 Ling Zhao
Ling Zhao Chen Hou1,2
Chen Hou1,2 Naihong Yan
Naihong Yan- 1Research Laboratory of Ophthalmology, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Ophthalmology, West China Hospital, Sichuan University, Chengdu, China
Retinitis pigmentosa (RP) is an important cause of irreversible blindness worldwide and lacks effective treatment strategies. Although mutations are the primary cause of RP, research over the past decades has shown that neuroinflammation is an important cause of RP progression. Due to the abnormal activation of immunity, continuous sterile inflammation results in neuron loss and structural destruction. Therapies targeting inflammation have shown their potential to attenuate photoreceptor degeneration in preclinical models. Regardless of variations in genetic background, inflammatory modulation is emerging as an important role in the treatment of RP. We summarize the evidence for the role of inflammation in RP and mention therapeutic strategies where available, focusing on the modulation of innate immune signals, including TNFα signaling, TLR signaling, NLRP3 inflammasome activation, chemokine signaling and JAK/STAT signaling. In addition, we describe epigenetic regulation, the gut microbiome and herbal agents as prospective treatment strategies for RP in recent advances.
1 Introduction
1.1 Retinal inflammation and RP
Retinitis pigmentosa (RP) is a category of inherited retinal dystrophies marked by vision loss and, ultimately, blindness. More than 3000 mutations in over 80 distinct genes or loci have been identified as causes of non-syndromic RP (1, 2). These mutations can be transmitted in an autosomal-dominant, autosomal-recessive, or X-linked manner. There are also syndromic forms of RP, such as Usher syndrome and Bardet-Biedl syndrome (3). The prevalence of RP is reported to be 1/3,000 to 1/5,000 (https://www.orpha.net/consor/cgi-bin/index.php?lng=EN). Early in the progression of RP, patients experience night blindness and difficulty with dark adaptation. They gradually lose peripheral vision and develop tunnel vision as the disease advances, indicating the loss of rod function. Cone involvement contributes to visual acuity decline over time, and finally, typically in middle age, central vision loss occurs (2). Due to its clinical and genetic heterogeneity, RP has limited therapeutic options. It has long been recognized that inflammation and immune responses are associated with RP, and this theme has recently gained increasing attention (4–6). Greater understanding of the molecular processes driving RP inflammation is expected to provide new therapeutic approaches independent of the genetic background.
Inflammation activation is a prominent feature of RP. In RP, inflammation is characterized by activation of the innate immune system, including dysfunction of the immune barrier, activation and infiltration of immune cells, and upregulation of topical and peripheral inflammatory factors. Bone-spicule pigmentation, attenuated retinal vessels and waxy pallor of the optic disc are typical clinical manifestations of RP. In addition, inflammatory cells are commonly observed in the vitreous due to the collapse of the blood−retina barrier (BRB). Higher cell density correlates with younger age and impaired visual function (6). In addition, it has been reported that increased aqueous flare in RP patients correlates closely with visual function and the extent of global retinal degeneration (7–11). Aqueous flare is generally seen in individuals with inflammatory ocular disorders, indicating deficits of the blood–aqueous barrier and inflammatory protein/cell leakage (12, 13). BRB disruption begins early in the disease. Prior to the infiltration of inflammatory cells and photoreceptor (PR) starvation, the tight junctions of the retinal pigment epithelium (RPE) and the retinal vasculature become leaky, thereby promoting the formation of an inflammatory milieu and the degeneration of PRs (14–17).
Microglia are essential components of the retinal innate immune system and play a pivotal role in retinal inflammatory responses. Gupta et al. reported that microglial activation is engaged in human RP. With thinning of the PR layer, microglia were observed to infiltrate degenerative foci; these microglia were enlarged amoeboid cells containing rhodopsin-positive cytoplasmic inclusions (18). Microglia promote retinal inflammation via infiltration, phagocytosis, and secretion of proinflammatory mediators, whereas genetic ablation or inhibition of microglial phagocytosis ameliorates PR degeneration in RP model mice (19).
Several studies (6, 20–22) have reported elevated levels of inflammatory factors in serum, vitreous, and aqueous humor, indicating a proactive inflammatory response in RP patients. Immunologic disorders observed in patients with RP are summarized in a previous work (23).
Animal models are essential for the study of RP (24, 25). Activation of the immune system has been detected in various rodent RP animal models (26–28). Due to this similarity, animal models are indispensable for clarifying the pathogenesis of RP and developing treatments. Clinically, synthetic corticosteroids with potent anti-inflammatory and immunosuppressive properties are used in treatments for RP-related cystoid macular edema (29). In RP models, it also works. In RCS and rhodopsin mutant model S334ter-4 rats, fluocinolone acetonide treatment markedly protects PR from degeneration and suppresses microglial activity (30, 31). In combination with polyamidoamine dendrimers, fluocinolone acetonide selectively targets the microglia localized in the outer retina where degeneration is ongoing (32). Dexamethasone administration to rd10 mice reduces retinal inflammation, restores cone structure and function, and preserves RPE integrity by preserving ZO-1 density (15, 33).
1.2 Microglia and Müller glia in RP retinas
Microglial activation is a sign of neuroinflammation. Retinal resident microglia arise from yolk sac erythromyeloid progenitors, comprising 85% of total retina macrophages (34–36). They colonize the developing retina during embryogenesis, shape the retina by secreting neurotrophic factors, engulf and eliminate unwanted neurons and synapses, and engage in vascular development of the eye (37–39). In postnatal retinas, microglia are maintained throughout life independent of circulating monocytes and by self-renewal (34, 40, 41).
Microglia express a variety of receptors (e.g., CX3CR1, TLRs, IL-1R, and TNFR) that allow them to detect environmental changes and initiate the inflammatory signal cascade (42). Microglia activation induces a robust inflammatory response, including the release of proinflammatory factors, phagocytosis, and inflammatory cell recruitment. Excessive microglial phagocytosis contributes to local inflammation and neurodegeneration (43, 44).
Classically, activated microglia were categorized into two groups: M1 (classically activated) and M2 (alternatively activated), with the belief that M1 microglia secrete proinflammatory factors such as TNFα, IL-1β, IL-6, and inducible nitric oxide synthase (iNOS) that fuel inflammation, whereas M2 microglia produce anti-inflammatory cytokines (e.g., IL-4, IL-10, IL-13, IL-18) that are beneficial for damage repair (45). Recent research, however, suggests that the microglial phenotype varies in response to environmental changes (46).
In healthy retinas, microglia tile the inner and outer plexiform layers without overlapping (37), where they are ramified cells responsible for immune surveillance and maintenance of synaptic structure and transmission (47–50). In response to insults, microglia rapidly change into an amoeboid appearance and migrate into lesion areas, removing dead/dying neurons and neuronal debris while concurrently releasing proinflammatory factors as well as protective cytokines and trophic factors to repair damage and restore homeostasis (51), after which microglia recover to the “resting” state; this process usually results in minimal retinal remodeling.
In RP retinas, initial mutation-driven PR degeneration increases extracellular signal molecules (e.g., ATP, HSPs, HMGB1, DNA, and many others) termed damage-associated molecular patterns (DAMPs) (52–56). The “eat-me” signal, phosphatidylserine, appears on stressed rods (19). Microglia proliferate and infiltrate the PR layer and subretinal space, where they function as reactive phagocytes, phagocytose dead and stressed PRs, secrete proinflammatory cytokines (e.g., TNFα and IL-1β) and chemokines (e.g., CCL2 and RANTES), and recruit infiltrating immune cells (19, 24, 57, 58). Due to this mutant genetic background, however, microglial activation persists, and the continuous production of inflammatory and cytotoxic factors exacerbates PR loss until the late stage, at which point PRs mostly die and the retinal structure is severely damaged (59).
Müller glia are another group of retinal cells engaged in degeneration. Müller glia are retinal macroglia that provide homeostasis, metabolism, and functional support for neurons (60). Depending on the severity, the Müller glial response to injury refers to reactive gliosis accompanied by Müller proliferation or not. Reactive gliosis can be beneficial because it releases protective factors such as neurotrophic factors, whereas prolonged gliosis is detrimental and generally results in neurodegeneration (61). Müller glia are potential modulators of retinal inflammation. Upon BRB disruption, Müller glia compensate for RPE deficiency by sealing the leaky choroid and inducing claudin-5 expression (14). Müller glia share characteristics with immune cells. Müller glia express multiple cytokine receptors and are a major source of cytokines and inflammatory factors (62). Proteomic evidence supports the capacity of Müller glia for antigen presentation and inflammatory signaling transduction in response to immune stimulation (63, 64). Müller glia contribute to the phagocytic clearance of dead PRs (65). Moreover, the interaction between Müller glia and microglia modulates retinal inflammation and degeneration (66–68).
As the predominant glial population of the retina, Müller glia are abundant and widely distributed. Müller glia traverse the thickness of the neuroretina structurally, allowing them to keep touch with all types of retinal cells. Due to its neurotrophic function and regeneration potential, the Müller cell has been studied in a variety of degenerative retinal disorders (69). In actuality, Müller glia are also intimately linked to retinal inflammation. For more information about how Müller glia interact with the innate immune system and monitor retinal inflammation, we refer the reader to this article (70).
Complicated mechanisms are involved in the regulation of retinal inflammation in RP. Microglia and Müller glia are major cellular populations that express and modulate these signaling pathways (Figure 1). Here, we review treatment strategies from the perspective of inflammation management (Table 1), focus on molecular mechanisms related to immunomodulation, and discuss new findings regarding epigenetic modification and the gut microbiome as novel therapies for RP.
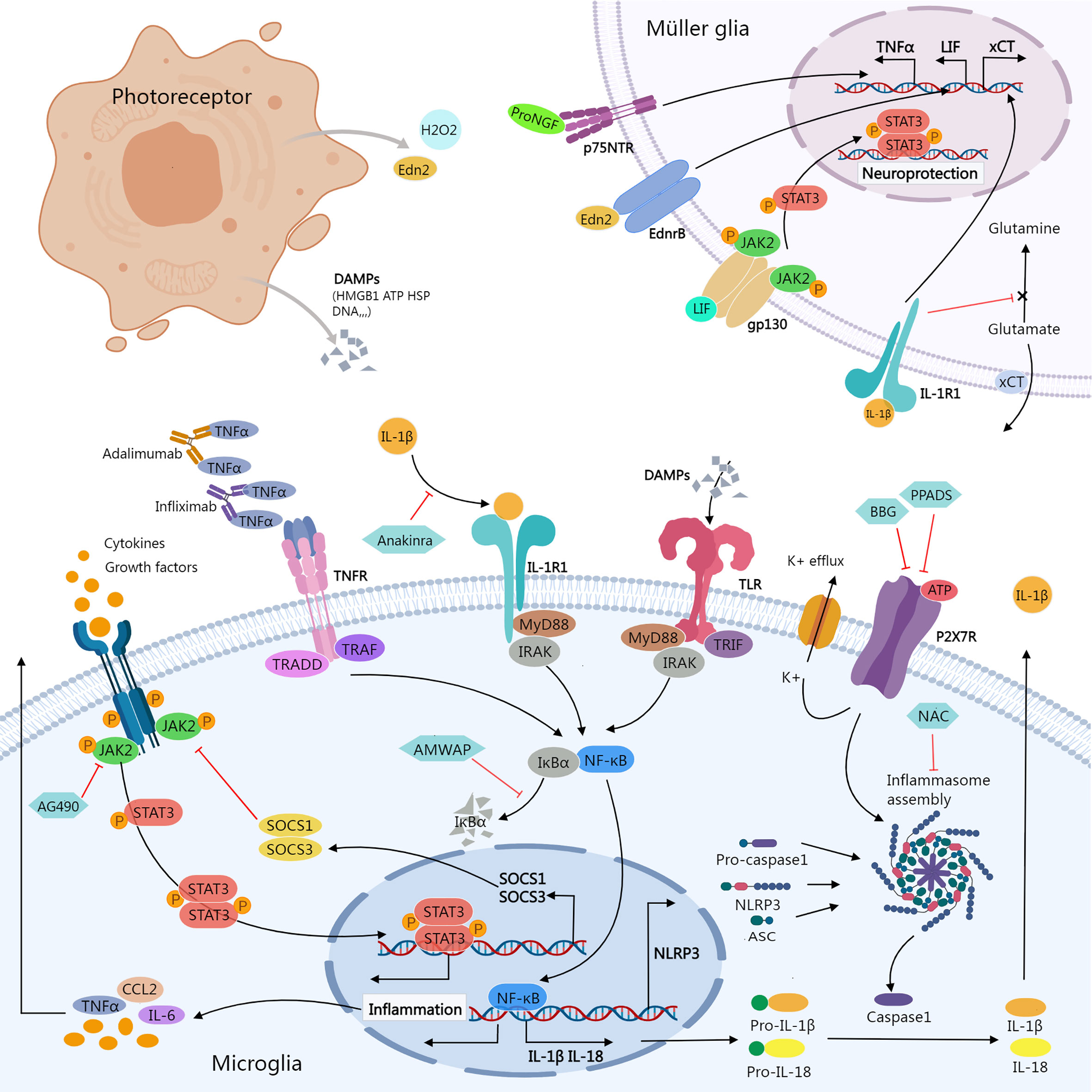
Figure 1 Inflammatory signals between photoreceptor, microglia, and Müller glia. Damaged or dying photoreceptors release signal molecules that stimulate the activation of microglia and Müller glia. In Müller glia, activation of NFGR promotes TNFα production; Edn2 binds to Ednrb promotes LIF transcription; LIF binding to gp130 activates the JAK2/STAT3 pathway, which promotes Müller glial neuroprotection; IL-1β binding to IL-1R1 interferes with glutamate conversion into glutamine, resulting in elevated extracellular glutamate concentration. In microglia, activation of JAK2/STAT3 signaling stimulates the release of inflammatory molecules. SOCS1/SOCS3 act as negative feedback factors of JAK/STAT pathway; AG490 specifically inhibits JAK2. Ligands-binding to TNFR, IL-1R, and TLR stimulates NF-κB signaling cascades, promoting transcription of inflammatory genes including NLRP3, pro-IL-1β, and pro-IL-18; ATP stimulates K+ efflux via P2X7R, promoting NLRP3 inflammasome activation and the release of active caspase-1, mature IL-1β, and IL-18. Biological agents like infliximab and adalimumab provide neuroprotection by neutralizing TNFα. Anakinra blocks the biologic activity of IL-1β. AMWAP prevents NF-κB translocating into the nucleus by inhibiting IκBα degradation. NAC suppresses the activation of NLRP3. BBG and PPADS reduce the activation of the NLRP3 inflammasome via inhibiting P2X7R signaling. AMWAP, activated microglia/macrophage WAP domain protein; ASC, apoptosis-associated speck-like protein containing a CARD; ATP, adenosine triphosphate; BBG, Brilliant Blue G; CCL2, C-C Motif Chemokine Ligand 2; DAMPs, damage-associated molecular patterns; Edn2, endothelin 2; Ednrb, endothelin receptor B; HMGB1, High-mobility group box-1; HSP, heat shock protein; JAK, janus kinase; LIF, leukemia inhibitory factor; MyD88, myeloid differentiation primary response 88; NAC, N-acetylcysteine; PPADS, pyridoxal-phosphate-6-azophenyl-2’,4’-disulfonic acid; proNGF, pro-nerve growth factor; p75NTR, p75 neurotrophin receptor; STAT, signal transducer and activator of transcription; TNFR, Tumor necrosis factor receptor; TRADD, TNFR1-associated death domain protein; TRAF, TNF receptor associated factor; TLR, Toll-like receptors; TRIF, TIR-domain-containing adaptor-inducing interferon-β; xCT, core subunit of the cystine/glutamate transporter system xc-; IRAK, Interleukin-1 receptor-associated kinase.
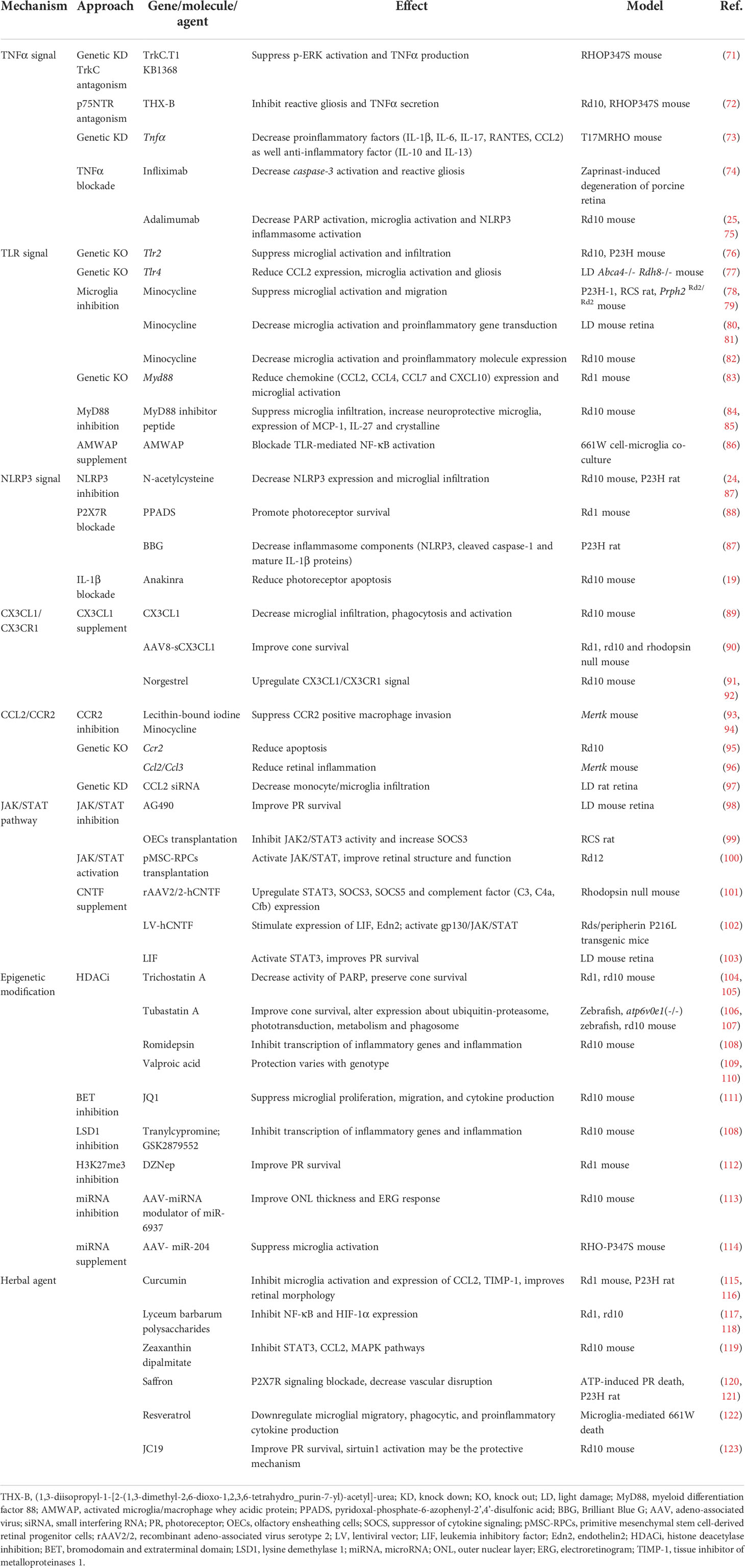
Table 1 Treatments for retinitis pigmentosa and related mechanisms.
2 Mechanisms related to inflammation in RP
2.1 TNFα signaling
Tumor necrosis factor α (TNFα) is a strong proinflammatory cytokine that plays vital roles in immune modulation, cell proliferation, differentiation, and apoptosis. TNFα is produced predominantly by T and innate immune cells and is initially synthesized as transmembrane protein (tmTNFα), a precursor that requires proteolytic cleavage by TNFα-converting enzyme (also ADAM17) to release a soluble form (sTNFα) (124). Both tmTNFα and sTNFα are implicated in the inflammatory response.
TNFα initiates a signal cascade by binding to its receptors, TNFR1 and TNFR2. TNFR1 is activated by both tmTNFα and sTNFα, whereas TNFR2 is proposed to be fully activated primarily by tmTNFα (125). Ligand binding to TNFR1 recruits the adaptor molecule TNFR1-associated death domain protein, which then leads to the assembly of several signaling complexes known as complexes I, IIa, IIb, and IIc. Complex I formation stimulates nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases (MAPKs). Complex IIa and IIb assembly activates caspase-8 and facilitates apoptosis, and complex IIc formation activates the mixed lineage kinase domain-like protein and induces necroptosis and inflammation. TNFR2 stimulation activates NF-κB, MAPKs, and protein kinase B (125).
TNFα is postulated to participate in the pathogenesis of RP (74). TNFα and TNFR expression levels are elevated in the retina of RP models and in the aqueous humor of RP patients (24, 74, 126–128); microglia (129) and Müller glia (130, 131) are the primary cellular sources of TNFα. TNFα signaling has been found to mediate PR death via RIP1/3-related necrosis and caspase3/7-dependent apoptosis, in addition to triggering proinflammatory signaling in the RP retina (73, 126).
2.1.1 NGF receptor and TNFα production
Increased TNFα expression in the retina is linked to nerve growth factor (NGF) receptor activation. Müller glia in rhodopsin mutant RP model RHOP347S mice upregulate the expression of TrkC. T1, a truncated TrkC receptor isoform, and its ligand NT-3. TrkC.T1 increases local TNFα production by activating MAPK/Erk, ultimately leading to PR death. This process can be reversed by genetic knockdown of TrkC.T1, TrkC antagonism, or MAPK/Erk inhibition (71). Similarly, TrkC.T1 knockout (KO) and TrkC inhibition increased retinal ganglion cell survival in a mouse model of glaucoma by reducing TNFα production (132), implying that TrkC.T1 is upstream of TNFα.
It has been reported that microglia-derived proNGF facilitates PR death via p75NTR (133), proNGF binding to p75NTR in Müller glia induces robust expression of TNFα and TNFα-dependent neuron death in rodent retina (131, 134), the expression levels of proNGF and p75NTR are increased in the retina of rd10 at early degenerative stages, pharmacological antagonism of p75NTR with THX-B ((1,3-diisopropyl-1-[2-(1,3-dimethyl-2,6-dioxo-1,2,3,6tetrahydro-purin-7-yl)-acetyl]-urea)) affords neuroprotection to PRs, and the treatment also mediates reduction of TNFα production, microglial activation, and reactive gliosis (72).
2.1.2 TNFα inhibition
TNFα knockdown in the T17M rhodopsin mutant mouse model reduces PR death and PR-related functional loss, and this neuroprotective effect is associated with reductions in proinflammatory cytokines (IL-1β, IL-6, IL-17, RANTES) and chemokines (CCL2) (73).
Infliximab and adalimumab are biological TNFα inhibitors approved for treating inflammatory disorders such as Crohn’s disease, ulcerative colitis, rheumatoid arthritis, plaque psoriasis, and uveitis (125, 135). By lowering the expression of TNFR1 and caspase-3 activity, infliximab alleviated retinal degeneration induced by PDE6 inhibition in cultured porcine retina (74, 127). Adalimumab administered intraperitoneally or topically improved PR survival while decreasing microglial activation and reactive gliosis in the rd10 retina. Inhibition of PARP and RIPK signaling, as well as NLRP3 inflammasome assembly, are mechanisms involved in this protective response (25, 75).
2.1.3 Protective effects of TNF signaling
Notably, TNFα KO retinas tend to express both pro- and anti-inflammatory factors at reduced levels when compared with controls (73), implying that removing TNFα would also damage the immune system’s defenses. Recent work by Kuhn et al. established that TNFα, in collaboration with TNFR1, TNFR2, and p75NTR, induces signals that are indispensable for neural development and that disturbances to TNFR family signaling result in unhealthy axonal development (136).
ADAM17 regulates the expression of TNFα as well as the receptor TNFR (124). Muliyil et al. reported that ADAM17 and soluble TNF mediate a novel cytoprotective pathway in Drosophila. Loss of ADAM17 or TNF/TNFR signaling drives the accumulation of lipid droplets and degeneration in the Drosophila retina, whereas restoration of ADAM17 or TNF/TNFR in glia is sufficient to rescue the degeneration phenotype. TNF and TNFR are explicitly needed in glia; loss of either in glia, but not neurons, leads to the accumulation of lipid droplets. Furthermore, inactivation of ADAM17 in human iPSC-derived microglia similarly induces aberrant lipid droplet accumulation and mitochondrial reactive oxygen species generation (137), indicating that comparable processes in which TNF works not as an inflammatory trigger but as a trophic survival factor (137) may also be involved in the mammalian retina.
Benoot et al. evaluated the numerous contradictory findings of TNFα application in lung cancer (138), bringing to our awareness the varied functions of various TNF family members and the positive impacts of TNF signaling. Modern genomic, transcriptomic, and proteomic techniques are useful for identifying signaling events and molecules in signal transduction (139, 140). Tanzer et al. (141) discussed in detail how modern proteomic approaches offer a novel perspective on TNF signaling.
Currently, the precise mechanisms of TNFα synthesis remain obscure. Future investigation of TNFα signaling requires the power of new technology, and the potential protective function of TNFα merits greater consideration.
2.1.4 TNFα and microglia-Müller glia interaction
Müller glia exposed to activated microglia modify the expression of a variety of signaling molecules, including (1) elevation of growth factors such as GDNF and leukemia inhibitory factor (LIF), (2) enhanced proinflammatory factor production, and (3) overexpression of chemokines and adhesion proteins (142). TNFα is the most prevalent cytokine produced by reactive microglia, and it stimulates LIF expression in Müller glia in a p38MAPK-dependent manner. Inhibition of p38 MAPK activity lowered LIF expression and accelerated PR mortality in light-damaged retinas (143), similar to previous reports that TNFα prevents cell death by activating the JAK/STAT3 pathway through the IL-6 receptor (144). When TNFα stimuli engage previously activated Müller glia, however, inducible cytokines consisting of more proinflammatory cytokines (TNFα, iNOS, IL-6) and less LIF are produced (145). In other words, depending on the type and degree of stimuli, Müller glia activation generates both neuroprotective and proinflammatory responses, and Müller glia under continuous stimulation are likely to exhibit a detrimental phenotype.
2.2 TLR signaling
Toll-like receptors (TLRs) are a class of pattern recognition receptors (PRRs) responsible for identifying pathogen-associated molecular patterns (PAMPs) and DAMPs and mediating immune responses; the generation of PAMPs or DAMPs prompts pathogen invasion or tissue injury. TLR expression is conserved among species, and to date, 10 TLRs (TLR1–10) in humans and 12 TLRs (TLR1–9 and TLR11–13) in mice have been described. TLRs are predominantly but not exclusively expressed on immune cells (146–148).
TLRs serve as the first line of defense for the innate immune system. Upon recognition of DAMPs or PAMPs, TLRs dimerize and initiate recruitment of Toll/IL-1 receptor (TIR) domain-containing adaptor molecules, including myeloid differentiation primary response 88 (MyD88), TIR-domain-containing adaptor-inducing interferon-β (TRIF), MyD88 adaptor-like protein (Mal), and TRIF-related adaptor molecule (TRAM), thereby initiating intracellular signaling cascades: the MyD88- or TRIF-dependent pathways (146). TLR activation facilitates the transduction of NF-κB and MAPK (149–151), as well as the release of proinflammatory cytokines (TNFα, IL-6, IL-1β, and IFNβ), chemokines, and cluster of differentiation 80 (CD80), CD86, CD40, and major histocompatibility complex class II (146).
Activation of TLR signaling has been shown to worsen inflammation and accelerate the course of RP (77, 152, 153). Microglia highly express TLRs (154), and TLR activation in the retina facilitates microglial activation and infiltration (77, 155). Moreover, microglia in the rd1 retina undergo RIP1/RIP3-dependent necroptosis mediated by TLR4 activation, which amplifies retinal inflammation and destruction with large amounts of proinflammatory cytokines (TNFα and CCL2) (152).
2.2.1 DAMPs activate TLRs in RP
High-mobility group box-1 (HMGB1) is a proinflammatory factor and DAMP released by dying cells or activated macrophages that mediates the immune response via PRRs (156–158). HMGB1 stimulates an inflammatory response in diabetic retinopathy through TLR4/NF-κB signaling (159).
Increased levels of HMGB1 were detected in the vitreous of patients with RP, along with the presence of necrotic enlarged cone cells (53). In cultured cone-like 661W cells, recombinant HMGB1 treatment induces apoptosis and upregulates the expression of IL-6 and TNFα (160), and external HMGB1 induces retinal ganglion cell death via TLR2/4 signaling (161), whereas HMGB1 inhibition or neutralization attenuates the inflammatory response and promotes retinal neuron survival (162, 163).
2.2.2 TLR blockade
Upregulation of Tlr2, Il1b, Myd88 and Tirap was found in RP model rd10 and P23H mice, demonstrating TLR activation involvement in RP-associated retinal degeneration. Genetic deletion of TLR2 alleviated PR loss and vision impairment in both models (76). Similarly, in a light-induced retinal degeneration model, genetic TLR4 deletion reduced retinal inflammation and degeneration (77). Minocycline is an effective microglial inhibitor. In inherited and induced RP models, minocycline administration decreased microglial infiltration and proinflammatory molecule expression and promoted PR survival and functional retention (78–82). Minocycline treatment suppresses MAPK and NF-κB signaling in LPS-stimulated microglia (164), and it has been ascertained that minocycline prevents microglial activation by inhibiting TLR2 (165, 166) and TLR4 (167) signaling.
2.2.3 MyD88
Most TLRs (except for TLR3) use MyD88 as a downstream adaptor protein; moreover, MyD88 is a component of the IL-1R signaling cascade (168, 169). MyD88 features a death domain and a TIR domain. Upon TLR/IL-1R ligation, MyD88 is recruited to the receptor and interacts with IRAK2/4 through their death domains, which activates NF-κB, activator protein-1, and interferon regulatory factors (169).
MyD88 KO mice display attenuated immune responses and are unable to produce normal levels of inflammatory cytokines (170). This diminished immune response preserved PR survival and retinal function during degeneration in rd1 mice lacking MyD88 (83). Similarly, pharmacologic inhibition of MyD88 in rd10 mice with MyD88 inhibitor peptide reduced PR apoptosis and improved rod-related function; treatment also lowered the number of microglia in the PR layer and increased microglia/macrophage expression of the neuroprotective marker Arg1 (84). Further proteomic analysis demonstrated that treatment with such MyD88 inhibitor peptides boosted crystalline expression, suggesting that MyD88 inhibition may also enhance intrinsic tissue-protective mechanisms (85).
2.2.4 AMWAP
Activated microglia/macrophage WAP domain protein (AMWAP), secreted by reactive microglia, is a hallmark of microglial activation. While AMWAP overexpression in microglia lowers the production of proinflammatory factors such as IL-6, iNOS, CCL2, CASP11, and TNFα, extracellular AMWAP endocytosed by microglia inhibits TLR2- and TLR4-induced NF-κB translocation by preventing IRAK-1 and IκBα proteolysis (86). AMWAP administration lowers the apoptosis of 661w cone-like cells treated with microglia-conditioned medium (86), indicating that AMWAP is a potential self-modulator of TLR signaling in microglia.
The TLR signaling pathway plays a fundamental role in inflammatory and immune responses. Molecules released from injured neurons induce an intracellular signaling cascade through TLR/MyD88, contributing to further retinal damage. Blockage of TLR/MyD88 alleviates RP by reducing inflammatory responses and enhancing protective effects.
2.3 NLRP3 inflammasome activation
Inflammasomes are cytosolic multiprotein complexes that facilitate the release of mature IL-1β, IL-18, and cleaved caspase-1. The intracellular PRRs, NOD-like receptors (NLRs), are important components of the inflammasome complex. Some NLRs oligomerize upon activation to form multiprotein complexes that function as caspase-1-activating scaffolds (171). NLRP3 is the most well-studied NLR; NLRP3 inflammasome assembly requires two signals: a priming signal that activates NF-κB, followed by transcription of NLRP3, pro-IL-1β, and pro-IL-18. A second activation signal facilitates the recruitment and oligomerization of NLRP3, adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), and pro-caspase-1. Once the inflammasome is assembled, it stimulates pro-caspase-1 self-cleavage and activation, and cleaved caspase-1 catalyzes pro-IL-1β and pro-IL-18 maturation and induces the release of their mature forms. The recognition of DAMPs or PAMPs that act through PRRs such as TLRs or cytokines that act through particular receptors (TNFR, IL-1R) exemplifies the priming signal. The activation signal encompasses a wide range of stimuli, including ion flux (K+, Cl-, Ca2+), lysosomal instability, mitochondrial dysfunction, reactive oxygen species generation, and trans-Golgi disassembly, with K+ efflux being the upstream event in almost all NLRP3 activations (172, 173).
Inflammasome activation initiates the host’s defense response to endogenous or external damaging stimuli and aids in homeostasis maintenance. Nevertheless, chronic inflammasome activation and the subsequent overproduction of caspase-1, IL-1β, and IL-18 can be detrimental.
Canine models of RP upregulate NLRP3 inflammasome-related genes (26). NLRP3 was detected in cone PRs and one-third of reactive microglia in P23H rhodopsin mutant retinas, which also upregulates the expression of mature IL-1β and IL-18, as well as cleaved caspase-1, indicating inflammasome activation during retinal degeneration. In rd10 mice, administration of the antioxidant N-acetylcysteine prevented PR loss and suppressed inflammatory factors and microglial activation (24). Studies conducted on P23H mice demonstrated that N-acetylcysteine lowered NLRP3 expression by 50% and decreased microglial infiltration, hence improving cone survival and retinal function (87).
2.3.1 P2X7R
The purinergic receptor P2X7R is an adenosine triphosphate (ATP)-gated ion channel and a well-known inflammasome activator that can enhance the expression of the NLRP3 inflammasome in microglia (174). By inducing K+ efflux, ATP-mediated P2X7R activation promotes NLRP3 inflammasome activation (173).
ATP is abundant in PRs as an energy source and neurotransmitter. During retinal degeneration, ATP leaches from dying PRs and activates P2X7R (175). Intravitreal injection of PPADS (pyridoxal-phosphate-6-azophenyl-2’,4’-disulfonic acid), a purinergic antagonist, lowers PR loss in rd1 mice (88). In contrast, intravitreal administration of ATP to WT (wild-type) retinas induces PR degeneration similar to that in the P23H RP model (176), whereas treatment with the selective P2X7R inhibitor BBG (Brilliant Blue G) protects against this ATP-mediated PR apoptosis (177). BBG therapy also reduced inflammasome components (NLRP3, cleaved caspase-1 and mature IL-1β proteins) in P23H retinas (87).
In the absence of extracellular ATP, P2X7R functions as a scavenger receptor that governs microglial clearance of extracellular debris, whereas P2X7R overactivation triggers NLRP3 inflammasome activation by provoking lysosomal instability. Lowering extracellular ATP levels may have the dual benefit of enhancing phagocytosis while decreasing inflammation (178).
2.3.2 IL-1β
IL-1β is a key product of NLRP3 inflammasome activation and a potent immunomodulation factor that orchestrates inflammatory and host defense responses (172, 179). IL-1β signals through IL-1R1. IL-1β binding to IL-1R1 stimulates pathways such as NF-κB, p38, JNKs, ERKs, and MAPKs, facilitating inflammatory cell recruitment and local/systemic inflammatory responses (180). Appropriate IL-1β/IL-1R1 signaling is required for a host’s defensive response to infections, whereas excessive IL-1β signaling is seen in a variety of hereditary and nonhereditary autoinflammatory disorders. IL-1β activity is endogenously regulated by IL-1R2 and IL-1Ra; IL-1R2 is a decoy receptor that sequesters the IL-1β signal, while IL-1Ra blocks IL-1β by competitively binding to IL-1R1 (180).
Intravitreal delivery of exogenous IL-1β triggered an immediate inflammatory response in the retina, including leukocyte recruitment and BRB destruction (181). However, IL-1β does not trigger PR death directly, as IL-1R1 expression is low in PRs. Through IL-1R1 expressed on Müller glia, IL-1β drives glutamate excitotoxicity-induced rod PR loss. The IL-1β/IL-1R1 signal disrupts the process of glutamate conversion into glutamine in Müller glia, resulting in an increased intracellular glutamate concentration, and upregulates xCT (the core subunit of the cystine/glutamate transporter system xc-) expression, which facilitates glutamate release into the extracellular space. Furthermore, IL-1β upregulates the expression of the ionotropic glutamate receptor in retinal neurons, which may increase neuronal vulnerability to glutamate excitotoxicity (182). Infiltrating microglia in the rd10 retina upregulate the expression of IL-1β (19) and block IL-1β signaling using anakinra, a commercially available recombinant IL-1Ra that is fully active in blocking IL-1R1 (183), which reduces PR apoptosis and preserves outer nuclear layer thickness in rd10 animals (19). In contrast, Todd et al. (184) demonstrated that IL-1β expressed by reactive microglia provides neuroprotection via IL-1R1 expressed on astrocytes in another mouse model of NMDA-induced retinal degeneration. Despite the use of different models, we were able to determine that IL-1β acts on the surface receptors of distinct glial cells and has varying effects on PR survival.
2.4 Chemokine signaling
2.4.1 CX3CL1/CX3CR1
CX3CR1 expression in the central nervous system (CNS) is considered to be restricted to microglia, and the expression of its sole ligand, CX3CL1 (also known as fractalkine), is confined to certain neurons (185). CX3CL1/CX3CR1 signaling facilitates the interaction between neurons and glia and plays a vital role in CNS neuroinflammation (186, 187).
CX3CL1/CX3CR1 signaling contributes to normal microglial and PR function. CX3CR1 signaling governs the dynamic activity of retinal microglia (188). Microglial ablation and repopulation in the mouse retina have shown that microglial recruitment is regulated by CX3CL1/CX3CR1 signaling (40), and Müller glia augment microglial migration and infiltration by increasing CX3CL1 secretion and microglial CX3CR1 expression (68). In addition, CX3CR1 signaling is required for retinal neuron growth, as CX3CR1-deficient retinas have shorter outer segments and diminished cone-related retinal function (189).
CX3CL1/CX3CR1 signaling affects microglial homeostasis by modulating the inflammatory response and phagocytosis. Increasing CX3CL1/CX3CR1 signaling in RP retinas could be beneficial. CX3CR1 deficiency impairs microglial phagocytic clearance of neurotoxic species. Reportedly, CX3CL1 signaling enhances microglial erythrophagocytosis through the CD163/HO-1 axis (190), whereas CX3CR1 KO weakens microglial phagocytosis to β-amyloid and mediates lysosomal dysfunction, resulting in an escalation of neuroinflammation due to β-amyloid accumulation (191).
CX3CR1 deficiency enhances the inflammatory response of microglia. CX3CR1-deficient microglia exhibit greater neurotoxicity (192), and CX3CR1-deficient microglia have an elevated amount of surface P2X7R, which increases IL-1β maturation and release (193). CX3CR1 deletion in microglia-like cells generated from human iPSCs induced enhanced inflammatory responses to LPS stimuli and phagocytic activity to fluorescent beads (194). Loss of CX3CR1 signaling in young animals resulted in a microglial transcriptome similar to that of aged mice, with dysregulated expression of genes related to immune function (195).
CX3CL1 expression is downregulated in rd10 retina before the onset of primary rod degeneration (196), and CX3CR1 KO in rd10 mice increases microglial infiltration and phagocytosis, as well as the generation of pro-inflammatory cytokines, which accelerates PR loss (82, 89), whereas exogenous CX3CL1 supplementation preserves morphology and function (89). CX3CL1 has been shown to deactivate microglia by blocking the NF-κB pathway and activating the Nrf2 pathway (197). A norgestrel-supplemented diet protected rd10 retinas from PR degeneration, and this protection was achieved by the upregulation of CX3CL1/CX3CR1 signaling and the reduction of proinflammatory cytokine production (91, 92).
Recent work by Wang et al. demonstrated that overexpression of soluble CX3CL1 via AAV8 prolongs cone survival and improves cone-related visual function in RP model rd1 and rd10 mice. This therapeutic effect is restricted to cone PRs, has no effect on microglial activity or inflammatory factor levels and is not even dependent on the presence of a normal number of microglia (90). In light of this, further research is needed to determine whether CX3CL1 action in the retina is limited to microglia or whether other pathways exist.
2.4.2 CCL2/CCR2
Part of the evidence suggests that CCL2/CCR2 signaling is detrimental, as inhibition of CCL2/CCR2 signaling attenuates microglial activity and degeneration in RP (95–97). CCL2 is highly expressed by stressed PRs, activated microglia, and Müller glia in degenerating retina (95, 198, 199). By binding to its receptor, CCR2, which is expressed on peripheral mononuclear phagocytes, mediates the influx of circulating monocytes into inflamed retinas (200). Using fluorescent protein-labeled Mertk (-/-) Cx3cr1 (GFP/+) Ccr2 (RFP/+) mice, Kohno H and colleagues demonstrated that both minocycline and lecithin-bound iodine (LBI) ameliorate PR death by inhibiting CCL2/CCR2 signaling (93, 94). Meanwhile, constitutive expression of CX3CR1 in the retina represses CCL2 expression and the recruitment of neurotoxic inflammatory CCR2+ monocytes (201).
However, there is evidence that CCL2 signaling may have a protective role in the degradation of RP. In a light-induced mouse model of degeneration, blocking CCL2/CCR2 signaling decreased infiltrating monocytes but had no effect on the rate of retinal thinning (198). Alde-Low EPCs (low aldehyde dehydrogenase activity endothelial progenitor cells) transplantation therapy rescued vasculature and PRs in rd1 mice, and CCL2 secreted by Alde-Low EPCs recruited a subpopulation of monocyte-derived macrophages that highly expressed CCR2 and the neuroprotective factors TGF-β, IGF-1 and IL-10 (202). In brief, induction of CCL2 expression by Alde-Low EPCs in rd1 retinas resulted in the recruitment of neuroprotective macrophages.
It is apparent that CCL2/CCR2 signaling mediates the recruitment of monocyte-derived macrophages in the degenerating retina, but it remains to be determined whether these recruited cells are beneficial or detrimental.
2.5 JAK/STAT signaling
The JAK/STAT signaling pathway is a ubiquitously expressed intracellular signal transduction system implicated in a wide range of biological functions. Various ligands, including cytokines, growth hormones, growth factors, and their receptors, can activate the JAK/STAT pathway (203). Briefly, ligand binding to specific receptors induces receptor multimerization and JAK activation, activated JAKs phosphorylate the receptors, activate and phosphorylate their primary substrate STAT, and phosphorylated STAT dimerizes and translocates into the nucleus, where it binds to particular regions to either activate or inhibit the transcription of target genes. Suppressor of cytokine signaling (SOCS) is a negative modulator of JAK/STAT signaling, and its expression is promoted by stimulation of JAK/STAT signaling (203, 204). Numerous studies on the JAK/STAT pathway have revealed its significance in neoplastic and inflammatory disorders (203, 205).
2.5.1 JAK/STAT and microglia-associated inflammation
Expression and activation of STAT proteins are implicated in the plasticity of the retina during embryonic and postnatal stages (206), and mice deficient in SOCS1/STAT1 develop severe ocular illnesses with massive inflammatory cell infiltration (207). STAT signaling plays a central role in the degeneration of the rd10 retina, as evidenced by proteomic profiling (208). Furthermore, activation of JAK/STAT signaling was also observed in the retinas of light-induced and inherited (rd1 and VPP mouse) RP animal models (98, 209).
AG490 is a JAK2-specific inhibitor that suppresses microglial activation and the production of inflammatory factors such as TNFα and IL-6 by reducing STAT3 phosphorylation (210). AG490 induces M2-type microglial polarization by blocking JAK2/STAT3 signaling in acute paraquat exposure-induced microglial activation (211). In light-damaged retinas, AG490 treatment decreased JAK and STAT phosphorylation as well as PR apoptosis (98).
Olfactory ensheathing cell (OEC) transplantation improved retinal function in RCS rats. OEC treatment dramatically reduced active resident microglia/infiltrated macrophages and the release of proinflammatory cytokines while increasing anti-inflammatory cytokines in the transplantation area. This neuroprotection appears to be mediated in part by increased SOCS3 expression and decreased JAK2/STAT3 activity. Coculture of OECs with the BV2 microglial cell line revealed a shift in microglial cytokine release toward an anti-inflammatory pattern (99). According to the literature, SOCS3-deficient microglia display increased phagocytic activity (212), whereas elevated SOCS3 expression in microglia decreases GM-CSF/IFN-γ-driven inflammatory responses by blocking the activities of JAK1 and JAK2 through its KIR domain (213). In addition, increasing SOCS1 signaling with SOCS1-KIR, a SOCS1 mimetic peptide, suppressed the recruitment of inflammatory cells into the retina and stimulated IL-10 production (214).
Multiple jakinibs (JAK inhibitors) are approved for the clinical management of malignancy, rheumatic, lymphoproliferative, and inflammatory diseases, and most recently, coronavirus disease 2019 (205), but their efficacy in the treatment of RP has not been evaluated.
2.5.2 JAK/STAT and Müller glial neuroprotection
Activation of JAK/STAT signaling has a protective effect on the RP retina. pMSC-derived retinal progenitor cell transplantation increased PR preservation in rd12 mice, and this protection was partially mediated by activation of the JAK/STAT pathway (100).
Application of ciliary neurotrophic factor (CNTF) in RP preclinical research has gained significant neuroprotection and has been employed in clinical trials (101, 215, 216).. CNTF therapy enhances the protective properties of Müller glia through LIF/gp130/STAT3 signaling, thereby preventing retinal degeneration. CNTF treatment elevates the expression of LIF and endothelin 2 (Edn2) (102), and LIF is essential for CNTF-elicited STAT3 activation (217). LIF belongs to the IL-6 cytokine family and signals through the gp130 receptor. In a mouse model of light-induced retinal degeneration, intravitreal delivery of LIF improved PR survival and retinal function by activating STAT3 in Müller glia and PR (103). Stressed PRs secrete signal molecules such as Edn2 and H2O2 that facilitate LIF induction in Müller glia; Edn2 triggers LIF transduction by binding to endothelin receptor B (Ednrb) localized to Müller glia; and H2O2 increases LIF transcript levels by stabilizing LIF mRNA via ILF3 (interleukin enhancer binding factor 3) (98, 218–220). LIF deficiency or Ednrb antagonism diminishes JAK/STAT activation and the amount of reactive Müller glia, resulting in accelerated degeneration; in contrast, LIF supplementation or Ednrb agonism improves PR survival in degenerating retina (218, 221).
Deletion of gp130 in either Müller glia or rod PRs severely dampened the activation of CNTF-triggered signaling as well as PR rescue (102), and when Müller glia were ablated, LIF no longer provided protection (222). However, other research suggests that gp130 deficiency in Müller glia decreases STAT3 phosphorylation but does not weaken the neuroprotection of exogenous LIF (223) because gp130 activation in PR presumably mediates a cell-autonomous protective mechanism with a general protective role independent of pathological stimulus (223, 224).
Modulation of JAK/STAT signaling results in contrary immunomodulatory effects in different retinal components. On the one hand, inhibition of JAK2/STAT3 in microglia contributes to inflammation mitigation. On the other hand, LIF-induced STAT3 signaling in Müller glia favors neuroprotection, which seems to be an endogenous protective mechanism. We speculate that this paradoxical outcome involves crosstalk between retinal microglia and Müller glia, which is not yet fully understood. Phosphorylated JAK also activates PI3K, so there may be synergy between JAK/STAT signaling and other pathways.
3 Epigenetic modulation in inflammation suppression
Epigenetic modifications, which include DNA methylation, histone modification, and noncoding RNAs, refer to changes in gene expression patterns without altering the genomic DNA sequence (225). Epigenetic modifications are implicated in aspects of individual growth and disease development, including gene expression, cell proliferation and differentiation, misfolded protein response, and cytoskeletal dynamics (226). Although the concept of curing diseases through epigenetic regulation is relatively new, it has demonstrated considerable therapeutic potential in research on cancer, autoimmune diseases, endocrine diseases, congenital disease and many others (227, 228). Epigenetic changes contribute to the development of RP, and remarkable progress has been made in the treatment of RP with epigenetic modification therapies.
3.1 Histone acetylation and methylation
Histone acetylation and methylation are the two most well-studied types of histone modification, with acetylation typically resulting in increased gene expression and methylation being related to either increased or decreased gene transcription. Histone acetylation is regulated by histone acetyltransferases and histone deacetylases (HDACs), while histone methylation is regulated by lysine methyltransferases and arginine methyltransferases and histone demethylation by histone demethylases. Enzymes that add or remove epigenetic marks on histones are known as “writers” and “erasers.” In addition, there are “readers” containing bromodomains, chromodomains, or Tudor domains that are able to decipher histone codes (229).
RP retinas exhibit excessive HDAC activity (104, 105, 230), and HDAC inhibition delays retinal degeneration in RP animal models (rd1 and rd10 mice and zebrafish) (104–107). In rd10 mice, the HDAC inhibitor romidepsin prevented rod degeneration and enhanced retinal function. Two molecular mechanisms contribute to this neuroprotective effect. First, by acting on histone targets in PRs, increasing chromatin accessibility and upregulating neuroprotective genes, and second, by acting on nonhistone targets in microglia and resident and invading immune cells, it suppresses inflammatory gene transcription and inflammation (108).
Microglial activity is related to histone methylation levels. LPS-activated microglia increase HDAC expression, which is accompanied by an increase in inflammatory gene expression (231). HDAC inhibition or knockdown promotes a protective microglial phenotype and reduces neuroinflammation (232–234).
Valproic acid is an HDAC inhibitor that reduces PR degeneration in rd1 and P23H RP models (109, 110). Valproic acid increases the expression of STAT1 by inhibiting HDAC3 expression; subsequently, acetylated STAT1 forms a complex with nuclear NF-κB p65, preventing NF-κB p65 DNA-binding activity (235).
Moreover, suppression of the “read” (bind) behavior to histone acetylation marks of bromodomain and extraterminal domain proteins by JQ1 ameliorated PR degeneration and maintained electroretinographic function in rd10 mice. This protection seems to be partially mediated by the inhibition of retinal microglial proliferation, migration, and cytokine production (111).
Several studies, including our previous report, have reported altered histone methylation in RP retinas (112, 236, 237). Lysine demethylase 1 inhibition attenuated PR degeneration in rd10 mice, in part by inhibiting microglial-related inflammation (108). DZNep (3-deazaneplanocin A) specifically inhibits Ezh2 (H3K27 trimethyltransferase) and mediates neuroprotective effects in rd1 mice by inhibiting H3K27me3 deposition (112). Ezh2 reportedly mediates TLR-induced inflammatory gene expression (238) and activation of multiple types of inflammasomes in microglia (239), hence promoting microglial-related pathologies.
3.2 MicroRNA
MicroRNAs (miRNAs) are small noncoding RNAs that modify gene expression post-transcriptionally by targeting messenger RNAs, long noncoding RNAs, and pseudogenes and circular RNAs. MiRNAs can be packed into exosomes or microvesicles to perform long-distance cell-to-cell communication. MiRNAs play a critical role in gene expression modulation and are therefore interesting candidates for the development of biomarkers and therapeutic targets (240). Throughout development, miRNAs are required for retinal neuron differentiation (241, 242). Dysregulated miRNAs were found in the retinas of mouse and canine models of RP (243, 244), indicating the involvement of miRNAs in the etiology of RP.
MiRNAs regulate microglial phenotypes, as evidenced by various studies on retinal and neurodegenerative disorders (245, 246). Inhibition of miR-6937-5p preserved the outer nuclear layer thickness and promoted the ERG wave response in rd10 mice (113), and AAV-miR-204 attenuated retinal degeneration in two different mouse models. By downregulating microglial activation and PR mortality, miR-204 alters the expression profiles of transgenic retinas toward those of healthy retinas (114). In addition, miR-223 is required for the regulation of microglial inflammation and the maintenance of normal retinal function (247).
3.3 DNA methylation and trained immunity: Epigenetic reprogramming of immunity phenotype
DNA methylation refers to the addition of a methyl group to the 5′-carbon of a cytosine (C) ring, resulting in the formation of 5-methylcytosine (5mC), which mainly occurs in the promoter regions. Typically, methylation modifications result in gene repression, and global genomic hypermethylation relates to heterochromatin formation and inhibits transcription (248). Aberrant regulation of DNA methylation results in PR degeneration and neuronal loss in the retina. In the absence of DNA methyltransferase 1, the initiation of PR differentiation is severely hindered (249). In RP retinas, binding sites of several important transcription factors for retinal physiology were hypermethylated (250). The role of DNA methylation in the development of retinitis pigmentosa has been reviewed in detail elsewhere (251) and will not be repeated here.
We argue that trained immunity regulates the microglial phenotype in RP by plasticizing microglial reactivity via epigenetic modification.
Trained immunity, also known as innate immune memory, refers to the phenomenon in which innate immunity modifies its function after an initial insult and reacts more vigorously to subsequent stimuli. Epigenic reprogramming determines the immune phenotype of immune cells and leads to long-lasting functional alterations (252, 253) (Figure 2). Using macrophages as an illustration, in the resting state, the promoter regions of inflammatory genes are enriched with repressive epigenetic marks, called epigenetic barriers, to prevent activation in the absence of stimuli. Upon stimulus, repressive epigenetic marks are removed, and activating epigenetic marks are introduced to the promoters and enhancers of specific genes in an attempt to encourage inflammatory molecule synthesis and phagocytosis to eliminate the insult. After stimulus elimination, activating epigenetic marks are partly retained (254). The innate immune system may become overly trained in chronic inflammatory diseases as a result of such mechanisms, resulting in pathological tissue damage.
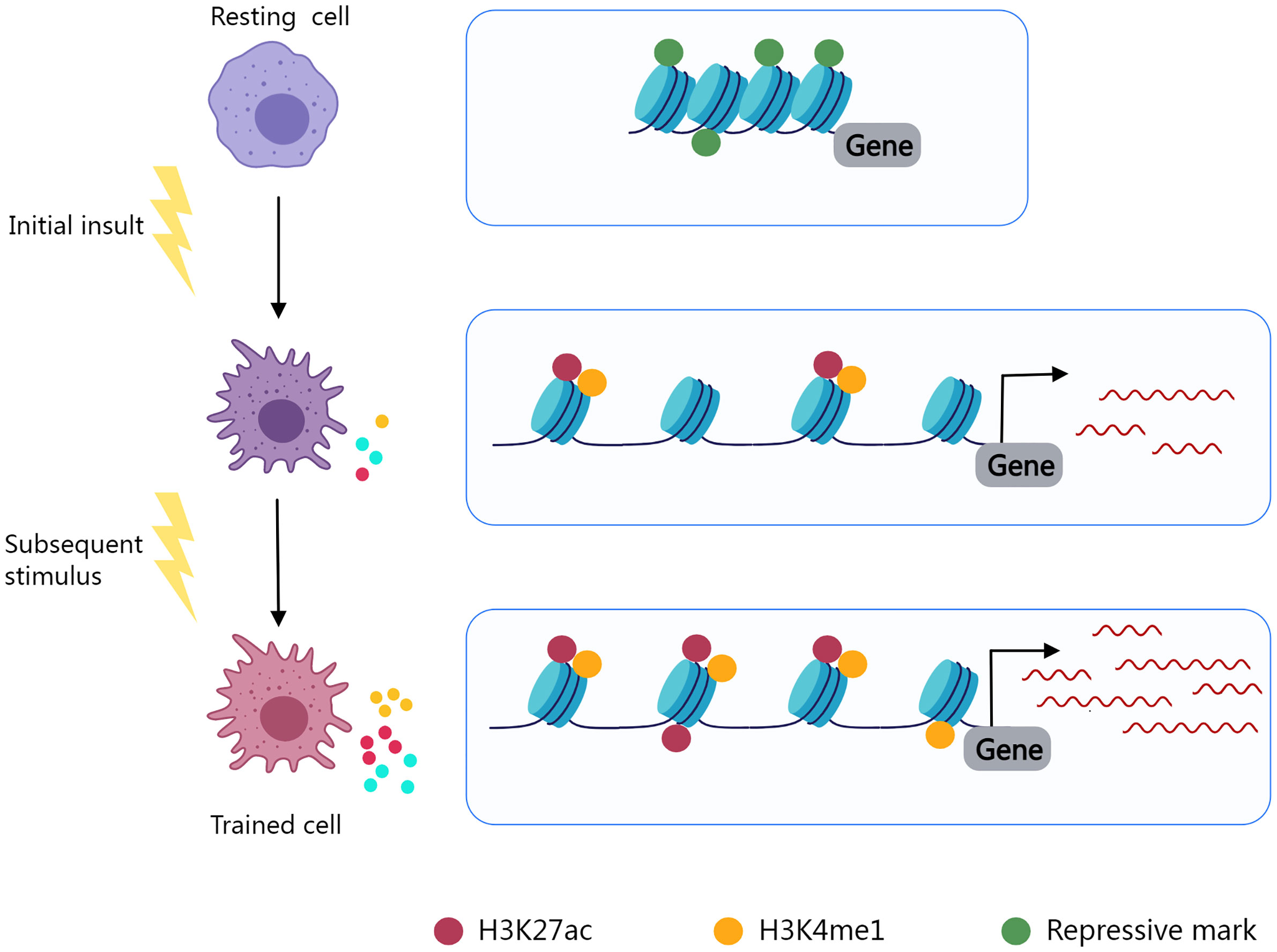
Figure 2 Epigenetic reprogramming determines the immune phenotype of trained cells. Resting cells contain inhibitory epigenetic marks in immune response-related gene regulation areas. Cell activation and epigenetic reprogramming are initiated by an initial insult. Repressive marks fade and epigenetic activating markers (H3K27ac, H3K4me1) are present. Activating markers are partially preserved after stimulus elimination, and trained cells exhibit enhanced immune responses to subsequent stimuli.
In the context of neurodegenerative disorders of the CNS, the relationship between trained immunity and microglial phenotype has been discussed (255, 256). Low-dose LPS intraperitoneally administered to mice induced long-lasting innate immune memory in brain microglia and exacerbated Alzheimer’s disease pathology. Activated microglia are enriched with the epigenetic marks H3K4me1 and H3K27ac, which define active enhancers (256). In RP model P23H rats, intraperitoneal injection of low-dose LPS increased microglial activation and the number of infiltrating microglia, as well as elevated the expression levels of several inflammation-related genes (257). In addition to the activation of retinal microglia, elevated levels of serum cytokines show the activation of peripheral immune cells in RP (22, 23). Recent work by Su et al. revealed that monocytes from patients with autosomal recessive RP exhibit a trained-like phenotype. Upon stimulation, these monocytes produce more TNF-α, IL-6, and IL-1β and upregulate inflammatory pathways such as NF-κB (258). Current evidence supports a role for trained immunity in RP pathogenesis by epigenetic reprogramming of microglia and peripheral macrophages to modulate the immune phenotype and trigger an active immune response, although many details remain to be confirmed.
4 Gut microbiome and microglial activity
The gut microbiome, which resides in the intestinal tract and performs nutrition metabolism, has recently been found to influence the maturation of the immune system. Components and metabolites of microbial cells engage in the modulation of immune recognition and immune tolerance through innate immune receptors on intestinal epithelial cells and influence the function of innate myeloid cells and lymphoid cells through diverse mechanisms (Figure 3). In addition, the microbiota’s make-up and function are subject to the innate immune system. Therefore, gut dysbiosis may induce immune system dysregulation and trigger disease emergence (259).
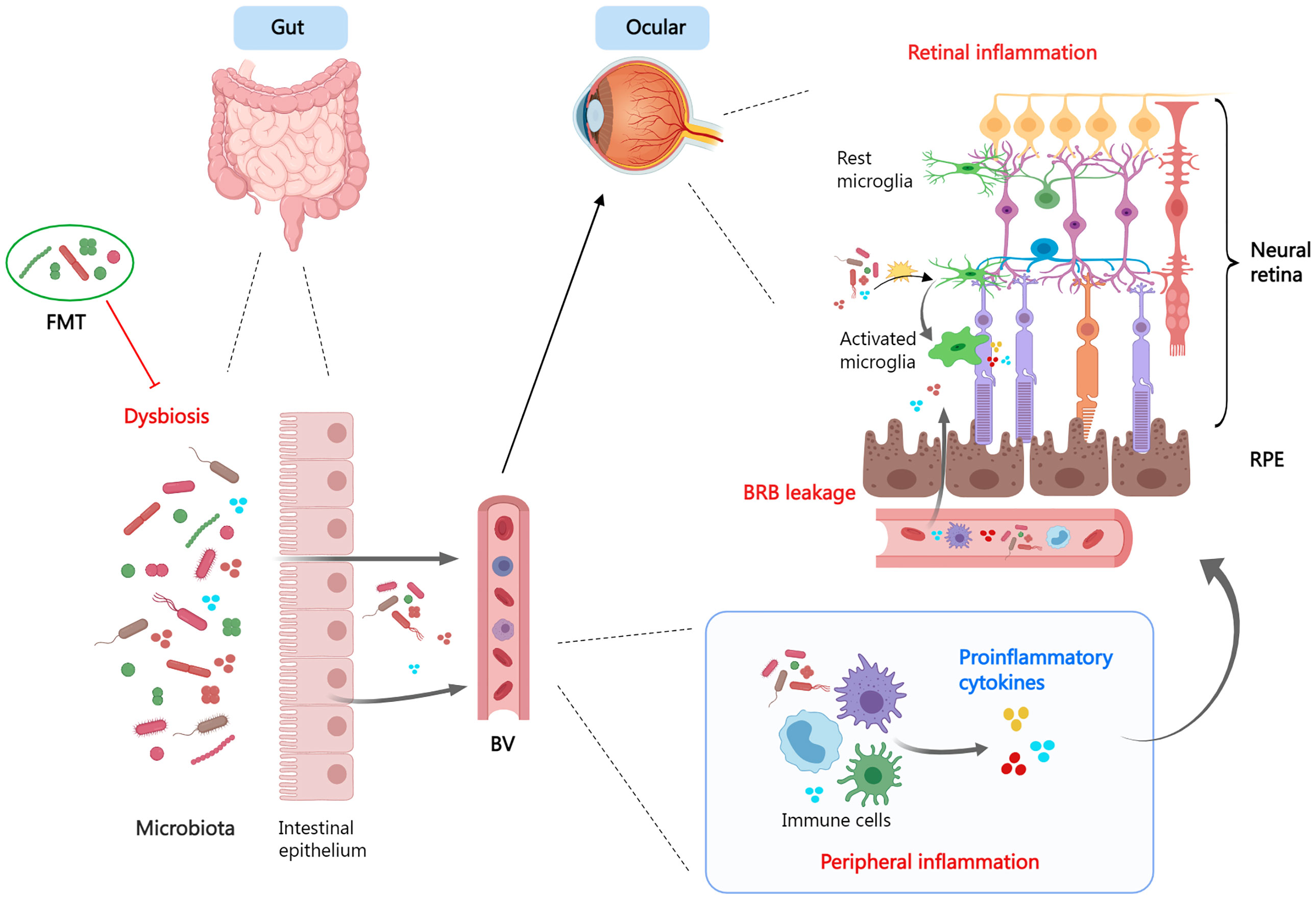
Figure 3 The gut-retina axis. When dysbiosis is present, the microbiota and its derivatives enter the circulation via the leaky intestinal mucosa and mediate systemic- and local- (ocular) inflammatory responses. Dysbiosis promotes a pro-inflammatory phenotype of microglia and exacerbates tissue damage. Fecal microbiota transplant is a viable therapeutic strategy. FMT, fecal microbiota transplant; BV, blood vessel; BRB, blood−retina barrier; RPE, retinal pigment epithelium.
The gut microbiome has been linked to retinal degenerative disorders such as age-related macular degeneration (260) and diabetic retinopathy (261). Using the rd10 RP mouse model, Kutsyr O. et al. (262) related alterations in the composition profiles of the gut microbiome to RP. Compared to healthy mice, the gut microbiome of rd10 mice had reduced ASV richness and α diversity. Rd10 mice, in particular, feature a high proportion of B. caecimuris, a species that is uncommon in healthy gut mice, but lack four species (Rikenella spp., Muribaculaceace spp., Prevotellaceae UCG-001 spp., and Bacilli spp.) that are common in the healthy gut microbiome (262). The gut microbiome is susceptible to dietary influences. Further research by the same group demonstrated that a short-term high-fat diet significantly modifies the gut flora, enhances retinal oxidative stress and inflammation, and ultimately accelerates the degeneration of the rd10 retina (263). Thus, dysbiosis in the gut contributes to retinal inflammation and constitutes the pathogenesis of RP.
By exchanging the intestinal microbiota (Fecal microbiota transplant, FMT) of young and aged mice, emerging evidence by Parker et al. (264) suggests that the gut microbiome is a modifier of retinal inflammation. Compared to young mice, aged mice exhibit increased systemic and tissue inflammation, as evidenced by elevated serum proinflammatory cytokines (TNFα, IL-6), microglial overactivation in the brain, and C3 accumulation at the RPE/Britch’s membrane interface. Transferring aged donor microbiota to young mice disrupts the intestinal epithelial barrier and triggers inflammation in the retina and brain, whereas transfer of aged mice with young donor microbiota could reverse age-related inflammation (264).
The evidence above supports the “diet-gut microbiome-retina axis” hypothesis in the pathogenesis of RP. Despite the fact that this work is still in its early stages, the gut microbiota is a promising therapeutic target for RP.
5 Herbal agents in inflammation suppression
Herbal compounds, or phytochemicals derived from plants, possess a wide range of biological activities and have been explored for the treatment of RP, demonstrating anti-inflammatory properties in RP investigations.
Curcumin is a polyphenolic compound produced from the spice turmeric. Curcumin provided morphological and functional protection in rd1 mice, P23H rats, and an MNU-induced RP model (115, 116, 265, 266). A single vitreous injection of curcumin reduced PR loss in rd1 mice by inhibiting microglial activation and modulating the expression of CCL2, TIMP-1 and VCAM-1 (115).
Lyceum barbarum polysaccharides and zeaxanthin dipalmitate are two main bioactive agents extracted from wolfberry. Lyceum barbarum polysaccharides protects against retinal degeneration by modifying inflammation and apoptosis through the inhibition of NF-κB and HIF-1α expression (117, 118). zeaxanthin dipalmitate acts through several pathways, including STAT3, CCL2 and MAPK, in parallel to inhibiting inflammation in the rd10 retina (119).
Saffron, widely used in traditional Chinese medicine for its anti-inflammatory and antioxidant properties, protects PRs exposed to environmental ATP by blocking P2X7R signaling (120). In P23H rats, saffron administration increased PR survival and functional retention while decreasing vascular disruption (121).
Resveratrol (3,40,5-trihydroxystilbene) is found in chocolate, fruits, and vegetables. Resveratrol treatment inhibited microglia-mediated death of 661W cells via downregulation of microglial migratory, phagocytic, and proinflammatory cytokine production (122). Subretinal injection of JC19 (3,4’-diglucosyl resveratrol), a resveratrol prodrug, reduced PR loss and improved functional performance in ERG tests of the rd10 retina. The author speculates that sirtuin1 activation is the underlying mechanism (123).
6 Conclusion and future perspectives
A large body of research conducted on the inflammatory processes during RP tries to discover common mechanisms that target multiple RP genotypes and develop appropriate therapeutic options. However, after reviewing the existing literature, we discovered that no single treatment is appropriate for all types of RP, and the application of valproic acid is a prime example, with treatment effects significantly varying between models with different genetic backgrounds and even exhibiting detrimental effects. This raises the prospect that a link between genetics and RP inflammation needs more investigation. To date, genetic mutations remain the only identified risk factor for RP. Different permutations of inheritance pattern, genotype, and the number of mutations lead to variations in the phenotype and pathological progression of RP. Similarly, we anticipate that the multiple phenotypes of inflammatory activation in RP are closely related to the genetic background. Nevertheless, the relationship between genetic background and inflammation is currently unclear due to the lack of corresponding evidence.
Both RP patients and animal models have a more susceptible immune system and are prone to developing inflammation. This abnormal immune system may depend heavily on the genetic background. The gut microbiota play a critical role in the maturation of the innate immune system after birth, and trained immunity is implicated in this process; however, the influence of genetic background on the maturation of the immune system has not been investigated. Therefore, long-term clinical observation and family tracing of the RP population are necessary. What needs to be documented should include, but is not limited to, macroscopic clinical manifestations, structural and functional measurements, and monitoring of local and peripheral inflammation levels. And appropriate follow-up criteria need to be established to ensure consistency of measurements and to obtain usable information.
Inflammation is an important feature of RP, and the present review highlights the role of immunomodulation in RP treatment. There has been significant interest in modulating the inflammatory response as a strategy to treat RP, and an increasing number of studies have proven the effectiveness of immunomodulation in ameliorating and perhaps reversing retinal degeneration. Therapeutic strategies based on immunomodulation are a potential treatment for RP, and deepening the understanding of immune modulation is helpful in establishing suitable therapies. As with immunotherapies already carried out, artificial regulation of immunity will bring inevitable side effects. It is challenging to regulate immunity accurately and to enhance the beneficial effects and minimize the harmful ones concurrently. Many of these specific mechanisms need to be further studied, especially the interactions between these pathways.
Author contributions
LZ wrote the manuscript and painted the figure. NY and CH reviewed and modified the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Science and Technology Department of Sichuan Province (2020YFS0205).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet (2013) 84(2):132–41. doi: 10.1111/cge.12203
2. Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, et al. Non-syndromic retinitis pigmentosa. Prog In Retin Eye Res (2018) 66:157–86. doi: 10.1016/j.preteyeres.2018.03.005
3. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet (2006) 368(9549):1795–809. doi: 10.1016/S0140-6736(06)69740-7
4. Brinkman CJ, Pinckers AJ, Broekhuyse RM. Immune reactivity to different retinal antigens in patients suffering from retinitis pigmentosa. Invest Ophthalmol Vis Sci (1980) 19(7):743–50.
5. Heredia CD, Vich JM, Huguet J, Garcia-Calderon JV, Garcia-Calderon PA. Altered cellular immunity and suppressor cell activity in patients with primary retinitis pigmentosa. Br J Ophthalmol (1981) 65(12):850–4. doi: 10.1136/bjo.65.12.850
6. Yoshida N, Ikeda Y, Notomi S, Ishikawa K, Murakami Y, Hisatomi T, et al. Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology (2013) 120(1):100–5. doi: 10.1016/j.ophtha.2012.07.006
7. Murakami Y, Yoshida N, Ikeda Y, Nakatake S, Fujiwara K, Notomi S, et al. Relationship between aqueous flare and visual function in retinitis pigmentosa. Am J Ophthalmol (2015) 159(5):958–63.e1. doi: 10.1016/j.ajo.2015.02.001
8. Fujiwara K, Ikeda Y, Murakami Y, Tachibana T, Funatsu J, Koyanagi Y, et al. Aqueous flare and progression of visual field loss in patients with retinitis pigmentosa. Invest Ophthalmol Vis Sci (2020) 61(8):26. doi: 10.1167/iovs.61.8.26
9. Fujiwara K, Ikeda Y, Murakami Y, Nakatake S, Tachibana T, Yoshida N, et al. Association between aqueous flare and epiretinal membrane in retinitis pigmentosa. Invest Ophthalmol Vis Sci (2016) 57(10):4282–6. doi: 10.1167/iovs.16-19686
10. Nishiguchi KM, Yokoyama Y, Kunikata H, Abe T, Nakazawa T. Correlation between aqueous flare and residual visual field area in retinitis pigmentosa. Br J Ophthalmol (2019) 103(4):475–80. doi: 10.1136/bjophthalmol-2018-312225
11. Nagasaka Y, Ito Y, Ueno S, Terasaki H. Increased aqueous flare is associated with thickening of inner retinal layers in eyes with retinitis pigmentosa. Sci Rep (2016) 6:33921. doi: 10.1038/srep33921
12. Strobbe E, Cellini M, Fresina M, Campos EC. Et-1 plasma levels, aqueous flare, and choroidal thickness in patients with retinitis pigmentosa. J Ophthalmol (2015) 2015:292615. doi: 10.1155/2015/292615
13. Kuchle M, Nguyen NX, Martus P, Freissler K, Schalnus R. Aqueous flare in retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol (1998) 236(6):426–33. doi: 10.1007/s004170050101
14. Ivanova E, Alam NM, Prusky GT, Sagdullaev BT. Blood-retina barrier failure and vision loss in neuron-specific degeneration. JCI Insight (2019) 5(8):e126747. doi: 10.1172/jci.insight.126747
15. Napoli D, Biagioni M, Billeri F, Di Marco B, Orsini N, Novelli E, et al. Retinal pigment epithelium remodeling in mouse models of retinitis pigmentosa. Int J Mol Sci (2021) 22(10):5381. doi: 10.3390/ijms22105381
16. Vinores SA, Kuchle M, Derevjanik NL, Henderer JD, Mahlow J, Green WR, et al. Blood-retinal barrier breakdown in retinitis pigmentosa: Light and electron microscopic immunolocalization. Histol Histopathol (1995) 10(4):913–23.
17. Wang W, Kini A, Wang Y, Liu T, Chen Y, Vukmanic E, et al. Metabolic deregulation of the blood-outer retinal barrier in retinitis pigmentosa. Cell Rep (2019) 28(5):1323–34.e4. doi: 10.1016/j.celrep.2019.06.093
18. Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res (2003) 76(4):463–71. doi: 10.1016/S0014-4835(02)00332-9
19. Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, et al. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med (2015) 7(9):1179–97. doi: 10.15252/emmm.201505298
20. ten Berge JC, Fazil Z, van den Born I, Wolfs RCW, Schreurs MWJ, Dik WA, et al. Intraocular cytokine profile and autoimmune reactions in retinitis pigmentosa, age-related macular degeneration, glaucoma and cataract. Acta Ophthalmol (2019) 97(2):185–92. doi: 10.1111/aos.13899
21. Lu B, Yin H, Tang Q, Wang W, Luo C, Chen X, et al. Multiple cytokine analyses of aqueous humor from the patients with retinitis pigmentosa. Cytokine (2020) 127:154943. doi: 10.1016/j.cyto.2019.154943
22. Okita A, Murakami Y, Shimokawa S, Funatsu J, Fujiwara K, Nakatake S, et al. Changes of serum inflammatory molecules and their relationships with visual function in retinitis pigmentosa. Invest Ophth Vis Sci (2020) 61(11):30. doi: 10.1167/iovs.61.11.30
23. McMurtrey JJ. Tso MOM. a review of the immunologic findings observed in retinitis pigmentosa. Surv Ophthalmol (2018) 63(6):769–81. doi: 10.1016/j.survophthal.2018.03.002
24. Yoshida N, Ikeda Y, Notomi S, Ishikawa K, Murakami Y, Hisatomi T, et al. Laboratory evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology (2013) 120(1):E5–12. doi: 10.1016/j.ophtha.2012.07.008
25. de la Camara CMF, Hernandez-Pinto AM, Olivares-Gonzalez L, Cuevas-Martin C, Sanchez-Arago M, Hervas D, et al. Adalimumab reduces photoreceptor cell death in a mouse model of retinal degeneration. Sci Rep (2015) 5:11764. doi: 10.1038/srep11764
26. Appelbaum T, Santana E, Aguirre GD. Strong upregulation of inflammatory genes accompanies photoreceptor demise in canine models of retinal degeneration. PloS One (2017) 12(5):e0177224. doi: 10.1371/journal.pone.0177224
27. Rohrer B, Demos C, Frigg R, Grimm C. Classical complement activation and acquired immune response pathways are not essential for retinal degeneration in the Rd1 mouse. Exp Eye Res (2007) 84(1):82–91. doi: 10.1016/j.exer.2006.08.017
28. Uren PJ, Lee JT, Doroudchi MM, Smith AD, Horsager A. A profile of transcriptomic changes in the Rd10 mouse model of retinitis pigmentosa. Mol Vis (2014) 20:1612–28.
29. Park UC, Park JH, Ma DJ, Cho IH, Oh BL, Yu HG. A randomized paired-eye trial of intravitreal dexamethasone implant for cystoid macular edema in retinitis pigmentosa. Retina (2020) 40(7):1359–66. doi: 10.1097/IAE.0000000000002589
30. Glybina IV, Kennedy A, Ashton P, Abrams GW, Iezzi R. Photoreceptor neuroprotection in rcs rats Via low-dose intravitreal sustained-delivery of fluocinolone acetonide. Invest Ophthalmol Vis Sci (2009) 50(10):4847–57. doi: 10.1167/iovs.08-2831
31. Glybina IV, Kennedy A, Ashton P, Abrams GW, Iezzi R. Intravitreous delivery of the corticosteroid fluocinolone acetonide attenuates retinal degeneration in S334ter-4 rats. Invest Ophthalmol Vis Sci (2010) 51(8):4243–52. doi: 10.1167/iovs.09-4492
32. Iezzi R, Guru BR, Glybina IV, Mishra MK, Kennedy A, Kannan RM. Dendrimer-based targeted intravitreal therapy for sustained attenuation of neuroinflammation in retinal degeneration. Biomaterials (2012) 33(3):979–88. doi: 10.1016/j.biomaterials.2011.10.010
33. Guadagni V, Biagioni M, Novelli E, Aretini P, Mazzanti CM, Strettoi E. Rescuing cones and daylight vision in retinitis pigmentosa mice. FASEB J (2019) 33(9):10177–92. doi: 10.1096/fj.201900414R
34. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi: 10.1126/science.1194637
35. O'Koren EG, Yu C, Klingeborn M, Wong AYW, Prigge CL, Mathew R, et al. Microglial function is distinct in different anatomical locations during retinal homeostasis and degeneration. Immunity (2019) 50(3):723–37.e7. doi: 10.1016/j.immuni.2019.02.007
36. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-Sac-Derived erythro-myeloid progenitors. Nature (2015) 518(7540):547–51. doi: 10.1038/nature13989
37. Hume DA, Perry VH, Gordon S. Immunohistochemical localization of a macrophage-specific antigen in developing mouse retina: Phagocytosis of dying neurons and differentiation of microglial cells to form a regular array in the plexiform layers. J Cell Biol (1983) 97(1):253–7. doi: 10.1083/jcb.97.1.253
38. Silverman SM, Wong WT. Microglia in the retina: Roles in development, maturity, and disease. Annu Rev Vis Sci (2018) 4:45–77. doi: 10.1146/annurev-vision-091517-034425
39. Dixon MA, Greferath U, Fletcher EL, Jobling AI. The contribution of microglia to the development and maturation of the visual system. Front Cell Neurosci (2021) 15:659843. doi: 10.3389/fncel.2021.659843
40. Zhang Y, Zhao L, Wang X, Ma W, Lazere A, Qian HH, et al. Repopulating retinal microglia restore endogenous organization and function under Cx3cl1-Cx3cr1 regulation. Sci Adv (2018) 4(3):eaap8492. doi: 10.1126/sciadv.aap8492
41. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain cns microglia maintenance and function throughout adult life. Nat Neurosci (2007) 10(12):1538–43. doi: 10.1038/nn2014
42. Younger D, Murugan M, Rama Rao KV, Wu LJ, Chandra N. Microglia receptors in animal models of traumatic brain injury. Mol Neurobiol (2019) 56(7):5202–28. doi: 10.1007/s12035-018-1428-7
43. Butler CA, Popescu AS, Kitchener EJA, Allendorf DH, Puigdellivol M, Brown GC. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J Neurochem (2021) 158(3):621–39. doi: 10.1111/jnc.15327
44. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci (2014) 15(4):209–16. doi: 10.1038/nrn3710
45. Zhou T, Huang ZJ, Sun XW, Zhu XW, Zhou LL, Li M, et al. Microglia polarization with M1/M2 phenotype changes in Rd1 mouse model of retinal degeneration. Front Neuroanat (2017) 11:77. doi: 10.3389/fnana.2017.00077
46. Ochocka N, Kaminska B. Microglia diversity in healthy and diseased brain: Insights from single-cell omics. Int J Mol Sci (2021) 22(6):3027. doi: 10.3390/ijms22063027
47. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron (2012) 74(4):691–705. doi: 10.1016/j.neuron.2012.03.026
48. Wang X, Zhao L, Zhang J, Fariss RN, Ma W, Kretschmer F, et al. Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. J Neurosci (2016) 36(9):2827–42. doi: 10.1523/JNEUROSCI.3575-15.2016
49. Ransohoff RM, Perry VH. Microglial physiology: Unique stimuli, specialized responses. Annu Rev Immunol (2009) 27:119–45. doi: 10.1146/annurev.immunol.021908.132528
50. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308(5726):1314–8. doi: 10.1126/science.1110647
51. Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain (2009) 132(Pt 2):288–95. doi: 10.1093/brain/awn109
52. Rohrer B, Pinto FR, Hulse KE, Lohr HR, Zhang L, Almeida JS. Multidestructive pathways triggered in photoreceptor cell death of the Rd mouse as determined through gene expression profiling. J Biol Chem (2004) 279(40):41903–10. doi: 10.1074/jbc.M405085200
53. Murakami Y, Ikeda Y, Nakatake S, Tachibana T, Fujiwara K, Yoshida N, et al. Necrotic enlargement of cone photoreceptor cells and the release of high-mobility group box-1 in retinitis pigmentosa. Cell Death Discovery (2015) 1:15058. doi: 10.1038/cddiscovery.2015.58
54. Vallazza-Deschamps G, Cia D, Gong J, Jellali A, Duboc A, Forster V, et al. Excessive activation of cyclic nucleotide-gated channels contributes to neuronal degeneration of photoreceptors. Eur J Neurosci (2005) 22(5):1013–22. doi: 10.1111/j.1460-9568.2005.04306.x
55. Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat Rev Genet (2010) 11(4):273–84. doi: 10.1038/nrg2717
56. Mahaling B, Low SWY, Beck M, Kumar D, Ahmed S, Connor TB, et al. Damage-associated molecular patterns (Damps) in retinal disorders. Int J Mol Sci (2022) 23(5):2591. doi: 10.3390/ijms23052591
57. Blank T, Goldmann T, Koch M, Amann L, Schon C, Bonin M, et al. Early microglia activation precedes photoreceptor degeneration in a mouse model of Cngb1-linked retinitis pigmentosa. Front Immunol (2017) 8:1930. doi: 10.3389/fimmu.2017.01930
58. Zeiss CJ, Johnson EA. Proliferation of microglia, but not photoreceptors, the outer nuclear layer of the Rd-1 mouse. Invest Ophth Vis Sci (2004) 45(3):971–6. doi: 10.1167/iovs.03-0301
59. Zhang L, Cui X, Jauregui R, Park KS, Justus S, Tsai YT, et al. Genetic rescue reverses microglial activation in preclinical models of retinitis pigmentosa. Mol Ther (2018) 26(8):1953–64. doi: 10.1016/j.ymthe.2018.06.014
60. Reichenbach A, Bringmann A. Glia of the human retina. Glia (2020) 68(4):768–96. doi: 10.1002/glia.23727
61. Goldman D. Muller Glial cell reprogramming and retina regeneration. Nat Rev Neurosci (2014) 15(7):431–42. doi: 10.1038/nrn3723
62. Eastlake K, Banerjee PJ, Angbohang A, Charteris DG, Khaw PT, Limb GA. Muller Glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia (2016) 64(4):495–506. doi: 10.1002/glia.22942
63. Schmalen A, Lorenz L, Grosche A, Pauly D, Deeg CA, Hauck SM. Proteomic phenotyping of stimulated Muller cells uncovers profound pro-inflammatory signaling and antigen-presenting capacity. Front Pharmacol (2021) 12:771571. doi: 10.3389/fphar.2021.771571
64. Lorenz L, Hirmer S, Schmalen A, Hauck SM, Deeg CA. Cell surface profiling of retinal Muller glial cells reveals association to immune pathways after lps stimulation. Cells (2021) 10(3):711. doi: 10.3390/cells10030711
65. Sakami S, Imanishi Y, Palczewski K. Muller Glia phagocytose dead photoreceptor cells in a mouse model of retinal degenerative disease. FASEB J (2019) 33(3):3680–92. doi: 10.1096/fj.201801662R
66. Di Pierdomenico J, Martinez-Vacas A, Hernandez-Munoz D, Gomez-Ramirez AM, Valiente-Soriano FJ, Agudo-Barriuso M, et al. Coordinated intervention of microglial and Muller cells in light-induced retinal degeneration. Invest Ophthalmol Vis Sci (2020) 61(3):47. doi: 10.1167/iovs.61.3.47
67. Arroba AI, Alvarez-Lindo N, van Rooijen N, de la Rosa EJ. Microglia-Muller glia crosstalk in the Rd10 mouse model of retinitis pigmentosa. Adv Exp Med Biol (2014) 801:373–9. doi: 10.1007/978-1-4614-3209-8_47
68. Zhang S, Zhang SS, Gong WQ, Zhu GP, Wang ST, Wang YL, et al. Muller Cell regulated microglial activation and migration in rats with n-Methyl-N-Nitrosourea-Lnduced retinal degeneration. Front Neurosci (2018) 12:890. doi: 10.3389/fnins.2018.00890
69. Devoldere J, Peynshaert K, De Smedt SC, Remaut K. Muller Cells as a target for retinal therapy. Drug Discov Today (2019) 24(8):1483–98. doi: 10.1016/j.drudis.2019.01.023
70. Chen Y, Xia Q, Zeng Y, Zhang Y, Zhang M. Regulations of retinal inflammation: Focusing on Muller glia. Front Cell Dev Biol (2022) 10:898652. doi: 10.3389/fcell.2022.898652
71. Galan A, Jmaeff S, Barcelona PF, Brahimi F, Sarunic MV, Saragovi HU. In retinitis pigmentosa Trkc.T1-dependent vectorial erk activity upregulates glial tnf-alpha, causing selective neuronal death. Cell Death Dis (2017) 8(12):3222. doi: 10.1038/s41419-017-0074-8
72. Platon-Corchado M, Barcelona PF, Jmaeff S, Marchena M, Hernandez-Pinto AM, Hernandez-Sanchez C, et al. P75(Ntr) antagonists attenuate photoreceptor cell loss in murine models of retinitis pigmentosa. Cell Death Dis (2017) 8(7):e2922. doi: 10.1038/cddis.2017.306
73. Rana T, Kotla P, Fullard R, Gorbatyuk M. Tnfa knockdown in the retina promotes cone survival in a mouse model of autosomal dominant retinitis pigmentosa. Biochim Biophys Acta Mol Basis Dis (2017) 1863(1):92–102. doi: 10.1016/j.bbadis.2016.10.008
74. de la Camara CMF, Olivares-Gonzalez L, Hervas D, Salom D, Millan JM, Rodrigo R. Infliximab reduces zaprinast-induced retinal degeneration in cultures of porcine retina. J Neuroinflamm (2014) 11:172. doi: 10.1186/s12974-014-0172-9
75. Olivares-Gonzalez L, Velasco S, Millan JM, Rodrigo R. Intravitreal administration of adalimumab delays retinal degeneration in Rd10 mice. FASEB J (2020) 34(10):13839–61. doi: 10.1096/fj.202000044RR
76. Sanchez-Cruz A, Mendez AC, Lizasoain I, de la Villa P, de la Rosa EJ, Hernandez-Sanchez C. Tlr2 gene deletion delays retinal degeneration in two genetically distinct mouse models of retinitis pigmentosa. Int J Mol Sci (2021) 22(15):7815. doi: 10.3390/ijms22157815
77. Kohno H, Chen Y, Kevany BM, Pearlman E, Miyagi M, Maeda T, et al. Photoreceptor proteins initiate microglial activation Via toll-like receptor 4 in retinal degeneration mediated by all-Trans-Retinal. J Biol Chem (2013) 288(21):15326–41. doi: 10.1074/jbc.M112.448712
78. Di Pierdomenico J, Scholz R, Valiente-Soriano FJ, Sanchez-Migallon MC, Vidal-Sanz M, Langmann T, et al. Neuroprotective effects of Fgf2 and minocycline in two animal models of inherited retinal degeneration. Invest Ophth Vis Sci (2018) 59(11):4392–403. doi: 10.1167/iovs.18-24621
79. Hughes EH, Schlichtenbrede FC, Murphy CC, Broderick C, Van Rooijen N, Ali RR, et al. Minocycline delays photoreceptor death in the rds mouse through a microglia-independent mechanism. Exp Eye Res (2004) 78(6):1077–84. doi: 10.1016/j.exer.2004.02.002
80. Zhang C, Lei B, Lam TT, Yang F, Sinha D, Tso MO. Neuroprotection of photoreceptors by minocycline in light-induced retinal degeneration. Invest Ophthalmol Vis Sci (2004) 45(8):2753–9. doi: 10.1167/iovs.03-1344
81. Scholz R, Sobotka M, Caramoy A, Stempfl T, Moehle C, Langmann T. Minocycline counter-regulates pro-inflammatory microglia responses in the retina and protects from degeneration. J Neuroinflamm (2015) 12:209. doi: 10.1186/s12974-015-0431-4
82. Peng B, Xiao J, Wang K, So KF, Tipoe GL, Lin B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J Neurosci (2014) 34(24):8139–50. doi: 10.1523/jneurosci.5200-13.2014
83. Syeda S, Patel AK, Lee T, Hackam AS. Reduced photoreceptor death and improved retinal function during retinal degeneration in mice lacking innate immunity adaptor protein Myd88. Exp Neurol (2015) 267:1–12. doi: 10.1016/j.expneurol.2015.02.027
84. Garces K, Carmy T, Illiano P, Brambilla R, Hackam AS. Increased neuroprotective microglia and photoreceptor survival in the retina from a peptide inhibitor of myeloid differentiation factor 88 (Myd88). J Mol Neurosci (2020) 70(6):968–80. doi: 10.1007/s12031-020-01503-0
85. Carmy-Bennun T, Myer C, Bhattacharya SK, Hackam AS. Quantitative proteomic analysis after neuroprotective Myd88 inhibition in the retinal degeneration 10 mouse. J Cell Mol Med (2021) 25(20):9533–42. doi: 10.1111/jcmm.16893
86. Aslanidis A, Karlstetter M, Scholz R, Fauser S, Neumann H, Fried C, et al. Activated Microglia/Macrophage whey acidic protein (Amwap) inhibits nfkappab signaling and induces a neuroprotective phenotype in microglia. J Neuroinflamm (2015) 12:77. doi: 10.1186/s12974-015-0296-6
87. Viringipurampeer IA, Metcalfe AL, Bashar AE, Sivak O, Yanai A, Mohammadi Z, et al. Nlrp3 inflammasome activation drives bystander cone photoreceptor cell death in a P23h rhodopsin model of retinal degeneration. Hum Mol Genet (2016) 25(8):1501–16. doi: 10.1093/hmg/ddw029
88. Puthussery T, Fletcher E. Extracellular atp induces retinal photoreceptor apoptosis through activation of purinoceptors in rodents. J Comp Neurol (2009) 513(4):430–40. doi: 10.1002/cne.21964
89. Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, et al. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by Cx3cl1-Cx3cr1 signaling in a mouse model of retinitis pigmentosa. Glia (2016) 64(9):1479–91. doi: 10.1002/glia.23016
90. Wang SK, Xue YL, Rana P, Hong CM, Cepko CL. Soluble Cx3cl1 gene therapy improves cone survival and function in mouse models of retinitis pigmentosa. Proc Natl Acad Sci U.S.A. (2019) 116(20):10140–9. doi: 10.1073/pnas.1901787116
91. Roche SL, Wyse-Jackson AC, Gomez-Vicente V, Lax P, Ruiz-Lopez AM, Byrne AM, et al. Progesterone attenuates microglial-driven retinal degeneration and stimulates protective fractalkine-Cx3cr1 signaling. PloS One (2016) 11(11):e0165197. doi: 10.1371/journal.pone.0165197
92. Roche SL, Wyse-Jackson AC, Ruiz-Lopez AM, Byrne AM, Cotter TG. Fractalkine-Cx3cr1 signaling is critical for progesterone-mediated neuroprotection in the retina. Sci Rep (2017) 7:43067. doi: 10.1038/srep43067
93. Kohno H, Terauchi R, Watanabe S, Ichihara K, Watanabe T, Nishijima E, et al. Effect of lecithin-bound iodine treatment on inherited retinal degeneration in mice. Trans Vision Sci Technol (2021) 10(13):8. doi: 10.1167/tvst.10.13.8
94. Terauchi R, Kohno H, Watanabe S, Saito S, Watanabe A, Nakano T. Minocycline decreases Ccr2-positive monocytes in the retina and ameliorates photoreceptor degeneration in a mouse model of retinitis pigmentosa. PloS One (2021) 16(4):e0239108. doi: 10.1371/journal.pone.0239108
95. Guo CR, Otani A, Oishi A, Kojima H, Makiyama Y, Nakagawa S, et al. Knockout of Ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp Eye Res (2012) 104:39–47. doi: 10.1016/j.exer.2012.08.013
96. Kohno H, Maeda T, Perusek L, Pearlman E, Maeda A. Ccl3 production by microglial cells modulates disease severity in murine models of retinal degeneration. J Immunol (2014) 192(8):3816–27. doi: 10.4049/jimmunol.1301738
97. Rutar M, Natoli R, Provis JM. Small interfering rna-mediated suppression of Ccl2 in Muller cells attenuates microglial recruitment and photoreceptor death following retinal degeneration. J Neuroinflamm (2012) 9:221. doi: 10.1186/1742-2094-9-221
98. Samardzija M, Wenzel A, Aufenberg S, Thiersch M, Remé C, Grimm C. Differential role of jak-stat signaling in retinal degenerations. FASEB J (2006) 20(13):2411–3. doi: 10.1096/fj.06-5895fje
99. Xie J, Li YJ, Dai JM, He Y, Sun DY, Dai C, et al. Olfactory ensheathing cells grafted into the retina of rcs rats suppress inflammation by down-regulating the Jak/Stat pathway. Front Cell Neurosci (2019) 13:341. doi: 10.3389/fncel.2019.00341
100. Brown C, Agosta P, McKee C, Walker K, Mazzella M, Alamri A, et al. Human primitive mesenchymal stem cell-derived retinal progenitor cells improved neuroprotection, neurogenesis, and vision in Rd12 mouse model of retinitis pigmentosa. Stem Cell Res Ther (2022) 13(1):148. doi: 10.1186/s13287-022-02828-w
101. Lipinski DM, Barnard AR, Singh MS, Martin C, Lee EJ, Davies WIL, et al. Cntf gene therapy confers lifelong neuroprotection in a mouse model of human retinitis pigmentosa. Mol Ther (2015) 23(8):1308–19. doi: 10.1038/mt.2015.68
102. Rhee KD, Nusinowitz S, Chao K, Yu F, Bok D, Yang XJ. Cntf-mediated protection of photoreceptors requires initial activation of the cytokine receptor Gp130 in müller glial cells. Proc Natl Acad Sci U.S.A. (2013) 110(47):E4520–9. doi: 10.1073/pnas.1303604110
103. Ueki Y, Wang J, Chollangi S, Ash JD. Stat3 activation in photoreceptors by leukemia inhibitory factor is associated with protection from light damage. J Neurochem (2008) 105(3):784–96. doi: 10.1111/j.1471-4159.2007.05180.x
104. Sancho-Pelluz J, Alavi MV, Sahaboglu A, Kustermann S, Farinelli P, Azadi S, et al. Excessive hdac activation is critical for neurodegeneration in the Rd1 mouse. Cell Death Dis (2010) 1(2):e24. doi: 10.1038/cddis.2010.4
105. Samardzija M, Corna A, Gomez-Sintes R, Jarboui MA, Armento A, Roger JE, et al. Hdac inhibition ameliorates cone survival in retinitis pigmentosa mice. Cell Death Differ (2021) 28(4):1317–32. doi: 10.1038/s41418-020-00653-3
106. Sundaramurthi H, Roche SL, Grice GL, Moran A, Dillion ET, Campiani G, et al. Selective histone deacetylase 6 inhibitors restore cone photoreceptor vision or outer segment morphology in zebrafish and mouse models of retinal blindness. Front Cell Dev Biol (2020) 8:689. doi: 10.3389/fcell.2020.00689
107. Leyk J, Daly C, Janssen-Bienhold U, Kennedy BN, Richter-Landsberg C. Hdac6 inhibition by tubastatin a is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis (2017) 8(8):e3028. doi: 10.1038/cddis.2017.415
108. Popova EY, Kawasawa YI, Zhang SSM, Barnstable CJ. Inhibition of epigenetic modifiers Lsd1 and Hdac1 blocks rod photoreceptor death in mouse models of retinitis pigmentosa. J Neurosci (2021) 41(31):6775–92. doi: 10.1523/jneurosci.3102-20.2021
109. Mitton KP, Guzman AE, Deshpande M, Byrd D, DeLooff C, Mkoyan K, et al. Different effects of valproic acid on photoreceptor loss in Rd1 and Rd10 retinal degeneration mice. Mol Vis (2014) 20:1527–44.
110. Vent-Schmidt RYJ, Wen RH, Zong Z, Chiu CN, Tam BM, May CG, et al. Opposing effects of valproic acid treatment mediated by histone deacetylase inhibitor activity in four transgenic X. Laevis Models Retinitis Pigmentosa J Neurosci (2017) 37(4):1039–54. doi: 10.1523/jneurosci.1647-16.2016
111. Zhao L, Li J, Fu YM, Zhang MX, Wang BW, Ouellette J, et al. Photoreceptor protection Via blockade of bet epigenetic readers in a murine model of inherited retinal degeneration. J Neuroinflamm (2017) 14(1):14. doi: 10.1186/s12974-016-0775-4
112. Zheng SJ, Xiao LR, Liu Y, Wang YJ, Cheng L, Zhang JJ, et al. Dznep inhibits H3k27me3 deposition and delays retinal degeneration in the Rd1 mice. Cell Death Dis (2018) 9(3):310. doi: 10.1038/s41419-018-0349-8
113. Anasagasti A, Lara-López A, Milla-Navarro S, Escudero-Arrarás L, Rodríguez-Hidalgo M, Zabaleta N, et al. Inhibition of microrna 6937 delays photoreceptor and vision loss in a mouse model of retinitis pigmentosa. Pharmaceutics (2020) 12(10):913. doi: 10.3390/pharmaceutics12100913
114. Karali M, Guadagnino I, Marrocco E, De Cegli R, Carissimo A, Pizzo M, et al. Aav-Mir-204 protects from retinal degeneration by attenuation of microglia activation and photoreceptor cell death. Mol Ther Nucleic Acids (2020) 19:144–56. doi: 10.1016/j.omtn.2019.11.005
115. Wang YH, Yin ZY, Gao LX, Sun DY, Hu XS, Xue LY, et al. Curcumin delays retinal degeneration by regulating microglia activation in the retina of Rd1 mice. Cell Physiol Biochem (2017) 44(2):479–93. doi: 10.1159/000485085
116. Vasireddy V, Chavali VRM, Joseph VT, Kadam R, Lin JH, Jamison JA, et al. Rescue of photoreceptor degeneration by curcumin in transgenic rats with P23h rhodopsin mutation. PloS One (2011) 6(6):e21193. doi: 10.1371/journal.pone.0021193
117. Liu F, Zhang J, Xiang Z, Xu D, So KF, Vardi N, et al. Lycium barbarum polysaccharides protect retina in Rd1 mice during photoreceptor degeneration. Invest Ophthalmol Vis Sci (2018) 59(1):597–611. doi: 10.1167/iovs.17-22881
118. Wang K, Xiao J, Peng B, Xing F, So KF, Tipoe GL, et al. Retinal structure and function preservation by polysaccharides of wolfberry in a mouse model of retinal degeneration. Sci Rep (2014) 4:7601. doi: 10.1038/srep07601
119. Liu F, Liu XB, Zhou YM, Yu YK, Wang K, Zhou ZQ, et al. Wolfberry-derived zeaxanthin dipalmitate delays retinal degeneration in a mouse model of retinitis pigmentosa through modulating Stat3, Ccl2 and mapk pathways. J Neurochem (2021) 158(5):1131–50. doi: 10.1111/jnc.15472
120. Corso L, Cavallero A, Baroni D, Garbati P, Prestipino G, Bisti S, et al. Saffron reduces atp-induced retinal cytotoxicity by targeting P2x7 receptors. Purinergic Signal (2016) 12(1):161–74. doi: 10.1007/s11302-015-9490-3
121. Fernandez-Sanchez L, Lax P, Esquiva G, Martin-Nieto J, Pinilla I, Cuenca N. Safranal, a saffron constituent, attenuates retinal degeneration in P23h rats. PloS One (2012) 7(8):e43074. doi: 10.1371/journal.pone.0043074
122. Wiedemann J, Rashid K, Langmann T. Resveratrol induces dynamic changes to the microglia transcriptome, inhibiting inflammatory pathways and protecting against microglia-mediated photoreceptor apoptosis. Biochem Biophys Res Commun (2018) 501(1):239–45. doi: 10.1016/j.bbrc.2018.04.223
123. Valdes-Sanchez L, Garcia-Delgado AB, Montero-Sanchez A, de la Cerda B, Lucas R, Penalver P, et al. The resveratrol prodrug Jc19 delays retinal degeneration in Rd10 mice. Retin Degener Dis: Mech Exp Ther (2019) 1185:457–62. doi: 10.1007/978-3-030-27378-1_75
124. Bradley JR. Tnf-mediated inflammatory disease. J Pathol (2008) 214(2):149–60. doi: 10.1002/path.2287
125. Kalliolias GD, Ivashkiv LB. Tnf biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol (2016) 12(1):49–62. doi: 10.1038/nrrheum.2015.169
126. Sato K, Li SH, Gordon WC, He JB, Liou GI, Hill JM, et al. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J Neurosci (2013) 33(44):17458–68. doi: 10.1523/jneurosci.1380-13.2013
127. de la Camara CMF, Sequedo MD, Gomez-Pinedo U, Jaijo T, Aller E, Garcia-Tarraga P, et al. Phosphodiesterase inhibition induces retinal degeneration, oxidative stress and inflammation in cone-enriched cultures of porcine retina. Exp Eye Res (2013) 111:122–33. doi: 10.1016/j.exer.2013.03.015
128. Hollingsworth TJ, Wang XD, White WA, Simpson RN, Jablonski MM. Chronic proinflammatory signaling accelerates the rate of degeneration in a spontaneous polygenic model of inherited retinal dystrophy. Front Pharmacol (2022) 13:839424. doi: 10.3389/fphar.2022.839424
129. Zeng HY, Zhu XA, Zhang C, Yang LP, Wu LM, Tso MO. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in Rd mice. Invest Ophthalmol Vis Sci (2005) 46(8):2992–9. doi: 10.1167/iovs.05-0118
130. deKozak Y, Cotinet A, Goureau O, Hicks D, ThillayeGoldenberg B. Tumor necrosis factor and nitric oxide production by resident retinal glial cells from rats presenting hereditary retinal degeneration. Ocular Immunol Inflamm (1997) 5(2):85–94. doi: 10.3109/09273949709085056
131. Lebrun-Julien F, Bertrand MJ, De Backer O, Stellwagen D, Morales CR, Di Polo A, et al. Prongf induces tnfalpha-dependent death of retinal ganglion cells through a P75ntr non-Cell-Autonomous signaling pathway. Proc Natl Acad Sci U.S.A. (2010) 107(8):3817–22. doi: 10.1073/pnas.0909276107
132. Bai Y, Shi Z, Zhuo Y, Liu J, Malakhov A, Ko E, et al. In glaucoma the upregulated truncated Trkc.T1 receptor isoform in glia causes increased tnf-alpha production, leading to retinal ganglion cell death. Invest Ophthalmol Vis Sci (2010) 51(12):6639–51. doi: 10.1167/iovs.10-5431
133. Srinivasan B, Roque CH, Hempstead BL, Al-Ubaidi MR, Roque RS. Microglia-derived pronerve growth factor promotes photoreceptor cell death Via P75 neurotrophin receptor. J Biol Chem (2004) 279(40):41839–45. doi: 10.1074/jbc.M402872200
134. Bai Y, Dergham P, Nedev H, Xu J, Galan A, Rivera JC, et al. Chronic and acute models of retinal neurodegeneration trka activity are neuroprotective whereas P75ntr activity is neurotoxic through a paracrine mechanism. J Biol Chem (2010) 285(50):39392–400. doi: 10.1074/jbc.M110.147801
135. Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, et al. The role of tumor necrosis factor alpha (Tnf-alpha) in autoimmune disease and current tnf-alpha inhibitors in therapeutics. Int J Mol Sci (2021) 22(5):2719. doi: 10.3390/ijms22052719
136. Kuhn KD, Edamura K, Bhatia N, Cheng I, Clark SA, Haynes CV, et al. Molecular dissection of tnfr-tnf alpha bidirectional signaling reveals both cooperative and antagonistic interactions with P75 neurotrophic factor receptor in axon patterning. Mol Cell Neurosci (2020) 103:103467. doi: 10.1016/j.mcn.2020.103467
137. Muliyil S, Levet C, Dusterhoft S, Dulloo I, Cowley SA, Freeman M. Adam17-triggered tnf signalling protects the ageing drosophila retina from lipid droplet-mediated degeneration. EMBO J (2020) 39(17):e104415. doi: 10.15252/embj.2020104415
138. Benoot T, Piccioni E, De Ridder K, Goyvaerts C. Tnfα and immune checkpoint inhibition: Friend or foe for lung cancer? Int J Mol Sci (2021) 22(16):8691. doi: 10.3390/ijms22168691
139. Tanzer MC, Bludau I, Stafford CA, Hornung V, Mann M. Phosphoproteome profiling uncovers a key role for cdks in tnf signaling. Nat Commun (2021) 12(1):6053. doi: 10.1038/s41467-021-26289-6
140. Dichtl S, Sanin DE, Koss CK, Willenborg S, Petzold A, Tanzer MC, et al. Gene-selective transcription promotes the inhibition of tissue reparative macrophages by tnf. Life Sci Alliance (2022) 5(4):e202101315. doi: 10.26508/lsa.202101315
141. Tanzer MC. A proteomic perspective on tnf-mediated signalling and cell death. Biochem Soc Trans (2022) 50(1):13–20. doi: 10.1042/BST20211114
142. Wang M, Ma W, Zhao L, Fariss RN, Wong WT. Adaptive müller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J Neuroinflamm (2011) 8:173. doi: 10.1186/1742-2094-8-173
143. Agca C, Gubler A, Traber G, Beck C, Imsand C, Ail D, et al. P38 mapk signaling acts upstream of lif-dependent neuroprotection during photoreceptor degeneration. Cell Death Dis (2013) 4(9):e785. doi: 10.1038/cddis.2013.323
144. Coelho-Santos V, Goncalves J, Fontes-Ribeiro C, Silva AP. Prevention of methamphetamine-induced microglial cell death by tnf-alpha and il-6 through activation of the jak-stat pathway. J Neuroinflamm (2012) 9:103. doi: 10.1186/1742-2094-9-103
145. Hu X, Xu MX, Zhou H, Cheng S, Li F, Miao Y, et al. Tumor necrosis factor-alpha aggravates gliosis and inflammation of activated retinal müller cells. Biochem Biophys Res Commun (2020) 531(3):383–9. doi: 10.1016/j.bbrc.2020.07.102
146. Federico S, Pozzetti L, Papa A, Carullo G, Gemma S, Butini S, et al. Modulation of the innate immune response by targeting toll-like receptors: A perspective on their agonists and antagonists. J Med Chem (2020) 63(22):13466–513. doi: 10.1021/acs.jmedchem.0c01049
147. Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (Tlrs) and nod-like receptors (Nlrs) in inflammatory disorders. Semin Immunol (2009) 21(4):242–53. doi: 10.1016/j.smim.2009.06.005
148. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
149. Jiang L, Xu F, He WJ, Chen LF, Zhong HB, Wu Y, et al. Cd200fc reduces Tlr4-mediated inflammatory responses in lps-induced rat primary microglial cells Via inhibition of the nf-kappa b pathway. Inflamm Res (2016) 65(7):521–32. doi: 10.1007/s00011-016-0932-3
150. Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte Tlr4 activation induces a proinflammatory environment through the interplay between Myd88-dependent nfkappab signaling, mapk, and Jak1/Stat1 pathways. Glia (2011) 59(2):242–55. doi: 10.1002/glia.21094
151. Swaroop S, Sengupta N, Suryawanshi AR, Adlakha YK, Basu A. Hsp60 plays a regulatory role in il-1beta-Induced microglial inflammation Via Tlr4-P38 mapk axis. J Neuroinflamm (2016) 13:27. doi: 10.1186/s12974-016-0486-x
152. Huang Z, Zhou T, Sun X, Zheng Y, Cheng B, Li M, et al. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through Tlr4 activation. Cell Death Differ (2018) 25(1):180–9. doi: 10.1038/cdd.2017.141
153. Ko MK, Saraswathy S, Parikh JG, Rao NA. The role of Tlr4 activation in photoreceptor mitochondrial oxidative stress. Invest Ophth Vis Sci (2011) 52(8):5824–35. doi: 10.1167/iovs.10-6357
154. Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, et al. Tlr signaling tailors innate immune responses in human microglia and astrocytes. J Immunol (2005) 175(7):4320–30. doi: 10.4049/jimmunol.175.7.4320
155. Chinnery HR, Naranjo Golborne C, Leong CM, Chen W, Forrester JV, McMenamin PG. Retinal microglial activation following topical application of intracellular toll-like receptor ligands. Invest Ophthalmol Vis Sci (2015) 56(12):7377–86. doi: 10.1167/iovs.15-17587
156. Pisetsky DS, Gauley J, Ullal AJ. Hmgb1 and microparticles as mediators of the immune response to cell death. Antioxid Redox Signal (2011) 15(8):2209–19. doi: 10.1089/ars.2010.3865
157. Das N, Dewan V, Grace PM, Gunn RJ, Tamura R, Tzarum N, et al. Hmgb1 activates proinflammatory signaling Via Tlr5 leading to allodynia. Cell Rep (2016) 17(4):1128–40. doi: 10.1016/j.celrep.2016.09.076
158. Lin F, Shan W, Zheng Y, Pan L, Zuo Z. Toll-like receptor 2 activation and up-regulation by high mobility group box-1 contribute to post-operative neuroinflammation and cognitive dysfunction in mice. J Neurochem (2021) 158(2):328–41. doi: 10.1111/jnc.15368
159. Zhu SH, Liu BQ, Hao MJ, Fan YX, Qian C, Teng P, et al. Paeoniflorin suppressed high glucose-induced retinal microglia mmp-9 expression and inflammatory response Via inhibition of Tlr4/Nf-kappab pathway through upregulation of Socs3 in diabetic retinopathy. Inflammation (2017) 40(5):1475–86. doi: 10.1007/s10753-017-0571-z
160. Bohm MR, Schallenberg M, Brockhaus K, Melkonyan H, Thanos S. The pro-inflammatory role of high-mobility group box 1 protein (Hmgb-1) in photoreceptors and retinal explants exposed to elevated pressure. Lab Invest (2016) 96(4):409–27. doi: 10.1038/labinvest.2015.156
161. Sakamoto K, Mizuta A, Fujimura K, Kurauchi Y, Mori A, Nakahara T, et al. High-mobility group box-1 is involved in nmda-induced retinal injury the in rat retina. Exp Eye Res (2015) 137:63–70. doi: 10.1016/j.exer.2015.06.003
162. Liu L, Jiang Y, Steinle JJ. Glycyrrhizin protects the diabetic retina against permeability, neuronal, and vascular damage through anti-inflammatory mechanisms. J Clin Med (2019) 8(7):957. doi: 10.3390/jcm8070957
163. Tonner H, Hunn S, Auler N, Schmelter C, Beutgen VM, von Pein HD, et al. A monoclonal anti-Hmgb1 antibody attenuates neurodegeneration in an experimental animal model of glaucoma. Int J Mol Sci (2022) 23(8):4107. doi: 10.3390/ijms23084107
164. Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and ikappabalpha degradation in a stimulus-specific manner in microglia. J Neurochem (2006) 96(2):314–23. doi: 10.1111/j.1471-4159.2005.03520.x
165. Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, et al. Minocycline attenuates lipopolysaccharide (Lps)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflamm (2008) 5:15. doi: 10.1186/1742-2094-5-15
166. Hu F, Ku MC, Markovic D, Dzaye O, Lehnardt S, Synowitz M, et al. Glioma-associated microglial Mmp9 expression is upregulated by Tlr2 signaling and sensitive to minocycline. Int J Cancer (2014) 135(11):2569–78. doi: 10.1002/ijc.28908
167. Halder SK, Matsunaga H, Ishii KJ, Akira S, Miyake K, Ueda H. Retinal cell type-specific prevention of ischemia-induced damages by lps-Tlr4 signaling through microglia. J Neurochem (2013) 126(2):243–60. doi: 10.1111/jnc.12262
168. Ku CA, Chiodo VA, Boye SL, Hayes A, Goldberg AFX, Hauswirth WW, et al. Viral-mediated vision rescue of a novel Aipl1 cone-rod dystrophy model. Hum Mol Genet (2015) 24(3):670–84. doi: 10.1093/hmg/ddu487
169. Chen L, Zheng L, Chen P, Liang G. Myeloid differentiation primary response protein 88 (Myd88): The central hub of Tlr/Il-1r signaling. J Med Chem (2020) 63(22):13316–29. doi: 10.1021/acs.jmedchem.0c00884
170. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of Myd88-deficient mice to endotoxin. Immunity (1999) 11(1):115–22. doi: 10.1016/s1074-7613(00)80086-2
171. Guo H, Callaway JB, Ting JP. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
172. Weber A, Wasiliew P, Kracht M. Interleukin-1beta (Il-1beta) processing pathway. Sci Signal (2010) 3(105):cm2. doi: 10.1126/scisignal.3105cm2
173. Swanson KV, Deng M, Ting JP. The Nlrp3 inflammasome: Molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
174. Zhang Y, Xu Y, Sun Q, Xue S, Guan H, Ji M. Activation of P2x7r- Nlrp3 pathway in retinal microglia contribute to retinal ganglion cells death in chronic ocular hypertension (Coh). Exp Eye Res (2019) 188:107771. doi: 10.1016/j.exer.2019.107771
175. Fletcher EL. Advances in understanding the mechanisms of retinal degenerations. Clin Exp Optom (2020) 103(6):723–32. doi: 10.1111/cxo.13146
176. Vessey KA, Greferath U, Aplin FP, Jobling AI, Phipps JA, Ho T, et al. Adenosine triphosphate-induced photoreceptor death and retinal remodeling in rats. J Comp Neurol (2014) 522(13):2928–50. doi: 10.1002/cne.23558
177. Notomi S, Hisatomi T, Kanemaru T, Takeda A, Ikeda Y, Enaida H, et al. Critical involvement of extracellular atp acting on P2rx7 purinergic receptors in photoreceptor cell death. Am J Pathol (2011) 179(6):2798–809. doi: 10.1016/j.ajpath.2011.08.035
178. Campagno KE, Mitchell CH. The P2x7 receptor in microglial cells modulates the endolysosomal axis, autophagy, and phagocytosis. Front Cell Neurosci (2021) 15:645244. doi: 10.3389/fncel.2021.645244
179. Basu A, Krady JK, Levison SW. Interleukin-1: A master regulator of neuroinflammation. J Neurosci Res (2004) 78(2):151–6. doi: 10.1002/jnr.20266
180. Gabay C, Lamacchia C, Palmer G. Il-1 pathways in inflammation and human diseases. Nat Rev Rheumatol (2010) 6(4):232–41. doi: 10.1038/nrrheum.2010.4
181. Bamforth SD, Lightman SL, Greenwood J. Ultrastructural analysis of interleukin-1 beta-induced leukocyte recruitment to the rat retina. Invest Ophthalmol Vis Sci (1997) 38(1):25–35.
182. Charles-Messance H, Blot G, Couturier A, Vignaud L, Touhami S, Beguier F, et al. Il-1beta induces rod degeneration through the disruption of retinal glutamate homeostasis. J Neuroinflamm (2020) 17(1):1. doi: 10.1186/s12974-019-1655-5
183. Dinarello CA. Overview of the il-1 family in innate inflammation and acquired immunity. Immunol Rev (2018) 281(1):8–27. doi: 10.1111/imr.12621
184. Todd L, Palazzo I, Suarez L, Liu X, Volkov L, Hoang TV, et al. Reactive microglia and Il1beta/Il-1r1-Signaling mediate neuroprotection in excitotoxin-damaged mouse retina. J Neuroinflamm (2019) 16(1):118. doi: 10.1186/s12974-019-1505-5
185. Wolf Y, Yona S, Kim KW, Jung S. Microglia, seen from the Cx3cr1 angle. Front Cell Neurosci (2013) 7:26. doi: 10.3389/fncel.2013.00026
186. Luo P, Chu SF, Zhang Z, Xia CY, Chen NH. Fractalkine/Cx3cr1 is involved in the cross-talk between neuron and glia in neurological diseases. Brain Res Bull (2019) 146:12–21. doi: 10.1016/j.brainresbull.2018.11.017
187. Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: Fractalkine and Cx3cr1 take centre stage. Open Biol (2013) 3(12):130181. doi: 10.1098/rsob.130181
188. Liang KJ, Lee JE, Wang YD, Ma W, Fontainhas AM, Fariss RN, et al. Regulation of dynamic behavior of retinal microglia by Cx3cr1 signaling. Invest Ophthalmol Vis Sci (2009) 50(9):4444–51. doi: 10.1167/iovs.08-3357
189. Jobling AI, Waugh M, Vessey KA, Phipps JA, Trogrlic L, Greferath U, et al. The role of the microglial Cx3cr1 pathway in the postnatal maturation of retinal photoreceptors. J Neurosci (2018) 38(20):4708–23. doi: 10.1523/JNEUROSCI.2368-17.2018
190. You M, Long C, Wan Y, Guo H, Shen J, Li M, et al. Neuron derived fractalkine promotes microglia to absorb hematoma Via Cd163/Ho-1 after intracerebral hemorrhage. Cell Mol Life Sci (2022) 79(5):224. doi: 10.1007/s00018-022-04212-6
191. Puntambekar SS, Moutinho M, Lin PB, Jadhav V, Tumbleson-Brink D, Balaji A, et al. Cx3cr1 deficiency aggravates amyloid driven neuronal pathology and cognitive decline in alzheimer's disease. Mol Neurodegener (2022) 17(1):47. doi: 10.1186/s13024-022-00545-9
192. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci (2006) 9(7):917–24. doi: 10.1038/nn1715
193. Hu SJ, Calippe B, Lavalette S, Roubeix C, Montassar F, Housset M, et al. Upregulation of P2rx7 in Cx3cr1-deficient mononuclear phagocytes leads to increased interleukin-1beta secretion and photoreceptor neurodegeneration. J Neurosci (2015) 35(18):6987–96. doi: 10.1523/JNEUROSCI.3955-14.2015
194. Murai N, Mitalipova M, Jaenisch R. Functional analysis of Cx3cr1 in human induced pluripotent stem (Ips) cell-derived microglia-like cells. Eur J Neurosci (2020) 52(7):3667–78. doi: 10.1111/ejn.14879
195. Gyoneva S, Hosur R, Gosselin D, Zhang B, Ouyang Z, Cotleur AC, et al. Cx3cr1-deficient microglia exhibit a premature aging transcriptome. Life Sci Alliance (2019) 2(6):e201900453. doi: 10.26508/lsa.201900453
196. Zieger M, Ahnelt PK, Uhrin P. Cx3cl1 (Fractalkine) protein expression in normal and degenerating mouse retina: In vivo studies. PloS One (2014) 9(9):e106562. doi: 10.1371/journal.pone.0106562
197. Jiang M, Xie H, Zhang C, Wang T, Tian H, Lu L, et al. Enhancing Fractalkine/Cx3cr1 signalling pathway can reduce neuroinflammation by attenuating microglia activation in experimental diabetic retinopathy. J Cell Mol Med (2022) 26(4):1229–44. doi: 10.1111/jcmm.17179
198. Karlen SJ, Miller EB, Wang XL, Levine ES, Zawadzki RJ, Burns ME. Monocyte infiltration rather than microglia proliferation dominates the early immune response to rapid photoreceptor degeneration. J Neuroinflamm (2018) 15(1):344. doi: 10.1186/s12974-018-1365-4
199. Feng C, Wang X, Liu T, Zhang M, Xu G, Ni Y. Expression of Ccl2 and its receptor in activation and migration of microglia and monocytes induced by photoreceptor apoptosis. Mol Vis (2017) 23:765–77.
200. Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, et al. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in cc chemokine receptor 2. Proc Natl Acad Sci U.S.A. (1997) 94(22):12053–8. doi: 10.1073/pnas.94.22.12053
201. Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, et al. Ccr2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med (2013) 5(11):1775–93. doi: 10.1002/emmm.201302692
202. Fukuda S, Nagano M, Yamashita T, Kimura K, Tsuboi I, Salazar G, et al. Functional endothelial progenitor cells selectively recruit neurovascular protective monocyte-derived F4/80(+)/Ly6c(+) macrophages in a mouse model of retinal degeneration. Stem Cells (2013) 31(10):2149–61. doi: 10.1002/stem.1469
203. Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, et al. The role of Jak/Stat signaling pathway and its inhibitors in diseases. Int Immunopharmacol (2020) 80:106210. doi: 10.1016/j.intimp.2020.106210
204. Darnell JE Jr., Kerr IM, Stark GR. Jak-stat pathways and transcriptional activation in response to ifns and other extracellular signaling proteins. Science (1994) 264(5164):1415–21. doi: 10.1126/science.8197455
205. Luo Y, Alexander M, Gadina M, O'Shea JJ, Meylan F, Schwartz DM. Jak-stat signaling in human disease: From genetic syndromes to clinical inhibition. J Allergy Clin Immunol (2021) 148(4):911–25. doi: 10.1016/j.jaci.2021.08.004
206. Zhang SS, Wei JY, Li C, Barnstable CJ, Fu XY. Expression and activation of stat proteins during mouse retina development. Exp Eye Res (2003) 76(4):421–31. doi: 10.1016/s0014-4835(03)00002-2
207. Yu CR, Mahdi RM, Liu XB, Zhang A, Naka T, Kishimoto T, et al. Socs1 regulates Ccr7 expression and migration of Cd4(+) T cells into peripheral tissues. J Immunol (2008) 181(2):1190–8. doi: 10.4049/jimmunol.181.2.1190
208. Ly A, Merl-Pham J, Priller M, Gruhn F, Senninger N, Ueffing M, et al. Proteomic profiling suggests central role of stat signaling during retinal degeneration in the Rd10 mouse model. J Proteome Res (2016) 15(4):1350–9. doi: 10.1021/acs.jproteome.6b00111
209. Lange C, Thiersch M, Samardzija M, Grimm C. The differential role of Jak/Stat signaling in retinal degeneration. Adv Exp Med Biol (2010) 664:601–7. doi: 10.1007/978-1-4419-1399-9_69
210. Chen S, Dong Z, Cheng M, Zhao Y, Wang M, Sai N, et al. Homocysteine exaggerates microglia activation and neuroinflammation through microglia localized Stat3 overactivation following ischemic stroke. J Neuroinflamm (2017) 14(1):187. doi: 10.1186/s12974-017-0963-x
211. Fan Z, Zhang W, Cao Q, Zou L, Fan X, Qi C, et al. Jak2/Stat3 pathway regulates microglia polarization involved in hippocampal inflammatory damage due to acute paraquat exposure. Ecotoxicol Environ Saf (2022) 234:113372. doi: 10.1016/j.ecoenv.2022.113372
212. Korovina I, Neuwirth A, Sprott D, Troullinaki M, Poitz DM, Deussen A, et al. Myeloid Socs3 deficiency regulates angiogenesis Via enhanced apoptotic endothelial cell engulfment. J Innate Immun (2020) 12(3):248–56. doi: 10.1159/000502645
213. Zhang X, He B, Li H, Wang Y, Zhou Y, Wang W, et al. Socs3 attenuates gm-Csf/Ifn-Γ-Mediated inflammation during spontaneous spinal cord regeneration. Neurosci Bull (2020) 36(7):778–92. doi: 10.1007/s12264-020-00493-8
214. He C, Yu CR, Mattapallil MJ, Sun L, Larkin J, Egwuagu CE. Socs1 mimetic peptide suppresses chronic intraocular inflammatory disease (Uveitis). Mediators Inflammation (2016) 2016:2939370. doi: 10.1155/2016/2939370
215. Birch DG, Bennett LD, Duncan JL, Weleber RG, Pennesi ME. Long-term follow-up of patients with retinitis pigmentosa receiving intraocular ciliary neurotrophic factor implants. Am J Ophthalmol (2016) 170:10–4. doi: 10.1016/j.ajo.2016.07.013
216. Birch DG, Weleber RG, Duncan JL, Jaffe GJ, Tao W. Ciliary neurotrophic factor retinitis pigmentosa study g. randomized trial of ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for retinitis pigmentosa. Am J Ophthalmol (2013) 156(2):283–92.e1. doi: 10.1016/j.ajo.2013.03.021
217. Kirsch M, Trautmann N, Ernst M, Hofmann HD. Involvement of Gp130-associated cytokine signaling in müller cell activation following optic nerve lesion. Glia (2010) 58(7):768–79. doi: 10.1002/glia.20961
218. Joly S, Lange C, Thiersch M, Samardzija M, Grimm C. Leukemia inhibitory factor extends the lifespan of injured photoreceptors in vivo. J Neurosci (2008) 28(51):13765–74. doi: 10.1523/JNEUROSCI.5114-08.2008
219. Rattner A, Nathans J. The genomic response to retinal disease and injury: Evidence for endothelin signaling from photoreceptors to glia. J Neurosci (2005) 25(18):4540–9. doi: 10.1523/JNEUROSCI.0492-05.2005
220. Agca C, Boldt K, Gubler A, Meneau I, Corpet A, Samardzija M, et al. Expression of leukemia inhibitory factor in müller glia cells is regulated by a redox-dependent mrna stability mechanism. BMC Biol (2015) 13:30. doi: 10.1186/s12915-015-0137-1
221. Burgi S, Samardzija M, Grimm C. Endogenous leukemia inhibitory factor protects photoreceptor cells against light-induced degeneration. Mol Vis (2009) 15:1631–7.
222. Coorey NJ, Shen W, Zhu L, Gillies MC. Differential expression of il-6/Gp130 cytokines, jak-stat signaling and neuroprotection after müller cell ablation in a transgenic mouse model. Invest Ophthalmol Vis Sci (2015) 56(4):2151–61. doi: 10.1167/iovs.14-15695
223. Ueki Y, Chollangi S, Le YZ, Ash JD. Gp130 activation in müller cells is not essential for photoreceptor protection from light damage. Adv Exp Med Biol (2010) 664:655–61. doi: 10.1007/978-1-4419-1399-9_75
224. Ueki Y, Le YZ, Chollangi S, Muller W, Ash JD. Preconditioning-induced protection of photoreceptors requires activation of the signal-transducing receptor Gp130 in photoreceptors. Proc Natl Acad Sci U.S.A. (2009) 106(50):21389–94. doi: 10.1073/pnas.0906156106
225. Skvortsova K, Iovino N, Bogdanovic O. Functions and mechanisms of epigenetic inheritance in animals. Nat Rev Mol Cell Biol (2018) 19(12):774–90. doi: 10.1038/s41580-018-0074-2
226. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet (2009) 10(1):32–42. doi: 10.1038/nrg2485
227. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol (2010) 28(10):1057–68. doi: 10.1038/nbt.1685
228. Zhang L, Lu Q, Chang C. Epigenetics in health and disease. Adv Exp Med Biol (2020) 1253:3–55. doi: 10.1007/978-981-15-3449-2_1
229. Alaskhar Alhamwe B, Khalaila R, Wolf J, von Bulow V, Harb H, Alhamdan F, et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin Immunol (2018) 14:39. doi: 10.1186/s13223-018-0259-4
230. Trifunovic D, Petridou E, Comitato A, Marigo V, Ueffing M, Paquet-Durand F. Primary rod and cone degeneration is prevented by hdac inhibition. Retin Degener Dis: Mech Exp Ther (2018) 1074:367–73. doi: 10.1007/978-3-319-75402-4_45
231. Kannan V, Brouwer N, Hanisch UK, Regen T, Eggen BJ, Boddeke HW. Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J Neurosci Res (2013) 91(9):1133–42. doi: 10.1002/jnr.23221
232. Patnala R, Arumugam TV, Gupta N, Dheen ST. Hdac inhibitor sodium butyrate-mediated epigenetic regulation enhances neuroprotective function of microglia during ischemic stroke. Mol Neurobiol (2017) 54(8):6391–411. doi: 10.1007/s12035-016-0149-z
233. Durham BS, Grigg R, Wood IC. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J Neurochem (2017) 143(2):214–24. doi: 10.1111/jnc.14144
234. Yang H, Ni W, Wei P, Li S, Gao X, Su J, et al. Hdac inhibition reduces white matter injury after intracerebral hemorrhage. J Cereb Blood Flow Metab (2021) 41(5):958–74. doi: 10.1177/0271678X20942613
235. Chen S, Ye J, Chen X, Shi J, Wu W, Lin W, et al. Valproic acid attenuates traumatic spinal cord injury-induced inflammation Via Stat1 and nf-kappab pathway dependent of Hdac3. J Neuroinflamm (2018) 15(1):150. doi: 10.1186/s12974-018-1193-6
236. Iwagawa T, Watanabe S. Molecular mechanisms of H3k27me3 and H3k4me3 in retinal development. Neurosci Res (2019) 138:43–8. doi: 10.1016/j.neures.2018.09.010
237. Mbefo M, Berger A, Schouwey K, Gerard X, Kostic C, Beryozkin A, et al. Enhancer of zeste homolog 2 (Ezh2) contributes to rod photoreceptor death process in several forms of retinal degeneration and its activity can serve as a biomarker for therapy efficacy. Int J Mol Sci (2021) 22(17):9331. doi: 10.3390/ijms22179331
238. Zhang X, Wang Y, Yuan J, Li N, Pei S, Xu J, et al. Macrophage/Microglial Ezh2 facilitates autoimmune inflammation through inhibition of Socs3. J Exp Med (2018) 215(5):1365–82. doi: 10.1084/jem.20171417
239. Yuan J, Zhu Q, Zhang X, Wen Z, Zhang G, Li N, et al. Ezh2 competes with P53 to license lncrna Neat1 transcription for inflammasome activation. Cell Death Differ (2022) 29(10):2009–23. doi: 10.1038/s41418-022-00992-3
240. Chen L, Heikkinen L, Wang C, Yang Y, Sun H, Wong G. Trends in the development of mirna bioinformatics tools. Brief Bioinform (2019) 20(5):1836–52. doi: 10.1093/bib/bby054
241. Reh TA, Hindges R. Micrornas in retinal development. Annu Rev Vis Sci (2018) 4:25–44. doi: 10.1146/annurev-vision-091517-034357
242. Xiang L, Chen XJ, Wu KC, Zhang CJ, Zhou GH, Lv JN, et al. Mir-183/96 plays a pivotal regulatory role in mouse photoreceptor maturation and maintenance. Proc Natl Acad Sci U.S.A. (2017) 114(24):6376–81. doi: 10.1073/pnas.1618757114
243. Anasagasti A, Ezquerra-Inchausti M, Barandika O, Muñoz-Culla M, Caffarel MM, Otaegui D, et al. Expression profiling analysis reveals key microrna-mrna interactions in early retinal degeneration in retinitis pigmentosa. Invest Ophthalmol Vis Sci (2018) 59(6):2381–92. doi: 10.1167/iovs.18-24091
244. Genini S, Guziewicz KE, Beltran WA, Aguirre GD. Altered mirna expression in canine retinas during normal development and in models of retinal degeneration. BMC Genomics (2014) 15:172. doi: 10.1186/1471-2164-15-172
245. Chen Y, Lin J, Schlotterer A, Kurowski L, Hoffmann S, Hammad S, et al. Microrna-124 alleviates retinal vasoregression Via regulating microglial polarization. Int J Mol Sci (2021) 22(20):11068. doi: 10.3390/ijms222011068
246. Burgaletto C, Platania CBM, Di Benedetto G, Munafo A, Giurdanella G, Federico C, et al. Targeting the mirna-155/Tnfsf10 network restrains inflammatory response in the retina in a mouse model of alzheimer's disease. Cell Death Dis (2021) 12(10):905. doi: 10.1038/s41419-021-04165-x
247. Fernando N, Wong JHC, Das S, Dietrich C, Aggio-Bruce R, Cioanca AV, et al. Microrna-223 regulates retinal function and inflammation in the healthy and degenerating retina. Front Cell Dev Biol (2020) 8:516. doi: 10.3389/fcell.2020.00516
248. Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M. DNA Methylation dynamics in aging: How far are we from understanding the mechanisms? Mech Ageing Dev (2018) 174:3–17. doi: 10.1016/j.mad.2017.12.002
249. Rhee KD, Yu J, Zhao CY, Fan G, Yang XJ. Dnmt1-dependent DNA methylation is essential for photoreceptor terminal differentiation and retinal neuron survival. Cell Death Dis (2012) 3:e427. doi: 10.1038/cddis.2012.165
250. Farinelli P, Perera A, Arango-Gonzalez B, Trifunovic D, Wagner M, Carell T, et al. DNA Methylation and differential gene regulation in photoreceptor cell death. Cell Death Dis (2014) 5(12):e1558. doi: 10.1038/cddis.2014.512
251. Dvoriantchikova G, Lypka KR, Ivanov D. The potential role of epigenetic mechanisms in the development of retinitis pigmentosa and related photoreceptor dystrophies. Front Genet (2022) 13:827274. doi: 10.3389/fgene.2022.827274
252. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Science (2016) 352(6284):aaf1098. doi: 10.1126/science.aaf1098
253. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
254. Rodriguez RM, Suarez-Alvarez B, Lopez-Larrea C. Therapeutic epigenetic reprogramming of trained immunity in myeloid cells. Trends Immunol (2019) 40(1):66–80. doi: 10.1016/j.it.2018.11.006
255. Neher JJ, Cunningham C. Priming microglia for innate immune memory in the brain. Trends Immunol (2019) 40(4):358–74. doi: 10.1016/j.it.2019.02.001
256. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature (2018) 556(7701):332–8. doi: 10.1038/s41586-018-0023-4
257. Noailles A, Maneu V, Campello L, Lax P, Cuenca N. Systemic inflammation induced by lipopolysaccharide aggravates inherited retinal dystrophy. Cell Death Dis (2018) 9(3):350. doi: 10.1038/s41419-018-0355-x
258. Su H, Liang Z, Weng S, Sun C, Huang J, Zhang T, et al. Mir-9-5p regulates immunometabolic and epigenetic pathways in B-Glucan-Trained immunity Via Idh3α. JCI Insight (2021) 6(9):e144260. doi: 10.1172/jci.insight.144260
259. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature (2016) 535(7610):65–74. doi: 10.1038/nature18847
260. Zysset-Burri DC, Keller I, Berger LE, Largiader CR, Wittwer M, Wolf S, et al. Associations of the intestinal microbiome with the complement system in neovascular age-related macular degeneration. NPJ Genom Med (2020) 5:34. doi: 10.1038/s41525-020-00141-0
261. Khan R, Sharma A, Ravikumar R, Parekh A, Srinivasan R, George RJ, et al. Association between gut microbial abundance and sight-threatening diabetic retinopathy. Invest Ophthalmol Vis Sci (2021) 62(7):19. doi: 10.1167/iovs.62.7.19
262. Kutsyr O, Maestre-Carballa L, Lluesma-Gomez M, Martinez-Garcia M, Cuenca N, Lax P. Retinitis pigmentosa is associated with shifts in the gut microbiome. Sci Rep (2021) 11(1):6692. doi: 10.1038/s41598-021-86052-1
263. Kutsyr O, Noailles A, Martinez-Gil N, Maestre-Carballa L, Martinez-Garcia M, Maneu V, et al. Short-term high-fat feeding exacerbates degeneration in retinitis pigmentosa by promoting retinal oxidative stress and inflammation. Proc Natl Acad Sci U.S.A. (2021) 118(43):e2100566118. doi: 10.1073/pnas.2100566118
264. Parker A, Romano S, Ansorge R, Aboelnour A, Le Gall G, Savva GM, et al. Fecal microbiota transfer between young and aged mice reverses hallmarks of the aging gut, eye, and brain. Microbiome (2022) 10(1):68. doi: 10.1186/s40168-022-01243-w
265. Scott PA, Kaplan HJ, McCall MA. Prenatal exposure to curcumin protects rod photoreceptors in a transgenic Pro23his swine model of retinitis pigmentosa. Trans Vision Sci Technol (2015) 4(5):5. doi: 10.1167/tvst.4.5.5
Keywords: retinal inflammation, innate immune, gut microbiome, trained immunity, epigenetic modification, retinitis pigmentosa
Citation: Zhao L, Hou C and Yan N (2022) Neuroinflammation in retinitis pigmentosa: Therapies targeting the innate immune system. Front. Immunol. 13:1059947. doi: 10.3389/fimmu.2022.1059947
Received: 02 October 2022; Accepted: 17 October 2022;
Published: 27 October 2022.
Edited by:
Inderjeet Kaur, L V Prasad Eye Institute, IndiaReviewed by:
Nivedita Chatterjee, Vision Research Foundation, IndiaChitra Kannabiran, L V Prasad Eye Institute, India
Copyright © 2022 Zhao, Hou and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naihong Yan, eWFubmFpaG9uZ0AxMjYuY29t