
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 09 January 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1076167
This article is part of the Research Topic Insights in Autoimmune and Autoinflammatory Disorders: 2022 View all 14 articles
 Claudia Grossi1†
Claudia Grossi1† Nagaja Capitani2,3†
Nagaja Capitani2,3† Marisa Benagiano2
Marisa Benagiano2 Cosima Tatiana Baldari3
Cosima Tatiana Baldari3 Chiara Della Bella2
Chiara Della Bella2 Paolo Macor4
Paolo Macor4 Francesco Tedesco1
Francesco Tedesco1 Maria Orietta Borghi1,5
Maria Orietta Borghi1,5 Norma Maugeri6
Norma Maugeri6 Mario Milco D’Elios7*
Mario Milco D’Elios7* Pier Luigi Meroni1*
Pier Luigi Meroni1*Antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterized by recurrent vascular thrombosis and miscarriages in the absence of known causes. Antibodies against phospholipid-binding proteins (aPL) are pathogenic players in both clotting and pregnancy APS manifestations. There is sound evidence that antibodies specific for beta2 glycoprotein I (β2GPI) trigger thrombotic and pregnancy complications by interacting with the molecule on the membranes of different cell types of the coagulation cascade, and in placenta tissues. In addition to the humoral response against β2GPI, both peripheral and tissue CD4+ β2GPI-specific T cells have been reported in primary APS as well as in systemic lupus erythematosus (SLE)-associated APS. While adaptive immunity plays a clear role in APS, it is still debated whether innate immunity is involved as well. Acute systemic inflammation does not seem to be present in the syndrome, however, there is sound evidence that complement activation is crucial in animal models and can be found also in patients. Furthermore, neutrophil extracellular traps (NETs) have been documented in arterial and venous thrombi with different etiology, including clots in APS models. Keeping in mind that β2GPI is a pleiotropic glycoprotein, acting as scavenger molecule for infectious agents and apoptotic/damaged body constituents and that self-molecules externalized through NETs formation may become immunogenic autoantigens, we demonstrated β2GPI on NETs, and its ability to stimulate CD4+β2GPI-specific T cells. The aim of this review is to elucidate the role of β2GPI in the cross-talk between the innate and adaptive immunity in APS.
Anti-Phospholipid Syndrome (APS) is an autoantibody-mediated vasculopathy characterized by recurrent arterial/venous thrombosis and/or miscarriages (1). There is evidence that antibodies against phospholipid-binding proteins (aPL) are pathogenic in addition of being diagnostic/classification markers (2). In particular, antibodies specific for beta2 glycoprotein I (β2GPI) may trigger clotting and pregnancy complications by interacting with the molecule on the membranes of cells of the coagulation cascade and of placenta tissues. In addition to the humoral response against β2GPI, both peripheral and tissue CD4+ β2GPI-specific T cells have been reported in primary APS as well as in systemic lupus erythematosus (SLE)-associated APS (3). Besides their helper role for B cell activation, these cells have been involved in the formation of the atherosclerotic plaques associated with APS and in modulating their instability (4, 5).
While adaptive immunity plays a clear role in APS, innate immunity is emerging as another player. For example, complement activation is crucial in animal models and can be demonstrated also in patients (6). Despite this finding, acute systemic inflammation does not seem to be the main characteristic of the syndrome (7), and the cross-talk between complement and the coagulation cascade was suggested to be more important in APS thrombotic vasculopathy (8, 9). The lack of a clear inflammatory signature in placentas from obstetric APS is consistent with a similar non inflammatory pathogenesis (10). The description of the involvement of cells of the innate immunity, such as monocytes, and their increased tissue factor (TF) membrane expression in APS is consistent with the thrombophilic state in these patients (11, 12). Moreover, β2GPI is naturally present on neutrophils, more than on monocytes, both in APS patients and healthy controls, and neutrophil extracellular traps (NETs) have been recently shown to contribute to the coagulation cascade and to be an integral component of arterial and venous thrombi with different etiology, including APS clots (13–15).
In this review, we will discuss the state of the art and we will present new data on the role of neutrophils as innate immunity effector cells in APS.
Β2GPI is an abundant plasma protein highly conserved across the animal kingdom, suggesting important biological functions (16, 17). Despite the involvement of the molecule in several biological pathways (Table 1), the specific biological role of β2GPI is still a matter of research (17, 18, 25). Nevertheless, β2GPI is widely accepted as the main antigenic target for aPL both in APS laboratory diagnostic assays and pathogenic mechanisms (2, 17).
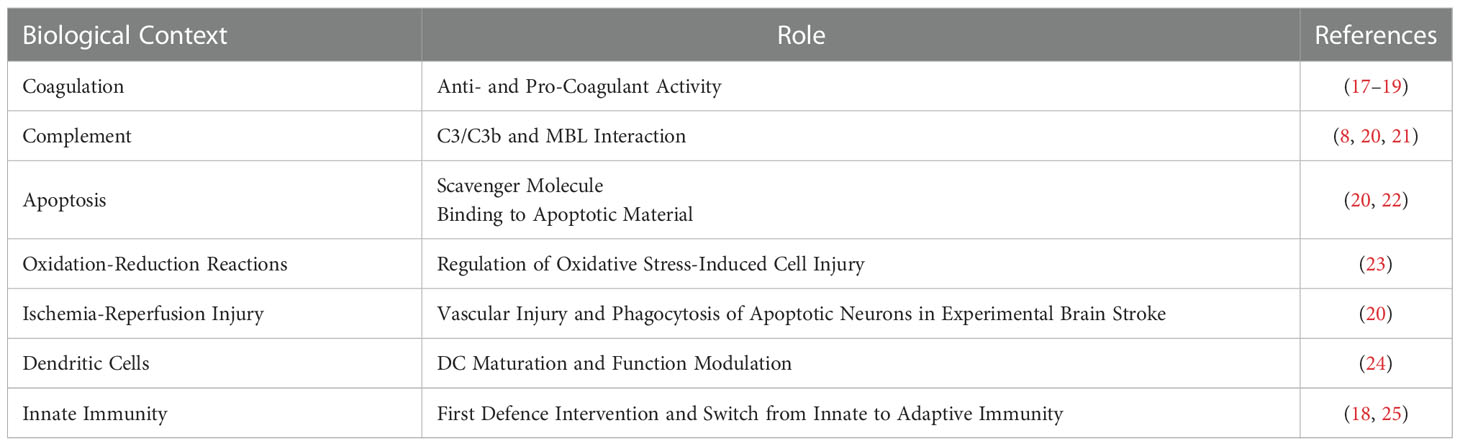
Table 1 β2GPI pleiotropic role.
B2GPI binds to several receptors expressed on different cell types involved in the coagulation cascade and placentation, and can be recognized by circulating aPL. Once bound, the antibodies may crosslink the β2GPI-receptor complex and trigger intracellular signaling leading to cell responses crucial for the clinical manifestations (2). For example, the typical vascular APS is characterized by thrombotic events in well-localized medium/large vessels. There is sound evidence that β2GPI-dependent aPL induce an endothelial perturbation with the expression of a pro-inflammatory and pro-coagulant phenotype suggesting that endothelial perturbation/activation is likely the initiating step. Once activated, the endothelium may involve other circulating cells downstream such as monocytes, neutrophils, and platelets, all contributing to clotting. Complement activation is a hallmark of APS with increased circulating levels of activation products and complement tissue deposition (6). Despite this, an inflammatory signature is not characteristic of vascular APS supporting the idea that the crosstalk of complement with the coagulation cascade is playing a more critical role (26).
On the other hand, the catastrophic APS (CAPS), being systemic thrombotic microangiopathy, displays different pathogenesis and affects small vessels in several anatomical sites with massive production of pro-inflammatory cytokines and the involvement of platelets and neutrophils (27).
Β2GPI-dependent aPL are also associated with obstetric APS manifestations and their pathogenic role is supported by several in vitro and in vivo models. B2GPI can be found on placental and decidual tissues, it binds to its cell membrane receptors and in addition, its cationic PL-binding site in the fifth domain interacts with the anionic charges of the syncytiotrophoblast cell membranes (1). The β2GPI-receptor complex is recognized by maternal β2GPI-dependent aPL which mediate defective placentation eventually responsible for miscarriages (1).
As a result of intense work over the past decade, the molecular basis of immunothrombosis in APS is starting to be elucidated (28). Neutrophils are an essential component of innate immunity. Phagocytosis, the production of reactive oxygen species, the release of proteases and the generation of extracellular traps (NETs) are effector mechanisms that neutrophils rely on to prevent infections, eliminate pathogens and control bystander inflammatory vascular damage (29, 30). NETs however have been documented in arterial and venous thrombi with different etiology (31–33).
NETs consist of lattices of extracellular DNA, decorated with citrullinated histones and neutrophil proteases (29, 34, 35). Various components might be involved in the pro-coagulant effects of NETs. Cell-free DNA activates the coagulation cascade through the intrinsic pathway acting on factors XII and XI (36, 37). Extracellular histones promote thrombin generation by activating platelets (38) and by impairing thrombomodulin-dependent protein C activation (39). The extrinsic pathway of coagulation seems to be activated by the presence of bioactive tissue factor along NETs (40).
The pro-thrombotic action of NETs could be relevant for the pathogenesis of APS. An increase in the plasma concentration of NET by-products has in fact been documented in patients with vascular APS (41). The increase may reflect a higher NET generation, a lower NET degradation rate or both (42).
The presence of neutrophils directly involved in the generation of NETs in APS patients has been suggested by the detection of “Low-Density Granulocyte” (LDG), a population originally described in SLE patients as responsible for spontaneous NET release (43, 44). Furthermore, classical “high-density” neutrophils from APS patients are more efficient than those from healthy subjects in generating NETs (45, 46), probably indicating that a previous in vivo priming occurred.
The three-dimensional structure offered by NETs absorbs and traps exogenous molecules, including the humoral innate immune response players. This has been well documented for complement (28). There is evidence that β2GPI is also expressed by circulating phagocytes regardless of their state of activation (13). As such it could represent a target antigen recognized by anti-β2GPI antibodies. The interaction of the aPL with the β2GPI at the neutrophil membrane could determine an activation signal, stimulating and amplifying the generation of NETs (13, 15, 47)
One model of fetal loss induced by the passive infusion of very high amounts of aPL raised the potential role of neutrophil recruitment and activation by C5a and/or Tumor Necrosis Factor (TNF) in aPL-mediated placenta damage (48). This finding may suggest increased NETosis which in turn can contribute to damage of trophoblast, decidual and endothelial cells. A recent study by Lu et al. (14) reported elevated circulating levels of cell-free DNA and NETs in sera of pregnant women with APS indirectly supporting the possible pathogenic role of NETosis. However, a direct histological demonstration of increased NETosis in APS placentas is still lacking, even though Marder et al. (49) reported that the number of neutrophils, neutrophil NETs and the ratio of NETs/total neutrophils are increased in placentas from SLE patients with miscarriages, in particular, if aPL are present.
The potential interaction NETs-β2GPI in vascular and obstetric APS is outlined in Supplementary Table 1 and in Supplementary Figure 1.
Figure 1A shows that β2GPI binds to NETs obtained from neutrophils purified from healthy donors and cultured in presence of human serum as a source of β2GPI. The binding of β2GPI to NETs was assessed by indirect immunofluorescence (IIF) and confocal microscopy using MBB2 monoclonal IgG specific for the D1 of the molecule (50) and antibodies against elastase or citrullinated histone H3 as NET markers. Moreover, β2GPI containing NETs significantly stimulate the proliferation of SLE-APS derived β2GPI-specific CD4+ T cell clones, more efficiently than native human purified β2GPI (Figure 1B). The reactivity of MBB2 monoclonal IgG supports the exposure of D1 immunodominant epitope of β2GPI, as reported when the molecule binds to anionic structures (17). We previously reported that β2GPI-specific CD4+ T cell clones from PAPS atherosclerotic plaques are more reactive to D1 than to D4,5 (4). The data presented are consistent with this finding and support that the β2GPI co-localizing with NETs offers immunodominant epitopes.
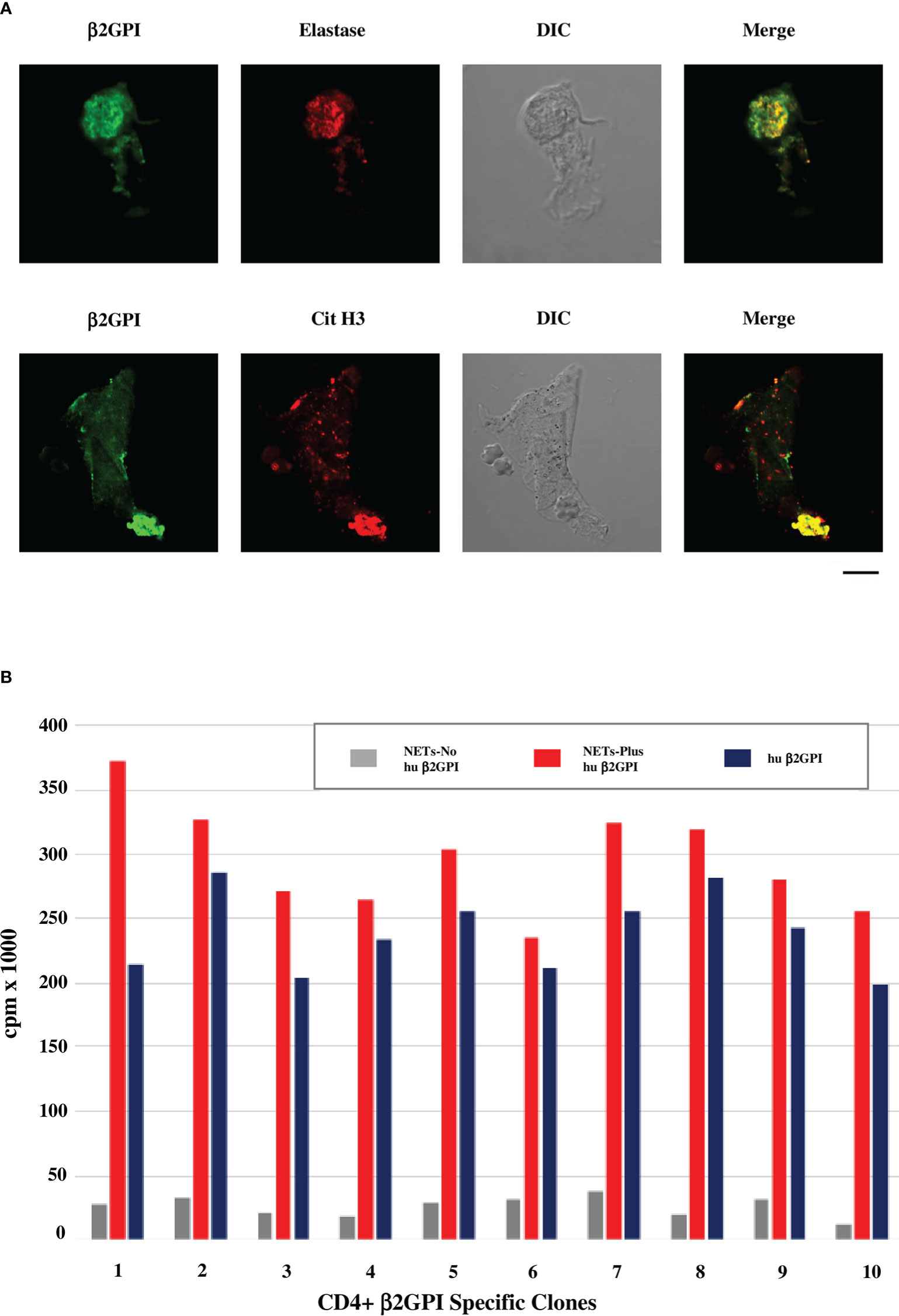
Figure 1 β2GPI co-localizes with NET markers and β2GPI-containing NETs stimulate proliferation of SLE-APS derived T cell clones. (A) Confocal microscopic analysis of human neutrophils cultured in media with human AB serum and stimulated with PMA for production of NETs. Green staining identifies β2GPI, detected by MBB2 antibody (50), red staining identifies elastase by rabbit polyclonal anti-neutrophil elastase antibodies (Abcam) in the upper panel or citrullinated histone H3 (Cit H3) by IgG monoclonal anti-citrullinated histone H3 antibodies (LS-Bio) in the lower panel. Differential Interference Contrast (DIC) was applied as a non-destructive contrast enhancing technique to locate a specific area of interest in the specimen. Briefly, human neutrophils were purified from peripheral blood of healthy donors (EasySep Human Neutrophil isolation kit, Stemcell Technologies) and cultured in medium RPMI 1640 with 5% human serum (Sigma Aldrich) as source of β2GPI, or in β2GPI-free medium (RPMI 1640 in the absence of any source of β2GPI). The cells were stimulated by 100nM PMA for 2h at 37°C for the production of NETs and analyzed for the presence of β2GPI by IF, using the human MBB2 MoAb IgG against D1-β2GPI (50). Co-localization of β2GPI with NET markers, such as elastase and histone H3, was investigated by confocal microscope (Zeiss LSM700 from Carl Zeiss). Alexa Fluor 488- and 555-labeled secondary antibodies were used: Alexa Fluor™ 488-Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody and Alexa Fluor™ 555-Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody (Life Technologies). Scale bar 10 mm. (B) Proliferative response of SLE-APS patient derived β2GPI-specific T-cell clones to NETs, containing or not human β2GPI, or to human purified β2GPI alone (10 nM). Human β2GPI was purified as previously described (51). T-cell clones were derived from the atherosclerotic plaques of the patients, as previously described (4, 5). Further methodological details are reported in the Supplementary Material.
The study was approved by the Ethics Committee at Istituto Auxologico Italiano (22_07_2010) and the patients gave their informed consent.
Under physiological conditions NETs degrade rapidly, an event crucial to quench their potential to damage bystander tissues. Endogenous DNases play an important role in the dismantling of NETs (29, 42, 52). However, DNases only cleave NETs, allowing the release of proteases and histones. In the circulation these molecules, although isolated from the NETs, will retain their ability to damage the vascular wall (29, 53). Another mechanism of NET elimination is phagocytosis by macrophages. The extent of phagocytosis could be accelerated by resolvins, thus contributing to the action of this group of molecules in terminating inflammation (54).
Defective NET degradation could further support the endothelial perturbation leading to the thrombophilic phenotype in APS. Preliminary data showed that NET degradation is impaired in APS patients and this effect is related to antibodies against NETs, which do not correlate with aPL (55).
Strategies specifically designed to interfere with NETs are not yet available. It is however possible that the benefit associated with some anti-inflammatory treatments is at least partially due to the modulation of NET production or biological activity. Low molecular weight heparins (LMWH) are currently used, mostly on an empirical basis, in conditions in which NETs play a critical role. The clinical benefit, that is independent of the anticoagulant action, has been demonstrated in controlled studies in patients with asthma, chronic obstructive pulmonary disease, inflammatory bowel disease, and COVID-19 (56, 57). LMWH has also emerged as an option for the prevention of complications of pregnancy in women at high risk of recurrence (58–60) and is a cornerstone in the treatment of pregnancy complications associated to APS (58, 61).
Several mechanisms are likely to be involved in the anti-inflammatory action of LMWH, including its effect on neutrophils and neutrophil interaction with the vessel wall and platelets (62). Moreover, LMWH can dismantle NETs that have already been organized (63) while inducing a profound change in the ability of human neutrophils to generate NETs and to mobilize the content of the primary granules in response to unrelated inflammatory stimuli (64–66). Moreover, heparin was shown to displace tissue-bound β2GPI because of its stronger avidity for the molecule (67). This last pharmacological mechanism could represent an additional direct effect of heparin on the availability of β2GPI on NETs.
The pathogenesis of APS is becoming more and more complex in the last years involving different mechanisms that are likely to play a diverse role depending on the clinical variants of the syndrome. In particular, evidence of the interplay between adaptive and innate immunity is emerging.
Vascular APS is characterized by clotting in localized areas of the arterial or the venous tree at variance with CAPS, in which systemic thrombotic microangiopathy is responsible for the clinical picture. Endothelial perturbation is apparently the initial pathogenic step and it may explain the clinical manifestations associated with classical vascular APS. However, endothelial activation involves other cells downstream, particularly platelets and neutrophils.
A lot of attention has been recently paid to the role of neutrophils in APS as an example of the interplay with innate immunity. We have previously discussed the evidence for the involvement of neutrophil activation and NET formation in APS from both animal models and from studies in patients (13–15).
We describe in this paper the ability of β2GPI to interact with NETs and demonstrate that the bound molecule displays the epitope characteristics showed by β2GPI complexed with anionic structures (17). Moreover, β2GPI-NETs complexes stimulate β2GPI-specific CD4+ T cells more efficiently than the circulating naïve molecule. This mechanism may be responsible for offering self β2GPI to autoreactive β2GPI specific T helper cells eventually leading to loss of tolerance, β2GPI specific B cell clonal expansion and aPL production in genetically predisposed subjects. Infections and/or inflammatory events are relatively frequent, may induce neutrophil activation and NETosis and ultimately can be responsible for β2GPI binding to NETs.
Once the anti-β2GPI response is started, aPL can react with β2GPI on neutrophils as described (13, 15, 47). This may activate neutrophils directly, boost inflammation and force neutrophils to act as antigen-presenting cells via MHC II-mediated pathways, further stimulating the adaptive response against β2GPI (68). In addition, neutrophils can be indirectly activated via complement activation leading to further NET formation and β2GPI binding. The mutual interactions among the complement and coagulation systems may eventually contribute to thrombotic events in vascular APS (69).
Mitochondrial NETosis offers another potential linkage between β2GPI and NETs. Mitochondria exert a peculiar role in neutrophils, being ejected outside the cell engaging bacteria, an early event in the antimicrobial response (70). The mitochondrial membrane contains cardiolipin (CL) as a unique phospholipid that plays a central role in mitochondrial function and dynamics (71). We may speculate that mitochondria in the extracellular milieu could lead to unusual CL exposure and allow the interaction between CL and β2GPI, naturally present on neutrophils (13).
The presence of circulating aPL may affect all these systems. For example, NETs could not be regularly cleared by β2GPI-dependent scavenging mechanisms because of aPL interference. The reported impaired NET degradation in APS patients associated with the presence of antibodies against NETs is consistent with this effect (55).
It is likely that apoptotic blebs and NETs may interplay at the sites of inflammation.
Their clearance systems partially overlap (72). β2GPI plays a role in scavenging LPS and apoptotic blebs, and at the same time is able to bind nucleic acids via the cationic binding site in the fifth domain, these being good starting points for a candidate protein involved in NET clearance (20, 22, 30, 72). Once again, β2GPI-dependent aPL may interfere with these mechanisms. An additional pro-thrombotic effect of NETs has been recently associated with activated protein C resistance in APS (73).
In conclusion, there is emerging evidence that β2GPI is at crossroads between adaptive and innate immunity in APS pathogenesis and may represent a potential target for new therapies. Consistently, synthetic peptides competing with the PL-binding site and able to inhibit the binding of β2GPI to physiological targets showed a protective effect in animal models (74–76).
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The study was approved by the Ethics Committee at Istituto Auxologico Italiano (22_07_2010). The patients/participants provided their written informed consent to participate in this study.
MMD'E, PLM contributed to conception and design of the study, NC, MB, CTB, CDB performed the experiments and organized the database, CG, PM, FT, prepared purified β2GPI and MBB2. CG, MOB, NM, PLM wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by grants from Italian Ministry of Health, IRCCS Istituto Auxologico Italiano, Ricerca Corrente (PLM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1076167/full#supplementary-material
1. Meroni PL, Borghi MO, Grossi C, Chighizola CB, Durigutto P, Tedesco F. Obstetric and vascular antiphospholipid syndrome: Same antibodies but different diseases? Nat Rev Rheumatol (2018) 14:433–40. doi: 10.1038/s41584-018-0032-6
2. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat Rev Rheumatol (2011) 7:330–9. doi: 10.1038/nrrheum.2011.52
3. Ludwig RJ, Pamela Blanco L, Amedei A, Rauch J, Salem D, Subang R, et al. β2-glycoprotein I-reactive T cells in autoimmune disease. Front Immunol (2018) 9:2836. doi: 10.3389/fimmu.2018.02836
4. Benagiano M, Gerosa M, Romagnoli J, Mahler M, Borghi MO, Grassi A, et al. β2 glycoprotein I recognition drives Th1 inflammation in atherosclerotic plaques of patients with primary antiphospholipid syndrome. J Immunol (2017) 198:2640–8. doi: 10.4049/jimmunol.1600305
5. Benagiano M, Borghi MO, Romagnoli J, Mahler M, Della Bella C, Grassi A, et al. Interleukin-17/Interleukin-21 and interferon-γ producing T cells specific for β2 glycoprotein I in atherosclerosis inflammation of systemic lupus erythematosus patients with antiphospholipid syndrome. Haematologica (2019) 104:2519–27. doi: 10.3324/haematol.2018.209536
6. Tedesco F, Borghi MO, Gerosa M, Chighizola CB, Macor P, Lonati PA, et al. Pathogenic role of complement in antiphospholipid syndrome and therapeutic implications. Front Immunol (2018) 9:1388. doi: 10.3389/fimmu.2018.01388
7. de Groot PG, Urbanus RT. Antiphospholipid syndrome-not a noninflammatory disease. Semin Thromb Hemost (2015) 41:607–14. doi: 10.1055/s-0035-1556725
8. Durigutto P, Macor P, Pozzi N, Agostinis C, Bossi F, Meroni PL, et al. Complement activation and thrombin generation by MBL bound to β2-glycoprotein I. J Immunol (2020) 205:1385–92. doi: 10.4049/jimmunol.2000570
9. Rawish E, Sauter M, Sauter R, Nording H, Langer HF. Complement, inflammation and thrombosis. Br J Pharmacol (2021) 178:2892–904. doi: 10.1111/bph.15476
10. Viall CA, Chamley LW. Histopathology in the placentae of women with antiphospholipid antibodies: A systematic review of the literature. Autoimmun Rev (2015) 14:446–71. doi: 10.1016/j.autrev.2015.01.008
11. López-Pedrera C, Buendía P, Aguirre MA, Velasco F, Cuadrado MJ. Antiphospholipid syndrome and tissue factor: A thrombotic couple. Lupus (2006) 15:161–6. doi: 10.1191/0961203306lu2276rr
12. Adams M. Novel considerations in the pathogenesis of the antiphospholipid syndrome: Involvement of the tissue factor pathway of blood coagulation. Semin Thromb Hemost (2008) 34:251–5. doi: 10.1055/s-0028-1082268
13. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Nunez-Alvarez CN, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol (2015) 67:2990–3003. doi: 10.1002/art.39247
14. Lu Y, Dong Y, Zhang Y, Shen D, Wang X, Ge R, et al. Antiphospholipid antibody-activated NETs exacerbate trophoblast and endothelial cell injury in obstetric antiphospholipid syndrome. J Cell Mol Med (2020) 24:6690–703. doi: 10.1111/jcmm.15321
15. Tambralli A, Gockman K, Knight JS. NETs in APS: Current knowledge and future perspectives. Curr Rheumatol Rep (2020) 22:67. doi: 10.1007/s11926-020-00936-1
16. Weaver JC, Krilis SA, Giannakopoulos B. Oxidative post-translational modification of βeta 2-glycoprotein I in the pathophysiology of the anti-phospholipid syndrome. Free Radic Biol Med (2018) 125:98–103. doi: 10.1016/j.freeradbiomed.2018.03.048
17. McDonnell T, Wincup C, Buchholz I, Pericleous C, Giles I, Ripoll V, et al. The role of beta-2-glycoprotein I in health and disease associating structure with function: More than just APS. Blood Rev (2020) 39:100610. doi: 10.1016/j.blre.2019.100610
18. Matsuura E, Lopez LR, Shoenfeld Y, Ames PRJ. β2-glycoprotein I and oxidative inflammation in early atherogenesis: A progression from innate to adaptive immunity? Autoimmun Rev (2012) 12:241–9. doi: 10.1016/j.autrev.2012.04.003
19. Alluri RK, Forbes GL, Kundu S, Schmaier AH, McCrae KR. Mice deficient in ß2-glycoprotein I have a delayed time to thrombosis. Blood (2019) 134:2423–3. doi: 10.1182/blood-2019-130521
20. Grossi C, Artusi C, Meroni PL, Borghi MO, Neglia L, Lonati PA, et al. β2 glycoprotein I participates in phagocytosis of apoptotic neurons and in vascular injury in experimental brain stroke. J Cereb Blood Flow Metab (2021) 41:2038–53. doi: 10.1177/0271678X20984551
21. Gropp K, Weber N, Reuter M, Micklisch S, Kopka I, Hallström T, et al. β2-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood (2011) 118:2774–83. doi: 10.1182/blood-2011-02-339564
22. Maiti SN, Balasubramanian K, Ramoth JA, Schroit AJ. β-2-glycoprotein 1-dependent macrophage uptake of apoptotic cells: Binding to lipoprotein receptor-related protein receptor family members. J Biol Chem (2008) 283:3761–6. doi: 10.1074/jbc.M704990200
23. Ioannou Y, Zhang JY, Passam FH, Rahgozar S, Qi JC, Giannakopoulos B, et al. Naturally occurring free thiols within β2-glycoprotein I in vivo: Nitrosylation, redox modification by endothelial cells, and regulation of oxidative stress-induced cell injury. Blood (2010) 116:1961–70. doi: 10.1182/blood-2009-04-215335
24. Buttari B, Profumo E, Capozzi A, Facchiano F, Saso L, Sorice M, et al. Advanced glycation end products of human β2 glycoprotein I modulate the maturation and function of DCs. Blood (2011) 117:6152–61. doi: 10.1182/blood-2010-12-325514
25. Ağar Ç, de Groot PG, Mörgelin M, Monk SDDC, van Os G, Levels JHM, et al. β2-glycoprotein I: A novel component of innate immunity. Blood (2011) 117:6939–47. doi: 10.1182/blood-2010-12-325951
26. Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood (2019) 133:906–18. doi: 10.1182/blood-2018-11-882993
27. Sciascia S, Lopez-Pedrera C, Roccatello D, Cuadrado MJ. Catastrophic antiphospholipid syndrome (CAPS). Best Pract Res Clin Rheumatol (2012) 26:535–41. doi: 10.1016/j.berh.2012.07.005
28. Knight JS, Kanthi Y. Mechanisms of immunothrombosis and vasculopathy in antiphospholipid syndrome. Semin Immunopathol (2022) 44:347–62. doi: 10.1007/s00281-022-00916-w
29. Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood (2019) 133:2178–85. doi: 10.1182/blood-2018-11-844530
30. Manfredi AA, Covino C, Rovere-Querini P, Maugeri N. Instructive influences of phagocytic clearance of dying cells on neutrophil extracellular trap generation. Clin Exp Immunol (2015) 179:24–9. doi: 10.1111/cei.12320
31. Maugeri N, Campana L, Gavina M, Covino C, de Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemostasis (2014) 12:2074–88. doi: 10.1111/jth.12710
32. Genchi A, Semerano A, Gullotta GS, Strambo D, Schwarz G, Bergamaschi A, et al. Cerebral thrombi of cardioembolic etiology have an increased content of neutrophil extracellular traps. J Neurol Sci (2021) 423:117355. doi: 10.1016/j.jns.2021.117355
33. Thålin C, Hisada Y, Lundström S, Mackman N, Wallén H. Neutrophil extracellular traps: Villains and targets in arterial, venous, and cancer-associated thrombosis. Arterioscler Thromb Vasc Biol (2019) 39:1724–38. doi: 10.1161/ATVBAHA.119.312463
34. Grayson PC, Kaplan MJ. At The bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J Leukoc Biol (2016) 99:253–64. doi: 10.1189/jlb.5bt0615-247r
35. Manfredi AA, Ramirez GA, Rovere-Querini P, Maugeri N. The neutrophil’s choice: Phagocytose vs make neutrophil extracellular traps. Front Immunol (2018) 9:288. doi: 10.3389/fimmu.2018.00288
36. Gould TJ, Vu TT, Stafford AR, Dwivedi DJ, Kim PY, Fox-Robichaud AE, et al. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arterioscler Thromb Vasc Biol (2015) 35:2544–53. doi: 10.1161/ATVBAHA.115.306035
37. Swystun LL, Mukherjee S, Liaw PC. Breast cancer chemotherapy induces the release of cell-free DNA, a novel procoagulant stimulus. J Thromb Haemost. (2011) 9:2313–21. doi: 10.1111/j.1538-7836.2011.04465.x
38. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
39. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein c activation. J Thromb Haemost (2011) 9:1795–803. doi: 10.1111/j.1538-7836.2011.04422.x
40. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J (2015) 36:1405–14. doi: 10.1093/eurheartj/ehv105
41. Mazetto BM, Hounkpe BW, da Silva Saraiva S, Vieira-Damiani G, dos Santos APR, Jacinto BC, et al. Association between neutrophil extracellular traps (NETs) and thrombosis in antiphospholipid syndrome. Thromb Res (2022) 214:132–7. doi: 10.1016/j.thromres.2022.05.001
42. Boeltz S, Amini P, Anders H-J, Andrade F, Bilyy R, Chatfield S, et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ (2019) 26:28. doi: 10.1038/s41418-018-0261-x
43. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
44. Carmona-Rivera C, Zhao W, Yalavarthi S. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis (2015) 74:1417–24. doi: 10.1136/annrheumdis-2013-204837
45. Knight JS, Meng H, Coit P, Yalavarthi S, Sule G, Gandhi AA, et al. Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target. JCI Insight (2017) 2:e93897. doi: 10.1172/JCI.INSIGHT.93897
46. Mauracher LM, Krall M, Roiß J, Hell L, Koder S, Hofbauer TM, et al. Neutrophil subpopulations and their activation potential in patients with antiphospholipid syndrome and healthy individuals. Rheumatol (Bulgaria) (2021) 60:1687–99. doi: 10.1093/rheumatology/keaa532
47. Ali RA, Estes SK, Gandhi AA, Yalavarthi S, Hoy CK, Shi H, et al. Defibrotide inhibits antiphospholipid antibody–mediated neutrophil extracellular trap formation and venous thrombosis. Arthritis Rheumatol (2022) 74:902–7. doi: 10.1002/art.42017
48. Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest (2003) 112:1644–54. doi: 10.1172/jci18817
49. Marder W, Knight JS, Kaplan MJ, Somers EC, Zhang X, O’Dell AA, et al. Placental histology and neutrophil extracellular traps in lupus and pre-eclampsia pregnancies. Lupus Sci Med (2016) 3:e000134. doi: 10.1136/lupus-2015-000134
50. Agostinis C, Durigutto P, Sblattero D, Borghi MO, Grossi C, Guida F, et al. A non-complement-fixing antibody to β2 glycoprotein i as a novel therapy for antiphospholipid syndrome. Blood (2014) 123:3478–87. doi: 10.1182/blood-2013-11-537704
51. Agostinis C, Biffi S, Garrovo C, Durigutto P, Lorenzon A, Bek A, et al. In vivo distribution of β2 glycoprotein I under various pathophysiologic conditions. Blood (2011) 118:4231–8. doi: 10.1182/blood-2011-01-333617
52. Jiménez-Alcázar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Sci (1979) (2017) 358:1202–6. doi: 10.1126/science.aam8897
53. Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun (2015) 6:6673. doi: 10.1038/ncomms7673
54. Chiang N, Sakuma M, Rodriguez AR, Spur BW, Irimia D, Serhan CN. Resolvin T-series reduce neutrophil extracellular traps. Blood (2022) 139:1222–33. doi: 10.1182/blood.2021013422
55. Zuo Y, Yalavarthi S, Gockman K, Madison JA, Gudjonsson JE, Kahlenberg JM, et al. Anti-neutrophil extracellular trap antibodies and impaired neutrophil extracellular trap degradation in antiphospholipid syndrome. Arthritis Rheumatol (2020) 72:2130–5. doi: 10.1002/art.41460
56. Saithong S, Saisorn W, Tovichayathamrong P, Filbertine G, Torvorapanit P, Wright HL, et al. Anti-inflammatory effects and decreased formation of neutrophil extracellular traps by enoxaparin in COVID-19 patients. Int J Mol Sci (2022) 23:4805. doi: 10.3390/ijms23094805
57. Baccellieri D, Bertoglio L, Apruzzi L, Ardita V, D’Angelo A, Bossi M, et al. Incidence of deep venous thrombosis in COVID-19 hospitalized patients during the first peak of the Italian outbreak. Phlebology (2021) 36:375–83. doi: 10.1177/0268355520975592
58. Cervera R, Balasch J. Autoimmunity and recurrent pregnancy losses. Clin Rev Allergy Immunol (2010) 39:148–52. doi: 10.1007/s12016-009-8179-1
59. Ruffatti A, Tonello M, Visentin MS, Bontadi A, Hoxha A, de Carolis S, et al. Risk factors for pregnancy failure in patients with anti-phospholipid syndrome treated with conventional therapies: A multicentre, case-control study. Rheumatology (2011) 50:1684–9. doi: 10.1093/rheumatology/ker139
60. Dodd JM, Mcleod A, Windrim RC, Kingdom J. Antithrombotic therapy for improving maternal or infant health outcomes in women considered at risk of placental dysfunction. Cochrane Database Syst Rev (2013) 6:CD006780. doi: 10.1002/14651858.CD006780.pub3
61. Girardi G, Redecha P, Salmon JE. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med (2004) 10:1222–6. doi: 10.1038/nm1121
62. Evangelista V, Piccardoni P, Maugeri N, de Gaetano G, Cerletti C. Inhibition by heparin of platelet activation induced by neutrophil-derived cathepsin G. Eur J Pharmacol (1992) 216:401–5. doi: 10.1016/0014-2999(92)90437-9
63. Panda R, Castanheira FVS, Schlechte JM, Surewaard BGJ, Shim HB, Zucoloto AZ, et al. A functionally distinct neutrophil landscape in severe COVID-19 reveals opportunities for adjunctive therapies. JCI Insight (2022) 7:e152291. doi: 10.1172/jci.insight.152291
64. Feldman M, Ginsburg I. A novel hypothetical approach to explain the mechanisms of pathogenicity of rheumatic arthritis. Mediterr J Rheumatol (2021) 32:112–7. doi: 10.31138/mjr.32.2.112
65. Sohrabipour S, Muniz VS, Sharma N, Dwivedi DJ, Liaw PC. Mechanistic studies of DNase I activity: Impact of heparin variants and PAD4. Shock (2021) 56:975–87. doi: 10.1097/SHK.0000000000001804
66. Manfredi AA, Rovere-Querini P, D’Angelo A, Maugeri N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol Res (2017) 123:146–56. doi: 10.1016/j.phrs.2016.08.008
67. di Simone N, Meroni PL, D’Asta M, di Nicuolo F, D’Alessio MC, Caruso A. Pathogenic role of anti-β2-glycoprotein I antibodies on human placenta: Functional effects related to implantation and roles of heparin. Hum Reprod Update (2007) 13:189–96. doi: 10.1093/humupd/dml051
68. Polak D, Bohle B. Neutrophils-typical atypical antigen presenting cells? Immunol Lett (2022) 247:52–8. doi: 10.1016/j.imlet.2022.04.007
69. de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol (2019) 16:19–27. doi: 10.1038/s41423-018-0024-0
70. Dunham-Snary KJ, Surewaard BG, Mewburn JD, Bentley RE, Martin AY, Jones O, et al. Mitochondria in human neutrophils mediate killing of staphylococcus aureus: Mitochondria-mediated killing of s. aureus. Redox Biol (2022) 49:102225. doi: 10.1016/j.redox.2021.102225
71. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: Molecular and pharmacological aspects. Cells (2019) 8:728. doi: 10.3390/cells8070728
72. Radic M. Clearance of apoptotic bodies, NETs, and biofilm DNA: Implications for autoimmunity. Front Immunol (2014) 5:365. doi: 10.3389/fimmu.2014.00365
73. Foret T, Dufrost V, Salomon Du Mont L, Costa P, Lakomy C, Lagrange J, et al. A new pro-thrombotic mechanism of neutrophil extracellular traps in antiphospholipid syndrome: Impact on activated protein c resistance. Rheumatol (Bulgaria) (2022) 61:2993–8. doi: 10.1093/rheumatology/keab853
74. Martinez de la Torre Y, Pregnolato F, D’Amelio F, Grossi C, di Simone N, Pasqualini F, et al. Anti-phospholipid induced murine fetal loss: Novel protective effect of a peptide targeting the β2 glycoprotein I phospholipid-binding site Implications for human fetal loss. J Autoimmun (2012) 38:J209–15. doi: 10.1016/j.jaut.2011.11.009
75. di Simone N, D’Ippolito S, Marana R, di Nicuolo F, Castellani R, Pierangeli SS, et al. Antiphospholipid antibodies affect human endometrial angiogenesis: Protective effect of a synthetic peptide (TIFI) mimicking the phospholipid binding site of β2glycoprotein I. Am J Reprod Immunol (2013) 70:299–308. doi: 10.1111/aji.12130
Keywords: β2GPI, neutrophils, T cells, NETs, APS, aPL, inflammation, thrombosis
Citation: Grossi C, Capitani N, Benagiano M, Baldari CT, Della Bella C, Macor P, Tedesco F, Borghi MO, Maugeri N, D’Elios MM and Meroni PL (2023) Beta 2 glycoprotein I and neutrophil extracellular traps: Potential bridge between innate and adaptive immunity in anti-phospholipid syndrome. Front. Immunol. 13:1076167. doi: 10.3389/fimmu.2022.1076167
Received: 21 October 2022; Accepted: 20 December 2022;
Published: 09 January 2023.
Edited by:
Marie-Agnes DRAGON-DUREY, Université Paris Cité, FranceReviewed by:
Ian P Giles, University College London, United KingdomCopyright © 2023 Grossi, Capitani, Benagiano, Baldari, Della Bella, Macor, Tedesco, Borghi, Maugeri, D’Elios and Meroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pier Luigi Meroni, cGllcmx1aWdpLm1lcm9uaUB1bmltaS5pdA==; cC5tZXJvbmlAYXV4b2xvZ2ljby5pdA==; Mario Milco D’Elios, ZGVsaW9zQHVuaXNpLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.