 Vilasha Bulleeraz
Vilasha Bulleeraz Michelle Goy
Michelle Goy Faiza Basheer1,2
Faiza Basheer1,2 Clifford Liongue
Clifford Liongue Alister C. Ward
Alister C. Ward- 1School of Medicine, Deakin University, Geelong, VIC, Australia
- 2Institute for Mental and Physical Health and Clinical Translation (IMPACT), Deakin University, Geelong, VIC, Australia
Introduction: The granulocyte colony-stimulating factor receptor (G-CSFR), encoded by the CSF3R gene, is involved in the production and function of neutrophilic granulocytes. Somatic mutations in CSF3R leading to truncated G-CSFR forms are observed in acute myeloid leukemia (AML), particularly those subsequent to severe chronic neutropenia (SCN), as well as in a subset of patients with other leukemias.
Methods: This investigation introduced equivalent mutations into the zebrafish csf3r gene via genome editing and used a range of molecular and cellular techniques to understand the impact of these mutations on immune cells across the lifespan.
Results: Zebrafish harboring truncated G-CSFRs showed significantly enhanced neutrophil production throughout successive waves of embryonic hematopoiesis and a neutrophil maturation defect in adults, with the mutations acting in a partially dominant manner.
Discussion: This study has elucidated new insights into the impact of G-CSFR truncations throughout the life-course and created a bone fide zebrafish model for further investigation.
1. Introduction
The granulocyte colony-stimulating factor receptor (G-CSFR) is a key regulator of neutrophil production, or granulopoiesis, by impacting on the proliferation and differentiation of precursors as well as enhancing neutrophil survival (1, 2). The G-CSFR is activated by the cytokine G-CSF, which is produced developmentally (3), but also in response to injury and infection to stimulate so-called ‘emergency’ granulopoiesis (2). This cytokine is also used clinically in situations where neutrophil numbers are low (4).
Acquired somatic mutations in CSF3R, the gene encoding G-CSFR, have been found in a variety of leukemias (5). These include nonsense and frameshift mutations within exon 17 that serve to truncate the G-CSFR intracellular domain that are observed in acute myeloid leukemia (AML), particularly subsequent to severe chronic neutropenia (SCN) (6, 7), but also de novo and relapsed forms of the disease (8–10). Similar mutations have also been identified in chronic neutrophilic leukemia (CNL), atypical chronic myelogenous leukemia (aCML) and chronic myelomonocytic leukemia (CMML) patients (9, 11–14), although these are usually co-incident with alternative constitutively-activating G-CSFR mutations (15). The locations of the receptor truncations are quite variable, being found from position 738 through to 819 (5, 7–14). They lead to hyperresponsiveness to G-CSF, enhanced signaling particular of STAT5, and increased proliferation (16–18), with the ability to mediate leukemic transformation in vitro (10). Various mouse models of G-CSFR truncation mutations have been generated by different gene targeting approaches (19–21). While all exhibited enhanced responsiveness to G-CSF and altered receptor internalization, they were variable with respect to their effects on steady-state neutrophil levels.
Zebrafish has proven to be a robust experimental platform for the study of blood and immune cell development and its perturbation in disease (22). Zebrafish granulopoiesis occurs in multiple waves like mammals (23). This includes a primitive wave generating myeloid precursors in the rostral blood island that differentiate into neutrophils as they migrate across the yolk sac and a definitive wave that ultimately produces neutrophils from the kidney marrow, the equivalent of mammalian bone marrow (24). Zebrafish has been shown to have a structurally and functionally conserved csf3r gene (3), which contributes to neutrophil production throughout the life-course (25, 26). The zebrafish csf3r gene was targeted with CRISPR-Cas9 to generate truncating hyperresponsive G-CSFR mutations based on those observed in leukemia. These csf3r mutant fish possessed enhanced numbers of neutrophils during primitive and definitive hematopoiesis, but in adulthood neutrophil numbers were normal but maturation was reduced. Together these data identify on-going impacts of G-CSFR truncations on neutrophil production, and describe a useful in vivo model for further studies.
2. Materials and methods
2.1. Fish husbandry
Wild-type and Tg(mpo::GFP) zebrafish, in which neutrophils are fluorescently marked (27), were maintained using standard husbandry practices (28) in a Techniplast aquarium system at 28.5°C with pH 7.0, ammonia <1.5 ppm, nitrite <3 ppm, nitrate <30 ppm and conductivity at 500 µS on a 14 h/10 h light/dark cycle and fed twice daily. Embryos were manually spawned and maintained at 28.5°C in petri dishes containing aquarium water and 0.00005% (w/v) methylene blue. At 8 h post-fertilization (hpf) this was replaced with aquarium water containing 0.003% (w/v) 1-phenyl-2-thio-urea (PTU) to inhibit pigmentation and maintain embryo transparency.
2.2. Genetic manipulation and analysis
Wild-type embryos at the 1 cell stage were injected with 12.5 ng/μL CRISPR guide RNA (gRNA) that targeted a sequence within csf3r exon 16 (CTGTTAGCAGGAGACGAGCC) in order to recapitulate human truncation mutations and 100 pg/nL Cas9 in sterile nuclease-free water along with 1:16 vol:vol 1% (w/v) phenol red and raised to adulthood. These were out-crossed with wild-type fish and carriers of relevant mutant alleles identified from fin clips obtained under anesthesia with benzocaine. Genomic DNA was isolated with QuickExtract following the manufacturer’s instructions, and subjected to PCR with csf3r-specific primers for High Resolution Melt (HRM) analysis (29) with primers 5’-ATTCCTCCAACCTCCAGC and 5’-CAGAGAAGCGGTTCAGTGC using Precision Melt Supermix and Analysis Software (BioRad) to identify potential mutants that were confirmed by Sanger sequencing using the primers 5’-CAGTGCTGGTGTATCTGTCCC and 5’-GCGAGTTAGATGTGATTGACC at the Australian Genome Research Facility. These founder fish were further out-crossed before being in-crossed to generate homozygous mutant fish. One allele was also crossed onto the Tg(mpo::GFP) (27) background. In some experiments in vitro transcribed csf3a mRNA was injected into 1-8 cell stage embryos to stimulate the G-CSFR as described (30), since the encoded ligand appears more potent than that produced by the alternate orthologue csf3b (31).
2.3. Whole-mount in situ hybridization
Embryos were collected at appropriate time points reflecting primitive (22 hpf) and definitive (5 dpf) waves of hematopoiesis (24) and anesthetized with 0.4 mg/mL benzocaine before fixation with 4% (w/v) paraformaldehyde in phosphate-buffered saline. Fixed embryos were stored at 4°C for at least 1 day, after which embryos were dehydrated with 100% (v/v) methanol for long-term storage at -20°C. Rehydrated embryos were subjected to WISH with anti-sense DIG-labeled probes specific for blood and immune cell lineages as described (32, 33). Stained embryos were mounted in 2% w/v methylcellulose and visualized using MVX10 monozoom microscope with a 1 × MVXPlan Apochromat lens (NA = 0.25) with an Olympus DP74 camera. Quantitation was achieved by enumeration of individual cells stained with the probe or measuring the area of staining using ImageJ software in a blind fashion on images taken on a dissecting microscope. Data were analyzed for significance with a Student’s t-test, using Welch’s correction where necessary.
2.4. Reverse-transcription polymerase chain reaction
Total RNA was extracted from 20-30 pooled zebrafish embryos with RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol, and subjected to quantitative real-time reverse-transcriptase PCR (qRT2-PCR) with the primers for actb (5’-TGGCATCACACCTTCTAC and 5’- AGACCATCACCAGAGTCC), and csf3r (5’- CAGAGAAGCGGTTCAGTGC and 5’-ATTCCTCCAATCCTCCAGC). Data was normalized relative to actb and fold-change in csf3r expression calculated using the ΔΔCT method (34).
2.5. Ex vivo analyses
Adult zebrafish were euthanized with benzocaine and blood and kidney collected. Kidneys from fish on the Tg(mpo::GFP) background were placed in ice-cold phosphate-buffered saline supplemented with 1 mM EDTA and 2% (v/v) fetal calf serum and passage through a 40 μm sieve. These isolated kidney cells were analyzed using a BD FACSCantoII analyzer with lineages identified in a SSC/FSC plot, and GFP+ neutrophils identified in the FITC channel, while apoptosis was assessed using a PE-Annexin V/7-AAD apoptosis detection kit (Biolegend). A minimum of 100,000 events were collected for each sample using FACSCanto II flow cytometer (BD Biosciences) and analyzed using BD FACSDiva software (v6.0). Neutrophils were sorted on a FACSAriaIII (BD Biosciences) for further analysis. Cytospin preparations of blood and sorted GFP+ neutrophils were stained with Giemsa (Sigma) and slides viewed on a Leica DME stereomicroscope and imaged with a DP70 camera and differential counts performed, with overall cell and nuclear morphology used to stage neutrophils. Data were analyzed for significance with a Student’s t-test.
3. Results
3.1. Generation of zebrafish csf3r mutants based on human hyperresponsive truncation mutations
Truncation mutations affecting the human G-CSFR intracellular domain have been observed in AML, commencing from position 739 through 819 (5, 8–10), and in other leukemias from 738 through 791 (9, 11–14), with truncations across this range able to mediate leukemic transformation in vitro (10) (Figure 1A). The intracellular domain is largely conserved in the zebrafish G-CSFR (Figure 1A), and so exon 16 of the zebrafish csf3r gene was targeted by genome editing using CRISPR/Cas9 (35) to recapitulate the human mutations (Figure 1B). A guide RNA (gRNA) was designed targeting sequences encoding a di-leucine motif shown to play an important role in G-CSFR internalization (17) (Figure 1C) that lies roughly in the middle of the AML mutants (Figure 1C). Wild-type embryos injected with this gRNA and Cas9-encoding mRNA at the one cell stage were raised to adulthood, with their progeny subjected to HRM and Sanger sequencing to identify potential csf3r mutations. A total of four csf3r mutant alleles were identified with fish carrying these alleles out-crossed before the resultant carrier progeny were in-crossed with the mutations clearly evident upon sequencing of homozygote carriers of each allele. Two alleles were chosen: mdu26 containing a complex indel (1 bp insertion and 8 bp deletion) and mdu27 containing a 5 bp insertion (Figure 1C). Both mutations caused a frameshift followed by a stop codon rendering the intracellular domain truncated, specifically P751fs23* for mdu26 and L752fs2* for mdu27, including the complete or partial loss of the di-leucine motif in each case. Of the other alleles, one had a 1 bp deletion leading to P751fs25* and so almost identical to mdu26, while the other had a 17 bp insertion and 2 bp deletion resulting in a 5 amino acid insertion but no truncation, and so mdu26 and mdu27 were chosen for further study. The mdu27 allele was crossed with Tg(mpo:GFP) fish in which neutrophils are fluorescently marked (27). Expression of csf3r was found to be similar in wild-type and mutant embryos at several timepoints (Figure 1D), ruling out any significant nonsense-mediated decay (36).
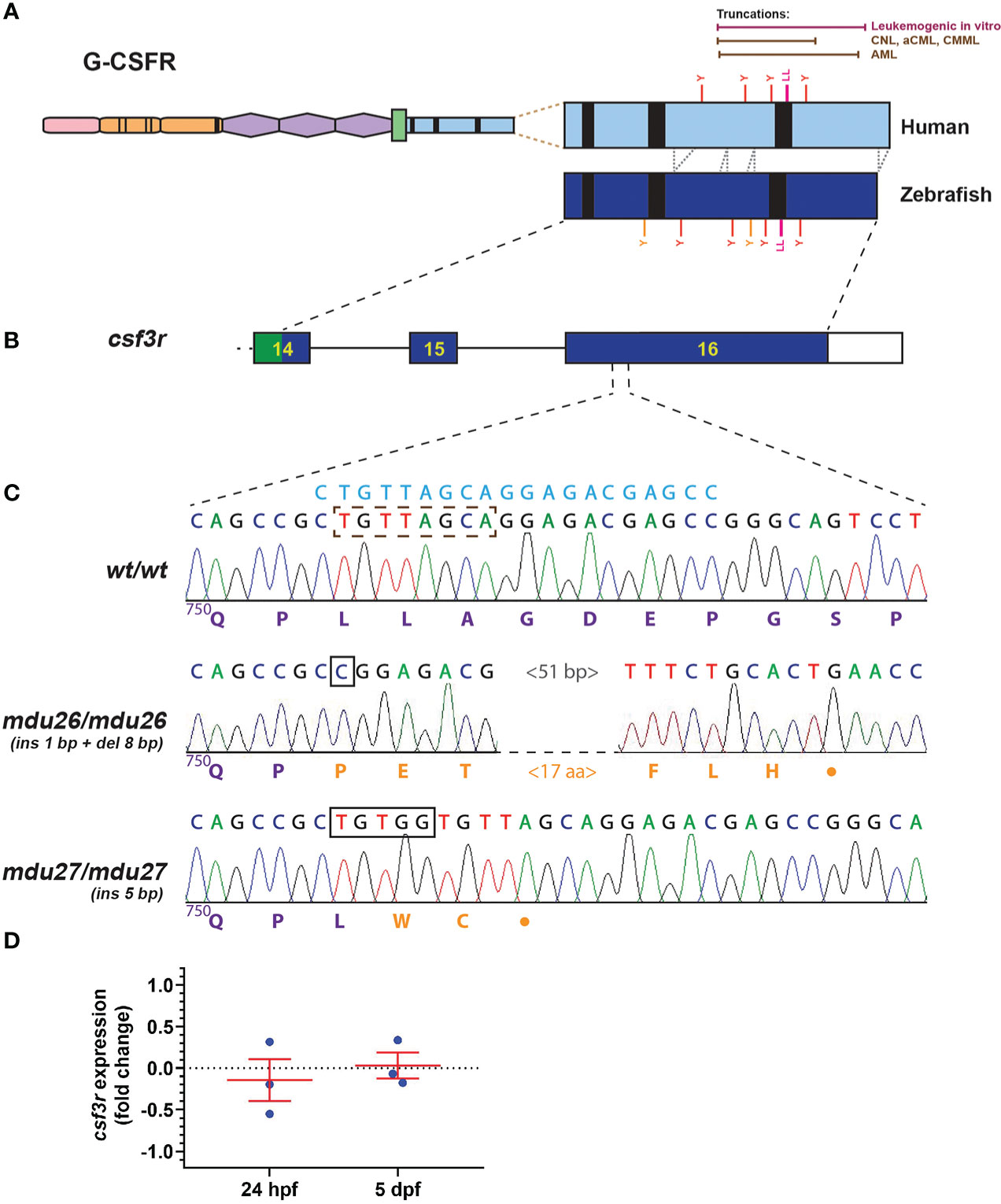
Figure 1 Generation of leukemia-derived G-CSFR truncation mutants. Schematic diagram of the G-CSFR showing the extracellular immunoglobulin domain (pink), cytokine receptor homology domain (orange) and fibronectin type III-like domains (purple), transmembrane region (green), and intracellular region (blue) containing Box 1-3 (black rectangles). The intracellular region is expanded to show tyrosine (Y) residues and a di-leucine motif (LL) in both human and zebrafish proteins, with the relative positions of truncation mutations found in various human leukemias or demonstrated to be leukemogenic in vitro shown above (A). Exons 14-16 of the zebrafish csf3r gene encoding the intracellular region presented as numbered boxes with connecting lines depicting introns (B). Sequence traces of the indicated part of csf3r showing homozygous wild-type (wt/wt) and mutant zebrafish, with nucleotide sequence above and encoded protein sequences below, in purple for native and orange for de novo sequence (C). The gRNA target site is shown above in blue, with deleted nucleotide sequences indicated with a brown dotted box and inserted sequences shown with black boxes. The csf3rmdu26 (mdu26) allele represents a complex 1 bp insertion/8 bp deletion and the csf3rmdu27 (mdu27) allele a 5 bp insertion both of which cause a frameshift resulting in translation from an alternative reading frame followed by a stop codon. Gene expression analysis of csf3r in pooled WT and mutant embryos at the indicated timepoints presented as fold-change (log2) relative to actb (D), showing mean and SEM (not significant, n=3).
3.2. Impact of G-CSFR truncation mutation on embryonic primitive hematopoiesis
Primitive hematopoiesis commences around 12 hpf in zebrafish and is well established by 22 hpf (37). Both heterozygous csf3rwt/mdu26 and homozygous csf3rmdu26/mdu26 embryos showed increased numbers of cells expressing mpo, a marker of mature neutrophils (38), compared to csf3rwt/wt embryos (Figures 2A–D). A similar increase in mpo was observed in embryos homozygous for the other allele (csf3rmdu27/mdu27) compared to csf3rwt/wt embryos (Supplementary Figures 1A–C). In contrast, no differences were seen in the number of cells expressing spi1b, a marker of myeloid precursors (39) (Figures 2E–H) or the area of expression for gata1a, a marker of erythrocyte precursors (40) (Figures 2I–L).
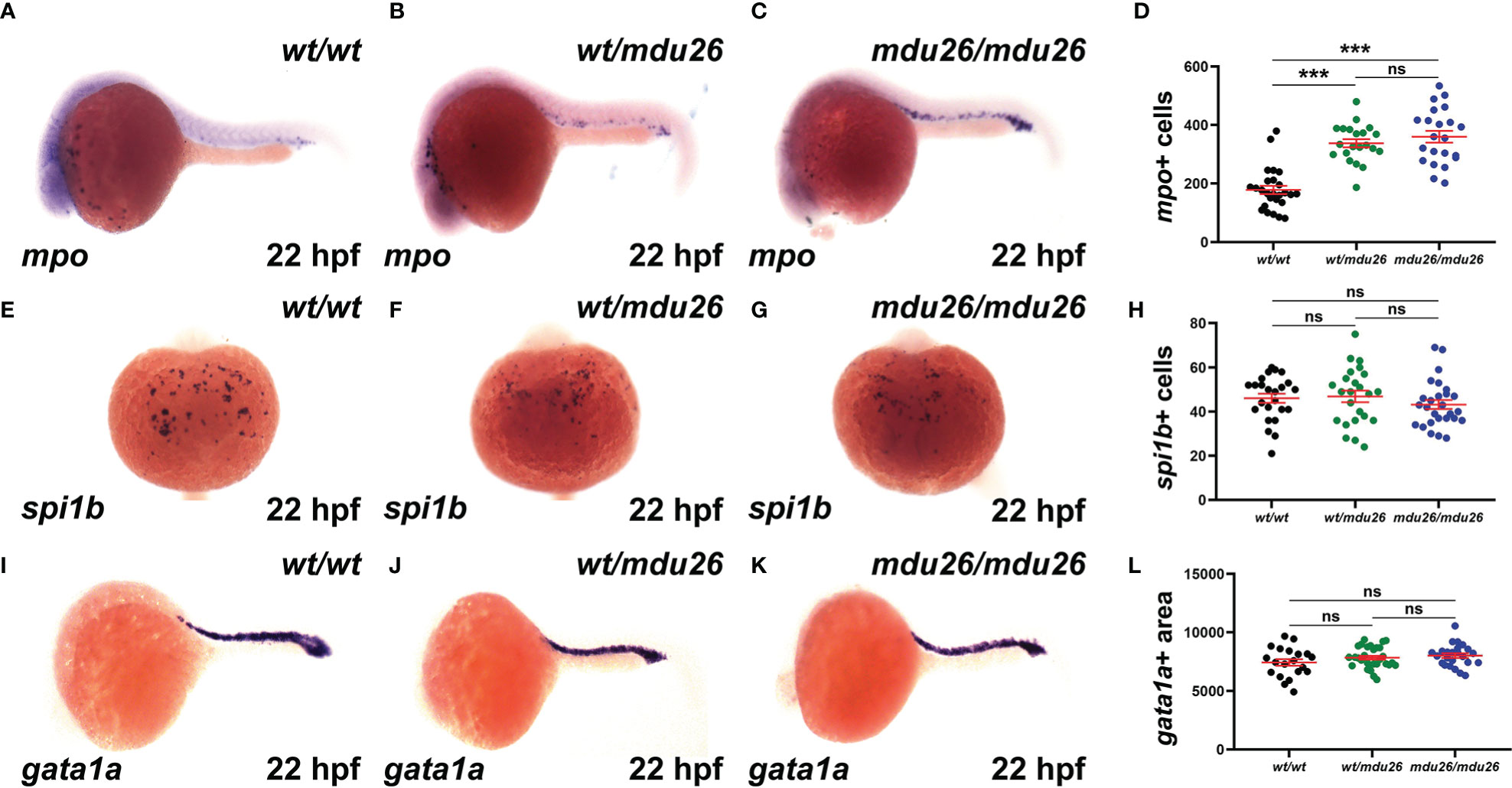
Figure 2 Effect of G-CSFR truncation mutation on primitive hematopoiesis. Wild-type (wt/wt), heterozygous (wt/mdu26) and homozygous (mdu26/mdu26) mutant csf3r embryos were subjected to WISH at 22 hpf with mpo (A–C), spi1b (E–G) and gata1a (I–K), with representative images shown. Individual embryos were assessed for the number of mpo+ (D) or spi1b+ (H) cells or the area of gata1a (L) staining, with mean and SEM in red and level of statistical significance indicated (***: p < 0.001, ns: not significant; n=20-30).
3.3. Impact of G-CSFR truncation mutation on embryonic definitive hematopoiesis
Definitive hematopoiesis commences during the second day post-fertilization (dpf) and fully supplants primitive hematopoiesis by 5 dpf (22). At this time point, a significant increase in mpo+ cells was observed in homozygous csf3rmdu26/mdu26 compared to csf3rwt/wt embryos, but there was no longer a difference between heterozygous csf3rwt/mdu26 and csf3rwt/wt embryos (Figures 3A–D). A similar elevation in mpo+ cells was seen in homozygous csf3rmdu27/mdu27 compared to csf3rwt/wt embryos (Supplementary Figures 1D–F). In contrast, there were no differences in the number of cells positive for mpeg1.1, a marker of macrophages (41) (Figures 3E–H), or the area of staining for rag1, a marker of T cells (42) (Figures 3I–L), or of hbbe1.1, a marker of mature erythrocytes (43) (Figures 3M–P).
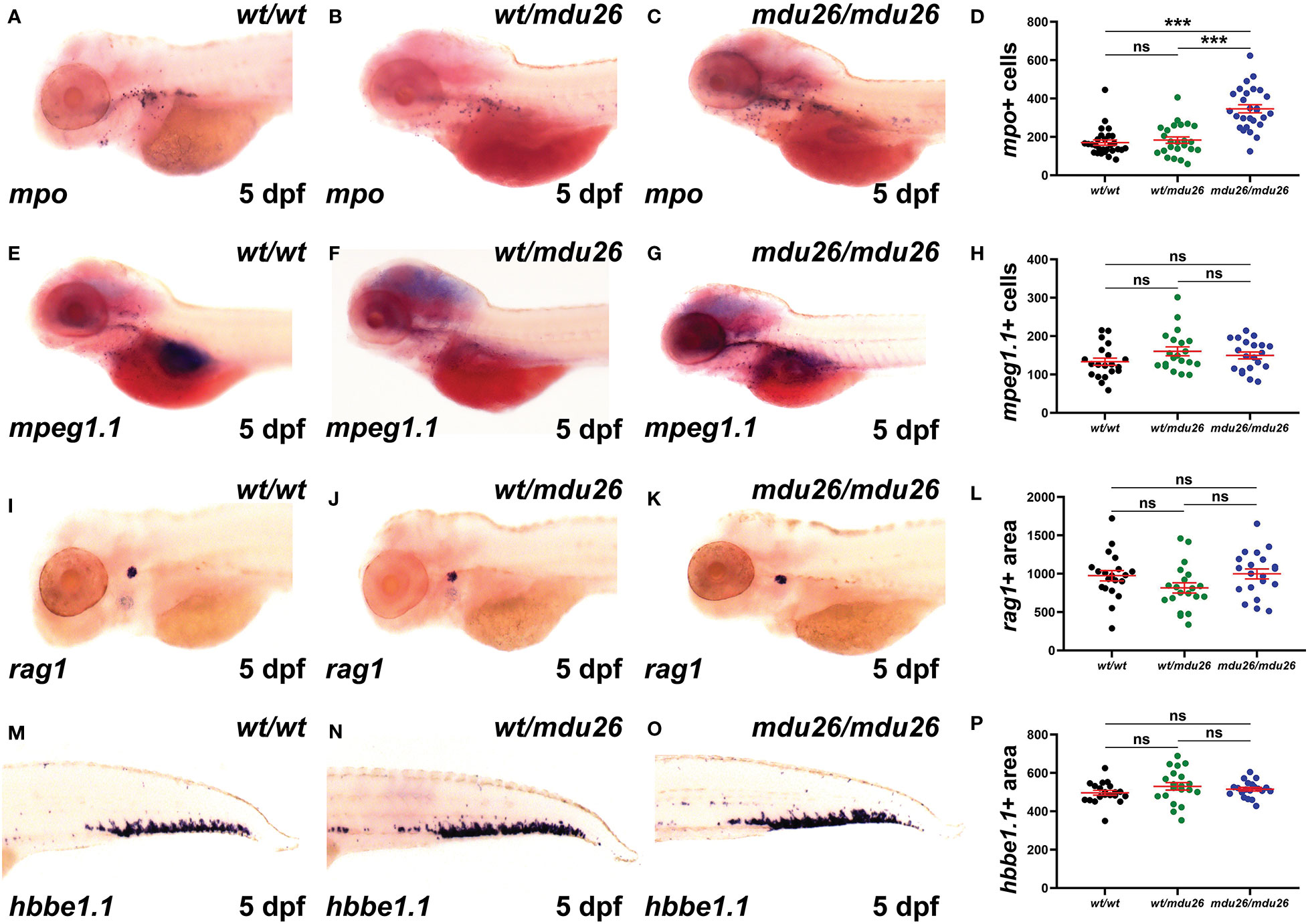
Figure 3 Effect of G-CSFR truncation mutation on definitive hematopoiesis. Wild-type (wt/wt), heterozygous (wt/mdu26) and homozygous (mdu26/mdu26) mutant csf3r embryos were subjected to WISH at 5 dpf with mpo (A–C), mpeg1.1 (E–G), rag1 (I–K) and hbbe1.1 (M–O), with representative images shown. Individual embryos were assessed for the number of mpo+ (D) or mpeg1.1+ (H) cells or the area of staining for rag1 (L) or hbbe1.1 (P), with mean and SEM shown in red and level of statistical significance indicated (***: p < 0.001, ns: not significant; n=20-28).
3.4. Impact of G-CSFR truncation mutation on adult steady-state hematopoiesis
Adult blood samples were subjected to histological analysis, which was quantified using differential counting. No significant differences were observed between csf3rmdu26/mdu26, csf3rwt/mdu26 and csf3rwt/wt fish with regard to specific blood populations (Figures 4A–D), which was confirmed in csf3rmdu27/mdu27 and csf3rwt/mdu27 adults (Supplementary Figures 1G–J). The adult kidney marrow was further analyzed in csf3rmdu27/mdu27 fish on the Tg(mpo::GFP) background using FACS. The SSC/FSC plot revealed a significant increase in the relative number of myeloid cells in, with other populations concomitantly decreased (Figures 4E–H). However, specific analysis of the GFP+ neutrophil population revealed no change in overall neutrophil numbers (Figures 4I–L). Moreover, histological analysis of sorted GFP+ cells revealed a decrease in relative maturity of neutrophils in both csf3rwt/mdu27 and csf3rmdu27/mdu27 compared to csf3rwt/wt fish (Figures 4M–P).
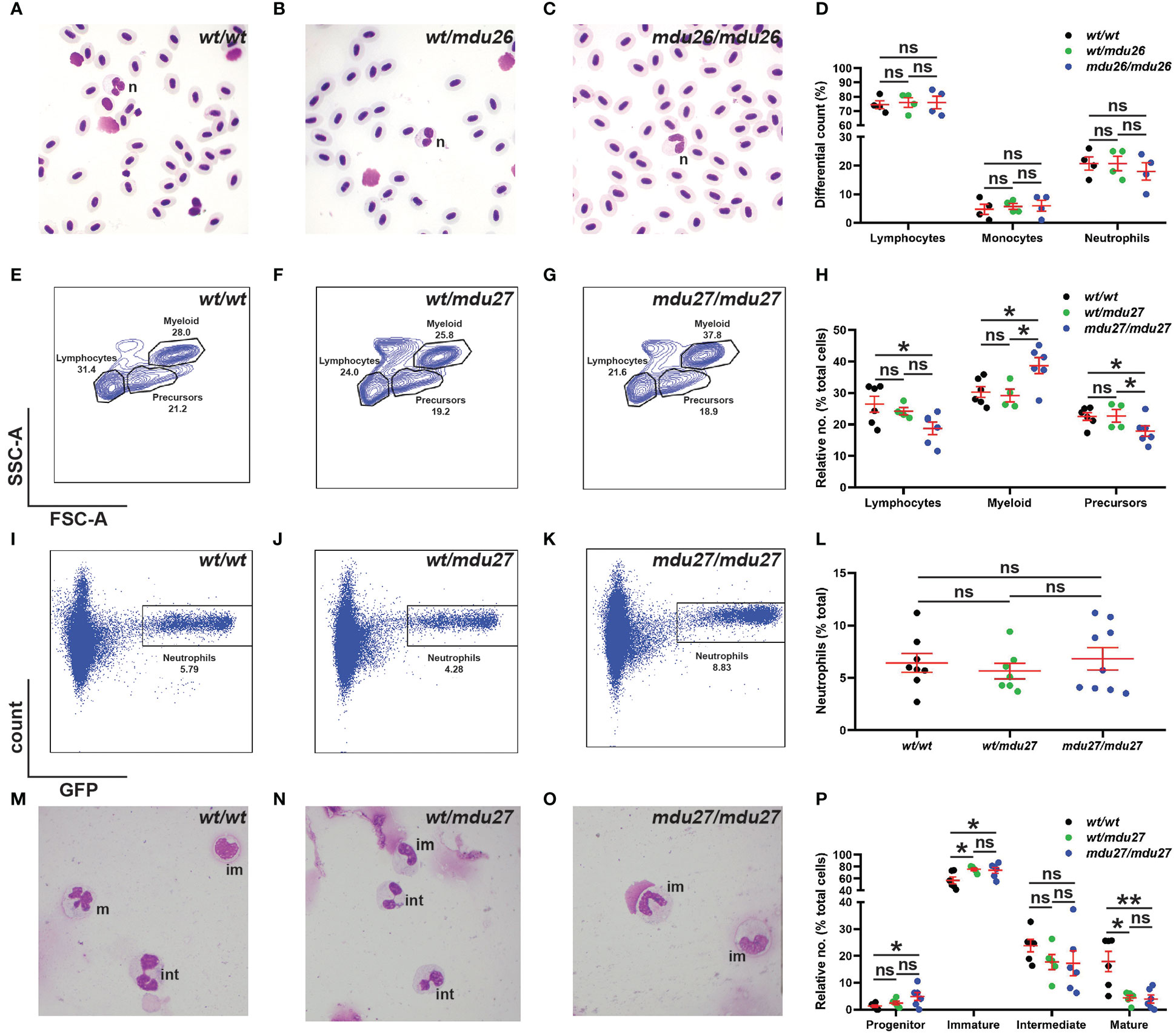
Figure 4 Effect of G-CSFR truncation mutation on adult hematopoiesis. (A–D). Adult blood cells from wild-type (wt/wt), heterozygous (wt/mdu26) and homozygous (mdu26/mdu26) mutant csf3r fish were subjected to Giemsa-staining (A–C), along with differential quantitation of the indicated blood cell populations for individual fish (D), with mean and SEM shown in red and level of statistical significance indicated (ns: not significant; n=4). Abbreviation: n: neutrophil. (E–P). Adult kidney cells from wild-type (wt/wt), heterozygous (wt/mdu27) and homozygous (mdu27/mdu27) mutant csf3r fish on a Tg(mpo::GFP) background were subjected to FACS analysis using SSC/FSC (E–G) and GFP fluorescence (I–K), along with quantitation of indicated cell populations (H) and GFP+ neutrophils (L), with sorted GFP+ cells subjected to Giemsa-staining (M–O), along with differential quantitation of relative differentiation (P). Panels (H, L, P) display results for individual fish, with mean and SEM shown in red and level of statistical significance indicated (*: p < 0.05, ns: not significant; n=4-9). im, immature; int; intermediate; m, mature.
Since G-CSFR signaling has been implicated in cell survival (2), it was of interest to assess if this might explain the altered granulopoiesis. However, no difference was observed for early (AnnexinV+/7AAD-) and late apoptotic (AnnexinV+/7AAD+) populations between csf3rmdu27/mdu27 and csf3rwt/wt fish in either the total myeloid (Supplementary Figures 2A–D) or GFP+ neutrophil (Supplementary Figures 2E–H) populations.
3.5. Impact of truncating G-CSFR mutants on emergency granulopoiesis
G-CSF is known for its key role in ‘emergency’ neutrophil production such as during an infection (1, 2). Moreover, G-CSF is directly administered to SCN patients to alleviate neutropenia (44). Therefore, it was important to identify the effects of G-CSFR intracellular truncation mutations on emergency hematopoiesis stimulated by G-CSF. Both csf3rmdu27/mdu27 and csf3rwt/wt embryos were injected with csf3a mRNA encoding a zebrafish G-CSF or left uninjected, as previously described (3). WISH analysis revealed a significant increase in mpo+ cells at 23 hpf following enforced G-CSF expression in csf3rwt/wt embryos (Figures 5A, B, E), as expected (3). Both uninjected and injected csf3rmdu27/mdu27 mutants had significantly more neutrophils than the corresponding csf3rwt/wt embryos (Figures 5A–E), but there was no significant difference in neutrophil numbers in injected versus uninjected csf3rmdu27/mdu27 embryos (Figures 5C–E). A significant increase in mpo+ cells in injected compared to uninjected csf3rwt/wt embryos at 5 dpf was again evident (Figures 5F, G, J) and between uninjected csf3rmdu27/mdu27 and both uninjected csf3rwt/wt (Figures 5F, H, J) and injected csf3rmdu27/mdu27 embryos (Figures 5H–J), but not between the injected csf3rmdu27/mdu27 and injected csf3rwt/wt embryos (Figures 5G, I, J). This was confirmed in csf3rmdu27/mdu27 and csf3rwt/wt embryos on the Tg(mpo::GFP) background (Supplementary Figure 3).
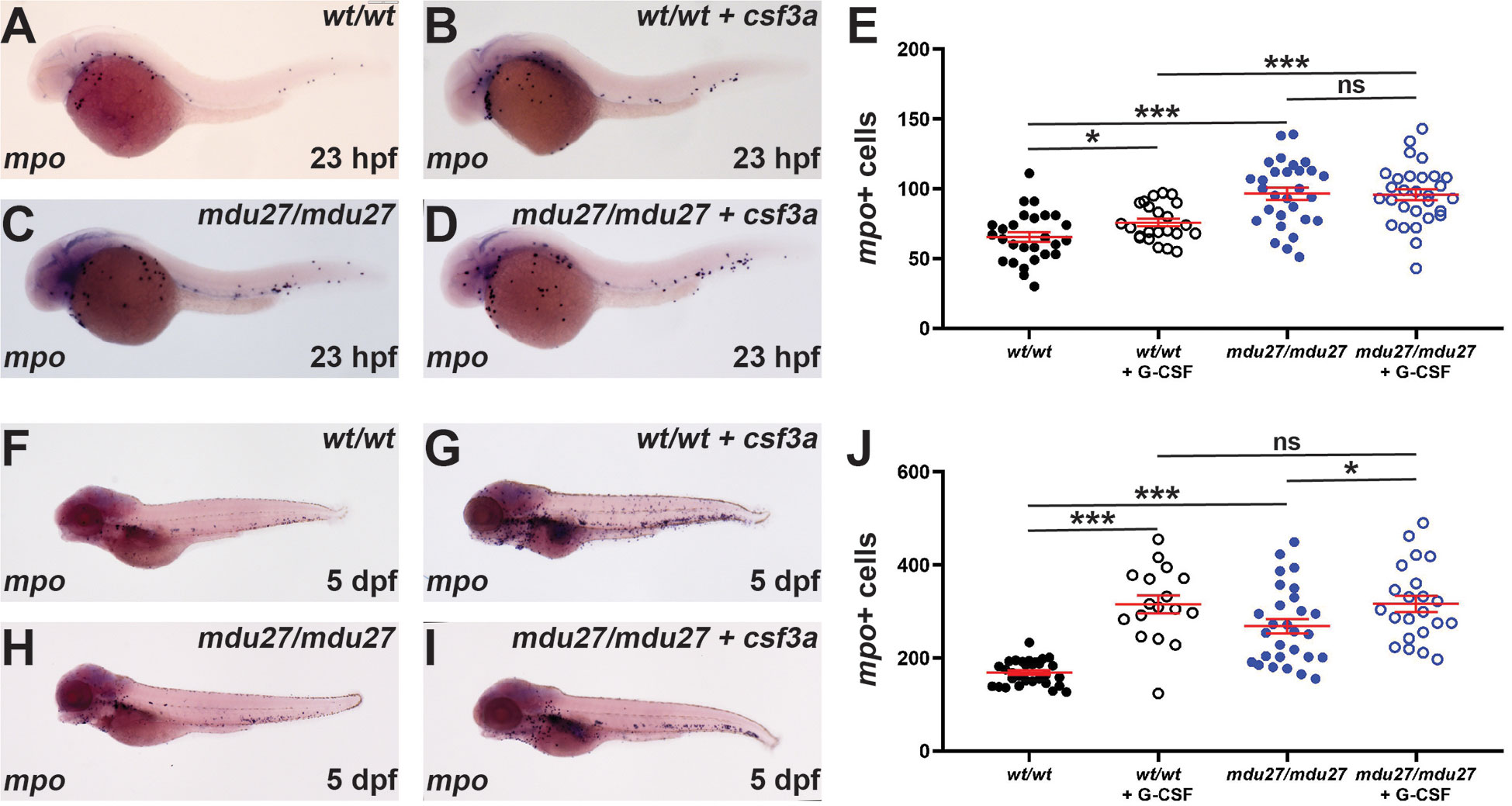
Figure 5 Effect of G-CSFR truncation mutation on emergency hematopoiesis. Wild-type (wt/wt) and homozygous (mdu27/mdu27) mutant csf3r embryos, either uninjected or injected with mRNA encoding G-CSF (+ csf3a) were subjected to WISH with mpo during primitive (A–D) and early definitive (F–I) hematopoiesis, with representative images shown. Individual embryos were assessed for the number of mpo+ cells for primitive (E) and definitive (J) hematopoiesis, with mean and SEM shown in red and level of statistical significance indicated (***: p < 0.001, *: p < 0.05, ns: not significant; n=17-30).
4. Discussion
The G-CSFR has a pivotal function in the regulation of neutrophil production and function, as well as hematopoietic stem cell mobilization, particularly in ‘emergency’ situations (1, 2). Mutations that render the G-CSFR non-functional have been shown to result in profound neutropenia (45–47), which has been verified in mice (19, 48). In contrast, mutations that enhance G-CSFR signaling are associated with leukemias and other myeloproliferative disorders (11, 12). These include acquired truncation mutations first identified in SCN patients with a strong predisposition to the development of AML (5, 49), but also described in de novo and recurrent AML, as well as CNL, aCML and CMML patients (8–14). These mutations serve to truncate the G-CSFR intracellular domain, resulting in hyperresponsiveness to G-CSF (5, 18, 49). Previous studies have shown zebrafish possess a conserved G-CSFR, including key residues critical for intracellular signaling (3), with inactivating mutations of zebrafish G-CSFR able to reproduce the sustained neutrophil deficiency observed in SCN (25, 26). This study aimed to create truncating G-CSFR mutations in zebrafish using CRISPR-Cas9-mediated genome editing and to characterize their impact on primitive, definitive and emergency hematopoiesis.
Three mouse models of truncated ‘hyperresponsive’ G-CSFR mutants have been generated. One in which the endogenous mouse gene was mutated (19) and another in which a truncated human G-CSFR was expressed transgenically (21) showed decreased peripheral neutrophils, although the bone marrow contained normal neutrophil numbers and an elevated number of immature myeloid cells. In contrast, an alternate mouse model with targeted mutation of the endogenous gene showed normal circulating neutrophil numbers, but elevation of band and segmented neutrophils as well as progenitors in the bone marrow (20). The results in zebrafish adults are consistent with this last mouse model, with the number of peripheral neutrophils not significantly different between genotypes, but those in the kidney marrow exhibiting reduced maturation. This zebrafish study has allowed for the first time to assess the effect of hyperresponsive truncating G-CSFR mutations on primitive and early definitive hematopoiesis. Strikingly, during primitive hematopoiesis, neutrophil numbers were elevated. While this is clearly different to what is observed in adult mice and zebrafish, it should be noted that G-CSF/G-CSFR signaling in zebrafish has been shown to be particularly important for primitive neutrophil production, with ablation of either leading to an almost complete loss of this population (3, 26). This developmental stage is therefore perhaps more comparable to the situation when adult mice are injected with G-CSF, when significant increases in neutrophils are observed. In contrast, during early definitive zebrafish hematopoiesis the effects of G-CSFR truncation were less marked, consistent with previous studies showing the G-CSFR ablation has a more modest impact on zebrafish neutrophils during this phase (3, 26). All mouse models noted hyperresponsive to exogenous G-CSF resulting in increased production of cells along the neutrophil lineage (19–21). This collectively illustrates the complex regulation of neutrophil production mediated by G-CSFR.
Truncated G-CSFR mutants have been described extensively as exerting a ‘dominant-negative’ effect on wild-type receptors (5). However, the data presented here indicated that the truncating csf3r allele was not strictly dominant over the wild-type allele. Notably, heterozygous mutants were similar to homozygous mutants with respect to primitive hematopoiesis, but more like wild-type fish with respect to early definitive hematopoiesis and variable in adults. Moreover, close examination reveals that this is also the case in other studies. For example, while heterozygous and homozygous mutant mice showed equivalent initial responses to G-CSF injection, these were more sustained in homozygote animals (50). In other studies, cells expressing both normal and truncated G-CSFR exhibited intermediate phenotypes or responses, including with respect to sustained signaling and impaired internalization (17, 50). Thus, truncating G-CSFR mutations should be more accurately described as ‘co-dominant’. Indeed, there are good biochemical rationale as to why this may be. Firstly, since functional receptor complexes consist of multimers, a relevant cell in a heterozygous individual will express a mixture of receptor forms on their surface, a proportion of which will contain only wild-type receptors able to signal normally. Secondly, those receptor complexes that contain at least one wild-type receptor chain will still interact with the normal regulatory machinery. Thirdly, the enhanced signaling in heterozygote cells is likely associated with increased expression of SOCS3, a key negative regulator of the G-CSFR (51), enhancing its ability to act on those receptor complexes carrying a wild-type G-CSFR chain, since it retains the requisite SOCS3 binding site (52).
Zebrafish expressing truncated G-CSFRs did not exhibit overt leukemia, which is also true of mouse models (19–21), confirming that co-operating genes are absolutely required. In patients who have acquired AML subsequent to SCN, a number of common genetic lesions have been identified, including partial or complete loss of chromosome 7 and activating RAS mutations (53, 54). Inactivating mutations in RUNX1 (55, 56) and CEBPA (57) as well as the PML-RARa oncoprotein fusion (58) have been reported to cooperate with truncated hyperresponsive G-CSFR mutations. In addition, STAT5, which shows particularly strong sustained activation by truncated G-CSFRs (17), has been implicated in mediating the effects of truncated G-CSFRs (59). The zebrafish model that was generated in this study will be highly valuable in analyzing additional co-operating genes.
A range of other G-CSFR mutations have been identified that are associated a variety of leukemias and myeloproliferative disorders (5). These include activating mutations such as T618I in CNL and related disorders (11, 55, 60), and a E785K polymorphism associated with MDS (61). These exert distinctive pathological effects, activate different (and sometimes overlapping) pathways (11, 60), and have been demonstrated to respond differentially to pharmacological agents. For example, the JAK2 inhibitor ruxolitinib has been shown to decrease proliferative signaling and neutrophil count in patients with G-CSFR-T6181 mutations whereas the SRC inhibitor dasatinib (but not ruxolitinib) was able to selectively inhibit proliferation of cells expressing truncated G-CSFRs (11). Analyzing the array of pathogenic G-CSFR mutants in zebrafish provides an opportunity to explore the specificity of their action, downstream pathways and therapeutic sensitivity in the context of a whole animal, which is an exciting prospect. Finally, the truncating mutations often occur concurrently with these (and other) G-CSFR mutations (11, 55, 62), which could also be investigated in zebrafish.
This study has successfully created and characterized a zebrafish model of hyperresponsive G-CSFR truncations. Significantly enhanced neutrophil production was observed throughout successive waves of embryonic hematopoiesis, but a defect in neutrophil maturation was also evident in adult kidney marrow. The G-CSFR truncation also displayed partial dominance. Collectively, this has shed new light on the impact of such mutations across the lifespan and generated a bone fide zebrafish model to facilitate further clinically-relevant studies to explore the role of co-operating mutations and genes, as well as downstream effector pathways that may inform new approaches to therapy, which could be readily trialed in this model.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by Deakin University Animal Ethics Committee.
Author contributions
AW conceived the project. FB and VB generated the zebrafish G-CSFR mutants and VB and MG performed most of the in vivo and in vitro characterization. AW and CL co-supervised VB, MG and FB and contributed to on-going experimental design. All authors contributed to the article and approved the submitted version.
Funding
The authors recognize the support of an International Research Scholarship (FB) and project costs from Deakin University. The funder had no role in study design, data collection and interpretation or manuscript preparation.
Acknowledgments
The authors would like to thank the Deakin University Animal House staff for superb aquarium management and Somayyeh Heidary for technical expertise.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1095453/full#supplementary-material
Abbreviations
aCML, atypical chronic myeloid leukemia; AML, acute myeloid leukemia; Cas9, CRISPR-associated 9; CMML, chronic myelomonocytic leukemia; CNL, chronic neutrophilic leukemia; CRISPR, clustered regularly interspaced short palindromic repeats; CSF3(R), colony-stimulating factor 3 (receptor); dpf, days post fertilization; FSC, forward scatter; G-CSF(R), granulocyte colony-stimulating factor (receptor); GFP, green fluorescent protein; gRNA, guide RNA; hpf, hours post fertilization; HRM, high resolution melt; MDS, myelodysplastic syndrome; SCN, severe chronic neutropenia; SSC, side scatter; WISH, whole mount in situ hybridization; Y, tyrosine.
References
1. Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and 'emergency' hematopoiesis. Cytokine. (2008) 42(3):277–88. doi: 10.1016/j.cyto.2008.03.002
2. Liongue C, Wright C, Russell AP, Ward AC. Granulocyte colony-stimulating factor receptor: stimulating granulopoiesis and much more. Int J Biochem Cell Biol (2009) 41:2372–5. doi: 10.1016/j.biocel.2009.08.011
3. Liongue C, Hall C, O'Connell B, Crozier P, Ward AC. Zebrafish granulocyte colony-stimulating factor receptor signalling promotes myelopoiesis and myeloid cell migration. Blood. (2009) 113:2535–46. doi: 10.1182/blood-2008-07-171967
4. Dale DC, Bolyard AA, Schwinzer BG, Pracht G, Bonilla MA, Boxer LA, et al. The severe congenital neutropenia international registry: 10-year follow-up report. Support Cancer Ther (2006) 3:220–31. doi: 10.3816/SCT.2006.n.020
5. Liongue C, Ward AC. Granulocyte colony-stimulating factor receptor mutations in myeloid malignancy. Front Oncol (2014) 4:93. doi: 10.3389/fonc.2014.00093
6. Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: results of a long-term survey. Blood. (2007) 109:93–9. doi: 10.1182/blood-2006-02-004275
7. Touw IP, Palande K, Beekman R. Granulocyte colony-stimulating factor receptor signaling: implications for G-CSF responses and leukemic progression in severe congenital neutropenia. Hematology/oncology Clinics North America. (2013) 27(1):61–73. doi: 10.1016/j.hoc.2012.10.002
8. Maxson JE, Ries RE, Wang YC, Gerbing RB, Kolb EA, Thompson SL, et al. CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood. (2016) 127(24):3094–8. doi: 10.1182/blood-2016-04-709899
9. Zhang H, Reister Schultz A, Luty S, Rofelty A, Su Y, Means S, et al. Characterization of the leukemogenic potential of distal cytoplasmic CSF3R truncation and missense mutations. Leukemia. (2017) 31(12):2752–60. doi: 10.1038/leu.2017.126
10. Zhang Y, Wang F, Chen X, Zhang Y, Wang M, Liu H, et al. CSF3R mutations are frequently associated with abnormalities of RUNX1, CBFB, CEBPA, and NPM1 genes in acute myeloid leukemia. Cancer. (2018) 124(16):3329–38. doi: 10.1002/cncr.31586
11. Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med (2013) 368(19):1781–90. doi: 10.1056/NEJMoa1214514
12. Beekman R, Valkhof M, van Strien P, Valk PJ, Touw IP. Prevalence of a new auto-activating colony stimulating factor 3 receptor mutation (CSF3R-T595I) in acute myeloid leukemia and severe congenital neutropenia. Haematologica. (2013) 98(5):e62–3. doi: 10.3324/haematol.2013.085050
13. Sano H, Ohki K, Park MJ, Shiba N, Hara Y, Sotomatsu M, et al. CSF3R and CALR mutations in paediatric myeloid disorders and the association of CSF3R mutations with translocations, including t(8; 21). Br J Haematol (2015) 170(3):391–7. doi: 10.1111/bjh.13439
14. Adam FC, Szybinski J, Halter JP, Cantoni N, Wenzel F, Leonards K, et al. Co-Occurring CSF3R W791* germline and somatic T618I driver mutations induce early CNL and clonal progression to mixed phenotype acute leukemia. Curr Oncol (2022) 29(2):805–15. doi: 10.3390/curroncol29020068
15. Maxson JE, Tyner JW. Genomics of chronic neutrophilic leukemia. Blood. (2017) 129(6):715–22. doi: 10.1182/blood-2016-10-695981
16. Hunter MG, Avalos BR. Deletion of a critical internalization domain in the G-CSFR in acute myelogenous leukemia preceded by severe congenital neutropenia. Blood. (1999) 93:440–6. doi: 10.1182/blood.V93.2.440
17. Ward AC, van Aesch YM, Schelen AM, Touw IP. Defective internalization and sustained activation of truncated granulocyte colony-stimulating factor receptor found in severe congenital neutropenia/acute myeloid leukemia. Blood. (1999) 93:447–58. doi: 10.1182/blood.V93.2.447
18. Maxson JE, Luty SB, MacManiman JD, Abel ML, Druker BJ, Tyner JW. Ligand independence of the T618I mutation in the colony-stimulating factor 3 receptor (CSF3R) protein results from loss of O-linked glycosylation and increased receptor dimerization. J Biol Chem (2014) 289(9):5820–7. doi: 10.1074/jbc.M113.508440
19. Hermans MHA, Ward AC, Antonissen C, Karis A, Lowenberg B, Touw IP. Perturbed granulopoiesis in mice with a targeted mutation in the granulocyte colony-stimulating factor receptor gene associated with severe chronic neutropenia. Blood. (1998) 92:32–9. doi: 10.1182/blood.V92.1.32.413k42_32_39
20. McLemore ML, Poursine-Laurent J, Link DC. Increased granulocyte colony-stimulating factor responsiveness but normal resting granulopoiesis in mice carrying a targeted granulocyte colony-stimulating factor receptor mutation derived from a patient with severe congenital neutropenia. J Clin Invest. (1998) 102:483–92. doi: 10.1172/JCI3216
21. Mitsui T, Watanabe S, Taniguchi Y, Hanada S, Ebihara Y, Sato T, et al. Impaired neutrophil maturation in truncated murine G-CSF receptor transgenic mice. Blood. (2003) 101:2990–5. doi: 10.1182/blood.V101.8.2990
22. Rasighaemi P, Basheer F, Liongue C, Ward AC. Zebrafish as a model for leukemia and other hematopoietic disorders. J Hematol Oncol (2015) 8:29. doi: 10.1186/s13045-015-0126-4
23. Chen AT, Zon LI. Zebrafish blood stem cells. J Cell Biochem (2009) 108(1):35–42. doi: 10.1002/jcb.22251
24. Xu J, Du L, Wen Z. Myelopoiesis during zebrafish early development. J Genet Genomics (2012) 39(9):435–42. doi: 10.1016/j.jgg.2012.06.005
25. Pazhakh V, Clark S, Keightley MC, Lieschke GJ. A GCSFR/CSF3R zebrafish mutant models the persistent basal neutrophil deficiency of severe congenital neutropenia. Sci Rep (2017) 7:44455. doi: 10.1038/srep44455
26. Basheer F, Rasighaemi P, Liongue C, Ward AC. Zebrafish granulocyte colony-stimulating factor receptor maintains neutrophil number and function throughout the life span. Infect Immun (2019) 87(2):e00793–18. doi: 10.1128/IAI.00793-18
27. Renshaw SA, Loynes CA, Trushell DM, Elworthy S, Ingham PW, Whyte MK. A transgenic zebrafish model of neutrophilic inflammation. Blood. (2006) 108(13):3976–8. doi: 10.1182/blood-2006-05-024075
28. Lawrence C. The husbandry of zebrafish (Danio rerio): a review. Aquaculture. (2007) 269(1-4):1–20. doi: 10.1016/j.aquaculture.2007.04.077
29. Garritano S, Gemignani F, Voegele C, Nguyen-Dumont T, Le Calvez-Kelm F, De Silva D, et al. Determining the effectiveness of high resolution melting analysis for SNP genotyping and mutation scanning at the TP53 locus. BMC Genet (2009) 10:5. doi: 10.1186/1471-2156-10-5
30. Meier AB, Basheer F, Sertori R, Laird M, Liongue C, Ward AC. Granulocyte colony-stimulating factor mediated regulation of early myeloid cells in zebrafish. Front Biosci (Landmark Ed). (2022) 27(4):110. doi: 10.31083/j.fbl2704110
31. Stachura DL, Svoboda O, Campbell CA, Espin-Palazon R, Lau RP, Zon LI, et al. The zebrafish granulocyte colony-stimulating factors (Gcsfs): 2 paralogous cytokines and their roles in hematopoietic development and maintenance. Blood. (2013) 122(24):3918–28. doi: 10.1182/blood-2012-12-475392
32. Schulte-Merker S, Ho RK, Herrmann BG, Nusslein-Volhard C. The protein product of the zebrafish homologue of the mouse T gene is expressed in nuclei of the germ ring and the notochord of the early embryo. Development. (1992) 116(4):1021–32. doi: 10.1242/dev.116.4.1021
33. Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc (2008) 3(1):59–69. doi: 10.1038/nprot.2007.514
34. Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. (2001) 25(4):386–401. doi: 10.1006/meth.2001.1261
35. Sertori R, Trengove M, Basheer F, Ward AC, Liongue C. Genome editing in zebrafish: a practical overview. Brief Funct Genomics (2016) 15(4):322–30. doi: 10.1093/bfgp/elv051
36. Karousis ED, Muhlemann O. The broader sense of nonsense. Trends Biochem Sci (2022) 47(11):921–35. doi: 10.1016/j.tibs.2022.06.003
37. Jagannathan-Bogdan M, Zon LI. Hematopoiesis. Development. (2013) 140(12):2463–7. doi: 10.1242/dev.083147
38. Bennett CM, Kanki JP, Rhodes J, Liu TX, Paw BH, Kieran MW, et al. Myelopoiesis in the zebrafish, danio rerio. Blood. (2001) 98(3):643–51. doi: 10.1182/blood.V98.3.643
39. Lieschke GJ, Oates AC, Paw BH, Thompson MA, Hall NE, Ward AC, et al. Zebrafish SPI-1(PU.1) marks a site of myeloid development independent of primitive erythropoiesis: implications for axial patterning. Dev Biol (2002) 246(2):274–95. doi: 10.1006/dbio.2002.0657
40. Long Q, Meng A, Wang H, Jessen JR, Farrell MJ, Lin S. GATA-1 expression pattern can be recapitulated in living transgenic zebrafish using GFP reporter gene. Development. (1997) 124(20):4105–11. doi: 10.1242/dev.124.20.4105
41. Ellett F, Pase L, Hayman JW, Andrianopoulos A, Lieschke GJ. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood. (2011) 117(4):E49–56. doi: 10.1182/blood-2010-10-314120
42. Willett CE, Cherry JJ, Steiner L. Characterization and expression of the recombination activating genes (rag1 and rag2) of zebrafish. Immunogenetics. (1997) 45(6):394–404. doi: 10.1007/s002510050221
43. Brownlie A, Hersey C, Oates AC, Paw BH, Falick AM, Witkowska HE, et al. Characterization of embryonic globin genes of the zebrafish. Dev Biol (2003) 255(1):48–61. doi: 10.1016/S0012-1606(02)00041-6
44. Dale DC. How I diagnose and treat neutropenia. Curr Opin Hematol (2016) 23(1):1–4. doi: 10.1097/MOH.0000000000000208
45. Ward AC, van Aesch YM, Gits J, Schelen AM, de Koning JP, van Leeuwen D, et al. Novel point mutation in the extracellular domain of the granulocyte colony-stimulating factor (G-CSF) receptor in a case of severe congenital neutropenia hyporesponsive to G-CSF treatment. J Exp Med (1999) 190:497–507. doi: 10.1084/jem.190.4.497
46. Triot A, Jarvinen PM, Arostegui JI, Murugan D, Kohistani N, Dapena Diaz JL, et al. Inherited biallelic CSF3R mutations in severe congenital neutropenia. Blood. (2014) 123(24):3811–7. doi: 10.1182/blood-2013-11-535419
47. Sinha S, Zhu QS, Romero G, Corey SJ. Deletional mutation of the external domain of the human granulocyte colony-stimulating factor receptor in a patient with severe chronic neutropenia refractory to granulocyte colony-stimulating factor. J Pediatr Hematol Oncol (2003) 25(10):791–6. doi: 10.1097/00043426-200310000-00010
48. Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity. (1996) 5:491–501. doi: 10.1016/S1074-7613(00)80504-X
49. Dwivedi P, Greis KD. Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Exp Hematol (2017) 46:9–20. doi: 10.1016/j.exphem.2016.10.008
50. Hermans MHA, Antonissen C, Ward AC, Mayen AEM, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med (1999) 189:683–92. doi: 10.1084/jem.189.4.683
51. Kimura A, Kinjyo I, Matsumura Y, Mori H, Mashima R, Harada M, et al. SOCS3 is a physiological negative regulator for granulopoiesis and granulocyte colony-stimulating factor receptor signaling. J Biol Chem (2004) 279:6905–10. doi: 10.1074/jbc.C300496200
52. van de Geijn GJ, Gits J, Aarts LH, Heijmans-Antonissen C, Touw IP. G-CSF receptor truncations found in SCN/AML relieve SOCS3-controlled inhibition of STAT5 but leave suppression of STAT3 intact. Blood. (2004) 104:667–74. doi: 10.1182/blood-2003-08-2913
53. Kalra R, Dale D, Freedman M, Bonilla MA, Weinblatt M, Ganser A, et al. Monosomy 7 and activating RAS mutations accompany malignant transformation in patients with congenital neutropenia. Blood. (1995) 86:4579–86. doi: 10.1182/blood.V86.12.4579.bloodjournal86124579
54. Freedman MH, Alter BP. Risk of myelodysplastic syndrome and acute myeloid leukemia in congenital neutropenias. Semin Hematol (2002) 39(2):128–33. doi: 10.1053/shem.2002.31912
55. Beekman R, Valkhof MG, Sanders MA, van Strien PM, Haanstra JR, Broeders L, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood. (2012) 119(22):5071–7. doi: 10.1182/blood-2012-01-406116
56. Skokowa J, Steinemann D, Katsman-Kuipers JE, Zeidler C, Klimenkova O, Klimiankou M, et al. Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood. (2014) 123(14):2229–37. doi: 10.1182/blood-2013-11-538025
57. Lavallee VP, Krosl J, Lemieux S, Boucher G, Gendron P, Pabst C, et al. Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood. (2016) 127(24):3054–61. doi: 10.1182/blood-2016-03-705053
58. Kunter G, Woloszynek JR, Link DC. A truncation mutant of Csf3r cooperates with PML-RARalpha to induce acute myeloid leukemia in mice. Exp Hematol (2011) 39(12):1136–43. doi: 10.1016/j.exphem.2011.08.013
59. Liu F, Kunter G, Krem MM, Eades WC, Cain JC, Tomasson MH, et al. Csf3r mutations in mice confer a strong clonal HSC advantage via activation of Stat5. J Clin Invest. (2008) 118:946–55. doi: 10.1172/JCI32704
60. Mehta HM, Glaubach T, Long A, Lu H, Przychodzen B, Makishima H, et al. Granulocyte colony-stimulating factor receptor T595I (T618I) mutation confers ligand independence and enhanced signaling. Leukemia. (2013) 27(12):2407–10. doi: 10.1038/leu.2013.164
61. Wolfler A, Erkeland SJ, Bodner C, Valkhof M, Renner W, Leitner C, et al. A functional single-nucleotide polymorphism of the G-CSF receptor gene predisposes individuals to high-risk myelodysplastic syndrome. Blood. (2005) 105(9):3731–6. doi: 10.1182/blood-2004-06-2094
Keywords: cytokine receptors, G-CSFR, leukemia, neutropenia, zebrafish
Citation: Bulleeraz V, Goy M, Basheer F, Liongue C and Ward AC (2023) Leukemia-associated truncation of granulocyte colony-stimulating factor receptor impacts granulopoiesis throughout the life-course. Front. Immunol. 13:1095453. doi: 10.3389/fimmu.2022.1095453
Received: 11 November 2022; Accepted: 20 December 2022;
Published: 10 January 2023.
Edited by:
Sabyasachi Das, Emory University, United StatesReviewed by:
Ivo Paul Touw, Erasmus University Rotterdam, NetherlandsSeth Corey, Cleveland Clinic, United States
Copyright © 2023 Bulleeraz, Goy, Basheer, Liongue and Ward. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alister C. Ward, YXdhcmRAZGVha2luLmVkdS5hdQ==