 Yue Qin
Yue Qin Guotai Xu
Guotai Xu- 1National Institute of Biological Sciences, Beijing, China
- 2Tsinghua Institute of Multidisciplinary Biomedical Research, Tsinghua University, Beijing, China
Chimeric antigen receptor (CAR) T-cell therapy, belonging to adoptive immune cells therapy, utilizes engineered immunoreceptors to enhance tumor-specific killing. By now new generations of CAR T-cell therapies dramatically promote the effectiveness and robustness in leukemia cases. However, only a few CAR T-cell therapies gain FDA approval till now, which are applied to hematologic cancers. Targeting solid tumors through CAR T-cell therapies still faces many problems, such as tumor heterogeneity, antigen loss, infiltration inability and immunosuppressive micro-environment. Recent advances provide new insights about the mechanisms of CAR T-cell therapy resistance and give rise to potential reversal therapies. In this review, we mainly introduce existing barriers when treating solid tumors with CAR T-cells and discuss the methods to overcome these challenges.
Introduction
Immune therapies are powerful for cancer treatment and, regardless of the drug types used (chemotherapy, radiotherapy or chemical compounds with specific targets), tumor regression usually involves participation of the immune system (1, 2). Immune checkpoint blockage (ICB) can unleash the inhibition of immune response and enhance antitumor effect (3, 4). However, the expression of immune checkpoint is not tumor unique and may result in strong side effects (5, 6). Furthermore, ICB usually could not trigger an effective response due to few tumor-infiltrated lymphocytes (7). Recent studies showed that loss of some genes may elevate PD-1/PD-L1 in tumor niche and, however, enhanced immune cell infiltration, while the overall result of such gene loss tends to be reduced tumor growth which indicates the power of immune cells (8–12).
Proposed in 1989, CAR T-cells have evolved for three decades and usually employ the TCR activation intracellular domain fused with antibody variable domains to kill specific antigen bearing tumor cells (13, 14). Comparing with traditional adoptive cellular therapies that are composed of unmodified host T-cells, CAR T-cells become more sensitive and precise (15). Besides, many attempts gradually improve the effectiveness by utilizing different activation domains or combining costimulatory domains (Figure 1). The 1st generation of CAR contains single chain variable fragment (scFv) and a single CD3ζ subunit which is the intracellular part of CD3 complex that mediates the MHC-TCR signaling transduction to activate T-cells but showed poor efficacy, while adding one costimulatory signaling domain from CD28-CD3ζ or 4-1BB-CD3ζ is now commonly and successfully applied, called 2nd CAR. Furthermore, 3rd CARs that combined both CD28 and 4-1BB give the T cell stronger activation signals (16, 17). The FDA has approved six 2nd CAR T-cell therapies since 2017 as the result of positive clinical cases for anti-blood cancer treatment including acute lymphoblastic leukemia (ALL), non-Hodgkin lymphoma (NHL), follicular lymphoma, mantle cell lymphoma (MCL) and multiple myeloma (18–21).
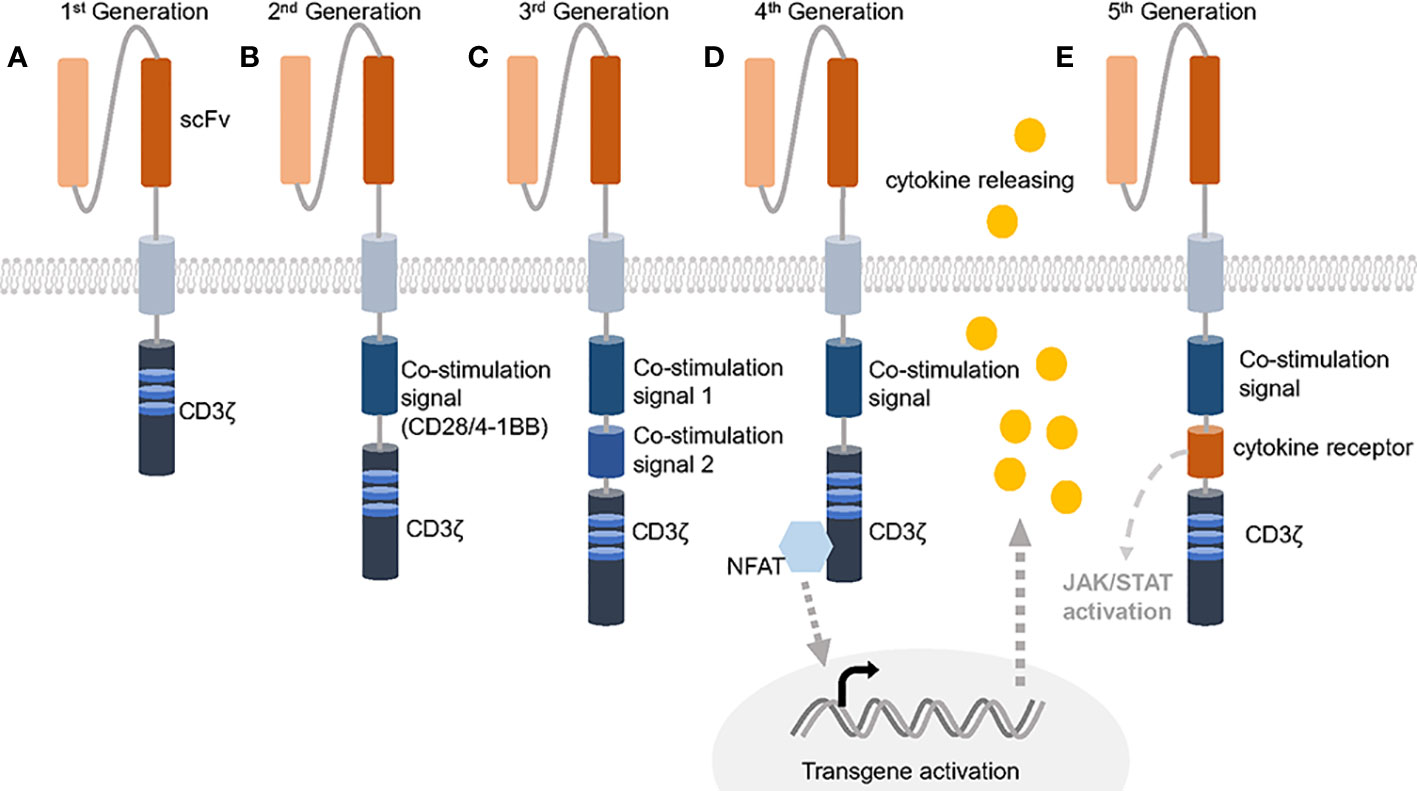
Figure 1 Five generations of CARs. (A) The 1st generation of CAR only contains the extracellular scFv and intracellular CD3ζ activation domine, which could not trigger enough active response. (B) 2nd generation of CAR has an additional co-stimulation domain based on 1st generation and obtained success against leukemia. (C) 3rd generation of CAR increases the number of co-stimulation domain and evoke stronger stimulation after antigen binding. (D, E) 4th generation of CAR is usually referred to TRUCKs that contains one more cytokine inducer compared with 2nd CAR to promote cytokine-mediated killing. 5th CAR can both activate the cytokine receptor dependent JAK/STAT signal and perform the normal function of a 2nd CAR.
Despite the encouraging results in leukemia, many CAR T-cell related clinical trials are still going on for solid tumors (Table 1) (22). Some shows positive response and demonstrate the safety of CAR T-cells. In 2016, the early successful case of CAR T-cells targeting solid tumors is reported and proved the feasibility (23). However, others face many problems such as antigen loss, T-cell exclusion and defects in T-cell infiltration (24–27). As engineered cells, CAR T-cells can be further improved to tackle the specific challenges (28, 29). Now, the 4th and the 5th generation of CARs are born as well as some other modifications on CARs like the dual-antigen targeting CARs that possess dual specificity and dual co-stimulation signal (30, 31). Such CARs help T-cells to sustain longer and act wider. Considering the complexity of solid tumor treatment and abundant state-of-the-art strategies, we divide this review into six topics containing targeting solid tumor antigen variability, enhancing tumor infiltration and T-cell activation-rest cycling, dealing with immunosuppressive microenvironment, controlling CAR T-cell epigenetics changes, choosing the suitable chassis for CAR T-cell production and utilizing combined therapies. In the review, we plan to explain the resistance mechanism and recent efforts to overcome the barriers.
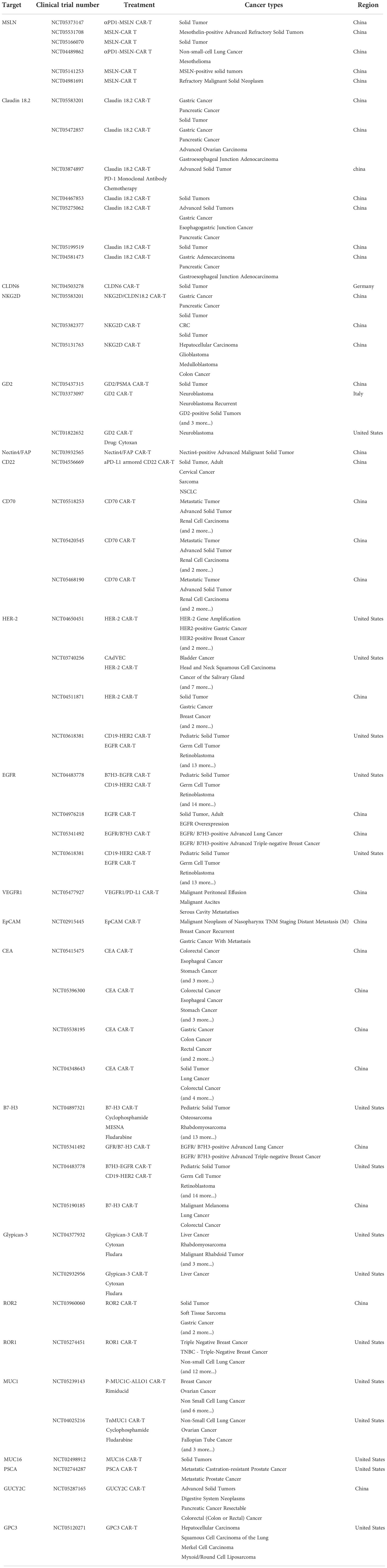
Table 1 Current ongoing CAR T therapy clinical trials in solid tumors.
Targeting solid tumor antigen variability
In fact, TCR T-cells against NY-ESO-1 positive synovial cell sarcoma and melanoma showed great potential in 2010 even before the first successful case that CD19 CAR T-cell cured an Acute Lymphocytic Leukemia (ALL) patient in 2013, which indicated the anti-solid tumor efficacy of engineered T-cell therapies (32). CAR-engineered T-cells targeting IL13Rα2 induced regression of glioblastoma and 3/11 neuroblastoma patients achieved complete remission after GD2-CAR T-cells infusion (23, 33). However, the antigen heterogeneity is a major concern when treating tumor using the concept of precision oncology. Biallelic loss was found when multiple myeloma treated by anti-BCMA CAR T-cells while EGFRvIII-directed CAR T-cells infiltrated glioblastoma and induced on-target tumor killing but the tumor gradually acquired resistance due to antigen loss suggesting the difficulty to identify a unique tumor associated antigen (TAA) (34, 35). Though the leukemia also lacks TAAs, blood system has its tissue-specific marker such as CD19 that represents the B lymphocytes (36). Importantly, the eradicated B-cells can be reloaded by stem cells, which makes it suitable for CAR T-cell therapies.
As for solid tumors, the tissues where they localized or derived are seldom capable of regeneration like the hematopoietic system (37). Thus, targets of CARs for solid tumors should be chosen more carefully and uniquely to avoid strong on-target, off-tumor cytotoxicity and properly for wider range of tumor cells to reduce antigen-loss effect (Figure 2). The ideal antigens would be the mutated oncogenes expressed on most tumor cell membrane but the proper neoantigens are rare and hard to determine (38, 39). B7-H3 is reported to be a checkpoint molecule highly expressed on brain tumors and antibody targeting B7-H3 results in potent antitumor activity (40). OR2H1 is an olfactory receptor widely expressed on ovarian cancers, non–small cell lung cancers as well as breast cancers but less detected on normal tissues. CAR T-cells targeting OR2H1 demonstrated antigen-specific cytotoxicity in vitro and in vivo suggesting it may be a promising target for epithelial cancers (41). Mesothelin (MSLN) is broadly overexpressed in many solid tumors including mesothelioma, lung adenocarcinomas and ovarian cancer while remains at a low level on mesothelial cells (42, 43). This target has been widely applied to immunotherapy, especially the CAR T therapy that resulted in successful clinical treatment (44). Beside epithelial cancer, pancreatic ductal adenocarcinoma (PDAC) is a highly lethal disease and many groups tried to find its specific targets. Using an antibody array screening, CD318, TSPAN8 and CD66c are identified to be proper PDAC antigen with limited expression off tumor (45). To validate the findings, CAR T-cells targeting these candidates are developed and exhibit anti-tumor response. CEACAM7 is another membrane protein strictly expressed on pancreas and colon and also proved to be an effective CAR T-cell target for PDAC (46). Recently, an B7-H3 CAR that has rear reactivity to B7-H3 -low tissues was developed and mediated clearance of abundant solid tumor xenografts (47).
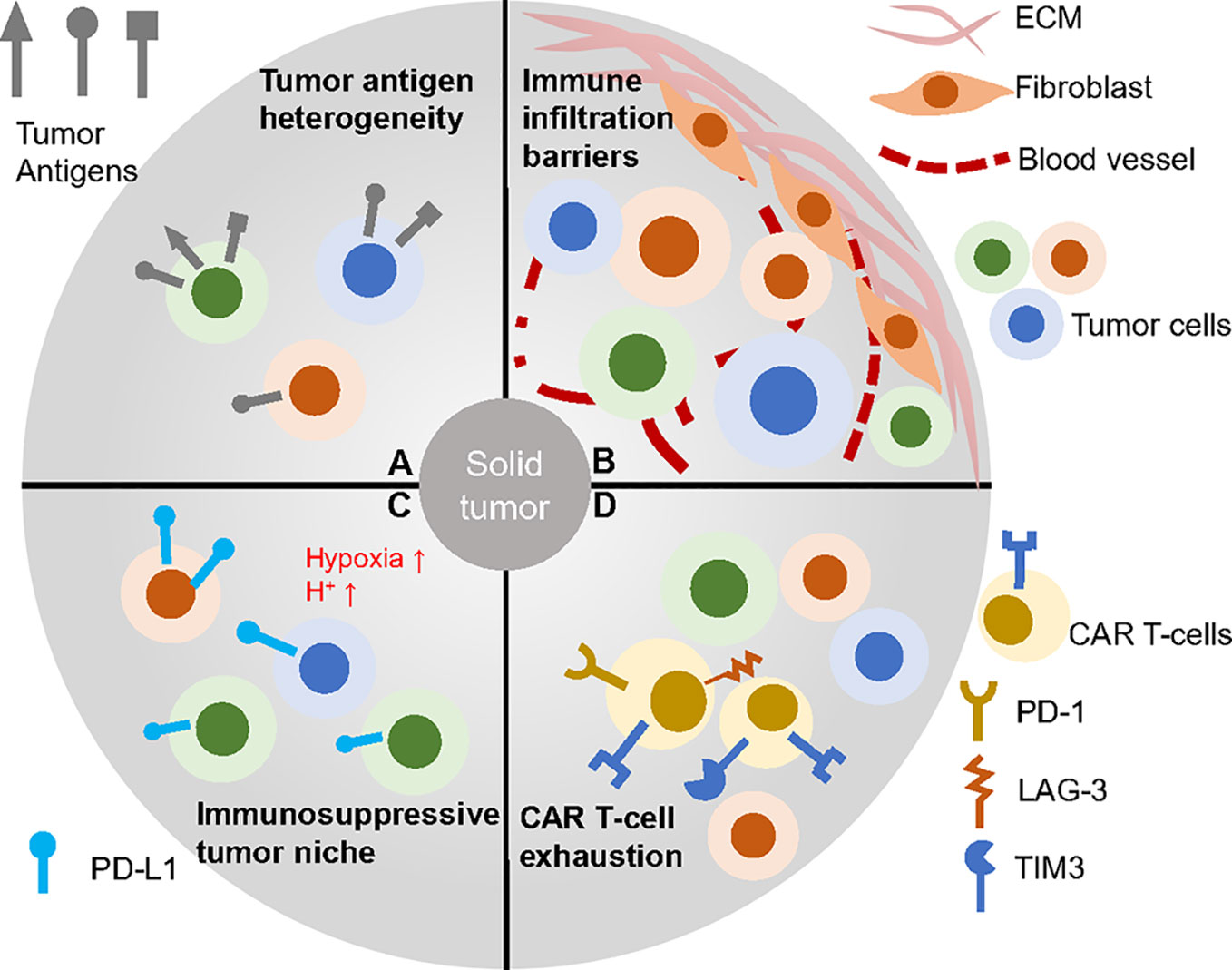
Figure 2 CAR T therapy faced challenges in solid tumors. (A) Solid tumor cells baring heterogeneous antigens are difficult to be targeted. (B) Complex tumor barriers inhibit immune infiltration. (C) Many factors contribution to the immunosuppressive environment such as low pH, hypoxia and immune check point blockage molecules. (D) CAR T-cells within the solid tumor often go exhausted, which impairs efficacy.
In addition to broad spectrum tumor antigens, multi-specific CAR T-cells were manufactured to cope with antigen loss. GD2.28ζ/B7-H3.BB expressing CAR T-cells control SH-SY5Y derived tumors without escape compared with either GD2.28ζ or B7-H3.BB only CAR T-cells (31). Furthermore, tumors bearing low levels of TAG-72, which is an adenocarcinoma maker, can also be effectively eliminated through TAG-72 and CD47 dual-targeting CAR T-cells. Also, anti-CD19 and CD22 CARs demonstrated the efficacy in phase I clinical trials for B-ALL (48). One group even measured the trivalent CAR T-cell (HER2IL, 13Rα2 and EphA2) against glioblastoma and gained significantly positive results (49). Besides, CART.BiTE is a novel design with a CAR specific for EGFRvIII as well as a secreted EGFR-specific bispecific T-cell engager (BiTE). CART.BiTE cells can thus targeting the EGFRvIII-positive or EGFR-positive glioblastoma while activating bystander T cells, which effectively represses tumor growth (50). All the studies illustrate muti-targeting CAR could be an alternative way to overcome tumor heterogeneity in some solid and blood tumors.
Recruiting and activating other immune cells can also help to clear the tumor cells without CAR specific antigens. Such engineered CAR T-cells include proinflammatory molecule CD40 ligand or IL-7 and CCL19 or CCL5 and CXCL9 expressing cell to stimulate antigen-presenting cells (51–53). Despite the spontaneous antigen loss due to stress, T-cell trogocytosis can mediate antigen transfer from tumor cells to T-cells and cause T-cell fratricide (54). Though it is unknown how much such mechanism accounts for tumor escape of CAR T-cell treated patients, the study provides us a new dimension about tumor resistance to immune system.
Enhancing tumor infiltration and T-cell activation-rest cycling
Solid tumors are enriched with extracellular matrix (ECM) and tumor associated fibroblasts and plenty of them are cold tumors, which means they lack complete blood vessels and proper conditions to recruit immune cells (55–57) (Figure 2). All of these characteristics block the infiltration of CAR T-cells and trap the intratumor immune cells. To tackle the problem, many research groups developed chemokine receptors bearing CAR T-cells that homes to the tumor bed as well as armed CAR T-cells against ECM. Chemokine receptor CCR2b mediates the CAR T-cells orientation to NSCLC tumors (58). CXCR1- or CXCR2-modified CAR T-cells also optimize the infiltration ability of T-cells in many tumor models (59). Except expressing chemokine receptors to facilitate homing, CAR T-cells are designed to seek specific antigens within tumors and therefore, tumor infiltrated CAR T-cells can act as lighthouses to recruit other immune cells, for example, IL-7 and CCL19 secreting CAR T-cells that attract other immune cells infiltration (52). Enzyme heparanase (HPSE) expressing CAR T-cells can effectively degrade ECM and enhance tumor infiltration with improved efficacy (60). VEGF is a cytokine important for angiogenesis of tumor cells. T-cells expressing anti-VEGF2 CARs inhibit multiple types of syngeneic tumors through increased immune infiltration (61). FAP-CAR T-cells are against fibroblast activation protein (FAP) expressing cancer-associated stromal cells. Although these cells are not intended to be direct tumor targeting, they destroy the tumor microenvironment and restrict solid tumor growth as well as desmoplasia (62).
Apart from the modification of CAR T-cells, combined therapies may facilitate CAR T-cells’ access to tumors. Docetaxel treatment upregulated the expression of CXCL11 and enhanced CD8+ T-cells recruitment (63). Low dose radiotherapies also can increase active T-cells in tumor microenvironment and normalize tumor blood vessels which may result from upregulation of several pro-angiogenic genes such as ANGPT2, TGFB2, FGF2 (64, 65). Intratumoral injection of CAR T-cells also directly exposes solid tumors to cytotoxicity and surmounts obstacles induced by tumor associated matrix and cells (43, 66). Prasad S. Adusumilli et al. found that regional delivery of M28z T-cells targeting MSLN showed superiority than the systemic route and Julia Tchou et al. proved the concept by intratumorally treating metastatic breast cancer with mRNA c-Met-CAR T-cell and showed improved tumor necroptosis (43, 67). One group compared regional delivery of CAR T-cells with intravenous injection in metastatic breast cancer and found improved effect using intracranial and intratumoral administration with complete tumor regression (68). Also, regional delivery of low dose anti-HER2 CAR T-cells induced durable regression of medulloblastoma (69). Though intratumor injection is a direct delivery method, this only helps CAR T-cells without other immune cells.
New materials can help the attraction of immune cells effectively. Recently, a kind of hydrogel without passive diffusion of cytokine was developed. Hydrogel containing cytokine and CAR T-cells can be injected near the tumor and create an inflammatory niche for improving CAR T- and immune cells efficacy (70). Similarly, fibrin gel accommodates CAR T-cells could effectively facilitate cell delivery either (71). Plenty of targets are discovered with the effect of enhanced immune cells infiltration. DDR1 is a collagen receptor whose expression is negative correlated with intratumoral T cells. DDR1 knockout tumors align collagen fibers and have more CD8+ T-cells. More importantly, tumor growth is significantly decreased without DDR1, from which CAR T-cell therapy can get benefit (72).
On the other hand, continuous antigen exposure changes the CAR T-cells to dysfunctional NK-like T-cells, which suggests that rest is vital for CAR T-cells’ normal cytotoxicity (73). Blood vessel is naturally born for cell traffic and thus when T-cells attack leukemia, they have chance to separate from the tumor antigen through blood flow and get rest (74, 75). In another way, CD8α-PILRα interactions mediated T-cell–myeloid cell cross-talk also maintains T-cell actively quiescence. Loss of the interactions may destroy naïve and memory CD8+ T-cell pools and results in poor T-cell function (76). However, solid tumors may lack the interaction but be enriched by antigen-CAR interaction. The CAR T-cells getting involved in these tumors could not have rest and therefore may become exhausted. In order to solve the challenge, inducible CARs and circuit CARs are developed for activation control and prove their efficacy (77). Synthetic intramembrane proteolysis receptors (SNIPRs) are a kind of inducible receptors based on the SynNotch module which is activated when binding with proper antigens (78). Such receptors can be coupled with CAR gene expression and only when the receptors counteract with the target antigen can the CARs be expressed on the cell membrane. This system provides a high signal-to-noise ratio for tumor specific killing. Another directed inducible CAR includes a protease controlling CAR degradation mechanism termed signal neutralization by an inhibitable protease (SNIP). Grazoprevir exposure causes protease inhibition and retains CAR function. The SNIP CAR shows significantly anti-tumor ability and more active T-cells without toxic effect (79). Ultrasound activation CAR provides precise T-cell activation that is restrict to the tumor site and may prevent always-on CAR T-cells as well as off-tumor cytotoxicity (80). Direct CAR signaling inhibitors such as dasatinib also help T-cells to rest and restores functionality (81).
Dealing with immunosuppressive microenvironment
As a “living drug”, CAR T-cells need to be nursed (26, 82). Nevertheless, solid tumors could hardly provide a cozy home for the T-cells like in the blood. Immunosuppressive microenvironment is a common problem faced by all immune therapies (29, 83). One group recently demonstrated quiescent cancer cells created a niche to inactivate both T-cells and dendritic cells through increased expression of chemoresistance, hypoxia, and glycolysis related genes especially HIF1a suggesting the necessary to modify tumor niche (84).
To target the hypoxic tumor microenvironment, hypoxia-inducible CAR T-cells provide tumor cytotoxicity only within the low oxygen condition and avoid on-target, off-tumor effect (85–87). Normalization of tumor vascularity can reverse hypoxia and benefit CAR T-cell therapies (88, 89). VEGF and ANG-1/2 are two major factors impact tumor vascularity. Blocking VEGF by anti-VEGF antibodies can induce prolonged vessel normalization and overcome immune therapy resistance. However, some tumors develop drug resistance after such treatment. Combined VEGF and ANG2 blockade may confer the resistance and promote anti-cancer immunity (90, 91).
Apart from incomplete vasculature, solid tumor niche forces the expression of many immune checkpoint factors such as PD-1, CTLA-4, TIGIT and LAG-3 on T-cells (92). These immune checkpoints inhibit T-cell normal function and promote T-cell exhaustion. Therefore, studies demonstrated LAG-3 disruption or PD-1 deletion through genetic modifications induced robust antigen-specific antitumor activity (93, 94). Xiaojun Liu et al. utilized the PD-1/PD-L1 interaction and created a PD1-CD28 switch which activates T-cell upon binding to PD-L1. This design augments CAR T-cell cytokine secretion and antitumor activity (95). Newly founded checkpoint PTP1B also limits the CAR T-cell therapy indicating that the complexity of checkpoint family needs further study (96). Some chemicals within tumor microenvironment also inhibit immune function. Tumor necrosis releases potassium ions interfere with mTOR signaling causing impaired function and T-cells overexpressing potassium channel can reverse the tumor resistance (97). The pH value in TME is lower than normal tissue such as blood due to tumor-derived lactic acid, which drives enhanced negative regulatory signals and higher activation thresholds of T-cells (98). Lots of drugs are developed to neutralize acidic environment and their combination with CAR T-cells may control tumor growth (99).
Besides, cancer-associated fibroblasts (CAFs) are now well studied and heavily heterogeneous. Most ECM is the product of CAFs including immunosuppressive fibroblast activation protein (FAP). Deletion of these cells provides opportunities for immune therapies (100). In particular, adoptive transfer of FAP-CAR T-cells reduces tumor growth in a FAP-dependent fashion and can be combined with Ad.E7 antitumor vaccine (101). Usually, fibroblasts secreted CXCL12 or CCL2 are also immunosuppressive and thus blockage therapies can be considered as an assistance of FAP-CAR T-cells (102). Regardless of the fibroblasts, immunosuppressive cells components such as myeloid-derived suppressor cells (MDSCs), regulatory T-cells (Tregs), tumor-associated macrophages (TAMs) in the solid tumor also inhibiting antitumor immune responses. MDSCs and Tregs within microenvironment inhibit the CAR T-cells by producing TGF-β and interleukin-10 (IL-10) while Tregs also express checkpoint molecules such as PD-1 and CTLA4 (102–105). Furthermore, Tregs recruitment is associated with TAMs and interleukin-6 (IL-6) secreted by MDSCs recruits Th17 cells which strengthen the immunosuppressive effect. Treg activation strongly inhibits CD8+ cell proliferation and it is reported that deleted the IL1R1+ Treg could improve T-cell ability in solid tumors (106). Tumor cells also release many cytokines including CCL2, CXCL12 and CSF1 that attract TAMs to help tumor escape by producing IL-10 and also directly inhibiting CAR T-cells via PD-1-PD-L1 interaction (107). Thus, TAMs density is severely related to patients’ survival in solid tumors (108). FRβ is a protein universally expressed on M2 TAMs and studies demonstrated solid tumors pretreated with CAR T-cells targeting FRβ improves pro-inflammatory monocytes enrichment and tumor-specific CAR T-cells efficacy (107). This study in another way proved that the combination of two kinds of CAR T-cells may acquire better responses against solid tumors. Reasonably, strategies to directly inhibit TGFβ, IL-10 and cytokine signaling can reverse the immunosuppression situation either. For example, Inhibition of CSF-1 Receptor by PLX3397 shows promising results to assist adoptive cell therapy in melanoma model and TGF-β-receptor kinase inhibitor SD-208 shelters the ROR1-CAR T-cells from suppression (109, 110). Also, combined therapies employing checkpoint blockade with CAR T therapy proved to be effective (111). One the other hand, the stimulatory factors can be utilized for CAR T-cell activation. T-cells redirected for universal cytokine-mediated killing (TRUCK) are thus established and eradicate ovarian tumors in vivo (112).
Controlling CAR T-cell epigenetics changes
As stem cells of hematopoietic system dividing and differentiating throughout the life, transcriptional regulators play an essential role to sustain various functions of daughter cells. CAR T-cells faces many stresses on the way to kill solid tumor cells, like persistent antigen exposure and inhibitory signals which promote the expression of exhaustion related genes such as LAG-3, TIM3, PD-1 (113). With the ongoing study of T-cell exhaustion, many evidences indicate the epigenetic changes are the major players and through manipulation of these changes T-cell can be more persistent. Direct down regulation of LAG-3, TIM-3, and PD-1 showed epigenetic changes in CAR T-cells which had enhanced tumor infiltration activity and CD56 expression, a marker of immune cell activation or cytotoxicity as well as cytokine secretion (113, 114). On the other hand, fast and continuous activation of CAR T-cells reduces pool of memory cells and results that the treatment could not last for long time (76). SOX4 and ID3 are two important factors involved in CAR T-cell exhaustion changes after long term of continuous antigen exposure. Knockout either of the two genes maintains the functional phenotypes by epigenetic modification (73). Another famous epigenetic modifier, SWI/SNF complex is related to many cancer types (115). cBAF is the most canonical sub complex of SWI/SNF and high cBAF activity determines the CD8+ T-cells to effector cells with decrease of memory T-cells (116, 117). Thus, cBAF activity deficient CAR T-cells improved therapy response in solid tumor models (117). Methylation of plasticity–associated genes also play a key role in T-cell dysfunction (118). Knockout DNA methyltransferase 3 alpha (DNMT3A) retained a stem-like epigenetic program in CAR T-cells and sustained cytokine expression during repeat antigen exposure (118). TET2 is another chromatin modifier that mainly activates gene expression and it is reported to activate T-cell proliferation (119). Disruption of TET2 promotes the formation of memory cells and altered T-cell differentiation that results in increased efficacy (120). TOX and TOX2 are two HMG-box transcription factors whose high expression are reported to enhance T-cell inhibitory receptors expression and their knockout greatly improved CD8+ T-cell function (121, 122). Like TOX, NR4A is a kind of NFAT response transcription factors including NR4A1, NR4A2 and NR4A3 with positive correlation to PD-1 and TIM3 expression. NR4A triple-knockout CAR T-cells promote tumor regression and prolong survival (123). Also, PI3Kδ/γ inhibitors help balancing CD4/CD8 ratios and increase CD8+ T-stem cell memory feature during CAR T-cell manufacturing process (124). In another way, CAR T-cells overexpressing c-Jun have the abilities to proliferate for long-term without exhaustion markers both in vivo and in vitro (125). One group combined the CAR with inducible activation of MyD88 and CD40 signaling pathways and showed their superior function in vivo and this method can be applied to other CAR T-cell enhancing factors like c-Jun activation CARs (126). All these reports suggest targeting epigenetic factors can prevent T-cell exhaustion and benefit CAR T-cell efficacy in solid tumors.
Choosing the suitable chassis for CAR T-cell production
CAR T-cell manufacture process is highly cost and may get failure sometimes because the T-cell activity and proliferation ability from the patients are viable which can result in poor function CAR T-cells or insufficient cell amount for solid tumor treatment (26, 127). Thus, plenty of research groups turn to develop stem cells beyond PBMC derived T-cells. Induced pluripotent stem cells (iPSCs) technology exhibits great power to make large-scaled less differentiated cells through reprogramming-factors cocktail, which was discovered by Shinya Yamanaka in 2006 (128). iPSCs derived T-cells are abundant source for CAR T therapies and differentiation processes are controllable. Groups established convenient method for conventional αβT-cell production (129–131). However, the process of iPSCs to T-cells are relative time consuming (about 40 days). The αβT-iPSCs have more possibility to differentiated to CD8+ T-cells rather than CD4+ T-cells (131, 132). Highly potential proliferate T-iPSCs could cause spontaneous cancer which makes the balance between safety and efficiency a dilemma. There is still a long way to go before the CAR T-iPSCs come into solid tumor treatment.
CD4+ and CD8+ αβT-cells are regarded as the directly antitumor population during tumor regression but these cells separated from patients may have disordered cytotoxicity (133). Moreover, innate T-cells including NK T-cells, mucosa-associated invariant T-cells (MAITs) and γδT-cells have shown promising results for CAR-engineered therapy for cancer. These cells are broadly presented in the tumor microenvironment and their antitumor abilities are characterized long ago (134–138). With the persistence in tumor microenvironment, innate T-cells become an ideal alternative cell chassis for CAR T-cells. Human NK T-cells are a mixed group of cells containing two major populations: type I NK T-cells bearing the Vα14Jα18 invariant TCR α-chain recognizes the glycosphingolipid α-galactosylceramide (α-GalCer) and analogues through CD1d molecules and type II NK T-cells are recognizing non-α-GalCer molecules but not well defined. Type I NK T-cells, also called invariant NK T-cells, mediated the secretion of IFN-γ and TNF-α that improves antitumor effect and IL-4, IL-10, and IL-13 modulating tumor niche (139). In recent works, Andras Heczey et al. developed anti-GD2 CAR-NK T-cells, which showed great tumor infiltration ability and expansion potential that mediated tumor regression response without dose-limiting toxicities (140). In hepatocellular carcinoma, Glypican-3-specific CAR-NK T-cells also mediated potent antitumor activity while the chondroitin sulfate proteoglycan 4 (CSPG4) CAR-NK T-cells demonstrated the efficacy against melanoma (141, 142). In addition to the CAR-NK T-cells, MAIT is another rise star for CAR T-cells, which expresses the Vα7.2–Jα33 invariant TCR α-chain in humans and can penetrate into solid tumors (132). Mikail Dogan et al. generated anti-Her2 CAR-MAIT that exhibited highly activity against breast tumors and also B cell lymphoma (143). Besides, γδT-cells are featured by the γδT-cell receptors that are not MHC restrict and thus could be used to treat solid tumor with less graft-versus-host disease (GvHD) risk. Also, they have the ability to mediate fast antigen-independent cytotoxicity which reduces the escape of heterogenous tumor cells. On the other hand, the cytokine released by innate γδT-cells, especially the low level of IL-6, are different from αβT-cells and less CSR side effects (139, 144). Due to the chemokine receptors on innate αβT-cells, they can find their way into solid tumors and complete highly efficient killing. GD2-targeting CAR γδT-cells are developed and retained antigen-specific neuroblastoma cells killing as well as low exhaustion phenotypes. Nevertheless, innate γδT-cells only account for 1-5% total CD3+ cells, which gives rise to the short of resource during producing CAR γδT-cells (145).
Immune system is a well-organized pool of different cells, in which each type of cells contributes to the overall function. The significance of crosstalk between multiple T-cells suggests that mixed population can achieve better anti-tumor efficacy (146–148). In summary, further knowledge about the function of different populations and study of the optimal T-cells combination may lead to the highly efficient CAR T-system rather than single kind CAR T-cells.
Utilizing combined therapies
Solid tumors have many approaches to escape anti-tumor therapies and therefore by no means we only take one treatment in the case that no obvious side effect is detected. Combined therapies can take out additive effect, synergistic effect or even synthetic lethality (149). Lots of clinical trials are going on to check out the proper secondary treatment with CAR T-cell therapies (Figure 3). Growing quantity of new combinations are now reported with improving immune attack of solid tumors.
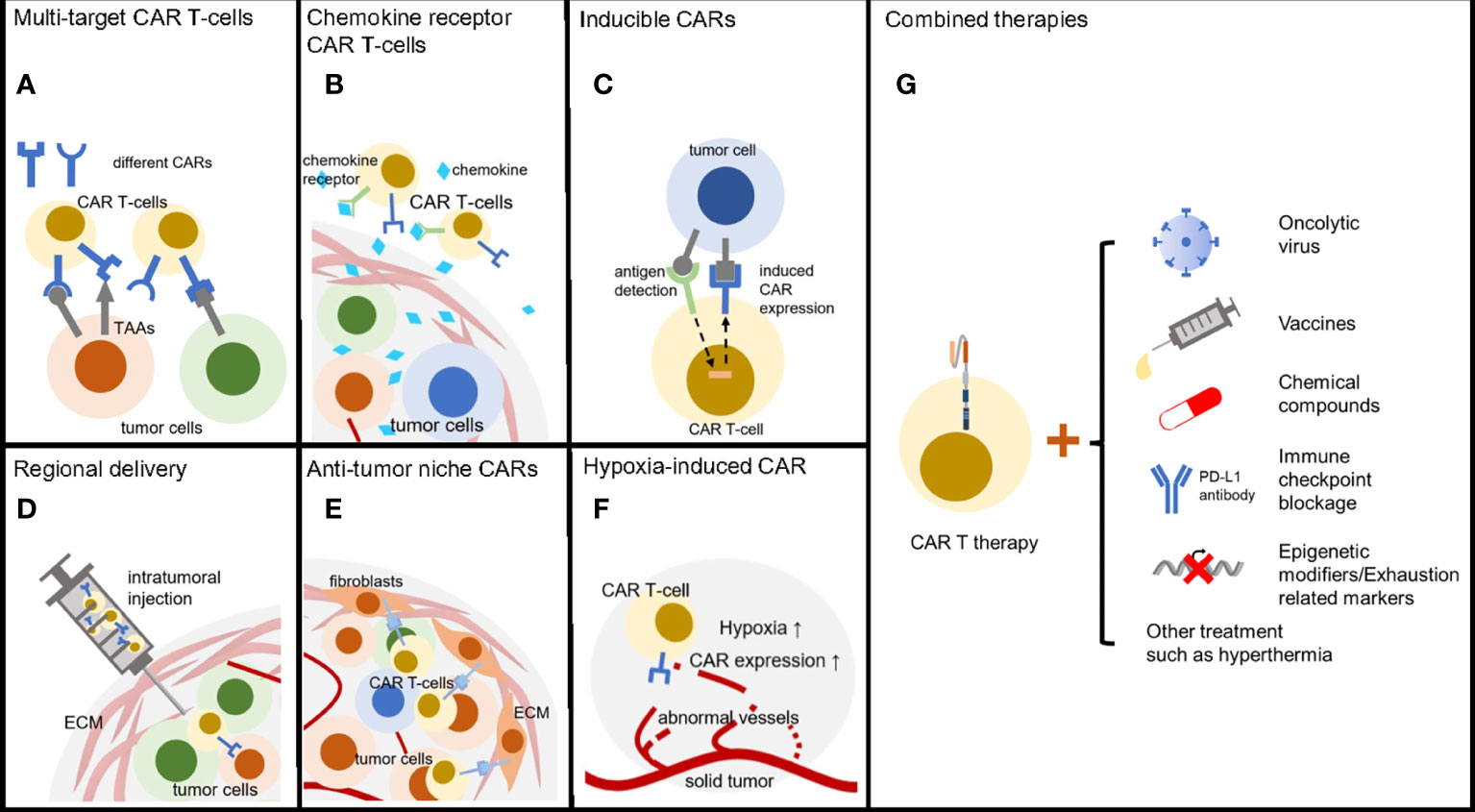
Figure 3 Methods to reverse CAR T therapy resistance. (A) T-cells armored with multi-specific CARs can broadly target heterogeneous tumor cells. (B) CAR T-cells with chemokine receptor are able to efficiently infiltrate solid tumors. (C) Inducible CARs enable T-cells precisely kill tumors cell with low exhaustion and off-tumor cytotoxicity. (D) Regional delivery directly transfers the CAR T-cells into solid tumors. (E) CARs targeting tumor niche can be an alternative anti-cancer choice through destroying of microenvironment. (F) Hypoxia inducible CARs have low off-target effect due to the regional activation. (G) Combined therapies including oncolytic virus, tumor vaccines, chemical drugs, ICB and epigenetic modifiers enhanced CAR T-cell treatment efficacy.
As mentioned above, solid tumors have few proper tumor-specific antigens to attract CAR T-cells. But oncolytic viruses can modify the tumor microenvironment and recruit activated T-cells. VSV-mIFNβ can promote a favorable chemokine profile for CAR T-cells and stimulate virus specific TCR activity which boosts memory CAR T-cells function and proliferation (150). This leads to better survival of subcutaneous melanoma and intracranial glioma tumors bearing mice (150). A study from the same group noticed the importance of administration schedule. Pre-treated with VSV-mIFNβ followed by CAR T-cell injection would induce virus-derived type I interferon that caused CAR T-cell death (151). Leyuan Ma et al. designed a kind of vaccine called amph-ligand that can be processed and presented on APCs (152). The amph-ligand contains a CAR ligand domain and CAR T-cells interacting with the APCs will be stimulated and expanses. This strategy is easy to apply to all CAR T-cell therapies and overcomes the low antigen expression of solid tumors. Tumor infiltrated CAR T-cells are ideal carriers of drug. Synthetic enzyme-armed killer (SEAKER) cells process small-molecule prodrug within the tumor and release the active drug such as 5’-O-Sulfamoyladenosine (AMS) resulting in strong antitumor response (153). It is known that mild hyperthermia can reduce solid tumor compact structure and promote immune cell infiltration. Recent study demonstrated photothermal ablation of the tumor can be combined with CAR T-cell therapies to maximize antitumor activity of melanoma (154). Solid tumors often ware abnormal glycosylation to mask the epitope of antigens and interfere with antitumor immunity (155, 156). One group recently combined 2-deoxy-D-glucose (2DG) treatment, a kind of glucose/mannose analog that can inhibit glycosylation, with CAR T-cell therapy and obtained promising efficacy against multiple solid carcinomas (157).
Checkpoint blockade and CAR T therapy co-treatment is another effective combined therapy considering the immunosuppressive tumor niche. Tumor infiltrated CAR T-cells are highly possible to express signs of phenotypic exhaustion such as PD-1 and reasonably blocking checkpoint could protect the CAR T-cells from immunosuppressive microenvironment (Figure 3 and Table 1) (111, 158, 159). Recent studies demonstrated the potential in both ovarian cancer and lung cancers. Additionally, the hydrogel containing CAR T-cells and anti-PD-L1-conjugated platelets developed by Quanyin Hu et al. provided a state-of-the-art way for solid tumor treatment (159). And now several clinical trials are going to study the efficacy of combined Checkpoint blockade and CAR T combined therapies.
Despite the existing therapeutic pairs, new druggable targets are rising. Robert T. Manguso et al. discovered a powerful immunotherapy target called phosphatase nonreceptor type 2 (PTPN2) through in vivo CRISPR screening (9). Deletion of PTPN2 results in enhanced T-cell stimulation and infiltration. Despite the tumor modification function, PTPN2 knockout also increased proliferative capacity of CD8+ T-cells (160). Thus, PTPN2 is suggested to be a potent mate with CAR T-cell therapies in solid tumors and it is valuable to develop PTPN2 inhibitors. PTPN22 is another immune modulator related to T-cell function (12, 161). Its activation plays a negative role in immune responses and thus inhibition of PTPN 22 may enhance antitumor response (8). However, it is controversial about how to combine this target with CAR T-cell therapies. PTPN 22 knockout only in CAR T-cells could not improve the efficacy but when systemically inhibited, it augments tumor repressing function (162). And one group has developed a PTPN22 inhibitor which greatly promotes anticancer immunity (8).
Though some combined therapies may be suitable for both hematological malignancy and solid tumors, we should be careful about the intrinsic differences between them. IFN-gamma signaling is well known for its importance to control tumor growth and mutation of IFN-gamma related component can result in resistance of immune therapies (163). One group compared the IFN-gamma signaling in solid and liquid tumors and found only killing solid tumors required IFN-gamma pathway (164). This result indicates the combination of IFN-gamma with CAR T therapies may not benefit blood tumor treatment. Accordingly, some promising targets combined with CAR T therapies in blood cancer could have no positive results in solid cancer due to their distinct reliance, which emphasizes more thorough researches and preclinical test.
Conclusions
After many years’ attempt, many researchers finally turn the concept of CAR T-cells into a powerful adoptive cell therapy and it acquired so much success in the past few years especially for blood malignancy. Advances learned from hematologic tumors treatment equip CAR T-cells with new-generation armors but there are still lots of considerations due to the complexity of the solid tumor itself, the microenvironment and immune system. Luckily, more and more fantastic ideas are placed into practice including CAR modifications, T-cell enhancement and collaborative therapies. These methods provided positive cases in many solid tumor models and even in patients. Beside the evolution on CAR T-cells, the differences between blood and solid tumors are getting clear and discovery of pan-immune system modulators such as PTPN family members strengthens the application of CAR T therapies. In short, CAR T-cells have great potential to be easily modified according to rising concepts to reverse specific tumor resistance. However, the remaining problem is how to optimize CAR T-cells through integrating the advantages of existing and new-born methods, which needs more research practice and clinical investigations.
On the other hand, few studies discuss the challenges of CAR T therapy during the CAR manufacture process and the component of CAR T-cells may also play an important role during solid tumor treatment such as the ratio of different T-cells. Furthermore, CAR T therapies induced side effect is a barrier restricting the effective cell dose during solid tumor treatment. Larger cell dose means both stronger opposite and side effect, so it is also valuable to explore the proper methods to reduce off-tumor effect. In all, CAR T-cells treating solid tumors are promising with the continuous efforts on the bench and in the hospital.
Author contributions
GX and YQ conceptualized the frame work. YQ wrote the manuscript and made the figures under the supervision of GX. YQ and GX contributed to the editing of the content. All authors reviewed the article and agreed to the submitted version.
Funding
The work was supported by the Ministry of Science and Technology of the People´s Republic of China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol (2009) 27(1):83–117. doi: 10.1146/annurev.immunol.021908.132544
2. Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest (2008) 118(6):1991–2001. doi: 10.1172/JCI35180
3. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
4. Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov (2022) 21(7):509–28. doi: 10.1038/s41573-021-00345-8
5. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer (2016) 54:139–48. doi: 10.1016/j.ejca.2015.11.016
6. Kubli SP, Berger T, Araujo DV, Siu LL, Mak TW. Beyond immune checkpoint blockade: emerging immunological strategies. Nat Rev Drug Discov (2021) 20(12):899–919. doi: 10.1038/s41573-021-00155-y
7. Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol (2021) 18(4):842–59. doi: 10.1038/s41423-020-00565-9
8. Ho WJ, Croessmann S, Lin J, Phyo ZH, Charmsaz S, Danilova L, et al. Systemic inhibition of PTPN22 augments anticancer immunity. J Clin Invest (2021) 131(17):e146950. doi: 10.1172/JCI146950
9. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature (2017) 547(7664):413–8. doi: 10.1038/nature23270
10. Luo N, Formisano L, Gonzalez-Ericsson PI, Sanchez V, Dean PT, Opalenik SR, et al. Melanoma response to anti-PD-L1 immunotherapy requires JAK1 signaling, but not JAK2. OncoImmunology (2018) 7(6):e1438106. doi: 10.1080/2162402X.2018.1438106
11. Wiede F, Tiganis T. PTPN2: a tumor suppressor you want deleted? Immunol Cell Biol (2017) 95(10):859–61. doi: 10.1038/icb.2017.70
12. Wiede F, Lu K-H, Du X, Liang S, Hochheiser K, Dodd GT, et al. PTPN2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J (2020) 39(2):e103637. doi: 10.15252/embj.2019103637
13. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-t-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci (1989) 86(24):10024–8. doi: 10.1073/pnas.86.24.10024
14. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer (2021) 21(3):145–61. doi: 10.1038/s41568-020-00323-z
15. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi: 10.1126/science.aaa4967
16. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel) (2016) 8(3):36. doi: 10.3390/cancers8030036
17. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. New Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
18. Mullard A. FDA Approves first CAR T therapy. Nat Rev Drug Discov (2017) 16(10):669–. doi: 10.1038/nrd.2017.196
19. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371(16):1507–17. doi: 10.1056/NEJMoa1407222
20. Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA Approval summary: Axicabtagene ciloleucel for relapsed or refractory Large b-cell lymphoma. Clin Cancer Res (2019) 25(6):1702–8. doi: 10.1158/1078-0432.CCR-18-2743
21. Safarzadeh Kozani P, Safarzadeh Kozani P, Ahmadi Najafabadi M, Yousefi F, Mirarefin SMJ, Rahbarizadeh F. Recent advances in solid tumor CAR-T cell therapy: Driving tumor cells from hero to zero? Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.795164
22. Schaft N. The landscape of CAR-T cell clinical trials against solid tumors–a comprehensive overview. Cancers (2020) 12(9):2567. doi: 10.3390/cancers12092567
23. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
24. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
25. Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol (2019) 19(2):73–4. doi: 10.1038/s41577-018-0119-y
26. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol (2019) 16(6):372–85. doi: 10.1038/s41571-019-0184-6
27. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in b-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med (2018) 24(1):20–8. doi: 10.1038/nm.4441
28. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
29. Morgan MA, Schambach A. Engineering CAR-T cells for improved function against solid tumors. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02493
30. Qu J, Mei Q, Chen L, Zhou J. Chimeric antigen receptor (CAR)-t-cell therapy in non-small-cell lung cancer (NSCLC): current status and future perspectives. Cancer Immunol Immunother (2021) 70(3):619–31. doi: 10.1007/s00262-020-02735-0
31. Hirabayashi K, Du H, Xu Y, Shou P, Zhou X, Fucá G, et al. Dual-targeting CAR-T cells with optimal co-stimulation and metabolic fitness enhance antitumor activity and prevent escape in solid tumors. Nat Cancer (2021) 2(9):904–18. doi: 10.1038/s43018-021-00244-2
32. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29(7):917–24. doi: 10.1200/JCO.2010.32.2537
33. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118(23):6050–6. doi: 10.1182/blood-2011-05-354449
34. Samur MK, Fulciniti M, Aktas Samur A, Bazarbachi AH, Tai YT, Prabhala R, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun (2021) 12(1):868. doi: 10.1038/s41467-021-21177-5
35. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
36. Tedder TF. CD19: a promising b cell target for rheumatoid arthritis. Nat Rev Rheumatol (2009) 5(10):572–7. doi: 10.1038/nrrheum.2009.184
37. Xia H, Li X, Gao W, Fu X, Fang RH, Zhang L, et al. Tissue repair and regeneration with endogenous stem cells. Nat Rev Mater (2018) 3(7):174–93. doi: 10.1038/s41578-018-0027-6
38. Wang Z, Cao YJ. Adoptive cell therapy targeting neoantigens: A frontier for cancer research. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00176
39. Bianchi V, Harari A, Coukos G. Neoantigen-specific adoptive cell therapies for cancer: Making T-cell products more personal. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01215
40. Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res (2012) 18(14):3834–45. doi: 10.1158/1078-0432.CCR-12-0715
41. Martin AL, Anadon CM, Biswas S, Mine JA, Handley KF, Payne KK, et al. Olfactory receptor OR2H1 is an effective target for CAR T cells in human epithelial tumors. Mol Cancer Ther (2022) 21(7):1184–94. doi: 10.1158/1535-7163
42. Lv J, Li P. Mesothelin as a biomarker for targeted therapy. biomark Res (2019) 7(1):18. doi: 10.1186/s40364-019-0169-8
43. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Trans Med (2014) 6(261):261ra151–261ra151. doi: 10.1126/scitranslmed.3010162
44. Adusumilli PS, Zauderer MG, Rusch VW, O'Cearbhaill RE, Zhu A, Ngai DA, et al. Abstract CT036: A phase I clinical trial of malignant pleural disease treated with regionally delivered autologous mesothelin-targeted CAR T cells: Safety and efficacy. Cancer Res (2019) 79(13_Supplement):CT036–CT. doi: 10.1158/1538-7445.AM2019-CT036
45. Schäfer D, Tomiuk S, Küster LN, Rawashdeh WA, Henze J, Tischler-Höhle G, et al. Identification of CD318, TSPAN8 and CD66c as target candidates for CAR T cell based immunotherapy of pancreatic adenocarcinoma. Nat Commun (2021) 12(1):1453. doi: 10.1038/s41467-021-21774-4
46. Raj D, Nikolaidi M, Garces I, Lorizio D, Castro NM, Caiafa SG, et al. CEACAM7 is an effective target for CAR T-cell therapy of pancreatic ductal adenocarcinoma. Clin Cancer Res (2021) 27(5):1538–52. doi: 10.1158/1078-0432.CCR-19-2163
47. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res (2019) 25(8):2560–74. doi: 10.1158/1078-0432.CCR-18-0432
48. Shu R, Evtimov VJ, Hammett MV, Nguyen NN, Zhuang J, Hudson PJ, et al. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol Ther Oncol (2021) 20:325–41. doi: 10.1016/j.omto.2021.01.002
49. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol (2018) 20(4):506–18. doi: 10.1093/neuonc/nox182
50. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol (2019) 37(9):1049–58. doi: 10.1038/s41587-019-0192-1
51. Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 ligand-modified chimeric antigen receptor T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell (2019) 35(3):473–88.e6. doi: 10.1016/j.ccell.2019.02.006
52. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol (2018) 36(4):346–51. doi: 10.1038/nbt.4086
53. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell (2019) 35(6):885–900.e10. doi: 10.1016/j.ccell.2019.05.004
54. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature (2019) 568(7750):112–6. doi: 10.1038/s41586-019-1054-1
55. Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci (2020) 6:160. doi: 10.3389/fmolb.2019.00160
56. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
57. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov (2019) 18(3):197–218. doi: 10.1038/s41573-018-0007-y
58. Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, et al. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Front Immunol (2021) 12:628906. doi: 10.3389/fimmu.2021.628906
59. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun (2019) 10(1):4016. doi: 10.1038/s41467-019-11869-4
60. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med (2015) 21(5):524–9. doi: 10.1038/nm.3833
61. Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest (2010) 120(11):3953–68. doi: 10.1172/JCI43490
62. Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res (2015) 75(14):2800–10. doi: 10.1158/0008-5472.CAN-14-3041
63. Gao Q, Wang S, Chen X, Cheng S, Zhang Z, Li F, et al. Cancer-cell-secreted CXCL11 promoted CD8(+) T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J Immunother Cancer (2019) 7(1):42. doi: 10.1186/s40425-019-0511-6
64. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24(5):589–602. doi: 10.1016/j.ccr.2013.09.014
65. Marques FG, Carvalho L, Sousa JS, Rino J, Diegues I, Poli E, et al. Low doses of ionizing radiation enhance angiogenesis and consequently accelerate post-embryonic development but not regeneration in zebrafish. Sci Rep (2020) 10(1):3137. doi: 10.1038/s41598-020-60129-9
66. Melero I, Castanon E, Alvarez M, Champiat S, Marabelle A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat Rev Clin Oncol (2021) 18(9):558–76. doi: 10.1038/s41571-021-00507-y
67. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res (2017) 5(12):1152–61. doi: 10.1158/2326-6066.CIR-17-0189
68. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2(+) breast cancer metastasis to the brain. Clin Cancer Res (2018) 24(1):95–105. doi: 10.1158/1078-0432.CCR-17-2041
69. Nellan A, Rota C, Majzner R, Lester-McCully CM, Griesinger AM, Mulcahy Levy JM, et al. Durable regression of medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer (2018) 6(1):30. doi: 10.1186/s40425-018-0340-z
70. Grosskopf AK, Labanieh L, Klysz DD, Roth GA, Xu P, Adebowale O, et al. Delivery of CAR-T cells in a transient injectable stimulatory hydrogel niche improves treatment of solid tumors. Sci Adv (2022) 8(14):eabn8264. doi: 10.1126/sciadv.abn8264
71. Ogunnaike EA, Valdivia A, Yazdimamaghani M, Leon E, Nandi S, Hudson H, et al. Fibrin gel enhances the antitumor effects of chimeric antigen receptor T cells in glioblastoma. Sci Adv (2021) 7(41):eabg5841. doi: 10.1126/sciadv.abg5841
72. Sun X, Wu B, Chiang HC, Deng H, Zhang X, Xiong W, et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature (2021) 599(7886):673–8. doi: 10.1038/s41586-021-04057-2
73. Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, Donahue G, et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell (2021) 184(25):6081–100.e26. doi: 10.1016/j.cell.2021.11.016
74. Hunter MC, Teijeira A, Halin C. T Cell trafficking through lymphatic vessels. Front Immunol (2016) 7. doi: 10.3389/fimmu.2016.00613
75. Donnadieu E, Dupré L, Pinho LG, Cotta-de-Almeida V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J Leukoc Biol (2020) 108(4):1067–79. doi: 10.1002/JLB.1MR0520-746R
76. Zheng L, Han X, Yao S, Zhu Y, Klement J, Wu S, et al. The CD8α-PILRα interaction maintains CD8(+) T cell quiescence. Science (2022) 376(6596):996–1001. doi: 10.1126/science.aaz8658
77. Labanieh L, Majzner RG, Mackall CL. Programming CAR-T cells to kill cancer. Nat Biomed Eng (2018) 2(6):377–91. doi: 10.1038/s41551-018-0235-9
78. Zhu I, Liu R, Garcia JM, Hyrenius-Wittsten A, Piraner DI, Alavi J, et al. Modular design of synthetic receptors for programmed gene regulation in cell therapies. Cell (2022) 185(8):1431–43.e16. doi: 10.1016/j.cell.2022.03.023
79. Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell (2022) 185(10):1745–63.e22. doi: 10.1016/j.cell.2022.03.041
80. Wu Y, Liu Y, Huang Z, Wang X, Jin Z, Li J, et al. Control of the activity of CAR-T cells within tumours via focused ultrasound. Nat BioMed Eng (2021) 5(11):1336–47. doi: 10.1038/s41551-021-00779-w
81. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science (2021) 372(6537):eaba1786. doi: 10.1126/science.aba1786
82. Liu D. CAR-T “the living drugs”, immune checkpoint inhibitors, and precision medicine: a new era of cancer therapy. J Hematol Oncol (2019) 12(1):113. doi: 10.1186/s13045-019-0819-1
83. Hou AJ, Chen LC, Chen YY. Navigating CAR-T cells through the solid-tumour microenvironment. Nat Rev Drug Discov (2021) 20(7):531–50. doi: 10.1038/s41573-021-00189-2
84. Baldominos P, Barbera-Mourelle A, Barreiro O, Huang Y, Wight A, Cho JW, et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell (2022) 185(10):1694–708.e19. doi: 10.1016/j.cell.2022.03.033
85. Kosti P, Opzoomer JW, Larios-Martinez KI, Henley-Smith R, Scudamore CL, Okesola M, et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep Med (2021) 2(4):100227. doi: 10.1016/j.xcrm.2021.100227
86. Liao Q, He H, Mao Y, Ding X, Zhang X, Xu J. Engineering T cells with hypoxia-inducible chimeric antigen receptor (HiCAR) for selective tumor killing. biomark Res (2020) 8(1):56. doi: 10.1186/s40364-020-00238-9
87. He H, Liao Q, Zhao C, Zhu C, Feng M, Liu Z, et al. Conditioned CAR-T cells by hypoxia-inducible transcription amplification (HiTA) system significantly enhances systemic safety and retains antitumor efficacy. J Immunother Cancer (2021) 9(10):e002755. doi: 10.1136/jitc-2021-002755
88. Yang T, Xiao H, Liu X, Wang Z, Zhang Q, Wei N, et al. Vascular normalization: A new window opened for cancer therapies. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.719836
89. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature (2017) 544(7649):250–4. doi: 10.1038/nature21724
90. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
91. Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci U S A (2016) 113(16):4476–81. doi: 10.1073/pnas.1525360113
92. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ, Guedan S. CAR-T cells hit the tumor microenvironment: Strategies to overcome tumor escape. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01109
93. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget (2017) 8(10):17002–11. doi: 10.18632/oncotarget.15218
94. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med (2017) 11(4):554–62. doi: 10.1007/s11684-017-0543-6
95. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res (2016) 76(6):1578–90. doi: 10.1158/0008-5472.CAN-15-2524
96. Wiede F, Lu KH, Du X, Zeissig MN, Xu R, Goh PK, et al. PTP1B is an intracellular checkpoint that limits T-cell and CAR T-cell antitumor immunity. Cancer Discovery (2022) 12(3):752–73. doi: 10.1158/2159-8290.CD-21-0694
97. Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature (2016) 537(7621):539–43. doi: 10.1038/nature19364
98. Boedtkjer E, Pedersen SF. The acidic tumor microenvironment as a driver of cancer. Annu Rev Physiol (2020) 82:103–26. doi: 10.1146/annurev-physiol-021119-034627
99. Zhong S, Jeong JH, Chen Z, Chen Z, Luo JL. Targeting tumor microenvironment by small-molecule inhibitors. Transl Oncol (2020) 13(1):57–69. doi: 10.1016/j.tranon.2019.10.001
100. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science (2010) 330(6005):827–30. doi: 10.1126/science.1195300
101. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi: 10.1158/2326-6066.CIR-13-0027
102. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
103. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19(6):369–82. doi: 10.1038/s41577-019-0127-6
104. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med (2016) 94(5):509–22. doi: 10.1007/s00109-015-1376-x
105. Endharti AT, Rifa IM, Shi Z, Fukuoka Y, Nakahara Y, Kawamoto Y, et al. Cutting edge: CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J Immunol (2005) 175(11):7093–7. doi: 10.4049/jimmunol.175.11.7093
106. Mair F, Erickson JR, Frutoso M, Konecny AJ, Greene E, Voillet V, et al. Extricating human tumour immune alterations from tissue inflammation. Nature (2022) 605(7911):728–35. doi: 10.1038/s41586-022-04718-w
107. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun (2021) 12(1):877. doi: 10.1038/s41467-021-20893-2
108. Zhang Q-w, Liu L, Gong C-y, Shi H-s, Zeng Y-h, Wang X-z, et al. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PloS One (2012) 7(12):e50946. doi: 10.1371/journal.pone.0050946
109. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res (2014) 74(1):153–61. doi: 10.1158/0008-5472.CAN-13-1816
110. Stüber T, Monjezi R, Wallstabe L, Kühnemundt J, Nietzer SL, Dandekar G, et al. Inhibition of TGF-β-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J Immunother Cancer (2020) 8(1):e000676. doi: 10.1136/jitc-2020-000676
111. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell (2021) 39(2):193–208.e10. doi: 10.1016/j.ccell.2020.11.005
112. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology (2015) 4(3):e994446. doi: 10.4161/2162402X.2014.994446
113. Lichtenegger FS, Rothe M, Schnorfeil FM, Deiser K, Krupka C, Augsberger C, et al. Targeting LAG-3 and PD-1 to enhance T cell activation by antigen-presenting cells. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00385
114. Zou F, Lu L, Liu J, Xia B, Zhang W, Hu Q, et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat Commun (2019) 10(1):4109. doi: 10.1038/s41467-019-11893-4
115. Xu G, Chhangawala S, Cocco E, Razavi P, Cai Y, Otto JE, et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat Genet (2020) 52(2):198–207. doi: 10.1038/s41588-019-0554-0
116. Mashtalir N, D'Avino AR, Michel BC, Luo J, Pan J, Otto JE, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell (2018) 175(5):1272–88.e20. doi: 10.1016/j.cell.2018.09.032
117. Guo A, Huang H, Zhu Z, Chen MJ, Shi H, Yuan S, et al. cBAF complex components and MYC cooperate early in CD8+ T cell fate. Nature (2022) 607(7917):135–41. doi: 10.1038/s41586-022-04849-0
118. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med (2021) 13(620):eabh0272. doi: 10.1126/scitranslmed.abh0272
119. Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature (2018) 554(7690):123–7. doi: 10.1038/nature25434
120. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature (2018) 558(7709):307–12. doi: 10.1038/s41586-018-0178-z
121. Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A (2019) 116(25):12410–5. doi: 10.1073/pnas.1905675116
122. Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol (2019) 20(7):890–901. doi: 10.1038/s41590-019-0403-4
123. Chen J, López-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature (2019) 567(7749):530–4. doi: 10.1038/s41586-019-0985-x
124. Funk CR, Wang S, Chen KZ, Waller A, Sharma A, Edgar CL, et al. PI3Kδ/γ inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood (2022) 139(4):523–37. doi: 10.1182/blood.2021011597
125. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-jun overexpression in CAR T cells induces exhaustion resistance. Nature (2019) 576(7786):293–300. doi: 10.1038/s41586-019-1805-z
126. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov (2017) 7(11):1306–19. doi: 10.1158/2159-8290.CD-17-0263
127. Hernandez I, Prasad V, Gellad WF. Total costs of chimeric antigen receptor T-cell immunotherapy. JAMA Oncol (2018) 4(7):994–6. doi: 10.1001/jamaoncol.2018.0977
128. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. doi: 10.1016/j.cell.2006.07.024
129. Wang Z, McWilliams-Koeppen HP, Reza H, Ostberg JR, Chen W, Wang X, et al. 3D-organoid culture supports differentiation of human CAR+ iPSCs into highly functional CAR T cells. Cell Stem Cell (2022) 29(4):515–27.e8. doi: 10.1016/j.stem.2022.02.009
130. Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell (2013) 12(1):114–26. doi: 10.1016/j.stem.2012.11.002
131. Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S-i, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell (2013) 12(1):31–6. doi: 10.1016/j.stem.2012.12.006
132. Zhou Y, Li M, Zhou K, Brown J, Tsao T, Cen X, et al. Engineering induced pluripotent stem cells for cancer immunotherapy. Cancers (Basel) (2022) 14(9):2266. doi: 10.3390/cancers14092266
133. van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer (2020) 20(4):218–32. doi: 10.1038/s41568-019-0235-4
134. Kabelitz D, Wesch D, Pitters E, Zöller M. Characterization of tumor reactivity of human V gamma 9V delta 2 gamma delta T cells in vitro and in SCID mice in vivo. J Immunol (2004) 173(11):6767–76. doi: 10.4049/jimmunol.173.11.6767
135. Moreno M, Molling JW, von Mensdorff-Pouilly S, Verheijen RH, Hooijberg E, Kramer D, et al. IFN-gamma-producing human invariant NKT cells promote tumor-associated antigen-specific cytotoxic T cell responses. J Immunol (2008) 181(4):2446–54. doi: 10.4049/jimmunol.181.4.2446
136. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J Clin Invest (2016) 126(6):2341–55. doi: 10.1172/JCI83476
137. Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-delta CAR-T cells show CAR-directed and independent activity against leukemia. Front Immunol (2020) 11:1347. doi: 10.3389/fimmu.2020.01347
138. Nishimoto KP, Barca T, Azameera A, Makkouk A, Romero JM, Bai L, et al. Allogeneic CD20-targeted γδ T cells exhibit innate and adaptive antitumor activities in preclinical b-cell lymphoma models. Clin Transl Immunol (2022) 11(2):e1373. doi: 10.1002/cti2.1373
139. Cortés-Selva D, Dasgupta B, Singh S, Grewal IS. Innate and innate-like cells: The future of chimeric antigen receptor (CAR) cell therapy. Trends Pharmacol Sci (2021) 42(1):45–59. doi: 10.1016/j.tips.2020.11.004
140. Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med (2020) 26(11):1686–90. doi: 10.1038/s41591-020-1074-2
141. Simon B, Wiesinger M, März J, Wistuba-Hamprecht K, Weide B, Schuler-Thurner B, et al. The generation of CAR-transfected natural killer T cells for the immunotherapy of melanoma. Int J Mol Sci (2018) 19(8):2365. doi: 10.3390/ijms19082365
142. Shaik RS, Rathi P, Courtney A, Schneller N, Guo L, Barragan G, et al. Glypican-3-specific CAR-NKT cells overexpressing BATF3 mediate potent antitumor activity against hepatocellular carcinoma. J Clin Oncol (2022) 40(16_suppl):e14521–e. doi: 10.1200/JCO.2022.40.16_suppl.e14521
143. Dogan M, Karhan E, Kozhaya L, Placek L, Chen X, Yigit M, et al. Engineering human MAIT cells with chimeric antigen receptors for cancer immunotherapy. J Immunol (2022) 209(8):1523–31. doi: 10.4049/jimmunol.2100856
144. Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol (2013) 13(2):88–100. doi: 10.1038/nri3384
145. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature (1995) 375(6527):155–8. doi: 10.1038/375155a0
146. Boulch M, Cazaux M, Loe-Mie Y, Thibaut R, Corre B, Lemaître F, et al. A cross-talk between CAR T cell subsets and the tumor microenvironment is essential for sustained cytotoxic activity. Sci Immunol (2021) 6(57):eabd4344. doi: 10.1126/sciimmunol.abd4344
147. Fisher J, Sharma R, Don DW, Barisa M, Hurtado MO, Abramowski P, et al. Engineering γδT cells limits tonic signaling associated with chimeric antigen receptors. Sci Signal (2019) 12(598):eaax1872. doi: 10.1126/scisignal.aax1872
148. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult b cell ALL patients. J Clin Invest (2016) 126(6):2123–38. doi: 10.1172/JCI85309
149. O'Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet (2017) 18(10):613–23. doi: 10.1038/nrg.2017.47
150. Evgin L, Kottke T, Tonne J, Thompson J, Huff AL, van Vloten J, et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci Transl Med (2022) 14(640):eabn2231. doi: 10.1126/scitranslmed.abn2231
151. Evgin L, Huff AL, Wongthida P, Thompson J, Kottke T, Tonne J, et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat Commun (2020) 11(1):3187. doi: 10.1038/s41467-020-17011-z
152. Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science (2019) 365(6449):162–8. doi: 10.1126/science.aav8692
153. Gardner TJ, Lee JP, Bourne CM, Wijewarnasuriya D, Kinarivala N, Kurtz KG, et al. Engineering CAR-T cells to activate small-molecule drugs in situ. Nat Chem Biol (2022) 18(2):216–25. doi: 10.1038/s41589-021-00932-1
154. Chen Q, Hu Q, Dukhovlinova E, Chen G, Ahn S, Wang C, et al. Photothermal therapy promotes tumor infiltration and antitumor activity of CAR T cells. Adv Mater (2019) 31(23):e1900192. doi: 10.1002/adma.201900192
155. Bartish M, Del Rincón SV, Rudd CE, Saragovi HU. Aiming for the sweet spot: Glyco-immune checkpoints and γδ T cells in targeted immunotherapy. Front Immunol (2020) 11:564499. doi: 10.3389/fimmu.2020.564499
156. Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin siglec-9. Nat Immunol (2016) 17(11):1273–81. doi: 10.1038/ni.3552
157. Greco B, Malacarne V, De Girardi F, Scotti GM, Manfredi F, Angelino E, et al. Disrupting n-glycan expression on tumor cells boosts chimeric antigen receptor T cell efficacy against solid malignancies. Sci Transl Med (2022) 14(628):eabg3072. doi: 10.1126/scitranslmed.abg3072
158. Schoutrop E, El-Serafi I, Poiret T, Zhao Y, Gultekin O, He R, et al. Mesothelin-specific CAR T cells target ovarian cancer. Cancer Res (2021) 81(11):3022–35. doi: 10.1158/0008-5472.CAN-20-2701
159. Hu Q, Li H, Archibong E, Chen Q, Ruan H, Ahn S, et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat Biomed Eng (2021) 5(9):1038–47. doi: 10.1038/s41551-021-00712-1
160. LaFleur MW, Nguyen TH, Coxe MA, Miller BC, Yates KB, Gillis JE, et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat Immunol (2019) 20(10):1335–47. doi: 10.1038/s41590-019-0480-4
161. Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem (2006) 281(16):11002–10. doi: 10.1074/jbc.M600498200
162. Du X, Darcy PK, Wiede F, Tiganis T. Targeting protein tyrosine phosphatase 22 does not enhance the efficacy of chimeric antigen receptor T cells in solid tumors. Mol Cell Biol (2022) 42(3):e0044921. doi: 10.1128/mcb.00449-21
163. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol (2018) 18(9):545–58. doi: 10.1038/s41577-018-0029-z
Keywords: CAR T-cell, solid tumor, antigen viability, microenvironment, tumor infitration, combined therapy
Citation: Qin Y and Xu G (2022) Enhancing CAR T-cell therapies against solid tumors: Mechanisms and reversion of resistance. Front. Immunol. 13:1053120. doi: 10.3389/fimmu.2022.1053120
Received: 25 September 2022; Accepted: 21 November 2022;
Published: 08 December 2022.
Edited by:
Zhaohua Hou, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Lili Yang, Ronald Reagan UCLA Medical Center, United StatesMehrdad Hefazi, Mayo Clinic, United States
Leonardo M. R. Ferreira, Medical University of South Carolina, United States
Copyright © 2022 Qin and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guotai Xu, eHVndW90YWlAbmlicy5hYy5jbg==