 Mengmeng Zhao
Mengmeng Zhao Zihui Zheng
Zihui Zheng Chenfei Li
Chenfei Li Jun Wan
Jun Wan Menglong Wang
Menglong Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 November 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1053175
This article is part of the Research Topic Targeting Signalling Pathways in Inflammatory Diseases View all 17 articles
Cardiovascular and metabolic diseases (CVMDs) are a leading cause of death worldwide and impose a major socioeconomic burden on individuals and healthcare systems, underscoring the urgent need to develop new drug therapies. Developmental endothelial locus-1 (DEL-1) is a secreted multifunctional domain protein that can bind to integrins and play an important role in the occurrence and development of various diseases. Recently, DEL-1 has attracted increased interest for its pharmacological role in the treatment and/or management of CVMDs. In this review, we present the current knowledge on the predictive and therapeutic role of DEL-1 in a variety of CVMDs, such as atherosclerosis, hypertension, cardiac remodeling, ischemic heart disease, obesity, and insulin resistance. Collectively, DEL-1 is a promising biomarker and therapeutic target for CVMDs.
A wide range of diseases that affect the heart and blood vessels are collectively referred to as cardiovascular diseases (CVDs), including atherosclerosis (AS), myocardial infarction (MI), hypertension, cardiac hypertrophy, and heart failure. Metabolic diseases, including diabetes, obesity and nonalcoholic fatty liver disease, are closely related to the occurrence and development of CVDs (1, 2). Cardiovascular and metabolic diseases (CVMDs) are the leading causes of death worldwide and result in a major socioeconomic burden on individuals and healthcare systems (3–6). These diseases are caused by a combination of multiple pathological factors, and their pathogenesis has not been fully elucidated. Although effective primary prevention and treatment strategies have reduced morbidity and mortality from CVMDs over the past 20 years, the prognosis of CVMDs remains unsatisfactory, and effective interventions are still lacking (7, 8).
Immune cells and inflammatory responses are involved in all stages of the occurrence and development of multiple CVMDs (9–11). The expression levels of various inflammatory mediators correlate with the clinical diagnosis and prognosis of CVMDs (12–18). Inflammation-related molecules such as interleukin-6 and growth differentiation factor 15 have been identified as biomarkers of CVDs (19). Regulation of immune function and the inflammatory response is an important strategy for the treatment of CVMDs (20–24). Increasing evidence shows that tissue-resident immune cells are involved in regulating the pathophysiological processes of CVMDs (25–28). Local tissues, such as vascular endothelium and adipose tissue, also have an important impact on the occurrence and development of CVMDs (29–32). Various local tissues in the human body are not only passive targets of immune and inflammatory responses but also active regulators of immunity (33). Local tissue signaling can regulate immune cell accumulation and functional plasticity and play a key role in immune-driven CVMDs (34, 35). Stromal and parenchymal cell-derived signals (including growth factors, cytokines, and other locally acting homeostatic factors) as well as intercellular adhesion interactions mediate local tissue-to-immune communication in CVMDs such as myocardial infarction (36–38). The compartmentalized expression of tissue signaling can facilitate optimal performance of cell-type-specific effects and spatial regulation of immune responses. Therefore, homeostatic molecules in the tissue microenvironment at different locations may be critical for CVMDs.
Developmental endothelial locus-1 (DEL-1) is a secreted multifunctional domain protein. As a local tissue signal, it exerts different regulatory functions in different expression regions (39). Endothelial cell-derived DEL-1 mainly regulates inflammation initiation by inhibiting neutrophil recruitment, while macrophage-derived DEL-1 promotes the resolution of inflammation by enhancing neutrophil apoptosis and macrophage efferocytosis (40). Increasing evidence has shown that the regulation of immune system homeostasis by DEL-1 plays an important role in CVMDs (41–43). In this article, we review the regulatory role of local tissue DEL-1 signaling in CVMDs and look forward to the future development of DEL-1 (Table 1).
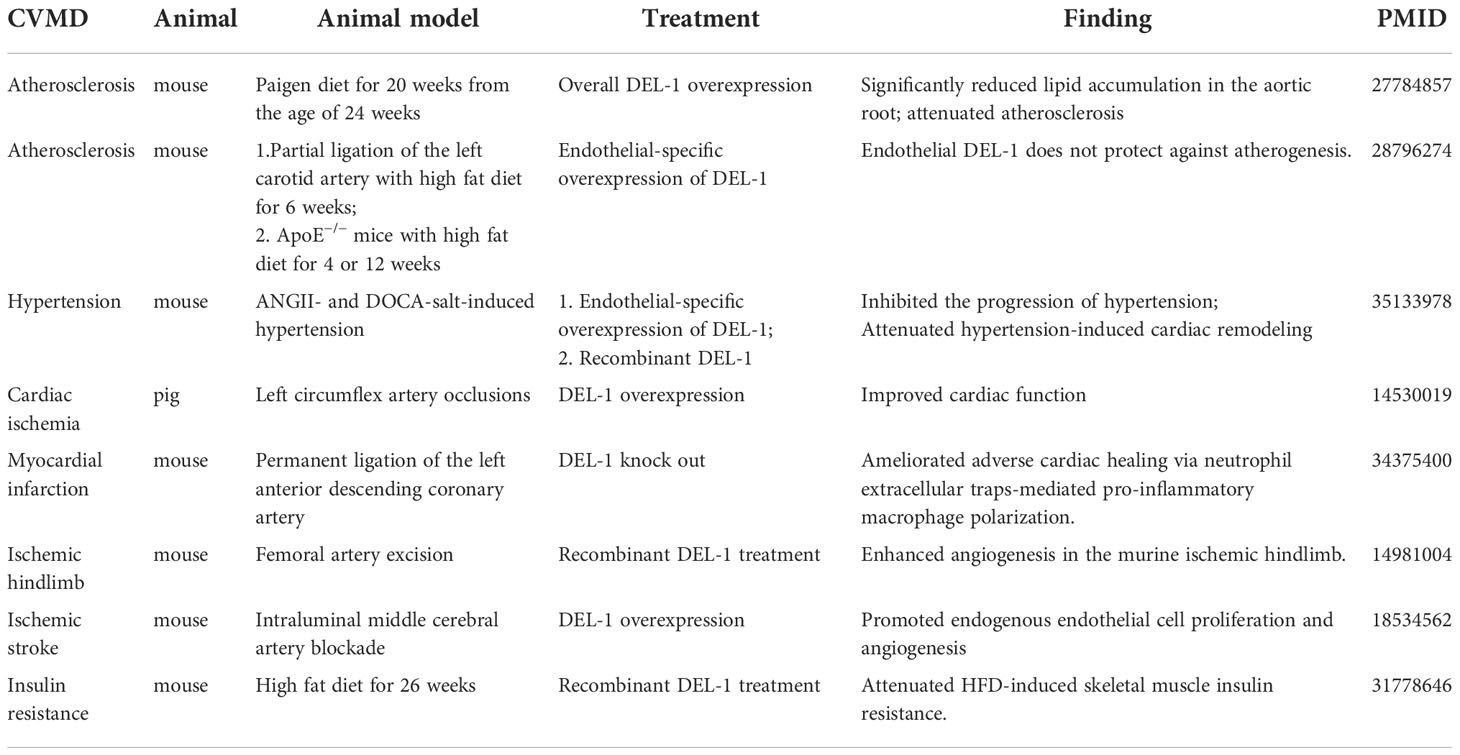
Table 1 Roles of DEL-1 in cardiovascular and metabolic diseases.
DEL-1 is a 52 KD multifunctional matrix protein encoded by EDIL3 (epidermal growth factor (EGF)-like repeats and discoidin domains 3), which was cloned and characterized in angioplasty cells and early endothelial cells as early as 1998 (44). Increasing evidence shows that DEL-1 is expressed in tissues such as the brain, lung, and gums (39, 45, 46). Some tissue-resident cells, such as mesenchymal stromal cells, macrophages, neuronal cells, osteoclasts and some hematopoietic microenvironment cells, can also secrete DEL-1 (39, 40, 47, 48).
The mechanism regulating DEL-1 expression in tissues has not been elucidated. The reciprocal regulatory role of IL-17 and DEL-1 is now widely recognized (Figure 1). IL-17 directly inhibits endothelial DEL-1 expression, thereby promoting lymphocyte function-associated antigen 1 (LFA-1)-dependent neutrophil recruitment, while DEL-1 counteracts IL-17 production and IL-17-dependent inflammation (45, 49). Mechanistically, IL-17 reduces DEL-1 expression in a glycogen synthase kinase 3β (GSK3β)-dependent process that inhibits the binding of the key transcription factor CCAAT/enhancer-binding protein β (C/EBPβ) to the EDIL3 promoter, thereby downregulating EDIL3 transcription. This inhibitory action of IL-17 can be reversed at the GSK-3β level by PI3K/Akt signaling induced by D-resolvins. Interestingly, DEL-1 expression is reduced in aged mice, which may be related to the increased expression level of IL-17 (39, 50).
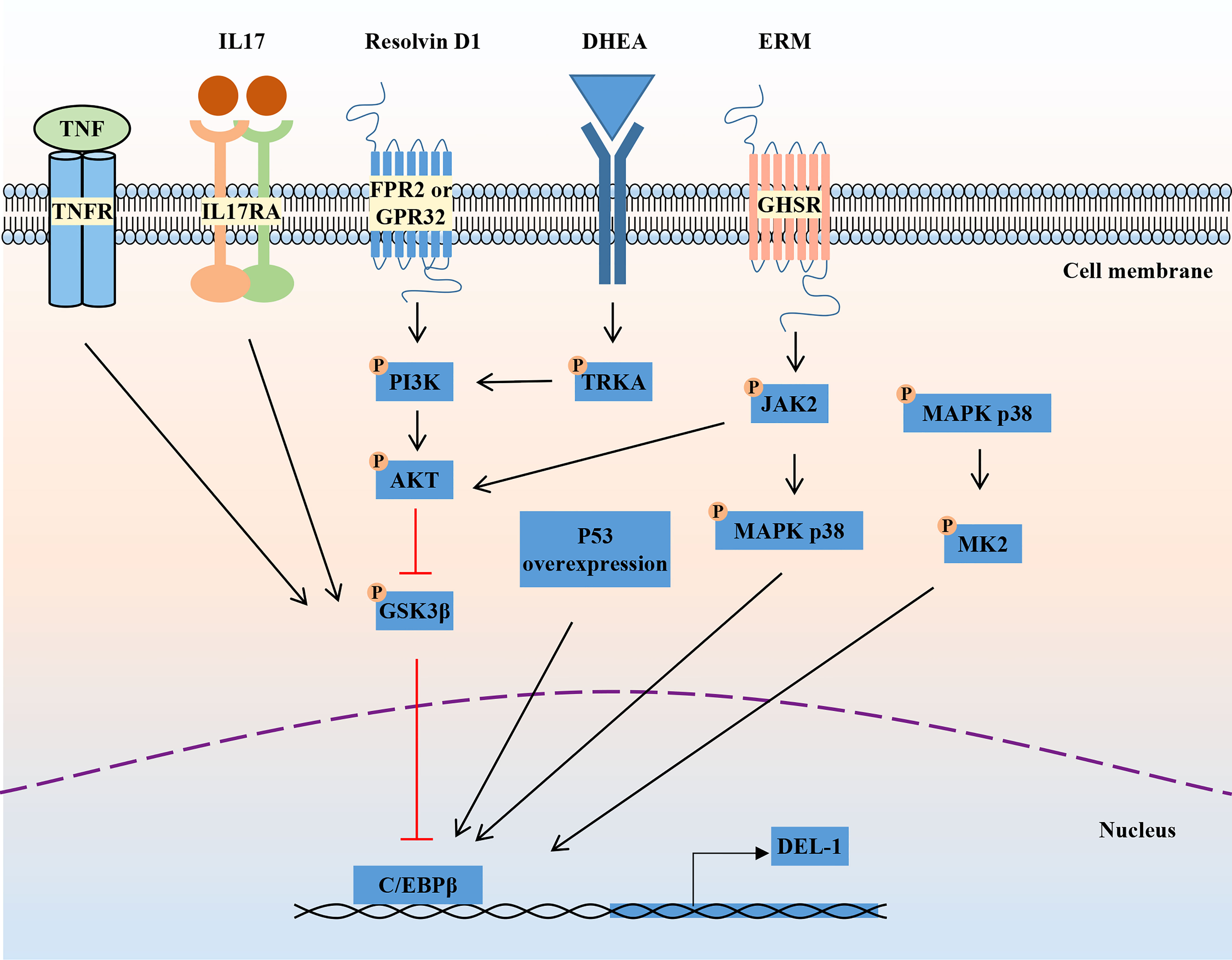
Figure 1 Regulation of DEL-1 expression. IL-17 and TNF reduce DEL-1 expression in a GSK3β-dependent process that inhibits the binding of the key transcription factor C/EBPβ to the EDIL3 promoter, thereby downregulating EDIL3 transcription. This inhibitory action of IL-17 can be reversed at the GSK-3β level by PI3K/AKT signaling induced by D-resolvins. Through interaction with GHSR, ERM activates JAK2 signaling, leading to DEL-1 transcription, which is MAPK p38-mediated and C/EBPβ dependent, as well as to PI3K/AKT-mediated reversal of the GSK3β-dependent inhibitory effect of IL-17 on DEL-1 expression. DHEA reduced DEL-1 expression and secretion in endothelial cells by activating TRKA and downstream PI3K/AKT signaling to restore C/EBPβ binding to the DEL-1 promoter. In addition, P53 overexpression and the activation of P38/MK2 signaling were reported to promote DEL-1 expression. DEL-1, developmental endothelial locus-1; GSK-3β, glycogen synthase kinase 3β; C/EBPβ, CCAAT/enhancer-binding protein β; PI3K, phosphoinositide 3-kinase; MAPK, mitogen-activated protein kinases; GHSR, growth hormone secretagogue receptor; ERM, erythromycin; JAK2, janus kinase 2; DHEA, dehydroepiandrosterone; TRKA, tropomyosin receptor kinase A.
Another pro-inflammatory cytokine, TNF, can also reduce DEL-1 expression and secretion in endothelial cells by reducing C/EBPβ binding to the DEL-1 promoter, while the steroid hormone dehydroepiandrosterone (DHEA) increased DEL-1 expression and secretion in endothelial cells by activating tropomyosin receptor kinase A (TRKA) and downstream PI3K/AKT signaling to counteract the inhibitory effect of TNF and restore C/EBPβ binding to the DEL-1 promoter (51). Recently, erythromycin was reported to reverse the inhibitory effect of IL-17 on DEL-1 expression by binding to growth hormone secretagogue receptor (GHSR) and activating JAK2/MAPK p38 signaling (52). Furthermore, another independent research group found that overexpression of the p53 response element enhanced the transcriptional activity of EDIL3 (53). Primary endothelial cells isolated from p53 knockdown mice showed decreased DEL-1 mRNA expression (53). In melanoma cells, inhibition of p38/MK2 signaling reduced DEL-1 expression, suggesting that DEL-1 may be a downstream target of MK2 (54). In conclusion, the regulatory mechanism of DEL-1 expression is still unclear and needs to be further explored.
DEL-1 comprises three N-terminal EGF-like repeats (E1, E2 and E3) and two C-terminal discoidin I-like domains (C1 and C2) (44, 55). The RGD (Arg–Gly–Asp) motif in the second EGF-like repeat (E2) allows DEL-1 to interact with different integrins, including the β2 (e.g., αLβ2 and αMβ2) and β3 (e.g., αVβ3) integrins (44, 56, 57). The discoidin I-like domain and glycosaminoglycan mediate the interaction of DEL-1 with phosphatidylserine (PS) (40, 58). These interactions in turn confer important functions of DEL-1 in regulating immunity that have a major impact on the initiation and resolution of inflammation, suggesting that DEL-1 may be a promising therapeutic target (39). Specifically, the interaction of DEL-1 with αLβ2 or αMβ2 blocks the binding of the latter to its endothelial counterreceptor intercellular adhesion molecule-1 (ICAM-1), thereby inhibiting leukocyte adhesion and recruitment to sites of inflammation (46, 59). With its anti-inflammatory properties, DEL-1 can prevent a variety of inflammation-related conditions, such as multiple sclerosis and lung inflammation (45–48, 60, 61). DEL-1 can capture platelet microparticles by linking with PS and promote endothelial cell clearance of microparticles in an αVβ3 integrin-dependent manner (62). In addition, DEL-1 can act as a bridging molecule to bind PS on apoptotic cells and αVβ3 integrin on macrophages at both ends, mediating the burial of apoptotic cells and promoting inflammation resolution (40, 63). Collectively, DEL-1 exerts anti-inflammatory effects by inhibiting neutrophil recruitment and migration, promoting inflammatory resolution by accelerating macrophage reprogramming, and regulating myelopoiesis (Figure 2). These functions are discussed in detail in the review by Hajishengallis et al. (39, 64). Experiments with various deletion mutants of DEL-1 showed that fragments containing the C-terminus of C1 with a lectin-like structure were deposited directly in the ECM (58). The deposition efficiency varied according to the presence of other domains in DEL-1. The fragment containing E3 and C1 had the strongest deposition activity, while the fragment containing C2 was highly homologous to C1 and had low deposition activity (58). These data suggest that the discoidin domain of the DEL-1 protein contributes to its deposition and function in the extracellular matrix.
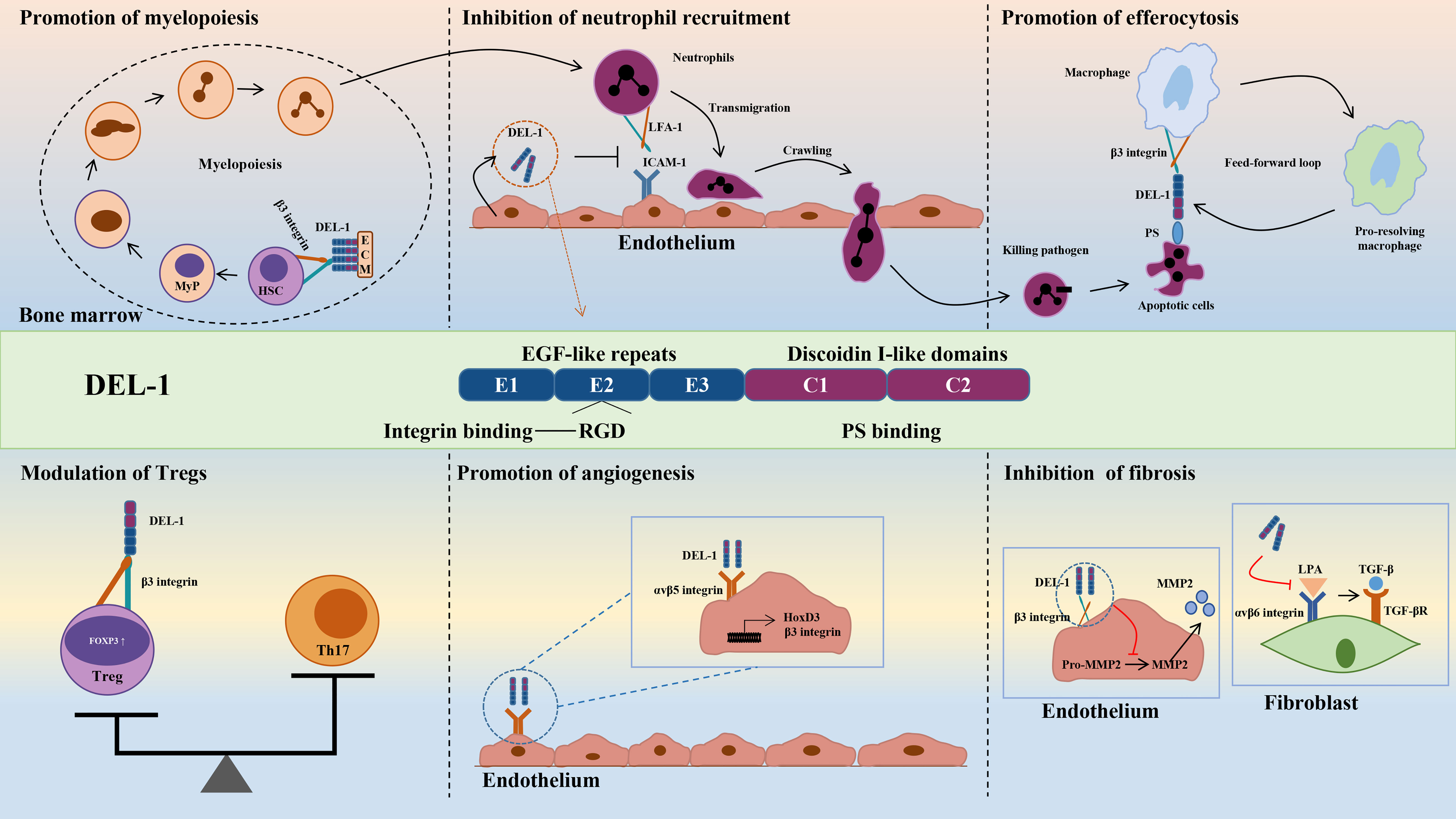
Figure 2 Structure and biological roles of DEL-1. The figure shows the multidomain structure of DEL-1 as well as six major regulatory activities of this protein, namely, promoting myelopoiesis, inhibiting neutrophil recruitment, promoting efferocytosis, modulating Tregs, promoting angiogenesis and inhibiting fibrosis. DEL-1, developmental endothelial locus-1; ECM, extracellular matrix; HSC, hematopoietic stem cell; MyP, myeloid progenitors; ICAM-1, intercellular adhesion molecule-1; LFA-1, lymphocyte function-associated antigen-1; PS, phosphatidyl serine; Th17, T helper 17 cell; Treg, T regulatory cell; FOXP3, forkhead box P3; HoxD3, homeobox D3; MMP2, matrix metallopeptidase 2; LPA, latency-associated peptide; TGF-β, transforming growth factor-β.
Genetic knockout or overexpression of DEL-1 in mice is an important tool in studying the function of DEL-1. EDIL3-/- mice have a specific phenotype characterized by increased development of spontaneous periodontitis (45). DEL-1 deficiency promoted neutrophil infiltration and inflammatory bone loss in mice with periodontitis (45). In experimental allergic encephalomyelitis (EAE), DEL-1 deficiency increased immune cell infiltration and inflammatory responses in the central nervous system, leading to increased disease severity (47). DEL-1-deficient mice exhibit increased neutrophil infiltration and inflammatory responses during lung inflammation (46). In postoperative peritoneal adhesion (PPA) mice, EDIL3-/- mice had a higher incidence of PPA and an increased inflammatory response, resulting in more severe PPA (65). Myelopoiesis in EDIL3-/- mice was suppressed in hematopoietic stem cells (HSCs) (66). The position of DEL-1 expression critically determines its regulatory function. In the future, the application of different transgenic mice with tissue- or cell-specific knockout or overexpression of DEL-1 may better help us to study its function.
As a lipid-driven chronic inflammatory disease that underlies various CVDs, such as ischemic heart disease (IHD) (67–69), AS is caused by the accumulation and oxidative modification of low-density lipoprotein (LDL) in the arterial intima (70). As the tissue microenvironment changes, endothelial cells release chemokines and adhesion molecules, which promote the recruitment and migration of monocytes on the endothelium; monocytes subsequently differentiate into macrophages to phagocytose oxidized low-density lipoprotein (oxLDL), while the excessive accumulation of oxLDL eventually leads to the transformation of macrophages into foam cells and initiates the secretion of inflammatory cytokines to promote the development of AS plaques; moreover, smooth muscle cells migrate to the subendothelial space to form fibrous caps and stabilize the plaques. Finn et al. found that the serum level of DEL-1 in patients with coronary heart disease (3.9 ± 0.2 pg/mg total protein) was significantly higher than that in healthy subjects (2.9 ± 0.1 pg/mg total protein) (71). However, there is still a lack of clinical evidence to prove that DEL-1 is related to the occurrence and development of AS.
In vitro evidence showed that DEL-1 can not only directly bind to oxLDL but also inhibit the uptake of oxLDL in cells transfected with multiple scavenger receptor genes in a dose-dependent manner, such as lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), scavenger receptor A (SR-A), scavenger receptor class B type I (SR-BI), and the cluster of differentiation 36 (CD36) (72). DEL-1 inhibited the uptake of oxLDL by human coronary artery endothelial cells (HCAECs) and macrophages. Furthermore, the oxLDL-induced increase in monocyte chemotactic protein-1 (MCP-1) and intercellular adhesion molecule-1 (ICAM−1) expression in HCAECs was significantly inhibited by DEL-1, which has the potential to alleviate monocyte adhesion. OxLDL-induced endothelin-1 secretion in HCAECs was also significantly inhibited by DEL-1 (72). Therefore, Del-1 not only inhibited the binding of oxLDL to the receptors but also inhibited the cellular response to oxLDL (Figure 3).
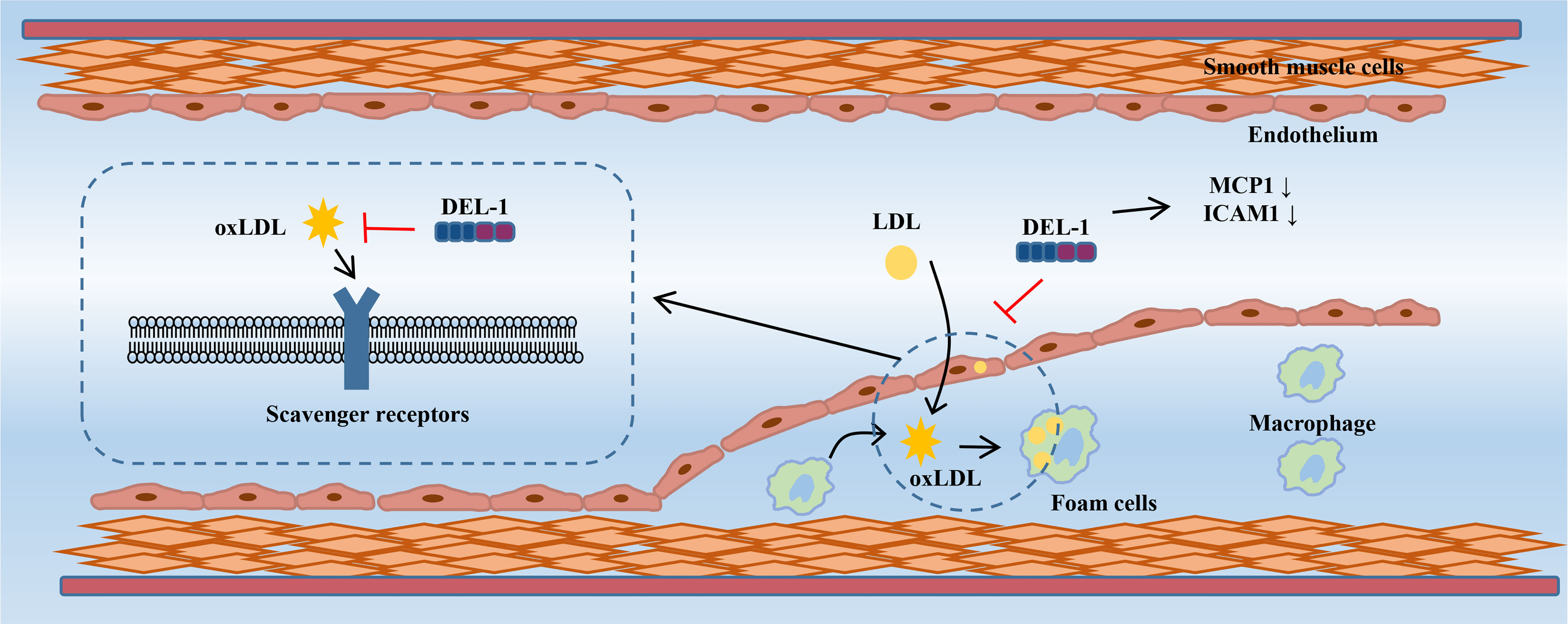
Figure 3 DEL-1 binds directly to oxLDL to block its binding to scavenger receptors and exert significant anti-atherogenic effects. In addition, DEL-1 reduced the expression of MCP1 and ICAM1 in the endothelial cell. DEL-1, developmental endothelial locus-1; oxLDL, oxidized low-density lipoprotein; MCP1, monocyte chemotactic protein-1; ICAM1, intercellular adhesion molecule-1.
In a mouse model of AS, DEL-1 overexpression inhibited the receptor-binding activity of a modified LDL in serum, reduced the expression of adhesion molecules MCP-1 and ICAM-1 in the aorta, and reduced the oil red O-positive atherosclerotic area at the aortic roots (72). These results suggest that DEL-1 overexpression inhibits the occurrence of AS. However, in contrast to the above results, Subramanian et al. constructed an AS model by partially ligating the left carotid artery in ApoE−/− mice and found that endothelial cell-specific overexpression of DEL-1 had no significant effect on the development and cellular composition of AS plaques (73). These researchers fed ApoE−/− mice a high-fat diet for 4 or 12 weeks to study early or late lesions and found that endothelial cell-specific overexpression of DEL-1 did not affect early or late stages of AS and did not prevent AS (73). The apparent discrepancy between the results of this study and those of Kakino et al. may be due to the following: 1. The transgenic mice in Kakino et al.’s study overexpressed DEL-1 in all cell types. In addition to the mechanism mediated by endothelial cell-derived DEL-1, other mechanisms may also play a role, such as macrophages. 2. Differences in experimental methods between the two studies may also lead to conflicting results, such as differences in the background of ApoE−/− mice, differences in high fat diets, differences in modeling methods, and so on. In the future, transgenic mice with macrophage-specific expression may help to further elucidate the role of DEL-1 in AS.
Intercellular signaling plays a key role in AS formation, affecting the occurrence and progression of CHD, and circulating microRNAs (miRNAs) may be involved in this process (74). There were clear differences in circulating miRNA transport between CHD patients and healthy subjects, especially the reduction in miRNA enrichment in microparticles (MPs) (71, 75). Furthermore, MPs from CHD patients were less efficient at transferring miRNAs to cultured HUVECs, suggesting that MP uptake is impaired in the disease state. DEL-1 can mediate the uptake of MPs by endothelial cells by binding to PS on the external surface of MPs (62, 63). Although circulating levels of DEL-1 are increased in CHD patients, these patients have less DEL-1 binding to MPs (71). Therefore, Finn et al. suggested that DEL-1 binding to MPs was impaired in the serum of individuals with CHD, thereby altering circulating miRNA transport and affecting CHD initiation and progression. In the future, in addition to regulating the expression of DEL-1, regulating the function of DEL-1 may be an important aspect in the treatment of AS.
Hypertension refers to a clinical syndrome characterized by increased systemic arterial blood pressure (systolic and/or diastolic blood pressure), which may be accompanied by functional or organic damage to organs such as the heart, brain, and kidneys (76). Hypertension is the most common chronic disease and the main risk factor for cardiovascular and cerebrovascular diseases (77). Although the pathophysiological mechanisms of hypertension are not fully understood, strong evidence suggests that immune hyperactivation and chronic inflammatory responses play a crucial direct role in the development of hypertension (78). Our team’s previous clinical and animal studies also proved that immune microenvironment disturbances are closely related to hypertension (79–83). Activated T lymphocytes and proinflammatory cytokines such as IL-17 are involved in the occurrence and development of angiotensin II (ANGII) and deoxycorticosterone acetate-salt (DOCA-salt)-induced hypertension (84–89). Gene knockout or neutralization with antibodies against IL-17 limited the progression of hypertension (86, 88, 90). DEL-1 can inhibit inflammation through various anti-inflammatory effects to alleviate IL-17-mediated conditions, such as inflammatory bone loss and multiple sclerosis, suggesting that DEL-1 may be a potential target for the treatment of hypertension (45, 48). Furthermore, DEL-1 promoted vascular smooth muscle cell (VSMC) adhesion, migration and proliferation in a dose-dependent manner, and this process was mediated through αVβ3 integrin (91). These data suggested that DEL-1 has a paracrine role in vascular remodeling.
Recently, Failer et al. found that endothelial DEL-1-overexpressing mice had less adventitial collagen, lower medial thickness, and more elastin, suggesting that DEL-1 overexpression prevents ANGII-induced aortic remodeling (41). DEL-1 overexpression also prevented the progression of ANGII-induced hypertension, endothelial dysfunction and aortic fibrosis. DEL-1 overexpression alleviated the infiltration of CD45 leukocytes, TCR-β T cells and CD45IL-17 leukocytes in the aorta after ANGII infusion. Moreover, DEL-1 overexpression inhibited the expression of proinflammatory cytokines induced by ANGII and increased the expression level of the anti-inflammatory cytokine IL-10. In addition to inflammation, DEL-1 overexpression inhibits the activity of matrix metallopeptidase 2 (MMP2) in the aorta, whose increase critically contributes to aortic remodeling in hypertension (92, 93).
Failer et al. next investigated the preventive and therapeutic effects of recombinant DEL-1-FC on ANGII-induced hypertension. Intervention with recombinant DEL-1-FC administered before or after hypertension prevented or eliminated ANGII-induced aortic remodeling, hypertension, arterial stiffness, and inflammation (41). Recombinant DEL-1-FC also inhibited the activity of MMP2 in the aorta while promoting the infiltration of anti-inflammatory Tregs. Failer et al. also found that the mutation of the RGE part of DEL-1 abolished the protective effect of DEL-1-FC, suggesting that RGE is involved in the pathophysiological process of DEL-1 inhibiting the occurrence and development of hypertension. In a DOCA salt-induced hypertension model, recombinant DEL-1 treatment similarly attenuated aortic remodeling, hypertension, and inflammatory progression and promoted Treg infiltration (41).
A series of in vitro experiments further demonstrated that DEL-1 overexpression and recombinant DEL-1 treatment inhibited ANGII-induced activation of MMP2 in human and mouse vascular tissues, which was αVβ3 integrin-dependent (41, 94). Correspondingly, RGE mediates the binding of αVβ3 integrin to DEL-1, which may explain the abolition of the protective effect of DEL-1 by RGE mutation (41, 56). In conclusion, the findings of Failer et al. fully demonstrate the protective role of DEL-1 in the occurrence and development of hypertension and suggest this molecule may become a potential drug for the treatment of hypertension in the future (Figure 4).
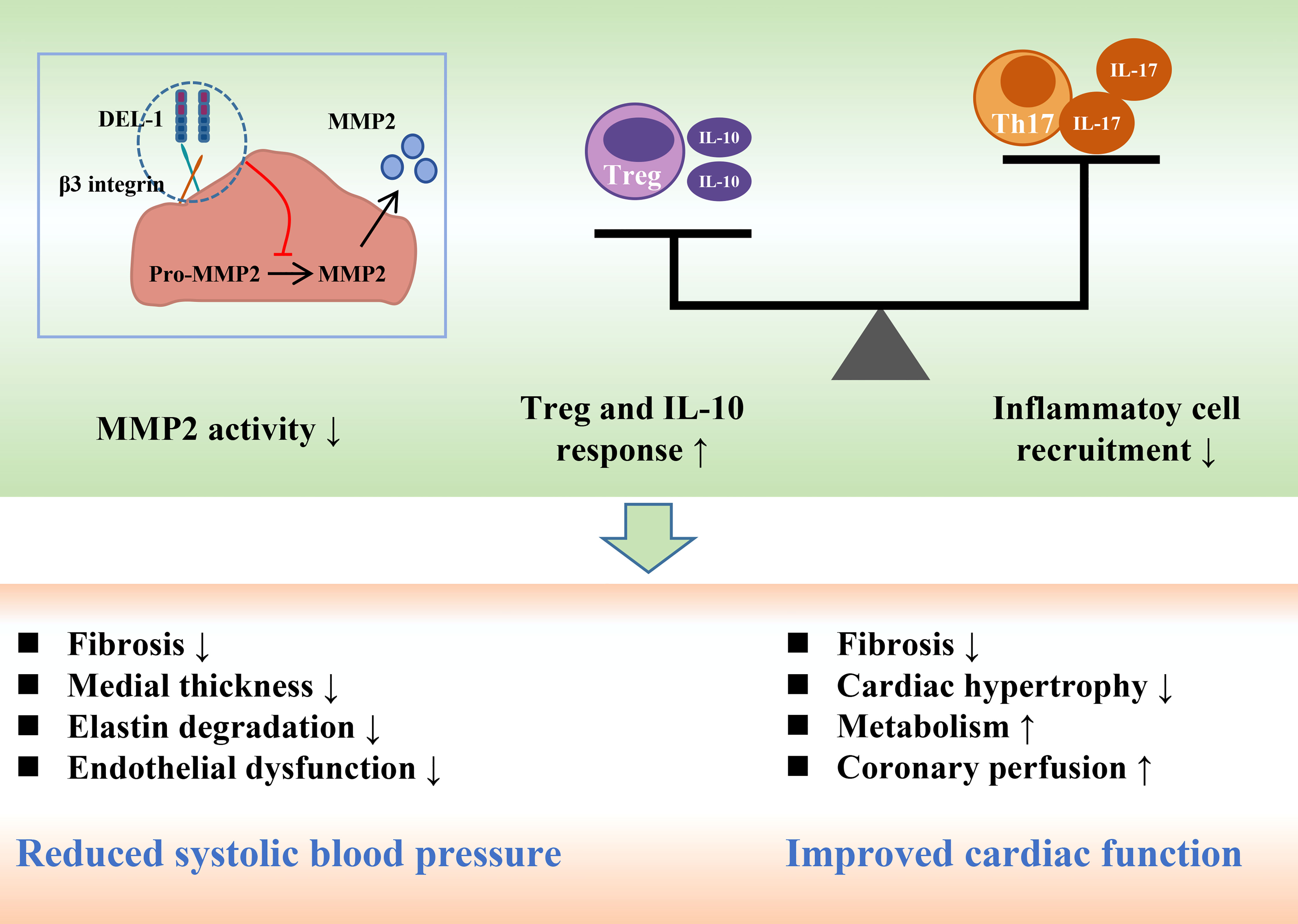
Figure 4 DEL-1 reduces blood pressure and maintains cardiac function by inhibiting MMP2 activity, reducing inflammatory cell infiltration and enhancing Treg and IL-10 responses. DEL-1, developmental endothelial locus-1; MMP2, matrix metallopeptidase 2; Th17, T helper 17 cell; Treg, T regulatory cell; IL, interleukin.
Cardiac remodeling is an independent risk factor for heart failure, arrhythmias, and sudden death and is a key determinant of the clinical course and long-term prognosis of patients with CVDs (95). Pathological cardiac remodeling is characterized by cardiomyocyte hypertrophy and interstitial fibrosis under various cardiac stresses, such as hypertension and MI, resulting in increased myocardial stiffness and impaired cardiac contractility (96, 97). Cardiac remodeling is associated with fibrosis, capillary sparseness, increased production of proinflammatory cytokines, cellular dysfunction (impaired signaling, inhibition of autophagy, and abnormal cardiomyocyte/noncardiomyocyte interactions), and adverse epigenetic alterations (95). Our previous studies further shed light on the pathogenesis of cardiac remodeling, suggesting that inhibition of cardiac remodeling by pharmacological or genetic approaches significantly improves cardiac dysfunction and survival (21, 98–101).
In mice, fibroblasts constituted 27% of all cardiac cells, contributing to the maintenance of homeostasis under physiological conditions and regulating tissue remodeling in response to stress (95, 102, 103). Pathological fibrosis results from abnormal regulation of extracellular matrix (ECM) production in tissues or organs, including collagen (97). Compared with that in normal lung tissue, the expression level of DEL-1 was decreased in lung fibrous tissue, suggesting that DEL-1 may be associated with pulmonary fibrosis (60). DEL-1 deficiency promoted collagen synthesis and secretion by regulating transforming growth factor (TGF-β), thereby aggravating bleomycin-induced pulmonary fibrosis (60, 104). Yan et al. found that DEL-1-deficient mice had a higher incidence of postoperative peritoneal adhesions, accompanied by enhanced collagen production (65). In contrast, DEL-1 supplementation reduced the incidence and severity of postoperative peritoneal adhesions. In vitro studies have demonstrated that DEL-1 inhibits TGF-β activation in 293T cells and RAW264.7 mouse macrophages by binding to αVβ6 integrin (104). These data suggest that DEL-1 plays an important role in the initiation and progression of tissue fibrosis.
The immune system and inflammatory response mediate pathological cardiac remodeling (97). Immunomodulation may be an important strategy to alleviate cardiac remodeling. Failer et al. found that endothelial DEL-1 overexpression or recombinant DEL-1 treatment inhibited AGNII or DOCA salt-induced inflammation and MMP2 activation in the heart, thereby reducing cardiac hypertrophy, fibrosis, and dysfunction (41). However, cardiac remodeling in this study belongs to target organ damage caused by hypertension, and the regulation of DEL-1 on blood pressure may indirectly affect cardiac remodeling. Therefore, this study may have certain limitations. Future studies on cardiac remodeling may help us further understand the function of DEL-1.
Ischemic heart disease (IHD), mainly caused by coronary atherosclerosis and its complications, can induce congestive HF and life-threatening arrhythmias and is the leading cause of death worldwide (105, 106). Acute myocardial infarction (AMI) is the most serious IHD with the highest mortality rate (106). In a pig model of cardiac ischemia induced by left circumflex artery ligation, DEL-1 treatment improved cardiac function (107). Wei et al. found that DEL-1 levels were decreased in severe AMI patients, which is consistent with the finding thar WT mice with MI showed low levels of cardiac DEL-1 (42). Compared with WT mice, DEL-1-/- mice showed significantly improved cardiac function and alleviated cardiac remodeling post-MI. Mechanistically, the protective effect of DEL-1 deficiency in MI was associated with enhanced neutrophil recruitment and expansion of proinflammatory monocyte-derived macrophages (42). Injection of a neutrophil-specific C-X-C motif chemokine receptor 2 (CXCR2) antagonist impaired macrophage polarization, increased cellular debris and exacerbated adverse cardiac remodeling, thereby abrogating the protective effect of DEL-1 deficiency. Inhibition of neutrophil extracellular trap (NET) formation by treatment with a neutrophil elastase inhibitor or DNase I abrogated differences in macrophage polarization and cardiac function between WT and DEL-1-/- mice after MI. Collectively, these data suggest that DEL-1 is a key regulator of neutrophil recruitment and macrophage polarization during cardiac remodeling after MI (Figure 5).

Figure 5 Deletion of DEL-1 promotes neutrophil infiltration and formation of NETs, thereby promoting macrophage efferocytosis and alleviating cardiac fibrosis and cardiac dysfunction after MI. DEL-1, developmental endothelial locus-1; NETs, neutrophil extracellular trap; MI, myocardial infarction.
Increasing evidence has shown that healing of MI involves a series of delicately regulated inflammatory responses (108). Following MI, injured cardiomyocytes release damage-associated molecular patterns (DAMPs), cytokines, and chemokines, leading to substantial recruitment of neutrophils and monocytes/macrophages to the myocardium (109, 110). These neutrophils and monocytes contribute to the removal of debris and dead cells, as well as the activation of repair pathways. Furthermore, recruited monocytes give rise to proinflammatory or repairing macrophages. Proinflammatory macrophages produce cytokines, release MMPs to promote extracellular matrix destruction, and clear cellular debris, while repairing macrophages promotes fibroblast-to-myofibroblast transformation and enhances collagen deposition, leading to the formation of crosslinked collagen (111). A scar is formed to protect the left ventricle (LV) from rupture of the heart. Wei et al. confirmed the integral role of inflammation in the healing process (42). However, excessive inflammation may exacerbate MI-induced myocardial injury (109, 111). DEL-1 has anti-inflammatory and proresolving effects, and the lack of DEL-1 may inhibit inflammatory resolution, leading to an excessive inflammatory response and exacerbating tissue damage (40, 60, 112, 113). Therefore, the extent of the increased inflammation caused by DEL-1 deficiency in Wei et al.’s study requires further scrutiny.
The study by Wei et al. is the only report of amelioration of DEL-1 deficiency (42). In previous reports, inhibition of neutrophil recruitment improved cardiac dysfunction and cardiac remodeling after MI (114–116). Inhibition of neutrophils by DEL-1 also exerted protective effects in other diseases, which seems to contradict the study by Wei et al. (46, 48, 117). Multiple actions of DEL-1, such as anti- and proinflammatory resolution (39), coronary vasodilation (41), inhibition of MMP2 activity (41) and promotion of angiogenesis (118, 119), may protect the heart from MI-induced injury. The study by Wei et al. has certain limitations, such as the lack of cell-specific gene-edited mice and the lack of analysis of preventive or therapeutic effects of recombinant DEL-1 (42). The future use of endothelial or macrophage-specific DEL-1 transgenic mice and recombinant DEL-1 may help us further understand the role and mechanism of DEL-1 in IHD.
DEL-1 was found to regulate vascular morphogenesis or remodeling in embryonic development as early as 1998 when it was first cloned and characterized (44). DEL-1 provides a unique autocrine angiogenic pathway for the embryonic endothelium, which is mediated in part by integrin αvβ3 (120). DEL-1 mediates VSMC adhesion, migration and proliferation through interaction with integrin αvβ3, which may regulate vascular wall development and remodeling (91). Aoka et al. found that DEL-1 accelerates tumor growth by promoting enhanced angiogenesis (121). Expression of endogenous DEL-1 protein is increased in ischemic hindlimbs (122). DEL-1 binding to αvβ5 upregulated the expression of the transcription factor Hox D3 and integrin αvβ3, thereby promoting angiogenesis and functional recovery in a hindlimb ischemia model (57). Exogenous intramuscular administration of DEL-1 significantly enhanced angiogenesis in ischemic hindlimbs in mice, suggesting that DEL-1 may be a novel therapeutic agent for ischemic patients (122). A clinical study compared VLTS-589 (a plasmid encoding Del-1 conjugated to poloxamer 188) with the poloxamer 188 control in the treatment of intermittent claudication in patients with moderate to severe peripheral arterial disease (123). Intramuscular delivery of a plasmid expressing DEL-1 and the control significantly improved baseline exercise capacity at 30, 90, and 180 days, but there was no difference in outcome measures between the two groups. DEL-1-mediated angiogenesis has also been reported in many other diseases, such as ischemia models, lung adenocarcinoma, retinopathy, squamous cell carcinoma, and psoriasis (119, 124–129). Taken together, these data suggest that DEL-1-regulated angiogenesis may be a target for many diseases, but its clinical value requires further clinical trials.
Similar to MI, strokes are also caused by vascular or microvascular diseases that disrupt the blood supply to the brain, leading to brain dysfunction (130). The number of new vessels generated in ischemic brain tissue is associated with decreased morbidity and longer survival in stroke patients, suggesting that restoration of cerebral microvascular circulation is important for functional recovery after ischemic attacks (131). DEL-1 expression was increased in the ischemic cortical peri-infarct area after ischemic stroke (118). DEL-1 gene transfer induced cerebral angiogenesis and may be a novel and effective method for stimulating cerebral angiogenesis after stroke (118). Electroconvulsive seizures (ECSs) have been shown to treat major depression by modulating neurotrophy and angiogenesis (132, 133). Newton et al. found that ECS treatment increased DEL-1 expression in brain tissue and promoted angiogenesis in the adult rat hippocampus (134). In conclusion, DEL-1-mediated angiogenesis may be one of the targets for the treatment of cerebrovascular diseases.
The prevalence of metabolic diseases, including diabetes, is increasing, while the westernization of dietary habits has led to an increase in obesity (135). Obesity-related chronic low-grade inflammation has been reported to cause insulin resistance in muscle, liver, and adipose tissue (136). Insulin resistance refers to the decrease in the efficiency of insulin to promote glucose uptake and utilization for various reasons, and the compensatory secretion of excessive insulin produces hyperinsulinemia to maintain the stability of serum glucose levels (137). Insulin resistance predisposes patients to metabolic syndrome and type 2 diabetes. DEL-1 ameliorates palmitate-induced endoplasmic reticulum (ER) stress and insulin resistance in the mouse skeletal muscle cell line C2C12 via SIRT1/SERCA2-related signaling (43). In vivo experiments showed that DEL-1 administration increased the expression of SIRT1 and SERCA2, thereby ameliorating insulin resistance in skeletal muscle of high fat diet (HFD)-fed mice and improving HFD-impaired glucose tolerance and insulin sensitivity (43). These results suggest that DEL-1 may be a novel therapeutic target for the management of insulin resistance and type 2 diabetes.
Regular exercise is the treatment of choice for obesity and obesity-mediated metabolic disorders such as insulin resistance, type 2 diabetes, atherosclerosis, and hypertension (138). Compared with that in healthy subjects, DEL-1 mRNA expression was decreased in the muscle of obese and diabetic patients (139). Exercise increases DEL-1 mRNA expression levels in obese/diabetic patients in a time-dependent manner (139). DEL-1 secreted by exercising skeletal muscle can affect various tissues through the bloodstream, including adipose tissue (140). In vitro experiments showed that DEL-1 attenuated palmitate-induced inflammation and insulin signaling impairment in adipocytes by regulating AMPK/HO-1 signaling (139). In addition, DEL-1 treatment promoted AMPK phosphorylation and enhanced adipocyte thermogenesis but did not affect intracellular lipid accumulation (139).
In another endometrial cancer (EC) cohort study, Cobb et al. found an association between patient BMI and increased DEL-1 expression in cancer tissue (141). Furthermore, HFD increased the expression of DEL-1 in tumors compared with a low-fat diet in EC model mice (141). These data suggest that DEL-1 may serve as a novel obesity-driving target that should be further explored in future research work.
Thus, DEL-1-mediated anti-inflammatory and proresolving effects provide a basis for the amelioration of metabolic diseases. DEL-1 is involved in the regulation of obesity and insulin resistance. However, the current relevant evidence is still insufficient, and more research is needed in the future to reveal the role of DEL-1 in metabolic diseases.
DEL-1 has received considerable attention since it was first cloned and characterized as a factor promoting embryonic angiogenesis (44). DEL-1 is widely expressed in different tissues, such as the brain, lung and blood vessels, to maintain tissue homeostasis. As a secreted protein, the serum level of DEL-1 may be related to the diagnosis and prognosis of various diseases, such as MI, sepsis and osteoarthritis (42, 142, 143). As a local tissue signal, DEL-1 exerts anti-inflammatory and proresolving effects in different tissues and stages, thereby ameliorating a variety of inflammation-related diseases (39). Emerging studies over the past few years have convincingly demonstrated that DEL-1 has a therapeutic effect on a variety of CVMDs, including AS, hypertension, cardiac remodeling, and insulin resistance. This review summarizes the potential involvement of DEL-1 in cardiovascular and metabolic homeostasis, thereby defining DEL-1 as a promising biomarker and therapeutic target for CVMDs.
Despite our detailed understanding of the role of DEL-1 in various pathophysiological processes, several questions remain to be answered. We propose some solutions to these questions in this review. First, systemic overexpression rather than endothelial cell-specific overexpression of DEL-1 inhibited the occurrence and development of AS, and the mechanism remains unclear (72, 73). Other cells, such as macrophage-specific overexpression mice, may help us understand the role of DEL-1 in AS. Future basic research on the use of recombinant DEL-1 in the treatment of AS can provide a reference for its clinical application. Second, although Wei et al. found that DEL-1 treatment attenuated hypertension-induced cardiac remodeling, this protective effect may be attributable to reduced blood pressure (41). More direct evidence for the treatment of DEL-1 in cardiac remodeling is lacking. The application of other cardiac remodeling models could better reveal the therapeutic effect of DEL-1 on cardiac remodeling. In vitro experiments can also help us further understand the mechanism by which DEL-1 treatment improves cardiac remodeling. Third, DEL-1 deficiency ameliorated cardiac dysfunction and remodeling in MI by promoting inflammation (42). Although the data in this study are sufficient, we remain concerned about the extent of increased inflammation caused by DEL-1 deficiency, as excessive inflammation is damaging. Future treatment with DEL-1 overexpression or recombinant protein may help us to further understand the role and mechanism of DEL-1 in MI.
The protective effect of DEL-1 in CVMDs has important clinical value. There is currently only one phase II, multicenter, double-blind, placebo-controlled study of DEL-1 in the treatment of intermittent claudication, which combined a plasmid encoding DEL-1 with poloxamer 188 to form VLTS-589 and delivered this treatment intramuscularly (123). Although the outcomes of DEL-1 plasmid-treated patients did not change compared with the controls, this was an important attempt at clinical application of DEL-1. Some researchers have also used DEL-1 for tissue engineering to promote angiogenesis (119, 124). On the one hand, we can use gene therapy that promotes the expression of DEL-1 by constructing plasmids for clinical experiments, and on the other hand, we can also use nanomaterials and other technologies to deliver recombinant DEL-1 protein or plasmids to target tissues, such as the heart and brain. In addition, well-designed, large-scale, high-quality, and multicenter clinical trials are needed to evaluate the safety, toxicological profile, and clinical utility of DEL-1 in human patients with CVMDs.
Collectively, DEL-1 is a promising biomarker and therapeutic target for CVMDs.
JW and MW participated in the design of the project. MZ and CL were responsible for drafting the manuscript. ZZ was responsible for the figures of this review. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (82070436, 82100292, 82270454).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bonora E. The metabolic syndrome and cardiovascular disease. Ann Med (2006) 38(1):64–80. doi: 10.1080/07853890500401234
2. Kasper P, Martin A, Lang S, Kütting F, Goeser T, Demir M, et al. NAFLD and cardiovascular diseases: a clinical review. Clin Res Cardiol (2021) 110(7):921–37. doi: 10.1007/s00392-020-01709-7
3. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics-2018 update: A report from the American heart association. Circulation (2018) 137(12):e67–e492. doi: 10.1161/CIR.0000000000000558
4. Timmis A, Townsend N, Gale C, Grobbee R, Maniadakis N, Flather M, et al. European Society of cardiology: Cardiovascular disease statistics 2017. Eur Heart J (2018) 39(7):508–79. doi: 10.1093/eurheartj/ehx628
5. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep (2018) 20(2):12. doi: 10.1007/s11906-018-0812-z
6. Townsend N, Nichols M, Scarborough P, Rayner M. Cardiovascular disease in Europe–epidemiological update 2015. Eur Heart J (2015) 36(40):2696–705. doi: 10.1093/eurheartj/ehv428
7. Roth GA, Forouzanfar MH, Moran AE, Barber R, Nguyen G, Feigin VL, et al. Demographic and epidemiologic drivers of global cardiovascular mortality. N Engl J Med (2015) 372(14):1333–41. doi: 10.1056/NEJMoa1406656
8. Mechanick JI, Farkouh ME, Newman JD, Garvey WT. Cardiometabolic-based chronic disease, adiposity and dysglycemia drivers: JACC state-of-the-Art review. J Am Coll Cardiol (2020) 75(5):525–38. doi: 10.1016/j.jacc.2019.11.044
9. Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity (2017) 47(3):406–20. doi: 10.1016/j.immuni.2017.08.009
10. Ilatovskaya DV, Halade GV, DeLeon-Pennell KY. Adaptive immunity-driven inflammation and cardiovascular disease. Am J Physiol Heart Circ Physiol (2019) 317(6):H1254–h1257. doi: 10.1152/ajpheart.00642.2019
11. Pedicino D, Giglio AF, Ruggio A, Massaro G, D'Aiello A, Trotta F, et al. Lymphocytes and innate-adaptive immunity crosstalk: Role in cardiovascular disease and therapeutic perspectives. Thromb Haemost (2018) 118(8):1352–69. doi: 10.1055/s-0038-1666860
12. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of CANTOS and beyond. J Am Coll Cardiol (2017) 70(18):2278–89. doi: 10.1016/j.jacc.2017.09.028
13. Danesh J, Kaptoge S, Mann AG, Sarwar N, Wood A, Angleman SB, et al. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PloS Med (2008) 5(4):e78. doi: 10.1371/journal.pmed.0050078
14. Ridker PM. From c-reactive protein to interleukin-6 to interleukin-1: Moving upstream to identify novel targets for atheroprotection. Circ Res (2016) 118(1):145–56. doi: 10.1161/CIRCRESAHA.115.306656
15. Chen YL, Qiao YC, Pan YH, Xu Y, Huang YC, Wang YH, et al. Correlation between serum interleukin-6 level and type 1 diabetes mellitus: A systematic review and meta-analysis. Cytokine (2017) 94:14–20. doi: 10.1016/j.cyto.2017.01.002
16. Atieh MA, Faggion CM Jr., Seymour GJ. Cytokines in patients with type 2 diabetes and chronic periodontitis: A systematic review and meta-analysis. Diabetes Res Clin Pract (2014) 104(2):e38–45. doi: 10.1016/j.diabres.2014.02.002
17. Wang T, He C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev (2018) 44:38–50. doi: 10.1016/j.cytogfr.2018.10.002
18. Zhang J, Xu Y, Ding W, Zhao M, Liu J, Ye J, et al. Increased expression of IL-20 is associated with ischemic cardiomyopathy and acute myocardial infarction. biomark Med (2021) 15(17):1641–50. doi: 10.2217/bmm-2020-0529
19. Lyngbakken MN, Myhre PL, Røsjø H, Omland T. Novel biomarkers of cardiovascular disease: Applications in clinical practice. Crit Rev Clin Lab Sci (2019) 56(1):33–60. doi: 10.1080/10408363.2018.1525335
20. Ruscitti P, Masedu F, Alvaro S, Airò P, Battafarano N, Cantarini L, et al. Anti-interleukin-1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): A multicentre, open-label, randomised controlled trial. PloS Med (2019) 16(9):e1002901. doi: 10.1371/journal.pmed.1002901
21. Zhao M, Zhang J, Xu Y, Liu J, Ye J, Wang Z, et al. Selective inhibition of NLRP3 inflammasome reverses pressure overload-induced pathological cardiac remodeling by attenuating hypertrophy, fibrosis, and inflammation. Int Immunopharmacol (2021) 99:108046. doi: 10.1016/j.intimp.2021.108046
22. Unamuno X, Gómez-Ambrosi J, Ramírez B, Rodríguez A, Becerril S, Valentí V, et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell Mol Immunol (2021) 18(4):1045–57. doi: 10.1038/s41423-019-0296-z
23. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med (2019) 380(8):752–62. doi: 10.1056/NEJMoa1809798
24. Lutgens E, Atzler D, Döring Y, Duchene J, Steffens S, Weber C. Immunotherapy for cardiovascular disease. Eur Heart J (2019) 40(48):3937–46. doi: 10.1093/eurheartj/ehz283
25. Chen X, Wu Y, Wang L. Fat-resident tregs: an emerging guard protecting from obesity-associated metabolic disorders. Obes Rev (2013) 14(7):568–78. doi: 10.1111/obr.12033
26. Stolley JM, Masopust D. Tissue-resident memory T cells live off the fat of the land. Cell Res (2017) 27(7):847–8. doi: 10.1038/cr.2017.49
27. Zaman R, Hamidzada H, Kantores C, Wong A, Dick SA, Wang Y, et al. Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity (2021) 54(9):2057–2071.e6. doi: 10.1016/j.immuni.2021.07.006
28. Wong NR, Mohan J, Kopecky BJ, Guo S, Du L, Leid J, et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity (2021) 54(9):2072–2088.e7. doi: 10.1016/j.immuni.2021.07.003
29. Gimbrone MA Jr., García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res (2016) 118(4):620–36. doi: 10.1161/CIRCRESAHA.115.306301
30. Tombor LS, John D, Glaser SF, Luxán G, Forte E, Furtado M, et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun (2021) 12(1):681. doi: 10.1161/CIRCRESAHA.115.306301
31. Horckmans M, Bianchini M, Santovito D, Megens RTA, Springael JY, Negri I, et al. Pericardial adipose tissue regulates granulopoiesis, fibrosis, and cardiac function after myocardial infarction. Circulation (2018) 137(9):948–60. doi: 10.1161/CIRCULATIONAHA.117.028833
32. Burhans MS, Hagman DK, Kuzma JN, Schmidt KA, Kratz M. Contribution of adipose tissue inflammation to the development of type 2 diabetes mellitus. Compr Physiol (2018) 9(1):1–58. doi: 10.1002/cphy.c170040
33. Matzinger P, Kamala T. Tissue-based class control: the other side of tolerance. Nat Rev Immunol (2011) 11(3):221–30. doi: 10.1038/nri2940
34. Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol (2011) 12(11):1035–44. doi: 10.1038/ni.2109
35. Hajishengallis G, Chavakis T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol (2013) 34(1):1–6. doi: 10.1016/j.it.2012.08.003
36. Gonçalves LM. Angiogenic growth factors: potential new treatment for acute myocardial infarction? Cardiovasc Res (2000) 45(2):294–302. doi: 10.1016/S0008-6363(99)00358-2
37. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol (2015) 5(4):1841–75. doi: 10.1002/cphy.c150006
38. Daseke MJ 2nd, Tenkorang MAA, Chalise U, Konfrst SR, Lindsey ML. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol (2020) 91-92:109–16. doi: 10.1016/j.matbio.2020.03.010
39. Hajishengallis G, Chavakis T. DEL-1-Regulated immune plasticity and inflammatory disorders. Trends Mol Med (2019) 25(5):444–59. doi: 10.1016/j.molmed.2019.02.010
40. Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, et al. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol (2019) 20(1):40–9. doi: 10.1038/s41590-018-0249-1
41. Failer T, Amponsah-Offeh M, Neuwirth A, Kourtzelis I, Subramanian P, Mirtschink P, et al. Developmental endothelial locus-1 protects from hypertension-induced cardiovascular remodeling via immunomodulation. J Clin Invest (2022) 132(6):e126155. doi: 10.1172/JCI126155.
42. Wei X, Zou S, Xie Z, Wang Z, Huang N, Cen Z, et al. EDIL3 deficiency ameliorates adverse cardiac remodeling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc Res (2021) 118(9):2179–95. doi: 10.1093/cvr/cvab269.
43. Sun JL, Park J, Lee T, Jeong JH, Jung TW. DEL-1 ameliorates high-fat diet-induced insulin resistance in mouse skeletal muscle through SIRT1/SERCA2-mediated ER stress suppression. Biochem Pharmacol (2020) 171:113730. doi: 10.1016/j.bcp.2019.113730
44. Hidai C, Zupancic T, Penta K, Mikhail A, Kawana M, Quertermous EE, et al. Cloning and characterization of developmental endothelial locus-1: an embryonic endothelial cell protein that binds the alphavbeta3 integrin receptor. Genes Dev (1998) 12(1):21–33. doi: 10.1101/gad.12.1.21
45. Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, et al. The leukocyte integrin antagonist del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol (2012) 13(5):465–73. doi: 10.1038/ni.2260
46. Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, et al. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science (2008) 322(5904):1101–4. doi: 10.1126/science.1165218
47. Choi EY, Lim JH, Neuwirth A, Economopoulou M, Chatzigeorgiou A, Chung KJ, et al. Developmental endothelial locus-1 is a homeostatic factor in the central nervous system limiting neuroinflammation and demyelination. Mol Psychiatry (2015) 20(7):880–8. doi: 10.1038/mp.2014.146
48. Shin J, Maekawa T, Abe T, Hajishengallis E, Hosur K, Pyaram K, et al. DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci Transl Med (2015) 7(307):307ra155. doi: 10.1126/scitranslmed.aac5380
49. Shin J, Hosur KB, Pyaram K, Jotwani R, Liang S, Chavakis T, et al. Expression and function of the homeostatic molecule del-1 in endothelial cells and the periodontal tissue. Clin Dev Immunol (2013) 2013:617809. doi: 10.1155/2013/617809
50. Folwaczny M, Karnesi E, Berger T, Paschos E. Clinical association between chronic periodontitis and the leukocyte extravasation inhibitors developmental endothelial locus-1 and pentraxin-3. Eur J Oral Sci (2017) 125(4):258–64. doi: 10.1111/eos.12357
51. Ziogas A, Maekawa T, Wiessner JR, Le TT, Sprott D, Troullinaki M, et al. DHEA inhibits leukocyte recruitment through regulation of the integrin antagonist DEL-1. J Immunol (2020) 204(5):1214–24. doi: 10.4049/jimmunol
52. Maekawa T, Tamura H, Domon H, Hiyoshi T, Isono T, Yonezawa D, et al. Erythromycin inhibits neutrophilic inflammation and mucosal disease by upregulating DEL-1. JCI Insight (2020) 5(15):e136706. doi: 10.1172/jci.insight.136706
53. Kim H, Lee SH, Lee MN, Oh GT, Choi KC, Choi EY. p53 regulates the transcription of the anti-inflammatory molecule developmental endothelial locus-1 (Del-1). Oncotarget (2013) 4(11):1976–85. doi: 10.18632/oncotarget.1318
54. Wenzina J, Holzner S, Puujalka E, Cheng PF, Forsthuber A, Neumüller K, et al. Inhibition of p38/MK2 signaling prevents vascular invasion of melanoma. J Invest Dermatol (2020) 140(4):878–890.e5. doi: 10.1016/j.jid.2019.08.451
55. Goris A, Sawcer S, Vandenbroeck K, Carton H, Billiau A, Setakis E, et al. New candidate loci for multiple sclerosis susceptibility revealed by a whole genome association screen in a Belgian population. J Neuroimmunol (2003) 143(1-2):65–9. doi: 10.1016/j.jneuroim.2003.08.013
56. Schürpf T, Chen Q, Liu JH, Wang R, Springer TA, Wang JH. The RGD finger of del-1 is a unique structural feature critical for integrin binding. FASEB J (2012) 26(8):3412–20. doi: 10.1096/fj.11-202036
57. Zhong J, Eliceiri B, Stupack D, Penta K, Sakamoto G, Quertermous T, et al. Neovascularization of ischemic tissues by gene delivery of the extracellular matrix protein del-1. J Clin Invest (2003) 112(1):30–41. doi: 10.1172/JCI17034
58. Hidai C, Kawana M, Kitano H, Kokubun S. Discoidin domain of Del1 protein contributes to its deposition in the extracellular matrix. Cell Tissue Res (2007) 330(1):83–95. doi: 10.1007/s00441-007-0456-9
59. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol (2015) 15(11):692–704. doi: 10.1038/nri3908
60. Kang YY, Kim DY, Lee SH, Choi EY. Deficiency of developmental endothelial locus-1 (Del-1) aggravates bleomycin-induced pulmonary fibrosis in mice. Biochem Biophys Res Commun (2014) 445(2):369–74. doi: 10.1016/j.bbrc.2014.02.009
61. Kourtzelis I, Kotlabova K, Lim JH, Mitroulis I, Ferreira A, Chen LS, et al. Developmental endothelial locus-1 modulates platelet-monocyte interactions and instant blood-mediated inflammatory reaction in islet transplantation. Thromb Haemost (2016) 115(4):781–8. doi: 10.1160/th15-05-0429
62. Dasgupta SK, Le A, Chavakis T, Rumbaut RE, Thiagarajan P. Developmental endothelial locus-1 (Del-1) mediates clearance of platelet microparticles by the endothelium. Circulation (2012) 125(13):1664–72. doi: 10.1161/CIRCULATIONAHA.111.068833
63. Hanayama R, Tanaka M, Miwa K, Nagata S. Expression of developmental endothelial locus-1 in a subset of macrophages for engulfment of apoptotic cells. J Immunol (2004) 172(6):3876–82. doi: 10.4049/jimmunol.172.6.3876
64. Hajishengallis G, Chavakis T. DEL-1: a potential therapeutic target in inflammatory and autoimmune disease? Expert Rev Clin Immunol (2021) 17(6):549–52. doi: 10.1080/1744666X.2021.1915771
65. Fu Y, Tsauo J, Sun Y, Wang Z, Kim KY, Lee SH, et al. Developmental endothelial locus-1 prevents development of peritoneal adhesions in mice. Biochem Biophys Res Commun (2018) 500(3):783–9. doi: 10.1016/j.bbrc.2018.04.158
66. Mitroulis I, Chen LS, Singh RP, Kourtzelis I, Economopoulou M, Kajikawa T, et al. Secreted protein del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J Clin Invest (2017) 127(10):3624–39. doi: 10.1172/JCI92571
67. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci (Lond) (2018) 132(12):1243–52. doi: 10.1042/CS20180306
68. Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev (2013) 93(3):1317–542. doi: 10.1152/physrev.00004.2012
69. Libby P. The changing landscape of atherosclerosis. Nature (2021) 592(7855):524–33. doi: 10.1038/s41586-021-03392-8
71. Finn NA, Eapen D, Manocha P, Al Kassem H, Lassegue B, Ghasemzadeh N, et al. Coronary heart disease alters intercellular communication by modifying microparticle-mediated microRNA transport. FEBS Lett (2013) 587(21):3456–63. doi: 10.1016/j.febslet.2013.08.034
72. Kakino A, Fujita Y, Nakano A, Horiuchi S, Sawamura T. Developmental endothelial locus-1 (Del-1) inhibits oxidized low-density lipoprotein activity by direct binding, and its overexpression attenuates atherogenesis in mice. Circ J (2016) 80(12):2541–9. doi: 10.1253/circj.CJ-16-0808
73. Subramanian P, Prucnal M, Gercken B, Economopoulou M, Hajishengallis G, Chavakis T. Endothelial cell-specific overexpression of developmental endothelial locus-1 does not influence atherosclerosis development in ApoE(-/-) mice. Thromb Haemost (2017) 117(10):2003–5. doi: 10.1160/th17-03-0160
74. Burnier L, Fontana P, Angelillo-Scherrer A, Kwak BR. Intercellular communication in atherosclerosis. Physiol (Bethesda) (2009) 24:36–44. doi: 10.1152/physiol.00036.2008
75. Boulanger CM, Dignat-George F. Microparticles: an introduction. Arterioscler Thromb Vasc Biol (2011) 31(1):2–3. doi: 10.1161/ATVBAHA.110.220095
76. Benetos A, Petrovic M, Strandberg T. Hypertension management in older and frail older patients. Circ Res (2019) 124(7):1045–60. doi: 10.1161/circresaha.118.313236
77. Cooper LL, Rong J, Benjamin EJ, Larson MG, Levy D, Vita JA, et al. Components of hemodynamic load and cardiovascular events: the framingham heart study. Circulation (2015) 131(4):354–61; discussion 361. doi: 10.1161/circulationaha.114.011357
78. Madhur MS, Elijovich F, Alexander MR, Pitzer A, Ishimwe J, Van Beusecum JP, et al. Hypertension: Do inflammation and immunity hold the key to solving this epidemic? Circ Res (2021) 128(7):908–33. doi: 10.1161/circresaha.121.318052
79. Ye J, Que B, Huang Y, Lin Y, Chen J, Liu L, et al. Interleukin-12p35 knockout promotes macrophage differentiation, aggravates vascular dysfunction, and elevates blood pressure in angiotensin II-infused mice. Cardiovasc Res (2019) 115(6):1102–13. doi: 10.1093/cvr/cvy263
80. Ye J, Wang Y, Wang Z, Ji Q, Huang Y, Zeng T, et al. Circulating Th1, Th2, Th9, Th17, Th22, and treg levels in aortic dissection patients. Mediators Inflammation (2018) 2018:5697149. doi: 10.1155/2018/5697149
81. Ye J, Wang Y, Wang Z, Lin Y, Liu L, Zhou Q, et al. Circulating IL-37 levels are elevated in patients with hypertension. Exp Ther Med (2021) 21(6):558. doi: 10.3892/etm.2021.9990
82. Ye J, Wang Y, Wang Z, Liu L, Yang Z, Wang M, et al. The expression of IL-12 family members in patients with hypertension and its association with the occurrence of carotid atherosclerosis. Mediators Inflammation (2020) 2020:2369279. doi: 10.1155/2020/2369279
83. Ye J, Ji Q, Liu J, Liu L, Huang Y, Shi Y, et al. Interleukin 22 promotes blood pressure elevation and endothelial dysfunction in angiotensin II-treated mice. J Am Heart Assoc (2017) 6(10): e005875. doi: 10.1161/JAHA.117.005875
84. Kirabo A, Fontana V, Faria AP de, Loperena R, Galindo CL, Wu J, et al. DC Isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest (2014) 124(10):4642–56. doi: 10.1172/JCI74084
85. Itani HA, McMaster WG Jr., Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, et al. Activation of human T cells in hypertension: Studies of humanized mice and hypertensive humans. Hypertension (2016) 68(1):123–32. doi: 10.1161/HYPERTENSIONAHA.116.07237
86. Saleh MA, Norlander AE, Madhur MS. Inhibition of interleukin 17-a but not interleukin-17F signaling lowers blood pressure and reduces end-organ inflammation in angiotensin II-induced hypertension. JACC Basic Transl Sci (2016) 1(7):606–16. doi: 10.1016/j.jacbts.2016.07.009
87. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension (2010) 55(2):500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094
88. Li Y, Wu Y, Zhang C, Li P, Cui W, Hao J, et al. γδT cell-derived interleukin-17A via an interleukin-1β-dependent mechanism mediates cardiac injury and fibrosis in hypertension. Hypertension (2014) 64(2):305–14. doi: 10.1161/HYPERTENSIONAHA.113.02604
89. Basting T, Lazartigues E. DOCA-salt hypertension: an update. Curr Hypertens Rep (2017) 19(4):32. doi: 10.1007/s11906-017-0731-4
90. Amador CA, Barrientos V, Peña J, Herrada AA, González M, Valdés S, et al. Spironolactone decreases DOCA-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension (2014) 63(4):797–803. doi: 10.1161/HYPERTENSIONAHA.113.02883
91. Rezaee M, Penta K, Quertermous T. Del1 mediates VSMC adhesion, migration, and proliferation through interaction with integrin alpha(v)beta(3). Am J Physiol Heart Circ Physiol (2002) 282(5):H1924–32. doi: 10.1152/ajpheart.00921.2001
92. Barhoumi T, Fraulob-Aquino JC, Mian MOR, Ouerd S, Idris-Khodja N, Huo KG, et al. Matrix metalloproteinase-2 knockout prevents angiotensin II-induced vascular injury. Cardiovasc Res (2017) 113(14):1753–62. doi: 10.1093/cvr/cvx115
93. Diaz-Canestro C, Puspitasari YM, Liberale L, Guzik TJ, Flammer AJ, Bonetti NR, et al. MMP-2 knockdown blunts age-dependent carotid stiffness by decreasing elastin degradation and augmenting eNOS activation. Cardiovasc Res (2021) 118(10):2385–96. doi: 10.1093/cvr/cvab300.
94. Liu P, Sun M, Sader S. Matrix metalloproteinases in cardiovascular disease. Can J Cardiol (2006) 22 Suppl B(Suppl B):25b–30b. doi: 10.1016/s0828-282x(06)70983-7
95. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol (2018) 15(7):387–407. doi: 10.1038/s41569-018-0007-y
96. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther (2010) 128(1):191–227. doi: 10.1016/j.pharmthera.2010.04.005
97. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol (2016) 97:245–62. doi: 10.1016/j.yjmcc.2016.06.001
98. Wang M, Zhao M, Yu J, Xu Y, Zhang J, Liu J, et al. MCC950, a selective NLRP3 inhibitor, attenuates adverse cardiac remodeling following heart failure through improving the cardiometabolic dysfunction in obese mice. Front Cardiovasc Med (2022) 9:727474. doi: 10.3389/fcvm.2022.727474
99. Wang Z, Xu Y, Wang M, Ye J, Liu J, Jiang H, et al. TRPA1 inhibition ameliorates pressure overload-induced cardiac hypertrophy and fibrosis in mice. EBioMedicine (2018) 36:54–62. doi: 10.1016/j.ebiom.2018.08.022
100. Ye J, Liu L, Ji Q, Huang Y, Shi Y, Shi L, et al. Anti-Interleukin-22-Neutralizing antibody attenuates angiotensin II-induced cardiac hypertrophy in mice. Mediators Inflammation (2017) 2017:5635929. doi: 10.1155/2017/5635929
101. Wang Z, Ye D, Ye J, Wang M, Liu J, Jiang H, et al. The TRPA1 channel in the cardiovascular system: Promising features and challenges. Front Pharmacol (2019) 10:1253. doi: 10.3389/fphar.2019.01253
102. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res (2016) 118(3):400–9. doi: 10.1161/CIRCRESAHA.115.307778
103. Vliegen HW, van der Laarse A, Cornelisse CJ, Eulderink F. Myocardial changes in pressure overload-induced left ventricular hypertrophy. a study on tissue composition, polyploidization and multinucleation. Eur Heart J (1991) 12(4):488–94. doi: 10.1093/oxfordjournals.eurheartj.a059928
104. Kim DY, Lee SH, Fu Y, Jing F, Kim WY, Hong SB, et al. Del-1, an endogenous inhibitor of TGF-β activation, attenuates fibrosis. Front Immunol (2020) 11:68. doi: 10.3389/fimmu.2020.00068
105. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res (2015) 116(4):674–99. doi: 10.1161/CIRCRESAHA.116.305348
106. Moran AE, Forouzanfar MH, Roth GA, Mensah GA, Ezzati M, Flaxman A, et al. The global burden of ischemic heart disease in 1990 and 2010: the global burden of disease 2010 study. Circulation (2014) 129(14):1493–501. doi: 10.1161/CIRCULATIONAHA.113.004046
107. Kown MH, Suzuki T, Koransky ML, Penta K, Sakamoto G, Jahncke CL, et al. Comparison of developmental endothelial locus-1 angiogenic factor with vascular endothelial growth factor in a porcine model of cardiac ischemia. Ann Thorac Surg (2003) 76(4):1246–51. doi: 10.1016/S0003-4975(03)00721-5
108. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204(12):3037–47. doi: 10.1084/jem.20070885
109. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol (2014) 11(5):255–65. doi: 10.1038/nrcardio.2014.28
110. Frodermann V, Nahrendorf M. Neutrophil-macrophage cross-talk in acute myocardial infarction. Eur Heart J (2017) 38(3):198–200. doi: 10.1093/eurheartj/ehw085
111. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res (2012) 110(1):159–73. doi: 10.1161/CIRCRESAHA.111.243162
112. Wang H, Li X, Kajikawa T, Shin J, Lim JH, Kourtzelis I, et al. Stromal cell-derived DEL-1 inhibits tfh cell activation and inflammatory arthritis. J Clin Invest (2021) 131(19): e150578. doi: 10.1172/JCI150578
113. Li X, Colamatteo A, Kalafati L, Kajikawa T, Wang H, Lim JH, et al. The DEL-1/β3 integrin axis promotes regulatory T cell responses during inflammation resolution. J Clin Invest (2020) 130(12):6261–77. doi: 10.1172/JCI137530
114. Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol (2010) 11(4):328–34. doi: 10.1038/ni.1854
115. Salio M, Chimenti S, De Angelis N, Molla F, Maina V, Nebuloni M, et al. Cardioprotective function of the long pentraxin PTX3 in acute myocardial infarction. Circulation (2008) 117(8):1055–64. doi: 10.1161/CIRCULATIONAHA.107.749234
116. Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med (2011) 17(5):581–8. doi: 10.1038/nm.2354
117. Li R, Zeng J, Ren T. Expression of DEL-1 in alveolar epithelial cells prevents lipopolysaccharide-induced inflammation, oxidative stress, and eosinophil recruitment in acute lung injury. Int Immunopharmacol (2022) 110:108961. doi: 10.1016/j.intimp.2022.108961
118. Fan Y, Zhu W, Yang M, Zhu Y, Shen F, Hao Q, et al. Del-1 gene transfer induces cerebral angiogenesis in mice. Brain Res (2008) 1219:1–7. doi: 10.1016/j.brainres.2008.05.003
119. Ciucurel EC, Vlahos AE, Sefton MV. Using del-1 to tip the angiogenic balance in endothelial cells in modular constructs. Tissue Eng Part A (2014) 20(7-8):1222–34. doi: 10.1089/ten.tea.2013.0241
120. Penta K, Varner JA, Liaw L, Hidai C, Schatzman R, Quertermous T. Del1 induces integrin signaling and angiogenesis by ligation of alphaVbeta3. J Biol Chem (1999) 274(16):11101–9. doi: 10.1074/jbc.274.16.11101
121. Aoka Y, Johnson FL, Penta K, Hirata Ki K, Hidai C, Schatzman R, et al. The embryonic angiogenic factor Del1 accelerates tumor growth by enhancing vascular formation. Microvasc Res (2002) 64(1):148–61. doi: 10.1006/mvre.2002.2414
122. Ho HK, Jang JJ, Kaji S, Spektor G, Fong A, Yang P, et al. Developmental endothelial locus-1 (Del-1), a novel angiogenic protein: its role in ischemia. Circulation (2004) 109(10):1314–9. doi: 10.1161/01.CIR.0000118465.36018.2D
123. Grossman PM, Mendelsohn F, Henry TD, Hermiller JB, Litt M, Saucedo JF, et al. Results from a phase II multicenter, double-blind placebo-controlled study of del-1 (VLTS-589) for intermittent claudication in subjects with peripheral arterial disease. Am Heart J (2007) 153(5):874–80. doi: 10.1016/j.ahj.2007.01.038
124. Ciucurel EC, Sefton MV. Del-1 overexpression in endothelial cells increases vascular density in tissue-engineered implants containing endothelial cells and adipose-derived mesenchymal stromal cells. Tissue Eng Part A (2014) 20(7-8):1235–52. doi: 10.1089/ten.tea.2013.0242
125. Klotzsche-von Ameln A, Cremer S, Hoffmann J, Schuster P, Khedr S, Korovina I, et al. Endogenous developmental endothelial locus-1 limits ischaemia-related angiogenesis by blocking inflammation. Thromb Haemost (2017) 117(6):1150–63. doi: 10.1160/TH16-05-0354
126. Jeong D, Ban S, Oh S, Jin Lee S, Yong Park S, Koh YW. Prognostic significance of EDIL3 expression and correlation with mesenchymal phenotype and microvessel density in lung adenocarcinoma. Sci Rep (2017) 7(1):8649. doi: 10.1038/s41598-017-08851-9
127. Shen W, Zhu S, Qin H, Zhong M, Wu J, Zhang R, et al. EDIL3 knockdown inhibits retinal angiogenesis through the induction of cell cycle arrest in vitro. Mol Med Rep (2017) 16(4):4054–60. doi: 10.3892/mmr.2017.7122
128. Niu X, Han Q, Liu Y, Li J, Hou R, Li J, et al. Psoriasis-associated angiogenesis is mediated by EDIL3. Microvasc Res (2020) 132:104056. doi: 10.1016/j.mvr.2020.104056
129. Kitano H, Mamiya A, Ishikawa T, Fujiwara Y, Masaoka Y, Miki T, et al. An epidermal growth factor motif of developmental endothelial locus 1 protein inhibits efficient angiogenesis in explanted squamous cell carcinoma In vivo. Rev Invest Clin (2020) 73(1):039–51. doi: 10.24875/ric.20000375
130. Kalani A, Kamat PK, Kalani K, Tyagi N. Epigenetic impact of curcumin on stroke prevention. Metab Brain Dis (2015) 30(2):427–35. doi: 10.1007/s11011-014-9537-0
131. Cramer SC, Nelles G, Benson RR, Kaplan JD, Parker RA, Kwong KK, et al. A functional MRI study of subjects recovered from hemiparetic stroke. Stroke (1997) 28(12):2518–27. doi: 10.1161/01.STR.28.12.2518
132. Altar CA, Laeng P, Jurata LW, Brockman JA, Lemire A, Bullard J, et al. Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. J Neurosci (2004) 24(11):2667–77. doi: 10.1523/JNEUROSCI.5377-03.2004
133. Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, et al. Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. J Neurosci (2003) 23(34):10841–51. doi: 10.1523/JNEUROSCI.23-34-10841.2003
134. Newton SS, Girgenti MJ, Collier EF, Duman RS. Electroconvulsive seizure increases adult hippocampal angiogenesis in rats. Eur J Neurosci (2006) 24(3):819–28. doi: 10.1111/j.1460-9568.2006.04958.x
135. Zhou B, Lu Y, Hajifathalian K, Bentham J, Cesare M, Danaei G, et al. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet (2016) 387(10027):1513–30. doi: 10.1016/S0140-6736(16)00618-8
136. Stöhr R, Federici M. Insulin resistance and atherosclerosis: convergence between metabolic pathways and inflammatory nodes. Biochem J (2013) 454(1):1–11. doi: 10.1042/bj20130121
137. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity (2022) 55(1):31–55. doi: 10.1016/j.immuni.2021.12.013
138. Stefani L, Galanti G. Physical exercise prescription in metabolic chronic disease. Adv Exp Med Biol (2017) 1005:123–41. doi: 10.1007/978-981-10-5717-5_6
139. Kwon CH, Sun JL, Kim MJ, Abd El-Aty AM, Jeong JH, Jung TW. Clinically confirmed DEL-1 as a myokine attenuates lipid-induced inflammation and insulin resistance in 3T3-L1 adipocytes via AMPK/HO-1- pathway. Adipocyte (2020) 9(1):576–86. doi: 10.1080/21623945.2020.1823140
140. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
141. Cobb LP, Siamakpour-Reihani S, Zhang D, Qin X, Owzar K, Zhou C, et al. Obesity and altered angiogenic-related gene expression in endometrial cancer. Gynecol Oncol (2021) 163(2):320–6. doi: 10.1016/j.ygyno.2021.08.010
142. Kim WY, Lee SH, Kim DY, Ryu HJ, Chon GR, Park YY, et al. Serum developmental endothelial locus-1 is associated with severity of sepsis in animals and humans. Sci Rep (2019) 9(1):13005. doi: 10.1038/s41598-019-49564-5
Keywords: DEL-1, cardiovascular diseases, metabolic diseases, inflammation resolution, anti-inflammation
Citation: Zhao M, Zheng Z, Li C, Wan J and Wang M (2022) Developmental endothelial locus-1 in cardiovascular and metabolic diseases: A promising biomarker and therapeutic target. Front. Immunol. 13:1053175. doi: 10.3389/fimmu.2022.1053175
Received: 25 September 2022; Accepted: 11 November 2022;
Published: 28 November 2022.
Edited by:
Uzma Saqib, Indian Institute of Technology Indore, IndiaReviewed by:
Evelyn Mendoza, Universidad Libre, ColombiaCopyright © 2022 Zhao, Zheng, Li, Wan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Menglong Wang, d2h1d2FuZ21lbmdsb25nQDE2My5jb20=; Jun Wan, d2FuanVuQHdodS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.