastatic microenvironment (14). In the B16 melanoma model, FRCs in tumor draining LN(dLNs) alter the secretion of ECM components, cytokines, and chemokines, and assist immune cell recruitment, activation, and differentiation, facilitating the establishment of immunosuppressive niches (15). In addition, TRCs in tumor dLNs restrain the expression of CCL21 and IL-7, thus limiting CD4+ T cell priming (15).
FRCs can support immune homeostasis and protect tissues from autoimmune responses (5). Homeostasis of T regulatory (Treg) cells, which have critical roles in balancing immune responses in mouse LNs, is maintained by MHC class II FRCs (16, 17). In addition, LN-TRCs elicit CD4+ T cell differentiation through the trans-presentation of CD25 to naïve CD4+ T cells. CD25-deficient LN-TRCs induce deterioration of autoimmune diseases by enhancing IL-17-producing T helper type 17 (Th17) cell differentiation (18).
IL-33 constitutively produced by FRCs can assist in the anti-viral CD8+ T cell responses in acute and chronic lymphocytic choriomeningitis virus (LCMV)-infected animal models (19, 20). In addition, FRCs present antigen to naïve LCMV-specific CD8+ and CD4+ T cells by MHCI and MHCII, respectively (6). In acute LCMV models, the absence of type I interferon-α receptor on FRC impaired the expansion of LCMV specific CD8+ T cells and the acquisition of effector phenotypes (21). Taken together, LN-FRCs play diverse roles in the immune system by controlling the homeostasis, proliferation, and function of T cells.
The tumor microenvironment (TME) is mainly composed of tumor cells, immune cells, cancer-associated mesenchymal cells, and endothelial cells, and it plays an instrumental role in tumorigenesis and immune responses (1, 22, 23). CAFs, which are important mesenchymal cells in the TME, have heterogeneous cell phenotypes and functions. The roles of CAFs in TME have been widely described, and CAFs have been shown to facilitate tumor progression in most cancers through the production of cytokines, chemokines, and growth factors (Figure 1) (1, 22).
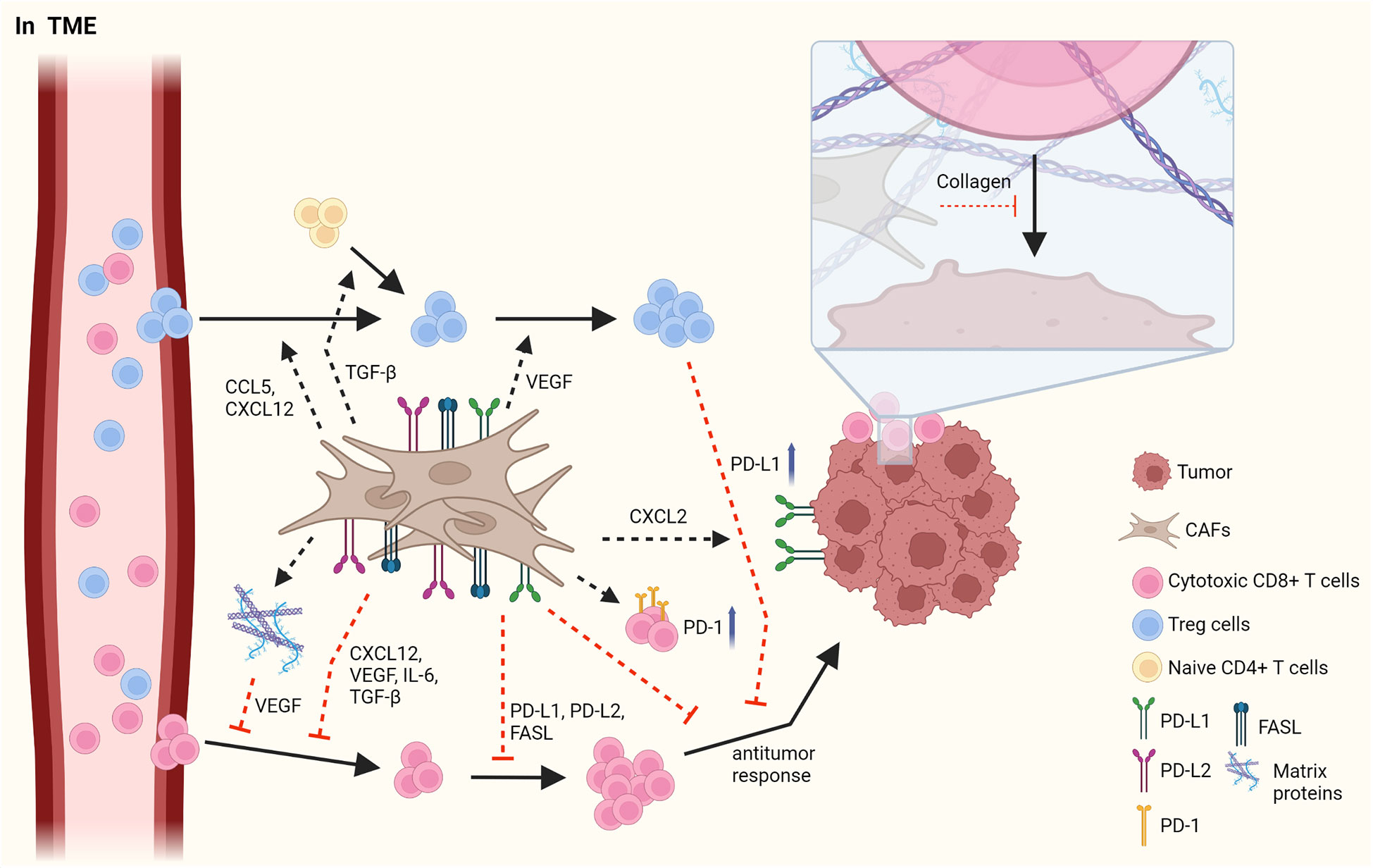
Figure 1 The interaction between CAFs and T cells in TME. CAFs have immunosuppressive roles by affecting Treg cells and cytotoxic CD8+ T cells. CAFs elicit the infiltration of Treg cells by secretion of CCL5 and CXCL12 and increase the proliferation of them through VEGF. In addition, CAFs facilitate the skewing of naïve CD4+ T cells to Treg cells. However, CAFs obstruct the migration of CD8+ T cells via various cytokines and chemokines such as CXCL12, VEGF, IL-6, and TGF-β. CAFs decrease the frequency and the activation of CD8+ T cells by PD-L1, PD-L2 and FASL. In addition, CAFs suppress an anti-tumor response of CD8+ T cells by upregulating PD-1 expression on the surface of T cells and by increasing PD-L1 expression on tumor through CXCL2. LRRC15+ CAFs induced by TGF-β signaling regulate anti-tumor response of CD8+ T cells. Furthermore, the accumulation of matrix protein, which is produced by CAFs can obstruct the migration of T cells and collagen can block the access of T cells to tumor, limiting the anti-tumor activity of T cells. Figure is generated with biorender (www.biorender.com).
Distinct subsets of T cells, such as cytotoxic CD8+ T cells, Th cells, and Treg cells, play essential roles in regulating anti-tumor responses, and interactions between CAFs and T cells in the TME have been reported. Infiltrating Treg cells orchestrate immunosuppressive responses. CAFs facilitate the migration of Treg cells into the TME via CCL5 or CXCL12, thus increasing the frequency of these cells (24, 25). Infiltration of Treg cells is also enhanced by CCL17 and CCL22, which is induced by decreased expression of CD68 in CAFs and is also maintained by CAF-derived vascular endothelial growth factor-A (26–28). Moreover, Treg cells are differentiated from naïve T cells by inducing Foxp3 expression, which is facilitated by TGF-β from CAFs (29). Contrarily, the proliferation of CD4+Foxp3+ Treg cells has been reported to have improved due to myofibroblast exhaustion in pancreatic ductal adenocarcinoma (PDAC) (28). These results indicate that CAFs play an opposite role to Treg cells in the TME.
CD8+ T cells have anti-tumor responses by promoting the apoptosis of tumor cells. In contrast to their cytotoxic activities, CAFs suppress the infiltration, growth, and anti-tumor response of CD8+ T cells. Pancreatic stellate cells(PSCs), which can be altered into activated CAFs by TGF-β and platelet-derived growth factor (PDGF), reduces the infiltration of CD8+ T cells into tumor sites by attracting CD8+ T cells toward PSCs through CXCL12 expression (30). In response to hypoxia induced by CAF-mediated ECM modification, the angiogenic factor VEGF secreted by CAFs hinders the migration of CD8+ T cells into the TME (31, 32). In addition, CAFs induce the reduced CD8+ T cell infiltration by secreting IL-6 and TGF-β, leading to a decreased anti-tumor activity. Clinical trials on the blockade of IL-6 have shown enhanced T cell function and improved prognosis in patients (33, 34). In tumorigenesis of pancreatic adenocarcinoma, the formation of leucine-rich-repeat-containing protein 15(LRRC15)+ CAF is increased by TGF-β signaling (35). The depletion of LRRC15+ CAF induces the anti-tumor response of CD8+ T cells with increased expression of TNF and IFN-γ and also enhances the efficacy of anti-PD-L1 therapy (35).
When expressed on the surface of T cells and tumor cells, immune checkpoint molecules prevent the function of T cells. CAFs can disturb the anti-tumor activity of effector T cells by inducing the expression of immune checkpoint molecules, such as FAS/FASL and PD-1/PD-L1 or PD-L2, on their surface, followed by decreased CD8+ T cell frequency and activation and, in contrast, increased tumor cell viability (36, 37). CAFs are also able to increase immune checkpoint molecules in tumor and immune cells by secreting diverse cytokines (37, 38). CAFs upregulate PD-1, CTLA-4, TIM-3, and LAG-3 expression on both CD8+ and CD4+ T cells, resulting in hampered proliferation of T cells and impaired recognition of tumor cells in pancreatic cancer (39).
CAFs can inhibit T cell recruitment into the TME by having ECM proteins as a physical barrier, thus restricting their involvement in anti-tumor responses. A previous report indicated that the increase of collagen in neighboring tumor cells obstructs the interaction between T cells and tumor cells in lung and pancreatic cancers (40). Hypoxia-derived VEGF, caused by the accumulation of various matrix proteins in the ECM, can also decrease the migration of T cells into the TME (31, 32). Overall, CAFs promote the initiation and progression of cancer through several mechanisms, such as an increase in immunosuppressive responses of Treg cells and suppression of anti-tumor responses of CD8+ T cells.
Synoviocytes, mesenchymal cells of the synovium, have distinct characteristics in RA compared with osteoarthritis (OA) and dermal fibroblasts in RA (Figure 2) (1, 41). Synoviocytes in RA show increased proliferation with defective expression of the tumor suppressor p53. Researchers have highlighted the mutations in the gene encoding p53 in synoviocytes of patients with RA (42). In addition, the expression of anti-apoptotic proteins, such as sentrin and synoviolin, is increased in the synoviocytes of patients with RA (43, 44). Synoviocytes consistently interact with infiltrating immune cells, especially T cells, thereby increasing the recruitment, differentiation, activation, and survival of T cells (45, 46). CD34- Thy1+ fibroblasts, which is highly expanded in RA patients, induce T cell infiltration into synovium, thereby increasing tissue inflammation (47). Increased migration of T cells to the synovium promotes interaction between T cells and synoviocytes, consequently contributing to disease chronicity (48). Infiltrating T cells are retained in the synovium by synoviocyte-derived CXCL12 and TGF-β (49, 50). Pro-inflammatory cytokines, such as IL-1β, TNF, and IL-17, produced by immune cells increase CCL20 secretion by synoviocytes, subsequently enabling CCR6+ Th17 cell recruitment (51–53).
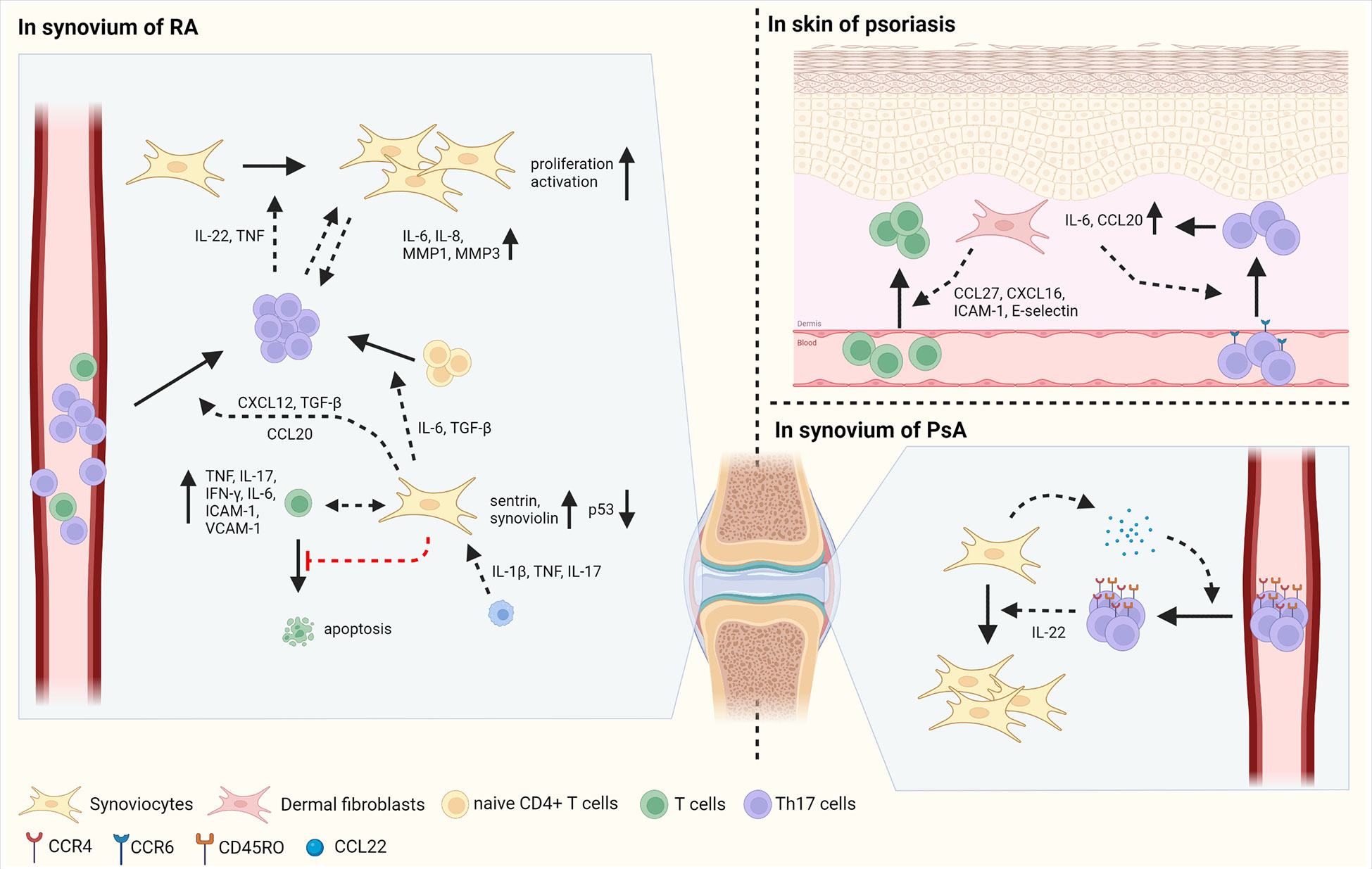
Figure 2 The interaction between fibroblasts and T cells in autoimmune diseases. Fibroblasts play inflammatory roles in autoimmune disorders such as RA, psoriasis, and PsA. In synovium of RA, synoviocytes have the mutation of p53, a tumor suppressor and increased expression of sentrin and synoviolin, which are anti-apoptotic proteins, inducing proliferation of synoviocytes. Synoviocytes elicit the differentiation of Th17 cells from naïve CD4+ T cells by IL-6 and TGF-β production. In addition, synoviocytes enhance the migration of Th17 cells via CXCL12 and TGF-β. IL-1β, TNF and IL-17 secreted by immune cells facilitate the production of CCL20 in synoviocytes, increasing the infiltration of Th17 cells into synovium. Infiltrated Th17 cells secret IL-22 and TNF, consequently improving the proliferation and activation of synoviocytes. Moreover, the contact between Th17 cells and synoviocytes induces the secretion of IL-6, IL-8, MMP-1, and MMP3 in synoviocytes. In cell-cell contact manner, synoviocytes increase the expression of TNF, IL-17, IFN-γ, IL-6, ICAM-1, and VCAM-1 in activated T cells. Besides, synoviocytes suppress the apoptosis of T cells by increasing survival signals and decreasing death signals. In psoriasis, fibroblasts produce CCL27, CXCL16, ICAM-1, and E-selectin, enabling to increased migration of T cells into skin. In addition, CCL20 and IL-6 produced by fibroblasts facilitate the infiltration of CCR6+ Th17 cells, followed by increased expression of IL-6 and CCL20 in fibroblasts. In PsA, synoviocytes elicit the migration of CD45RO+ Th17 cells by the secretion of CCL22. Then, infiltrated Th17 cells promote the proliferation of synoviocytes via IL-22 production. Figure is generated with biorender (www.biorender.com).
Synoviocytes also regulate T cell differentiation via the production of diverse cytokines. For example, TGF-β and IL-6 produced by synoviocytes facilitate Th17 differentiation (54, 55). Synoviocytes from RA patients also contribute to the expansion of CD4+ T cells and increase the proportion of T cells expressing TNF, IFN-γ, and IL-17 in the synoviocyte-T cell co-culture system (56, 57). In addition, synoviocytes suppress T cell apoptosis by sustaining survival-related signals and restraining cell death signals (58, 59). T cells influence synoviocytes, increasing their proliferation, invasive capacity, and MMP secretion (57). In RA synovium, the production of IL-22, mainly by Th17 cells, is promoted and enhances the proliferation of synoviocytes. In addition, activated T cells can promote synoviocyte activation via TNF. Interactions between synoviocytes and activated T cells improve the production of TNF, IL-17, IFN-γ, IL-6, ICAM-1, and VCAM-1 by synoviocytes in a cell-contact-dependent manner (60). Th17 cells, but not Th1 cells, act as inducers of IL-6, IL-8, MMP1, and MMP3 production when co-cultured with synoviocytes of patients with RA, implying that Th17 cells are crucial subtypes in a pro-inflammatory feedback loop with synoviocytes (61). Thus, highly proliferative synoviocytes influence the function of T cells by expressing cytokines, chemokines, and cell adhesion molecules that promote inflammatory responses in RA.
Distinct types of cells are present in the skin, including keratinocytes and skin fibroblasts in the epidermis and dermis, respectively. Fibroblasts in psoriasis and PsA have similar characteristics (3); they have pro-inflammatory effects by interacting with T cells (Figure 2). Initiation of inflammation induces T cell migration into inflamed skin via CCL27 and CXCL16 and adhesion molecules, such as ICAM-1 and E-selectin (3). Fibroblasts present in psoriatic lesions produce higher levels of CCL27 than those in non-lesioned or healthy individuals. Moreover, in the lesions of patients with psoriasis, CCR10+ cells are more abundant than compared in healthy individuals, facilitating T cell-mediated inflammation via CCL27-CCR10 interaction (62).
Interactions between T cells and mesenchymal cells increase the production of IL-17, a pivotal cytokine in the pathogenesis of psoriasis and PsA (63). IL-17 activates skin mesenchymal cells, including fibroblasts and keratinocytes, and induces the production of the pro-inflammatory cytokine IL-6 and chemokine CCL20 in mesenchymal cells (64, 65). CCL20 elicits the recruitment of CCR6+ Th17 to the lesional skin, consequently contributing to the progression of psoriasis (65, 66).
In the synovium of patients with PsA, the levels of ICAM-1 and VCAM-1 expression and the number of T cells are similar to those observed in the synovium of patients with RA (67). CCR4 and CCL22, which are necessary for T cell infiltration into the skin, are abundant in the synovial fluid of patients with PsA (68, 69). Most CCR4+ CD4+ T cells in the synovium of patients with PsA also express CD45RO, a memory T cell marker, implying that the CCL22-CCR4 axis attracts memory T cells to the joint (69). Compared to patients with OA, patients with PsA have a higher concentration of IL-22 in the synovial fluid. T cells from the synovial fluid of patients with PsA produce large amounts of IL-22. In vitro studies have revealed that IL-22 facilitates increased proliferation of PsA synoviocytes, leading to deteriorated development of PsA (70). Further studies are required to compare the characteristics of PsA synoviocytes with those of RA synoviocytes, and their disease-specific interactions with infiltrated T cells. Overall, fibroblasts facilitate inflammatory responses by recruiting and activating Th17 cells through the secretion of cytokines and chemokines and increased expression of cell adhesion molecules in the skin of psoriasis patients and in the synovium of patients with PsA.
As mentioned above, fibroblasts interacting with T cells participate in the pathogenesis of cancers and autoimmune disorders by producing cytokines and chemokines and promoting the survival and proliferation of T cells (1, 3). Therefore, targeting these interactions has received attention as a potential therapeutic option. Numerous approaches have been developed to date, such as direct targeting of cells and indirect targeting of the cytokine-receptor required for cell interaction (1, 3). To directly target disease-specific fibroblasts in different diseases, identification of the specific markers of fibroblasts in distinct disease-associated environments is required; however, it is difficult to determine selective markers for fibroblasts. As fibroblast activation protein (FAP) is one of the proteins that are highly expressed on CAFs in various cancers, FAP+ CAF is efficiently depleted by diverse approaches like immunotoxin, antibodies, DNA vaccines, and chimeric antigen receptor T cells, thereby contributing to the attenuation of tumor growth (71–73). The elimination of FAP+ CAFs increases T cell infiltration and enhances the efficacy of immune checkpoint inhibitors (74). Nevertheless, because FAP is also expressed in normal tissues, it can cause side effects, such as cachexia and anemia. In addition, depletion of α smooth muscle actin in CAFs facilitates undifferentiated tumor growth and subsequently decreases patient survival (28). Another approach to the direct killing of fibroblasts is the induction of apoptotic signals. To that end, delivery of PUMA, a pro-apoptotic gene, via viral vectors facilitates extensive cell death of synoviocytes in vitro and in the joints of rats with adjuvant-induced arthritis, resulting in decreased joint inflammation and destruction (75). Treatment of RA synoviocytes with cadmium enhances cell apoptosis and inhibits the production of pro-inflammatory cytokines. This effect was observed in rats with adjuvant-induced arthritis, whose clinical scores were alleviated, and joint destruction was protected (76).
An alternative strategy to indirectly target cell interactions has also been investigated. For instance, the CXCR4 inhibitor AMD3100 can restrict CXCL12-mediated T cell exclusion and promote T cell infiltration into the TME (77). In RA, psoriasis, and PsA, the infliximab (against TNF) reduces the infiltration of T cells into the synovium, and consequently decreases T cell-mesenchymal cell interactions (78–80). In addition, tocilizumab, a monoclonal antibody against the IL-6 receptor, diminishes the frequency of Th17 cells and production of cytokines secreted by Th17 cells in RA (81). Similar to RA treatment, tofacitinib, a JAK inhibitor, blocks the TNF-induced production of chemokines by synoviocytes and consequently limits immune cell infiltration. Peficitinib, another JAK inhibitor, decreases the production of pro-inflammatory mediators and inhibits the migration and proliferation of synoviocytes (82, 83).
Recently, the involvement of fibroblasts in the regulation of immune responses by interacting with immune cells has been investigated. Although the heterogeneous and multifaceted characteristics of these cells have made it difficult to identify their roles in health and diseases, recent studies have elicited their potential to control immune networks using advanced technologies. Here, we have discussed the crosstalk between fibroblasts and T cells, the modulation of immune balance, and the pathogenesis of inflammatory diseases. Fibroblasts crucially participate in immune responses by interacting with T cells in secondary lymphoid organs and disease sites. Owing to their importance, studies on the interaction between fibroblasts and T cells as therapeutic approaches have been constantly investigated. Nevertheless, further research is needed to understand the interaction between fibroblasts and T cells, which might improve the therapeutic advantages for various diseases.
BL drafted the manuscript, figures. S-HL and KS contributed to study conception and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2022R1C1C1007391).
Author S-HL is employed by GenoFocus Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Davidson S, Coles M, Thomas T, Kollias G, Ludewig B, Turley S, et al. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat Rev Immunol (2021) 21(11):704–17. doi: 10.1038/s41577-021-00540-z
2. Correa-Gallegos D, Jiang D, Rinkevich Y. Fibroblasts as confederates of the immune system. Immunol Rev (2021) 302(1):147–62. doi: 10.1111/imr.12972
3. Noack M, Miossec P. Importance of lymphocyte-stromal cell interactions in autoimmune and inflammatory rheumatic diseases. Nat Rev Rheumatol (2021) 17(9):550–64. doi: 10.1038/s41584-021-00665-4
4. Krishnamurty AT, Turley SJ. Lymph node stromal cells: Cartographers of the immune system. Nat Immunol (2020) 21(4):369–80. doi: 10.1038/s41590-020-0635-3
5. Li L, Wu J, Abdi R, Jewell CM, Bromberg JS. Lymph node fibroblastic reticular cells steer immune responses. Trends Immunol (2021) 42(8):723–34. doi: 10.1016/j.it.2021.06.006
6. Ng CT, Nayak BP, Schmedt C, Oldstone MB. Immortalized clones of fibroblastic reticular cells activate virus-specific T cells during virus infection. Proc Natl Acad Sci USA (2012) 109(20):7823–8. doi: 10.1073/pnas.1205850109
7. Li L, Shirkey MW, Zhang T, Piao W, Li X, Zhao J, et al. Lymph node fibroblastic reticular cells preserve a tolerogenic niche in allograft transplantation through laminin A4. J Clin Invest (2022) 132(13):e156994. doi: 10.1172/jci156994
8. Fletcher AL, Baker AT, Lukacs-Kornek V, Knoblich K. The fibroblastic T cell niche in lymphoid tissues. Curr Opin Immunol (2020) 64:110–6. doi: 10.1016/j.coi.2020.04.007
9. Pikor NB, Cheng HW, Onder L, Ludewig B. Development and immunological function of lymph node stromal cells. J Immunol (2021) 206(2):257–63. doi: 10.4049/jimmunol.2000914
10. Siegert S, Huang HY, Yang CY, Scarpellino L, Carrie L, Essex S, et al. Fibroblastic reticular cells from lymph nodes attenuate T cell expansion by producing nitric oxide. PloS One (2011) 6(11):e27618. doi: 10.1371/journal.pone.0027618
11. Yu M, Guo G, Zhang X, Li L, Yang W, Bollag R, et al. Fibroblastic reticular cells of the lymphoid tissues modulate T cell activation threshold during homeostasis Via hyperactive cyclooxygenase-2/Prostaglandin E(2) axis. Sci Rep (2017) 7(1):3350. doi: 10.1038/s41598-017-03459-5
12. Knoblich K, Cruz Migoni S, Siew SM, Jinks E, Kaul B, Jeffery HC, et al. The human lymph node microenvironment unilaterally regulates T-cell activation and differentiation. PloS Biol (2018) 16(9):e2005046. doi: 10.1371/journal.pbio.2005046
13. Brown FD, Sen DR, LaFleur MW, Godec J, Lukacs-Kornek V, Schildberg FA, et al. Fibroblastic reticular cells enhance T cell metabolism and survival Via epigenetic remodeling. Nat Immunol (2019) 20(12):1668–80. doi: 10.1038/s41590-019-0515-x
14. Simon T, Li L, Wagner C, Zhang T, Saxena V, Brinkman CC, et al. Differential regulation of T-cell immunity and tolerance by stromal laminin expressed in the lymph node. Transplantation (2019) 103(10):2075–89. doi: 10.1097/tp.0000000000002774
15. Riedel A, Shorthouse D, Haas L, Hall BA, Shields J. Tumor-induced stromal reprogramming drives lymph node transformation. Nat Immunol (2016) 17(9):1118–27. doi: 10.1038/ni.3492
16. Baptista AP, Roozendaal R, Reijmers RM, Koning JJ, Unger WW, Greuter M, et al. Lymph node stromal cells constrain immunity via mhc class ii self-antigen presentation. Elife (2014) 3:e04433. doi: 10.7554/eLife.04433
17. Nadafi R, Gago de Graça C, Keuning ED, Koning JJ, de Kivit S, Konijn T, et al. Lymph node stromal cells generate antigen-specific regulatory T cells and control autoreactive T and b cell responses. Cell Rep (2020) 30(12):4110–23.e4. doi: 10.1016/j.celrep.2020.03.007
18. Kim D, Kim M, Kim TW, Choe YH, Noh HS, Jeon HM, et al. Lymph node fibroblastic reticular cells regulate differentiation and function of Cd4 T cells Via Cd25. J Exp Med (2022) 219(5):e20200795. doi: 10.1084/jem.20200795
19. Aparicio-Domingo P, Cannelle H, Buechler MB, Nguyen S, Kallert SM, Favre S, et al. Fibroblast-derived il-33 is dispensable for lymph node homeostasis but critical for Cd8 T-cell responses to acute and chronic viral infection. Eur J Immunol (2021) 51(1):76–90. doi: 10.1002/eji.201948413
20. Tan Z, Sun B. Il-33/St2 signaling in liver transplantation. Cell Mol Immunol (2021) 18(3):761–3. doi: 10.1038/s41423-020-0418-7
21. Perez-Shibayama C, Islander U, Lütge M, Cheng HW, Onder L, Ring SS, et al. Type I interferon signaling in fibroblastic reticular cells prevents exhaustive activation of antiviral Cd8(+) T cells. Sci Immunol (2020) 5(51):e20200795. doi: 10.1126/sciimmunol.abb7066
22. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol Cancer (2021) 20(1):131. doi: 10.1186/s12943-021-01428-1
23. Zhang J, Zhang N, Fu X, Wang W, Liu H, McKay MJ, et al. Bioinformatic analysis of cancer-associated fibroblast related gene signature as a predictive model in clinical outcomes and immune characteristics of gastric cancer. Ann Transl Med (2022) 10(12):698. doi: 10.21037/atm-22-2810
24. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell (2018) 33(3):463–79.e10. doi: 10.1016/j.ccell.2018.01.011
25. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature (2007) 449(7162):557–63. doi: 10.1038/nature06188
26. Bourhis M, Palle J, Galy-Fauroux I, Terme M. Direct and indirect modulation of T cells by vegf-a counteracted by anti-angiogenic treatment. Front Immunol (2021) 12:616837. doi: 10.3389/fimmu.2021.616837
27. Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res (2009) 29(3):881–8.
28. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
29. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral Cd4+Cd25- naive T cells to Cd4+Cd25+ regulatory T cells by tgf-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
30. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester Cd8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology (2013) 145(5):1121–32. doi: 10.1053/j.gastro.2013.07.025
31. Bellone M, Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front Oncol (2013) 3:231. doi: 10.3389/fonc.2013.00231
32. De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A, Maggiolini M. Hif-1α/Gper signaling mediates the expression of vegf induced by hypoxia in breast cancer associated fibroblasts (Cafs). Breast Cancer Res (2013) 15(4):R64. doi: 10.1186/bcr3458
33. Kato T, Noma K, Ohara T, Kashima H, Katsura Y, Sato H, et al. Cancer-associated fibroblasts affect intratumoral Cd8(+) and Foxp3(+) T cells Via Il6 in the tumor microenvironment. Clin Cancer Res (2018) 24(19):4820–33. doi: 10.1158/1078-0432.Ccr-18-0205
34. Thomas DA, Massagué J. Tgf-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
35. Krishnamurty AT, Shyer JA, Thai M, Gandham V, Buechler MB, Yang YA, et al. Lrrc15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature (2022) 611(7934):148–54. doi: 10.1038/s41586-022-05272-1
36. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of Cd8 (+) T cells to protect tumour cells. Nat Commun (2018) 9(1):948. doi: 10.1038/s41467-018-03347-0
37. Eskandari-Malayeri F, Rezaei M. Immune checkpoint inhibitors as mediators for immunosuppression by cancer-associated fibroblasts: A comprehensive review. Front Immunol (2022) 13:996145. doi: 10.3389/fimmu.2022.996145
38. Goehrig D, Nigri J, Samain R, Wu Z, Cappello P, Gabiane G, et al. Stromal protein Big-H3 reprogrammes tumour microenvironment in pancreatic cancer. Gut (2019) 68(4):693–707. doi: 10.1136/gutjnl-2018-317570
39. Gorchs L, Fernández Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human pancreatic carcinoma-associated fibroblasts promote expression of Co-inhibitory markers on Cd4(+) and Cd8(+) T-cells. Front Immunol (2019) 10:847. doi: 10.3389/fimmu.2019.00847
40. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest (2012) 122(3):899–910. doi: 10.1172/jci45817
41. Komatsu N, Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis - immune cell-Fibroblast-Bone interactions. Nat Rev Rheumatol (2022) 18(7):415–29. doi: 10.1038/s41584-022-00793-5
42. Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the P53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA (1997) 94(20):10895–900. doi: 10.1073/pnas.94.20.10895
43. Franz JK, Pap T, Hummel KM, Nawrath M, Aicher WK, Shigeyama Y, et al. Expression of sentrin, a novel antiapoptotic molecule, at sites of synovial invasion in rheumatoid arthritis. Arthritis Rheum (2000) 43(3):599–607. doi: 10.1002/1529-0131(200003)43:3<599::Aid-anr17>3.0.Co;2-t
44. Toh ML, Gonzales G, Koenders MI, Tournadre A, Boyle D, Lubberts E, et al. Role of interleukin 17 in arthritis chronicity through survival of synoviocytes Via regulation of synoviolin expression. PloS One (2010) 5(10):e13416. doi: 10.1371/journal.pone.0013416
45. Pap T, Müller-Ladner U, Gay RE, Gay S. Fibroblast biology. role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res (2000) 2(5):361–7. doi: 10.1186/ar113
46. Vallejo AN, Yang H, Klimiuk PA, Weyand CM, Goronzy JJ. Synoviocyte-mediated expansion of inflammatory T cells in rheumatoid synovitis is dependent on Cd47-thrombospondin 1 interaction. J Immunol (2003) 171(4):1732–40. doi: 10.4049/jimmunol.171.4.1732
47. Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun (2018) 9(1):789. doi: 10.1038/s41467-018-02892-y
48. Yoshitomi H. Regulation of immune responses and chronic inflammation by fibroblast-like synoviocytes. Front Immunol (2019) 10:1395. doi: 10.3389/fimmu.2019.01395
49. Bradfield PF, Amft N, Vernon-Wilson E, Exley AE, Parsonage G, Rainger GE, et al. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (Cxcl12), which supports distinct patterns and rates of Cd4+ and Cd8+ T cell migration within synovial tissue. Arthritis Rheum (2003) 48(9):2472–82. doi: 10.1002/art.11219
50. Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seisdedos F, et al. Persistent induction of the chemokine receptor Cxcr4 by tgf-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol (2000) 165(6):3423–9. doi: 10.4049/jimmunol.165.6.3423
51. Chabaud M, Page G, Miossec P. Enhancing effect of il-1, il-17, and tnf-alpha on macrophage inflammatory protein-3alpha production in rheumatoid arthritis: Regulation by soluble receptors and Th2 cytokines. J Immunol (2001) 167(10):6015–20. doi: 10.4049/jimmunol.167.10.6015
52. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of Ccr6-expressing Th17 cells to inflamed joints Via Ccl20 in rheumatoid arthritis and its animal model. J Exp Med (2007) 204(12):2803–12. doi: 10.1084/jem.20071397
53. Tanida S, Yoshitomi H, Nishitani K, Ishikawa M, Kitaori T, Ito H, et al. Ccl20 produced in the cytokine network of rheumatoid arthritis recruits Ccr6+ mononuclear cells and enhances the production of il-6. Cytokine (2009) 47(2):112–8. doi: 10.1016/j.cyto.2009.05.009
54. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor rorgammat directs the differentiation program of proinflammatory il-17+ T helper cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
55. Kobayashi S, Watanabe T, Suzuki R, Furu M, Ito H, Ito J, et al. Tgf-B induces the differentiation of human Cxcl13-producing Cd4(+) T cells. Eur J Immunol (2016) 46(2):360–71. doi: 10.1002/eji.201546043
56. Mori M, Hashimoto M, Matsuo T, Fujii T, Furu M, Ito H, et al. Cell-Contact-Dependent activation of Cd4(+) T cells by adhesion molecules on synovial fibroblasts. Mod Rheumatol (2017) 27(3):448–56. doi: 10.1080/14397595.2016.1220353
57. Petrasca A, Phelan JJ, Ansboro S, Veale DJ, Fearon U, Fletcher JM. Targeting bioenergetics prevents Cd4 T cell-mediated activation of synovial fibroblasts in rheumatoid arthritis. Rheumatol (Oxford) (2020) 59(10):2816–28. doi: 10.1093/rheumatology/kez682
58. Bartok B, Firestein GS. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol Rev (2010) 233(1):233–55. doi: 10.1111/j.0105-2896.2009.00859.x
59. Nanki T, Hayashida K, El-Gabalawy HS, Suson S, Shi K, Girschick HJ, et al. Stromal cell-derived factor-1-Cxc chemokine receptor 4 interactions play a central role in Cd4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol (2000) 165(11):6590–8. doi: 10.4049/jimmunol.165.11.6590
60. Bombara MP, Webb DL, Conrad P, Marlor CW, Sarr T, Ranges GE, et al. Cell contact between T cells and synovial fibroblasts causes induction of adhesion molecules and cytokines. J Leukoc Biol (1993) 54(5):399–406. doi: 10.1002/jlb.54.5.399
61. van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17a production. Arthritis Rheum (2011) 63(1):73–83. doi: 10.1002/art.30093
62. Homey B, Alenius H, Müller A, Soto H, Bowman EP, Yuan W, et al. Ccl27-Ccr10 interactions regulate T cell-mediated skin inflammation. Nat Med (2002) 8(2):157–65. doi: 10.1038/nm0202-157
63. Noack M, Ndongo-Thiam N, Miossec P. Role of podoplanin in the high interleukin-17a secretion resulting from interactions between activated lymphocytes and psoriatic skin-derived mesenchymal cells. Clin Exp Immunol (2016) 186(1):64–74. doi: 10.1111/cei.12830
64. Gao Y, Yao X, Zhai Y, Li L, Li H, Sun X, et al. Single cell transcriptional zonation of human psoriasis skin identifies an alternative immunoregulatory axis conducted by skin resident cells. Cell Death Dis (2021) 12(5):450. doi: 10.1038/s41419-021-03724-6
65. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 cytokines stimulate Ccl20 expression in keratinocytes in vitro and in vivo: Implications for psoriasis pathogenesis. J Invest Dermatol (2009) 129(9):2175–83. doi: 10.1038/jid.2009.65
66. Furue K, Ito T, Tsuji G, Nakahara T, Furue M. The Ccl20 and Ccr6 axis in psoriasis. Scand J Immunol (2020) 91(3):e12846. doi: 10.1111/sji.12846
67. Veale D, Yanni G, Rogers S, Barnes L, Bresnihan B, Fitzgerald O. Reduced synovial membrane macrophage numbers, elam-1 expression, and lining layer hyperplasia in psoriatic arthritis as compared with rheumatoid arthritis. Arthritis Rheum (1993) 36(7):893–900. doi: 10.1002/art.1780360705
68. Campbell JJ, O'Connell DJ, Wurbel MA. Cutting edge: Chemokine receptor Ccr4 is necessary for antigen-driven cutaneous accumulation of Cd4 T cells under physiological conditions. J Immunol (2007) 178(6):3358–62. doi: 10.4049/jimmunol.178.6.3358
69. Flytlie HA, Hvid M, Lindgreen E, Kofod-Olsen E, Petersen EL, Jørgensen A, et al. Expression of Mdc/Ccl22 and its receptor Ccr4 in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Cytokine (2010) 49(1):24–9. doi: 10.1016/j.cyto.2009.10.005
70. Mitra A, Raychaudhuri SK, Raychaudhuri SP. Functional role of il-22 in psoriatic arthritis. Arthritis Res Ther (2012) 14(2):R65. doi: 10.1186/ar3781
71. Duperret EK, Trautz A, Ammons D, Perales-Puchalt A, Wise MC, Yan J, et al. Alteration of the tumor stroma using a consensus DNA vaccine targeting fibroblast activation protein (Fap) synergizes with antitumor vaccine therapy in mice. Clin Cancer Res (2018) 24(5):1190–201. doi: 10.1158/1078-0432.Ccr-17-2033
72. Fang J, Xiao L, Joo KI, Liu Y, Zhang C, Liu S, et al. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int J Cancer (2016) 138(4):1013–23. doi: 10.1002/ijc.29831
73. Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest (2006) 116(7):1955–62. doi: 10.1172/jci26532
74. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting Cxcl12 from fap-expressing carcinoma-associated fibroblasts synergizes with anti-Pd-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
75. Hong SS, Marotte H, Courbon G, Firestein GS, Boulanger P, Miossec P. Puma gene delivery to synoviocytes reduces inflammation and degeneration of arthritic joints. Nat Commun (2017) 8(1):146. doi: 10.1038/s41467-017-00142-1
76. Bonaventura P, Courbon G, Lamboux A, Lavocat F, Marotte H, Albarède F, et al. Protective effect of low dose intra-articular cadmium on inflammation and joint destruction in arthritis. Sci Rep (2017) 7(1):2415. doi: 10.1038/s41598-017-02611-5
77. Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, et al. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci (2015) 106(2):134–42. doi: 10.1111/cas.12584
78. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res (2018) 6:15. doi: 10.1038/s41413-018-0016-9
79. Rendon A, Schäkel K. Psoriasis pathogenesis and treatment. Int J Mol Sci (2019) 20(6):1475. doi: 10.3390/ijms20061475
80. Smeets TJ, Kraan MC, van Loon ME, Tak PP. Tumor necrosis factor alpha blockade reduces the synovial cell infiltrate early after initiation of treatment, but apparently not by induction of apoptosis in synovial tissue. Arthritis Rheum (2003) 48(8):2155–62. doi: 10.1002/art.11098
81. Guggino G, Giardina AR, Raimondo S, Giardina G, Sireci G, Dieli F, et al. Targeting il-6 signalling in early rheumatoid arthritis is followed by Th1 and Th17 suppression and Th2 expansion. Clin Exp Rheumatol (2014) 32(1):77–81.
82. Caporali R, Zavaglia D. Real-world experience with tofacitinib for the treatment of rheumatoid arthritis. Clin Exp Rheumatol (2019) 37(3):485–95.
Keywords: fibroblasts, T cells, cancer, autoimmune disease, immune response, inflammation
Citation: Lee B, Lee S-H and Shin K (2023) Crosstalk between fibroblasts and T cells in immune networks. Front. Immunol. 13:1103823. doi: 10.3389/fimmu.2022.1103823
Received: 21 November 2022; Accepted: 20 December 2022;
Published: 09 January 2023.
Edited by:
Chao Li, China Medical University, ChinaReviewed by:
Yuan Yao, Guangzhou First People’s Hospital, ChinaCopyright © 2023 Lee, Lee and Shin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seung-Hyo Lee, c2wxMzEzNDVAa2Fpc3QuYWMua3I=; Kihyuk Shin, dGVyaWFraWxsZXJAaGFubWFpbC5uZXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.