 Tianyue Xu†
Tianyue Xu† Liwen Huang
Liwen Huang Xiaowei Liu
Xiaowei Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 December 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1057850
This article is part of the Research Topic Community Series in Novel Insights into Immunotherapy Targeting Tumor Microenvironment in Ovarian Cancer, volume I View all 13 articles
With encouraging antitumor effects, immunotherapy represented by immune checkpoint blockade has developed into a mainstream cancer therapeutic modality. However, only a minority of ovarian cancer (OC) patients could benefit from immunotherapy. The main reason is that most OC harbor a suppressive tumor immune microenvironment (TIME). Emerging studies suggest that M2 tumor-associated macrophages (TAMs), T regulatory cells (Tregs), myeloid-derived suppressor cells (MDSCs), and cancer-associated fibroblasts (CAFs) are enriched in OC. Thus, reversing the suppressive TIME is considered an ideal candidate for improving the efficiency of immunotherapy. Nanoparticles encapsulating immunoregulatory agents can regulate immunocytes and improve the TIME to boost the antitumor immune response. In addition, some nanoparticle-mediated photodynamic and photothermal therapy can directly kill tumor cells and induce tumor immunogenic cell death to activate antigen-presenting cells and promote T cell infiltration. These advantages make nanoparticles promising candidates for modulating the TIME and improving OC immunotherapy. In this review, we analyzed the composition and function of the TIME in OC and summarized the current clinical progress of OC immunotherapy. Then, we expounded on the promising advances in nanomaterial-mediated immunotherapy for modulating the TIME in OC. Finally, we discussed the obstacles and challenges in the clinical translation of this novel combination treatment regimen. We believe this resourceful strategy will open the door to effective immunotherapy of OC and benefit numerous patients.
Ovarian cancer (OC) has a high lethality rate and is the second primary cause of death from gynecologic cancer worldwide (1). Currently, the major treatments for OC are surgery, chemotherapy, and radiotherapy (2, 3). Although patients can achieve short-term remission with these approaches, five-year survival rates are only approximately 30% (4). Recently, immunotherapy has received increasing attention, especially immune checkpoint blockade (ICB), which has emerged as an effective strategy for OC therapy. The anti-PD-1 antibody pembrolizumab has received regulatory approval to treat OC (5).
Although ICB holds tremendous potential for cancer therapy, the current clinical data on OC immunotherapy is not ideal. In general, the limited efficacy of ICB is mainly due to four reasons (1): tumor antigen deficiency (2), insufficient T lymphocyte infiltration, (3) defective tumor antigen processing and presentation mechanisms, and (4) the suppressive tumor immune microenvironment (TIME). Notably, the suppressive TIME is a significant barrier to the immunotherapy of OC. Ovarian tumors contain a large number of immunosuppressive cells, such as M2 tumor-associated macrophages (TAMs), CD4+ regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and cancer-associated fibroblasts (CAFs), which inhibit the immune response.
In recent years, nanoparticles have been expected to play a significant role in regulating the TIME and improving the efficacy of OC immunotherapy. On the one hand, nanotechnology-mediated photothermal therapy (PTT) and photodynamic therapy (PDT) can induce immunogenic cell death (ICD) of tumor cells, promote antigen presentation, and enhance tumor T cell infiltration (6). For instance, copper sulfide nanoparticles remodeled the TIME by inducing ICD, thus improving the efficiency of immune checkpoint inhibitors (ICIs) in OC. On the other hand, nanoparticles can be used as excellent drug carriers, which can load immunomodulators, such as adjuvants, cytokines, and siRNA, to regulate immunosuppressive cells and inhibit immune checkpoints. For example, Kang Yanan et al. prepared liposomes containing toll-like receptor (TLR) agonists and successfully repolarized M2 TAMs in OC (7).
Above all, nanoparticle-mediated immunotherapy holds great promise in modulating the TIME of OC and improving the treatment outcome. Here, we summarized the composition and function of the TIME in OC and discussed recent advances in immunotherapy to treat OC in preclinical and clinical settings. Moreover, the advantages and progress of nanoparticle-mediated immunotherapy in regulating the TIME and boosting the antitumor immunity of OC are also summarized. Finally, the current limitations and future development strategies in clinical translation of this nanoparticle-mediated immunotherapy have also been discussed.
Approximately 90% of ovarian tumors originate in epithelial cells and are called epithelial ovarian cancer (EOC). EOC has been categorized into different subtypes according to histology. The prevalent histologic subtype is high-grade serous ovarian cancer (HGSOC), which accounts for about 80% of cases. Other rarer subtypes include low-grade serous, mucinous, clear cell, and endometrioid tumors. With the development of genomics and single-cell technology, the understanding of the TIME in OC has been deepened (8–12). It has been found that different OC subtypes have distinct macrophage polarization (13). The results of single-cell RNA sequencing revealed that ascites cells in different HGSOC patients differ in composition and functional program including diverse fibroblasts and macrophages (10). In addition, CD8+ and CD4+ T cells have distinct infiltration levels in differentially growing metastases within a single individual (9). Therefore, the TIME of OC is very complex and heterogeneous, which is primarily made up of CD8+ T cells, CD4+ T cells, NK cells, macrophages, MDSCs, etc. Based on their function, these cells can be categorized as activated and suppressive immune cells. Activated immune cells mainly include CD8+ T cells and NK cells. Suppressive immune cells mainly include Tregs, M2 macrophages, MDSCs, etc. In the TIME, activated immune cells play a role in tumor growth inhibition and tumor immunosurveillance. In contrast, suppressive immune cells dampen the function of activated immune cells and promote the growth of tumors (Figure 1).
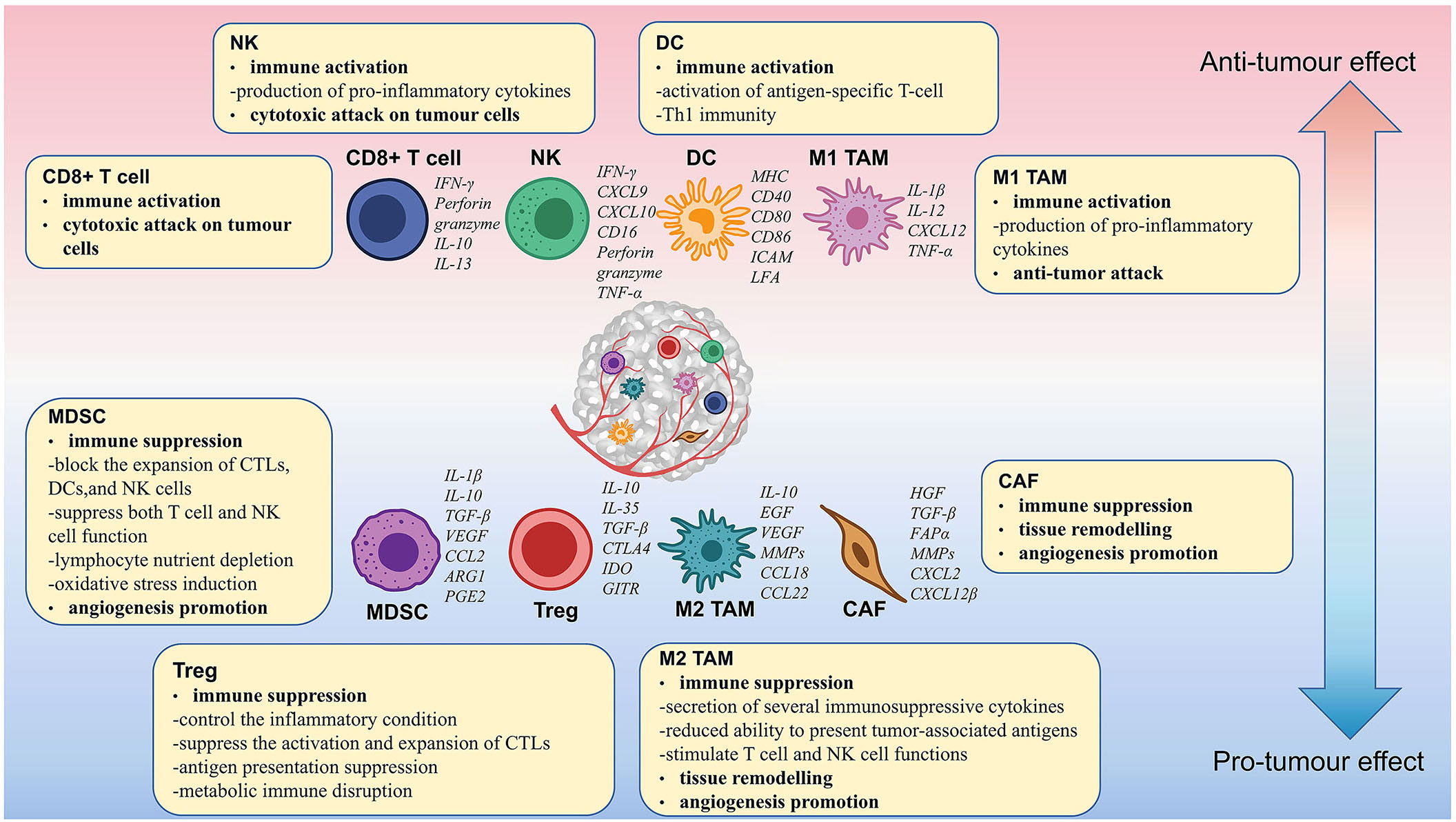
Figure 1 Immune cell functions and their interactions in the ovarian cancer tumor microenvironment. M2 TAMs, Tregs, MDSCs, and CAFs suppress the immune response and promote the proliferation, growth, and metastasis of OC. CD8+ T cells, NK cells, mature DCs, and M1 TAMs enhanced the immune response and suppressed tumor growth.
T lymphocytes are the main component of the TIME and are central to adaptive immunity. Mature T cells are classified as CD3+ CD8+ T cells and CD3+ CD4+ T cells, according to their marker gene (14). CD8+ T cells are the prime activated immune cells and are also known as cytotoxic T cells (CTLs). The T cell receptor (TCR) on CD8+ T cells binds to the MHC-I compound on tumor cells, resulting in the production of cytolytic factors (e.g., perforin and granzyme) and inflammatory cytokines (e.g., IL-2 and IL-12) that directly kill tumor cells (15). The mechanism of ICB and adoptive cell therapy (ACT) is to activate CD8+ T cells. An essential prerequisite for the PD-L1 blockade response in OC patients is sufficient T-cell infiltration. Higher infiltrating levels of CD8+ T cells in the TIME indicate a better prognosis in OC patients (16). However, high levels of TGFβ in OC inhibit the function of CTLs (17). Recently, it has been found that the infiltration level of CD8+ T cells in OC is regulated by CXCL9 expressed in antigen presentation cells (APCs) and CCL5 expressed in tumor cells (18).
In the suppressive TIME of OC, dysfunction of CD8+ T cells is another significant cause of immune dysfunction. T-cell dysfunction is caused by inhibiting T-cell mitochondrial biogenesis and the inability to produce sufficient energy intermediates (19). Activation of the IRE1α-XBP1 pathway regulates the T-cell mitochondrial activity and reduces T-cell infiltration and IFN-γ expression. Cytokine IFN-γ levels are linked to TIL infiltration, and increased IFN-γ levels can improve OC patient survival. Downregulation of XBP1 or control of endoplasmic reticulum stress enhances T-cell activity and metabolic adaptation (20). T-cell proliferation is also disturbed by lipid metabolites secreted by tumor cells, including 9-HODE, 5-HETE, and PGD2, which bind to T-cell PPAR and inhibit cell cycle protein E (21).
NK cells are innate lymphoid-like cells with potent natural cytotoxicity against tumor cells. NK cells participate in immune regulation via a variety of mechanisms, including (1) the expression of CD16 to exert antibody-dependent cytotoxicity (ADCC) and detect target cells encapsulated in antibodies; (2) the production of perforin and granzyme to induce apoptosis in tumor cells directly; and (3) the release of antitumor cytokines such as TNF-α and IFN-γ (22). Various studies have demonstrated the effectiveness of NK cell therapy in patients with OC. Poznanski et al. found that expansion of patient-derived CD56superbright CD16+ NK cells could exert potent cytotoxicity against autologous tumors in an autologous-derived xenograft mouse model of OC patients (23). Recently, Sun et al. showed that intravenous injection of NK cells isolated from the peripheral blood of OC patients inhibited the systemic metastasis of OC and increased the survival rate (24).
The most critical cytotoxic receptors for NK cells involved in immune surveillance are the NKG2D receptor, the CD16 receptor, and natural cytotoxic receptors for NKG receptors, such as NKp30 (25). In contrast, the proinflammatory cytokine MIF transcriptionally downregulates the NK cytotoxicity receptor NKG2D and decreases the cytotoxicity of NK cells (26). Furthermore, chronic receptor-ligand interactions reduce the expression of NK-cell surface receptors, impairing NK-cell cytolytic function and IFN-γ secretion ability (27). Greppi et al. found that OC cells released B7-H6 suppressed the expression of NKp30 on NK cells (28). TGF-β also inhibits NKp30 expression and dampens NK-cell-induced dendritic cell (DC) killing (27). In addition, OC ascites contain high levels of IL-18 and TGF-β, which can suppress the expression of CD16 and ADCC in NK cells (22). In OC, NK cells can also affect T cells to interfere with tumor progression. NK cells promote CD8+ T-cell recruitment in OC by upregulating CCL5, CXCL9, and CXCL10 via the CCR5 mechanism (29).
Tregs are a heterogeneous subpopulation of CD4+ T cells that express CD25, CTL antigen 4 (CTLA-4), and the transcription factor FoxP3 (30). Tregs are classical immunosuppressive cells that exert immunosuppressive effects and maintain immune self-tolerance in vivo. Treg to CD8+ T cell ratios in tumors negatively correlate with survival in OC patients (31). Blocking Treg differentiation, migration, or immunosuppressive functions can reinforce the antitumor immune responses. Moreover, macrophage-secreted CCL22 in the peritoneal cavity can promote Treg migration (32). The hypoxic tumor microenvironment (TME) also favors the metabolic reprogramming of Tregs leading to Treg proliferation. Accumulated Tregs upregulate the secretion levels of IL-10 and promote angiogenesis and immune tolerance of tumors (33).
Tregs can establish a suppressive TIME through multiple mechanisms. On the one hand, Tregs can release immunosuppressive cytokines, such as IL-35, IL-10, and transforming growth factor β (TGF-β), which inhibit the function of CD8+ T cells and promote tumor cell growth (34). On the other hand, Tregs inhibit the TCR signaling pathway of CD4+ CD25- conventional T cells (Tcons), suppress calcium (Ca2+) signaling in Tcons, and reduce the activation of NFAT and NF-κB in Tcons (34). Moreover, the perforin and granzyme released from Tregs directly kill other immune cells, such as DCs, monocytes, and CD8+ T cells (34). Multiple receptors expressed on Tregs isolated from OC are associated with TCR involvement, including PD-1, ICOS, and 4-1BB. These receptors make Tregs more sensitive to anti-CD3/anti-CD28 stimuli and have a more potent inhibitory capacity (35). Treg-expressed CD73 and CD39 convert pro-inflammatory ATP to adenosine to mediate immunosuppression (36). CD4+ Tregs can differentiate into CD4+ effector T cells upon the activation of glucocorticoid-induced tumor necrosis factor receptor family-related receptor (GITR). Therefore, stimulation of GITR is expected to eliminate Treg-mediated suppression (37).
The secretion of high levels of proinflammatory cytokines (e.g., TNF and IL-6) in OC malignant ascites promoted high expression of tumor necrosis factor receptor 2 (TNFR2) in Tregs. TNFR2+ Tregs enhanced the expression of immunosuppressive molecules, including TGF-β, CD39, CD73, GARP PD-L1, and CTLA-4 (38, 39). The upregulated CTLA-4 in Tregs inhibits the activation and proliferation of effector T cells (40). Tregs in OC induce B7-H4 expression, deliver inhibitory activity to APCs, and blunt the antitumor immune response (41). Abnormal hyperactivation of signal transducer and activator of transcription-3 (STAT3) in tumor-infiltrating immune cells positively regulates the number of Tregs and MDSCs. Therefore, targeting the IL6/JAK/STAT3 signaling pathway is a feasible strategy to alleviate the immunosuppressive TME (42).
TAMs are the largest immune cell population in the TIME of OC, accounting for 39% of the immune cellular repertoire. There are two main types of TAMs based on phenotype: proinflammatory M1-like and anti-inflammatory M2-like (43). M2 macrophages are associated with tumor immunosuppression in OC. In a study of 140 OC patients, Macciò and his colleagues found that a high density of M2 macrophages led to poor overall survival (OS) and prognosis. OS and the M1/M2 ratio were positively associated (13). Polarization and recruitment of M2 macrophages are key factors in OC progression and metastasis. The TIME can shift macrophages from the M1 to the M2 phenotype, creating a suppressive TIME. Ying et al. found that MiR-222-3p in the exosomes of EOC cells induces M2 phenotypic polarization through activation of the STAT3 pathway (44). Collagen triple helix repeat containing 1 (CTHRC1) is an OC secretory protein that induces M2-like polarization in TAMs by activating the STAT6 signaling pathway (45). Another reason for the poor survival and prognosis of OC is the recruitment of M2 macrophages. OC overexpressed UBR5, an E3 ligase, can recruit and activate TAMs by regulating multiple cytokines and chemokines, such as CCL2 and colony-stimulating factor 1 (CSF-1) (46). CSF-1 is a major macrophage survival factor. Targeting TAMs with anti-CSF-1R antibodies is a new therapeutic strategy for OC (47–49). Multiple cell signaling pathways can induce protumor and immunosuppressive properties in M2 TAMs, such as JNK, IL-13, IL-4, AMPK, PPARγ, IRF-3, IRF-4, and C/EBPβ (50).
The most common metastatic route for OC cells is the body cavity route, forming spheroids for metastasis. M2 macrophages are enriched in the omentum, which is the primary site of choice for OC metastasis. M2 macrophages secrete EGF to activate tumor cell EGFR and upregulate the VEGF/VEGFR signaling pathway to promote tumor cell proliferation and migration. In addition, EGF upregulates the expression of ICAM-1 and ɑMβ2 integrin in TAMs and facilitates the interaction between TAMs and tumor cells to form spheroids (51, 52). TAMs also secrete multiple cytokines and chemokines to reshape the suppressive TIME of OC and promote OC progression. El-Arabey et al. reported that TAMs promote the growth, migration, chemoresistance, and epithelial-mesenchymal transition (EMT) of TP53-mutated HGSOC cell lines by exosomes releasing GATA3 (43). Macrophage secretion of TNF-α induces MIF and EMMPRIN into tumor cells in an NF-κB- and JNK-dependent manner. Subsequently, macrophages release various MMPs to enhance tumor invasion, migration, and vascularization (53). TAMs also secrete IL-6 and IL-10, which activate the STAT3 pathway and promote tumor proliferation (54). TAMs secrete several chemokines, including CCL17, CCL22, and CCL18. These chemokines recruit Tregs and Th2 subsets and promote T-cell differentiation toward a Th2 phenotype (55).
MDSCs are a heterogeneous group of nonterminally differentiated myeloid cells with immunosuppressive properties. Consistent with other immunosuppressive cells, the infiltration of MDSCs is related to shorter OS in OC patients (56). High concentrations of several cytokines (e.g., IL-6, IL-10, IL-1β, VEGF, PGE2, and TNF-α) in the ascites of OC patients induce the accumulation of MDSCs (57). Growth factors G-CSF and GM-CSF promote the production of MDSCs by activating STAT3 and STAT5 signaling pathways and downregulating IRF-8 (58). Multiple chemokines (e. g, CCL1, CCL5, CCL7, CXCL8, and CXCL12) drive the recruitment of MDSCs to OC tumor sites via the CCR2, CXCR4, and CCR5 axes. Triggering of the CXCL12-CXCR4 pathway is controlled by the tumor-associated inflammatory mediator PGE2, and targeting PGE2 has the potential to block the migration of MDSCs into ascites (59).
Notably, MDSCs can increase the stem cell-like properties of OC cells. Li et al. found that induction of the CSF2/p-STAT3 signaling pathway by MDSCs could enhance the stemness of EOC cells (60). Cui et al. demonstrated that MDSCs induced microRNA101 expression and suppressed CtBP2, thereby enhancing the stem cell-like properties of OC (61). The immunosuppressive properties of MDSCs are dependent on PGE2-induced DNA methyltransferase 3 alpha (DNMT3A) upregulation and hypermethylation of myeloid genes (62). MDSCs have re-editable properties similar to those of macrophages. STAT3 inhibition and TLR signaling modulation can repolarize MDSCs and activate their immune function (63).
MDSCs generate a suppressive TIME by inhibiting the activities of activated immune cells. Previous studies found that MDSCs inhibit the activity and proliferation of NK cells and block the antigenic expression of DCs (56). In addition, MDSCs produce TGF-β, IDO, IL-10, and nitric oxide (NO) to reduce the proliferation and cytotoxicity of NK cells and exert immunosuppressive functions (64). The effects of MDSCs on NK cells were manifested by downregulating the expression of surface natural cytotoxicity receptors (NCRs), NKG2D, and DNAM-1 (65). Meanwhile, MDSCs can polarize M1 macrophages to the M2 phenotype and induce Treg amplification. MDSCs induce the activation and accumulation of M2 macrophages and stimulate the production of more IL-10. In turn, IL-10 can upregulate immunosuppressive factors, such as PD-L1 and Arg-1, inducing the activation and amplification of MDSCs (66). HIF-1 in the hypoxic environment of OC redifferentiates MDSCs into TAMs and promotes tumor progression (67). In addition, MDSCs secreted TGF-β and IL-10 can stimulate Treg migration and differentiation through CD40-CD40 L interactions (68).
MDSCs suppress the T-cell-mediated antitumor immune responses in the TIME of OC by multiple mechanisms (1): Depleting nutrients required by lymphocytes: MDSCs inhibit T-cell proliferation by upregulating ARG-1 to consume arginine and isolate 1-cysteine (69); (2) Restricting T-cell recruitment and inducing T-cell apoptosis by expressing Galectin9, AMPKα-1, and ADAM17 (70); (3) Regulating NO and ROS production and stimulate oxidative stress; (4) Production of peroxynitrite (PNT) inhibits the TCR signaling pathway: MDSCs produce peroxynitrite, which nitrates complexes in the TCR-CD8 complex when direct contact with T cells. Meanwhile, MDSCs disrupt the binding of CD8+ T cells to specific peptide-major histocompatibility complex (pMHC) dimers and inhibit T cell antigen recognition (71). (5) Secreting TGF-β enhances T-cell immunosuppressive phenotypic differentiation, such as promoting the differentiation of Th17, Th2, and Tregs (72). (6) Enhancing the expression of PD-L1 on tumor cells by the AKT/mTOR signaling pathway in a PGE2-dependent manner (73).
CAFs are another major cell subpopulation in OC masses and play a crucial role in OC progression (74). Tumor cells are protected from immune surveillance by an extracellular layer of dense extracellular matrix (ECM) rich in fibronectin. However, this protective shell is produced by abnormal remodeling of the ECM and excessive deposition of fibroblasts. OC cells construct a strong protective barrier by reprogramming fibroblasts with miRNAs, mainly downregulating miR-214 and miR-31 and upregulating miR-155 (75). Overexpression of STAT4 in EOC cells depends on tumor-derived Wnt7a to induce the production of CAFs (76). NNMT regulates CAF differentiation, reduces histone methylation and s-adenosylmethionine, and supports OC proliferation, growth, and metastasis (77).
Fibroblasts are a key component of the basement membrane and peritoneum of the greater omentum in OC. CAFs recruit ascites tumor cells expressing high levels of alpha5-integrin (ITGA5) to form heterogeneous spheroids called MUs. Additionally, CAFs secrete EGF to maintain the MU structure by maintaining ITGA5 expression, which aids in the trans-somatic metastasis and OC peritoneal dissemination of HGSOC (78). Many markers activate CAFs, such as the extremely heterogeneous αSMA and FAP, and these markers help us develop new therapeutic targets for cancer (79, 80).
CAFs can remodel the ECM by secreting various cytokines as well as produce multiple paracrine signals with OC cells to induce OC cell growth, migration, and invasion. CAFs secrete DKK3 and activate YAP/TAZ and β-linked protein, the former inducing CAF tumorigenesis and the latter promoting OC invasion (81). CAF-derived POSTN promotes EMT by activating the PI3K/AKT pathway. It also promoted TGF-β1-induced activation of fibroblasts and invasion and migration of OC cells (82). TGF-β receptor type II and SMAD signaling upregulate VCAN and activate the NF-kB signaling pathway in CAFs. Alterations in these signaling pathways increase matrix metalloproteinase 9, CD44, and hyaluronic acid-mediated motor receptor expression in CAFs and promote the progression of advanced OC (83). The hepatocyte growth factor (HGF) is a key growth factor derived from CAFs. HGF stimulates OC cell growth and drug resistance by activating the c-Met/PI3K/Akt and GRP78 pathways (84). Fibroblast growth factor-1 (FGF-1) is another crucial factor in CAFs. FGF-1 regulates tumor progression by phosphorylating FGF-4, increasing the expression of Snail1 and MMP3, and activating the MAPK/ERK pathway (85). In addition, CAFs promote OC metastasis by secreting VEGF-A and tenascin-c (86).
Moreover, CAFs recruit immune cells and remodel the TIME via several cytokines and chemokines. Taki et al. found that CAFs produce the chemokines CXCL1 and CXCL2, which recruit MDSCs in OC (59). In addition, CXCL12β expression in CAF cells promotes the migration and differentiation of Tregs (87). CAFs interact with multiple immune components to regulate the immune activity of innate and adaptive cells and suppress antitumor immunity. Interleukin (IL)-1β is a major immunosuppressant in the TME and is significantly associated with CAF-expressed PS1. PS1 positively correlates with IL-1β levels under the regulation of the WNT/β-linked protein pathway. Inhibition of PS1 expression increases the proliferation and migration of CTLs and DCs (88). Browning et al. found that CAFs produce IL-6, a cytokine with protumorigenic function, which leads to severely poor prognosis and chemoresistance in OC patients (54).
During the past years, various immunotherapies, including ICB, cancer vaccines, ACT, and cytokines, have been approved for OC treatment (Table 1). In this section, we will expatiate the progress of these treatments in OC.
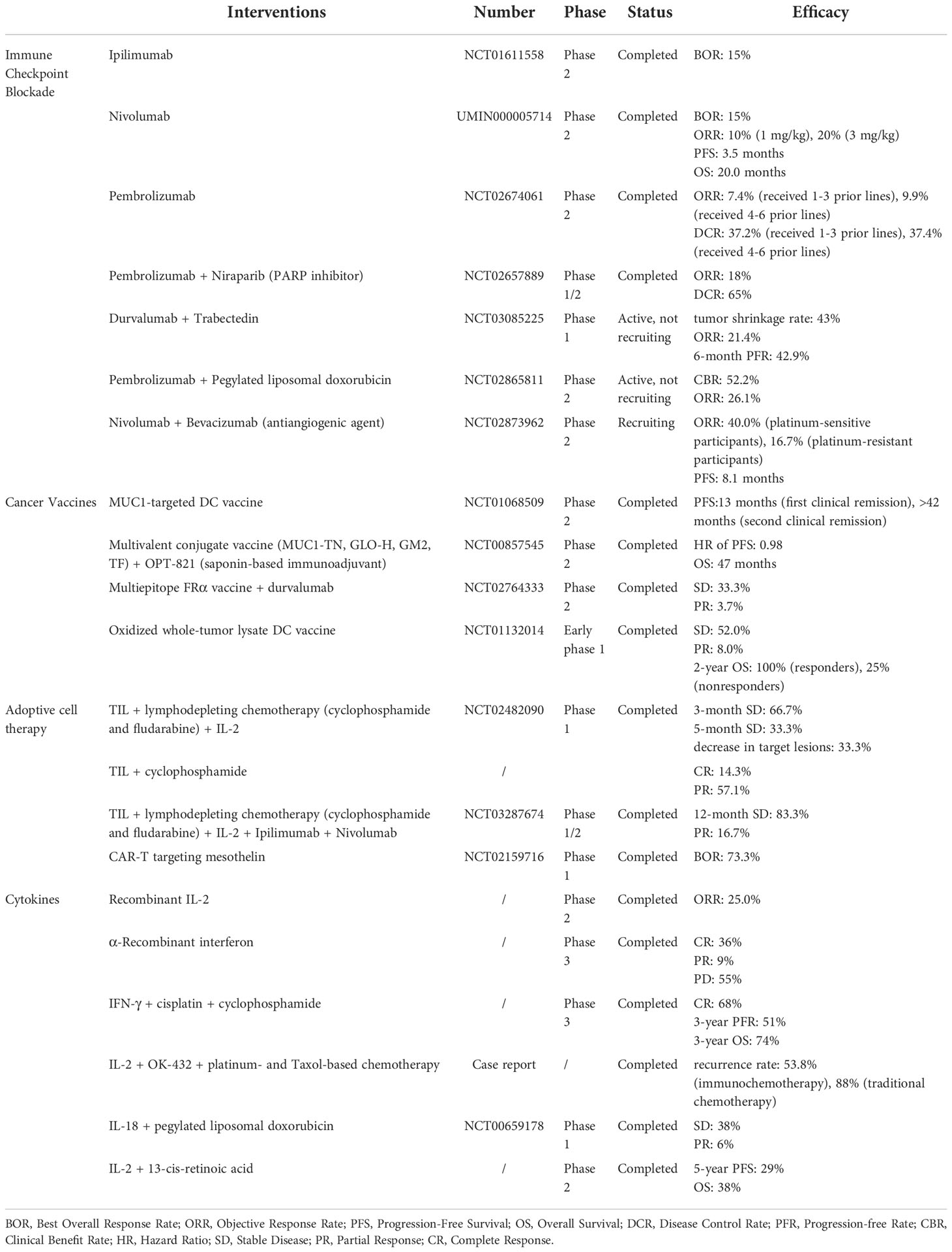
Table 1 Clinical trials of immunotherapy for ovarian cancer.
Effective immunotherapy relies on antigen presentation, inhibition of immunosuppressive cells, and activation of effector T cells. Among them, T-cell-mediated immune responses are crucial and modulated by inhibitory and stimulatory signals. Immune checkpoints regulate T cell activities and are closely related to tumor immunity. Currently, ICIs targeting CTLA-4 and programmed cell death protein 1 (PD-1) or PD-1 ligand (PD-L1) have achieved breakthrough results in clinical trials (89). Anti-PD-L1 antibodies (avelumab, durvalumab, and atezolizumab), anti-PD-1 antibodies (nivolumab and pembrolizumab), and anti-CTLA-4 antibodies (ipilimumab) have received FDA approval for the treatment of several malignancies represented by melanoma and non-small cell lung cancer (5, 90). However, the objective response rates (ORR) for single-agent ICIs in OC are only 6-15% (91). For example, in a phase II study of ipilimumab for patients with platinum-sensitive OC, the best overall response rate (BOR) was just 15% (NCT01611558). In addition, the BOR for platinum-resistant OC patients treated with nivolumab was 15%. In this clinical trial, 40% of the patients had grade 3 or 4 treatment-related side events (92). Besides, in a phase II clinical study of pembrolizumab in advanced recurrent OC (NCT02674061), the ORR was also less than 10% (93).
Due to the unsatisfactory efficacy of single-agent ICIs in OC, combination therapy has recently received much attention. Several studies have combined ICB with polyadenosine diphosphate ribose polymerase (PARP) inhibition, chemotherapy, and antiangiogenic therapy to improve the efficacy of OC immunotherapy (91). For example, pembrolizumab was combined with the PARP inhibitor niraparib for recurrent OC treatment. In this clinical trial, the ORR was 18%, and the illness control rate was 65% (94). Besides, the ORR for OC patients treated with durvalumab plus the anticancer drug trabectedin was 21.4% (95). In addition, patients with platinum-resistant OC responded well to pembrolizumab plus pegylated liposomal doxorubicin. In a clinical trial, 52.2% of the patients achieved a clinical benefit, and 26.1% experienced an overall response (96). Moreover, a phase II trial of nivolumab plus bevacizumab in recurrent OC patients was conducted. The results showed that platinum-sensitive patients had an ORR of 40.0% and platinum-resistant patients had an ORR of 16.7% (97). However, although combination therapy represents an approach to improving the efficacy of ICB, ICIs still demonstrated limited clinical activity for OC patients. The core reason is the suppressive TIME in OC, which leads to insufficient CTL activity. According to Grzywa TM et al., TAMs can decrease the amount of L-arginine in the TME and decrease T cell activation (98).
Cancer vaccines can promote antigen presentation by APCs and enhance the anti-tumor activities of antigen-specific CTLs. They have additional advantages in establishing immune memory and preventing tumor recurrence (99). At present, cancer vaccines, represented by DC vaccines, have achieved successful clinical results in the immunotherapy of various malignancies, including OC, melanoma, and prostatic cancer (100). DC vaccines that target MUC1 and NY-ESO-1 have been used to treat patients with OC. In a phase II clinical study of the MUC1-targeted DC vaccine for EOC patients, MUC1 T-cell-specific responses were observed but did not result in substantially increased progression-free survival (PFS) (101). Multiple antigens have been incorporated into cancer vaccines considering the negative effects of immune escape. However, this strategy still does not improve clinical outcomes for OC. For example, combining a multivalent conjugate vaccine (MUC1-TN, GLO-H, GM2, TF) with an adjuvant for patients with OC in the second or third clinical complete remission following chemotherapy did not prolong OS or PFS compared to adjuvant alone (102).
Taken together, although cancer vaccines can induce strong immune responses, their current clinical outcomes in OC have not been satisfactory. The main reason is the weak immunogenicity and suppressive TIME in OC. According to Schumacher et al., neoantigen recognition is uncertain in OCs, because of the inadequate mutational load and tumor heterogeneity (89, 103). At present, some strategies have been proposed to solve this dilemma. On the one hand, cancer vaccines can be combined with other treatment strategies, such as ICB. For example, patients with advanced platinum-resistant OC treated with the multiepitope FRα vaccine plus durvalumab achieved durable survival, with partial response rates of 3.7% and stable disease (SD) rates of 33.3% (104). On the other hand, incorporating as many tumor antigens as possible into cancer vaccines is a promising strategy to solve the heterogeneity of OC and improve treatment effectiveness (105). Tanyi JL et al. constructed an oxidized whole-tumor lysate DC vaccine for treating patients with platinum-treated recurrent OC. After administration, the vaccine stimulates T-cell responses and patients experience prolonged survival (106).
ACT is an immunotherapeutic regimen that harnesses autologous or allogeneic anticancer lymphocytes to promote tumor regression (105). ACT is mainly divided into three types: expanded natural TILs, chimeric antigen receptor T cells (CAR-T), and T-cell receptor engineered T cells (TCR-T) (107). ACT has achieved striking clinical success in various cancers, such as B-cell leukemias and melanoma. For example, patients with advanced melanoma responded favorably to ACT, with complete tumor shrinkage (108). However, despite several attempts, ACT has not achieved the desired effect for OC patients. Patients with recurrent or advanced EOC were treated with TILs following a single intravenous injection of cyclophosphamide. The results show that only 14.3% of the patients experienced a complete response, and 57.1% experienced a partial response (109). Besides, in a phase I clinical study of CAR-T targeting mesothelin (CAR-T-meso) for patients with OC, the CAR-T-meso cells showed limited clinical activity and short persistence (110).
The poor antitumor activity of ACT in OC is largely associated with the suppressive TIME. At present, several immunotherapies, such as ICB and cytokines, which can regulate the TIME, have been combined with ACT for OC to improve therapeutic activity. The combination of IL-2 with TILs was used to treat six patients with progressive platinum-resistant metastatic OC. There were 4 patients with SD for 3 months and 2 patients for 5 months (111). In another clinical trial, 83.3% of patients with late-stage metastatic HGSOC who received TILs, IL-2, ipilimumab, and nivolumab had SD for up to 12 months. The addition of ipilimumab improved T-cell proliferation positively impacted the T-cell phenotype and boosted CD8 T-cell tumor reactivity (112).
In conclusion, ACT has shown excellent potential in OC treatment, but its successful clinical application still faces obstacles. The physical barriers in OC limit the accessibility of CAR-T cells to tumor cells. Local CAR-T cell administration will offer solutions to this problem and improve antitumor efficiency (113). In addition, the small number of targeted antigens and their heterogeneous expression in ovarian tumors predispose them to antigen escape. Novel CAR-T cells simultaneously targeting multiple TAAs may increase the effectiveness of ACT in OC patients (114).
A large class of tiny biomolecules known as cytokines plays a crucial role in cell signaling. Among them, IFNs, ILs, and chemokines have all been extensively utilized as immunomodulators to treat cancer (115). For instance, IFN-α has achieved FDA approval and is used to treat leukemia in clinical settings (115). In addition, IL-2 can cause complete and long-lasting tumor regression in patients with metastatic melanoma and renal cancer (116). Recently, cytokine-mediated immunotherapy has been evaluated in OC clinical studies but has not yet achieved excellent outcomes. A clinical study found that OC patients had a low response rate to a single intravenous injection of recombinant IL-12 (117). Besides, the overall response rate for platinum-resistant OC patients receiving intraperitoneal administration of IL-2 was 25% (118). Moreover, intraperitoneal injection of α-recombinant interferon (rIFN-α2) resulted in complete remission in 36% of patients with EOC but also induced significant toxic side effects (119). In addition, an adenoviral vector expressing IFN-β was used to treat two OC patients. One of the patients with distant metastasis and malignant pleural effusion achieved a complete response (120).
At present, cytokine-based immunotherapy has been combined with various antitumor therapies such as chemotherapy and antiangiogenic therapy to improve clinical efficacy. In OC patients, combining IFN-γ with first-line chemotherapy improved PFS while causing acceptable toxicity (121). IL-2 was combined with picibanil (OK-432) and traditional chemotherapy drugs for patients with advanced OC. These patients had a lower recurrence rate than patients receiving chemotherapy alone (122). In a clinical trial, patients with OC were treated with IL-18 plus pegylated doxorubicin liposomes. The results show that 6% of patients had a partial objective response, and 38% had an SD (123). In addition, the combination of 13-cis retinoic acid, which has antiangiogenic activity, with low-dose IL-2 was used to treat advanced OC patients. The patients had a 5-year PFS rate of 29% and an OS rate of 38%, with an increased number of lymphocytes and NK cells (124).
Above all, cytokines as excellent immunomodulators have shown exciting potential in combination therapy of OC. However, their low stability and short half-life essentially limit their application in the clinic. These issues are considered to be overcome by utilizing nanoparticle-based drug delivery systems.
During the past decades, multiple nanoparticles have been applied in OC treatment or synergized immunotherapy, such as liposomes, polymeric micelles, silica-based nanoparticles (SNPs), and metal-based nanomaterials (Table 2). Nanoparticles not only serve as a vehicle to carry anticancer drugs but also as a regulator to modulate the TIME (Figure 2). In this section, we will discuss the role of nanoparticles in remodeling the TIME of OC and improving the efficacy of immunotherapy.

Table 2 Nanoparticles for regulating TIME and improving immunotherapy.
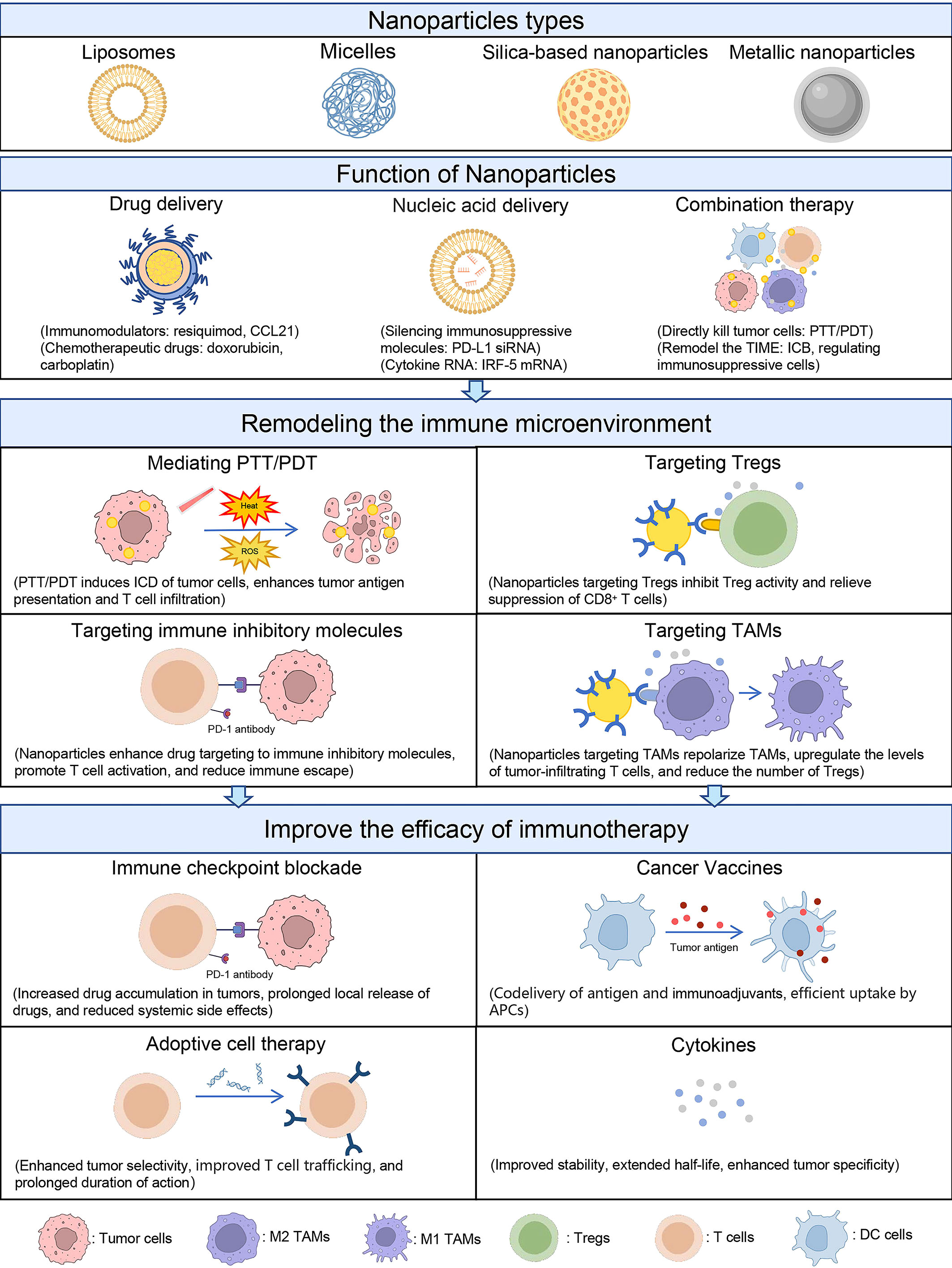
Figure 2 Schematic of nanoparticle-mediated immunotherapy regulating the TIME. Nanoparticles are mainly classified as liposomes, micelles, SNPs, and metallic nanoparticles. These nanoparticles have the functions of delivering drugs, delivering nucleic acids, and mediating combination therapy. Based on these functions, nanoparticles can regulate TIME in four ways: (1) mediating PTT and PDT to induce ICD in tumor cells; (2) improving drug targeting to immunosuppressive molecules; (3) targeting Tregs; and (4) targeting TAMs. By reversing the immunosuppressive state of TIME, nanoparticles can enhance the efficacy of immunotherapy such as ICB, ACT, tumor vaccines, and cytokines.
Liposomes are spherical bilayer nanoparticles composed of cholesterol and phospholipids, which have been used as drug vehicles due to their excellent encapsulation efficiency, targeting ability, biosafety, and biocompatibility (137). The remarkable properties of liposomes result in their FDA approval for use in clinical cancer treatment (138). In recent years, immunomodulators, such as adjuvants, photosensitizers, and tumor antigens, have been encapsulated in liposomes to regulate the TIME. For example, Xiong W et al. encapsulated the photosensitizer IR775 and metformin into liposomes. Under laser irradiation, the photosensitizer IR775 generates reactive oxygen species, which induce ICD in bladder and colon cancer cells and enhance antigen presentation (139). After PDT, the upregulation of IFN-γ amplifies the expression of PD-L1 on tumor cells (140, 141). The coencapsulation component metformin mediates the downregulation of PD-L1, which alleviates T cell exhaustion, synergistically enhancing the antitumor effect of PDT.
Since TAMs are the main immune cell population in OC, remodeling TAMs is a prospective strategy to improve the poor clinical outcomes of OC immunotherapy. TLR 7 and TLR 8 agonists, such as liquimod and resiquimod, serve as strong immunostimulatory molecules and have the ability to remodel TAMs (142). However, these small molecule drugs have serious toxicities when administered systemically. As an excellent target drug-delivery system, liposomes provide a way to deliver drugs into TAMs and remodel the TIME. For example, by loading resiquimod into liposomes, the drugs are efficiently delivered into TAMs and transformed M2 macrophages to the M1-type. Under treatment, the levels of tumor-infiltrating T cells were upregulated, while the percentage of Tregs in the TIME was reduced. When combined with PD-1 blockade, resiquimod-loaded liposomes significantly improve the antitumor efficiency of anti-PD-1 antibodies in OC (7).
Reducing the number of Tregs in the TIME is beneficial for promoting antigen presentation as well as T-cell recruitment and proliferation. Indoleamine 2,3-dioxygenase (IDO) is a metabolic immune regulator that can induce the expansion of Tregs (143). Kuo-Ching Mei et al. co-deliver cholesterol-conjugated indoximod prodrug, an inhibitor of the IDO-1 pathway, and chemotherapeutic agent mitoxantrone by liposomes into tumors. As a result, the number of Foxp3+ Tregs was obviously decreased, which in conjunction with chemotherapeutic drug-induced ICD, significantly boosted the immunotherapy response in multiple solid tumors (144). Therefore, liposomes encapsulating IDO pathway inhibitors can effectively reprogram the TIME by reducing the number of Tregs. This strategy is also expected to be successful in OC immunotherapy. Because a study has shown that IDO is widely expressed in 56% of ovarian tumors and is associated with decreased TIL numbers (125).
Polymeric micelles generally consist of a lipophilic core and a hydrophilic outer shell. Micelles have become widely used as drug carriers due to their biosafety, biocompatibility, surface modification, tumor targeting, and environmental responsiveness (145). These excellent biological properties have enabled micelles to be FDA-approved for the delivery of anticancer drugs (146). Recently, polymeric micelles have been used for drug delivery, bioimaging, and immunomodulation. Various immunomodulators, such as immunostimulants, immunoadjuvants, photosensitizers, and nucleic acids, have been entrapped into micelles to modulate the TIME (147). For example, FA-modified poly (ethylene glycol)-chitooligosaccharide lactate (COL) micelles were used as HIF-1a siRNA carriers. The micelles are efficiently taken up by cells via receptor-mediated endocytosis and significantly induce the transfection and gene knockout of HIF-1a in vitro, which effectively inhibits the proliferation of OC (148).
ICB can sensitize T cell-mediated tumor killing and has shown advantages in OC treatment. Genetic interventions, such as PD-L1 siRNA, are emerging as an effective strategy to suppress immune checkpoint signaling. However, the low transfection efficiency of gene therapy restrains its application as promising immunotherapy (126, 149). Cationic polymer micelles as excellent nucleic acid delivery vectors offer an attractive approach to boost genetic immunotherapy and improve the ICB response. Recently, Teo, P. Y. et al. loaded PD-L1 siRNA into folate (FA) or FA-polyethylene glycol (PEG)-modified PEI nanoparticles. The positively charged cationic polymer micelles facilitate the uptake of PD-L1 siRNA by interacting with negatively charged cell membranes. After administration, PD-L1 siRNA successfully transfected OC cells, effectively blocked PD-1/PD-L1 interaction, and enhanced the efficiency of ICB for OC (150). Recently, Ling, Xiang et al. constructed a pH-responsive nanocoordination polymer to deliver siPD-L1. This micelle was endocytosed into endocytic vesicles and ruptured when the endocytic vesicles transform into acidic endolysosomal, disrupting the organelle membrane and releasing siPD-L1 into the cytoplasm. Then PD-L1 was successfully knocked out for immune checkpoint inhibition, which remodeled the TIME and enhanced immune activation in OC (151).
Repolarizing M2 TAMs to the M1 phenotype is an effective strategy to remodel the TIME in OC and enhance antitumor immunity. Although various immunomodulators have been shown to repolarize TAMs, there are still many difficulties in repolarizing TAMs, such as poor targeting and the instability of immunomodulators. Due to the good surface modification, the polymeric micelles can be chemically bonded with diversified active targeting ligands to achieve specific targeting to TAMs. For example, mannose-modified polymeric micelles were used to deliver mRNA encoding IRF-5 and its activating kinase IKKβ. These polymeric micelles target mannose receptors overexpressed in M2 TAMs, delivering their payload exclusively to M2 TAMs. Following treatment, the immunosuppressive and tumor-promoting effects of M2 TAMs were successfully reversed (152). In addition, Yin Wen and his colleagues loaded the TLR7/8 agonist resiquimod and cisplatin onto poly(l-glutamate)-graft-methoxy polyethylene glycol (PLG-g-mPEG) nanoparticles. Benefiting from the protective function of these micelles, TLR agonists were successfully delivered and induced repolarization of macrophages, resulting in a synergistic anticancer effect of chemotherapy and macrophages in OC (153).
SNPs are one of the most important nanomaterials applied in biomedical applications because of their excellent biocompatibility, biosafety, easy synthesis, and surface modification (154, 155). SNPs are mainly divided into three types: spheres, core-shells, and mesoporous SNPs (MSNPs) (156). Typically, MSNPs have many empty pores and large surface areas, which endow them as good candidates for drug delivery, bioimaging, and immune regulation. Immunomodulators, such as adjuvants, photosensitizers, cytokines, and siRNA, can be loaded into SNPs to regulate the TIME. For example, LyP-1 peptide-modified SNP-loaded siRNA against PI3K-γ can target TAMs and significantly knock down PI3K-γ expression (the knockdown efficiency is 81%), which leads to TAM polarization and remodels the TIME of OC (157). In addition, the surface modifiability of SNPs endows them can be wrapped with polymers and tumor-targeted peptides. These modified SNPs can enhance the drug delivery ability and avoid the toxicity of anticancer drugs. In our previous study, we developed tumor cell-targeted MSNPs by conjugating the indicated PAA and PEG on their surface (154). The MSNPs can selectively deliver MEK inhibitors into tumor cells instead of T-cells. MSNP encapsulation avoids the cytotoxicity of MEK inhibition on T cells and improves the antitumor efficiency of anti-PD-1 antibodies. The results suggest that MSNPs can avoid small molecule drug-induced immune toxicity and coordinate tumor-targeted therapy and immunotherapy.
Cytokines are classical immunoregulators that have been applied to treat OC. However, their rapid biodegradation and short half-life limit their clinical application. Uniform and large pore diameters as well as the easy surface modification ability endow MSNPs with high loading capacity, making them a candidate vehicle for carrier cytokines. In addition, the enhanced permeability and retention (EPR) effect of nanoparticles causes the carried cytokines to accumulate in TME and enhances their antitumor efficiency. Wimalachandra DC et al. developed an FA-modified SNP to load CCL21. Upon injection, CCL21-loaded SNPs accumulated at the TME of OC, which further recruited immune cells into the tumor tissue (127). Haber et al. found that negatively charged SNPs with a particle size larger than 100 nm administered intraperitoneally selectively accumulated in TAMs in mouse ovarian tumors (128). These results demonstrated that SNPs could serve as a candidate drug delivery system to remodel TAMs and enhance the anti-OC immune response by loading immune regulation agents.
ICB has been demonstrated to be an effective immunotherapy strategy and approved for the clinical treatment of OC. However, systemic toxicity and low local concentrations still need to be addressed. MSNPs have extremely high drug loading and can achieve controlled drug release by surface modification, making them ideal candidates for the delivery of ICIs. Bongseo Choi and his colleagues loaded an anti-PD-1 antibody into the pores of MSNPs and blocked the pores with iron oxide ferumoxytol, finally realizing the sustained release of PD-1 at the tumor site. This MSNP-mediated local ICB treatment after chemotherapy effectively promotes T cell infiltration and reduces Treg numbers in the TIME. This result indicates that MSNPs can achieve sustained release of ICIs and improve the duration of action and tumor specificity of ICIs (129).
Metallic nanoparticles are a kind of novel nanomaterial composed of pure metals (e.g., gold, silver, copper, iron, platinum, etc.) or their compounds (e.g., hydroxides, oxides, sulfides, etc.) (158). In recent years, metallic nanoparticles have been widely used for bioimaging and cancer treatment because of their excellent optical polarizability, electrical conductivity, biocompatibility, chemical properties, and potent photothermal properties induced by near-infrared (NIR) lasers (159). Metallic nanoparticles can mediate tumor PTT and PDT. PTT/PDT is an effective strategy to remodel the TIME and improve the efficiency of immunotherapy (160, 161). The mechanism of metallic nanoparticle-mediated PTT is that metal elements absorb light energy and convert it into heat to destroy malignant cells. Moreover, metallic nanoparticle-mediated PTT and PDT induce ICD in tumor cells, releasing tumor antigens and damage-associated molecular patterns (DAMPs) to stimulate the tumor-specific immune response and enhance immunotherapy (130). For example, gold nanoparticle-mediated PTT has been widely used alone or in combination with immunotherapy, chemotherapy, and targeted therapy to treat malignant tumors. In our previous study, we constructed a MAPK pathway inhibitor-loaded silica-modified gold nanocage (AuNCs) for synergistic melanoma therapy with an anti-PD-1 antibody. AuNC-mediated PTT along with MAPK-targeted therapy effectively kills tumor cells and enhances T-cell infiltration. This treatment regimen significantly improved the antitumor efficiency of PD-1 immunotherapy in the immune “cold” tumor and abscopal tumor models (131).
Although metallic nanoparticles mediated PTT/PDT has achieved gratifying antitumor efficiency in shallow tumors (e.g., melanoma), it has not reached the desired therapeutic effect in OC. In an OC mouse model, PTT alone did not inhibit tumor growth or prolong survival (132). The reason is that (1) the complex suppressive TIME and (2) the OC tumors located in a deep part of the human body prevent a laser from irradiating the tumor. Recently, Qizhen Cao and his colleagues developed copper monosulfide (CuS) nanoparticles to mediate a pulsed wave (PW) laser that can treat OC. CuS nanoparticles mediate photothermolysis, resulting in tumor cell death and improving the antitumor efficiency of PD-1 immunotherapy by promoting antigen presentation and T-cell infiltration (133). In addition, Xiong J et al. developed Fe3O4 nanoparticles coated with hybrid biomimetic membranes, which were formed by the fusion of red blood cell membranes and mouse-derived ID8 OC cell membranes. These metallic nanoparticles can extend the circulation half-life as well as homologous target ID8 OC cells and show synergistic PTT. Under this treatment, the tumor-specific antigens were released, which further improved the efficiency of immunotherapy by activating CD8+ CTLs and decreasing Foxp3+ Tregs (162).
Since the TIME in OC is also an important reason for limiting the effectiveness of PTT, combining strategies to modulate the TIME is a promising strategy to improve the effectiveness of PTT. The surface modifiability of metallic nanoparticles allows them to be coated with polymers and serve as vehicles for immunomodulators, such as adjuvants, cytokines, and siRNA, to regulate the TIME. For example, João Conde et al. constructed targeting peptide-modified gold nanoparticles encapsulated with siRNA against vascular endothelial growth factor (VEGF). Gold nanoparticles can selectively silence VEGF expression in tumor cells and TAMs, inhibiting immunosuppressive M2 TAMs (163). This strategy to remodel the TIME is expected to improve the antitumor effectiveness of PTT in OC.
Besides the nanoparticles introduced above, many other nanoparticles, such as carbon-based nanomaterials (CNMs) and metal-organic frameworks (MOFs), have also been reported to enhance cancer immunotherapy. CNMs, including carbon nanotubes, carbon dots, and carbon nanohorns, have gained attention in biological applications (164). Among them, carbon nanotubes have been explored as photothermal transduction agents and drug delivery carriers due to their surface modification, enhanced cellular internalization, electronic and optical properties, and biocompatibility (165). Carbon nanotubes can mediate PTT and induce ICD in tumor cells, and serve as delivery vehicles for tumor antigens and immunoadjuvants (166). For example, Yong Li et al. constructed annexin A5- modified single-walled carbon nanotubes for synergistic metastatic breast cancer therapy with an anti-CTLA-4 antibody. The nanoparticle-mediated PTT enhanced the abscopal response of ICB and increased the 100-day survival of tumor-bearing mice (167).
MOFs are novel nanoparticles composed of metal ions or clusters and organic ligands (134). The properties of MOFs, such as high porosity, large surface areas, surface modification, and luminescence characteristics, endow them as good candidates for drug delivery and diagnosis agents (135). Recently, various immunomodulators, including immunoadjuvants, tumor antigens, photothermal agents, and sonosensitizers, have been encapsulated in MOFs to modulate the TIME and synergize with immunotherapy (136, 161, 168, 169). For instance, Jiali Luo et al. developed cancer cell membrane-coated triphenylphosphonium decorated MOFs. The MOF-loaded sonosensitizer facilitated antigen presentation by mediating sonodynamic therapy. Co-delivered TLR agonist R387 promoted DC maturation. When combined with ICB, this nanoplatform finally reversed the suppressive TIME and enhanced the antitumor efficacy of immunotherapy (168).
With a more in-depth understanding of the TIME, immunotherapy, especially ICB, tumor vaccines, ACT, and cytokines, has gained extensive attention in the treatment of OC. Although these immunotherapies have achieved excellent results in a variety of tumors, they are not ideal for the treatment of OC. This is mainly due to the suppressive TIME in OC, including Tregs, TAMs, MDSCs, and CAFs, which inhibit the antitumor immune response. Therefore, using smart strategies to transform the suppressive TIME into an antitumor state is of great significance for increasing the effectiveness of OC immunotherapy and extending patient survival.
In recent years, with the development of nanomedicine, immunotherapy combined with nanomaterials to modulate immune stimulation has achieved excellent preclinical and clinical efficacy. Mainstream nanoparticles include liposomes, micelles, SNPs, and metallic nanoparticles. Based on the excellent properties of nanoparticles in biocompatibility, drug loading, targeting capability, surface modification, and photothermal conversion, they have been widely used for regulating immune response and immunotherapy. On the one hand, nanoparticles can deliver immunomodulators that regulate the TIME of OC. On the other hand, photosensitizer-loaded nanoparticles or metallic nanoparticles can mediate PDT/PTT to induce the ICD of tumor cells, promoting antigen presentation and T-cell infiltration. These advantages make nanoparticles promising candidates for modulating the TIME and improving OC immunotherapy.
However, the toxicity, specific tumor targeting, and effectiveness of nanoparticles also need to be considered in clinical translation. In terms of toxicity, nanoparticles can not only interact with organism’s cells or blood cells but also produce toxic ions by the dissolution of nanomaterials. Through these mechanisms, nanoparticles exert toxicity leading to the damage of cells or vital enzymatic functions (170). Several strategies hold promise for reducing these toxic effects of nanoparticles. The nanoparticle can carrier negative surface charges and attenuate nanoparticle-cell interaction through modification with ligands, such as PEG (171). Covering the shell material or minimizing the surface area can reduce the dissolution of toxic ions (170). For specific tumor targeting, the poor manifestation of the EPR effect in the clinic and the nanoparticle-protein complex formed in systemic circulation cause off-target effects. Specifically, the differentials between animal models and human tumors and the complex TEM contribute to the failure of EPR-mediated targeting delivery in clinical translation. Several strategies, including enhancement of vascular permeability and depletion of tumor extracellular matrix, provide a chance to minimize the gaps between theoretical expectation and clinical outcome (172). Otherwise, serum proteins and opsonins are easily adsorbed on nanoparticle surfaces, which is probable to mask targeting ligands. Based on a deeper understanding of the interactions between nanoparticles and organisms, consideration of the protein corona effect when designing nanoparticles could improve the targeting efficiency (173). In terms of effectiveness, inefficient and unstable drug loading results in low drug concentrations in the TME and insufficient therapy. The limited light penetration depth also restricts the anti-tumor effects of photosensitive nanoparticle -mediated PTT. To improve the antitumor efficiency, nanoparticles with high pore volume and novel loading strategies have been developed that are beneficial for the enrichment of drugs at tumor sites (174). Some other approaches, including improving the photothermal conversion efficiency and developing NIR-II window PTT, are promising strategies to enhance the efficiency of PTT (175).
In this review, we discussed the impact of the TIME in OC on immunotherapy and mentioned the role of nanoparticles in modulating the TIME and improving the immunotherapeutic efficacy of OC. Using multiple approaches to overcome current shortcomings, we expect to leverage nanoparticle-based drug delivery systems to provide opportunities for the clinical application of OC immunotherapy.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was funded by the National Natural Science Foundation of China (No. 22105137 and No. 82172634), Key Program of the Science and Technology Bureau of Sichuan (No. 2021YFSY0007), China Postdoctoral Science Foundation (No. 2020M683324 and No. 2022T150449), 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (No. ZYYC20013).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Le Saux O, Ray-Coquard I, Labidi-Galy SI eds. Challenges for immunotherapy for the treatment of platinum resistant ovarian cancer. In: Seminars in cancer biology. (2021) 77:127–43. doi: 10.1016/j.semcancer.2020.08.017
2. Nero C, Ciccarone F, Pietragalla A, Duranti S, Daniele G, Salutari V, et al. Ovarian cancer treatments strategy: focus on PARP inhibitors and immune check point inhibitors. Cancers. (2021) 13(6):1298. doi: 10.3390/cancers13061298
3. Coward JI, Middleton K, Murphy F. New perspectives on targeted therapy in ovarian cancer. Int J Womens Health (2015) 7:189–203. doi: 10.2147/IJWH.S52379
4. Baldwin LA, Huang B, Miller RW, Tucker T, Goodrich ST, Podzielinski I, et al. Ten-year relative survival for epithelial ovarian cancer. Obstet Gynecol. (2012) 120(3):612–8. doi: 10.1097/AOG.0b013e318264f794
5. Coukos G, Tanyi J, Kandalaft L. Opportunities in immunotherapy of ovarian cancer. Ann Oncol (2016) 27:i11–i5. doi: 10.1093/annonc/mdw084
6. Sau S, Alsaab HO, Bhise K, Alzhrani R, Nabil G, Iyer AK. Multifunctional nanoparticles for cancer immunotherapy: A groundbreaking approach for reprogramming malfunctioned tumor environment. J Controlled Release. (2018) 274:24–34. doi: 10.1016/j.jconrel.2018.01.028
7. Kang Y, Flores L, Ngai HW, Cornejo YR, Haber T, McDonald M, et al. Large, Anionic liposomes enable targeted intraperitoneal delivery of a TLR 7/8 agonist to repolarize ovarian tumors’ microenvironment. Bioconjugate Chem (2021) 32(8):1581–92. doi: 10.1021/acs.bioconjchem.1c00139
8. Olalekan S, Xie B, Back R, Eckart H, Basu A. Characterizing the tumor microenvironment of metastatic ovarian cancer by single-cell transcriptomics. Cell Rep (2021) 35(8):109165. doi: 10.1016/j.celrep.2021.109165
9. Jiménez-Sánchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. (2017) 170(5):927–38.e20. doi: 10.1016/j.cell.2017.07.025
10. Izar B, Tirosh I, Stover EH, Wakiro I, Cuoco MS, Alter I, et al. A single-cell landscape of high-grade serous ovarian cancer. Nat Med (2020) 26(8):1271–9. doi: 10.1038/s41591-020-0926-0
11. Oda K, Hamanishi J, Matsuo K, Hasegawa K. Genomics to immunotherapy of ovarian clear cell carcinoma: Unique opportunities for management. Gynecologic Oncol (2018) 151(2):381–9. doi: 10.1016/j.ygyno.2018.09.001
12. Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. (2021) 184(3):792–809.e23. doi: 10.1016/j.cell.2021.01.010
13. Maccio A, Gramignano G, Cherchi MC, Tanca L, Melis L, Madeddu C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci Rep (2020) 10(1):6096. doi: 10.1038/s41598-020-63276-1
14. Germain RN. T-Cell development and the CD4-CD8 lineage decision. Nat Rev Immunol (2002) 2(5):309–22. doi: 10.1038/nri798
15. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. (2011) 35(2):161–8. doi: 10.1016/j.immuni.2011.07.010
16. Paijens S, Vledder A, Loiero D, Duiker E, Bart J, Hendriks A, et al. Prognostic image-based quantification of CD8CD103 T cell subsets in high-grade serous ovarian cancer patients. Oncoimmunology. (2021) 10(1):1935104. doi: 10.1136/ijgc-2021-ESGO.370
17. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFbeta enhance the efficacy of cancer immunotherapy. Nat Commun (2018) 9(1):741. doi: 10.1038/s41467-017-02696-6
18. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell (2019) 35(6):885–900.e10. doi: 10.1016/j.ccell.2019.05.004
19. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. (2016) 45(2):374–88. doi: 10.1016/j.immuni.2016.07.009
20. Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. (2018) 562(7727):423–8. doi: 10.1038/s41586-018-0597-x
21. Siu KT, Rosner MR, Minella AC. An integrated view of cyclin e function and regulation. Cell Cycle (2012) 11(1):57–64. doi: 10.4161/cc.11.1.18775
22. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582
23. Poznanski SM, Nham T, Chew MV, Lee AJ, Hammill JA, Fan IY, et al. Expanded CD56(superbright)CD16(+) NK cells from ovarian cancer patients are cytotoxic against autologous tumor in a patient-derived xenograft murine model. Cancer Immunol Res (2018) 6(10):1174–85. doi: 10.1158/2326-6066.CIR-18-0144
24. Sun Y, Yao Z, Zhao Z, Xiao H, Xia M, Zhu X, et al. Natural killer cells inhibit metastasis of ovarian carcinoma cells and show therapeutic effects in a murine model of ovarian cancer. Exp Ther Med (2018) 16(2):1071–8. doi: 10.3892/etm.2018.6342
25. Worzfeld T, Pogge von Strandmann E, Huber M, Adhikary T, Wagner U, Reinartz S, et al. The unique molecular and cellular microenvironment of ovarian cancer. Front Oncol (2017) 7:24. doi: 10.3389/fonc.2017.00024
26. Krockenberger M, Dombrowski Y, Weidler C, Ossadnik M, Honig A, Hausler S, et al. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J Immunol (2008) 180(11):7338–48. doi: 10.4049/jimmunol.180.11.7338
27. Greppi M, Tabellini G, Patrizi O, Candiani S, Decensi A, Parolini S, et al. Strengthening the AntiTumor NK cell function for the treatment of ovarian cancer. Int J Mol Sci (2019) 20(4):890. doi: 10.3390/ijms20040890
28. Pesce S, Tabellini G, Cantoni C, Patrizi O, Coltrini D, Rampinelli F, et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology. (2015) 4(4):e1001224. doi: 10.1080/2162402X.2014.1001224
29. Wong JL, Berk E, Edwards RP, Kalinski P. IL-18-primed helper NK cells collaborate with dendritic cells to promote recruitment of effector CD8+ T cells to the tumor microenvironment. Cancer Res (2013) 73(15):4653–62. doi: 10.1158/0008-5472.CAN-12-4366
30. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
31. Wilson AL, Moffitt LR, Wilson KL, Bilandzic M, Wright MD, Gorrell MD, et al. DPP4 inhibitor sitagliptin enhances lymphocyte recruitment and prolongs survival in a syngeneic ovarian cancer mouse model. Cancers. (2021) 13(3):487. doi: 10.3390/cancers13030487
32. Wertel I, Surówka J, Polak G, Barczyński B, Bednarek W, Jakubowicz-Gil J, et al. Macrophage-derived chemokine CCL22 and regulatory T cells in ovarian cancer patients. Tumor Biol (2015) 36(6):4811–7. doi: 10.1007/s13277-015-3133-8
33. Sasidharan Nair V, Saleh R, Toor SM, Cyprian FS, Elkord E. Metabolic reprogramming of T regulatory cells in the hypoxic tumor microenvironment. Cancer immunology Immunother (2021) 70(8):2103–21. doi: 10.1007/s00262-020-02842-y
34. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol (2012) 3:51. doi: 10.3389/fimmu.2012.00051
35. Toker A, Nguyen LT, Stone SC, Yang SYC, Katz SR, Shaw PA, et al. Regulatory T cells in ovarian cancer are characterized by a highly activated phenotype distinct from that in melanoma. Clin Cancer Res (2018) 24(22):5685–96. doi: 10.1158/1078-0432.CCR-18-0554
36. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
37. Amoozgar Z, Kloepper J, Ren J, Tay RE, Kazer SW, Kiner E, et al. Targeting treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat Commun (2021) 12(1):1–16. doi: 10.1038/s41467-021-22885-8
38. Govindaraj C, Scalzo-Inguanti K, Madondo M, Hallo J, Flanagan K, Quinn M, et al. Impaired Th1 immunity in ovarian cancer patients is mediated by TNFR2+ tregs within the tumor microenvironment. Clin Immunol (2013) 149(1):97–110. doi: 10.1016/j.clim.2013.07.003
39. Kampan NC, Madondo MT, McNally OM, Stephens AN, Quinn MA, Plebanski M. Interleukin 6 present in inflammatory ascites from advanced epithelial ovarian cancer patients promotes tumor necrosis factor receptor 2-expressing regulatory T cells. Front Immunol (2017) 8:1482. doi: 10.3389/fimmu.2017.01482
40. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
41. Kryczek I, Wei S, Zhu G, Myers L, Mottram P, Cheng P, et al. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res (2007) 67(18):8900–5. doi: 10.1158/0008-5472.CAN-07-1866
42. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol (2018) 15(4):234–48. doi: 10.1038/nrclinonc.2018.8
43. El-Arabey AA, Denizli M, Kanlikilicer P, Bayraktar R, Ivan C, Rashed M, et al. GATA3 as a master regulator for interactions of tumor-associated macrophages with high-grade serous ovarian carcinoma. Cell Signal (2020) 68:109539. doi: 10.1016/j.cellsig.2020.109539
44. Ying X, Wu Q, Wu X, Zhu Q, Wang X, Jiang L, et al. Epithelial ovarian cancer-secreted exosomal miR-222-3p induces polarization of tumor-associated macrophages. Oncotarget. (2016) 7(28):43076–87. doi: 10.18632/oncotarget.9246
45. Bai Y, Yin K, Su T, Ji F, Zhang S. CTHRC1 in ovarian cancer promotes M2-like polarization of tumor-associated macrophages via regulation of the STAT6 signaling pathway. Onco Targets Ther (2020) 13:5743–53. doi: 10.2147/OTT.S250520
46. Song M, Yeku OO, Rafiq S, Purdon T, Dong X, Zhu L, et al. Tumor derived UBR5 promotes ovarian cancer growth and metastasis through inducing immunosuppressive macrophages. Nat Commun (2020) 11(1):1–16. doi: 10.1038/s41467-020-20140-0
47. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. (2017) 5(1):53. doi: 10.1186/s40425-017-0257-y
48. Moughon DL, He H, Schokrpur S, Jiang ZK, Yaqoob M, David J, et al. Macrophage blockade using CSF1R inhibitors reverses the vascular leakage underlying malignant ascites in late-stage epithelial ovarian cancer. Cancer Res (2015) 75(22):4742–52. doi: 10.1158/0008-5472.CAN-14-3373
49. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25(6):846–59. doi: 10.1016/j.ccr.2014.05.016
50. Cheng H, Wang Z, Fu L, Xu T. Macrophage polarization in the development and progression of ovarian cancers: An overview. Front Oncol (2019) 9:421. doi: 10.3389/fonc.2019.00421
51. Yeung TL, Leung CS, Yip KP, Au Yeung CL, Wong ST, Mok SC. Cellular and molecular processes in ovarian cancer metastasis. a review in the theme: Cell and molecular processes in cancer metastasis. Am J Physiol Cell Physiol (2015) 309(7):C444–56. doi: 10.1152/ajpcell.00188.2015
52. Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S, et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest. (2016) 126(11):4157–73. doi: 10.1172/JCI87252
53. Hagemann T, Wilson J, Kulbe H, Li NF, Leinster DA, Charles K, et al. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa b and JNK. J Immunol (2005) 175(2):1197–205. doi: 10.4049/jimmunol.175.2.1197
54. Browning L, Patel MR, Horvath EB, Tawara K, Jorcyk CL. IL-6 and ovarian cancer: inflammatory cytokines in promotion of metastasis. Cancer Manag Res (2018) 10:6685–93. doi: 10.2147/CMAR.S179189
55. Goswami KK, Ghosh T, Ghosh S, Sarkar M, Bose A, Baral R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol (2017) 316:1–10. doi: 10.1016/j.cellimm.2017.04.005
56. Mabuchi S, Sasano T, Komura N. Targeting myeloid-derived suppressor cells in ovarian cancer. Cells. (2021) 10(2):329. doi: 10.3390/cells10020329
57. Wu L, Deng Z, Peng Y, Han L, Liu J, Wang L, et al. Ascites-derived IL-6 and IL-10 synergistically expand CD14(+)HLA-DR-/low myeloid-derived suppressor cells in ovarian cancer patients. Oncotarget. (2017) 8(44):76843–56. doi: 10.18632/oncotarget.20164
58. Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. (2013) 123(10):4464–78. doi: 10.1172/JCI68189
59. Taki M, Abiko K, Baba T, Hamanishi J, Yamaguchi K, Murakami R, et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat Commun (2018) 9(1):1685. doi: 10.1038/s41467-018-03966-7
60. Li X, Wang J, Wu W, Gao H, Liu N, Zhan G, et al. Myeloid-derived suppressor cells promote epithelial ovarian cancer cell stemness by inducing the CSF2/p-STAT3 signalling pathway. FEBS J (2020) 287(23):5218–35. doi: 10.1111/febs.15311
61. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. (2013) 39(3):611–21. doi: 10.1016/j.immuni.2013.08.025
62. Rodriguez-Ubreva J, Catala-Moll F, Obermajer N, Alvarez-Errico D, Ramirez RN, Company C, et al. Prostaglandin E2 leads to the acquisition of DNMT3A-dependent tolerogenic functions in human myeloid-derived suppressor cells. Cell Rep (2017) 21(1):154–67. doi: 10.1016/j.celrep.2017.09.018
63. Safarzadeh E, Mohammadi A, Mansoori B, Duijf PH, Hashemzadeh S, Khaze V, et al. STAT3 silencing and TLR7/8 pathway activation repolarize and suppress myeloid-derived suppressor cells from breast cancer patients. Front Immunol (2021) 11:613215. doi: 10.3389/fimmu.2020.613215
64. Tumino N, Di Pace AL, Besi F, Quatrini L, Vacca P, Moretta L. Interaction between MDSC and NK cells in solid and hematological malignancies: impact on HSCT. Front Immunol (2021) 12:638841. doi: 10.3389/fimmu.2021.638841
65. Zhang J, Han X, Hu X, Jin F, Gao Z, Yin L, et al. IDO1 impairs NK cell cytotoxicity by decreasing NKG2D/NKG2DLs via promoting miR-18a. Mol Immunol (2018) 103:144–55. doi: 10.1016/j.molimm.2018.09.011
66. Weber R, Groth C, Lasser S, Arkhypov I, Petrova V, Altevogt P, et al. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol (2021) 359:104254. doi: 10.1016/j.cellimm.2020.104254
67. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
68. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res (2010) 70(1):99–108. doi: 10.1158/0008-5472.CAN-09-1882
69. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol (2018) 9:398. doi: 10.3389/fimmu.2018.00398
70. Trillo-Tinoco J, Sierra RA, Mohamed E, Cao Y, de Mingo-Pulido A, Gilvary DL, et al. AMPK alpha-1 intrinsically regulates the function and differentiation of tumor myeloid-derived suppressor cells. Cancer Res (2019) 79(19):5034–47. doi: 10.1158/0008-5472.CAN-19-0880
71. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13(7):828–35. doi: 10.1038/nm1609
72. Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci (2019) 76(10):1901–18. doi: 10.1007/s00018-019-03048-x
73. Komura N, Mabuchi S, Shimura K, Yokoi E, Kozasa K, Kuroda H, et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol Immunother. (2020) 69(12):2477–99. doi: 10.1007/s00262-020-02628-2
74. Zhang M, Chen Z, Wang Y, Zhao H, Du Y. The role of cancer-associated fibroblasts in ovarian cancer. Cancers (Basel). (2022) 14(11):2637. doi: 10.3390/cancers14112637
75. Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME, et al. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discovery (2012) 2(12):1100–8. doi: 10.1158/2159-8290.CD-12-0206
76. Zhao L, Ji G, Le X, Luo Z, Wang C, Feng M, et al. An integrated analysis identifies STAT4 as a key regulator of ovarian cancer metastasis. Oncogene. (2017) 36(24):3384–96. doi: 10.1038/onc.2016.487
77. Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature. (2019) 569(7758):723–8. doi: 10.1038/s41586-019-1173-8
78. Gao Q, Yang Z, Xu S, Li X, Yang X, Jin P, et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J Exp Med (2019) 216(3):688–703. doi: 10.1084/jem.20180765
79. Kanzaki R, Pietras K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci (2020) 111(8):2708–17. doi: 10.1111/cas.14537
80. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int J Cancer. (2020) 146(4):895–905. doi: 10.1002/ijc.32193
81. Ferrari N, Ranftl R, Chicherova I, Slaven ND, Moeendarbary E, Farrugia AJ, et al. Dickkopf-3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat Commun (2019) 10(1):130. doi: 10.1038/s41467-018-07987-0
82. Yue H, Li W, Chen R, Wang J, Lu X, Li J. Stromal POSTN induced by TGF-beta1 facilitates the migration and invasion of ovarian cancer. Gynecol Oncol (2021) 160(2):530–8. doi: 10.1016/j.ygyno.2020.11.026
83. Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, et al. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res (2013) 73(16):5016–28. doi: 10.1158/0008-5472.CAN-13-0023
84. Deying W, Feng G, Shumei L, Hui Z, Ming L, Hongqing W. CAF-derived HGF promotes cell proliferation and drug resistance by up-regulating the c-Met/PI3K/Akt and GRP78 signalling in ovarian cancer cells. Biosci Rep (2017) 37(2):BSR20160470. doi: 10.1042/BSR20160470
85. Sun Y, Fan X, Zhang Q, Shi X, Xu G, Zou C. Cancer-associated fibroblasts secrete FGF-1 to promote ovarian proliferation, migration, and invasion through the activation of FGF-1/FGFR4 signaling. Tumour Biol (2017) 39(7):1010428317712592. doi: 10.1177/1010428317712592
86. O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF-a and tenascin-c produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci U S A. (2011) 108(38):16002–7. doi: 10.1073/pnas.1109493108
87. Givel AM, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. miR200-regulated CXCL12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun (2018) 9(1):1056. doi: 10.1038/s41467-018-03348-z
88. Zhang H, Jiang R, Zhou J, Wang J, Xu Y, Zhang H, et al. CTL attenuation regulated by PS1 in cancer-associated fibroblast. Front Immunol (2020) 11:999. doi: 10.3389/fimmu.2020.00999
89. Yang C, Xia B-R, Zhang Z-C, Zhang Y-J, Lou G, Jin W-L. Immunotherapy for ovarian cancer: adjuvant, combination, and neoadjuvant. Front Immunol (2020) 11:577869. doi: 10.3389/fimmu.2020.577869
90. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol (2015) 33(17):1974. doi: 10.1200/JCO.2014.59.4358
91. Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine ovarian cancer model. Cancer Immunol Res (2015) 3(9):1030–41. doi: 10.1158/2326-6066.CIR-15-0073
92. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol (2015) 33(34):4015–22. doi: 10.1200/JCO.2015.62.3397
93. Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol (2019) 30(7):1080–7. doi: 10.1093/annonc/mdz135
94. Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol (2019) 5(8):1141–9. doi: 10.1001/jamaoncol.2019.1048
95. Toulmonde M, Brahmi M, Giraud A, Chakiba C, Bessede A, Kind M, et al. Trabectedin plus durvalumab in patients with advanced pretreated soft tissue sarcoma and ovarian carcinoma (TRAMUNE): an open-label, multicenter phase ib study. Clin Cancer Res (2022) 28(9):1765–72. doi: 10.1158/1078-0432.CCR-21-2258
96. Lee EK, Xiong N, Cheng SC, Barry WT, Penson RT, Konstantinopoulos PA, et al. Combined pembrolizumab and pegylated liposomal doxorubicin in platinum resistant ovarian cancer: A phase 2 clinical trial. Gynecol Oncol (2020) 159(1):72–8. doi: 10.1016/j.ygyno.2020.07.028
97. Liu JF, Herold C, Gray KP, Penson RT, Horowitz N, Konstantinopoulos PA, et al. Assessment of combined nivolumab and bevacizumab in relapsed ovarian cancer: a phase 2 clinical trial. JAMA Oncol (2019) 5(12):1731–8. doi: 10.1001/jamaoncol.2019.3343
98. Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, et al. Myeloid cell-derived arginase in cancer immune response. Front Immunol (2020) 11:938. doi: 10.3389/fimmu.2020.00938
99. Odunsi K. Immunotherapy in ovarian cancer. Ann Oncol (2017) 28:viii1–7. doi: 10.1093/annonc/mdx444
100. Yang Y, Guo X, Hu B, He P, Jiang X, Wang Z, et al. Generated SecPen_NY-ESO-1_ubiquitin-pulsed dendritic cell cancer vaccine elicits stronger and specific T cell immune responses. Acta Pharm Sin B (2021) 11(2):476–87. doi: 10.1016/j.apsb.2020.08.004
101. Gray HJ, Benigno B, Berek J, Chang J, Mason J, Mileshkin L, et al. Progression-free and overall survival in ovarian cancer patients treated with CVac, a mucin 1 dendritic cell therapy in a randomized phase 2 trial. J Immunother Cancer. (2016) 4:34. doi: 10.1186/s40425-016-0137-x
102. O'Cearbhaill RE, Deng W, Chen LM, Lucci JA 3rd, Behbakht K, Spirtos NM, et al. A phase II randomized, double-blind trial of a polyvalent vaccine-KLH conjugate (NSC 748933 IND# 14384) + OPT-821 versus OPT-821 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer who are in second or third complete remission: An NRG Oncology/GOG study. Gynecol Oncol (2019) 155(3):393–9. doi: 10.1016/j.ygyno.2019.09.015
103. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. (2014) 14(2):135–46. doi: 10.1038/nrc3670
104. Zamarin D, Walderich S, Holland A, Zhou Q, Iasonos AE, Torrisi JM, et al. Safety, immunogenicity, and clinical efficacy of durvalumab in combination with folate receptor alpha vaccine TPIV200 in patients with advanced ovarian cancer: a phase II trial. J immunotherapy Cancer (2020) 8(1):e000829. doi: 10.1136/jitc-2020-000829
105. Chester C, Dorigo O, Berek JS, Kohrt H. Immunotherapeutic approaches to ovarian cancer treatment. J immunotherapy cancer. (2015) 3(1):1–10. doi: 10.1186/s40425-015-0051-7
106. Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Trans Med (2018) 10(436):eaao5931. doi: 10.1126/scitranslmed.aao5931
107. Met Ö, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathology. (2018) 41(1):49–58. doi: 10.1007/s00281-018-0703-z
108. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. New Engl J Med (1988) 319(25):1676–80. doi: 10.1056/NEJM198812223192527
109. Aoki Y, Takakuwa K, Kodama S, Tanaka K, Takahashi M, Tokunaga A, et al. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res (1991) 51(7):1934–9.
110. Haas AR, Tanyi JL, O’Hara MH, Gladney WL, Lacey SF, Torigian DA, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther (2019) 27(11):1919–29. doi: 10.1016/j.ymthe.2019.07.015
111. Pedersen M, Westergaard MCW, Milne K, Nielsen M, Borch TH, Poulsen LG, et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: a pilot study. OncoImmunology (2018) 7(12):e1502905. doi: 10.1080/2162402X.2018.1502905
112. Kverneland AH, Pedersen M, Westergaard MCW, Nielsen M, Borch TH, Olsen LR, et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget. (2020) 11(22):2092. doi: 10.18632/oncotarget.27604
113. Nizzero S, Shen H, Ferrari M, Corradetti B. Immunotherapeutic transport oncophysics: space, time, and immune activation in cancer. Trends Cancer. (2020) 6(1):40–8. doi: 10.1016/j.trecan.2019.11.008
114. Jaspers JE, Brentjens RJ. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol Ther (2017) 178:83–91. doi: 10.1016/j.pharmthera.2017.03.012
115. Lim S, Park J, Shim MK, Um W, Yoon HY, Ryu JH, et al. Recent advances and challenges of repurposing nanoparticle-based drug delivery systems to enhance cancer immunotherapy. Theranostics. (2019) 9(25):7906. doi: 10.7150/thno.38425
116. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discovery (2019) 18(3):175–96. doi: 10.1038/s41573-018-0006-z
117. Hurteau JA, Blessing JA, DeCesare SL, Creasman WT. Evaluation of recombinant human interleukin-12 in patients with recurrent or refractory ovarian cancer: a gynecologic oncology group study. Gynecol Oncol (2001) 82(1):7–10. doi: 10.1006/gyno.2001.6255
118. Vlad AM, Budiu RA, Lenzner DE, Wang Y, Thaller JA, Colonello K, et al. A phase II trial of intraperitoneal interleukin-2 in patients with platinum-resistant or platinum-refractory ovarian cancer. Cancer immunology Immunother (2010) 59(2):293–301. doi: 10.1007/s00262-009-0750-3
119. Berek JS, Hacker NF, Lichtenstein A, Jung T, Spina C, Knox RM, et al. Intraperitoneal recombinant α-interferon for “salvage” immunotherapy in stage III epithelial ovarian cancer: a gynecologic oncology group study. Cancer Res (1985) 45(9):4447–53.
120. Sterman DH, Recio A, Haas AR, Vachani A, Katz SI, Gillespie CT, et al. A phase I trial of repeated intrapleural adenoviral-mediated interferon-β gene transfer for mesothelioma and metastatic pleural effusions. Mol Ther (2010) 18(4):852–60. doi: 10.1038/mt.2009.309
121. Windbichler G, Hausmaninger H, Stummvoll W, Graf A, Kainz C, Lahodny J, et al. Interferon-gamma in the first-line therapy of ovarian cancer: a randomized phase III trial. Br J cancer. (2000) 82(6):1138–44. doi: 10.1054/bjoc.1999.1053
122. Wang YL, Peng HH, Su SY, Lin CT. Combined immunotherapy (OK-432, IL-2) with chemotherapy decrease the recurrence rate in advanced ovarian cancer. Reprod Sci (2019) 26(2):244–9. doi: 10.1177/1933719118768684
123. Simpkins F, Flores A, Chu C, Berek JS, Lucci J 3rd, Murray S, et al. Chemoimmunotherapy using pegylated liposomal doxorubicin and interleukin-18 in recurrent ovarian cancer: a phase I dose-escalation study. Cancer Immunol Res (2013) 1(3):168–78. doi: 10.1158/2326-6066.CIR-13-0098
124. Recchia F, Di Orio F, Candeloro G, Guerriero G, Piazze J, Rea S. Maintenance immunotherapy in recurrent ovarian cancer: Long term follow-up of a phase II study. Gynecologic Oncol (2010) 116(2):202–7. doi: 10.1016/j.ygyno.2009.09.042
125. Inaba T, Ino K, Kajiyama H, Yamamoto E, Shibata K, Nawa A, et al. Role of the immunosuppressive enzyme indoleamine 2, 3-dioxygenase in the progression of ovarian carcinoma. Gynecologic Oncol (2009) 115(2):185–92. doi: 10.1016/j.ygyno.2009.07.015
126. Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC, et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev (2017) 46(14):4218–44. doi: 10.1039/C6CS00636A
127. Wimalachandra DC, Li Y, Liu J, Shikha S, Zhang J, Lim Y-C, et al. Microfluidic-based immunomodulation of immune cells using upconversion nanoparticles in simulated blood vessel–tumor system. ACS Appl materials interfaces. (2019) 11(41):37513–23. doi: 10.1021/acsami.9b15178
128. Haber T, Cornejo YR, Aramburo S, Flores L, Cao P, Liu A, et al. Specific targeting of ovarian tumor-associated macrophages by large, anionic nanoparticles. Proc Natl Acad Sci U S A. (2020) 117(33):19737–45. doi: 10.1073/pnas.1917424117
129. Choi B, Jung H, Yu B, Choi H, Lee J, Kim DH. Sequential MR image-guided local immune checkpoint blockade cancer immunotherapy using ferumoxytol capped ultralarge pore mesoporous silica carriers after standard chemotherapy. Small. (2019) 15(52):1904378. doi: 10.1002/smll.201904378
130. Wen Y, Chen X, Zhu X, Gong Y, Yuan G, Qin X, et al. Photothermal-chemotherapy integrated nanoparticles with tumor microenvironment response enhanced the induction of immunogenic cell death for colorectal cancer efficient treatment. ACS Appl Mater Interfaces. (2019) 11(46):43393–408. doi: 10.1021/acsami.9b17137
131. Liu X, Feng Y, Xu J, Shi Y, Yang J, Zhang R, et al. Combination of MAPK inhibition with photothermal therapy synergistically augments the anti-tumor efficacy of immune checkpoint blockade. J Controlled Release. (2021) 332:194–209. doi: 10.1016/j.jconrel.2021.02.020
132. Cao Q, Wang W, Zhou M, Huang Q, Wen X, Zhao J, et al. Induction of antitumor immunity in mice by the combination of nanoparticle-based photothermolysis and anti-PD-1 checkpoint inhibition. Nanomedicine. (2020) 25:102169. doi: 10.1016/j.nano.2020.102169
133. Assuncao E, Williams S. Comparison of continuous wave and pulsed wave laser welding effects. Optics Lasers Engineering. (2013) 51(6):674–80. doi: 10.1016/j.optlaseng.2013.01.007
134. Lau J, Trojniak AE, Maraugha MJ, VanZanten AJ, Osterbaan AJ, Serino AC, et al. Conformal ultrathin film metal–organic framework analogues: Characterization of growth, porosity, and electronic transport. Chem Materials. (2019) 31(21):8977–86. doi: 10.1021/acs.chemmater.9b03141
135. Mendes RF, Figueira F, Leite JP, Gales L, Paz FAA. Metal–organic frameworks: a future toolbox for biomedicine? Chem Soc Rev (2020) 49(24):9121–53. doi: 10.1039/D0CS00883D
136. Fan Z, Liu H, Xue Y, Lin J, Fu Y, Xia Z, et al. Reversing cold tumors to hot: an immunoadjuvant-functionalized metal-organic framework for multimodal imaging-guided synergistic photo-immunotherapy. Bioactive Materials. (2021) 6(2):312–25. doi: 10.1016/j.bioactmat.2020.08.005
137. Liu X, Li Z, Wang X, Chen Y, Wu F, Men K, et al. Novel antimicrobial peptide-modified azithromycin-loaded liposomes against methicillin-resistant staphylococcus aureus. Int J nanomedicine. (2016) 11:6781–94. doi: 10.2147/IJN.S107107
138. Beltrán-Gracia E, López-Camacho A, Higuera-Ciapara I, Velázquez-Fernández JB, Vallejo-Cardona AA. Nanomedicine review: Clinical developments in liposomal applications. Cancer Nanotechnology. (2019) 10(1):1–40. doi: 10.1186/s12645-019-0055-y
139. Xiong W, Qi L, Jiang N, Zhao Q, Chen L, Jiang X, et al. Metformin liposome-mediated PD-L1 downregulation for amplifying the photodynamic immunotherapy efficacy. ACS Appl Materials Interfaces. (2021) 13(7):8026–41. doi: 10.1021/acsami.0c21743
140. Showalter A, Limaye A, Oyer JL, Igarashi R, Kittipatarin C, Copik AJ, et al. Cytokines in immunogenic cell death: applications for cancer immunotherapy. Cytokine. (2017) 97:123–32. doi: 10.1016/j.cyto.2017.05.024
141. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest (2017) 127(8):2930–40. doi: 10.1172/JCI91190
142. Wagner TL, Ahonen CL, Couture AM, Gibson SJ, Miller RL, Smith RM, et al. Modulation of TH1 and TH2 cytokine production with the immune response modifiers, r-848 and imiquimod. Cell Immunol (1999) 191(1):10–9. doi: 10.1006/cimm.1998.1406
143. Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The indoleamine 2, 3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol (2008) 181(8):5396–404. doi: 10.4049/jimmunol.181.8.5396
144. Mei K-C, Liao Y-P, Jiang J, Chiang M, Khazaieli M, Liu X, et al. Liposomal delivery of mitoxantrone and a cholesteryl indoximod prodrug provides effective chemo-immunotherapy in multiple solid tumors. ACS nano. (2020) 14(10):13343–66. doi: 10.1021/acsnano.0c05194
145. Ghosh B, Biswas S. Polymeric micelles in cancer therapy: State of the art. J Controlled Release. (2021) 332:127–47. doi: 10.1016/j.jconrel.2021.02.016
146. Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. (2011) 3(3):1377–97. doi: 10.3390/polym3031377
147. Yang F, Shi K, Jia Y-p, Hao Y, Peng J-r, Qian Z-y. Advanced biomaterials for cancer immunotherapy. Acta Pharmacologica Sinica. (2020) 41(7):911–27. doi: 10.1038/s41401-020-0372-z
148. Li TS, Yawata T, Honke K. Efficient siRNA delivery and tumor accumulation mediated by ionically cross-linked folic acid-poly(ethylene glycol)-chitosan oligosaccharide lactate nanoparticles: for the potential targeted ovarian cancer gene therapy. Eur J Pharm Sci (2014) 52:48–61. doi: 10.1016/j.ejps.2013.10.011
149. Watts JK, Corey DR. Silencing disease genes in the laboratory and the clinic. J pathology. (2012) 226(2):365–79. doi: 10.1002/path.2993
150. Teo PY, Yang C, Whilding LM, Parente-Pereira AC, Maher J, George AJ, et al. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: strategies to enhance T cell killing. Adv Healthc Mater (2015) 4(8):1180–9. doi: 10.1002/adhm.201500089
151. Ling X, Han W, Jiang X, Chen X, Rodriguez M, Zhu P, et al. Point-source burst of coordination polymer nanoparticles for tri-modality cancer therapy. Biomaterials (2021) 270:120690. doi: 10.1016/j.biomaterials.2021.120690
152. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun (2019) 10(1):3974. doi: 10.1038/s41467-019-11911-5
153. Yin W, Qian S. Delivery of cisplatin and resiquimod in nanomicelles for the chemoimmunotherapy of ovarian cancer. Cancer Nanotechnology. (2022) 13(1):1–17. doi: 10.1186/s12645-021-00094-8
154. Liu X, Feng Y, Xu G, Chen Y, Luo Y, Song J, et al. MAPK-targeted drug delivered by a pH-sensitive MSNP nanocarrier synergizes with PD-1 blockade in melanoma without T-cell suppression. Advanced Functional Materials (2019) 29(12):1806916. doi: 10.1002/adfm.201806916
155. Peng F, Su Y, Zhong Y, Fan C, Lee S-T, He Y. Silicon nanomaterials platform for bioimaging, biosensing, and cancer therapy. Accounts Chem Res (2014) 47(2):612–23. doi: 10.1021/ar400221g
156. Yang Y, Zhang M, Song H, Yu C. Silica-based nanoparticles for biomedical applications: from nanocarriers to biomodulators. Accounts Chem Res (2020) 53(8):1545–56. doi: 10.1021/acs.accounts.0c00280
157. Kim B, Sun S, Varner JA, Howell SB, Ruoslahti E, Sailor MJ. Securing the payload, finding the cell, and avoiding the endosome: Peptide-targeted, fusogenic porous silicon nanoparticles for delivery of siRNA. Advanced Materials. (2019) 31(35):1902952. doi: 10.1002/adma.201902952
158. Alavi M, Kamarasu P, McClements DJ, Moore MD. Metal and metal oxide-based antiviral nanoparticles: Properties, mechanisms of action, and applications. Adv Colloid Interface Sci (2022) 306:102726. doi: 10.1016/j.cis.2022.102726
159. Evans ER, Bugga P, Asthana V, Drezek R. Metallic nanoparticles for cancer immunotherapy. Materials Today (2018) 21(6):673–85. doi: 10.1016/j.mattod.2017.11.022
160. Zhao Y, Liu X, Liu X, Yu J, Bai X, Wu X, et al. Combination of phototherapy with immune checkpoint blockade: Theory and practice in cancer. Front Immunol (2022) 13:955920. doi: 10.3389/fimmu.2022.955920
161. Zhao Y, Jiang X, Liu X, Liu X, Liu Z, Liu X. Application of photo-responsive metal-organic framework in cancer therapy and bioimaging. Front bioengineering Biotechnol (2022) 10:1031986. doi: 10.3389/fbioe.2022.1031986
162. Xiong J, Wu M, Chen J, Liu Y, Chen Y, Fan G, et al. Cancer-erythrocyte hybrid membrane-camouflaged magnetic nanoparticles with enhanced photothermal-immunotherapy for ovarian cancer. ACS Nano. (2021) 15(12):19756–70. doi: 10.1021/acsnano.1c07180
163. Conde J, Bao C, Tan Y, Cui D, Edelman ER, Azevedo HS, et al. Dual targeted immunotherapy via in vivo delivery of biohybrid RNAi-peptide nanoparticles to tumor-associated macrophages and cancer cells. Advanced Funct materials. (2015) 25(27):4183–94. doi: 10.1002/adfm.201501283
164. Sargazi S, Simge E, Mobashar A, Gelen SS, Rahdar A, Ebrahimi N, et al. Aptamer-conjugated carbon-based nanomaterials for cancer and bacteria theranostics: A review. Chemico-Biological Interactions. (2022) 361:109964. doi: 10.1016/j.cbi.2022.109964
165. Li Y, Li X, Doughty A, West C, Wang L, Zhou F, et al. Phototherapy using immunologically modified carbon nanotubes to potentiate checkpoint blockade for metastatic breast cancer. Nanomedicine: Nanotechnology Biol Med (2019) 18:44–53. doi: 10.1016/j.nano.2019.02.009
166. Hassan HA, Diebold SS, Smyth LA, Walters AA, Lombardi G, Al-Jamal KT. Application of carbon nanotubes in cancer vaccines: Achievements, challenges and chances. J Controlled release. (2019) 297:79–90. doi: 10.1016/j.jconrel.2019.01.017
167. McKernan P, Virani NA, Faria GN, Karch CG, Prada Silvy R, Resasco DE, et al. Targeted single-walled carbon nanotubes for photothermal therapy combined with immune checkpoint inhibition for the treatment of metastatic breast cancer. Nanoscale Res letters. (2021) 16(1):1–9. doi: 10.1186/s11671-020-03459-x
168. Luo J, Wang X, Shi Z, Zeng Y, He L, Cao J, et al. Enhancement of antitumor immunotherapy using mitochondria-targeted cancer cell membrane-biomimetic MOF-mediated sonodynamic therapy and checkpoint blockade immunotherapy. J Nanobiotechnology. (2022) 20(1):1–17. doi: 10.1186/s12951-022-01453-2
169. Zhong X, Zhang Y, Tan L, Zheng T, Hou Y, Hong X, et al. An aluminum adjuvant-integrated nano-MOF as antigen delivery system to induce strong humoral and cellular immune responses. J Controlled Release. (2019) 300:81–92. doi: 10.1016/j.jconrel.2019.02.035
170. Buchman JT, Hudson-Smith NV, Landy KM, Haynes CL. Understanding nanoparticle toxicity mechanisms to inform redesign strategies to reduce environmental impact. Accounts Chem Res (2019) 52(6):1632–42. doi: 10.1021/acs.accounts.9b00053
171. Xu L, Yang J, Xue B, Zhang C, Shi L, Wu C, et al. Molecular insights for the biological interactions between polyethylene glycol and cells. Biomaterials. (2017) 147:1–13. doi: 10.1016/j.biomaterials.2017.09.002
172. Zi Y, Yang K, He J, Wu Z, Liu J, Zhang W. Strategies to enhance drug delivery to solid tumors by harnessing the EPR effects and alternative targeting mechanisms. Advanced Drug Delivery Rev (2022) 188:114449. doi: 10.1016/j.addr.2022.114449
173. Wheeler KE, Chetwynd AJ, Fahy KM, Hong BS, Tochihuitl JA, Foster LA, et al. Environmental dimensions of the protein corona. Nat Nanotechnology. (2021) 16(6):617–29. doi: 10.1038/s41565-021-00924-1
174. Liu Y, Yang G, Jin S, Xu L, Zhao CX. Development of high-drug-loading nanoparticles. ChemPlusChem. (2020) 85(9):2143–57. doi: 10.1002/cplu.202000496
Keywords: immunotherapy, ovarian cancer, tumor immune microenvironment, nanoparticles, drug delivery system
Citation: Xu T, Liu Z, Huang L, Jing J and Liu X (2022) Modulating the tumor immune microenvironment with nanoparticles: A sword for improving the efficiency of ovarian cancer immunotherapy. Front. Immunol. 13:1057850. doi: 10.3389/fimmu.2022.1057850
Received: 30 September 2022; Accepted: 21 November 2022;
Published: 01 December 2022.
Edited by:
Haitao Wang, Center for Cancer Research (NIH), United StatesReviewed by:
Eswari Dodagatta-Marri, University of California, San Francisco, United StatesCopyright © 2022 Xu, Liu, Huang, Jing and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaowei Liu, eGlhb3dlaWxpdTMxMkAxNjMuY29t; Jing Jing, ampfemN5QHZpcC4xNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.