 Stephen C. Frederico
Stephen C. Frederico Xiaoran Zhang2
Xiaoran Zhang2 Baoli Hu
Baoli Hu Gary Kohanbash
Gary Kohanbash
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 09 January 2023
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1092399
This article is part of the Research Topic Immune Microenvironment and Immunotherapy in Malignant Brain Tumors View all 23 articles
Gliomas have an extremely poor prognosis in both adult and pediatric patient populations as these tumors are known to grow aggressively and respond poorly to standard of care treatment. Currently, treatment for gliomas involves surgical resection followed by chemoradiation therapy. However, some gliomas, such as diffuse midline glioma, have more limited treatment options such as radiotherapy alone. Even with these interventions, the prognosis for those diagnosed with a glioma remains poor. Immunotherapy is highly effective for some cancers and there is great interest in the development of effective immunotherapies for the treatment of gliomas. Clinical trials evaluating the efficacy of immunotherapies targeted to gliomas have largely failed to date, and we believe this is partially due to the poor choice in pre-clinical mouse models that are used to evaluate these immunotherapies. A key consideration in evaluating new immunotherapies is the selection of pre-clinical models that mimic the glioma-immune response in humans. Multiple pre-clinical options are currently available, each one with their own benefits and limitations. Informed selection of pre-clinical models for testing can facilitate translation of more promising immunotherapies in the clinical setting. In this review we plan to present glioma cell lines and mouse models, as well as alternatives to mouse models, that are available for pre-clinical glioma immunotherapy studies. We plan to discuss considerations of model selection that should be made for future studies as we hope this review can serve as a guide for investigators as they choose which model is best suited for their study.
In adults, malignant brain tumors account for approximately one-third of all CNS tumors with glioblastoma (GBM) and diffuse low-grade gliomas (LGG) being the most common subtypes (1). In children, brain tumors are the most common form of solid malignancy and account for the majority of cancer mortality (1, 2). Brainstem tumors account for 10% of all pediatric tumors within the CNS with diffuse midline glioma (DMG) being the most common subtype. The prognosis for patients diagnosed with DMG is extremely poor as greater than 90% of patients die within 2 years of their initial diagnosis (2, 3). Typical treatment of malignant gliomas involves surgical resection (in surgically accessible tumors), as well as chemotherapy and radiation therapy in lesions that are deemed higher risk (4). Unfortunately, outcomes remain poor despite this multi-modal approach and there is a dire need for new therapeutic modalities (4–6).
Neoplastic cells are constantly generated throughout a person’s lifetime, most of which are inevitably removed by the host immune system through anti-tumor immunity. The few neoplastic cells that manage to escape anti-tumor immunity eventually become a tumor (7). The concept of immunotherapy is the promotion of immune recognition, activation, and elimination of neoplastic cells. Immunotherapy in the form of immune checkpoint inhibitors (ICI) have radically transformed the treatment paradigm of cancer. ICIs are able to induce dramatic and durable response in many solid tumors and have now become the first-line treatment for the treatment of melanoma, colorectal cancer, and non-small cell lung cancer (8). Other immunotherapy approaches include adoptive cell transfer, cytokine/chemokine-based therapies, and tumor vaccination. Given the lack of effective therapies in malignant gliomas and the effectiveness of immunotherapy for other solid malignancies, immunotherapy for malignant gliomas has become an area of great interest.
Pre-clinical studies in animal models of malignant gliomas have yielded many promising immunotherapy candidates, many of which have eventually failed in clinical trials (9, 10). This discrepancy between pre-clinical and clinical results, points to the failure of pre-clinical models of malignant gliomas at recapitulating the tumor immune cell interactions within the tumor microenvironment. Many pre-clinical options are currently available, each one with their own benefits and limitations. Informed selection of pre-clinical models for testing can facilitate translation of more promising immunotherapies in the clinical setting. Here, we review commonly used and recently developed glioma cell lines, mouse models, as well as alternative animal models, in an effort to highlight which of these may be best suited for immunotherapy studies.
Immunotherapy is a catch-all term that includes a wide variety of approaches of manipulating the host immune system to eliminate cancer. As such, there is no one perfect pre-clinical model to evaluate the different immunotherapy approaches in gliomas. We propose several key considerations that investigators should take when selecting pre-clinical models of glioma for evaluation of immunotherapy.
The first and perhaps the most impactful decision the investigator has to make is the origin of the tumor. They can be from the same species as the model animal (allogeneic) or patient-derived (xenograft). Allogeneic tumors can be generated from spontaneously occurring tumors, carcinogen mutagenesis, genetic engineering, and transposon mutagenesis. Xenograft tumors are patient-derived cell lines and cancer stem cells (CSCs). Allogeneic tumors can be implanted on immunocompetent mice whereas xenograft models can only be implanted in immunocompromised or humanized mice.
Therapeutic approaches such as CAR-T and tumor vaccines require that animal models express some of the same tumor neo-antigens as the human tumor. In this respect, genetically engineered mouse models (GEMMs) of gliomas are not always the best choice. GEMM does a good job at recapitulating driver mutations, however, these tumors poorly express tumor neoantigens that are expressed by gliomas, limiting the usefulness of this model when evaluating immune therapies.
For some tumors residing outside the CNS, it has been observed that tumors having a higher mutational burden are better candidates for immunotherapy. This higher mutational burden often leads to the production of more tumor neoantigens which can be targeted by the immune system. This observation has been made in colorectal cancer, as well as other cancers outside the CNS (11, 12). However, the opposite has been observed in gliomas, where a higher tumor mutational burden is often associated with worse survival (12, 13). These findings highlight the need for paying close attention to tumor mutational burden when choosing a pre-clinical model for glioma immunotherapy studies as models having very high tumor mutational burden may not be best suited for immunotherapy studies.
Gliomas are known as aggressive malignancies that are known to grow quickly. It has been reported that GBM specifically has a median specific growth rate of 1.4% per day, with an equivalent volume doubling time of 49.6 days (14). Choosing a pre-clinical model that has a high growth rate is crucial for immunotherapy studies as these tumors grow quickly in patients. Additionally, GBM as well as other gliomas, grow in an infiltrative manner unlike most CNS tumors (15). Given these findings, it is crucial for investigators performing glioma immunotherapy studies to choose pre-clinical models that possess high growth rates and closely parallel glioma growth patterns as this will best replicate what is observed in patients.
GL261 is an allogeneic tumor cell line that was originally created by intracranially injecting C57BL/6 mice with a known carcinogen, that being, 3-methylcholantrene (16). Small pieces of the tumor were taken and subjected to serial passaging over time which is believed to be one of the reasons that GL261 lacks important glial differentiation markers (17). The growth of intracranial GL261 tumors has been described in the literature as rapid with a slightly invasive growth pattern. Additionally, it has been noted that lymphocyte infiltration is extremely low in these tumors. Szatmári and colleagues found that after intracranially implanting 1 × 105, 1 × 104, 1 × 103 and 1 × 102 GL261 cells into immunocompetent mice, the mean survival time was 25, 27, 36 and 55 days respectively (16). It has been noted that a higher level of MHC1 antigens can be detected in wildtype GL261 tumors when compared to healthy brain, and it has been noted that MHC1 is upregulated in cells exposed to interferon-gamma (16). Compared to other tumor lines, GL261 has a higher mutational burden as whole exome sequencing has shown in vitro GL261 to have 212 frameshift and 4766 missense mutations (18). In the same study, it was shown that in vitro SB28 had 67 frameshift and 41 missense mutations (18). Commonly, GL261 cells are administered to mice via intracranial injection, but these tumors can also be grown in the subcutaneous space by injecting mice with GL261 cells in the flank.
After H. Fraser and colleagues observed the first incidences of mice developing spontaneous gliomas, Serano and colleagues developed the SMA-560 cell line after performing a serial transplantation of spontaneous murine astrocytoma (19). Specifically, tumor tissue underwent homogenization, in vitro culturing, and subsequent transplantation into VM/Dk mice (19). The median survival for animals bearing SMA-560 tumors following injection with 1×104 tumor cells/5 μl has been reported to be approximately 26 days (20). Notably, SMA-560 has high expression of glial fibrillary acid protein (GFAP) and the astrocyte marker glutamine synthetase, and low expression of S-100 proteins (21, 22). Additionally, it has been noted that while MHC1 expression is low in SMA-560 at baseline, it is upregulated in cells exposed to interferon-gamma (23). In a study by Johanns and colleagues, it was observed that SMA-560 had 2171 non-synonymous exome mutations as compared to 4,932 for GL261 (24). SMA-560 cells can be administered to mice via intracranial injection, but these tumors can also be grown in the subcutaneous space by injecting mice with SMA-560 cells in the flank.
CT-2A is an allogeneic cell line that was generated from a malignant astrocytoma that was formed in C57BL/6J mice that were injected in the cerebrum with a known carcinogen, that being, 20-methylcholanthrene (25). These cells have a high tumorigenicity as mice have a median survival of 20 days after intracranial injection with 1x104 cells (23). This tumor is known to have high levels of complex gangliosides and very low distribution of GM3 (monosialodihexosylganglioside) which has been classified as an anti-angiogenic ganglioside (26, 27). Additionally, CT-2A cells are known to be deficient in the tumor suppressor PTEN and are wild-type for p53. These tumors have a high mitotic index and unlike many other tumors, demonstrate high levels of microvascular proliferation (25, 28). Commonly, CT-2A cells are administered to mice via intracranial injection, but these tumors can also be grown in the subcutaneous space by injecting mice with CT-2A cells in the flank.
SB28 is an allogeneic cell line that was generated by using sleeping beauty transposons to insert constructs capable of targeting the P53, RAS and PDGF pathways. These sleeping beauty transposon flanked pT2/CAG-NRasV12 and pT2/shp53/mPDGF constructs were then injected into the right ventricle of C57L/6 mice (23). It has been noted that SB28 bearing mice have extremely low MHC1 expression and limited CD8 T cell infiltration posing a challenge for immunotherapy-based studies using this cell line (18). The median overall survival for mice injected with 1x104 SB28 cells is 19 days and whole exome sequencing has demonstrated these cells have just 108 mutations as compared to the over 4900 mutations present in GL261 (18, 24). This cell line can be injected intracranially, but these tumors can also be grown in the subcutaneous space by injecting mice with SB28 cells in the flank.
U251 is a xenograft cell line that was derived from a glioblastoma multiforme using explant technique (29). These cells must be injected into immunocompromised mice and the median overall survival for tumor bearing mice is 22 days (30). It has been noted in previous studies that B7-H4 expression is upregulated in U251 glioma stem-like cells and while U251 cells do not carry an IDH1 mutation, these cells do carry mutations in hTERT, PTEN and p53 (31). Additionally, these cells have a methylated MGMT status (31). These cells must be injected into immunocompromised mice, limiting their usability in immunotherapy-based studies.
U87 is a xenograft cell line that was derived from a GBM in a female patient. Immunocompromised mice bearing U87 tumors have a median survival of 28.6 days following tumor implantation (30). Interestingly, it has been observed that U87 and U251 tumors only grow to kill their hosts when 1,000,000 or 1,500,000 cells, respectively, are injected in the striatum of nude mice (30). It has been shown that injecting less cells leads to a lack of tumor growth and avoidance of death of the host. It has been noted that U87 cells possess hTERT, ATRX and PTEN mutations, however, these cells do not carry p53 or IDH1 mutations (31). Additionally, these cells have a methylated MGMT status (32). These cells must be injected into immunocompromised mice and can be injected intracranially or into the flank region.
QPP is an immunocompetent murine spontaneous GBM model, in which three common patient-relevant tumor suppressor genes, Quaking (Qk in mouse and QKI in human), Trp53, and PTEN, were deleted (33). The tumors that were derived from this model displayed histopathological and transcriptomic heterogeneity, which can manifest the subtypes of GBM (33). The cell line QPP7, isolated from this model, was used to establish the syngeneic orthotopic glioma in C57L/6 mice with genetic manipulations in previous research (34). Importantly, this syngeneic mouse glioma demonstrated the landscape of the tumor immune microenvironment, including M1/M2-like macrophages, T cells, NK cells, and myeloid-derived suppressor cells (MDSCs) (34). Based on immune profiling and single-cell sequencing analyses, a most recent study reported that both implanted and spontaneous QPP models recapitulate the immunosuppressive myeloid dominant nature of the tumor microenvironment of human gliomas (35).
The Cre-LoxP system allows for the targeting of tumor genes in mouse brain tissue of interest which provides for insight into the genetic drivers of GBM and the differences in genetic drivers between primary and secondary GBMs (36). To create this model, a mouse strain known as the “Cre driver strain” which has Cre recombinase with a promoter, and a mouse strain known as the “LoxP floxed strain” that has LoxP floxed exons of the target gene, were bred together (36). By breeding these strains together this would result in a deletion of the floxed region and a subsequent inactivation of the gene in the desired brain tissue of interest, leaving the target gene active in tissues outside this region (36). Specifically, this model has been used to test the role of p53 and PTEN function in GFAP positive GBM. In a study by Zheng and colleagues, the research team created a p53 and PTEN double knockout mouse where this knockout was targeted to astrocytes specifically (37). The research team found that a loss of both p53 and PTEN would regulate Myc levels and subsequently control NSC self-renewal and differentiation (37, 38). The Cre-LoxP mouse model is extremely valuable for testing immunotherapy applications as this model can activate or inactivate genes that can impact the tumor microenvironment, and this system has also been used to control the cytotoxic potential of CAR-T cells (39).
The HGMM model was developed by Huang and colleagues to better understand the role of CCL18 which is expressed in humans but not in rodents (40). Specifically, the research team developed this model to study the interaction of human glioma cells with human microglia. To create this model, the research team depleted intrinsic microglia from murine organotypic brain slices and then injected either human glioma cells into these slices, or injected both human glioma cells and human stem cell derived microglial cells into these slices (40). Interestingly, the research team found that in slices injected with both human glioma cells and human stem cell derived microglial cells, human stem cell derived microglial cells showed higher levels of sphericity and cell body volume highlighting how the tumor microenvironment impacts the morphology of these cells (40). Additionally, the research team assessed whether the presence of human stem cell derived microglial cells with human glioma cells created an increase in the expression level of genes known to be upregulated by the glioma environment. The research team found that there was an upregulation in IL-10, Osteopontin, MMP14, VEGF, TGF β, and CCL18 in samples containing both human stem cell derived microglial cells with human glioma cells, as opposed to samples containing human glioma cells alone (40).
Ultimately, this model highlights that the presence of human stem cell derived microglial cells with human glioma cells results in not only larger tumors but also an upregulation of genes similar to those known to be upregulated in certain gliomas (40–45). Recent findings have begun to shed light on how GBM can use microglia to induce immunosuppression within the tumor microenvironment. This model will be valuable in developing a deeper understanding of this hijacking and potentially enable therapeutic exploitation of this mechanism (46).
While glioma mouse models are beneficial to use in immunotherapy studies due to their low cost and availability, these models certainly have their limitations (47–49). Some of the limitations of mouse models include the lack of a highly immunosuppressive glioma microenvironment that is commonly observed in human gliomas, and that patient derived xenografts often must be transplanted into immunocompromised rodents (47, 48, 50). These limitations along with others highlight the need for creating alternative models that can be used in pre-clinical glioma immunotherapy studies.
Drosophila melanogaster is one alternative to glioma mouse models as 75% of human genes share functional orthologs with drosophila (50, 51). This finding makes drosophila a useful model for studying gliomagenesis as gliomas can be induced in this model using the GAL4/upstream activation sequence system (50, 52, 53). Additionally, this model is valuable for studying centromere dysfunction which has been shown to lead to tumor development as a result of perturbation of stem cell division (50, 54). However, it is important to note that this model lacks an adaptive immune system and relies on humoral and cell-mediated innate immunity for its defense against pathogens, limiting this models role in immunotherapy-based studies (55–57).
Canine brain tumor models are an exciting large animal model that have recently been developed for neuro-oncology studies (58). It has been demonstrated that intracranial gliomas spontaneously arise in canines and that these tumors share similar morphological and immunological characteristics with human gliomas (50, 59). Additionally, molecular characterization of canine gliomas has shown that these tumor share similar somatic alterations that are known drivers of human gliomas such as mutations in Tp53 and IDH (60–62). Immunotherapy studies in glioma-bearing canines are limited at this time as this model is still new to the neuro-oncology space and a limited number of canines develop gliomas on an annual basis (63). However, the limited number of studies that have occurred in glioma-bearing canines have highlighted the promise associated with this model (63–66).
Danio rerio (zebrafish) is a final alternative model that should be considered for glioma immunotherapy studies as zebrafish lack an adaptive immune system until six weeks of age, allowing for the implantation of human glioma cells that lead to an invasive glioma (50, 67, 68). Additionally, this model has a similar microenvironment with regards to density, to what is observed in the human brain (50). Limitations of this model include differences in the tumor microenvironment compared to that of humans, and that the optimal temperature for human cells is 37°C compared to fish cells which is 28°C (50, 69). Recently, a zebrafish model has been developed that can engraft human tumors at 37°C (70). It will be interesting to observe whether this model can be used in future immunotherapy studies.
In this manuscript we present glioma cell lines and mouse models, as well as alternative glioma animal models that can be used in immunotherapy studies. Additionally, we discuss some of the benefits and limitations associated with these animal models (see Table 1). When choosing a model we believe it is first crucial to assess where the tumor is derived from. Specifically, some allogeneic models such as GL261, CT2A, etc. were created by administering known carcinogens to mice, which is believed to be dissimilar to how gliomas arise within humans. Models created via carcinogens do not replicate the developmental biology of gliomas to the fullest extent and as such may not be best suited for immunotherapy studies as these models do not entirely possess the immunosuppressive mechanisms observed in human gliomas (9, 10, 91).
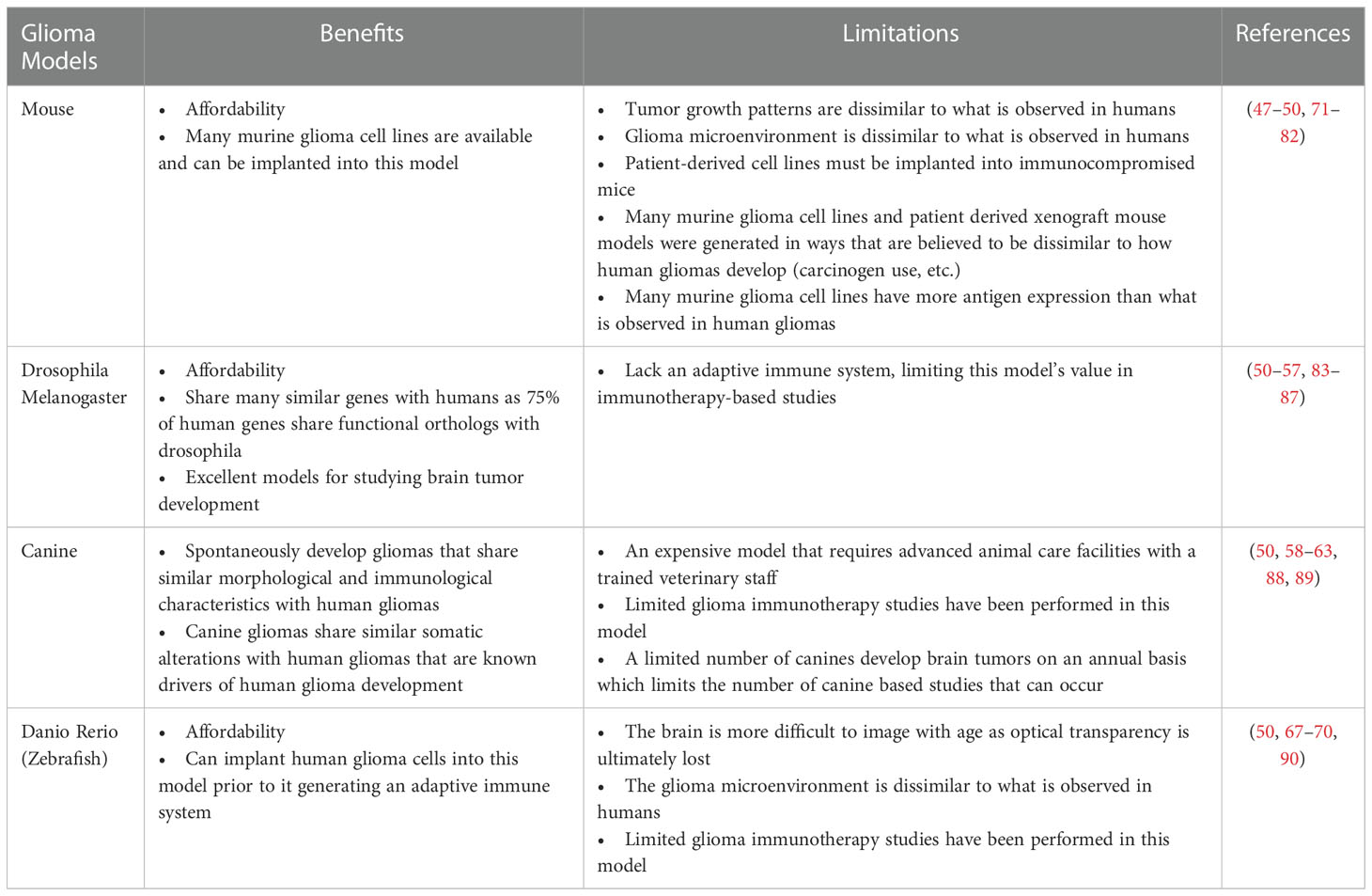
Table 1 Benefits and limitations associated with animal models of glioma.
Tumor antigen expression and mutational burden are also important considerations when choosing a brain tumor model for pre-clinical brain tumor immunotherapy studies. Many gliomas, especially GBM and DMG, are known as “immunologically cold” tumors as these malignancies express few antigens and have low mutational burdens. Many allogeneic mouse models and some patient derived xenografts such as U87 have increased antigen expression and/or tumor mutational burden. This is problematic for pre-clinical immunotherapy studies as findings showing efficacy in these pre-clinical models may largely be due to there being more antigenic targets than there should be and/or an increased mutational burden, suggesting that a therapy is fit for clinical trial when it truly is not. Canine and zebrafish models may be beneficial in future glioma immunotherapy studies as canines spontaneously generate gliomas, and zebrafish can undergo transplantation with patient derived xenografts prior to their generation of an adaptive immune system. Ultimately however, we believe that selecting a model that is both patient derived, and “immunologically cold” is crucial for future pre-clinical glioma immunotherapy studies as we believe this will help reduce the number of failed immunotherapy clinical trials observed in the neuro-oncology space.
SF, XZ, and GK contributed to the conception and design of the review. The first draft was written by SF, XZ, and BH. GK supervised the writing of the manuscript and is the corresponding author. All authors contributed to the manuscript and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
CNS, Central Nervous System; GBM, Glioblastoma Multiforme; LGG, Low-Grade Glioma; DMG, Diffuse Midline Glioma; IDH1, Isocitrate Dehydrogenase 1; IDH2, Isocitrate Dehydrogenase 2; ICI, Immune Checkpoint Inhibitors; CSC, Cancer Stem Cell; GEMM, Genetically Engineered Mouse Model; GFAP, Glial Fibrillary Acid Protein.
1. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2014-2018. Neuro Oncol (2021) 23(12 Suppl 2):iii1–iii105. doi: 10.1093/neuonc/noab200
2. Johnson KJ, Cullen J, Barnholtz-Sloan JS, Ostrom QT, Langer CE, Turner MC, et al. Childhood brain tumor epidemiology: A brain tumor epidemiology consortium review. Cancer Epidemiol Biomarkers Prev (2014) 23(12):2716–36. doi: 10.1158/1055-9965.EPI-14-0207
3. Frederico S, Sneiderman C, Pollack I, Kohanbash G. 222 developing an adoptive cell transfer immunotherapy for pediatric high-grade gliomas. J ImmunoTher Cancer (2022) 10(Suppl 2):A236–A. doi: 10.1136/jitc-2022-SITC2022.0222
4. Sharma A, Graber JJ. Overview of prognostic factors in adult gliomas. Ann Palliative Med (2020) 10(1):863–74. doi: 10.21037/apm-20-640
5. Claus EB, Walsh KM, Wiencke JK, Molinaro AM, Wiemels JL, Schildkraut JM, et al. Survival and low-grade glioma: the emergence of genetic information. Neurosurg Focus (2015) 38(1):E6. doi: 10.3171/2014.10.FOCUS12367
6. Liang J, Lv X, Lu C, Ye X, Chen X, Fu J, et al. Prognostic factors of patients with gliomas – an analysis on 335 patients with glioblastoma and other forms of gliomas. BMC Cancer (2020) 20(1):35. doi: 10.1186/s12885-019-6511-6
7. Akindona FA, Frederico SC, Hancock JC, Gilbert MR. Exploring the origin of the cancer stem cell niche and its role in anti-angiogenic treatment for glioblastoma. Front Oncol (2022) 12:947634. doi: 10.3389/fonc.2022.947634
8. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5
9. Frederico SC, Hancock JC, Brettschneider EES, Ratnam NM, Gilbert MR, Terabe M. Making a cold tumor hot: The role of vaccines in the treatment of glioblastoma. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.672508
10. Ratnam NM, Frederico SC, Gonzalez JA, Gilbert MR. Clinical correlates for immune checkpoint therapy: significance for CNS malignancies. Neuro-Oncol Advances (2020) 3(1). doi: 10.1093/noajnl/vdaa161
11. Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, Ross JS, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol (2019) 30(7):1096–103. doi: 10.1093/annonc/mdz134
12. McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol (2021) 32(5):661–72. doi: 10.1016/j.annonc.2021.02.006
13. Wang L, Ge J, Lan Y, Shi Y, Luo Y, Tan Y, et al. Tumor mutational burden is associated with poor outcomes in diffuse glioma. BMC Cancer (2020) 20(1):213. doi: 10.1186/s12885-020-6658-1
14. Stensjøen AL, Solheim O, Kvistad KA, Håberg AK, Salvesen Ø, Berntsen EM. Growth dynamics of untreated glioblastomas in vivo. Neuro Oncol (2015) 17(10):1402–11. doi: 10.1093/neuonc/nov029
15. Seker-Polat F, Pinarbasi Degirmenci N, Solaroglu I, Bagci-Onder T. Tumor cell infiltration into the brain in glioblastoma: From mechanisms to clinical perspectives. Cancers (2022) 14(2). doi: 10.3390/cancers14020443
16. Szatmári T, Lumniczky K, Désaknai S, Trajcevski S, Hídvégi EJ, Hamada H, et al. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci (2006) 97(6):546–53. doi: 10.1111/j.1349-7006.2006.00208.x
17. Weiner NE, Pyles RB, Chalk CL, Balko MG, Miller MA, Dyer CA, et al. A syngeneic mouse glioma model for study of glioblastoma therapy. J Neuropathol Exp Neurol (1999) 58(1):54–60. doi: 10.1097/00005072-199901000-00007
18. Genoud V, Marinari E, Nikolaev SI, Castle JC, Bukur V, Dietrich PY, et al. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology. (2018) 7(12):e1501137. doi: 10.1080/2162402X.2018.1501137
19. Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, et al. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Trans Med (2014) 12(1):107. doi: 10.1186/1479-5876-12-107
20. Heimberger AB, Crotty LE, Archer GE, McLendon RE, Friedman A, Dranoff G, et al. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J Neuroimmunol (2000) 103(1):16–25. doi: 10.1016/S0165-5728(99)00172-1
21. Pilkington GJ, Lantos PL, Darling JL, Thomas DG. Three cell lines from a spontaneous murine astrocytoma show variation in astrocytic differentiation. Neurosci Lett (1982) 34(3):315–20. doi: 10.1016/0304-3940(82)90194-X
22. Bradford R, Darling JL, Thomas DG. The in-vitro chemosensitivity of three cell lines derived from the VM/DK spontaneous murine astrocytoma. J Neurol Neurosurg Psychiatry (1986) 49(12):1361–6. doi: 10.1136/jnnp.49.12.1361
23. Haddad AF, Young JS, Amara D, Berger MS, Raleigh DR, Aghi MK, et al. Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neurooncol Adv (2021) 3(1):vdab100. doi: 10.1093/noajnl/vdab100
24. Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, et al. Endogenous neoantigen-specific CD8 T cells identified in two glioblastoma models using a cancer immunogenomics approach. Cancer Immunol Res (2016) 4(12):1007–15. doi: 10.1158/2326-6066.CIR-16-0156
25. Binello E, Qadeer ZA, Kothari HP, Emdad L, Germano IM. Stemness of the CT-2A immunocompetent mouse brain tumor model: Characterization in vitro. J Cancer (2012) 3:166–74. doi: 10.7150/jca.4149
26. Seyfried TN, Mukherjee P. Ganglioside GM3 is antiangiogenic in malignant brain cancer. J Oncol (2010) 2010:961243. doi: 10.1155/2010/961243
27. Cotterchio M, Seyfried TN. Serum gangliosides in mice with metastatic and non-metastatic brain tumors. J Lipid Res (1994) 35(1):10–4. doi: 10.1016/S0022-2275(20)40115-4
28. Martínez-Murillo R, Martínez A. Standardization of an orthotopic mouse brain tumor model following transplantation of CT-2A astrocytoma cells. Histol Histopathol (2007) 22(12):1309–26. doi: 10.14670/HH-22.1309
29. Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol (2011) 14(14):Unit 14.16. doi: 10.1002/0471141755.ph1416s52
30. Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE, et al. Intracranial glioblastoma models in preclinical neuro-oncology: Neuropathological characterization and tumor progression. J Neurooncol (2007) 85(2):133–48. doi: 10.1007/s11060-007-9400-9
31. Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarié A, et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PloS One (2015) 10(4):e0123721. doi: 10.1371/journal.pone.0123721
32. Patil V, Pal J, Somasundaram K. Elucidating the cancer-specific genetic alteration spectrum of glioblastoma derived cell lines from whole exome and RNA sequencing. Oncotarget. (2015) 6(41):43452–71. doi: 10.18632/oncotarget.6171
33. Shingu T, Ho AL, Yuan L, Zhou X, Dai C, Zheng S, et al. Qki deficiency maintains stemness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation. Nat Genet (2017) 49(1):75–86. doi: 10.1038/ng.3711
34. Chen A, Jiang Y, Li Z, Wu L, Santiago U, Zou H, et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J Clin Invest (2021) 131(16). doi: 10.1172/JCI147552
35. Zamler DB, Shingu T, Kahn LM, Huntoon K, Kassab C, Ott M, et al. Immune landscape of a genetically engineered murine model of glioma compared with human glioma. JCI Insight (2022) 7(12). doi: 10.1172/jci.insight.148990
36. Jin F, Jin-Lee HJ, Johnson AJ. Mouse models of experimental glioblastoma. In: Debinski W, editor. Gliomas. Brisbane (AU: Exon Publications (2021).
37. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. (2008) 455(7216):1129–33. doi: 10.1038/nature07443
38. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple ras-dependent phosphorylation pathways regulate myc protein stability. Genes Dev (2000) 14(19):2501–14. doi: 10.1101/gad.836800
39. Tristán-Manzano M, Justicia-Lirio P, Maldonado-Pérez N, Cortijo-Gutiérrez M, Benabdellah K, Martin F. Externally-controlled systems for immunotherapy: From bench to bedside. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.02044
40. Huang Y, Motta E, Nanvuma C, Kuhrt LD, Yuan Y, Xia P, et al. Microglia/macrophage-derived human CCL18 promotes glioma progression via CCR8-ACP5 axis analyzed in humanized slice model. Cell Rep (2022) 39(2):110670. doi: 10.1016/j.celrep.2022.110670
41. Widodo SS, Dinevska M, Furst LM, Stylli SS, Mantamadiotis T. IL-10 in glioma. Br J Cancer (2021) 125(11):1466–76. doi: 10.1038/s41416-021-01515-6
42. Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest (2019) 129(1):137–49. doi: 10.1172/JCI121266
43. Ulasov I, Yi R, Guo D, Sarvaiya P, Cobbs C. The emerging role of MMP14 in brain tumorigenesis and future therapeutics. Biochim Biophys Acta (2014) 1846(1):113–20. doi: 10.1016/j.bbcan.2014.03.002
44. Plate KH, Breier G, Weich HA, Mennel HD, Risau W. Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer (1994) 59(4):520–9. doi: 10.1002/ijc.2910590415
45. Chao M, Liu N, Sun Z, Jiang Y, Jiang T, Xv M, et al. TGF-β signaling promotes glioma progression through stabilizing Sox9. Front Immunol (2021) 11. doi: 10.3389/fimmu.2020.592080
46. Maas SLN, Abels ER, Van De Haar LL, Zhang X, Morsett L, Sil S, et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J Neuroinflamm (2020) 17(1):120. doi: 10.1186/s12974-020-01797-2
47. Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM, Sakariassen P, et al. In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol (2012) 14(8):979–93. doi: 10.1093/neuonc/nos135
48. Gould SE, Junttila MR, de Sauvage FJ. Translational value of mouse models in oncology drug development. Nat Med (2015) 21(5):431–9. doi: 10.1038/nm.3853
49. Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol (2019) 16(8):509–20. doi: 10.1038/s41571-019-0177-5
50. Akter F, Simon B, de Boer NL, Redjal N, Wakimoto H, Shah K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochim Biophys Acta Rev Cancer (2021) 1875(1):188458. doi: 10.1016/j.bbcan.2020.188458
51. Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in drosophila melanogaster. Genome Res (2001) 11(6):1114–25. doi: 10.1101/gr.169101
52. Read RD, Cavenee WK, Furnari FB, Thomas JB. A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PloS Genet (2009) 5(2):e1000374. doi: 10.1371/journal.pgen.1000374
53. Witte HT, Jeibmann A, Klämbt C, Paulus W. Modeling glioma growth and invasion in drosophila melanogaster. Neoplasia. (2009) 11(9):882–8. doi: 10.1593/neo.09576
54. Castellanos E, Dominguez P, Gonzalez C. Centrosome dysfunction in drosophila neural stem cells causes tumors that are not due to genome instability. Curr Biol (2008) 18(16):1209–14. doi: 10.1016/j.cub.2008.07.029
55. Salminen TS, Vale PF. Drosophila as a model system to investigate the effects of mitochondrial variation on innate immunity. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00521
56. Govind S. Innate immunity in drosophila: Pathogens and pathways. Insect Sci (2008) 15(1):29–43. doi: 10.1111/j.1744-7917.2008.00185.x
57. Irving P, Troxler L, Hetru C. Is innate enough? the innate immune response in drosophila. Comptes Rendus Biologies (2004) 327(6):557–70. doi: 10.1016/j.crvi.2004.03.007
58. LeBlanc AK, Mazcko C, Brown DE, Koehler JW, Miller AD, Miller CR, et al. Creation of an NCI comparative brain tumor consortium: informing the translation of new knowledge from canine to human brain tumor patients. Neuro Oncol (2016) 18(9):1209–18. doi: 10.1093/neuonc/now051
59. Hicks J, Platt S, Kent M, Haley A. Canine brain tumours: A model for the human disease? Vet Comp Oncol (2017) 15(1):252–72. doi: 10.1111/vco.12152
60. Amin SB, Anderson KJ, Boudreau CE, Martinez-Ledesma E, Kocakavuk E, Johnson KC, et al. Comparative molecular life history of spontaneous canine and human gliomas. Cancer Cell (2020) 37(2):243–57.e7. doi: 10.1016/j.ccell.2020.01.004
61. Alsaihati BA, Ho K-L, Watson J, Feng Y, Wang T, Dobbin KK, et al. Canine tumor mutational burden is correlated with TP53 mutation across tumor types and breeds. Nat Commun (2021) 12(1):4670. doi: 10.1038/s41467-021-24836-9
62. Kawakami S, Michishita M, Sakaue M, Morimatsu M, Uemura M, Kashiwagi N, et al. Novel canine isocitrate dehydrogenase 1 mutation Y208C attenuates dimerization ability. Oncol Lett (2020) 20(6):351. doi: 10.3892/ol.2020.12214
63. Hicks WH, Bird CE, Pernik MN, Haider AS, Dobariya A, Abdullah KG, et al. Large Animal models of glioma: Current status and future prospects. Anticancer Res (2021) 41(11):5343–53. doi: 10.21873/anticanres.15347
64. Chambers MR, Foote JB, Bentley RT, Botta D, Crossman DK, Della Manna DL, et al. Evaluation of immunologic parameters in canine glioma patients treated with an oncolytic herpes virus. J Transl Genet Genom (2021) 5(4):423–42. doi: 10.20517/jtgg.2021.31
65. Olin MR, Ampudia-Mesias E, Pennell CA, Sarver A, Chen CC, Moertel CL, et al. Treatment combining CD200 immune checkpoint inhibitor and tumor-lysate vaccination after surgery for pet dogs with high-grade glioma. Cancers (Basel) (2019) 11(2). doi: 10.3390/cancers11020137
66. Hubbard ME, Arnold S, Bin Zahid A, McPheeters M, Gerard O’Sullivan M, Tabaran AF, et al. Naturally occurring canine glioma as a model for novel therapeutics. Cancer Invest (2018) 36(8):415–23. doi: 10.1080/07357907.2018.1514622
67. Novoa B, Figueras A. Zebrafish: model for the study of inflammation and the innate immune response to infectious diseases. Adv Exp Med Biol (2012) 946:253–75. doi: 10.1007/978-1-4614-0106-3_15
68. Trede NS, Langenau DM, Traver D, Look AT, Zon LI. The use of zebrafish to understand immunity. Immunity. (2004) 20(4):367–79. doi: 10.1016/S1074-7613(04)00084-6
69. Vittori M, Motaln H, Turnšek TL. The study of glioma by xenotransplantation in zebrafish early life stages. J Histochem Cytochem (2015) 63(10):749–61. doi: 10.1369/0022155415595670
70. Yan C, Brunson DC, Tang Q, Do D, Iftimia NA, Moore JC, et al. Visualizing engrafted human cancer and therapy responses in immunodeficient zebrafish. Cell. (2019) 177(7):1903–14.e14. doi: 10.1016/j.cell.2019.04.004
71. Anderson RC, Elder JB, Brown MD, Mandigo CE, Parsa AT, Kim PD, et al. Changes in the immunologic phenotype of human malignant glioma cells after passaging in vitro. Clin Immunol (2002) 102(1):84–95. doi: 10.1006/clim.2001.5152
72. Nigro JM, Misra A, Zhang L, Smirnov I, Colman H, Griffin C, et al. Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma. Cancer Res (2005) 65(5):1678–86. doi: 10.1158/0008-5472.CAN-04-2921
73. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell (2006) 9(5):391–403. doi: 10.1016/j.ccr.2006.03.030
74. Vaubel RA, Tian S, Remonde D, Schroeder MA, Mladek AC, Kitange GJ, et al. Genomic and phenotypic characterization of a broad panel of patient-derived xenografts reflects the diversity of glioblastoma. Clin Cancer Res (2020) 26(5):1094–104. doi: 10.1158/1078-0432.CCR-19-0909
75. Cho SY. Patient-derived xenografts as compatible models for precision oncology. Lab Anim Res (2020) 36:14. doi: 10.1186/s42826-020-00045-1
76. Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. (2007) 317(5836):337. doi: 10.1126/science.1142596
77. Clément V, Dutoit V, Marino D, Dietrich PY, Radovanovic I. Limits of CD133 as a marker of glioma self-renewing cells. Int J Cancer (2009) 125(1):244–8. doi: 10.1002/ijc.24352
78. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res (2004) 64(19):7011–21. doi: 10.1158/0008-5472.CAN-04-1364
79. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. (2004) 432(7015):396–401. doi: 10.1038/nature03128
80. Wang J, Sakariassen P, Tsinkalovsky O, Immervoll H, Bøe SO, Svendsen A, et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer (2008) 122(4):761–8. doi: 10.1002/ijc.23130
81. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell (2010) 17(4):362–75. doi: 10.1016/j.ccr.2009.12.049
82. Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA, et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery. (2008) 62(2):505–14. doi: 10.1227/01.neu.0000316019.28421.95
83. McClung C, Hirsh J. Stereotypic behavioral responses to free-base cocaine and the development of behavioral sensitization in drosophila. Curr Biol (1998) 8(2):109–12. doi: 10.1016/S0960-9822(98)70041-7
84. Read RD, Fenton TR, Gomez GG, Wykosky J, Vandenberg SR, Babic I, et al. A kinome-wide RNAi screen in drosophila glia reveals that the RIO kinases mediate cell proliferation and survival through TORC2-akt signaling in glioblastoma. PloS Genet (2013) 9(2):e1003253. doi: 10.1371/journal.pgen.1003253
85. Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in drosophila neural stem cells. Cell. (2006) 124(6):1241–53. doi: 10.1016/j.cell.2006.01.038
86. Mukherjee S, Tucker-Burden C, Zhang C, Moberg K, Read R, Hadjipanayis C, et al. Drosophila brat and human ortholog TRIM3 maintain stem cell equilibrium and suppress brain tumorigenesis by attenuating notch nuclear transport. Cancer Res (2016) 76(8):2443–52. doi: 10.1158/0008-5472.CAN-15-2299
87. Awasaki T, Lee T. New tools for the analysis of glial cell biology in drosophila. Glia. (2011) 59(9):1377–86. doi: 10.1002/glia.21133
88. Boss MK. Canine comparative oncology for translational radiation research. Int J Radiat Biol (2022) 98(3):496–505. doi: 10.1080/09553002.2021.1987572
89. Young JS, Bernal G, Polster SP, Nunez L, Larsen GF, Mansour N, et al. Convection-enhanced delivery of polymeric nanoparticles encapsulating chemotherapy in canines with spontaneous supratentorial tumors. World Neurosurg (2018) 117:e698–704. doi: 10.1016/j.wneu.2018.06.114
90. Zeng A, Ye T, Cao D, Huang X, Yang Y, Chen X, et al. Identify a blood-brain barrier penetrating drug-TNB using zebrafish orthotopic glioblastoma xenograft model. Sci Rep (2017) 7(1):14372. doi: 10.1038/s41598-017-14766-2
Keywords: glioma, GBM, immunotherapy, brain, tumor, pre-clinical, mouse model
Citation: Frederico SC, Zhang X, Hu B and Kohanbash G (2023) Pre-clinical models for evaluating glioma targeted immunotherapies. Front. Immunol. 13:1092399. doi: 10.3389/fimmu.2022.1092399
Received: 08 November 2022; Accepted: 20 December 2022;
Published: 09 January 2023.
Edited by:
Liangxue Zhou, Sichuan University, ChinaReviewed by:
Zhenyu Li, Department of Pathology, Chongqing University, ChinaCopyright © 2023 Frederico, Zhang, Hu and Kohanbash. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary Kohanbash, Z2FyeS5rb2hhbmJhc2gyQGNocC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.