 Jin Gong
Jin Gong Wei Tu
Wei Tu Dean Tian
Dean Tian
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 January 2023
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1083780
This article is part of the Research Topic Targeting Signalling Pathways in Inflammatory Diseases View all 17 articles
Hepatocytes, the major parenchymal cells in the liver, are responsible for a variety of cellular functions including carbohydrate, lipid and protein metabolism, detoxification and immune cell activation to maintain liver homeotasis. Recent studies show hepatocytes play a pivotal role in liver inflammation. After receiving liver insults and inflammatory signals, hepatocytes may undergo organelle damage, and further respond by releasing mediators and expressing molecules that can act in the microenvironment as well as initiate a robust inflammatory response. In this review, we summarize how the hepatic organelle damage link to liver inflammation and introduce numerous hepatocyte-derived pro-inflammatory factors in response to chronic liver injury.
Chronic liver disease is characterized by hepatocyte injury and inflammation that lead to the development of cirrhosis and liver cancer, accounting for approximately 2 million deaths every year worldwide (1). Multiple etiologies include chronic HBV and HCV infection, nonalcoholic steatohepatitis (NASH), alcoholic liver disease, and autoimmune liver disease cause the global burden of liver disease. Hepatocytes comprise the majority (~85%) of the liver mass, and play a role in various biochemical and metabolic functions (2). Traditional concepts viewed hepatocytes as targets of immune or insults mediated injury, resulting in hepatocyte death which identified as a typical pathological feature in liver disease. However, recent studies have emphasized a role for hepatocyte as active drivers in liver inflammation and fibrosis through intercellular communication (3). Organelle damage, including mitochondria, lysosome, endoplasmic reticulum may determine the severity of hepatocyte injury (4). It is widely accepted that sterile hepatocyte death leads to the release of damage-associated molecular patterns (DAMPs), which are recognized by the innate immune system through pattern recognition receptors, and exaggerate inflammatory response in liver (5). What’s more, stressed hepatocytes engage in liver inflammation as well, for they can change their phenotype, make an adaptation to the microenvironment and alter their surrounding cell populations (2). Substantial evidence show that hepatocytes constitutively produce and secrete a variety of mediators that play important roles in immune regulation and fibrosis (6, 7). In this review, we will provide current literature investigating the adaptive and maladaptive alterations of hepatocytes during the initiation of liver injury, and how the stressed hepatocytes interact with the surrounding cells to trigger a proinflammatory microenvironment in chronic liver disease.
Endoplasmic reticulum (ER) is the major site of secretory and transmembrane protein folding, calcium homeostasis and lipid synthesis. Upon the accumulation of misfolded proteins in the ER, unfolded protein response (UPR) is activated by three ER-transmembrane sensors, namely PKR like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol requiring enzyme 1 (IRE1), coordinately through downstream factors including X-box binding protein 1 (XBP1), α-subunit of eukaryotic initiation factor 2 (eIF2α), C/EBP homologous protein (CHOP), activating Transcription Factor 4 (ATF4), to resolve the protein folding defect (8). Sustained or massive ER stress leads to hepatocyte steatosis and apoptosis (9) (Figure 1).
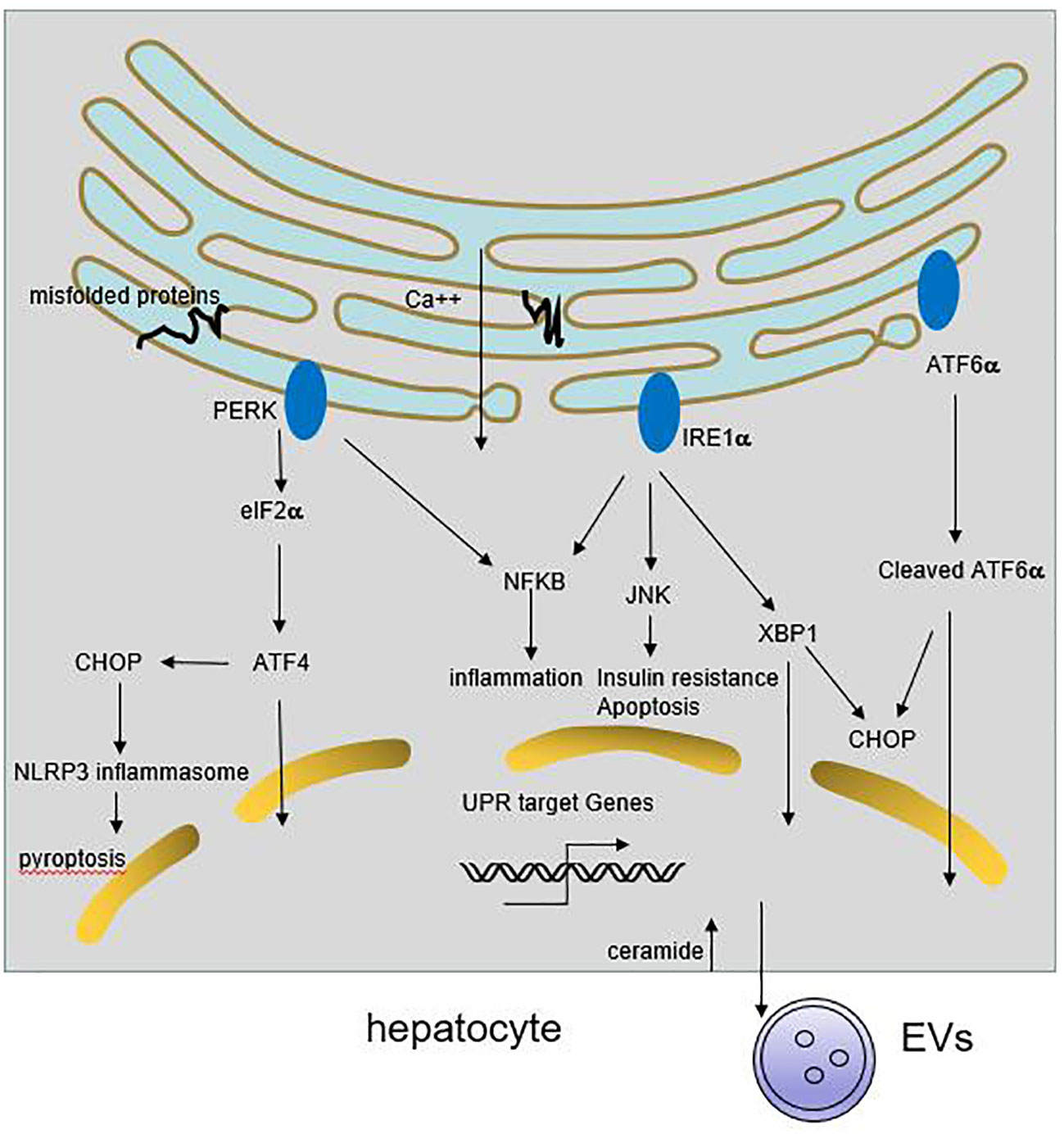
Figure 1 Role of ER stress in liver inflammation. Multiple stimuli lead to the activation of UPR response in hepatocyte. The three ER transmembrane sensors, PERK, IRE1 and ATF6, coordinately through downstream signaling cascades to resolve the protein folding defect and promote cell survival. If the adaptive UPR is overwhelmed by sustained or massive ER stress, it leads to hepatocyte steatosis and death. Meanwhile, ER stress may trigger NFκB and JNKs activation, resulting in release of proinflammatory cytokines. On the other hand, ER stress can induce CHOP-dependent NLRP3 inflammasome activation in hepatocytes. Besides, activation of IRE1A in hepatocytes promotes the release of inflammatory extracellular vesicles (EVs), thereby accummulating immune cells infiltration.
ER stress is observed in many chronic liver diseases. Chronic ER stress plays a causative role in NAFLD progression by promoting lipogenesis, disturbing mitochondrial function and modulating insulin signaling (10). ER stress markers are shown to decline in livers of obese patients following weight loss after bariatric surgery (11). It has confirmed that impaired autophagic flux is associated with increased ER stress in livers from patients with biopsy-proven NASH during the development of NAFLD (12). Various HBV and HCV proteins localize inside the ER lumen and are undergo envelopment. HBV infection can cause ER stress, which enhance HBV viral replication by initiating autophagy (13). Moreover, chronic HCV infection induce ER stress and the minimal expression of UPR target genes, which confers hepatocytes adaptation and resistance to liver injury (14–16). Hepatic PHLDA3 regulates ER stress-induced hepatocyte death through Akt inhibition in HCV hepatitis (17). Besides, it is reported that interferon regulatory factor 3 (IRF3) is activated by ER stress and induce hepatocyte apoptosis in early alcoholic liver disease (18).
Under chronic ER stress, UPR is linked to the activation of several inflammatory response pathways including NFκB, JNK, ROS, IL-6, TNF-α (8, 19). Activated IRE1α induces JNKs activation, and subsequent implicates in cell pro-inflammatory and pro-apoptotic pathways. Knockdown of JNK1 gene protects mice from the development of obesity and insulin resistance (20). Enhanced ER stress can trigger NFκB activation through IRE1α and PERK pathway, followed by the secretion of inflammatory and chemotactic cytokines in hepatocytes (21, 22). Some HCV and HBV protein accumulate at the ER membranes which cause a deregulation of Ca2+ flux, generation of reactive oxygen and nitrogen species, and the resulting ER stress could induce IL-8 transcription (10, 23, 24). ER stress also induces CHOP-dependent NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome activation in hepatocytes, potentially causing pyroptotic death and hepatic inflammation in patients with HBV-associated liver failure and NAFLD (25, 26). Recent study shows that activation of IRE1A in hepatocytes promotes the release of inflammatory extracellular vesicles (EVs), which recruit macrophages to liver, resulting in liver inflammation and injury in steatohepatitis (27). Therefore, chronic ER stress cause inflammation and the deregulation of lipid metabolism, that further exacerbate liver diseases.
Autophagy is a catabolic lysosomal process responsible for clearing damaged proteins, dysfunction organelles and lipid droplets. It is considered as a cellular response to maintain energy balance and in reaction to multiple of cellular stress, such as starvation, hypoxia, and viral infection (28).
Autophagy generally plays a protective role in hepatocytes, since they can protect against steatosis and hepatocyte death. It is reported autophagy can selectively degrades lipid droplets, termed lipophagy, as evidenced by the increase in lipid accumulation upon inhibition of autophagy in hepatocytes (29). Recent studies with specific genetic inhibition of autophagy have established that hepatocytes are more susceptible to various liver injury, such as alcohol, toxic agents, lipotoxic metabolites, and pro-inflammatory factors. Autophagy may promote cell survival by clearing misfolded proteins, lipids and damaged mitochondria (30–33).
Studies show that regulation of autophagy links to the progression of chronic liver diseases. Impaired autophagic flux links to steatosis and progression to NASH in NAFLD patients and mouse models by genetic or phamacological inhibition of autophagy (12). Shen et al. have uncovered pathogenesis of IL-1β-induced liver injury in steatohepatitis by finding that IL-1β becomes cytotoxic and pro-inflammatory to hepatocytes when inhibition of autophagy, leading to cell necrosis and liver inflammation (34). Although autophagy can alleviate hepatocyte apoptosis and steatosis in acute alcohol liver disease (35), decrease autophagic flux in hepatocyte is observed in models of chronic alcohol exposure (36, 37). A significant decrease in UQCRC2 protein expression cause impaired mitophagy, which may aggravate MLKL-mediated hepatocyte necroptosis and inflammation in alcoholic liver disease (38, 39). Furthermore, early autophagy enhance HBV infection and envelopment (40). Inhibition of autophagy by liver-specific knockout of Atg5 in HBV transgenic mice can obviously reduce HBV DNA level (41). Additionally, autophagy plays an important role in HBV-mediated immune response (40). GAL9, a type I IFN-stimulated gene, exerts effect on direct autophagic degradation of HBc in HBV-infected hepatocytes (42). ATG12 is required for HBV replication and impediment of the IFN signaling pathway, as evidence by decreased levels of IFN-α, IFN-β in ATG12-knockdown hepatocytes (43). Autophagy inhibition also abrogates HBx-induced activation of nuclear factor-κB (NF-κB) and production of interleukin-6 (IL-6), IL-8, and CXCL2 (44). Similarly, autophagy is required to promote HCV replication, partly through suppression of innate immunity (45, 46). HCV-induced autophagy can suppress host innate immune response through autophagic degradation of TRAF6, which is an important signaling molecule that mediates the activation of NF-kB and expression of cytokines and interferons (47). Meanwhile, loss of autophagy signaling upregulates HCV-induced cytoplasmic RIG-I signaling and IFN-β–mediated antiviral responses (48). Interference of HCV-induced mitophagy by Drp1 silencing enhances innate immune signaling (49). The correlation between AIH and autophagy in hepatocyte is not clear. It has been observed increased LC3 and p62 expression in hepatocytes of AIH patients, and p62 level is strongly correlated with necroinflammatory grade, which indicates that decreasing autophagic activity may be linked to severity of inflammation in AIH (50).
Mitochondria are abundant in the liver and required for lipid metabolism and energy production. They can directly or indirectly influence other cellular components such as the lysosomes, the endoplasmic reticulum (ER), and cytosolic pathway, to meet the cellular demands and alleviate mitochondrial dysfunction (51). Generally, mitochondria maintain normal morphology and homeostasis by the way of mitochondrial quality control, including the regulation of mitochondrial fusion, fission, biogenesis, and mitophagy (52). When they fail to adapt to various stress, they can release mitochondrial DNA (mtDNA) in the cytosol or circulation, which could induce cGAS-STING-dependent type I interferon (IFN) response. Furthermore, mtDNA synthesis can activate the NLRP3 inflammasome which initiates inflammation (53). In addition, mitochondrial dysfunction can generate excessive reactive oxygen species (ROS), which stimulate synthesis of cytokines to amplify the inflammatory cascade reaction and cause apoptosis and necrosis of hepatocytes (52) (Figure 2).
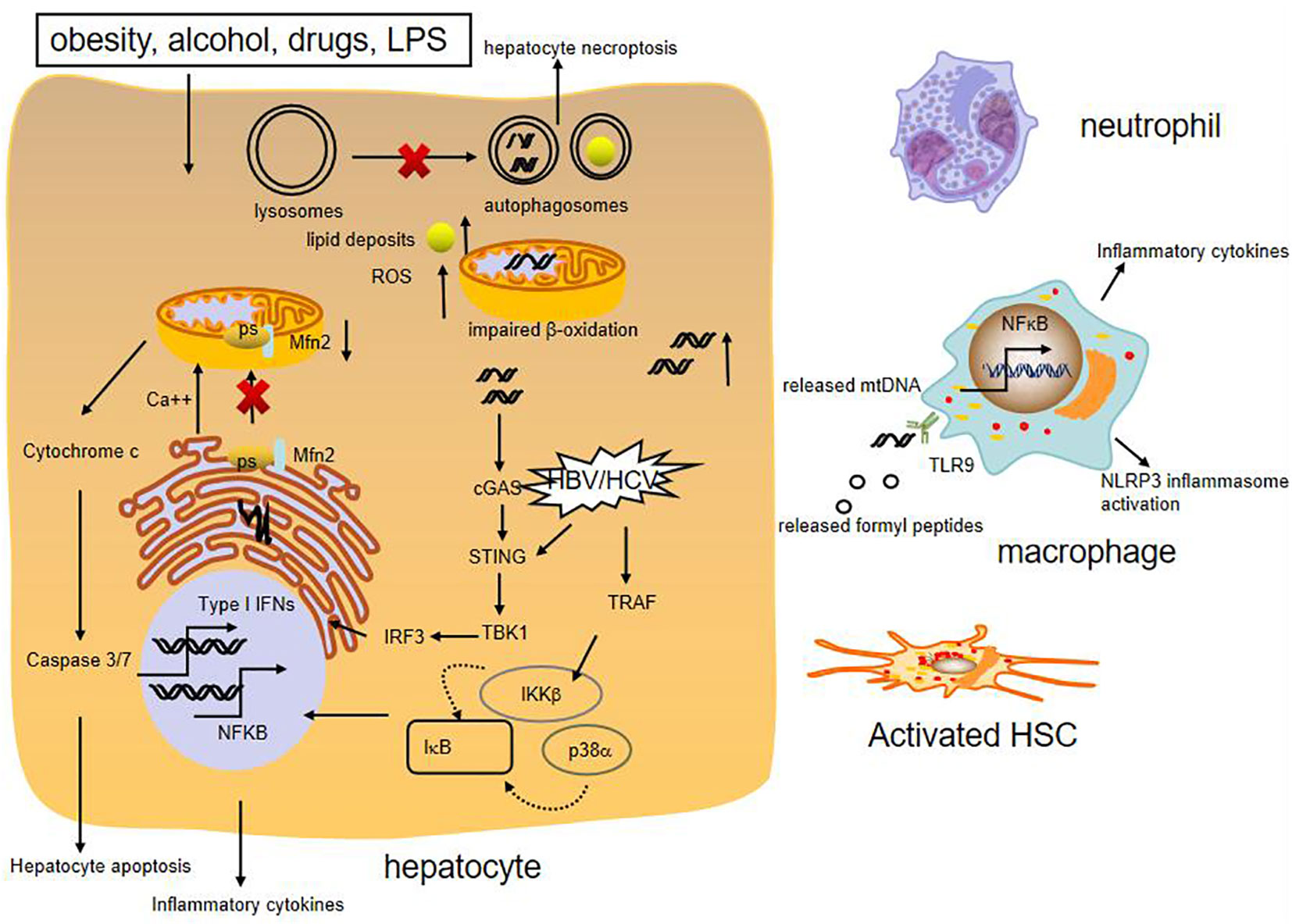
Figure 2 Role of mitochondrial damage in liver inflammation. Various liver injury impair mitochondrial respiration and increase ROS formation, cause mtDNA damage. High levels of ROS can increase synthesis of cytokines, which cause apoptosis and necrosis of hepatocytes. The presence of mtDNA in the cytosol or circulation can trigger proinflammatory and type I IFN responses. Moreover, release of mitochondria-derived danger signals, such as mtDNA, formylated proteins, can attract macrophage and neutrohphils, resulting in activation of NFκB and NLRP3 inflammasome. MtDNA also promotes fibrogenic activation of HSCs. Besides, reduced expression of mitochondrial protein Mfn2 leads to deficient ER-mitochondrial phosphatidylserine transfer, which provokes liver inflammation. HBV and HCV can activate innate immune antiviral signaling and inflammatory pathways through induction of type I interferons and expression of inflammatory cytokines by NFκB.
Emerging evidence shows that mitochondria dysfunction, especially mitochondria-derived immunogenic components (including its DNA) have profound impacts on the development of various chronic liver diseases. It is reported that NASH patients produce high mitochondrial levels of ROS and ROS-mediated mtDNA damage (54). Moreover, mtDNA is elevated in the serum of NASH patients and in association with histological degree of hepatic fibrosis. The mtDNA released from injured hepatocyte mitochondria could directly activate hepatic stellate cells (HSCs) and promote inflammation through binding to endosomal TLR9 of Kupffer cells (55, 56). Besides, Mitochondrial protein mitofusion 2 (Mfn2) plays an important role in connecting ER membranes to mitochondria and mitochondrial fusion, studies show that hepatic mfn2 deficiency impairs ER-mitochondrial phosphatidylserine transfer and mitochondrial function, leading to ER stress and liver inflammation in NAFLD (57, 58). Mitochondrial dysregulation is also observed in hepatocytes of patients with AIH and experimental mouse model with immune-mediated liver injury. Blockade of dynamin-related protein 1(Drp1)-mediated mitochondrial fission protects mice from concanavalin A (ConA)-induced liver injury (59). In addition, hepatic ATF4 plays a pathological role in alcohol-induced mitochondrial dysfunction and liver injury by repressing TFAM expression, while AMPK protects against alcohol-induced liver injury through up-regulating mitophagy (39, 60). Apart from the above, chronic HBV and HCV infection could induce mitochondrial oxidative stress and mitochondrial antiviral signaling-mediated innate immune signaling as well (61, 62).
During chronic liver injury, stressed hepatocytes can release mediators that involved in crosstalk between hepatocytes and surrounding cell populations. Besides, hepatocytes serve as liver-resident nonprofessional antigen presenting cells (APCs), resulting in a bias toward immune tolerance.
Extracellular vesicles (EVs) are homogeneous vesicles containing lipid, nuclear acid, proteins, which can be secreted by various cell types to the extracellular space and circulation. EVs include microvesicles, exosomes and apoptotic bodies depending on their source and molecular structure.
A growing body of evidence have identified EVs as a conveyor mediating intercellular communication in liver diseases (63) (List in Table 1). Hepatocyte-derived EVs as pathogenic mediators play a role in NASH (77). Hepatocyte-derived exosomes from early onset obese mice promote insulin sensitivity through miR-3075 (64). The increase in plasma mtDNA contained in EVs of hepatocyte origin could drive NASH development by activation of TLR9 (56). EVs are also shown as mediators of toxic lipid-induced intercellular signaling. Lipotoxic activation of hepatocytes induce release of EVs enriched in ceramide, CXCL10, miR-192-5p, which trigger chemotaxis and inflammatory phenotype switch of macrophages (65–68). Besides, EVs mediate cell-to-cell communication in alcoholic liver disease. In patients with alcoholic hepatitis, the number of circulating EVs is reported higher than those in healthy individuals, and the EVs contain elevated levels of miR122, miR192 and miR309 (69). Hepatocyte-derived EVs modulate activation of liver marcophages by transferring miRNA-122 and CD40ligand after alcohol exposure (70, 71). In addition, it is reported that exosomes isolated from sera of chronic HBV and HCV infected patients or supernatants of those hepatocytes contain viral RNA, which can mediate viral transmission to naive hepatocytes (72, 75). These hepatic derived-exosomes involve in host innate immune response and virus-mediated immunosuppression. HCV-associated exosomes can transfer immunomodulatory viral RNA from infected cells to neighboring immune cells and trigger myeloid-derived suppressor cell expansion (73). EVs from hepatitis C virus-infected cells stimulate monocytes to produce galectin-9, which induces apoptosis of hepatitis C virus-specific T cells and increases inhibitory regulatory T cells (74). Similarly, HBV components are observed to be transmitted into NK cells by exosomes, resulting in NK-cell dysfunction (75). Exosomes also can regulate innate immune response against HBV through inducing NKG2D ligand expression in macrophages, which stimulates IFN-γ from NK cells, and suppressing IL-12p35 mRNA expression to counteract he host innate immune response (76). In a word, EVs exert a crucial role on the crosstalk between hepatocytes and nonparenchymal liver cells.
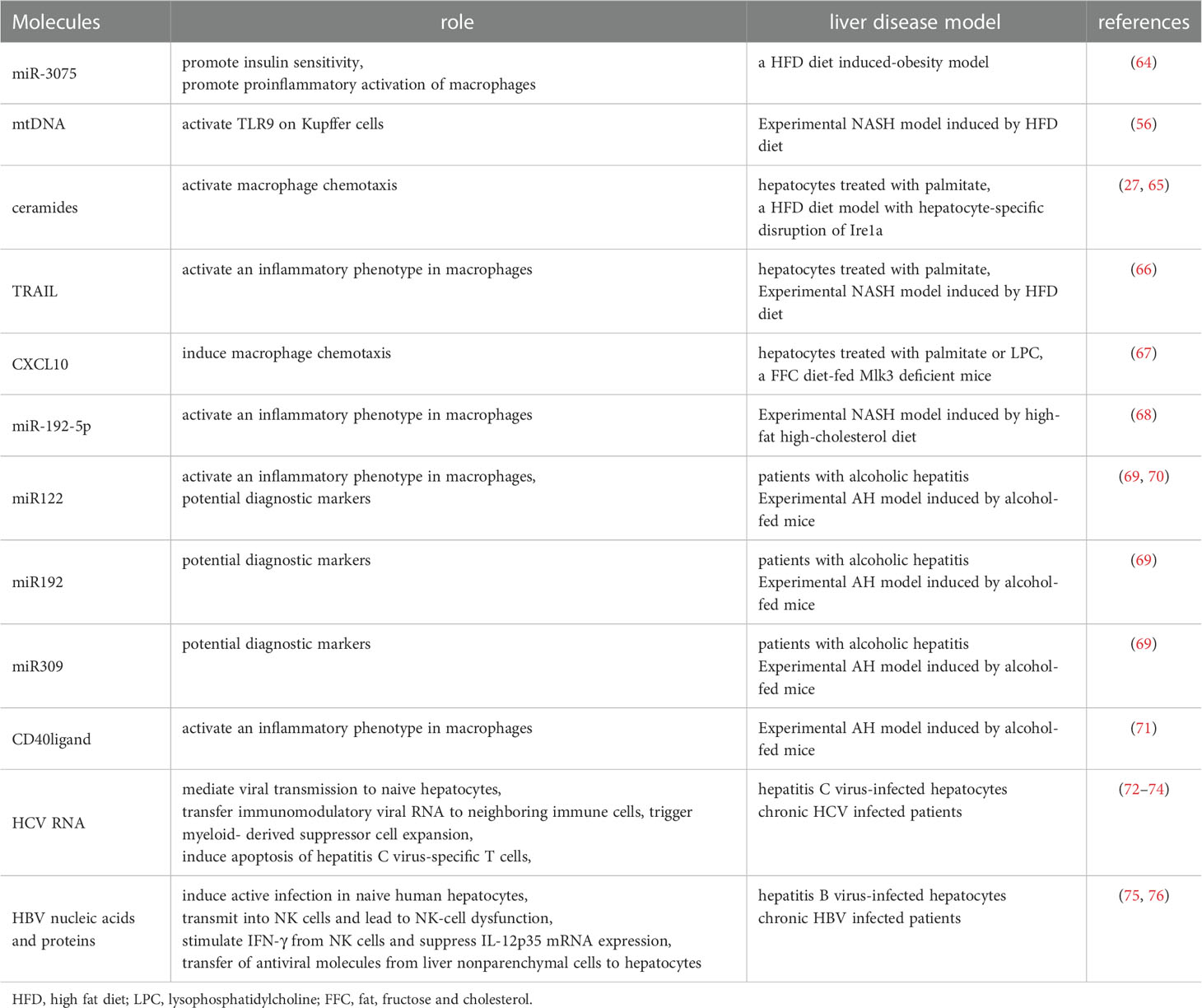
Table 1 Biosynthesis of secreted extracellular vesicles by hepatocytes.
Hepatocytes can produce diverse cytokines to regulate liver injury, repair, and inflammation in liver injury. Here, we make a summary of cytokines that involved in the pathogenesis of chronic liver diseases below.
IL-6 can be synthesized by hepatocytes in response to specific stimuli to induce acute phase response, it implicates in the liver regeneration following partial hepatectomy and exerts antiviral effects on limiting the replication of HBV in hepatocytes (78, 79). Moreover, substantial studies show that IL-6 trans-signaling promotes inflammation in chronic liver diseases (80). Excessive lipid accumulation in hepatocytes stimulates IL11 protein secretion, autocrine IL11 activity drives lipotoxicity and underlies the transition from NAFLD to NASH (81). Interleukin 33 (IL-33) functions as an “alarmin” released from hepatocytes in response to tissue damages. It exerts protective effects on hepatocytes through the activation of autophagy and suppression of cell death, meanwhile, it regulates host innate immunity by recruitment and activation of ST2-positive target immune cells in the liver (82). Furthermore, it is responsible for repressing viral transcription, protein production and genome replication in HBV-infected hepatocytes (83). IL-32 is markedly induced in hepatocytes in various liver diseases. It plays an important role in inflammatory response by promoting proinflammatory cytokines such as IL-1β and tumor necrosis factor alpha (TNF-α) (84, 85). IL32 also has a critical role in the pathogenesis of NAFLD, partly due to its association with hepatocyte insulin resistance and cholesterol homeostasis (86, 87). Besides, it can suppress HBV transcription and replication (88). Hepatocyte also can produce several chemokines to attract immune cells in response to liver injury. For example, hepatocyte can express chemokine MCP-1, which recruits macrophages to promote liver steatosis and inflammation in alcoholic and non-alcoholic fatty liver disease. Moreover, hepatic MCP-1 expression is found to regulate fatty acid oxidation resulting in steatosis during chronic alcohol exposure (89, 90). Apart from the above, hepatocytes can secrete high amounts of CXCL1, leading to hepatic neutrophil infiltration through TLR2 and TLR9-dependent pathway in alcohol-mediated liver injury (91). Hepatocyte is the main source for necrotic cell-induced CXCL1 production, which dependent of NF-κB activation by Kupffer cells, resulting in neutrophils mobilization and finally clearing dead cells (92). Another study shows that hepatocyte-specific gp130 signaling is sufficient to induce CXCL1 expression, independent of NF-κB activation, triggering a robust systemic innate immune response (93). Steatotic hepatocytes also can stimulate IL-8 production, an active neutrophil chemoattractant, potentially contributing to hepatic inflammation (94).
In clinical hepatitis, viral or autoimmune especially, hepatocytes can directly modulate immune cells via cell-cell interactions. Hepatocytes could function as nonprofessional APCs because they express MHC class II during inflammation. MHC-II overexpressing hepatocytes are capable of activating CD4+ T-cells in vitro, but they only induce T helper cell (Th) 2 differentiation, which impair antiviral CD8 T-cell responses and viral clearance (95, 96). Hepatocytes appear to play a role in the liver tolerogenic effect. They can activate CD8+ T cells in a manner that leads to apoptosis of these cells since lack of either costimulatory signals or CD4+ T cell help (97). What’s more, the hepatocytes may endocytose and kill CD8+ T cells that recognize them, a process known as suicidal emperipolesis (98). In viral infection, virus-positive hepatocytes can be eliminated by activated circulating CD8+ T-cells through directly recognizing antigen on hepatocytes, leading to CD8+T-cell exhaustion (99). Among the underlying mechanism, Notch signaling may performed an important regulatory role in the interaction between hepatocytes and T cells activation. It is reported hepatocytes fine -tune liver inflammation by upregulation of Jagged1 and activation of Notch signaling in Th1 cells, resulting in induction of IL10-producing CD4+ T cells (100). Besides, Notch signaling contributes to liver inflammation by regulation of interleukin-22-producing cells in hepatitis B virus infection (101). In addition, hepatocytes may induce tolerance via Notch-mediated conversion of CD4(+) T cells into Foxp3(+) Tregs upon TCR stimulation (102). Apart from these, intercellular adhesion molecule 1 (ICAM-1) is involved in CD4+ T cell engulfment by hepatocytes and huh-7 cells by facilitating T cell early adhesion and internalization (103).
A growing number of evidences have demonstrated stressed hepatocytes exert a pivotal role on the development of inflammation and fibrosis via cell-cell interactions during liver injury. In this review, we summarize the role of hepatic organelle disorders in the pathogenesis of chronic liver diseases, especially, their links to liver inflammation. Furthermore, we introduce a wide variety of pro-inflammatory signals carried by hepatocyte derived-EVs that can deliver the message to neighbor target cells and in the circulation to modulate immune response. Besides, we conclude several cytokines and chemokines of hepatocyte origin which engage in chronic liver diseases. Finally, we address briefly antigen-presentation properties of hepatocytes in immune regulation. Understanding of the molecular mechanisms involved in the regulation of hepatic organelle damage, as well as role of hepatocyte in immune regulation may provide us novel insights of dysregulated inflammation during liver injury and identify new therapeutic targets for various liver diseases.
JG and JL contributed to select the topic of the manuscript. WT collected relevant literature. JG wrote the manuscript. JL and DT reviewed and edited the final version of manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the National Natural Science Foundation of China (No. 81900504).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol (2019) 70(1):20. doi: 10.1016/j.jhep.2018.09.014
2. Tu T, Calabro SR, Lee A, Maczurek AE, Budzinska MA, Warner FJ, et al. Hepatocytes in liver injury: Victim, bystander, or accomplice in progressive fibrosis? J Gastroenterol Hepatol (2015) 30(12):1696–704. doi: 10.1111/jgh.13065
3. Wree A, Holtmann TM, Inzaugarat ME, Feldstein AE. Novel drivers of the inflammatory response in liver injury and fibrosis. Semin Liver Dis (2019) 39(3):275–82. doi: 10.1055/s-0039-1685515
4. Rinella ME. Nonalcoholic fatty liver disease: A systematic review. Jama (2015) 313(22):2263–73. doi: 10.1001/jama.2015.5370
5. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology (2014) 147(4):765–83.e4. doi: 10.1053/j.gastro.2014.07.018
6. Zhou Z, Xu MJ, Gao B. Hepatocytes: A key cell type for innate immunity. Cell Mol Immunol (2016) 13(3):301–15. doi: 10.1038/cmi.2015.97
7. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology (2015) 61(3):1066–79. doi: 10.1002/hep.27332
8. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol (2011) 54(4):795–809. doi: 10.1016/j.jhep.2010.11.005
9. Mollica MP, Lionetti L, Putti R, Cavaliere G, Gaita M, Barletta A. From chronic overfeeding to hepatic injury: Role of endoplasmic reticulum stress and inflammation. Nutr Metab Cardiovasc Dis (2011) 21(3):222–30. doi: 10.1016/j.numecd.2010.10.012
10. Liu C, Zhou B, Meng M, Zhao W, Wang D, Yuan Y, et al. FOXA3 induction under endoplasmic reticulum stress contributes to non-alcoholic fatty liver disease. J Hepatol (2021) 75(1):150–62. doi: 10.1016/j.jhep.2021.01.042
11. Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes (2009) 58(3):693–700. doi: 10.2337/db08-1220
12. Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis (2014) 5:e1179. doi: 10.1038/cddis.2014.162
13. Wang X, Wei Z, Cheng B, Li J, He Y, Lan T, et al. Endoplasmic reticulum stress promotes HBV production by enhancing use of the autophagosome/multivesicular body axis. Hepatology (2022) 75(2):438–54. doi: 10.1002/hep.32178
14. Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N, et al. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PloS Pathog (2009) 5(2):e1000291. doi: 10.1371/journal.ppat.1000291
15. Merquiol E, Uzi D, Mueller T, Goldenberg D, Nahmias Y, Xavier RJ, et al. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PloS One (2011) 6(9):e24660. doi: 10.1371/journal.pone.0024660
16. Asselah T, Bieche I, Mansouri A, Laurendeau I, Cazals-Hatem D, Feldmann G, et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis c. J Pathol (2010) 221(3):264–74. doi: 10.1002/path.2703
17. Han CY, Lim SW, Koo JH, Kim W, Kim SG. PHLDA3 overexpression in hepatocytes by endoplasmic reticulum stress via IRE1-Xbp1s pathway expedites liver injury. Gut (2016) 65(8):1377–88. doi: 10.1136/gutjnl-2014-308506
18. Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci USA (2013) 110(41):16544–9. doi: 10.1073/pnas.1308331110
19. Li X, Wang Y, Wang H, Huang C, Huang Y, Li J. Endoplasmic reticulum stress is the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in liver fibrosis. Inflammation Res (2015) 64(1):1–7. doi: 10.1007/s00011-014-0772-y
20. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature (2002) 420(6913):333–6. doi: 10.1038/nature01137
21. Luedde T, Schwabe RF. NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol (2011) 8(2):108–18. doi: 10.1038/nrgastro.2010.213
22. Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol (2018) 69(4):927–47. doi: 10.1016/j.jhep.2018.06.008
23. Scrima R, Piccoli C, Moradpour D, Capitanio N. Targeting endoplasmic reticulum and/or mitochondrial Ca(2+) fluxes as therapeutic strategy for HCV infection. Front Chem (2018) 6:73. doi: 10.3389/fchem.2018.00073
24. Tsuge M, Hiraga N, Zhang Y, Yamashita M, Sato O, Oka N, et al. Endoplasmic reticulum-mediated induction of interleukin-8 occurs by hepatitis b virus infection and contributes to suppression of interferon responsiveness in human hepatocytes. Virology (2018) 525:48–61. doi: 10.1016/j.virol.2018.08.020
25. Han CY, Rho HS, Kim A, Kim TH, Jang K, Jun DW, et al. FXR inhibits endoplasmic reticulum stress-induced NLRP3 inflammasome in hepatocytes and ameliorates liver injury. Cell Rep (2018) 24(11):2985–99. doi: 10.1016/j.celrep.2018.07.068
26. Lebeaupin C, Proics E, de Bieville CH, Rousseau D, Bonnafous S, Patouraux S, et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis (2015) 6:e1879. doi: 10.1038/cddis.2015.248
27. Dasgupta D, Nakao Y, Mauer AS, Thompson JM, Sehrawat TS, Liao CY, et al. IRE1A stimulates hepatocyte-derived extracellular vesicles that promote inflammation in mice with steatohepatitis. Gastroenterology (2020) 159(4):1487–503.e17. doi: 10.1053/j.gastro.2020.06.031
28. Gual P, Gilgenkrantz H, Lotersztajn S. Autophagy in chronic liver diseases: The two faces of janus. Am J Physiol Cell Physiol (2017) 312(3):C263–C73. doi: 10.1152/ajpcell.00295.2016
29. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature (2009) 458(7242):1131–5. doi: 10.1038/nature07976
30. Shen Y, Malik SA, Amir M, Kumar P, Cingolani F, Wen J, et al. Decreased hepatocyte autophagy leads to synergistic IL-1beta and TNF mouse liver injury and inflammation. Hepatology (2020) 72(2):595–608. doi: 10.1002/hep.31209
31. Czaja MJ, Ding WX, Donohue TM Jr., Friedman SL, Kim JS, Komatsu M, et al. Functions of autophagy in normal and diseased liver. Autophagy (2013) 9(8):1131–58. doi: 10.4161/auto.25063
32. Zhou B, Kreuzer J, Kumsta C, Wu L, Kamer KJ, Cedillo L, et al. Mitochondrial permeability uncouples elevated autophagy and lifespan extension. Cell (2019) 177(2):299–314.e16. doi: 10.1016/j.cell.2019.02.013
33. Amir M, Zhao E, Fontana L, Rosenberg H, Tanaka K, Gao G, et al. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ (2013) 20(7):878–87. doi: 10.1038/cdd.2013.21
34. Francis H, Wu N, Alpini G, Meng F. Hepatocyte autophagy: Maintaining a toxic-free environment. Hepatology (2020) 72(2):371–4. doi: 10.1002/hep.31219
35. Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology (2010) 139(5):1740–52. doi: 10.1053/j.gastro.2010.07.041
36. Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol (2013) 58(5):993–9. doi: 10.1016/j.jhep.2013.01.011
37. Chao X, Wang S, Zhao K, Li Y, Williams JA, Li T, et al. Impaired TFEB-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology (2018) 155(3):865–79.e12. doi: 10.1053/j.gastro.2018.05.027
38. Zhou Y, Wu R, Wang X, Jiang Y, Xu W, Shao Y, et al. Activation of UQCRC2-dependent mitophagy by tetramethylpyrazine inhibits MLKL-mediated hepatocyte necroptosis in alcoholic liver disease. Free Radic Biol Med (2022) 179:301–16. doi: 10.1016/j.freeradbiomed.2021.11.008
39. Lu X, Xuan W, Li J, Yao H, Huang C, Li J. AMPK protects against alcohol-induced liver injury through UQCRC2 to up-regulate mitophagy. Autophagy (2021) 17(11):3622–43. doi: 10.1080/15548627.2021.1886829
40. Lin Y, Zhao Z, Huang A, Lu M. Interplay between cellular autophagy and hepatitis b virus replication: A systematic review. Cells (2020) 9(9). doi: 10.3390/cells9092101
41. Tian Y, Sir D, Kuo CF, Ann DK, Ou JH. Autophagy required for hepatitis b virus replication in transgenic mice. J Virol (2011) 85(24):13453–6. doi: 10.1128/JVI.06064-11
42. Miyakawa K, Nishi M, Ogawa M, Matsunaga S, Sugiyama M, Nishitsuji H, et al. Galectin-9 restricts hepatitis b virus replication via p62/SQSTM1-mediated selective autophagy of viral core proteins. Nat Commun (2022) 13(1):531. doi: 10.1038/s41467-022-28171-5
43. Kunanopparat A, Hirankarn N, Kittigul C, Tangkijvanich P, Kimkong I. Autophagy machinery impaired interferon signalling pathways to benefit hepatitis b virus replication. Asian Pac J Allergy Immunol (2016) 34(1):77–85. doi: 10.12932/AP0636.34.1.2016
44. Luo MX, Wong SH, Chan MT, Yu L, Yu SS, Wu F, et al. Autophagy mediates HBx-induced nuclear factor-kappaB activation and release of IL-6, IL-8, and CXCL2 in hepatocytes. J Cell Physiol (2015) 230(10):2382–9. doi: 10.1002/jcp.24967
45. Chu JYK, Ou JJ. Autophagy in HCV replication and protein trafficking. Int J Mol Sci (2021) 22(3). doi: 10.3390/ijms22031089
46. Chan ST, Ou JJ. Hepatitis c virus-induced autophagy and host innate immune response. Viruses (2017) 9(8). doi: 10.3390/v9080224
47. Chan ST, Lee J, Narula M, Ou JJ. Suppression of host innate immune response by hepatitis c virus via induction of autophagic degradation of TRAF6. J Virol (2016) 90(23):10928–35. doi: 10.1128/JVI.01365-16
48. Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis c virus infection suppresses innate antiviral immunity in vitro. J Clin Invest (2011) 121(1):37–56. doi: 10.1172/JCI41474
49. Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, et al. Hepatitis c virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci USA (2014) 111(17):6413–8. doi: 10.1073/pnas.1321114111
50. Szekerczes T, Gogl A, Illyes I, Mandl J, Borka K, Kiss A, et al. Autophagy, mitophagy and MicroRNA expression in chronic hepatitis c and autoimmune hepatitis. Pathol Oncol Res (2020) 26(4):2143–51. doi: 10.1007/s12253-020-00799-y
51. Mottis A, Herzig S, Auwerx J. Mitocellular communication: Shaping health and disease. Science (2019) 366(6467):827–32. doi: 10.1126/science.aax3768
52. Mansouri A, Gattolliat CH, Asselah T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology (2018) 155(3):629–47. doi: 10.1053/j.gastro.2018.06.083
53. Youle RJ. Mitochondria-striking a balance between host and endosymbiont. Science (2019) 365(6454). doi: 10.1126/science.aaw9855
54. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab (2015) 21(5):739–46. doi: 10.1016/j.cmet.2015.04.004
55. An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun (2020) 11(1):2362. doi: 10.1038/s41467-020-16092-0
56. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest (2016) 126(3):859–64. doi: 10.1172/JCI83885
57. Hou J, Zhang J, Cui P, Zhou Y, Liu C, Wu X, et al. TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J Clin Invest (2021) 131(4). doi: 10.1172/JCI135197
58. Hernandez-Alvarez MI, Sebastian D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, et al. Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell (2019) 177(4):881–95.e17. doi: 10.1016/j.cell.2019.04.010
59. He GW, Gunther C, Kremer AE, Thonn V, Amann K, Poremba C, et al. PGAM5-mediated programmed necrosis of hepatocytes drives acute liver injury. Gut (2017) 66(4):716–23. doi: 10.1136/gutjnl-2015-311247
60. Hao L, Zhong W, Dong H, Guo W, Sun X, Zhang W, et al. ATF4 activation promotes hepatic mitochondrial dysfunction by repressing NRF1-TFAM signalling in alcoholic steatohepatitis. Gut (2021) 70(10):1933–45. doi: 10.1136/gutjnl-2020-321548
61. Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis c virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA (2005) 102(49):17717–22. doi: 10.1073/pnas.0508531102
62. Waris G, Huh KW, Siddiqui A. Mitochondrially associated hepatitis b virus X protein constitutively activates transcription factors STAT-3 and NF-kappa b via oxidative stress. Mol Cell Biol (2001) 21(22):7721–30. doi: 10.1128/MCB.21.22.7721-7730.2001
63. Kostallari E, Valainathan S, Biquard L, Shah VH, Rautou PE. Role of extracellular vesicles in liver diseases and their therapeutic potential. Adv Drug Delivery Rev (2021) 175:113816. doi: 10.1016/j.addr.2021.05.026
64. Ji Y, Luo Z, Gao H, Dos Reis FCG, Bandyopadhyay G, Jin Z, et al. Hepatocyte-derived exosomes from early onset obese mice promote insulin sensitivity through miR-3075. Nat Metab (2021) 3(9):1163–74. doi: 10.1038/s42255-021-00444-1
65. Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J Lipid Res (2016) 57(2):233–45. doi: 10.1194/jlr.M063412
66. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology (2016) 150(4):956–67. doi: 10.1053/j.gastro.2015.12.037
67. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed lineage kinase 3 mediates release of c-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology (2016) 63(3):731–44. doi: 10.1002/hep.28252
68. Liu XL, Pan Q, Cao HX, Xin FZ, Zhao ZH, Yang RX, et al. Lipotoxic hepatocyte-derived exosomal MicroRNA 192-5p activates macrophages through Rictor/Akt/Forkhead box transcription factor O1 signaling in nonalcoholic fatty liver disease. Hepatology (2020) 72(2):454–69. doi: 10.1002/hep.31050
69. Momen-Heravi F, Saha B, Kodys K, Catalano D, Satishchandran A, Szabo G. Increased number of circulating exosomes and their microRNA cargos are potential novel biomarkers in alcoholic hepatitis. J Transl Med (2015) 13:261. doi: 10.1186/s12967-015-0623-9
70. Momen-Heravi F, Bala S, Kodys K, Szabo G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific miRNA-122 and sensitize monocytes to LPS. Sci Rep (2015) 5:9991. doi: 10.1038/srep09991
71. Verma VK, Li H, Wang R, Hirsova P, Mushref M, Liu Y, et al. Alcohol stimulates macrophage activation through caspase-dependent hepatocyte derived release of CD40L containing extracellular vesicles. J Hepatol (2016) 64(3):651–60. doi: 10.1016/j.jhep.2015.11.020
72. Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. Exosomes from hepatitis c infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PloS Pathog (2014) 10(10):e1004424. doi: 10.1371/journal.ppat.1004424
73. Wang L, Cao D, Wang L, Zhao J, Nguyen LN, Dang X, et al. HCV-associated exosomes promote myeloid-derived suppressor cell expansion via inhibiting miR-124 to regulate T follicular cell differentiation and function. Cell Discov (2018) 4:51. doi: 10.1038/s41421-018-0052-z
74. Harwood NM, Golden-Mason L, Cheng L, Rosen HR, Mengshol JA. HCV-infected cells and differentiation increase monocyte immunoregulatory galectin-9 production. J Leukoc Biol (2016) 99(3):495–503. doi: 10.1189/jlb.5A1214-582R
75. Yang Y, Han Q, Hou Z, Zhang C, Tian Z, Zhang J. Exosomes mediate hepatitis b virus (HBV) transmission and NK-cell dysfunction. Cell Mol Immunol (2017) 14(5):465–75. doi: 10.1038/cmi.2016.24
76. Kouwaki T, Fukushima Y, Daito T, Sanada T, Yamamoto N, Mifsud EJ, et al. Extracellular vesicles including exosomes regulate innate immune responses to hepatitis b virus infection. Front Immunol (2016) 7:335. doi: 10.3389/fimmu.2016.00335
77. Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut (2018) 67(5):963–72. doi: 10.1136/gutjnl-2017-315691
78. Fazel Modares N, Polz R, Haghighi F, Lamertz L, Behnke K, Zhuang Y, et al. IL-6 trans-signaling controls liver regeneration after partial hepatectomy. Hepatology (2019) 70(6):2075–91. doi: 10.1002/hep.30774
79. Palumbo GA, Scisciani C, Pediconi N, Lupacchini L, Alfalate D, Guerrieri F, et al. IL6 inhibits HBV transcription by targeting the epigenetic control of the nuclear cccDNA minichromosome. PloS One (2015) 10(11):e0142599. doi: 10.1371/journal.pone.0142599
80. Giraldez MD, Carneros D, Garbers C, Rose-John S, Bustos M. New insights into IL-6 family cytokines in metabolism, hepatology and gastroenterology. Nat Rev Gastroenterol Hepatol (2021) 18(11):787–803. doi: 10.1038/s41575-021-00473-x
81. Dong J, Viswanathan S, Adami E, Singh BK, Chothani SP, Ng B, et al. Hepatocyte-specific IL11 cis-signaling drives lipotoxicity and underlies the transition from NAFLD to NASH. Nat Commun (2021) 12(1):66. doi: 10.1038/s41467-020-20303-z
82. Arshad MI, Piquet-Pellorce C, Samson M. IL-33 and HMGB1 alarmins: sensors of cellular death and their involvement in liver pathology. Liver Int (2012) 32(8):1200–10. doi: 10.1111/j.1478-3231.2012.02802.x
83. Gao Z, Shen Z, Wu J, Song Y, Liu N, Deng Q, et al. Interleukin-33 mediates both immune-related and non-immune-related inhibitory effects against hepatitis b virus. Antiviral Res (2022) 206:105404. doi: 10.1016/j.antiviral.2022
84. Pan X, Cao H, Lu J, Shu X, Xiong X, Hong X, et al. Interleukin-32 expression induced by hepatitis b virus protein X is mediated through activation of NF-kappaB. Mol Immunol (2011) 48(12-13):1573–7. doi: 10.1016/j.molimm.2011.03.012
85. Moschen AR, Fritz T, Clouston AD, Rebhan I, Bauhofer O, Barrie HD, et al. Interleukin-32: A new proinflammatory cytokine involved in hepatitis c virus-related liver inflammation and fibrosis. Hepatology (2011) 53(6):1819–29. doi: 10.1002/hep.24285
86. Dali-Youcef N, Vix M, Costantino F, El-Saghire H, Lhermitte B, Callari C, et al. Interleukin-32 contributes to human nonalcoholic fatty liver disease and insulin resistance. Hepatol Commun (2019) 3(9):1205–20. doi: 10.1002/hep4.1396
87. Damen M, Dos Santos JC, Hermsen R, Adam van der Vliet J, Netea MG, Riksen NP, et al. Interleukin-32 upregulates the expression of ABCA1 and ABCG1 resulting in reduced intracellular lipid concentrations in primary human hepatocytes. Atherosclerosis (2018) 271:193–202. doi: 10.1016/j.atherosclerosis.2018.02.027
88. Kim DH, Park ES, Lee AR, Park S, Park YK, Ahn SH, et al. Intracellular interleukin-32gamma mediates antiviral activity of cytokines against hepatitis b virus. Nat Commun (2018) 9(1):3284. doi: 10.1038/s41467-018-05782-5
89. Obstfeld AE, Sugaru E, Thearle M, Francisco AM, Gayet C, Ginsberg HN, et al. C-c chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes (2010) 59(4):916–25. doi: 10.2337/db09-1403
90. Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: Regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology (2011) 54(6):2185–97. doi: 10.1002/hep.24599
91. Roh YS, Zhang B, Loomba R, Seki E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am J Physiol Gastrointest Liver Physiol (2015) 309(1):G30–41. doi: 10.1152/ajpgi.00031.2015
92. Su L, Li N, Tang H, Lou Z, Chong X, Zhang C, et al. Kupffer cell-derived TNF-alpha promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis (2018) 9(3):323. doi: 10.1038/s41419-018-0377-4
93. Schumacher N, Yan K, Gandrass M, Muller M, Krisp C, Hasler R, et al. Cell-autonomous hepatocyte-specific GP130 signaling is sufficient to trigger a robust innate immune response in mice. J Hepatol (2021) 74(2):407–18. doi: 10.1016/j.jhep.2020.09.021
94. Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, et al. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology (2007) 46(3):823–30. doi: 10.1002/hep.21752
95. Herkel J, Jagemann B, Wiegard C, Lazaro JF, Lueth S, Kanzler S, et al. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocyutes. Hepatology (2003) 37(5):1079–85. doi: 10.1053/jhep.2003.50191
96. Wiegard C, Wolint P, Frenzel C, Cheruti U, Schmitt E, Oxenius A, et al. Defective T helper response of hepatocyte-stimulated CD4 T cells impairs antiviral CD8 response and viral clearance. Gastroenterology (2007) 133(6):2010–8. doi: 10.1053/j.gastro.2007.09.007
97. Bertolino P, Trescol-Biemont MC, Rabourdin-Combe C. Hepatocytes induce functional activation of naive CD8+ T lymphocytes but fail to promote survival. Eur J Immunol (1998) 28(1):221–36. doi: 10.1002/(SICI)1521-4141(199801)28:01<221::AID-IMMU221>3.0.CO;2-F
98. Crispe IN. Hepatocytes as immunological agents. J Immunol (2016) 196(1):17–21. doi: 10.4049/jimmunol.1501668
99. Mehrfeld C, Zenner S, Kornek M, Lukacs-Kornek V. The contribution of non-professional antigen-presenting cells to immunity and tolerance in the liver. Front Immunol (2018) 9:635. doi: 10.3389/fimmu.2018.00635
100. Burghardt S, Erhardt A, Claass B, Huber S, Adler G, Jacobs T, et al. Hepatocytes contribute to immune regulation in the liver by activation of the notch signaling pathway in T cells. J Immunol (2013) 191(11):5574–82. doi: 10.4049/jimmunol.1300826
101. Wei X, Wang JP, Hao CQ, Yang XF, Wang LX, Huang CX, et al. Notch signaling contributes to liver inflammation by regulation of interleukin-22-Producing cells in hepatitis b virus infection. Front Cell Infect Microbiol (2016) 6:132. doi: 10.3389/fcimb.2016.00132
102. Burghardt S, Claass B, Erhardt A, Karimi K, Tiegs G. Hepatocytes induce Foxp3(+) regulatory T cells by notch signaling. J Leukoc Biol (2014) 96(4):571–7. doi: 10.1189/jlb.2AB0613-342RR
Keywords: hepatocyte, organelle damage, hepatic inflammation, extracellular vesicles, cytokines
Citation: Gong J, Tu W, Liu J and Tian D (2023) Hepatocytes: A key role in liver inflammation. Front. Immunol. 13:1083780. doi: 10.3389/fimmu.2022.1083780
Received: 29 October 2022; Accepted: 30 December 2022;
Published: 18 January 2023.
Edited by:
Chit Laa Poh, Sunway University, MalaysiaReviewed by:
Laurent Dubuquoy, U1286 Institute for Translational Research in Inflammation (INFINITE), FranceCopyright © 2023 Gong, Tu, Liu and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingmei Liu, NDY0MjAxMzY0QHFxLmNvbQ==; Dean Tian, ZGF0aWFuQHRqaC50am11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.