
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 December 2022
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1075813
This article is part of the Research Topic Repurposing Cancer Immunotherapies for Use in Autoimmunity and Transplantation View all 7 articles
 Farbod Ghobadinezhad1,2
Farbod Ghobadinezhad1,2 Nasim Ebrahimi3Fatemeh Mozaffari4Neda Moradi5
Nasim Ebrahimi3Fatemeh Mozaffari4Neda Moradi5 Sheida Beiranvand6Mehran Pournazari7Fatemeh Rezaei-Tazangi8Roya Khorram9Maral Afshinpour10Rob A. Robino11,12,13
Sheida Beiranvand6Mehran Pournazari7Fatemeh Rezaei-Tazangi8Roya Khorram9Maral Afshinpour10Rob A. Robino11,12,13 Amir Reza Aref14,15*
Amir Reza Aref14,15* Leonardo M. R. Ferreira11,12,13*
Leonardo M. R. Ferreira11,12,13*Autoimmune disease, caused by unwanted immune responses to self-antigens, affects millions of people each year and poses a great social and economic burden to individuals and communities. In the course of autoimmune disorders, including rheumatoid arthritis, systemic lupus erythematosus, type 1 diabetes mellitus, and multiple sclerosis, disturbances in the balance between the immune response against harmful agents and tolerance towards self-antigens lead to an immune response against self-tissues. In recent years, various regulatory immune cells have been identified. Disruptions in the quality, quantity, and function of these cells have been implicated in autoimmune disease development. Therefore, targeting or engineering these cells is a promising therapeutic for different autoimmune diseases. Regulatory T cells, regulatory B cells, regulatory dendritic cells, myeloid suppressor cells, and some subsets of innate lymphoid cells are arising as important players among this class of cells. Here, we review the roles of each suppressive cell type in the immune system during homeostasis and in the development of autoimmunity. Moreover, we discuss the current and future therapeutic potential of each one of these cell types for autoimmune diseases.
Protecting the body against foreign pathogenic agents and activating repair systems when tissue is damaged are the primary functions of our immune system. The immune system must constantly strike a balance between attacking harmful agents and preventing damage to the body. Disturbances in this balance can cause autoimmune diseases, in which the immune system attacks self-tissues. Although investigations have focused more on the role of adaptive immunity, both innate and adaptive immunity appear to be involved in the development of autoimmune diseases (1). Albeit individually rare, autoimmune disorders comprise a wide range of complex diseases that affect about 5% of the world’s population. Loss of self-tolerance leads to the production of autoantibodies against self-tissues and cells, as well as the emergence of autoreactive T cells (2). The incidence of autoimmune diseases is increasing worldwide (3). Within these, type 1 diabetes (T1D), multiple sclerosis (MS), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and Crohn’s disease (CD) are the most common types of autoimmune diseases, posing enormous health challenges (4). Currently, the main goal of autoimmune disease research is to find more effective treatment solutions. Present treatments can alleviate some autoimmune symptoms but lack specificity and need to be prescribed for long periods. In contrast, the use of living drugs, such as regulatory T (Treg) cells, a subset of T lymphocytes dedicated to inhibiting specific immune responses, holds the potential to be more specific, cause less side effects, and be more effective (5). Current drugs used to treat autoimmune disease include immunosuppressive drugs, such as ciclosporin (cytokine gene transcription inhibitor), anti-metabolite drugs, such as azathioprine (purine synthesis inhibitor), and biologic drugs, such as belimumab (human monoclonal antibody against B-cell activating factor). Cell therapy as another approach for treat autoimmune disease is assumed that are less toxic and can aim to help restore immune tolerance, as opposed to global non-specific immune suppression. Therefore, checking the accuracy of these assumptions is on the agenda of many ongoing studies (5). Notable examples of such living drugs include immune cells, such as regulatory B (Breg) and Treg cells. In general, the superior ability of immune cells, when compared to small molecules and biologicals, to either maintain or disrupt the body’s immune balance provides a unique opportunity to treat autoimmune disorders and accelerate the repair of deregulated nodes in the immune system (4). Many studies have been performed on cell-based therapy for various types of autoimmune diseases. Several types of immune cells, including Breg and Treg cells, regulatory dendritic cells (DCs), as well as mesenchymal stem cells, monocytes, and macrophages, have been shown to relieve inflammation and symptoms of autoimmune diseases (6, 7). Adoptive immune cell therapies have sparked great interest due to their advantages, including delivery convenience, capacity of naturally homing to target tissues, and the ability to significantly alter the course of disease, as supported by preclinical research and promising results from early clinical trials in autoimmune diseases and transplantation (8). Here, we review a variety of immune cell-based therapies for autoimmunity, including their prospects and potential for treating and reducing the burden of these diseases worldwide.
A subset of naïve T cells develops naturally in the immune system to maintain immune homeostasis and autoimmune tolerance (9). During 1970-1980, many efforts were made to detect CD4+ T cells able to suppress autoimmune diseases in rodents through reliable molecular markers (10). Finally, in the mid-1990s, it became clear that this group of CD4+ T cells, known as regulatory T cells (Treg cells), continuously express the α chain of the interleukin 2 receptor (IL-2), or CD25, at high levels (11). CD4+CD25+ cells comprise only 3-10% the peripheral CD4+ T cell population. Their key role in autoimmune diseases was clarified when their deletion was shown to result in a wide range of human-like autoimmune disorders in healthy mice, including type 1 diabetes, thyroiditis, and autoimmune gastritis. On the other hand, infusion of CD4+CD25+ T cells inhibited autoimmune disease development in mice (12). In 2003, it was reported that CD25+CD4+ T cells in rodents and humans uniquely express the transcription factor Foxp3 at high levels (13). Mutations in the Foxp3 gene in both mice and humans cause autoimmune and inflammatory diseases such as type 1 diabetes (T1D) and thyroiditis. In addition, the incidence of allergy and inflammatory bowel disease (IBD), which together cause IPEX syndrome (immune dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome), is caused by dysfunction in FOXP3+ T cells (14). In summary, regulatory T cells develop in the thymus, are characterized by high constitutive expression of Foxp3 in the nucleus and CD25 on the surface, and suppress excessive immune responses against environmental, microbial, and self-antigens (15).
Treg cells constitute a form of dominant tolerance, directly suppressing the activation, expansion, and function of effector immune cells. A number of mechanisms of Treg cell-mediated immune suppression have been revealed (Figure 1A), from depletion of IL-2 in the milieu via high surface expression of CD25 and secretion of anti-inflammatory cytokines, such as IL-10, IL-35, and TGF-β (contact-independent) to trogocytosis of CD80 and CD86 receptors in antigen presenting cells (APCs) via CTLA4 (contact-dependent) (16, 17). Treg cell function is classically characterized by two main tenets: infectious tolerance and bystander suppression. Infectious tolerance consists of the transfer of suppressive capacity from one cell population to another and is believed to occur mainly via anti-inflammatory cytokines that block dendritic cell (DC) maturation, bestowing DCs with a tolerogenic phenotype and drive naïve T cell differentiation into induced Tregs (18, 19), whereas bystander suppression refers to Tregs’ capacity to suppress immune responses specific for an antigen distinct from the one they recognize in the same milieu (20). Both phenomena are local, happening at the level of the immune microenvironment.
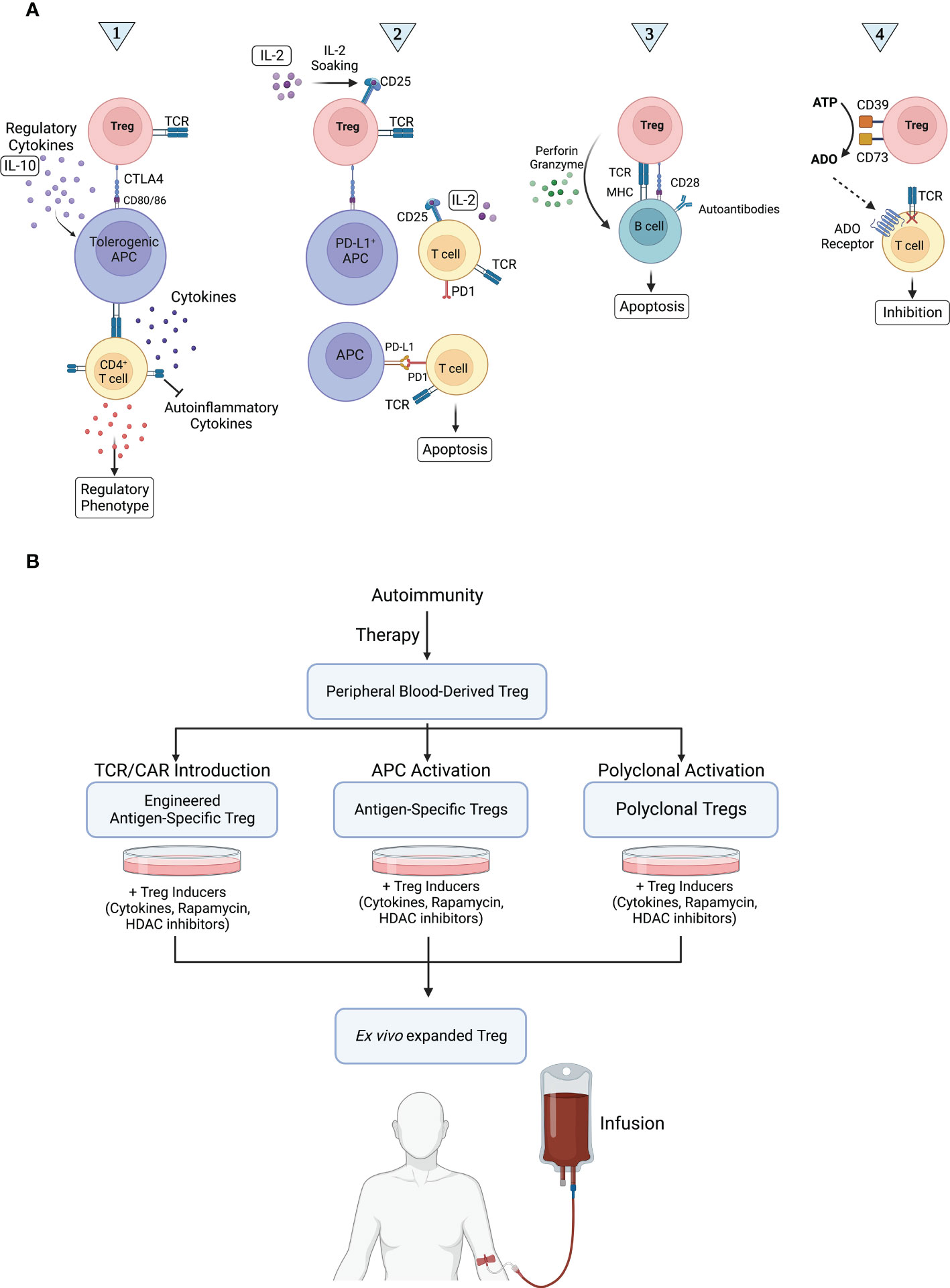
Figure 1 Treg cell mechanisms of action in autoimmunity and therapeutic application. (A) Treg cells are thought to work via four main routes. Route 1: Treg cells expand and secrete large amounts of IL-10, enhancing tolerogenic APC activity. Tolerogenic APCs in turn interact with CD4+ T cells, inhibiting their release of self-inflammatory cytokines. In addition, tolerogenic APCs stimulate the development of a regulatory phenotype in naïve T cells and induce the production of Treg cells, ultimately suppressing autoimmune reactions. Route 2: The release of IL-2 in the microenvironment is detected by CD25+ Treg cells, which interact with and induce PD-L1 expression in APCs. PD-L1+ APCs then induce apoptosis in activated PD-1+ Teff cells via PD-1/PD-L1 signaling, inhibiting immune responses, such as those targeting self-antigens. Route 3: The interaction of Treg cells with autoreactive B cells induces B cell apoptosis via perforin/granzyme-mediated cytotoxicity, preventing autoantibody production. Route 4: Treg cells convert extracellular ATP into adenosine (ADO), a potent immunosuppressant, using ectoenzymes CD39 and CD73. ADO binds to its receptor in Teff cells, inhibiting them. (B) Any disruption in routes 1, 2, 3, and 4 can lead to autoimmunity. In Treg cell-based therapy, Treg cells are collected from peripheral blood, activated, and expanded ex vivo. Finally, Treg cells are injected to suppress immune responses via routes 1, 2, 3, and 4.
Despite these hallmarks, heterogeneity of the Treg cell compartment is one of the main challenges of studying Tregs in autoimmune diseases and at large. Systematic characterization of Treg cell subsets can lead to a more precise identification of their roles in various autoimmune diseases and hence the development of more specific treatment strategies for treatment (21).
Treg cells can be divided based on their developmental origin. Thymic Tregs (tTreg) develop in the thymus. A small fraction of Treg cells is derived from conventional T cells (Tconv) and mature peripherally (pTreg) under certain conditions, including exposure to microbial antigens in the intestinal mucosa. Both tTregs and pTregs express Foxp3 and have suppressive function. On the other hand, ex vivo antigenic stimulation in the presence of cytokines TGF-β and IL-2 can induce Foxp3 expression in Tconv cells, which are phenotypically and functionally similar to tTreg and pTreg cells and are known as induced Treg cells (iTreg). However, iTreg cells differ in terms of functional stability and specificity and, therefore, come with different advantages and challenges in treating autoimmune diseases (22).
Type 1 regulatory T (Tr1) cells are a subtype of induced regulatory cells. Unlike Treg cells, Tr1 cells secrete immunosuppressive cytokines, such as IL-10 and TGF-β, but do not express CD25 or Foxp3. In addition, Foxp3+ Treg cells and Tr1 cells differ in their metabolic programs (23). For example, in vitro induced Tr1 cells display high rates of aerobic glycolysis, while Foxp3+ Treg cells prefer oxidative phosphorylation. Although both Foxp3+ Treg cells and Tr1 cell populations are present in the spleen, Peyers’ patches, and lymph nodes, Tr1 cells are more abundant in the small intestine, whereas Foxp3+ Treg cells are more abundant in the large intestine. Interestingly, mice lacking induced Foxp3+ Treg cells have a compensatory increase in Tr1 cells in the mesenteric lymph nodes (24). Transfer of Foxp3+ Treg cells, on the other hand, results in the generation of Tr1 cells and the development of antigen specific tolerance in recipient mice in an allogeneic pancreatic islet transplantation model. Overall, it appears that Foxp3+ Treg cells are essential for initiating tolerance induction at the site of inflammation, while Tr1 cells may be important for the long-term maintenance of immune tolerance in some settings (25). Consistent with a role for Tr1 cells in immune tolerance, Tr1 cell dysfunction has been reported in autoimmune patients, which also indicates their therapeutic potential. However, a high dose of Tr1 cells is needed to be effective in treatment. A Phase I trial (NCT03198234) testing a cell product, T-allo10, containing up to 15% CD49b+LAG-3+ Tr1 cells, is administered during HLA mismatched hematopoietic stem cell transplant (HSCT). T-allo10 is created by stimulating donor-derived CD4+ T cells with host-derived tolerogenic DCs (DC-10) in the presence of IL-10. The therapy is well tolerated and T-allo10 cells detectable in the peripheral blood of patients up to 1 year after transfer, but the effects on GvHD and long-term tolerance are still being studied (26).
Treg cells can also be classified based on their function. Curiously, comprehensive analysis of human peripheral blood Treg cells revealed the existence of subsets of T helper-like Treg cells, i.e. Treg cells that share chemokine receptor and transcription factor expression with T helper cells while being suppressive. For instance, Th1-Treg cells can be characterized as CXCR3+T-BET+FOXP3+ Treg cells, Th2-Treg cells as CCR8+GATA3+FOXP3+ cells, and-so-forth (27–29). Recent work by Levings and colleagues showed that it is possible to generate Treg cells in vitro that preferentially migrate to Th1-inflamed sites (30). In this study, interferon-gamma (IFN-γ) and IL-12 were added during Treg cell expansion in vitro, resulting in epigenetically stable Th1-like CXCR3+T-BET+FOXP3+ Treg cells (30). In a separate study, investigators discovered that a larger proportion of activated FOXP3hiCD45RAlo Treg cells in allogeneic hematopoietic stem cells was associated with less development of acute GvHD in bone marrow transplant patients (31). Utilizing tissue-specific and/or Th-like Treg cell subsets (29, 32) in therapies for autoimmune disorders requires more research. Yet, one can envision specific subsets of Treg cells being advantageous in scenarios where certain cytokine secretion patterns or tissue homing properties are instrumental for treatment efficaciousness.
Another category of Th-like Treg cells is follicular regulatory T (Tfr) cells, which inhibit follicular helper T (Tfh) cells. Tfr cells were first identified in mice and play a vital role in the germinal center response and antibody production (33). Tfr cells highly express CXCR5, and, similarly to Tfh cells, require CD28 and ICOS for their development and maintenance (34). In addition, while Tfh cells are derived from CD4+Foxp3- precursors, Tfr cells originate from CD25+Foxp3+ precursors. Recent studies have also shown that deficiencies in Tfr cells lead to antibody accumulation and the occurrence of a wide range of autoimmune diseases (35). Interestingly, when compared to healthy donors, coronavirus disease 2019 (COVID-19) convalescent patients with severe disease, which has been hypothesized to have an autoimmune-like component, had higher frequencies of effector memory Tfh cells and lower frequencies of central memory Tfh cells (36).
Based on the characteristics of the different subsets of Treg cells, different strategies can be adopted for the treatment of autoimmune diseases using them (Figure 1B). So far, results of treatments with Treg cells in the early stages of clinical trials for patients with transplant rejection, GvHD, and autoimmune disorders have been promising (37). However, one of the main challenges of these studies is the isolation of pure Treg cells and their expansion to an adequate amount for clinical applications, to reach the so-called clinical dose. Treg cell therapy is based on the idea that injecting of an efficient dose of Treg cells restores the balance between effector T cells and immune-regulatory cells (such as Treg cells) in favor of increasing immune tolerance. Some clinical studies have shown that Treg cell treatment can effectively and safely reduce autoimmune symptoms and organ transplant rejection (38). The first human clinical study for adaptive Treg transfer was reported by Trzonkowski and colleagues in 2009. Treg cells (CD4+CD25+CD127- cells) extracted from two donor families were expanded ex vivo and transferred to GvHD patients. Infusion of Treg cells significantly reduced disease symptoms in subjects with chronic GvHD and resulted in immunosuppression (39). However, in grade 4 acute GvHD, symptoms improved only temporarily. Recently, the adoptive transfer of Treg cells to SLE patients was investigated by Dall’Era and colleagues. Flow cytometry and whole transcriptome analyses revealed that accumulation of Treg cells in the skin subdued IFN-γ pathway and increased IL-17 pathway activity. This group reported the first case of adoptive transfer of Treg cells to SLE patients. In general, their results showed that this treatment led to an increase in Treg cells in the inflamed skin and a dynamically change in local immune response from Th1 to Th17 (40). Achieving good results using Treg cell adoptive transfer into patients requires methods to isolate and expand Treg cells from different sources with high efficiency and purity. In addition, there has been uncertainty about how well in vitro functional assays correlate with in vivo activity, and the complexity of modifying protocols to improve properties such as antigen specificity and homing receptor expression.
Treg cells are distributed almost throughout the body; peripheral blood and umbilical cord blood (UCB) are the most valuable sources for Treg cell isolation. Currently, the most common source for the production of autologous Treg products is peripheral blood. Yet, with the increased prevalence of cord blood banking, UCB may eventually become a more abundant and common source for autologous Treg cells (41). Indeed, using third-party UCB units as an allogeneic source of Treg cells for therapy is becoming more common. These products are enriched with naïve Treg cells, which have the potential to expand more than memory cells, with each UCB unit containing ca. 6 x 106 such cells (42). According to protocols laid out by Brunstein and colleagues, this number can be expanded in vitro more than 27,000 times to reach a clinical dose (43).
The pediatric thymus is another source of Treg cells that has recently received attention. Usually, the thymus is removed during pediatric cardiac surgery (44). The main advantage of using pediatric thymus is the high number of Treg cells in this organ, 100 times more than in a UCB unit, or about 500 x 106 cells. Furthermore, the almost complete absence of Tconv cells in the pediatric thymus has made it easier to purify Treg cells, significantly reducing the possibility of contamination with Tconv cells. Interestingly, thymus Treg cell (tTreg) immunosuppressive function is greater than that of Treg cells derived from peripheral blood or UCB, even in the presence of inflammation. Among other advantages of tTreg cells, it can be mentioned that they express an especially high level of CD25, which leads to high sensitivity to IL-2 stimulation even at low doses (45). As IL-2 also drives the activation of Tconv and natural killer cells (NK cells), stimulating a robust inflammatory response, it is advantageous that tTreg cells can be stimulated and expanded with IL-2 in levels at which Tconv cells and NK cells do not respond (46).
Other Treg sources include non-lymphatic tissues, namely intestines, lungs, joints, skin, and muscles. Although the purification methods for these Treg cells are laborious and low efficiency, efforts to expand tissue Tregs in the laboratory continue due to their unique characteristics (47). In parallel, efforts are underway to differentiate induced pluripotent stem cells (iPSC) into Tregs, as well as to convert Tconv cells into iTreg cells by inducing Foxp3 expression (48). For example, ectopic Foxp3 expression together with Notch signaling pathway activation by stromal cells made it possible to produce Treg cells from mouse iPSCs. This approach will most likely be used in gene therapy for IPEX (49). Alternatively, iTreg cells can be generated from CD4+CD25- Tconv cells in the presence of IL-2 and TGF-β. Currently, iTreg cells are in clinical trials (NCT 01634217) (50).
A critical point in cell therapy design is deciding whether to use autologous or allogeneic cells. With regards to autoimmune diseases and organ transplantation, all clinical trials thus far have used the patient’s autologous peripheral blood Treg cells. In contrast, allogeneic cells are commonly derived from UCB units with at least 4 HLA alleles in common with the recipient (51, 52). Although autologous Treg cells have the highest chance of being accepted by the recipient, their production can be very challenging. Autologous products must be manufactured uniquely for each patient, so manufacturing processes must be robust and reproducible despite the high variability between donors. On the other hand, the cost of producing autologous cells for each patient is very high; for this reason, attention has been drawn to allogeneic cell therapy products (51, 52). In addition, allogeneic cells can be derived from primary cell populations with less variability and, as a result, are more comprehensively under quality control to reduce patient risks. Studies on animal models have shown that allogeneic Treg cells have the same power as donor-derived Treg cells in preventing graft rejection (53). In humans, UCB-derived Treg cells were safe and reduced the incidence of acute and chronic GvHD in transplant patients (51, 52). Although allogeneic cell therapy products have only been investigated in immunocompromised people, their main limitation of allogeneic cells in healthy people is limited survival time due to rejection in the patient (54). In non-human primates, allogeneic Treg cells in the blood could not be detected by flow cytometry for more than 3 to 6 weeks after infusion (54). On the other hand, autologous Treg cells can be identified by mass spectrometry for more than a year after injection. Using a flow cytometry-based readout, the percentage of Tregs in circulation peaked 7-14 days post-transfer and fell near the detection limit within three months, similar to findings with allogeneic cells (40, 55). As a result, more research is needed to determine why the majority of infused Tregs appear to disappear from circulation in these various contexts.
One of the challenges is not determining the tolerance level for HLA mismatch in Treg cells. In studies of antiviral T therapy, third-party virus-specific T cells with the lowest HLA mismatch (one allele) reduced viral load, which can be exploited for the treatment of drug-resistant infections after hematopoietic stem cell transplantation (56). These results suggest that it is not necessary to fully match the HLAs to effectively transfer Treg cells. Of note, if the suppressive function of Treg cells is transferred to other cell populations, i.e. infectious tolerance, there is no need for the long-term survival of Treg cells (57). Allogeneic Treg products pose a risk of alloimmune sensitization, especially in GvHD patients and non-immunosuppressed patients. However, treatment with allogeneic specific Treg cell transfer after allogeneic hematopoietic stem cell transplantation did not lead to severe GvHD (58). Treg cells are generally less prone to allogeneic sensitization due to their role in suppressing the immune response compared to Tconv cell transfer. To increase the probability of allogeneic Treg cell survival in the patient, genetic modification can be used to remove or edit HLA molecules. Research in the regenerative medicine and stem cell field is driving the field of HLA editing, featuring strategies such as knocking out β-2 microglobulin to eliminate HLA class I surface expression) and/or CIIT2 to eliminate HLA class II (59). As such changes can render the cells susceptible to NK cell-mediated lysis, complementary strategies seek to increase the expression of non-classical HLA molecules to inhibit NK cells (59, 60).
Selecting between polyclonal Treg cells or antigen-specific Treg cells is another decision to be made when designing Treg cell therapies. Although producing polyclonal Treg cells requires less effort, a large number of them need to be injected for their effectiveness. On the other hand, only a small fraction of Treg cells is required for antigen-specific Treg cell strategy (61). Using antigen-specific Treg cells are reduced off-target suppression and increased potency. Putnam and co-workers expanded alloreactive Treg products by co-culturing recipient Treg cells with donor B cells (62). Clinical studies with alloreactive Tregs focus on solid organ transplantation and GvHD, namely NCT02711826, NCT01795573, NCT02188719, and NCT02244801.
To circumvent the rarity of antigen-specific Treg cells, methods of artificially generating antigen-specific Treg cells have been developed, including ectopic expression of a chimeric antigen receptor (CAR) or a T cell receptor (TCR), as discussed in later sections. TCR recognition is HLA-restricted and TCR affinity towards its cognate antigen is lower than that of CARs. Moreover, as recognition of antigens by CARs is not limited to HLA, CAR Treg cell therapy can target more diverse antigens, including protein, carbohydrate, and glycolipid antigens (63).
CD25 is present on all Treg cells, independently of their origin, and is a selective marker for Treg cell isolation (64). Instead of relying on CD25 expression alone, fluorescence-assisted cell sorting (FACS) CD45RA+CD25+CD127- naïve Treg cells increases Treg purity (65). Compared to magnetic selection, closed-system Good Manufacturing Practice (GMP) compatible FACS can simplify Treg cell isolation based on multiple markers. Miltenyi’s three-laser MACSQuant Tyto Cell Sorter allows for GMP-compatible FACS isolation of human Treg cells by utilizing single-use closed-cartridge systems, preventing contamination between samples and aerosol formation. Following isolation, Treg cells are expanded in vitro to reach clinical dose. Bead-immobilized antibodies, artificial antigen-presenting cells (APCs), and soluble antibody reagents are standard methods used to activate Treg cells in vitro.
Many Treg expansion protocols use magnetic beads covalently attached to anti-CD3 and anti-CD28 antibodies. However, removing the magnetic beads before infusion has limited their application due to cell loss and potential incomplete bead removal. With regards to artificial APCs, co-stimulatory molecules and an Fc receptor are expressed on the cell surface. Studies have shown that K562 cells expressing the co-stimulatory receptor CD86 and the high affinity Fc receptor CD64 and loaded with anti-CD3 antibody perform better than anti-CD3/CD28 beads to expand UCB Treg cells. K562 is a human myelogenous leukemia cell line that is devoid of HLA and CD80/CD86 and can be readily expanded. By loading these cells with anti-CD3 monoclonal antibody, the primary signals for Treg cell activation are triggered through binding to anti-CD3 sequestered by CD64 and to CD86. Because these artificial APCs are lethally irradiated, they gradually disappear from culture, obviating the need to eliminate them before injection (66). However, while artificial APCs are particularly successful in stimulating Treg growth, they complicate the cell production process by necessitating additional cell testing and batch validation. For ease of elimination before Treg administration to the patient, additional activation reagents are available in the soluble form. For instance, the T cell TransAct is a nanomatrix polymer conjugated to anti-CD3 and anti-CD28 antibodies. Since this reagent is soluble, it is easily removed by centrifugation (67). However, its use in clinical studies has not yet been reported. Overall, culture and expansion protocols for Treg cells for clinical use vary, with several research groups working towards reaching maximum efficiency (38).
Another critical point in the design of cellular therapy is the choice between fresh or cryopreserved Treg cells. Since cryopreservation provides several advantages, including the possibility of long-term storage of the product, increased time for injection, and hence more time for release testing, many researchers have turned to it. However, the reduction in the quality of Treg cells after cryopreservation is an important limitation of choosing this strategy for clinical applications. Studies on the decline of the quality of FOXP3+ Treg cells and CD25highCD127- Treg cells after cryopreservation have produced contradictory results (68). Although it has been reported that the expression of Foxp3, CD25, and the suppressive activity of Treg cells decreases after thawing, these characteristics can be restored after reactivation (69). Yet, few clinical trials using cryopreserved Treg cells have been reported due to uncertainty regarding the effects of freezing and thawing these cells (70). This and other parts of the Treg manufacturing process stand to benefit from advances in the production of Tconv cells for cancer immunotherapy (71).
One factor determining the effectiveness of Treg cells is their successful migration to both inflammation sites and lymph nodes (72). Therefore, many researchers are looking for protocols that, in addition to maximizing Treg stability, also increase their migration potential, which requires increasing the expression of homing receptors. For example, Hoeppli and colleagues reported that, by creating appropriate cell culture conditions, stable expression of homing receptors such as α4β7 and CXCR3 chemokine receptor can be induced (30). INF-γ and IL-2 addition during Treg cell expansion increased the expression of CXCR3, enabling Treg cell migration towards CXCL10 (30). Importantly, these cells maintained high expression levels of CXCR3 after injection into mice and in the absence of INF-γ and IL-2. Concomitant expression of CD62L and CCR7 further bestowed these cells will the potential to migrate to lymphoid tissues and the site of inflammation. Interestingly, Parmer and colleagues found that adding fucose to the surface of Treg cells induces sialyl-Lewis X moiety formation on the P-selectin ligand, increasing the viability of UCB Treg cells in GvHD likely by increasing the binding potential to E-selectin (73). Indeed, this strategy is being investigated in the NCT 02423915 phase I/II clinical trial, which assesses the effectiveness and safety of fucosylated Treg cells in reducing or preventing GvHD in humans.
In addition to optimizing culture conditions, two other strategies, creating antigen-specific Treg cells and increasing the expression of Treg-specific genes, can improve Treg cell function. For over a decade, studies have shown that antigen-specific Treg cells are more potent than their polyclonal counterparts. Infusing Treg cells with donor allospecific TCRs leads to long-term survival in mice with MHC-mismatched heart transplants (74). This notion is currently being utilized in the context of autoimmunity, where the restricted number of disease peptide-MHC complexes allows for the selection of a few TCRs to engineer Tregs compatible with a substantial proportion of afflicted people (75). One of the alternative approaches to TCRs is the engineering of Treg cells to express a CAR specific for the desired antigen. CARs are synthesized proteins with combined extracellular antigen binding domains, so-called single chain fragment variable (scFv), and intracellular signaling domains (63, 76). Preclinical studies in mice have demonstrated the promise of this strategy. For example, in the treatment of experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, CAR Treg successfully targeted the myelin glycoprotein of oligodendrocytes and ameliorated disease. Similar results were obtained in the treatment of colitis by targeting carcinoembryonic antigen (CEA). Further studies showed that human HLA-A2-specific CAR Treg cells perform better than polyclonal cells in preventing xenogeneic GvHD (77, 78).
Our increasing understanding of the role of Treg cells in autoimmune diseases has made Treg cell-targeted therapies promising strategies to alleviate these disorders. Two such strategies, low-dose interleukin-2 (IL-2) administration and Treg cell adoptive transfer, have received much attention in recent research and have been tested in clinical trials for several autoimmune diseases (40, 51, 52, 55, 79–82) (Table 1).
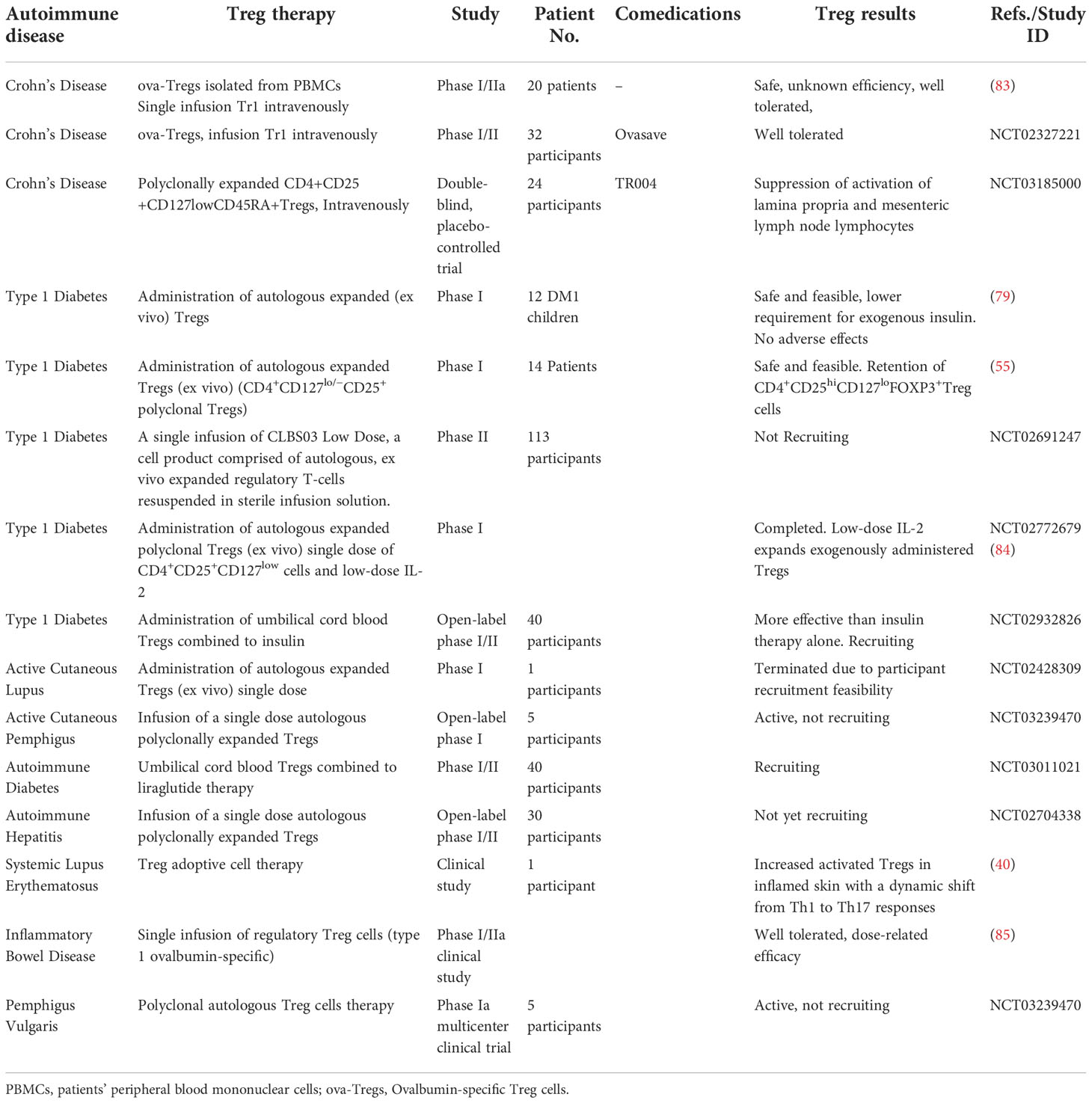
Table 1 Recent clinical studies on Treg cell therapy in autoimmune disease.
In its current form, Treg cell adoptive transfer strategy requires large Treg cell numbers to be clinically effective, in the order of 8 x 109 cells. For this purpose, Treg cells are collected from autologous peripheral blood (40, 79) or umbilical cord blood (51, 82) and expanded ex vivo using a number of protocols. As mentioned before, recent research points toward artificial APCs as key in increasing the number of tTreg cells obtained. In addition, FOXP3 expression and the suppressive function of tTreg cells are maintained by artificial APCs (86–88). Other research has shown the role of various biological compounds, including retinoic acid, IL-2, rapamycin, TGF-β (transforming growth factor-β), histone deacetylase (HDAC) inhibitors, DNA methyltransferase inhibitors, among others, in supplying Treg cells with functional and phenotypic stability (89–91). For instance, a study by Lu and colleagues determined that tTreg cells displayed higher expansion and suppressive function in vitro and in vivo following antagomir-mediated knockdown of miR-146b-5p (92). Another study elucidated the role of D-mannose in inducing FOXP3 expression and the conversion of naïve T cells into Treg cells (93). In addition, a systematic review by Dwivedi and colleagues indicates that prebiotics (substrates that support colonization by specific non-pathogenic bacteria) and probiotics (non-pathogenic bacteria or bacterial products found in food supplements) have a significant effect on the proliferation and induction of Treg cells in animal models and human cell cultures (94). More recently, Skartsis and colleagues showed that culturing human polyclonal Treg cells ex vivo in the presence of IL-6 and tumor necrosis factor alpha (TNF-α), two pleiotropic cytokines, in conjunction with CD28 super-agonist, dramatically boosts their proliferation while maintaining their phenotype and suppressive function in vitro and in vivo (95). Altogether, these and ongoing investigations provide practical methods to continue improving on the generation of adequate numbers of bona fide human Treg cells for adoptive cell therapy for the treatment of autoimmune diseases.
Treg cell adoptive therapy has been tested in various autoimmune diseases (5, 96). For instance, while SLE is characterized by unusual innate and adaptive immune responses, mounting evidence shows the essential role of Treg cells in this complex autoimmune disease, especially in its peak stages (97–100). In preclinical studies, Treg cell infusion in autoantibody-positive mice delayed renal complications and significantly increased their survival rate (101). In a case study reported by Dall’Era and colleagues, autologous Treg cells were amplified and administered to one SLE patient. The authors observed an increased percentage of activated Tregs in diseased skin (40). In phase I trials focused on T1D, patients treated with ex vivo expanded polyclonal Treg cells experienced minimal to no side effects. In one trial, the infused Treg cells were labeled with deuterium, allowing their tracking in peripheral blood. The authors found that a small fraction of the infused Tregs could be detected in peripheral blood up to one year after treatment and they maintained a Treg phenotype, suggesting that there was no Treg instability (55). Encouragingly, in some of the trials there was evidence that treatment of T1D patients with Treg cells can increase β cell survival and C-peptide levels, concomitantly reducing dependence on exogenous insulin (55, 79, 80, 102). Of note, these studies were informed by pioneering studies using human Treg cells for the treatment of graft-versus-host disease (GvHD), in which safety and efficacy were demonstrated over the years (39, 51, 81, 103, 104).
Several strategies to improve on the quality and induction of Treg cells used for therapy are currently being tested. For instance, Kasahara and colleagues (105) set out to optimize the generation of stable induced Tregs (iTregs) to prevent GvHD in mice, where traditional iTregs have been shown to be ineffective. Co-culturing naïve T cells with allogeneic dendritic cells in the presence of TGF-β and retinoic acid resulted in alloantigen-specific iTregs. Interestingly, vitamin C stabilized Foxp3 expression in adoptively transplanted iTregs in a GvHD environment. Indeed, vitamin C therapy triggered active DNA demethylation, particularly at the conserved non-coding sequence 2 (CNS2) enhancer of the Foxp3 gene locus, reducing iTreg conversion to pro-inflammatory ex-Treg cells. GvHD symptoms were suppressed more effectively in vitamin C-treated iTregs than in untreated iTregs. Importantly, Vitamin C also boosted the generation of FOXP3high iTreg population from human naïve T cells in vitro, which remained stable even when exposed to IL-6. Vitamin C treatment is thus a promising molecule in adoptive Treg cell immunotherapy (105). In another study, Kasagi and colleagues (106) focused on mice with experimental autoimmune encephalomyelitis (EAE, a model for MS) and autoimmune diabetes (nonobese diabetic – NOD mice) and identified a mechanism to create autoantigen-specific Treg cells in vivo. In brief, the authors used either systemic sublethal irradiation to cause immune cell death or monoclonal antibodies to deplete B and CD8+ T cells in animals with established autoimmune disorders, and then administered self-antigen-derived peptides. Interestingly, these peptides drove naïve CD4+ T cells into a Foxp3+ Treg cell fate instead of Teff cells. Mechanistically, apoptotic cells stimulated professional phagocytes to release TGF-β, which in turn promoted induced Treg cell generation. Strikingly, these de novo generated antigen-specific Treg cells reduced autoimmunity while maintaining immune responses to bacterial antigens. Independently, Sun and co-workers (107) developed a method for producing antigen-specific Treg cells by culturing murine CD4+ T cells with retinoic acid; adoptive transfer of these cells reversed the progression of collagen-induced arthritis (CIA) in mice by suppressing TNF-α. Further testing revealed that the Treg cells remained stable in vivo after infusion. Altogether, these techniques aimed at augmenting Treg cell numbers and/or function might one day be used in the treatment of autoimmunity in humans (Table 1).
Antigen-specific Treg cells perform better than polyclonal Treg cells in the prevention and treatment of autoimmune disease in animal models (108). This observation has catalyzed the development of various methods to generate antigen-specific Treg cells, such as overexpression of TCRs (109, 110), antigen-stimulated expansion (111, 112), and the utilization of CARs (77). Recent preclinical studies have shown the high potential of antigen-specific Treg cells in the treatment of various autoimmune diseases (83, 105–107). CAR Tregs, in particular, have received much attention for the immunotherapy of autoimmune disease (5). The first reports of engineered Tregs, in 2005, focused on EAE. Transgenic mice were generated expressing a chimeric receptor comprising an MHC complex bound to an EAE peptide, MBP89-101, linked to an intracellular CD3ζ chain. Adoptive transfer of engineered Tregs from these transgenic mice not only prevented MBP89-101-induced EAE, but also treated it one month post-induction, after epitope spreading had occurred, demonstrating the bystander suppression capacity of these cells (113). Follow-up studies by the same group elucidated that these engineered Tregs induced MBP89-101-specific T cells to secrete IL-10. Adoptive transfer of such non-transgenic MBP89-101-specific T cells prevented EAE in recipient mice, indicating that these engineered Tregs could also induce infectious tolerance (114). More than a decade after those initial mouse studies, human Tregs engineered with CARs were reported. Given the difficulties of modeling autoimmune disease in humanized mice, these studies targeted GvHD and organ transplant rejection instead. The first one, published in 2016, generated CAR Tregs redirected against HLA-A2 using a CAR comprising an extracellular domain with an anti-HLA-A2 scFv and an intracellular domain with a tandem CD28-CD3ζ signaling domain (77). In this study, HLA-A2 CAR Tregs successfully prevented xenogeneic GvHD (xGvHD) induction by co-injected HLA-A2-expressing PBMCs in NSG mice. Two subsequent studies by different groups confirmed this finding and further demonstrated the capacity of HLA-A2 CAR Tregs to suppress a mixed lymphocyte reaction (MLR) between HLA-A2-positive and HLA-A2-negative PBMCs in vivo (measured by mouse ear swelling) and protect HLA-A2-expressing human skin grafts from rejection by HLA-A2-negative PBMCs in NSG mice (115, 116).
The last two years have seen some engineered cell studies focused on T1D. In the NOD mouse, a different group engineered NOD CD4+ T cells by constitutively expressing Foxp3, making them phenotypically similar to Treg cells in vitro, and a CAR activated by insulin. Yet, despite being detectable up to 17 weeks post transfer in vivo, these “converted” insulin CAR Foxp3+ T cells did not prevent diabetes in NOD mice (117). Most recently, human Tregs bearing a humanized HLA-A2 CAR were shown to protect NSG mice from xGvHD when HLA-A2 was present either in the co-infused PBMCs or in the mouse host (HLA-A2 transgenic NSG mice), a closer recapitulation of the human disease (78). Moreover, these HLA-A2 CAR Tregs trafficked to HLA-A2-expressing mouse or human islets transplanted in the right kidney capsule of NSG mice rendered diabetic with streptozotocin (STZ) treatment and did not impair their function, whereas conventional T cells engineered in the same fashion rejected the transplanted islets in less than 2 weeks (78). Next steps include showing efficacious islet protection and diabetes prevention/reversal in NOD and more sophisticated human immune system (HIS) mouse models (118).
Low-dose IL-2 therapy can be seen as a strategy to fight various autoimmune diseases complementary to Treg cell infusion. CD25, the IL-2 receptor (IL-2R) α chain, is constitutively expressed at high levels by Treg cells. The IL-2R α chain, along with the β (CD122) and γ (CD132) chains, make up the high affinity IL-2R. In general, IL-2 is an essential factor for the growth of lymphocytes. Indeed, IL-2 has been FDA approved for use in cancer immunotherapy since the 1990s, to stimulate anti-tumor Teff cells and natural killer (NK) cells. Yet, the high doses of IL-2 required for tumor regression are commonly accompanied by severe side effects, such as vascular leak syndrome (119). In addition, IL-2 plays a particularly vital role in the division, activity, and stability of Treg cells. Importantly, unlike Teff cells, Treg cells do not secrete IL-2 and are thus fully dependent on exogenous sources of this cytokine (120–122). Encouragingly, low doses of IL-2, in the range of 0.33-4.5 x 106 international units (IU), selectively enhance the development and maintenance of Treg cells without Teff cell activation. These observations led investigators to posit that low-dose IL-2 would be an effective treatment for autoimmunity by boosting Treg cells (123). Enhancing Treg: Teff balance in lupus-prone mice treated with recombinant IL-2 reduced disease progression and increased survival rate (98). In a study by Johnson and colleagues, NOD mice treated with IL-2 experienced an increase in the number and proportion of Treg cells (124). In T1D patients, administration of low dose IL-2 has no serious side effects, and only mild to moderate side effects, such as inflammatory reactions at the injection site and influenza syndrome, have been reported (125–127). Although low doses of IL-2 increase Treg cells in T1D, they also increase the number of other cells expressing IL-2 receptor, such as eosinophils and NK cells (127). Indeed, a recent study by Dong and colleagues where T1D patients were treated with autologous polyclonal Treg cells followed by one or two courses of low-dose IL-2 revealed that, while IL-2 increased the numbers of both infused and endogenous Treg cells, it also boosted subsets of NK cells and CD8+ T cells (84). Hence, more research is needed to corroborate the use of this strategy to overcome autoimmune diseases. With regards to GvHD, numerous investigations showed that treatment with low-dose IL-2 is a beneficial regimen for achieving tolerance and reducing the risk of GvHD (128, 129). During this therapy strategy, however, NK cells were selectively expanded (121). Low-dose recombinant IL-2 treatment to active SLE patients resulted in substantial Treg cell growth, improved Treg function in peripheral blood, and clearly decreased disease activity (130–132). In a pilot study in 37 SLE patients, low-dose IL-2 increased the number of Treg cells and subsequently reduced the SLE disease activity index scores (133). A research in primary Sjogren’s syndrome reached the same outcome, confirming the unique therapeutic impact of low-dose IL-2 on immune-related diseases (134). In summary, low-dose IL-2 treatment, while promising, requires significant investigation before it can be safely employed in clinical practice. More studies are needed to determine the safe and effective dose range, including cumulative exposure, unwanted side effects, and whether there may be long-term complications. Also required are double-blind, placebo-controlled randomized trials, as well as basic research into the underlying mechanisms of IL-2 therapy. Efforts are underway to improve low-dose IL-2 therapy specificity. In one vein, investigators are mutating IL-2, generating muteins with reduced affinity to IL-2Rβγ, thus further increasing their selectivity towards cells expressing very high levels of CD25, i.e. Treg cells (135). Of note, recent intriguing work in the mouse aimed at circumventing the issue of specificity of IL-2 by generating an artificial molecule, ortho-IL-2, which only binds to an artificial receptor, ortho-IL-2R, allowing for the selective maintenance and expansion of Treg cells engineered to express ortho-IL-2R (136).
Understanding the mechanisms behind Treg cell function and number, and the pathways regulating them has yielded promising results for autoimmune disease therapy. T cell differentiation and function is highly dependent on the function of the mammalian target of rapamycin (mTOR) signaling pathway, the major nutrient-sensing regulator of cell growth, with rapamycin being a natural inhibitor of this pathway (90, 137). Studies have demonstrated that rapamycin can improve Treg cell lineage and functional stability. This stems in part from the differential regulation of the phosphatidylinositol 3-kinase (PI3K)/AKT pathway in conventional T (Tconv) cells vs. Treg cells: Tconv cells donwregulate phosphatase and tensin homologue on chromosome 10 (PTEN), a negative regulator of the PI3K/Akt pathway, which together with mTOR controls cell growth, whereas Treg cells do not. Indeed, genetically deleting PTEN specifically in Treg cells in the mouse results in Treg cell destabilization, loss of Foxp3 expression, and Th1-driven autoimmunity (138). Moreover, mTOR defects disrupt the differentiation of naïve T cells into Th1, Th2, and Th17 cells, thus favoring naïve T cell differentiation into Treg cells (139). Hence, rapamycin-mediated inhibition of mTOR disproportionally negatively affects the growth and proliferation of Tconv cells compared with Treg cells, resulting in the inclusion of rapamycin in many Treg cell ex vivo expansion protocols (70). Recent studies on the use of rapamycin in model animals with a variety of autoimmune diseases, such as T1D (140), autoimmune pancreatitis (141), and SLE (142), show significant expansion of Treg cells and improvement in disease symptoms. In humans, it should be noted that rapamycin treatment in SLE patients has been shown to be safe and effective (143). With regards to IPEX (144), rapamycin restores Treg cell function, including upregulation of immune-regulating proteins such as glucocorticoid-induced TNFR-related protein (GITR). Furthermore, rapamycin treatment positively impacts the histological and clinical progression of IPEX (145).
The epigenetic regulation of T-cell-mediated immunity is also beginning to be targeted in the context of Treg cells and autoimmune disease. For instance, histone deacetylase (HDAC) inhibitors have been shown to have anti-inflammatory effects and can be potential treatments for autoimmune disease patients (146), enhancing Treg cell function of Treg cells in vitro and in vivo (147). Studies have also shown that HDAC11 inhibition augments Treg suppressive function and prolongs long-term allograft survival in mice (148). Choi and colleagues reported that administration of vorinostat, an HDAC inhibitor, considerably increased the Treg cell number and FOXP3 expression in patients with hematological malignancies who received allogeneic hematopoietic stem cell transplantation (149). Severe reduction in GvHD incidence, increased Treg cell function, and inhibition of Th17 cell differentiation are the other effects of HDAC inhibitors. Interestingly, culturing RA patient-derived PBMCs with lipopolysaccharide (LPS) and HDAC inhibitors in vitro resulted in a greater percentage of induced Treg cells and higher IL-10 production (150), implying that HDAC inhibitors could be effective in the treatment of RA.
In a complex and dynamic process, by balancing between cell division and apoptosis, B cells develop in the bone marrow and mature into functional B cells, the central players in humoral immunity (151). At the turn of the 21st century, the concept of regulatory B cells (Breg cells) was described by Bhan and Mizoguchi, referring to a subset of B cells with regulatory characteristics (152). Breg cells, similarly to Treg cells, exert their function by secreting regulatory cytokines such as TGF-β and IL-10. In addition, Breg cells can express inhibitory molecules which suppress autoreactive B cells and pathogenic T cells in a cell contact-dependent manner (153). Under the appropriate stimulation conditions and times, all types of B cells can differentiate into Breg cells. Unsurprisingly, Breg cell populations are heterogeneous (154) and the suppressive functions of Breg cells are executed through different mechanisms in different autoimmune disease models (155). In addition, it has been shown that dynamic changes in Breg cells are correlated with the development of autoimmune diseases in humans (156, 157) (Figure 2).
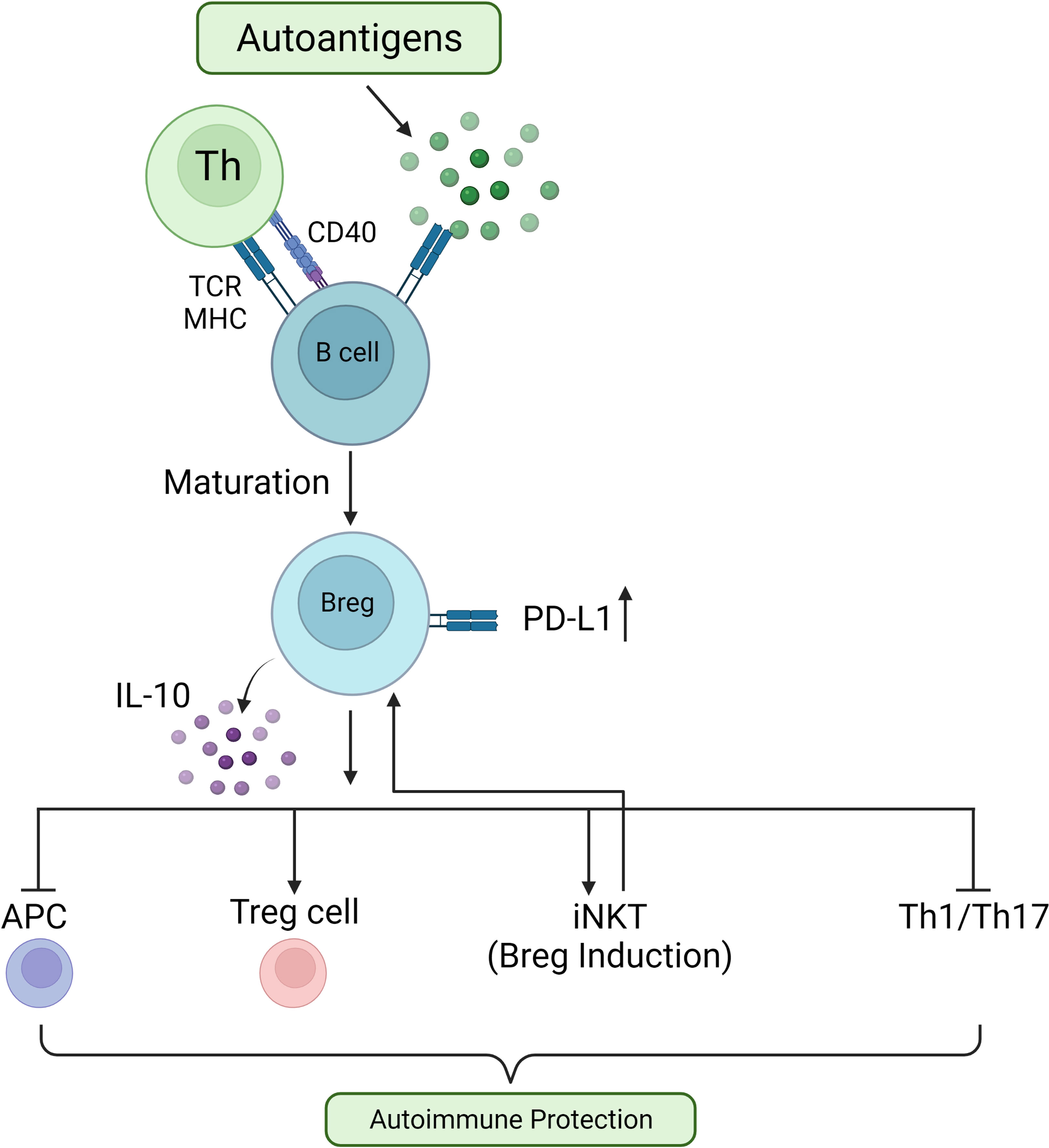
Figure 2 Role of Breg cells in preventing autoimmune reactions. In response to autoantigens, autoreactive B cells interact with T helper (Th) cells via TCR/MHC and CD40L/CD40, driving B cell differentiation into Breg cells. These IL-10-secreting Breg cells quell inflammatory responses by inhibiting APCs, increasing Treg cell activity, increasing invariant Natural Killer-like T (iNKT) cell numbers, and suppressing Th1 and Th17 cell activities.
So far, different mechanisms have been described through which Breg cells exert their impact on the immune response directly or indirectly. In mice, for example, two subsets of Breg cells have been identified. The first is a B10 cell subset (IL-10-producing B cells), characterized as CD19hiCD1dhiCD5+ cells, and the second is a T2-MZP cell subset (splenic transitional 2-marginal zone precursor), identified as CD19+CD23+CD21+CD1dhi cells (158, 159). Both Breg cell subsets secrete IL-10, inhibiting T-cell proliferation and Th1 cytokine production (IFN-γ and TNF-α) (159, 160). Studies in animal models have shown that transferring a small number of in vitro expanded Breg cells suffices to protect the recipient animals against various autoimmune diseases for a long period of time. This suggests that Breg cells either divide in vivo and/or can create an efficient immunosuppressive cascade with other immunosuppressive cells (161). In addition to suppressing Th1-mediated immune responses, B cells induce the generation of regulatory Tr1 cells from Teff cells (162, 163). Gray and colleagues reported that apoptotic cells (ACs) can induce T and B cells to secrete IL-10 (163). In addition, it has been shown that IL-10 derived from B cells is essential for the induction of IL-10-secreting T cells. Breg cell function is thus critical to maintaining immune homeostasis and tolerance (164). However, Breg cells make up only 10% of the circulating B cells in a healthy human (165). Because B cell-induced suppression is primarily mediated by IL-10 production, IL-10 is the main marker used to identify Breg cells. Yet, IL-10-producing B cells can be found as part of B cell populations with distinct surface markers that perform different functions (165). These subsets include CD24hiCD27+ B cells, IL-10-producing CD24hiCD38hi B cells, CD27intCD38hi plasmablasts, CD19+TIM1+ B cells, and CD38+CD1d+IgM+CD147+GrB+ B cells. All of these cells suppress pro-inflammatory responses (156, 157, 166). Interestingly, however, only approximately 20% of B cells in each of these B cell subgroups generate IL-10. For instance, IL-10-producing B cells were found to make up 21% of CD24hiCD27+ B cells, as compared to only 1% of non-CD24hiCD27+ B cells (167). Conversely, IL-10-producing Breg cells often express different levels of CD24, CD27, and CD1d.
Breg cells inhibit Th1 cell responses and Th17 cell differentiation. In addition, they convert naïve CD4+ T cells into Tr1 and Treg cells (156, 168). Though IL-10 is the main cytokine involved in Breg-mediated suppression, interaction with CD80 and CD86 on the surface of Breg cells improves inhibition of Th1 responses (156, 168). According to recent research, Breg cells are involved in immune responses to infections, cancer, and autoimmune diseases. For example, Breg1 cells, characterized as CD25hiCD71hiCD73lo B cells, maintain allergen tolerance by producing allergy-specific IgG4 antibodies and by suppressing allergen-specific T cell proliferation (169).
Importantly, Breg cells, similarly to other immunosuppressive cells, can arise via differentiation into induced Breg (iBreg) cells in response to various stimuli. T cells expressing cytotoxic T lymphocyte–associated protein 4 (CTLA-4) have been shown to promote the differentiation of a population of iBreg cells that modulate immune responses by producing TGF-β and indoleamine 2,3-dioxygenase (IDO), converting naïve T cells into TGF-β- and IL-10-producing Treg cells (170). Albeit the exact role of CTLA-4+ T cells in Breg differentiation is unknown, it is conceivable that this Breg cell expansion mechanism occurs in vivo to prevent severe inflammation. In addition, CD1d-mediated lipid antigen presentation by IL-10–producing B cells (CD24hiCD38hi Breg cells) is critical for preserving the number and activity of invariant Natural Killer-like T (iNKT) cells, a cell subset with immunosuppressive properties (171). CD39+CD73+ Breg cells are responsible for the transition from an adenosine triphosphate (ATP) driven pro-inflammatory environment to an adenosine-induced anti-inflammatory environment. Exogenous ATP can be hydrolyzed to adenosine 5′-monophosphate (AMP) and adenosine by the concerted action of the ectonucleotidases CD39 and CD73 (172). In vitro, CD39+CD73+ Breg cells limit CD4+ and CD8+ T cell proliferation by producing 5′-AMP following activation with CD40L and IL-4 (172). Interestingly, IL-35 has been demonstrated to be essential to murine Breg cell-mediated suppression. Mice deficient in IL-35 specifically in B cells exhibited worse illness and enhanced resistance to Salmonella infection in EAE (173). In a mouse model of experimental autoimmune uveitis, IL-35-induced Breg cells halted disease progression (174). Still, a role for IL-35 in Breg cells in humans is yet to be uncovered. Altogether, Breg cells are increasingly thought to have a complex function in immunological regulation, with a rising number of inhibitory mechanisms attributed to them.
Breg cells are an IL-10 producing anti-inflammatory B cell subset. However, there are no definitive cell surface markers or lineage-defining transcription factors that define Breg cells, potentially limiting the therapeutic potential of Breg cells. Although IL-10 expression has been beneficial in identifying populations of suppressive B cells in mice and humans, many surface markers used to classify Breg cells are down- or upnregulated during inflammatory responses, resulting in inherent problems in the description of different Breg cell subsets across different experimental settings, which may account for some of the discrepancies in described Breg cell subsets. As a consequence of the variety of Breg cell subsets, identifying a Breg-cell specific transcription factor, analogous to Foxp3 in Treg cells, has been a major difficulty in Breg cell - based therapies (175). The discovery of such a chemical would assist to resolve the phenotypic of Breg cells and address the question of whether these cells constitute a different lineage. So far, two models of Breg cell formation have been proposed. The first is that, like thymus-derived Treg cells, Breg cells are a distinct lineage of B cells in which a specific factor regulates the expression of genes responsible for their suppressive character. The second is that in response to certain stimuli, B cells adopt a regulatory phenotype in order to reduce local inflammation. Despite significant effort, no study using gene arrays on Breg cells in both mice and humans has clearly discovered a lineage specific marker analogous to Foxp3 (169, 173). The inability to find a distinct transcription factor, along with the variety of the phenotypic of Breg cells, supports the hypothesis that suppressor B cells are “reactive,” rather than lineage specific. In contrast to natural Treg cells, any B cell might possibly develop into a “Breg” cell in response to the correct environmental signals (176).
Abundant evidence now indicates that defects in the number and/or function of Breg cells are associated with various autoimmune diseases in mice, with severity of autoimmunity being inversely correlated with the number of Breg cells (156, 168, 171, 177, 178). Regarding autoimmune diseases, two main hypotheses are proposed for the role of Breg cells. First, inflammation is exacerbated by the lack of immunological suppression caused by Breg cells. A second possibility is that chronic inflammation is the cause of decreased Breg cell numbers and function. Currently, human studies on Breg cells are very limited, and evidence for both theories is largely derived from studies in animal models. When compared to wild-type (WT) mice, chimeric mice with an IL-10 deficiency specifically in B cells suffer from worse arthritis and EAE, accompanied by increased Th1 or Th17 cell responses, respectively (179, 180). Adoptive transfer of mouse Breg cell subgroups has also been proven to inhibit autoimmune disorders, such EAE, arthritis, and lupus (162, 180, 181). Of note, inflammatory cytokines, such as IL-6, IL-21, IL-1β, IFN-α, IFN-β and B cell-activating factor (BAFF), induce Breg cell expansion (160, 182–186). Activation of CD40 or TLR can increase this effect (182, 187, 188). Notwithstanding, chronic exposure of B cells to high doses of proinflammatory cytokines causes a decrease in functional Breg cells (182). Curiously, commensal bacteria are also involved in Breg cell expansion. In osteoarthritis mice, IL-1β and IL-6, whose secretion is influenced by the intestinal microbiota, directly increase Breg cell differentiation and IL-10 production. In agreement with a role for the microbiota, mice treated with antibiotics displayed a significant reduction in Breg cells (183).
New therapies, including those directly targeting Breg cells, are starting to address ambiguities regarding the role of Breg cells in autoimmune disease in humans. Suppressive B cells were first reported in MS patients. Helminth-infected MS patients showed a higher number of IL-10–producing CD19+CD1dhi cells that were associated with a better clinical outcome. T cell proliferation and IFN-γ production were inhibited by B cells derived from MS patients with helminth infections (189). Although it is not easily possible to determine the exact mechanism of action in human studies, it was suggested that the disease symptoms in MS patients were reduced due to the expansion of IL-10-producing B cells – Breg cells. In addition, there is a significant decrease in IL-10-producing B cells in patients with relapsing-remitting MS (RRMS) compared with recovering patients, as well as healthy individuals (177). It is noteworthy that treatment with IFN-β for RRMS patients expands CD24hiCD38hi Breg cells in these patients (177, 185). In EAE studies, while WT mice responded to IFN-β treatment, B cell-deficient mice did not respond to the treatment regimen, indicating that Breg cells are critical for the efficacy of IFN-β treatment also in the mouse (185). Other studies have reported the development of Breg cells in MS patients treated with alemtuzumab (an anti-CD52 therapy) and fingolimod (a sphingosine-1-phosphate modulator) (190–192). Immunomodulatory therapies in MS patients may thus operate, at least in part, by promoting a change in B cells toward an anti-inflammatory Breg cell phenotype.
Several studies on the role of Breg cells in SLE have revealed that SLE patients have defects in the number and function of circulating Breg cells, resulting from a lack of differentiation of immature CD19+CD24hiCD38hi cells into Breg cells (156, 182, 193, 194). Immature B cells in SLE patients do not respond properly to known signals for Breg cell differentiation: while healthy immature B cells differentiate into Breg cells upon CD40 stimulation, CD24hiCD38hi B cells isolated from SLE patients do not produce IL-10 following CD40 activation and are unable to suppress Th1 responses (156, 193). Moreover, SLE patients’ abundant CD19+FSChi polyclonally activated B cells (iBreg cells) have a considerably decreased capacity to inhibit Th cell responses when compared to the same B cell subset in healthy people (194). In addition, TLR9-activated pDCs cause a marked increase in immunosuppressive IL-10–producing CD24+CD38hi Breg cells in healthy people, but not in SLE patients (182).
A study by Borja and colleagues demonstrated that the number of CD24hiCD38hi Breg cells is lower in RA patients than in healthy individuals, rendering them unable to suppress Th17 cell responses and convert naïve CD4+ cells into Treg cells. Indeed, the number of Breg cells in RA patients is negatively correlated with the activity and severity of the disease. In the same vein, recent studies have shown that the levels of B10, IL-10+CD5+CD1dhi B cells, and IL-10+TIM1+ B cells are higher in healthy people than in RA patients (168). However, in a study by Kim and colleagues (195), the number of IL-10+ Breg cells in RA patients was higher than in healthy individuals. The discrepancies across the studies are most likely related to the stimuli utilized to induce IL-10 production by B cells in vitro. The research that showed a decrease in IL-10+ Breg cells employed either TLR or CD40 activation of B cells, whereas the study that showed an increase used CD40 ligation in conjunction with TLR activation. More research is thus needed to consolidate the role of Breg cells in RA.
CD24hiCD38hi B cells from individuals with pemphigus produced less IL-10 after long-term stimulation and had a worse capacity for inhibiting Th1 responses (196, 197). In contrast to untreated patients or patients who did not react to therapy, pemphigus patients who responded to treatment with the B cell-depleting agent rituximab had higher frequencies of CD24hiCD38hi B cells and IL-10 production (198). It is possible that Breg cells can help people with rituximab-resistant pemphigus regain tolerance. IL-10-producing Breg cells are considerably decreased in frequency in individuals with CD and ulcerative colitis when compared to healthy controls (199). Lower levels of suppressive IL-10–producing Breg cells have also been linked to disease development in individuals with psoriasis (178), systemic sclerosis (200, 201), and T1D (202). While more research is needed to fully comprehend the mechanisms of Breg cell-mediated suppression in these disorders, there is enough evidence to conclude that Breg cells are numerically deficient in autoimmune diseases, a factor likely contributing to loss of immunological tolerance (203). The few clinical studies on the role of Breg cells in treating autoimmune disorders are summarized in Table 2.

Table 2 Effect of Breg cells on autoimmune symptoms.
Unlike most current drugs for immune-related diseases, which targeting the symptoms of the disease and often cause widespread toxicity in the long term, cellular immunotherapy aims to accurately target and/or modify the immune cells that lead to disease progression. As this therapeutic strategy has been effective in treating several types of cancer, scientists are commencing to utilize it to treat autoimmune diseases (204). As discussed in the previous section, Breg cells seem to play a role in modulating immune responses and fostering immune tolerance. Therefore, strategies to isolate, expand, and infuse Breg cells, or otherwise expand endogenous Breg cells, can open new windows for autoimmune disease therapy (205). B cell-depletion drugs such as rituximab, an anti-CD20 monoclonal antibody, have yielded promising results in the treatment of autoimmune diseases (206, 207). For instance, the use of rituximab for treatment of experimental autoimmune vasculitis has demonstrated that it depletes B cells by inducing B cell apoptosis, enhancing Treg cells’ immunomodulatory capacity via IL-10 (208). However, removing all B cells also eliminates Breg cells that suppress inflammation. Hence, there is interest in targeting specific subsets of effector B (Beff) cells or Breg cells for depletion. However, although there are surface markers that can be used alone or in combination to enrich for IL-10-producing Breg cells, their specificity is not very reliable. The lack of specific markers for Breg cells is the main challenge to realize such treatment strategy (156, 157, 166, 209).
Dendritic cells (DCs), first described in the 1970s by Ralph Steinmann (210), are professional APCs (211). Conventional DCs stimulate naïve T cells and are often more potent APCs than macrophages or B cells. Yet, some subsets of DCs in central and peripheral lymphoid organs instead induce tolerance or antigen-specific unresponsiveness (212). These subsets of DCs are collectively known as tolerogenic or regulatory DCs (DCreg) (213). Low surface expression of major histocompatibility complex (MHC) and co-stimulatory molecules is the main feature of DCreg cells, which leads to their weak capacity to induce Teff cells. Indeed, DCreg cells’ roles are to induce autoreactive T cell anergy and Treg cell differentiation, contributing to the maintenance of immune tolerance (214). DCreg cells have been utilized to treat GvHD (215) and autoimmune diseases (216). Yet, as with Treg cell-based therapies, the lack of abundant and sustained sources of the required numbers of DCreg cells is a major obstacle to the clinical use of these cells. Therefore, recent research has focused on finding ways to produce more and purer DCreg cells (217).
As mentioned above, various DC subtypes play a vital role in maintaining immune homeostasis. The regulatory potential of DCreg cells relies on their immature status and is induced by signals from tissues (including the tumor microenvironment), apoptotic cells, and other immune cells (218). Immunosuppressive mediators, pathogenic stimuli, and genetic manipulation can also induce regulatory function in DCs. DCreg cells, although weakly, maintain the capacity to present antigen to T cells. In addition, they reduce co-stimulatory molecule expression (CD40, CD80, and CD86), as well as production of proinflammatory cytokines, such as IL-12. Simultaneously, DCreg cells increase their levels of inhibitory molecules (IDO, PD-L1, and CD95L) and anti-inflammatory cytokines (IL-10, TGF-β). Furthermore, DCreg cells are resistant to maturation signals (219). DCreg cells enhance immune tolerance through various mechanisms. These mechanisms include Treg cell generation, T cell apoptosis induction, T cell unresponsiveness induction (anergy), and inhibition of T cell responses (220).
Various strategies have been described to generate tolerogenic DCreg cells in mice and humans. Murine bone marrow precursors (221) and human peripheral blood monocytes (222) are the most common cell sources for DCreg cell production. Thus far, different conditions for the production of DCs have been described, with the combination of granulocyte macrophage colony-stimulating factor (GM-CSF) with IL-4 being the most frequent approach (223). In vitro, DC exposure to anti-inflammatory agents, either natural or pharmacological, leads to the acquisition of DCreg cell properties (Figure 3).
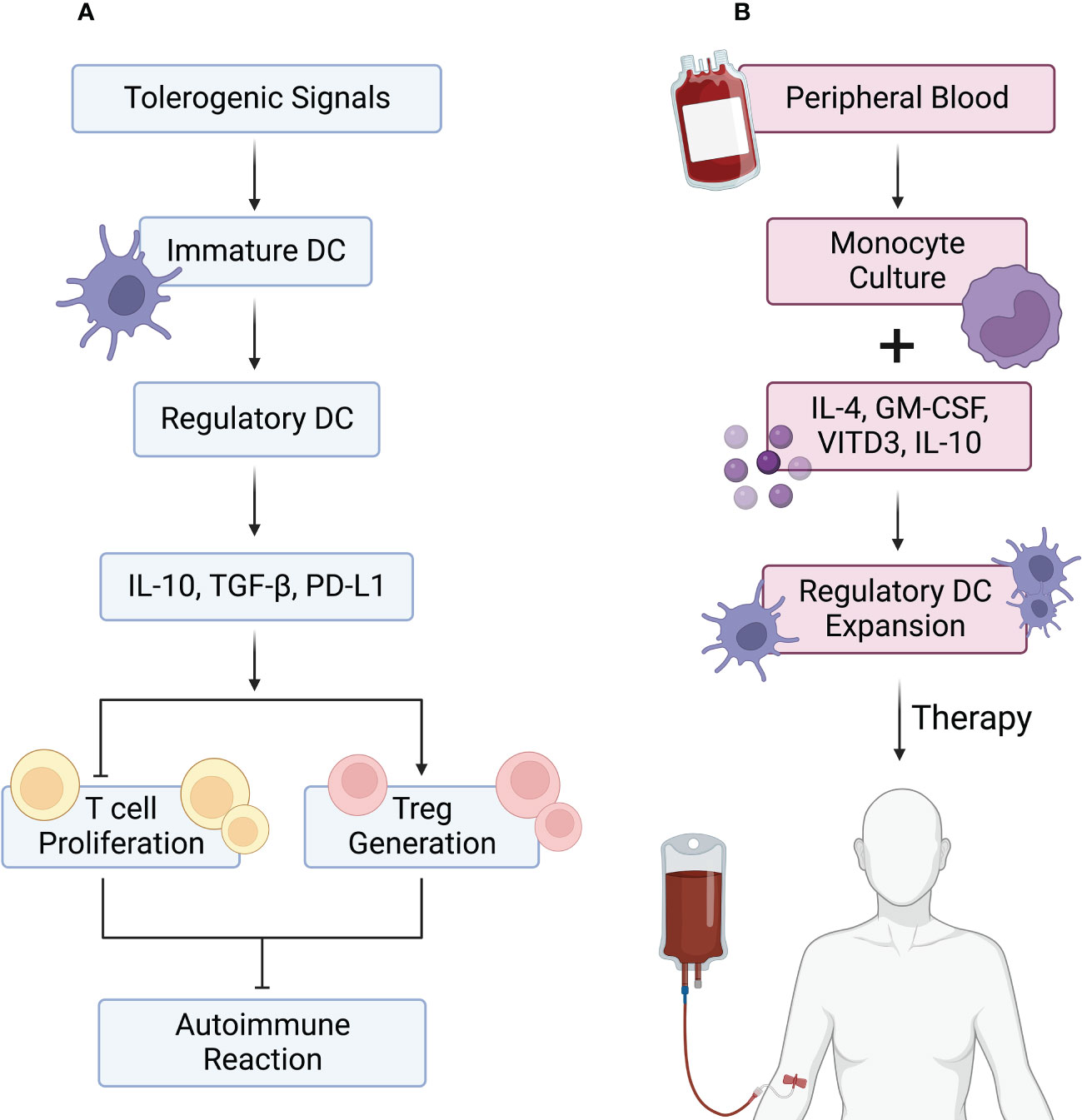
Figure 3 DCreg cells in autoimmune reactions and their use in autoimmune disease therapy. (A) Tolerogenic signals, including anti-inflammatory cytokines and apoptotic cells, lead to the differentiation of immature dendritic cells (DCs) into regulatory DCs (DCreg cells). DCreg cells are characterized by PD-L1 expression and IL-10 and TGF-β production. These properties allow DCreg cells to block effector T (Teff) cell proliferation while inducing regulatory T (Treg) cell differentiation, leading to inhibition of autoimmune reactions. (B) DCreg cells can be generated for therapy by isolating human peripheral blood monocytes and culturing them with IL-4, GM-CSF, vitamin D3 (vit D3), and IL-10. The resulting DCreg cells are then expanded in vitro and infused into patients.
DCreg cell differentiation can be induced in vitro by a variety of biomolecules encountered under tolerogenic circumstances in vivo. In models of organ allograft rejection, allergies, and GvHD, for example, incubating DCs with IL-10 bestows them the ability to induce Treg cell generation (224, 225). Interestingly, DCs remain immature with IL-10 signaling, even in the presence of maturation signals, allowing DCs to reduce the neuropathology associated with EAE (226, 227). Treatment of human DCs with pro-inflammatory stimulants and the active form of vitamin D (1,25-dihydroxyvitamin D3) forces them to express a variety of factors key to immune tolerance induction, including TRAIL (tumor necrosis factor (TNF) receptor apoptosis-inducing ligand), IDO, IL-10, inhibitory receptors CYP24A1 and CD300LF, TGF-β, and CCL2 (228). Numerous other factors, including vasoactive intestinal peptide (VIP), thymic stromal lymphopoietin (TSLP), estrogen, GM-CSF, binding immunoglobulin protein (BiP), TNF-α, and prostaglandin (PG) E2, are involved in inducing the ability of DCreg cells to produce Treg cells (229).
The use of pharmaceutical agents has also been successful in polarizing DCs towards a DCreg cell fate both in vitro and in disease models (230, 231). These include histamine, anti-inflammatory agents (e.g. acetylsalicylic acid), adenosine receptor agonists, as well as immunosuppressive drugs, namely cyclosporine A, rapamycin, corticosteroids, BAY-117085, mycophenolatemofetil (MMF), deoxyspergualin, and tacrolimus (FK506) (232). Prednisolone or dexamethasone treatment induces the development of DCreg cells with the ability to induce Treg cells by inhibiting the expression of molecules involved in antigen presentation, inflammatory cytokines, chemokines, as well as the NF-κB pathway (233). On the other hand, rapamycin increases DCreg cells which boost Treg cell expansion in vitro and in vivo by inhibiting the mechanistic target of rapamycin (mTOR) (233, 234). DCs treated with BAY-117085, an irreversible NF-κB inhibitor, induce Treg cells and suppress established experimental autoimmune arthritis in mice (232).
Another strategy is to genetically manipulate DCs to modulate their maturity (219), with liposomes and electroporation being the most effective gene delivery methods in these cells (235). In this method, DCreg cells are produced by ectopically expressing immunomodulatory genes, including PDL-1, TGF-β, IL-4, and CTLA-4. Moreover, antisense oligodeoxy-nucleotides and small interfering RNAs (siRNAs) can be applied to silence target genes in DCs, such as CD86, IL-12, and CD40 (236, 237). In mice, these engineered DCreg cells have been demonstrated to cause T cell hyporesponsiveness, extend allograft survival, stimulate Treg cell differentiation, and decrease autoimmune diabetes or delayed-type hypersensitivity (238–240).
While several strategies for generating DCreg cells have demonstrated promising outcomes in transplantation and autoimmune disease mouse models, there are notable differences in efficacy between mice and humans. Therefore, comprehensive investigations comparing various DCreg cell-generating methodologies are required. Naranjo-Gómez and colleagues (241), for example, evaluated various factors and agents to create human DCreg cells for clinical applications and found substantial variations in DCreg cell properties, underscoring the necessity of proper agent selection. More recently, Boks and colleagues (242) examined several agents for creating therapeutic grade DCreg cells and found that IL-10-treated DCs had the highest tolerogenic potency, holding clinical potential.
As mentioned above, DCs have a dual role in immune tolerance, having both inhibitory and activating roles in autoimmune reactions (243). In the mouse, eliminating DCs leads to widespread autoimmune disease and transferring bone marrow from DC-deficient mice into WT mice leads to autoimmunity in the recipient animals, confirming the central role of DCs in maintaining immune tolerance (244). In the presence of pro-inflammatory cytokines, DCs stimulate Teff cells and prevent the production of Treg cell production. In contrast, in the presence of anti-inflammatory cytokines, DCs induce T cell anergy and induce of Treg cell generation (245). Adjusting the production of autoantibodies indirectly through cross-talk between B cells and Th cells is yet another role of DCs so that any disturbance in PRR (pattern recognition receptor) signaling or abnormal cytokine or chemokine production in DCs can shift the balance between prevention and promotion of autoimmune disease (246). Furthermore, apoptotic cells that accumulate owing to DCs’ inability to completely absorb and remove apoptotic cells might emit danger signals, such as HMGB1 (High mobility group box protein 1) and accessible autoantigens, causing inflammatory reactions in DCs and driving autoimmunity (247). Of note, defects in DC apoptosis may also lead to an increase in DC numbers and autoimmunity. Indeed, a number of molecules involved in various stages of DC maturation, activation, differentiation, and migration have emerged as prospective anti-autoimmune targets (247, 248).
Recent work has demonstrated that dysregulation of DC function can lead to the disruption of intestinal immune homeostasis and concomitantly IBD. When PRRs activate intestinal DCs in response to infectious pathogens, they activate NF-κB signaling, which results in the production of TNF-α, IL-6, -12, and -23, as well as inflammasome signaling, which results in the secretion of IL-18 and IL-1β (249). Effector cytokines activate both innate immune (NK cells, macrophages, and γδ T cells) and adaptive immune cells (Th1, Th2, and Th17) involved in intestinal inflammation and IBD pathology (250, 251). Due to complex roles of intestinal DCs in both mediating intestinal immune homeostasis and promoting the development of IBD, much effort has gone into identifying conditioning factors that drive tolerization of intestinal DCs to prevent, alleviate, or even reverse colitis. Strikingly, DCreg cells derived using vasoactive intestinal peptide (DC-VIP) dramatically reduced the severity of TNBS-induced colitis in mice on both a clinical and histopathological level. Downregulation of Th1 cell inflammatory responses and generation of IL-10-producing Treg cells were linked to the therapeutic impact of DC VIP-DCreg cell injection. In addition, DCreg cells treated with dexamethasone and vitamin D3 or pulsed with enterobacterial extract have shown therapeutic effects in preventing colitis in several animal models (252, 253).
Recent animal studies have enhanced our understanding of the plasticity of DCs and DCreg cells in regulating inflammation, maintaining immune homeostasis, especially in autoimmune disease. The main challenge now is to translate our knowledge of DCs in mouse models to humans so that the therapeutic potential of DCs in autoimmune diseases can be assessed. The phenotypic differences between human and mouse DC subsets, as well as limited access to human samples, makes identifying functional parallels between mouse and human DC subsets exceedingly challenging. Various subsets of DCreg cells have been identified in humans that function as immune regulators, including CD1c+ cDCs in the blood and liver and CD141+CD14+ DCs in human skin (254, 255). Liver CDs (CD1c+) show high production of IL-10 upon LPS induction and induce IL-10-producing Treg cells and IL-4-producing Th2 cells (256). Recent studies have highlighted the specificity of DCreg cells in patients with autoimmune diseases and their potential for the treatment of these diseases. DCreg cells from RRMS patients conditioned with vitamin D3 exhibited a stable semi-mature phenotype and caused stable antigen-specific T cell hyporesponsiveness (256). In a study by Harry colleagues, DCs from RA patients were conditioned with clinical-grade dexamethasone and vitamin D3, which reduced stimulation of autologous antigen-specific T cells, inhibited mature DC-induced T cell activation, and made T cells hyporesponsive to subsequent stimulation (257). In a phase 1 clinical trial in patients with T1D, autologous DC therapy was performed. These DCs were generated ex vivo with antisense oligonucleotides targeting CD40, CD80, and CD86. Treatment with these modified DCs increased the frequency of B220+CD11c- B cell populations without any discernible side effects in diabetic patients (258). In another phase I trial, DCreg cells generated with NF-κB signaling inhibitors were produced in RA patients, and tolerance in these patients was increased with no serious side effects (259). These encouraging results provide prospective therapeutic methods for treating autoimmune disorders via injection of autologous tolerogenic DCs.
In recent years, several Phase I or I/II DCreg cell clinical trials have been concluded, with further trials currently recruiting participants or have not yet published their findings, including long-term trials in organ transplant patients (260). Because these were Phase I or I/II trials, they were designed to determine whether DCreg cell-based immunotherapy is safe and well-tolerated, rather than to test whether it is effective; the results showed that the vast great majority of patients did not experience serious side effects (258, 261–264). However, no significant illness improvement was observed in any of these protocols, even though a small subgroup of treatment group individuals in the RA (261, 265) and Crohn’s disease (262) studies reported reduced symptoms.
Despite the challenges in translating DCreg cell therapy from model animals to humans, technological innovations such as closed cell culture systems have helped to bring the use of DCreg cells into the clinic for the treatment of T1D and RA. The first clinical trial of DCreg cells was conducted in T1D patients. Autologous DCs were treated ex vivo with antisense oligonucleotides to reduce CD80, CD86, and CD40 expression. In this clinical trial, autologous DCreg cells were safe and did not cause side effects in T1D patients (258). In the clinical trial for RA, DCreg cells were developed by treatment with BAY11-7082 and, unlike in the T1D trial, also loaded with antigens. Improvement of clinical symptoms was observed in patients with severe RA. However, none of the participants presented serious side effects, a momentous step in the development of DCreg cell-based therapies (266, 267).
In another clinical trial (NCT01352858) for RA, monocyte-derived DCreg cells, were modified with vitamin D3 and dexamethasone and treated with TLR4 agonist and clinical-grade MPLA (Monophosphoryl lipid A) (268). The TLR4 ligand is essential for DCs to successfully process and display antigen on MHC class II molecules, and it may also give DCs the capacity to move to lymph nodes in a CCR7-dependent manner, where they can engage with T cells and induce autoantigen-specific Treg. Produced DCregs by this method show a high expression level of the MHC-II, while CD80 and 86 expression is intermediate, and the expression of CD40 is very low (268). Thus, despite the ability to present antigen, their capacity to stimulate T cells is lower than that of adult DCs. Furthermore, these DCreg cells had high TGF-β and IL-10 levels while having low IL-12, IL-23, and TNF-α levels. In a CIA mouse model, DCreg cells produced with LPS, vitamin D3, and dexamethasone greatly reduced the severity and development of arthritis (269). Human DCreg cells with the potential to suppress autoreactive T cells in the rheumatoid joint might be generated using a similar approach. DCreg cells present antigen in conjunction with low co-stimulatory molecule expression, causing memory T cells to become hyporesponsive while polarizing naïve T cells to produce anti-inflammatory cytokines (259). Table 3 summarizes clinical research on the use of DCs in the therapy of autoimmune disorders.
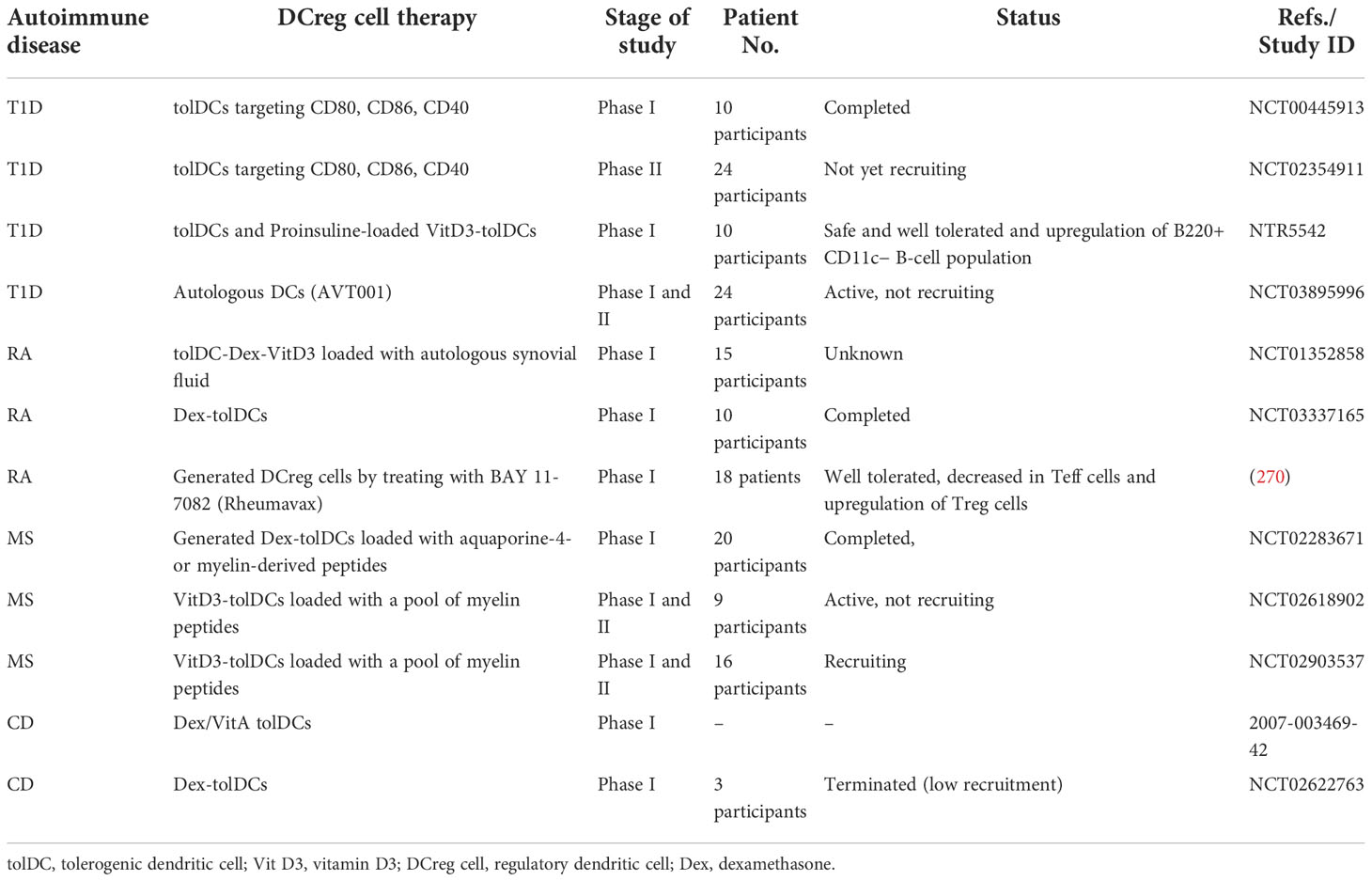
Table 3 Clinical trials using DCs in autoimmune disease therapy.
Myeloid-derived suppressor cells (MDSCs) were first described 30 years ago as myeloid cells with potent immunoregulatory capacity (271). Further studies have since clarified the physiological and pathological roles of MDSCs as universal immune regulators. There are two major groups of MDSCs: polymorphonuclear or granulocytic MDSCs (PMN-MDSCs), morphologically and phenotypically similar to neutrophils, and monocyte-like MDSCs, which resemble monocytes (M-MDSCs) (272). Immune suppression is one of the critical features of MDSCs, allowing these cells to be distinguishable from neutrophils and monocytes in blood (273). It has become clear that MDSCs are involved in a wide range of inflammatory disorders, including autoimmune diseases (274–276). MDSCs inhibit T cell responses, and induce Treg cells, thus playing a protective role in autoimmune diseases (277). Recently, many studies have focused on the role of MDSCs in autoimmunity. However, most of these studies were carried out using animal models and extensive investigations to explain the exact characteristics of MDSCs in autoimmune diseases in humans will need to be conducted. The role of MDSCs in suppressing the immune system shows how these cells can modulate the immune response and cause cancer cells to escape from the immune system (277). Unlike information about the role of MDSCs in tumor progression, our knowledge of their function and role in autoimmune diseases is controversial and limited. Recently, however, researchers have drawn attention to MDSCs’ role and their therapeutic potential in autoimmune diseases, such as T1D (278), SLE (275, 279), MS (280–282), RA (283) and IBD (284, 285)
Hematopoietic stem cells (HSCs) in bone marrow are the source of common myeloid progenitor cells, which eventually produce immature myeloid cells (IMCs). IMCs have no suppressive activity and are immunologically inactive (286). Under normal conditions in healthy individuals, IMCs differentiate into mature and functional macrophages, DCs, and granulocytes (287). In contrast, in pathological conditions, especially in the presence of inflammation, infection, autoimmune disease, cancer or trauma, IMC differentiation into immune cells is impaired. In these circumstances, IMCs become activated, proliferate in response to external and internal factors, and differentiate into MDSCs (288). IMC expansion leads to the production of MDSCs in peripheral tissues, which inhibit immune responses by producing suppressive signals (286). Several factors, including PGE2, macrophage colony-stimulating factor (M-CSF), IL-3, cyclooxygenase-2 (COX-2), vascular endothelial growth factor (VEGF), IL-6, stem cell factor (SCF)-1, and granulocyte/macrophage colony-stimulating factor (GM-CSF) play essential roles in MDSC growth (286, 289, 290), while IFN-γ, TGF-β, IL-13, and IL-4 are linked to MDSC activation (286, 291, 292).
MDSCs use various mechanisms to suppress the immune response, based on either cell-cell contact or soluble intermediates. Induced nitric oxide synthase (iNOS) and arginase-1 (Arg-1) are enzymes whose levels are increased in MDSCs (293). L-Arginine is the common substrate of iNOS and Arg-1; iNOS catalyzes the production of nitric oxide (NO), a type of ROS, from arginine, whereas Arg-1 catalyzes the hydrolysis of L-arginine into urea and ornithine. By limiting the amounts of L-arginine, a non-essential amino acid, in the microenvironment, these enzymes restrict T cell activation and proliferation (286, 294, 295). For example, CD11b+Gr-1+ MDSC accumulation was observed in the liver of an autoimmune hepatitis (AIH) mouse model, as well as in secondary lymphatic tissues and the spleen of IBD and EAE mice (296).
Notably, in all studies, CD11b+Gr-1+ MDSCs inhibited T cell proliferation through either NO or cell-cell contact. Moreover, CD11b+Gr-1low MDSCs were seen in lupus-prone MRL Faslpr mice that developed autoimmune organ damage. Ex vivo, these cells suppressed CD4+ T cell proliferation that could be blocked by an Arg-1 inhibitor, demonstrating that Arg-1 was the primary suppressive strategy used by MDSCs in this autoimmune condition (275). Ma and colleagues recently reported that CD11b+Ly6Chi monocytes sorted from peritoneal cells dramatically suppressed T cell proliferation ex vivo that was cell contact-dependent and involved NO and PGE2. Furthermore, these cells also limited Th1 cell differentiation while enhancing Treg cell formation in a pristane-induced lupus mouse model, indicating that these were monocytic MDSCs (276). Moreover, additional critical factors utilized by MDSCs to limit T cell proliferation and cytotoxicity include IL-10, TGF-β, COX-2, IDO, PD-L1, and PGE2 (297, 298). Of note, Treg cell-mediated suppression is also aided by MDSCs: Gr-1+CD115+ MDSCs promote Foxp3+ Treg cell expansion in murine tumor models (299). In addition, Ly6G+ phagocytic MDSCs can control cytokine secretion by B cells in the CNS, potentially playing a role in recovery rates in EAE (300). Nonetheless, further studies are required to elucidate the involvement of MDSCs in the regulation of Treg and B cells. Preclinical studies on MDSCs and their association with autoimmune disease models are summarized in Table 4.
Table 4 Preclinical studies on MDSCs in autoimmune disease.
Generally, studying the role and function of MDSCs in mouse autoimmunity models and in vitro has yielded conflicting results. While MDSCs extracted from autoimmune inflammatory sites inhibit T cell responses in vitro, in vivo endogenous MDSCs fail to reduce the severity of autoimmune diseases, such as EAE (282, 308). Interestingly, however, adoptive transfer of MDSCs induces immune tolerance to self-antigens and can reduce the severity of some autoimmune diseases in mice, such as T1D (278), IBD (285), and inflammatory eye disease (309). Moreover, adoptively transferred MDSCs generated ex vivo inhibit GvHD and prevent graft rejection in mice (310–312). For instance, these early successes in using exogenous MDSCs to suppress autoimmunity in animal models make them a promising cell-based immunotherapy target. Sources and processes for generating exogenous MDSCs are various, with growth factors and cytokines to expand exogenous MDSCs isolated from bone marrow or peripheral blood being one such process (310, 313, 314). HSCs and even embryonic stem cells have also been used as sources of exogenous MDSCs (311).
In humans, peripheral blood-derived monocytes can be differentiated and expanded in vitro in the presence of PGE2 to produce large amount of functional and stable MDSCs (315). Importantly, human MDSCs generated ex vivo suppress CD4+ and CD8+ T cell division via iNOS and Arg-1 (314). These methods have the potential to be used for cell-based immunotherapy of autoimmune diseases. However, there are certain dangers connected with using MDSCs in this context. On one hand, MDSCs would not be selective for antigen-specific T cells, potentially blocking both damaging T cell responses to self-antigens and beneficial immunological responses to pathogens or malignancies. On the other hand, controlling the migration and accumulation of the injected MDSCs would also be problematic and the injection of MDSCs may result in the release of inflammatory factors. As a result, more thorough studies in animal models are required before MDSC treatment can be safely tested in humans.
Innate lymphoid cells (ILCs) are a recently unveiled group of lymphocytes which, despite their lack of TCR expression, have many parallels with CD4+ T helper cells. ILCs are located at barrier surfaces and respond rapidly to environmental changes by local multiplication. As with other innate immune cells, ILCs are activated before adaptive immunity develops (316). Some studies report the association and interaction of ILCs with T cells in allergy and IBD patients. Indeed, ILCs can be harmful when dysregulated, contributing to chronic inflammation and autoimmune disorders such as asthma, IBD, GvHD, psoriasis, RA, and atopic dermatitis (317).
ILCs are mainly divided into 3 subgroups according to their function, the cytokines they produce, and their dependence on different transcription factors. Group 1 ILCs, which include ILC1s and NK cells; group 2 ILCs or ILC2s, and group 3 ILCs, encompassing lymphoid tissue inducer (LTi) cells and two subsets of ILC3s, categorized as natural cytotoxicity receptor (NCR)-positive and NCR-negative ILC3s. The absence of specific markers of B, T, and other hematopoietic cell lineages is usually used to identify ILCs, in addition to differential expression of markers. For instance, CD127 is expressed on most ILC1s, but not on mature NK cells. In the same vein, ILC2s express the prostaglandin D2 receptor CRTH2 and ILC3s express the natural cytotoxicity receptor NKp44 in humans. In mice, ILC2s express the IL-33 receptor ST2, whereas ILC3s express NKp46, allowing for the distinction of these two ILC subsets (318–320).
To date, very few studies have implicated ILCs and their cytokines in RA (321). All three groups of ILCs have been detected in the peripheral blood of healthy individuals (322). A study by Ren and colleagues indicated that the numbers of CD3−CD56+NKp44+CCR6+ cells in peripheral blood, as well as in synovial fluid, in RA patients were increased compared to healthy people of the same age (323). In this study, it was found that CD3−CD56+NKp44+CCR6+ cells were ILC3s, and their increase is positively associated with the patients’ 28-joint disease activity score (DAS28) (323). A study by a different group confirmed an increase in ILC3 cell numbers in the synovial fluid of RA patients (324). Patients with inflammatory arthritis have also been found to have ILC1-like cells in their synovial fluid and synovial tissue; this cohort of patients included mostly RA individuals, but also patients with juvenile idiopathic arthritis and psoriatic arthritis (PsA) (325). These cells produced IFN-γ in response to a combination of IL-2, -12, and -15 stimulation (325). Interestingly, ILC1-like cells are more sensitive to IL-12 and IL-18 activation and generate more IFN than their peripheral blood counterparts (326). It is thought that these CD3-CD16-CD56bright cells are either NK cells or non-cytotoxic ILC1s (327). In fact, recent research found that ILC1s predominate in the synovial fluid of RA patients (328).
The discovery of cytokine pathways implicated in autoimmune disorders, as well as the identification of ILCs as prolific makers of these cytokines, suggests that these cellular actors may play a role in the pathogenesis of these diseases. The IL−23–IL−17 cytokine axis is involved in the pathogenesis of various autoimmune diseases (329). For example, the anti−IL−17 monoclonal antibody secukinumab has been used as an IL-17 inhibitor in the treatment of AS and PsA (330). Initially, anti−IL−17 monoclonal antibodies were used to target Th17 cells. However, it is now known that ILC3s are also an important source of IL-17. Therefore, ILC3s are increasingly considered as additional cellular targets for therapeutic interventions in autoimmune diseases. In addition to producing IL-17, these cells also produce other disease-promoting cytokines, including lymphotoxin, GM-CSF, IL-22, and TNF-α. Utilizing selective inhibitors of ILC3s and LTi cells in autoimmune diseases may be a useful strategy (331, 332). Tertiary lymphoid organs (TLOs), also defined as ectopic lymphoid-like structures, are temporary lymphoid cell aggregates with structural features comparable to secondary lymphoid organs. Persistent immunological activation, which can be caused by microbial infection, chronic allograft rejection, or autoimmune illness, leads to the formation of such structures (333). This strategy has been effective in animal models of T1D and arthritis (332, 334). Specific treatments based on ILCs have not yet been introduced. Anti-IL-17, anti-IL-23 or anti-LTi cells target a wide range of cells. Clarifying characteristic pathways for ILCs and research into gene expression profiles could provide researchers with more specific targets. It has been shown that CD25-specific monoclonal antibody therapy preferentially depletes ILC3s in patients with MS (335). The plasticity of ILCs (336) opens up another avenue for developing specialized treatment options to take advantage of this group of immune cells and transdifferentiate IL17-producing ILC3s into non-harmful cells.
Success in treating autoimmune diseases with immune cell therapy will depend on developing treatment approaches with significant potency and effectiveness. These treatments should also have lasting effects without side effects. According to a paradigm put forward by Mous and colleagues, three aspects ought to be considered for immunotherapies to reach clinical efficacy: host immune system as the field of effect, growth factors as therapeutic enhancers, and immune cells as the effective factors. Potentially confounding biological factors, such as sex, comorbidities, age, and gut microbiota, must also be carefully evaluated given the significant variability among the human population. To modify and alter the treatment based on each patient’s unique and dynamic immunological state, frequent monitoring of effectiveness, safety, and validated biomarkers will be required. It appears that immune cell-based therapies for autoimmune diseases should be personalized according to the individual’s immune conditions and re-adjusted as the disease progresses or improves. Various types of immune regulatory cells, such as Treg cells, Breg cells, DCreg cells, MDSCs, and ILC subsets, have increasingly been the subject of extensive studies. Some of these cells, such as Treg and DCreg cells, have entered the clinical realm, while others, such as Breg cells, are in the preclinical stage and no clinical studies have yet been reported. MDSCs and ILC subsets, on the other hand, are still at the drawing board stage. However, the results obtained from applying these cell types towards autoimmune disease therapy have been promising. Continued comprehensive investigations on overcoming the challenges in using each one of these immune regulatory cell types are getting us closer to definitive treatments to several autoimmune diseases and will eliminate the suffering caused by them.
The manuscript was written by all authors. RAR generated the figures. LF and ARA contributed to and revised the manuscript, which was then reviewed, commented on, and accepted by all authors. All authors contributed to the article and approved the submitted version.
LF is supported by grants from the Human Islet Research Network (HIRN, U24DK104162-07), the American Cancer Society (ACS, IRG-19-137-20), the South Carolina Clinical and Translational Research Institute (SCTR, 1TL1TR001451-01), the Diabetes Research Connection (DRC, IPF 22-1224), the Integrated Islet Distribution Program (IIDP, 2UC4DK098085), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, 1R43DK133029-01).
This article is wholeheartedly dedicated to Nika Shahkarami, a 16-year-old Iranian girl who was brutally murdered following recent protests in Iran. Still, her voice will always resonate with the chant “woman, life, freedom”.
ARA was employed by Xsphera Biosciences. LF is an inventor in patents and patent applications pertaining to the use of immune regulatory cells for therapy.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhernakova A, Van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet (2009) 10(1):43–55. doi: 10.1038/nrg2489
2. Miller FW, Alfredsson L, Costenbader KH, Kamen DL, Nelson LM, Norris JM, et al. Epidemiology of environmental exposures and human autoimmune diseases: findings from a national institute of environmental health sciences expert panel workshop. J Autoimmun (2012) 39(4):259–71. doi: 10.1016/j.jaut.2012.05.002
3. Brauer M, Freedman G, Frostad J, Van Donkelaar A, Martin RV, Dentener F, et al. Ambient air pollution exposure estimation for the global burden of disease 2013. Environ Sci Technol (2016) 50(1):79–88. doi: 10.1021/acs.est.5b03709
4. Touitou I. New genetic interpretation of old diseases. Autoimmun Rev (2012) 12(1):5–9. doi: 10.1016/j.autrev.2012.07.014
5. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discovery (2019) 18(10):749–69. doi: 10.1038/s41573-019-0041-4
6. Jasim SA, Yumashev AV, Abdelbasset WK, Margiana R, Markov A, Suksatan W, et al. Shining the light on clinical application of mesenchymal stem cell therapy in autoimmune diseases. Stem Cell Res Ther (2022) 13(1):101.doi: 10.1186/s13287-022-02782-7
7. Su M, Zhao C, Luo S. Therapeutic potential of chimeric antigen receptor based therapies in autoimmune diseases. Autoimmun Rev (2022) 21(1):102931. doi: 10.1016/j.autrev.2021.102931
8. Hu X, Leak RK, Thomson AW, Yu F, Xia Y, Wechsler LR, et al. Promises and limitations of immune cell-based therapies in neurological disorders. Nat Rev Neurol (2018) 14(9):559–68. doi: 10.1038/s41582-018-0028-5
9. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
10. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122
11. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
12. Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, et al. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol (1999) 162(9):5317–26.
13. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
14. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci (2018) 1417(1):5–22. doi: 10.1111/nyas.13011
15. Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol (2012) 12(3):157–67. doi: 10.1038/nri3155
16. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol (2019) 20(3):158–72. doi: 10.1038/s41577-019-0232-6
17. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
19. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. “Infectious” transplantation tolerance. Science (1993) 259(5097):974–7. doi: 10.1126/science.8094901
20. Miller A, Lider O, Weiner HL. Antigen-driven bystander suppression after oral administration of antigens. J Exp Med (1991) 174(4):791–8. doi: 10.1084/jem.174.4.791
21. Zhang R, Miao J, Zhu P. Regulatory T cell heterogeneity and therapy in autoimmune diseases. Autoimmun Rev (2021) 20(5):102715. doi: 10.1016/j.autrev.2020.102715
22. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev (2014) 259(1):88–102. doi: 10.1111/imr.12160
23. Bacchetta R, Bigler M, Touraine JL, Parkman R, Tovo PA, Abrams J, et al. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J Exp Med (1994) 179(2):493–502. doi: 10.1084/jem.179.2.493
24. Gagliani N, Jofra T, Valle A, Stabilini A, Morsiani C, Gregori S, et al. Transplant tolerance to pancreatic islets is initiated in the graft and sustained in the spleen. Am J Transplant (2013) 13(8):1963–75. doi: 10.1111/ajt.12333
25. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N. The biology of T regulatory type 1 cells and their therapeutic application in immune-mediated diseases. Immunity (2018) 49(6):1004–19. doi: 10.1016/j.immuni.2018.12.001
26. Chen PP, Cepika AM, Agarwal-Hashmi R, Saini G, Uyeda MJ, Louis DM, et al. Alloantigen-specific type 1 regulatory T cells suppress through CTLA-4 and PD-1 pathways and persist long-term in patients. Sci Transl Med (2021) 13(617):eabf5264. doi: 10.1126/scitranslmed.abf5264
27. DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol (2016) 16(3):149–63. doi: 10.1038/nri.2015.18
28. Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol (2013) 13(6):461–7. doi: 10.1038/nri3464
29. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ treg cells that phenotypically mirror effector Th cells. Blood (2012) 119(19):4430–40. doi: 10.1182/blood-2011-11-392324
30. Hoeppli R, MacDonald K, Leclair P, Fung V, Mojibian M, Gillies J, et al. Tailoring the homing capacity of human tregs for directed migration to sites of Th1-inflammation or intestinal regions. Am J Transplantation (2019) 19(1):62–76. doi: 10.1111/ajt.14936
31. Roschupkina T, Juliusson G. Subpopulations of T regulatory cells in blood stem cell harvests influence development of acute graft versus host disease in allogeneic transplant recipients. Cytometry Part B: Clin Cytometry (2018) 94(2):264–9. doi: 10.1002/cyto.b.21404
32. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol (2018) 19(7):665–73. doi: 10.1038/s41590-018-0120-4
33. Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (2011) 187(9):4553–60. doi: 10.4049/jimmunol.1101328
34. Vaeth M, Müller G, Stauss D, Dietz L, Klein-Hessling S, Serfling E, et al. Follicular regulatory T cells control humoral autoimmunity via NFAT2-regulated CXCR5 expression. J Exp Med (2014) 211(3):545–61. doi: 10.1084/jem.20130604
35. Miles B, Connick E. Control of the germinal center by follicular regulatory T cells during infection. Front Immunol (2018) 9:2704. doi: 10.3389/fimmu.2018.02704
36. Gong F, Dai Y, Zheng T, Cheng L, Zhao D, Wang H, et al. Peripheral CD4+ T cell subsets and antibody response in COVID-19 convalescent individuals. J Clin Invest (2020) 130(12):6588–99. doi: 10.1172/JCI141054
37. Gliwiński M, Iwaszkiewicz-Grześ D, Trzonkowski P. Cell-based therapies with T regulatory cells. BioDrugs (2017) 31(4):335–47. doi: 10.1007/s40259-017-0228-3
38. MacDonald KN, Piret JM, Levings MK. Methods to manufacture regulatory T cells for cell therapy. Clin Exp Immunol (2019) 197(1):52–63. doi: 10.1111/cei.13297
39. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (Orlando Fla) (2009) 133(1):22–6. doi: 10.1016/j.clim.2009.06.001
40. Dall’Era M, Pauli ML, Remedios K, Taravati K, Sandova PM, Putnam AL, et al. Adoptive treg cell therapy in a patient with systemic lupus erythematosus. Arthritis Rheumatol (2019) 71(3):431–40. doi: 10.1002/art.40737
41. Seay HR, Putnam AL, Cserny J, Posgai AL, Rosenau EH, Wingard JR, et al. Expansion of human tregs from cryopreserved umbilical cord blood for GMP-compliant autologous adoptive cell transfer therapy. Mol Ther Methods Clin Dev (2017) 4:178–91. doi: 10.1016/j.omtm.2016.12.003
42. Fujimaki W, Takahashi N, Ohnuma K, Nagatsu M, Kurosawa H, Yoshida S, et al. Comparative study of regulatory T cell function of human CD25CD4 T cells from thymocytes, cord blood, and adult peripheral blood. Clin Dev Immunol (2008) 2008:305859. doi: 10.1155/2008/305859
43. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127(8):1044–51. doi: 10.1182/blood-2015-06-653667
44. Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplant (2016) 16(1):58–71. doi: 10.1111/ajt.13456
45. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol (2017) 18(2):173–83. doi: 10.1038/ni.3646
46. Tahvildari M, Dana R. Low-dose IL-2 therapy in transplantation, autoimmunity, and inflammatory diseases. J Immunol (2019) 203(11):2749–55. doi: 10.4049/jimmunol.1900733
47. Pesenacker AM, Broady R, Levings MK. Control of tissue-localized immune responses by human regulatory T cells. Eur J Immunol (2015) 45(2):333–43. doi: 10.1002/eji.201344205
48. Singh J, Zúñiga-Pflücker JC. Producing proT cells to promote immunotherapies. Int Immunol (2018) 30(12):541–50. doi: 10.1093/intimm/dxy051
49. Honaker Y, Hubbard N, Xiang Y, Fisher L, Hagin D, Sommer K, et al. Gene editing to induce FOXP3 expression in human CD4(+) T cells leads to a stable regulatory phenotype and function. Sci Transl Med (2020) 12(546):eaay6422. doi: 10.1126/scitranslmed.aay6422
50. MacMillan ML, Hippen KL, McKenna DH, Kadidlo D, Sumstad D, DeFor TE, et al. First-in-human phase 1 trial of induced regulatory T cells for graft-versus-host disease prophylaxis in HLA-matched siblings. Blood Adv (2021) 5(5):1425–36. doi: 10.1182/bloodadvances.2020003219
51. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood–derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. J Am Soc Hematol (2016) 127(8):1044–51. doi: 10.1182/blood-2015-06-653667
52. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. J Am Soc Hematol (2011) 117(3):1061–70. doi: 10.1182/blood-2010-07-293795
53. Steiner D, Brunicki N, Blazar BR, Bachar-Lustig E, Reisner Y. Tolerance induction by third-party “off-the-shelf” CD4+CD25+ treg cells. Exp Hematol (2006) 34(1):66–71. doi: 10.1016/j.exphem.2005.10.011
54. Zhang H, Guo H, Lu L, Zahorchak AF, Wiseman RW, Raimondi G, et al. Sequential monitoring and stability of ex vivo-expanded autologous and nonautologous regulatory T cells following infusion in nonhuman primates. Am J Transplant (2015) 15(5):1253–66. doi: 10.1111/ajt.13113
55. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
56. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-Shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein-Barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol: Off J Am Soc Clin Oncol (2017) 35(31):3547–57. doi: 10.1200/JCO.2017.73.0655
57. Gravano DM, Vignali DA. The battle against immunopathology: infectious tolerance mediated by regulatory T cells. Cell Mol Life Sci (2012) 69(12):1997–2008. doi: 10.1007/s00018-011-0907-z
58. Pierini A, Colonna L, Alvarez M, Schneidawind D, Nishikii H, Baker J, et al. Donor requirements for regulatory T cell suppression of murine graft-versus-Host disease. J Immunol (2015) 195(1):347–55. doi: 10.4049/jimmunol.1402861
59. Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, et al. HLA-e-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol (2017) 35(8):765–72. doi: 10.1038/nbt.3860
60. Torikai H, Cooper LJ. Translational implications for off-the-shelf immune cells expressing chimeric antigen receptors. Mol Ther (2016) 24(7):1178–86. doi: 10.1038/mt.2016.106
61. Tang Q, Lee K. Regulatory T-cell therapy for transplantation: how many cells do we need? Curr Opin Organ Transplant (2012) 17(4):349–54. doi: 10.1097/MOT.0b013e328355a992
62. Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant (2013) 13(11):3010–20. doi: 10.1111/ajt.12433
63. Radic M, Neeli I, Marion T. Prospects for CAR T cell immunotherapy in autoimmune diseases: clues from lupus. Expert Opin Biol Ther (2022) 22(4):499–507. doi: 10.1080/14712598.2022.2026921
64. Theil A, Tuve S, Oelschlägel U, Maiwald A, Döhler D, Oßmann D, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy (2015) 17(4):473–86. doi: 10.1016/j.jcyt.2014.11.005
65. Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood (2006) 108(13):4260–7. doi: 10.1182/blood-2006-06-027409
66. McKenna DH Jr., Sumstad D, Kadidlo DM, Batdorf B, Lord CJ, Merkel SC, et al. Optimization of cGMP purification and expansion of umbilical cord blood-derived T-regulatory cells in support of first-in-human clinical trials. Cytotherapy (2017) 19(2):250–62. doi: 10.1016/j.jcyt.2016.10.011
67. Xu J, Melenhorst JJ, Fraietta JA. Toward precision manufacturing of immunogene T-cell therapies. Cytotherapy (2018) 20(5):623–38. doi: 10.1016/j.jcyt.2017.12.007
68. Dawson NAJ, Lam AJ, Cook L, Hoeppli RE, Broady R, Pesenacker AM, et al. An optimized method to measure human FOXP3(+) regulatory T cells from multiple tissue types using mass cytometry. Eur J Immunol (2018) 48(8):1415–9. doi: 10.1002/eji.201747407
69. Gołąb K, Grose R, Placencia V, Wickrema A, Solomina J, Tibudan M, et al. Cell banking for regulatory T cell-based therapy: strategies to overcome the impact of cryopreservation on the treg viability and phenotype. Oncotarget (2018) 9(11):9728–40. doi: 10.18632/oncotarget.23887
70. Fraser H, Safinia N, Grageda N, Thirkell S, Lowe K, Fry LJ, et al. A rapamycin-based GMP-compatible process for the isolation and expansion of regulatory T cells for clinical trials. Mol Ther Methods Clin Dev (2018) 8:198–209. doi: 10.1016/j.omtm.2018.01.006
71. Iyer RK, Bowles PA, Kim H, Dulgar-Tulloch A. Industrializing autologous adoptive immunotherapies: Manufacturing advances and challenges. Front Med (2018) 5:150. doi: 10.3389/fmed.2018.00150
72. Lam AJ, Hoeppli RE, Levings MK. Harnessing advances in T regulatory cell biology for cellular therapy in transplantation. Transplantation (2017) 101(10):2277–87. doi: 10.1097/TP.0000000000001757
73. Parmar S, Liu X, Najjar A, Shah N, Yang H, Yvon E, et al. Ex vivo fucosylation of third-party human regulatory T cells enhances anti-graft-versus-host disease potency in vivo. Blood (2015) 125(9):1502–6. doi: 10.1182/blood-2014-10-603449
74. Sicard A, Levings MK, Scott DW. Engineering therapeutic T cells to suppress alloimmune responses using TCRs, CARs, or BARs. Am J Transplant (2018) 18(6):1305–11. doi: 10.1111/ajt.14747
75. Dawson NAJ, Vent-Schmidt J, Levings MK. Engineered tolerance: Tailoring development, function, and antigen-specificity of regulatory T cells. Front Immunol (2017) 8:1460. doi: 10.3389/fimmu.2017.01460
76. Beheshti SA, Shamsasenjan K, Ahmadi M, Abbasi B. CAR treg: A new approach in the treatment of autoimmune diseases. Int Immunopharmacol (2022) 102:108409. doi: 10.1016/j.intimp.2021.108409
77. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126(4):1413–24. doi: 10.1172/JCI82771
78. Muller YD, Ferreira LMR, Ronin E, Ho P, Nguyen V, Faleo G, et al. Precision engineering of an anti-HLA-A2 chimeric antigen receptor in regulatory T cells for transplant immune tolerance. Front Immunol (2021) 12:686439. doi: 10.3389/fimmu.2021.686439
79. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4+ CD25highCD127-regulatory T cells prolongs survival of pancreatic islets–results of one year follow-up. Clin Immunol (2014) 153(1):23–30. doi: 10.1016/j.clim.2014.03.016
80. Marek-Trzonkowska N, Myśliwiec M, Iwaszkiewicz-Grześ D, Gliwiński M, Derkowska I, Żalińska M, et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J Trans Med (2016) 14(1):1–11. doi: 10.1186/s12967-016-1090-7
81. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology (2016) 64(2):632–43. doi: 10.1002/hep.28459
82. Voskens CJ, Fischer A, Roessner S, Lorenz C, Hirschmann S, Atreya R, et al. Characterization and expansion of autologous GMP-ready regulatory T cells for TREG-based cell therapy in patients with ulcerative colitis. Inflammatory Bowel Dis (2017) 23(8):1348–59. doi: 10.1097/MIB.0000000000001192
83. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hébuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory crohn’s disease. Gastroenterology (2012) 143(5):1207–17.e2. doi: 10.1053/j.gastro.2012.07.116
84. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low-dose IL-2 and treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight (2021) 6(18):e147474. doi: 10.1172/jci.insight.147474
85. Jia X, Zhai T, Wang B, Yao Q, Li Q, Mu K, et al. Decreased number and impaired function of type 1 regulatory T cells in autoimmune diseases. J Cell Physiol (2019) 234(8):12442–50. doi: 10.1002/jcp.28092
86. Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-Scale in vitro expansion of polyclonal human CD4+ CD25high regulatory T cells. Blood (2004) 104(3):895–903. doi: 10.1182/blood-2004-01-0086
87. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (Tregs) with minimal loss of in vivo functional activity. Sci Trans Med (2011) 3(83):83ra41–1. doi: 10.1126/scitranslmed.3001809
88. Lam AJ, Lin DTS, Gillies JK, Uday P, Pesenacker AM, Kobor MS, et al. Optimized CRISPR-mediated gene knockin reveals FOXP3-independent maintenance of human treg identity. Cell Rep (2021) 36(5):109494. doi: 10.1016/j.celrep.2021.109494
89. Schmidt A, Eriksson M, Shang M-M, Weyd H, Tegnér J. Comparative analysis of protocols to induce human CD4+ Foxp3+ regulatory T cells by combinations of IL-2, TGF-beta, retinoic acid, rapamycin and butyrate. PloS One (2016) 11(2):e0148474. doi: 10.1371/journal.pone.0148474
90. Rossetti M, Spreafico R, Saidin S, Chua C, Moshref M, Leong JY, et al. Ex vivo–expanded but not in vitro–induced human regulatory T cells are candidates for cell therapy in autoimmune diseases thanks to stable demethylation of the FOXP3 regulatory T cell–specific demethylated region. J Immunol (2015) 194(1):113–24. doi: 10.4049/jimmunol.1401145
91. Miyara M, Chader D, Burlion A, Goldstein J, Sterlin D, Norol F, et al. Combination of IL-2, rapamycin, DNA methyltransferase and histone deacetylase inhibitors for the expansion of human regulatory T cells. Oncotarget (2017) 8(62):104733. doi: 10.18632/oncotarget.10914
92. Lu Y, Hippen KL, Lemire AL, Gu J, Wang W, Ni X, et al. miR-146b antagomir–treated human tregs acquire increased GVHD inhibitory potency. J Am Soc Hematol (2016) 128(10):1424–35. doi: 10.1182/blood-2016-05-714535
93. Zhang D, Chia C, Jiao X, Jin W, Kasagi S, Wu R, et al. D-mannose induces regulatory T cells and suppresses immunopathology. Nat Med (2017) 23(9):1036–45. doi: 10.1038/nm.4375
94. Dwivedi M, Kumar P, Laddha NC, Kemp EH. Induction of regulatory T cells: a role for probiotics and prebiotics to suppress autoimmunity. Autoimmun Rev (2016) 15(4):379–92. doi: 10.1016/j.autrev.2016.01.002
95. Skartsis N, Peng Y, Ferreira LMR, Nguyen V, Ronin E, Muller YD, et al. IL-6 and TNFα drive extensive proliferation of human tregs without compromising their lineage stability or function. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.783282
96. Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-cell therapy for autoimmune and autoinflammatory diseases: The next frontier. J Allergy Clin Immunol (2018) 142(6):1710–8. doi: 10.1016/j.jaci.2018.10.015
97. Scalapino KJ, Daikh DI. Suppression of glomerulonephritis in NZB/NZW lupus prone mice by adoptive transfer of ex vivo expanded regulatory T cells. PloS One (2009) 4(6):e6031. doi: 10.1371/journal.pone.0006031
98. Humrich JY, Morbach H, Undeutsch R, Enghard P, Rosenberger S, Weigert O, et al. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proc Natl Acad Sci (2010) 107(1):204–9. doi: 10.1073/pnas.0903158107
99. Comte D, Karampetsou MP, Kis-Toth K, Yoshida N, Bradley SJ, Kyttaris VC, et al. Brief report: CD4+ T cells from patients with systemic lupus erythematosus respond poorly to exogenous interleukin-2. Arthritis Rheumatol (2017) 69(4):808–13. doi: 10.1002/art.40014
100. Costa N, Marques O, Godinho S, Carvalho C, Leal B, Figueiredo A, et al. Two separate effects contribute to regulatory T cell defect in systemic lupus erythematosus patients and their unaffected relatives. Clin Exp Immunol (2017) 189(3):318–30. doi: 10.1111/cei.12991
101. Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in new Zealand Black/New Zealand white lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol (2006) 177(3):1451–9. doi: 10.4049/jimmunol.177.3.1451
102. Marek-Trzonkowska N, Myśliwec M, Siebert J, Trzonkowski P. Clinical application of regulatory T cells in type 1 diabetes. Pediatr Diabetes (2013) 14(5):322–32. doi: 10.1111/pedi.12029
103. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117(14):3921–8. doi: 10.1182/blood-2010-10-311894
104. Meyer EH, Laport G, Xie BJ, MacDonald K, Heydari K, Sahaf B, et al. Transplantation of donor grafts with defined ratio of conventional and regulatory T cells in HLA-matched recipients. JCI Insight (2019) 4(10):e127244. doi: 10.1172/jci.insight.127244
105. Kasahara H, Kondo T, Nakatsukasa H, Chikuma S, Ito M, Ando M, et al. Generation of allo-antigen-specific induced treg stabilized by vitamin c treatment and its application for prevention of acute graft versus host disease model. Int Immunol (2017) 29(10):457–69. doi: 10.1093/intimm/dxx060
106. Kasagi S, Zhang P, Che L, Abbatiello B, Maruyama T, Nakatsukasa H, et al. In vivo–generated antigen-specific regulatory T cells treat autoimmunity without compromising antibacterial immune response. Sci Trans Med (2014) 6(241):241ra78–ra78. doi: 10.1126/scitranslmed.3008895
107. Sun G, Hou Y, Gong W, Liu S, Li J, Yuan Y, et al. Adoptive induced antigen-specific treg cells reverse inflammation in collagen-induced arthritis mouse model. Inflammation (2018) 41(2):485–95. doi: 10.1007/s10753-017-0704-4
108. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199(11):1455–65. doi: 10.1084/jem.20040139
109. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
110. Brusko TM, Koya RC, Zhu S, Lee MR, Putnam AL, McClymont SA, et al. Human antigen-specific regulatory T cells generated by T cell receptor gene transfer. PloS One (2010) 5(7):e11726. doi: 10.1371/journal.pone.0011726
111. Izquierdo C, Ortiz AZ, Presa M, Malo S, Montoya A, Garabatos N, et al. Treatment of T1D via optimized expansion of antigen-specific tregs induced by IL-2/anti-IL-2 monoclonal antibody complexes and peptide/MHC tetramers. Sci Rep (2018) 8(1):1–14. doi: 10.1038/s41598-018-26161-6
112. Xiang Y, Ren M, Jiang H, Yang T, He Y, Ao D, et al. Ex vivo expansion of antigen-specific CD4+ CD25+ regulatory T cells from autologous naïve CD4+ T cells of multiple sclerosis patients as a potential therapeutic approach. Eur Rev Med Pharmacol Sci (2016) 20(24):5261–70.
113. Mekala DJ, Geiger TL. Immunotherapy of autoimmune encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Blood (2005) 105(5):2090–2. doi: 10.1182/blood-2004-09-3579
114. Mekala DJ, Alli RS, Geiger TL. IL-10-dependent infectious tolerance after the treatment of experimental allergic encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Proc Natl Acad Sci United States America (2005) 102(33):11817–22. doi: 10.1073/pnas.0505445102
115. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant (2017) 17(4):931–43. doi: 10.1111/ajt.14185
116. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant (2017) 17(4):917–30. doi: 10.1111/ajt.14175
117. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimmun (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
118. Khosravi-Maharlooei M, Madley R, Borsotti C, Ferreira LMR, Sharp RC, Brehm MA, et al. Modeling human T1D-associated autoimmune processes. Mol Metab (2021) 56:101417. doi: 10.1016/j.molmet.2021.101417
119. Wrangle JM, Patterson A, Johnson CB, Neitzke DJ, Mehrotra S, Denlinger CE, et al. IL-2 and beyond in cancer immunotherapy. J Interferon Cytokine Res (2018) 38(2):45–68. doi: 10.1089/jir.2017.0101
120. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12(3):180–90. doi: 10.1038/nri3156
121. Hirakawa M, Matos T, Liu H, Koreth J, Kim HT, Paul NE, et al. Low-dose IL-2 selectively activates subsets of CD4+ tregs and NK cells. JCI Insight (2016) 1(18):e89278. doi: 10.1172/jci.insight.89278
122. Abbas AK, Trotta E D, Marson A, Bluestone JA. Revisiting IL-2: Biology and therapeutic prospects. Sci Immunol (2018) 3(25):eaat1482. doi: 10.1126/sciimmunol.aat1482
123. Zhao Z, Zhang X, Su L, Xu L, Zheng Y, Sun J. Fine tuning subsets of CD4+ T cells by low-dosage of IL-2 and a new therapeutic strategy for autoimmune diseases. Int Immunopharmacol (2018) 56:269–76. doi: 10.1016/j.intimp.2018.01.042
124. Johnson MC, Garland AL, Nicolson SC, Li C, Samulski RJ, Wang B, et al. β-Cell–specific IL-2 therapy increases islet Foxp3+ treg and suppresses type 1 diabetes in NOD mice. Diabetes (2013) 62(11):3775–84. doi: 10.2337/db13-0669
125. Rosenzwajg M, Churlaud G, Mallone R, Six A, Dérian N, Chaara W, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun (2015) 58:48–58. doi: 10.1016/j.jaut.2015.01.001
126. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol (2013) 1(4):295–305. doi: 10.1016/S2213-8587(13)70113-X
127. Todd JA, Evangelou M, Cutler AJ, Pekalski ML, Walker NM, Stevens HE, et al. Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin-2: a non-randomised, open label, adaptive dose-finding trial. PloS Med (2016) 13(10):e1002139. doi: 10.1371/journal.pmed.1002139
128. Koreth J, Kim HT, Jones KT, Lange PB, Reynolds CG, Chammas MJ, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. J Am Soc Hematol (2016) 128(1):130–7. doi: 10.1182/blood-2016-02-702852
129. Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu M-F, Liu H, et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res (2014) 20(8):2215–25. doi: 10.1158/1078-0432.CCR-13-3205
130. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheumatic Dis (2016) 75(7):1407–15. doi: 10.1136/annrheumdis-2015-207776
131. Humrich JY, von Spee-Mayer C, Siegert E, Alexander T, Hiepe F, Radbruch A, et al. Rapid induction of clinical remission by low-dose interleukin-2 in a patient with refractory SLE. Ann Rheumatic Dis (2015) 74(4):791–2. doi: 10.1136/annrheumdis-2014-206506
132. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat Med (2016) 22(9):991–3. doi: 10.1038/nm.4148
133. Schmidt A, Rieger CC, Venigalla RK, Éliás S, Max R, Lorenz H-M, et al. Analysis of FOXP3+ regulatory T cell subpopulations in peripheral blood and tissue of patients with systemic lupus erythematosus. Immunol Res (2017) 65(2):551–63. doi: 10.1007/s12026-017-8904-4
134. Miao M, Hao Z, Guo Y, Zhang X, Zhang S, Luo J, et al. Short-term and low-dose IL-2 therapy restores the Th17/Treg balance in the peripheral blood of patients with primary sjögren’s syndrome. Ann Rheumatic Dis (2018) 77(12):1838–40. doi: 10.1136/annrheumdis-2018-213036
135. Peterson LB, Bell CJM, Howlett SK, Pekalski ML, Brady K, Hinton H, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun (2018) 95:1–14. doi: 10.1016/j.jaut.2018.10.017
136. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science (2018) 359(6379):1037–42. doi: 10.1126/science.aar3246
137. Sabatini DM. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc Natl Acad Sci United States America (2017) 114(45):11818–25. doi: 10.1073/pnas.1716173114
138. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in treg cells maintains homeostasis and lineage stability. Nat Immunol (2015) 16(2):188–96. doi: 10.1038/ni.3077
139. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30(6):832–44. doi: 10.1016/j.immuni.2009.04.014
140. Manirarora JN, Wei C-H. Combination therapy using IL-2/IL-2 monoclonal antibody complexes, rapamycin, and islet autoantigen peptides increases regulatory T cell frequency and protects against spontaneous and induced type 1 diabetes in nonobese diabetic mice. J Immunol (2015) 195(11):5203–14. doi: 10.4049/jimmunol.1402540
141. Schwaiger T, van den Brandt C, Fitzner B, Zaatreh S, Kraatz F, Dummer A, et al. Autoimmune pancreatitis in MRL/Mp mice is a T cell-mediated disease responsive to cyclosporine a and rapamycin treatment. Gut (2014) 63(3):494–505. doi: 10.1136/gutjnl-2012-303635
142. Gu Z, Tan W, Ji J, Feng G, Meng Y, Da Z, et al. Rapamycin reverses the senescent phenotype and improves immuno-regulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY) (2016) 8(5):1102. doi: 10.18632/aging.100925
143. Lai Z-W, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet (2018) 391(10126):1186–96. doi: 10.1016/S0140-6736(18)30485-9
144. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
145. Passerini L, Barzaghi F, Curto R, Sartirana C, Barera G, Tucci F, et al. Treatment with rapamycin can restore regulatory T-cell function in IPEX patients. J Allergy Clin Immunol (2020) 145(4):1262–71.e13. doi: 10.1016/j.jaci.2019.11.043
146. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov (2014) 13(9):673–91. doi: 10.1038/nrd4360
147. Nijhuis L, Peeters JGC, Vastert SJ, van Loosdregt J. Restoring T cell tolerance, exploring the potential of histone deacetylase inhibitors for the treatment of juvenile idiopathic arthritis. Front Immunol (2019) 10:151. doi: 10.3389/fimmu.2019.00151
148. Huang J, Wang L, Dahiya S, Beier UH, Han R, Samanta A, et al. Histone/protein deacetylase 11 targeting promotes Foxp3+ treg function. Sci Rep (2017) 7(1):1–11. doi: 10.1038/s41598-017-09211-3
149. Choi SW, Braun T, Chang L, Ferrara JL, Pawarode A, Magenau JM, et al. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol (2014) 15(1):87–95. doi: 10.1016/S1470-2045(13)70512-6
150. Oh BR, Suh D-H, Bae D, Ha N, Choi YI, Yoo HJ, et al. Therapeutic effect of a novel histone deacetylase 6 inhibitor, CKD-l, on collagen-induced arthritis in vivo and regulatory T cells in rheumatoid arthritis in vitro. Arthritis Res Ther (2017) 19(1):1–16. doi: 10.1186/s13075-017-1357-2
151. Welner RS, Pelayo R, Kincade PW. Evolving views on the genealogy of b cells. Nat Rev Immunol (2008) 8(2):95–106. doi: 10.1038/nri2234
152. Mizoguchi A, Bhan AK. A case for regulatory b cells. J Immunol (2006) 176(2):705–10. doi: 10.4049/jimmunol.176.2.705
153. Lundy SK. Killer b lymphocytes: the evidence and the potential. Inflammation Res (2009) 58(7):345–57. doi: 10.1007/s00011-009-0014-x
154. Gray D, Gray M. What are regulatory b cells? Eur J Immunol (2010) 40(10):2677–9. doi: 10.1002/eji.201040961
155. DiLillo DJ, Matsushita T, Tedder TF. B10 cells and regulatory b cells balance immune responses during inflammation, autoimmunity, and cancer. Ann New York Acad Sci (2010) 1183(1):38–57. doi: 10.1111/j.1749-6632.2009.05137.x
156. Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19+ CD24hiCD38hi b cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity (2010) 32(1):129–40. doi: 10.1016/j.immuni.2009.11.009
157. Iwata Y, Matsushita T, Horikawa M, DiLillo DJ, Yanaba K, Venturi GM, et al. Characterization of a rare IL-10–competent b-cell subset in humans that parallels mouse regulatory B10 cells. J Am Soc Hematol (2011) 117(2):530–41. doi: 10.1182/blood-2010-07-294249
158. Yanaba K, Bouaziz J-D, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory b cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity (2008) 28(5):639–50. doi: 10.1016/j.immuni.2008.03.017
159. Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, et al. Novel suppressive function of transitional 2 b cells in experimental arthritis. J Immunol (2007) 178(12):7868–78. doi: 10.4049/jimmunol.178.12.7868
160. Yang M, Sun L, Wang S, Ko K-H, Xu H, Zheng B-J, et al. Cutting edge: novel function of b cell-activating factor in the induction of IL-10–producing regulatory b cells. J Immunol (2010) 184(7):3321–5. doi: 10.4049/jimmunol.0902551
161. Mauri C. Regulation of immunity and autoimmunity by b cells. Curr Opin Immunol (2010) 22(6):761–7. doi: 10.1016/j.coi.2010.10.009
162. Blair PA, Chavez-Rueda KA, Evans JG, Shlomchik MJ, Eddaoudi A, Isenberg DA, et al. Selective targeting of b cells with agonistic anti-CD40 is an efficacious strategy for the generation of induced regulatory T2-like b cells and for the suppression of lupus in MRL/lpr mice. J Immunol (2009) 182(6):3492–502. doi: 10.4049/jimmunol.0803052
163. Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory b cells. Proc Natl Acad Sci (2007) 104(35):14080–5. doi: 10.1073/pnas.0700326104
164. Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, et al. Mice lacking endogenous IL-10–producing regulatory b cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol (2011) 186(10):5569–79. doi: 10.4049/jimmunol.1100284
165. Mauri C. CD19 CD24 hi CD38 hi b cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity (2010) 32:129–40. doi: 10.1016/j.immuni.2009.11.009
166. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity (2014) 41(6):1040–51. doi: 10.1016/j.immuni.2014.10.016
167. Zha B, Wang L, Liu X, Liu J, Chen Z, Xu J, et al. Decrease in proportion of CD19+ CD24(hi) CD27+ b cells and impairment of their suppressive function in graves’ disease. PloS One (2012) 7(11):e49835. doi: 10.1371/journal.pone.0049835
168. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+ CD24hiCD38hi b cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Trans Med (2013) 5(173):173ra23–ra23. doi: 10.1126/scitranslmed.3005407
169. van de Veen W, Stanic B, Yaman G, Wawrzyniak M, Söllner S, Akdis DG, et al. IgG4 production is confined to human IL-10–producing regulatory b cells that suppress antigen-specific immune responses. J Allergy Clin Immunol (2013) 131(4):1204–12. doi: 10.1016/j.jaci.2013.01.014
170. Nouel A, Pochard P, Simon Q, Segalen I, Le Meur Y, Pers J, et al. B-cells induce regulatory T cells through TGF-β/IDO production in a CTLA-4 dependent manner. J Autoimmun (2015) 59:53–60. doi: 10.1016/j.jaut.2015.02.004
171. Bosma A, Abdel-Gadir A, Isenberg DA, Jury EC, Mauri C. Lipid-antigen presentation by CD1d+ b cells is essential for the maintenance of invariant natural killer T cells. Immunity (2012) 36(3):477–90. doi: 10.1016/j.immuni.2012.02.008
172. Saze Z, Schuler PJ, Hong C-S, Cheng D, Jackson EK, Whiteside TL. Adenosine production by human b cells and b cell–mediated suppression of activated T cells. J Am Soc Hematol (2013) 122(1):9–18. doi: 10.1182/blood-2013-02-482406
173. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing b cells are critical regulators of immunity during autoimmune and infectious diseases. Nature (2014) 507(7492):366–70. doi: 10.1038/nature12979
174. Wang R-X, Yu C-R, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory b cells that suppress autoimmune disease. Nat Med (2014) 20(6):633–41. doi: 10.1038/nm.3554
175. Jansen K, Cevhertas L, Ma S, Satitsuksanoa P, Akdis M, van de Veen W. Regulatory b cells, a to z. Allergy (2021) 76(9):2699–715. doi: 10.1111/all.14763
176. Mavropoulos A, Zafiriou E, Dardiotis E, Sakkas LI, Bogdanos DP. B regulatory cells in patients with autoimmune diseases: Pathogenic significance and therapeutic potential. Trans Autoimmun: Elsevier (2022) p:37–53. doi: 10.1016/B978-0-12-824390-9.00021-9
177. Knippenberg S, Peelen E, Smolders J, Thewissen M, Menheere P, Tervaert JWC, et al. Reduction in IL-10 producing b cells (Breg) in multiple sclerosis is accompanied by a reduced naive/memory breg ratio during a relapse but not in remission. J Neuroimmunol (2011) 239(1-2):80–6. doi: 10.1016/j.jneuroim.2011.08.019
178. Hayashi M, Yanaba K, Umezawa Y, Yoshihara Y, Kikuchi S, Ishiuji Y, et al. IL-10-producing regulatory b cells are decreased in patients with psoriasis. J Dermatol Sci (2016) 81(2):93–100. doi: 10.1016/j.jdermsci.2015.11.003
179. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3(10):944–50. doi: 10.1038/ni833
180. Carter NA, Rosser EC, Mauri C. Interleukin-10 produced by b cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res Ther (2012) 14(1):1–9. doi: 10.1186/ar3736
181. Matsushita T, Yanaba K, Bouaziz J-D, Fujimoto M, Tedder TF. Regulatory b cells inhibit EAE initiation in mice while other b cells promote disease progression. J Clin Invest (2008) 118(10):3420–30. doi: 10.1172/JCI36030
182. Menon M, Blair PA, Isenberg DA, Mauri C. A regulatory feedback between plasmacytoid dendritic cells and regulatory b cells is aberrant in systemic lupus erythematosus. Immunity (2016) 44(3):683–97. doi: 10.1016/j.immuni.2016.02.012
183. Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, et al. Regulatory b cells are induced by gut microbiota–driven interleukin-1β and interleukin-6 production. Nat Med (2014) 20(11):1334–9. doi: 10.1038/nm.3680
184. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory b cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491(7423):264–8. doi: 10.1038/nature11501
185. Schubert RD, Hu Y, Kumar G, Szeto S, Abraham P, Winderl J, et al. IFN-β treatment requires b cells for efficacy in neuroautoimmunity. J Immunol (2015) 194(5):2110–6. doi: 10.4049/jimmunol.1402029
186. Rafei M, Hsieh J, Zehntner S, Li M, Forner K, Birman E, et al. A granulocyte-macrophage colony–stimulating factor and interleukin-15 fusokine induces a regulatory b cell population with immune suppressive properties. Nat Med (2009) 15(9):1038–45. doi: 10.1038/nm.2003
187. Genestier L, Taillardet M, Mondiere P, Gheit H, Bella C, Defrance T. TLR agonists selectively promote terminal plasma cell differentiation of b cell subsets specialized in thymus-independent responses. J Immunol (2007) 178(12):7779–86. doi: 10.4049/jimmunol.178.12.7779
188. Grammer AC, Lipsky PE. CD154–CD40 interactions mediate differentiation to plasma cells in healthy individuals and persons with systemic lupus erythematosus. Arthritis Rheumatism: Off J Am Coll Rheumatol (2002) 46(6):1417–29. doi: 10.1002/art.10287
189. Correale J, Farez M, Razzitte G. Helminth infections associated with multiple sclerosis induce regulatory b cells. Ann Neurol (2008) 64(2):187–99. doi: 10.1002/ana.21438
190. Grützke B, Hucke S, Gross CC, Herold MV, Posevitz-Fejfar A, Wildemann BT, et al. Fingolimod treatment promotes regulatory phenotype and function of b cells. Ann Clin Trans Neurol (2015) 2(2):119–30. doi: 10.1002/acn3.155
191. Thompson SA, Jones JL, Cox AL, Compston DAS, Coles AJ. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol (2010) 30(1):99–105. doi: 10.1007/s10875-009-9327-3
192. Heidt S, Hester J, Shankar S, Friend PJ, Wood K. B cell repopulation after alemtuzumab induction–transient increase in transitional b cells and long-term dominance of naïve b cells. Am J Transplantation (2012) 12(7):1784–92. doi: 10.1111/j.1600-6143.2012.04012.x
193. Heinemann K, Wilde B, Hoerning A, Tebbe B, Kribben A, Witzke O, et al. Decreased IL-10+ regulatory b cells (Bregs) in lupus nephritis patients. Scandinavian J Rheumatol (2016) 45(4):312–6. doi: 10.3109/03009742.2015.1126346
194. Gao N, Dresel J, Eckstein V, Gellert R, Störch H, Venigalla RK, et al. Impaired suppressive capacity of activation-induced regulatory b cells in systemic lupus erythematosus. Arthritis Rheumatol (2014) 66(10):2849–61. doi: 10.1002/art.38742
195. Kim J, Lee HJ, Yoo IS, Kang SW, Lee JH. Regulatory b cells are inversely associated with disease activity in rheumatoid arthritis. Yonsei Med J (2014) 55(5):1354–8. doi: 10.3349/ymj.2014.55.5.1354
196. Zhu HQ, Xu RC, Chen YY, Yuan HJ, Cao H, Zhao XQ, et al. Impaired function of CD 19+ CD 24hi CD 38hi regulatory b cells in patients with pemphigus. Br J Dermatol (2015) 172(1):101–10. doi: 10.1111/bjd.13192
197. Amagai M. Pemphigus: autoimmunity to epidermal cell adhesion molecules. Adv Dermatol (1996) 11:319–52.
198. Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, Lim A, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein b cell response. Sci Trans Med (2013) 5(175):175ra30–ra30. doi: 10.1126/scitranslmed.3005166
199. Bing X, Linlang L, Keyan C. Decreased Breg/Th17 ratio improved the prognosis of patients with ulcerative colitis. Can J Gastroenterol Hepatol (2018) 2018:5760849–. doi: 10.1155/2018/5760849
200. Mavropoulos A, Simopoulou T, Varna A, Liaskos C, Katsiari CG, Bogdanos DP, et al. Breg cells are numerically decreased and functionally impaired in patients with systemic sclerosis. Arthritis Rheumatol (2016) 68(2):494–504. doi: 10.1002/art.39437
201. Matsushita T, Hamaguchi Y, Hasegawa M, Takehara K, Fujimoto M. Decreased levels of regulatory b cells in patients with systemic sclerosis: association with autoantibody production and disease activity. Rheumatology (2016) 55(2):263–7. doi: 10.1093/rheumatology/kev331
202. Kleffel S, Vergani A, Tezza S, Nasr MB, Niewczas MA, Wong S, et al. Interleukin-10+ regulatory b cells arise within antigen-experienced CD40+ b cells to maintain tolerance to islet autoantigens. Diabetes (2015) 64(1):158–71. doi: 10.2337/db13-1639
203. Oka A, Ishihara S, Mishima Y, Tada Y, Kusunoki R, Fukuba N, et al. Role of regulatory b cells in chronic intestinal inflammation: association with pathogenesis of crohn’s disease. Inflammatory Bowel Dis (2014) 20(2):315–28. doi: 10.1097/01.MIB.0000437983.14544.d5
204. Mauri C, Menon M. Human regulatory b cells in health and disease: therapeutic potential. J Clin Invest (2017) 127(3):772–9. doi: 10.1172/JCI85113
205. Miyagaki T, Fujimoto M, Sato S. Regulatory b cells in human inflammatory and autoimmune diseases: from mouse models to clinical research. Int Immunol (2015) 27(10):495–504. doi: 10.1093/intimm/dxv026
206. Ramos-Casals M, Soto M, Cuadrado M, Khamashta M. Rituximab in systemic lupus erythematosusA systematic review of off-label use in 188 cases. Lupus (2009) 18(9):767–76. doi: 10.1177/0961203309106174
207. Aguiar R, Araújo C, Martins-Coelho G, Isenberg D. Use of rituximab in systemic lupus erythematosus: a single center experience over 14 years. Arthritis Care Res (2017) 69(2):257–62. doi: 10.1002/acr.22921
208. Gan PY, Dick J, O’Sullivan KM, Oudin V, Cao Le A, Koo Yuk Cheong D, et al. Anti-CD20 mAb-induced b cell apoptosis generates T cell regulation of experimental myeloperoxidase ANCA-associated vasculitis. J Am Soc Nephrol (2021) 32(5):1071–83. doi: 10.1681/ASN.2020060834
209. Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory b cell subset characterized by CD1d upregulation. Immunity (2002) 16(2):219–30. doi: 10.1016/S1074-7613(02)00274-1
210. Steinman RM, Witmer MD. Lymphoid dendritic cells are potent stimulators of the primary mixed leukocyte reaction in mice. Proc Natl Acad Sci (1978) 75(10):5132–6. doi: 10.1073/pnas.75.10.5132
211. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu Y-J, et al. Immunobiology of dendritic cells. Annu Rev Immunol (2000) 18(1):767–811. doi: 10.1146/annurev.immunol.18.1.767
212. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol (2003) 21(1):685–711. doi: 10.1146/annurev.immunol.21.120601.141040
213. Schmidt SV, Nino-Castro AC, Schultze JL. Regulatory dendritic cells: there is more than just immune activation. Front Immunol (2012) 3:274. doi: 10.3389/fimmu.2012.00274
214. Wu J, Horuzsko A. Expression and function of immunoglobulin-like transcripts on tolerogenic dendritic cells. Hum Immunol (2009) 70(5):353–6. doi: 10.1016/j.humimm.2009.01.024
215. Ezzelarab M, Thomson AW. Tolerogenic dendritic cells and their role in transplantation. Semin Immunol. (2011) 23(4):252–63. doi: 10.1016/j.smim.2011.06.007
216. Lo J, Clare-Salzler MJ. Dendritic cell subsets and type I diabetes: focus upon DC-based therapy. Autoimmun Rev (2006) 5(6):419–23. doi: 10.1016/j.autrev.2005.12.001
217. Kalantari T, Kamali-Sarvestani E, Zhang G-X, Safavi F, Lauretti E, Khedmati M-E, et al. Generation of large numbers of highly purified dendritic cells from bone marrow progenitor cells after co-culture with syngeneic murine splenocytes. Exp Mol Pathol (2013) 94(2):336–42. doi: 10.1016/j.yexmp.2012.12.001
218. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature (2007) 449(7161):419–26. doi: 10.1038/nature06175
219. Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol (2007) 7(8):610–21. doi: 10.1038/nri2132
220. Manicassamy S, Pulendran B. Dendritic cell control of tolerogenic responses. Immunol Rev (2011) 241(1):206–27. doi: 10.1111/j.1600-065X.2011.01015.x
221. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med (1992) 176(6):1693–702. doi: 10.1084/jem.176.6.1693
222. Goxe B, Latour N, Chokri M, Abastado J, Salcedo M. Simplified method to generate large quantities of dendritic cells suitable for clinical applications. Immunol Investigations (2000) 29(3):319–36. doi: 10.3109/08820130009060870
223. Romani N, Gruner S, Brang D, Kämpgen E, Lenz A, Trockenbacher B, et al. Proliferating dendritic cell progenitors in human blood. J Exp Med (1994) 180(1):83–93. doi: 10.1084/jem.180.1.83
224. Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol (2010) 108:111–65. doi: 10.1016/B978-0-12-380995-7.00004-5
225. Jonuleit H, Schmitt E, Steinbrink K, Enk AH. Dendritic cells as a tool to induce anergic and regulatory T cells. Trends Immunol (2001) 22(7):394–400. doi: 10.1016/S1471-4906(01)01952-4
226. Corinti S, Albanesi C, la Sala A, Pastore S, Girolomoni G. Regulatory activity of autocrine IL-10 on dendritic cell functions. J Immunol (2001) 166(7):4312–8. doi: 10.4049/jimmunol.166.7.4312
227. Laouar Y, Town T, Jeng D, Tran E, Wan Y, Kuchroo VK, et al. TGF-β signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci (2008) 105(31):10865–70. doi: 10.1073/pnas.0805058105
228. Széles L, Keresztes G, Töröcsik D, Balajthy Z, Krenács L, Póliska S, et al. 1,25-dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. J Immunol (2009) 182(4):2074–83. doi: 10.4049/jimmunol.0803345
229. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol (2002) 3(7):673–80. doi: 10.1038/ni805
230. Hackstein H, Thomson AW. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat Rev Immunol (2004) 4(1):24–34. doi: 10.1038/nri1256
231. Adorini L, Giarratana N, Penna G. Pharmacological induction of tolerogenic dendritic cells and regulatory T cells. Semin Immunol (2004) 16(2):127–34. doi: 10.1016/j.smim.2003.12.008
232. Martin E, Capini C, Duggan E, Lutzky VP, Stumbles P, Pettit AR, et al. Antigen-specific suppression of established arthritis in mice by dendritic cells deficient in NF-kappaB. Arthritis Rheumatism (2007) 56(7):2255–66. doi: 10.1002/art.22655
233. Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol (2009) 39(11):3147–59. doi: 10.1002/eji.200839103
234. Fischer R, Turnquist HR, Taner T, Thomson AW. Use of rapamycin in the induction of tolerogenic dendritic cells. Handb Exp Pharmacol (2009) 188:215–32. doi: 10.1007/978-3-540-71029-5_10
235. Humbert J-M, Halary F. Viral and non-viral methods to genetically modify dendritic cells. Curr Gene Ther (2012) 12(2):127–36. doi: 10.2174/156652312800099580
236. Fjose A, Ellingsen S, Wargelius A, Seo H-C. RNA Interference: mechanisms and applications. Biotechnol Annu Rev (2001) 7:31–57. doi: 10.1016/s1387-2656(01)07032-6
237. Hilkens CM, Isaacs JD, Thomson AW. Development of dendritic cell-based immunotherapy for autoimmunity. Int Rev Immunol (2010) 29(2):156–83. doi: 10.3109/08830180903281193
238. Yang D-F, Qiu W-H, Zhu H-F, Lei P, Wen X, Dai H, et al. CTLA4-ig-modified dendritic cells inhibit lymphocyte-mediated alloimmune responses and prolong the islet graft survival in mice. Transplant Immunol (2008) 19(3-4):197–201. doi: 10.1016/j.trim.2008.05.005
239. Ruffner MA, Robbins PD. Dendritic cells transduced to express interleukin 4 reduce diabetes onset in both normoglycemic and prediabetic nonobese diabetic mice. PloS One (2010) 5(7):e11848. doi: 10.1371/journal.pone.0011848
240. Tomasoni S, Aiello S, Cassis L, Noris M, Longaretti L, Cavinato RA, et al. Dendritic cells genetically engineered with adenoviral vector encoding dnIKK2 induce the formation of potent CD4+ T-regulatory cells. Transplantation (2005) 79(9):1056–61. doi: 10.1097/01.TP.0000161252.17163.31
241. Naranjo-Gómez M, Raich-Regue D, Onate C, Grau-Lopez L, Ramo-Tello C, Pujol-Borrell R, et al. Comparative study of clinical grade human tolerogenic dendritic cells. J Trans Med (2011) 9(1):1–14. doi: 10.1186/1479-5876-9-89
242. Boks MA, Kager-Groenland JR, Haasjes MS, Zwaginga JJ, van Ham SM, ten Brinke A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction–a comparative study of human clinical-applicable DC. Clin Immunol (2012) 142(3):332–42. doi: 10.1016/j.clim.2011.11.011
243. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol (2013) 13(8):566–77. doi: 10.1038/nri3477
244. Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med (2009) 206(3):549–59. doi: 10.1084/jem.20082394
245. Lang KS, Burow A, Kurrer M, Lang PA, Recher M. The role of the innate immune response in autoimmune disease. J Autoimmun (2007) 29(4):206–12. doi: 10.1016/j.jaut.2007.07.018
246. O’Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol (2002) 2(1):37–45. doi: 10.1038/nri702
247. Lleo A, Selmi C, Invernizzi P, Podda M, Gershwin ME. The consequences of apoptosis in autoimmunity. J Autoimmun (2008) 31(3):257–62. doi: 10.1016/j.jaut.2008.04.009
248. Schett G, Elewaut D, McInnes IB, Dayer J-M, Neurath MF. How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy. Nat Med (2013) 19(7):822–4. doi: 10.1038/nm.3260
249. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature (2011) 474(7351):298–306. doi: 10.1038/nature10208
250. Pierik M, Joossens S, Van Steen K, Van Schuerbeek N, Vlietinck R, Rutgeerts P, et al. Toll-like receptor-1,-2, and-6 polymorphisms influence disease extension in inflammatory bowel diseases. Inflammatory Bowel Dis (2006) 12(1):1–8. doi: 10.1097/01.MIB.0000195389.11645.ab
251. Hugot J-P, Chamaillard M, Zouali H, Lesage S, Cézard J-P, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to crohn’s disease. Nature (2001) 411(6837):599–603. doi: 10.1038/35079107
252. Pedersen AE, Schmidt EGW, Gad M, Poulsen SS, Claesson MH. Dexamethasone/1α-25-dihydroxyvitamin D3-treated dendritic cells suppress colitis in the SCID T-cell transfer model. Immunology (2009) 127(3):354–64. doi: 10.1111/j.1365-2567.2008.02996.x
253. Pedersen A, Gad M, Kristensen N, Haase C, Nielsen C, Claesson M. Tolerogenic dendritic cells pulsed with enterobacterial extract suppress development of colitis in the severe combined immunodeficiency transfer model. Immunology (2007) 121(4):526–32. doi: 10.1111/j.1365-2567.2007.02600.x
254. Kassianos AJ, Hardy MY, Ju X, Vijayan D, Ding Y, Vulink AJ, et al. Human CD 1c (BDCA-1)+ myeloid dendritic cells secrete IL-10 and display an immuno-regulatory phenotype and function in response to e scherichia coli. Eur J Immunol (2012) 42(6):1512–22. doi: 10.1002/eji.201142098
255. Qian C, Cao X. Naturally occurring CD 1c+ human regulatory dendritic cells: Immunoregulators that are expanded in response to e. Coli Infect Eur J Immunol (2012) 42(6):1388–92. doi: 10.1002/eji.201242632
256. Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, et al. Human liver dendritic cells promote T cell hyporesponsiveness. J Immunol (2009) 182(4):1901–11. doi: 10.4049/jimmunol.0803404
257. Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheumatic Dis (2010) 69(11):2042–50. doi: 10.1136/ard.2009.126383
258. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34(9):2026–32. doi: 10.2337/dc11-0472
259. Hilkens C, Isaacs J. Tolerogenic dendritic cell therapy for rheumatoid arthritis: where are we now? Clin Exp Immunol (2013) 172(2):148–57. doi: 10.1111/cei.12038
260. Thomson AW, Ezzelarab MB. Generation and functional assessment of nonhuman primate regulatory dendritic cells and their therapeutic efficacy in renal transplantation. Cell Immunol (2020) 351:104087. doi: 10.1016/j.cellimm.2020.104087
261. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype–positive rheumatoid arthritis patients. Sci Trans Med (2015) 7(290):290ra87–ra87. doi: 10.1126/scitranslmed.aaa9301
262. Jauregui-Amezaga A, Cabezón R, Ramírez-Morros A, España C, Rimola J, Bru C, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory crohn’s disease: a phase I study. J Crohn’s Colitis (2015) 9(12):1071–8. doi: 10.1093/ecco-jcc/jjv144
263. Zubizarreta I, Flórez-Grau G, Vila G, Cabezón R, España C, Andorra M, et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc Natl Acad Sci (2019) 116(17):8463–70. doi: 10.1073/pnas.1820039116
264. Macedo C, Tran LM, Zahorchak AF, Dai H, Gu X, Ravichandran R, et al. Donor-derived regulatory dendritic cell infusion results in host cell cross-dressing and T cell subset changes in prospective living donor liver transplant recipients. Am J Transplantation (2021) 21(7):2372–86. doi: 10.1111/ajt.16393
265. Bell G, Anderson A, Diboll J, Reece R, Eltherington O, Harry R, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheumatic Dis (2017) 76(1):227–34. doi: 10.1136/annrheumdis-2015-208456
266. Martin E, Capini C, Duggan E, Lutzky VP, Stumbles P, Pettit AR, et al. Antigen-specific suppression of established arthritis in mice by dendritic cells deficient in NF-κB. Arthritis Rheumatism: Off J Am Coll Rheumatol (2007) 56(7):2255–66. doi: 10.1002/art.22655
267. Thomas R. Dendritic cells and the promise of antigen-specific therapy in rheumatoid arthritis. Arthritis Res Ther (2013) 15(1):1–10. doi: 10.1186/ar4130
268. Stoop JN, Robinson JH, Hilkens CM. Developing tolerogenic dendritic cell therapy for rheumatoid arthritis: what can we learn from mouse models? Ann Rheumatic Dis (2011) 70(9):1526–33. doi: 10.1136/ard.2011.151654
269. Stoop JN, Harry RA, von Delwig A, Isaacs JD, Robinson JH, Hilkens CM. Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheumatism (2010) 62(12):3656–65. doi: 10.1002/art.27756
270. Thomas R, Street S, Ramnoruth N. Safety and preliminary evidence of efficacy in a phase I clinical trial of autologous tolerising dendritic cells exposed to citrullinated peptides (Rheumavax) in patients with rheumatoid arthritis. Ann Rheum Dis (2011) 70(Suppl 3):169.
271. Gabrilovich DI, Bronte V, Chen S-H, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425–. doi: 10.1158/0008-5472.CAN-06-3037
272. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
273. Dufait I, Schwarze JK, Liechtenstein T, Leonard W, Jiang H, Escors D, et al. Ex vivo generation of myeloid-derived suppressor cells that model the tumor immunosuppressive environment in colorectal cancer. Oncotarget (2015) 6(14):12369. doi: 10.18632/oncotarget.3682
274. Nicholson LB, Raveney BJ, Munder M. Monocyte dependent regulation of autoimmune inflammation. Curr Mol Med (2009) 9(1):23–9. doi: 10.2174/156652409787314499
275. Iwata Y, Furuichi K, Kitagawa K, Hara A, Okumura T, Kokubo S, et al. Involvement of CD11b+ GR-1 low cells in autoimmune disorder in MRL-fas lpr mouse. Clin Exp Nephrol (2010) 14(5):411–7. doi: 10.1007/s10157-010-0309-9
276. Wegner A, Verhagen J, Wraith DC. Myeloid-derived suppressor cells mediate tolerance induction in autoimmune disease. Immunology (2017) 151(1):26–42. doi: 10.1111/imm.12718
277. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer (2019) 120(1):16–25. doi: 10.1038/s41416-018-0333-1
278. Hu C, Du W, Zhang X, Wong FS, Wen L. The role of Gr1+ cells after anti-CD20 treatment in type 1 diabetes in nonobese diabetic mice. J Immunol (2012) 188(1):294–301. doi: 10.4049/jimmunol.1101590
279. Park MJ, Lee SH, Kim EK, Lee EJ, Park SH, Kwok SK, et al. Myeloid-derived suppressor cells induce the expansion of regulatory b cells and ameliorate autoimmunity in the sanroque mouse model of systemic lupus erythematosus. Arthritis Rheumatol (2016) 68(11):2717–27. doi: 10.1002/art.39767
280. Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK, et al. CD11b+ ly-6Chi suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol (2007) 179(8):5228–37. doi: 10.4049/jimmunol.179.8.5228
281. Iacobaeus E, Douagi I, Jitschin R, Marcusson-Ståhl M, Andrén AT, Gavin C, et al. Phenotypic and functional alterations of myeloid-derived suppressor cells during the disease course of multiple sclerosis. Immunol Cell Biol (2018) 96(8):820–30. doi: 10.1111/imcb.12042
282. King IL, Dickendesher TL, Segal BM. Circulating ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. J Am Soc Hematol (2009) 113(14):3190–7. doi: 10.1182/blood-2008-07-168575
283. Fujii W, Ashihara E, Hirai H, Nagahara H, Kajitani N, Fujioka K, et al. Myeloid-derived suppressor cells play crucial roles in the regulation of mouse collagen-induced arthritis. J Immunol (2013) 191(3):1073–81. doi: 10.4049/jimmunol.1203535
284. Kurmaeva E, Bhattacharya D, Goodman W, Omenetti S, Merendino A, Berney S, et al. Immunosuppressive monocytes: possible homeostatic mechanism to restrain chronic intestinal inflammation. J Leukocyte Biol (2014) 96(3):377–89. doi: 10.1189/jlb.3HI0613-340RR
285. Haile LA, Von Wasielewski R, Gamrekelashvili J, Krüger C, Bachmann O, Westendorf AM, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: a new immunoregulatory pathway. Gastroenterology (2008) 135(3):871–81.e5. doi: 10.1053/j.gastro.2008.06.032
286. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
287. Ribechini E, Greifenberg V, Sandwick S, Lutz MB. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol (2010) 199(3):273–81. doi: 10.1007/s00430-010-0151-4
288. Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget (2017) 8(2):3649. doi: 10.18632/oncotarget.12278
289. Fujio K, Okamura T, Sumitomo S, Yamamoto K. Regulatory T cell-mediated control of autoantibody-induced inflammation. Front Immunol (2012) 3:28. doi: 10.3389/fimmu.2012.00028
290. Hammerich L, Warzecha KT, Stefkova M, Bartneck M, Ohl K, Gassler N, et al. Cyclic adenosine monophosphate–responsive element modulator alpha overexpression impairs function of hepatic myeloid-derived suppressor cells and aggravates immune-mediated hepatitis in mice. Hepatology (2015) 61(3):990–1002. doi: 10.1002/hep.27571
291. Bromberg J. Stat proteins and oncogenesis. J Clin Invest (2002) 109(9):1139–42. doi: 10.1172/JCI0215617
292. Levy DE, Darnell J. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 3(9):651–62. doi: 10.1038/nrm909
293. Haverkamp JM, Crist SA, Elzey BD, Cimen C, Ratliff TL. In vivo suppressive function of myeloid-derived suppressor cells is limited to the inflammatory site. Eur J Immunol (2011) 41(3):749–59. doi: 10.1002/eji.201041069
294. Ohl K, Tenbrock K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
295. Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, et al. Myeloid cell-derived arginase in cancer immune response. Front Immunol (2020) 11:938. doi: 10.3389/fimmu.2020.00938
296. Cripps JG, Wang J, Maria A, Blumenthal I, Gorham JD. Type 1 T helper cells induce the accumulation of myeloid-derived suppressor cells in the inflamed Tgfb1 knockout mouse liver. Hepatology (2010) 52(4):1350–9. doi: 10.1002/hep.23841
297. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J Immunol (2009) 182(1):240–9. doi: 10.4049/jimmunol.182.1.240
298. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi: 10.1038/nri1457
299. Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+ CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299
300. Knier B, Hiltensperger M, Sie C, Aly L, Lepennetier G, Engleitner T, et al. Myeloid-derived suppressor cells control b cell accumulation in the central nervous system during autoimmunity. Nat Immunol (2018) 19(12):1341–51. doi: 10.1038/s41590-018-0237-5
301. Chou H-S, Hsieh C-C, Charles R, Wang L, Wagner T, Fung JJ, et al. Myeloid-derived suppressor cells (MDSC) protect islet transplants via B7-H1 mediated enhancement of T regulatory cells. Transplantation (2012) 93(3):272. doi: 10.1097/TP.0b013e31823ffd39
302. Kurkó J, Vida A, Ocskó T, Tryniszewska B, Rauch TA, Glant TT, et al. Suppression of proteoglycan-induced autoimmune arthritis by myeloid-derived suppressor cells generated in vitro from murine bone marrow. PloS One (2014) 9(11):e111815. doi: 10.1371/journal.pone.0111815
303. Wang W, Jiao Z, Duan T, Liu M, Zhu B, Zhang Y, et al. Functional characterization of myeloid-derived suppressor cell subpopulations during the development of experimental arthritis. Eur J Immunol (2015) 45(2):464–73. doi: 10.1002/eji.201444799
304. Nishimura K, Saegusa J, Matsuki F, Akashi K, Kageyama G, Morinobu A. Tofacitinib facilitates the expansion of myeloid-derived suppressor cells and ameliorates arthritis in SKG mice. Arthritis Rheumatol (2015) 67(4):893–902. doi: 10.1002/art.39007
305. Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Müller M, et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med (2010) 207(7):1453–64. doi: 10.1084/jem.20091474
306. Lourenço EV, Wong M, Hahn BH, Palma-Diaz MF, Skaggs BJ. Laquinimod delays and suppresses nephritis in lupus-prone mice and affects both myeloid and lymphoid immune cells. Arthritis Rheumatol (2014) 66(3):674–85. doi: 10.1002/art.38259
307. Trigunaite A, Khan A, Der E, Song A, Varikuti S, Jørgensen TN. Gr-1(high) CD11b+ cells suppress b cell differentiation and lupus-like disease in lupus-prone male mice. Arthritis Rheumatism (2013) 65(9):2392–402. doi: 10.1002/art.38048
308. Ma H, Wan S, Xia CQ. Immunosuppressive CD11b+ Ly6Chi monocytes in pristane-induced lupus mouse model. J Leukocyte Biol (2016) 99(6):1121–9. doi: 10.1189/jlb.3A0415-158R
309. Tu Z, Li Y, Smith D, Doller C, Sugita S, Chan C-C, et al. Myeloid suppressor cells induced by retinal pigment epithelial cells inhibit autoreactive T-cell responses that lead to experimental autoimmune uveitis. Invest Ophthalmol Visual Sci (2012) 53(2):959–66. doi: 10.1167/iovs.11-8377
310. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1–dependent mechanism that is up-regulated by interleukin-13. J Am Soc Hematol (2010) 116(25):5738–47. doi: 10.1182/blood-2010-06-287839
311. Zhou Z, French DL, Ma G, Eisenstein S, Chen Y, Divino CM, et al. Development and function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells (2010) 28(3):620–32. doi: 10.1002/stem.301
312. MacDonald KP, Rowe V, Clouston AD, Welply JK, Kuns RD, Ferrara JL, et al. Cytokine expanded myeloid precursors function as regulatory antigen-presenting cells and promote tolerance through IL-10-producing regulatory T cells. J Immunol (2005) 174(4):1841–50. doi: 10.4049/jimmunol.174.4.1841
313. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity (2010) 32(6):790–802. doi: 10.1016/j.immuni.2010.05.010
314. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol (2010) 185(4):2273–84. doi: 10.4049/jimmunol.1000901
315. Obermajer N, Kalinski P. Generation of myeloid-derived suppressor cells using prostaglandin e 2. Transplant Res (2012) 1(1):1–6. doi: 10.1186/2047-1440-1-15
316. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517(7534):293–301. doi: 10.1038/nature14189
317. Shikhagaie MM, Germar K, Bal SM, Ros XR, Spits H. Innate lymphoid cells in autoimmunity: emerging regulators in rheumatic diseases. Nat Rev Rheumatol (2017) 13(3):164–73. doi: 10.1038/nrrheum.2016.218
318. Klose CS, Diefenbach A. Transcription factors controlling innate lymphoid cell fate decisions. Curr Top Microbiol Immunol (2014) 381:215–55. doi: 10.1007/82_2014_381
319. Klose CS, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157(2):340–56. doi: 10.1016/j.cell.2014.03.030
320. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol (2012) 30:647–75. doi: 10.1146/annurev-immunol-020711-075053
321. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi: 10.1038/nri3365
322. Teunissen MB, Munneke JM, Bernink JH, Spuls PI, Te Velde A, Cheuk S, et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR+ ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol (2014) 134(9):2351–60. doi: 10.1038/jid.2014.146
323. Ren J, Feng Z, Lv Z, Chen X, Li J. Natural killer-22 cells in the synovial fluid of patients with rheumatoid arthritis are an innate source of interleukin 22 and tumor necrosis factor-α. J Rheumatol (2011) 38(10):2112–8. doi: 10.3899/jrheum.101377
324. Koo J, Kim S, Jung WJ, Lee YE, Song GG, Kim K-S, et al. Increased lymphocyte infiltration in rheumatoid arthritis is correlated with an increase in LTi-like cells in synovial fluid. Immune Netw (2013) 13(6):240–8. doi: 10.4110/in.2013.13.6.240
325. Dalbeth N, Callan MF. A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheumatism (2002) 46(7):1763–72. doi: 10.1002/art.10410
326. Dalbeth N, Gundle R, Davies RJ, Lee YG, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol (2004) 173(10):6418–26. doi: 10.4049/jimmunol.173.10.6418
327. Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells–how did we miss them? Nat Rev Immunol (2013) 13(2):75–87. doi: 10.1038/nri3349
328. Leijten EF, van Kempen TS, Boes M, Michels-van Amelsfort JM, Hijnen D, Hartgring SA, et al. Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol (2015) 67(10):2673–8. doi: 10.1002/art.39261
329. van Nieuwenhuijze A, Koenders M, Roeleveld D, Sleeman MA, van den Berg W, Wicks IP. GM-CSF as a therapeutic target in inflammatory diseases. Mol Immunol (2013) 56(4):675–82. doi: 10.1016/j.molimm.2013.05.002
330. van der Heijde D, Landewé RB, Mease PJ, McInnes IB, Conaghan PG, Pricop L, et al. Brief report: Secukinumab provides significant and sustained inhibition of joint structural damage in a phase III study of active psoriatic arthritis. Arthritis Rheumatol (2016) 68(8):1914–21. doi: 10.1002/art.39685
331. Fava RA, Kennedy SM, Wood SG, Bolstad AI, Bienkowska J, Papandile A, et al. Lymphotoxin-beta receptor blockade reduces CXCL13 in lacrimal glands and improves corneal integrity in the NOD model of sjögren’s syndrome. Arthritis Res Ther (2011) 13(6):R182. doi: 10.1186/ar3507
332. Gatumu MK, Skarstein K, Papandile A, Browning JL, Fava RA, Bolstad AI. Blockade of lymphotoxin-beta receptor signaling reduces aspects of sjögren’s syndrome in salivary glands of non-obese diabetic mice. Arthritis Res Ther (2009) 11(1):R24–R. doi: 10.1186/ar2617
333. Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol (2006) 7(4):344–53. doi: 10.1038/ni1330
334. Fava RA, Notidis E, Hunt J, Szanya V, Ratcliffe N, Ngam-Ek A, et al. A role for the lymphotoxin/LIGHT axis in the pathogenesis of murine collagen-induced arthritis. J Immunol (2003) 171(1):115–26. doi: 10.4049/jimmunol.171.1.115
335. Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med (2012) 4(145):145ra06. doi: 10.1126/scitranslmed.3004140
Keywords: autoimmunity, regulatory immune cells, immune suppressive cells, adoptive cell therapy, regulatory T cell, tolerogenic dendritic cell, IL-10-producing cell, regulatory B cell
Citation: Ghobadinezhad F, Ebrahimi N, Mozaffari F, Moradi N, Beiranvand S, Pournazari M, Rezaei-Tazangi F, Khorram R, Afshinpour M, Robino RA, Aref AR and Ferreira LMR (2022) The emerging role of regulatory cell-based therapy in autoimmune disease. Front. Immunol. 13:1075813. doi: 10.3389/fimmu.2022.1075813
Received: 20 October 2022; Accepted: 28 November 2022;
Published: 14 December 2022.
Edited by:
Ryusuke Yoshimi, Yokohama City University, JapanReviewed by:
Shuji Sumitomo, Kobe City Medical Center General Hospital, JapanCopyright © 2022 Ghobadinezhad, Ebrahimi, Mozaffari, Moradi, Beiranvand, Pournazari, Rezaei-Tazangi, Khorram, Afshinpour, Robino, Aref and Ferreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonardo M. R. Ferreira, ZmVycmVpcmxAbXVzYy5lZHU=; Amir Reza Aref, YW1pckB4c3BoZXJhYmlvLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.