 Zhukai Cong
Zhukai Cong Cui Yang
Cui Yang Zhaojin Zeng1
Zhaojin Zeng1 Han Xiao
Han Xiao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 January 2023
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1090773
This article is part of the Research Topic Inflammation in Respiratory and Neurological Diseases and the immune-interaction of the lung-brain axis View all 17 articles
Introduction: Catecholamines such as norepinephrine or epinephrine have been reported to participate in the development of acute respiratory distress syndrome (ARDS) by activating adrenergic receptors (ARs). But the role of α1-AR in this process has yet to be elucidated.
Methods: In this study, ARDS mouse model was induced by intratracheal instillation of lipopolysaccharide. After treatment with α1-AR agonist phenylephrine or antagonist prazosin, lung pathological injury, alveolar barrier disruption and inflammation, and haemodynamic changes were evaluated. Cytokine levels and cell viability of alveolar macrophages were measured in vitro. Nuclear factor κB (NF-κB), mitogen-activated protein kinase, and Akt signalling pathways were analysed by western blot.
Results: It showed that α1-AR activation alleviated lung injuries, including reduced histopathological damage, cytokine expression, and inflammatory cell infiltration, and improved alveolar capillary barrier integrity of ARDS mice without influencing cardiovascular haemodynamics. In vitro experiments suggested that α1-AR stimulation inhibited secretion of TNF-α, IL-6, CXCL2/MIP-2, and promoted IL-10 secretion, but did not affect cell viability. Moreover, α1-AR stimulation inhibited NF-κB and enhanced ERK1/2 activation without significantly influencing p38, JNK, or Akt activation.
Discussion: Our studies reveal that α1-AR stimulation could ameliorate lipopolysaccharide-induced lung injury by inhibiting NF-κB and promoting ERK1/2 to suppress excessive inflammatory responses of alveolar macrophages.
Acute respiratory distress syndrome (ARDS) is a common critical illness characterized by acute hypoxic respiratory insufficiency or failure, often requiring hospitalisation in an intensive care unit (1). The life-threatening illness can be caused by a variety of non-cardiogenic factors, including pneumonia, sepsis, and trauma (2). Because of its multifactorial aetiology and complex pathogenesis, ARDS shows great heterogeneity across different subpopulations of patients (3). Although prior studies have made considerable progress in understanding the pathogenesis of ARDS, no effective drug interventions are currently available (4) and the morbidity and mortality rates remain high (5, 6) . This is especially important with the outbreak of Corona Virus Disease 2019 (COVID-19), the severe stage of which can lead to ARDS, bringing tremendous challenges to clinical treatment and basic research (7). Therefore, it is urgent to further explore the pathogenesis of ARDS and identify feasible therapeutic strategy.
An uncontrolled inflammatory response is generally regarded as the core mechanism resulting in diffuse alveolar damage and lung oedema (8). Clinical evidence suggests that cytokines and inflammatory cells in plasma or bronchoalveolar lavage fluid (BALF) of patients with ARDS are usually increased, and associated with mortality (9). Accordingly, regulation of the immune inflammatory response is considered a potential treatment strategy for ARDS (8).
In addition to its established role as a regulator of the cardiovascular system, a growing body of evidence indicates that sympathetic nervous system (SNS) is an integrative interface between the nervous system and the immune system (10). SNS dysfunction is common during sepsis (11, 12) and sepsis-induced complication, like ARDS (13), and can influence disease progression (14). Norepinephrine (NE), an important neurotransmitter released from the SNS, is an essential vasoactive agent used clinically to treat septic shock (15). Recent evidence suggests that NE could regulates inflammatory response of immune cells and participate in the development of ARDS. And our previous studies showed that NE could inhibit activation of alveolar macrophages and alleviate lung inflammation in ARDS mice induced by lipopolysaccharide (LPS) (16), however, the underlying mechanism is unclear. Adrenergic receptors (ARs) including α1-AR, α2-AR, and β-AR mediate the effects of NE (17). Previous studies showed that blockade of α2-AR (16, 18, 19) or stimulation of β-AR (20–22) could alleviate lung injury by reducing inflammation. However, the role and mechanism of α1-AR in ARDS is still not fully understood. In vivo experiments indicate that phenylephrine (PE), a specific agonist of α1-AR, is favourable for protecting the structural and functional integrity of alveolar-capillary barriers (23, 24). Other in vitro studies suggest that α1-AR stimulation could influence cytokines expression of inflammatory cells (25, 26). Therefore, we explored the effect of α1-AR activation on lung inflammation in a mouse model of ARDS. We hypothesized that the beneficial effect of α1-AR for ARDS arise from its ability to alleviate inflammation through effects on alveolar macrophages.
Male C57BL/6J mice (8–12 weeks old) were purchased from and maintained in the Department of Laboratory Animal Science at Peking University Health Science Centre (Peking, China). Mice were kept on a 12-h light/dark cycle with ad libitum access to standard diet and water. All animal experimental procedures were approved by the Animal Care and Scientific Committee of Peking University Health Science Centre (Approval No: SA2020336).
Mice were anaesthetized by intraperitoneal administration of 1% pentobarbital sodium (70 mg/kg, Sigma-Aldrich, St. Louis, MO, USA). As shown in Figure 1A, after anaesthesia, the ARDS mouse model was established by intratracheal instillation of 2 mg/kg LPS (Escherichia coli 0111:B4, Sigma-Aldrich) in 50 µL of phosphate-buffered saline (PBS). To examine the effect of α1-AR on lung injury of ARDS mice, animal experiments were divided into two parts. In the first part, 20 min before LPS stimulation, varying concentrations of the α1-AR agonist PE (10-7–10-5 M, Selleck Chemicals, Houston, TX, USA) in 50 µL of PBS were injected into the tracheae of ARDS mice. In the second part, 20 min before PE (10-5 M) intervention, the α1-AR antagonist prazosin (PRA, 10-5 M) in 50 µL of PBS was injected into the tracheae of ARDS mice. Simultaneously, control groups were treated with 50 µL of PBS. Mice were sacrificed 24 h after LPS stimulation, and lung tissues and BALF were obtained.
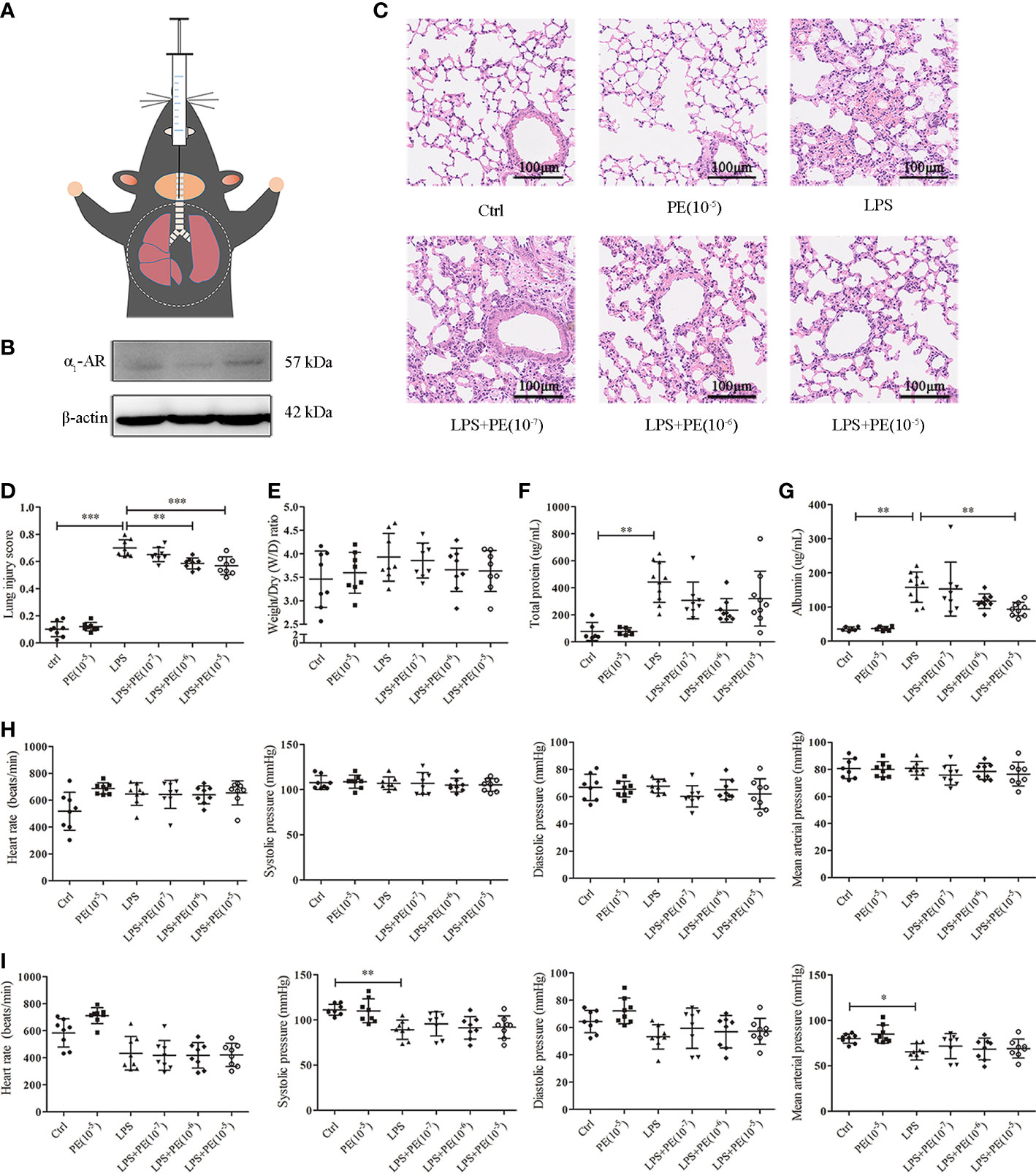
Figure 1 Phenylephrine (PE) attenuated lung pathological injury and alveolar capillary barrier disruption without influencing the cardiovascular haemodynamics of ARDS mice. Mice were given an intratracheal instillation of 2 mg/kg lipopolysaccharide (LPS) to induce ARDS, and PE was injected into trachea 20 min before LPS stimulation. Lung tissues and bronchoalveolar lavage fluid (BALF) were collected 24 h after LPS stimulation. (A) Method for intratracheal instillation of LPS or PE in mice. (B) Expression of α1-AR in lung tissues of mice. (C) Haematoxylin and eosin staining of lung slices. Scale bar = 100 μM. (D) Histology scores of lungs were judged according to guidelines of the American Thoracic Society. (E) Wet/dry weight ratio of lung tissues. (F) Levels of total protein in BALF. (G) Levels of albumin in BALF. (H) Heart rate (HR), systolic blood pressure (SP), diastolic blood pressure (DP), and mean artery pressure (MAP) of mice were monitored before establishing ARDS model. (I) HR, SP, DP, and MAP of mice were monitored 24 h after establishing ARDS model. Data are represented as the mean ± SD, n = 6–10 per group. *p < 0.05, **p < 0.01, ***p < 0.001.
Mice were placed in a noninvasive blood pressure monitor (Softron Biotechnology, Beijing, China) for 10 min daily to become accustomed to the environment for 3 days before establishing the ARDS model. Heart rate (HR), systolic blood pressure (SP), diastolic blood pressure (DP), and mean artery pressure (MAP) of mice were monitored before and 24 h after establishing ARDS model to estimate haemodynamic changes.
Bronchoalveolar lavage was carried out with 1 mL of ice-cold PBS three times. The collected BALF was centrifuged at 1000 × g at 4°C for 5 min. BALF supernatant was stored at -80°C for subsequent detection. One part of the cell pellet was used for total cell counts with a haemocytometer, while the other part was used for differential counts of inflammatory cells by Wright-Giemsa staining (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
The inferior lobe of the right lung from mice without bronchoalveolar lavage was fixed in 4% paraformaldehyde for 24–48 h, dehydrated in an ascending gradient of alcohol, embedded in paraffin, and sliced into 5-µm sections. After staining with haematoxylin and eosin, lung tissues were scanned by a digital pathology microscope (Hamamatsu Photonics, Hamamatsu City, Japan). Histological changes were assessed by a blinded investigator according to a standardized histology scoring system published by American Thoracic Society (27).
The left lung lobe from mice without bronchoalveolar lavage was weighed to record the wet weight. Next, the lung lobe was dried in an oven at 65°C for 48 h until all moisture was removed, and the dry weight was measured. The wet/dry weight ratio was calculated as a measure of the severity of pulmonary oedema.
The murine alveolar macrophage cell line MH-S was purchased from Bio-Rad Laboratories (Hercules, CA, USA). Cells were cultured in RPMI-1640 medium (Biological Industries, Kibbutz Beit Haemek, Israel) with 10% foetal bovine serum (Biological Industries) at 37°C in the presence of 5% CO2.
For LPS activation, MH-S cells were stimulated with 100 ng/mL LPS (Sigma-Aldrich). In accordance with the animal experiment design, during the first part, MH-S cells were incubated with PE (10-8–10-5 M) for 30 min and then stimulated with LPS for 6 h. In the second part, cells were pre-incubated with PRA (10-5 M) for 30 min, followed by PE (10-5 M) for 30 min, and then stimulated with LPS for 6 h. Cell supernatants and pellets were collected for evaluation.
Inflammatory cytokines tumour necrosis factor α (TNF-α), interleukin (IL)-6, IL-10, and chemokine (C-X-C motif) ligand 2/macrophage inflammatory protein 2 (CXCL2/MIP-2) in BALF and cell supernatant were detected by enzyme-linked immunosorbent assay (ELISA) duoset kits (R&D Systems, Minneapolis, MN, USA) according to the kit manufacturer’s instructions. The concentration of albumin in BALF was also measured by ELISA (Elabscience, Wuhan, China).
Cell Counting Kit-8 (CCK-8) and calcein-AM/PI double staining assays (Yeasen Biotechnology, Shanghai, China) were used to measure cell viability. For CCK-8, MH-S cells were seeded into 96-well plates at a density of 5 × 103 cells/well. After treatment with PE, PRA, or LPS, cells were incubated with 10 μL of CCK-8 reagent for 2 h, and the absorbance value of each well was measured using an Automatic Microplate Reader (Thermo Fisher Scientific, Waltham, MA, USA) at 450 nm. For calcein-AM/PI double staining, MH-S cells were seeded into 48-well plates at a density of 2 × 105 cells/well. After identical administration of PE, PRA, or LPS, a mixture of calcein-AM and PI were added into each well for 15 min. Finally, live cells (yellow-green fluorescence) and dead cells (red fluorescence) were simultaneously observed at a 490 ± 10 nm excitation wavelength under a fluorescence microscope (Leica, Wetzlar, Germany).
MH-S cells were seeded into 15-mm glass bottom dishes (Nest Biotechnology, Jiangsu, China) at a density of 2 × 105 cells. After fixation with 4% paraformaldehyde, cells were permeabilized with 0.5% Triton X-100, blocked in blocking buffer, and incubated overnight with an anti-α1-AR antibody (1:100; Abcam, Cambridge, UK) at 4°C. The following day, cells were incubated with a fluorochrome-conjugated secondary antibody [1:500; Cell Signaling Technology (CST), Danvers, MA, USA] for 1 h at room temperature protected from light. DAPI reagent (Solarbio, Beijing, China) was used for nuclei staining. Finally, specimens were observed under a fluorescence microscope (Leica).
Lung tissues were first ground into a single-cell suspension. Obtained lung cells and MH-S cells were lysed in RIPA buffer (Applygen, Beijing, China) for protein extraction. After determining protein concentrations with a bicinchoninic acid colorimetric assay kit (Applygen), protein samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then transferred to polyvinylidene fluoride membranes (MilliporeSigma, Burlington, MA, USA). Membranes were blocked in 1× Tris-buffered saline containing Tween with 5% w/v non-fat dry milk for 1 h at room temperature and incubated overnight at 4°C with primary antibodies against p-p65 (1:1,000; CST), p65 (1:1,000; CST), p-p38 (1:1,000; CST), p38 (1:1,000; CST), p-ERK1/2 (1:1,000; CST), ERK1/2 (1:1,000; CST), p-JNK (1:1,000; CST), JNK (1:1,000; CST), p-Akt (1:1,000; CST), Atk (1:1,000; CST), α1-AR (1:1,000; Abcam), and β-actin (1:5,000; Applygen). After three washes, membranes were incubated with secondary antibodies for 1 h at room temperature. Protein bands were visualized using a chemiluminescence image analysis system (Tanon Science and Technology, Shanghai, China).
All experiments were repeated at least five times, and statistical analysis was conducted using SPSS 22.0 software (IBM SPSS, Chicago, IL, USA). Data conforming to a normal distribution are presented as mean ± standard deviation (SD), and were analysed with one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test or Welch’s ANOVA followed by Dunnett’s T3 post hoc test. Experimental data not conforming to a normal distribution are presented as median (25%, 75%), and were analysed by non-parametric test. P values < 0.05 were considered statistically significant.
Histopathological lesions provide intuitive and reliable results (28). To evaluate the effect of α1-AR on ARDS, expression of α1-AR was first confirmed in lung tissues of mice without intervention (Figure 1B). Evaluation of histopathological lesions provide intuitive and reliable results (28). Histology showed that intratracheal instillation of LPS induced evident lung injury, including alveolar septum thickening, fusion of alveoli, interstitial oedema, and inflammatory cell infiltration (Figure 1C). However, treatment with the α1-AR specific agonist PE mitigated these changes. Compared with the LPS group, PE at both of the concentration of 10-6 and 10-5 M treatment significantly improved the degree of lung injury (Figure 1D), as assessed by the standardized histology scoring system (27). Alveolar capillary barrier disruption, another key indicator of ARDS, is usually evaluated by measuring the wet/dry weight ratio of lung tissues, as well as total protein and/or albumin concentration in BALF. Our results show that LPS stimulation produced no evident changes in the wet/dry weight ratio of lung tissues (Figure 1E), however it increased total protein and albumin concentration in BALF, suggesting the occurrence of lung injury (Figures 1E, G). Although PE had no significant effect on total protein concentrations in BALF, albumin concentrations (an indicator with higher sensitivity) were evidently reduced following intervention with PE (10- 5 M).
Clinically, PE serves as a vasoactive agent typically used to increase patient blood pressure (29). Therefore, cardiovascular haemodynamic parameters were monitored in this study. As shown in Figure 1H, we first observed HR, SP, DP, and MAP of mice before establishing the ARDS model in mice. As expected, no changes were observed in the group without treatment, indicating that our monitoring method was stable and reliable. After stimulation with LPS, SP and MAP were significantly reduced compared with those in the control group, although there were no differences in HR or DP. PE treatment did not influence any of these indicators (Figure 1I).
Excessive inflammatory responses are generally considered the major underlying pathogenesis of ARDS (2). Therefore, we detected the number of exudated inflammatory cells and cytokine levels in BALF. As shown in Figure 2A, LPS induced infiltration of large numbers of inflammatory cells (Figure 2B), especially neutrophils (Figure 2C), into lung tissues. Treatment with PE (10-6 M) obviously decreased the number of total cells and neutrophils in BALF. Similarly, inflammatory cytokines TNF-α (Figure 2D), IL-6 (Figure 2E), and CXCL-2/MIP-2 (Figure 2F) in BALF were dramatically increased after LPS stimulation, but inhibited by treatment with PE.
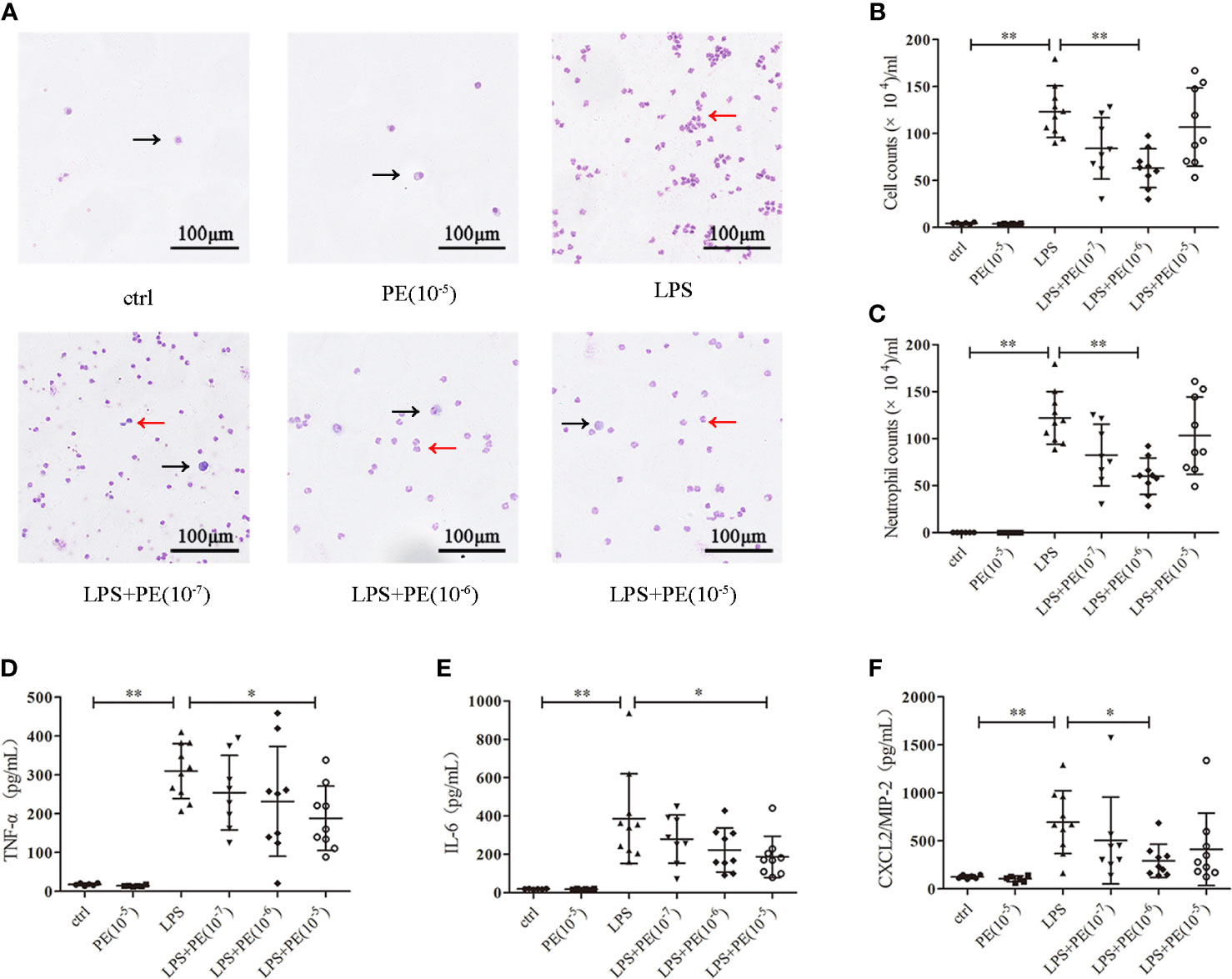
Figure 2 Phenylephrine (PE) ameliorated lung inflammation in ARDS mice. Mice were given an intratracheal instillation of 2 mg/kg lipopolysaccharide (LPS) to induce ARDS, and PE was injected into trachea 20 min before LPS stimulation. Lung tissues and bronchoalveolar lavage fluid (BALF) were collected 24 h after stimulation with LPS. (A) Inflammatory cells in BALF stained by Wright-Giemsa stain. Scale bar = 100 μM. (B) Counts of total inflammatory cells in BALF. (C) Counts of neutrophils in BALF. Levels of TNF-α (D), IL-6 (E), and CXCL2/MIP-2 (F) in BALF. Data are represented as the mean ± SD, n = 6–10 per group. *p < 0.05, **p < 0.01.
Alveolar macrophages are a primary cell type in the lung inflammatory processes (30). Interestingly, we detected α1-AR expression in alveolar macrophages without intervention (Figures 3A, B). To determine whether the effect of PE on lung inflammation involved the effects on alveolar macrophages, we examined cytokine levels in the murine alveolar macrophage line MH-S following LPS stimulation and intervention with PE. The results show that PE inhibited the secretion of TNF-α (Figure 3C) at the concentration of 10-7, 10-6 and 10-5 M, while concentrations of 10-6 and 10-5 M PE inhibited the secretion of IL-6 (Figure 3D) and CXCL2/MIP-2 (Figure 3E), and significantly promoted the secretion of IL-10 (Figure 3F). In addition, we observed the effect of PE on cell viability by calcein-AM/PI double staining and CCK-8 assay. The results show that there were no significant differences among the groups (Figures 3G, H), suggesting that the anti-inflammatory effect of PE did not involve effects on cell viability.
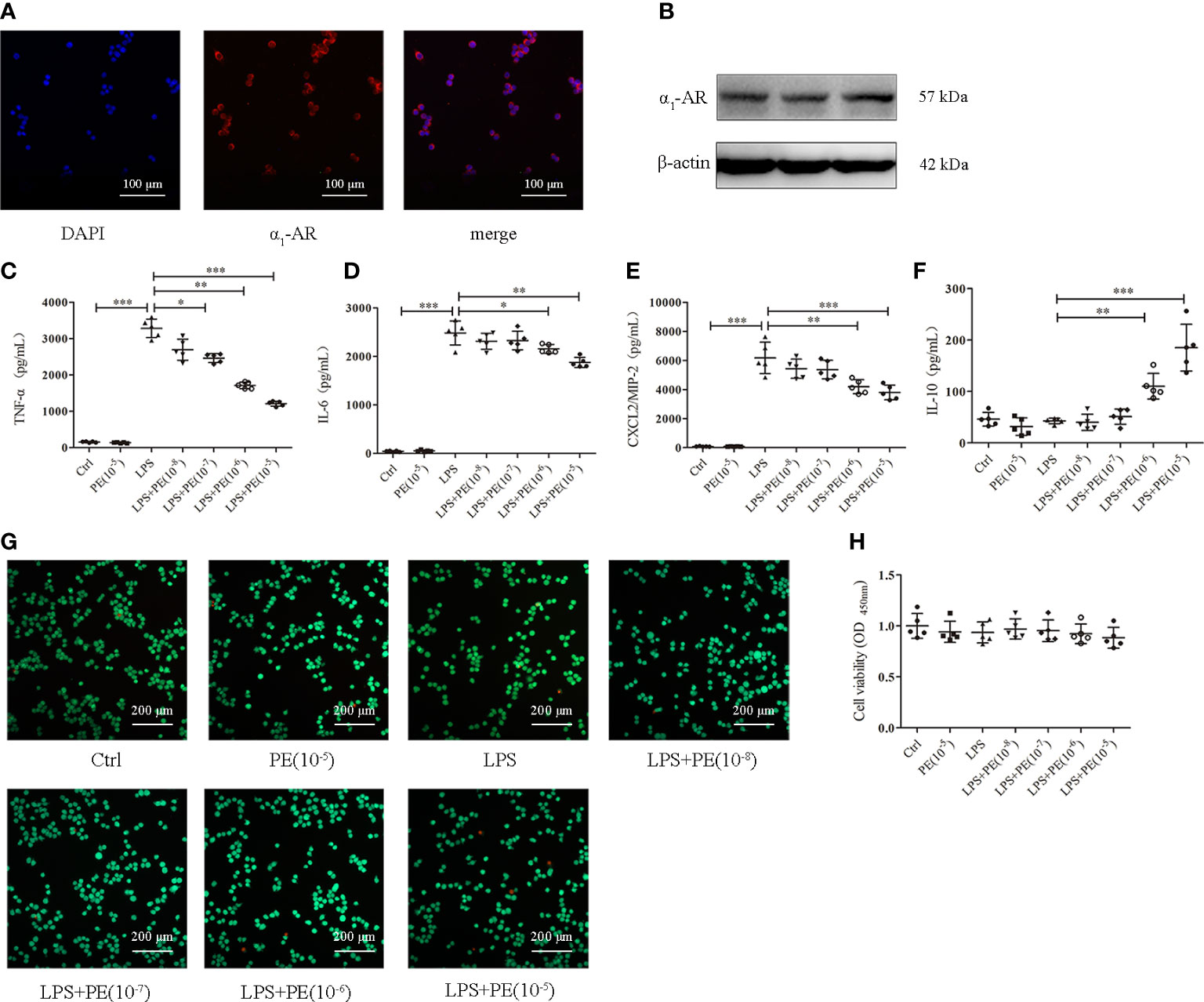
Figure 3 Phenylephrine (PE) inhibited inflammation in lipopolysaccharide (LPS)-activated murine alveolar macrophages without influencing cell viability. MH-S cells were incubated with PE (10-8-10-5 M) for 30 min, followed by LPS (100 ng/mL) for 6 h Expression of α1-AR in MH-S cells was detected by immunofluorescence (A) and western blotting (B). Levels of TNF-α (C), IL-6 (D), CXCL2/MIP-2 (E), and IL-10 (F) released from MH-S cells. (G) Live and dead cells were stained by calcein-AM and propidium iodide, respectively. Scale bar = 200 μM. (H) Cell viability was measured by CCK-8 assay. Data are represented as the mean ± SD, n = 5 per group. *p < 0.05, **p < 0.01, ***p < 0.001.
NF-κB, Akt, and MAPK signalling pathways are important for regulating inflammatory responses (31). Increased phosphorylation levels of p65 and Akt represent activation of NF-κB and Akt signaling pathways respectively (31). The MAPKs in mammals include p38, ERK and JNK which are serine-threonine protein kinases that regulate various cellular functions including inflammation (32). As shown in Figure 4, LPS stimulation caused the activation of NF-κB, Akt, and p38, ERK1/2 and JNK. However, PE (10-5M) treatment could inhibit NF-κB activation (Figure 4B) and further promote ERK1/2 activation (Figure 4E), but had no effect on activation of Akt (Figure 4C), p38 (Figure 4D), or JNK (Figure 4F) by LPS.
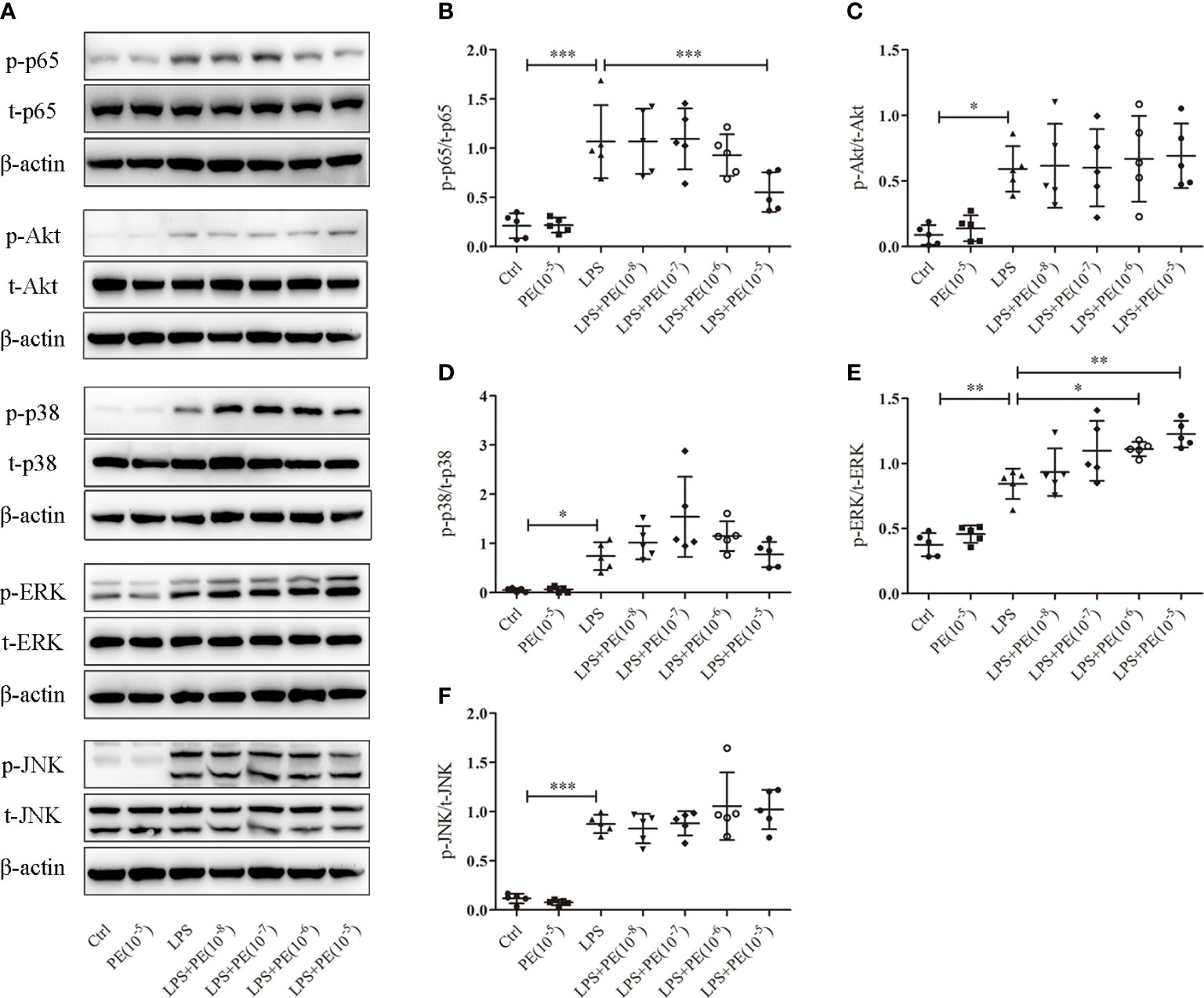
Figure 4 Phenylephrine (PE) suppressed NF-κB activation and enhanced ERK1/2 activation in alveolar macrophages stimulated by lipopolysaccharide (LPS). MH-S cells were incubated with PE (10-8-10-5 M) for 30 min, followed by LPS (100 ng/mL) for 30 min. (A) Western blotting was used to evaluate activation of p65, Akt, p38, ERK1/2, and JNK. Phosphorylation of p65 (B), Akt (C), p38 (D), ERK1/2 (E), and JNK (F) were analysed according to grey values. Data are represented as the mean ± SD, n = 5 per group. *p < 0.05, **p < 0.01, ***p < 0.001.
To further determine whether the protective effect of PE on lung injury of ARDS mice occurred through activation of α1-AR, we administered the α1-AR specific blocker PRA to ARDS mice along with PE intervention. A concentration of 10-5 M PE was chosen for these animal experiments, because it could more thoroughly alleviate lung injury according to experimental results described above. Although no significant differences were observed in lung injury scores (Figure 5B), W/D ratios (Figure 5C), or total protein levels (Figure 5D) between LPS+PE and LPS+PE+PRA groups, PRA aggravated alveolar septum thickening and the fusion of alveoli (Figure 5A), and reversed the inhibitory effect of PE on leakage of albumin into alveoli (Figure 5E). These results suggest that PRA reversed the effect of PE on lung injury of ARDS mice to some extent.
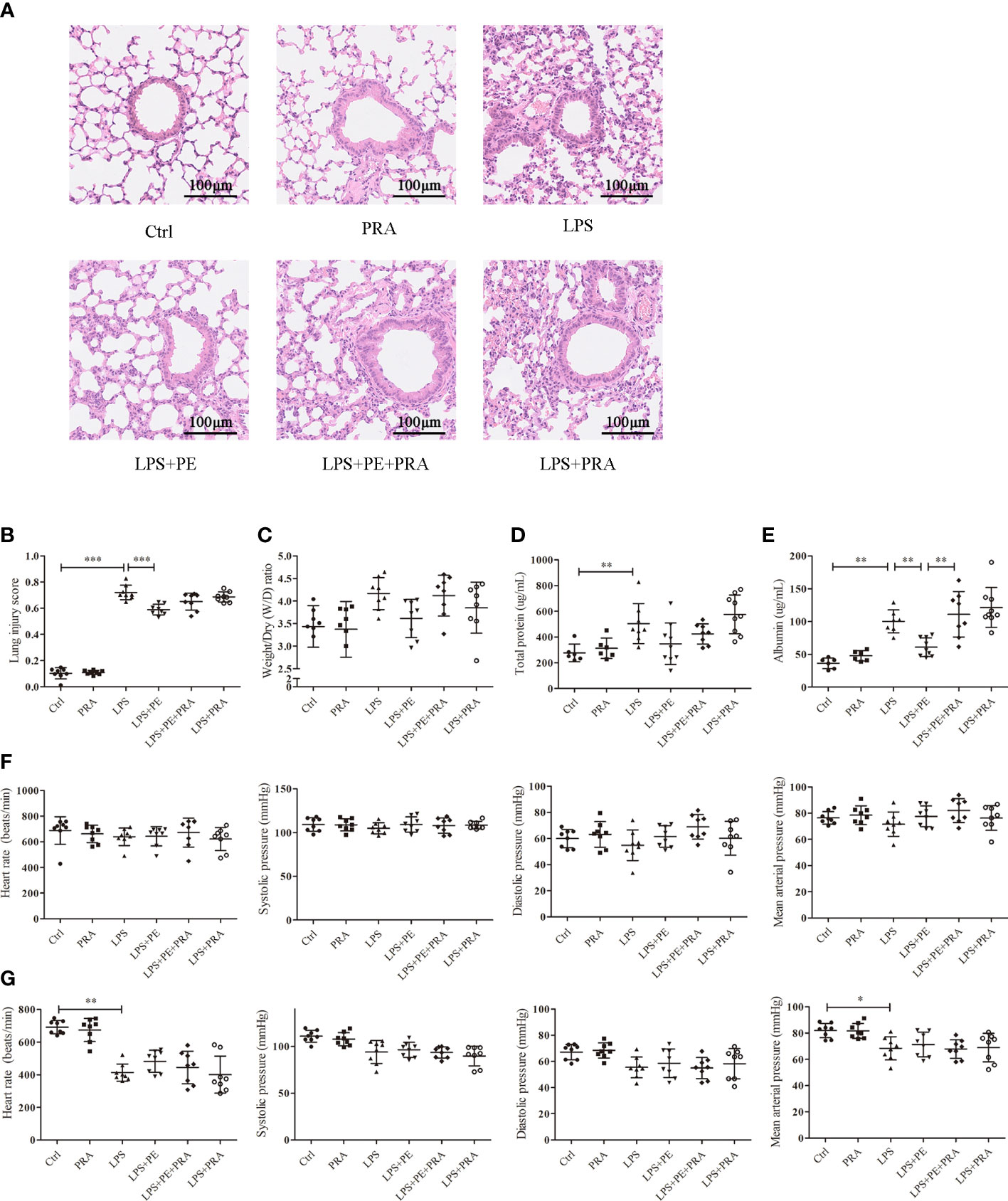
Figure 5 Prazosin (PRA) reversed the effect of phenylephrine (PE) on lung pathological injury and alveolar capillary barrier disruption without influencing the cardiovascular haemodynamic of ARDS mice. Mice were given an intratracheal instillation of 2 mg/kg lipopolysaccharide (LPS) to induce ARDS. PRA and PE were also injected into the trachea 40 min or 20 min before LPS stimulation. Lung tissues and bronchoalveolar lavage fluid (BALF) were collected 24 h after LPS stimulation. (A) Haematoxylin and eosin staining of lung slices. Scale bar = 100 μM. (B) Histology scores of lungs were judged according to guidelines of the American Thoracic Society. (C) Wet/dry weight ratio of lung tissues. (D) Levels of total protein in BALF. (E) Levels of albumin in BALF. (F) Heart rate (HR), systolic blood pressure (SP), diastolic blood pressure (DP), and me an artery pressure (MAP) of mice were monitored before establishing ARDS model. (G) HR, SP, DP, and MAP of mice were monitored 24 h after establishing ARDS model. Data are represented as the mean ± SD, n = 6–9 per group. *p < 0.05, **p < 0.01, ***p < 0.001.
PRA works as a specific blocker of α1-AR and is used to treat high blood pressure (33). Therefore, we monitored its effect on the cardiovascular haemodynamics of ARDS mice. Consistent with previous results (Figure 1H, I), HR, SP, DP, and MAP of mice before establishing ARDS model were unchanged without any treatment (Figure 5F). After stimulation with LPS, HR and MAP were significantly reduced compared with those in the control group, although there were no differences in SP or DP. Indeed, neither PE nor PRA influenced these indicators (Figure 5G).
The effect of PRA on lung inflammation was also evaluated. From the staining assay, further treatment of PRA increased numbers of inflammatory cells in the BALF of ARDS mice compared with the LPS+PE group (Figure 6A). Although image analysis revealed a similar trend, the difference was not statistically significant (Figure 6B, C). However, our results show that PRA could reverse the inhibitory effect of PE on expression of TNF-α (Figure 6D) and IL-6 (Figure 6E) in lung tissues of ARDS mice. Consistent with previous results (Figure 2F), neither PE (10-6 M) nor PRA affected expression of CXCL-2/MIP-2 (Figure 6F).
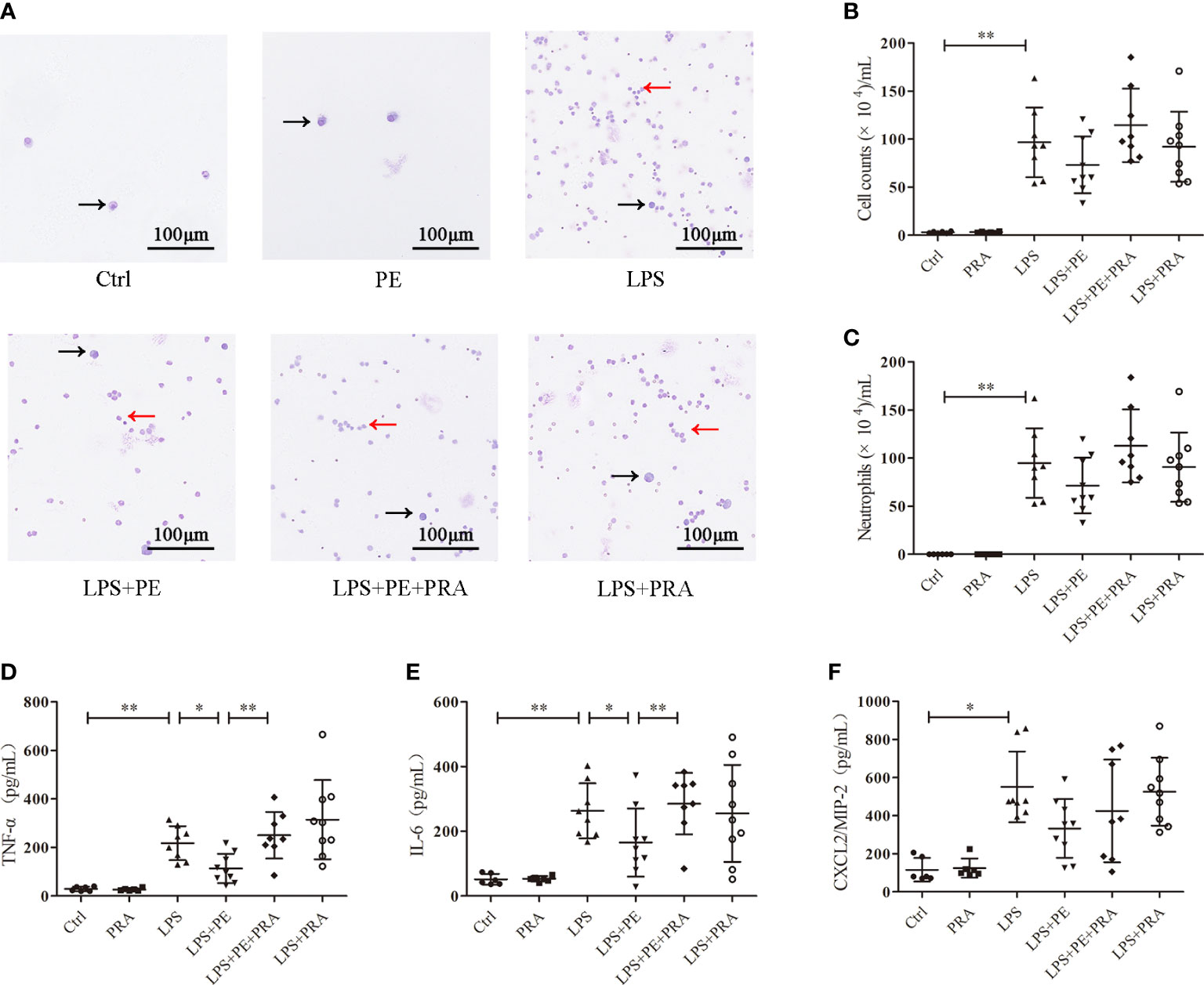
Figure 6 Prazosin (PRA) reversed the effect of phenylephrine (PE) on lung inflammation in ARDS mice. Mice were given an intratracheal instillation of 2 mg/kg lipopolysaccharide (LPS) to induce ARDS. PRA (10-5 M) and PE (10-5 M) were also injected into the trachea 40 min or 20 min before LPS stimulation. Lung tissues and bronchoalveolar lavage fluid (BALF) were collected 24 h after LPS stimulation. (A) Inflammatory cells in BALF stained by Wright-Giemsa stain. Scale bar = 100 μM. (B) Counts of total inflammatory cells in BALF. (C) Counts of neutrophils in BALF. Levels of TNF-α (D), IL-6 (E), and CXCL2/MIP-2 (F) in BALF. Data are represented as the mean ± SD, n = 6–10 per group. *p < 0.05, **p < 0.01.
We also observed the effect of PRA on inflammatory responses of alveolar macrophages. As shown in Figures 7A–D, PRA partially reversed the effect of PE on secretion of TNF-α, IL-6, CXCL2/MIP-2, and IL-10 from alveolar macrophages after stimulation with LPS. Calcein-AM/PI double-staining and CCK-8 assays were used to rule out of the possibility that the effect of PRA on inflammatory responses of alveolar macrophages did not result from influences on cell viability (Figures 7E, F).
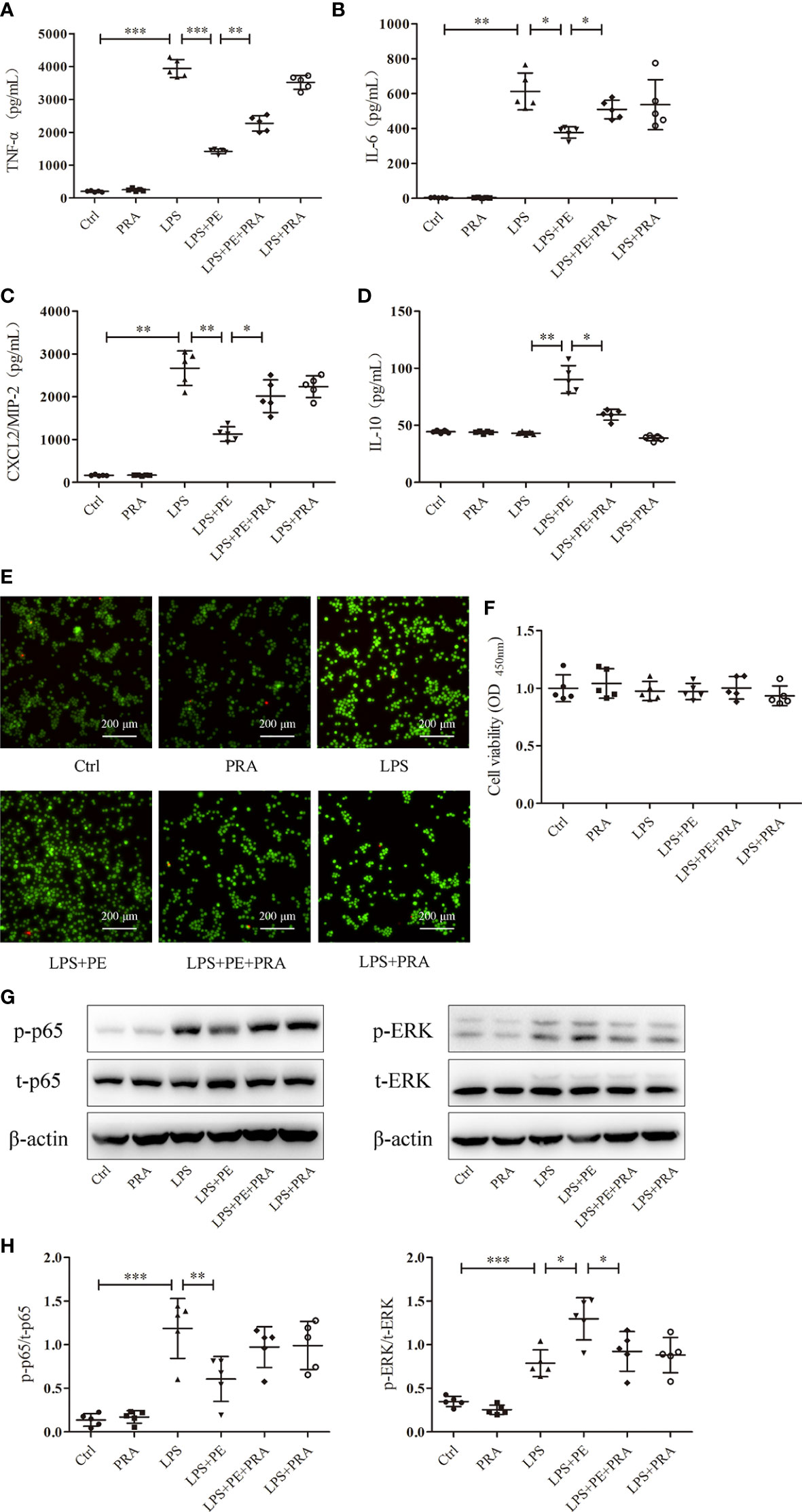
Figure 7 Prazosin (PRA) reversed the effect of phenylephrine (PE) on inflammation in lipopolysaccharide (LPS)-activated murine alveolar macrophages without influencing cell viability by suppressing NF-κB activation and enhancing ERK1/2 activation. MH-S cells were incubated with PRA (10-5 M) for 30 min, followed by PE (10-5 M) treatment for 30 min, and then LPS (100 ng/mL) for 6 h Levels of TNF-α (A), IL-6 (B), CXCL2/MIP-2 (C), and IL-10 (D) released from MH-S cells. (E) Live and dead cells were stained by calcein-AM and propidium iodide, respectively. Scale bar = 200 μM. (F) Cell viability was measured by CCK-8 assay. (G) Western blotting was used to evaluate activation of p65 and ERK1/2. (H) Phosphorylation of p65 and ERK1/2 were analysed according to grey values. Data are represented as the mean ± SD, n = 5 per group. *p < 0.05, **p < 0.01, ***p < 0.001.
Although we detected many signal molecules related to inflammation, only NF-κB and ERK1/2 exhibited changes following intervention with PE. To further determine whether this phenomenon was attributed to activation of α1-AR by PE, the activation of NF-κB and ERK1/2 was observed following treatment with PRA. As shown in Figures 7G, H, PE promoted the activation of ERK1/2, while further treatment with PRA partially reversed the effect of PE. Although there was no significant statistical difference in NF-κB activation between LPS+PE and LPS+PE+PRA groups, PRA treatment still increased expression of p-p65 to some degree (according to the protein band).
ARDS is a life-threatening condition with high morbidity and mortality rates for patients in the intensive care unit (1). Although massive efforts have been made, current treatment is still limited to mechanical ventilation (34), fluid management (35), and other supportive therapies (36, 37); at present, there is no proven pharmacotherapy (4). To counter the problems above, preliminary exploration was carried out through animal and cell experiments in this study. The results show that α1-AR stimulation alleviated lung injury of ARDS mice, including inhibition of lung inflammation and improvement of alveolar capillary barrier integrity, without influencing cardiovascular haemodynamics. Cell experiments suggest that the protective effect of α1-AR on lung injury might arise from its suppression of inflammation through alveolar macrophages by a mechanism likely related to activation of NF-κB and ERK.
According to clinical practice, ARDS is a complication of a variety of diseases including intrapulmonary factors (e.g., infectious pneumonia and aspiration of gastric contents) and extrapulmonary factors (e.g., sepsis and haemorrhagic shock) (27). Intratracheal instillation of LPS, a method that mimics infectious pneumonia, was used to induce ARDS in mice in this study (38). Infectious pneumonia is the most prevalent cause of ARDS, accounting for 59.4% of cases (39). Moreover, studies have demonstrated that the pathological injury and functional indexes induced by this method are more consistent with experimental requirements compared with those induced by other models (40). Our results show that significant pathological damage, alveolar-capillary barrier destruction, and inflammatory response occurred in the lung tissue of ARDS mice, consistent with criteria established by the American Thoracic Society (27).
At present, limited information is available about the effect of α1-AR on lung injury induced by infectious stimuli such as LPS. Our results show that lung pathological damage and albumin levels in BALF were gradually mitigated with increased concentrations of PE. Moreover, this effect of PE could be partially reversed by further treatment with PRA, an α1-AR blocker. Previously, Satoshi Fukuda et al. explored the effect of PE on burn and smoke inhalation-induced acute lung injury of goats (24). Although there are differences in these animal models, their results showed that aerosol inhalation of PE could reduce pulmonary vascular permeability. Similarly, there was no significant difference in W/D ratio in their study either. Nai-Jing Li et al. found that intrapulmonary instillation of PE increased alveolar fluid clearance in ventilator-induced lung injury rats, and PRA abolished this effect too (23). Collectively, these studies and our findings indicate that α1-AR stimulation exerts an important protective effect on lung injury induced by various factors, and this protective effect is mainly related to barrier permeability and fluid balance in the lungs.
Before exploring protective mechanisms of α1-AR, the physiological function of α1-AR should first be considered. α1-AR is widely known as an important sympathetic neurotransmitter receptor capable of constricting blood vessels (41). PE is usually used to treat septic shock (42), episodes of paroxysmal supraventricular tachycardia, and hypotension during general anaesthesia and spinal anaesthesia (29). Notably, sympathetic overstimulation can lead to neurogenic pulmonary oedema – a disease similar to ARDS (43). Therefore, PE and PRA were delivered by intratracheal injection to avoid affecting the circulatory system as much as possible, and cardiovascular haemodynamics were monitored in this study. Our results show that LPS (2 mg/kg) disrupted haemodynamic stability, consistent with clinical practice, while neither PE nor PRA affected HR, SP, DP, or MAP of ARDS mice. These results suggest that LPS, a powerful toxin produced by bacteria, caused circulatory system dysfunction. Furthermore, intratracheal injection of PE or PRA appeared to mainly affect lung tissues without entering the bloodstream, thereby minimizing the impact on heart and blood vessels. Similarly, it was reported that PE intratracheal infusion had no effect on haemodynamics in goats with burn and smoke inhalation-induced lung injury at multiple time points (from 3 to 48 hours) (24). Thus, the beneficial effect of PE was unlikely associated with its effects on haemodynamics.
Damage of alveolar-capillary barriers caused by uncontrolled inflammation is considered a central mechanism of ARDS (44). Many studies aimed to reduce lung injury by inhibiting excessive inflammatory responses. Unfortunately, few reported effects of α1-AR on lung inflammation; indeed, only Satoshi Fukuda’s results show that nebulized PE tended to decrease IL-8 concentrations in BALF (24). We further found that α1-AR stimulation reduced numbers of inflammatory cells and levels of TNF-α, IL-6, and CXCL2/MIP-2 in BALF of ARDS mice. It should be noted that there were far more neutrophils than macrophages in the BALF of ARDS mice, although both alveolar macrophages and neutrophils are crucial inflammatory cells related to its occurrence. Under normal conditions (as shown for the control group in in Figure 2A), only a small number of macrophages (average seven per alveolar) were observed (45). Following LPS stimulation, there was an inflammatory response in the lung that caused recruitment of neutrophils, mainly under the induction of chemokines and cytokines secreted by macrophages. Because increased neutrophil numbers are considered one of the most relevant features of ARDS (27), total cells and neutrophils in BALF were detected in this study. In addition, previous studies suggested that PE treatment could decrease TNF-α and IL-6 levels in the plasma and alleviate myocarditis of sepsis rats (46, 47). Combined with the results described above, it seems likely that the lung protective effect of α1-AR derives from its ability to promote proper immune homeostasis by suppressing inflammatory responses.
Although macrophages, neutrophils, alveolar epithelium, pulmonary microvascular endothelium, and other cells participate in the formation of excessive inflammation during the development of ARDS (48), alveolar macrophages play a predominant role (49). Alveolar macrophages are intrinsic resident cells in alveoli and their depletion has been shown to improve IgG immune complex-induced lung injury by attenuating inflammation (50). MH-S, a continuous alveolar macrphage cell line from mice established by Mbawuike and Herscowitz in 1989 (51), is widely used in studies of bacterial pneumonia (52), chronic obstructive pulmonary disease (53), asthma (54) and ARDS (55) because it retains complete functional characteristics from parent alveolar macrophage. Thus, the alveolar macrophage cell line MH-S was used to detect the anti-inflammatory effect of α1-AR in vitro. Our results show that α1-AR stimulation suppressed the release of TNF-α, IL-6, and CXCL2/MIP-2, and promoted the release of IL-10 from LPS-stimulated alveolar macrophages. To determine whether this phenomenon was related to the effect of α1-AR stimulation on cell viability, we measured cytotoxicity with a CCK-8 kit and live/dead staining. The results show that cell viability was not affected. Previously, Laurel A. Grisanti et al. found that PE could significantly reduce expression of TNF-α, IL-8, and MIP-1β in human THP-1 monocytes stimulated with LPS, consistent with our research (25). However, their results also showed that PE could promote expression of IL-1β, an important proinflammatory cytokine with wide biological effects (26). We wondered if the same change in expression of IL-1β occurred in our study, but found that LPS did not induce IL-1β release from MH-S cells (data not shown). In addition, Hongmei Li et al. used PE and PRA to verify that α1-AR stimulation could inhibit TNF-α secretion from cardiomyocytes in LPS-stimulated rats (56), which further confirmed the reliability of our findings. Interestingly, a previous study showed that PE dose-dependently attenuated TNF-α production and enhanced IL-10 release in isolated primary human monocytes (57). In contrast to the current findings, β-AR rather than α1-AR was thought to mediate the potent anti-inflammatory effects of PE. However, what must be emphasized is that PE is considered as a specific α1-AR agonist in clincal and basic research (58). Thus, we chose PE to study the effects of α1-AR on ARDS. Moreover, PRA (a apecific of α1-AR antagonist) reversed the effect of PE in our study, further confirming a role for α1-AR in this process. Importantly, its potential to activate other receptors (α2 or β), is worth exploring.
The occurrence of uncontrolled lung inflammation involves a complex signalling pathway network connected by important nodes and hubs. NF-κB is a known core molecule, while MAPKs and Akt are focal points (59). All of these signalling pathways may be involved in the regulation of inflammatory responses by α1-AR (26, 46, 47, 56). Accordingly, to clarify the specific mechanism by which the anti-inflammatory effect of α1-AR occurs in alveolar macrophages, we detected all the molecules described in the studies above under the condition of different concentrations of PE. Finally, according to our results, we concluded that α1-AR stimulation inhibited NF-κB activation and enhanced ERK1/2 activation without significantly influencing p38, JNK, or Akt activation in LPS-stimulated MH-S cells. Research by Laurel A. Grisanti showed that PE increases IL-1β expression in LPS-stimulated human monocytes and macrophages by promoting activation of p38 rather than ERK1/2 or JNK (26). Other reports suggest that α1-AR stimulation suppressed inflammation by inhibiting p38 and NF-κB activation, but enhanced activation of ERK1/2 in LPS-stimulated cardiomyocytes (46) and Akt in myocardium from septic animals (47). According to these studies, aspects of inflammatory regulation and molecular mechanisms related to α1-AR may completely differ across various cells, tissues, and disease conditions. In addition, we also noticed that there was a higher dose of PE was used in the research of Laurel A. Grisanti than that of other researches which might lead to different results. Overall, the reported activation of ERK1/2 and NF-κB in previous studies was consistent with our results; in particular, NF-κB is a prototypical proinflammatory transcription factor that plays important roles in the expression of numerous cytokines and chemokines, and many studies have demonstrated its involvement in the occurrence and development of ARDS (60, 61). The observed activation of NF-κB in this study is consistent with changes of cytokines, suggesting that α1-AR activation may alleviate lung injury by inhibiting NF-κB activation. The ERK1/2 pathway was previously reported to participate in marcophage activation, although its effect in this process was debatable (62, 63). What is clear is that stimulation of α1-AR could alleviate sepsis-induced cardiomyocyte apoptosis and inflammation by activating ERK1/2 pathway (46, 56). Moreover, evidence suggests that ERK1/2 activation could participate in M2-like macrophages polarization (64). Collectively, these studies identify a relationship between α1-AR and ERK1/2 pathway. Our current study has provided preliminary information showing that NF-κB and ERK1/2 in the protective effect of α1-AR on lung injury.
Here we conducted a comprehensive and systematic study to evaluate the role of α1-AR from several aspects, including pathological damage, barrier disruption, inflammatory responses, and molecular mechanism in ARDS. The results further expanded our understanding of the effect of sympathetic nervous system in ARDS and provided basic research clues for its clinical treatment. Certainly, it is undeniable that there are still many limitations of this study that need to be solved in the future. First, haemodynamics were only monitored before establishing ARDS model and sacrificing of mice, and did not show evident changes after treatment with PE or PRA. If continuous and effective monitoring was carried out, these results would be more convincing. Second, the observed changes of cytokines imply that there might be a relationship between α1-AR and macrophage polarization. Because TNF-α, IL-6, CXCL2/MIP-2, and IL-10 are phenotypical markers of M1 or M2 macrophages, our results suggest that α1-AR stimulation could potentially transform alveolar macrophages from an M1 to M2 subtype. To verify this conjecture, we also detected other typical markers of macrophage polarization, including CD86 and iNOS for the M1 subtype, and CD206 and Arg-1 for the M2 subtype. Confusingly, none of the molecules were detected in MH-S cells after stimulation with LPS (data not shown). We considered that this might be related to our experimental conditions, such as the LPS concentration or time-period of stimulation. In addition, detection of number and type of macrophages in BALF may also provide a more accurate reflection of the role of macrophages in the protective effect of PE against LPS. However, the number of macrophages in BALF was too few to be detected in some samples. Finally, and more importantly, the molecular mechanisms underlying the effects of α1-AR in lung injury need to be further explored. At present, no other research has verified the involvement of NF-κB, Akt, and MAPK signalling pathways in alveolar macrophages with activated α1-AR. Although we confirmed that NF-κB and ERK1/2 participated in the mechanism of the protective effect of α1-AR, many other factors could also be involved.
In summary, our study demonstrated that α1-AR stimulation ameliorates LPS-induced lung injury by suppressing excessive inflammatory responses in lung tissues without affecting haemodynamics. The mechanism of anti-inflammation elicited by α1-AR stimulation may be associated with its ability to inhibit alveolar macrophage activation by suppressing NF-κB activation and promoting ERK1/2 activation.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Animal Care and Scientific Committee of Peking University Health Science Centre (Approval No: SA2020336).
ZC designed and performed the experiments, collected and analyzed data, and drafted and approved the final version. CY, ZZ, FZ, and ZS performed experiments and data analysis. CW designed the experiments and revised the article. XZ and HX supervised the research design, revised the article, and approved the final version. All authors contributed to the article and approved the submitted version.
This study was funded by Beijing Municipal Natural Science Foundation (Grant/Award 7212130), National Natural Science Foundation of China (Grant/Award 82172166), Capital Health Research and Development of Special Fund (Grant/Award 2020-2-4094) and Peking University Third Hospital Cohort Study Project (BYSYDL2021010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gorman EA, O'Kane CM, McAuley DF. Acute respiratory distress syndrome in adults: diagnosis, outcomes, long-term sequelae, and management. LANCET (2022) 400:1157–1170. doi: 10.1016/S0140-6736(22)01439-8
2. Bos L, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. LANCET (2022) 400:1145–1156. doi: 10.1016/S0140-6736(22)01485-4
3. Matthay MA, Arabi YM, Siegel ER, Ware LB, Bos L, Sinha P, et al. Phenotypes and personalized medicine in the acute respiratory distress syndrome. Intensive Care Med (2020) 46:2136–52. doi: 10.1007/s00134-020-06296-9
4. Beitler JR, Thompson BT, Baron RM, Bastarache JA, Denlinger LC, Esserman L, et al. Advancing precision medicine for acute respiratory distress syndrome. Lancet Respir Med (2022) 10:107–20. doi: 10.1016/S2213-2600(21)00157-0
5. Pham T, Rubenfeld GD. Fifty years of research in ARDS. the epidemiology of acute respiratory distress syndrome. a 50th birthday review. Am J Respir Crit Care Med (2017) 195:860–70. doi: 10.1164/rccm.201609-1773CP
6. Liu L, Yang Y, Gao Z, Li M, Mu X, Ma X, et al. Practice of diagnosis and management of acute respiratory distress syndrome in mainland China: a cross-sectional study. J Thorac Dis (2018) 10:5394–404. doi: 10.21037/jtd.2018.08.137
7. Lamers MM, Haagmans BL. SARS-CoV-2 pathogenesis. Nat Rev Microbiol (2022) 20:270–84. doi: 10.1038/s41579-022-00713-0
8. De Virgiliis F, Di Giovanni S. Lung innervation in the eye of a cytokine storm: neuroimmune interactions and COVID-19. Nat Rev Neurol (2020) 16:645–52. doi: 10.1038/s41582-020-0402-y
9. Morrell ED, Bhatraju PK, Mikacenic CR, Radella FN, Manicone AM, Stapleton RD, et al. Alveolar macrophage transcriptional programs are associated with outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med (2019) 200:732–41. doi: 10.1164/rccm.201807-1381OC
10. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev (2000) 52:595–638.
11. Stolk RF, van der Poll T, Angus DC, van der Hoeven JG, Pickkers P, Kox M. Potentially inadvertent immunomodulation: Norepinephrine use in sepsis. Am J Respir Crit Care Med (2016) 194:550–8. doi: 10.1164/rccm.201604-0862CP
12. Carrara M, Ferrario M, Bollen PB, Herpain A. The autonomic nervous system in septic shock and its role as a future therapeutic target: a narrative review. Ann Intensive Care (2021) 11:80. doi: 10.1186/s13613-021-00869-7
13. Liu Y, Tao T, Li W, Bo Y. Regulating autonomic nervous system homeostasis improves pulmonary function in rabbits with acute lung injury. BMC PULM Med (2017) 17:98. doi: 10.1186/s12890-017-0436-0
14. Baranski GM, Sifri ZC, Cook KM, Alzate WD, Livingston DH, Mohr AM. Is the sympathetic system involved in shock-induced gut and lung injury? J Trauma Acute Care Surg (2012) 73:343–50. doi: 10.1097/TA.0b013e31825a785a
15. Permpikul C, Tongyoo S, Viarasilpa T, Trainarongsakul T, Chakorn T, Udompanturak S. Early use of norepinephrine in septic shock resuscitation (CENSER). a randomized trial. Am J Respir Crit Care Med (2019) 199:1097–105. doi: 10.1164/rccm.201806-1034OC
16. Cong Z, Li D, Lv X, Yang C, Zhang Q, Wu C, et al. alpha2A-adrenoceptor deficiency attenuates lipopolysaccharide-induced lung injury by increasing norepinephrine levels and inhibiting alveolar macrophage activation in acute respiratory distress syndrome. Clin Sci (Lond) (2020) 134:1957–71. doi: 10.1042/CS20200586
17. Karemaker JM. An introduction into autonomic nervous function. Physiol MEAS (2017) 38:R89–R118. doi: 10.1088/1361-6579/aa6782
18. Cong Z, Li D, Tao Y, Lv X, Zhu X. alpha2A -AR antagonism by BRL-44408 maleate attenuates acute lung injury in rats with downregulation of ERK1/2, p38MAPK, and p65 pathway. J Cell Physiol (2020) 235:6905–14. doi: 10.1002/jcp.29586
19. Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. NATURE (2007) 449:721–5. doi: 10.1038/nature06185
20. Bosmann M, Grailer JJ, Zhu K, Matthay MA, Sarma JV, Zetoune FS, et al. Anti-inflammatory effects of beta2 adrenergic receptor agonists in experimental acute lung injury. FASEB J (2012) 26:2137–44. doi: 10.1096/fj.11-201640
21. Maris NA, de Vos AF, Dessing MC, Spek CA, Lutter R, Jansen HM, et al. Antiinflammatory effects of salmeterol after inhalation of lipopolysaccharide by healthy volunteers. Am J Respir Crit Care Med (2005) 172:878–84. doi: 10.1164/rccm.200503-451OC
22. McAuley DF, Frank JA, Fang X, Matthay MA. Clinically relevant concentrations of beta2-adrenergic agonists stimulate maximal cyclic adenosine monophosphate-dependent airspace fluid clearance and decrease pulmonary edema in experimental acid-induced lung injury. Crit Care Med (2004) 32:1470–6. doi: 10.1097/01.CCM.0000129489.34416.0E
23. Li NJ, Gu X, Li W, Li Y, Li SQ, He P. Effect of phenylephrine on alveolar fluid clearance in ventilator-induced lung injury. Chin Med Sci J (2013) 28:1–6. doi: 10.1016/S1001-9294(13)60011-5
24. Fukuda S, Lopez E, Ihara K, Niimi Y, Andersen CR, Jacob S, et al. Superior effects of nebulized epinephrine to nebulized albuterol and phenylephrine in burn and smoke inhalation-induced acute lung injury. SHOCK (2020) 54:774–82. doi: 10.1097/SHK.0000000000001590
25. Grisanti LA, Perez DM, Porter JE. Modulation of immune cell function by alpha(1)-adrenergic receptor activation. Curr Top MEMBR (2011) 67:113–38. doi: 10.1016/B978-0-12-384921-2.00006-9
26. Grisanti LA, Woster AP, Dahlman J, Sauter ER, Combs CK, Porter JE. alpha1-adrenergic receptors positively regulate toll-like receptor cytokine production from human monocytes and macrophages. J Pharmacol Exp Ther (2011) 338:648–57. doi: 10.1124/jpet.110.178012
27. Kulkarni HS, Lee JS, Bastarache JA, Kuebler WM, Downey GP, Albaiceta GM, et al. Update on the features and measurements of experimental acute lung injury in animals: An official American thoracic society workshop report. Am J Respir Cell Mol Biol (2022) 66:e1–e14. doi: 10.1165/rcmb.2021-0531ST
28. Tomashefski JJ. Pulmonary pathology of acute respiratory distress syndrome. Clin CHEST Med (2000) 21:435–66. doi: 10.1016/S0272-5231(05)70158-1
29. Khan E, Wong M, Ibrahim M, Babazade R, Simon M, Mendonca R, et al. Effect of phenylephrine infusion and spinal anesthesia on cardiac output during cesarean section by point of care echocardiogram: A case series. J Clin Anesth (2021) 75:110474. doi: 10.1016/j.jclinane.2021.110474
30. Soni S, Wilson MR, O'Dea KP, Yoshida M, Katbeh U, Woods SJ, et al. Alveolar macrophage-derived microvesicles mediate acute lung injury. THORAX (2016) 71:1020–9. doi: 10.1136/thoraxjnl-2015-208032
31. Xu Z, Chu M. Advances in immunosuppressive agents based on signal pathway. Front Pharmacol (2022) 13:917162. doi: 10.3389/fphar.2022.917162
32. Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: an update. Arch Toxicol (2015) 89:867–82. doi: 10.1007/s00204-015-1472-2
33. Docherty JR. The pharmacology of alpha1-adrenoceptor subtypes. Eur J Pharmacol (2019) 855:305–20. doi: 10.1016/j.ejphar.2019.04.047
34. Grieco DL, Maggiore SM, Roca O, Spinelli E, Patel BK, Thille AW, et al. Non-invasive ventilatory support and high-flow nasal oxygen as first-line treatment of acute hypoxemic respiratory failure and ARDS. Intensive Care Med (2021) 47:851–66. doi: 10.1007/s00134-021-06459-2
35. Vignon P, Evrard B, Asfar P, Busana M, Calfee CS, Coppola S, et al. Fluid administration and monitoring in ARDS: which management? Intensive Care Med (2020) 46:2252–64. doi: 10.1007/s00134-020-06310-0
36. Weiss TT, Cerda F, Scott JB, Kaur R, Sungurlu S, Mirza SH, et al. Prone positioning for patients intubated for severe acute respiratory distress syndrome (ARDS) secondary to COVID-19: a retrospective observational cohort study. Br J Anaesth (2021) 126:48–55. doi: 10.1016/j.bja.2020.09.042
37. Moss M, Huang DT, Brower RG, Ferguson ND, Ginde AA, Gong MN, et al. Early neuromuscular blockade in the acute respiratory distress syndrome. N Engl J Med (2019) 380:1997–2008. doi: 10.1056/NEJMoa1901686
38. Reiss LK, Uhlig U, Uhlig S. Models and mechanisms of acute lung injury caused by direct insults. Eur J Cell Biol (2012) 91:590–601. doi: 10.1016/j.ejcb.2011.11.004
39. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA (2016) 315:788–800. doi: 10.1001/jama.2016.0291
40. Chen H, Bai C, Wang X. The value of the lipopolysaccharide-induced acute lung injury model in respiratory medicine. Expert Rev Respir Med (2010) 4:773–83. doi: 10.1586/ers.10.71
41. Scanzano A, Cosentino M. Adrenergic regulation of innate immunity: a review. Front Pharmacol (2015) 6:171. doi: 10.3389/fphar.2015.00171
42. Arishi H, AlQahtani S, Tamim H, Sadat M, Alenezi FZ, Bin HF, et al. Combination of norepinephrine with phenylephrine versus norepinephrine with vasopressin in critically ill patients with septic shock: A retrospective study. J Crit Care (2022) 72:154121. doi: 10.1016/j.jcrc.2022.154121
43. Busl KM, Bleck TP. Neurogenic pulmonary edema. Crit Care Med (2015) 43:1710–5. doi: 10.1097/CCM.0000000000001101
44. Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol (2013) 75:593–615. doi: 10.1146/annurev-physiol-030212-183756
45. Robbe P, Draijer C, Borg TR, Luinge M, Timens W, Wouters IM, et al. Distinct macrophage phenotypes in allergic and nonallergic lung inflammation. Am J Physiol Lung Cell Mol Physiol (2015) 308:L358–67. doi: 10.1152/ajplung.00341.2014
46. Yu X, Jia B, Wang F, Lv X, Peng X, Wang Y, et al. alpha(1) adrenoceptor activation by norepinephrine inhibits LPS-induced cardiomyocyte TNF-alpha production via modulating ERK1/2 and NF-kappaB pathway. J Cell Mol Med (2014) 18:263–73. doi: 10.1111/jcmm.12184
47. Li HM, Li KY, Xing Y, Tang XX, Yang DM, Dai XM, et al. Phenylephrine attenuated sepsis-induced cardiac inflammation and mitochondrial injury through an effect on the PI3K/Akt signaling pathway. J Cardiovasc Pharmacol (2019) 73:186–94. doi: 10.1097/FJC.0000000000000651
48. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers (2019) 5:18. doi: 10.1038/s41572-019-0069-0
49. Fan E, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res (2018) 19:50. doi: 10.1186/s12931-018-0756-5
50. Lentsch AB, Czermak BJ, Bless NM, Van Rooijen N, Ward PA. Essential role of alveolar macrophages in intrapulmonary activation of NF-kappaB. Am J Respir Cell Mol Biol (1999) 20:692–8. doi: 10.1165/ajrcmb.20.4.3414
51. Mbawuike IN, Herscowitz HB. MH-s, a murine alveolar macrophage cell line: morphological, cytochemical, and functional characteristics. J Leukoc Biol (1989) 46:119–27. doi: 10.1002/jlb.46.2.119
52. Li QR, Tan SR, Yang L, He W, Chen L, Shen FX, et al. Mechanism of chlorogenic acid in alveolar macrophage polarization in klebsiella pneumoniae-induced pneumonia. J Leukoc Biol (2022) 112:9–21. doi: 10.1002/JLB.3HI0721-368R
53. Le Y, Wang Y, Zhou L, Xiong J, Tian J, Yang X, et al. Cigarette smoke-induced HMGB1 translocation and release contribute to migration and NF-kappaB activation through inducing autophagy in lung macrophages. J Cell Mol Med (2020) 24:1319–31. doi: 10.1111/jcmm.14789
54. Huang C, Du W, Ni Y, Lan G, Shi G. The effect of short-chain fatty acids on M2 macrophages polarization in vitro and in vivo. Clin Exp Immunol (2022) 207:53–64. doi: 10.1093/cei/uxab028
55. Xia L, Zhang C, Lv N, Liang Z, Ma T, Cheng H, et al. AdMSC-derived exosomes alleviate acute lung injury via transferring mitochondrial component to improve homeostasis of alveolar macrophages. THERANOSTICS (2022) 12:2928–47. doi: 10.7150/thno.69533
56. Li H, Xing Y, Yang D, Tang X, Lu D, Wang H. Alpha-1 adrenergic receptor agonist phenylephrine inhibits sepsis-induced cardiomyocyte apoptosis and cardiac dysfunction via activating ERK1/2 signal pathway. SHOCK (2019) 52:122–33. doi: 10.1097/SHK.0000000000001205
57. Stolk RF, Naumann F, van der Pasch E, Schouwstra J, Bressers S, van Herwaarden AE, et al. Phenylephrine impairs host defence mechanisms to infection: a combined laboratory study in mice and translational human study. Br J Anaesth (2021) 126:652–64. doi: 10.1016/j.bja.2020.11.040
58. Chhatar S, Lal G. Role of adrenergic receptor signalling in neuroimmune communication. Curr Res Immunol (2021) 2:202–17. doi: 10.1016/j.crimmu.2021.11.001
59. Englert JA, Bobba C, Baron RM. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight (2019) 4:e124061. doi: 10.1172/jci.insight.124061
60. Millar MW, Fazal F, Rahman A. Therapeutic targeting of NF-kappaB in acute lung injury: A double-edged sword. CELLS-BASEL (2022) 11:3317. doi: 10.3390/cells11203317
61. Christman JW, Sadikot RT, Blackwell TS. The role of nuclear factor-kappa b in pulmonary diseases. CHEST (2000) 117:1482–7. doi: 10.1378/chest.117.5.1482
62. Somensi N, Rabelo TK, Guimaraes AG, Quintans-Junior LJ, de Souza AA, Moreira J, et al. Carvacrol suppresses LPS-induced pro-inflammatory activation in RAW 264.7 macrophages through ERK1/2 and NF-kB pathway. Int IMMUNOPHARMACOL (2019) 75:105743. doi: 10.1016/j.intimp.2019.105743
63. Goral J, Choudhry MA, Kovacs EJ. Acute ethanol exposure inhibits macrophage IL-6 production: role of p38 and ERK1/2 MAPK. J Leukoc Biol (2004) 75:553–9. doi: 10.1189/jlb.0703350
Keywords: acute respiratory distress syndrome, α1 adrenergic receptor, alveolar macrophage, inflammation, NF-κb
Citation: Cong Z, Yang C, Zeng Z, Wu C, Zhao F, Shen Z, Xiao H and Zhu X (2023) α1-adrenoceptor stimulation ameliorates lipopolysaccharide-induced lung injury by inhibiting alveolar macrophage inflammatory responses through NF-κB and ERK1/2 pathway in ARDS. Front. Immunol. 13:1090773. doi: 10.3389/fimmu.2022.1090773
Received: 06 November 2022; Accepted: 19 December 2022;
Published: 06 January 2023.
Edited by:
Ping Yuan, Tongji University, ChinaReviewed by:
Hong-Da Zhang, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaCopyright © 2023 Cong, Yang, Zeng, Wu, Zhao, Shen, Xiao and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Zhu, eGl6aHVjY21AMTYzLmNvbQ==; Han Xiao, eGlhb2hhbkBiam11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.