 Xiaoqing Xu1,2,3†Yuxi Zhang1,2,3†Zhaobing Pan1,2,3Xiaojing Zhang1,2,3Xiaonan Liu1,2,3
Xiaoqing Xu1,2,3†Yuxi Zhang1,2,3†Zhaobing Pan1,2,3Xiaojing Zhang1,2,3Xiaonan Liu1,2,3 Lili Tang1,2,3Xiaoguang Zhang4*
Lili Tang1,2,3Xiaoguang Zhang4* Fusheng Zhou1,2,3,5*Hui Cheng1,2,3*
Fusheng Zhou1,2,3,5*Hui Cheng1,2,3*- 1Department of Dermatology, the First Affiliated Hospital, Anhui Medical University, Hefei, Anhui, China
- 2Institute of Dermatology, Anhui Medical University, Hefei, Anhui, China
- 3Key Laboratory of Dermatology (Anhui Medical University), Ministry of Education, Hefei, Anhui, China
- 4Department of Dermatology, The Second Hospital of Hebei Medical University, Shijiazhuang, Hebei, China
- 5Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Hefei, Anhui, China
Introduction: Munro's microabscess is a typical pathological feature in the early psoriatic lesion, mainly characterized by the accumulation of neutrophils in the epidermis. DNA methylation microenvironment of Munro's microabscess and the crosstalk with transcription and its effect on neutrophils have not yet been revealed.
Methods: Performed genome-wide DNA methylation analysis and further differential methylation analysis of psoriatic skin lesions with and without Munro's microabscess from two batch samples consisting of 114 former samples in the discovery stage and 21 newly-collected samples in the validation stage. Utilized GO, MEME, and other tools to conduct downstream analysis on differentially methylated sites (DMSs). Correlation analysis of methylation level and transcriptome data was also conducted.
Results: We observed 647 overlapping DMSs associated with Munro's microabscess. Subsequently, GO pathway analysis revealed that DNA methylation might affect the physical properties associated with skin cells through focal adhesion and cellsubstrate junction and was likely to recruit neutrophils in the epidermis. Via the MEME tool, used to investigate the possible binding transcription factors (TFs) of 20 motifs around the 647 DMSs, it was found that DNA methylation regulated the binding of AP1 family members and the recruitment of neutrophils in the epidermis through the TGF-beta pathway and the TH17 pathway. Meanwhile, combined with our earlier transcriptome data, we found DNA methylation would regulate the expressions of CFDP, SIRT6, SMG6, TRAPPC9, HSD17B7, and KIAA0415, indicating these genes would potentially promote the process of Munro's microabscess.
Discussion: In conclusion, DNA methylation may affect the course of psoriasis by regulating the progression of Munro's microabscess in psoriatic skin lesions.
Introduction
Psoriasis is an immune-mediated, chronic, and inflammatory skin disease characterized by symmetrically well-defined erythema, covered with silvery scales, involving the elbows, knees, torso, and scalp (1, 2). Typical histopathological traits of psoriasis include hyperkeratosis with parakeratosis, immune cell infiltration, Munro’s microabscess, acanthosis thickening, vascular dilatation congestion, elongation of rete pegs, and granulosa thinning (1). In our previous studies, we explored the transcriptome (3) and chromatin accessibility patterns (4) in psoriatic lesions, and further data mining suggested that each pathological feature might be affected by both transcription and chromatin accessibilities, thus supporting the notion that these kinds of molecular changes would be involved in modulating the epidermal microenvironment (5). Munro’s microabscess is a characteristic pathological hallmark of early psoriasis, with inflammation encompassing polymorphonuclear leukocytes and forming within the skin’s epidermal layer, which can further trigger the following immune response in the surrounding microenvironment (6–8). Researchers have demonstrated that IL-1 signaling is vital for neutrophil recruitment and controlling chemokine expression in Munro’s microabscess formation in mouse psoriasiform imiquimod-induced skin inflammation (6). The pathogenesis of psoriasis is a complex cascade reaction. Therefore, it is necessary to investigate the emergence and regulation of the local microenvironment of Munro’s microabscess.
Genomic DNA methylation underlies the vast majority of biological inheritance. Healthy monozygotic twins of the same sex and age and nearly identical genomes exhibit indistinguishable methylomes at a young age and, over time, experience disparities and methylation differences through exposure to different environmental factors (9). Aberrant DNA methylation is associated with various inflammatory diseases and malignant tumors (10–12). Meanwhile, antitumor epigenetic drugs targeting DNA methylation have been developed and serve as a hotspot in the cancer treatment community (13).
As a systemic immune disorder, the occurrence and development of psoriasis involve both genetic and environmental factors (1). Furthermore, epigenetic modifications, such as DNA methylation, microRNA, chromatin remodeling, and other forms of non-DNA sequence changes, mediate the crosstalk between genetics and environmental factors (9, 10, 14). DNA methylation is an essential component of epigenetics and is associated with gene expression repression (15). All of the modifications in the genome would regulate the gene expression and further bring the corresponding phenotype. Earlier, we performed genome-wide DNA methylation for psoriatic peripheral blood mononuclear cell samples and psoriatic lesions, identified nine differentially methylated sites (DMSs) strongly related to psoriasis, and interrogated transcriptome to verify the expression of DMS-related genes (16). In the following research, we found differences in chromatin accessibility and gene expression in different pathological changes of psoriasis (5). At the same time, a study shows that DNA methylation analysis combined with gene expression displayed a discordant difference in monozygotic twins psoriasis (17). Another joint analysis study explored the DNA methylation patterns of local microenvironment and their functional regulation of gene expression among psoriatic lesions, non-lesions and healthy controls, which investigated skin biomarkers for psoriasis onset (18). Meanwhile, the cardinal pathological features of psoriasis are likely regulated by DNA methylation (19), but so far, the mechanism behind psoriasis pathological changes has not been fully studied by DNA methylation combined transcriptome. Therefore, the characteristics of methylation in the key pathological changes of psoriasis and its relationship with gene expression are worthy of further study.
To explore the methylation profiles of Munro’s microabscess, we first performed data mining of previously 114 psoriatic lesions with and without Munro’s microabscess. In order to confirm these results, we further collected 21 psoriatic tissue samples, including 4 with and 17 without microabscess, and assayed them using the same genome-wide methylation arrays. A total of 647 overlapped DMSs were subjected to further analysis, and we integrated and analyzed the DNA methylation and transcriptome. Overall, we explored the regulatory mechanism of methylation in Munro’s microabscess, a key pathological change in psoriatic skin lesions, and possibly a therapeutic target in psoriasis.
Materials and methods
Subjects
Skin tissue samples were collected from the Institute of Dermatology, Anhui Medical University. DNA methylation data from 114 samples and transcriptome data from 20 samples, and their detailed clinical features had been described previously (3, 16). The re-collected 21 DNA methylation samples follow the same procedures. All the patients in the study were included according to the following criteria: (I) more than one demarcated, erythematous, scaly lesion identified by at least two dermatologists; (II) the identification of each lesion by clinical histopathology; (III) before the skin biopsy, no systemic antipsoriatic therapy for two weeks; and (IV) before the biopsy, no topical antipsoriatic treatment within one week. Even if these standards are fully followed, it is difficult to avoid the long-term effects of previous drug use on skin lesions. Without other secondary lesions, skin biopsies were performed at the edge of the psoriatic area. All lesions were quickly frozen in liquid nitrogen immediately after removal. After 21 psoriatic lesions were collected, DNA was immediately isolated using the FlexiGene DNA Kit (Qiagen, Hilden, Germany).
Histopathological analysis
The collected skin tissue was fixed, embedded in paraffin, and sliced, and then the overall histopathological abnormalities were evaluated by Hematoxylin–Eosin staining, including Munro’s microabscess, a characteristic feature of psoriasis. Finally, the differential methylation site analysis of samples with or without Munro’s microabscess was performed.
Methylation analysis and identifying Munro’s microabscess-related differentially methylated sites
Genome-wide DNA methylation sequencing processing the DNA samples using the Infinium Human-Methylation450 array (Illumina 450K). The R software was used for bioinformatics analysis. Using the R package ChAMP (http://www.bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html), the locusby-locus nonparametric Wilcoxon ranked test was used to Munro’s microabscess-related differentially methylated sites (20). Differential methylation data were analyzed via Benjamini–Hochberg-adjusted t-tests with a P-value of 0.05. The detailed data processing steps can be found in our previous studies (16). The hypermethylation loci were defined as the methylation levels were higher in Munro’s microabscess group than that in without-Munro’s microabscess group, and vice versa, as hypomethylation.
MEME analysis
We used the MEME suite (https://meme-suite.org/meme/doc/cite.html) to define motifs that can be bound by TFs of DNA sequences around the 20 bp nearby the hypermethylated and hypomethylated loci and further predict TFs.
eFORGE analysis
The eFORGE v2.0 program (http://https://eforge.altiusinstitute.org/) is a web-based tool for analysis and interpretation via EWAS data. eFORGE identifies cell-type-specific regulatory components of a set of EWAS-recognized differentially methylated positions. For the 647 unique DMSs from the overlapping two sets, we assessed the enrichment and depletion of overlap with tissue-specific or cell-type-specific regulatory features, including DNase 1 hypersensitive sites (DHS), all 15 state chromatin marks, and all five H3 histone marks (H3K27me3, H3K4me1, H3K4me3, H3K36me3, H3K9me3) using eFORGE v2.0. The set of 647 DMSs was entered as input in eFORGE and tested separately for enrichment and depletion of overlap with each of the three putative functional elements (DHS, all 15 state chromatin marks, and all five H3 marks) compared to the respective data from consolidated ROADMAP epigenomics. –log10 binomial p-value was used to evaluate the significance. The Benjamini-Yekutieli (BY)-corrected false discovery rate was used to correct multiple independent tests. Enrichment or depletion was considered statistically significant at P < 0.05.
GO and KEGG pathway analysis and transcribed data heatmap plotted
The data were all implemented and plotted using https://www.bioinformatics.com.cn, a free online platform for data analysis and visualization.
Statistical analysis
Differential methylation statistical analyses were performed on the R (version 4.1.1) platform (https://www.R-project.org/). GraphPad Prism, Version 7.04 for Windows, was used to construct a volcano plot. The dmlTest and t-test were used to evaluate the significance. The correlation analysis was performed using SPSS. P < 0.05 was considered statistically significant.
Results
The genome-wide methylation profile of Munro’s microabscess in 114 tissue samples
We previously assayed genome-wide DNA methylation for psoriasis using the Infinium Human-Methylation450 array (Illumina 450K) (16). In that study, the 114 psoriatic skin samples could be subgrouped into 30 with and 84 without Munro’s microabscess. Figure 1A describes the main procedures in the current project. In brief, we extensively assayed the DNA methylation, gene expression, and their integration to reveal the main molecular features of Munro’s microabscess.
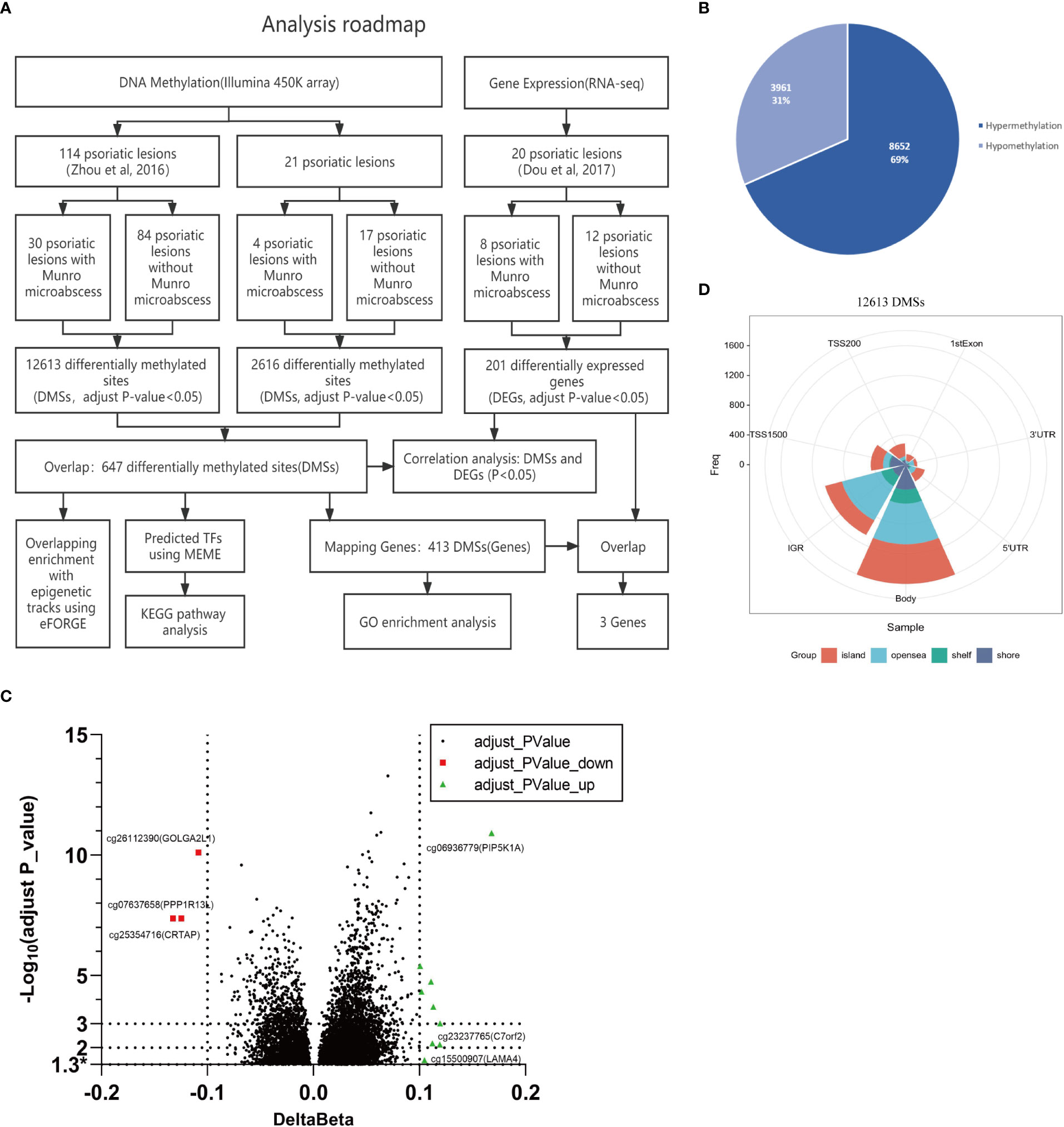
Figure 1 Idea map and differential methylation characteristics of 114 samples with and without Munro’s microabscess. (A) The whole analysis roadmap. (B) The number and proportion of hypermethylated and hypomethylated probes at 12,613 DMSs with or without Munro’s microabscess in 114 samples. (C) Volcano plot using -Log10(adjust P_value) and delta Beta at 12,613 DMSs.* stands adjust P_valve < 0.05. (D) Distribution of 12613 DMSs in CpG regions and on the genome.
Firstly, we evaluated the genome-wide methylation differences between 30 samples with and 84 samples without Munro’s microabscess in psoriatic skin tissue. With a Benjamini–Hochberg-adjusted t-test P-value of 0.05, we found 12,613 DMSs, including 8,652 (69%) hypermethylation loci and 3,961 (31%) hypomethylation loci (Munro’s microabscess minus no Munro’s microabscess, Figure 1B). Additionally, with absolute delta beta difference (ΔBeta) > 0.05, we obtained 993 DMSs, with 833(84%) hypermethylation loci and 160(16%) hypomethylation loci. For the 15 most significantly hypomethylated DMSs, including cg26112390 (ΔBeta=-0.11, adjusted P-value= 7.77*10-11), cg25354716 (ΔBeta=-0.12, adjusted P-value=4.2*10-8), and cg07637658 (ΔBeta=-0.13, adjusted P-value=4.2*10-8), the mapping genes are GOLGA2L1, CRTAP, and PPP1R13L, and for the 15 most remarkably hypermethylated DMSs, including cg06936779 (ΔBeta=0.17, adjusted P-value=1.21*10-11), cg06662428 (ΔBeta=0.12, adjusted P-value=9.89*10-4), and cg23237765 (ΔBeta=0.12, adjusted P-value=7.38*10-3), the mapping genes are PIP5K1A, PAOX, and C7orf20 (Figure 1C; Supplementary Table 1).
We explored the gene contexts of these 12,613 DMSs, 36% (1604/12,613) of which are located on the gene body, followed by IGR with 27% (1074/12,613) (Figure 1D). A similar methylation pattern can be observed for both hypermethylated and hypomethylated loci (Supplementary Figure 1, Supplementary Table 1). These results suggest that there may be some abnormal methylation of the targeted gene body and IGR to regulate the occurrence of Munro’s microabscess in psoriasis patients.
The verification of DMSs in 21 fresh tissue samples
In order to confirm the above findings, we re-collected 21 psoriatic lesions, including 4 with and 17 without Munro’s microabscess, to profile genome-wide DNA methylation using the same methylation array (Figure 1A). In all samples, after filtering the ambiguous and duplicate probes, we detected 345,753 sites. With the same statistical strategy, we obtained 2,616 DMSs, containing 89% (2,317/2616) hypermethylated and 11% (299/2616) hypomethylated sites (Figure 1A, Figure 2A). Most of these DMSs were located on the gene body 43% (1,112/2,616) and IGR 30% (758/2,616) (Figure 2A). By overlapping the 12,613 DMSs generated from the 114 samples, we obtained 647 loci, including 526 (81%) hypermethylated and 121 (19%) hypomethylated loci (Figure 1A). The gene location context and hyper-/hypomethylation status of the 647 DMSs in the two sets are consistent (Figure 2B, C). Meanwhile, 5/15 of the most significant cases of hypomethylation are compatible with 114 psoriatic lesions’ data, with mapping genes including GOLGA2L1, CASKIN2, and HSPA1L; 12/15 are associated with hypermethylation, for which the mapping genes include PIP5K1A, PAOX, and C7orf2 (Figure 2D; Supplementary Table 1).
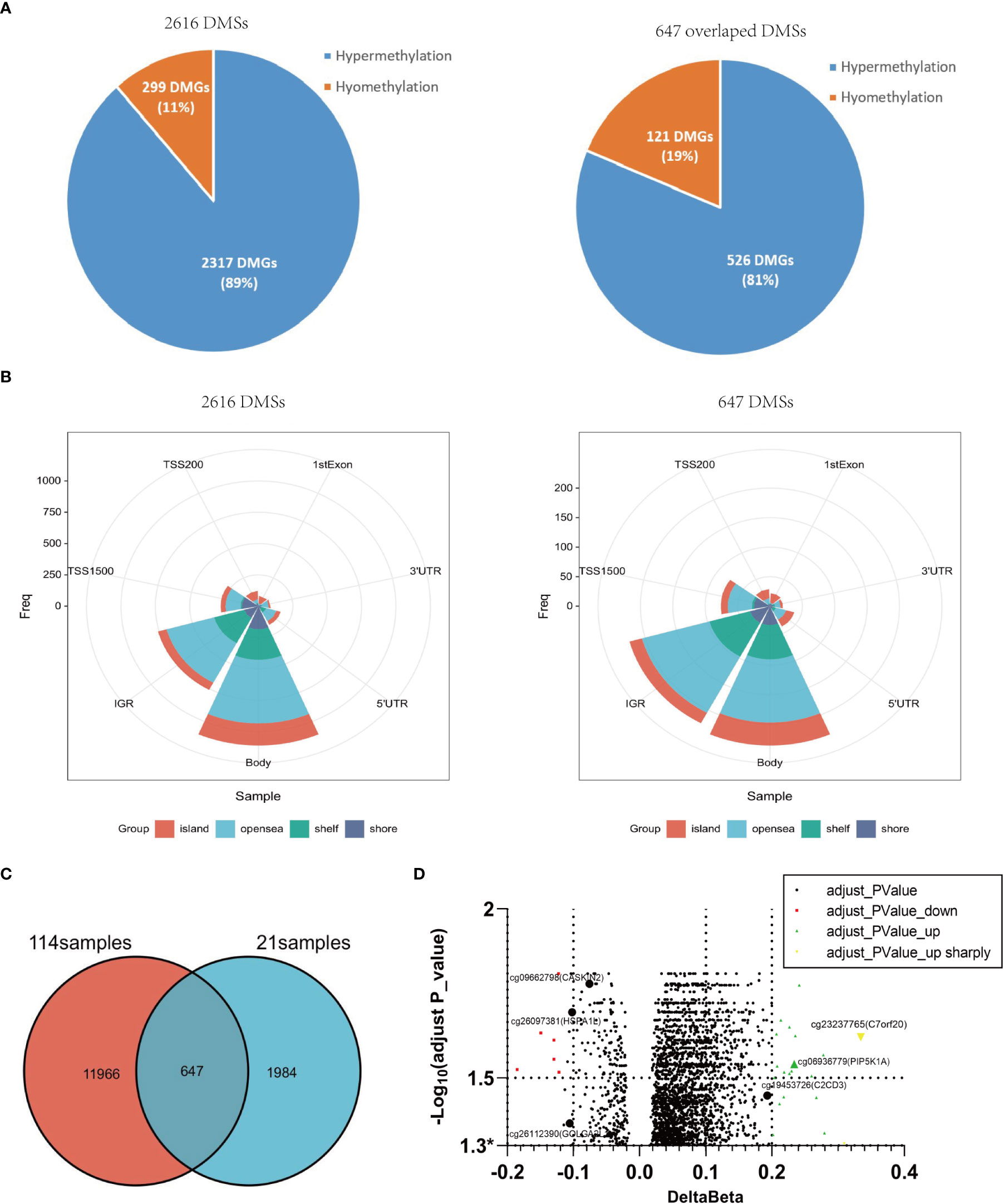
Figure 2 Differential methylation characteristics of 21 samples with and without Munro’s microabscess and overlapping with 114samples. (A): The number, proportion, and distribution of 2,616 DMSs with or without Munro’s microabscess in 21 samples. (B) The overlapped DMSs between 114 samples and 21 samples. (C) The number, proportion, and distribution of 647 overlapped DMSs with or without Munro’s microabscess. (D) Volcano plot using -Log10(adjust P_value) and delta Beta at 2616 DMSs.* represents adjusted P-valve<0.05.
To reveal the potential roles of these loci, we performed GO enrichment analysis on 413 genes that 647 DMSs mapped. Interestingly, 19 genes, such as PIP5K1A, CAV3, and CLASP1, were enriched in both focal adhesion and cell-substrate junctions (Supplementary Table 2). Furthermore, the methylation levels of 2/19 were hypomethylated, while 17 were hypermethylated (Supplementary Table 3). In addition, 13 of 19 sites were positioned within the gene body, and nine sites were in the open sea (Supplementary Table 4). These findings indicate that the 19 genes might chemotactically recruit leukocytes through focal adhesion and cell-substrate junctions, via changes in DNA methylation at the gene body, contributing to the formation of Munro’s microabscess.
Predicted transcription factors on 647 overlapped DMSs
To further define whether DMSs would affect the binding of transcription factors (TFs), we performed Multiple Em for Motif Elicitation (MEME) motif analyses of DNA sequences around the 20 bp nearby the hypermethylated and hypomethylated loci, respectively (21). For the hypermethylated loci, five binding motifs were enriched around the GTGGCTCACG, CCTGTAATC, AGTTCGAGAC, AGCTACTCGG, TCTGTCGCCC (Figure 3). The most binding motif was GTGGCTCACG (P = 2.5*10-141). This motif can be bound by the leucine zipper family, covering FOS, FOSB, JUND, FOSL1, JUN, JUNB, FOSL2, BACH1, and BACH2. In addition, the second significant binding motif was CCTGTAATC (P = 2.7*10-104), which can be bound by TGIF1, TEAD4, TEAD1, TF7L2, and NR2C2 (Figure 3). The other three motifs can be bound by a series of TFs, including BHA15, PRD14, ZN121, SMAD4, BATF, and others (Figure 3). A total of 47 TFs were identified for the five types of binding motifs. Subsequently, further KEGG pathway analysis showed that the 47 TFs were enriched in seven signaling pathways, such as human t-cell leukemia virus one injection, the cell cycle, the TGF-beta signaling pathway, and Th17 cell differentiation (P<0.0001, Supplementary Figure 2). The TGF-beta signaling pathway and Th17 cell differentiation are not only related to the pathogenesis of psoriasis but also the chemotaxis of neutrophils (22–25).
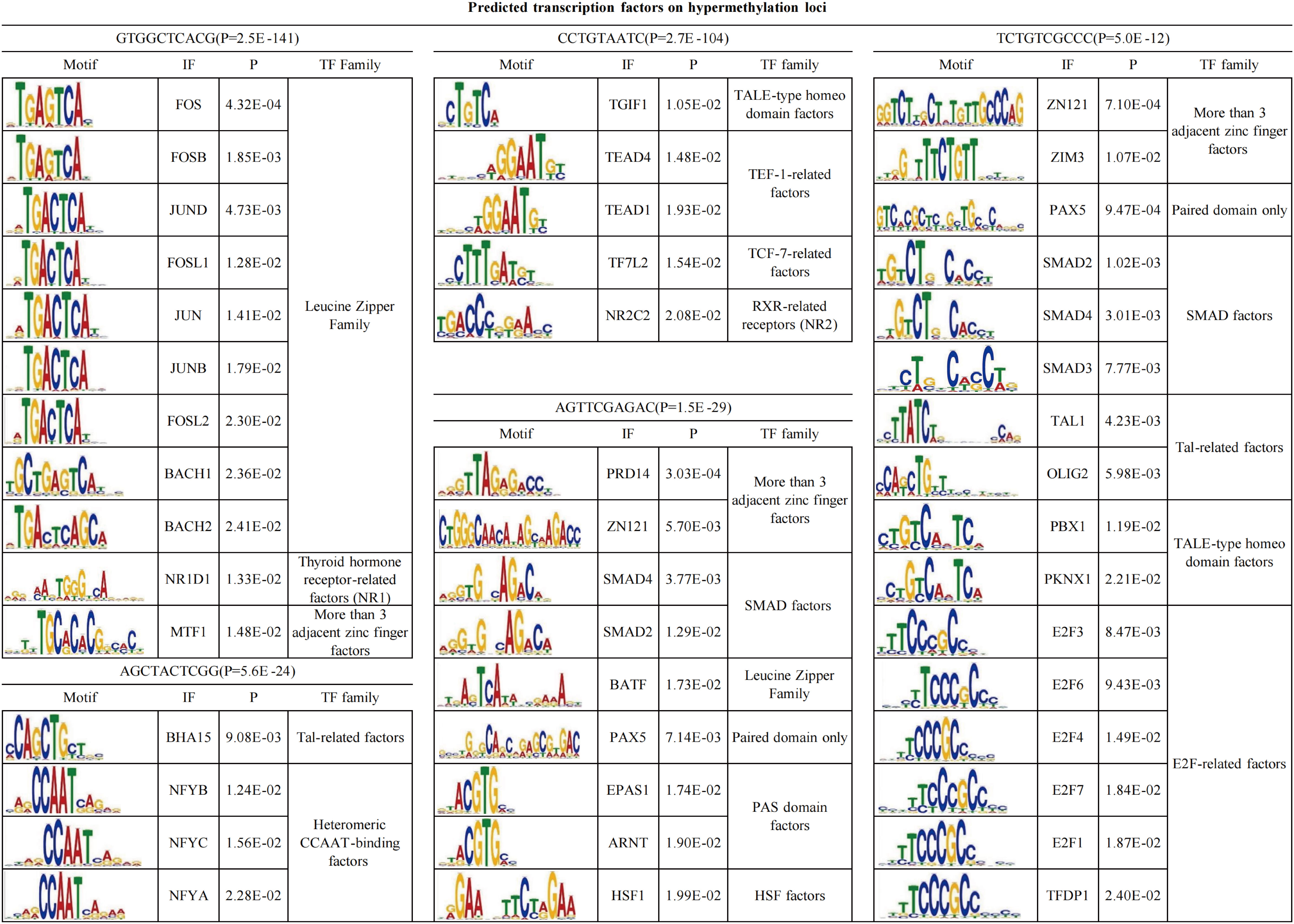
Figure 3 Predicted transcription factors on hypermethylation loci using MEME.
For hypomethylated sites, only one motif, TTYTCCGKYT, was observed. The motif can be bound by E2F-related factors, Ets-related factors, MECP2, IRF, RUNX3, and OLIG2 (Supplementary Figure 3). E2F-related factors, including TFDP1, E2F6, E2F7, E2F1, E2F4, and E2F3, are crucial for the cell cycle, cell fate, and cell differentiation, including those of neutrophils (26, 27). In summary, DMSs would modulate the motif binding of several TFs and thus be involved in cellular biological processes during the accumulation of neutrophils in Munro’s microabscess.
DNA methylation would regulate gene expression in Munro’s microabscess
The potential roles of DNA methylation can be annotated by other types of epigenetic modifications, such as DNaseI hotspots, five histone marks (H3K4me1, H3K4me3, H3K27me3, H3K9me3, and H3K36me3), and 15 chromatin states (Figure 4A). To functionally check whether the 647 DMSs could be annotated with other epigenetic tracks, we introduced the eFORGE algorithm, a useful tool to analyze and interpret cell-type-specific DNA methylation and epigenome data (28). The 1 kb surrounding the selected probes was searched as a proximity filter, and eFORGE utilizes data from the Roadmap Epigenomics, ENCODE, and BLUEPRINT projects, to calculate the enrichment scores and cell types functional annotation by comparing the overlap of DMSs and 1000 random probes with selected epigenetic markers, respectively. We found that some of the 647 DMSs could be annotated by histone modifications (Supplementary Figure 4A), DNaseI hotspots (Supplementary Figure 4B), and/or 15 chromatin states (Figure 4A). For example, the DMSs showed significant overlapping with H3K36me3 in monocytes from PBMC and primary T from cord blood, and with H3k9me3 in primary hematopoietic stem cells G-CSF-mobilization (Supplementary Figure 4A). The tracks of 15 chromatin states revealed a high enrichment of both blood and skin (Figure 4A). Moreover, the DMS signals are concentrated in various lymphocytes and neutrophils, potentially impacting gene transcription processes. At the same time, these DMSs were related to keratinocytes and other skin parenchyma cells (Figure 4A).
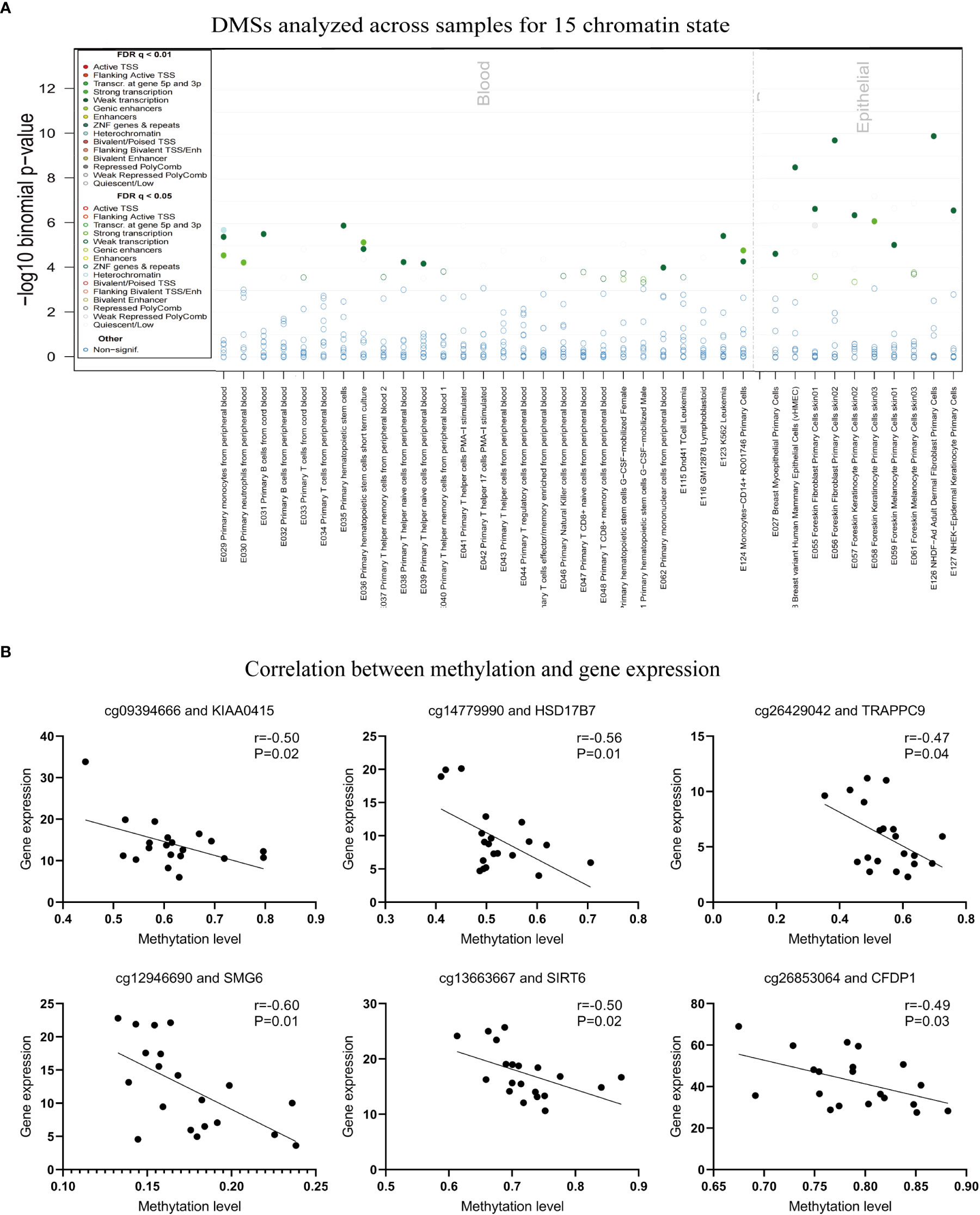
Figure 4 DNA methylation can be annotated by other types of epigenetic modifications via eForge, and the correlation between methylation and transcript expression in DMSs. (A) The Annotation of 15 chromatin states for 647 DMSs in blood and epithelial cells. (B) Correlation between methylation and gene expression.
As a critical regulator of cell biology, DNA methylation is of great importance for regulating gene transcription (12). Based on our RNA-seq dataset previously generated from the same samples, we extracted the gene expression levels and compared the expression in patients in the presence and absence of Munro’s microabscess (3). Then, we obtained 201 differentially expressed genes (DEGs, t-test, p<0.05, Figure 1A). Based on these DEGs, 95% (20/21) of samples could be clearly separated, except for one sample without- been wrongly grouped into with-Munro’s microabscess (Supplementary Figure 5A). Additionally, the following genes showed the most remarkably increased expression in Munro’s microabscess: COL5A2, CDCA7L, and DPP3 (Supplementary Figure 5B). In contrast, the most remarkably decreased expression genes in Munro’s microabscess were DYNLRB2, C9orf103, and RFESD (Supplementary Figure 5C). However, When 201 DEGs overlapped with 413 genes mapped by the 647 DMSs, we obtained only three genes, PEMT, BAI2, and ACACA, that were implicated in both methylation and expression analysis (Supplementary Figure 5D). Compared with the group without Munro’s microabscess, the PEMT gene expression was reduced, while the methylation level of the corresponding DMS was increased.
To further explore the role of these specific DMSs in gene expression, we correlated the level of methylation and gene expression level and found that cg26853064, cg13663667, cg12946690, cg26429042, cg14779990, and cg09394666 were negatively correlated with the expression of the mapping genes CFDP1, SIRT6, SMG6, TRAPPC9, HSD17B7, and KIAA0415 (Figure 4B; Supplementary Table 3). In summary, these findings indicate that DNA methylation would potentially affect the formation of Munro’s microabscess and is probably closely related to neutrophils.
Discussion
The onset of psoriasis is associated with genetic, immune, and environmental factors. The environment can interact with the genome through epigenetic modifications, including DNA methylation, affecting gene biological behavior and phenotype (15, 29). DNA methylation is involved in the pathogenesis of psoriasis, and the levels of 5-mC in psoriasis lesions are positively correlated with disease severity (15, 30, 31). Different pathological features of psoriasis tissue have distinguishing methylation patterns (19). Neutrophilic infiltrations that appear with Munro’s microabscess are hallmarks of the early psoriatic tissue reaction (1, 6). Our data show that Munro’s-related DMSs were more likely to appear in the gene body. DNA methylation at promoters is implicated in gene silencing, while the function of the gene body remains unclear. Studies have shown that Dnmt3b-dependent DNA methylation in the gene body prevents abnormal transcription initiation events and ensures mRNA transcription initiation fidelity (32, 33). Loss of transcription initiation fidelity may affect multiple biological processes, impairing molecular mechanisms such as gene expression regulation, miRNA targeting, and truncated protein production (33). A study showed abnormal RNA transcription in cancer is closely connected with gene body DNA methylation (34). To some extent, there is a positive correlation between DNA methylation and gene expression (35). Therefore, we speculate that Munro’s microabscess in early psoriasis may be linked with abnormal DNA methylation in the gene body.
Only 2,616 DMSs were obtained from 21 validation samples, far less than 12,613 DMSs in 114 samples. We reasoned this discrepancy would be due to insufficient sample size, as we saw only 4 Munro’s microabscess samples in the validation stage; this might dramatically decrease the statistical power to identify DMSs. And the functions of the genes matched by most significant hypomethylated DMSs are related to the composition of the intrinsic components of the cell, such as GOLGA2L1 is involved in Golgi organization and spindle assembly, CASKIN2 is a scaffold protein, HSPA1L lead to mitochondrial dysfunction (36, 37). In the meantime, PIP5K1A is the most significant hypermethylated DMSs and is related to the fundamental processes for cell migration, invasion, and metastasis in cancer (38). Those genes indicate that Munro’s microabscess skin cells may be abnormalities in cellular structure and function that lead to disease progression.
The GO enrichment analysis for genes corresponding to 647 DMSs indicated that 19 genes were enriched in two cellular components: focal adhesion and cell-substrate junctions. Interestingly, an abnormal condition regarding focal adhesion and cell-substrate junctions causes their reduced maturation and faster turnover and further affects the induction of cell adhesion, morphology, migration, and differentiation (39). We speculate that DNA methylation is mainly involved in the process of focal adhesion and cell-substrate function, and via the changes in biomechanics in skin cells, it favors neutrophil recruitment in the epidermis and further leads to the formation of Munro’s microabscess.
Then, the investigation of TFs that bind to 20 motifs around the hypermethylation probes produced five meaningful binding motifs. Interestingly, the leucine zipper family, including most AP1 family members, can bind to the most significant motif. A study using methylated binding microarrays showed that DNA methylation inhibited the binding of the leucine zipper family TFs (40). Deletion of AP1 family members, such as FOS, FOSB, and JUND, promotes inflammation characterized by neutrophil infiltration (41, 42). Accordingly, the most prominent motifs enriched at DNA hypermethylation sites in psoriatic Munro’s microabscess, favoring the promotion of the recruitment of neutrophils predominantly, can be bound by AP1-family TFs. Consequently, the results for all 47 TFs binding to hypermethylation loci showed seven signaling pathways. The human t-cell leukemia virus 1 injection pathway has the largest number of TFs enriched, and this suggests that there is a common partial mechanism or an unknown interaction between virus injection and Munro’s microabscess formed in psoriasis. The regulation of cell cycle signaling undergoes a transformation in the neutrophil population in the stress response status (43), as does the appearance of Munro’s microabscess. The osteoclast differentiation signaling pathway and IL-17 signaling pathway have a strong correlation with psoriatic arthritis (44–47). Interestingly, AP1 family members, FOS, FOSB, JUND, FOSL1, JUN, JUNB, and FOSL2 are enriched in osteoclast differentiation, while the first 5 TFs are also enriched in the IL-17 signaling pathway. In other words, the neutrophil aggregation in psoriasis with Munro’s microabscess may be combined with AP1 binding suppression, resulting from the DNA hypermethylation of AP1 binding sites. We hypothesize that DNA hypermethylation mediates the binding of the AP1 family to further contribute to Munro’s microabscess, and it may have a specific correlation with psoriatic arthritis.
At the same time, TGIF1, SMAD4, SMAD2, SMAD3, E2F4, and TFDP1 binding to the hypermethylation site motif is related to the TGF-beta signaling pathway, which can directly regulate the biological activities of neutrophils (23, 48). In addition, interestingly, the absence or inhibition of SMAD4 activity accounts for increased neutrophil infiltration (49). Therefore, hypermethylation of the SMAD-binding motif inhibits its binding to DNA and promotes neutrophil infiltration in the epidermis by regulating the TGF signaling pathway.
Various epigenetic modifications are crucial to regulating psoriasis pathogenesis—not only DNA methylation but also chromatin modification, histone modification, etc. (50). eFORGE is a web-based tool used to analyze and enrich overlapping with DNase I hypersensitive sites, five histone marks, and 15 chromatin states to determine the cell-type-specific regulatory component of a set of EWAS-identified differentially methylated positions (51). We performed eFORGE to overlap the 1 kb region around 647 DMSs and found some remarkable functional enrichment related to the chromatin state and histone modification. This suggests that Munro’s microabscess has an altered chromatin status. The signal of histone modification and chromatin state enrichment showed the functional enrichment of blood neutrophil-related signals, suggesting that neutrophils play a vital role in the onset of Munro’s microabscess. Histone modification also indicates the blood’s enrichment of monocytes and T-cell signals. Therefore, we speculate that interactions between various inflammatory cells may exist during Munro’s microabscess; there may also be a repetitive methylation profile in activating these inflammatory cells.
Interestingly, by comparing the transcriptome data from the same batch of samples with the 647 DMSs for GO pathway analysis, it was found that the enriched pathways, focal adhesion, and cell-substrate junction signaling pathway were consistent with the 647 DMSs. This shows the reliability of our data. Meanwhile, correlation analysis between DNA methylation levels and gene expression showed that six probes and their mapping genes’ expression have a meaningful negative correlation. It is reported that the knockdown of SIRT6 refreshes the corresponding recruitment of macrophages and neutrophils (52). A study found that circ-SMG6 may deteriorate myocardial I/R injury by promoting neutrophil recruitment (53). Hypermethylation levels of the cg26853064 and cg12946660 probes matched to SIRT6 and SMG6, repressed gene expression, and were beneficial for neutrophil recruitment. The gene function of HSD17B7 is controlled by altering epigenetic gene silencing in reverting cholesterol auxotrophy (54). TRAPPC9-related CPG methylation correlates with breastfeeding and early-life growth trajectories (55). The area near KIAA0415 is differentially methylated in the orofacial cleft (56). However, there is not enough evidence for CFDP1, TRAPPC9, HSD17B7, and KIAA0415’s effects on psoriasis and neutrophils. Based on the available data, we can only speculate that the DNA methylation of these genes affects their expressionlevels and may regulate psoriatic Munro’s microabscess development.
In conclusion, this study defines a unique pattern of DNA methylation in the development of psoriatic Munro’s microabscess, characterized by changes in gene body methylation. It thus provides insight into possible DNA methylation within the local microenvironment of skin lesions in psoriasis onset. We also explored the possibility that the hypermethylation of AP1 family members and other transcription factor binding regions affects their binding on neutrophils through TGF-beta, TL-17, and other signaling pathways, thereby leading to the occurrence of Munro’s microabscess. The effects of other epigenetic modifications on 647 DMSs were excluded. At the same time, combined with the transcriptome data of psoriatic skin lesions from the same samples, it was explained that changes in methylation levels further regulate gene expression to affect the occurrence of Munro’s microabscess. The epigenetic changes in the local microenvironment of psoriasis skin lesions affect related signaling pathways to regulate the biological behavior of corresponding cells and the expression of associated genes, to then influence the occurrence and development of psoriasis pathology, and these results provide new insights into local, specific treatment for psoriasis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by The Institutional Review Board and local Ethics Committee of the First Affiliated Hospital of Anhui Medical University. The patients/participants provided their written informed consent to participate in this study.
Author contributions
HC, FZ, XX, YZ, and XGZ, carried out the design and bioinformatics analysis of this study. Samples were collected by ZP, XJZ, XL, and LT. XX and YZ writing the original draft preparation. HC, FZ, and XGZ reviewed and revised this manuscript. All authors have read and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Anhui colleges (KJ2021A0286); Anhui Provincial Natural Science Foundation for Distinguished Young Scholars (2208085J46); Fund Project of National Natural Science Foundation of China (31671307); the Youth Fund Project of National Natural Science Foundation of China (82103723).
Acknowledgments
We are very grateful to all the volunteer subjects who participated in the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1057839/full#supplementary-material
References
1. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet (2021) 397(10281):1301–15. doi: 10.1016/S0140-6736(20)32549-6
2. Griffiths CEM, Barker JNWN. Pathogenesis and clinical features of psoriasis. Lancet (2007) 370(9583):263–71. doi: 10.1016/S0140-6736(07)61128-3
3. Dou J, Zhang L, Xie X, Ye L, Yang C, Wen L, et al. Integrative analyses reveal biological pathways and key genes in psoriasis. Br J Dermatol (2017) 177(5):1349–57. doi: 10.1111/bjd.15682
4. Tang L, Wang M, Shen C, Wen L, Li M, Wang D, et al. Assay for transposase-accessible chromatin using sequencing analysis reveals a widespread increase in chromatin accessibility in psoriasis. J Invest Dermatol (2021) 141(7):1745–1753. doi: 10.1016/j.jid.2020.12.031
5. Xu X, Tang X, Zhang Y, Pan Z, Wang Q, Tang L, et al. Chromatin accessibility and transcriptome integrative analysis revealed AP-1-mediated genes potentially modulate histopathology features in psoriasis. Clin Epigenetics (2022) 14(1):38. doi: 10.1186/s13148-022-01250-6
6. Uribe-Herranz M, Lian LH, Hooper KM, Milora KA, Jensen LE. IL-1R1 signaling facilitates munro's microabscess formation in psoriasiform imiquimod-induced skin inflammation. J Invest Dermatol (2013) 133(6):1541–9. doi: 10.1038/jid.2012.512
7. Steffen C. William John Munro And munro's abscess, and Franz kogoj and kogoj's spongiform pustule. Am J Dermatopathol (2002) 24(4):364–8. doi: 10.1097/00000372-200208000-00016
8. Christophers E, Metzler G, Röcken M. Bimodal immune activation in psoriasis. Br J Dermatol (2014) 170(1):59–65. doi: 10.1111/bjd.12631
9. Planterose Jimenez B, Liu F, Caliebe A, Montiel Gonzalez D, Bell JT, Kayser M, et al. Equivalent DNA methylation variation between monozygotic co-twins and unrelated individuals reveals universal epigenetic inter-individual dissimilarity. Genome Biol (2021) 22(1):18. doi: 10.1186/s13059-020-02223-9
10. Surace AEA, Hedrich CM. The role of epigenetics in Autoimmune/Inflammatory disease. Front Immunol (2019) 10:1525. doi: 10.3389/fimmu.2019.01525
11. Mervis JS, McGee JS. DNA Methylation and inflammatory skin diseases. Arch Dermatol Res (2020) 312(7):461–6. doi: 10.1007/s00403-019-02005-9
12. Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet (2021) 37(11):1012–27. doi: 10.1016/j.tig.2021.05.002
13. Yang B, Wang JQ, Tan Y, Yuan R, Chen ZS, Zou C. RNA Methylation and cancer treatment. Pharmacol Res (2021) 174:105937. doi: 10.1016/j.phrs.2021.105937
14. Boskovic A, Rando OJ. Transgenerational epigenetic inheritance. Annu Rev Genet (2018) 52:21–41. doi: 10.1146/annurev-genet-120417-031404
15. Luo Y, Qu K, Kuai L, Ru Y, Huang K, Yan X, et al. Epigenetics in psoriasis: perspective of DNA methylation. Mol Genet Genomics (2021) 296(5):1027–40. doi: 10.1007/s00438-021-01804-y
16. Zhou F, Wang W, Shen C, Li H, Zuo X, Zheng X, et al. Epigenome-wide association analysis identified nine skin DNA methylation loci for psoriasis. J Invest Dermatol (2016) 136(4):779–87. doi: 10.1016/j.jid.2015.12.029
17. Gervin K, Vigeland MD, Mattingsdal M, Hammero M, Nygard H, Olsen AO, et al. DNA Methylation and gene expression changes in monozygotic twins discordant for psoriasis: identification of epigenetically dysregulated genes. PloS Genet (2012) 8(1):e1002454. doi: 10.1371/journal.pgen.1002454
18. Verma D, Ekman AK, Bivik Eding C, Enerbäck C. Genome-wide DNA methylation profiling identifies differential methylation in uninvolved psoriatic epidermis. J Invest Dermatol (2018) 138(5):1088–93. doi: 10.1016/j.jid.2017.11.036
19. Chandra A, Senapati S, Roy S, Chatterjee G, Chatterjee R. Epigenome-wide DNA methylation regulates cardinal pathological features of psoriasis. Clin Epigenetics (2018) 10(1):108. doi: 10.1186/s13148-018-0541-9
20. Wang D, Yan L, Hu Q, Sucheston LE, Higgins MJ, Ambrosone CB, et al. IMA: an r package for high-throughput analysis of illumina's 450K infinium methylation data. Bioinformatics (2012) 28(5):729–30. doi: 10.1093/bioinformatics/bts013
21. Bailey TL, Johnson J, Grant CE, Noble WS. The MEME suite. Nucleic Acids Res (2015) 43(W1):W39–49. doi: 10.1093/nar/gkv416
23. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
24. Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol (2014) 32:227–55. doi: 10.1146/annurev-immunol-032713-120225
25. Minns D, Smith KJ, Alessandrini V, Hardisty G, Melrose L, Jackson-Jones L, et al. The neutrophil antimicrobial peptide cathelicidin promotes Th17 differentiation. Nat Commun (2021) 12(1):1285. doi: 10.1038/s41467-021-21533-5
26. Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer (2019) 19(6):326–38. doi: 10.1038/s41568-019-0143-7
27. Li L, Wang S, Zhang Y, Pan J. The E2F transcription factor 2: What do we know? Biosci Trends (2021) 15(2):83–92. doi: 10.5582/bst.2021.01072
28. Breeze CE, Reynolds AP, van Dongen J, Dunham I, Lazar J, Neph S, et al. eFORGE v2.0: updated analysis of cell type-specific signal in epigenomic data. Bioinf (2019) 35(22):4767–9. doi: 10.1093/bioinformatics/btz456
29. Moore LD, Le T, Fan G. DNA Methylation and its basic function. Neuropsychopharmacology (2013) 38(1):23–38. doi: 10.1038/npp
30. Dopytalska K, Ciechanowicz P, Wiszniewski K, Szymanska E, Walecka I. The role of epigenetic factors in psoriasis. Int J Mol Sci (2021) 22(17):9294. doi: 10.3390/ijms22179294
31. Zhang P, Su Y, Chen H, Zhao M, Lu Q. Abnormal DNA methylation in skin lesions and PBMCs of patients with psoriasis vulgaris. J Dermatol Sci (2010) 60(1):40–2. doi: 10.1016/j.jdermsci
32. Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature (2017) 543(7643):72–7. doi: 10.1038/nature21373
33. Teissandier A, Bourc'his D. Gene body DNA methylation conspires with H3K36me3 to preclude aberrant transcription. EMBO J (2017) 36(11):1471–3. doi: 10.15252/embj.201796812
34. Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell (2014) 26(4):577–90. doi: 10.1016/j.ccr.2014.07.028
35. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet (2012) 13(7):484–92. doi: 10.1038/nrg3230
36. Lee JH, Yoon YM, Song KH, Noh H, Lee SH. Melatonin suppresses senescence-derived mitochondrial dysfunction in mesenchymal stem cells via the HSPA1L-mitophagy pathway. Aging Cell (2020) 19(3):e13111. doi: 10.1111/acel.13111
37. Bencsik N, Pusztai S, Borbély S, Fekete A, Dülk M, Kis V, et al. Dendritic spine morphology and memory formation depend on postsynaptic caskin proteins. Sci Rep (2019) 9(1):16843. doi: 10.1038/s41598-019-53317-9
38. Peng JM, Lin SH, Yu MC, Hsieh SY. CLIC1 recruits PIP5K1A/C to induce cell-matrix adhesions for tumor metastasis. J Clin Invest (2021) 131(1):e133525. doi: 10.1172/JCI133525
39. Allahyari Z, Gaborski TR. Engineering cell-substrate interactions on porous membranes for microphysiological systems. Lab Chip (2022) 22(11):2080–9. doi: 10.1039/d2lc00114d
40. Mann IK, Chatterjee R, Zhao J, He X, Weirauch MT, Hughes TR, et al. CG methylated microarrays identify a novel methylated sequence bound by the CEBPB|ATF4 heterodimer that is active in vivo. Genome Res (2013) 23(6):988–97. doi: 10.1101/gr.146654.112
41. Cao S, Schnelzer A, Hannemann N, Schett G, Soulat D, Bozec A. The transcription factor FRA-1/AP-1 controls lipocalin-2 expression and inflammation in sepsis model. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.701675
42. Diaz-Cañestro C, Reiner MF, Bonetti NR, Liberale L, Merlini M, Wüst P, et al. AP-1 (Activated protein-1) transcription factor JunD regulates Ischemia/Reperfusion brain damage via IL-1β (Interleukin-1β). Stroke (2019) 50(2):469–77. doi: 10.1161/STROKEAHA.118.023739
43. Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity (2018) 48(2):364–79.e8. doi: 10.1016/j.immuni.2018.02.002
44. Kovács OT, Tóth E, Ozohanics O, Soltész-Katona E, Marton N, Buzás EI, et al. Proteomic changes of osteoclast differentiation in rheumatoid and psoriatic arthritis reveal functional differences. Front Immunol (2022) 13:892970. doi: 10.3389/fimmu.2022.892970
45. Lembo C, Raimondo A, de Paulis A, Mormile I, Rossi FW, Lembo S, et al. Clinical predictors of psoriatic arthritis and osteoclast differentiation. Exp Dermatol (2021) 30(12):1834–7. doi: 10.1111/exd.14424
46. McGonagle DG, McInnes IB, Kirkham BW, Sherlock J, Moots R. The role of IL-17A in axial spondyloarthritis and psoriatic arthritis: recent advances and controversies. Ann Rheum Dis (2019) 78(9):1167–78. doi: 10.1136/annrheumdis-2019-215356
47. Vecellio M, Hake VX, Davidson C, Carena MC, Wordsworth BP, Selmi C. The IL-17/IL-23 axis and its genetic contribution to psoriatic arthritis. Front Immunol (2020) 11:596086. doi: 10.3389/fimmu.2020.596086
48. Gallucci RM. Transforming growth factor-β and psoriasis: a new link. Br J Dermatol (2017) 177(6):1480–1. doi: 10.1111/bjd.16028
49. Häger M, Pedersen CC, Larsen MT, Andersen MK, Hother C, Grønbæk K, et al. MicroRNA-130a-mediated down-regulation of Smad4 contributes to reduced sensitivity to TGF-β1 stimulation in granulocytic precursors. Blood (2011) 118(25):6649–59. doi: 10.1182/blood-2011-03-339978
50. Shao S, Gudjonsson JE. Epigenetics of psoriasis. Adv Exp Med Biol (2020) 1253:209–21. doi: 10.1007/978-981-15-3449-2_8
51. Breeze CE, Paul DS, van Dongen J, Butcher LM, Ambrose JC, Barrett JE, et al. eFORGE: A tool for identifying cell type-specific signal in epigenomic data. Cell Rep (2016) 17(8):2137–50. doi: 10.1016/j.celrep.2016.10.059
52. Fu W, Li H, Fu H, Zhao S, Shi W, Sun M, et al. The SIRT3 and SIRT6 promote prostate cancer progression by inhibiting necroptosis-mediated innate immune response. J Immunol Res (2020) 2020:8820355. doi: 10.1155/2020/8820355
53. Huang C, Qu Y, Feng F, Zhang H, Shu L, Zhu X, et al. Cardioprotective effect of circ_SMG6 knockdown against myocardial Ischemia/Reperfusion injury correlates with miR-138-5p-Mediated EGR1/TLR4/TRIF inactivation. Oxid Med Cell Longev (2022) 2022:1927260. doi: 10.1155/2022/1927260
54. Seth G, Ozturk M, Hu WS. Reverting cholesterol auxotrophy of NS0 cells by altering epigenetic gene silencing. Biotechnol Bioeng (2006) 93(4):820–7. doi: 10.1002/bit.20720
55. Briollais L, Rustand D, Allard C, Wu Y, Xu J, Rajan SG, et al. DNA Methylation mediates the association between breastfeeding and early-life growth trajectories. Clin Epigenetics (2021) 13(1):231. doi: 10.1186/s13148-021-01209-z
Keywords: psoriasis, DNA methylation, Munro’s microabscess, pathology, neutrophil
Citation: Xu X, Zhang Y, Pan Z, Zhang X, Liu X, Tang L, Zhang X, Zhou F and Cheng H (2022) Genome-wide DNA methylation of Munro’s microabscess reveals the epigenetic regulation in the pathogenesis of psoriasis. Front. Immunol. 13:1057839. doi: 10.3389/fimmu.2022.1057839
Received: 30 September 2022; Accepted: 25 November 2022;
Published: 08 December 2022.
Edited by:
Steven O’Reilly, STipe Therapeutics, DenmarkReviewed by:
Xianyong Yin, University of Michigan, Ann Arbor, United StatesMaría Carmen Ovejero-Benito, Universidad San Pablo CEU, Spain
Copyright © 2022 Xu, Zhang, Pan, Zhang, Liu, Tang, Zhang, Zhou and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Cheng, YWhtdS5jaDgxM0AxNjMuY29t; Fusheng Zhou, YmlvemhvdWZzQDE2My5jb20=; Xiaoguang Zhang, emhhbmd4aWFvZ3VhbmcwNzhAMTYzLmNvbQ==
†These authors have contributed equally to this work