 Xiaokang Wang
Xiaokang Wang Jingqian Zhao
Jingqian Zhao Yuanqing Li1
Yuanqing Li1 Jiaoyu Rao
Jiaoyu Rao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 December 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1090989
This article is part of the Research TopicCommunity Series in Epigenetics of the Immune Component of Inflammation, Volume IIView all 10 articles
Proteinuria or nephrotic syndrome are symptoms of podocytopathies, kidney diseases caused by direct or indirect podocyte damage. Human health worldwide is threatened by diabetic nephropathy (DN), the leading cause of end-stage renal disease (ESRD) in the world. DN development and progression are largely dependent on inflammation. The effects of podocyte damage on metabolic disease and inflammatory disorders have been documented. Epigenetic and endoplasmic reticulum (ER) stress are also evident in DN. Targeting inflammation pathway and ER stress in podocytes may be a prospective therapy to prevent the progression of DN. Here, we review the mechanism of epigenetics and ER stress on podocyte inflammation and apoptosis, and discuss the potential amelioration of podocytopathies by regulating epigenetics and ER stress as well as by targeting inflammatory signaling, which provides a theoretical basis for drug development to ameliorate DN.
Diabetes mellitus (DM) is characterized by systemic hyperglycemia and can lead to a wide range of complications. It has seriously threatened the quality of life of millions worldwide. Although intensive measures have been taken to control blood glucose, the number of people with DM worldwide was half a billion in 2021 (1). Around 30–40% of type 1 or 2 DM patients are, unfortunately, prognosed with diabetic nephropathy (DN) and eventually develop end-stage renal disease (ESRD) (2, 3), imposing an enormous economic burden on individuals, families, and societies. Therefore, novel treatments for DN are urgently needed.
The roles of various pathways in the pathogenesis of DN have been established. Activation of the renin-angiotensin-aldosterone system (RAAS) (4), reactive oxygen species (ROS) (5) and inflammation (6) are related to the onset and development of DN (7). Although treatment with angiotensin-converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARB) slows the progression of diabetic nephropathy to ESRD, the risk of ESRD remains high and is associated with residual proteinuria (8), suggesting that better strategies are needed to prevent DN. There has been a growing consensus that inflammation pathways are important in DN progression. New therapeutic strategies could be developed by identifying novel inflammatory factors (9). In patients with diabetic kidney disease (DKD), glomerular macrophage accumulation correlates strongly with progression of kidney impairment (10). During infiltration, macrophages release cytokines such as TNF, IL-1, and IFN-γ, accelerating the progression of DKD (11). By altering the viability of podocytes, macrophages contribute directly to DKD in bone-marrow chimeric mice (12). Genetic deficiency or pharmacological blockade of C-C chemokine receptor type 2 impairs macrophage recruitment to DKD in mouse models, reducing albuminuria and urea nitrogen levels as well as improving histological parameters. Treatment with the CCR2 inhibitor CCX140-B for 52 weeks significantly reduced urinary albumin excretion in T2DM patients with proteinuria (13). Angiotensinogen and cytokines are implicated in DKD—NF-κB initiates inflammation and stimulates the transcription of genes encoding cytokines and adhesion molecules (14). T2DM patients with NF-κB activation in muscle are likely to develop peripheral-tissue insulin resistance (15). DKD patients with bardoxolone have shown renoprotective effects with inhibitors of NF-κB (16). Therefore, research on small molecular targets of inflammatory signaling pathways in DN is needed.
Epigenetic modifications play a role in DKD, by affecting gene expression in response to environmental factors (17, 18). Epigenetic modifications enable mitotically and/or meiotically heritable changes in gene function without altering the underlying DNA sequence (19). Most disease-associated loci and single-nucleotide polymorphisms (SNPs) are found in non-coding regions of the genome, including regulatory regions such as enhancers and promoters, as well as in non-coding RNAs (20), which can affect gene expression by altering transcription factor binding and chromatin accessibility.
Podocytopathies is a key factor in DN development. And podocytopathies can lead to destruction of the glomerular basement membrane (GBM), inducing proteinuria (21). Mice with podocyte-specific conditional disruption of slit molecule genes suffer severe proteinuria and sclerosis (22). Proteinuria is closely associated with foot process effacement in minimal change disease (23). Moreover, albuminuria is diagnostic of early DN. Although podocyte injury is associated with the progression of DN (24), the underlying mechanisms are unclear. Interestingly, ER stress accelerates DN progression by injuring podocytes, endothelial cells, and mesangial cells (25, 26). Hyperglycemia and proteinuria (27) reportedly induce ER stress in DN. Persistent ER stress could affect ER function and induce a maladaptive unfolded protein response (UPR), activating apoptotic signaling pathways and causing podocyte apoptosis (28).
Taken together, the interaction of epigenetic modifications and ER stress with inflammation and apoptosis in podocytopathies is unclear. Studies on the epigenetic mechanism of DN (17) have attenuated ER stress by targeting long non-coding RNA (lncRNA) and/or microRNA. And epigenetics may regulate diabetes-related diseases under inflammatory conditions. A review of recent research on epigenetic mechanisms and endoplasmic reticulum stress in inflammation or apoptosis in podocytopathy presented here. Looking forward to provide new drug treatment strategies for podocytopathy during DN progression.
The glomerular filtration barrier is maintained by podocytes, highly specialized epithelial cells that contain critical molecules required for selective permeability (29). Loss of large numbers of podocytes leads to proteinuria, mesangial expansion, and glomerular sclerosis. Diabetic patients’ podocytes are susceptible to injury due to apoptosis, which is the most common mode of podocyte loss due to glucose-induced oxidative stress and advanced glycation end products (28, 30). However, little is known of the underlying mechanisms (31).
Podocyte injury is characterized by hypertrophy, epithelial mesenchymal transition (32), detachment, and apoptosis (33). Glomerular hyperfiltration is accompanied by podocyte hypertrophy and abnormal podocyte function (34). Moreover, hyperglycemia could decrease the attachment of podocytes to the GBM—integrin α3β1 is an important receptor for podocyte–GBM attachment (35). The levels of podocyte cytoskeleton proteins, such as synaptopodin, podocin, and nephrin, were significantly reduced in diabetic kidneys (36). That is, a disordered podocyte cytoskeleton hampers adhesion, increasing GBM permeability and inducing proteinuria. Therefore, injury and depletion of podocytes is a crucial pathologic mechanism of albuminuria in DN. Proteinuria is also associated with a decrease in podocyte density and number in type 1 and type 2 diabetes. Indeed, podocytes in urine are an earlier marker of DN than proteinuria (37) (Figure 1).
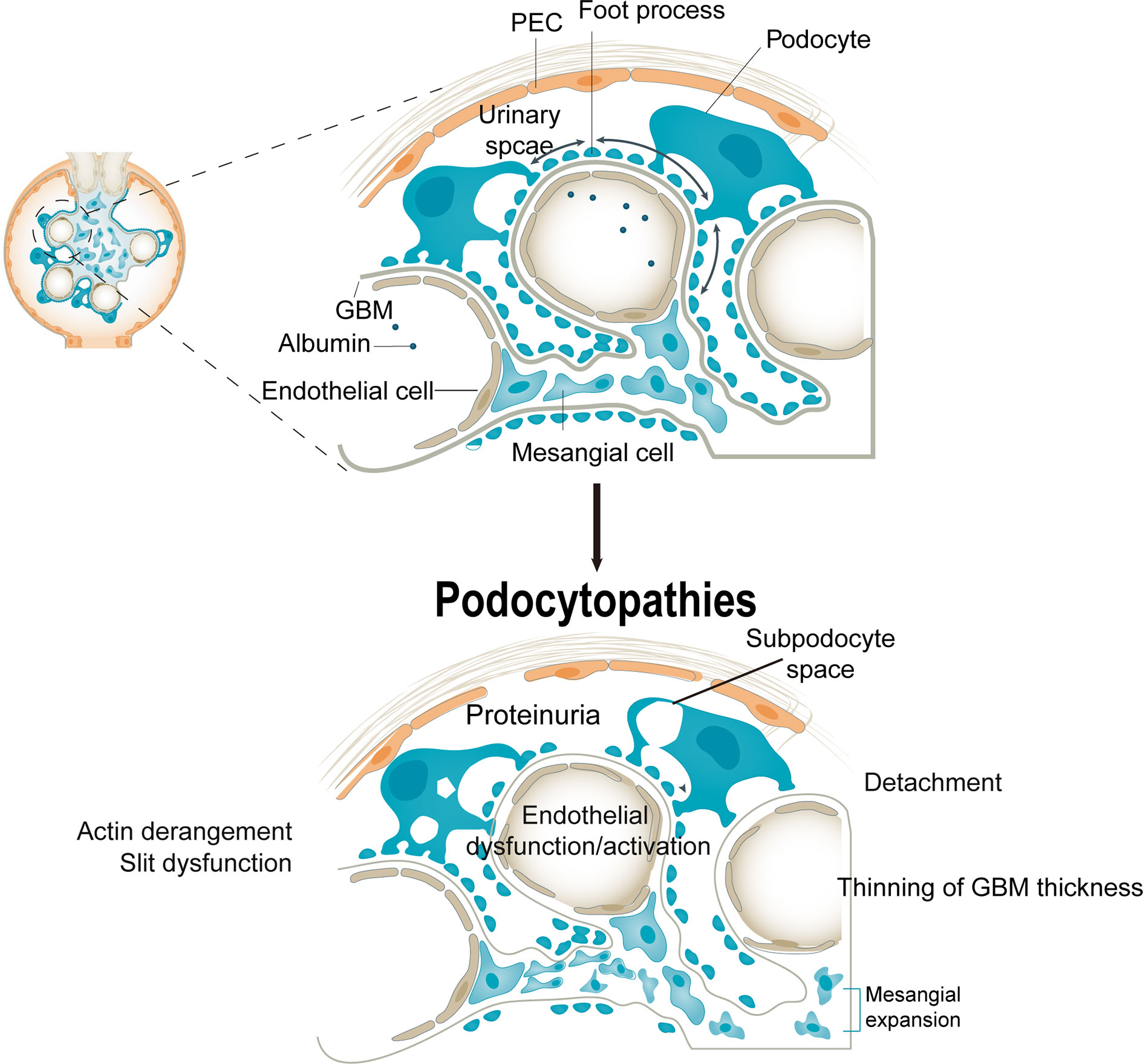
Figure 1 Consequences of podocytopathies. Podocyte injury causes cellular and structural responses in the glomerulus. An injured podocyte has cytoplasmic blebs, protein droplets, and an expanded subpodocyte space. Foot process effacement is associated with actin derangement, which induces dysfunction of slit molecules. The injured podocyte detaches from the glomerular basement membrane, which is associated with abnormalities of adhesion molecules, coagulation cascade, chemokine receptor expression, and lipid peroxidation. These changes may depend on podocyte detachment but also on aberrant factors from injured podocytes. PECs, parietal epithelial cells; VEGF, vascular endothelial growth factor.
Podocyte apoptosis contributes to filtration dysfunction in DN. As highly differentiated cells, podocytes have limited self-renewal via mitosis under adverse conditions, rendering them vulnerable to damage by stress (28, 38). Therefore, podocytes are dependent on clearance mechanisms such as the adaptive UPR and autophagy to restore intracellular hemostasis (38). Podocyte autophagy deficiency triggers proteinuria and glomerulosclerosis, and maintaining a certain level of autophagy ensures normal physiological function (39). In aldosterone-induced ER stress and podocyte injury, inhibition and induction of ER stress suppress and enhance, respectively, autophagy (40). Hence, restoration of podocyte structure and function after injury is important.
Histone modifications (acetylation, methylation, phosphorylation, ubiquitination, and carbonylation) are important in epigenetics; histone acetylation and methylation predominate. Inhibitors of histone deacetylases (HDACs) (such as valproic acid) and SAHA (such as pan-HDAC inhibitors) reduce proteinuria and glomerulosclerosis, thereby increasing survival (41). Epigenetics are implicated in the progression of various kidney diseases, including DN. Histone acetyltransferases’ acetylation activities are balanced by HDACs, which modulate physiological and pathological gene transcription. A class III NAD+-dependent HDAC, sirtuin6 (Sirt6), belongs to the sirtuin family, which consists of Sirt1 to Sirt7 that differ in their cellular and tissue distributions. Genes involved in glucose metabolism, DNA repair, telomerase activity, genomic stability, and cellular senescence are regulated by Sirt6-dependent deacetylation of H3K9 or H3K56. Several phenotypes of premature aging have been observed in mice lacking Sirt6. Podocytopathies were associated with a reduced level of Sirt6 in renal biopsies and a reduced glomerular filtration rate (42). In a DN mouse model, Sirt6 deletion exacerbated podocyte injury and proteinuria. Sirt6 suppresses Notch1 and Notch4 transcription, negatively regulating Notch signaling. Therefore, Sirt6 has potential as a therapeutic target for proteinuric kidney disease. In addition, many cellular biological processes regulated by Sirt1 are involved in kidney diseases, such as autophagy and apoptosis (43). Enhanced Sirt1 function in podocytes protects diabetic kidneys (44). Hence, histone modifications protect podocytes in hyperglycemia.
Differences in DNA methylation are associated with predisposition to disease or treatment response. Inter-individual epigenetic differences among individuals can be used as predictive biomarkers of disease susceptibility (45). Thus, DNA methylation levels can be used to distinguish diabetic ESRD and renal complications without diabetes. The METTL3-mediated m6A modification contributes to podocyte injury in DN and targeting m6A by METTL3 has therapeutic potential (46) (Figure 2). Sirt1 mRNA was degraded in podocytosis by METTL14-mediated m6A modification. By upregulating Sirt1, podocytes activate autophagy and reduce apoptosis and inflammation, thereby alleviating proteinuria and delaying the progression of podocytopathies. Dysregulation of the RNA m6A modification may be mediated by METTL14 and may be a therapeutic target for podocytopathies (Figure 2).
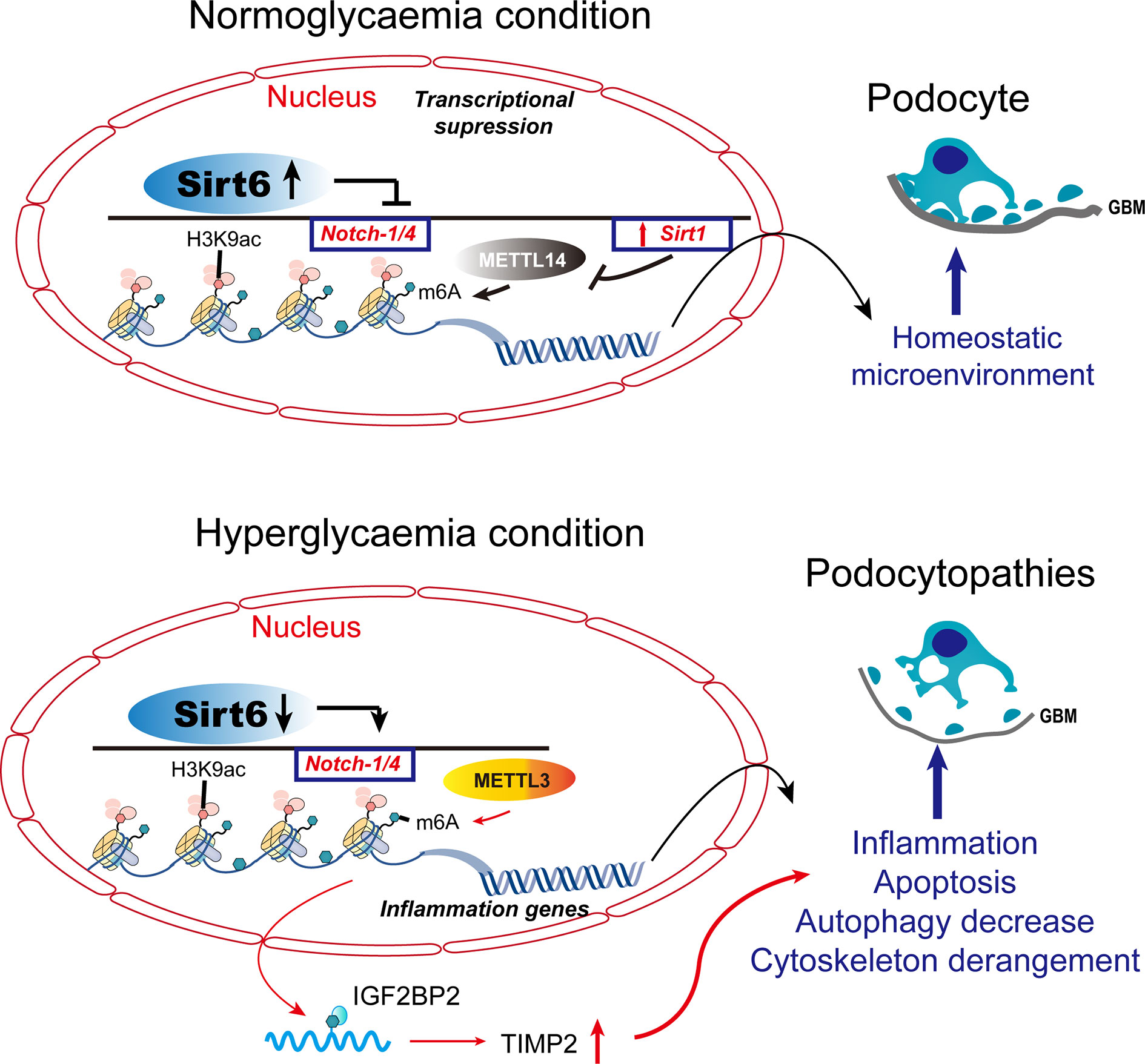
Figure 2 Histone modifications and DNA methylation in podocytopathies. Under normal conditions, Sirt6 inhibits the transcription of Notch1 and Notch4 by decreasing H3K9ac levels in the promoter regions of Notch1 and Notch4. METTL14 deficiency inhibits Sirt1 mRNA m6A modification, thereby increasing Sirt1 levels in normal podocytes. METTL3 modulates the m6A modification of TIMP2 in an IGF2BP2-dependent manner and exerts pro-inflammatory and -apoptotic effects. TIMP2, tissue inhibitor of metalloproteinase 2; IGF2BP2, insulin-like growth factor 2 mRNA binding protein 2.
When Sirt6 is reduced under pathological conditions, H3K9ac levels increase in the promoters of Notch1 and Notch4, thereby increasing transcription. Sirt1 mRNA m6A modification and degradation are elevated when podocyte injury induces the expression of METTL14. As a result of Notch-signaling activation, podocyte injury is triggered by inflammation, apoptosis, actin-cytoskeleton degeneration, and autophagy inhibition.
Aberrant epigenetic alterations are implicated in the pathogenesis of DKD and modifying the podocyte injury response. DACH1 and Pax transactivation-domain interacting protein (PTIP) target genes are similar. PTIP is recruited by Pax proteins to promoter Pax response elements (PREs), increasing the levels of H3K4Me3 and activating transcription (47). PTIP is also recruited by sequence-specific binding of DACH1 to its target gene promoters, which suppresses transcription and reduces promoter H3K4Me3 levels. When podocyte DACH1 expression is reduced, PTIP is less likely to be recruited to target gene promoters by DACH1. Decreased DACH1 expression in podocytes under hyperglycemic conditions reduces the expression of multiple target genes and downstream signals, thereby damaging podocytes. Downregulation is caused by reduced recruitment of PTIP by the DACH1 target gene promoter, leading to excessive podocyte epigenome activity. Therapeutic interventions aimed at improving podocyte DACH1 activity will correct multiple dysfunctional signals simultaneously. Therefore, testing and development of drugs for DACH1 should be prioritized (Figure 3).
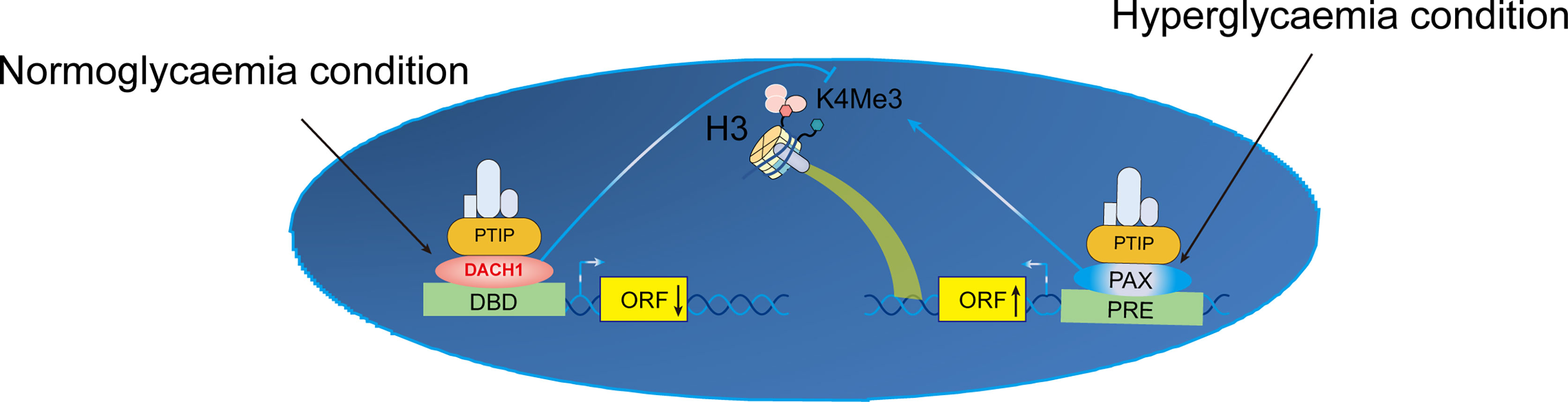
Figure 3 Proposed mechanism of transcriptional activation of K4Me in podocytes. Crosstalk between normal and hyperglycemic conditions in DN. ORF, open reading frame; DBD, DNA-binding domain; H3K4Me3, trimethylation of lysine 4 on histone H3 protein subunit; PRE, Pax response elements; DACH1, Dachshund homolog 1; PTIP, Pax transactivation-domain interacting protein.
Epigenetic determinants such as DNA methylation, histone modification, or RNA can be transferred to the next generation through meiotic products (gametes). DNA methylation and chromatin histone modifications are epigenetic mechanisms, but lncRNAs and microRNAs can also be modified post-transcriptionally (17). MicroRNAs are endogenous non-coding single-stranded RNAs of about 20-24 nucleotides, which can degrade or silence the translation of target gene mRNA by base-pairing with the 3’-UTR sequence (48). MicroRNA regulation of podocyte apoptosis has been a focus of research (48, 49). MicroRNAs regulate autophagy genes, affect the level of autophagy, and are implicated in DN (50, 51). Various miRNA regulatory mechanisms are involved in DN physiological and pathological processes, affecting podocyte apoptosis (49), and are targets for DN prevention and treatment (Figure 4).
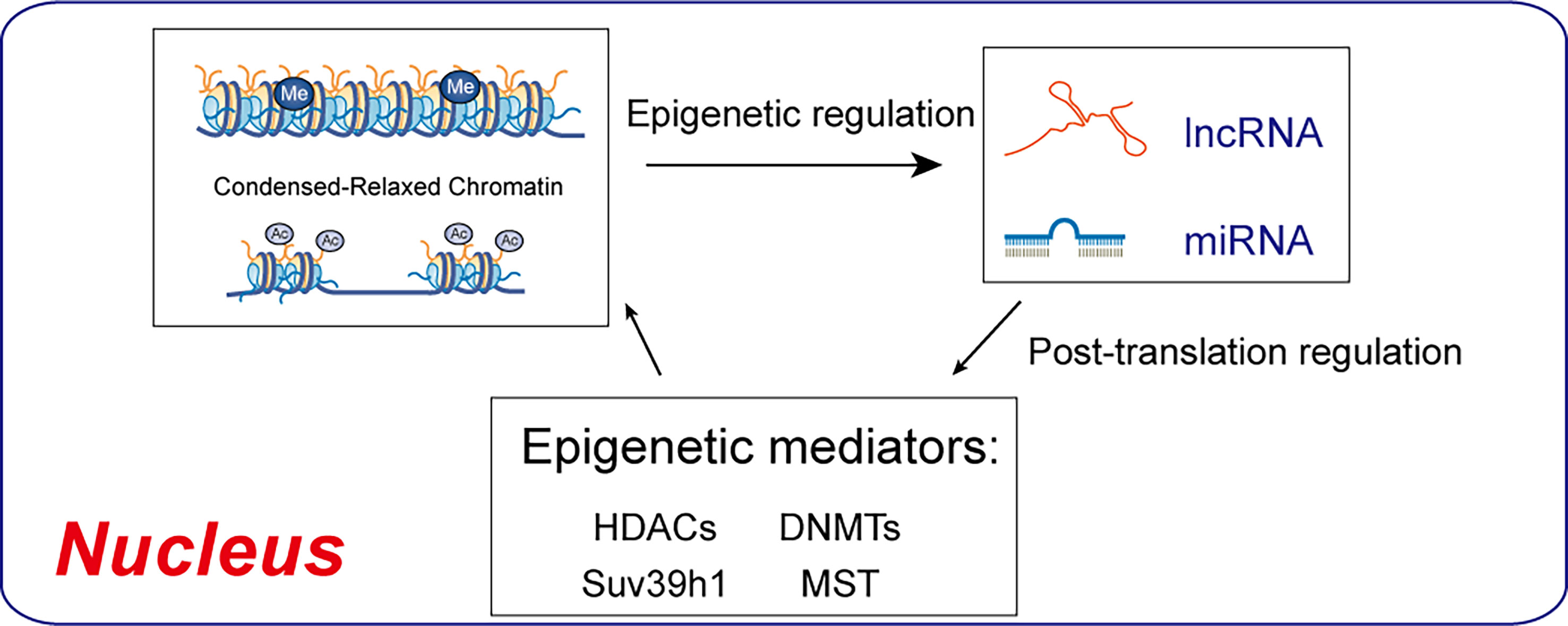
Figure 4 Regulatory interactions between epigenetic machinery and non-coding RNAs. DNMTs and HDACs are epigenetic mediators of DNA methylation and histone acetylation, which result in condensed-relaxed chromatin. lncRNAs and miRNAs modulate epigenetic mediators, thus regulating chromatin remodeling.
Long noncoding RNAs (lncRNAs), as competing endogenous RNAs (ceRNAs), confirm the interaction and competitive regulatory mechanisms between lncRNA and microRNA (52, 53, Wang et al., 2022). Enzymes including DNMTs regulate a variety of miRNAs associated with Alzheimer Disease development and progression, including miR-34 a/b/c, miR-107, miR-124, miR-125b, and miR-137 (54). Upregulation of about 40 miRNAs in a lncRNA in the kidneys of diabetic mice promoted DN. lncRNAs have not been systematically studied in podocytopathy to date. miRNAs and epigenetic enzymes play important roles in a variety of diseases.
Although podocytes play a crucial role in glomerular filtration by forming a slit diaphragm between interdigitating foot processes, the molecular details of protein folding and degradation in the ER are unclear. The ER plays a vital role in maintaining protein synthesis and hemostasis. Two studies have evaluated the pathophysiological roles of the ER in kidney dysfunction (55, 56). ER stress is characterized by the UPR. Sustained ER stress causes cell death, inflammation, and autophagy in the renal tubules. Therefore, inhibiting ER stress may have therapeutic potential for DN (57).
The upregulation of ER stress markers and downstream signaling molecules has been reported in cells, animals, and individuals with DN (55). CHOP, a key regulator of ER stress, was significantly increased in renal biopsies of type 2 DN patients (58, 59). High glucose induces ER stress, podocyte phenotypic switch, and podocyte loss in rat, effects inhibited by exogenous ER molecular chaperones (60, 61). ER stress also triggers podocyte apoptosis. ER markers such as GRP78 are upregulated in diabetic rats’ podocytes (55, 62). Expression of GRP78 and other factors related to ER is increased significantly in HG-induced podocytes, leading to apoptosis (60). A newly hallmark, RTN1A, was found to be increased in the context of ER stress (58). Therefore, ER stress may be a key regulator of podocyte death. Here, based on the three arms of the UPR, we discuss the role of ER stress in inflammation and podocyte apoptosis.
Epigenetic modifications can affect endoplasmic reticulum stress response, resulting in disease risk. MicroRNA expression and DNA and histone methylation patterns are epigenetic phenomena associated with ER stress genes. Some results suggest that the methylation characteristics of leukocyte ER regulatory genes may be related to abdominal/central obesity markers (waist circumference), dyslipidemia and insulin resistance (63). However, the finding provides insight into the relationship between disorders and epigenetics as well as complications resulting from endoplasmic reticulum stress, further research is needed to confirm this.
TLR4 knockout mice showed reduced albuminuria, renal dysfunction, inflammation, and fibrosis after STZ treatment. Diabetes-induced podocyte loss was reduced by deletion of Tlr4 despite tubular injury suppression (64, 65). High glucose upregulated TLR4 expression in cultured podocytes by triggering ROS production and NF-κB activation (66). By activating the protein kinase C pathway and NADPH oxidase (NOX), high glucose increases TLR2 and TLR4 expression in monocytes (67). High glucose-induced upregulation of IL-6 and CCL2 was inhibited by silencing TLR4, while TLR2-deficient tubular cells showed no significant reduction in cytokine production. High glucose induced the expression of TLR4 but not of TLR2 in mesangial cells (68). GIT27, an anti-TLR agent, showed an anti-proteinuric effect in db/db mice treated with an immunomodulator that targets macrophages by blocking TLR2, TLR4, and TLR6 signals (69). Additionally, GIT27 inhibited cytokine production induced by glucose and free fatty acids in cultured podocytes (11). Therefore, TLR4 signaling is implicated in inflammation in hyperglycemic patients.
Akin to TLRs, NLRs play an important role in innate immunity by triggering pro-inflammatory cascades when pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) trigger innate immunity (70, 71). A variety of human diseases are caused by the NLRP3 inflammasome complex, including cancer, liver disease, and DKD. NLRP3 inflammasome activation is a two-step process. First, priming by PAMP- or DAMP-induced activation of TLR signaling, resulting in NF-κB-dependent expression of NLRP3, pro-IL-1β, and pro-IL-18. Second, various cellular mechanisms, such as potassium efflux, pore-forming toxins, calcium influx, mitochondrial dysfunction, and intracellular ROS production, trigger the formation of the NLRP3 complex and activation of CASP1 (70). Nod-like receptor protein 3 (NLRP3) inflammasome activation is responsible for the accumulation of intracellular or plasma S-adenosyl-homocysteine, promoting hyperglycemia-induced podocyte injury (72). Nephropathy in db/db mice was preceded by inflammasome activation in podocytes and endothelial cells (73). Diabetes-induced hyperglycemia increased thioredoxin-interacting protein (TXNIP) expression in the kidneys of diabetic mice, resulting in NOX and inflammasome activation (3, 74), podocyte loss and the onset of albuminuria (75). In mice, angiotensin II also activated NLRP3 inflammasomes, produced mitochondrial dysfunction, and led to nephron and podocin loss, as well as albuminuria (76). Therefore, NLRP3 inflammasome activation induces podocyte apoptosis and mitochondrial dysfunction, resulting in proteinuria. Additionally, NLRP3-knockout mice with STZ-induced DKD had improved renal function, glomerulosclerosis, tubulointerstitial inflammation, and renal fibrosis (77).
The ER chaperone immunoglobulin-binding protein (BiP) or glucose-regulated protein 78 (GRP78) mediates the UPR signaling network. The UPR consists of three major branches, which are initiated by the activation of three protein sensors—inositol-requiring enzyme 1α (IRE1α), activating transcription factor 6 (ATF6), and protein kinase RNA (PKR)-like ER kinase (78, 79) (Figures 5, 6). Under physiological conditions, the transducers are bound to BiP and are inactive. Accumulation of misfolded or unfolded proteins under stress detaches BiP from the transducers, simultaneously activating them (55). Subsequently, spliced Xbp1 (XBP1s), which encodes a potent transcriptional activator, is cleaved by IRE1-mediated sequence-specific cleavage (Figure 5A). Moreover, ER stress reduces insulin signaling during the development of diabetes via activation of JNK, plus it induces pancreatic apoptosis, which worsens diabetes complications (80). Insulin supplementation can inhibit the transcriptional regulation of XBP1s and thereby alleviate inflammation (Figure 6A).
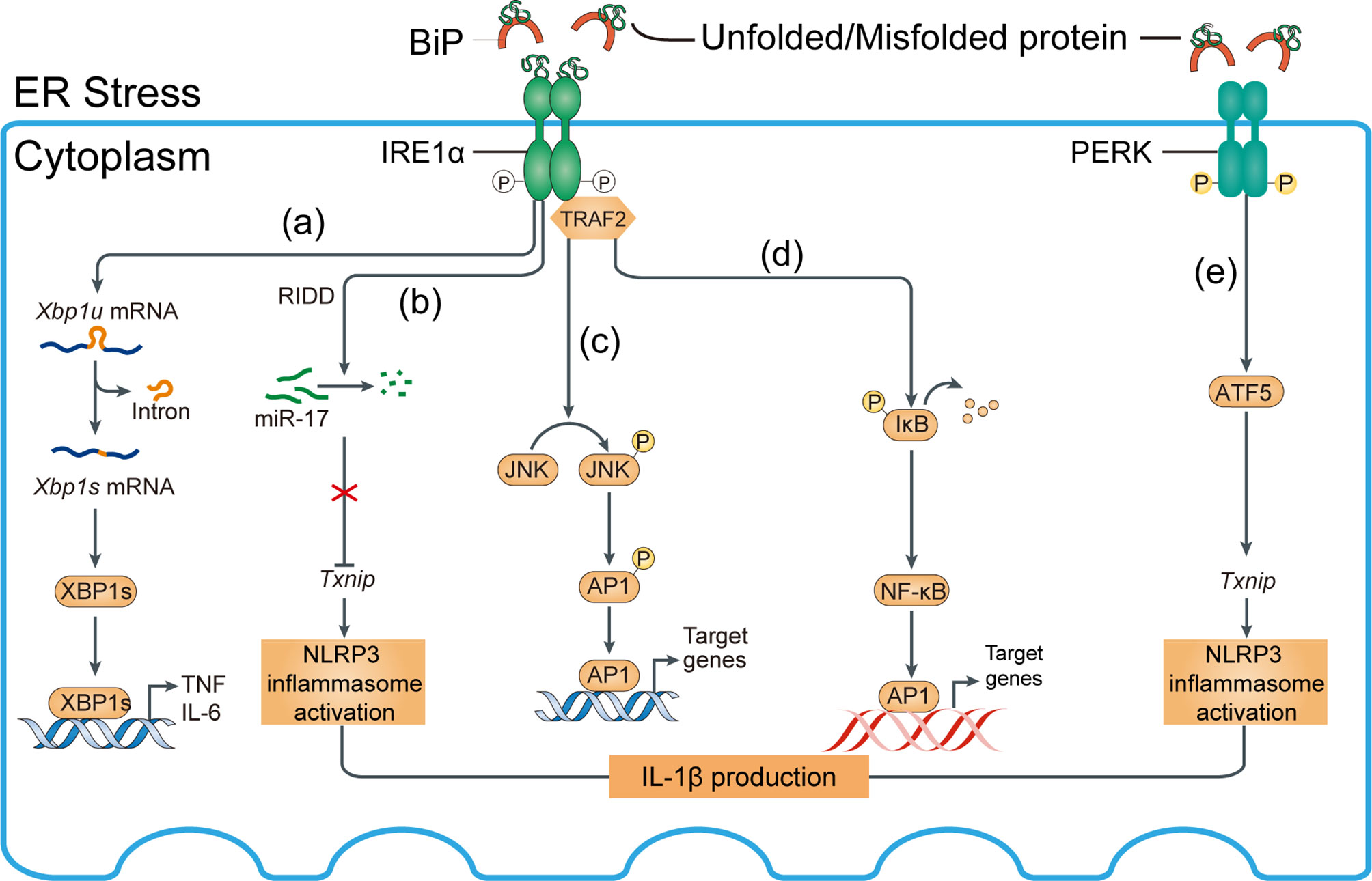
Figure 5 Mechanisms of ER stress-induced inflammation. Inositol-requiring enzyme 1α (IRE1α) and PKR-like ER kinase (PERK) activation induce inflammasome activation. (A) IRE1α activation and subsequent splicing of X-box binding protein 1 (Xbp1) produces the transcription factor XBP1s, which directly binds the promoters of tumor necrosis factor (TNF) and interleukin-6 (IL-6). (B) Regulated IRE1α-dependent decay (RIDD)-dependent degradation of miR-17, which in unstressed conditions represses thioredoxin-interacting protein (Txnip), increases the Txnip level, NLRP3 inflammasome activation, and IL-1β expression. (C) Activated IRE1α forms a complex with TNF receptor-associated factor 2 (TRAF2) to induce phosphorylation of Jun N-terminal kinase (JNK) and upregulation of proinflammatory genes via activated activator protein 1 (AP1). (D) IRE1α–TRAF2 complex recruits IκB kinase (IKK); subsequent phosphorylation and degradation of IκB releases nuclear factor-κB (NF-κB) for nuclear translocation. BiP, binding immunoglobulin protein. (E) Txnip can be induced by the PKR-like ER kinase (PERK)-activating transcription factor 5 (ATF5) pathway to induce inflammasome activation.
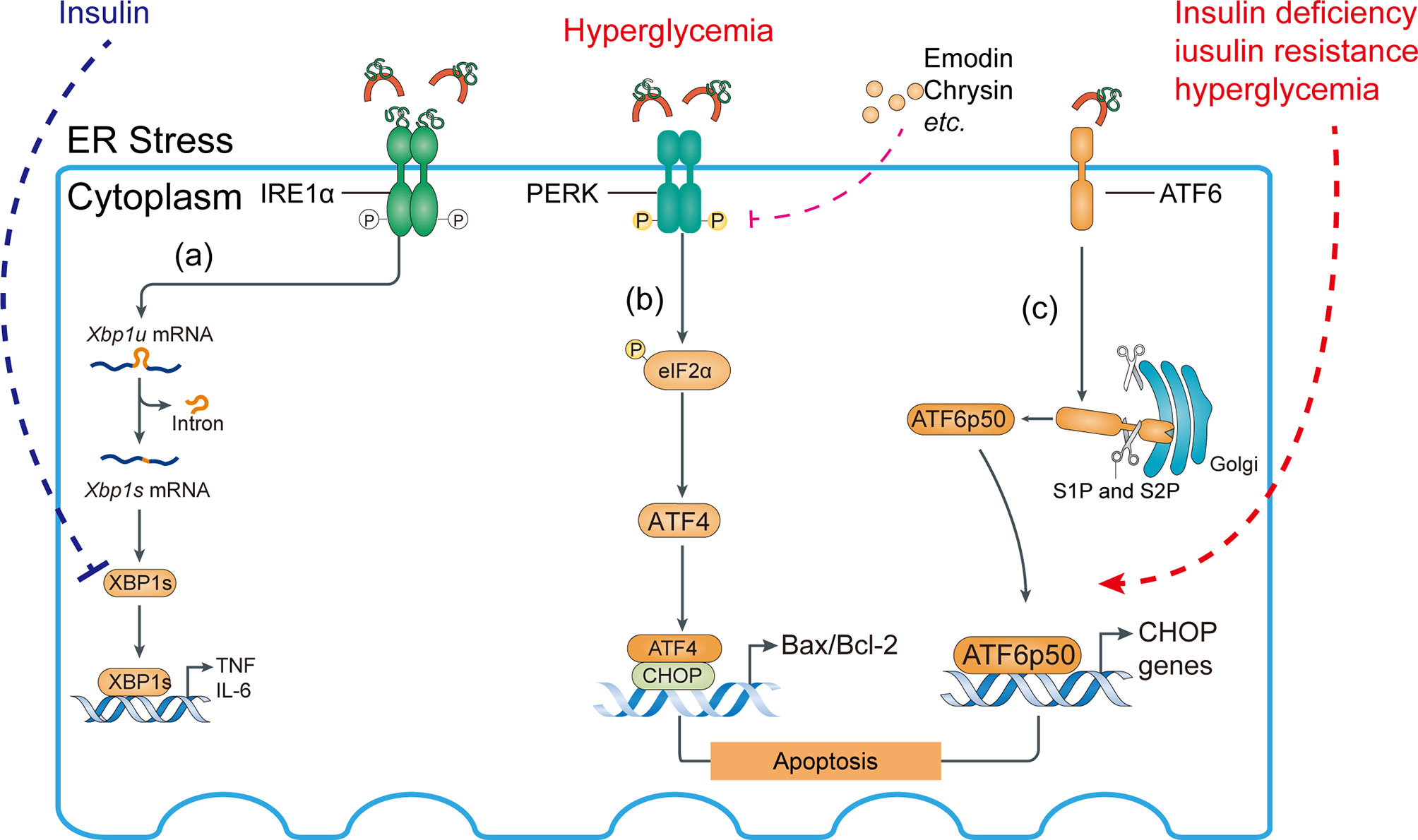
Figure 6 Activation of ATF6 pathway and abnormal IRE1 pathway may aggravate ER stress response. (A) Role of the IRE1 pathway in the ER stress response. Insulin signaling inhibits the interaction of XBP1s with the phosphatidylinositol 3-kinase (PI3K) regulatory subunits p-85α and p-85β, suppressing proinflammatory cytokine production. (B) Compounds such as emodin and chrysin inhibit PERK activation and decrease the Bax/Bcl-2 ratio in hyperglycemia, preventing podocyte apoptosis. (C) Insulin deficiency, insulin resistance, and hyperglycemia aggravate site 1 protease (S1P)- and S2P-mediated cleavage of ATF6α, allowing their fragments to translocate to the nucleus.
After separating from BiP, UP-mediated phosphorylation of protein kinase RNA-like ER kinase (PERK) activates downstream activating transcript factor 5 (ATF5) (81–83) and its target genes such as Txnip, thus inducing inflammation of kidney cells, such as podocytes (84) (Figure 5E). PERK is also implicated in macrophage immunosuppression and its downstream targets include ATF4, ATF5, and TXNIP (85).
Eukaryotic translation initiation factor-2α (eIF2α) phosphorylation reduces the translation of most mRNAs by inhibiting the delivery of the initiator Met tRNAi to the initiation complex, leading to reduced protein translation and protein loading into the ER, thereby alleviating the early stage of ER stress (86–88). Acute diseases typically benefit from an adaptive UPR, but chronic diseases (such as hyperglycemia) may be adversely affected by long-term or continuous UPR activation (89). In fact, eIF2α promotes the translation of a number of mRNAs containing short upstream open reading frames such as ATF4, activating downstream factors (e.g., CHOP, Bax/Bcl-2 transcriptional expression) and inducing apoptosis (86). Unfortunately, the mechanism responsible for the transition of the UPR from an adaptive to a cytotoxic response is unclear (Figure 6B).
In the Golgi apparatus, proteins are continuously cleaved to release cytosolic effectors or proteins from the membrane and allow them to enter the nucleus. Under ER stress, the transport of ATF6 from the ER to the Golgi apparatus is mediated by Golgi-resident site 1 protease (S1P) and S2P in an intramembrane region to release the cytoplasmic DNA binding protein portion, the ATF6 fragment. The ATF6 fragment translocates to the nucleus to induce gene expression (90), including UPR target genes (Figure 6C).
All branches of the UPR may be linked to podocyte apoptosis. The UPR is activated by increased expression of BiP, p-IRE1α, p-eIF2α, CHOP, ATF-6, and XBP1s in hyperglycemic podocytes in vitro and in vivo (91). ER stress activates the PERK pathway, upregulating the expression of downstream factors related to apoptosis such as Bax, leading to podocyte apoptosis. Increased phosphorylated PERK (p-PERK), phosphorylated eIF2α (p-eIF2α), ATF4 and CHOP increased the Bax/Bcl-2 ratio, promoting podocyte apoptosis (92, 93). By contrast, there are reversed results after using shRNA or knockdown PERK directly and thus podocyte apoptosis was controlled. Inhibition of PERK maintained the structure and function of the glomerular filtration barrier by increasing the production of the slit-diaphragm proteins podocin/nephrin (92). However, there is no information on the other two UPR branches. Confusingly, suppression of the PERK pathway under ER stress exacerbated β-cell apoptosis in thapsigargin-induced primary human islets, mouse islets, and INS-1 beta cells (94). It is possible that activated PERK induces phosphorylation of eIF2α, suppressing protein translation and loading into the ER and thereby alleviating ER stress. Therefore, PERK is implicated in podocyte apoptosis in DN, but the mechanism warrants further research.
Histone modification and DNA methylation status are altered by hyperglycemia, causing chromatin structure to change from concentration (K9Me and K27Me) to relaxation (K4Me), activating the expression of pathogenic genes (Figure 4) (95). Various mechanisms can enable transcription factors and cofactors to access promoters and enhancers through histone acetylation, to enhance gene expression (96). However, insulin or anti-diabetic drugs continue to promote the expression of pathogenic genes after normoglycemia. The use of epigenetic drugs or induction of site-specific changes, such as locus modification or DNA methylation, may eliminate podocytopathies.
The structure-activity relationship (SAR) approach based on the resveratrol structure showed BF175 to be an effective SIRT1 agonist, activated by the downstream transcription factor PGC-1α. The protective effect of BF175 on diabetes podocytes was SIRT1 dependent (44). METTL14 expression was highest in mice with Adriamycin-induced DNA and an elevated m6A RNA level. Furthermore, METTL14 levels were increased in renal biopsies from diabetics, models of DN, and human podocytes cultured with advanced glycation end products. Mice with podocyte-specific METTL14 deletions had improved glomerular function and alleviated podocyte injury, which was characterized by activation of autophagy and inhibition of apoptosis and inflammation (97).
There is an increase in METTL3 in podocytes from renal biopsy samples from patients with DN, which is associated with renal damage, in addition to METTL14 (46). METTL3 Flox/Flox (METTL3fl/fl) mouse lines have been used as METTL3-deficient models (97). Further, STZ-induced diabetic mice with METTL3 podocyte-conditional knockouts showed significant improvement of podocyte injury and albuminuria. As a result of METTL3, TAB3 was modified by m6A, and its stability increased in a manner dependent on IGF2BP2. Genetic and pharmacological inhibition of METTL3 can attenuate renal injury and inflammation, suggesting that the METTL3/TAB3 axis is a potential therapeutic target in podocytopathies (98).
TXNIP-mediated oxidative stress and NLRP3 inflammasome activation has been decribed previously. A study by Yunjun Xiao et al. has shown that H3K27me3 is reduced trimethylated and its occupancy at promoters of early growth response 1 (EGR1) is enhanced by histone methyltransferase enhancer of zeste homolog 2 (EZH2). S-adenosylhomocysteine (SAH) is hydrolyzed by SAH hydrolase (SAHH) to homocysteine and adenosine. Adenosine dialdehyde (ADA), an inhibitor of SAHH, accumulates intracellular or plasma levels of the SAH levels and may exert as a regulator to hyperglycaemia-induced podocytopathy (72).
KLF4 epigenetics regulate podocyte phenotype and function, and the podocyte epigenome can be used to ameliorate proteinuria. A ChIP analysis of DNA methyltransferases (DNMTs) showed that DNMT1 binding to the nephrin promoter region was significantly reduced in KLF4-overexpressing podocytes, but its binding to the vimentin promoter was unaffected (99). To confirm that the transient upregulation of KLF4 has an extended effect on podocyte function, the Tet-On gene induction system induced by doxycycline was used to construct mice with KLF4-inducible and podocyte-specific overexpression. Doxycycline increased the expression of KLF4, and KLF4 was co-located with the podocyte marker nephrin, but not with the endothelial or mesangial marker desmin. Therefore, doxycycline may play an important role in the epigenetic regulation of KLF4 in podocytes.
Hyperglycemia suppresses RCAN1 expression in cultured podocytes, which is alleviated by pretreatment with the DNA methyltransferase inhibitor 5-Aza-2’-deoxycytidine. Mechanistically, increased expression of RCAN1 decreased hyperglycemia-induced nuclear factor of activated T cells (NFAT) transcriptional activity. Increased RCAN1 expression also stabilized actin cytoskeleton organization, consistent with inhibition of the calcineurin pathway (100). Hence, 5-Aza-20-deoxycytidine may suppress the expression of CXCL2, NFAT, and Wnt6 in hyperglycemic podocytes (Table 1).
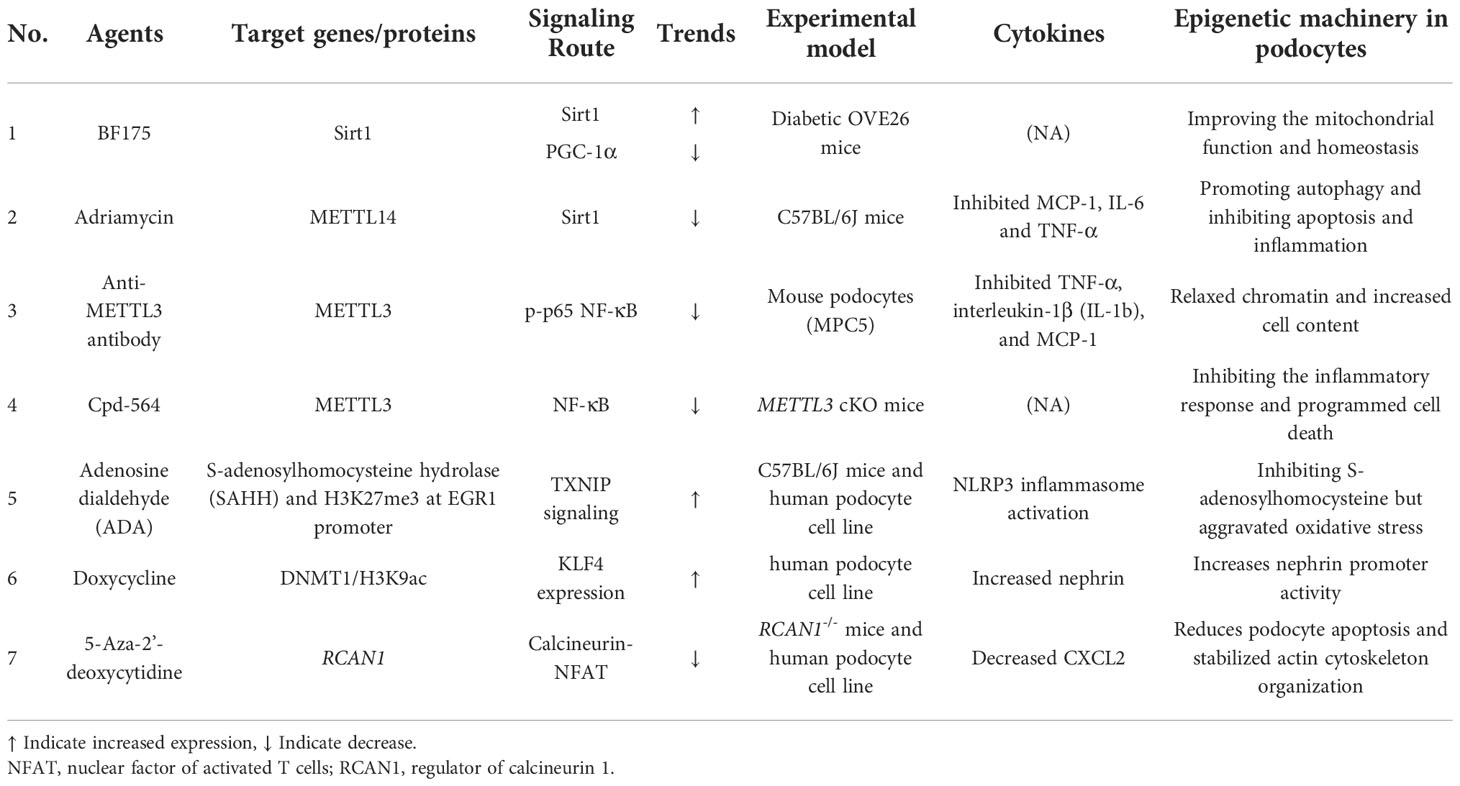
Table 1 Reports of epigenetic drug treatment on podocytes during DN progression.
miRNA-mediated epigenetic regulation of inflammatory gene expression is implicated in diabetes complications (101). MicroRNA-10 targets the NLRP3 inflammasome to suppress inflammation in diabetic kidneys. Patients with DKD and diabetic mice with miR-10a/b had low levels of miR-10a and -10b, which are expressed predominantly in podocytes. When miR-10a and b were delivered to the kidneys, NLRP3 inflammasomes were not activated and renal inflammation was prevented. By contrast, miR-10a/b knockdown increased activation of the NLRP3 inflammasome. Thus, upregulation of miR-10a/b could prevent podocytopathy in hyperglycemics (102) (Figure 7).
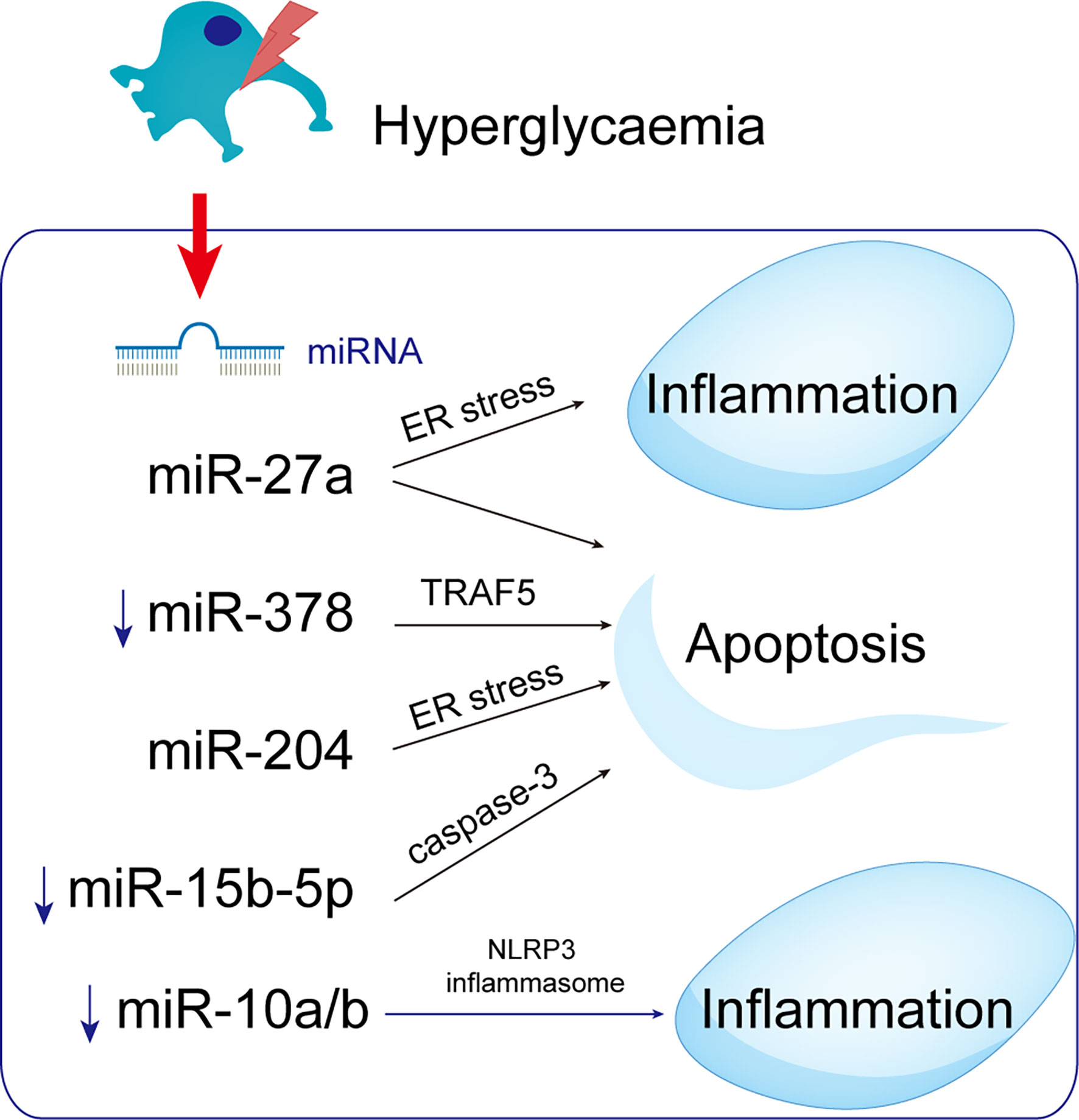
Figure 7 MicroRNAs with potential roles in podocytopathies. Hyperglycemia up- or down-regulates miRNAs in podocytes, resulting in podocytopathies. ↑ Indicate increased expression, ↓ Indicate decrease.
miRNA-27a is upregulated in podocytes of DN patients, possibly enhancing podocyte migration, diffusion, and apoptosis (103). Furthermore, miRNA-27a causes podocyte injury and apoptosis by targeting ER stress (52). The expression of miR-378 was downregulated in DN rats and high glucose-mediated podocytes, promoting podocyte apoptosis, which was reversed by inhibiting the expression of tumor-necrosis factor receptor associated factor 5 (TRAF5) by miR-378 (104). Moreover, miRNAs may be related to DN pathogenesis because they affect the expression of genes in the UPR signaling pathways. MiR-204 directly inhibits its target PERK under ER stress, exacerbating ER stress-induced apoptosis (105). MiR-204 targets the insulin transcription factor MAFA and thereby inhibits insulin translation and synthesis, indirectly activating ER stress (105–107). MiR-15b-5p was downregulated in patients with DN and could cause increased apoptosis in human kidney cells supported by elevated active caspase-3 and decreased viability and proliferation (108), linking miRNAs to the pathogenesis of DN.
As a competitive RNA (ceRNA), lncRNAs interact with and competitively regulate microRNAs (52, 53, 109). When lncRNA MALAT1 is decreased, microRNA let-7f is upregulated and KLF5 is inhibited, reducing podocyte damage in DN (110). miR-130a-3p competes with TLR4 as an endogenous sponge, as well as TLR4 as a miR-130a-3p target gene. As a result of miR-130a-3p/TLR4 crosstalk, MIAT knockdown protected podocytes from hyperglycemia-induced damage. Reduced MIAT expression restores slit-diaphragm integrity, attenuates foot process effacement, prevents dedifferentiation, and suppresses mitogenic catastrophe in podocytes during hyperglycemia. In response to ER stress, lnc-MGC and miR-379 cluster miRNAs are increased, and DN phenotypes (hypertrophy and fibrosis) are aggravated (111). Those findings show that miRNAs and lncRNAs are related to ER stress.
The lncRNA plasmacytoma variant translocation 1 (PVT1; 1.9 kb) is linked to kidney disease and encodes many alternative transcripts but not a protein. Silencing of PVT1 inhibits podocyte damage and apoptosis via the forkhead box A1 (FOXA1) in DN, which is of clinical importance (112). On human chromosome 20, small nucleolar RNA host gene 17 (SNHG17) is highly expressed in colorectal cancer tissues (113). SNHG17 controls mitophagy-induced apoptosis in diabetic podocytes. lncRNA SNHG17 knockdown promotes Parkin-dependent mitophagy and reduces apoptosis of podocytes by regulating the degradation of mammalian sterile 20-like kinase 1 (MST1) (114).
The AKT/mTOR pathway inhibits autophagy in a variety of cell types, including cancer cells, cardiomyocytes, and podocytes (115). The SPAG5/AKT/mTOR pathway inhibits podocyte autophagy and aggravates apoptosis. Therefore, SPAG5-AS1/SPAG5 has therapeutic potential for podocytopathies and DN (116) (Figure 8).
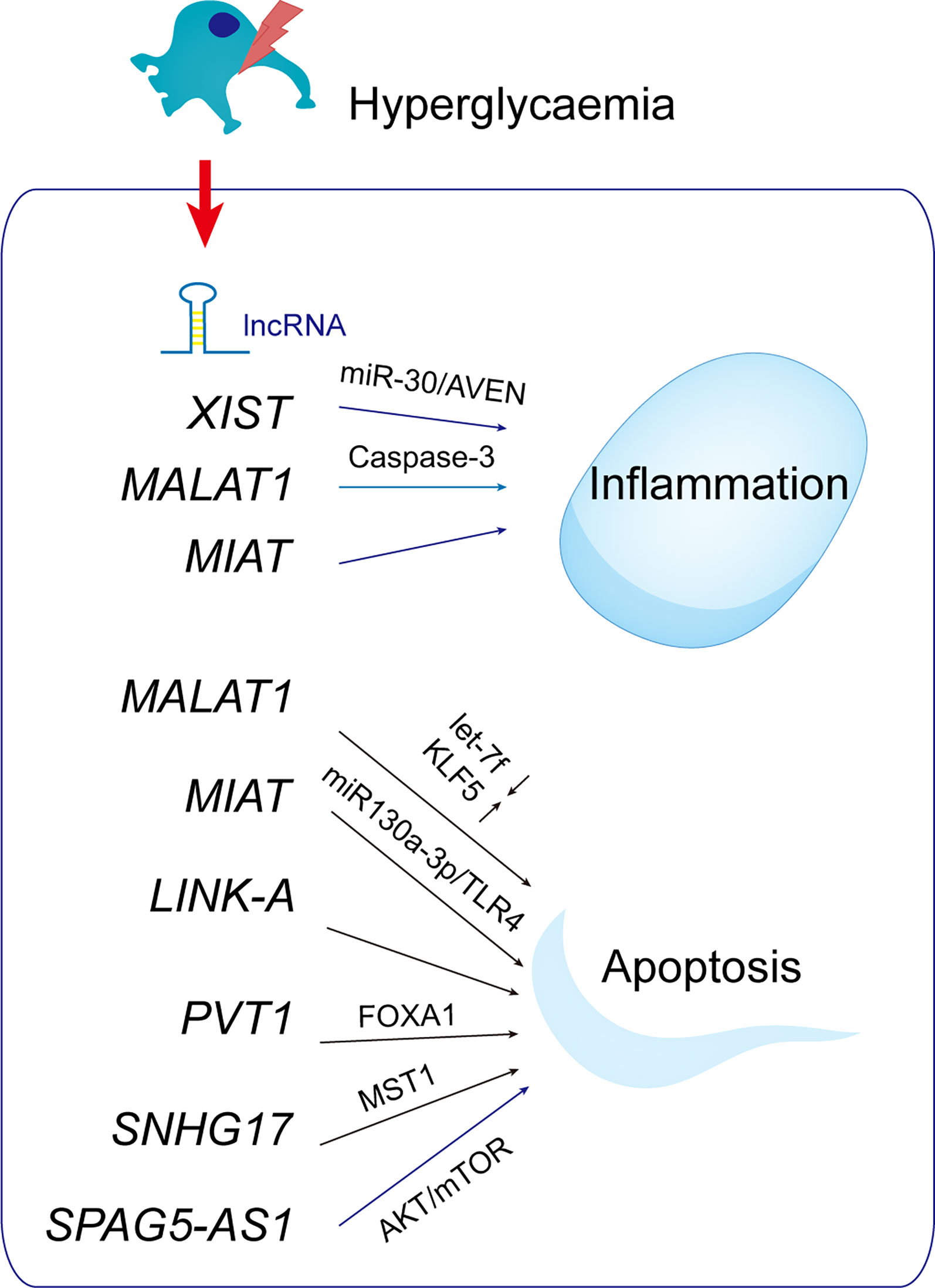
Figure 8 LncRNAs with potential roles in podocytopathies. Hyperglycemia up- or down-regulates lncRNAs in podocytes, resulting in podocytopathies.
There has been considerable interest in targeting ER stress as a therapy to improve DN. A variety of natural compounds inhibit ER stress and so may have therapeutic value for podocytopathies.
Natural products that modulate ER stress are listed in Table 2. ER stress can be regulated by inhibiting UPR sensors and their downstream factors. Emodin (93), chrysin (92), berberine (119), huaiqihuang (118) and epigallocatechin 3-gallate (EGCG) (122) inhibit the PERK signaling pathway to alleviate ER stress and improve DN. MC-TG (117) reduces ER stress and inflammation, possibly by inhibiting IRE1/NF-κB. Also, OA and NAC (120) inhibit the three UPR sensors to suppress ER stress. ER stress is triggered by impaired SERCA2 activity or expression, hampering Ca2+ homeostasis (124). AS-IV may alleviate ER stress by increasing SERCA2 expression, thus restoring intracellular Ca2+ homeostasis (121). ER chaperones such as 4-phenylbutyrate (4-PBA) and ursodeoxycholic acid (UDCA) enhance the folding capacity of the ER (91, 123). Therefore, natural compounds have potential for inhibiting ER stress and thus attenuating DN.
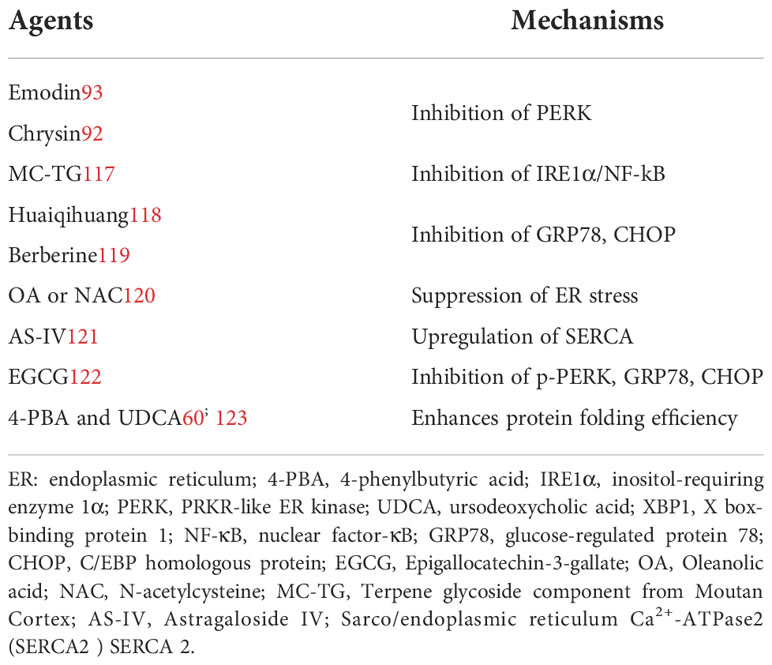
Table 2 Therapies targeting ER stress.
Preventing ER stress can improve DN and attenuate oxidative stress. The effect of hyperglycemia-mediated oxidative stress on podocytes suggests that podocyte oxidation is activated by upregulating ER markers after exposure to high glucose for 24 hours, whereas inhibition of ER stress by ER inhibitors diminishes oxidative stress and exerts a renoprotective effect (60). Similarly, crosstalk between ER stress and oxidative stress mediated by aldosterone contributes to podocyte injury, which can be ameliorated by berberine (119). Therefore, novel therapeutic strategies aimed at attenuating mitochondrial dysfunction may ameliorate palmitic acid-induced podocyte injury.
Autophagy delivers proteins and damaged organelles to lysosomes for degradation and recycling to maintain intercellular hemostasis. Autophagic self-repair is important in neurons, podocytes, and other cells in the anaphase of division because their differentiation and proliferation are limited (125). Resveratrol attenuated hyperglycemia-induced apoptosis by activating autophagy in db/db mice and podocytes (51). miR-383-5p overexpression significantly inhibited autophagy and enhanced apoptosis, effects reversed by resveratrol. Therefore, modulation of autophagy may be a novel therapeutic approach for DN. Activated eIF2α upregulates the mRNA level of ATF4, which transcriptionally regulates autophagy factors to induce autophagy. Also, phosphorylation-dependent selective translation upregulates Atg12 expression and stimulates Atg5-Atg12-Atg16 complex formation, thereby initiating autophagy. This could promote cell survival via autophagic decomposition of cellular components to generate energy under adverse conditions (126). Therefore, modulation of the crosstalk between ER stress and autophagy may have therapeutic potential for DN.
A hyperglycemic environment can induce podocytopathy. As podocyte function declines, so does glomerular function. Podocyte inflammation and apoptosis are the main causes and characteristics of early proteinuria in DN. Although epigenetic mediators (e.g., HDACs and DNMTs) are not genes, they regulate inflammation and apoptosis, and modulate the inflammatory response and apoptosis induced by ER stress in hyperglycemia. Therefore, protecting podocytes from deleterious effects can maintain the filter membrane, and thereby prevent DN. Natural drugs may also be useful for treating podocytopathy by transcriptionally regulating downstream factors of ER stress. We reviewed the epigenetic mechanisms and natural substances that regulate ER stress and inflammatory signaling pathways in podocytosis, with the aim of identifying new therapeutic targets in progressive DN.
XW and JR reviewed the literature and drafted the manuscript. JZ and YL participated in the conception and interpretation of the relevant literature for the manuscript. The final version of this review has been edited, revised critically, and approved by all authors.
This work was supported by research grants from the Project of Guangdong Administration of Traditional Chinese Medicine (No. 20221273), Shenzhen Longhua District Science and Technology Innovation Fund Projects (Nos. 2022045, 2020036, and 2020152).
We would like to give our sincere gratitude to all the reviewers for their helpful comments on this paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF diabetes atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Gnudi L, Coward RJM, Long DA. Diabetic nephropathy: Perspective on novel molecular mechanisms. Trends Endocrinol Metab (2016) 11:820–30. doi: 10.1016/j.tem.2016.07.002
3. Wang X, Wu T, Ma H, Huang X, Huang K, Ye C, et al. VX-765 ameliorates inflammation and extracellular matrix accumulation by inhibiting the NOX1/ROS/NF-κB pathway in diabetic nephropathy. J Pharm Pharmacol (2022) 3:377–86. doi: 10.1093/jpp/rgab112
4. Majewski C, Bakris GL. Has RAAS blockade reached its limits in the treatment of diabetic nephropathy? Curr Diab Rep (2016) 4:24. doi: 10.1007/s11892-016-0713-y
5. Eisa NH, Khodir AE, El-Sherbiny M, Elsherbiny NM, Said E. Phenethyl isothiocyanate attenuates diabetic nephropathy via modulation of glycative/oxidative/inflammatory signaling in diabetic rats. Biomed Pharmacother (2021) 142:111666. doi: 10.1016/j.biopha.2021.111666
6. Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol (2011) 6:327–40. doi: 10.1038/nrneph.2011.51
7. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: Challenges, progress, and possibilities. Clin J Am Soc Nephrol (2017) 12:2032–45. doi: 10.2215/CJN.11491116
8. Roscioni SS, Heerspink HJL, de Zeeuw D. The effect of RAAS blockade on the progression of diabetic nephropathy. Nat Rev Nephrol (2014) 2:77–87. doi: 10.1038/nrneph.2013.251
9. Liu X-Q, Jiang L, Lei L, Nie Z-Y, Zhu W, Wang S, et al. Carnosine alleviates diabetic nephropathy by targeting GNMT, a key enzyme mediating renal inflammation and fibrosis. Clin Sci (2020) 23:3175–93. doi: 10.1042/CS20201207
10. Calle P, Hotter G. Macrophage phenotype and fibrosis in diabetic nephropathy. Int J Mol Sci (2020) 8:2806–20. doi: 10.3390/ijms21082806
11. Tang SCW, Yiu WH. Innate immunity in diabetic kidney disease. Nat Rev Nephrol (2020) 4:206–22. doi: 10.1038/s41581-019-0234-4
12. You H, Gao T, Cooper TK, Brian Reeves W, Awad AS. Macrophages directly mediate diabetic renal injury. Am J Physiol Renal Physiol (2013) 12:F1719–27. doi: 10.1152/ajprenal.00141.2013
13. de Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al. The effect of CCR2 inhibitor CCX140-b on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol (2015) 9:687–96. doi: 10.1016/S2213-8587(15)00261-2
14. Yi H, Peng R, Zhang L-Y, Sun Y, Peng H-M, Liu H-D, et al. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis (2017) 2:e2583. doi: 10.1038/cddis.2016.451
15. Sriwijitkamol A, Christ-Roberts C, Berria R, Eagan P, Pratipanawatr T, DeFronzo RA, et al. Reduced skeletal muscle inhibitor of kappaB beta content is associated with insulin resistance in subjects with type 2 diabetes: reversal by exercise training. Diabetes (2006) 3:760–7. doi: 10.2337/diabetes.55.03.06.db05-0677
16. de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med (2013) 26:2492–503. doi: 10.1056/NEJMoa1306033
17. Kato M, Natarajan R. Diabetic nephropathy–emerging epigenetic mechanisms. Nat Rev Nephrol (2014) 9:517–30. doi: 10.1038/nrneph.2014.116
18. Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet (2011) 8:529–41. doi: 10.1038/nrg3000
19. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev (2009) 7:781–3. doi: 10.1101/gad.1787609
20. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (2012) 6099:1190–5. doi: 10.1126/science.1222794
21. Maezawa Y, Takemoto M, Yokote K. Cell biology of diabetic nephropathy: Roles of endothelial cells, tubulointerstitial cells and podocytes. J Diabetes Investig (2015) 1:3–15. doi: 10.1111/jdi.12255
22. Unnersjö-Jess D, Butt L, Höhne M, Witasp A, Kühne L, Hoyer PF, et al. A fast and simple clearing and swelling protocol for 3D in-situ imaging of the kidney across scales. Kidney Int (2021) 4:1010–20. doi: 10.1016/j.kint.2020.10.039
23. Nagata M. Podocyte injury and its consequences. Kidney Int (2016) 6:1221–30. doi: 10.1016/j.kint.2016.01.012
24. Ke G, Chen X, Liao R, Xu L, Zhang L, Zhang H, et al. Receptor activator of NF-κB mediates podocyte injury in diabetic nephropathy. Kidney Int (2021) 2:377–90. doi: 10.1016/j.kint.2021.04.036
25. Marciniak SJ, Chambers JE, Ron D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat Rev Drug Discovery (2022) 2:115–40. doi: 10.1038/s41573-021-00320-3
26. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
27. Lindenmeyer MT, Rastaldi MP, Ikehata M, Neusser MA, Kretzler M, Cohen CD, et al. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol (2008) 11:2225–36. doi: 10.1681/ASN.2007121313
28. Dai H, Liu Q, Liu B. Research progress on mechanism of podocyte depletion in diabetic nephropathy. J Diabetes Res (2017) 2615286:2615286–297. doi: 10.1155/2017/2615286
29. Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev (2003) 1:253–307. doi: 10.1152/physrev.00020.2002
30. Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol (2015) 4:F287–97. doi: 10.1152/ajprenal.00533.2014
31. Lan X, Kumar V, Jha A, Aslam R, Wang H, Chen K, et al. EDA2R mediates podocyte injury in high glucose milieu. Biochimie (2020) 174:74–83. doi: 10.1016/j.biochi.2020.04.003
32. Loeffler I, Wolf G. Epithelial-to-Mesenchymal transition in diabetic nephropathy: Fact or fiction? Cells (2015) 4:631–52. doi: 10.3390/cells4040631
33. Lu C-C, Wang G-H, Lu J, Chen P-P, Zhang Y, Hu Z-B, et al. Role of podocyte injury in glomerulosclerosis. Adv Exp Med Biol (2019) 1165:195–232. doi: 10.1007/978-981-13-8871-2_10
34. Jo HA, Kim J-Y, Yang SH, Han SS, Joo KW, Kim YS, et al. The role of local IL6/JAK2/STAT3 signaling in high glucose-induced podocyte hypertrophy. Kidney Res Clin Pract (2016) 4:212–8. doi: 10.1016/j.krcp.2016.09.003
35. Pozzi A, Zent R. Integrins in kidney disease. J Am Soc Nephrol (2013) 7:1034–9. doi: 10.1681/ASN.2013010012
36. Jim B, Ghanta M, Qipo A, Fan Y, Chuang PY, Cohen HW, et al. Dysregulated nephrin in diabetic nephropathy of type 2 diabetes: a cross sectional study. PloS One (2012) 5:e36041. doi: 10.1371/journal.pone.0036041
37. Vogelmann SU, Nelson WJ, Myers BD, Lemley KV. Urinary excretion of viable podocytes in health and renal disease. Am J Physiol Renal Physiol (2003) 1:F40–8. doi: 10.1152/ajprenal.00404.2002
38. Xin W, Li Z, Xu Y, Yu Y, Zhou Q, Chen L, et al. Autophagy protects human podocytes from high glucose-induced injury by preventing insulin resistance. Metabolism (2016) 9:1307–15. doi: 10.1016/j.metabol.2016.05.015
39. Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest (2010) 4:1084–96. doi: 10.1172/JCI39492
40. Yuan Y, Xu X, Zhao C, Zhao M, Wang H, Zhang B, et al. The roles of oxidative stress, endoplasmic reticulum stress, and autophagy in aldosterone/mineralocorticoid receptor-induced podocyte injury. Lab Invest (2015) 12:1374–86. doi: 10.1038/labinvest.2015.118
41. Tang Y, Lin Y-H, Ni H-Y, Dong J, Yuan H-J, Zhang Y, et al. Inhibiting histone deacetylase 2 (HDAC2) promotes functional recovery from stroke. J Am Heart Assoc (2017) 10:7236–65. doi: 10.1161/JAHA.117.007236
42. Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting notch signaling. Nat Commun (2017) 1:413. doi: 10.1038/s41467-017-00498-4
43. Morigi M, Perico L, Benigni A. Sirtuins in renal health and disease. J Am Soc Nephrol (2018) 7:1799–809. doi: 10.1681/ASN.2017111218
44. Hong Q, Zhang L, Das B, Li Z, Liu B, Cai G, et al. Increased podocyte sirtuin-1 function attenuates diabetic kidney injury. Kidney Int (2018) 6:1330–43. doi: 10.1016/j.kint.2017.12.008
45. Sapienza C, Lee J, Powell J, Erinle O, Yafai F, Reichert J, et al. DNA Methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics (2011) 1:20–8. doi: 10.4161/epi.6.1.13362
46. Jiang L, Liu X, Hu X, Gao L, Zeng H, Wang X, et al. METTL3-mediated mA modification of TIMP2 mRNA promotes podocyte injury in diabetic nephropathy. Mol Ther (2022) 4:1721–40. doi: 10.1016/j.ymthe.2022.01.002
47. Muñoz IM, Rouse J. Control of histone methylation and genome stability by PTIP. EMBO Rep (2009) 3:239–45. doi: 10.1038/embor.2009.21
48. Sankrityayan H, Kulkarni YA, Gaikwad AB. Diabetic nephropathy: The regulatory interplay between epigenetics and microRNAs. Pharmacol Res (2019) 141:574–85. doi: 10.1016/j.phrs.2019.01.043
49. Wei B, Liu Y-S, Guan H-X. MicroRNA-145-5p attenuates high glucose-induced apoptosis by targeting the notch signaling pathway in podocytes. Exp Ther Med (2020) 3:1915–24. doi: 10.3892/etm.2020.8427
50. Deshpande S, Abdollahi M, Wang M, Lanting L, Kato M, Natarajan R. Reduced autophagy by a microRNA-mediated signaling cascade in diabetes-induced renal glomerular hypertrophy. Sci Rep (2018) 1:6954. doi: 10.1038/s41598-018-25295-x
51. Huang S-S, Ding D-F, Chen S, Dong C-L, Ye X-L, Yuan Y-G, et al. Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Sci Rep (2017) 7:45692. doi: 10.1038/srep45692
52. Bai X, Geng J, Li X, Wan J, Liu J, Zhou Z, et al. Long noncoding RNA LINC01619 regulates MicroRNA-27a/Forkhead box protein O1 and endoplasmic reticulum stress-mediated podocyte injury in diabetic nephropathy. Antioxid Redox Signal (2018) 4:355–76. doi: 10.1089/ars.2017.7278
53. Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet (2016) 5:272–83. doi: 10.1038/nrg.2016.20
54. Athanasopoulos D, Karagiannis G, Tsolaki M. Recent findings in Alzheimer disease and nutrition focusing on epigenetics. Adv Nutr (2016) 5:917–27. doi: 10.3945/an.116.012229
55. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol (2017) 11:681–96. doi: 10.1038/nrneph.2017.129
56. Gallazzini M, Pallet N. Endoplasmic reticulum stress and kidney dysfunction. Biol Cell (2018) 9:205–16. doi: 10.1111/boc.201800019
57. Shu S, Zhu J, Liu Z, Tang C, Cai J, Dong Z. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine (2018) 37:269–80. doi: 10.1016/j.ebiom.2018.10.006
58. Fan Y, Zhang J, Xiao W, Lee K, Li Z, Wen J, et al. Rtn1a-mediated endoplasmic reticulum stress in podocyte injury and diabetic nephropathy. Sci Rep (2017) 1:323. doi: 10.1038/s41598-017-00305-6
59. Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (2014) 8:629–40. doi: 10.1093/abbs/gmu048
60. Cao A-L, Wang L, Chen X, Wang Y-M, Guo H-J, Chu S, et al. Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Lab Invest (2016) 6:610–22. doi: 10.1038/labinvest.2016.44
61. Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest (2011) 6:2181–96. doi: 10.1172/JCI44771
62. Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, et al. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes (2005) 3:657–63. doi: 10.2337/diabetes.54.3.657
63. Ramos-Lopez O, Riezu-Boj JI, Milagro FI, Moreno-Aliaga MJ, Martinez JA. Endoplasmic reticulum stress epigenetics is related to adiposity, dyslipidemia, and insulin resistance. Adipocyte (2018) 2:137–42. doi: 10.1080/21623945.2018.1447731
64. Jialal I, Major AM, Devaraj S. Global toll-like receptor 4 knockout results in decreased renal inflammation, fibrosis and podocytopathy. J Diabetes Complications (2014) 6:755–61. doi: 10.1016/j.jdiacomp.2014.07.003
65. Lin M, Yiu WH, Wu HJ, Chan LYY, Leung JCK, Au WS, et al. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol (2012) 1:68–102. doi: 10.1681/ASN.2010111210
66. Wei M, Li Z, Xiao L, Yang Z. Effects of ROS-relative NF-κB signaling on high glucose-induced TLR4 and MCP-1 expression in podocyte injury. Mol Immunol (2015) 2 Pt A:261–71. doi: 10.1016/j.molimm.2015.09.002
67. Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes (2008) 11:3090–8. doi: 10.2337/db08-0564
68. Kaur H, Chien A, Jialal I. Hyperglycemia induces toll like receptor 4 expression and activity in mouse mesangial cells: relevance to diabetic nephropathy. Am J Physiol Renal Physiol (2012) 8:F1145–50. doi: 10.1152/ajprenal.00319.2012
69. Cha JJ, Hyun YY, Lee MH, Kim JE, Nam DH, Song HK, et al. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinology (2013) 6:2144–55. doi: 10.1210/en.2012-2080
70. Wang X, Rao J, Tan Z, Xun T, Zhao J, Yang X. Inflammatory signaling on cytochrome P450-mediated drug metabolism in hepatocytes. Front Pharmacol (2022) 13:1043836. doi: 10.3389/fphar.2022.1043836
71. Wang X, Ye C, Xun T, Mo L, Tong Y, Ni W, et al. Polysaccharide a ameliorates abnormal voriconazole metabolism accompanied with the inhibition of TLR4/NF-κB pathway. Front Pharmacol (2021) 12:663325. doi: 10.3389/fphar.2021.663325
72. Dai X, Liao R, Liu C, Liu S, Huang H, Liu J, et al. Epigenetic regulation of TXNIP-mediated oxidative stress and NLRP3 inflammasome activation contributes to SAHH inhibition-aggravated diabetic nephropathy. Redox Biol (2021) 102033:102033–47. doi: 10.1016/j.redox.2021.102033
73. Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int (2015) 87(1):74–84. doi: 10.1038/ki.2014.271
74. Gao P, He F-F, Tang H, Lei C-T, Chen S, Meng X-F, et al. NADPH oxidase-induced NALP3 inflammasome activation is driven by thioredoxin-interacting protein which contributes to podocyte injury in hyperglycemia. J Diabetes Res (2015) 87:504761. doi: 10.1155/2015/504761
75. Fakhruddin S, Alanazi W, Jackson KE. Diabetes-induced reactive oxygen species: Mechanism of their generation and role in renal injury. J Diabetes Res (2017) 2017:8379327. doi: 10.1155/2017/8379327
76. Zhao M, Bai M, Ding G, Zhang Y, Huang S, Jia Z, et al. Angiotensin II stimulates the NLRP3 inflammasome to induce podocyte injury and mitochondrial dysfunction. Kidney Dis (2018) 2:83–94. doi: 10.1159/000488242
77. Liu Y, Xu Z, Ma F, Jia Y, Wang G. Knockdown of TLR4 attenuates high glucose-induced podocyte injury via the NALP3/ASC/Caspase-1 signaling pathway. Biomed Pharmacother (2018) 107:1393–401. doi: 10.1016/j.biopha.2018.08.134
78. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest (2002) 10:1389–98. doi: 10.1172/JCI0216886
79. Packer M. Role of impaired nutrient and oxygen deprivation signaling and deficient autophagic flux in diabetic CKD development: Implications for understanding the effects of sodium-glucose cotransporter 2-inhibitors. J Am Soc Nephrol (2020) 5:907–19. doi: 10.1681/ASN.2020010010
80. Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell (2010) 140(3):338–48. doi: 10.1016/j.cell.2010.01.001
81. Manié SN, Lebeau J, Chevet E. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 3. orchestrating the unfolded protein response in oncogenesis: an update. Am J Physiol Cell Physiol (2014) 10:C901–7. doi: 10.1152/ajpcell.00292.2014
82. Ni L, Yuan C. The mitochondrial-associated endoplasmic reticulum membrane and its role in diabetic nephropathy. Oxid Med Cell Longev (2021) 2021:8054817. doi: 10.1155/2021/8054817
83. Yi H-S, Chang JY, Shong M. The mitochondrial unfolded protein response and mitohormesis: a perspective on metabolic diseases. J Mol Endocrinol (2018) 3:R91–105. doi: 10.1530/JME-18-0005
84. Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, et al. Identification of novel stress-induced genes downstream of chop. EMBO J (1998) 13:3619–30. doi: 10.1093/emboj/17.13.3619
85. Raines LN, Zhao H, Wang Y, Chen H-Y, Gallart-Ayala H, Hsueh P-C, et al. PERK is a critical metabolic hub for immunosuppressive function in macrophages. Nat Immunol (2022) 3:431–45. doi: 10.1038/s41590-022-01145-x
86. B’Chir W, Maurin A-C, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res (2013) 16:7683–99. doi: 10.1093/nar/gkt563
87. Chiang W-C, Hiramatsu N, Messah C, Kroeger H, Lin JH. Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest Ophthalmol Vis Sci (2012) 11:7159–66. doi: 10.1167/iovs.12-10222
88. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell (2018) 2:169–81. doi: 10.1016/j.molcel.2017.06.017
89. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 2:89–102. doi: 10.1038/nrm3270
90. Yoshida H, Nadanaka S, Sato R, Mori K. XBP1 is critical to protect cells from endoplasmic reticulum stress: evidence from site-2 protease-deficient Chinese hamster ovary cells. Cell Struct Funct (2006) 2:117–25. doi: 10.1247/csf.06016
91. Cao A, Wang L, Chen X, Guo H, Chu S, Zhang X, et al. Ursodeoxycholic acid ameliorated diabetic nephropathy by attenuating hyperglycemia-mediated oxidative stress. Biol Pharm Bull (2016) 8:1300–8. doi: 10.1248/bpb.b16-00094
92. Kang M-K, Park S-H, Kim Y-H, Lee E-J, Antika LD, Kim DY, et al. Chrysin ameliorates podocyte injury and slit diaphragm protein loss via inhibition of the PERK-eIF2α-ATF-CHOP pathway in diabetic mice. Acta Pharmacol Sin (2017) 8:1129–40. doi: 10.1038/aps.2017.30
93. Tian N, Gao Y, Wang X, Wu X, Zou D, Zhu Z, et al. Emodin mitigates podocytes apoptosis induced by endoplasmic reticulum stress through the inhibition of the PERK pathway in diabetic nephropathy. Drug Des Devel Ther (2018) 12:2195–211. doi: 10.2147/DDDT.S167405
94. Wiseman RL, Mesgarzadeh JS, Hendershot LM. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol Cell (2022) 8:1477–91. doi: 10.1016/j.molcel.2022.03.025
95. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol (2019) 6:327–45. doi: 10.1038/s41581-019-0135-6
96. Ryu H-Y, Hochstrasser M. Histone sumoylation and chromatin dynamics. Nucleic Acids Res (2021) 11:6043–52. doi: 10.1093/nar/gkab280
97. Lu Z, Liu H, Song N, Liang Y, Zhu J, Chen J, et al. METTL14 aggravates podocyte injury and glomerulopathy progression through n-methyladenosine-dependent downregulating of Sirt1. Cell Death Dis (2021) 10:881. doi: 10.1038/s41419-021-04156-y
98. Wang J-N, Wang F, Ke J, Li Z, Xu C-H, Yang Q, et al. Inhibition of attenuates renal injury and inflammation by alleviating m6A modifications via IGF2BP2-dependent mechanisms. Sci Transl Med (2022) 640:eabk2709. doi: 10.1126/scitranslmed.abk2709
99. Hayashi K, Sasamura H, Nakamura M, Azegami T, Oguchi H, Sakamaki Y, et al. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J Clin Invest (2014) 6:2523–37. doi: 10.1172/JCI69557
100. Li H, Zhang W, Zhong F, Das GC, Xie Y, Li Z, et al. Epigenetic regulation of RCAN1 expression in kidney disease and its role in podocyte injury. Kidney Int (2018) 6:1160–76. doi: 10.1016/j.kint.2018.07.023
101. Liu Y-J, Wang H, Zhong H-J, Chong C-M, Yang G-J. Editorial: Epigenetics of the immune component of inflammation. Front Immunol (2022) 13:1000836. doi: 10.3389/fimmu.2022.1000836
102. Ding H, Li J, Li Y, Yang M, Nie S, Zhou M, et al. MicroRNA-10 negatively regulates inflammation in diabetic kidney via targeting activation of the NLRP3 inflammasome. Mol Ther (2021) 7:2308–20. doi: 10.1016/j.ymthe.2021.03.012
103. Zhou Z, Wan J, Hou X, Geng J, Li X, Bai X. MicroRNA-27a promotes podocyte injury via PPARγ-mediated β-catenin activation in diabetic nephropathy. Cell Death Dis (2017) 3:e2658. doi: 10.1038/cddis.2017.74
104. Lei X, Zhang B-d, Ren J-G, Luo F-L. Astragaloside suppresses apoptosis of the podocytes in rats with diabetic nephropathy via miR-378/TRAF5 signaling pathway. Life Sci (2018) 2021:77–83. doi: 10.1016/j.lfs.2018.05.037
105. Xu G, Chen J, Jing G, Grayson TB, Shalev A. miR-204 targets PERK and regulates UPR signaling and β-cell apoptosis. Mol Endocrinol (2016) 8:917–24. doi: 10.1210/me.2016-1056
106. Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR, et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun (2015) 6:6496. doi: 10.1038/ncomms7496
107. Xu G, Chen J, Jing G, Shalev A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat Med (2013) 9:1141–6. doi: 10.1038/nm.3287
108. Shen H, Fang K, Guo H, Wang G. High glucose-induced apoptosis in human kidney cells was alleviated by miR-15b-5p mimics. Biol Pharm Bull (2019) 5:758–63. doi: 10.1248/bpb.b18-00951
109. Wang X, Wu S, Yang Y, Zhao J. LncRNA CARMN affects hepatocellular carcinoma prognosis by regulating the miR-192-5p/LOXL2 axis. Oxid Med Cell Longev (2022) 2022:9277360. doi: 10.1155/2022/9277360
110. Zhang H, Yan Y, Hu Q, Zhang X. LncRNA MALAT1/microRNA let-7f/KLF5 axis regulates podocyte injury in diabetic nephropathy. Life Sci (2021) 266:118794. doi: 10.1016/j.lfs.2020.118794
111. Kato M, Wang M, Chen Z, Bhatt K, Oh HJ, Lanting L, et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat Commun (2016) 7:12864–1280. doi: 10.1038/ncomms12864
112. Liu D-W, Zhang J-H, Liu F-X, Wang X-T, Pan S-K, Jiang D-K, et al. Silencing of long noncoding RNA PVT1 inhibits podocyte damage and apoptosis in diabetic nephropathy by upregulating FOXA1. Exp Mol Med (2019) 8:1–15. doi: 10.1038/s12276-019-0259-6
113. Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, et al. The PINK1-parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A (2013) 16:6400–5. doi: 10.1073/pnas.1221132110
114. Guo F, Wang W, Song Y, Wu L, Wang J, Zhao Y, et al. LncRNA SNHG17 knockdown promotes parkin-dependent mitophagy and reduces apoptosis of podocytes through Mst1. Cell Cycle (2020) 16:1997–2006. doi: 10.1080/15384101.2020.1783481
115. Jin J, Shi Y, Gong J, Zhao L, Li Y, He Q, et al. Exosome secreted from adipose-derived stem cells attenuates diabetic nephropathy by promoting autophagy flux and inhibiting apoptosis in podocyte. Stem Cell Res Ther (2019) 1:95. doi: 10.1186/s13287-019-1177-1
116. Xu J, Deng Y, Wang Y, Sun X, Chen S, Fu G. SPAG5-AS1 inhibited autophagy and aggravated apoptosis of podocytes via SPAG5/AKT/mTOR pathway. Cell Prolif (2020) 2:e12738. doi: 10.1111/cpr.12738
117. Chen J, Hou X-F, Wang G, Zhong Q-X, Liu Y, Qiu H-H, et al. Terpene glycoside component from moutan cortex ameliorates diabetic nephropathy by regulating endoplasmic reticulum stress-related inflammatory responses. J Ethnopharmacol (2016) 193:433–44. doi: 10.1016/j.jep.2016.09.043
118. Li T-X, Mao J-H, Huang L, Fu H-D, Chen S-H, Liu A-M, et al. Beneficial effects of huaiqihuang on hyperglycemia-induced MPC5 podocyte dysfunction through the suppression of mitochondrial dysfunction and endoplasmic reticulum stress. Mol Med Rep (2017) 2:1465–71. doi: 10.3892/mmr.2017.6753
119. Wang B, Xu X, He X, Wang Z, Yang M. Berberine improved aldo-induced podocyte injury via inhibiting oxidative stress and endoplasmic reticulum stress pathways both In vivo and In vitro. Cell Physiol Biochem (2016) 1:217–28. doi: 10.1159/000445618
120. Lee ES, Kim HM, Kang JS, Lee EY, Yadav D, Kwon M-H, et al. Oleanolic acid and n-acetylcysteine ameliorate diabetic nephropathy through reduction of oxidative stress and endoplasmic reticulum stress in a type 2 diabetic rat model. Nephrol Dial Transplant (2016) 3:391–400. doi: 10.1093/ndt/gfv377
121. Guo H, Cao A, Chu S, Wang Y, Zang Y, Mao X, et al. Astragaloside IV attenuates podocyte apoptosis mediated by endoplasmic reticulum stress through upregulating Sarco/Endoplasmic reticulum Ca-ATPase 2 expression in diabetic nephropathy. Front Pharmacol (2016) 27:500. doi: 10.3389/fphar.2016.00500
122. Kim H-S, Quon MJ, Kim J-A. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol (2014), 187–95. doi: 10.1016/j.redox.2013.12.022
123. Lee E-S, Aryal YP, Kim T-Y, Kim J-Y, Yamamoto H, An C-H, et al. Facilitation of reparative dentin using a drug repositioning approach with 4-phenylbutric acid. Front Physiol (2022) 13:885593. doi: 10.3389/fphys.2022.885593
124. Cardozo AK, Ortis F, Storling J, Feng Y-M, Rasschaert J, Tonnesen M, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes (2005) 2:452–61. doi: 10.2337/diabetes.54.2.452
125. Yang D, Livingston MJ, Liu Z, Dong G, Zhang M, Chen J-K, et al. Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol Life Sci (2018) 4:669–88. doi: 10.1007/s00018-017-2639-1
Keywords: epigenetics, endoplasmic reticulum, podocyte, diabetic nephropathy, inflammation signaling
Citation: Wang X, Zhao J, Li Y, Rao J and Xu G (2022) Epigenetics and endoplasmic reticulum in podocytopathy during diabetic nephropathy progression. Front. Immunol. 13:1090989. doi: 10.3389/fimmu.2022.1090989
Received: 06 November 2022; Accepted: 30 November 2022;
Published: 22 December 2022.
Edited by:
Guan-Jun Yang, Ningbo University, ChinaReviewed by:
Yongshu Li, Shenzhen University, ChinaCopyright © 2022 Wang, Zhao, Li, Rao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaokang Wang, a2FuZ3RhZV93b25AaS5zbXUuZWR1LmNu
†ORCID: Xiaokang Wang, orcid.org/0000-0002-4325-9907
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.