 Xiao Wang
Xiao Wang Yupei Yuan
Yupei Yuan Yihan Liu
Yihan Liu Leiliang Zhang
Leiliang Zhang- 1Department of Infectious Diseases, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, China
- 2Department of Pathogen Biology, School of Clinical and Basic Medical Sciences, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, Shandong, China
- 3Medical Science and Technology Innovation Center, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, Shandong, China
Rift Valley fever (RVF) is a zoonotic disease caused by Rift Valley fever virus (RVFV), an emerging arbovirus within the Phenuiviridae family of Bunyavirales that has potential to cause severe diseases in both humans and livestock. It increases the incidence of abortion or foetal malformation in ruminants and leads to clinical manifestations like encephalitis or haemorrhagic fever in humans. Upon virus invasion, the innate immune system from the cell or the organism is activated to produce interferon (IFN) and prevent virus proliferation. Meanwhile, RVFV initiates countermeasures to limit antiviral responses at transcriptional and protein levels. RVFV nonstructural proteins (NSs) are the key virulent factors that not only perform immune evasion but also impact the cell replication cycle and has cytopathic effects. In this review, we summarize the innate immunity host cells employ depending on IFN signal transduction pathways, as well as the immune evasion mechanisms developed by RVFV primarily with the inhibitory activity of NSs protein. Clarifying the arms race between host innate immunity and RVFV immune evasion provides new avenues for drug target screening and offers possible solutions to current and future epidemics.
1. Introduction
Rift Valley fever virus (RVFV), belonging to the Phlebovirus genus of the Phenuiviridae family from Bunyavirales (1), is an arthropod-borne virus that affects people and livestock. It was first discovered in 1930 when a fatal infectious disease broke out among sheep in the Rift Valley, Kenya (2). Since the 1950s, the RVF pandemic had regularly occurred throughout Africa (3). The infected area expanded to Yemen and Saudi Arabia in the Arabian Peninsula after 2000 (4, 5). Transmitted by Aedes and Culex mosquitoes, RVFV spreads in larger geographic ranges due to climate change, making its propagation a possible hazard to non-epidemic countries (6, 7). Instead of mosquito bites, most human cases are caused by contact with infected animal fluids or tissues (8). RVFV causes human diseases including mild flu-like symptoms, hepatitis, retinitis, lethal encephalitis, and hemorrhagic fever, and the overall mortality rate is 0.5 to 1% (9). Pregnant livestock, especially sheep, are highly susceptible to RVFV infection. It generates abortion storms in which almost all pregnant infected animals have miscarriages (10). It also incurs a high mortality rate among newborn lambs (11). Therefore, RVFV infection has severe economic and human health costs. RVFV is now classified as a Category A disease by the National Institute of Allergy and Infectious Diseases (NIAID) and the National Institutes of Health (NIH) because of its potential for purposeful aerosol transmission and the absence of FDA-approved antiviral therapies or licensed vaccinations for humans. RVFV is also a select agent by the Centers for Disease Control and Prevention (CDC) and the U.S. Department of Agriculture (USDA).
RVFV genome consists of tripartite negative-sense single-stranded RNA segments. The RNA-dependent RNA polymerase is encoded by the large (L) segment (Figure 1). The medium (M) segment encodes the nonstructural protein NSm and envelope glycoproteins Gn and Gc. NSm-1 and NSm-2 are expressed from alternative start codons (Figure 1) (12). The nucleocapsid protein (N) and the nonstructural proteins (NSs) are both encoded by the small (S) segment (Figure 1). These two proteins are expressed in an ambisense manner, which means the N gene is encoded in the negative-sense genome, and NSs gene is encoded in the positive-sense genome (13, 14).
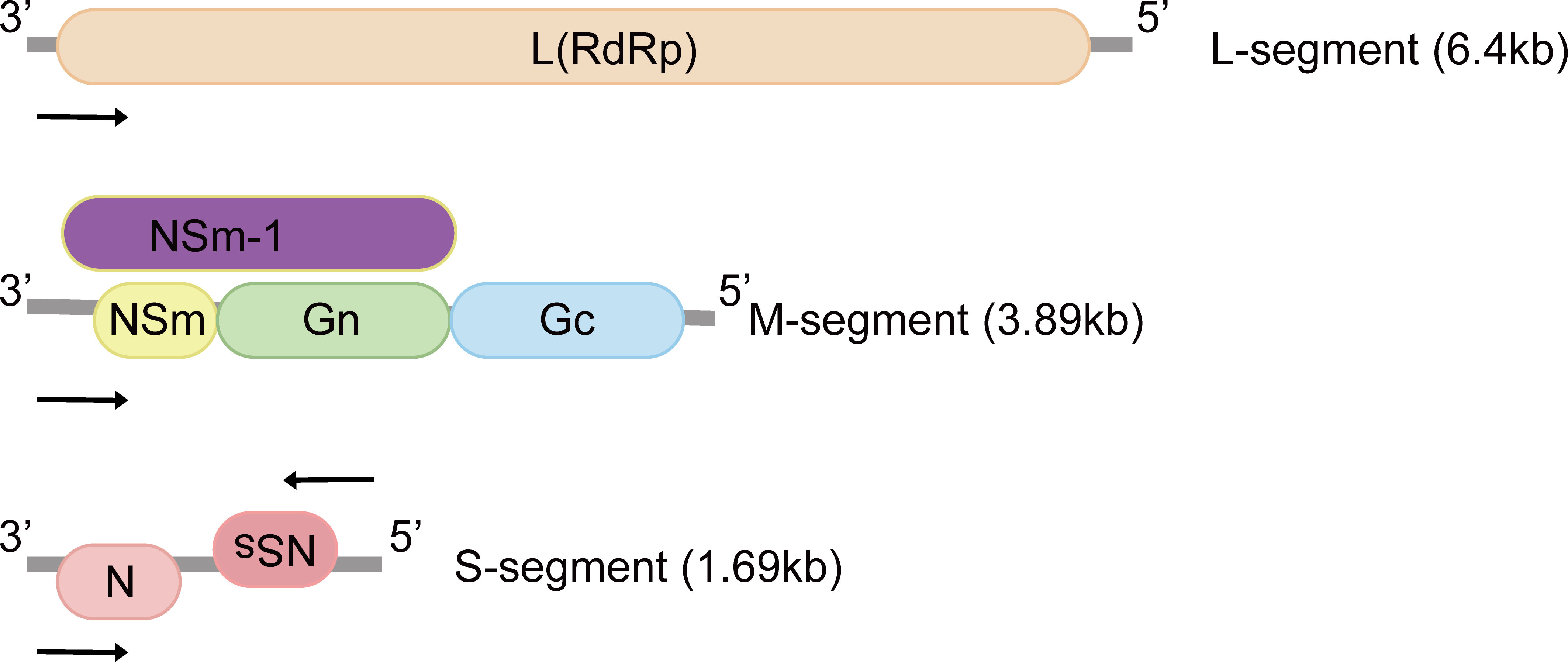
Figure 1 Genomic structure of Rift Valley fever virus. The tripartite negative-sense single-stranded RNA segments are named according to sizes: small (S), medium (M) and large (L), and proteins encoded by each segment are illustrated. The M-segment encodes at least four types of proteins: Gn, Gc, NSm and NSm-1. N and NSs are expressed in an ambisense manner. L, L protein; RdRp, RNA-dependent RNA polymerase; Gn, Gc: glycoproteins; NSm, non-structural protein M; N, nucleocapsid protein; NSs, non-structural protein S.
When host cells detect an RNA virus, a series of complicated innate immune responses are initiated to eliminate the virus, alert cells nearby, and assemble more specialized immune cells to the site of infection. Retinoic acid-inducible gene I (RIG-I), melanoma differentiation factor 5 (MDA5), and Toll-like-receptors (TLRs) are cytosol pattern recognition receptors (PRRs) capable of detecting RNA viruses (15). RIG-I can be recruited to the mitochondria where it associates with the mitochondrial antiviral signaling protein (MAVS) (16). This activated RIG-I/MAVS complex serves as the intersection of multiple innate immune pathways stimulated immediately, particularly the interferon (IFN) and nuclear factor κB (NF-κB) signals (17). However, RNA viruses have advanced procedures to avoid, exploit, or dysregulate these innate immune pathways, which can be seen in chikungunya virus (CHIKV) infection (18). The E1, E2, and nsP2 proteins of CHIKV potently inhibit the activity of MDA5/RIG-I, and nsP2 also suppresses the downstream phosphorylation of signal transducer and activator of transcription 1 (STAT1), blocking the IFN-induced JAK-STAT pathway (18). In this review, we summarize host innate immune responses to RVFV and how they are dysregulated by viral interference, which will offer possible insights into the vaccine and antiviral developments against RVFV.
2. Innate antiviral host defense: Interferon response as the crucial step
Interferon is a potent cytokine and a key component of the first line of defense against viral infection (19), which has immunological effects mainly through the direct induction of anti-pathogen molecules that inhibit viral replication (20). There are three types of IFNs involved in antiviral immunity, including IFN-I, IFN-II, and IFN-III. IFN-I and IFN-III share important antiviral properties and are expressed by cells with immunologic and tissue specificity (20, 21).
RNA-triggered intrinsic immunity of RVFV is initiated predominantly by recognition of RIG-I (22). RIG-I consists of a C-terminal domain, a DECH helicase, and N-terminal caspase activation and recruitment domains (CARDs). When cytoplasmic RIG-I is bound with viral RNA, its recruitment to MAVS is activated through the liberated CARDs (23). RIG-I/MAVS complex then catalyzes the combination of TANK-binding kinase 1(TBK1) and inhibitor of κB kinase ϵ (IKKϵ) to phosphorylate and dimerize interferon regulatory factor 3 (IRF3). The phosphorylated dimeric IRF3 could be transported into the nucleus to directly promote IFN-I transcription (24). IFN-I aims primarily to activate the JAK/STAT immune signals in autocrine and paracrine manners, which results in IFN-stimulated genes (ISGs) expression, eliciting subsequent adaptive immune responses (25).
2.1. MAVS is crucial for mounting IFN-I response
RIG-I is critical for IFN generation in a TLR-independent way by primary immune cells like macrophages and dendritic cells. That signaling through MAVS protects cells against mortality and mild morbidity during live RVFV mucosal infection (22). RVFV has been emerging as a noticeable neuropathogen (26, 27) and airborne transmission causes severe encephalitis. To understand the precise molecular mechanisms by which RVFV infection is controlled in the brain, a recent study found that microglia, the resident immune cell in the central nervous system acting as macrophages, strongly upregulated transcriptional levels of antiviral immune genes and increased levels of activation markers as well as cytokine secretion. This process was dependent on MAVS rather than TLR3 or TLR7 (28). MAVS-/- mice displayed IFN-I defects and lymphocyte infiltration dysregulation, leading to enhanced susceptibility to RVFV and higher mortality. This study defines a protective role for MAVS in propagating antiviral responses in the brain and suggests that signaling through MAVS may also be required for cerebral functional T and NK cell responses.
2.2. Intrinsic antiviral effect of exosomes
Exosomes belong to extracellular vesicles (EVs) and make contributions to cell–cell communication, immunomodulation, as well as infectivity enhancement during viral infections (29). The content of exosomes depends on the cellular origin and the type of infection (30, 31). They are thought to originate from late endosomes and then are secreted into the extracellular environment (32).
Although studies have shown the role of exosomes in viral infections (33), little is known about the mechanisms by which exosome exchanges control the immune response and impact the pathogenesis of RVFV. Researchers generated RVFV-resistant latent clones whose exosomes contain not only normal marker CD63 but also viral RNA and proteins like N and NSs (34). Some of the neighboring recipient cells showed drastically increased apoptosis via PARP cleavage and caspase 3 activation. Later, one study revealed how exosomes affect viral production and protect recipient cells in an innate immune manner (35). Exosomes that are purified from RVFV-infected cells carry RNA genome segments, which activate RIG-I to induce IFN-dependent activation of autophagy in naïve recipient cells like monocytes to suppress viral replication and dissemination.
2.3. Host cell metabolites and immune response
2.3.1. Polyamine depletion stimulates innate immune signal
To successfully infect a host cell, viruses need cellular metabolites, and there are different ways they can take over these molecules. One of the critical members of these metabolites is polyamines. They are small, positively charged host-derived molecules that play diverse roles in human cells (36), and polyamine-depleted mammalian cells maintain viability without significant toxicity (37). RNA viruses rely on polyamines for replication (38) and a recent study showed that diverse bunyaviruses, especially RVFV, La Crosse virus (LACV), and Keystone virus (KEYV), require polyamines for productive infection (39). Viral noninfectious particles can interfere with productive infection via binding cellular receptors or usurping cellular and viral machinery from infectious viruses (40). In polyamine-depleted cells, bunyaviruses produce a large number of noninfectious virions that are indistinguishable from infectious particles, but these particles could disrupt productive infection and stimulate antiviral signaling pathways like the IFN-I pathway. To conclude, polyamine depletion results in the accumulation of noninfectious particles that interfere with viral replication and stimulate innate immune signaling to limit infectivity. Later, researchers investigated how polyamines precisely function in RVFV infection and found that spermidine, a specific type of polyamine, is required for RVFV replication (41). Furthermore, RVFV also relies on polyamines for cholesterol synthesis to complete replication and form progeny virions, including the incorporation of cholesterol in virions (42). It will emphasize a promising method of targeting host polyamines to reduce virus replication.
2.3.2. AMPK inhibits fatty acid synthesis to restrict viral infection
Viruses also manipulate cellular lipids to form complex structures required for viral replication, many of which are dependent on de novo fatty acid synthesis (43). For example, envelope formation during viral assembly involves membrane lipid modifications (44). The energy regulator AMP-activated protein kinase (AMPK), which strongly inhibits fatty acid synthesis (45), could restrict infection of RVFV, and it relies on the upstream activator LKB1 (46). AMPK is activated during RVFV infection, leading to the phosphorylation and inhibition of acetyl-CoA carboxylase, the first rate-limiting enzyme in fatty acid synthesis. Therefore, the activation of AMPK both restricts infection and reduces lipid levels. Also, this pathway plays a broad role in the antiviral defense of various arboviruses. Taken together, AMPK is an important component of the host cell innate immune response that provides a novel antiviral therapeutic target associated with the suppression of fatty acid metabolism.
2.4. TCF/β-catenin regulates virus-induced IFN-β expression
Production of IFN-β plays a key role in the innate antiviral response. Using genome-wide RNA interference (RNAi) screening, canonical Wnt/β-catenin signaling was found to be an important host pathway during RVFV infection. It can regulate optimal cell cycle conditions and mediate the formation of the TCF/β-catenin complex to promote efficient viral replication (47). β-catenin can be found within a degradation complex associated with GSK-3, the Wnt/β-catenin pathway kinase. Inhibiting GSK-3 increases the amount of β-catenin and promotes its nuclear accumulation. β-catenin interacts with T-cell factor (TCF), rather than IRF3, to form the TCF/β-catenin complex, which can be recruited over the IFN-β promoter and increase the degree of constitutive IFN-β expression in uninfected cells (48). Additionally, raising the level of constitutive IFN-β is capable of conferring an effective antiviral state to naïve cells in order to promote subsequent virus-induced IFN-β expression. In RVFV infection, active TCF/β-catenin complexes are formed and the host Wnt/β-catenin pathway is targeted at the transcriptional and protein levels. NS protein is the major virulent factor to inhibit Wnt/β-catenin signaling by regulating relevant gene expression. Removal of NS protein from RVFV activates the Wnt/β-catenin pathway, forming a TCF/β-catenin complex, and TCF directly upregulates IFN-β expression.
3. Viral countermeasure and innate immune evasion
Host cells tend to take immediate measures to limit viral replication and propagation right after being infected, and simultaneously the virus initiates countermeasures to limit the cell’s antiviral responses. This includes suppressing the host innate immune pathway, and directly disrupting host gene expression.
3.1. Alternative splicing of RIOK3 during RVFV infection reverses its antiviral and anti-inflammatory effects
Transcriptome studies have revealed that viral invasion could change host splicing patterns (49). A significant post- and co-transcriptional regulatory mechanism known as alternative splicing (AS) affects the expression of more than 95% of the genes in the human genome and increases genetic coding capacity (50). Through its functional relationship with nonsense-mediated decay (NMD) to degrade premature termination selectively, AS enables the creation of structurally varied protein isoforms from a single gene and can help regulate gene expression (51, 52).
Atypical RIO Kinase 3 (RIOK3) has been demonstrated to play a significant role in promoting IFN-I production via PRR signaling mediated by RIG-I-like receptors to inhibit RVFV propagation (53). However, RIOK3 mRNA expression is distorted shortly after RVFV infection to produce alternatively spliced variants, RIOK3 X2, the truncated protein encoding premature termination codons to act as NMD substrate (54). This alternative splicing of the RIOK3 transcript reduces interferon expression conversely. Splicing factor TRA2-β is the key to regulating RIOK3 splicing isoforms (55). TRA2-β interaction with specific regions of RIOK3 pre-mRNA is essential for constitutive splicing of RIOK3 mRNA and RIOK3’s antiviral effect while lacking TRA2-β increases alternative splicing. TRA2-β mRNA is also alternatively spliced during RVFV infection, leading to a decrease in cellular TRA2-β levels. The roles of RIOK3 and its spliced isoform in both IFN and NF-κB pathways are intriguing (56). RIOK3 negatively regulates the inflammatory response, but RIOK3 X2 reverses the effects, mitigating the IFN response and increasing the inflammatory NF-κB response. Therefore, both RIOK3 and its X2 isoform have particular functions in separate RVFV-induced innate immune pathways (Figure 2).
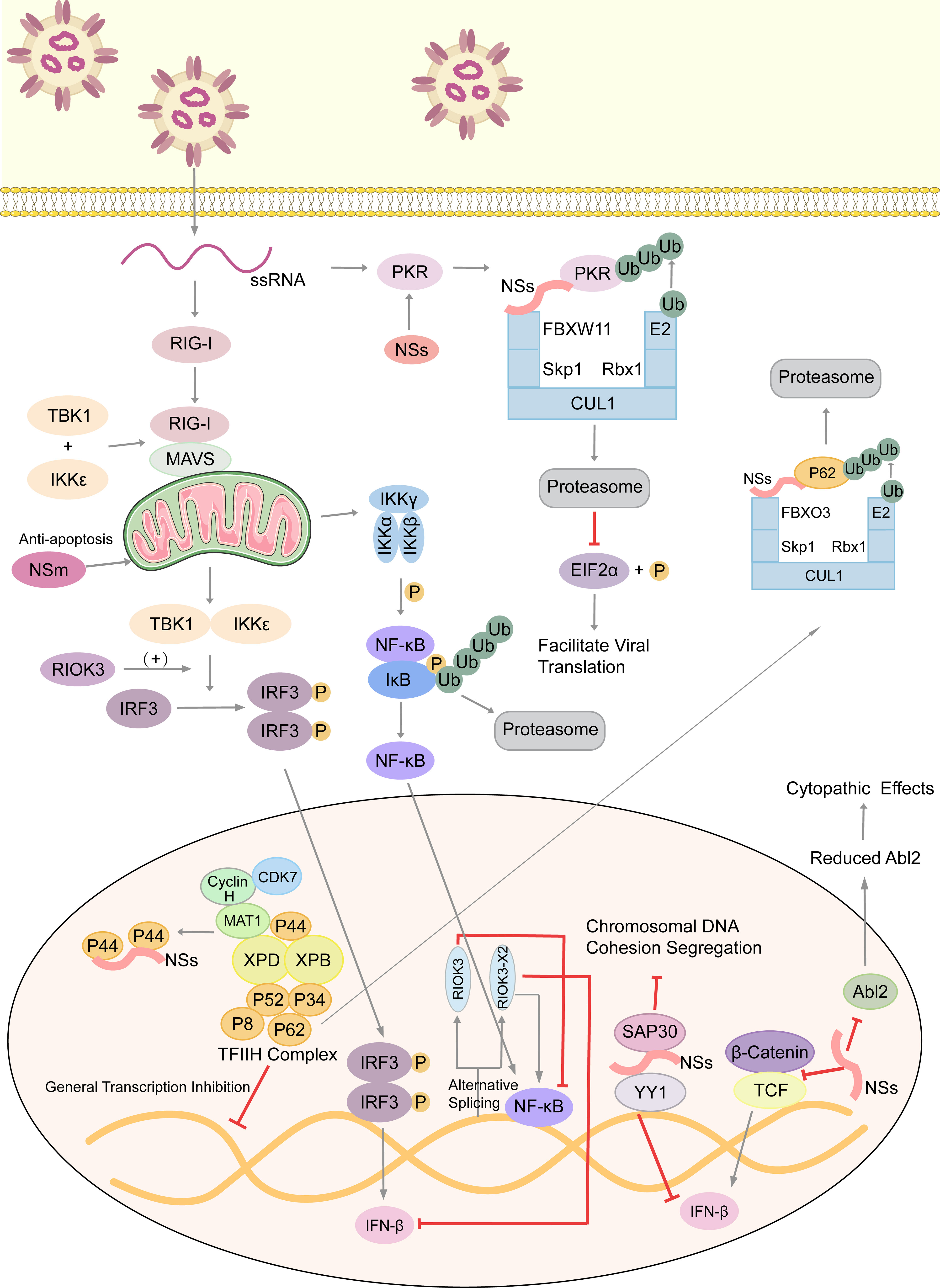
Figure 2 Arm race between RVFV and host, with emphasis on immune evasion by RVFV. RVFV ssRNA is recognized by cytosolic RIG-I. RIG-I associates with mitochondrial MAVS to activate multiple innate immune pathways. The kinase RIOK3 facilitates IFN expression and inhibits the inflammatory response pathway mediated by NF-κB, while the alternative splicing isoform RIOK3 X2 antagonizes these effects. RVFV infection also stimulates the Wnt pathway to produce IFNβ, but the NSs protein inhibits this process. NSs conduct the immune escape from several aspects. Those include inhibiting the aggregation of TFIIH complex to extensively inhibit host transcription, degrading kinase PKR to decrease eIF2α phosphorylation and promote the translation of viral proteins, and forming SAP30-NSs-YY1 co-repressor complex at the IFN promoter to block its transcription. Moreover, NSs affect the formation of cytoskeleton via suppressing Abl2 expression, which changes cell morphology and movement. Also, NSs could damage the host chromosomal DNA and disrupt mitosis. NSm, however, plays an anti-apoptotic role in mitochondria. ssRNA, single-stranded RNA; RIG-I, retinoic acid-inducible gene I; MAVS, mitochondrial antiviral signaling protein; TBK1, TANK-binding kinase 1; IKK, inhibitor of κB kinase; IκB, inhibitor of NF-κB; IRF3, interferon regulatory factor 3; TCF, T-cell factor; RIOK3, RIO Kinase 3; TFIIH, transcription factor IIH; CUL1, cullin 1; FBXO3, F-box protein 3; Skp1, S-phase kinase associated protein 1; FBXW11, F-box and WD repeat domain containing 11; Rbx-1, ring-box 1; PKR, protein kinase R; eIF2α, eukaryotic initiation factor 2α; SAP30, Sin3A associated protein 30; YY1, transcription factor Yin Yang 1; Abl2, Abelson murine leukemia viral oncogene 2; P, phosphate group; Ub, ubiquitin.
3.2. NSs protein: primary virulence factor inducing immune escape
3.2.1. Main functions of RVFV NSs protein
RVFV NSs accumulates in the nucleus and cytoplasm, while nuclear NSs forms a filamentous structure (57). Encoded on the S-segment of the RVFV genome, it is an important virulence factor which could potently suppress the innate immune response (58). NSs binds to Sin3A Associated Protein 30 (SAP30), and through interactions with the transcription factor Yin Yang 1(YY1) protein, the NSs-SAP30-YY1 complex blocks the activation of the IFN-β promoter (59). Viral evasion can occur with the contribution of NSs protein since RVFV lacking NSs is shown to induce abundant IFN-I in mice and no viremia is present (60).
Also, NSs generally inhibits host transcription and facilitates viral translation. Eukaryotic transcription factor IIH (TFIIH) is a general transcription factor for transcriptional initiation by eukaryotic RNA polymerase II and plays an important role in nucleotide excision DNA repair. TFIIH is comprised of ten subunits, including the core complex XPD, XPB, p44, p62, p8, p34 and p52. RVFV NSs could competitively bind to p44 and sequester it from binding with XPD (61). p62 is degraded by NSs in a post-translational way. Under the ubiquitin-proteasome pathway, NSs works as an adaptor protein in the cullin 1-Skp1-FBXO3 E3 ligase complex for p62 degradation (62, 63). These two methods disrupt the recruitment of the TFIIH complex in the nucleus, thus leading to the host transcriptional shutoff (Figure 2).
Similarly, RVFV NSs protein enhances the post-translational degradation of dsRNA-dependent protein kinase R(PKR) (64). PKR is a translation-inhibiting protein kinase. It can phosphorylate Eukaryotic initiation factor 2α (eIF2α), and then phosphorylated eIF2α inhibits the translation process. NSs participates in the formation of the E3 ligase complex, which consists of CUL1, Skp1, and FBXW11 (65, 66). This E3 ligase complex promotes the degradation of PKR via the ubiquitin-proteasome system as well. In consequence, PKR-mediated eIF2α phosphorylation is blocked and viral translation is facilitated effectively (Figure 2) (67).
Because of the genetic similarity of viruses within the Phenuiviridae family, NSs is also a key virulence factor of other phenuiviruses and its antiviral immune suppression in those phenuiviruses is worth investing in. NSs of Dabie bandavirus (severe fever with thrombocytopenia syndrome virus, SFTSV) has an intriguing mechanism to form granules in the cytoplasm, the inclusion bodies, to entrap factors involved in IFN-induced antiviral responses, like TBK1 and the E3 ubiquitin ligase TRIM25 that is essential for RIG-I activation (68). TBK1 is a critical regulator of not only IFN responses but also the NF-κB inflammatory pathway because it hinders the form of the IKK complex to limit the release and nuclear translocation of NF-κB. The inhibition of TBK1 during SFTSV infection leads to hyper-activation of NF-κB and inflammatory response (69). NSs of SFTSV could induce the cytokine storm, which leads to a high fatality rate of SFTS. Therefore, the different regulatory mechanisms of NS proteins in the innate immune system between RVFV and SFTSV have strong correspondence with their divergent pathogenicity and clinical manifestations.
3.2.2. NSs affects host cell replication
Nuclear abnormalities and a decreased mitotic rate observed in RVFV-infected cells, like micronuclei and lobulated nuclei, are largely because of the chromosomal cohesion and segregation defects (70). NSs filaments accumulating in the nucleus induce canonical DNA damage signaling, including checkpoint kinase 2 (Chk2), ataxia-telangiectasia mutated (ATM), and p53 (Figure 2). They also induce cell cycle arrest at the S phase or the G0/G1 phase (71). The SAP30-YY1 complex formed by NSs protein could affect not only IFN-β expression but also the cohesion and segregation of chromatin DNA. Through the SAP30-binding domain, RVFV NSs filaments interact with the pericentromeric major γ-satellite sequence, but not the centromeric minor α-satellite sequence. Also, YY1 could mediate the interaction between the NSs-SAP30 complex and the γ-satellite sequence DNA (70). It is assumed that through NSs-mediated DNA damage, erroneous host cell replication impairs normal tissue development and may contribute to fetal deformity in infected ruminants.
3.2.3. NSs and cytopathic effects
Besides functioning as the main virulence factor counteracting the host innate antiviral response to facilitate viral replication and spread, the role of NSs in RVFV-induced cytopathic effects was investigated (72). Abelson murine leukemia viral oncogene homolog 2 (Abl2) is a key regulator of the actin cytoskeleton, regulating cell morphology and mobility as well as cell-cell and cell-matrix adhesion (73, 74) via its tyrosine kinase domain and two filamentous actin binding domains (75). The impact of NSs expression on the actin cytoskeleton was examined when carrying out infections with the NSs-expressing virulent (ZH548) strain, the attenuated (MP12) strain, and the non-NSs-expressing (ZH548ΔNSs) strain, as well as following the ectopic expression of NSs. The upregulation of Abl2 expression in macrophages, fibroblasts, and hepatocytes, which would be identified as a component of antiviral responses, was blocked by NSs expression. In addition, ZH548-infected cells had increased mobility compared to ZH548ΔNSs-infected fibroblasts with substantial alterations in cell morphology, including the loss of lamellipodia, cell spreading, and distortion of adherens junctions. All these phenomena are similar to the ZH548-induced cytopathic effects seen in vivo. Taken together, NSs protein affects the actin cytoskeleton of host cells at the transcriptional and cellular levels, and the upregulation of Abl2 expression is proposed to be part of the host strategy to restrict virulence (Figure 2).
3.3. Anti-apoptotic role of NSm proteins
Like NSs protein, NSm is not essential for viral replication in cell cultures (76). A recent study screened and identified 9 host proteins that putatively interact with RVFV NSm, and three of them (Cpsf2, Ppil2, SNAP-25) are the most promising targets during viral infection (77). RVFV NSm was identified as the first Phlebovirus protein that has an anti-apoptotic function (78). The C-terminal region of NSm, which contains a basic amino acid cluster and a putative transmembrane domain, targets itself to the mitochondrial outer membrane to resist apoptosis (Figure 2) (79).
In comparison to RVFV arMP-12-infected cells, RVFV arMP-12-del21/384-infected cells which lacked NSm expression caused widespread cell death because of the cleavage of Caspase-3 and its downstream substrate poly(ADP-ribose) polymerase. And the initiator caspases, caspase-8 and -9, were all activated earlier. Further, NSm does not require other viral proteins to prevent cell apoptosis because NSm production prevents the staurosporine(STP)-induced activation of caspase-8 and-9. The P38-MAPK pathway is essential for cell survival, and RVFV NSm could also regulate the p38-MAPK response in mammalian cells (80). The specific host factors involved in the NSm-mediated anti-apoptosis are worth investigating, and whether NSm contributes to reaching a balance with the pro-apoptotic NSs protein is an interesting problem requiring further comparison of various environmental factors and mutual molecular mechanisms.
4. Conclusion and perspectives
As one of the most important bunyaviruses, RVFV has been responsible for significant human and ruminant outbreaks that have devastated local economies with increasing mortality and morbidity. Upon viral infection, the innate immune system is activated as the first line of defense. IFN response is induced by intricate upstream pathways. The MDA5 and RIG-I are RIG-I-like receptors (RLRs) sensing foreign RNA in the cytoplasm, which transduce a signaling cascade to induce downstream IFN production and subsequent antiviral responses. Lack of metabolites essential for the viral replication cycle and production of noninfectious particles are also methods against RVFV infection. RVFV evolves to evade immune attacks and in turn impairs cellular functions, which is mainly achieved by the powerful virulent NSs protein. These studies are critical to the development of RVFV attenuated vaccines that have been created so far, as well as the research on novel targets for effective viral inhibitors.
According to the effects of exosomes, it is verified that antiviral autophagy can be induced by IFN signals in RVFV infection. ERK1/2 Akt/mTOR signaling pathway might participate in the antiviral immunity since they have been in anti-tumor immunity (81). Meanwhile, IFN-β also activates caspase-dependent apoptosis, and autophagy could in turn decrease apoptosis to promote cell growth (81). Although IFN-induced innate immunity is TLR-independent, there still exists a Toll receptor-autophagy axis in RVFV infection with Toll-7 and Toll-like receptor adaptor MyD88 (82). Since NSs presents potent suppression of IFN-β transcription and induces the p53 signaling pathway to increase cell apoptosis (83), it is of great significance to discover other independent autophagy-activated systems to restrict viral replication in time. Further research on the precise mechanism in which autophagy is initiated by anti-RVFV innate immune responses and on the intrinsic correlation between IFN-induced autophagy and IFN-related apoptosis during RVFV infection is still needed.
TCF/β-catenin complexes can upregulate the level of IFN-β expression in response to RVFV infection, which is antagonized by the virulence factor NSs. In addition, Wnt/β-catenin signals are shown to regulate the polyamine metabolic pathway in aggressive prostate cancer, reducing the concentrations of citrate and spermine (84). β-catenin signals also promote fatty acid β-oxidation as energy resources for osteoblast metabolism (85). Given that polyamine depletion and fatty acid synthesis inhibition could shut off viral replication and stimulate IFN-induced innate immune responses, it is a promising strategy to target the Wnt/β-catenin pathway and produce a combined action to limit the progress of RVFV infection.
NSs is a key virulence factor of phenuiviruses as an antiviral immune antagonist, but NSs in these viruses have slight differences in anti-immune mechanisms and corresponding cellular effects. Unlike RVFV filamentous NSs in the nucleus, SFTSV, Toscana virus (TOSV) and Uukuniemi virus (UUKV) NSs proteins localize only in the cytoplasm, so they could not directly inhibit host transcription. Besides the unique cytoplasmic granules generated by SFTSV NSs to sequester numerous host factors, TOSV NSs degrades PKR to facilitate viral translation similarly with RVFV, but TOSV NSs could also degrade RIG-I to suppress IFN-β signal activation with its E3 ubiquitin ligase activity (86, 87). NSs of the Punta Toro virus (PTV) can inhibit host transcription, but the nuclear NSs does not form a filamentous structure (88). Sandfly fever virus (SFV) NSs blocks downstream IFN-I signals by inhibiting Jak1 phosphorylation (89). In general, despite the genetic diversity of NSs among different phenuiviruses, it is of great significance to find out their highly conserved IFN-inhibitory activity in immune evasion to drive the development of broad-spectrum drugs and effective vaccines.
From what has been discussed above, most studies have concentrated on how the virus works to counteract the innate immune response, which is the body’s initial and first line of defense. However, RVFV has also developed additional means of attacking various cellular processes, like the cytopathic effects and pro-apoptosis. These methods contribute to viral pathogenicity and should be further investigated in the future.
Author contributions
LZ conceived the study. XW wrote the first draft. YY and LZ revised the manuscript. XW and YL generated the Figures. All authors read and approved the final manuscript.
Funding
This work was supported by grants from National College Students innovation and entrepreneurship training program [202210439003], Taishan Scholars Program, and Academic promotion programme of Shandong First Medical University [2019LJ001].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, et al. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2017). Arch Virol (2017) 162:2505–38. doi: 10.1007/s00705-017-3358-5
2. Daubney R, Hudson JR, Garnham PCC. Enzootic hepatitis or rift valley fever. an undescribed virus disease of sheep cattle and man from east africa. J Pathol Bacteriol (1931) 34:545–79. doi: 10.1002/path.1700340418
3. McMillen CM, Hartman AL. Rift valley fever in animals and humans: Current perspectives. Antiviral Res (2018) 156:29–37. doi: 10.1016/j.antiviral.2018.05.009
4. Madani TA, Al-Mazrou YY, Al-Jeffri MH, Mishkhas AA, Al-Rabeah AM, Turkistani AM, et al. Rift valley fever epidemic in Saudi Arabia: epidemiological, clinical, and laboratory characteristics. Clin Infect Dis (2003) 37:1084–92. doi: 10.1086/378747
5. Nasher AAW, Shiban AK, Al-Eriyani M, Aly-Bourgy A, Al-Kohlani AH, Benbrake M, et al. Outbreak of rift valley fever–Yemen, august-October 2000. MMWR Morb Mortal Wkly Rep (2000) 49:1065–6.
6. Samy AM, Elaagip AH, Kenawy MA, Ayres CFJ, Peterson AT, Soliman DE. Climate change influences on the global potential distribution of the mosquito culex quinquefasciatus, vector of West Nile virus and lymphatic filariasis. PloS One (2016) 11:e0163863. doi: 10.1371/journal.pone.0163863
7. Iwamura T, Guzman-Holst A, Murray KA. Accelerating invasion potential of disease vector aedes aegypti under climate change. Nat Commun (2020) 11:2130. doi: 10.1038/s41467-020-16010-4
8. Nicholas DE, Jacobsen KH, Waters NM. Risk factors associated with human rift valley fever infection: systematic review and meta-analysis. Trop Med Int Health (2014) 19:1420–9. doi: 10.1111/tmi.12385
9. Ikegami T, Makino S. The pathogenesis of rift valley fever. Viruses (2011) 3:493–519. doi: 10.3390/v3050493
10. Coetzer JA. The pathology of rift valley fever. II. lesions occurring in field cases in adult cattle, calves and aborted foetuses. Onderstepoort J Vet Res (1982) 49:11–7.
11. Bird BH, Githinji JWK, Macharia JM, Kasiiti JL, Muriithi RM, Gacheru SG, et al. Multiple virus lineages sharing recent common ancestry were associated with a large rift valley fever outbreak among livestock in Kenya during 2006-2007. J Virol (2008) 82:11152–66. doi: 10.1128/JVI.01519-08
12. Kreher F, Tamietti C, Gommet C, Guillemot L, Ermonval M, Failloux A-B, et al. The rift valley fever accessory proteins NSm and P78/NSm-GN are distinct determinants of virus propagation in vertebrate and invertebrate hosts. Emerg Microbes Infect (2014) 3:e71. doi: 10.1038/emi.2014.71
13. Bouloy M, Weber F. Molecular biology of rift valley fever virus. Open Virol J (2010) 4:8–14. doi: 10.2174/1874357901004010008
14. Ikegami T. Molecular biology and genetic diversity of rift valley fever virus. Antiviral Res (2012) 95:293–310. doi: 10.1016/j.antiviral.2012.06.001
15. Brisse M, Ly H. Comparative structure and function analysis of the RIG-I-Like receptors: RIG-I and MDA5. Front In Immunol (2019) 10:1586. doi: 10.3389/fimmu.2019.01586
16. Sun Q, Sun L, Liu H-H, Chen X, Seth RB, Forman J, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity (2006) 24:633–42. doi: 10.1016/j.immuni.2006.04.004
17. Seth RB, Sun L, Ea C-K, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
18. Liu Y, Yuan Y, Zhang L. Innate immune evasion by alphaviruses. Front Immunol (2022) 13:1005586. doi: 10.3389/fimmu.2022.1005586
19. Sadler AJ, Williams BRG. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8:559–68. doi: 10.1038/nri2314
20. Stanifer ML, Pervolaraki K, Boulant S. Differential regulation of type I and type III interferon signaling. Int J Mol Sci (2019) 20:1445. doi: 10.3390/ijms20061445
21. Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res (2011) 17:2619–27. doi: 10.1158/1078-0432.CCR-10-1114
22. Ermler ME, Yerukhim E, Schriewer J, Schattgen S, Traylor Z, Wespiser AR, et al. RNA Helicase signaling is critical for type i interferon production and protection against rift valley fever virus during mucosal challenge. J Virol (2013) 87:4846–60. doi: 10.1128/JVI.01997-12
23. Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell (2011) 147:423–35. doi: 10.1016/j.cell.2011.09.039
24. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25:349–60. doi: 10.1016/j.immuni.2006.08.009
25. Nan Y, Wu C, Zhang Y-J. Interplay between janus Kinase/Signal transducer and activator of transcription signaling activated by type I interferons and viral antagonism. Front In Immunol (2017) 8:1758. doi: 10.3389/fimmu.2017.01758
26. Wiley CA. Emergent viral infections of the CNS. J Neuropathol Exp Neurol (2020) 79:823–42. doi: 10.1093/jnen/nlaa054
27. Connors KA, Hartman AL. Advances in understanding neuropathogenesis of rift valley fever virus. Annu Rev Virol (2022) 9:437–50. doi: 10.1146/annurev-virology-091919-065806
28. Hum NR, Bourguet FA, Sebastian A, Lam D, Phillips AM, Sanchez KR, et al. MAVS mediates a protective immune response in the brain to rift valley fever virus. PloS Pathog (2022) 18:e1010231. doi: 10.1371/journal.ppat.1010231
29. Sampey GC, Meyering SS, Zadeh MA, Saifuddin M, Hakami RM, Kashanchi F. Exosomes and their role in CNS viral infections. J Neurovirol (2014) 20:199–208. doi: 10.1007/s13365-014-0238-6
30. Hui WW, Hercik K, Belsare S, Alugubelly N, Clapp B, Rinaldi C, et al. Salmonella enterica serovar typhimurium alters the extracellular proteome of macrophages and leads to the production of proinflammatory exosomes. Infect Immun (2018) 86:e00386-17. doi: 10.1128/IAI.00386-17
31. Tucher C, Bode K, Schiller P, Claßen L, Birr C, Souto-Carneiro MM, et al. Extracellular vesicle subtypes released from activated or apoptotic T-lymphocytes carry a specific and stimulus-dependent protein cargo. Front In Immunol (2018) 9:534. doi: 10.3389/fimmu.2018.00534
32. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci (2018) 75:193–208. doi: 10.1007/s00018-017-2595-9
33. Fleming A, Sampey G, Chung M-C, Bailey C, van Hoek ML, Kashanchi F, et al. The carrying pigeons of the cell: exosomes and their role in infectious diseases caused by human pathogens. Pathog Dis (2014) 71:109–20. doi: 10.1111/2049-632X.12135
34. Ahsan NA, Sampey GC, Lepene B, Akpamagbo Y, Barclay RA, Iordanskiy S, et al. Presence of viral RNA and proteins in exosomes from cellular clones resistant to rift valley fever virus infection. Front In Microbiol (2016) 7:139. doi: 10.3389/fmicb.2016.00139
35. Alem F, Olanrewaju AA, Omole S, Hobbs HE, Ahsan N, Matulis G, et al. Exosomes originating from infection with the cytoplasmic single-stranded RNA virus rift valley fever virus (RVFV) protect recipient cells by inducing RIG-I mediated IFN-b response that leads to activation of autophagy. Cell Biosci (2021) 11:220. doi: 10.1186/s13578-021-00732-z
36. Mandal S, Mandal A, Johansson HE, Orjalo AV, Park MH. Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc Natl Acad Sci U.S.A. (2013) 110:2169–74. doi: 10.1073/pnas.1219002110
37. Simoneau AR, Gerner EW, Nagle R, Ziogas A, Fujikawa-Brooks S, Yerushalmi H, et al. The effect of difluoromethylornithine on decreasing prostate size and polyamines in men: results of a year-long phase IIb randomized placebo-controlled chemoprevention trial. Cancer Epidemiol Biomarkers Prev (2008) 17:292–9. doi: 10.1158/1055-9965.EPI-07-0658
38. Mounce BC, Cesaro T, Moratorio G, Hooikaas PJ, Yakovleva A, Werneke SW, et al. Inhibition of polyamine biosynthesis is a broad-spectrum strategy against RNA viruses. J Virol (2016) 90:9683–92. doi: 10.1128/JVI.01347-16
39. Mastrodomenico V, Esin JJ, Graham ML, Tate PM, Hawkins GM, Sandler ZJ, et al. Polyamine depletion inhibits bunyavirus infection via generation of noninfectious interfering virions. J Virol (2019) 93:e00530-19. doi: 10.1128/JVI.00530-19
40. Rezelj VV, Levi LI, Vignuzzi M. The defective component of viral populations. Curr Opin Virol (2018) 33:74–80. doi: 10.1016/j.coviro.2018.07.014
41. Mastrodomenico V, Esin JJ, Qazi S, Khomutov MA, Ivanov AV, Mukhopadhyay S, et al. Virion-associated polyamines transmit with bunyaviruses to maintain infectivity and promote entry. ACS Infect Dis (2020) 6:2490–501. doi: 10.1021/acsinfecdis.0c00402
42. Mastrodomenico V, LoMascolo NJ, Cruz-Pulido YE, Cunha CR, Mounce BC. Polyamine-linked cholesterol incorporation in rift valley fever virus particles promotes infectivity. ACS Infect Dis (2022) 8:1439–48. doi: 10.1021/acsinfecdis.2c00071
43. Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol (2008) 6:363–74. doi: 10.1038/nrmicro1890
44. Welsch S, Müller B, Kräusslich H-G. More than one door - budding of enveloped viruses through cellular membranes. FEBS Lett (2007) 581:2089–97. doi: 10.1016/j.febslet.2007.03.060
45. Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans (2002) 30:1064–70. doi: 10.1042/bst0301064
46. Moser TS, Schieffer D, Cherry S. AMP-activated kinase restricts rift valley fever virus infection by inhibiting fatty acid synthesis. PloS Pathog (2012) 8:e1002661. doi: 10.1371/journal.ppat.1002661
47. Harmon B, Bird SW, Schudel BR, Hatch AV, Rasley A, Negrete OA. A genome-wide RNA interference screen identifies a role for wnt/β-catenin signaling during rift valley fever virus infection. J Virol (2016) 90:7084–97. doi: 10.1128/JVI.00543-16
48. Marcato V, Luron L, Laqueuvre LM, Simon D, Mansuroglu Z, Flamand M, et al. Beta-catenin upregulates the constitutive and virus-induced transcriptional capacity of the interferon beta promoter through T-cell factor binding sites. Mol Cell Biol (2016) 36:13–29. doi: 10.1128/MCB.00641-15
49. Havranek KE, White LA, Lanchy J-M, Lodmell JS. Transcriptome profiling in rift valley fever virus infected cells reveals modified transcriptional and alternative splicing programs. PloS One (2019) 14:e0217497. doi: 10.1371/journal.pone.0217497
50. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature (2008) 456:470–6. doi: 10.1038/nature07509
51. Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci U.S.A. (2003) 100:189–92. doi: 10.1073/pnas.0136770100
52. Chang Y-F, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem (2007) 76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909
53. Feng J, De Jesus PD, Su V, Han S, Gong D, Wu NC, et al. RIOK3 is an adaptor protein required for IRF3-mediated antiviral type I interferon production. J Virol (2014) 88:7987–97. doi: 10.1128/JVI.00643-14
54. Havranek KE, White LA, Bisom TC, Lanchy JM, Lodmell JS. The atypical kinase RIOK3 limits RVFV propagation and is regulated by alternative splicing. Viruses (2021) 13:367. doi: 10.3390/v13030367
55. White LA, Bisom TC, Grimes HL, Hayashi M, Lanchy JM, Lodmell JS. Tra2beta-dependent regulation of RIO kinase 3 splicing during rift valley fever virus infection underscores the links between alternative splicing and innate antiviral immunity. Front Cell Infect Microbiol (2021) 11:799024. doi: 10.3389/fcimb.2021.799024
56. Bisom TC, White LA, Lanchy JM, Lodmell JS. RIOK3 and its alternatively spliced isoform have disparate roles in the innate immune response to rift valley fever virus (MP12) infection. Viruses (2022) 14:2064. doi: 10.3390/v14092064
57. Flick R, Bouloy M. Rift valley fever virus. Curr Mol Med (2005) 5:827–34. doi: 10.2174/156652405774962263
58. Ly HJ, Ikegami T. Rift valley fever virus NSs protein functions and the similarity to other bunyavirus NSs proteins. Virol J (2016) 13:118. doi: 10.1186/s12985-016-0573-8
59. Le May N, Mansuroglu Z, Léger P, Josse T, Blot G, Billecocq A, et al. A SAP30 complex inhibits IFN-beta expression in rift valley fever virus infected cells. PloS Pathog (2008) 4:e13. doi: 10.1371/journal.ppat.0040013
60. Bouloy M, Janzen C, Vialat P, Khun H, Pavlovic J, Huerre M, et al. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J Virol (2001) 75:1371–7. doi: 10.1128/JVI.75.3.1371-1377.2001
61. Le May N, Dubaele S, Proietti De Santis L, Billecocq A, Bouloy M, Egly J-M. TFIIH transcription factor, a target for the rift valley hemorrhagic fever virus. Cell (2004) 116:541–50. doi: 10.1016/S0092-8674(04)00132-1
62. Kalveram B, Lihoradova O, Ikegami T. NSs protein of rift valley fever virus promotes posttranslational downregulation of the TFIIH subunit p62. J Virol (2011) 85:6234–43. doi: 10.1128/JVI.02255-10
63. Kainulainen M, Habjan M, Hubel P, Busch L, Lau S, Colinge J, et al. Virulence factor NSs of rift valley fever virus recruits the f-box protein FBXO3 to degrade subunit p62 of general transcription factor TFIIH. J Virol (2014) 88:3464–73. doi: 10.1128/JVI.02914-13
64. Habjan M, Pichlmair A, Elliott RM, Overby AK, Glatter T, Gstaiger M, et al. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J Virol (2009) 83:4365–75. doi: 10.1128/JVI.02148-08
65. Mudhasani R, Tran JP, Retterer C, Kota KP, Whitehouse CA, Bavari S. Protein kinase r degradation is essential for rift valley fever virus infection and is regulated by SKP1-CUL1-F-box (SCF)FBXW11-NSs E3 ligase. PloS Pathog (2016) 12:e1005437. doi: 10.1371/journal.ppat.1005437
66. Kainulainen M, Lau S, Samuel CE, Hornung V, Weber F. NSs virulence factor of rift valley fever virus engages the f-box proteins FBXW11 and β-TRCP1 to degrade the antiviral protein kinase PKR. J Virol (2016) 90:6140–7. doi: 10.1128/JVI.00016-16
67. Ikegami T, Narayanan K, Won S, Kamitani W, Peters CJ, Makino S. Rift valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PloS Pathog (2009) 5:e1000287. doi: 10.1371/journal.ppat.1000287
68. Wu X, Qi X, Qu B, Zhang Z, Liang M, Li C, et al. Evasion of antiviral immunity through sequestering of TBK1/IKKϵ/IRF3 into viral inclusion bodies. JVirol (2014) 88:3067–76. doi: 10.1128/JVI.03510-13
69. Khalil J, Yamada S, Tsukamoto Y, Abe H, Shimojima M, Kato H, et al. The non-structural protein NSs of SFTSV causes cytokine storm through the hyper-activation of NF-κB. Mol Cell Biol (2020) 41:e00542-20. doi: 10.1128/MCB.00542-20
70. Mansuroglu Z, Josse T, Gilleron J, Billecocq A, Leger P, Bouloy M, et al. Nonstructural NSs protein of rift valley fever virus interacts with pericentromeric DNA sequences of the host cell, inducing chromosome cohesion and segregation defects. J Virol (2010) 84:928–39. doi: 10.1128/JVI.01165-09
71. Baer A, Austin D, Narayanan A, Popova T, Kainulainen M, Bailey C, et al. Induction of DNA damage signaling upon rift valley fever virus infection results in cell cycle arrest and increased viral replication. J Biol Chem (2012) 287:7399–410. doi: 10.1074/jbc.M111.296608
72. Bamia A, Marcato V, Boissière M, Mansuroglu Z, Tamietti C, Romani M, et al. The NSs protein encoded by the virulent strain of rift valley fever virus targets the expression of Abl2 and the actin cytoskeleton of the host, affecting cell mobility, cell shape, and cell-cell adhesion. J Virol (2020) 95:e01768-20. doi: 10.1128/JVI.01768-20
73. Zandy NL, Playford M, Pendergast AM. Abl tyrosine kinases regulate cell-cell adhesion through rho GTPases. Proc Natl Acad Sci U.S.A. (2007) 104:17686–91. doi: 10.1073/pnas.0703077104
74. Bradley WD, Koleske AJ. Regulation of cell migration and morphogenesis by abl-family kinases: emerging mechanisms and physiological contexts. J Cell Sci (2009) 122:3441–54. doi: 10.1242/jcs.039859
75. Khatri A, Wang J, Pendergast AM. Multifunctional abl kinases in health and disease. J Cell Sci (2016) 129:9–16. doi: 10.1242/jcs.175521
76. Won S, Ikegami T, Peters CJ, Makino S. NSm and 78-kilodalton proteins of rift valley fever virus are nonessential for viral replication in cell culture. J Virol (2006) 80:8274–8. doi: 10.1128/JVI.00476-06
77. Engdahl C, Näslund J, Lindgren L, Ahlm C, Bucht G. The rift valley fever virus protein NSm and putative cellular protein interactions. Virol J (2012) 9:139. doi: 10.1186/1743-422X-9-139
78. Won S, Ikegami T, Peters CJ, Makino S. NSm protein of rift valley fever virus suppresses virus-induced apoptosis. J Virol (2007) 81:13335–45. doi: 10.1128/JVI.01238-07
79. Terasaki K, Won S, Makino S. The c-terminal region of rift valley fever virus NSm protein targets the protein to the mitochondrial outer membrane and exerts antiapoptotic function. J Virol (2013) 87:676–82. doi: 10.1128/JVI.02192-12
80. Narayanan A, Popova T, Turell M, Kidd J, Chertow J, Popov SG, et al. Alteration in superoxide dismutase 1 causes oxidative stress and p38 MAPK activation following RVFV infection. PloS One (2011) 6:e20354. doi: 10.1371/journal.pone.0020354
81. Li Y, Zhu H, Zeng X, Fan J, Qian X, Wang S, et al. Suppression of autophagy enhanced growth inhibition and apoptosis of interferon-beta in human glioma cells. Mol Neurobiol (2013) 47:1000–10. doi: 10.1007/s12035-013-8403-0
82. Moy RH, Gold B, Molleston JM, Schad V, Yanger K, Salzano M-V, et al. Antiviral autophagy restrictsRift valley fever virus infection and is conserved from flies to mammals. Immunity (2014) 40:51–65. doi: 10.1016/j.immuni.2013.10.020
83. Narayanan A, Amaya M, Voss K, Chung M, Benedict A, Sampey G, et al. Reactive oxygen species activate NFkappaB (p65) and p53 and induce apoptosis in RVFV infected liver cells. Virology (2014) 449:270–86. doi: 10.1016/j.virol.2013.11.023
84. Casero RA Jr., Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat Rev Cancer (2018) 18:681–95. doi: 10.1038/s41568-018-0050-3
85. Moorer MC, Riddle RC. Regulation of osteoblast metabolism by wnt signaling. Endocrinol Metab (Seoul) (2018) 33:318–30. doi: 10.3803/EnM.2018.33.3.318
86. Kalveram B, Ikegami T. Toscana virus NSs protein promotes degradation of double-stranded RNA-dependent protein kinase. J Virol (2013) 87:3710–8. doi: 10.1128/JVI.02506-12
87. Gori Savellini G, Anichini G, Gandolfo C, Prathyumnan S, Cusi MG. Toscana virus non-structural protein NSs acts as E3 ubiquitin ligase promoting RIG-I degradation. PloS Pathog (2019) 15:e1008186. doi: 10.1371/journal.ppat.1008186
88. Lihoradova OA, Indran SV, Kalveram B, Lokugamage N, Head JA, Gong B, et al. Characterization of rift valley fever virus MP-12 strain encoding NSs of punta toro virus or sandfly fever Sicilian virus. PloS Negl Trop Dis (2013) 7:e2181. doi: 10.1371/journal.pntd.0002181
Keywords: Rift Valley fever virus, innate immunity, interferon, immune evasion, nonstructural proteins
Citation: Wang X, Yuan Y, Liu Y and Zhang L (2022) Arm race between Rift Valley fever virus and host. Front. Immunol. 13:1084230. doi: 10.3389/fimmu.2022.1084230
Received: 30 October 2022; Accepted: 12 December 2022;
Published: 22 December 2022.
Edited by:
Chenhe Su, Wistar Institute, United StatesReviewed by:
Chuanmin Zhou, Wuhan University, ChinaYuexiu Zhang, The Ohio State University, United States
Aarthi Narayanan, George Mason University, United States
Copyright © 2022 Wang, Yuan, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leiliang Zhang, YXJtemhhbmdAaG90bWFpbC5jb20=